the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 18 Mar 2022
| 18 Mar 2022
OH-initiated atmospheric degradation of hydroxyalkyl hydroperoxides: mechanism, kinetics, and structure–activity relationship
Long Chen
Yu Huang
Yonggang Xue
Zhihui Jia
Wenliang Wang
Hydroxyalkyl hydroperoxides (HHPs), formed in the reactions of Criegee intermediates (CIs) with water vapor, play essential roles in the formation of secondary organic aerosol (SOA) under atmospheric conditions. However, the transformation mechanisms for the OH-initiated oxidation of HHPs remain incompletely understood. Herein, the quantum chemical and kinetics modeling methods are applied to explore the mechanisms of the OH-initiated oxidation of the distinct HHPs (HOCH2OOH, HOCH(CH3)OOH, and HOC(CH3)2OOH) formed from the reactions of CH2OO, anti-CH3CHOO, and (CH3)2COO with water vapor. The calculations show that the dominant pathway is H-abstraction from the -OOH group in the initiation reactions of the OH radical with HOCH2OOH and HOC(CH3)2OOH. H-abstraction from the -CH group is competitive with that from the -OOH group in the reaction of the OH radical with HOCH(CH3)OOH. The barrier of H-abstraction from the -OOH group slightly increases when the number of methyl groups increase. In pristine environments, the self-reaction of the RO2 radical initially produces a tetroxide intermediate via oxygen-to-oxygen coupling, and then it decomposes into propagation and termination products through asymmetric two-step O–O bond scission, in which the rate-limiting step is the first O–O bond cleavage. The barrier height of the reactions of distinct RO2 radicals with the HO2 radical is not affected by the number of methyl substitutions. In urban environments, the reaction with O2 to form formic acid and the HO2 radical is the dominant removal pathway for the HOCH2O radical formed from the reaction of the HOCH2OO radical with NO. The β-site C–C bond scission is the dominant pathway in the dissociation of the HOCH(CH3)O and HOC(CH3)2O radicals formed from the reactions of NO with HOCH(CH3)OO and HOC(CH3)2OO radicals. These new findings deepen our understanding of the photochemical oxidation of hydroperoxides under realistic atmospheric conditions.
- Article
(5508 KB) - Full-text XML
-
Supplement
(2298 KB) - BibTeX
- EndNote





Hydroxyalkyl hydroperoxides (HHPs), formed in the reactions of Criegee intermediates (CIs) with water vapor and in the initiation OH-addition with subsequent HO2-termination reactions, play important roles in the formation of secondary organic aerosol (SOA; Qiu et al., 2019; Kumar et al., 2014). The CIs formed from the ozonolysis of alkenes are characterized by high reactivity and excess energy, which can either prompt the unimolecular decay to an OH radical or, after collisional stabilization, bimolecular reactions with various trace gases such as SO2, NO2, and H2O to produce sulfate, nitrate, and SOA, thereby influencing air quality and human health (Lester and Klippenstein, 2018; Chen et al., 2017, 2019; Liu et al., 2019; Chhantyal-Pun et al., 2017; Anglada and Solé, 2016; Gong and Chen, 2021). Among these reactions, the bimolecular reaction of CIs with water is regarded as the dominant chemical sink, since its concentration (1.3–8.3×1017 molecules cm−3) is several orders of magnitude greater than those of SO2 and NO2 (∼1012 molecules cm−3) in the atmosphere (Huang et al., 2015; Khan et al., 2018; Taatjes et al., 2013; Taatjes, 2017). The primary products of the reaction of CIs with water are highly oxygenated HHPs, which are difficult to detect and identify through the available analytical techniques because of their thermal instability (Qiu et al., 2019; Anglada and Solé, 2016; Chao et al., 2015; Chen et al., 2016a; Ryzhkov and Ariya, 2003).
Due to the presence of both hydroxyl and perhydroxy moieties, HHPs have relatively low volatility and contribute significantly to the formation of SOA (Qiu et al., 2019). The atmospheric degradation of HHPs initiated by an OH radical is expected to be one of the dominant loss processes because the OH radical is a powerful oxidizing agent (Gligorovski et al., 2015; Allen et al., 2018). The reaction with the OH radical includes three possible H-abstraction channels, namely (a) alkyl hydrogen, (b) -OH hydrogen, and (c) -OOH hydrogen, which are followed by further reactions to generate organic peroxy radicals (RO2) as reactive intermediates (Allen et al., 2018). On the basis of our current mechanistic understanding, RO2 radicals have three possible channels in pristine environments: (1) RO2 radicals can proceed self- and cross-reactions to generate the alkoxy radical RO, alcohol, carbonyl, and accretion products (Berndt et al., 2018; Zhang et al., 2012; Valiev et al., 2019); (2) RO2 radicals can react with the HO2 radical to form closed-shell hydroperoxide (ROOH), an RO radical, an OH radical, etc. (Dillon and Crowley, 2008; Iyer et al., 2018); (3) RO2 radicals can undergo autoxidation through intramolecular H-shift and alternating O2-addition steps to produce highly oxygenated organic molecules (HOMs), which have been identified as low volatility compounds that contribute to the formation of SOA (Crounse et al., 2013; Jokinen et al., 2014; Wang et al., 2018; Ehn et al., 2014; Iyer et al., 2021). In urban environments, RO2 radicals can react with NOx to generate peroxynitrate (RO2NO2), organic nitrate (RONO2), RO radicals, and other SOA precursors (Wang et al., 2017; Xu et al., 2014, 2020; Ma et al., 2021). The relative importance of distinct pathways is highly dependent on the nature of RO2 radicals and the concentrations of coreactants.
Hydroxymethyl hydroperoxide (HMHP, HOCH2OOH), the simplest HHP derived from the ozonolysis of all terminal alkenes in the presence of water, is observed in significant abundance in the atmosphere (Allen et al., 2018). The measured concentration of HMHP varies considerably depending on the location, season, and altitude, and its concentration has been reported to be as high as 5 ppbv in forested regions (Allen et al., 2018; Francisco and Eisfeld, 2009). In one study, the concentration of HMHP was measured during the summer of 2013 in the southeastern United States, and it was found that the average mixing ratio of HMHP was 0.25 ppbv with a maximum of 4.0 ppbv in the boundary layer (Allen et al., 2018). Allen et al. (2018) conducted the OH-initiated oxidation of HMHP in an environmental chamber and simulated the effect of HMHP oxidation on global formic acid production by applying the chemical transport model GEOS-Chem. They found that H-abstraction from the methyl group of HMHP results in the formation of formic acid and contributes approximately 1.7 Tg yr−1 to global formic acid production. Francisco and Eisfeld (2009) employed ab initio CCSD (T) and MP2 methods to study the atmospheric oxidation mechanism of HMHP initiated by the OH radical, and they also concluded that the degradation of HMHP can contribute to the formation of formic acid in the atmosphere. Additionally, the unimolecular decomposition of HMHP is another key removal process in the atmosphere. Chen et al. (2016b) found that the formation of CH2O and H2O2 is more preferable than the production of HCOOH and H2O. Kumar et al. (2014) also concluded that the aldehyde- or ketone-forming pathway is kinetically favored over the carboxylic acid-forming channel in the unimolecular decomposition of various HHPs. All of the above milestone investigations offer highly useful information on the decomposition of HHPs in the gas phase. However, to the best of our knowledge, there have been few studies on the subsequent transformations of the resulting H-abstraction products formed from the OH-initiated oxidation of larger HHPs. The effect of the size and number of substituents on the rates and outcomes of SOA precursors (e.g., ROOR, HOMs) is uncertain up to now. Therefore, it is necessary to evaluate the potential of larger HHPs and their oxidation products to substantial SOA formation under different NOx conditions.
In the present study, the mechanisms and kinetics of distinct HHPs oxidation initiated by OH radicals are investigated by employing quantum chemical and kinetics modeling methods. For the resulting H-abstraction products of RO2 radicals, the subsequent reactions involving self-reaction, isomerization, and reaction with HO2 radicals are considered in the absence of NO, while the subsequent reactions including addition, decomposition, and H-abstraction by O2 are considered in the presence of NO. The HHPs investigated in the present study are generated from the bimolecular reactions of distinct carbonyl oxides (CH2OO, anti-CH3CHOO, and (CH3)2COO) with water vapor.
2.1 Electronic structure and energy calculations
The equilibrium geometries of all open-shell species, including reactant (R), pre-reactive complex (RC), transition state (TS), post-reactive complex (PC), and product (P), are fully optimized at the unrestricted M06-2X/6-311+G(2df,2p) level of theory (UM06-2X; Zhao and Truhlar, 2006; Zheng and Truhlar, 2009), whereas all the closed-shell species are optimized at the restricted M06-2X/6-311+G(2df,2p) level of theory (RM06-2X). This is because the M06-2X functional has been proven to be reliable for describing thermochemistry, kinetics, and non-covalent interactions (Zhao and Truhlar, 2008). Harmonic vibrational frequencies are performed at the same level to verify that each stationary point is either a true minima (with no imaginary frequency) or a transition state (with one imaginary frequency). Zero-point vibrational energy (ZPVE) and Gibbs free-energy corrections (Gcorr) from harmonic vibrational frequencies are scaled by a factor of 0.98 (Zhao and Truhlar, 2006). Intrinsic reaction coordinate (IRC) calculations are performed to verify the connection between the transition state and the designated reactant and product (Fukui, 1981). The single-point energies are calculated at the (U/R)M06-2X/ma-TZVP level of theory (Zheng et al., 2011).
The tetroxide intermediate formed from the self-reaction of the RO2 radical proceeds through asymmetric two-step O–O bond scission to produce a caged tetroxide intermediate of overall singlet multiplicity comprising two same-spin alkoxyl radicals (spin down) and triplet oxygen (spin up). This type of reaction mechanism can be described by broken symmetry unrestricted DFT (UDFT) and multi-reference CASSCF methods (Lee et al., 2016; Bach et al., 2005). Previous studies have demonstrated that the UDFT method is suitable for identifying the minimum of a metastable singlet caged radical complex and the transition state of O–O bond homolysis, and the resulting energies obtained using the UDFT method are comparable to those obtained using the more accurate and expensive CASSCF method (Lee et al., 2016; Bach et al., 2005). In the present study, the UDFT method, which provides a balance between computational accuracy and efficiency, is selected to study asymmetric O–O bond scission. The broken symmetry UM06-2X/6-311+G(2df,2p) method is applied to generate the initial guesses for the geometries of the tetroxide intermediate and transition state with mixed HOMO and LUMO (S2≈1) by using the guess = mix keyword. The single-point energies are refined at the UM06-2X/ma-TZVP level of theory.
To further evaluate the reliability of the employed method for predicting the reaction mechanisms, the single-point energies of all stationary points involved in the initiation reactions of OH radicals with distinct HHPs are recalculated at the (U/R)CCSD(T)/6-311+G(2df,2p) level of theory based on the (U/R)M06-2X optimized geometries. Furthermore, the stability of the pre-reactive complexes is assessed by performing basis set superposition error (BSSE) using the counterpoise method (Boys and Bernardi, 1970). For simplicity, no prefix is used throughout this article. Herein, the Gibbs free energy (G) for each species is obtained by combining the single-point energy with the Gibbs correction (). The electronic energy () and free energy () barriers are defined as the difference in energy between the transition state and pre-reactive complex ( and ). The reaction free energy (ΔG) is referred to the difference in energy between the product and reactant (). The calculated and for the initiation H-abstraction pathways are summarized in Table S1 in the Supplement. As shown in Table S1, the mean absolute deviations (MADs) of and between the CCSD(T)/6-311+G(2df,2p) and M06-2X/ma-TZVP approaches are 0.43 and 0.45 kcal mol−1, respectively; the largest deviations of and are 1.2 and 1.1 kcal mol−1, respectively. These results reveal that the energies obtained using the M06-2X/ma-TZVP method are in good accord with those obtained using the gold-standard coupled-cluster approach CCSD (T) within the uncertainties of systematic errors. Therefore, the M06-2X/ma-TZVP method is selected to investigate the atmospheric degradation of HHP initiated by OH radicals under different conditions. In the following sections, is applied to construct the reaction profiles unless otherwise stated.
For the H-shift reactions of RO2 radicals, reactants, transition states, and products have multiple conformers. Previous literature has demonstrated that the reaction kinetics of multi-conformer involvement are more precise than that of single-conformer approximation (Møller et al., 2016, 2020). Herein, the multi-conformer treatment is performed to investigate the H-shift reactions of RO2 radicals. A conformer search within the Molclus program is employed to generate a pool of conformers for RO2 radicals (Lu, 2020). The selected conformers are further optimized at the M06-2X/6-311+G(2df,2p) level of theory, followed by single-point energy calculations at the M06-2X/ma-TZVP level of theory. On the basis of the calculated Gibbs free energies, the Boltzmann populations (wi) of each RO2 conformer are expressed as Eq. (1):
where ΔGi is the relative Gibbs free energy of conformer i, kB is Boltzmann's constant, and T is temperature in Kelvin. All the quantum chemical calculations are performed by using the Gaussian 09 program (Frisch et al., 2009).
2.2 Kinetics calculations
The rate coefficients of unimolecular reactions are calculated by using the Rice–Ramsperger–Kassel–Marcus theory coupled with the energy-grained master equation (RRKM-ME) method (Holbrook et al., 1996), and the rate coefficients of bimolecular reactions are determined by utilizing traditional transition state theory (TST; Fernández-Ramos et al., 2007). RRKM-ME calculations are performed by using the MESMER 6.0 program (Glowacki et al., 2012). N2 is used as the buffer gas. A single exponential down model is employed to simulate the collision energy transfer (〈ΔE〉down=200 cm−1). The collisional Lennard-Jones parameters are estimated by using an empirical formula proposed by Gilbert and Smith (1990). For the H-shift reactions of RO2 radicals, the rate coefficients are determined by employing the multi-conformer transition state theory (MC-TST) approach (Møller et al., 2016). The MC-TST rate coefficient kMC-TST is calculated by the sum of individual intrinsic reaction coordinate TST (IRC-TST) rate coefficient kIRC-TST, each weighted by the Boltzmann population of the corresponding RO2 conformer (Møller et al., 2016):
where kIRC-TSTi represents the rate coefficient of conformer i, and wi is the relative Boltzmann population of the corresponding reactant connected to TSi. The one-dimensional asymmetry Eckart model is employed to calculate the tunneling correction (Eckart, 1930). Considering the uncertainty in barrier heights (∼1.0 kcal mol−1 by the M06-2X method) and in tunneling corrections, the uncertainty of the calculated rate coefficient is about 1 order of magnitude in the present study.
3.1 Initiation reaction of HHPs with an OH radical
Previous literatures have proposed that the lifetime of CI with respect to the reaction with water vapor exhibits strong dependence on the nature of CI (Anglada and Solé, 2016; Taatjes et al., 2013; Anglada et al., 2011), and the primary product is HHPs in both the gas phase and air–water interfaces (Chao et al., 2015; Chen et al., 2016a; Smith et al., 2015; Zhu et al., 2016; Zhong et al., 2018). In the present study, we mainly consider three kinds of HHPs originating from the addition of water to CH2OO and methyl-substituted CI (anti-CH3CHOO and (CH3)2COO). The lowest-energy HHP conformers (HOCH2OOH, HOCH(CH3)OOH, and HOC(CH3)2OOH) are obtained from a previous study, as shown in Fig. 1 (Chen et al., 2019), and they are selected as the model system to investigate the atmospheric degradation mechanism of HHP initiated by the OH radical. Letters and numbers are used to label carbon, oxygen, and hydrogen atoms at different reaction sites.
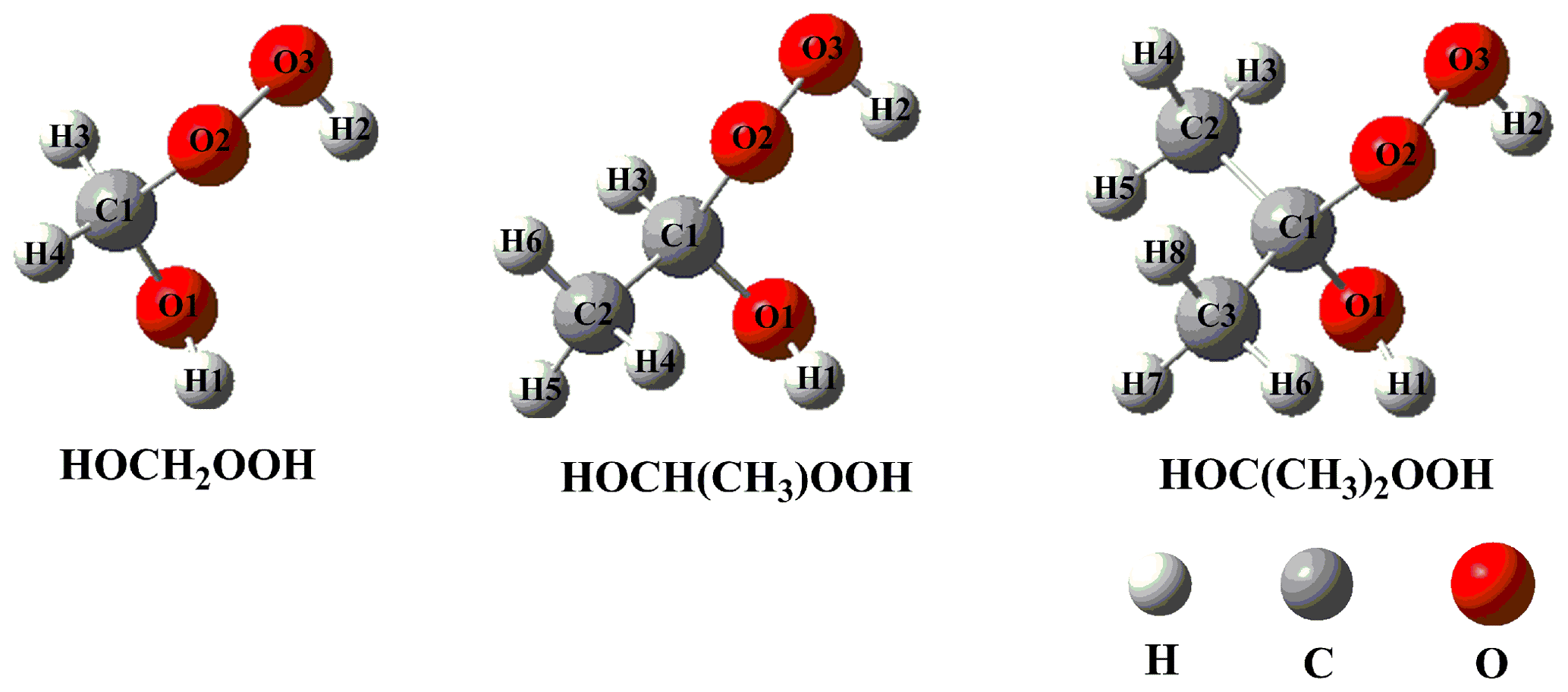
The free-energy and electronic-energy potential energy surfaces (PESs) for the initiation reactions of OH radicals with HOCH2OOH, HOCH(CH3)OOH, and HOC(CH3)2OOH are presented in Figs. 2–4 and S1–S3 in the Supplement, respectively. The optimized geometries of all stationary points are displayed in Figs. S6–S8 in the Supplement, respectively. As can be seen in Fig. 2, the reaction of HOCH2OOH with OH radicals proceeds through four pathways: H-abstraction from the -O1H1 (R1), -C1H3 (R2), -C1H4 (R3), and -O2O3H2 (R4) groups. For each pathway, a pre-reactive complex with a six- or seven-membered ring structure is formed in the entrance channel, which is stabilized by hydrogen bond interactions between the oxygen atom of an OH radical and the abstraction hydrogen atom of HOCH2OOH, and between the remnant hydrogen atom of an OH radical and one of the oxygen atoms of HOCH2OOH (Fig. S6). Then, it surmounts the modest barrier that is higher in energy than the reactants to react. The reaction barriers decrease in the order of 6.4 (R1) > 5.8 (R2) ≈ 5.4 (R3) > 1.5 (R4) kcal mol−1, indicating that H-abstraction from the -O2O3H2 group (R4) is more preferable than those from the -O1H1, -C1H3 and -C1H4 groups (R1–R3). The same conclusion is also derived from the energy barriers that R4 is the most favorable H-abstraction pathway (Fig. S1). The difference in barrier heights can be attributed to the bond dissociation energy (BDE) of the multiple types of bonds in the HOCH2OOH molecule. BDE decreases in the order of 103.7 (O1–H1) > 98.2 (C1–H3) ≈ 97.4 (C1–H4) > 87.2 (O3–H2) kcal mol−1, and this order is consistent with that of the barrier heights of H-abstraction reactions. Their reaction free-energy values indicate that the exothermicity of R4 is the largest among these four H-abstraction reactions. Based on the above discussions, it is concluded that H-abstraction from the -O2O3H2 group resulting in the formation of an HOCH2OO radical (R4) is feasible both thermodynamically and kinetically.
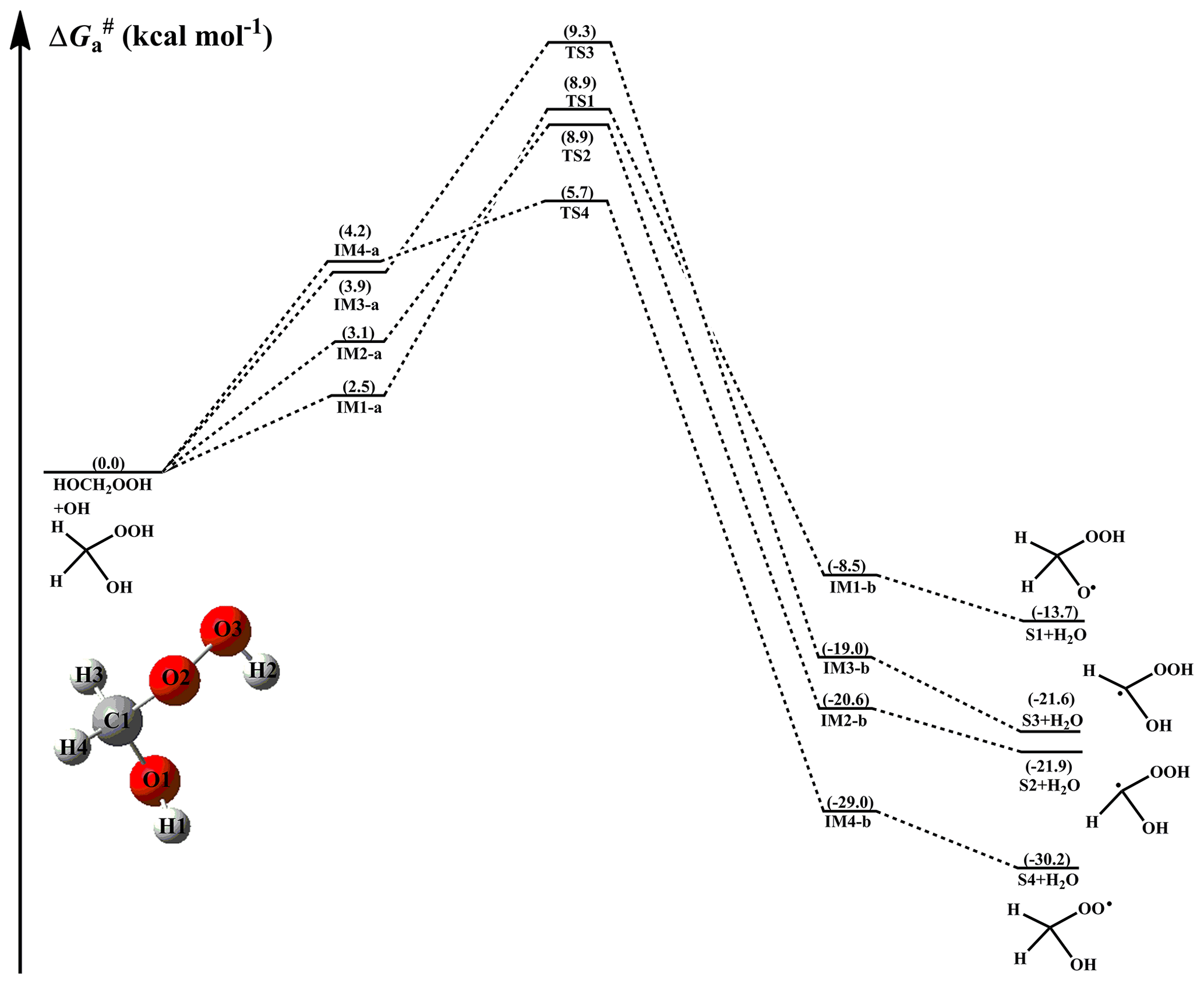
Figure 2PES () for the OH-initiated reactions of HOCH2OOH from the CH2OO+H2O reaction predicted at the M06-2X/ma-TZVP//M06-2X/6-311+G(2df,2p) level of theory (a and b represent the pre-reactive and post-reactive complexes).
Given the multiple reaction sites of hydrogen atoms, the atmospheric transformation of HOCH(CH3)OOH from the anti-CH3CHOO+H2O reaction should have six possible H-abstraction pathways as presented in Fig. 3. As shown in Fig. 3, each H-abstraction reaction begins with the formation of a weakly bound hydrogen bonded pre-reactive complex with a six- or seven-membered ring structure in the entrance channel (Fig. S7). Then it immediately transforms into the respective product via the corresponding transition state. The of H-abstraction from the -C1H3 (R6) and -O2O3H2 (R8) groups are 2.2 and 1.7 kcal mol−1, respectively, which are ∼4–5 kcal mol−1 lower than those from the -O1H1 (R5) and -CH3 (R7) groups. This result shows that R6 and R8 have nearly identical importance in the atmosphere. Compared with the barriers of H-abstraction at the Cα-position (R6) and Cβ-position (R7), it can be found that the former case is more favourable than the latter case. This conclusion is further supported by the Jara-Toro (2017, 2018) studies regarding the reactions of an OH radical with linear saturated alcohols (methanol, ethanol, and n-propanol) that H-abstraction at the Cα-position is predominant.

Figure 3PES () for the OH-initiated reactions of HOCH(CH3)OOH from the anti-CH3CHOO+H2O reaction predicted at the M06-2X/ma-TZVP//M06-2X/6-311+G(2df,2p) level of theory (a and b represent the pre-reactive and post-reactive complexes).
For the OH-initiated oxidation of HOCH(CH3)OOH from the syn-CH3CHOO+H2O reaction, the corresponding free-energy and electronic-energy PESs are displayed in Figs. S4 and S5 in the Supplement, respectively. From Fig. S4, it can be seen that H-abstraction by OH radicals from HOCH(CH3)OOH has six possible pathways. For each pathway, a per-reactive complex is formed prior to the corresponding transition state, and then it overcomes a modest barrier to reaction. The of R6′ and R8′ are 2.3 and 1.8 kcal mol−1, respectively, which are about 5 kcal mol−1 lower than those of R5′ and R7′. The result shows that H-abstraction from the -CH (R6′) and -OOH (R8′) groups is preferable kinetically. The same conclusion is also derived from the energy barriers : the R6′ and R8′ are the most favourable H-abstraction pathways (Fig. S5). It should be noted that although the barriers of R6′ and R8′ are comparable, the exoergicity of R6′ is significantly lower than that of R8′. The aforementioned conclusions are consistent with the results derived from the OH-initiated oxidation of HOCH(CH3)OOH from the anti-CH3CHOO+H2O reaction. Zhou et al. (2019) demonstrated that the bimolecular reaction of syn-CH3CHOO with water leading to the formation of HOCH(CH3)OOH is of less importance in the atmosphere, while the unimolecular decay to OH radicals is the major loss process of syn-CH3CHOO. Therefore, in the present study, we mainly focus on the subsequent mechanism of intermediates generated from OH-initiated oxidation of HOCH(CH3)OOH from the anti-CH3CHOO+H2O reaction.
From Fig. 4, it can be seen that H-abstraction from HOC(CH3)2OOH has eight possible H-abstraction pathways. All the H-abstraction reactions are strongly exothermic and spontaneous, suggesting that they are thermodynamically feasible under atmospheric conditions. It deserves mentioning that the release of energy of R12 is significantly greater than that of R9–R11. For each H-abstraction pathway, an RC with a six- or seven-membered ring structure is formed prior to the corresponding TS, which is more stable than the separate reactants due to the hydrogen bond interactions between HOC(CH3)2OOH and the OH radical. Then, the RC overcomes a modest barrier to reaction. The of H-abstraction from the -O2O3H2 group (R12) is 2.7 kcal mol−1, which is the lowest among these eight H-abstraction reactions. This result again shows that H-abstraction from the -O2O3H2 group is the dominant pathway.
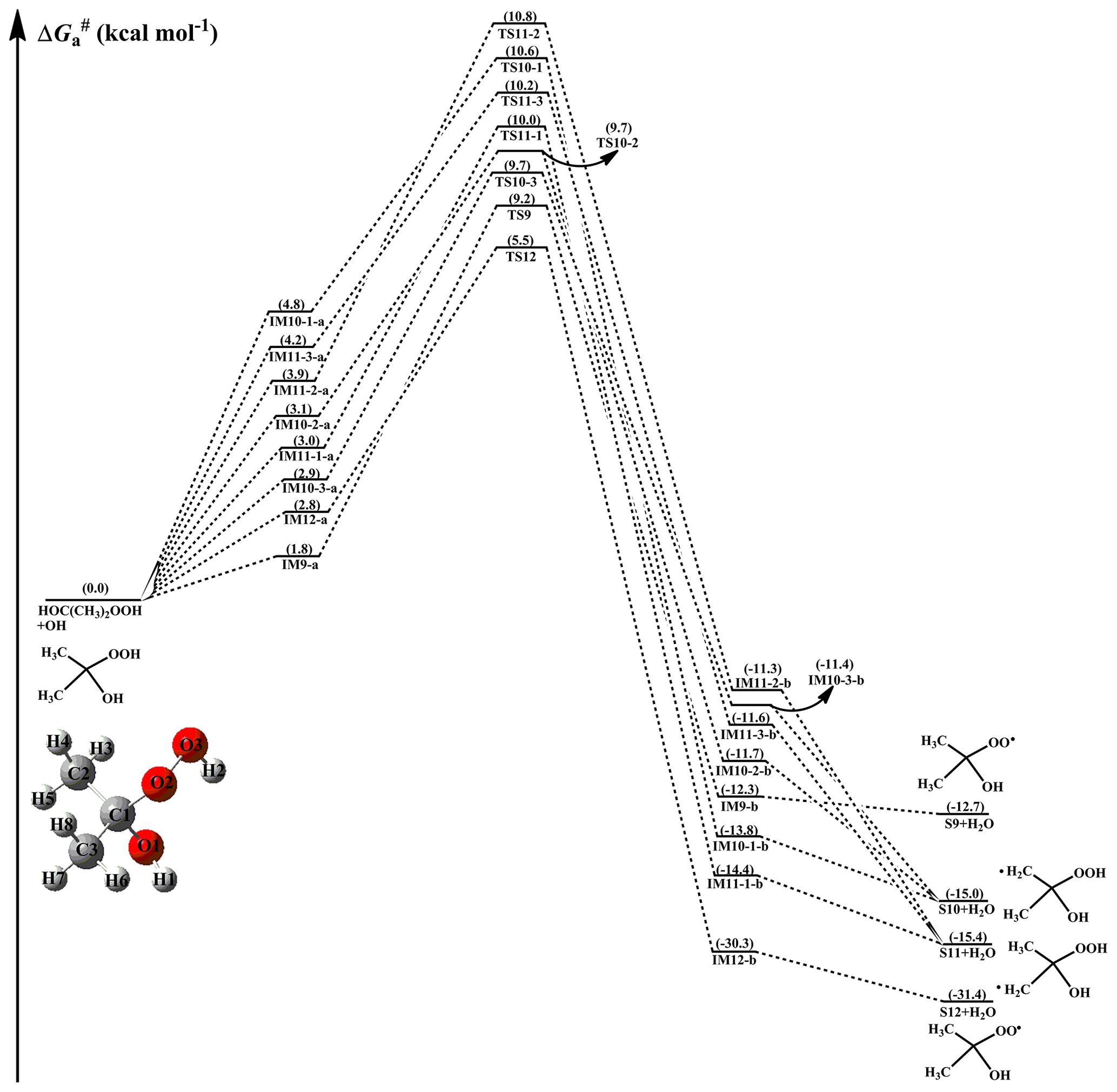
Figure 4PES () for the OH-initiated reactions of HOC(CH3)2OOH from the (CH3)2COO+H2O reaction predicted at the M06-2X/ma-TZVP//M06-2X/6-311+G(2df,2p) level of theory (a and b represent the pre-reactive and post-reactive complexes).
The rate coefficients of each H-abstraction pathway involved in the initiation reactions of distinct HHPs with OH radicals are estimated over the temperature range from 273 to 400 K, as summarized in Tables S2–S4 and Figs. S9–S11 in the Supplement. As shown in Table S2, the total rate coefficients ktot of HOCH2OOH reaction with OH radicals decrease slightly with increasing temperature. At ambient temperature, ktot is estimated to be , which is a factor of ∼5 greater than that ( , at 295 K) reported by Allen et al. (2018), who derived the result from the reaction of HMHP with OH radicals through CF3O− chemical ionization mass spectrometry (CIMS) and laser-induced fluorescence (LIF). This discrepancy can be attributed to the uncertainties associated with the barrier height and tunneling correction. kR4(O3-H2) is 1 to 2 orders of magnitude greater than kR1(O1-H1), kR2(C1-H3), and kR3(C1-H4) in the whole temperature range, indicating that R4 is the most favorable H-abstraction pathway. For example, kR4(O3-H2) is calculated to be at 298 K, which is higher than kR1(O1-H1) (), kR2(C1-H3) (), and kR3(C1-H4) () by 161, 29, and 15 times, respectively.
From Table S3, it can be seen that the total rate coefficients of the HOCH(CH3)OOH reaction with OH radicals decrease in the range between (273 K) and (400 K) with increasing temperature, and they exhibit a slightly negative temperature dependence. kR8(O3-H2) is approximately identical to in the full temperature range, and it is greater than kR5(O1-H1), kR6(C1-H3), kR7-1(C2-H4), kR7-2(C2-H5), and kR7-3(C2-H6) by 1 to 2 orders of magnitude. The result also demonstrates in that H-abstraction from the -OOH group (R8) is preferable kinetically. It should be noted that although the barriers of R8 and R6 are comparable, kR8(O3-H2) is greater than kR6(C1-H3) by approximately 1 order of magnitude over the temperature range studied. The most likely reason is the stability of pre-reactive complexes that IM8-a is more stable than IM6-a in energy. A similar conclusion is derived from the rate coefficients of the HOC(CH3)2OOH+OH reaction in that H-abstraction from the -OOH group (R12) is favorable kinetically (Table S4). The atmospheric lifetimes of HOCH2OOH, HOCH(CH3)OOH, and HOC(CH3)2OOH reactivity toward OH radicals are estimated to be 0.58–1.74, 0.60–1.79, and 1.23–3.69 h, respectively, at room temperature under typical OH radical concentrations of 5–15×106 molecules cm−3 during daylight (Long et al., 2017).
In summary, the dominant pathway is H-abstraction from the -OOH group in the initiation reactions of OH radicals with HOCH2OOH. H-abstraction from the -CH group is competitive with that from the -OOH group in the reaction of OH radicals with HOCH(CH3)OOH. Compared with the barriers of H-abstraction from the -OOH and -CH2 groups in the reaction of OH radicals with HOCH2OOH, the barrier of H-abstraction from the -CH group is reduced by 3.6 kcal mol−1, whereas the barrier of H-abstraction from the -OOH group is increased by 0.2 kcal mol−1 when a methyl group substitution occurs at the C1-position of HOCH2OOH. The dominant pathway is H-abstraction from the -OOH group in the reaction of OH radicals with HOC(CH3)2OOH, and its barrier height is increased by 1.2 kcal mol−1 compared with the OH+HOCH2OOH system. The barrier of H-abstraction from the -OOH group slightly increases when the number of methyl groups increases. It is interesting to compare the rate coefficient of the dominant pathway in the OH+HOCH2OOH system with those of the analogous reactions in the OH+HOCH(CH3)OOH and OH+HOC(CH3)2OOH reactions. It can be found that the rate coefficient is almost identical when a methyl group substitution occurs at the C1-position, whereas the rate coefficient decreases by a factor of 2–5 when two methyl groups are introduced at the C1-position.
3.2 Subsequent reactions of H-abstraction products of RO2 radicals in pristine environments
In principle, the H-abstraction products of RO2 radicals have three possible fates in pristine environments: (1) the self-reactions of RO2 radicals can produce (propagation channel), or generate or produce ROOR + O2 (termination channel), which is recognized as an important SOA precursor (Berndt et al., 2018; Zhang et al., 2012); (2) the RO2 radicals react with HO2 radicals to form hydroperoxide ROOH, alcohol, OH, and other products (Winiberg et al., 2016; Chen et al., 2021); (3) the RO2 radicals undergo autoxidation through intramolecular H-shift and alternating O2-addition steps to generate HOMs (Ehn et al., 2014; Bianchi et al., 2019; Nozière and Vereecken, 2019; Rissanen et al., 2014). The three aforementioned reactions are discussed in further detail in the subsequent subsections.
3.2.1 Reaction mechanism for the self-reaction of RO2 radicals
Self-reaction is a dominant removal pathway for RO2 radicals under low concentrations of NO and high concentrations of RO2 radicals. The self-reaction of RO2 radicals usually follows the Russell mechanism (Russell, 1957), and has four main possible pathways: (1) ; (2) ; (3) ; and (4) (Atkinson and Arey, 2003). The relative importance of different pathways varies considerably depending on the nature of RO2 radicals (Valiev et al., 2019; Lee et al., 2016). A schematic PES for the self-reaction of HOCH2OO radicals is drawn in Fig. 5. As can be seen in Fig. 5, the self-reaction of HOCH2OO radicals starts with the formation of tetroxide complexes IM13-a and IM14-a in the entrance channel, with 2.9 and 3.4 kcal mol−1 stability. Then they fragment into dimer S13 + 1O2 (R13) and HOCH2OOH + HOCHOO (R14) via TS13 and TS14 with the barriers of 43.3 and 51.5 kcal mol−1, respectively. However, the barriers of R13 and R14 are extremely high, making them irrelevant in the atmosphere.

Figure 5PES ( and , in italics) for the self-reaction of HOCH2OO radicals predicted at the M06-2X/ma-TZVP//M06-2X/6-311+G(2df,2p) level of theory.
From Fig. 5, it is seen that the self-reaction of HOCH2OO radicals proceeds via oxygen-to-oxygen coupling leading to the formation of tetroxide intermediate S14 with the electronic-energy and free-energy barriers of 7.3 and 19.6 kcal mol−1. Kumar and Francisco reported an electronic-energy barrier of 14.0 kcal mol−1 for the gas-phase decomposition of the HOCH2OO radical, which may be a new source of HO2 radicals in the troposphere (Kumar and Francisco, 2015, 2016). Compared with the electronic-energy barriers of the unimolecular dissociation of the HOCH2OO radical and its self-reaction, it can be found that the self-reaction of the HOCH2OO radical resulting in the formation of S14 is significantly feasible. The formed S14 can fragment into HOCH2O + HCOOH + HO2 via a concerted process of O2–O3 and O5–O6 bond rupture and O3–H6 bond formation with a barrier of 29.8 kcal mol−1. Alternatively, S14 can convert into the caged tetroxide intermediate S16 through asymmetric two-step O2–O3 and O5–O6 bond scission with the barriers of 19.1 and 3.1 kcal mol−1, respectively. The result shows that the latter pathway is preferred over the former channel owing to its lower barrier. The overall spin multiplicity of S16 is singlet, in which the O2 moiety maintains the triplet ground state (spin up) and is very loosely bound. In order to preserve overall singlet multiplicity, the two HOCH2O radical pairs (3(HOCH2O⋯HOCH2O)) must have triplet multiplicity (spin down). S16 can be regarded as the ground state 3O2 moving away from the two HOCH2O radical pairs that keep interacting. Due to the difficulty in performing the constrained optimization for the dissociation of S16, the 3O2 moiety is considered as a leaving moiety away from two HOCH2O radical pairs, and merely the dissociation of 3(HOCH2O⋯HOCH2O) is taken into consideration in the present study. It has three types of pathways: (1) it yields HOCH2OH and excited-state 3HCOOH through an alpha hydrogen transfer with a barrier of 14.0 and 10.2 kcal mol−1 exothermicity, followed by the excited 3HCOOH to go back to the ground-state 1HCOOH; (2) it generates two HOCH2O radicals via a barrierless process with an exoergicity of 16.9 kcal mol−1; (3) it produces dimer S17 via an intersystem crossing (ISC) step with an exoergicity of 32.1 kcal mol−1. Based on the calculated reaction barriers, the rate-limiting step is the cleavage of the O2–O3 bond (R17) in the unimolecular decay processes of S14. This conclusion coincides with the previous result obtained from the dissociation of di-t-butyl tetroxide that the rate-controlling step is the rupturing of a single O–O bond (Lee et al., 2016). Valiev et al. (2019) proposed that the ISC rate of the ROOR dimer formed from various (RO⋯R′O) systems is extremely high (>108 s−1) and exhibits a strong stereoselectivity.
Figure 6 depicts a schematic PES for the self-reaction of the HOCH(CH3)OO radical. As shown in Fig. 6, the self-reaction of the HOCH(CH3)OO radical can produce either dimer S18 and 1O2 via TS20 with a barrier of 44.4 kcal mol−1, or HOCH(CH3)OOH and HOC(CH3)OO through TS21 with a barrier of 54.3 kcal mol−1. But the barriers of R20 and R21 are considerably high, making them of less importance in the atmosphere. Alternatively, the self-reaction of the HOCH(CH3)OO radical proceeds via oxygen-to-oxygen coupling to form the tetroxide intermediate S19 with a barrier of 19.9 kcal mol−1 (Fig. 6). The formed S19 proceeds through asymmetric two-step O2–O3 and O5–O6 bond scission to produce the caged tetroxide intermediate S21 of overall singlet multiplicity comprising two same-spin alkoxyl radicals (spin down) and triplet oxygen (spin up). These two processes overcome the barriers of 21.4 and 1.3 kcal mol−1. Then, S21 decomposes into the propagation (2HOCH(CH3)O + 3O2) and termination products (HOCH(CH3)OH + 3CH3OOH + 3O2 and dimer S22 + 3O2) with an exoergicity of 12.5, 11.7, and 33.0 kcal mol−1. The rate-determining step is the rupturing of O2–O3 bond (R24) in the dissociation processes of S19.

Figure 6PES ( and , in italics) for the self-reaction of HOCH(CH3)OO radicals predicted at the M06-2X/ma-TZVP//M06-2X/6-311+G(2df,2p) level of theory.
As shown in Fig. 7, the dominant pathway for the self-reaction of the HO(CH3)2COO radical begins with the formation of tetroxide intermediate S24 via oxygen-to-oxygen coupling transition state TS28 with a barrier of 20.4 kcal mol−1; then it transforms into the caged tetroxide intermediate S26 of overall singlet spin multiplicity through asymmetric two-step O–O bond cleavage with the barriers of 22.0 and 3.4 kcal mol−1; finally, S26 can produce either two HO(CH3)2CO radicals with an exoergicity of 10.3 kcal mol−1, or dimer S27 with an exoergicity of 31.5 kcal mol−1. Compared with the self-reactions of HOCH2OO and HOCH(CH3)OO radicals, the termination product of the self-reaction of the HOC(CH3)2OO radical is exclusively dimer S27 because of the absence of an alpha hydrogen atom in the latter. Compared with the barrier of the rate-determining route R17 in the self-reaction of the HOCH2OO radical, the barrier of the rate-limiting step R29 is increased by about 3.0 kcal mol−1 when two methyl substitutions are introduced at the C1-position of the HOCH2OO radical. The reason might be attributed to the cage escape of alkoxyl radicals. Therefore, tertiary RO2 radicals have great opportunities to react with the HO2 radical or undergo autoxidation in pristine environments.
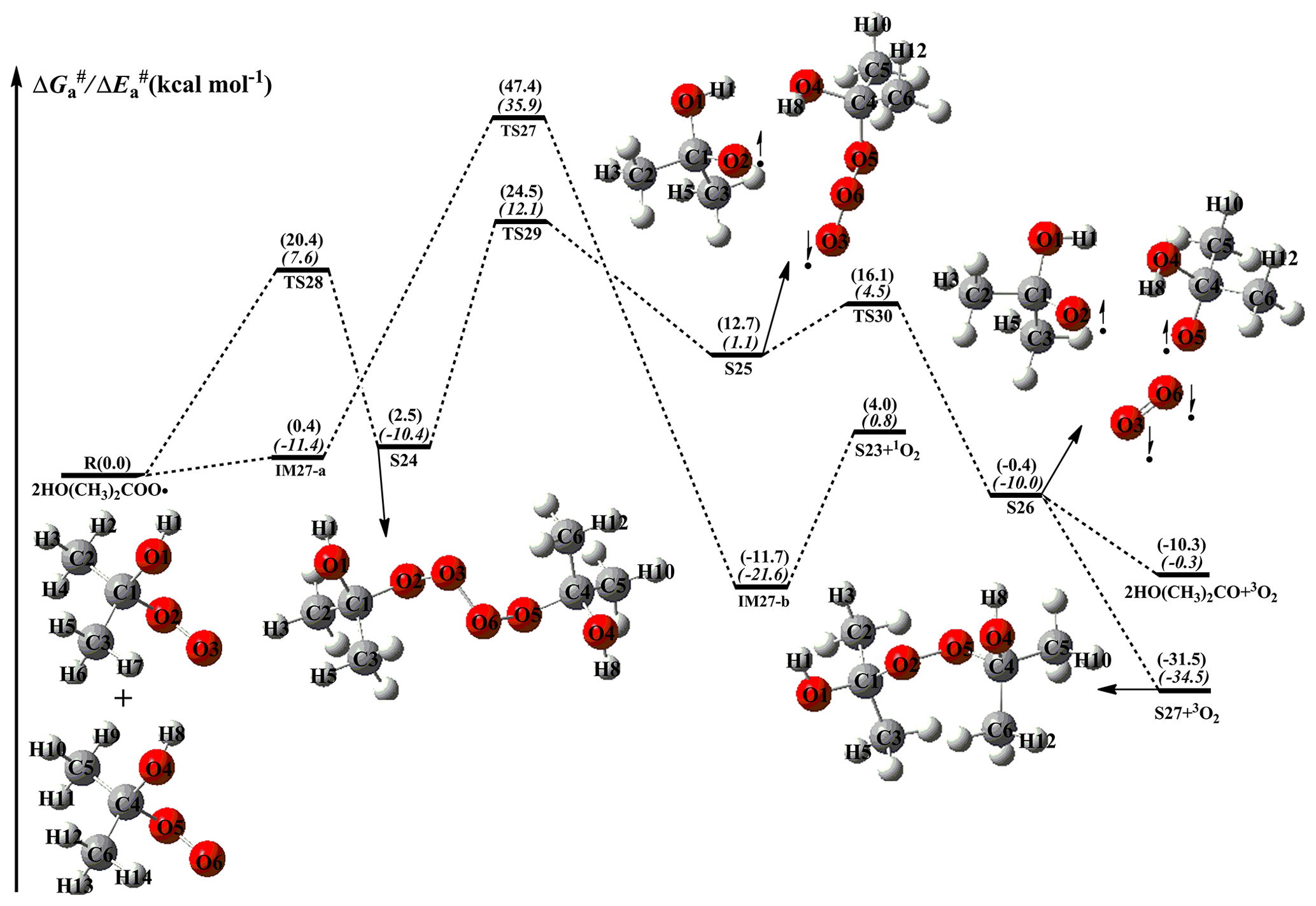
Figure 7PES ( and , in italics) for the self-reaction of HO(CH3)2COO radicals predicted at the M06-2X/ma-TZVP//M06-2X/6-311+G(2df,2p) level of theory.
3.2.2 Reaction mechanism for the reaction of RO2 radicals with an HO2 radical
When NO is present in low concentrations, the bimolecular reaction of RO2 radicals with the HO2 radical is generally expected to be the dominant pathway. The primary sources of HO2 radicals include the photo-oxidation of oxygenated volatile organic compounds (OVOCs) and the ozonolysis reaction, as well as the secondary sources include the reactions of the OH radical with CO, ozone, and VOCs, the reaction of the alkoxy radical RO with O2, and the red-light-induced decomposition of the α-hydroxy methylperoxy radical OHCH2OO (Kumar and Francisco, 2015; Stone et al., 2012; Hofzumahaus et al., 2009). The atmospheric concentration of HO2 radicals is 1.5–10×108 molecules cm−3 at the ground level in polluted urban environments (Stone et al., 2012). A schematic PES for the reactions of distinct RO2 radicals with HO2 the radical is presented in Fig. 8. As shown in Fig. 8, all the reactions are strongly exothermic and spontaneous, indicating that they are thermodynamically feasible in the atmosphere. The reaction of HOCH2OO with HO2 (R31) starts with the formation of a pre-reactive complex IM31-a in the entrance channel, which is more stable than the separate reactants by 3.8 kcal mol−1. Then, IM31-a converts into HOCH2OOH and O2 via a hydrogen atom transfer from the HO2 radical to the terminal oxygen atom of the HOCH2OO radical with a barrier of 2.0 kcal mol−1. The mechanisms of HOCH(CH3)OO + HO2 (R32) and HO(CH3)2COO + HO2 (R33) reactions are similar to that of the HOCH2OO + HO2 system. In order to avoid redundancy, a detailed discussion of the aforementioned mechanisms is not provided in the present study. Compared with the barrier of the HOCH2OO + HO2 reaction, the barrier height is lower by only 0.1 kcal mol−1 when one or two methyl substitutions occur at the C1-position of the HOCH2OO radical. This result suggests that the barrier height is not influenced by the number of methyl substitutions. The rate coefficients of the reactions of distinct RO2 radicals with the HO2 radical are summarized in Table S5 and Fig. S12 in the Supplement. As shown in Table S5, the rate coefficients kR31 of the HOCH2OO + HO2 reaction vary from (273 K) to (400 K), and they exhibit a negative temperature dependence. A similar conclusion is also obtained from the rate coefficients kR32 and kR33 in that they decrease with the temperature increasing. Notably, the rate coefficient slightly increases when the number of methyl groups increases. At ambient temperature, kR31 is estimated to be , which is consistent with the value of for the reaction of acyl peroxy radicals with the HO2 radical (Wennberg et al., 2018). The typical atmospheric concentrations of HO2 radicals are 5, 20, and 50 pptv in urban, rural, and forest environments, respectively (Bianchi et al., 2019), which translate into the pseudo-first-order rate constants [HO2] of , and s−1, respectively.
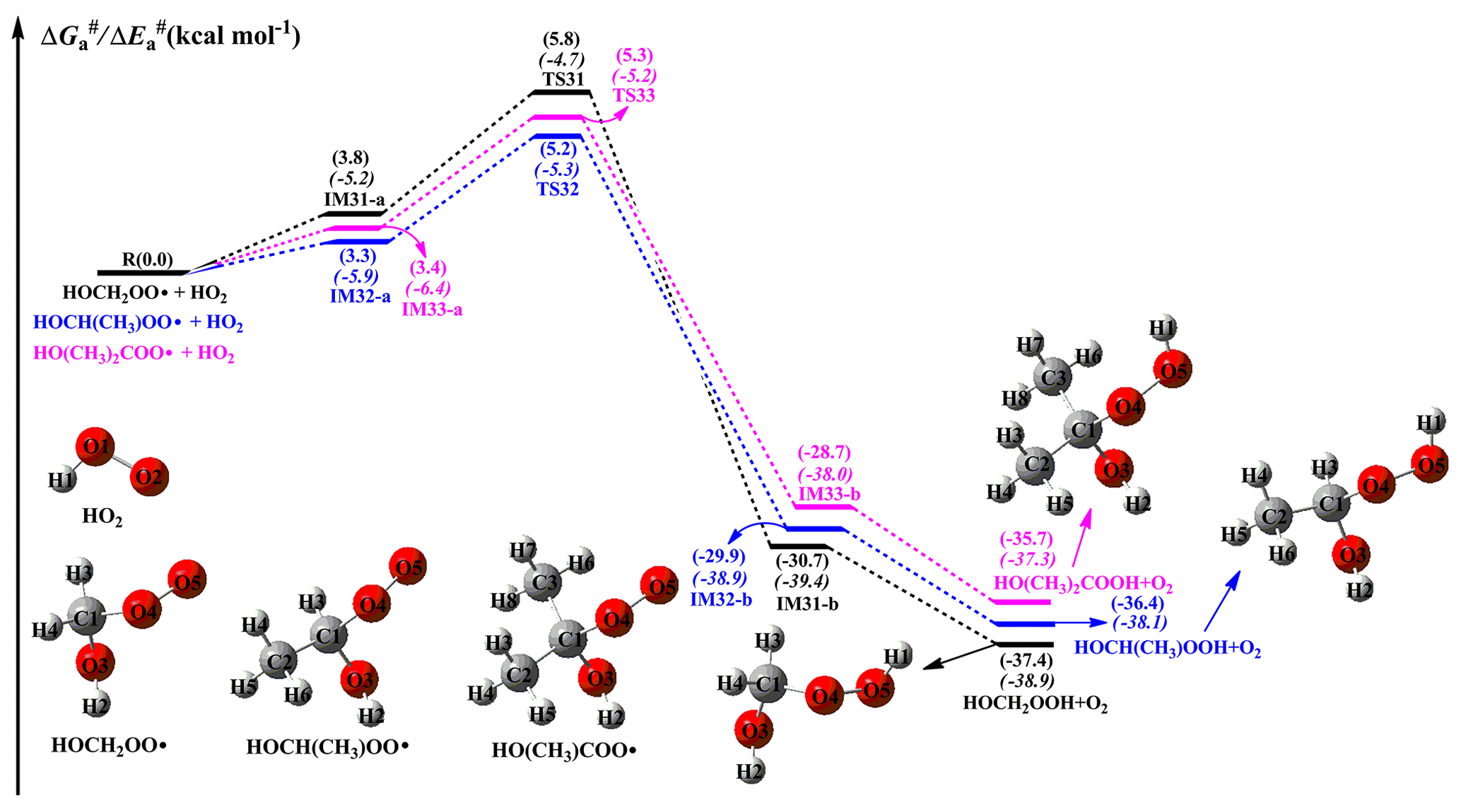
Figure 8PES ( and , in italics) for the reactions of distinct RO2 radicals with the HO2 radical predicted at the M06-2X/ma-TZVP//M06-2X/6-311+G(2df,2p) level of theory.
3.2.3 Reaction mechanism for the isomerization of RO2 radicals
The autoxidation of RO2 radicals is known to play an important role in the (re)generation of HOx radicals and the formation of HOMs (Xu et al., 2014; Bianchi et al., 2019; Rissanen et al., 2014; Ehn et al., 2017). The autoxidation mechanism includes an intramolecular H-shift from the -CH3 or -CH2- groups to the -OO site, leading to the formation of a hydroperoxyalkyl radical QOOH, followed by O2-addition to form a new peroxy radical (HOOQO2), one after the other, resulting in the formation of HOMs (Rissanen et al., 2014; Berndt et al., 2015). For the H-shift reactions of RO2 radicals, reactants, transition states and products have multiple conformers due to the effect of the degree of freedom for internal rotation. The calculated results show that the HOCH2OO radical has four energetically similar conformers (HOCH2OO-a, HOCH2OO-b, HOCH2OO-c, and HOCH2OO-d). The relative free energy and Boltzmann population (wi) of each individual conformer are listed in Table S6 in the Supplement, which indicate that the Boltzmann populations of these four conformers are 46.39 %, 46.31 %, 2.99 %, and 4.32 %, respectively.
A schematic PES for the H-shift reaction of an HOCH2OO radical is displayed in Fig. 9. As can be seen in Fig. 9, the lowest-energy conformer HOCH2OO-a can proceed via a 1,3-H shift from the -CH2 group to the terminal oxygen leading to the formation of S28-a (HOCHOOH) with a barrier of 41.6 kcal mol−1. HOCH2OO-b can isomerize to S28-b1 and S28-b2 via the four-membered ring transition states TS34-b1 and TS34-b2 (1,3-H shifts) with the barriers of 41.6 and 45.0 kcal mol−1, respectively. However, the 1,3-H shift reactions have comparatively high barriers, making them irrelevant in the atmosphere. Despite many attempts, the transition states of the H-shift reactions of HOCH2OO-c and HOCH2OO-d could not be located. The result suggests that the H-shift reactions of these two conformers are inhibited, which is consistent with the previous study that not all reactants will be in a conformation with a path across the barrier to reaction in the H-shift reactions of RO2 radicals (Møller et al., 2016). Similar to the case of the HOCH2OO radical, the isomerization of the HOCH(CH3)OO radical proceeds via the 1,3- and 1,4-H shifts from the -CH or -CH3 groups to the terminal oxygen, resulting in the formation of hydroperoxyalkyl radicals (Fig. S13 in the Supplement). These 1,3- and 1,4-H shift reactions are accompanied by extremely high barriers (>37.9 kcal mol−1), indicating that they are of less importance in the atmosphere. A similar conclusion is also derived from the isomerization of the HO(CH3)2COO radical in that 1,4-H shift reactions are unfavourable kinetically (Fig. S14 in the Supplement). The high barriers of the 1,3- and 1,4-H shifts can be interpreted as the result of the larger ring strain energy (RSE) in the cyclic transition state geometries. Consequently, the isomerization reactions of HOCH2OO, HOCH(CH3)OO, and HO(CH3)2COO radicals are unlikely to proceed in the atmosphere. This conclusion is further supported by previous studies, which found that the intramolecular H-shift isomerizations are important only for RO2 radicals with large carbon structures (Crounse et al., 2013; Jokinen et al., 2014; Rissanen et al., 2014).
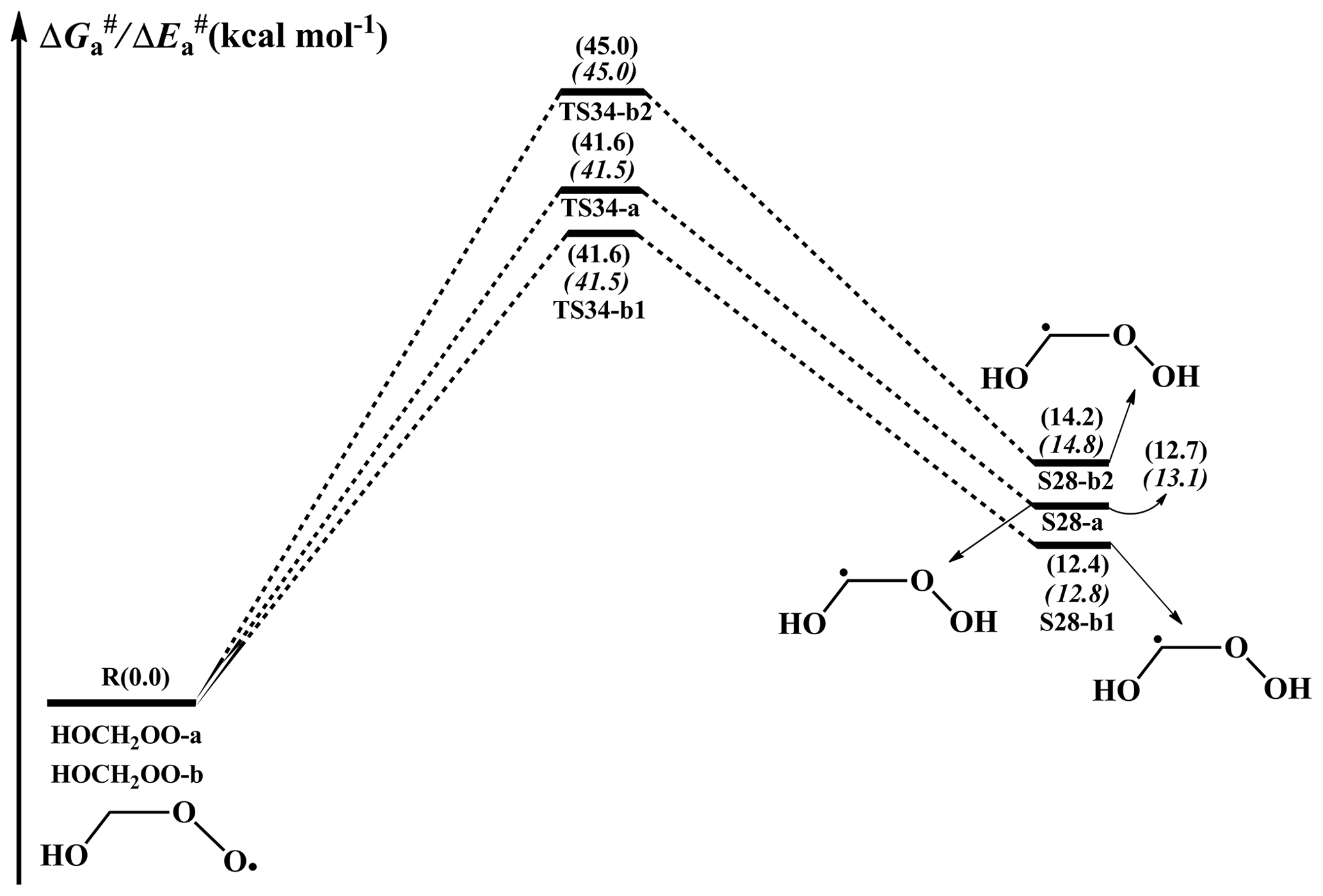
Figure 9PES ( and , in italics) for the isomerization of an HOCH2OO radical predicted at the M06-2X/ma-TZVP//M06-2X/6-311+G(2df,2p) level of theory.
The single-conformer rate coefficients (kIRC-TST) and multi-conformer rate coefficients (kMC-TST) of the isomerization of HOCH2OO, HOCH(CH3)OO, and HOC(CH3)2OO radicals are calculated over the temperature range of 273–400 K as listed in Table S9–S11 in the Supplement. As can be seen in Table S9, kIRC-TST of each conformer exhibits a marked positive temperature dependence over the temperature range studied. kMC-TST increases significantly with rising temperature, suggesting that a temperature increase promotes the isomerization of the HOCH2OO radical. A similar conclusion is also obtained for the isomerization of HOCH(CH3)OO and HOC(CH3)2OO radicals (Tables S10–S11). Notably, kMC-TST increases rapidly when the number of methyl groups increases. For example, the room temperature kMC-TST of HOCH2OO radical isomerization is calculated to be s−1, which is lower than those of the HOCH(CH3)OO ( s−1) and HO(CH3)2COO ( s−1) radicals' isomerization by 660 and 6820 times, respectively.
3.3 Subsequent reactions of H-abstraction products of RO2 radicals in urban environments
NOx is present in high concentrations in urban environments, and reaction with NO is the dominant chemical sink for RO2 radicals (Atkinson and Arey, 2003; Orlando and Tyndall, 2012; Perring et al., 2013). The main pathways for this type of reaction lead to the formation of NO2, RO radicals, organic nitrites, and organic nitrates at yields that are highly dependent on the nature of the R group (Orlando and Tyndall, 2012). The formation of NO2 through subsequent photolysis (λ<420 nm) produces ozone and NO, increasing the concentrations of near-surface ozone and propagating the NOx chain (Orlando and Tyndall, 2012). The schematic PES for the reactions of distinct RO2 radicals with NO are displayed in Figs. 10–12. As shown in Fig. 10, the bimolecular reaction of an HOCH2OO radical with NO initially leads to the formation of nitrite adduct S31 via the barrierless addition of NO to the terminal oxygen atom O3 of the HOCH2OO radical. The formed S31 exists as two isomers: S31-cis refers to O2 and O4 on the same side () with respect to the O3–N1 bond, whereas S31-trans refers to O2 and O4 on the opposite side () with respect to the O3–N1 bond. The calculations show that S31-cis is more stable than S31-trans by 1.1 kcal mol−1 in energy. Tautomerization between S31-cis and S31-trans proceeds through the rotation of the O3–N1 bond with a barrier of 14.4 kcal mol−1, implying that they can be regarded as separate atmospheric species. According to the Boltzmann-weighted distribution, the predicted proportions of S31-cis and S31-trans at room temperature are 86.5 % and 13.5 %, respectively. This result suggests that the dominant product of the reaction of an HOCH2OO radical with NO is S31-cis, so it is selected as a model compound in order to gain insight into the mechanism of secondary reactions in the following sections.
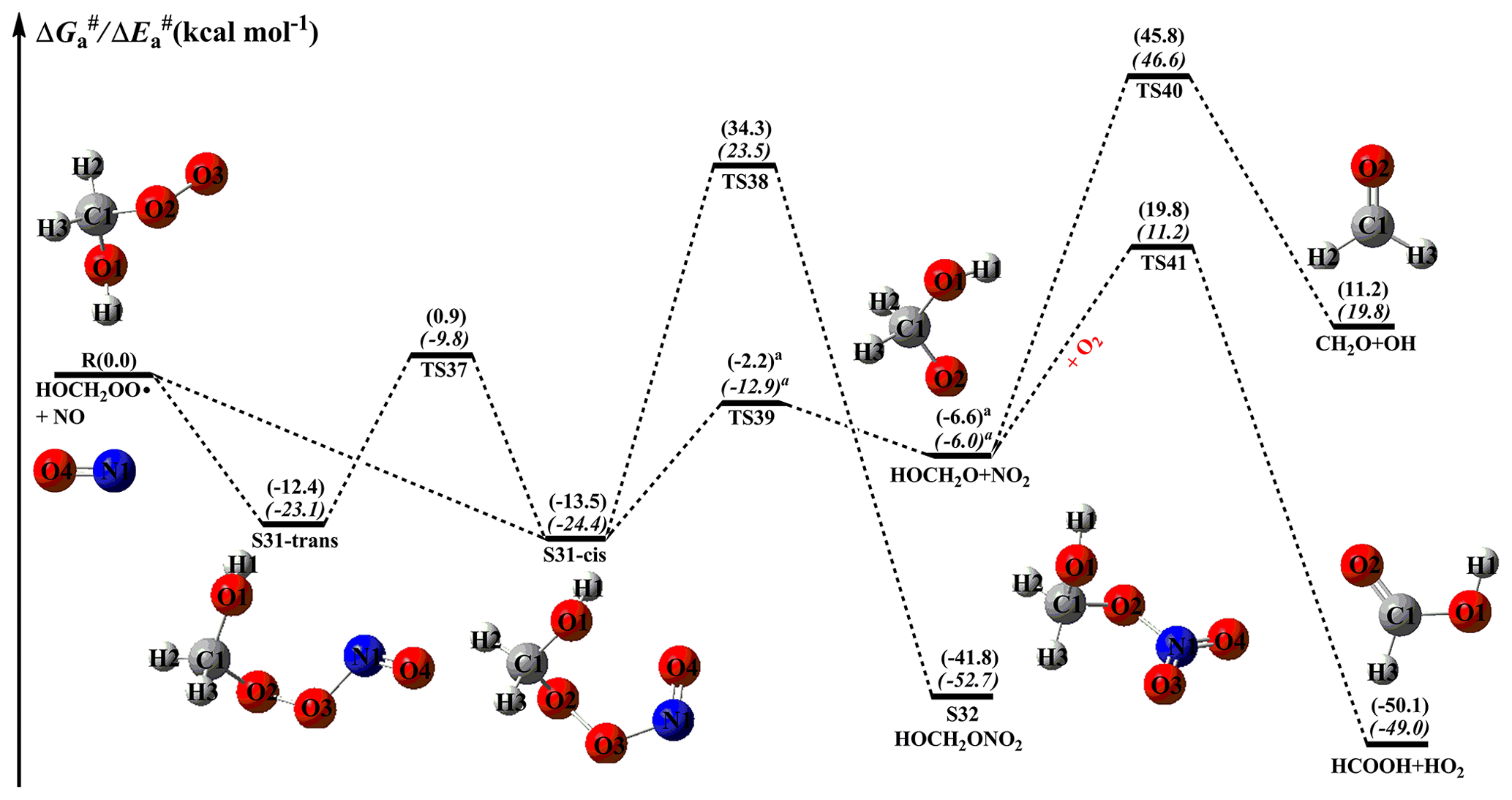
Figure 10PES ( and , in italics) for the reaction of an HOCH2OO radical with NO predicted at the M06-2X/ma-TZVP//M06-2X/6-311+G(2df,2p) level of theory (the superscript a is calculated at the MP2/ma-TZVP//MP2/6-311+G(2df,2p) level).

Figure 11PES ( and , in italics) for the reaction of an HOCH(CH3)OO radical with NO predicted at the M06-2X/ma-TZVP//M06-2X/6-311+G(2df,2p) level of theory (the superscript a is calculated at the MP2/ma-TZVP//MP2/6-311+G(2df,2p) level).
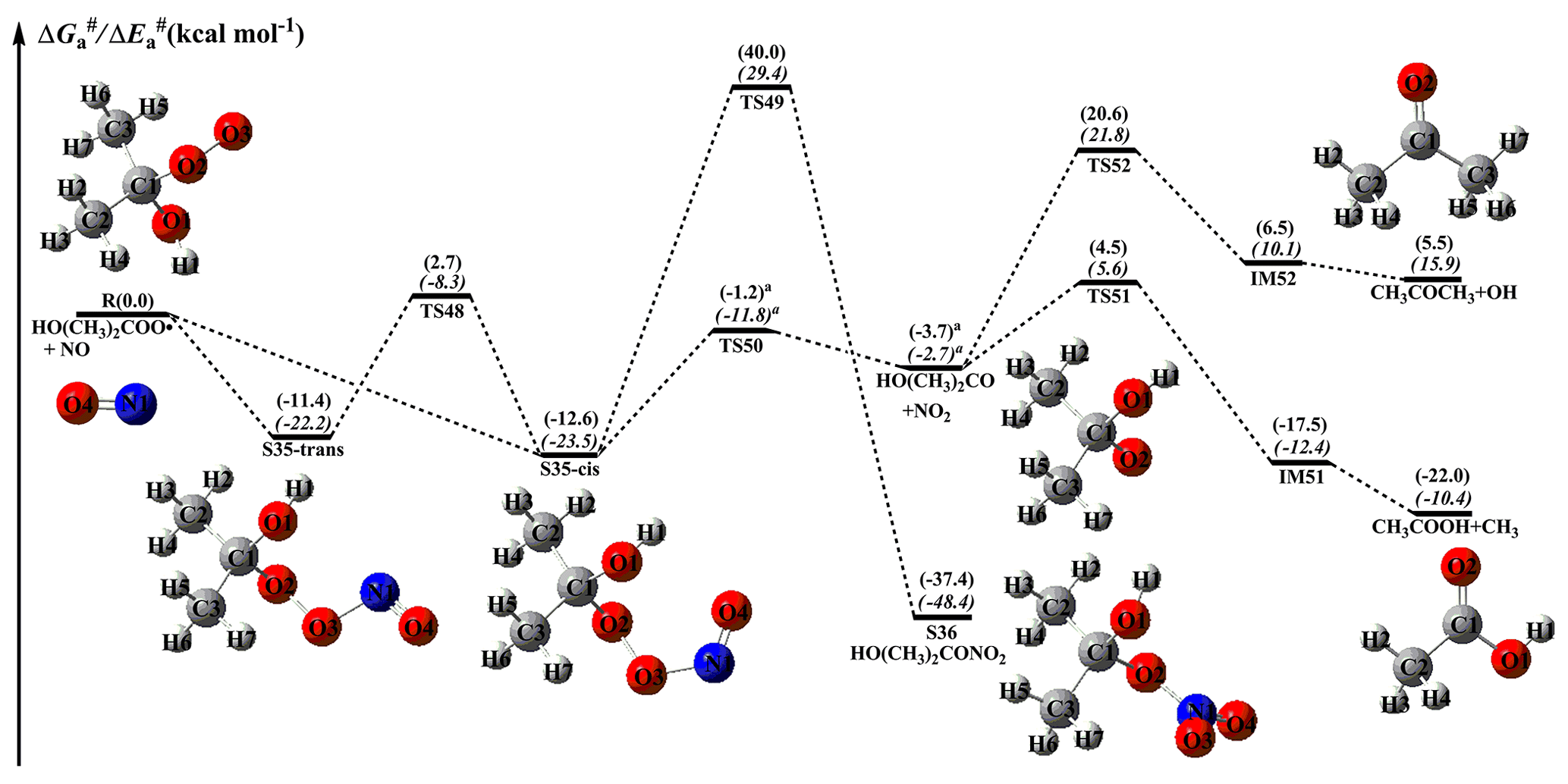
Figure 12PES ( and , in italics) for the reaction of an HO(CH3)2COO radical with NO predicted at the M06-2X/ma-TZVP//M06-2X/6-311+G(2df,2p) level of theory (the superscript a is calculated at the MP2/ma-TZVP//MP2/6-311+G(2df,2p) level).
S31-cis can either isomerize to organic nitrate S32 (R38) via a concerted process of O2–O3 bond breaking and O2–N1 bond formation with a barrier of 47.8 kcal mol−1, or decompose into an HOCH2O radical and NO2 (R39) via the cleavage of the O2–O3 bond with a barrier of 11.3 kcal mol−1. The result shows that the latter pathway is more favourable than the former channel. A similar conclusion is also obtained from the reactions of NO with HOCH(CH3)OO and HO(CH3)2COO radicals in that the formation of organic nitrate is of minor importance in the atmosphere. This result is further supported by prior studies, which found that the direct formation of organic nitrate from peroxy nitrites is a minor channel for the reactions of isoprene-derived RO2 radicals with NO (Piletic et al., 2017; Zhang et al., 2002). It should be noted that the transition state TS39 is not located using the M06-2X functional, but it is located at the MP2/6-311+G(2df,2p) level of theory and is verified using IRC calculations. The HOCH2O radical formed has two possible pathways: (1) it directly decomposes into CH2O and an OH radical (R40) via β-site C1–O1 bond scission with a barrier of 52.4 kcal mol−1; (2) it converts into HCOOH and an HO2 radical (R41) through H-abstraction by O2 with a barrier of 26.4 kcal mol−1. This result reveals that R41 is the most feasible channel in the fragmentation of the HOCH2O radical.
From Fig. 11, it can be seen that the addition of NO to an HOCH(CH3)OO radical leading to the formation of S33-cis is barrierless. Then, it decomposes into an HOCH(CH3)O radical and NO2 (R44) via the cleavage of the O2–O3 bond with a barrier of 11.5 kcal mol−1. The resulting HOCH(CH3)O radical has three possible pathways. The first is β-site C1–C2 bond scission leading to the formation of HCOOH + CH3 (R45) with a barrier of 8.3 kcal mol−1. The second is β-site C1–O1 bond cleavage resulting in the formation of CH3COH + OH (R46) with a barrier of 26.7 kcal mol−1. The third is H-abstraction by O2 leading to CH3COOH + HO2 (R47) with a barrier of 26.2 kcal mol−1. On the basis of the calculated reaction barriers, β-site C1–C2 bond scission is the dominant pathway in the fragmentation of the HOCH(CH3)O radical. This conclusion is further supported by the previous experimental result that β-hydroxy intermediates primarily undergo decomposition rather than react with O2 in the presence of NO (Aschmann et al., 2000). Equivalent to the HOCH(CH3)OO + NO reaction, the bimolecular reaction of HO(CH3)2COO radicals with NO has similar transformation pathways (Fig. 12). The reaction of HO(CH3)2COO with NO initially proceeds via a barrierless addition leading to S35-cis with a binding energy of 12.6 kcal mol−1. Then, S35-cis fragments into an HO(CH3)2CO radical and NO2 (R50) via the cleavage of the O2–O3 bond with a barrier of 11.4 kcal mol−1. The formed HO(CH3)2CO radical can either dissociate into CH3COOH + CH3 (R51) via the scission of the C1–C3 bond with a barrier of 8.2 kcal mol−1, or decompose into CH3COCH3+OH (R52) through the cleavage of the C1–O1 bond with a barrier of 24.3 kcal mol−1. This result also shows that β-site C–C bond scission is the dominant pathway.
The typical atmospheric concentrations of NO are around 10, 1, and 20 pptv in urban, rural, and forest environments, respectively (Bianchi et al., 2019). The rate coefficient of HOCH2OO radical reaction with NO is calculated to be at room temperature, resulting in the pseudo-first-order rate constants [NO] of , , and , respectively, in urban, rural, and forest environments. It is of interest to assess the relative importance for the H-shift reaction of HOCH2OO radicals and bimolecular reactions with HO2 radicals and NO based on the calculated kMC-TST, , and . It is found that the H-shift reaction is of less importance, the HO2 radical reaction is favorable in forest environments, and the NO reaction is predominant in urban and rural environments. A similar conclusion was also obtained from the cases of HOCH(CH3)OO and HO(CH3)2CHOO radicals.
The rate coefficients of the dominant pathways of HOCH2O, HOCH(CH3)O, and HO(CH3)2CHO radical fragmentations are summarized in Table S12 in the Supplement. As can be seen in Table S12, kR41 increases slightly with increasing temperature, and the discrepancy is about a factor of 12 at the two extremes of the studied temperature range. At the ground level with molecule cm−3, the pseudo-first-order rate constant [O2] is estimated to be 38.0 s−1 at room temperature. kR45 varies significantly from 2.0×106 (273 K) to 3.1×108 (400 K) s−1, and it exhibits a marked positive temperature dependence. A similar phenomenon is observed for kR51: it increases significantly with increasing temperature. kR51 is greater than kR45 by a factor of ∼1.3, suggesting that the rate coefficient of β-site C–C bond scission increases slightly when the number of methyl groups increases.
The detailed mechanisms and kinetic properties of OH-initiated oxidation of distinct HHPs and the subsequent transformation of resulting H-abstraction products are investigated using quantum chemical and kinetics modeling methods. The main conclusions are summarized as follows.
(a) The dominant pathway is H-abstraction from the -OOH group in the initiation reactions of OH radicals with HOCH2OOH and HOC(CH3)2OOH. H-abstraction from the -CH group is competitive with that from the -OOH group in the reaction of OH radicals with HOCH(CH3)OOH. The barrier of H-abstraction from the -OOH group slightly increases when the number of methyl groups increases. Compared with the rate coefficient of the dominant pathway in the parent system, it is almost identical when a methyl group substitution occurs at the C1-position, whereas it reduces by a factor of 2–5 when two methyl groups are introduced at the C1-position. The atmospheric lifetimes of HOCH2OOH, HOCH(CH3)OOH, and HOC(CH3)2OOH reactivity toward OH radicals are estimated to be 0.58–1.74, 0.60–1.79, and 1.23–3.69 h, respectively, at room temperature under the typical OH radical concentrations of 5–15×106 molecules cm−3 during daylight.
(b) The self-reaction of RO2 radicals initially produces a tetroxide intermediate via oxygen-to-oxygen coupling, and then it decomposes into propagation and termination products through asymmetric two-step O–O bond scission. The rate-limiting step is the first O–O bond cleavage, and the barrier increases when the number of methyl groups increases. This finding contributes toward the understanding of the self-reaction of complex RO2 radicals.
(c) The bimolecular reactions of distinct RO2 radicals with HO2 radicals lead to the formation of hydroperoxide ROOH as the main product, and the barrier height is not affected by the number of methyl substitutions. Compared with the rate coefficient for the HOCH2OO + HO2 reaction, the rate coefficients increase by a factor of 2–5 when one or two methyl groups are introduced at the C1-position. Using an HO2 radical concentration of ∼50 pptv in forest environments, the pseudo-first-order rate constants of the reactions of distinct RO2 radicals with an HO2 radical vary from 1 to s−1.
(d) The isomerization reactions of HOCH2OO, HOCH(CH3)OO, and HO(CH3)2COO radicals are unlikely to proceed in the atmosphere because the intramolecular H-shift steps have considerably high barriers and are strongly endergonic. The result suggests that the isomerization of RO2 radicals with small carbon structures is of less importance in the atmosphere.
(e) The reaction with O2 to form formic acid and an HO2 radical is the dominant removal pathway for HOCH2O radicals formed from the reaction of an HOCH2OO radical with NO. β-site C–C bond scission is the dominant pathway in the dissociation of HOCH(CH3)O and HOC(CH3)2O radicals formed from the reactions of NO with HOCH(CH3)OO and HOC(CH3)2OO radicals. The result suggests that methyl-substituted alkoxyl radicals primarily proceed via β-site C–C bond scission to produce aldehyde or carbonyl.
The data are accessible by contacting the corresponding author (huangyu@ieecas.cn).
The following information is provided in the Supplement: Y//X (Y = M06-2X, CCSD (T), X = 6-311+G(2df,2p), ma-TZVP) calculated energy barrier (, ) for the OH + HHPs reactions; rate coefficients of each elementary pathway involved in the initiation reactions of OH radicals with HOCH2OOH, HOCH(CH3)OOH, and HO(CH3)2COOH; rate coefficients of HO2 radical reactions with HOCH2OO, HOCH(CH3)OO, and HO(CH3)2COO radicals; the relative free energy and Boltzmann populations (wi) of the conformer of HOCH2OO, HOCH(CH3)OO, and HO(CH3)2COO radicals; the single-conformer rate coefficients (kIRC-TST) and multi-conformer rate coefficients (kMC-TST) of HOCH2OO, HOCH(CH3)OO, and HO(CH3)2COO radicals; rate coefficients of dominant pathways in the HOCH2OO• + NO, HOCH(CH3)OO• + NO, and HO(CH3)2CHOO• + NO reactions; PESs () for the OH-initiated reactions of HOCH2OOH, HOCH(CH3)OOH, and HOC(CH3)2OOH; geometries of all stationary points; plots of the rate coefficients of each elementary pathway versus temperature; PESs ( and , in italics) for the isomerization of HOCH(CH3)OO and HO(CH3)2COO radicals. The supplement related to this article is available online at: https://doi.org/10.5194/acp-22-3693-2022-supplement.
LC designed the study. LC and YH wrote the paper. LC performed theoretical calculation. YX, ZJ, and WW analyzed the data. All authors reviewed and commented on the paper.
The contact author has declared that neither they nor their co-authors have any competing interests
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This work was supported by the National Natural Science Foundation of China (grant nos. 42175134, 41805107, and 22002080). It was also partially supported as Key Projects of Chinese Academy of Sciences, China (grant no. ZDRW-ZS-2017-6), Strategic Priority Research Program of the Chinese Academy of Sciences, China (grant nos. XDA23010300 and XDA23010000), Key Project of International Cooperation of the Chinese Academy of Sciences, China (grant no. GJHZ1543), Research Grants Council of Hong Kong, China (grant no. PolyU 152083/14E), CAS “Light of West China” Program (XAB2019B01), and the General Project of Shaanxi Province (2020JQ-432).
This research has been supported by the National Natural Science Foundation of China (grant nos. 42175134, 41805107, and 22002080). It was also partially supported as Key Projects of the Chinese Academy of Sciences, China (grant no. ZDRW-ZS-2017-6), Strategic Priority Research Program of the Chinese Academy of Sciences, China (grant nos. XDA23010300 and XDA23010000), Key Project of International Cooperation of the Chinese Academy of Sciences, China (grant no. GJHZ1543), Research Grants Council of Hong Kong, China (grant no. PolyU 152083/14E), CAS “Light of West China” Program (grant no. XAB2019B01), and the General Project of Shaanxi Province (grant no. 2020JQ-432).
This paper was edited by Arthur Chan and reviewed by three anonymous referees.
Allen, H. M., Crounse, J. D., Bates, K. H., Teng, A. P., Krawiec-Thayer, M. P., Rivera-Rios, J. C., Keutsch, F. N., Clair, J. M. S., Hanisco, T. F., Møller, K. H., Kjaergaard, H. G., and Wennberg, P. O.: Kinetics and product yields of the OH initiated oxidation of hydroxymethyl hydroperoxide, J. Phys. Chem. A, 122, 6292–6302, https://doi.org/10.1021/acs.jpca.8b04577, 2018.
Anglada, J. M. and Solé, A.: Impact of the water dimer on the atmospheric reactivity of carbonyl oxides, Phys. Chem. Chem. Phys., 18, 17698–17712, https://doi.org/10.1039/C6CP02531E, 2016.
Anglada, J. M., González, J., and Torrent-Sucarrat, M.: Effects of the substituents on the reactivity of carbonyl oxides. A theoretical study on the reaction of substituted carbonyl oxides with water, Phys. Chem. Chem. Phys., 13, 13034–13045, https://doi.org/10.1039/c1cp20872a, 2011.
Aschmann, S. M., Arey, J., and Atkinson, R.: Formation of β-hydroxycarbonyls from the OH radical-initiated reactions of selected alkenes, Environ. Sci. Technol., 34, 1702–1706, https://doi.org/10.1021/es991125a, 2000.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003.
Bach, R. D., Dmitrenko, O., and Estévez, C. M.: Chemical behavior of the biradicaloid (HO⋯ONO) singlet states of peroxynitrous acid. The oxidation of hydrocarbons, sulfides, and selenides, J. Am. Chem. Soc., 127, 3140–3155, https://doi.org/10.1021/ja044245d, 2005.
Berndt, T., Richters, S., Kaethner, R., Voigtländer, J., Stratmann, F., Sipilä, M., Kulmala, M., and Herrmann, H.: Gas-phase ozonolysis of cycloalkenes: formation of highly oxidized RO2 radicals and their reactions with NO, NO2, SO2, and other RO2 radicals, J. Phys. Chem. A, 119, 10336–10348, https://doi.org/10.1021/acs.jpca.5b07295, 2015.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion product formation from self- and cross-reactions of RO2 radicals in the atmosphere, Angew. Chem. Int. Edit., 57, 3820–3824, https://doi.org/10.1002/anie.201710989, 2018.
Bianchi, F., Kurten, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly oxygenated organic molecules (HOM) from gas-phase autoxidation involving peroxy radicals: a key contributor to atmospheric aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Boys, S. F. and Bernardi, F.: The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors, Mol. Phys., 19, 553–566, https://doi.org/10.1080/00268977000101561, 1970.
Chao, W., Hsieh, J. T., Chang, C. H., and Lin, J. J. M.: Direct kinetic measurement of the reaction of the simplest Criegee intermediate with water vapor, Science, 347, 751–754, https://doi.org/10.1126/science.1261549, 2015.
Chen, L., Wang, W., Zhou, L., Wang, W., Liu, F., Li, C., and Lü, J.: Role of water clusters in the reaction of the simplest Criegee intermediate CH2OO with water vapour, Theor. Chem. Acc., 135, 252–263, https://doi.org/10.1007/s00214-016-1998-2, 2016a.
Chen, L., Wang, W., Wang, W., Liu, Y., Liu, F., Liu, N., and Wang, B.: Water-catalyzed decomposition of the simplest Criegee intermediate CH2OO, Theor. Chem. Acc., 135, 131–143, https://doi.org/10.1007/s00214-016-1894-9, 2016b.
Chen, L., Huang, Y., Xue, Y., Cao, J., and Wang, W.: Competition between HO2 and H2O2 reactions with in the oligomer formation: a theoretical perspective, J. Phys. Chem. A, 121, 6981–6991, https://doi.org/10.1021/acs.jpca.7b05951, 2017.
Chen, L., Huang, Y., Xue, Y., Shen, Z., Cao, J., and Wang, W.: Mechanistic and kinetics investigations of oligomer formation from Criegee intermediate reactions with hydroxyalkyl hydroperoxides, Atmos. Chem. Phys., 19, 4075–4091, https://doi.org/10.5194/acp-19-4075-2019, 2019.
Chen, L., Huang, Y., Xue, Y., Jia, Z., and Wang, W.: Atmospheric oxidation of 1-butene initiated by OH radical: Implications for ozone and nitrous acid formations, Atmos. Environ., 244, 118010–118021, https://doi.org/10.1016/j.atmosenv.2020.118010, 2021.
Chhantyal-Pun, R., Welz, O., Savee, J. D., Eskola, A. J., Lee, E. P. F., Blacker, L., Hill, H. R., Ashcroft, M., Khan, M. A. H., Lloyd-Jones, G. C., Evans, L., Rotavera, B., Huang, H., Osborn, D. L., Mok, D. K. W., Dyke, J. M., Shallcross, D. E., Percival, C. J., Orr-Ewing, A. J., and Taatjes, C. A.: Direct measurements of unimolecular and bimolecular reaction kinetics of the Criegee intermediate (CH3)2COO, J. Phys. Chem. A, 121, 4–15, https://doi.org/10.1021/acs.jpca.6b07810, 2017.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of organic compounds in the atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013.
Dillon, T. J. and Crowley, J. N.: Direct detection of OH formation in the reactions of HO2 with CH3C(O)O2 and other substituted peroxy radicals, Atmos. Chem. Phys., 8, 4877–4889, https://doi.org/10.5194/acp-8-4877-2008, 2008.
Eckart, C.: The penetration of a potential barrier by electrons, Phys. Rev., 35, 1303–1309, https://doi.org/10.1103/PhysRev.35.1303, 1930.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I. H., Rissanen, M., Jokinen,T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V. M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Ehn, M., Berndt, T., Wildt, J., and Mentel, T.: Highly oxygenated molecules from atmospheric autoxidation of hydrocarbons: a prominent challenge for chemical kinetics studies, Int. J. Chem. Kinet., 49, 821–831, https://doi.org/10.1002/kin.21130, 2017.
Fernández-Ramos, A., Ellingson, B. A., Meana-Pañeda, R., Marques, J. M. C., and Truhlar, D. G.: Symmetry numbers and chemical reaction rates, Theor. Chem. Acc., 118, 813–826, https://doi.org/10.1007/s00214-007-0328-0, 2007.
Francisco, J. S. and Eisfeld, W.: Atmospheric oxidation mechanism of hydroxymethyl hydroperoxide, J. Phys. Chem. A, 113, 7593–7600, https://doi.org/10.1021/jp901735z, 2009.
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Montgomery, J. A. Jr., Vreven, T., Kudin, K. N., Burant, J. C., Millam, J. M., Iyengar, S. S., Tomasi, J., Barone, V., Mennucci. B., Cossi, M., Scalmani, G., Rega, N., Petersson, G. A., Nakatsuji, H., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Klene, M., Li, X., Knox, J. E., Hratchian, H. P., Cross, J. B., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R. E., Yazyev, O., Austin, A. J., Cammi, R., Pomelli, C., Ochterski, J. W., Ayala, P. Y., Morokuma, K., Voth, G. A., Salvador, P., Dannenberg, J. J., Zakrzewski, V. G., Dapprich, S., Daniels, A. D., Strain, M. C., Farkas, O., Malick, D. K., Rabuck, A. D., Raghavachari, K., Foresman, J. B., Ortiz, J. V., Cui, Q., Baboul, A. G., Clifford, S., Cioslowski, J., Stefanov, B. B., Liu, G., Liashenko, A., Piskorz, P., Komaromi, I., Martin, R. L., Fox, D. J., Keith, T., Al-Laham, M. A., Peng, C. Y., Nanayakkara, A., Challacombe, M., Gill, P. M. W., Johnson, B., Chen, W., Wong, M. W., Gonzalez, C., and Pople, J. A.: Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, http://www.gaussian.com (last access: 9 February 2022), 2009.
Fukui, K.: The path of chemical reactions – the IRC approach, Accounts Chem. Res., 14, 363–368, https://doi.org/10.1021/ar00072a001, 1981.
Gilbert, R. G. and Smith, S. C.: Theory of unimolecular and recombination reactions, Blackwell Scientific, Carlton, Australia, ISBN 0632027495, 1990.
Gligorovski, S., Strekowski, R., Barbati, S., and Vione, D.: Environmental implications of hydroxyl radicals (•OH), Chem. Rev., 115, 13051–13092, https://doi.org/10.1021/cr500310b, 2015.
Glowacki, D. R., Liang, C. H., Morley, C., Pilling, M. J., and Robertson, S. H.: MESMER: an open-source master equation solver for multi-energy well reactions, J. Phys. Chem. A, 116, 9545–9560, https://doi.org/10.1021/jp3051033, 2012.
Gong, Y. and Chen, Z.: Quantification of the role of stabilized Criegee intermediates in the formation of aerosols in limonene ozonolysis, Atmos. Chem. Phys., 21, 813–829, https://doi.org/10.5194/acp-21-813-2021, 2021.
Hofzumahaus, A., Rohrer, F., Lu, K., Bohn, B., Brauers, T., Chang, C. C., Fuchs, H., Holland, F., Kita, K., Kondo, Y., Li, X., Lou, S., Shao, M., Zeng, L., Wahner, A., and Zhang, Y.: Amplified trace gas removal in the troposphere, Science, 324, 1702–1704, https://doi.org/10.1126/science.1164566, 2009.
Holbrook, K. A., Pilling, M. J., Robertson, S. H., and Robinson, P. J.: Unimolecular reactions, 2nd edn., Wiley, New York, ISBN 0-471-92268-4, 1996.
Huang, H. L., Chao, W., and Lin, J. J. M.: Kinetics of a Criegee intermediate that would survive high humidity and may oxidize atmospheric SO2, P. Natl. Acad. Sci. USA, 112, 10857–10862, https://doi.org/10.1073/pnas.1513149112, 2015.
Iyer, S., Reiman, H., Møller, K. H., Rissanen, M. P., Kjaergaard, H. G., and Kurtén, T.: Computational investigation of RO2+HO2 and RO2+RO2 reactions of monoterpene derived first-generation peroxy radicals leading to radical recycling, J. Phys. Chem. A, 122, 9542–9552, https://doi.org/10.1021/acs.jpca.8b09241, 2018.
Iyer, S., Rissanen, M. P., Valiev, R., Barua, S., Krechmer, J. E., Thornton, J., Ehn, M., and Kurtén, T.: Molecular mechanism for rapid autoxidation in α-pinene ozonolysis, Nat. Commun., 12, 878–883, https://doi.org/10.1038/s41467-021-21172-w, 2021.
Jara-Toro, R. A., Hernández, F. J., Taccone, R. A., Lane, S. I., and Pino, G. A.: Water catalysis of the reaction between methanol and OH at 294 K and the atmospheric implications, Angew. Chem. Int. Edit., 56, 2166–2170, https://doi.org/10.1002/anie.201612151, 2017.
Jara-Toro, R. A., Hernández, F. J., Garavagno, M. A., Taccone, R. A., and Pino, G. A.: Water catalysis of the reaction between hydroxyl radicals and linear saturated alcohols (ethanol and n-propanol) at 294 K, Phys. Chem. Chem. Phys., 20, 27885–27896, https://doi.org/10.1039/C8CP05411H, 2018.
Jokinen, T., Sipilä, M., Richters, S., Kerminen, V. M., Paasonen, P., Stratmann, F., Worsnop, D., Kulmala, M., Ehn, M., Herrmann, H., and Berndt, T.: Rapid autoxidation forms highly oxidized RO2 radicals in the atmosphere, Angew. Chem. Int. Edit., 53, 14596–14600, https://doi.org/10.1002/anie.201408566, 2014.
Khan, M. A. H., Percival, C. J., Caravan, R. L., Taatjes, C. A., and Shallcross, D. E.: Criegee intermediates and their impacts on the troposphere, Environ. Sci.-Proc. Imp., 20, 437–453, https://doi.org/10.1039/C7EM00585G, 2018.
Kumar, M. and Francisco, J. S.: Red-light-induced decomposition of an organic peroxy radical: a new source of the HO2 radical, Angew. Chem. Int. Edit., 54, 15711–15714, https://doi.org/10.1002/anie.201509311, 2015.
Kumar, M. and Francisco, J. S.: Red-light initiated decomposition of α-hydroxy methylperoxy radical in the presence of organic and inorganic acids: implications for the HOx formation in the lower stratosphere, J. Phys. Chem. A, 120, 2677–2683, https://doi.org/10.1021/acs.jpca.6b01515, 2016.
Kumar, M., Busch, D. H., Subramaniam, B., and Thompson, W. H.: Role of tunable acid catalysis in decomposition of α-hydroxyalkyl hydroperoxides and mechanistic implications for tropospheric chemistry, J. Phys. Chem. A, 118, 9701–9711, https://doi.org/10.1021/jp505100x, 2014.
Lee, R., Gryn'ova, G., Ingold, K. U., and Coote, M. L.: Why are sec-alkylperoxyl bimolecular self-reactions orders of magnitude faster than the analogous reactions of tert-alkylperoxyls? The unanticipated role of CH hydrogen bond donation, Phys. Chem. Chem. Phys., 18, 23673–23679, https://doi.org/10.1039/C6CP04670C, 2016.
Lester, M. I. and Klippenstein, S. J.: Unimolecular decay of Criegee intermediates to OH radical products: prompt and thermal decay processes, Accounts Chem. Res., 51, 978–985, https://doi.org/10.1021/acs.accounts.8b00077, 2018.
Liu, L., Bei, N., Wu, J., Liu, S., Zhou, J., Li, X., Yang, Q., Feng, T., Cao, J., Tie, X., and Li, G.: Effects of stabilized Criegee intermediates (sCIs) on sulfate formation: a sensitivity analysis during summertime in Beijing–Tianjin–Hebei (BTH), China, Atmos. Chem. Phys., 19, 13341–13354, https://doi.org/10.5194/acp-19-13341-2019, 2019.
Long, B., Bao, J. L., and Truhlar, D. G.: Reaction of SO2 with OH in the atmosphere, Phys. Chem. Chem. Phys., 19, 8091–8100, https://doi.org/10.1039/C7CP00497D, 2017.
Lu, T.: Molclus program, Version 1.9.3, http://www.keinsci.com/research/molclus.html, last access: 10 February 2020.
Ma, F., Guo, X., Xia, D., Xie, H. B., Wang, Y., Elm, J., Chen, J., and Niu, J.: Atmospheric chemistry of allylic radicals from isoprene: a successive cyclization-driven autoxidation mechanism, Environ. Sci. Technol., 55, 4399–4409, https://doi.org/10.1021/acs.est.0c07925, 2021.
Møller, K. H., Otkjær, R. V., Hyttinen, N., Kurtén, T., and Kjaergaard, H. G.: Cost-effective implementation of multiconformer transition state theory for peroxy radical hydrogen shift reactions, J. Phys. Chem. A, 120, 10072–10087, https://doi.org/10.1021/acs.jpca.6b09370, 2016.
Møller, K. H., Berndt, T., and Kjaergaard, H. G.: Atmospheric autoxidation of amines, Environ. Sci. Technol., 54, 11087–11099, https://doi.org/10.1021/acs.est.0c03937, 2020.
Nozière, B. and Vereecken, L.: Direct observation of aliphatic peroxy radical autoxidation and water effects: an experimental and theoretical study, Angew. Chem. Int. Edit., 58, 13976–13982, https://doi.org/10.1002/anie.201907981, 2019.
Orlando, J. J. and Tyndall, G. S.: Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 41, 6294–6317, https://doi.org/10.1039/c2cs35166h, 2012.
Perring, A. E., Pusede, S. E., and Cohen, R. C.: An observational perspective on the atmospheric impacts of alkyl and multifunctional nitrates on ozone and secondary organic aerosol, Chem. Rev., 113, 5848–5870, https://doi.org/10.1021/cr300520x, 2013.
Piletic, I. R., Edney, E. O., and Bartolotti, L. J.: Barrierless reactions with loose transition states govern the yields and lifetimes of organic nitrates derived from isoprene, J. Phys. Chem. A, 121, 8306–8321, https://doi.org/10.1021/acs.jpca.7b08229, 2017.
Qiu, J. T., Ishizuka, S., Tonokura, K., Colussi, A. J., and Enami, S.: Water dramatically accelerates the decomposition of α-hydroxyalkyl-hydroperoxides in aerosol particles, J. Phys. Chem. Lett., 10, 5748–5755, https://doi.org/10.1021/acs.jpclett.9b01953, 2019.
Rissanen, M. P., Kurtén, T., Sipilä, M., Thornton, J. A., Kangasluoma, J., Sarnela, N., Junninen, H., Jørgensen, S., Schallhart, S., Kajos, M. K., Taipale, R., Springer, M., Mentel, T. F., Ruuskanen, T., Petäjä, T., Worsnop, D. R., Kjaergaard, H. G., and Ehn, M.: The formation of highly oxidized multifunctional products in the ozonolysis of cyclohexene, J. Am. Chem. Soc., 136, 15596–15606, https://doi.org/10.1021/ja507146s, 2014.
Russell, G. A.: Deuterium-isotope effects in the autoxidation of aralkyl hydrocarbons. Mechanism of the interaction of peroxy radicals, J. Am. Chem. Soc., 79, 3871–3877, https://doi.org/10.1021/ja01571a068, 1957.
Ryzhkov, A. B. and Ariya, P. A.: A theoretical study of the reactions of carbonyl oxide with water in atmosphere: the role of water dimer, Chem. Phys. Lett., 367, 423–429, https://doi.org/10.1016/S0009-2614(02)01685-8, 2003.
Smith, M. C., Chang, C. H., Chao, W., Lin, L. C., Takahashi, K., Boering, K. A., and Lin, J. J. M.: Strong negative temperature dependence of the simplest Criegee intermediate CH2OO reaction with water dimer, J. Phys. Chem. Lett., 6, 2708–2713, https://doi.org/10.1021/acs.jpclett.5b01109, 2015.
Stone, D., Whalley, L. K., and Heard, D. E.: Tropospheric OH and HO2 radicals: field measurements and model comparisons, Chem. Soc. Rev., 41, 6348–6404, https://doi.org/10.1039/c2cs35140d, 2012.
Taatjes, C. A.: Criegee intermediates: what direct production and detection can teach us about reactions of carbonyl oxides, Annu. Rev. Phys. Chem., 68, 183–207, https://doi.org/10.1146/annurev-physchem-052516-050739, 2017.
Taatjes, C. A., Welz, O., Eskola, A. J., Savee, J. D., Scheer, A. M., Shallcross, D. E., Rotavera, B., Lee, E. P. F., Dyke, J. M., Mok, D. K. W., Osborn, D. L., and Percival, C. J.: Direct measurements of conformer-dependent reactivity of the Criegee intermediate CH3CHOO, Science, 340, 177–180, https://doi.org/10.1126/science.1234689, 2013.
Valiev, R. R., Hasan, G., Salo, V. T., Kubečka, J., and Kurten, T.: Intersystem crossings drive atmospheric gas-phase dimer formation, J. Phys. Chem. A, 123, 6596–6604, https://doi.org/10.1021/acs.jpca.9b02559, 2019.
Wang, S., Wu, R., Berndt, T., Ehn, M., and Wang, L.: Formation of highly oxidized radicals and multifunctional products from the atmospheric oxidation of alkylbenzenes, Environ. Sci. Technol., 51, 8442–8449, https://doi.org/10.1021/acs.est.7b02374, 2017.
Wang, S., Riva, M., Yan, C., Ehn, M., and Wang, L.: Primary formation of highly oxidized multifunctional products in the OH-initiated oxidation of isoprene: a combined theoretical and experimental study, Environ. Sci. Technol., 52, 12255–12264, https://doi.org/10.1021/acs.est.8b02783, 2018.
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., Clair, J. M. S., Teng, A. P., Zhang, X., and Seinfeld, J. H.: Gas-phase reactions of isoprene and its major oxidation products, Chem. Rev., 118, 3337–3390, https://doi.org/10.1021/acs.chemrev.7b00439, 2018.
Winiberg, F. A. F., Dillon, T. J., Orr, S. C., Groß, C. B. M., Bejan, I., Brumby, C. A., Evans, M. J., Smith, S. C., Heard, D. E., and Seakins, P. W.: Direct measurements of OH and other product yields from the HO2+CH3C(O)O2 reaction, Atmos. Chem. Phys., 16, 4023–4042, https://doi.org/10.5194/acp-16-4023-2016, 2016.
Xu, L., Kollman, M. S., Song, C., Shilling, J. E., and Ng, N. L.: Effects of NOx on the volatility of secondary organic aerosol from isoprene photooxidation, Environ. Sci. Technol., 48, 2253–2262, https://doi.org/10.1021/es404842g, 2014.
Xu, L., Møller, K. H., Crounse, J. D., Kjaergaard, H. G., and Wennberg, P. O.: New insights into the radical chemistry and product distribution in the OH-initiated oxidation of benzene, Environ. Sci. Technol., 54, 13467–13477, https://doi.org/10.1021/acs.est.0c04780, 2020.
Zhang, D., Zhang, R., Park, J., and North, S. W.: Hydroxy peroxy nitrites and nitrates from OH initiated reactions of isoprene, J. Am. Chem. Soc., 124, 9600–9605, https://doi.org/10.1021/ja0255195, 2002.
Zhang, P., Wang, W., Zhang, T., Chen, L., Du, Y., Li, C., and Lv, J.: Theoretical study on the mechanism and kinetics for the self-reaction of C2H5O2 radicals, J. Phys. Chem. A, 116, 4610–4620, https://doi.org/10.1021/jp301308u, 2012.
Zhao, Y. and Truhlar, D. G.: A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions, J. Chem. Phys., 125, 194101–194119, https://doi.org/10.1063/1.2370993, 2006.
Zhao, Y. and Truhlar, D. G.: The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 120, 215–241, https://doi.org/10.1007/s00214-007-0310-x, 2008.
Zheng, J. and Truhlar, D. G.: Direct dynamics study of hydrogen-transfer isomerization of 1-pentyl and 1-hexyl radicals, J. Phys. Chem. A, 113, 11919–11925, https://doi.org/10.1021/jp903345x, 2009.
Zheng, J., Xu, X., and Truhlar, D. G.: Minimally augmented Karlsruhe basis sets, Theor. Chem. Acc., 128, 295–305, https://doi.org/10.1007/s00214-010-0846-z, 2011.
Zhong, J., Kumar, M., Francisco, J. S., and Zeng, X. C.: Insight into chemistry on cloud/aerosol water surfaces, Accounts Chem. Res., 51, 1229–1237, https://doi.org/10.1021/acs.accounts.8b00051, 2018.
Zhou, X., Liu, Y., Dong, W., and Yang, X.: Unimolecular reaction rate measurement of syn-CH3CHOO, J. Phys. Chem. Lett., 10, 4817–4821, https://doi.org/10.1021/acs.jpclett.9b01740, 2019.
Zhu, C., Kumar, M., Zhong, J., Li, L., Francisco, J. S., and Zeng, X. C.: New mechanistic pathways for Criegee-water chemistry at the air/water interface, J. Am. Chem. Soc., 138, 11164–11169, https://doi.org/10.1021/jacs.6b04338, 2016.