the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 10 Mar 2023
| 10 Mar 2023
Comparison of isoprene chemical mechanisms under atmospheric night-time conditions in chamber experiments: evidence of hydroperoxy aldehydes and epoxy products from NO3 oxidation
Philip T. M. Carlsson
Luc Vereecken
Anna Novelli
François Bernard
Steven S. Brown
Bellamy Brownwood
Changmin Cho
John N. Crowley
Patrick Dewald
Peter M. Edwards
Nils Friedrich
Juliane L. Fry
Mattias Hallquist
Luisa Hantschke
Thorsten Hohaus
Sungah Kang
Jonathan Liebmann
Alfred W. Mayhew
Thomas Mentel
David Reimer
Franz Rohrer
Justin Shenolikar
Ralf Tillmann
Epameinondas Tsiligiannis
Rongrong Wu
Andreas Wahner
Astrid Kiendler-Scharr
The gas-phase reaction of isoprene with the nitrate radical (NO3) was investigated in experiments in the outdoor SAPHIR chamber under atmospherically relevant conditions specifically with respect to the chemical lifetime and fate of nitrato-organic peroxy radicals (RO2). Observations of organic products were compared to concentrations expected from different chemical mechanisms: (1) the Master Chemical Mechanism, which simplifies the NO3 isoprene chemistry by only considering one RO2 isomer; (2) the chemical mechanism derived from experiments in the Caltech chamber, which considers different RO2 isomers; and (3) the FZJ-NO3 isoprene mechanism derived from quantum chemical calculations, which in addition to the Caltech mechanism includes equilibrium reactions of RO2 isomers, unimolecular reactions of nitrate RO2 radicals and epoxidation reactions of nitrate alkoxy radicals. Measurements using mass spectrometer instruments give evidence that the new reactions pathways predicted by quantum chemical calculations play a role in the NO3 oxidation of isoprene. Hydroperoxy aldehyde (HPALD) species, which are specific to unimolecular reactions of nitrate RO2, were detected even in the presence of an OH scavenger, excluding the possibility that concurrent oxidation by hydroxyl radicals (OH) is responsible for their formation. In addition, ion signals at masses that can be attributed to epoxy compounds, which are specific to the epoxidation reaction of nitrate alkoxy radicals, were detected. Measurements of methyl vinyl ketone (MVK) and methacrolein (MACR) concentrations confirm that the decomposition of nitrate alkoxy radicals implemented in the Caltech mechanism cannot compete with the ring-closure reactions predicted by quantum chemical calculations. The validity of the FZJ-NO3 isoprene mechanism is further supported by a good agreement between measured and simulated hydroxyl radical (OH) reactivity. Nevertheless, the FZJ-NO3 isoprene mechanism needs further investigations with respect to the absolute importance of unimolecular reactions of nitrate RO2 and epoxidation reactions of nitrate alkoxy radicals. Absolute concentrations of specific organic nitrates such as nitrate hydroperoxides would be required to experimentally determine product yields and branching ratios of reactions but could not be measured in the chamber experiments due to the lack of calibration standards for these compounds. The temporal evolution of mass traces attributed to product species such as nitrate hydroperoxides, nitrate carbonyl and nitrate alcohols as well as hydroperoxy aldehydes observed by the mass spectrometer instruments demonstrates that further oxidation by the nitrate radical and ozone at atmospheric concentrations is small on the timescale of one night (12 h) for typical oxidant concentrations. However, oxidation by hydroxyl radicals present at night and potentially also produced from the decomposition of nitrate alkoxy radicals can contribute to their nocturnal chemical loss.
- Article
(11154 KB) - Full-text XML
- BibTeX
- EndNote





Isoprene (C5H8) is an unsaturated compound and the most emitted non-methane hydrocarbon in the atmosphere. Circa 500 Tg yr−1 of isoprene is emitted by plants as a co-product of photosynthesis activity (Guenther et al., 2012). The high reactivity of isoprene towards the most important daytime oxidant, the hydroxyl radical (OH), results in a chemical lifetime of a few hours for typical atmospheric conditions, so the majority of isoprene is oxidized during the day. However, isoprene can also be present in significant quantities after sunset, when the production rate of OH radicals is low, so oxidation by the nitrate radical (NO3) or ozone can gain in importance (Brown et al., 2009; Edwards et al., 2017).
Oxidants add preferentially to the C=C double bonds in isoprene, initiating a cascade of radical reactions. Theoretical studies of the OH-initiated oxidation of isoprene have shown that the primary organic peroxy radicals (RO2) formed after the OH addition are in thermal equilibrium with the alkyl radical through oxygen elimination and re-addition reactions at a timescale that is short relative to the chemical lifetimes of the RO2 radicals under atmospheric conditions (Peeters et al., 2009, 2014). As a consequence, fast H-shift reactions of RO2 isomers can constitute a large loss process for the entire RO2 pool. This applies to the 1,6-H-migration reactions of the Z-δ-RO2 isomers produced from the isoprene + OH reaction (Peeters et al., 2014). These H migrations lead eventually to the regeneration of OH radicals. Because this type of radical regeneration does not require the presence of nitric oxide (NO), it can significantly enhance radical concentrations in forested environments (Novelli et al., 2020). The OH-initiated oxidation of isoprene has been investigated in laboratory (Crounse et al., 2011; Berndt et al., 2019) and simulation chamber (Fuchs et al., 2013; Novelli et al., 2020) studies, which have contributed to the refinement of the chemical mechanism proposed by the theoretical studies. The results can partly explain high OH radical concentrations observed in field experiments in rainforests (Lelieveld et al., 2008; Whalley et al., 2011).
In contrast to the daytime, the loss of RO2 radicals due to the reaction with NO does not play a role at night in the absence of nearby emission sources because NO production from the photolysis of NO2 is stopped and NO is rapidly titrated to NO2 by the reaction with ozone. In some situations, ozone can also be locally completely consumed in the night if there are high NO emissions, for example from traffic or from power plants. In this case, NO can accumulate. However, for these conditions, the nitrate radical is rapidly lost in the reaction with NO. Therefore, it is unlikely that nitrate RO2 radicals and NO exist simultaneously. Thus, nitrate RO2 from the reaction of NO3 with organic compounds is expected to react mainly with hydroperoxy radicals (HO2), other organic peroxy radicals or the nitrate radical, or they may undergo unimolecular reactions.
In previous chamber and laboratory studies investigating the reaction of isoprene with NO3, the fate of RO2 was often dominated by RO2 self- and cross-reactions and RO2 reactions with NO3 due to high reactant concentrations (Barnes et al., 1990; Kwok et al., 1996; Perring et al., 2009; Kwan et al., 2012). A chamber study by Schwantes et al. (2015) focussed on the product distribution from the reaction of nitrate RO2 with HO2 because this reaction pathway is generally the dominant loss path in the atmosphere. Chamber studies by Rollins et al. (2009) and Ng et al. (2008) were also designed to reproduce atmospheric chemical conditions, for which the nitrate RO2 reacts along various pathways.
Near-explicit chemical mechanisms such as the Master Chemical Mechanism (Jenkin et al., 2015) and the isoprene mechanism developed by Wennberg et al. (2018) (called the Caltech mechanism in this work) were partly built by using results from these studies. In addition, it has been proposed that the nitrate RO2 radicals formed from the reaction of the nitrate radical with isoprene can interconvert at ambient temperature (Wennberg et al., 2018; Vereecken et al., 2021). This can enhance the importance of unimolecular reactions of specific RO2 if the chemical lifetime of the RO2 radicals is long enough for concentrations to re-equilibrate.
Furthermore, a theoretical study by Vereecken et al. (2021) revealed that unimolecular reactions of alkoxy radicals formed in the radical reaction chain subsequent to the addition of NO3 to isoprene lead to the production of epoxide RO2, influencing the distribution of organic products. This newly identified chemistry is included only in the FZJ-NO3 isoprene mechanism published by Vereecken et al. (2021).
The aim of this study is to compare the NO3 isoprene chemistry of different available explicit mechanisms (MCM, Caltech and FZJ-NO3) with respect to the fate of nitrato-organic peroxy radicals and the distribution of organic products for a series of chamber experiments performed under atmospherically relevant night-time conditions.
2.1 Experiments in the SAPHIR chamber
The experiments discussed in this work were performed in the atmospheric simulation chamber SAPHIR (Rohrer et al., 2005) at Forschungszentrum Jülich in 2018. The chamber is a 270 m3 double-wall reactor. It is operated at a slight overpressure of 35 Pa to prevent ambient air from leaking into the chamber. The space between the two films is continuously flushed with pure nitrogen to prevent contamination of the inner chamber. The walls are made of Teflon FEP film and are thus chemically inert while the full solar spectrum is transmitted into the chamber (Bohn and Zilken, 2005). Night-time can be simulated by a shutter system that covers the chamber. Synthetic air used for flushing the chamber and for replenishing losses due to sampling of instruments and leakage is produced from evaporating and mixing high-purity liquid nitrogen and oxygen (purity: 99.9999 %, Linde). Inside the chamber, two fans are operated to ensure homogeneous mixing of air. The temperature inside the chamber is similar to ambient temperature and ranges between 291 and 308 K with maximum values in the afternoon for the experiments in this work.
Reactive trace gases added to the chamber in the experiments were ozone produced by a silent discharge ozonizer (Ozonia), isoprene (C5H8, purity: 99 %, Sigma-Aldrich), propene (purity: 99.8 %, Linde), CO (purity: 99.997 %, Linde) and NO2 (purity: 99.2 %, 519 ppmv in nitrogen, Linde). Addition of gaseous species was controlled by calibrated mass flow controllers. Isoprene was injected as a liquid with a syringe into a hot volume, and the vapour was flushed into the chamber together with the replenishment flow of zero air.
Four experiments performed on 9, 10, 12 and 13 August 2018 (Experiments 1, 2, 3, 4) are analysed in this work (Table 1 and Figs. 1, 2, A1 and A2). Before each experiment, the chamber was flushed overnight with a high flow of zero air so that concentrations of trace gases from previous experiments were below the limit of detection of instruments. The chamber roof was always closed to simulate night-time conditions. Experiments were performed in dry synthetic air. NO3 was produced by the reaction of NO2 and O3. Typical mixing ratios after the injection were 5 ppbv NO2 and 100 ppbv O3. NO3 production rates ranged between 0.9 and 11 ppbv h−1. The highest NO3 production rates were reached in the experiment on 13 August 2018 (Experiment 4) and the lowest rates in the experiment on 10 August 2018 (Experiment 2).
Fuchs et al. (2018a)Fuchs et al. (2018b)Fuchs et al. (2018c)Fuchs et al. (2018d)Table 1Chemical conditions in the experiments in this work. Experiments analysed in this work were performed in dry air. Mixing ratios of trace gases give the range of values reached immediately after their injection.
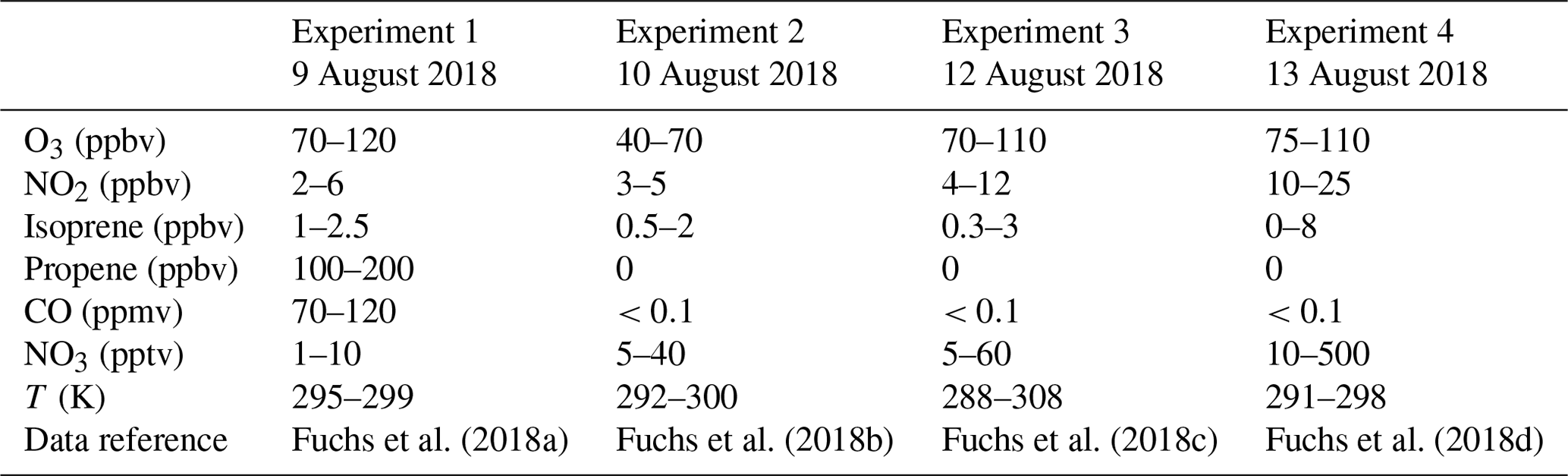
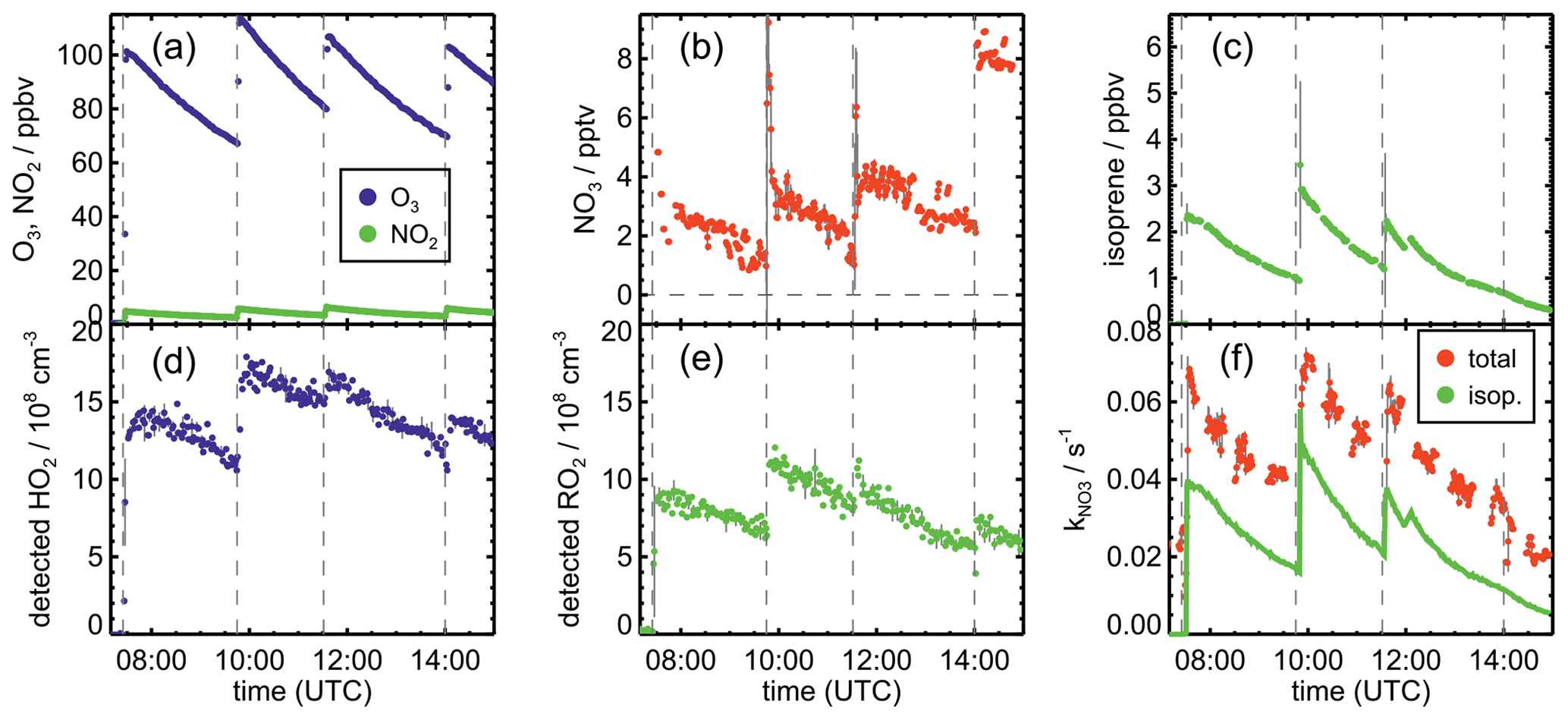
Figure 1Measurements of radical and trace gas concentrations and NO3 reactivity in the experiment on 9 August 2018 (Experiment 1) investigating the oxidation of isoprene (isop.) by NO3. Between 100 and 200 ppmv propene was present to produce HO2 radicals by its ozonolysis. OH radicals, which are produced in the ozonolysis reaction, are rapidly converted to HO2 in the reaction with 70 to 120 ppmv CO that was injected at the start of the experiment. OH reactivity was dominated by the high CO concentration and is not shown. NO3 reactivity does not include reactivity from organic radicals and NO2. NO3 reactivity from isoprene is calculated from measured isoprene concentrations and reaction rate constants recommended in the literature (Mellouki et al., 2021). The difference between measured reactivity and reactivity from isoprene can be attributed to propene in this experiment. Observed RO2 radicals only include a fraction of the total RO2 because the laser-induced fluorescence (LIF) instrument cannot detect all RO2 species formed in the reaction of isoprene with NO3 (Vereecken et al., 2021).
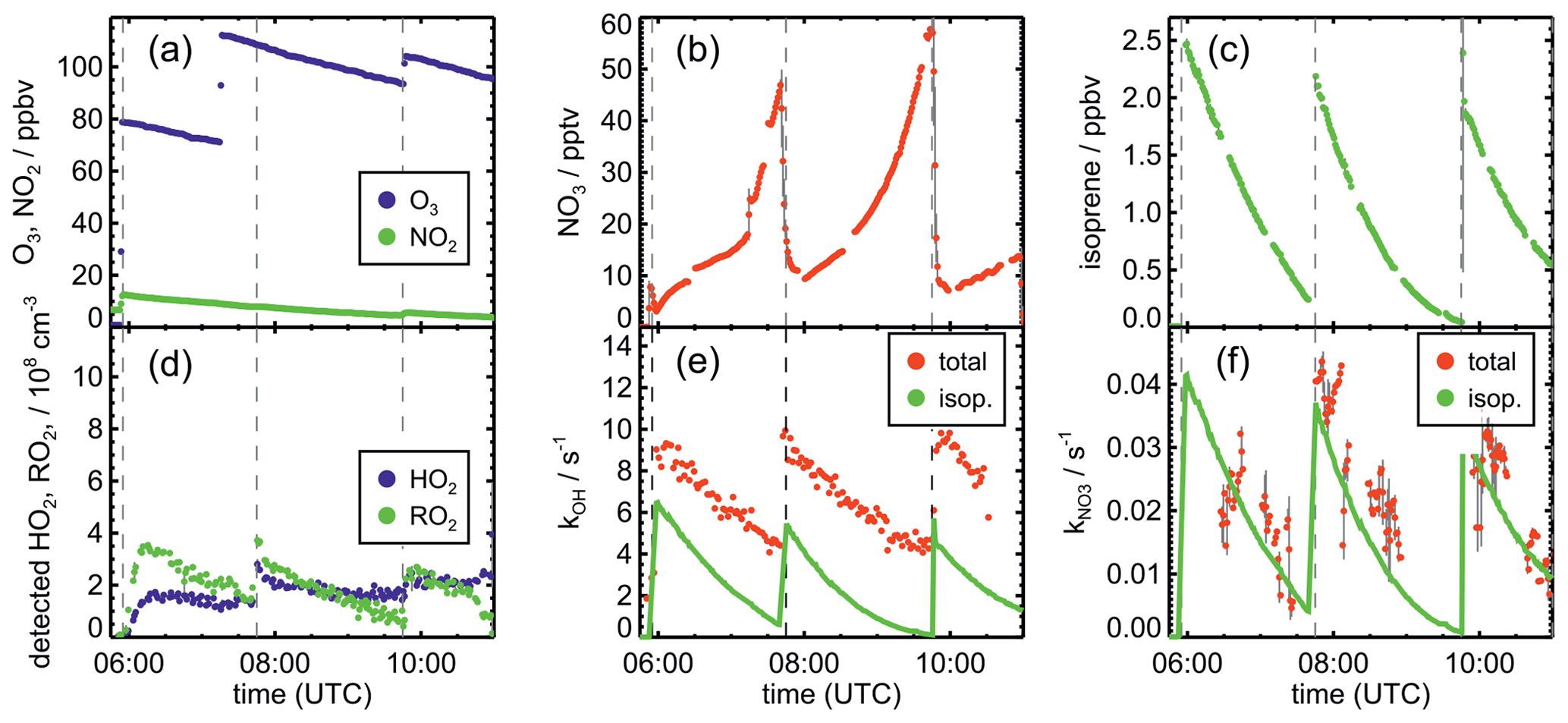
Figure 2Measurements of radical and trace gas concentrations and OH and NO3 reactivity in the experiment on 13 August 2018 (Experiment 4) investigating the oxidation of isoprene by NO3, when the total amount of oxidized isoprene was highest. OH and NO3 reactivity from isoprene is calculated from measured isoprene concentrations and reaction rate constants recommended in the literature (Mellouki et al., 2021). NO3 reactivity does not include reactivity from organic radicals and NO2. Observed RO2 radicals only include a fraction of the total RO2 because the LIF instrument cannot detect all RO2 species formed in the reaction of isoprene with NO3 (Vereecken et al., 2021).
After NO3 production started, isoprene was added. The injection of all three species was repeated after a few hours, when most of the isoprene had been consumed. Only NO2 and O3 were re-injected to enhance NO3 production in the last part of the experiments, except for the experiment on 10 August 2018 (Experiment 2). In the experiment on 9 August 2018 (Experiment 1), propene was injected to enhance HO2 concentrations by radical production via its ozonolysis. Excess CO was additionally injected to convert OH radicals to HO2.
In the experiments in this work, no measurable secondary organic aerosol was formed, so loss of product species on aerosol did not play a role (Brownwood et al., 2021).
The total amount of isoprene that was consumed by NO3 was (3.2 ± 0.5), (2.5 ± 0.5), (4.8 ± 0.5) and (11.6 ± 1.2) ppbv in the experiments on 9, 10, 12 and 13 August 2018 (Experiments 1, 2, 3, 4), respectively (Brownwood et al., 2021). Approximately 10 % of the total isoprene consumed in the experiment reacted with ozone except for the experiment on 9 August 2018 (Experiment 1), when 25 % to 30 % of isoprene was lost in the reaction with ozone due to the low NO3 and high ozone concentration. In addition, measurements of OH radicals suggest that up to 10 % of isoprene reacted with OH in the experiments without an OH scavenger. However, OH concentration measurements were close to the limit of detection of the instrument, so the fraction of isoprene that reacted with OH is rather uncertain. Overall, the dominant loss for isoprene was due to the reaction with NO3 radicals (80 % to 90 % of the total loss in most of the experiments).
The chemical conditions in the experiments were chosen such that the chemical loss of nitrated RO2 radicals differed between the experiments (Table 1). Similarly to the case for typical night-time conditions in the nocturnal residual layer in the absence of nearby sources, nitric oxide concentrations were zero, so RO2 reacted only with HO2, RO2 or NO3 or was re-arranged in unimolecular RO2 reactions (Vereecken et al., 2021).
In the experiments, the concentrations of NO3 precursor species, HO2, O3 and NO2, and of isoprene were varied. As a consequence, RO2 concentrations and therefore also the relative importance of RO2 loss reactions differed between these experiments.
2.2 Instrumentation
A large suite of instruments detected inorganic and organic species during the experiments. Isoprene and its oxidation products were measured by a proton transfer reaction time-of-flight mass spectrometer (Vocus PTR-MS, Aerodyne; Krechmer et al., 2018). The instrument was calibrated for isoprene, methyl vinyl ketone and methacrolein. The sensitivity of the instrument to isoprene was higher by a factor of 1.4 in dry air than in humid air in which calibration measurements were performed (Brownwood et al., 2021). Measured concentrations were corrected for this humidity effect. No calibration standards were available for organic nitrate products such as nitrated alcohols, carbonyls, hydroperoxides and epoxides.
Organic compounds were also detected by two other chemical ionization mass spectrometer (CIMS) instruments that used either Br− (Albrecht et al., 2019; Wu et al., 2021) or I− as reagent ions (Tsiligiannis et al., 2022). These instruments detected various oxygenated organic product species but were not calibrated to provide concentrations. Details of the measurements by the Br− CIMS instrument can be found in Wu et al. (2021) and those by the I− CIMS instrument in Tsiligiannis et al. (2022).
The high resolution of the mass spectrometer instruments allowed us to attribute the ion mass signals () to sum formulas of organic compounds (Table A1). In this work, ion signals that were the highest among all signals are discussed, most of which can be attributed to products of the isoprene oxidation (Wu et al., 2021; Tsiligiannis et al., 2022). Compared to the CIMS instruments, the precision of measurements by the Vocus PTR-MS instrument was higher for organic compounds that contain few oxygens. In general, the sensitivity of CIMS instruments can be different for different isomers and functional groups, so a change in the distribution of isomers could partly explain observed differences between instruments (B. H. Lee et al., 2014; Xiong et al., 2015, 2016). In addition, changes in the operational conditions of the instrument such as the temperature of the ionization region can lead to a variability in the instrument's sensitivity (Robinson et al., 2022).
The total organic nitrate concentration was measured by two instruments, in which the total NO2 concentration was detected by either a custom-built (Sobanski et al., 2016) or a commercial cavity ring-down instrument (Keehan et al., 2020) after thermal dissociation of nitrate compounds in a heated inlet (thermal dissociation–cavity ring-down spectrometer – TD-CRDS). A common data set from both instruments was created for this campaign. Details of these measurements can be found in Brownwood et al. (2021). These instruments also measured NO2 in the sampled air in a separate mode or second measurement channel. In addition, NO2 concentrations were measured by another custom-built cavity ring-down instrument (Liebmann et al., 2018) and a commercial chemiluminescence instrument combined with a blue-light converter (Eco Physics). NO2 concentration measurements from all instruments were combined into one common, quality-checked data set (Brownwood et al., 2021). Ozone concentrations were measured by a commercial instrument using UV absorption (Ansyco).
NO3 and N2O5 concentrations were measured with two custom-built instruments applying cavity-ring-down spectroscopy (Wagner et al., 2011; Sobanski et al., 2016). NO3 was detected at 662 nm and the sum of NO3 and N2O5 in a second channel, in which the inlet and cavity are heated to thermally decompose N2O5. Measurements were combined into one data set, also taking into account that NO3 and N2O5 can be expected to be in thermal equilibrium for conditions of the experiments in this work.
HO2, OH and RO2 radical concentrations were determined by a laser-induced fluorescence (LIF) instrument (Fuchs et al., 2011, 2012; Cho et al., 2021). OH radicals are excited at 308 nm in a low-pressure cell, and their fluorescence is measured by gated single-photon counting. The fluorescence cell for the detection of only OH radicals was equipped with a chemical modulation reactor (CMR), which allows us to account for potential interferences in the measurements (Cho et al., 2021). In another fluorescence cell, HO2 radicals are chemically converted to OH in their reaction with NO. RO2 radicals are converted eventually to OH in a third measurement channel (ROxLIF) that consists of an RO2 converter, in which RO2 and OH radicals are firstly converted to HO2 in the presence of NO and CO, and a fluorescence cell downstream of the converter, in which the sum of all radicals is detected by OH fluorescence after HO2 has reacted with excess NO. Recent studies confirmed that not all nitrate RO2 radicals can be detected by the ROxLIF method as they do not form HO2 or OH radicals after reacting with NO (Ashbourn et al., 1998; Novelli et al., 2021; Vereecken et al., 2021).
OH reactivity (kOH, the inverse of the chemical lifetime of the OH radical) was determined by a laser flash photolysis instrument, in which the time-resolved decay of artificially produced OH radicals is observed (Fuchs et al., 2017). If, as in this work, the OH reactivity from inorganic compounds is known, the contribution from organic compounds can be derived and compared to values based on the measurements of single compounds (Tan et al., 2021; Hantschke et al., 2021). In general, differences between measured and calculated OH reactivity can be used to determine if the detection of organic products that are reactive towards OH is complete.
The NO3 reactivity was also measured in this work (Liebmann et al., 2017; Dewald et al., 2020). The concentration of artificially produced NO3 is measured by cavity ring-down spectroscopy after reaction with either ambient or zero air in a flow tube. The NO3 reactivity can then be calculated from the relative change in NO3 concentrations between the two modes. In order to obtain the NO3 reactivity from organic compounds, the contribution of NO2 and NO3 losses in the flow tube was accounted for. NO3 reactivity from HO2 and RO2 radicals is not detected by the instrument due to loss of radicals in the inlet system (Dewald et al., 2020).
2.3 Modelling of trace gas concentrations
Trace gas concentrations were calculated using a chemical box model. In this work, three near-explicit chemical models have been applied: (1) the Master Chemical Mechanism version 3.3.1 (MCM) (Jenkin et al., 1997, 2015; Saunders et al., 2003), (2) the isoprene oxidation mechanism as introduced in the review article by Wennberg et al. (2018) and available at Bates and Wennberg (2017) (Caltech), and (3) the NO3 isoprene mechanism based on theoretical calculations by Vereecken et al. (2021) and detailed in the supplement of Vereecken et al. (2021) (FZJ-NO3 mechanism).
The Caltech mechanism includes reactions of isoprene and isoprene product species but does not include further reactions of organic products that are not specific products from the oxidation of isoprene such as glyoxal or methyl glyoxal. In this work, the Caltech mechanism is therefore extended with chemistry from the MCM for those species.
The FZJ-NO3 mechanism only includes the reaction steps subsequent to the initial addition of NO3 to isoprene, but the chemistry of organic products was not investigated in Vereecken et al. (2021). The chemistry of the trace gases not considered in Vereecken et al. (2021) is taken from the Caltech mechanism. The isoprene OH oxidation scheme is applied as described in the work by Novelli et al. (2020), where the OH oxidation of isoprene was investigated in chamber experiments. Further chemistry of organic products that are not specific to the oxidation of isoprene is taken from the MCM. Chemical loss of first-generation organic products which are not included in either the Caltech or the MCM models is estimated from similarities to other organic products.
In the model runs, the injections of trace gases in the experiments were implemented as source reactions, which are effective during the short period of time during the injection. The rates are adjusted such that the concentration change of the injected trace gas matches the observed increase in the concentration at the time of the injection. Physical parameters such as temperature and pressure were constrained to measured values. NO3 was also constrained to measured values in order to decouple its modelled concentrations from wall reactions of NO3 and N2O5, which are dependent on the chemical conditions of the experiment and hence hard to characterize accurately (Dewald et al., 2020). With NO3 concentrations constrained to measurements, the measured decay of isoprene, which is dominated by the reaction with NO3 for most of the time, is well described by the model, confirming that measured NO3 concentrations are consistent with the chemical loss of isoprene.
The initial reaction steps in the oxidation of isoprene by NO3 (Vereecken et al., 2021) are similar to those of the oxidation by OH. H-atom abstraction from isoprene by NO3 is estimated to be at least 2 orders of magnitude slower than NO3 addition, based on the available literature data on aliphatic and allylic H-abstraction reactions (Canosa-Mas et al., 1991; Atkinson et al., 2006), and is therefore not further considered in this work.
NO3 adds to either of the C=C double bonds, leading to allyl-resonance-stabilized alkyl radicals. Reversible oxygen addition and elimination reactions produce three different RO2 stereoisomers each from the addition of NO3 on carbon C1 and C4 (Fig. 3). The different RO2 isomers rapidly reach equilibrium concentrations. NO3 adds preferably on carbon C1 (yield of 87 %). The yield is higher in comparison to the corresponding OH addition (yield of 61 %). The additions on the inner carbons (C2 and C3) are expected to be of minor importance (Vereecken et al., 2021) and are not further considered in this work.
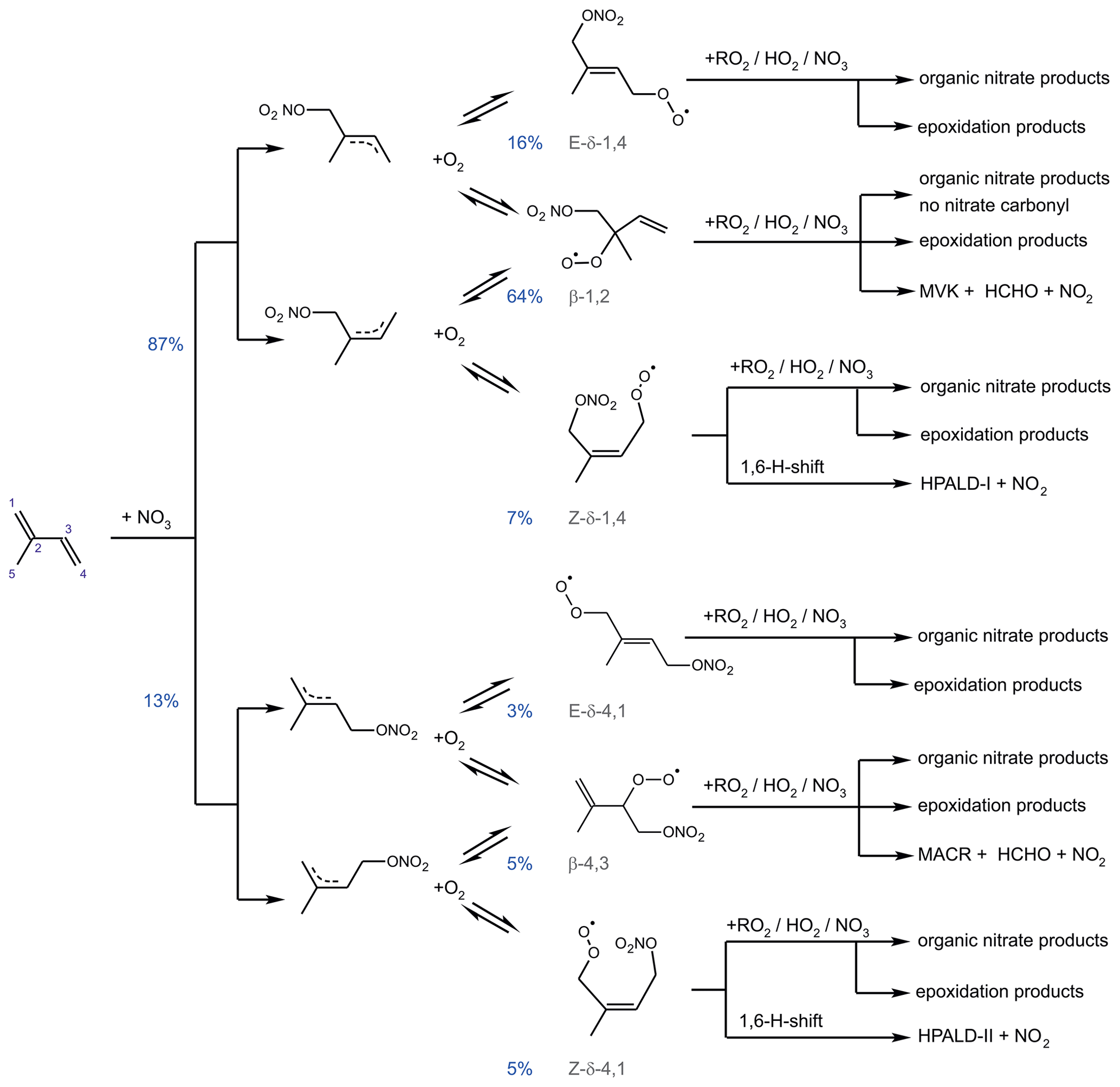
Figure 3Schematic reaction mechanism of the reaction of isoprene with NO3 as described in Vereecken et al. (2021). This includes fast interconversion of nitrate RO2 isomers by oxygen addition and elimination reactions. Only RO2 isomerization reactions (Vereecken et al., 2021) which can compete with bimolecular reactions for typical night-time conditions are shown. Percentage values given next to the structure of RO2 radicals are yields when equilibrium concentrations are established for typical night-time conditions such as in the experiments in this work. HPALD: hydroperoxy aldehyde; MVK: methyl vinyl ketone; MACR: methacrolein.
The isoprene NO3 mechanisms investigated in this work differ significantly in the treatment of the initially formed RO2. The FZJ-NO3 mechanism includes six RO2 isomers formed subsequently to the NO3 addition (Fig. 3). Specifically, the Z- and E-RO2 isomers of the δ-RO2 isomers are distinguished. In contrast, the Caltech mechanisms only treats δ- and β-RO2 isomers separately and does not include the equilibrium reactions between RO2 isomers. The MCM simplifies the addition of NO3 to isoprene even more by only considering the addition of NO3 on carbon C1 leading to the δ-RO2 radical.
It is important to distinguish between Z- and E-RO2 isomers because isomer-specific unimolecular H-shift reactions need to be considered. Competitive unimolecular H-shift reactions only occur for Z-δ-RO2 (Vereecken et al., 2021), leading to the formation of hydroperoxy aldehyde (HPALD) species (Fig. 3). Due to the re-equilibration reactions between RO2 isomers, these reaction channels can gain in importance if the rate of this RO2 loss reaction (0.01 to 0.05 s−1) is faster than the chemical loss due to bimolecular RO2 reactions. This will often be the case for night-time conditions, when mainly slow bimolecular RO2 reactions with NO3, HO2 and other RO2 radicals occur.
The distribution of organic products from the NO3 oxidation of isoprene depends highly on the competition between the different RO2 loss reactions. The bimolecular reaction of nitrate RO2 with HO2 radicals leads to the formation of nitrate hydroperoxide (NISOPOOH). Whereas one NISOPOOH isomer is the exclusive product of the RO2 + HO2 reaction in the MCM, the Caltech and FZJ-NO3 mechanisms include not only different isomers but also the decomposition of the initially formed HO2–RO2 reaction complex into an OH radical and a nitrate alkoxy radical with a yield of approximately 50 % for nitrate β-RO2 radicals.
Nitrate alkoxy radicals can also be the product of RO2 + RO2 reactions, but this reaction channel competes with the production of nitrate carbonyls (NC4CHO) and nitrate alcohols (ISOPCNO3). Alkoxy radicals are additionally formed from the reaction of nitrate RO2 with NO3 accompanied by the production of NO2. The nitrate alkoxy radicals are expected to rapidly decompose (Novelli et al., 2021; Vereecken et al., 2021). In the MCM, the decomposition leads exclusively to the formation of one isomer of the nitrate carbonyl product (NC4CHO) together with an HO2 radical. A similar mechanism is implemented in the Caltech and FZJ-NO3 mechanisms for most of the various nitrate alkoxy radical species except for those radicals produced from the most abundant β-1,2-RO2 isomer, from which nitrate carbonyl species cannot be formed.
The yield of dimerised peroxide compounds (ROOR) from the gas-phase reaction of RO2 + RO2 radicals is expected to be small. Due to their low volatility, however, ROOR compounds are important for the formation of secondary organic aerosol (SOA; Ng et al., 2008).

Figure 4Comparison of results from model calculations applying the different isoprene NO3 chemistry mechanisms for the experiment on 9 August 2018 (Experiment 1), when HO2 concentrations were enhanced and excess CO was present as an OH scavenger. MVK, MACR, NISOPOOH, ISOPCNO3 and NC4CHO are produced from all mechanisms, whereas the other species are only produced from either 1,6-H-shift reactions or ring-closure reactions of nitrate alkoxy radicals implemented only in the FZJ-NO3 mechanism. Grey and black dots are measured values. Measured organic peroxy radical concentrations only include part of the total RO2 because the LIF instrument cannot detect a fraction of nitrate RO2 (Vereecken et al., 2021). Organic products were detected by the Vocus PTR-MS instrument, which was only calibrated for MVK and MACR. All other traces are scaled to match best the results from the FZJ-NO3 mechanism.
In the Caltech mechanism, decomposition of these nitrate alkoxy radicals leads instantly to the formation of methyl vinyl ketone (MVK) or methacrolein (MACR) together with formaldehyde and NO2. This was determined from chamber experiments reported in Schwantes et al. (2015), in which a high yield of MVK was found, when nitrate RO2 mainly reacted with HO2. The fate of nitrate alkoxy radicals was also investigated by Vereecken et al. (2021). Quantum chemical calculations show that the decomposition reaction is slower than the ring-closure reactions leading to epoxide products. In contrast, four-membered ring closure (barrier ∼25 kcal mol−1; Vereecken, 2022) requires breaking the planar double bond to bring the radical O atom into an appropriate position for bonding. Five- to six-membered ring closure (barrier ∼13–29 kcal mol−1; Vereecken et al., 2021) is also favourable.
Differences between the chemical mechanisms also exist concerning the type of chemical loss reactions of first-generation stable organic products. Reactions with OH are considered in all mechanisms applying similar reaction rate constants. In addition, the MCM includes loss of isoprene organic nitrates due to ozonolysis reactions.
Results of the model calculations are shown in Fig. 4 for the experiment on 9 August 2018 (Experiment 1), when high HO2 concentrations were present, and therefore the main loss path for RO2 was the reaction with HO2. Figure 5 shows results for the experiment on 13 August 2018 (Experiment 4), when RO2 loss was distributed among all pathways that are relevant during the night-time (Brownwood et al., 2021) and the amount of oxidized isoprene was highest. Results from the other experiments are shown in the Appendix (Figs. A5 and A6). In all figures, ion mass signals of the Vocus PTR-MS instrument for which no calibration was available were scaled to concentrations predicted by the FZJ-NO3 model.
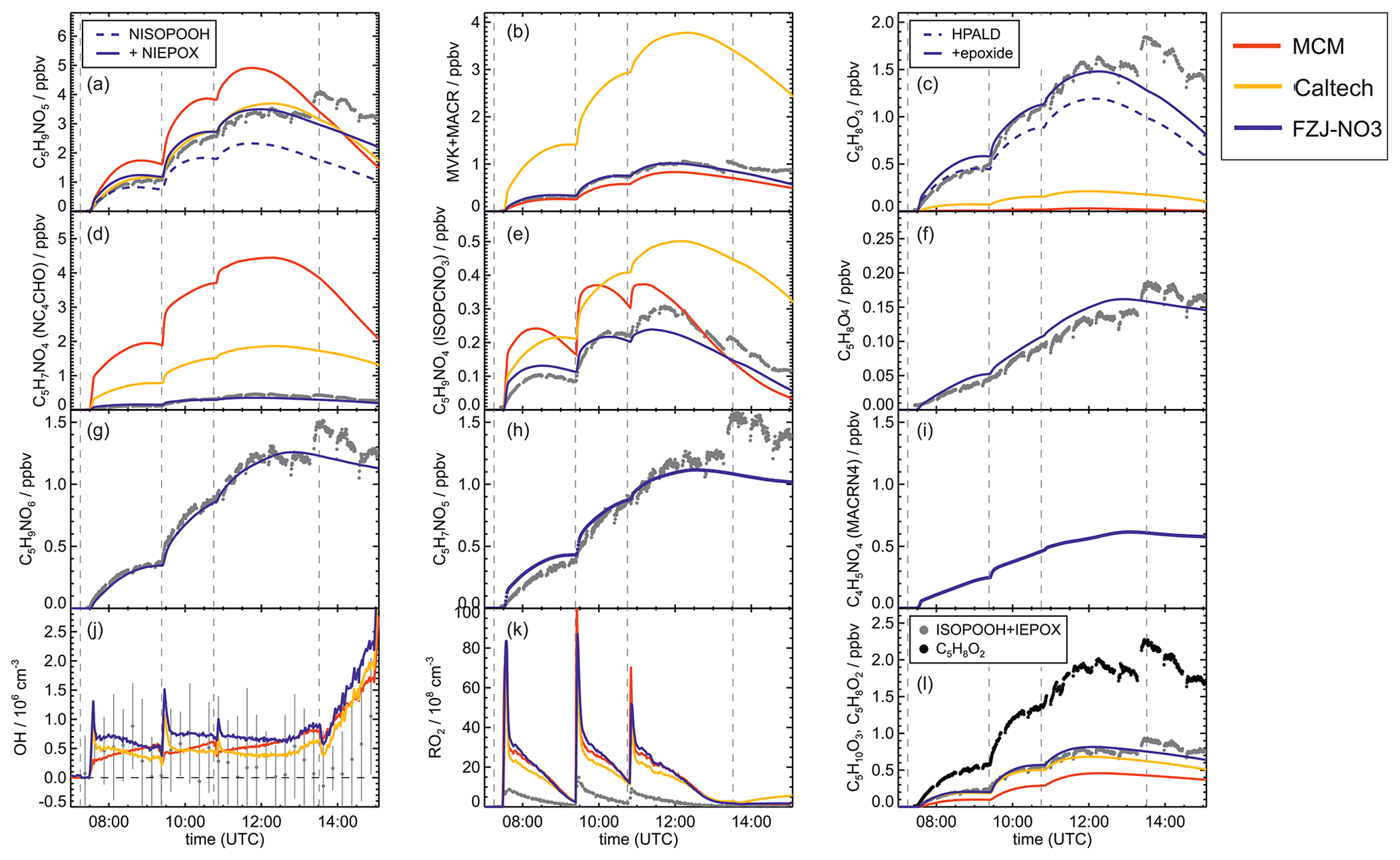
Figure 5Comparison of results from model calculations applying the different isoprene NO3 chemistry mechanisms for the experiment on 13 August 2018 (Experiment 4), when the amount of oxidized isoprene was highest. MVK, MACR, NISOPOOH, ISOPCNO3 and NC4CHO are produced from all mechanisms, whereas the other species are only produced from either 1,6-H-shift reactions or ring-closure reactions of nitrate alkoxy radicals only implemented in the FZJ-NO3 mechanism. Grey and black dots are measured values. Measured organic peroxy radical concentrations only include part of the total RO2 because the LIF instrument cannot detect a fraction of nitrate RO2 (Vereecken et al., 2021). Organic products were detected by the Vocus PTR-MS instrument, which was only calibrated for MVK and MACR. All other traces are scaled to match best the results from the FZJ-NO3 mechanism.
The highest HO2 concentrations of up to 17×108 cm−3 were measured in the experiment on 9 August 2018 (Experiment 1), when HO2 was enhanced by production of OH radicals in the ozonolysis of propene, and were rapidly converted to HO2 in the presence of excess CO (Fig. 1d). In the other experiments, measured HO2 concentrations were between 1×108 and 5×108 cm−3 with the highest values in the experiment on 13 August 2018 (Experiment 4). As discussed in Vereecken et al. (2021), the measured HO2 concentrations are much higher than predicted by model calculations for experiments in this work (up to a factor of 10) except for the experiment on 9 August 2018 (Experiment 1). Although it is possible that part of the measured HO2 radicals is due to an interference (Vereecken et al., 2021), the HO2 radical concentrations predicted by the model are too low to explain observed OH radical concentrations, for example during the last part of the experiment on 13 August 2018 (Experiment 4) (Sect. 5.5). Therefore, the measured HO2 radical concentrations are used in the further analysis in this work.
A large fraction of nitrate RO2 radicals cannot be detected by the LIF instrument used in this work (Novelli et al., 2021; Vereecken et al., 2021) because the detection scheme of the instruments requires that HO2 or OH radicals are formed subsequent to the reaction of RO2 with NO. However, this is only the case for some of the nitrate RO2 radicals from the reaction of isoprene with NO3 (Sect. 2.1). Therefore, measured RO2 concentrations, which are maximum around 1×109 cm−3 (Figs. 1 and 2d), need to be regarded as lower limits.
In all experiments, significant amounts (up to 1 ppbv) of methyl vinyl ketone (MVK) and methacrolein (MACR) were detected by the Vocus PTR-MS instrument.
Vocus PTR-MS, Br− CIMS and I− CIMS instruments also recorded ion signals from oxygenated organic compounds in the experiments that can be attributed to the sum formulas of a number of other product species including non-nitrate (HPALD: C5H8O3) and nitrate organic compounds (NISOPOOH: C5H9NO5; NC4CHO: C5H7NO4; ISOPCNO3: C5H9NO4) and epoxide products that are expected to be formed subsequent to the ring-closure reaction of alkoxy radicals (Reactions R9 and R17; C5H8O4, C5H8O3, C5H9NO6, C5H9NO5, C5H7NO5; Fig. 6).
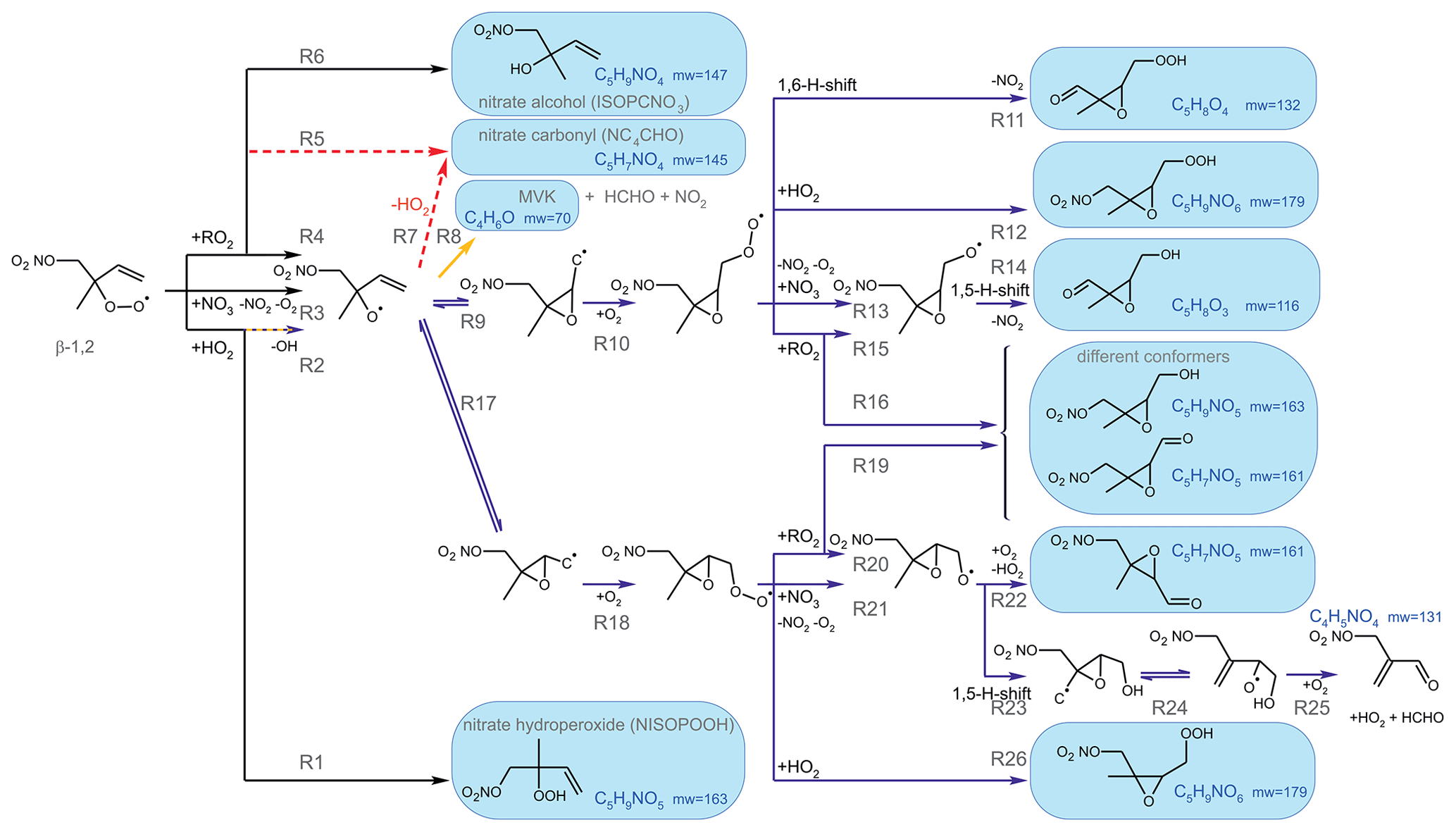
Figure 6Loss reactions of the most abundant β-1,2-RO2 species. Coloured arrows indicate the preferred reaction channel for the nitrate alkoxy radical in the different chemical models (yellow: Caltech; blue: FZJ-NO3). Dashed red arrows indicate corresponding reactions of the δ-RO2 species, which is the only RO2 represented in the MCM. Coloured boxes indicate species that were observed by the Vocus PTR-MS instrument and their molecular weight (mw). Though nitrate carbonyl products (NC4CHO) cannot be formed from this specific nitrate β-RO2 from isoprene, they are formed from other nitrate radicals, and thus nitrate carbonyls were also observed by the Vocus PTR-MS instrument.
Ion signals shown in Figs. 4, 5, A5 and A6 were the highest signals observed in the mass spectrometer instruments except for the ion signal corresponding to a C4H7NO5 compound observed by the I−- and Br− CIMS instruments. A species with this sum formula cannot be attributed to a major product species expected from the chemical mechanism. This is discussed in detail in Tsiligiannis et al. (2022).
Signals at the mass corresponding to NISOPOOH were the highest among all product signals observed by the Vocus PTR-MS instrument. The signal can include nitrate epoxides that are produced from the ring-closure reactions of alkoxy radicals (Sect. 5.3) and from the reaction of NISOPOOH with OH. However, the contribution of nitrate epoxides from the ring-closure reactions to the sum of product concentrations from both reactions is expected to be low in the experiments in this work, specifically in the experiment on 9 August 2018 (Experiment 1), when HO2 concentrations favoured RO2 + HO2 reactions and an OH scavenger was present (Fig. 4a). Fragmentation, however, may reduce the sensitivity of the Vocus PTR-MS instrument to NISOPOOH at the corresponding mass as shown by Li et al. (2022) for other hydroperoxide species.
Signals from all three mass spectrometry instruments (Figs. A7–A10) can be compared by scaling them to best match modelled concentrations of organic products applying the FZJ-NO3 chemical mechanism.
The relative behaviour of signals is similar for all instruments with a few exceptions: (1) in the experiment on 9 August 2018 (Experiment 1), the signals of the Br− CIMS instrument appear to be systematically lower after 10:00 UTC for unknown reasons (Fig. A7). (2) In the experiment on 13 August 2018 (Experiment 4), the loss rate of C5H9NO4 compounds appears to be slower in the signal of the Br− CIMS instrument than in the other mass spectrometer instruments (Fig. A10c) and expected from model calculations. This could be explained if other (fragments of) products were detected at that mass by the Br− CIMS instrument but not by the other instruments. (3) The loss rate of C5H10O3 compounds observed by the I− CIMS instrument appears to be faster than observed by the Vocus PTR-MS instrument and expected from model calculations in the experiment on 13 August 2018 (Experiment 4) (Fig. A10f). The difference in the observed temporal evolution of C5H10O3 compounds could be explained if the sensitivity of the instrument were lower for the hydroperoxide species than for the epoxide species, both of which are detected at the same mass (Sect. 5.5). Differences would become most obvious during this part of the experiment because these compounds have vastly different chemical lifetimes with respect to the reaction with OH, which was likely the dominant loss process for this part of the experiment. In some parts of the experiments, measurements by the I− CIMS instrument exhibited an oscillating behaviour, which is most likely an instrumental artefact (for example Fig. A10b).
Some species produced from different loss pathways can be structurally different but have the same sum formula. These isomers cannot be distinguished by the mass spectrometers (Fig. 6): (1) nitrate hydroperoxide (NISOPOOH) species have the same mass as some nitrate epoxide species (Reaction R16). This applies not only to nitrate epoxides formed from the reaction of OH with NISOPOOH, which does not play a major role in conditions of the experiments, but also to specific nitrate epoxide products formed subsequently to the ring-closure reaction of nitrate alkoxy radicals predicted by the FZJ-NO3 mechanism (Vereecken et al., 2021). (2) Hydroperoxy aldehyde (HPALD) species produced from unimolecular 1,6-H-shift reactions of the nitrate Z-δ-RO2 isomers have the same mass as one epoxide product also formed from the ring-closure reaction of nitrate alkoxy radicals (sum formula C5H8O3, Reaction R14). NO2 is eliminated, so these products do not contain nitrate functional groups.
The temporal behaviour of products depends on their production and destruction rates. They are formed from the same pool of nitrate RO2 radicals from the reaction of isoprene with NO3, which is the rate-limiting step for their production. The temporal evaluation of their concentrations at later times of the experiment when isoprene had been consumed is determined by the rate of loss processes, which can be chemical loss and dilution in these experiments.
Mainly measurements by the Vocus PTR-MS instrument are discussed in the next sections. However, the conclusions do not depend on the choice of the instrument as can be seen by the overall good agreement in time series of ion signals at the same mass of instruments (Figs. A7–A10). Results are also independent of the choice of scaling measured ion mass signals of the Vocus PTR-MS instrument to the model results of the FZJ-NO3 mechanism (Fig. A11).
5.1 Chemical lifetime of nitrate RO2 radicals
Using the RO2 chemistry as implemented in the FZJ-NO3 mechanism and measured HO2 concentrations results in overall loss rates of nitrate RO2 of around 0.035, 0.005, 0.008 and 0.014 s−1 in the experiments on 9, 10, 12 and 13 August 2018 (Experiments 1, 2, 3, 4). This implies chemical lifetimes of between 30 s and several minutes, which are similar to values under atmospheric night-time conditions. RO2 loss rates are 20 % to 50 % lower if the chemistry implemented in the Caltech mechanism or MCM is applied.
Overall, differences in the RO2 loss rates derived from the three mechanisms are mainly related to differences in the distribution of nitrate RO2 isomers, for which chemical lifetimes vary. In addition, implementation of unimolecular RO2 reactions shortens their chemical lifetime in the FZJ-NO3 mechanism (Figs. 3 and 6). Differences in RO2 loss rates between the chemical mechanisms are lowest for the experiment on 9 August 2018 (Experiment 1), in which the RO2 loss is dominated by the reaction with HO2 (Figs. 7 and A3). In this experiment, the overall loss rate was the highest, so unimolecular RO2 reactions implemented in the FZJ-NO3 mechanism were less competitive.
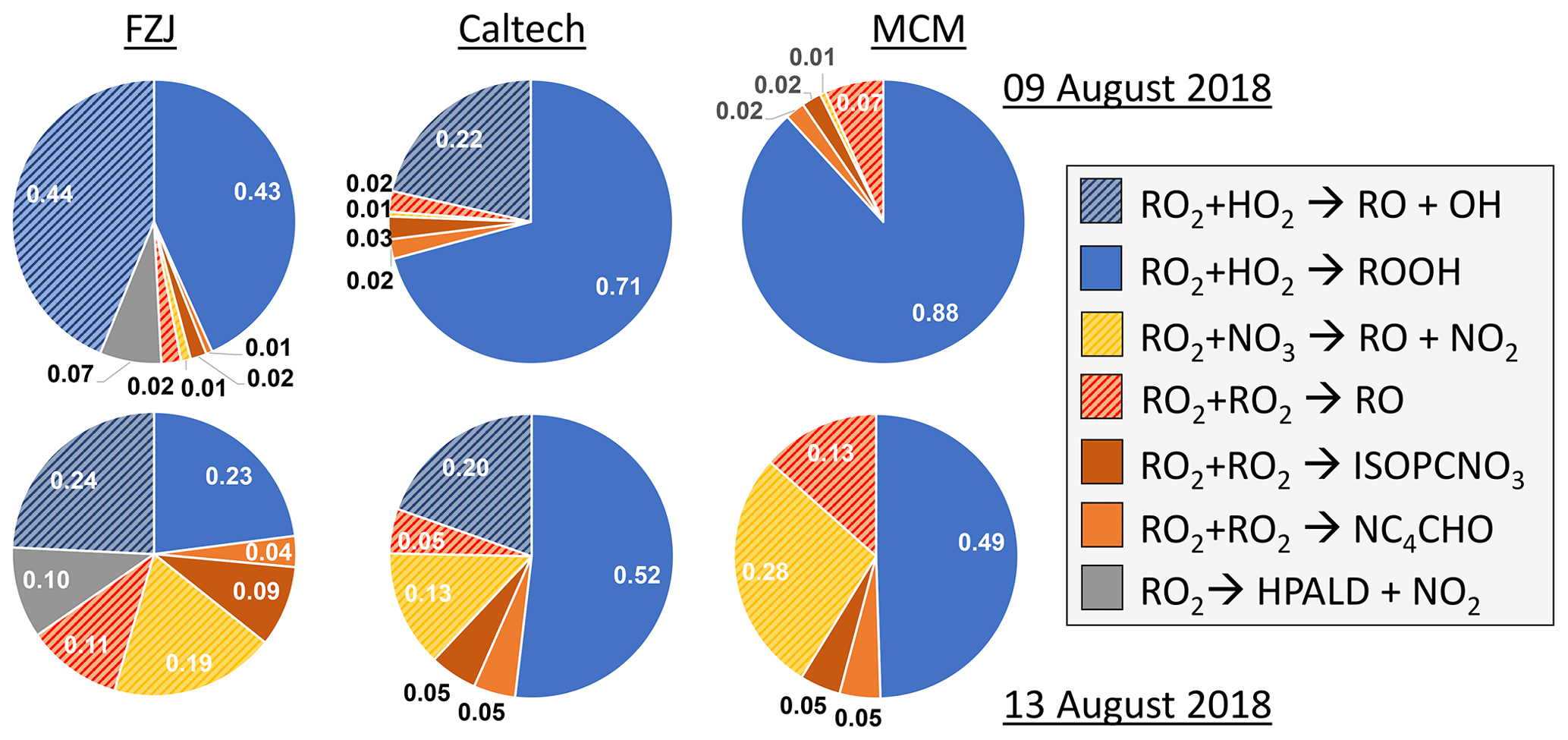
Figure 7Relative distribution of loss rates of nitrate RO2 for the experiment on 9 August 2018 (Experiment 1), when HO2 concentrations were enhanced, and for the experiment on 13 August 2018 (Experiment 4), when the amount of oxidized isoprene was highest. The total RO2 loss rate was 0.035 and 0.014 s−1 in the experiments on 9 August 2018 (Experiment 1) and 13 August 2018 (Experiment 4), respectively. Calculations of the loss rates of RO2 radicals in bimolecular reactions make use of measured HO2 and NO3 concentrations. Total RO2 concentrations and concentrations of speciated nitrate RO2 were derived from model calculations applying the FZJ-NO3 mechanism, Caltech mechanism or MCM. The chemical mechanisms differ with respect to the number of nitrate RO2 isomers that are considered, the type of RO2 loss reactions and products of loss reactions (Figs. 3 and 6). Reactions leading to nitrate alkoxy radicals are indicated by a striped pattern.
If HO2 concentrations are used as derived from model calculations, the total RO2 loss rates are lower by 30 % to 50 % compared to what is shown here due to the low predicted HO2 concentrations (Vereecken et al., 2021). The contribution of the different RO2 loss channels shifts towards higher contributions from RO2 reactions with other RO2 radicals and with NO3 (Fig. A4). In addition, unimolecular reactions further gain in importance due to the longer chemical lifetime of RO2 radicals.
5.2 Production of nitrate alkoxy radicals
Alkoxy radicals play an important role in determining the differences in the concentrations of organic products, obtained by model calculations applying the three mechanisms (Figs. 4, 5, A5 and A6). These differences are not only due to differences in the fate of alkoxy radicals but also due to differences in the formation rates of alkoxy radicals which are formed from nitrate RO2 radicals reacting with NO3, RO2 and HO2 radicals.
In all three mechanisms, the initial product from the reaction between nitrate RO2 and NO3 is a nitrate alkoxy radical and NO2 (Fig. 6, Reaction R3). Dewald et al. (2020) analysed NO3 reactivity measurements performed in the same experiments and concluded that the reaction rate constant of the reaction of nitrate RO2 with NO3 would need to be around cm3 s−1, which is nearly a factor of 2 higher than the generic reaction RO2 + NO3 rate constant based on the measured rate constant for CH3O2 + NO3 used in the MCM and the Caltech mechanism. With this rate constant, the loss rate of nitrate RO2 in the reaction with NO3 is between 0.001 and 0.003 s−1 in the experiments on 10, 12 and 13 August 2018 (Experiments 2, 3, 4), contributing between 5 % and 20 % of the total nitrate RO2 loss rate if the FZJ-NO3 mechanism is applied (Figs. 7 and A3).
Rate constants of RO2 + RO2 reactions for nitrate RO2 in the Caltech mechanism were derived from the measurement of isomer-specific product distributions in the experiments of Schwantes et al. (2015). From their findings, a reaction rate constant of cm3 s−1 for the self- and cross-reaction of the most abundant nitrate β-1,2-RO2 radical was found. As this rate refers to a tertiary radical instead of a primary one, it is significantly slower than the rate constant used in the MCM of cm3 s−1. Rate constants for other nitrate RO2 were estimated in the Caltech mechanism to be in the range of 10−12 to 10−13 cm3 s−1. In the FZJ-NO3 mechanism, all the rates for the nitrate RO2 self- and cross-reactions were calculated from a structure–activity relationship (Jenkin et al., 2019), resulting in an even lower rate constant for the self- and cross-reaction of the tertiary β-1,2-RO2 of only cm3 s−1 and for the cross-reactions of this radical with other primary nitrate RO2 of 2 to cm3 s−1. The rates of the reactions within the pool of the other nitrate RO2 are on the same order of magnitude as the values in the Caltech mechanism.
Only RO2 concentrations derived from model calculations can be used to estimate the loss rate of nitrate RO2 in RO2 + RO2 reactions (i.e. the alkoxy radical production rate) because the instrument detecting RO2 could only measure a lower limit of concentrations (Vereecken et al., 2021). This gives average RO2 loss rates of between 0.0005 and 0.002 s−1. The contribution to the total loss rate is less than 10 % in the experiments on 9, 10 and 12 August 2018 (Experiments 1, 2, 3, 4) but increased to up to 20 % in the experiment on 13 August 2018 (Experiment 4), when the production rate of nitrate RO2 was also highest (Fig. 7).
A yield of 60 % for the formation of alkoxy radicals (Fig. 6, Reaction R4) is generally applied for RO2 + RO2 radical reactions for primary and secondary RO2 (Jenkin et al., 2019). In the case of the most abundant nitrato-organic peroxy radical (tertiary β-1,2-RO2) from the reaction of isoprene with NO3, however, the yield is nearly 100 % for its self-reaction and 80 % if this nitrate RO2 reacts with other RO2 because the formation of a nitrate carbonyl product (NC4CHO) is not possible (Fig. 6, Reaction R5). Formation of peroxides (ROOR) is considered in the Caltech and FZJ-NO3 mechanisms with a small yield of 3.5 %. The MCM does not distinguish between nitrate RO2 isomers. Therefore, this increase in the yield of alkoxy radicals is only implemented in the Caltech and FZJ-NO3 mechanisms. With respect to the total yield of alkoxy radicals, the high yield for β-RO2 is partly compensated for by the lower rate constants of RO2 + RO2 radical reactions in the FZJ-NO3 and Caltech mechanisms than that applied in the MCM.
As discussed in Schwantes et al. (2015), reactions of nitrate β-RO2 and HO2 can also result in the formation of nitrate alkoxy radicals together with an OH radical (Fig. 6, Reaction R2). A yield of 50 % is assumed in the Caltech and FZJ-NO3 mechanisms (Sect. 5.3).
Overall, the total yield of alkoxy radicals produced in the reactions of nitrate RO2 differs significantly between the three mechanisms. In the FZJ-NO3 mechanism, the total yield is around 50 %. The value is similar in all experiments analysed in this work, but the type of reactions producing the alkoxy radicals shifts depending on the availability of reaction partners (Fig. 7). Alkoxy radicals yields are between 25 % and 40 % lower in the Caltech mechanism than in the FZJ-NO3 mechanism. The value is mainly due to the shift in the RO2 isomer distribution towards δ-RO2 isomers. The lowest total yields of alkoxy radicals between 7 % and 40 % are obtained if the MCM is applied because the MCM does not include alkoxy radical production from the reaction of nitrate RO2 with HO2.
5.3 Fate of nitrate alkoxy radicals
The fate of the alkoxy radicals is very different between the three mechanisms, which impacts the distribution of organic products. In the MCM, the only pathway for nitrate alkoxy radicals produced from isoprene is their decomposition forming a nitrate carbonyl (NC4CHO) together with an HO2 radical (Fig. 6, Reaction R7). This pathway is not possible for the alkoxy radical from the β-RO2 radicals, which are absent in the MCM but included in the FZJ-NO3 and Caltech mechanisms. Therefore, the overall yield of nitrate carbonyls (NC4CHO) from the subsequent chemistry of nitrate alkoxy radicals is highest if the MCM is applied in comparison to the results from the other two mechanisms.
In the Caltech mechanism, alkoxy radicals from β-RO2 radicals decompose exclusively to MVK or MACR together with a formaldehyde and an NO2 molecule (Fig. 6, Reaction R8; Wennberg et al., 2018). Therefore, nitrate carbonyl concentrations predicted by the Caltech model are at least a factor of 4 lower than calculated when applying the MCM. Small concentrations of nitrate carbonyls are also produced from reactions of nitrate δ-RO2 radicals.
Vereecken et al. (2021) calculated that ring-closure reactions leading to the formation of nitrate epoxy alkyl radicals are much faster than the decomposition reaction for the nitrate β-RO alkoxy isomer (Fig. 6; Reactions R9 and R17), so MVK and MACR production from this reaction is suppressed. Products from the epoxide pathway are discussed in Sect. 5.4. Differences between NC4CHO concentrations predicted by the FZJ-NO3 and Caltech mechanism are due to differences in the initial distribution of nitrate RO2 isomers. The FZJ-NO3 mechanism favours the β-1,2-RO2 radicals (Sect. 3) that do not produce NC4CHO and overall react more slowly with other RO2 than with the other nitrate RO2 radicals.
The Vocus PTR-MS instrument detected ion signals at the expected mass of NC4CHO with the sum formula C5H7NO4 in all experiments. Due to the lack of calibration, this measurement cannot be used to test the validity of any of the three chemical mechanisms. However, NC4CHO concentrations would be roughly consistent with predictions by the Caltech and FZJ-NO3 mechanisms if a sensitivity similar to that for ketones without nitrate functional groups (acetone, MVK, pentanone, nopinone) were assumed.
MVK and MACR are formed in all three mechanisms from the oxidation of isoprene by OH and ozone. Yields from the ozonolysis of isoprene are 0.17 and 0.41 for MVK and MACR, respectively (Nguyen et al., 2016). In the absence of NO as in typical night-time conditions, MVK and MACR are produced from the reaction of OH-derived RO2 radicals with other RO2 or HO2 radicals. The overall yield of MVK from the OH oxidation of isoprene in experiments in this work depends on the fate of RO2 radicals, but it is expected to be small due to the slow RO2 + RO2 reaction rate and small yields in the range of a few percent from the RO2 + HO2 reaction (Wennberg et al., 2018). In addition to the production from OH and O3 reactions, the Caltech mechanism includes a strong source for MVK through the decomposition of nitrate β-1,2-RO2 radicals produced from the NO3 oxidation.
In all experiments analysed in this work, measured MVK and MACR concentrations are consistent with predictions by the MCM and FZJ-NO3 mechanism (Figs. 4 and 5). In contrast, predictions by the Caltech mechanism are up to a factor of 2 to 4 higher than measured values. Discrepancies are highest in experiments in which a high fraction of the nitrate alkoxy radicals are formed from the reaction of nitrate RO2 with NO3 with an alkoxy radical yield of 1 (13 August 2018, Fig. 5) and are lowest in the experiment in which nitrate RO2 mainly reacted with HO2 (9 August 2018, Fig. 4). The good model–measurement agreement for MVK + MACR concentrations obtained using the FZJ-NO3 mechanism and MCM confirms that the decomposition of the nitrate alkoxy radicals is negligible as predicted by Vereecken et al. (2021) and unlike what is predicted by the Caltech mechanism.
5.4 Epoxide products from ring-closure reactions of nitrate alkoxy radicals
Epoxide formation from ring-closure reactions of nitrate alkoxy radicals leading to epoxy-RO2 radicals is implemented only in the FZJ-NO3 mechanism (Fig. 6, Reactions R9 and R17; Vereecken et al., 2021).
Nitrate epoxides can be formed from bimolecular reactions of epoxy-RO2 radicals with RO2 and HO2 (Fig. 6 – Reactions R12, R16, R19 and R26) and from nitrate epoxy alkoxy radicals produced by the reaction of epoxy-RO2 radicals with NO3 (Fig. 6, Reactions R13 and R21). One of the epoxy-RO radicals exclusively undergoes a 1,5-H-shift reaction for conditions of the experiments and decomposes into an epoxide and NO2 (Fig. 6, Reaction R14). Another epoxy-RO radical can decompose into a C5 nitrate epoxide, releasing HO2 (Fig. 6, Reaction R22). This reaction competes with a 1,5-H-shift reaction, in which a C4 nitrate and an HO2 radical and formaldehyde (HCHO) are formed (Fig. 6, Reaction R23).
Epoxy RO2 can also undergo unimolecular reactions (Vereecken et al., 2021) that compete with bimolecular reactions. The fastest unimolecular reaction is a 1,6-H-shift reaction with a rate constant of s−1 at room temperature, leading to a C5 epoxy product (C5H8O4) together with NO2 (Fig. 6, Reaction R11). This loss rate is lower than the loss rate due to bimolecular reactions, which are on the order of 10−2 s−1 for conditions of the experiments in this work but are high enough for low concentrations of this epoxide product to be formed (Fig. 5f).
The mass spectrometer instruments cannot distinguish between hydroxy nitrate epoxides formed from the reaction of epoxy-RO2 radicals with other RO2 radicals and nitrate hydroperoxide (NISOPOOH) species because they have the same sum formula, C5H9NO5. The concentration of epoxide C5H9NO5 species is expected to be at most 30 % to 40 % of the concentration of NISOPOOH in the experiment on 13 August 2018 (Fig. 5), when RO2 concentrations were highest. Their concentration is expected to be less than 10 % of that of NISOPOOH in the experiment on 9 August 2018 (Fig. 4a), when RO2 reactions with HO2 dominated the overall RO2 loss. Therefore, ion mass signals corresponding to C5H9NO5 species cannot be used to estimate the importance of the epoxidation reaction pathways.
Bimolecular reactions of epoxy RO2 can also lead to the formation of products with sum formulas that are specific to the epoxidation chemistry. Different isomers of nitrate carbonyls with the sum formula C5H7NO5 are produced from reactions of epoxy RO2 with other RO2 radicals or with NO3 (Fig. 6, Reactions R19 and R22). In addition, C5H9NO6 compounds are formed from reactions of nitrate epoxy RO2 with HO2 (Fig. 6, Reactions R12 and R26). Mixing ratios of these epoxides are predicted to be highest with mixing ratios of 1 ppbv in the experiment on 13 August 2018 (Experiment 4), when the total isoprene consumption by NO3 reactions was highest. Values are similar to mixing ratios of other products obtained in this experiment (Fig. 5g).
The mass spectrum measured by the Vocus PTR-MS instrument shows clear signals at the masses of epoxy nitrate compounds. The count rates are much lower than signals of other products, although expected concentrations are in the same range. This could be due to a lower sensitivity of the instrument to nitrate epoxides than to other organic nitrates. However, this could also indicate a lower-than-assumed production rate of alkoxy radicals, for example from the reaction of nitrate RO2 with HO2 (Sect. 5.5).
A C4 nitrate with the sum formula C4H5NO4 produced subsequent to the 1,5-H reaction of the nitrate alkoxy radical (Fig. 6, Reactions R23–R25) was not detected by the Vocus PTR-MS instrument in the experiments in this work, though significant mixing ratios of up to 0.6 ppbv are calculated by the FZJ-NO3 mechanism in the experiment on 13 August 2018 (Experiment 4), when the amount of oxidized isoprene was highest (Fig. 5i). There is no obvious reason why the sensitivity of the instrument to this compound would be lower than to other compounds. Only the I− CIMS instrument detected a very small signal (fewer than 30 counts) at the corresponding mass, which is at least a factor of 100 smaller than ion signals of masses at other products shown in Fig. A10.
The formation of this compound competes with the decomposition of the epoxy alkoxy radical, leading to an epoxy-C5 compound with the sum formula C5H7NO5 that is observed in the mass spectrum of the Vocus PTR-MS instrument (Fig. 6, Reaction R22). The fact that the C4 nitrate is not observed in the mass spectrum could indicate that the 1,5-H reaction is not competitive or that the branching ratio of two epoxy alkyl radicals from the nitrate alkoxy radical disfavours the epoxy alkyl radical that eventually leads to the formation of the C4 nitrate (Fig. 6, Reaction R17). Rate constants of the epoxidation chemistry calculated in Vereecken et al. (2021) have an uncertainty of a factor of 2 to 4. Therefore, low rate constants that weaken the formation of the C4 nitrate are within the uncertainty of calculations.
The other epoxy compound without a nitrate functional group is produced from a 1,6-H-shift reaction of one of the nitrate epoxy-RO2 radicals (Fig. 6, Reaction R11). Due to the relatively low reaction rate constant, only small mixing ratios of maximum 0.15 ppbv of this compound with the sum formula C5H8O4 are modelled for the experiment on 13 August 2018 (Experiment 4) (Fig. 5f). Nevertheless, a corresponding signal is observed in the mass spectrum of the Vocus PTR-MS instrument.
5.5 Reaction of nitrate RO2 with HO2
The chemical loss rate of nitrate RO2 towards reaction with HO2 was 0.032 s−1 (90 % of the total loss rate) in the experiment with high HO2 concentrations (9 August 2018, Experiment 1). The contribution to the total loss rate was 40 % to 50 % with loss rates between 0.002 and 0.007 s−1 in the other experiments (Fig. 6). In general, this reaction can proceed via two reaction pathways (Rollins et al., 2009; Kwan et al., 2012; Schwantes et al., 2015):
Nitrate hydroperoxide (NISOPOOH) is the only product in the MCM (Reaction R1) and a major product in the Caltech and FZJ-NO3 chemical mechanisms (Fig. 6, Reaction R1). The Caltech and FZJ-NO3 mechanisms assume that the yield of nitrate alkoxy radicals is approximately 0.5 if nitrate β-RO2 radicals react with HO2 (Reaction R2). The fate of nitrate alkoxy radicals is discussed above (Sect. 5.3). Predictions of NISOPOOH concentrations by the three mechanisms differ significantly. NISOPOOH concentrations predicted by the FZJ-NO3 mechanism are approximately half of the concentration calculated by the MCM, and concentrations predicted by the Caltech mechanism are between both values. This is mainly due to the different distribution of nitrate β- and δ-RO2 radicals in the FZJ-NO3 and Caltech mechanisms.
The Vocus PTR-MS instrument was not calibrated for NISOPOOH, so its concentrations could not be determined. The high count rate observed by this instrument and the two other CIMS instruments and the uncertainty in the branching ratio of Reactions (R1) and (R2) appear to support a high yield of NISOPOOH from the reaction of HO2 with nitrate RO2.
Alkoxy radical formation from the reaction of nitrate RO2 with HO2 is accompanied by the formation of OH (Fig. 6 and Reaction R2), which can be responsible for the formation of products that are specific to the OH oxidation of isoprene as observed in experiments designed to investigate the NO3 oxidation mechanism of isoprene.
OH concentrations were measured in the experiments in this work, but concentrations were around the limit of detection of the instrument (a few hundred thousands per cubic centimetre) in most experiments. Model calculations for the experiment on 13 August 2018 (Experiment 4), when reactant concentrations were highest, result in significant OH concentrations between 5×105 and 8×105 cm−3, and also model results indicate that OH concentrations could have been in the range of a few hundred thousands per cubic centimetre (Fig. 5j). A large fraction of OH, however, is produced by the reaction of HO2 with NO3, both of which are constrained to measured values in the model calculations. As discussed in Vereecken et al. (2021), model calculations without constraining HO2 to measured values cannot reproduce measured HO2 concentration, suggesting shortcomings of the model in describing HO2 source and/or sink reactions.
This is further analysed by comparing results of model runs, in which either HO2 concentrations are constrained to measurements or HO2 is calculated by the model (Fig. 8). In the unconstrained case, modelled HO2 concentrations are much lower than measurements. This reduces the OH concentration by a factor of 3 due to the lower production of OH from the reaction of HO2 with NO3. During the part of the experiment when isoprene is oxidized by NO3, differences between measured and modelled OH concentrations tend to be smaller if HO2 is not constrained to measured values. At later times of the experiment after 13:30 UTC, when isoprene had been consumed and NO3 concentrations were enhanced by additional injections of NO2 and O3 (Fig. 2b), measurements showed a steeper increase in OH concentrations than model calculations with unconstrained HO2. This further indicates that modelled HO2 concentrations might be too low.
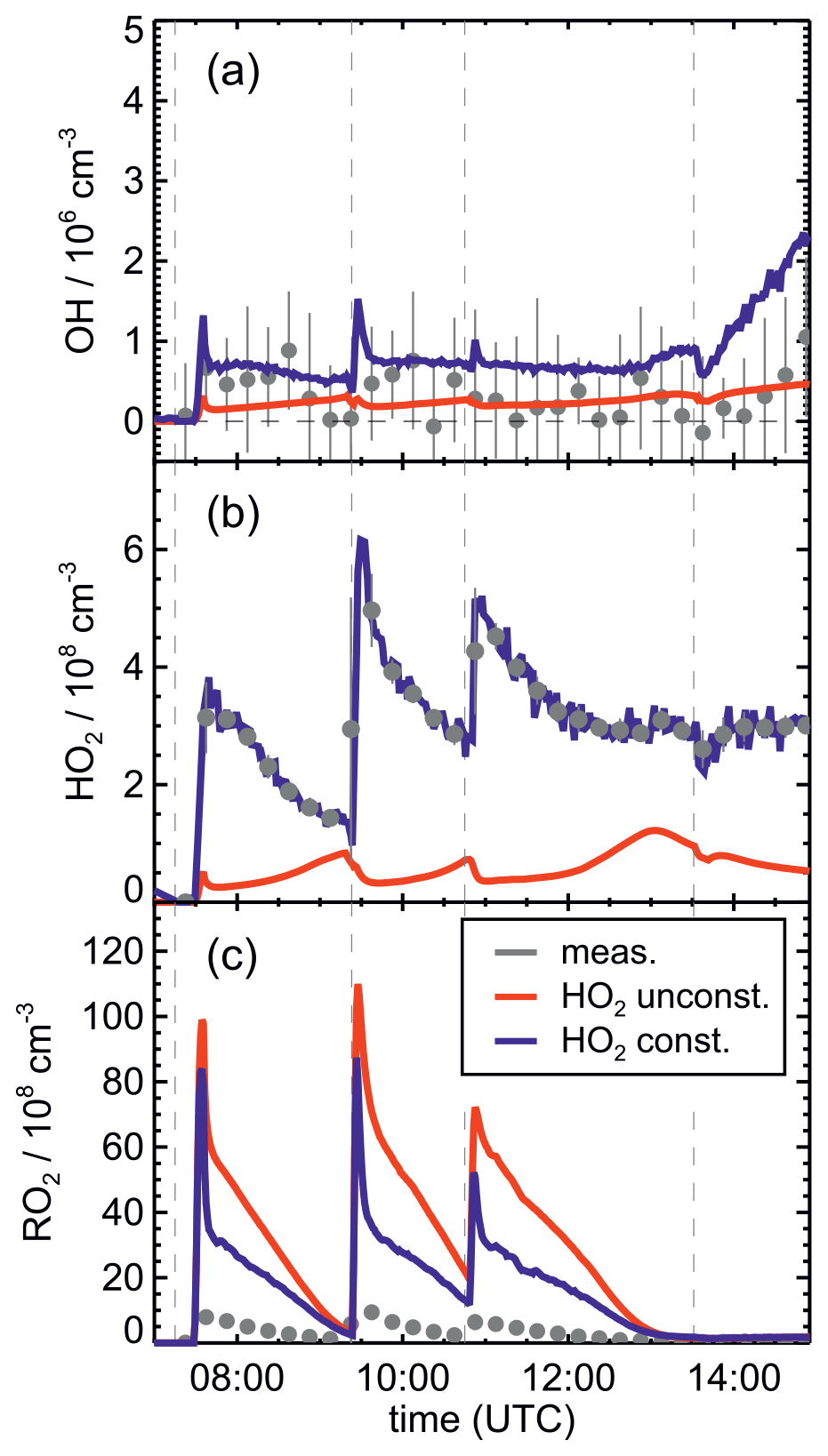
Figure 8Comparison of results from model calculations applying the FZJ-NO3 mechanism for the experiment on 13 August 2018 (Experiment 4) with HO2 concentrations being either constrained (const.) or unconstrained (unconst.) to measurements. A large fraction of OH is produced from the reaction of HO2 with NO3, so lower-than-measured HO2 concentrations in the unconstrained model run lead to low OH concentrations. Because HO2 + RO2 reactions contribute significantly to the total loss of RO2, modelled RO2 concentrations are higher in the unconstrained model run. RO2 measurements by the LIF instrument do not include all RO2 radicals (Vereecken et al., 2021), so measured concentrations are lower than modelled values.
If the yield of alkoxy radicals and therefore also of OH from the reaction of nitrate RO2 with HO2 were lower than 50 % as assumed in the Caltech and FZJ-NO3 mechanisms, modelled OH concentrations would be even lower. Sensitivity model runs show that modelled OH concentrations would only decrease by 1×105 to 3×105 cm−3 directly after the isoprene injections, when nitrate RO2 concentrations are also highest. However, such differences are in the range of the accuracy of measurements, which was a few hundred thousands per cubic centimetre due to the subtraction of an OH background signal that was determined by using a chemical modulation system (Cho et al., 2021).
Overall, considering the uncertainties in the measured OH concentrations and in the modelled OH due to the uncertainty in the OH production from the HO2 + NO3 reaction, differences between model results and measured values are too small to draw conclusions about the yield of alkoxy radicals from model–measurement comparison of OH concentrations.
5.6 Production of hydroperoxy aldehyde (HPALD) species from nitrate RO2 isomerization reactions
Only the FZJ-NO3 mechanism includes unimolecular loss reactions of nitrate RO2 (Fig. 3). The reaction rate constants of the 1,6-H shift reactions of the Z-δ-RO2 isomers have a strong temperature dependence (Vereecken et al., 2021). Values range between 0.016 and 0.023 s−1 for the Z-δ-1,4-RO2 isomer and 0.045 and 0.06 s−1 for the Z-δ-4,1-RO2 isomer for temperatures experienced in the experiments in this work.
The Z-δ-RO2 isomer fraction of the total RO2 concentration is only between 5 % and 6 %, and the Z-δ-4,1-RO2 isomer fraction is between 1 % and 2 %. The overall bulk RO2 isomerization rate is around 0.002 s−1, making the 1,6-H-shift reaction competitive with bimolecular reactions in all experiments except for the one with high HO2 concentrations (9 August 2018, Experiment 1). The contribution of unimolecular reactions to the overall loss rate is expected to be between 10 % and 30 % depending on the total RO2 loss rate (Fig. 7). This is similar to or even higher than the case for analogous, much faster 1,6-H-shift reactions in the OH-initiated isoprene oxidation (k(298 K)≈0.5 s−1; Peeters et al., 2014) due to significantly longer RO2 lifetimes during the night than during the day.
HPALD concentrations predicted by the model applying the FZJ-NO3 mechanism are between 0.1 and 1.2 ppbv, depending on the chemical conditions with different availability of reaction partners for competing bimolecular reactions. HPALD mixing ratios are calculated to be highest in the experiment on 13 August 2018 (Experiment 4), when the total concentration of oxidized isoprene was high. Approximately 10 % to 15 % of the HPALD that is predicted by the FZJ-NO3 mechanism is due to OH oxidation of isoprene also producing HPALD from 1,6-H-shift reactions. The modelled HPALD concentration from the OH reaction might be lower, however, due to the uncertainty in the modelled OH concentration (Sect. 5.5). It is worth noting that the fast 1,6-H-shift reaction rate of Z-δ-RO2 isomers from the OH oxidation of isoprene (bulk loss rate ≈ 0.006 s−1) makes these reactions very competitive with bimolecular reactions for night-time conditions (loss rate in the experiments in this work of 0.005 to 0.014 s−1, Sect. 5.1).
Although the absolute importance of HPALD formation from H-shift reactions of nitrate RO2 radicals is uncertain, HPALD is clearly formed from the oxidation of isoprene by NO3. This is demonstrated by the observation of a signal at the mass of HPALD in the experiment on 9 August 2018 (Experiment 1), when an OH scavenger was present, so HPALD could not be produced by OH reactions. In this experiment, HO2 + RO2 reactions were favoured, so formation of the epoxides with the same mass is also expected to be small (Fig. 4c). Therefore, the signal on the mass of HPALD can be attributed to HPALD formation from the oxidation of isoprene by NO3 in this experiment.
The relative importance of HPALD formation is expected to be highest for conditions of the experiment on 10 August 2018 (Experiment 2), when the total loss rate of RO2 due to bimolecular reaction is between 0.005 and 0.006 s−1. In this case, approximately 25 % to 30 % of the isoprene consumed by NO3 would form HPALD. Brownwood et al. (2021) calculated the yield of total organic nitrates from measurements for the same experiments analysed in this work and found a yield of (94 ± 20) % for this experiment. Values ranged between (112 ± 13) % and (140 ± 24) % in the other experiments. The lowest yield of organic nitrates is obtained in the experiment with the longest RO2 lifetime (10 August 2018, Experiment 2), supporting the finding that more non-nitrate organic products such as HPALD are formed in this experiment than in the other experiments. The signal of the Vocus PTR-MS instrument, however, does not clearly scale with the expected differences in the HPALD concentrations predicted for the experiments in this work. This and the overall high yields of organic nitrates indicate that the impact of unimolecular reactions producing HPALD might be overestimated in the FZJ-NO3 mechanism. Uncertainties in the quantum chemical calculations, from which reaction rates are taken, are a factor of 2 to 3.
Overall, experiments in this work and previous chamber experiments demonstrate that HPALD formation from 1,6-H shift reactions of Z-δRO2 isomers play a role in atmospheric night-time conditions.
5.7 Night-time loss rate of organic nitrate products and hydroperoxy aldehyde (HPALD) species
Chamber experiments in this work were designed to also investigate further oxidation of the organic products. This was achieved by re-injecting O3 and NO2 to enhance NO3 production after most of the isoprene had reacted away (Figs. 2 and 1a). Highest product concentrations were achieved in the experiment on 13 August 2018 (Experiment 4), when the amount of isoprene that was oxidized was highest. Therefore, the further discussion concentrates on this experiment (Fig. 5).
Reaction rate constants of nitrate products from the oxidation of isoprene with OH and O3 implemented in the Caltech mechanism are based on laboratory experiments with synthetic standards of isoprene hydroxy nitrate isomers (Wennberg et al., 2018; L. Lee et al., 2014). Values are assumed to be applicable to other organic nitrates such as nitrate carbonyls and nitrate hydroperoxides. Only part of the loss reactions listed in Wennberg et al. (2018) are implemented in the code of the Caltech mechanism (Bates and Wennberg, 2017) that is applied in model calculations in this work.
Rate constants for the reaction of the first-generation organic nitrates with ozone (Reactions R28, R31, R34 and R37) are in the range of 10−17 to 10−19 cm3 s−1 in L. Lee et al. (2014), with rates being relevant for only δ-nitrate alcohols and δ-hydroperoxides for typical oxidant concentrations during the night and too slow for β species. As only δ species are implemented in the MCM, the overall relevance of ozonolysis loss reactions is overestimated under atmospheric conditions in the MCM (Table 2).
Table 2Reaction rate constants for the reaction of first-generation major organic products from the reaction of isoprene with NO3 with OH, O3 and NO3 implemented in the MCM, Caltech mechanism and FZJ-NO3 mechanism. For simplicity, rate constants are given for a temperature of T=298 K and only for the organic nitrate that is produced from the most abundant β-1,2-RO2 radical, except for the MCM, where the δ-1,4-RO2 is solely present. For the nitrate carbonyl (NC4CHO), which cannot be produced from this RO2 isomer, the value for the E-δ-1,4-RO2 isomer is given instead. In the FZJ-NO3 mechanism, loss rates due to reactions that lead to loss rates much lower than the dilution rate of the chamber were set to upper-limit values that equal the loss rate due to dilution. Chemical lifetimes (τ) are calculated for the presence of 1×106 cm−3 OH, 100 ppbv O3 and 50 pptv NO3, which can be regarded as upper-limit concentrations for typical night-time conditions. The code of the Caltech mechanism (Bates and Wennberg, 2017) includes fewer loss reactions implemented as described in Wennberg et al. (2018). Chemical loss of nitrate epoxides is not implemented in the chemical mechanisms.
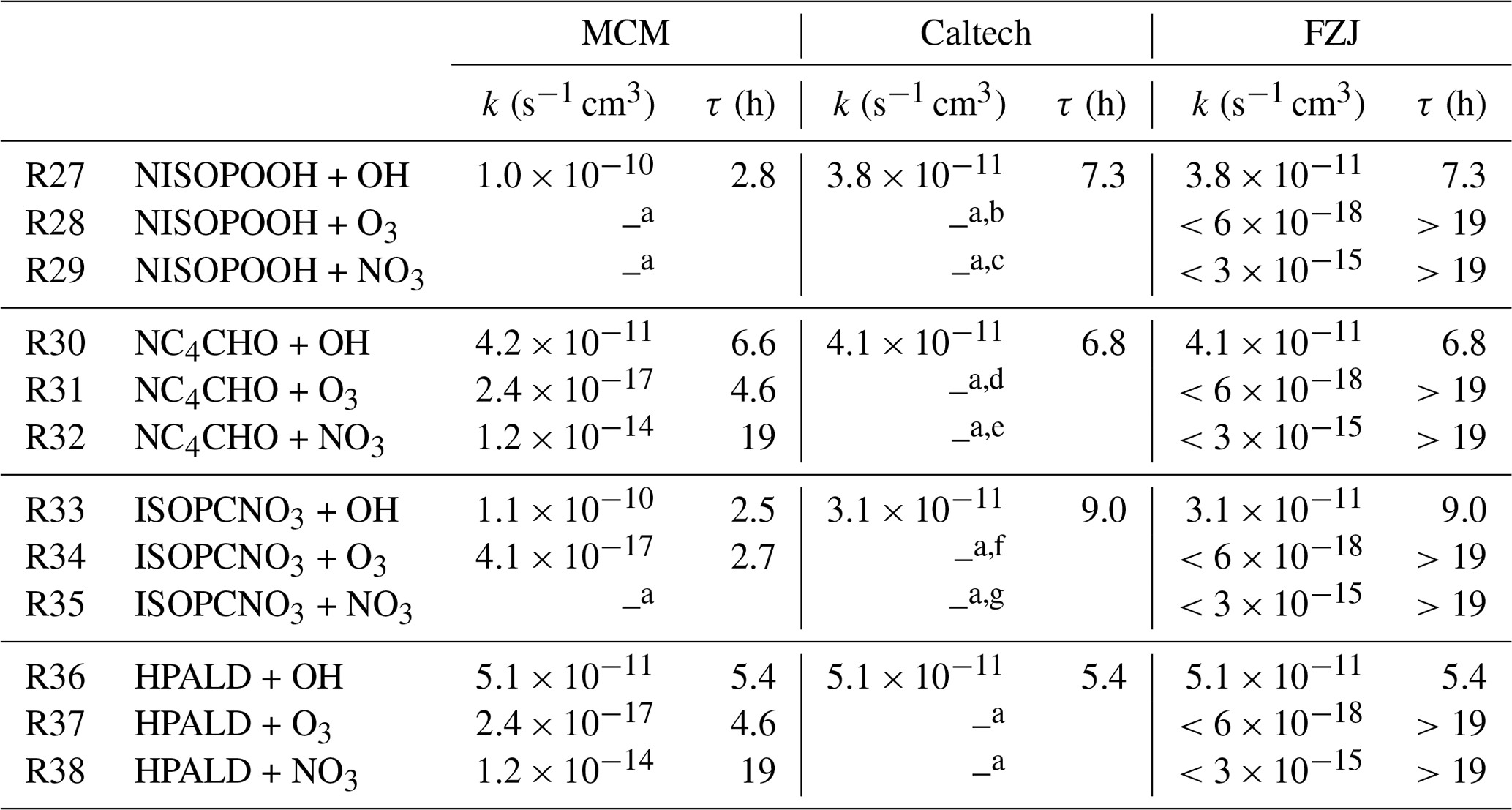
a Not implemented; b s−1 cm3, Wennberg et al. (2018); c s−1 cm3, Wennberg et al. (2018); d s−1 cm3, Wennberg et al. (2018); e s−1 cm3, Wennberg et al. (2018); f s−1 cm3, Wennberg et al. (2018); g s−1 cm3, Wennberg et al. (2018).
In the FZJ-NO3 mechanism, reaction rate constants of organic nitrates with OH radicals (Reactions R27, R30, R33 and R36) are taken from the Caltech mechanism, but rate constants with ozone and NO3 are optimized to best describe the temporal behaviour of the signals observed by the Vocus PTR-MS instrument at the respective mass (Table 2). Reaction rate constants of loss reactions that lead to loss rates much lower than the dilution rate of the chamber are set to upper-limit values that equal the loss rate due to dilution ( s−1). Reaction rate constants are likely even lower because doubling the loss rate from dilution would already worsen the model–measurement agreement of the temporal behaviour of products.
Chemical loss of NISOPOOH by reactions with NO3 (Reaction R29) and O3 (Reaction R28) is expected not to be relevant for atmospheric conditions in all mechanisms. This is consistent with the slow decay of the total signal for C5H9NO5 observed by the Vocus PTR-MS instrument in the experiment on 9 August 2018 (Experiment 1), when OH oxidation was suppressed by the presence of an OH scavenger (Fig. 4a). In this case, the loss rate is consistent with the dilution rate in the experiment.
In the MCM, the rate of the reaction of OH with hydroperoxide species, NISOPOOH, is assumed to be fast with a rate coefficient of 10−10 s−1 cm3. In contrast, the Caltech and FZJ-NO3 mechanisms assume a smaller rate coefficient for this reaction, by a factor of 3, which can account for the faster decay of NISOPOOH in the MCM than in the Caltech and FZJ-NO3 mechanisms.
In the MCM, products of the NISOPOOH + OH reaction (Reaction R27) are a nitrate alkoxy radical together with an OH radical, leading to a zero net loss of OH. In addition, the alkoxy radical produces a nitrate carbonyl (NC4CHO) together with an HO2 (Sect. 5.3). In contrast, in the Caltech and FZJ-NO3 mechanisms, a large fraction of the predicted products are epoxide products (yield: 0.37 to 1.0 depending on the precursor RO2 isomer; Schwantes et al., 2015) together with OH analogous to the formation of epoxides in the OH oxidation of isoprene (Paulot et al., 2009).
Nitrate epoxides have the same sum formula as NISOPOOH (C5H9NO5), so the Vocus PTR-MS instrument cannot distinguish between both compounds. The reaction of OH radicals with nitrate epoxides is expected to be much slower than their reaction with NISOPOOH due to the lack of C=C double bonds. Therefore, the time series of the sum of both compounds is affected by their different temporal behaviour in the Caltech and FZJ-NO3 mechanisms. The loss rate of C5H9NO5 compounds in the MCM is only determined by the fast loss of NISOPOOH because no epoxides are formed.
For the experiment on 13 August 2018 (Experiment 4) (Fig. 5a), the temporal behaviour of the total ion signal corresponding to C5H9NO5 species observed by the Vocus PTR-MS instrument fits best the modelled trace of the FZJ-NO3 mechanism with the low OH reaction rate of NISOPOOH. In addition, the low chemical loss rate of epoxides contributes to the slow decay of the ion signal at that mass, improving the model–measurement agreement. This demonstrates that OH reaction rate constants measured in L. Lee et al. (2014) for nitrate alcohols can be applied to NISOPOOH as implemented in the Caltech and FZJ-NO3 mechanisms. In contrast, the fast OH reaction rate constant for NISOPOOH implemented in the MCM cannot describe the observations.
If the MCM is used, a significant fraction of nitrate carbonyls, NC4CHO, that are produced from nitrate RO2 + RO2 reactions and from the decomposition of specific nitrate alkoxy radicals is expected to be consumed on the timescale of the experiment for the experiment on 13 August 2018 (Experiment 4) (Fig. 5d). For conditions of this experiment, reactions of NC4CHO not only with OH (Reaction R30) but also with NO3 (Reaction R32) for high NO3 concentrations can be relevant if reaction rate constants of the MCM are applied (Table 2). The loss of NC4CHO calculated using the MCM is faster than calculated using the Caltech and FZJ-NO3 mechanisms because of the fast OH reaction rate constants. In addition, the MCM overestimates the loss of NC4CHO by the reaction with ozone as discussed above.
The temporal behaviour of the modelled NC4CHO concentrations is in good agreement with the corresponding signal observed by the Vocus PTR-MS instrument for the Caltech and FZJ-NO3 mechanisms. This confirms that only a small fraction of NC4CHO is expected to be chemically lost for typical night-time conditions.
In addition, a fast loss rate due to the reaction with NO3 (Reaction R32) as suggested in Wennberg et al. (2018) would lead to a chemical lifetime of NC4CHO of less than 30 min in the last phase of the experiment on 13 August 2018 (Experiment 4), when NO3 mixing ratios increased to several hundreds of parts per trillion by volume (Fig. 2b), but this is not observed (Fig. 5d). Though not fully applicable, the structure–activity relationship in Kerdouci et al. (2014) gives reaction rate constants lower than 10−16 s−1 cm3, supporting the finding of a low loss rate due to the addition of NO3. Overall, further oxidation of nitrate carbonyls from isoprene is of minor importance for typical night-time conditions as experienced in these experiments.
Similar differences between model predictions like for NC4CHO are seen for nitrate alcohols (ISOPCNO3, Reactions R33–R35): the MCM predicts a significantly faster chemical loss than the Caltech and FZJ-NO3 mechanisms. A large part of the discrepancy is explained by the fast loss due to the reaction with ozone implemented in the MCM that is not applicable as discussed above. In addition, the reaction rate constant of the reaction of ISOPCNO3 with OH (Reaction R33) is up to 3 times faster in the MCM than in the Caltech and FZJ-NO3 mechanisms (Table 2). The signal of the Vocus PTR-MS instrument at the mass of ISOPCNO3 also confirms the low reaction rate constants with OH determined experimentally in L. Lee et al. (2014).
HPALD formation from the reaction of isoprene with NO3 is only implemented in the FZJ-NO3 mechanism. Wolfe et al. (2012) investigated the photo-oxidation of a closely related compound of HPALD to constrain photolysis rates and reaction rate constants in the reaction with OH (Reaction R36) and O3 (Reaction R37). A fast OH reaction rate constant of s−1 cm3 was found. This value is implemented in the MCM, Caltech mechanism and FZJ-NO3 mechanism (Table 2). The reaction rate constant of HPALD with ozone was determined in Wolfe et al. (2012) to be s−1 cm3, making the ozone reaction irrelevant for typical atmospheric conditions. There are no experimental values for the reaction rate constant of HPALD with NO3. The structure–activity relationship (SAR) described in Kerdouci et al. (2014) cannot be applied because the effect of a COOH substituent in the β position of the C=C double bond where the NO3 radical addition takes place is not considered. Omitting this substituent results in a reaction rate constant similar to the value in the MCM, indicating that a COOH substituent further lowers the reaction rate constant.
In the MCM, a fast reaction rate constant of HPALD with ozone (Reaction R37) is implemented, which would lead to a short chemical lifetime of 4.6 h for conditions of the experiment in this work (100 ppbv O3). In addition, the MCM assumes that HPALD reacts with NO3 (Reaction R38) with a fast reaction rate constant of s−1 cm3, which would lead to a significant loss of HPALD in the last part of the experiment on 13 August 2018 (Experiment 4). This is inconsistent with the temporal behaviour of the signal observed by the Vocus PTR-MS instrument, which is explained by the loss of HPALD by only its reaction with OH (Fig. 5c). In the experiment on 9 August 2018 (Experiment 1), when OH reactions were suppressed by the presence of the OH scavenger, the temporal behaviour of the HPALD signal is fully consistent with the only loss being due to dilution (Fig. 4c).
The further oxidation of epoxides produced from ring-closure reactions of nitrate alkoxy radicals calculated in Vereecken et al. (2021) has not been investigated so far. The temporal behaviour of signals measured by the Vocus PTR-MS instrument suggests that their loss rate can be explained by only the dilution rate in the experiments, indicating that chemical loss was not significant even in the presence of several hundreds of parts per trillion by volume of NO3, several hundreds of parts per billion of O3 and presumably several hundred thousands per cubic centimetre of OH in the last period of the experiment on 13 August 2018 (Experiment 4) (Fig. 5j). An upper-limit value of the reaction rate constant of the reaction of epoxides with OH of s−1 cm3 (T=298 K) can be assumed to be similar to the value found for epoxides produced from the OH reaction of hydroperoxides derived from isoprene (Bates et al., 2014), making chemical loss a minor loss pathway for typical conditions during the night-time.
In the presence of an aerosol surface, epoxides could be lost by particle uptake, but this was not relevant in the experiments analysed in this work due to the absence of seed aerosol. Loss to the Teflon surface of the chamber was not significant as demonstrated by the consistency of the loss rate with the dilution rate in the experiments.
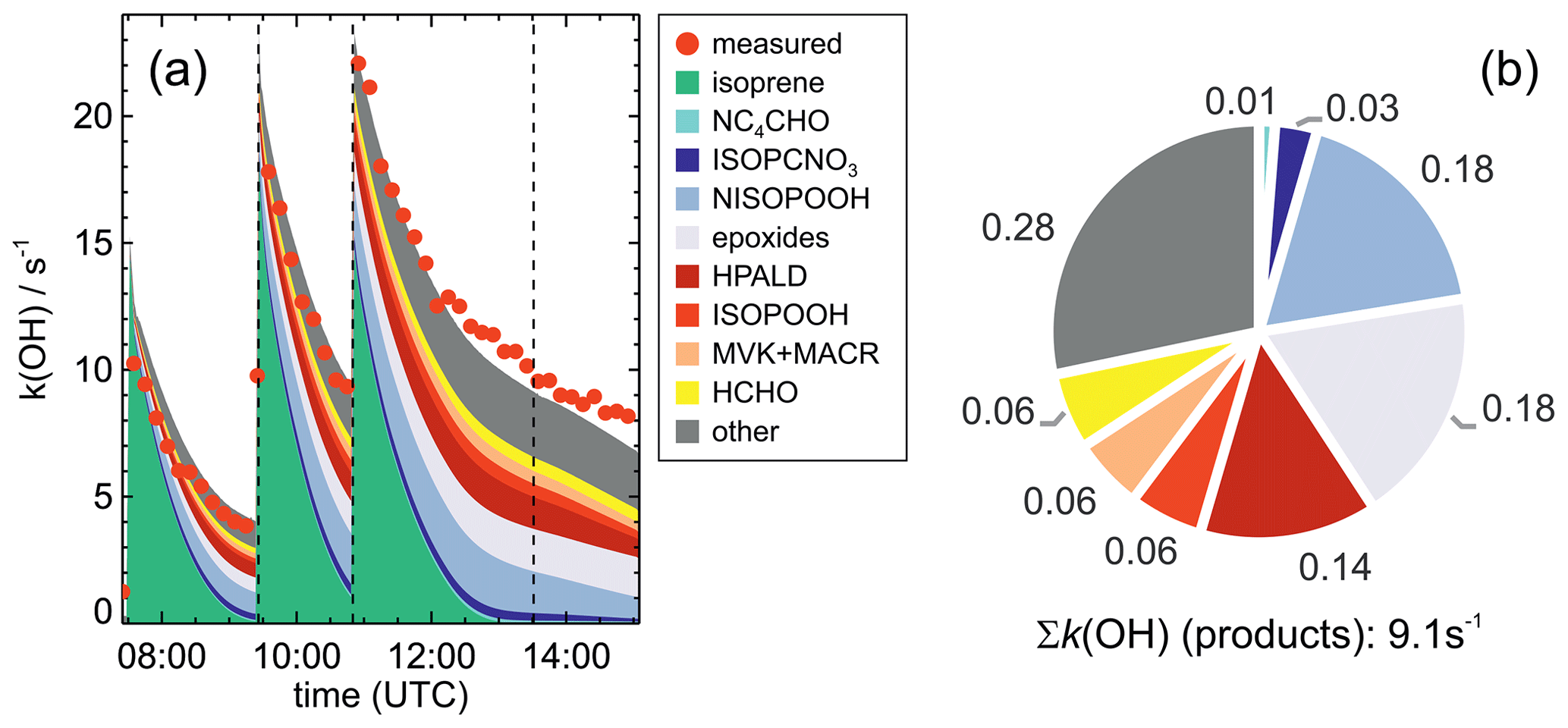
Figure 9Comparison of measured OH reactivity from organic compounds and OH reactivity (a) calculated from concentrations of organic compounds modelled applying the FZJ-NO3 chemical mechanism for the experiment on 13 August 2018 (Experiment 4). The reactivity from nitrated and non-nitrated hydroperoxide compounds (NISOPOOH, ISOPOOH) is partly invisible for the laser photolysis LIF instrument because these species produce OH radicals after reacting with it. The OH yield is rather uncertain but is expected to be less than 10 %, for example in the Caltech mechanism. In addition, the relative distribution of OH reactivity from organic products is shown (b). OH reactivity from organic compounds is derived by subtracting the reactivity via NO2 and O3 calculated using measured concentrations from the measured total OH reactivity. “Other” compounds include a high number of organic compounds that are produced in the reaction of isoprene with OH, O3 and NO3 and for which loss by the reaction with OH is implemented in the FZJ-NO3 mechanism. Dashed vertical lines indicate times when isoprene, NO2 and O3 were re-injected. The last injection included only NO2 and O3.
5.8 OH and NO3 reactivity from products
Overall, night-time oxidation of products from the reaction of isoprene with NO3 appears to be of minor importance. This is further supported by measurements of total OH and NO3 reactivity in the experiments in this work. In Fig. 9, measured OH reactivity from organic compounds (Sect. 2.1) is compared to values calculated from modelled concentrations of products for the experiment on 13 August 2013 (Experiment 4), when the total consumption of isoprene by NO3 was highest. Reaction rate constants for the reactions of organic compounds with OH are applied from the FZJ-NO3 mechanism.
OH reactivity is dominated by isoprene immediately after each injection (Fig. 9). After isoprene has reacted away, OH reactivity is only approximately 30 % of the initial reactivity, demonstrating the much lower reactivity from products than from isoprene. The major organic nitrate and epoxides produced from the reaction of NO3 with isoprene explain approximately 50 % of the total reactivity of organic products. Hydroperoxy aldehyde (HPALD) species, which are partly also produced from the OH oxidation of isoprene, contribute approximately 15 % to the OH reactivity from products. A similar contribution is obtained from compounds that are formed from the oxidation of isoprene by OH and O3, ISOPOOH, HCHO, MVK, and MACR. At the end of the experiment, 25 % of the total reactivity is due to a high number of organic compounds that are produced from minor reaction pathways or secondary oxidation.
The good agreement in the temporal behaviour of the observed and calculated OH reactivity is consistent with the low loss rate of products due to further oxidation reactions. In addition, measured OH reactivity values are consistent with OH reaction rate constants implemented in the FZJ-NO3 mechanism, so further OH oxidation of products is small for night-time conditions, when OH concentrations are typically a maximum of a few hundred thousands per cubic centimetre (Stone et al., 2012, 2014; Lu et al., 2014; Tan et al., 2017).
OH oxidation of nitrate hydroperoxides is the most important due to their fast reaction rate constant and their high concentrations for typical night-time conditions, when HO2 + RO2 reactions can dominate the loss of RO2. However, part of the reactivity from hydroperoxides is invisible for the OH reactivity instrument because OH is partly produced in their reactions with OH. Approximately 90 % of the reactivity is detected assuming an OH yield of 10 % as implemented in the Caltech and FZJ-NO3 mechanisms. In contrast, an OH yield of 100 % is assumed for NISOPOOH in the MCM, which is likely too high as formation of epoxide products is expected to be a major reaction pathway.
OH oxidation of HPALD produced from unimolecular reactions of nitrate RO2 can be significant because of the fast reaction of HPALD with OH.
In contrast, the absolute values of OH reactivity as well as its temporal behaviour calculated from model calculations using the MCM with high OH reaction rate constants and high yields of NISOPOOH and NC4CHO lead to results that are inconsistent with the observed OH reactivity (Fig. A12). This confirms that the MCM does not reproduce the product distribution and loss rates of products.
Dewald et al. (2020) discussed the NO3 reactivity measured in the experiments also investigated in this work. Consistent with conclusions above that the chemical loss of products by NO3 was not relevant, the authors found that the NO3 reactivity could be fully explained by the reactivity from isoprene and propene in these experiments. This confirms that loss of organic products from the reaction of isoprene with NO3 due to further NO3 oxidation is small compared to the dilution rate in the chamber experiments.
The high yield of MVK and MACR from the decomposition of β-RO radicals in the Caltech mechanism was derived from chamber experiments in Schwantes et al. (2015). In their experiments, 54 % to 74 % of the nitrate RO2 reacted with HO2, so the majority of alkoxy radicals were formed from this reaction. MVK and MACR concentrations, however, were only measured in two experiments in Schwantes et al. (2015), one of which was used to determine the MVK and MACR yields from the reaction of HO2 + RO2. The overall yield of the sum of MVK and MACR was relatively low, with a value of approximately 15 %. In order to determine the yield of MVK and MACR from the decomposition of alkoxy radicals from the RO2 + HO2 reactions, production from the isoprene oxidation by OH and O3 and from the potential decomposition of alkoxy radicals produced from other reaction channels needed to be subtracted. The authors used model calculations to estimate the actual OH concentration. Uncertainties in these calculations may explain the high MVK and MACR yield in Schwantes et al. (2015).
MVK and MACR concentrations were also measured in an experiment in the SAPHIR chamber reported by Rollins et al. (2009), in which low reactant concentrations were present as in this work (10 ppbv isoprene, 20 to 30 ppbv NO2, 40 to 60 ppbv O3). According to model calculations in Rollins et al. (2009), using MCM 3.2, the fate of nitrate RO2 radicals from isoprene with NO3 was dominated by their reactions with HO2. Measured MVK and MACR concentrations were consistent with the production of MVK and MACR mainly from the ozonolysis of isoprene. Therefore, this result supports the finding that MVK and MACR are not produced from the decomposition of alkoxy radicals from β-RO2 radicals. This is further supported by other experiments investigating the reaction of isoprene with NO3 at high reactant concentrations (Barnes et al., 1990; Kwok et al., 1996; Perring et al., 2009) and also by chamber experiments of Kwan et al. (2012).
Similarly to the experiments in this work, products that have the sum formulas of nitrate epoxide products expected to be formed in the FZJ-NO3 mechanism were observed in the experiments in Kwan et al. (2012) and Schwantes et al. (2015): (1) C5H9NO5 compounds, which appear at the same mass as NISOPOOH; (2) C5H7NO5 compounds from epoxy RO2 + RO2 reactions; and (3) C5H9NO6 compounds from epoxy RO2 + HO2 reactions.
In Kwan et al. (2012) and Schwantes et al. (2015), it is suggested that the product with the sum formula C5H9NO6 is a hydroxy hydroperoxy nitrate and that the product with the sum formula C5H7NO5 is a hydroxy carbonyl nitrate from a 1,5-H-shift reaction of δ-nitrate alkoxy radicals. Vereecken et al. (2021) calculated a reaction rate of 2.2×106 s−1 (T=298 K), which makes the 1,5-H-shift reaction too low to compete with the ring-closure reaction forming epoxy alkyl radicals (1.2×108 s−1, T=298 K) and subsequent O2 addition. It is worth noting that compounds suggested by Kwan et al. (2012) and Schwantes et al. (2015) would only be produced from nitrate δ-RO2 radicals that have small yields, whereas the nitrate epoxy products in the FZJ-NO3 mechanism are also produced from the most abundant nitrate β-RO2 radicals. This may also explain why compounds with these sum formulas were clearly detected in the experiments in all studies.
In the experiments in Kwan et al. (2012) and Schwantes et al. (2015), a C5H8O3 compound without a nitrate functional group was observed, which is consistent with observations in this work. Because HPALD appears at the same mass and HPALD is also produced from OH oxidation, the authors concluded that C5H8O3 is a product from the reaction of isoprene with OH. Nevertheless, their observations of C5H8O3 compounds could also be partly due to the production of epoxy species from the oxidation of isoprene by NO3 as described in the FZJ-NO3 mechanism.
The other product without a nitrate group that is produced from the ring-closure pathway of nitrate alkoxy radicals in the FZJ-NO3 mechanism, C5H8O4, was not observed in the experiments in Kwan et al. (2012) and Schwantes et al. (2015). The reason for this could be that the chemical lifetime of RO2 radicals was too short in the experiments in Kwan et al. (2012), in which high concentrations of reactants were present, so the 1,6-H-shift reaction of the epoxy-RO2 radical producing the C5H8O4 compound could not compete with bimolecular reactions. Similarly, RO2 reactions with HO2 were favoured in the experiments in Schwantes et al. (2015), so the 1,6-H reaction may have not been competitive.
Interestingly, similarly to the experiments in this work, no organic nitrate with the sum formula C4H5NO4 that is expected to be formed from the ring-closure reactions of nitrate alkoxy radicals (Fig. 6) was observed in the experiments in Kwan et al. (2012) and Schwantes et al. (2015). This further suggests that there is no significant production of this compound.
NISOPOOH has been detected by mass spectrometer instruments in previous chamber studies by Ng et al. (2008), Kwan et al. (2012) and Schwantes et al. (2015). Similarly to this work, the instruments were not calibrated for NISOPOOH, but the sensitivity of the instrument was calibrated for nitrate alcohols (ISOPCNO3). The sensitivity to other organic nitrates such as NISOPOOH was estimated from calculations of the dipole moment and polarizability (Ng et al., 2008; Kwan et al., 2012; Schwantes et al., 2015).
In the experiments of Schwantes et al. (2015), HO2 concentrations were enhanced. NISOPOOH yields were between 0.32 and 0.41, when 54 % and 76 % of the nitrate RO2 were calculated to react with HO2. The authors calculated that these yields are consistent with a 50 % branching ratio of the reaction of nitrate RO2 with HO2 (Reaction R2) to form alkoxy radicals. An uncertainty of ±20 % of the measured NISOPOOH concentration is stated. The uncertainty in the alkoxy radical yield, however, could be higher because the calculation also requires knowledge of the fraction of isoprene that reacted with NO3 and the fraction of RO2 that reacted with HO2, both of which are uncertain because NO3 and HO2 concentrations were not measured. Therefore, a NISOPOOH yield of the reaction of nitrate RO2 with HO2 higher than 50 % may also be consistent with the experimental results in Schwantes et al. (2015).
Ng et al. (2008) quantified NISOPOOH concentrations in their chamber experiment, which was performed at high concentrations of reactants (800 ppbv isoprene, 120 ppbv N2O5). They determined that 50 % of the reacted isoprene resulted in the formation of NISOPOOH, but the fraction of nitrate RO2 that reacted with HO2 could not be determined to calculate yields from specific reactions. Therefore, their experiments cannot be used to derive information about potential alkoxy radical formation from the reaction of RO2 with HO2. HO2 concentrations in experiments in Kwan et al. (2012) were presumably small because high reactant concentrations were used. This explains the relatively small overall NISOPOOH formation of 10 % from the reaction of isoprene with NO3.
Kwan et al. (2012) assumed that specific C5 organic compounds (HPALD, ISOPOOH, C5 hydroxy carbonyl C5H8O2) and MVK and MACR, all of which were quantified in their chamber experiments, were exclusively formed from OH radicals that are formed as a co-product of alkoxy radicals. In this case, the yield of nitrate alkoxy radical formation competing with the formation of NISOPOOH in the reaction of nitrate RO2 with HO2 is 38 % to 58 %. Although the experiments were performed in the absence of ozone, so OH was not produced by ozonolysis reactions, this approach gives only an upper limit of the yield because OH as well as some of the organic products may not have been exclusively produced by this assigned reaction pathway. For example, HPALD can also be produced from the oxidation of isoprene by NO3 from 1,6-H reactions of nitrate RO2 (Vereecken et al., 2021; Fig. 3; Sect. 5.6).
HPALD was also observed in chamber experiments in Kwan et al. (2012) and Schwantes et al. (2015). The authors attributed the observations to the OH oxidation of isoprene, but their observations could also indicate HPALD formation from nitrate RO2. Specifically in the experiments in Schwantes et al. (2015), the total loss rate of nitrate RO2 was calculated to be in the range of 0.03 to 0.13 s−1, so 1,6-H shift reactions with rates between 0.02 and 0.05 s−1 (T=298 K) calculated in Vereecken et al. (2021) can compete with bimolecular loss reactions.
Tsiligiannis et al. (2022) showed that a C4 nitrate with the sum formula C4H7NO5 was observed by the I− CIMS instrument in the experiments in this work and also in several field campaigns in which isoprene oxidation by NO3 was important. This compound was also detected in the chamber experiments by the Br− CIMS instrument (Wu et al., 2021), but signals observed by the Vocus PTR-MS instrument at the respective mass were below the limit of detection. Yields of C4H7NO5 determined in previous chamber experiments in Schwantes et al. (2015) were below 1 %.
Mayhew et al. (2022) applied the three chemical models investigated in this work to field observations in an urban location in Beijing in June 2017. Differences between model results were calculated similarly to in this work. The comparison of modelled data with measurements, however, is more complex for field experiments than for chamber experiments because trace gas concentrations are impacted not only by the chemical process but also by transport. In the field campaign in Beijing, organic nitrates from isoprene were detected by an I− CIMS instrument. The instrument was not specifically calibrated for those compounds, but the same sensitivity as that to isoprene epoxide (IEPOX) species was assumed. In general, concentrations of measured isoprene-derived organic nitrates were lower than calculations for all three models in the night (Mayhew et al., 2022). As pointed out by the authors, the potential loss of epoxide nitrates due to particle uptake could not entirely explain the model–measurement discrepancies.
Overall, results in the experiments in this work appear to be consistent with results in previous experiments, supporting the validity of the FZJ-NO3 mechanism.
The oxidation of isoprene by the nitrate radical, NO3, was investigated in chamber experiments covering different atmospherically relevant chemical regimes. The chemical lifetimes of RO2 radicals formed in the initial reaction of isoprene with NO3 were in the range of atmospheric lifetimes, with values of between 30 s and several minutes due to atmospheric concentrations of reaction partners (RO2, HO2 and NO3). In one experiment, RO2 + HO2 reactions were favoured by producing HO2 and OH radicals in the ozonolysis of propene in the presence of excess CO for the conversion of OH to HO2 radicals. Results from calculations of three near-explicit chemical models (MCM, Caltech, FZJ-NO3) were compared to measurements.
A critical difference between the three chemical mechanisms is the fate of nitrate alkoxy radicals formed in the radical reaction chain, which mainly undergo ring-closure reactions in the FZJ-NO3 mechanisms, whereas decomposition into MVK and MACR is not competitive. Measured concentrations of MVK and MACR in the experiments in this work are consistent with their production from only O3 and OH reactions with isoprene, in agreement with results in previous chamber experiments in Rollins et al. (2009) and Kwan et al. (2012).
Mass signals of most of the organic products expected from the ring-closure reactions of the nitrate alkoxy radicals were detected by the Vocus PTR-MS instrument, demonstrating that the reactions calculated in Vereecken et al. (2021) may indeed be relevant pathways. Signals at the same masses have been observed by chemical ionization mass spectrometry in previous chamber experiments (Kwan et al., 2012; Schwantes et al., 2015). One product of the ring-closure reaction of nitrate alkoxy radicals, which has the sum formula C4H5NO4 and was calculated by Vereecken et al. (2021) to be produced, could not be detected by the Vocus PTR-MS instrument in the experiments in this work and has also not been observed in experiments in Kwan et al. (2012) and Schwantes et al. (2015). Therefore, the reaction pathway leading to this product is likely less important than how it was implemented in the FZJ-NO3 mechanism, but this is within the uncertainty of the calculations in Vereecken et al. (2021).
The formation of hydroperoxy aldehyde (HPALD) species from 1,6-H-shift reactions of nitrate Z-δ-RO2 isomers is only implemented in the FZJ-NO3 mechanism (Vereecken et al., 2021).
A clear signal at the mass of HPALD was detected by the Vocus PTR-MS instrument in all experiments in this work. This was also the case in the experiment when an OH scavenger was present (9 August 2018, Experiment 1), demonstrating that HPALD was formed from the reaction of isoprene with NO3 and that the HPALD was not only formed from the small fraction of isoprene reacting with OH radicals and ozone in the experiments. This is also consistent with previous chamber experiments by Kwan et al. (2012) and Schwantes et al. (2015), in which HPALD formation was observed but attributed to the production from the reaction of OH with isoprene. Measurements of total organic nitrates in Brownwood et al. (2021) for the same experiments, however, give high yields of organic nitrates, hinting that reaction rate constants of 1,6-H-shift reactions might be lower than calculated by Vereecken et al. (2021).
In the night, the fate of nitrate RO2 includes bimolecular reactions with HO2 radicals, other RO2 radicals and NO3 radicals, all of which are significant for atmospheric conditions.
None of the current chemical models can predict C4H7NO5 yields estimated in Tsiligiannis et al. (2022). They could be formed from further oxidation of first-generation C5 nitrates by OH (Wennberg et al., 2018), but the expected yields in the experiments in this work are small due to the low OH concentrations. In addition, C4H7NO5 compounds were also detected in the experiment, when OH concentrations were suppressed by an OH scavenger, demonstrating that they are also formed from other reaction pathways. Further investigations are required to quantify the importance of C4H7NO5 in the NO3 isoprene oxidation scheme.
In the nocturnal atmosphere, not only is isoprene oxidized by NO3 but also a significant fraction reacts with ozone depending on the availability of nitrogen oxides and ozone (Edwards et al., 2017). It is worth noting that due to the fast reaction rate constant of isoprene with OH, reaction with OH could also contribute to the overall loss of isoprene in the night. Part of the OH radicals can be produced in the subsequent reaction chain of the NO3 oxidation of isoprene (Kwan et al., 2012; Vereecken et al., 2021). Fast unimolecular reactions of RO2 from the reaction of isoprene with OH (Peeters et al., 2014) can further gain in importance during the night compared to the daytime (Novelli et al., 2020) because of the long chemical lifetime of RO2 radicals in the range of minutes in the absence of NO, which is often the most important reaction partner for RO2 radicals during the day. Therefore, the yield of HPALD produced from the OH reactions with isoprene can be high in the night despite low OH concentrations. HPALD photolysis could then contribute to OH production the next day (Wolfe et al., 2012).
Only a small fraction of first-generation organic products are further oxidized for atmospheric night-time conditions but are most likely chemically processed by photolysis and reaction with OH the next day. Reaction rate constants of the reactions of nitrate carbonyl, nitrate alcohol and epoxides with NO3 and O3 give chemical lifetimes which are longer than a night for typical concentrations of NO3 and O3. Also HPALD does not react efficiently with NO3 and O3. Reaction rate constants of these reactions as implemented in chemical models such as the MCM, which lead to short chemical lifetimes in the range of hours, need to be revised.
Overall, results from experiments in this work demonstrate that the FZJ-NO3 mechanism for isoprene (Vereecken et al., 2021) gives a more complete and accurate description than previous chemical mechanisms of the nocturnal oxidation of isoprene. New reaction pathways in Vereecken et al. (2021) can have consequences for the nocturnal loss of reactive nitrogen and formation of secondary organic aerosol. However, large uncertainties still exist in the exact distribution of the different RO2 isomers formed in the reaction of isoprene with NO3 and their fate. Specifically, the yield of alkoxy radicals from the reaction of nitrate RO2 with HO2 is uncertain. Calibration of instruments detecting organic nitrate products for specific reaction pathways is urgently needed in future experiments in order to determine the absolute importance of these reaction pathways for atmospheric conditions.
Table A1Organic products expected to be produced from the oxidation of isoprene in this work and the ion mass () at which they are detected by the mass spectrometry instruments which undertook measurements in the experiments. Evaluation of the ion mass signals of the Br− CIMS instrument includes both major isotopes of Br (separated by a slash). Only the chemical structure of one isomer of the same compound is shown.
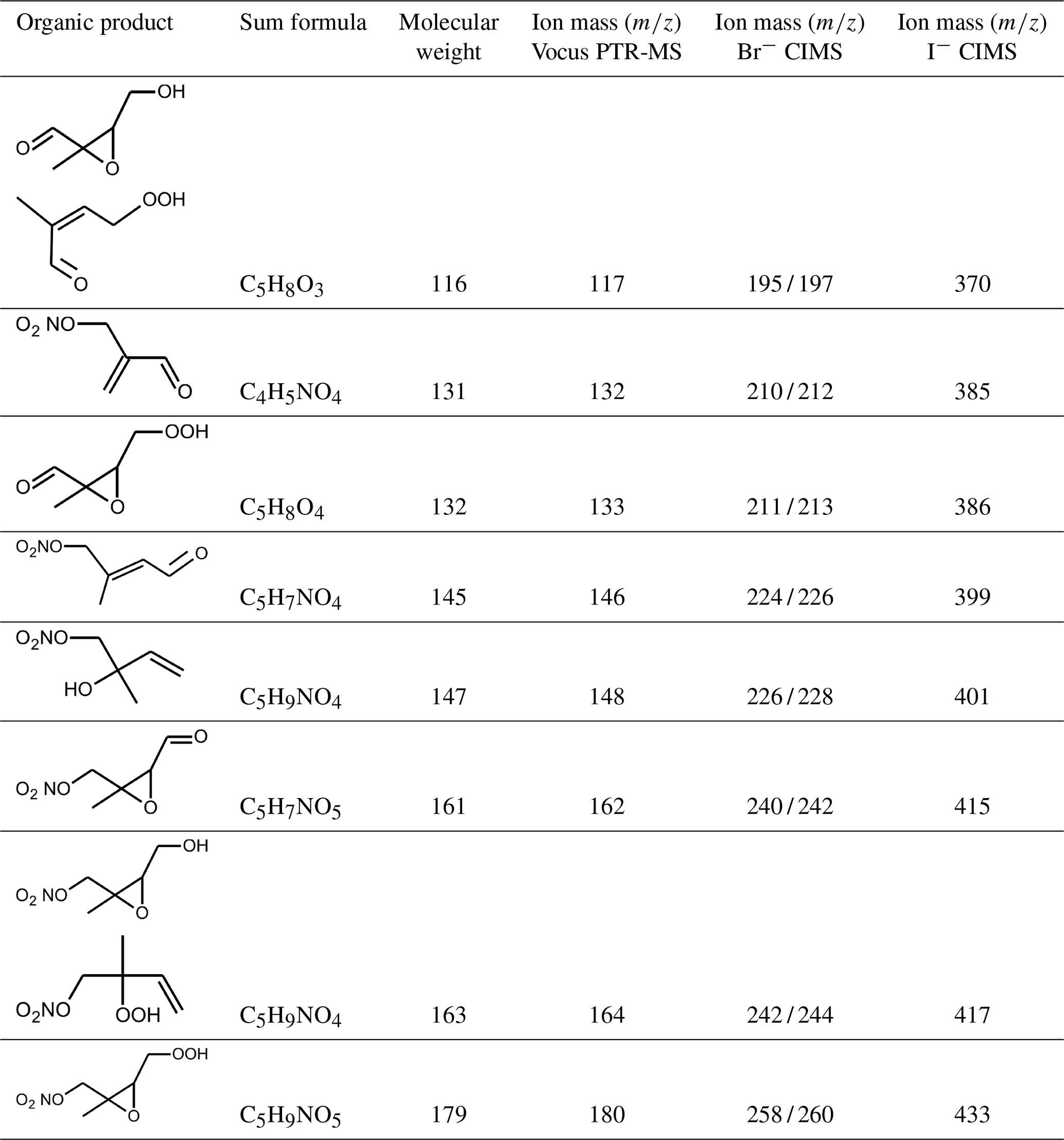
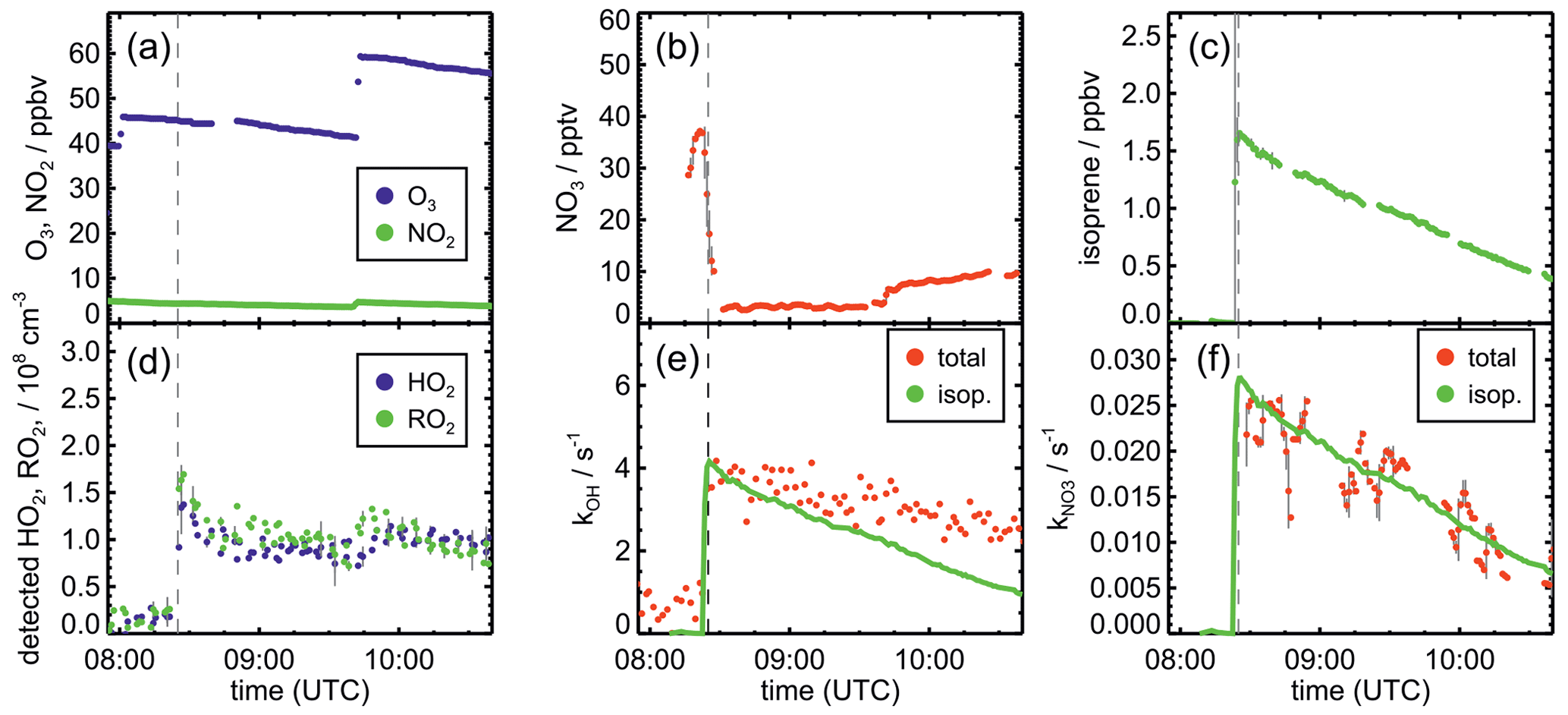
Figure A1Measurements of radical and trace gas concentrations and OH and NO3 reactivity in the experiment on 10 August 2018 (Experiment 2) investigating the oxidation of isoprene by NO3. NO3 reactivity does not include reactivity from organic radicals and NO2. OH and NO3 reactivity from isoprene is calculated from measured isoprene concentrations and reaction rate constants recommended in the literature (Mellouki et al., 2021). Observed RO2 radicals only include a fraction of the total RO2 because the LIF instrument cannot detect all RO2 radicals formed in the reaction of isoprene with NO3 (Vereecken et al., 2021).
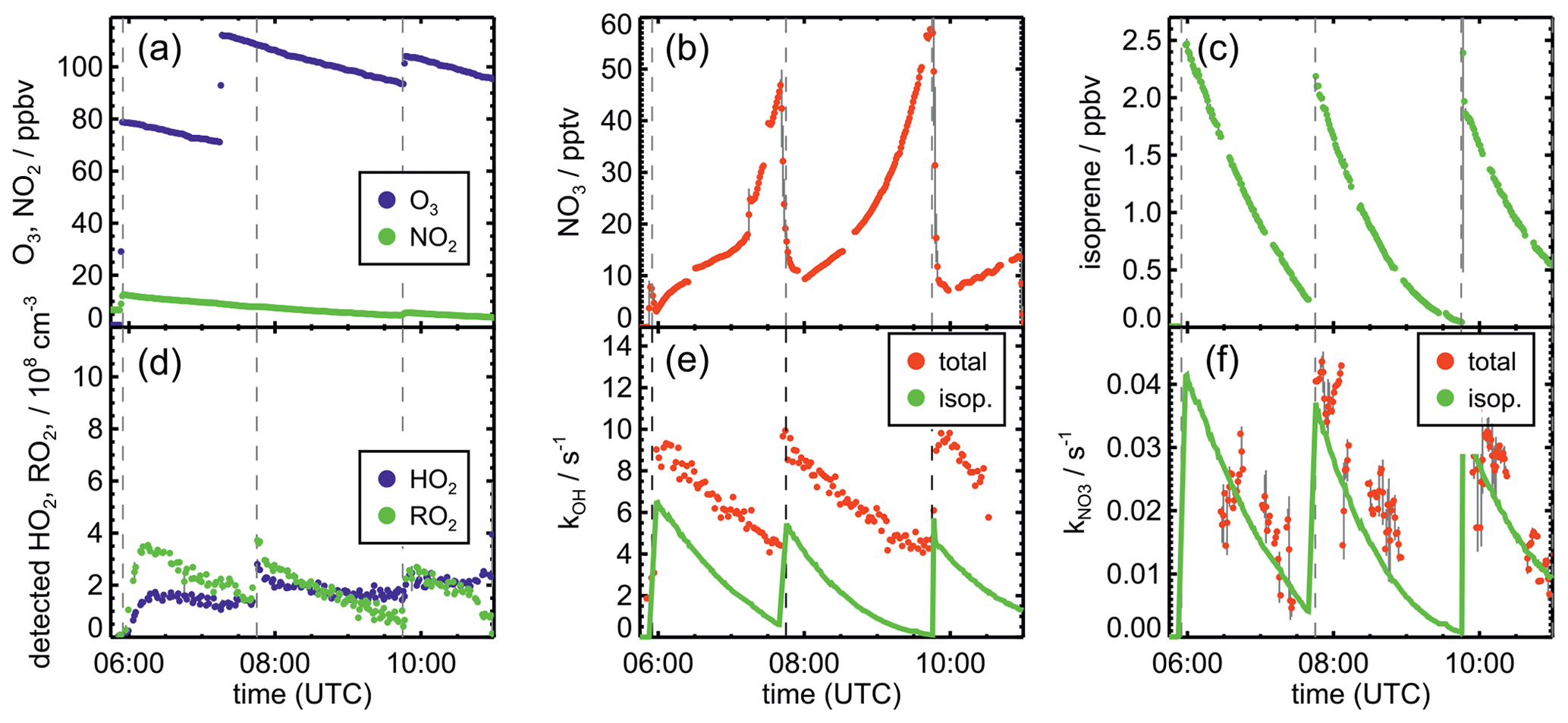
Figure A2Measurements of radical and trace gas concentrations and OH and NO3 reactivity in the experiment on 12 August 2018 (Experiment 3) investigating the oxidation of isoprene by NO3. NO3 reactivity does not include reactivity from organic radicals and NO2. OH and NO3 reactivity from isoprene is calculated from measured isoprene concentrations and reaction rate constants recommended in the literature (Mellouki et al., 2021). Observed RO2 radicals only include a fraction of the total RO2 because the LIF instrument cannot detect all RO2 radicals formed in the reaction of isoprene with NO3 (Vereecken et al., 2021).
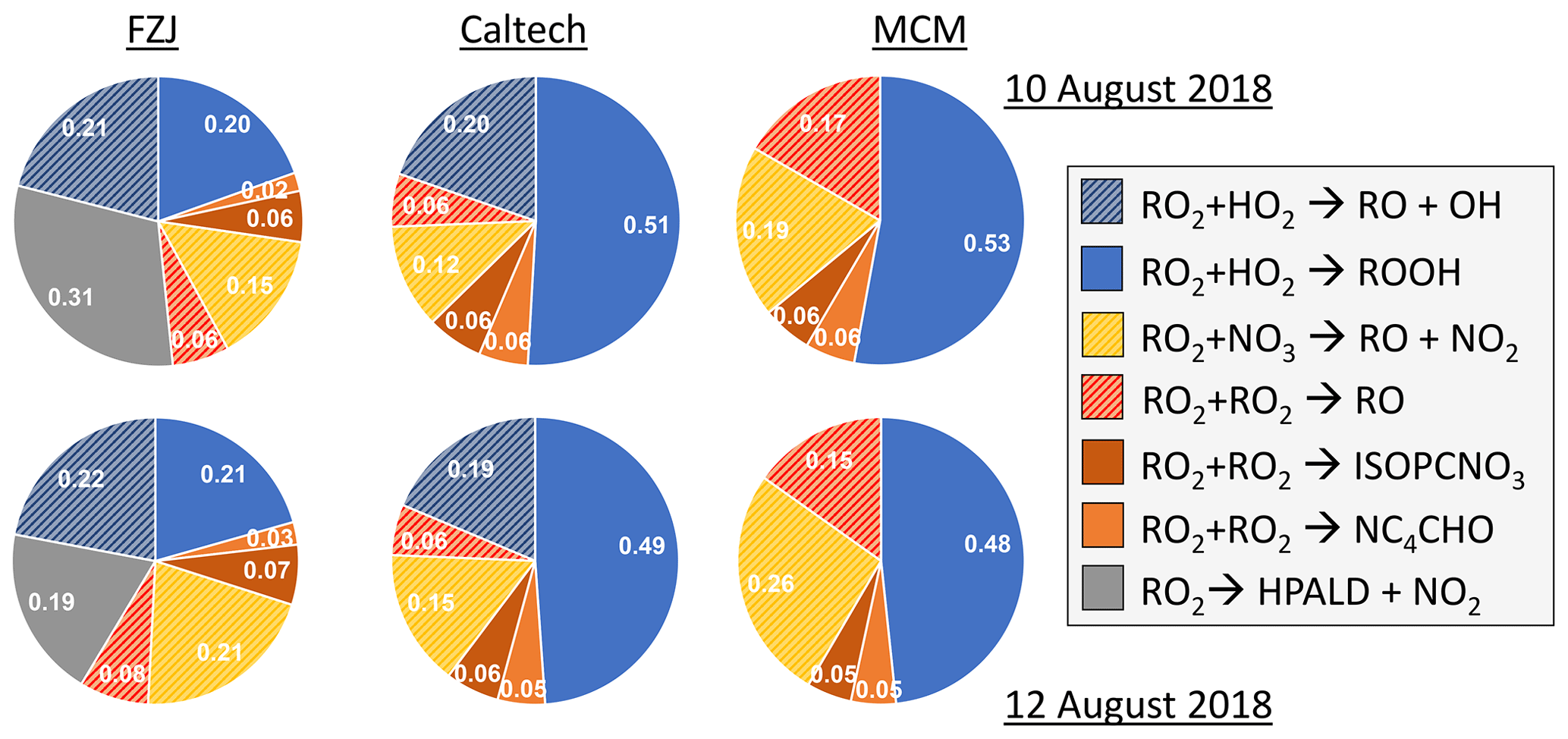
Figure A3Relative distribution of loss rates of nitrate RO2 for the experiment on 10 August 2018 (Experiment 2) and on 12 August 2018 (Experiment 3). The total RO2 loss rate was 0.005 and 0.008 s−1 in the experiment on 10 August 2018 (Experiment 2) and 12 August 2018 (Experiment 3), respectively. Calculations of the loss rates of RO2 radicals in bimolecular reactions make use of measured HO2 and NO3 concentrations. Total RO2 concentrations and concentrations of speciated nitrate RO2 were taken from model calculations applying the FZJ-NO3 mechanism, Caltech mechanism or MCM. The chemical mechanisms differ with respect to the number of nitrate RO2 isomers that are considered, the type of RO2 loss reactions and products of loss reactions (Figs. 3 and 6). Therefore, the distributions of nitrate RO2 radicals and RO2 concentrations differ between the model runs.
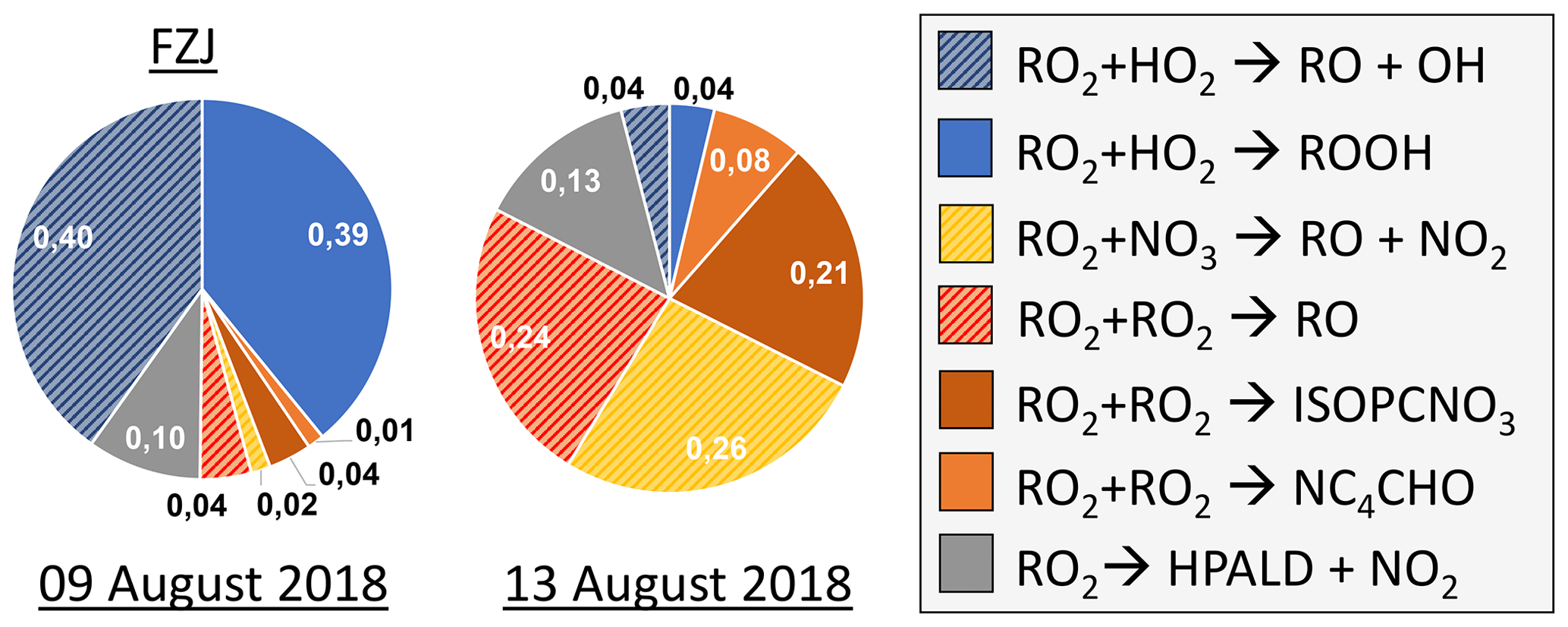
Figure A4Relative distribution of loss rates of nitrate RO2 for the experiments on 9 and 13 August 2018 (Experiments 1 and 4) if the FZJ-NO3 mechanism is applied and HO2 is not constrained to measured values. Total RO2 concentrations and concentrations of speciated nitrate RO2 were taken from model calculations.
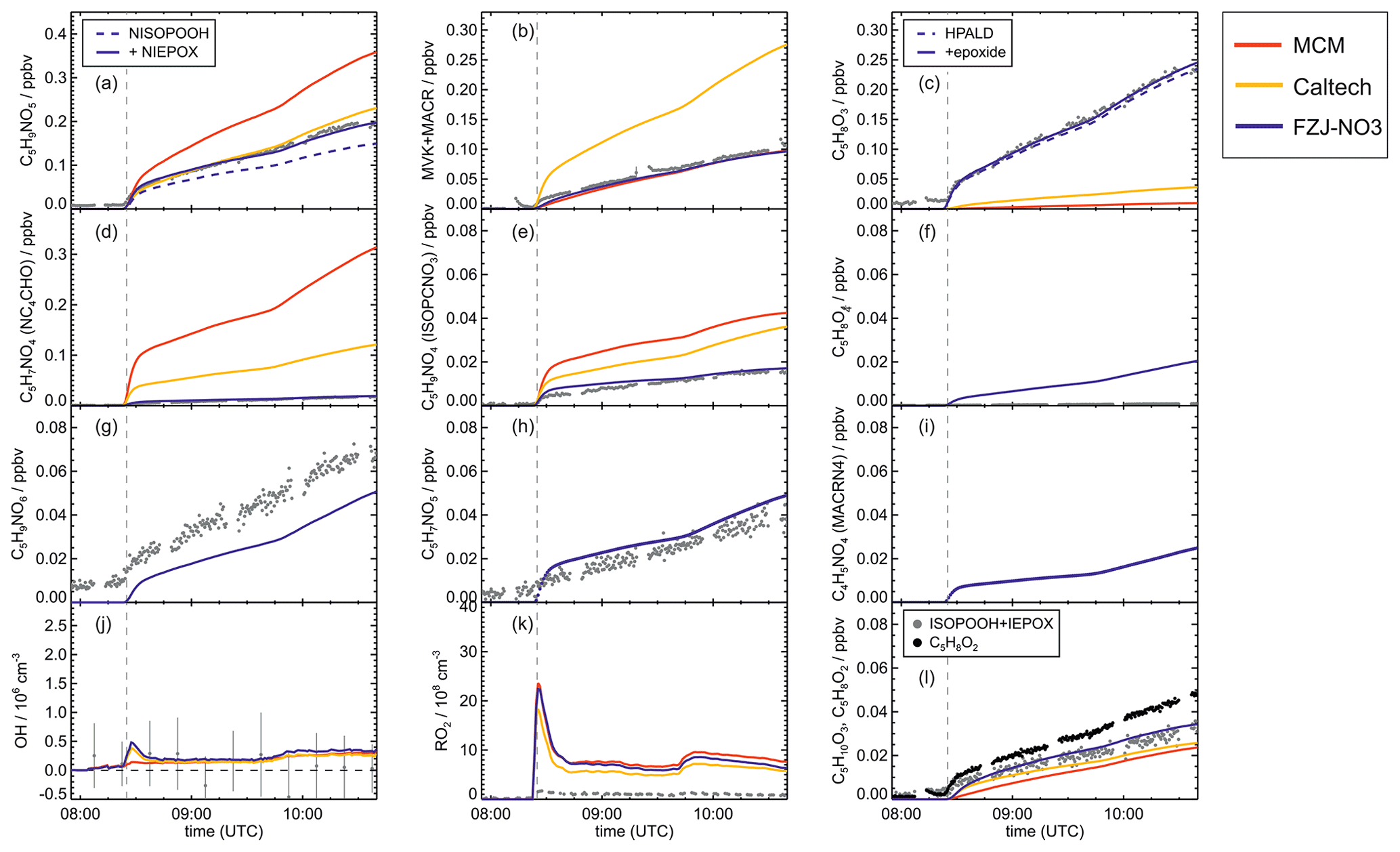
Figure A5Comparison of results from model calculations applying the different isoprene NO3 chemistry mechanisms for the experiment on 10 August 2018 (Experiment 2). MVK, MACR, NISOPOOH, ISOPCNO3 and NC4CHO are produced from all mechanisms, whereas the other compounds are only produced from either 1,6-H-shift reactions or ring-closure reactions of nitrate alkoxy radicals, which are only implemented in the FZJ-NO3 mechanism. Grey and black dots are measured values. Measured organic peroxy radical concentrations only include part of the total RO2 because the LIF instrument cannot detect a fraction of nitrate RO2 (Vereecken et al., 2021). Organic products were detected by the Vocus PTR-MS instrument, which was only calibrated for MVK and MACR. All other traces are scaled to match best the results from the FZJ-NO3 mechanism.
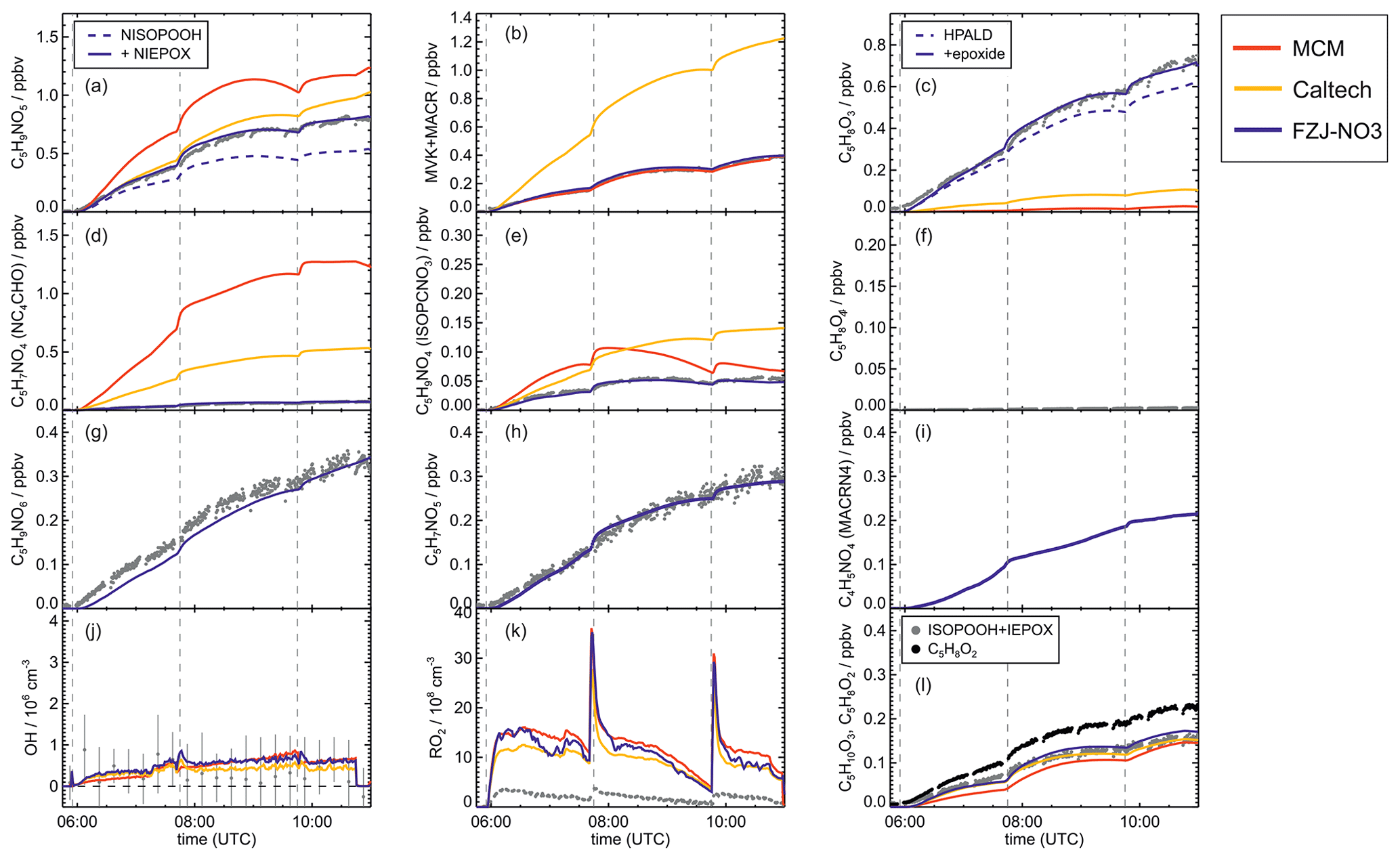
Figure A6Comparison of results from model calculations applying the different isoprene NO3 chemistry mechanisms for the experiment on 12 August 2018 (Experiment 3). MVK, MACR, NISOPOOH, ISOPCNO3 and NC4CHO are produced from all mechanisms, whereas the other compounds are only produced from either 1,6-H-shift reactions or ring-closure reactions of nitrate alkoxy radicals, which are only implemented in the FZJ-NO3 mechanism. Grey and black dots are measured values. Measured organic peroxy radical concentrations only include part of the total RO2 because the LIF instrument cannot detect a fraction of nitrate RO2 (Vereecken et al., 2021). Organic products were detected by the Vocus PTR-MS instrument, which was only calibrated for MVK and MACR. All other traces are scaled to match best the results from the FZJ-NO3 mechanism.
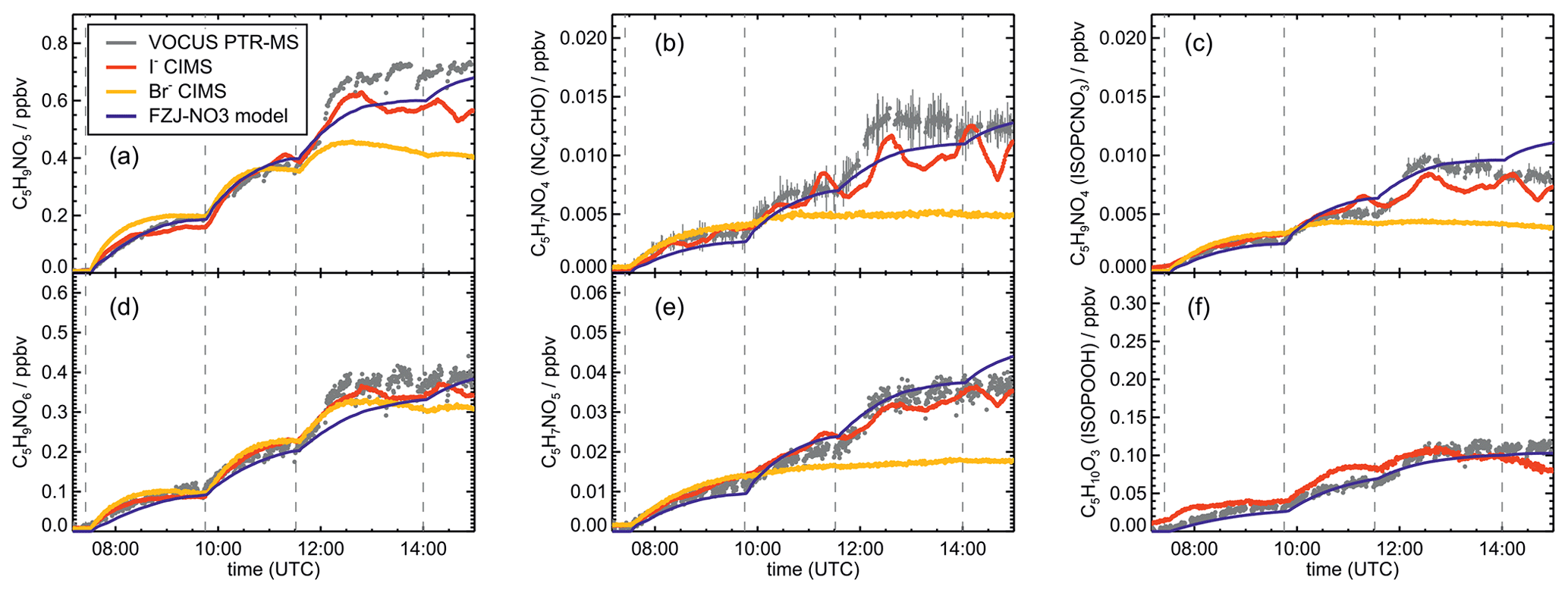
Figure A7Comparison of reported signals from three mass spectrometer instruments applying different ionization methods (Vocus PTR-MS, Br− CIMS, I− CIMS) and measuring organic products in the experiment on 9 August 2018 (Experiment 1). All signals are scaled to match best the concentrations resulting from model calculations applying the FZJ-NO3 chemical mechanism.
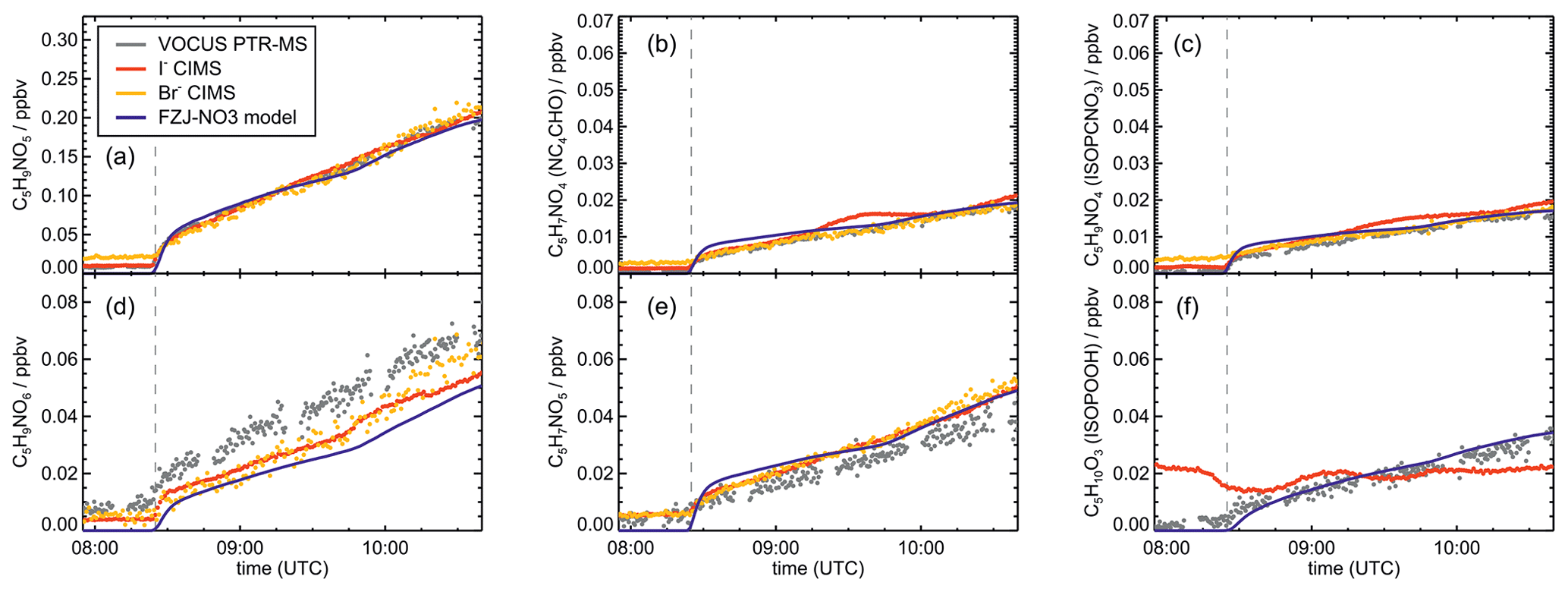
Figure A8Comparison of reported signals from three mass spectrometer instruments applying different ionization methods (Vocus PTR-MS, Br− CIMS, I− CIMS) and measuring organic products in the experiment on 10 August 2018 (Experiment 2). All signals are scaled to match best the concentrations resulting from model calculations applying the FZJ-NO3 chemical mechanism.
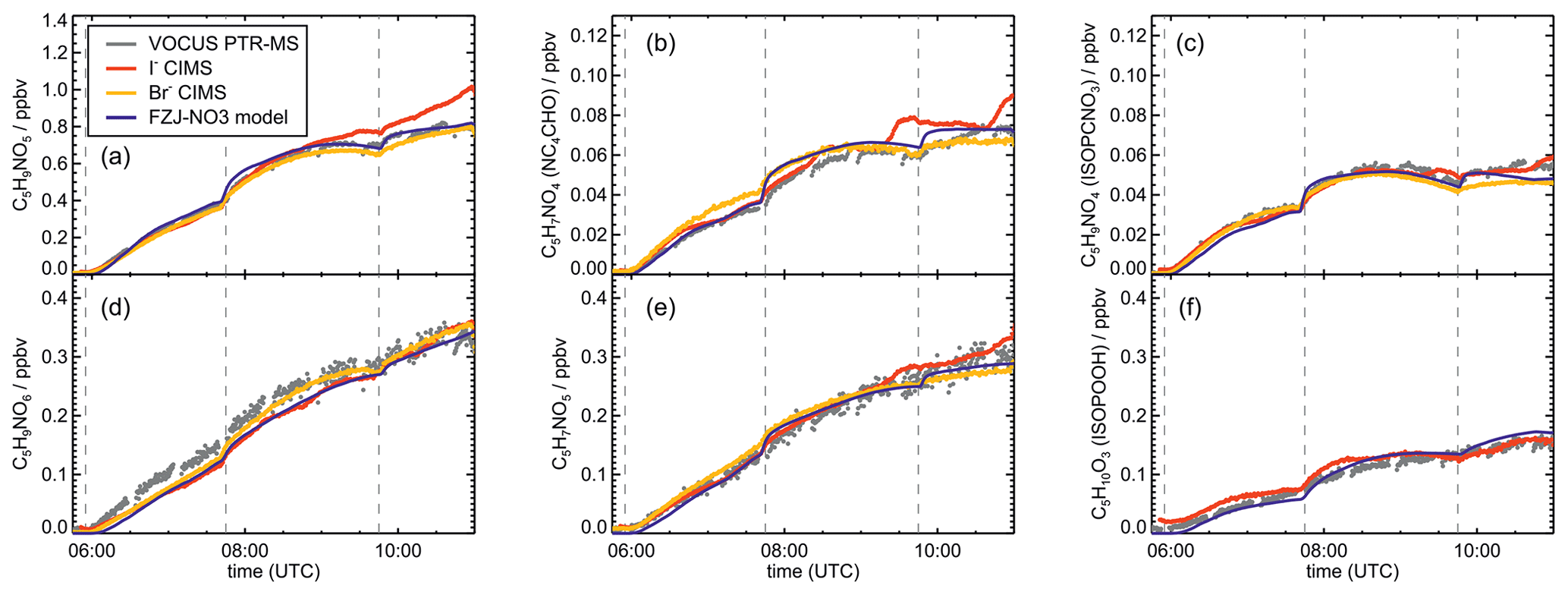
Figure A9Comparison of reported signals from three mass spectrometer instruments applying different ionization methods (Vocus PTR-MS, Br− CIMS, I− CIMS) and measuring organic products in the experiment on 12 August 2018 (Experiment 3). All signals are scaled to match best the concentrations resulting from model calculations applying the FZJ-NO3 chemical mechanism.

Figure A10Comparison of reported signals from three mass spectrometer instruments applying different ionization methods (Vocus PTR-MS, Br− CIMS, I− CIMS) and measuring organic products in the experiment on 13 August 2018 (Experiment 4). All signals are scaled to match best the concentrations resulting from model calculations applying the FZJ-NO3 chemical mechanism.

Figure A11Ion mass signal of the Vocus PTR-MS instrument scaled to the model results from the MCM, Caltech and FZJ-NO3 models in the experiment on 13 August 2018 (Experiment 4). Only species for which the instrument was not calibrated and which are produced in all models are shown.
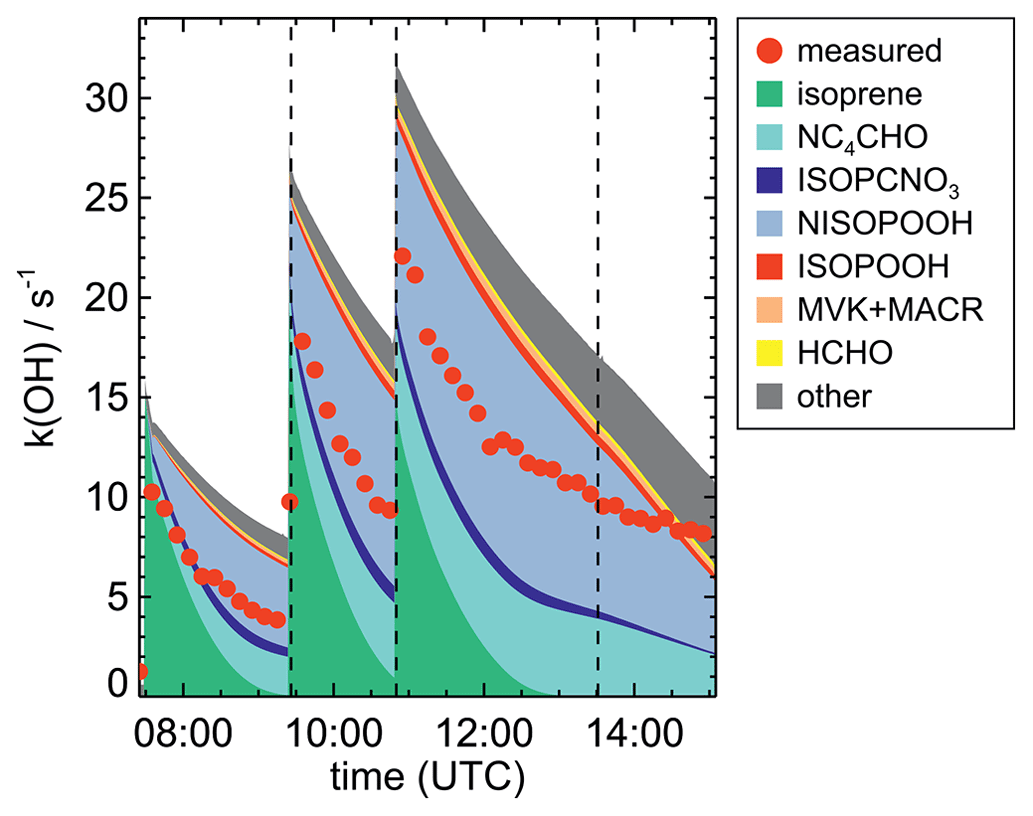
Figure A12Comparison of measured OH reactivity from organic compounds and OH reactivity calculated from concentrations of organic compounds modelled applying the MCM. Up to 10 % of the reactivity from hydroperoxide compounds (NISOPOOH, ISOPOOH) is invisible for the LP-LIF instrument because these species partly produce OH in their reaction with OH. The exact OH yield is uncertain. A 100 % yield is assumed in the MCM. OH reactivity from organic compounds is derived by subtracting the reactivity via NO2 and O3 calculated using measured concentrations from the measured total OH reactivity. “Other” compounds include a high number of organic compounds that are produced in the reaction of isoprene with OH, O3 and NO3 and for which loss by the reaction with OH is implemented in the MCM.
Data from the experiments in the SAPHIR chamber used in this work are available on the EUROCHAMP database web page (https://data.eurochamp.org/data-access/chamber-experiments/, last access: 9 March 2023). Data for each experiment are available as follows: experiment on 9 August 2018 (Experiment 1), Fuchs et al. (2018a) (https://doi.org/10.25326/PZ5Q-9X18); experiment on 10 August 2018 (Experiment 2), Fuchs et al. (2018b) (https://doi.org/10.25326/YZHF-T659); experiment on 12 August 2018 (Experiment 3), Fuchs et al. (2018c) (https://doi.org/10.25326/JCST-0Y45); and experiment on 13 August 2018 (Experiment 4), Fuchs et al. (2018d) (https://doi.org/10.25326/BSA7-WX31).
PTMC and HF wrote the manuscript, analysed the data and did model calculations of the experiments. SSB, MH, JLF, AN and HF designed and executed the experiments. LV provided insights into the chemical mechanisms. LH, TH, SK, TM, RT, DR, FR, RW, BB, JL, ET, JNC, PD, NF, JLF, JS and FB were responsible for measurements used in this work. All authors intensively discussed the manuscript and thereby contributed to the writing.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This research has been supported by H2020 Excellent Science (grant no. 681529), H2020 Research Infrastructures (grant no. 730997), Horizon 2020 (FORCeS (grant no. 821205)), the Vetenskapsrådet (grant nos. 2014-05332 and 2018-04430) and the Svenska Forskningsrådet Formas (grant nos. 2015-1537 and 2019-586).
The article processing charges for this open-access publication were covered by the Forschungszentrum Jülich.
This paper was edited by Gabriele Stiller and reviewed by two anonymous referees.
Albrecht, S. R., Novelli, A., Hofzumahaus, A., Kang, S., Baker, Y., Mentel, T., Wahner, A., and Fuchs, H.: Measurements of hydroperoxy radicals (HO2) at atmospheric concentrations using bromide chemical ionisation mass spectrometry, Atmos. Meas. Tech., 12, 891–902, https://doi.org/10.5194/amt-12-891-2019, 2019. a
Ashbourn, S. F. M., Jenkin, M. E., and Clemitshaw, K. C.: Laboratory studies of the response of a peroxy radical chemical amplifier to HO2 and a series of organic peroxy radicals, J. Atmos. Chem., 29, 233–266, https://doi.org/10.1023/A:1005992316512, 1998. a
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006. a
Barnes, I., Bastian, V., Becker, K. H., and Tong, Z.: Kinetics and products of the reactions of nitrate radical with monoalkenes, dialkenes, and monoterpenes, J. Phys. Chem., 94, 2413–2419, https://doi.org/10.1021/j100369a041, 1990. a, b
Bates, K. H. and Wennberg, P.: Isoprene oxidation model (Version 5), CaltechDATA [data set], https://doi.org/10.22002/D1.247, 2017. a, b, c
Bates, K. H., Crounse, J. D., Clair, J. M. S., Bennett, N. B., Nguyen, T. B., Seinfeld, J. H., Stoltz, B. M., and Wennberg, P. O.: Gas phase production and loss of isoprene epoxydiols, J. Phys. Chem. A, 118, 1237–1246, https://doi.org/10.1021/jp4107958, 2014. a
Berndt, T., Hyttinen, N., Herrmann, H., and Hansel, A.: First oxidation products from the reaction of hydroxyl radicals with isoprene for pristine environmental conditions, Com. Chem., 2, 21, https://doi.org/10.1038/s42004-019-0120-9, 2019. a
Bohn, B. and Zilken, H.: Model-aided radiometric determination of photolysis frequencies in a sunlit atmosphere simulation chamber, Atmos. Chem. Phys., 5, 191–206, https://doi.org/10.5194/acp-5-191-2005, 2005. a
Brown, S. S., deGouw, J. A., Warneke, C., Ryerson, T. B., Dubé, W. P., Atlas, E., Weber, R. J., Peltier, R. E., Neuman, J. A., Roberts, J. M., Swanson, A., Flocke, F., McKeen, S. A., Brioude, J., Sommariva, R., Trainer, M., Fehsenfeld, F. C., and Ravishankara, A. R.: Nocturnal isoprene oxidation over the Northeast United States in summer and its impact on reactive nitrogen partitioning and secondary organic aerosol, Atmos. Chem. Phys., 9, 3027–3042, https://doi.org/10.5194/acp-9-3027-2009, 2009. a
Brownwood, B., Turdziladze, A., Hohaus, T., Wu, R., Mentel, T. F., Carlsson, P. T. M., Tsiligiannis, E., Hallquist, M., Andres, S., Hantschke, L., Reimer, D., Rohrer, F., Tillmann, R., Winter, B., Liebmann, J., Brown, S. S., Kiendler-Scharr, A., Novelli, A., Fuchs, H., and Fry, J. L.: Gas-particle partitioning and SOA yields of organonitrate products from NO3-initiated oxidation of isoprene under varied chemical regimes, Earth Space Chem., 5, 785–800, https://doi.org/10.1021/acsearthspacechem.0c00311, 2021. a, b, c, d, e, f, g, h
Canosa-Mas, C. E., Smith, S. J., Waygood, S. J., and Wayne, R. P.: Study of the temperature dependence of the reaction of the nitrate radical with propene, J. Chem. Soc. Faraday T., 87, 3473–3478, https://doi.org/10.1039/FT9918703473, 1991. a
Cho, C., Hofzumahaus, A., Fuchs, H., Dorn, H.-P., Glowania, M., Holland, F., Rohrer, F., Vardhan, V., Kiendler-Scharr, A., Wahner, A., and Novelli, A.: Characterization of a chemical modulation reactor (CMR) for the measurement of atmospheric concentrations of hydroxyl radicals with a laser-induced fluorescence instrument, Atmos. Meas. Tech., 14, 1851–1877, https://doi.org/10.5194/amt-14-1851-2021, 2021. a, b, c
Crounse, J. D., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Peroxy radical isomerization in the oxidation of isoprene, Phys. Chem. Chem. Phys., 13, 13607–13613, https://doi.org/10.1039/C1CP21330J, 2011. a
Dewald, P., Liebmann, J. M., Friedrich, N., Shenolikar, J., Schuladen, J., Rohrer, F., Reimer, D., Tillmann, R., Novelli, A., Cho, C., Xu, K., Holzinger, R., Bernard, F., Zhou, L., Mellouki, W., Brown, S. S., Fuchs, H., Lelieveld, J., and Crowley, J. N.: Evolution of NO3 reactivity during the oxidation of isoprene, Atmos. Chem. Phys., 20, 10459–10475, https://doi.org/10.5194/acp-20-10459-2020, 2020. a, b, c, d, e
Edwards, P. M., Aikin, K. C., Dube, W. P., Fry, J. L., Gilman, J. B., de Gouw, J. A., Graus, M. G., Hanisco, T. F., Holloway, J., Hübler, G., Kaiser, J., Keutsch, F. N., Lerner, B. M., Neuman, J. A., Parrish, D. D., Peischl, J., Pollack, I. B., Ravishankara, A. R., Roberts, J. M., Ryerson, T. B., Trainer, M., Veres, P. R., Wolfe, G. M., Warneke, C., and Brown, S. S.: Transition from high- to low-NOx control of night-time oxidation in the southeastern US, Nat. Geosci., 10, 490, https://doi.org/10.1038/ngeo2976, 2017. a, b
Fuchs, H., Bohn, B., Hofzumahaus, A., Holland, F., Lu, K. D., Nehr, S., Rohrer, F., and Wahner, A.: Detection of HO2 by laser-induced fluorescence: calibration and interferences from RO2 radicals, Atmos. Meas. Tech., 4, 1209–1225, https://doi.org/10.5194/amt-4-1209-2011, 2011. a
Fuchs, H., Dorn, H.-P., Bachner, M., Bohn, B., Brauers, T., Gomm, S., Hofzumahaus, A., Holland, F., Nehr, S., Rohrer, F., Tillmann, R., and Wahner, A.: Comparison of OH concentration measurements by DOAS and LIF during SAPHIR chamber experiments at high OH reactivity and low NO concentration, Atmos. Meas. Tech., 5, 1611–1626, https://doi.org/10.5194/amt-5-1611-2012, 2012. a
Fuchs, H., Hofzumahaus, A., Rohrer, F., Bohn, B., Brauers, T., Dorn, H.-P., Häseler, R., Holland, F., Kaminski, M., Li, X., Lu, K., Nehr, S., Tillmann, R., Wegener, R., and Wahner, A.: Experimental evidence for efficient hydroxyl radical regeneration in isoprene oxidation, Nat. Geosci., 6, 1023–1026, https://doi.org/10.1038/NGEO1964, 2013. a
Fuchs, H., Novelli, A., Rolletter, M., Hofzumahaus, A., Pfannerstill, E. Y., Kessel, S., Edtbauer, A., Williams, J., Michoud, V., Dusanter, S., Locoge, N., Zannoni, N., Gros, V., Truong, F., Sarda-Esteve, R., Cryer, D. R., Brumby, C. A., Whalley, L. K., Stone, D., Seakins, P. W., Heard, D. E., Schoemaecker, C., Blocquet, M., Coudert, S., Batut, S., Fittschen, C., Thames, A. B., Brune, W. H., Ernest, C., Harder, H., Muller, J. B. A., Elste, T., Kubistin, D., Andres, S., Bohn, B., Hohaus, T., Holland, F., Li, X., Rohrer, F., Kiendler-Scharr, A., Tillmann, R., Wegener, R., Yu, Z., Zou, Q., and Wahner, A.: Comparison of OH reactivity measurements in the atmospheric simulation chamber SAPHIR, Atmos. Meas. Tech., 10, 4023–4053, https://doi.org/10.5194/amt-10-4023-2017, 2017. a
Fuchs, H., Novelli, A., Cho, C., Rohrer, F., Tillmann, R., Reimer, D., Hohaus, T., Turdziladze, A., Dewald, P., Liebmann, J., Friedrich, N., Shenolikar, J., Schuladen, J., Crowley, J., Brown, S. S., Bernard, F., Zhou, L., Mentel, T., Wu, R., and Hamilton, J. F.: Atmospheric simulation chamber study: isoprene + NO3 – Gas-phase oxidation – product study, AERIS [data set], https://doi.org/10.25326/PZ5Q-9X18, 2018a. a, b
Fuchs, H., Novelli, A., Cho, C., Rohrer, F., Tillmann, R., Reimer, D., Hohaus, T., Turdziladze, A., Dewald, P., Liebmann, J., Friedrich, N., Shenolikar, J., Schuladen, J., Crowley, J., Brown, S. S., Bernard, F., Zhou, L., Mentel, T., Wu, R., and Hamilton, J. F.: Atmospheric simulation chamber study: isoprene + NO3 – Gas-phase oxidation – product study, AERIS [data set], https://doi.org/10.25326/YZHF-T659, 2018b. a, b
Fuchs, H., Novelli, A., Cho, C., Rohrer, F., Tillmann, R., Reimer, D., Hohaus, T., Turdziladze, A., Dewald, P., Liebmann, J., Friedrich, N., Shenolikar, J., Schuladen, J., Crowley, J., Brown, S. S., Bernard, F., Zhou, L., Mentel, T., Wu, R., and Hamilton, J. F.: Atmospheric simulation chamber study: isoprene + NO3 – Gas-phase oxidation – product study, AERIS [data set], https://doi.org/10.25326/JCST-0Y45, 2018c. a, b
Fuchs, H., Novelli, A., Cho, C., Rohrer, F., Tillmann, R., Reimer, D., Hohaus, T., Turdziladze, A., Dewald, P., Liebmann, J., Friedrich, N., Shenolikar, J., Schuladen, J., Crowley, J., Brown, S. S., Bernard, F., Zhou, L., Mentel, T., Wu, R., and Hamilton, J. F.: Atmospheric simulation chamber study: isoprene + NO3 – Gas-phase oxidation – product study, AERIS [data set], https://doi.org/10.25326/BSA7-WX31, 2018d. a, b
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012. a
Hantschke, L., Novelli, A., Bohn, B., Cho, C., Reimer, D., Rohrer, F., Tillmann, R., Glowania, M., Hofzumahaus, A., Kiendler-Scharr, A., Wahner, A., and Fuchs, H.: Atmospheric photooxidation and ozonolysis of Δ3-carene and 3-caronaldehyde: rate constants and product yields, Atmos. Chem. Phys., 21, 12665–12685, https://doi.org/10.5194/acp-21-12665-2021, 2021. a
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: A protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997. a
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015. a, b
Jenkin, M. E., Valorso, R., Aumont, B., and Rickard, A. R.: Estimation of rate coefficients and branching ratios for reactions of organic peroxy radicals for use in automated mechanism construction, Atmos. Chem. Phys., 19, 7691–7717, https://doi.org/10.5194/acp-19-7691-2019, 2019. a, b
Keehan, N. I., Brownwood, B., Marsavin, A., Day, D. A., and Fry, J. L.: A thermal-dissociation–cavity ring-down spectrometer (TD-CRDS) for the detection of organic nitrates in gas and particle phases, Atmos. Meas. Tech., 13, 6255–6269, https://doi.org/10.5194/amt-13-6255-2020, 2020. a
Kerdouci, J., Picquet-Varrault, B., and Doussin, J.-F.: Structure–activity relationship for the gas-phase reactions of NO3 radical with organic compounds: Update and extension to aldehydes, Atmos. Environ., 84, 363–372, https://doi.org/10.1016/j.atmosenv.2013.11.024, 2014. a, b
Krechmer, J., Lopez-Hilfiker, F., Koss, A., Hutterli, M., Stoermer, C., Deming, B., Kimmel, J., Warneke, C., Holzinger, R., Jayne, J., Worsnop, D., Fuhrer, K., Gonin, M., and de Gouw, J.: Evaluation of a new reagent-ion source and focusing ion–molecule reactor for use in proton-transfer-reaction mass spectrometry, Anal. Chem., 90, 12011–12018, https://doi.org/10.1021/acs.analchem.8b02641, 2018. a
Kwan, A. J., Chan, A. W. H., Ng, N. L., Kjaergaard, H. G., Seinfeld, J. H., and Wennberg, P. O.: Peroxy radical chemistry and OH radical production during the NO3-initiated oxidation of isoprene, Atmos. Chem. Phys., 12, 7499–7515, https://doi.org/10.5194/acp-12-7499-2012, 2012. a, b, c, d, e, f, g, h, i, j, k, l, m, n, o, p, q, r, s, t
Kwok, E. S. C., Aschmann, S. M., Arey, J., and Atkinson, R.: Product formation from the reaction of the NO3 radical with isoprene and rate constants for the reactions of methacrolein and methyl vinyl ketone with the NO3 radical, Int. J. Chem. Kinet., 28, 925–934, 1996. a, b
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurtén, T., Worsnop, D. R., and Thornton, J. A.: An iodide-adduct high-resolution time-of-flight chemical-ionization mass spectrometer: Application to atmospheric inorganic and organic compounds, Environ. Sci. Technol., 48, 6309–6317, https://doi.org/10.1021/es500362a, 2014. a
Lee, L., Teng, A. P., Wennberg, P. O., Crounse, J. D., and Cohen, R. C.: On rates and mechanisms of OH and O3 reactions with isoprene-derived hydroxy nitrates, J. Phys. Chem. A, 118, 1622–1637, https://doi.org/10.1021/jp4107603, 2014. a, b, c, d
Lelieveld, J., Butler, T. M., Crowley, J. N., Dillon, T. J., Fischer, H., Ganzeveld, L., Harder, H., Lawrence, M. G., Martinez, M., Taraborrelli, D., and Williams, J.: Atmospheric oxidation capacity sustained by a tropical forest, Nature, 452, 737–740, https://doi.org/10.1038/nature06870, 2008. a
Li, H., Almeida, T. G., Luo, Y., Zhao, J., Palm, B. B., Daub, C. D., Huang, W., Mohr, C., Krechmer, J. E., Kurtén, T., and Ehn, M.: Fragmentation inside proton-transfer-reaction-based mass spectrometers limits the detection of ROOR and ROOH peroxides, Atmos. Meas. Tech., 15, 1811–1827, https://doi.org/10.5194/amt-15-1811-2022, 2022. a
Liebmann, J. M., Schuster, G., Schuladen, J. B., Sobanski, N., Lelieveld, J., and Crowley, J. N.: Measurement of ambient NO3 reactivity: design, characterization and first deployment of a new instrument, Atmos. Meas. Tech., 10, 1241–1258, https://doi.org/10.5194/amt-10-1241-2017, 2017. a
Liebmann, J. M., Muller, J. B. A., Kubistin, D., Claude, A., Holla, R., Plass-Dülmer, C., Lelieveld, J., and Crowley, J. N.: Direct measurements of NO3 reactivity in and above the boundary layer of a mountaintop site: identification of reactive trace gases and comparison with OH reactivity, Atmos. Chem. Phys., 18, 12045–12059, https://doi.org/10.5194/acp-18-12045-2018, 2018. a
Lu, K. D., Rohrer, F., Holland, F., Fuchs, H., Brauers, T., Oebel, A., Dlugi, R., Hu, M., Li, X., Lou, S. R., Shao, M., Zhu, T., Wahner, A., Zhang, Y. H., and Hofzumahaus, A.: Nighttime observation and chemistry of HOx in the Pearl River Delta and Beijing in summer 2006, Atmos. Chem. Phys., 14, 4979–4999, https://doi.org/10.5194/acp-14-4979-2014, 2014. a
Mayhew, A. W., Lee, B. H., Thornton, J. A., Bannan, T. J., Brean, J., Hopkins, J. R., Lee, J. D., Nelson, B. S., Percival, C., Rickard, A. R., Shaw, M. D., Edwards, P. M., and Hamilton, J. F.: Evaluation of isoprene nitrate chemistry in detailed chemical mechanisms, Atmos. Chem. Phys., 22, 14783–14798, https://doi.org/10.5194/acp-22-14783-2022, 2022. a, b
Mellouki, A., Ammann, M., Cox, R. A., Crowley, J. N., Herrmann, H., Jenkin, M. E., McNeill, V. F., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: volume VIII – gas-phase reactions of organic species with four, or more, carbon atoms (≥C4), Atmos. Chem. Phys., 21, 4797–4808, https://doi.org/10.5194/acp-21-4797-2021, 2021. a, b, c, d
Ng, N. L., Kwan, A. J., Surratt, J. D., Chan, A. W. H., Chhabra, P. S., Sorooshian, A., Pye, H. O. T., Crounse, J. D., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol (SOA) formation from reaction of isoprene with nitrate radicals (NO3), Atmos. Chem. Phys., 8, 4117–4140, https://doi.org/10.5194/acp-8-4117-2008, 2008. a, b, c, d, e
Nguyen, T. B., Tyndall, G. S., Crounse, J. D., Teng, A. P., Bates, K. H., Schwantes, R. H., Coggon, M. M., Zhang, L., Feiner, P., Milller, D. O., Skog, K. M., Rivera-Rios, J. C., Dorris, M., Olson, K. F., Koss, A., Wild, R. J., Brown, S. S., Goldstein, A. H., de Gouw, J. A., Brune, W. H., Keutsch, F. N., Seinfeld, J. H., and Wennberg, P. O.: Atmospheric fates of Criegee intermediates in the ozonolysis of isoprene, Phys. Chem. Chem. Phys., 18, 10241–10254, https://doi.org/10.1039/C6CP00053C, 2016. a
Novelli, A., Vereecken, L., Bohn, B., Dorn, H.-P., Gkatzelis, G. I., Hofzumahaus, A., Holland, F., Reimer, D., Rohrer, F., Rosanka, S., Taraborrelli, D., Tillmann, R., Wegener, R., Yu, Z., Kiendler-Scharr, A., Wahner, A., and Fuchs, H.: Importance of isomerization reactions for OH radical regeneration from the photo-oxidation of isoprene investigated in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 20, 3333–3355, https://doi.org/10.5194/acp-20-3333-2020, 2020. a, b, c, d
Novelli, A., Cho, C., Fuchs, H., Hofzumahaus, A., Rohrer, F., Tillmann, R., Kiendler-Scharr, A., Wahner, A., and Vereecken, L.: Experimental and theoretical study on the impact of a nitrate group on the chemistry of alkoxy radicals, Phys. Chem. Chem. Phys., 23, 5474–5495, https://doi.org/10.1039/D0CP05555G, 2021. a, b, c
Paulot, F., Crounse, J. D., Kjaergaard, H. G., Kurten, A., St. Clair, J. M., Seinfeld, J. H., and Wennberg, P. O.: Unexpected epoxide formation in the gas-phase photooxidation of isoprene, Science, 325, 730–733, https://doi.org/10.1126/science.1172910, 2009. a
Peeters, J., Nguyen, T. L., and Vereecken, L.: HOX radical regeneration in the oxidation of isoprene, Phys. Chem. Chem. Phys., 11, 5935–5939, https://doi.org/10.1039/b908511d, 2009. a
Peeters, J., Müller, J.-F., Stavrakou, T., and Nguyen, V. S.: Hydroxyl radical recycling in isoprene oxidation driven by hydrogen bonding and hydrogen tunneling: The upgraded LIM1 mechanism, J. Phys. Chem. A, 118, 8625–8643, https://doi.org/10.1021/jp5033146, 2014. a, b, c, d
Perring, A. E., Wisthaler, A., Graus, M., Wooldridge, P. J., Lockwood, A. L., Mielke, L. H., Shepson, P. B., Hansel, A., and Cohen, R. C.: A product study of the isoprene + NO3 reaction, Atmos. Chem. Phys., 9, 4945–4956, https://doi.org/10.5194/acp-9-4945-2009, 2009. a, b
Robinson, M. A., Neuman, J. A., Huey, L. G., Roberts, J. M., Brown, S. S., and Veres, P. R.: Temperature-dependent sensitivity of iodide chemical ionization mass spectrometers, Atmos. Meas. Tech., 15, 4295–4305, https://doi.org/10.5194/amt-15-4295-2022, 2022. a
Rohrer, F., Bohn, B., Brauers, T., Brüning, D., Johnen, F.-J., Wahner, A., and Kleffmann, J.: Characterisation of the photolytic HONO-source in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 5, 2189–2201, https://doi.org/10.5194/acp-5-2189-2005, 2005. a
Rollins, A. W., Kiendler-Scharr, A., Fry, J. L., Brauers, T., Brown, S. S., Dorn, H.-P., Dubé, W. P., Fuchs, H., Mensah, A., Mentel, T. F., Rohrer, F., Tillmann, R., Wegener, R., Wooldridge, P. J., and Cohen, R. C.: Isoprene oxidation by nitrate radical: alkyl nitrate and secondary organic aerosol yields, Atmos. Chem. Phys., 9, 6685–6703, https://doi.org/10.5194/acp-9-6685-2009, 2009. a, b, c, d, e
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003. a
Schwantes, R. H., Teng, A. P., Nguyen, T. B., Coggon, M. M., Crounse, J. D., St. Clair, J. M., Zhang, X., Schilling, K. A., Seinfeld, J. H., and Wennberg, P. O.: Isoprene NO3 oxidation products from the RO2 + HO2 pathway, J. Phys. Chem. A, 119, 10158–10171, https://doi.org/10.1021/acs.jpca.5b06355, 2015. a, b, c, d, e, f, g, h, i, j, k, l, m, n, o, p, q, r, s, t, u, v, w, x, y, z
Sobanski, N., Schuladen, J., Schuster, G., Lelieveld, J., and Crowley, J. N.: A five-channel cavity ring-down spectrometer for the detection of NO2, NO3, N2O5, total peroxy nitrates and total alkyl nitrates, Atmos. Meas. Tech., 9, 5103–5118, https://doi.org/10.5194/amt-9-5103-2016, 2016. a, b
Stone, D., Whalley, L. K., and Heard, D. E.: Tropospheric OH and HO2 radicals: field measurements and model comparisons, Chem. Soc. Rev., 41, 6348–6404, https://doi.org/10.1039/C2CS35140D, 2012. a
Stone, D., Evans, M. J., Walker, H., Ingham, T., Vaughan, S., Ouyang, B., Kennedy, O. J., McLeod, M. W., Jones, R. L., Hopkins, J., Punjabi, S., Lidster, R., Hamilton, J. F., Lee, J. D., Lewis, A. C., Carpenter, L. J., Forster, G., Oram, D. E., Reeves, C. E., Bauguitte, S., Morgan, W., Coe, H., Aruffo, E., Dari-Salisburgo, C., Giammaria, F., Di Carlo, P., and Heard, D. E.: Radical chemistry at night: comparisons between observed and modelled HOx, NO3 and N2O5 during the RONOCO project, Atmos. Chem. Phys., 14, 1299–1321, https://doi.org/10.5194/acp-14-1299-2014, 2014. a
Tan, Z., Fuchs, H., Lu, K., Hofzumahaus, A., Bohn, B., Broch, S., Dong, H., Gomm, S., Häseler, R., He, L., Holland, F., Li, X., Liu, Y., Lu, S., Rohrer, F., Shao, M., Wang, B., Wang, M., Wu, Y., Zeng, L., Zhang, Y., Wahner, A., and Zhang, Y.: Radical chemistry at a rural site (Wangdu) in the North China Plain: observation and model calculations of OH, HO2 and RO2 radicals, Atmos. Chem. Phys., 17, 663–690, https://doi.org/10.5194/acp-17-663-2017, 2017. a
Tan, Z., Hantschke, L., Kaminski, M., Acir, I.-H., Bohn, B., Cho, C., Dorn, H.-P., Li, X., Novelli, A., Nehr, S., Rohrer, F., Tillmann, R., Wegener, R., Hofzumahaus, A., Kiendler-Scharr, A., Wahner, A., and Fuchs, H.: Atmospheric photo-oxidation of myrcene: OH reaction rate constant, gas-phase oxidation products and radical budgets, Atmos. Chem. Phys., 21, 16067–16091, https://doi.org/10.5194/acp-21-16067-2021, 2021. a
Tsiligiannis, E., Wu, R., Lee, B. H., Salvador, C. M. G., Priestley, M., Carlsson, P. T. M., Novelli, S. K. A., Vereecken, L., Fuchs, H., Mayhew, A. W., Hamilton, J. F., Edwards, P. M., Fry, J. L., Brownwood, B., Brown, S. S., Wild, R. J., Bannan, T. J., Coe, H., Allan, J., Surrat, J. D., Bacak, A., Artaxo, P., Percival, C., Guo, S., Hu, M., Wang, T., Mentel, T. F., Thornton, J. A., and Hallquist, M.: A four carbon organonitrate as a significant product of secondary isoprene chemistry, Geophys. Res. Lett., 49, e2021GL097366, https://doi.org/10.1029/2021GL097366, 2022. a, b, c, d, e, f
Vereecken, L.: Replication Data for: Comparison of isoprene chemical mechanisms at atmospheric night-time conditions in chamber experiments: Evidence of hydroperoxy aldehydes and epoxy products from NO3 oxidation, Jülich Data [data set], https://doi.org/10.26165/JUELICH-DATA/YWB5P1, 2022. a
Vereecken, L., Carlsson, P. T. M., Novelli, A., Bernard, F., Brown, S. S., Cho, C., Crowley, J. N., Fuchs, H., Mellouki, W., Reimer, D., Shenolikar, J., Tillmann, R., Zhou, L., Kiendler-Scharr, A., and Wahner, A.: Theoretical and experimental study of peroxy and alkoxy radicals in the NO3-initiated oxidation of isoprene, Phys. Chem. Chem. Phys., 23, 5496–5515, https://doi.org/10.1039/D0CP06267G, 2021. a, b, c, d, e, f, g, h, i, j, k, l, m, n, o, p, q, r, s, t, u, v, w, x, y, z, aa, ab, ac, ad, ae, af, ag, ah, ai, aj, ak, al, am, an, ao, ap, aq, ar, as, at, au, av, aw, ax, ay
Wagner, N. L., Dubé, W. P., Washenfelder, R. A., Young, C. J., Pollack, I. B., Ryerson, T. B., and Brown, S. S.: Diode laser-based cavity ring-down instrument for NO3, N2O5, NO, NO2 and O3 from aircraft, Atmos. Meas. Tech., 4, 1227–1240, https://doi.org/10.5194/amt-4-1227-2011, 2011. a
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., St Clair, J. M., Teng, A. P., Zhang, X., and Seinfeld, J. H.: Gas-Phase reactions of isoprene and its major oxidation products, Chem. Rev., 118, 3337–3390, https://doi.org/10.1021/acs.chemrev.7b00439, 2018. a, b, c, d, e, f, g, h, i, j, k, l, m, n, o, p
Whalley, L. K., Edwards, P. M., Furneaux, K. L., Goddard, A., Ingham, T., Evans, M. J., Stone, D., Hopkins, J. R., Jones, C. E., Karunaharan, A., Lee, J. D., Lewis, A. C., Monks, P. S., Moller, S. J., and Heard, D. E.: Quantifying the magnitude of a missing hydroxyl radical source in a tropical rainforest, Atmos. Chem. Phys., 11, 7223–7233, https://doi.org/10.5194/acp-11-7223-2011, 2011. a
Wolfe, G. M., Crounse, J. D., Parrish, J. D., St. Clair, J. M., Beaver, M. R., Paulot, F., Yoon, T., Wennberg, P. O., and Keutsch, F. N.: Photolysis, OH reactivity and ozone reactivity of a proxy for isoprene-derived hydroperoxyenals, Phys. Chem. Chem. Phys., 14, 7276–7286, https://doi.org/10.1039/C2CP40388A, 2012. a, b, c
Wu, R., Vereecken, L., Tsiligiannis, E., Kang, S., Albrecht, S. R., Hantschke, L., Zhao, D., Novelli, A., Fuchs, H., Tillmann, R., Hohaus, T., Carlsson, P. T. M., Shenolikar, J., Bernard, F., Crowley, J. N., Fry, J. L., Brownwood, B., Thornton, J. A., Brown, S. S., Kiendler-Scharr, A., Wahner, A., Hallquist, M., and Mentel, T. F.: Molecular composition and volatility of multi-generation products formed from isoprene oxidation by nitrate radical, Atmos. Chem. Phys., 21, 10799–10824, https://doi.org/10.5194/acp-21-10799-2021, 2021. a, b, c, d
Xiong, F., McAvey, K. M., Pratt, K. A., Groff, C. J., Hostetler, M. A., Lipton, M. A., Starn, T. K., Seeley, J. V., Bertman, S. B., Teng, A. P., Crounse, J. D., Nguyen, T. B., Wennberg, P. O., Misztal, P. K., Goldstein, A. H., Guenther, A. B., Koss, A. R., Olson, K. F., de Gouw, J. A., Baumann, K., Edgerton, E. S., Feiner, P. A., Zhang, L., Miller, D. O., Brune, W. H., and Shepson, P. B.: Observation of isoprene hydroxynitrates in the southeastern United States and implications for the fate of NOx, Atmos. Chem. Phys., 15, 11257–11272, https://doi.org/10.5194/acp-15-11257-2015, 2015. a
Xiong, F., Borca, C. H., Slipchenko, L. V., and Shepson, P. B.: Photochemical degradation of isoprene-derived 4,1-nitrooxy enal, Atmos. Chem. Phys., 16, 5595–5610, https://doi.org/10.5194/acp-16-5595-2016, 2016. a