the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 07 Feb 2022
| 07 Feb 2022
Are reactive oxygen species (ROS) a suitable metric to predict toxicity of carbonaceous aerosol particles?
Zhi-Hui Zhang
Elena Hartner
Battist Utinger
Benjamin Gfeller
Andreas Paul
Martin Sklorz
Hendryk Czech
Bin Xia Yang
Xin Yi Su
Gert Jakobi
Jürgen Orasche
Jürgen Schnelle-Kreis
Seongho Jeong
Thomas Gröger
Michal Pardo
Thorsten Hohaus
Thomas Adam
Astrid Kiendler-Scharr
Yinon Rudich
Ralf Zimmermann
Markus Kalberer
It is being suggested that particle-bound or particle-induced reactive oxygen species (ROS), which significantly contribute to the oxidative potential (OP) of aerosol particles, are a promising metric linking aerosol compositions to toxicity and adverse health effects. However, accurate ROS quantification remains challenging due to the reactive and short-lived nature of many ROS components and the lack of appropriate analytical methods for a reliable quantification. Consequently, it remains difficult to gauge their impact on human health, especially to identify how aerosol particle sources and atmospheric processes drive particle-bound ROS formation in a real-world urban environment.
In this study, using a novel online particle-bound ROS instrument (OPROSI), we comprehensively characterized and compared the formation of ROS in secondary organic aerosols (SOAs) generated from organic compounds that represent anthropogenic (naphthalene, SOANAP) and biogenic (β-pinene, SOAβPIN) precursors. The SOA mass was condensed onto soot particles (SP) under varied atmospherically relevant conditions (photochemical aging and humidity) to mimic the SOA formation from a mixing of traffic-related carbonaceous primary aerosols and volatile organic compounds (VOCs). We systematically analyzed the ability of the aqueous extracts of the two aerosol types (SOANAP-SP and SOAβPIN-SP) to induce ROS production and OP. We further investigated cytotoxicity and cellular ROS production after exposing human lung epithelial cell cultures (A549) to extracts of the two aerosols. A significant finding of this study is that more than 90 % of all ROS components in both SOA types have a short lifetime, highlighting the need to develop online instruments for a meaningful quantification of ROS. Our results also show that photochemical aging promotes particle-bound ROS production and enhances the OP of the aerosols. Compared to SOAβPIN-SP, SOANAP-SP elicited a higher acellular and cellular ROS production, a higher OP, and a lower cell viability. These consistent results between chemical-based and biological-based analyses indicate that particle-bound ROS quantification could be a feasible metric to predict aerosol particle toxicity and adverse human effects. Moreover, the cellular ROS production caused by SOA exposure not only depends on aerosol type but is also affected by exposure dose, highlighting a need to mimic the process of particle deposition onto lung cells and their interactions as realistically as possible to avoid unknown biases.
- Article
(1279 KB) - Full-text XML
- BibTeX
- EndNote





Ambient particulate air pollution has been identified by the World Health Organization (WHO) as the most severe and urgent public health issue worldwide, mainly affecting people in urban areas (WHO, 2016). It is unknown which particle sources and properties are mainly responsible for their negative health effects, but particle-bound or particle-induced reactive oxygen species (ROS), which significantly contribute to the oxidative potential (OP) of aerosol particles, are considered central for a mechanistic understanding of many adverse health effects from particle exposure (e.g., Øvrevik, 2019; Daellenbach et al., 2020). It is therefore being suggested that ROS or OP may be a promising metric linking aerosol compositions to toxicity and adverse health effects (e.g., Hellack et al., 2017; Bates et al., 2019; Øvrevik, 2019).
ROS covers a variety of reactive species, including hydrogen peroxide (H2O2), organic peroxides, superoxide radicals (O), hydroxyl radicals (•OH), alkoxyl radicals (RO•), carbon-centered radicals (C•) and singlet oxygen (1O2) (Fuller et al., 2014; Hellack et al., 2017). Particle-bound ROS can be generated from primary emission sources and can also be formed from photochemical and heterogeneous reactions in the atmosphere (called exogenous ROS or acellular ROS), which can be introduced into the body via particle inhalation. In addition, ROS can also be formed endogenously in the lung lining layer or in lung tissue cells, after aerosol particles have been deposited onto the lung surface (e.g., Hellack et al., 2017). The ability of particles to generate ROS after deposition onto the lung tissue is often defined as “oxidative potential” (OP). Several chemical assays (i.e., acellular assays) were developed over the last years to either measure the ROS in particles directly through the application of high-performance liquid chromatography (HPLC), spin trapping or electron spin resonance spectroscopy (ESR) and fluorescence-based method (e.g., 2'7-dichlorofluorescein (DCFH)/horseradish peroxidase (HRP) assay), or to quantify the particle OP indirectly through measuring the consumption of antioxidants naturally present in human lungs (e.g., ascorbic acid, AA; reduced glutathione, GSH) or surrogates (e.g., dithiothreitol, DTT) (Hedayat et al., 2015). DCFH/HRP and DTT assays are two of the most popular assays used for ROS and OP measurement, respectively (e.g., Bates et al., 2019). The DCFH/HRP assay has been shown to be sensitive to a broad range inorganic and organic of particle-bound ROS components, especially peroxides, largely due to the ease of abstraction of the hydrogen atom located at the 9′ position of the DCFH molecule (Bates et al., 2019). Besides exogenous ROS, DCFH assay has also recently adapted to quantify endogenous ROS production after exposing cells to particles (e.g., Tuet et al., 2017a; Chowdhury et al., 2019). The rate of DTT reactions with aerosol extracts, a surrogate for the cellular antioxidant nicotinamide adenine dinucleotide phosphate (NADPH) and oxidation potential in general, has been extensively used in recent years to characterize particle OP (e.g., Tuet et al., 2017b; Wang et al., 2018; Bates et al., 2019).
Most ROS analysis techniques mentioned above use offline filter-based methods, where aerosols are analyzed hours (h) or days after collection and therefore likely severely underestimate the true particle-bound, exogenous ROS concentrations because the reactive and short-lived components of ROS such as radicals or some peroxides decay before analysis (Fuller et al., 2014; Krapf et al., 2016; Zhou et al., 2018a; Bates et al., 2019). This makes it difficult to gauge their impact on human health, especially to identify which particle sources contribute to ROS and what atmospheric conditions affect ROS formation in a real-world urban environment. However, appropriate analytical chemical methods, which also capture the short-lived fraction of ROS, for accurate ROS quantification are largely lacking. Moreover, these different chemical-based ROS or OP assays are sensitive to different ROS species, which is in turn relevant to the aerosol particle sources and chemical composition (Zhou et al., 2019; Campbell et al., 2021). Because of the highly complex composition and multiple sources of atmospheric aerosols, little has been done to establish the associations between chemical-based ROS/OP measurements in particles and cellular ROS production and other adverse biological effects induced by the particles. Therefore, to date, it is unknown which of the chemical assays is the most appropriate metric to predict the toxicity and adverse health effects of aerosol particles.
A large, sometimes dominant, aerosol fraction in the lower atmosphere is secondary organic aerosols (SOAs), which is formed via oxidation of anthropogenic and biogenic volatile organic compounds (AVOCsBVOCs) by atmospheric oxidants such as ozone, nitrate and hydroxyl radicals. In typical urban environments, traffic emission is one of the major sources of soot particles (SP). The oxidation of a complex mixture of AVOCs and BVOCs and exhaust emissions by vehicles play a significant role for the formation of SOA and contribute prominently to the urban SOA burden (e.g., Gentner et al., 2017). Previous studies have shown that the organic coating thickness of SP and their degree of oxygenation increased with photochemical age or oxidant levels in the atmosphere (Cappa et al., 2012; Liu et al., 2015). How SOA, generated from different AVOCs and BVOCs under varied atmospheric conditions, affects the particle-bound ROS formation has not been investigated yet. Therefore, the relative toxicological potentials of anthropogenic and biogenic SOA and the possible methods to predict their adverse health effects are unknown.
We recently developed and built a novel online instrument to quantify particle-bound ROS (OPROSI) based on the DCFH/HRP assay with high time resolution of about 5 min (Wragg et al., 2016). Using this unique instrument, we conducted a series of oxidation flow reactor (OFR) experiments to track the formation of ROS in SOA. Naphthalene and β-pinene were chosen as the representative anthropogenic and biogenic SOA precursors, respectively. SP was generated from a miniature combustion aerosol standard (mini-CAST) soot generator. SOA from the two precursors condensed on SP inside the OFR with varied atmospherically relevant conditions (UV intensity and humidity) to form particles from a photochemically aged (OH-dominated) mixture of primary combustion SP and naphthalene (SOANAP-SP) or β-pinene (SOAβPIN-SP). We also simultaneously collected filter samples to quantify ROS with conventional offline methods using the same chemical assay as the online ROS analysis (i.e., DCFH/HRP) and to determine OP using the DTT assay. We further systematically evaluated the changes in cell viability and of cellular ROS production after exposing human lung epithelial cell cultures (A549) to extracts of the two aerosol types. Our results indicate that ROS could be a feasible metric that can be used to link aerosol particle toxicity and adverse human effects, as well as highlight the importance of developing online instruments to quantify ROS. This is the first study to comprehensively investigate ROS production from individual SOA using our novel setup and to address the feasibility of utilizing chemical-based ROS or OP measurement to predict the adverse health effects of aerosol toxicity closely linking chemical and biological analyses.
2.1 Aerosol generation, online characterization and filter sample collection
We established a novel aerosol generation, aging and characterization setup to simulate ambient aerosols in the laboratory under well-controlled and reproducible conditions. The schematic of the experimental system is given in Fig. 1. The setup is explained here only briefly, but more details can be found in Offer et al. (2022). A miniature combustion aerosol standard (mini-CAST) soot generator (model 5201C, Jing Ltd, Switzerland) was used to produce combustion soot particles with a geometric mean diameter of about 100 nm and consisting mainly of elemental carbon (EC) and a very small fraction of primary organic matter (POM, less than 5 %). The soot particles were diluted in a custom-made porous tube diluter and further mixed with either naphthalene (Sigma-Aldrich, 147141-25G, 99 %, Germany) or β-pinene (Sigma-Aldrich, 402753-10G, ≥ 99 %, Germany) through an automated VOC dosing system. The mixture of soot and VOC was then humidified with a humidified dilution air flow in an ejector diluter (model VKL 10, Palas GmbH, Germany) and subsequently introduced into a commercial OFR (potential aerosol mass (PAM), Kang et al., 2007) to generate a photochemically aged SOA coating of the soot particles (i.e., SOANAP-SP or SOAβPIN-SP) under varied aging time and relative humidity (RH) conditions. The PAM allows us to reach an equivalent aging time comparable to average residence times of particles in the atmosphere. For each condition, 0.25 mg m−3 SP and 1 mg m−3 VOC were introduced into the PAM. A known concentration of deuterated butanol (D9-butanol, 98 % isotopic purity; Sigma Aldrich, Germany) was also added to the PAM together with soot and VOC, and the changes of the concentration of D9-butanol were monitored and used as a photochemical aging clock (Barmet et al., 2012). Besides soot particles coated with SOA, we additionally investigated the possible effects of aging on pure soot particles, i.e., SP passing through the PAM with no VOCs added. A KNO2-coated ceramic ozone denuder (BCE Special Ceramics GmbH, Germany) was installed downstream of the PAM, where an ozone analyzer (model APOA 350E, Horiba, Japan) monitored the ozone concentration. The purpose is to ensure that almost all ozone formed in the PAM was removed prior to conducting online composition measurements and offline filter sample collection to rule out any sampling artifacts caused by O3.
Several online instruments were used for a comprehensive gas- and particle-phase characterization during the aerosol formation and aging processes. The decay of D9-butanol in the PAM was quantified with a quadruple proton-transfer-reaction mass spectrometer (PTR-MS, Ionicon, Austria) to determine equivalent photochemical aging times (Barmet et al., 2012; Offer et al., 2022). More details on this calculation can be found in Barmet et al. (2012) and Offer et al. (2022). We generated SOA with a photochemical aging equivalent to 2, 3, 4, 5 and 9 d at ambient OH concentration of 1×106 molecules cm−3. The effects of relative humidity (40 % and 70 % RH) on SOA characterization were also evaluated. A high-resolution time-of-flight aerosol mass spectrometer (HR-TOF-AMS, Aerodyne Inc., Billerica, MA, USA) provided chemical aerosol mass loading information and aerosol elemental composition (e.g., O:C, H:C and N:C ratios) using the method described in Canagaratna et al. (2015), based on which the average carbon oxidation state (–H:C) was calculated following the method of Kroll et al. (2011). An ozone analyzer (ANSYCO GmbH, model O3-41M, Karlsruhe, Germany) was used to quantify the ozone concentration inside the PAM. A scanning mobility particle sizer (SMPS), which is composed of an electrostatic classifier (TSI, model 3082, USA) and a condensation particle counter (CPC, TSI model 3750, USA), was used to monitor particle number and size distribution.
For each experimental condition, we collected aerosol samples in parallel on 47 mm pre-baked (500 ∘C, 5 h) quartz fiber filters (Pall Corporation, USA) for offline analyses. The sampling flow rate was fixed at 20 L min−1, and the sampling duration was about 1 h. The filter samples were then kept in Analyslide petri dishes (Pall Corporation, USA), wrapped with aluminum foil and stored in a freezer at −20 ∘C until analyses.
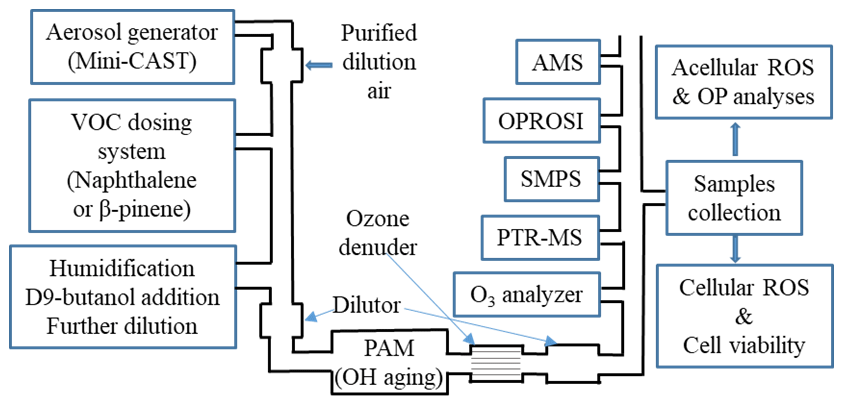
Figure 1A schematic of the experimental setup for SOA generation and characterization. SOA was generated from naphthalene and β-pinene condensed on soot particles (SP) from a miniature combustion aerosol standard (mini-CAST) soot generator under varied atmospheric conditions (UV lamp intensity and humidity). Several online instruments were used for a comprehensive gas- and particle-phase characterization (including the online ROS instrument OPROSI). Filter samples were collected for various chemical and biological offline analyses.
2.2 Online acellular ROS analysis
An online particle-bound ROS instrument (OPROSI) previously developed in our research group based on the DCFH/HRP assay (Fuller et al., 2014; Wragg et al., 2016) was further optimized and was used to quantify ROS formation and evolution during SOA formation and aging. A detailed description of the ROS analyzer can be found in Fuller et al. (2014) and Wragg et al. (2016) and is briefly summarized in Sect. 3.2. The instrument was calibrated with known concentrations of aqueous hydrogen peroxide (H2O2) solutions, which were prepared from a concentrated solution of H2O2 (3 % in water, biological grade, Sigma Aldrich), and all results are given as equivalent H2O2 concentration. The ROS concentration under each test condition was continually monitored for about 1 h. Blank measurements were taken for each particle collection time by placing a HEPA filter in-line in front of the OPROSI, and blank values were subtracted from the sample measurements. Four honeycomb charcoal denuders (Ionicon Analytic GmbH, Austria) were installed in series in front of the inlet of the OPROSI to rule out potential gas-phase artifacts in the OPROSI. The particle loss inside the denuders was quantified by simultaneously monitoring the particle number concentrations prior to and after the denuders, and results presented here were corrected for these losses.
2.3 Offline acellular ROS analysis
In parallel to online ROS measurement, we quantified water-soluble ROS contents in the collected aerosol samples using an offline DCFH/HRP method, where we used the same concentrations of DCFH and HRP as for the online ROS measurement. The detailed protocol has been given elsewhere (Fuller et al., 2014; Campbell et al., 2021) and is described only briefly here. A small round punch (6 mm diameter) of the filter was first extracted with Milli-Q water (> 18 MΩ cm; Millipore, Bedford, MA, USA) for 3 min using a vortex mixer (FB15024, Fisher Scientific). The extracts were then filtered through a polytetrafluoroethylene (PTFE) syringe membrane filter (pore size, 0.45 µm; Sartorius AG, Goettingen, Germany). A total of 500 µL extract was added to 500 µL of 20 µM of DCFH in 20 % phosphate buffered saline (PBS, pH 7.4, sigma Aldrich) and 1000 µL of 1.0 unit mL−1 HRP in 10 % PBS. This mixing dilutes both reactants to the same concentrations of DCFH (5 µM) and HRP (0.5 unit mL−1) as used in the online method. The concentrations of SOA extracts in the final solution corresponded between 0.3 and 0.45 µg mL−1 SOA, which was well within the linear range of the calibration curves. The filter extracts with DCFH and HRP were incubated in a water bath (model WB05, PolyScience) at 37 ∘C for 15 min and then transferred into a quartz cuvette (CV10Q1400F, Thorlabs, Germany), where excitation light from a light emitting diode (LED) (470 nm, Luxeon Rebel Star on CoolBase) was delivered via an optical fiber (Ocean Insight, USA), and finally their fluorescent emission at 522 nm was acquired using a QE Pro high-performance spectrometer (Ocean Insight, USA). The fluorescence response was calibrated with known concentration of H2O2 solutions (3 % in water, biological grade, Sigma Aldrich) in the range of 0–0.8 µM, and quantitative ROS concentrations are reported as “[H2O2] equivalents”. For each sample, three replicate analyses were conducted. The blank values were obtained by measuring the fluorescent spectra from blank filter extracts, and these were subtracted from filter sample measurements.
2.4 Particle OP analysis
We established a DTT assay protocol that follows the methods developed by Charrier and Anastasio (2012) and Lin and Yu (2019) with minor modifications. Briefly, the loss of 100 µM of DTT in 0.1 M phosphate buffer (77.8 mM NaH2PO4 and 22.2 mM KHPO4; pH 7.4) was measured over time at 37 ∘C. At time zero 2.5 mL of particle extracts was added to 2.5 mL of 200 µM of DTT in 0.2 M phosphate solution in a 5 mL falcon tube. The reaction mixture was maintained at 37 ∘C on a digital hotplate (Stuart, SD160). At known times (0, 10, 20, 30 and 40 min), a 0.6 mL aliquot of the reaction mixture was removed and added to 0.6 mL of 10 % trichloroacetic acid (99 %, Acros Organics) to stop the reaction. A total of 60 µL of 10.0 mM DTNB (≥ 98 %, Sigma Aldrich) (made in 0.10 M phosphate buffer; pH 7.4) was added in each tube, mixed well (vortex 5 s) and allowed to react with the remaining DTT for 5 min. A total of 0.8 mL of 0.67 M Tris base (pH 8.9; ≥ 99.9; Sigma Aldrich) with 33.3 mM of EDTA (Sigma Aldrich) was subsequently added to inhibit possible artifacts from metal-induced ROS formation. After waiting for 15 min, the absorbance of a given sample (0.9 mL) was quantified using a 1 cm path length quartz cuvette (CV10Q1400F, Thorlabs, USA) coupled to the QE Pro high-performance spectrometer (Ocean Insight, USA). The absorption was measured at 412 nm with 70 ms integration time. A solution of 0.5 mM Cu (copper(II) sulfate pentahydrate, Acros Organics) was used as the positive control and was tested on each experimental day. The quantity of DTT consumed at a certain incubation time (µM DTT) is calculated with the following Eq. (1):
where A0 is the absorbance of the sample at t=0 and At is the absorbance of the sample at a certain incubation time t (min), and CDTT is the initial DTT concentration in the reaction solution (µM).
To keep DTT loss rates comparable and to allow for comparable results, the mass of extracted particles was kept constant at 0.8 and 1.8 µg mL−1 in the reaction solution for naphthalene SOA and β-pinene SOA, respectively. It was ensured that less than 20 % of DTT was consumed by the particle extracts during the reaction. Under our experimental conditions, the absorbance is linearly proportional to the DTT concentration in the reaction solution (R2 > 0.99). The DTT loss rate ΔDTT (µM DTT min−1) is obtained from the slope of the plot of ΔDTTt versus time. To reduce background oxidation, we treated the Milli-Q water and the phosphate buffer with Chelex 100 resin (sodium form, Bio-Rad) to remove transition metals. We also washed all plastic ware (e.g., falcon tubes, pipette tips) using Milli-Q water after immersion in 5 % HNO3 solution for 3 d. For each sample, three replicate analyses were conducted. A blank filter with the same size as the analyzed samples was extracted and subsequently analyzed following the same procedures as the samples. The averaged value obtained from these blank filters was used as the blank signal. The ΔDTT of the blank filter was quantified, and results presented here were corrected for these losses. Blank filters and samples were all run in triplicate.
2.5 Cell viability and cellular ROS production
For the cell viability test, the filter samples were extracted in HBSS buffer (Hanks' buffered salt solution) with 10 % FBS (fetal bovine serum) by vortexing for 3 min, subsequently filtered using 0.45 mm PTFE syringe filters (Fisherbrand™) and then diluted for dose-response analyses. The human type-II cell alveolar epithelial cell line A549 (American Type Culture Collection) was cultured in DMEM (Dulbecco's Modified Eagle Medium, Gibco) with 10 % FBS at 37∘ in 5 % CO2. The A549 cells were seeded into transparent 96-well plates (Greiner Bio-One) at an optimized concentration of 2×105 cells mL−1 and cultured for 24 h to allow adherence and proliferation. The medium was then replaced, and the cells were exposed to 100 µL of particle extracts in HBSS for another 24 h. The concentrations of particle extracts added to the cell culture wells corresponded to about 3, 6, 12, 24, 48 and 96 µg mL−1 of SOA, respectively. At the end of the treatments, 10 µL of Cell Counting Kit-8 assay (CCK8; Dojindo, Kumamoto, Japan) at a final concentration of 20 µM in HBSS buffer was added to each well and incubated for another 2 h. Finally, the absorbance value in each well was measured at 450 nm using a microplate reader (Synergy HT, BioTek). Each sample was tested in six replicate wells, and the data on cell viability are reported as the relative percentage of the control, considered as 100 % of viable cells.
The cellular ROS was detected using the 2',7'-Dichlorodihydrofluorescein Diacetate (DCFH-DA) cellular ROS detection assay kit (ab113851, Abcam, Cambridge, UK) according to the manufacturer's instructions. Briefly, the particulate samples were extracted in HBSS buffer with 10 % FBS by vortexing for 3 min. The extracts were then filtered and were finally diluted to obtain a dose-response curve. The final concentrations of particle extracts in the cell culture wells are the same as for the cell viability test. The A549 cell line was cultured following the same protocol as the cell viability test. The A549 cells were seeded into 96-well black flat-bottom plates (Greiner Bio-One) at an optimized concentration of 2×105 cells mL−1 and cultured for 24 h to allow adherence and proliferation. The medium was then replaced, the cells were washed twice with HBSS buffer, and 100 µL of HBSS buffer together with 20 µM DCFH-DA assay was then added to each well and incubated for 30 min at 37 ∘C and 5 % CO2. After the DCFH-DA assay loading, the probe solution was removed, the cells were washed twice with the HBSS buffer again and 100 µL of aerosol extracts was added into each well. The cells were exposed to particle extracts for 2, 4, 8 and 24 h, respectively. Fluorescence was then measured (exem: 485528 nm) using a microplate reader (Synergy HT, BioTek). Positive controls included tert-butyl hydrogen peroxide (TBHP, 50 µM), and H2O2 (100 µM) was included in each plate. Negative controls including blank filter extract (extracted by using the same protocol as the filter samples) and control cells (i.e., probe-treated cells exposed to buffer only, no stimulants) were also included in each plate. In addition, the cellular ROS production induced by different concentration of H2O2 was also measured. The ROS production for each filter sample extract was expressed as a fold increase in fluorescence over the negative control (blank filter extract).
2.6 QA/QC and statistical analysis
The lower limit of detection (LOD), the linearity of calibration and the relative standard deviation (RSD) of replicate analyses were considered for the analysis of the online/offline acellular ROS data. LODs and RSD of replicate analyses were used to assess the data of particle OP, changes of cell viability and fold change of cellular ROS production. The LODs for online ROS and offline ROS were below 18 nM [H2O2] equivalent per cubic meter of air and 0.01 nM [H2O2] equivalent per cubic microgram of SOA, respectively. The RSD of duplicate analyses was lower than 5.8 % for online acellular ROS, below 6.2 % for offline acellular ROS, below 8.6 % for particle OP, and below 9.8 % for cellular ROS, respectively. A five-point calibration for H2O2 standard solution was performed, and the correlation coefficients (R2) obtained from the linear regression of the calibration curves was larger than 0.99. In this study, all analytical data were corrected for the average value of the blanks and are reported as the mean values. The significance of the differences of all tested parameters between the two aerosol types (i.e., SOANAP-SP or SOAβPIN-SP) under the same photooxidation aging and humidity conditions was assessed by Student's t test at p<0.05. The results of of the particles are reported as the mean values, with the relative standard deviations ranging from 2.5 to 3.2 %. With the exception of of the particles, all other reported values are presented as mean ± SD (standard deviations).
In this study, we quantified online and offline ROS, as well as OP from the aqueous extracts of fresh (i.e., not aged in the PAM) and aged soot particles generated with the mini-CAST soot generator but without addition of SOA. All the aqueous extracts of soot particles (fresh, aged, analyzed online or offline) showed no detectable ROS concentrations and no significant OP compared to blank filters. We therefore normalized ROS production and OP of the SOANAP-SP and SOAβINP-SP to organic matter (OM) mass concentration in aerosols quantified online using AMS rather than the total aerosol particle mass concentration. In the data presented below we defined an OM mass-based ROS content (nM [H2O2] equivalent per microgram of OM) and particle OP (nM min−1 µg−1 OM) to avoid biases due to the mass of soot, which contributes to particle mass but induces little to no ROS production or OP.
3.1 Online measurements of particle-bound ROS during SOA formation and aging
Figure 2 shows that online ROS content in anthropogenic SOANAP-SP increased significantly with an increase in photochemical age from 2 to 9 d, demonstrating that atmospheric aging dominated by reactions of OH radicals promotes more particle-bound ROS formation. A significant but much slower increase was observed for the ROS content in SOAβPIN-SP when aged for 2 to 4 d. However, no significant increase in the ROS content in SOAβPIN-SP was seen for further aging of 5 and 9 d. We observed that the online ROS content in SOANAP-SP was in the range of 3.0–9.4 and 2.9–9.6 nM [H2O2] equivalent per microgram of OM at 40 % RH and 70 % RH, respectively. The corresponding online ROS content in SOAβPIN-SP was in the range of 0.7–1.6 and 1.0–2.1 nM [H2O2] equivalent per microgram of OM. For each aging condition, SOANAP-SP showed a 2.8 to 5.8 times higher ROS content than that of SOAβPIN-SP. In addition, a change in RH from 40 % to 70 % showed marginal effects on ROS content in both aerosol types. This may be because naphthalene SOA and β-pinene SOA do not contain as highly oxidized compounds as other SOA types, which leads generally to a modest hygroscopicity (Chhabra et al., 2010; Chen et al., 2015), and thus the aerosol phase reactions might not be affected much by RH. These observations indicate that the ROS content is driven mainly by both the nature of the SOA precursor and the photochemical aging. Our observations are consistent with previous reports that photochemical oxidation processes producing SOA from vehicle exhausts or wood combustion promoted ROS formation (Platt et al., 2014; Zhou et al., 2018b). Using the same OPROSI, previous studies in our laboratory observed a ROS content of 0.58 nM [H2O2] equivalent per microgram of SOA generated from ozonolysis of oleic acid in a flow tube (Fuller et al., 2014) and of 0.4 nM [H2O2] equivalent per microgram of SOA formed from ozonolysis of limonene in an atmospheric simulation chamber (Gallimore et al., 2017). Together, these studies suggest that naphthalene SOA has a higher ROS content than biogenic SOA. This might be explained by the formation of quinones and semiquinone in the photooxidation of naphthalene as found in Offer et al. (2022), which can react with oxygen in water to generate superoxide radial and therefore contribute to ROS production. Such quinone-type compounds do not form from terpene oxidation (Offer et al., 2022). Since naphthalene is mainly emitted from anthropogenic activities in urban areas, including road traffic and biomass burning (Jia and Batterman, 2010), controlling aerosol particle and gaseous organic emissions from these sources might be an efficient measure to reduce ROS concentrations in ambient aerosol particles. We observed that a few days of photothermal aging of anthropogenic SOA significantly enhance particle-bound ROS formation. Such a time lag between emission and the highest ROS formation might especially affect downwind areas of urban centers where particle-bound ROS burden and particle toxicity could be enhanced due to photooxidation processes as demonstrated here.

Figure 2ROS content in anthropogenic SOANAP-SP increased significantly with an increase in photochemical age from 2 to 9 d. A significant but much slower increase was observed for the ROS content in SOAβPIN-SP when aged for 2 to 4 d. However, no significant increase in the ROS content in SOAβPIN-SP was seen for further aging of 5 and 9 d. A change in relative humidity (RH) from 40 % to 70 % showed marginal effects on ROS content in both aerosol types. Measurements were performed with the online instrument OPROSI. ROS was also quantified in uncoated soot (fresh and aged), but no ROS content was measured in uncoated soot; see text for details.
Previous studies have shown that peroxides, including highly oxidized organic molecules (HOMs), play an important role in the formation and evolution of SOA, including from naphthalene-derived SOA (e.g., Kautzman et al., 2010; Tu et al., 2016; Molteni et al., 2018), which likely contributes significantly to the formation of ROS (Tong et al., 2018, 2019; Wang et al., 2018; Chowdhury et al., 2018, 2019; Wei et al., 2021). Chowdhury et al. (2019) showed that SOA generated from photooxidation of both naphthalene and α-pinene in a PAM reactor contains a significant fraction of peroxides. Tong et al. (2018, 2019) observed a positive correlation between the total peroxide concentration and ROS yield from SOA generated from ozonolysis of β-pinene in a PAM and suggested that HOMs are important constituents of biogenic SOA to generate ROS radicals. Wei et al. (2021) recently demonstrated the formation of superoxide from the decomposition of organic hydroperoxides in SOA generated from the photochemical reaction of β-pinene. Based on these previous observations, we therefore suggest that organic peroxides in SOANAP-SP and SOAβINP-SP contribute significantly to the overall content of ROS in SOA as observed in this study. On the one hand, photooxidation aging on the timescale studied here of SOA is likely to increase in the formation of peroxides in both anthropogenic and biogenic SOA (e.g., Chowdhury et al., 2019) and therefore could form higher ROS contents in SOA. On the other hand, upon further oxidation, some early-generation ROS products can decompose rapidly due to their photolabile and thermal instability (Krapf et al., 2016; Wang et al., 2018). The thermal decomposition of peroxides was shown to be more prominent in β-pinene SOA compared to naphthalene-derived SOA (Krapf et al., 2016). These opposing factors could be a reason for the different trends of ROS evolution over time in different SOA systems as observed in this study. Despite this, our observations showed that photochemical aging processes tend to promote more particle-bound ROS formation in SOA. It is worth noting that other studies reported that naphthalene SOA contains a smaller fraction of organic peroxides than α-pinene or β-pinene SOA (Wang et al., 2018; Tong et al., 2018). This seems to be inconsistent with our finding as we found a higher ROS content in naphthalene-derived SOA compared to β-pinene-derived SOA. These differences might be explained by the oxidation regimes used (i.e., photooxidation vs. ozonolysis) in different studies and also the different contributions of other known and unknown ROS species in SOA.
Interestingly, we observed significant positive correlations between carbon oxidation state () of the particle bulk composition as measured with AMS and online ROS content, as evident from Fig. 3. The is also positively correlated to the OH aging time (not shown) and thus indicates that a high degree of oxidation induced by photooxidation aging is linked to higher ROS production. Under each test condition, Fig. 3 also showed a higher from SOANAP-SP compared to SOAβPIN-SP, consistent with a higher ROS content as also shown in Fig. 2. Zhou et al. (2019) recently measured particle-bound ROS content in the ambient atmosphere in Beijing (China) and Bern (Switzerland) using an online ROS instrument. A statistical analysis of their results showed that oxygenated organic aerosol (OOA) explained the highest fraction of the ROS concentration variance at both locations. Since ROS content is highly associated with the oxidation degree of organic aerosol generated from both laboratory-generated and atmospheric aerosols, including different precursors under different formation conditions, it is plausible that all the other effects (i.e., precursor identity, humidity) are ultimately an effect of the degree of oxidation of aerosols. This finding has notable implications for future studies because atmospheric photochemical aging leads to increases in aerosol oxidation (Jimenez et al., 2009; Ng et al., 2011 and Fig. 3 here) and aerosols have an atmospheric lifetime of approximately 1–2 weeks, during which these processes can occur. Finally, the observed correlation might suggest that values of observed from online AMS may be utilized to estimate ROS levels and vice versa.
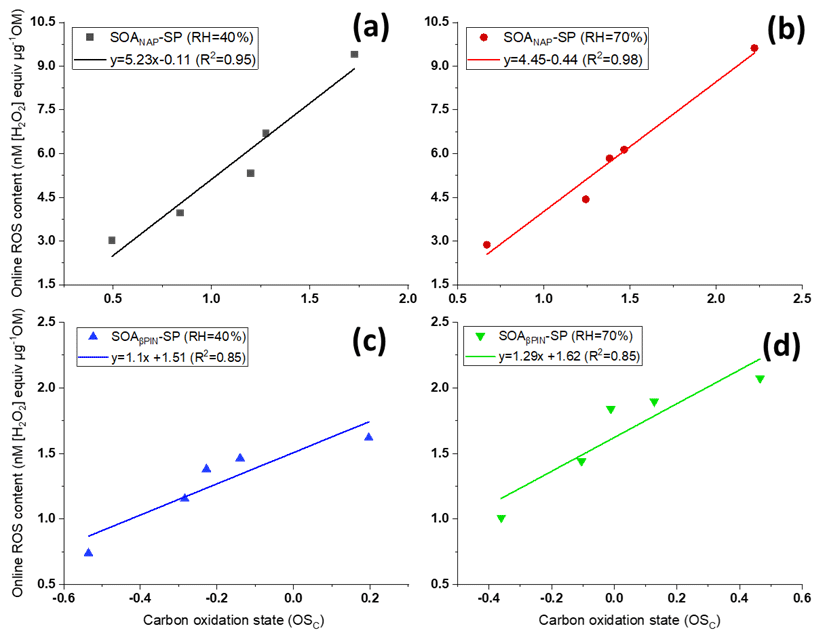
Figure 3Positive correlations between carbon oxidation state () of the particles and their ROS content using the online instrument OPROSI (in each of the panels, the data points from left to right correspond to 2, 3, 4, 5 and 9 d photochemical aging) for SOANAP-SP at 40 % and 70 % relative humidity, (a) and (b), respectively, and for SOAβPIN-SP at 40 % and 70 % relative humidity, (c) and (d), respectively. The data show that longer aging times increase the carbon oxidation state () and the corresponding ROS content in SOA. The line indicates linear regressions between and online ROS content.
3.2 Short-lived versus long-lived ROS
In our online ROS instrument, water-soluble ROS components react with HRP (the first reaction of the DCFH/HRP assay) seconds after the particles enter the instrument. This ensures that also short-lived ROS can be quantified by the assay before reacting with other aerosol components or the extraction solvent. Thus, our online method is likely to capture the most or all of the total ROS components, including short- and long-lived ROS. To determine the ratio of short- versus long-lived ROS, we collected aerosol samples on a quartz filter during all online measurements, which allow us to quantify the long-lived ROS fraction because there was a significant delay between aerosol collection and ROS analysis that causes the short-lived ROS to decay before analysis. Filter samples were collected for about 1 h at room temperature (allowing for a potential decay of short-lived ROS) and then stored at −20 ∘C for about 6 months before offline analysis using the same DCFH/HRP assay as the online ROS measurement. As shown in Fig. 4, a similar effect of photochemical aging and humidity on the offline (i.e., long-lived, stable) ROS content in both aerosol types was observed as for the online ROS content (see Fig. 2). However, the ROS concentrations per OM mass from offline filter samples were more than 1 order of magnitude lower than those from the online ROS measurements (Fig. 5). Specifically, the ratio of offline online ROS was 6.6 %–7.6 % and 6.7 %–7.3 % for SOANAP-SP produced at 40 % RH and 70 % RH, respectively. The corresponding ratio of offline online ROS for SOAβPIN-SP was 3.7 %–5.2 % and 3.8 %–4.9 %. Our observation demonstrates that the vast majority of ROS components are short-lived. On average 92.9 ± 0.4 % of ROS from SOANAP-SP and 95.8 ± 0.5 % of ROS from SOAβPIN-SP were composed of short-lived ROS components, which were lost during sampling and filter storage, highlighting the importance of a fast, online instrument as used in this study for a meaningful quantification of ROS. We also observed that the ratio of long-lived total ROS from SOAβPIN-SP was generally lower than that from SOANAP-SP, indicating that ROS components in SOAβPIN-SP are more labile.
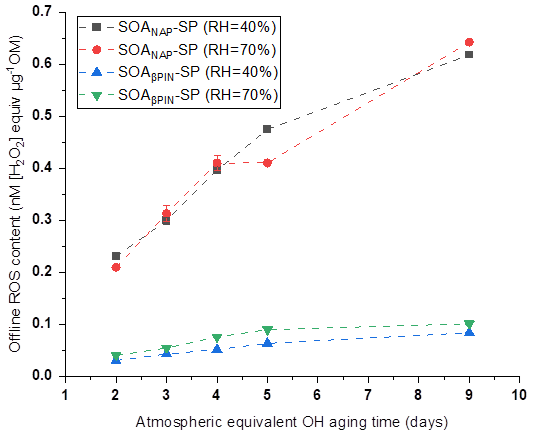
Figure 4ROS concentrations in SOA particles quantified offline from filter samples as a function of aging time. A similar effect of photochemical aging and relative humidity (RH) on the offline (i.e., long-lived, stable) ROS content in both aerosol types was observed as for the online ROS content (see Figs. 2 and 5).
A previous study in our group showed that approximately 75 % of the total ROS has a half-life of a few minutes when we compared offline and online ROS analyses in SOA formed from ozonolysis of oleic acid aerosol in a flow tube (Fuller et al., 2014). Similarly, Wang et al. (2011) reported that the H2O2 yield (a significant component of ROS) from both α-pinene and β-pinene SOA was about 70 % smaller after storing the filter samples in petri dishes in the dark at room temperature for 20 h. In the data shown here (Fig. 5), we observed an even higher fraction of short-lived ROS than Fuller et al. (2014) and Wang et al. (2011) reported. Such differences in short-lived/total ROS ratios are likely explained by the different SOA precursor and oxidation conditions: Fig. 5 clearly shows that two types of SOA, i.e., from naphthalene and β-pinene, have up to a factor of 2 different offline online ROS ratios. Zhou et al. (2018b, 2019) evaluated the ROS decay behavior in ambient aerosols collected in Bern and during wood combustion experiments through comparison of offline and online ROS measurements using the DCFH/HRP assay. They showed that a large fraction of ROS decomposes rapidly with a half-life of 1.7 ± 0.4 h and that on average 60 ± 20 % of the ROS decayed during filter storage time for about 4 months. Together, these results emphasize that although ROS from different sources may contain different ROS components, all studies highlight that the majority of total ROS has a short lifetime of minutes to hours. The findings indicate offline techniques, where there is often a delay of hours to months between particle collection and ROS analysis, may severely underestimate true ROS concentrations and that fast, online techniques are necessary for a reliable total ROS quantification in atmospheric aerosol particles and for a meaningful correlation with health outcomes.
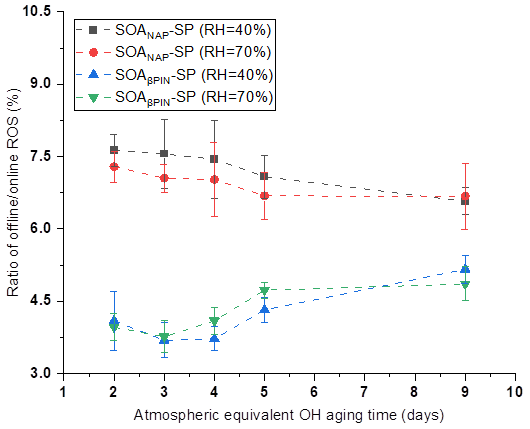
Figure 5Ratio of offline online ROS as a function of particle aging time. On average 92.9 ± 0.4 % of ROS from SOANAP-SP and 95.8 ± 0.5 % of ROS from SOAβPIN-SP were composed of short-lived ROS components.
As discussed in Sect. 3.1, peroxides, including HOMs, contribute significantly to the overall concentration of ROS in SOA. Therefore, their fate after partitioning into aerosol particles may reflect the fate of ROS. Krapf et al. (2016) reported that particle-bound HOMs, which account for 30 % of the initial SOA mass generated from α-pinene ozonolysis, are thermodynamically unstable with half-lives shorter than 1 h under dark conditions. Chowdhury et al. (2018) found that although a higher organic peroxide concentration was observed in aged naphthalene-derived SOA than in fresh naphthalene-derived SOA, the peroxide levels in the aged SOA were comparable to that in fresh SOA after 1 h sampling, and the peroxide levels in both SOAs decomposed drastically to reach the blank level after 4 h sampling. We note that all the filter sampling techniques for ROS and peroxide quantification have disadvantages such as the evaporation of semi-volatile species or decomposition of labile compounds during sampling and storage.
3.3 Aerosol oxidative potential (OP)
OP was evaluated from offline filter samples after storage of about 6 months at −20 ∘C based on the mass-normalized DTT decay rate (OP-DTT), with a higher DTT decay rate indicating a higher OP. As shown in Fig. 6, SOANAP-SP with a higher photochemical age has a higher OP. This trend is similar but not as pronounced as for ROS, measured by the DCFH/HRP assay (Fig. 2). Specifically, the OP of SOANAP-SP was in the range of 0.15–0.26 nM min−1 µg−1 at 40 % RH and of 0.14–0.21 nM min−1 µg−1 at 70 % RH. A similar range of OP from naphthalene SOA was reported in previous studies. For example, McWhinney et al. (2013) and Tuet et al. (2017b) reported 0.118 ± 0.014 and 0.210 nM min−1 µg−1 for naphthalene SOA, respectively, formed via photooxidation in a smog chamber. Wang et al. (2018) observed OP of 0.100–0.129 nM min−1 µg−1 for naphthalene SOA generated from photooxidation in a flow tube. The slightly different values among these studies are most likely due to differences in the photooxidation intensity and SOA formation environment (i.e., smog chamber vs. flow reactor). To the best of our knowledge, the OP of SOAβPIN-SP generated from photooxidation of β-pinene as observed in this study is the first reported in the literature. Unlike SOANAP-SP, photochemical aging of SOAβPIN-SP resulted in no significant change in its OP. This does not necessarily mean that photochemical aging does not affect the oxidative activity of SOAβPIN-SP but might indicate that OP in SOAβPIN-SP is also divided in a short- and long-lived fraction (similar to ROS measured with the DCFH/HRP assay) and that only a long-lived OP fraction is captured with the DTT assay. The finding is consistent with the results of Jiang and Jang (2018), who found that the DTT-reactive compounds decomposed after photochemical aging at ambient sunlight. Figure 6 also shows that under each test condition, SOAβPIN-SP had a lower OP than that SOANAP-SP. The trend is similar to that of ROS content measured by the DCFH/HRP assay as shown in Fig. 2. This finding is also consistent with a previous report that OP values for biogenic SOA are lower than those for anthropogenic SOA (Tuet et al., 2017a). Redox-active quinones can efficiently oxidize DTT and have been recognized as the major contributors to the OP of aromatic SOA (Charrier and Anastasio, 2012; McWhinney et al. 2013; Jiang and Jing, 2018). Therefore, one of the reasons for the high OP exhibited by SOANAP-SP as observed in this study is likely the abundance of naphthoquinones in naphthalene SOA, which have been shown to account for approximately 21 % and 30 % of the DTT activity for naphthalene SOA (Charrier and Anastasio, 2012 and McWhinney et al., 2013, respectively). The remaining DTT activity may arise from other quinone-like compounds that have not been identified or from other catalytic and/or noncatalytic oxidants in naphthalene SOA. This is supported by Wang et al. (2018) and Jiang and Jing (2018), who found that, besides quinones, other more oxygenated products (peroxides and hydroperoxides) and electron-deficient alkenes could also contribute to the high DTT activities of naphthalene SOA. Compared to naphthalene-derived SOA, β-pinene SOA is expected to contain a negligible amount of quinones, but peroxides are suggested to contribute significantly to the OP of β-pinene SOA (Wang et al., 2018; Jiang and Jing, 2018). Previous studies found that SOA formed from photooxidation of α-pinene consists of a substantial fraction of organic hydroperoxides (Wang et al., 2018; Jiang and Jing, 2018). This might also explain the lower OP of SOAβPIN-SP compared to SOANAP-SP as found in this study. During the photochemical aging, the molecular weight and the oxidation state of aerosol products increase continually owing to the addition of oxygenated functional groups (Offer et al., 2022). But the existing organic peroxides possibly also decompose during aging. These two opposing factors may be the reason why no significant change in OP of SOAβPIN-SP is observed with varied photochemical aging conditions.
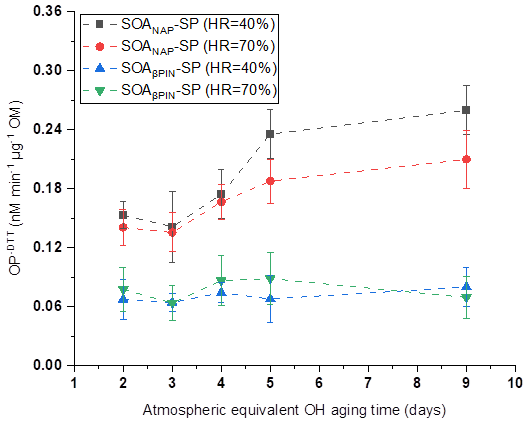
Figure 6Oxidative potential (OP) of the particles as a function of particle aging time. SOANAP-SP with a higher photochemical age has a higher OP, whereas photochemical aging of SOAβPIN-SP resulted in no significant change in its OP.
As can be seen from Fig. 7, we further found that OP and offline ROS measurements for SOANAP-SP are clearly positively correlated, while there is a weaker correlation between OP and offline ROS for SOAβPIN-SP. The positive correlation between OP and offline ROS content indicates that some redox compounds in SOANAP-SP are detected by the DTT as well as DCFH/HRP assay and/or may reflect a co-evolution of compounds sensitive to DTT and DCFH/HRP, respectively, during aging. For example, previous studies reported that SOA generated from photooxidation of naphthalene contains substantial fraction of organic peroxides (Tong et al., 2018; Chowdhury et al., 2019) and quinone and semiquinone compounds (McWhinney et al., 2013). Organic peroxides can react with the DCFH/HRP assay, and quinone and semiquinone compounds can react with the DTT assay. Furthermore, in aqueous phase, the decomposition of organic peroxides forms H2O2, and the reaction of quinone and semiquinone compounds with dissolved O2 forms O and H2O2 (Tong et al., 2018; Wei et al., 2021), which can be detected by DCFH/HRP and which lead to DTT consumption. Although the responses of DCFH and DTT may be caused by different compounds, the concentrations of all these compounds increase in SOA with increasing photooxidation aging. Conversely, our observation that there is no strong correlation between OP and offline ROS in SOAβPIN-SP indicates that OP and ROS respond to different compounds in SOA type, which emphasizes the importance of discerning which of the assays is relevant to health.
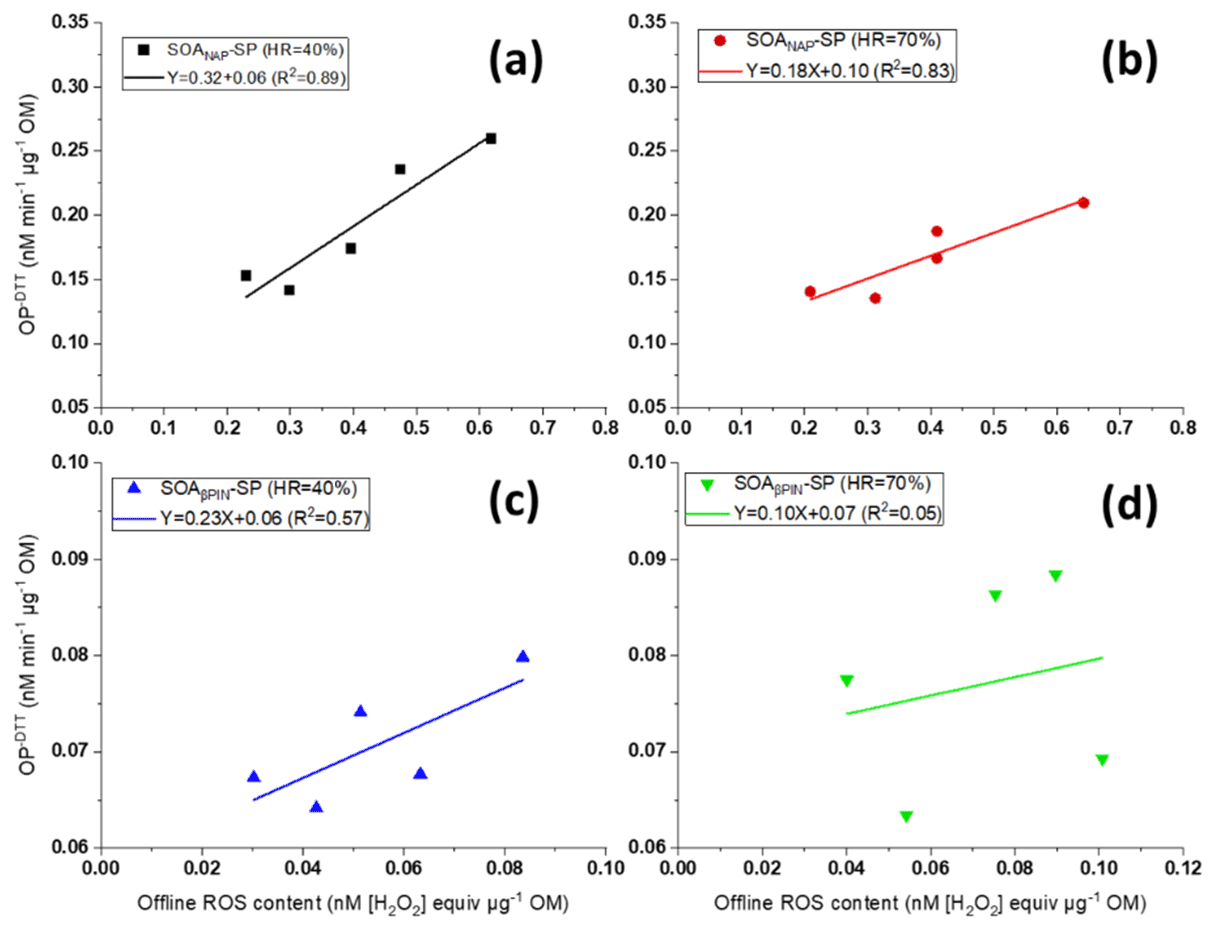
Figure 7Correlation between offline ROS content and the (offline) oxidative potential (OP) of particles (the data points from left to right correspond to 2, 3, 4, 5 and 9 d photochemical aging) for SOANAP-SP at 40 % and 70 % relative humidity, (a) and (b), respectively, and for SOAβPIN-SP at 40 % and 70 % relative humidity, (c) and (d), respectively. OP and offline ROS of SOANAP-SP are clearly positively correlated, while there is a weaker correlation between OP and offline ROS of SOAβPIN-SP.
3.4 Cell viability and cellular ROS production
The results shown in Sect. 3.1–3.3 were obtained from SOA mass produced from a mixture of 0.25 mg m−3 soot particles and 1 mg m−3 precursor VOC. We also evaluated the oxidative characteristics of aerosols produced from the mixture of 1 mg m−3 soot particles and 4 mg m−3 precursor VOCs under 3 d equivalent photochemical aging at 40 % RH for the biological studies discussed here. A comparison of the oxidative characteristics of these aerosols is given in Table 1. The of the particles generated from the mixture at high concentrations was significantly lower than from the mixture at low concentrations. This can be explained by the number of OH radicals per VOC molecule available, which is lower at the high naphthalene or β-pinene concentration, leading to a lower overall degree of oxidation. This change in is reflected differently in the ROS and OP responses of two SOA systems. The ROS concentration (in nM per µg SOA mass) in the SOAβPIN-SP is about the same at both precursor concentrations. However, it increases strongly for SOANAP-SP at higher SOA masses. Using the oxidation argument above, this indicates that some ROS components in the naphthalene oxidation scheme only partition from the gas phase into the particle phase under very high particle concentrations but would usually be present in the gas phase under ambient conditions. The same is observed for the DTT activity in both SOA types. We used the particles formed from high concentration of soot and VOC precursor mixture as model SOA for further biological study because their ROS concentrations were higher than under low-concentration conditions. We assume that although the higher particle concentrations had some effect on particle ROS content (Table 1), the overall trends discussed below (Figs. 8–11) are reflecting biological effects that would also occur at lower particle concentrations.
We further measured the cell viability and fold changes of cellular ROS caused by extracts of fresh (i.e., not aged) and aged soot, respectively, without SOA, compared to responses of cells without any material exposure (negative control). We observed for neither a significant change in cell vitality nor a significant amount of cellular ROS production from the aqueous extracts of fresh or aged soot particles compared to blank filter. We therefore believe that the biological responses of SOABPIN-SP and SOANAP-SP exclusively are caused by the SOA fraction in these particles.
Table 1Comparison of the oxidative characteristics of particles generated from different concentrations of soot and precursor VOCs under photooxidation aging conditions equivalent to 3 d of aging at 40 % RH.
Figures 8 and 9 show the variation of cell viability and fold change of cellular ROS production after exposing A549 cells to aqueous extracts of the two aerosol types, respectively, compared to effects of blank filter extracts. As shown in Fig. 8, exposure of the cells to particle extracts larger than about 20 µg mL−1 of OM led to a significantly lower cell viability than those to a lower exposure dose. Under the same exposure doses, the cell viability from SOANAP-SP was significantly lower than that from SOAβPIN-SP, indicating that the naphthalene-derived SOA has a higher toxicity than β-pinene-derived SOA. Figure 9 shows that exposure of the A549 cells in the particle extracts led to a significant increase in the fold change of cellular ROS production compared to blank filter extracts. At a similar exposure dose, SOANAP-SP SOA led to a much higher fold change of cellular ROS production than that of SOAβPIN-SP. These findings are consistent with the results obtained from chemical assays, which together indicate that SOANAP-SP is more toxic than of SOAβPIN-SP.

Figure 8Variation of cell viability caused by exposing A549 cells to particle extracts. Exposure of the cells to particle extract concentrations larger than 20 µg mL−1 of OM led to a significantly lower cell viability than exposures to a lower dose. Under the same exposure doses, the cell viability from SOANAP-SP was significantly lower than that from SOAβPIN-SP, indicating that the naphthalene-derived SOA has a higher toxicity than β-pinene-derived SOA.
In this study, acellular ROS concentration in cells exposed to SOA was calculated based on ROS content in OM as measured by offline DCFH/HRP assay (See Table 1). Figure 10 shows a positive correlation between the concentration of acellular ROS and the fold change of cellular ROS production, indicating that DCFH assay could serve as a screening metric for cellular responses associated with aerosol particle toxicity. Interestingly, we found that under a similar acellular ROS concentration in the cell exposure SOAβPIN-SP exhibited a high ability of cellular ROS production compared to SOANAP-SP as evident from the higher slope of the polynomial fit. This is in contrast to the higher ROS and OP concentration per particle mass in SOANAP-SP compared to SOAβPIN-SP as shown in Figs. 2, 4 and 6. In addition, an apparent quadratic relationship between acellular and cellular ROS as indicated by the fits shown in Fig. 10 could reflect how compensatory and non-linear effects of the cellular antioxidant defense systems affect cellular ROS production.
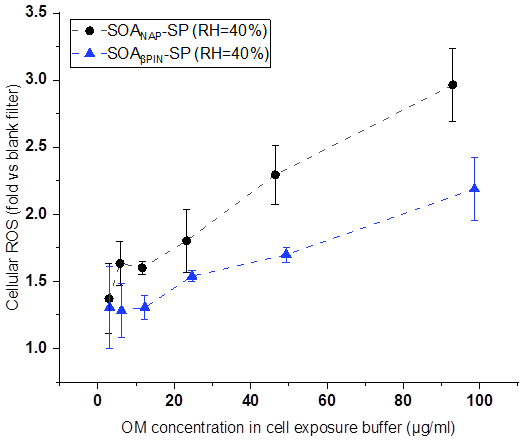
Figure 9Fold changes of cellular ROS production in A549 cells after exposure to particle extracts. Exposure of the A549 cells in the particle extracts led to a significant increase in the fold change of cellular ROS production compared to blank filter extracts. At a similar exposure dose, SOANAP-SP SOA led to a much higher fold change of cellular ROS production than SOAβPIN-SP SOA.
ROS components, the concentration of which was measured by the chemical DCFH/HRP assay, could diffuse into cells and hence may be measured by the cellular ROS assay as we used the same assay as for the acellular ROS measurement. For instance, Liu et al. (2020) recently investigated cellular ROS production when exposing murine alveolar macrophages to naphthalene SOA. They concluded that diffusion of hydrogen peroxides into the cells could be a main pathway contributing to cellular ROS production. Therefore, a high offline acellular ROS yield obtained from SOANAP-SP as observed in this study may be one reason leading to a high cellular ROS production. This view is supported by our observation that higher acellular ROS concentration in cell exposures led to a higher cellular ROS production as evident from Fig. 9. Besides H2O2, the presence of quinones in naphthalene SOA may also contribute to the cellular ROS production (Bolton et al., 2000; Valavanidis et al., 2005). However, as shown in Fig. 10, under the same acellular ROS concentration in the cell exposure experiments, fold changes of cellular ROS caused by the two SOA types is different, indicating that the cellular ROS production may be caused by different ROS components in naphthalene SOA and β-pinene SOA or that in addition to DCFH- and DTT-based ROS and OP, other SOA components and/or the interaction between compounds and the biological system may also affect cellular ROS production. For example, the cytotoxicity of SOA may be attributed to the presence of other oxygenated moieties that were also detected in laboratory and ambient particles. Nevertheless, our observation shows that naphthalene SOA simultaneously leads to a higher cellular ROS production and a cytotoxicity (cell viability) compared to β-pinene SOA, indicating that ROS could be one major reason that leads to cell toxicity.
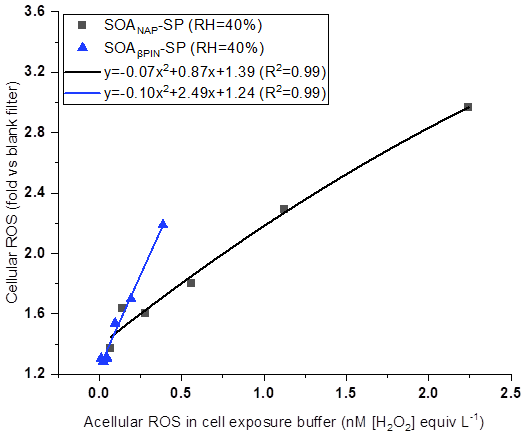
Figure 10A positive correlation between acellular ROS concentration in cell exposure experiments and fold change of cellular ROS production. Under a similar acellular ROS concentration, SOAβPIN-SP exhibited a high ability of cellular ROS production compared to SOANAP-SP as evident from the higher slope of the polynomial fit.
Chowdhury et al. (2019) observed that cells exposed to SOA generated from photochemical aging of naphthalene and α-pinene can generate cellular ROS and cellular superoxide. However, their results showed an association but not a clear correlation between total peroxide in the particles and cellular ROS production and between the superoxide levels generated by the SOA and superoxide measured within the exposed cells. This may indicate that apart from peroxide and superoxide, other ROS components may also contribute to cellular ROS. Moreover, it was recently shown that the amount of H2O2 and radicals, such as the superoxide anion, varies widely depending on the SOA precursor and its atmospheric aging processes (Tong et al., 2018; Chowdhury et al., 2019; Wei et al., 2021). All these observations could explain why under a similar concentration of acellular ROS in the cell exposure, fold changes of cellular ROS caused by the two SOA is different as observed in Fig. 10.

Figure 11Fold changes of ROS production caused by exposing A549 cells (a) to H2O2 and (b) to particle extracts. Panel (a) shows that the fold change of cellular ROS increased with an increase in the H2O2 concentration but decreased with increasing exposure time (e.g., from 2 to 24 h the cellular ROS production decreased by a factor of 2 at 100 µM H2O2 exposure). In contrast, panel (b) shows that the fold change of cellular ROS production, caused by exposing cells with particle extracts, increased with an increase in exposure time from 4 to 24 h.
A recent study from Liu et al. (2020) reported that H2O2 is a main reason that led to cellular ROS production. This can be further supported by previous studies reported by Wang et al. (2011) and Tong et al. (2018) that aqueous extract of naphthalene SOA contains a higher H2O2 concentration than that from β-pinene-derived SOA. H2O2 can penetrate biological membranes and therefore lead to cellular ROS production. However, we emphasize that although we used the metric of H2O2 equivalence to quantify the ROS content in particles, the cells were exposed to SOA extracts in our experiments, and this causes a different cellular response compared to H2O2 as demonstrated by Fig. 11. Specifically, Fig. 11a shows that the fold change of cellular ROS increased with an increase in the H2O2 concentration in cell exposure but decreased with increasing exposure time (e.g., from 2 to 24 h the cellular ROS production decreased by a factor of 2 at 100 µM H2O2 exposure). In contrast, Fig. 11b shows that the fold change of cellular ROS production, caused by exposing cells with particle extracts, increased with an increase in exposure time. This may indicate that the cells have the ability (i.e., via enzymes) to remove H2O2 efficiently, but they may not be well equipped to remove oxidizing SOA components. Our observations highlight that in addition to the diffusion of existing ROS in aqueous extracts into cells, ROS formed endogenously by cells initiated by the presence of particle extracts during cell exposure as well as the interaction between cells and particle extracts could affect the cellular ROS significantly.
This study reveals that photochemical aging of both anthropogenic and biogenic SOA increases particle-bound ROS production and particle OP, which correlates with an increase in the overall oxidation of the SOA mass. A few recent studies showed that not only SOA but also aging of primary anthropogenic emissions, including wood and coal combustion emissions, vehicular emissions, and biomass burning tends to increase particle-bound ROS formation and particle OP (e.g., Zhou et al., 2018b, 2019). The sum of these results highlights the impacts of photochemical processes on the toxicity of aerosols, an important aspect for the design of future air pollution mitigation strategies. Specifically, our results highlight that naphthalene SOA has a higher oxidative ability than β-pinene SOA. Since SOA that originates from anthropogenic pollutants might dominate the organic aerosol burden in the atmosphere of many urban areas, the results of this study underscore the need to better understand the effects of photochemical alteration on anthropogenic SOA toxicity and human health.
The comparison between online and offline ROS measurements demonstrated that offline methods seriously underestimate the total ROS concentration due to the fast degradation of the majority of ROS components, which are highly reactive. This highlights that it is crucial to develop novel online instruments for a meaningful quantification of ROS. The correlations of ROS and OP obtained from different methods as well as of acellular ROS and cellular ROS indicate that one chemical-based assay may not be entirely representative of physiological ROS responses and toxicity in a broader sense (as illustrated by the cell viability tests). At low aerosol particle exposure dose, the two types of aerosols (anthropogenic and biogenic SOA) led to a similar cellular ROS production although they contain different ROS concentrations. This highlights that it is necessary to mimic the process of particle deposition onto lung cells and their interactions as realistically as possible to avoid unknown biases. Further studies are needed to understand the formation and mechanisms of photochemical reactions that generate ROS. In addition, more in vitro and in vivo work should be performed to establish the relationship between the particle oxidation activity obtained by the assays used here or similar metrics and adverse health outcomes.
In a recent accompanying study, Offer et al. (2022) used the same aerosols as discussed in this study to expose lung cells to particles directly at the air–liquid interface (ALI) without prior particle collection on filters and extraction. In addition to cellular ROS and cell viability, the study by Offer et al. (2022) also characterized DNA damage, secretion of malondialdehyde and interleukin 8 (i.e., indicating higher oxidative stress) under more realistic exposure conditions. All these additional biological endpoints also indicate that SOANAP-SP causes higher negative biological responses than SOAβPIN-SP (i.e., higher DNA damage and higher secretion of malondialdehyde and interleukin 8). The consistent results obtained from these different particle exposure methods (ALI cell exposure vs. filter extracts) may be a positive indication for extrapolatory vitro toxicity studies, although more aerosol systems and multiple biological responses must be investigated first to confirm these conclusions.
Some limitations of the current study should be mentioned here. First, aerosol systems considered in this study were single SOA precursors. Ambient aerosol comprises SOA from multiple precursors, which may interact with each other and result in a different response level. Interactions between SOA and other compounds present in ambient aerosol (e.g., metals and other organic species or inorganic salts) are also likely to alter the response level. The roles of multiple metal species present in the ambient aerosols for ROS production and the toxicity of particles should be assessed in the future. Second, for the cell exposure, the doses investigated in this study are not accurately representing real-world exposures, i.e., with respect to exposure routes and potential recovery effects related to clearance. In addition, experiments with different cell lines and the observation of multiple biological responses are needed in future studies. Particle extracts used in this study also differ from real-world exposures. Additionally, our findings that aqueous extracts of the soot particles (fresh, aged, analyzed online or offline) showed no detectable ROS concentrations and no significant OP compared to blank filters do not necessarily mean that the insoluble part of soot particles is not a health-relevant part of ambient aerosol particles but indicate that no significant water-soluble fraction exists in the soot produced here that leads to ROS production and OP. Therefore, more research is warranted to identify and quantity if and how the insoluble fraction of soot particles causes a change in cytotoxicity and cellular ROS production.
The aerosol data as well as data of cellular analyses performed during this study are available upon request.
HC, TH, AKS, YR, RZ and MK conceived and supervised the study. ZHZ, EH, AP, MK, HC, GJ, JSK, SJ and TG designed and performed the experiments for aerosol generation, online gas and particle characterization, and filter sample collection. ZHZ, BU and BG analyzed filter samples for acellular ROS and OP. ZHZ, BXY and XYS designed and performed the cytotoxicity and cellular ROS analysis. AKS, YR, RZ and MK provided funding. ZHZ and MK analyzed the data and wrote the paper with input and approval from all the authors.
At least one of the (co-)authors is a member of the editorial board of Atmospheric Chemistry and Physics. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This work was supported by the Swiss National Science Foundation (200021_192192/1) and the Helmholtz International Laboratory aeroHEALTH (Interlabs-0005). Zhi-Hui Zhang also received funding from Horizon 2020 Marie Skłodowska-Curie Actions (MSCA; grant no. 792746).
This paper was edited by Alexander Laskin and reviewed by three anonymous referees.
Barmet, P., Dommen, J., DeCarlo, P. F., Tritscher, T., Praplan, A. P., Platt, S. M., Prévôt, A. S. H., Donahue, N. M., and Baltensperger, U.: OH clock determination by proton transfer reaction mass spectrometry at an environmental chamber, Atmos. Meas. Tech., 5, 647–656, https://doi.org/10.5194/amt-5-647-2012, 2012.
Bates, J. T., Fang, T., Verma, V., Zeng, L., Weber, R. J., Tolbert, P. E., Abrams, J. Y., Sarnat, S. E., Klein, M., Mulholland, J. A., and Russell, A. G.: Review of acellular assays of ambient particulate matter oxidative potential: methods and relationships with composition, sources, and health effects, Environ. Sci. Technol., 53, 4003–4019, https://doi.org/10.1021/acs.est.8b03430, 2019.
Bolton, J. L., Trush, M. A., Penning, T. M., Dryhurst, G., and Monks, T. J.: Role of quinones in toxicology, Chem. Res. Toxicol., 13, 135–160, https://doi.org/10.1021/tx9902082, 2000.
Campbell, S. J., Wolfer, K., Utinger, B., Westwood, J., Zhang, Z.-H., Bukowiecki, N., Steimer, S. S., Vu, T. V., Xu, J., Straw, N., Thomson, S., Elzein, A., Sun, Y., Liu, D., Li, L., Fu, P., Lewis, A. C., Harrison, R. M., Bloss, W. J., Loh, M., Miller, M. R., Shi, Z., and Kalberer, M.: Atmospheric conditions and composition that influence PM2.5 oxidative potential in Beijing, China, Atmos. Chem. Phys., 21, 5549–5573, https://doi.org/10.5194/acp-21-5549-2021, 2021.
Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R.: Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 15, 253–272, https://doi.org/10.5194/acp-15-253-2015, 2015.
Cappa, C. D., Onasch, T. B., Massoli, P., Worsnop, D. R., Bates, T. S., Cross, E. S., Davidovits, P., Hakala, J., Hayden, K. L., Jobson, B. T., Kolesar, K. R., Lack, D. A., Lerner, B. M., Li, S. M., Mellon, D., Nuaaman, I., Olfert, J. S., Petaja, T., Quinn, P. K., Song, C., Subramanian, R., Williams, E. J., and Zaveri, R. A.: Radiative absorption enhancements due to the mixing state of atmospheric black carbon, Science, 337, 1078–1081, https://doi.org/10.1126/science.1223447, 2012.
Charrier, J. G. and Anastasio, C.: On dithiothreitol (DTT) as a measure of oxidative potential for ambient particles: evidence for the importance of soluble transition metals, Atmos. Chem. Phys., 12, 9321–9333, https://doi.org/10.5194/acp-12-9321-2012, 2012.
Chen, Q., Heald, C. L., Jimenez, J. L., Canagaratna, M. R., Zhang, Q., He, L. Y., Huang, X. F., Campuzano-Jost, P., Palm, B. B., Poulain, L., Kuwata, M., Martin, S. T., Abbatt, J. P. D., Lee, A. K. Y., and Liggio, J.: Elemental composition of organic aerosol: The gap between ambient and laboratory measurements, Geophys. Res. Lett., 42, 4182–4189, https://doi.org/10.1002/2015GL063693, 2015.
Chhabra, P. S., Flagan, R. C., and Seinfeld, J. H.: Elemental analysis of chamber organic aerosol using an aerodyne high-resolution aerosol mass spectrometer, Atmos. Chem. Phys., 10, 4111–4131, https://doi.org/10.5194/acp-10-4111-2010, 2010.
Chowdhury, P. H., He, Q., Lasitza Male, T. L., Brune, W. H., Rudich, Y., and Pardo, M.: Exposure of lung epithelial cells to photochemically aged secondary organic aerosol shows increased toxic effects, Environ. Sci. Technol. Lett., 5, 424–430, https://doi.org/10.1021/acs.estlett.8b00256, 2018.
Chowdhury, P. H., He, Q., Carmieli, R., Li, C., Rudich, Y., and Pardo, M.: Connecting the oxidative potential of secondary organic aerosols with reactive oxygen species in exposed lung cells, Environ. Sci. Technol., 53: 13949–13958, https://doi.org/10.1021/acs.est.9b04449, 2019.
Daellenbach, K. R., Uzu, G., Jiang, J., Cassagnes, L.-E., Leni, Z., Vlachou, A., Stefenelli, G., Canonaco, F., Weber, S., Segers, A., Kuenen, J. J. P., Schaap, M., Favez, O., Albinet, A., Aksoyoglu, S., Dommen, J., Baltensperger, U., Geiser, M., El Haddad, I., Jaffrezo, J.-L., and Prévôt, A. S. H.: Sources of particulate-matter air pollution and its oxidative potential in Europe, Nature, 587, 414–419, https://doi.org/10.1038/s41586-020-2902-8, 2020.
Fuller, S. J., Wragg, F. P. H., Nutter, J., and Kalberer, M.: Comparison of on-line and off-line methods to quantify reactive oxygen species (ROS) in atmospheric aerosols, Atmos. Environ., 92, 97–103, https://doi.org/10.1016/j.atmosenv.2014.04.006, 2014.
Gallimore, P. J., Mahon, B. M., Wragg, F. P. H., Fuller, S. J., Giorio, C., Kourtchev, I., and Kalberer, M.: Multiphase composition changes and reactive oxygen species formation during limonene oxidation in the new Cambridge Atmospheric Simulation Chamber (CASC), Atmos. Chem. Phys., 17, 9853–9868, https://doi.org/10.5194/acp-17-9853-2017, 2017.
Gentner, D. R., Jathar, S. H., Gordon, T. D., Bahreini, R., Day, D. A., El-Haddad, I., Hayes, P. L., Pieber, S. M., Platt, S. M., Gouw, J., Goldstein, A. H., Harley, R. A., Jimenez, J. L., André S. H. Prévôt, A. S., and Robinson, A.: Review of urban secondary organic aerosol formation from gasoline and diesel motor vehicle emissions, Environ. Sci. Technol., 51, 1074–1093, https://doi.org/10.1021/acs.est.6b04509, 2017.
Hedayat, F., Stevanovic, S., Miljeivc, B., Bottle, S., and Ristovski, Z. D.: Review– evaluating the molecular assays for measuring the oxidative potential of particulate matter, Chem. Ind. Chem. Eng., 21, 201–210, https://doi.org/10.2298/CICEQ140228031H, 2015.
Hellack, B., Nickel, C., Albrecht, C., Kuhlbusch, T. A. J., Boland, S., Baeza-Squiban, A., Wohlleben, W., and Schin, R. P. F.: Analytical methods to assess the oxidative potential of nanoparticles: a review, Environ. Sci. Nano., 4, 1920–1934, 2017.
Jia, C. and Batterman, S.: A critical review of naphthalene sources and exposures relevant to indoor and outdoor air, Int. J. Env. Res. Pub. He., 7, 2903–2939, https://doi.org/10.3390/ijerph7072903, 2010.
Jiang, H. and Jang, M.: Dynamic oxidative potential of atmospheric organic aerosol under ambient sunlight, Environ. Sci. Technol., 52, 7496–7504, https://doi.org/10.1021/acs.est.8b00148, 2018.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of organic aerosols in the atmosphere, Science., 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Kang, E., Root, M. J., Toohey, D. W., and Brune, W. H.: Introducing the concept of Potential Aerosol Mass (PAM), Atmos. Chem. Phys., 7, 5727–5744, https://doi.org/10.5194/acp-7-5727-2007, 2007.
Kautzman, K. E., Surratt, J. D., Chan, M. N., Chan, A. W. H., Hersey, S. P., Chhabra, P. S., Dalleska, N. F., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Chemical composition of gas- and aerosol-phase products from the photooxidation of naphthalene, J. Phys. Chem. A, 114, 913–934, 2010.
Krapf, M., Haddad, I. E., Bruns, E. A., Molteni, U., Daellenbach, K. R., Prévôt, A. S. H., Baltensperger, U., and Dommen, J.: Labile peroxides in secondary organic aerosol, Chem, 1, 603–616, https://doi.org/10.1016/j.chempr.2016.09.007, 2016.
Kroll, J. H., Donahue, N. M., Jimenez, J. L., Kessler, S. H., Canagaratna, M. R., Wilson, K. R., Altieri, K. E., Mazzoleni, L. R., Wozniak, A. S., Bluhm, H., Mysak, E. R., Smith, J. D., Kolb, C. E., and Worsnop, D. R.: Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol, Nat. Chem., 3, 133–139, 2011.
Lin, M. and Yu, J. Z.: Dithiothreitol (DTT) concentration effect and its implications on the applicability of DTT assay to evaluate the oxidative potential of atmospheric aerosol samples, Environ. Pollut., 251, 938–944, https://doi.org/10.1016/j.envpol.2019.05.074, 2019.
Liu, F., Saavedra, M. G., Champion, J. A., Griendling, K. K., and Ng, N. L.: Prominent contribution of hydrogen peroxide to intracellular reactive oxygen species generated upon exposure to naphthalene secondary organic aerosols, Environ. Sci. Technol. Lett., 7, 171–177, https://doi.org/10.1021/acs.estlett.9b00773, 2020.
Liu, S., Aiken, A. C., Gorkowski, K., Dubey, M. K., Cappa, C. D., Williams, L. R., Herndon, S. C., Massoli, P., Fortner, E. C., Chhabra, P. S., Brooks, W. A., Onasch, T. B., Jayne, J. T., Worsnop, D. R., China, S., Sharma, N., Mazzoleni, C., Xu, L., Ng, N. L., Liu, D., Allan, J. D., Lee, J. D., Fleming, Z. L., Mohr, C., Zotter, P., Szidat, S., and Prevot, A. S. H.: Enhanced light absorption by mixed source black and brown carbon particles in UK winter, Nat. Commun., 6, 184–188, https://doi.org/10.1038/ncomms9435, 2015.
McWhinney, R. D., Zhou, S., and Abbatt, J. P. D.: Naphthalene SOA: redox activity and naphthoquinone gas–particle partitioning, Atmos. Chem. Phys., 13, 9731–9744, https://doi.org/10.5194/acp-13-9731-2013, 2013.
Molteni, U., Bianchi, F., Klein, F., El Haddad, I., Frege, C., Rossi, M. J., Dommen, J., and Baltensperger, U.: Formation of highly oxygenated organic molecules from aromatic compounds, Atmos. Chem. Phys., 18, 1909–1921, https://doi.org/10.5194/acp-18-1909-2018, 2018.
Ng, N. L., Canagaratna, M. R., Jimenez, J. L., Chhabra, P. S., Seinfeld, J. H., and Worsnop, D. R.: Changes in organic aerosol composition with aging inferred from aerosol mass spectra, Atmos. Chem. Phys., 11, 6465–6474, https://doi.org/10.5194/acp-11-6465-2011, 2011.
Offer, S., Hartner, E., Bucchianico, D. S., Bisig, C., Bauer, S., Pantzke, J., Zimmermann, E. J., Cao, X., Binder, S., Kuhn, E., Huber, A., Jeong, S., Käfer, U., Martens, P., Mesceriakovas, A., Bendl, J., Brejcha, R., Buchholz, A., Gat, D., Hohaus, T., Rastak, N., Jakobi, G., Kalberer, M., Kanashova, T., Hu, Y., Ogris, C., Marsico, A., Theis, F., Pardo, M., Gröger, T., Öder, S., Orasche, J., Paul, A., Ziehm, T., Zhang, Z. J., Adam, T., Sippula, O., Sklorz, M., Schnelle-Kreis, J., Czech, H., Kiendler-Scharr, A., Rudich, Y., Zimmermann, R.: Effect of atmospheric ageing on soot particle toxicity in lung cell models at the air-liquid interface: Differential toxicological impacts of biogenic and anthropogenic secondary organic aerosols (SOAs), Environ. Health. Persp., in review, 2022.
Øvrevik, J.: Oxidative potential versus biological effects: a review on the relevance of cell-free/abiotic assays as predictors of toxicity from airborne particulate matter, Int. J. Mol. Sci., 20, 4772, https://doi.org/10.3390/ijms20194772, 2019.
Platt, S. M., Haddad, I. E., Pieber, S. M., Huang, R. J., Zardini, A. A., Clairotte, M., Suarez-Bertoa, R., Barmet, P., Pfaffenberger, L., Wolf, R., Slowik, J. G., Fuller, S. J., Kalberer, M., Chirico, R., Dommen, J., Astorga, C., Zimmermann, R., Marchand, N., Hellebust, S., Temime-Roussel, B., Baltensperger, U., and Prévôt, A. S. H.: Two-stroke scooters are a dominant source of air pollution in many cities, Nat. Commun., 5, 1–7, https://doi.org/10.1038/ncomms4749, 2014.
Tong, H., Lakey, P. S. J., Arangio, A. M., Socorro, J., Shen, F., Lucas, K., Brune, W. H., Pöschl, U., and Shiraiwa, M.: Reactive oxygen species formed by secondary organic aerosols in water and surrogate lung fluid, Environ. Sci. Technol., 52, 11642–11651, https://doi.org/10.1021/acs.est.8b03695, 2018.
Tong, H., Zhang, Y., Filippi, A., Wang, T., Li, C., Liu, F., Leppla, D., Kourtchev, I., Wang, K., Keskinen, H. M., Levula, J. T., Arangio, A. M., Shen, F., Ditas, F., Martin, S. T., Artaxo, P., Godoi, R. H. M., Yamamoto, C. I., de Souza, R. A. F., Huang, R. J., Berkemeier, T., Wang, Y., Su, H., Cheng, Y., Pope, F. D., Fu, P., Yao, M., Pöhlker, C., Petäjä, T., Kulmala, M., Andreae, M. O., Shiraiwa, M., Pöschl, U., Hoffmann, T., and Kalberer, M.: Radical formation by fine particulate matter associated with highly oxygenated molecules, Environ. Sci. Technol., 53, 12506–12518,https://doi.org/10.1021/acs.est.9b05149, 2019.
Tu, P., Hall, W. A., and Johnston, M. V.: Characterization of highly oxidized molecules in fresh and aged biogenic secondary organic aerosol, Anal. Chem., 88, 4495–4501, https://doi.org/10.1021/acs.analchem.6b00378, 2016.
Tuet, W. Y., Chen, Y., Xu, L., Fok, S., Gao, D., Weber, R. J., and Ng, N. L.: Chemical oxidative potential of secondary organic aerosol (SOA) generated from the photooxidation of biogenic and anthropogenic volatile organic compounds, Atmos. Chem. Phys., 17, 839–853, https://doi.org/10.5194/acp-17-839-2017, 2017a.
Tuet, W. Y., Chen, Y., Fok, S., Champion, J. A., and Ng, N. L.: Inflammatory responses to secondary organic aerosols (SOA) generated from biogenic and anthropogenic precursors, Atmos. Chem. Phys., 17, 11423–11440, https://doi.org/10.5194/acp-17-11423-2017, 2017b.
Valavanidis, A., Fiotakis, K., Bakeas, E., and Vlahogianni, T.: Electron paramagnetic resonance study of the generation of reactive oxygen species catalysed by transition metals and quinoid redox cycling by inhalable ambient particulate matter, Redox. Rep., 10, 37–51, https://doi.org/10.1179/135100005X21606, 2005.
Wang, S., Ye, J., Soong, R., Wu, B., Yu, L., Simpson, A. J., and Chan, A. W. H.: Relationship between chemical composition and oxidative potential of secondary organic aerosol from polycyclic aromatic hydrocarbons, Atmos. Chem. Phys., 18, 3987–4003, https://doi.org/10.5194/acp-18-3987-2018, 2018.
Wang, Y., Kim, H., and Paulson, S. E.: Hydrogen peroxide generation from α- and β-pinene and toluene secondary organic aerosols, Atmos. Environ., 45, 3149–3156, https://doi.org/10.1016/j.atmosenv.2011.02.060, 2011.
Wei, J., Fang, T., Wong, C., Lakey, P. S. J., Nizkorodov, S. A., and Shiraiwa, M.: Superoxide Formation from Aqueous Reactions of Biogenic Secondary Organic Aerosols, Environ. Sci. Technol. 55, 260–270, https://doi.org/10.1021/acs.est.0c07789, 2021.
World Health Organisation: Ambient Air Pollution: A global assessment of exposure and burden of disease, report, available at: https://www.who.int/phe/publications/_air-pollution-global-assessment/en/ (last access: 26 March 2021), 2016.
Wragg, F. P. H., Fuller, S. J., Freshwater, R., Green, D. C., Kelly, F. J., and Kalberer, M.: An automated online instrument to quantify aerosol-bound reactive oxygen species (ROS) for ambient measurement and health-relevant aerosol studies, Atmos. Meas. Tech., 9, 4891–4900, https://doi.org/10.5194/amt-9-4891-2016, 2016.
Zhou, J., Bruns, E. A., Zotter, P., Stefenelli, G., Prévôt, A. S. H., Baltensperger, U., El-Haddad, I., and Dommen, J.: Development, characterization and first deployment of an improved online reactive oxygen species analyzer, Atmos. Meas. Tech., 11, 65–80, https://doi.org/10.5194/amt-11-65-2018, 2018a.
Zhou, J., Zotter, P., Bruns, E. A., Stefenelli, G., Bhattu, D., Brown, S., Bertrand, A., Marchand, N., Lamkaddam, H., Slowik, J. G., Prévôt, A. S. H., Baltensperger, U., Nussbaumer, T., El-Haddad, I., and Dommen, J.: Particle-bound reactive oxygen species (PB-ROS) emissions and formation pathways in residential wood smoke under different combustion and aging conditions, Atmos. Chem. Phys., 18, 6985–7000, https://doi.org/10.5194/acp-18-6985-2018, 2018b.
Zhou, J., Elser, M., Huang, R.-J., Krapf, M., Fröhlich, R., Bhattu, D., Stefenelli, G., Zotter, P., Bruns, E. A., Pieber, S. M., Ni, H., Wang, Q., Wang, Y., Zhou, Y., Chen, C., Xiao, M., Slowik, J. G., Brown, S., Cassagnes, L.-E., Daellenbach, K. R., Nussbaumer, T., Geiser, M., Prévôt, A. S. H., El-Haddad, I., Cao, J., Baltensperger, U., and Dommen, J.: Predominance of secondary organic aerosol to particle-bound reactive oxygen species activity in fine ambient aerosol, Atmos. Chem. Phys., 19, 14703–14720, https://doi.org/10.5194/acp-19-14703-2019, 2019.