the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 31 Jan 2022
| 31 Jan 2022
Secondary organic aerosol and organic nitrogen yields from the nitrate radical (NO3) oxidation of alpha-pinene from various RO2 fates
Kelvin H. Bates
Guy J. P. Burke
James D. Cope
The reaction of α-pinene with NO3 is an important sink of both α-pinene and NO3 at night in regions with mixed biogenic and anthropogenic emissions; however, there is debate on its importance for secondary organic aerosol (SOA) and reactive nitrogen budgets in the atmosphere. Previous experimental studies have generally observed low or zero SOA formation, often due to excessive [NO3] conditions. Here, we characterize the SOA and organic nitrogen formation from α-pinene + NO3 as a function of nitrooxy peroxy (nRO2) radical fates with HO2, NO, NO3, and RO2 in an atmospheric chamber. We show that SOA yields are not small when the nRO2 fate distribution in the chamber mimics that in the atmosphere, and the formation of pinene nitrooxy hydroperoxide (PNP) and related organonitrates in the ambient atmosphere can be reproduced. Nearly all SOA from α-pinene + NO3 chemistry derives from the nRO2+ RO2 pathway, which alone has an SOA mass yield of 56 (±7) %. Molecular composition analysis shows that particulate nitrates are a large (60 %–70 %) portion of the SOA and that dimer formation is the primary mechanism of SOA production from α-pinene + NO3 under simulated nighttime conditions. Synergistic dimerization between nRO2 and RO2 derived from ozonolysis and OH oxidation also contribute to SOA formation and should be considered in models. We report a 58 (±20) % molar yield of PNP from the nRO2+ HO2 pathway. Applying these laboratory constraints to model simulations of summertime conditions observed in the southeast United States (where 80 % of α-pinene is lost via NO3 oxidation, leading to 20 % nRO2+ RO2 and 45 % nRO2+ HO2), we estimate yields of 11 % SOA and 7 % particulate nitrate by mass and 26 % PNP by mole from α-pinene + NO3 in the ambient atmosphere. These results suggest that α-pinene + NO3 significantly contributes to the SOA budget and likely constitutes a major removal pathway of reactive nitrogen from the nighttime boundary layer in mixed biogenic–anthropogenic areas.
- Article
(3092 KB) - Full-text XML
-
Supplement
(3751 KB) - BibTeX
- EndNote





Monoterpenes (C10H16) are a major class of biogenic hydrocarbons. Although less abundant than isoprene in terms of absolute emission flux of non-methane hydrocarbons, they have a disproportionate importance for the formation of secondary organic aerosol (SOA), accounting for half of the total fine aerosol globally (Zhang et al., 2018), and for nitrogen oxide (NO, NO2, NO3) sequestration through the formation of gaseous and particle-phase organic nitrates (Pye et al., 2015). Thus, monoterpene chemistry plays a prevailing role in aerosol–climate interactions and atmospheric air quality. Of the monoterpenes, α-pinene is the most abundant globally (Sindelarova et al., 2014). This is especially notable over boreal coniferous forests where the α-pinene emission flux alone can overtake isoprene and the combined flux of all other monoterpenes during the summer season (Hakola et al., 2003). The atmospheric abundance, fast reaction rates, and nighttime emission profile of α-pinene conspire for it to dominate the fate of the nitrate radical (NO3) in the dark and also to play a significant role in the daytime (Ayres et al., 2015). The reaction of α-pinene + NO3, thus, is one of the most prevalent reactions observed in the summer in mixed biogenic–anthropogenic sites, such as the southeastern United States.
NO3 reacts with α-pinene by addition to the double bond, mainly producing a nitrooxy alkyl radical in the tertiary position, which is rapidly converted to a nitrooxy peroxy radical (nRO2) upon collisions with molecular oxygen (Fig. 1). Prominent field observations of a monoterpene nitrooxy hydroperoxide ((O2NO)ROOH) further suggest that the nRO2 reacts with HO2 significantly (Nguyen et al., 2015). However, laboratory research on α-pinene + NO3 has so far not demonstrated nitrooxy hydroperoxide formation, except for one experiment during the FIXCIT chamber studies where the pinene nitrooxy hydroperoxide (PNP) was abundantly produced from experiments using parts-per-million-level concentrations of formaldehyde to produce dark HO2 via the slow reaction of CH2O + NO3→ HO2+ HNO3 (Nguyen et al., 2014a). Although we now appreciate that the nRO2 from α-pinene can form monoterpene nitrooxy hydroperoxide, its absolute yield is unknown; furthermore, such high concentrations of formaldehyde in the exploratory FIXCIT experiment will affect SOA formation, and thus, aerosol yields were not extractable.
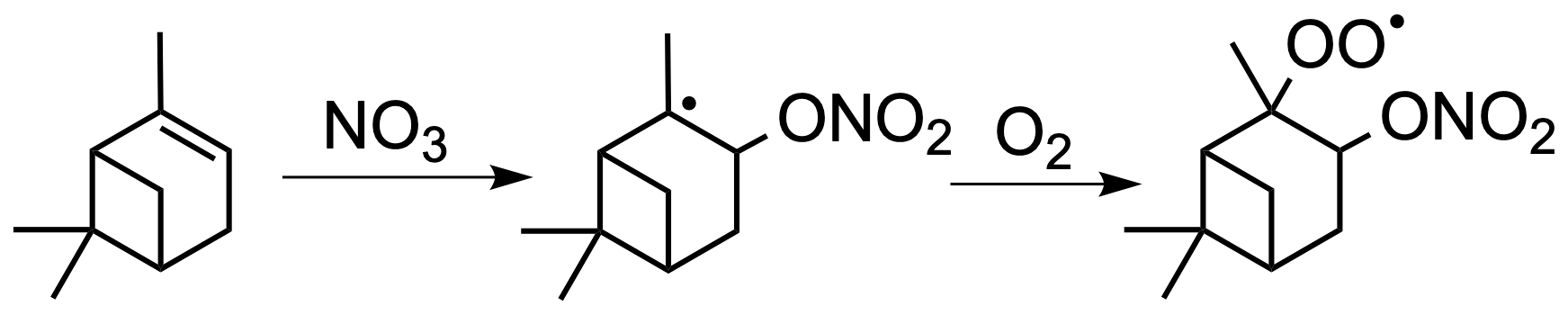
Figure 1The NO3-initiated oxidation of α-pinene produces a nitrated RO2 radical (nRO2). The major isomer is shown.
Determinations of SOA yields of α-pinene + NO3 in the literature have also suffered from high uncertainty, although it has generally been accepted that the yields are lower than those of many other monoterpenes and, thus, often not considered for modeling SOA and organic nitrogen formation (Pye et al., 2015; Ayres et al., 2015). Fry et al. (2014) reported an SOA yield of exactly zero from this reaction from experiments using N2O5 as an NO3 precursor. Hallquist et al. (1999), Moldanova et al. (2000), Bell et al. (2021), and Mutzel et al. (2021) also reported low mass yields (0.3 %–7 %, 0.3 %–6.9 %, 3 %–11 %, and 5.9 %–6.4 % respectively, depending on precursor concentrations) with reactions performed similarly. Spittler et al. (2006) performed the experiment with slow and minute introductions of N2O5 and reported higher mass yields, although they depend on whether the seed particle chosen was ammonium sulfate (9 %) or organic (16 %). Nah et al. (2016) and Kurtén et al. (2017) also observed minimal SOA formation (mass yields of 3.6 % and < 1 %, respectively) using NO2+ O3 as a source of NO3 and formaldehyde to promote HO2 chemistry. From these observations, it is clear that chamber reaction conditions are highly influential in the observed SOA yields and that previous studies may have each probed different nRO2 fates. Thus, a systematic investigation of how nRO2 fates dictate reaction outcomes will enable reconciliation of past results and accurate representation of this reaction in atmospheric models. The high initial NO3 concentrations (tens of parts per billion) used in some previous studies, derived from the decomposition of N2O5, will cause nRO2+ NO3 to dominate in the chamber, when it is negligible in the field ([NO3] is persistently at or below the detection limit of 1 pptv in the rural southeast United States; Ayres et al., 2015). Kurtén and coworkers further illuminated the role of alkoxy radical scission in SOA formation and predicted low SOA yields from the α-pinene + NO3 reaction when the RO2 radical is reduced to RO via bimolecular reaction with NO3, RO2, and even HO2 (Kurtén et al., 2017).
Given the high relevance of the α-pinene + NO3 reaction, it is critical to place tighter constraints on how this reaction contributes to SOA and organic nitrogen in the ambient environment. In light of the emerging appreciation for the importance of RO2 radical fate in designing chamber experiments (Nguyen et al., 2014a; Xu et al., 2019; Boyd et al., 2015; Teng et al., 2017; Crounse et al., 2013), we reinvestigate this reaction to probe the SOA yield and organic nitrate formation from α-pinene + NO3 from each relevant nRO2 reaction channel. While a chamber experiment may never truly replicate the field, and ours certainly are no exception, the nRO2 fate distribution in this work was designed to approach those expected in the ambient nighttime (Ayres et all., 2015; Romer et al., 2018), including any reaction synergies that may occur (Kenseth et al., 2018; Inomata, 2021). Finally, relatively little information is available for the nRO2 compared to their hydroxylated counterparts; this work also constrains the rate coefficients and branching ratios of the α-pinene nRO2 through a combination of chamber reactions and modeling. We demonstrate a new HO2 formation route in the dark chamber that does not require carbon inputs and thus enables SOA yields to be more accurately measured when probing the RO2+ HO2 pathway from the α-pinene + NO3 reaction.
2.1 Chamber reactions
Experiments were performed in a 10 m3 FEP Teflon atmospheric chamber in the dark and at a temperature of ∼ 22 ∘C, consistent with the average nighttime temperatures in the southeastern United States in the summer (Hidy et al., 2014). Experiments were performed dry to reduce uncertainty from variable wall loss corrections, to avoid rapid hydrolysis of tertiary nitrates (Vasquez et al., 2020) and the effects of gas–particle partitioning of H2O2 and N2O5, and because relative humidity (RH) is not expected to influence radical reactions. Temperature and RH were monitored continuously by a membrane probe (Vaisala Inc.) calibrated with saturated salt solutions. NOx and O3 mixing ratios were quantified with commercial chemiluminescence (Thermo 42i) and photometric (Thermo 49i) analyzers. The chemiluminescence analyzer was calibrated with a NO2 primary standard (200 ppmv, Air Liquide) diluted to desired concentrations using ultra-high-purity (UHP) N2 and zeroed with UHP N2. The photometric analyzer was cross calibrated for O3, produced by an ozone generator (A2Ozone Inc.) from ultra zero air (Air Liquide), with a Fourier-transform infrared spectrometer (FT-IR, Shimadzu Scientific Inst. Inc., IR-Tracer 100) using a 10 m gas cell. α-Pinene mixing ratios were quantified with a gas chromatograph coupled to a flame ionization detector (GC-FID) using a PLOT-Q column (Agilent Inc.) and a custom pneumatically injected method. The GC-FID was calibrated with a NIST-traceable 40 ppmv α-pinene primary standard in N2 (Matheson Gas) diluted to several concentrations with UHP N2 using calibrated mass flow controllers (SEC, Horiba Inst. Inc.). The mass flow controllers were calibrated using a primary flow calibrator (AP Buck, Inc.).
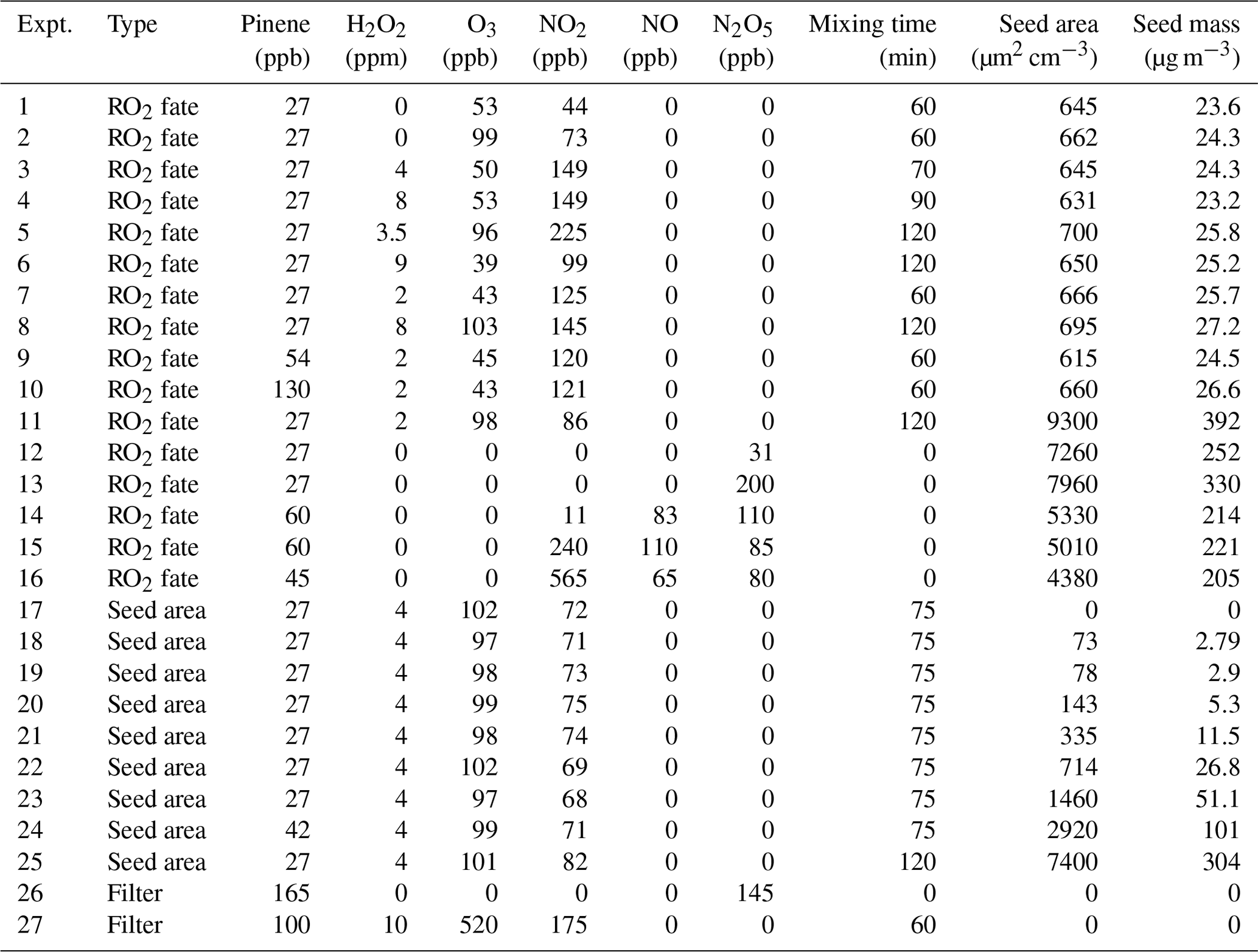
Initial conditions for all chamber experiments are described in Table 1. The chamber was cleaned prior to each experiment by continually flushing with custom-filtered zero air, quantified to be below the detection limits of all available analyzers, for at least 12 h (> 7 full air exchanges). Two types of experiments were conducted: those using O3+ NO2 as an NO3 source and those using N2O5 as an NO3 source. For the former, variable concentrations of NO2 (diluted from 2042 ppm ± 1 % in N2, Praxair) and O3 were injected to initiate the formation of low levels of NO3, according to the desired nRO2 fate. H2O2 (50 wt % in H2O, Aldrich) was then injected by flowing 4 L min−1 of ultra zero air through a bubbler warmed to 40 ∘C in a water bath. After the inorganic gas-phase reactants were introduced, the chamber was allowed to mix for 1–2 h, during which time secondary formation of NO3 (from O3+ NO2) and HO2 (from NO3+ H2O2) could proceed. Seed particles were introduced to the chamber during mixing by atomizing a solution of 0.06 M ammonium sulfate ((NH4)2SO4, ≥ 99 %, Aldrich) through a 210Po neutralizer. Lastly, liquid standards of α-pinene (Sigma Aldrich, > 99 %) were injected with gas-tight syringes into an airtight glass bulb and introduced to the chamber by a 4 L min−1 flow of ultra zero air. The α-pinene reacts quickly and thus was mixed rapidly from pulsed injections of high pressure ultra zero air (100 psi) for 2 min in order to initiate the reaction. The ozonolysis of α-pinene occurs concurrently with its NO3-initiated oxidation with this experiment design. Ozonolysis of α-pinene produces OH, which reacts with H2O2 to be an additional dark formation source of HO2, as well as with α-pinene itself. Control reactions were performed to accompany each experiment listed in Table 1 that includes ozone, using the same conditions minus NO2, in order to subtract out the SOA and other product formation from the purely ozonolytic reaction.
Table 1Initial conditions for chamber experiments performed in this work. Controls to subtract SOA yields from background chemistry (not listed here) were conducted for experiments 1–11 and 17–25 using identical conditions but omitting NO2.
For experiments that used N2O5 as the NO3 source, injections of α-pinene and other desired inorganic reactants, including seed particles, NO2, and NO (200 ppm ± 1 % in N2, Praxair), were conducted first. The reactions were then initiated by the rapid injection of gas-phase N2O5, which was previously evaporated into an evacuated 500 mL glass bulb to the desired pressure and backfilled to room pressure with N2. N2O5 was synthesized according to Claflin and Ziemann (2018), verified using FT-IR, and stored in the dark at −20 ∘C prior to use. In these experiments, the decomposition equilibrium of N2O5 ⇆ NO2+ NO3 was manipulated via injections of NO2 in order to slow NO3 formation and thus control the nRO2 fate.
During experiments, mixing ratios of oxygenated gas-phase organics were quantified with a custom-built triple-quadrupole chemical ionization mass spectrometer (CIMS) using CF3O− as the reagent ion. Instrumental details, including humidity-dependent calibration methods, have been described in detail previously (Crounse et al., 2006; St. Clair et al., 2010; Nguyen et al., 2014b; Praske et al., 2015). The CIMS detects PNP and other polar analytes predominantly without fragmentation as clusters with CF3O−. Although authentic standards of PNP are not available for direct calibration in CIMS, the analytical sensitivity of synthesized organic nitrates of different carbon length and neighboring groups in the CF3O− CIMS were found to be different from each other by a factor of 20 %–30 % (Lee et al., 2014; Teng et al., 2017); thus, sensitivity of PNP and pinene hydroxy-nitrate was assumed to be the same as isoprene hydroxynitrates with 30 % uncertainty.
A scanning mobility particle sizer (SMPS), comprised of an electrostatic classifier (TSI 3080) and a condensation particle counter (TSI 3772), was used to measure particle size distributions between 15 and 670 nm. Control experiments monitoring the dark decay of ammonium sulfate seed aerosol concentrations in the chamber were used to determine diameter-dependent particle wall loss rates (Schwantes et al., 2019), which were then used to correct experiment particle concentrations. Calculations of particle mass from measured aerodynamic diameter assume a density of 1.2 g cm−3.
2.1.1 Kinetic modeling
We use a kinetic model (Mechanism S1 in the Supplement) to simulate gas-phase chemistry for each experiment in the environmental chamber. The mechanism uses reaction parameters from the JPL Chemical Kinetics and Photochemical Data Evaluation (Burkholder et al., 2019) and is run on MATLAB (MathWorks, Inc.). We also include reactions of α-pinene with OH, O3, and NO3 and isomer-specific reactions of the subsequently produced peroxy radicals with HO2, NO, NO3, and other RO2 radicals (individually represented). Product yields from each pathway, along with rates of RO2 + RO2 reactions, are adjusted to fit the experimental data. We initialize simulations with the inorganic species listed in Table 1 and allow the model to run for the allotted mixing time before instantaneously adding α-pinene. The model does not include any wall deposition of vapors or gas–particle interactions and is used only to estimate the concentrations of gas-phase species and the contributions of each peroxy radical reactive pathway.
2.2 SOA composition analysis by high-resolution mass spectrometry (HRMS)
SOA was collected for composition analysis using Omnipore hydrophilic Teflon filters (0.2 µm diameter pore, Millipore Corp.) that are compatible with polar and non-polar organics. The filters were gently extracted using LC-MS grade acetonitrile (Optima, Fisher Scientific) to mass concentrations on the order of 100 µg mL−1, depending on the experiment, by several ultrasound pulses 1 s in duration in order to limit cavitation in the ultrasonic bath that may alter analyte compositions. Filter extracts were directly infused into a linear-trap-quadrupole (LTQ) Orbitrap XL mass spectrometer (Thermo Instrument Corp.) using positive and negative ion mode electrospray ionization (ESI) at 4 kV spray voltage and a mass resolving power of 60 000 at 400. An external calibration was performed in both ion modes using commercial mass standards in the range of 100–2000 (Pierce™ LTQ ESI Positive and Pierce™LTQ ESI Negative calibration solutions, Fisher Scientific), and the data were recalibrated until the mass accuracy obtained from standard solutions was < 1 ppm.
The data analysis was performed similarly to our previous works (Nguyen et al., 2010, 2011). Briefly, the raw data were de-convoluted using Decon2LS (freeware from Pacific Northwest National Laboratory) and background subtracted for peaks present in the solvent. The peaks were assigned to molecular formulas (CcHhOoNn) based on a custom MATLAB script that applies Lewis and Senior rules (Kind and Fiehn, 2007) and a Kendrick mass defect analysis (base CH2; Roach et al., 2011) that have been demonstrated on SOA mixtures. The prevalent ionization mechanism for this specific analyte mixture was found to be sodium cluster formation (M + Na+) in the positive mode, which occurs preferentially for carbonyls (Kruve et al., 2013), and nitrate cluster formation (M + NO) in the negative mode, which is efficient for organic nitrates, alcohols, and other functional groups (Sisco and Forbes, 2015; Mathis and McCord, 2005). The nitrate anion, prominently detected in the mass analyzer at 61.988, was not purposefully introduced but likely formed either from in-source fragmentation of organic nitrates or from HNO3 produced during the hydrolysis of tertiary organonitrates from the aqueous LC solvents and fortuitously acted as a reagent ion for chemical-assisted electrospray. While peak heights correlate well with concentration in direct-infusion ESI HRMS when the analyte matrix is similar (Chan et al., 2020), the correlation coefficients are unknown for each analyte in the mixture; thus, the HRMS data are qualitative. Furthermore, due to the labile -ONO2 groups, the organonitrate observations from HRMS likely represent a lower limit.
3.1 SOA and PNP yields from different nRO2 fates
To investigate the dependence of aerosol and gaseous PNP yields on the nRO2 reaction partner, we performed a series of environmental chamber experiments (experiments 1–16, Table 1) with various starting conditions designed to isolate or maximize the contributions of each RO2 reaction pathway. Example time profiles from three representative experiments are shown in Fig. 2. nRO2+ NO3, nRO2+ NO, and nRO2+ RO2 chemistries were isolated in experiments using N2O5 as the NO3
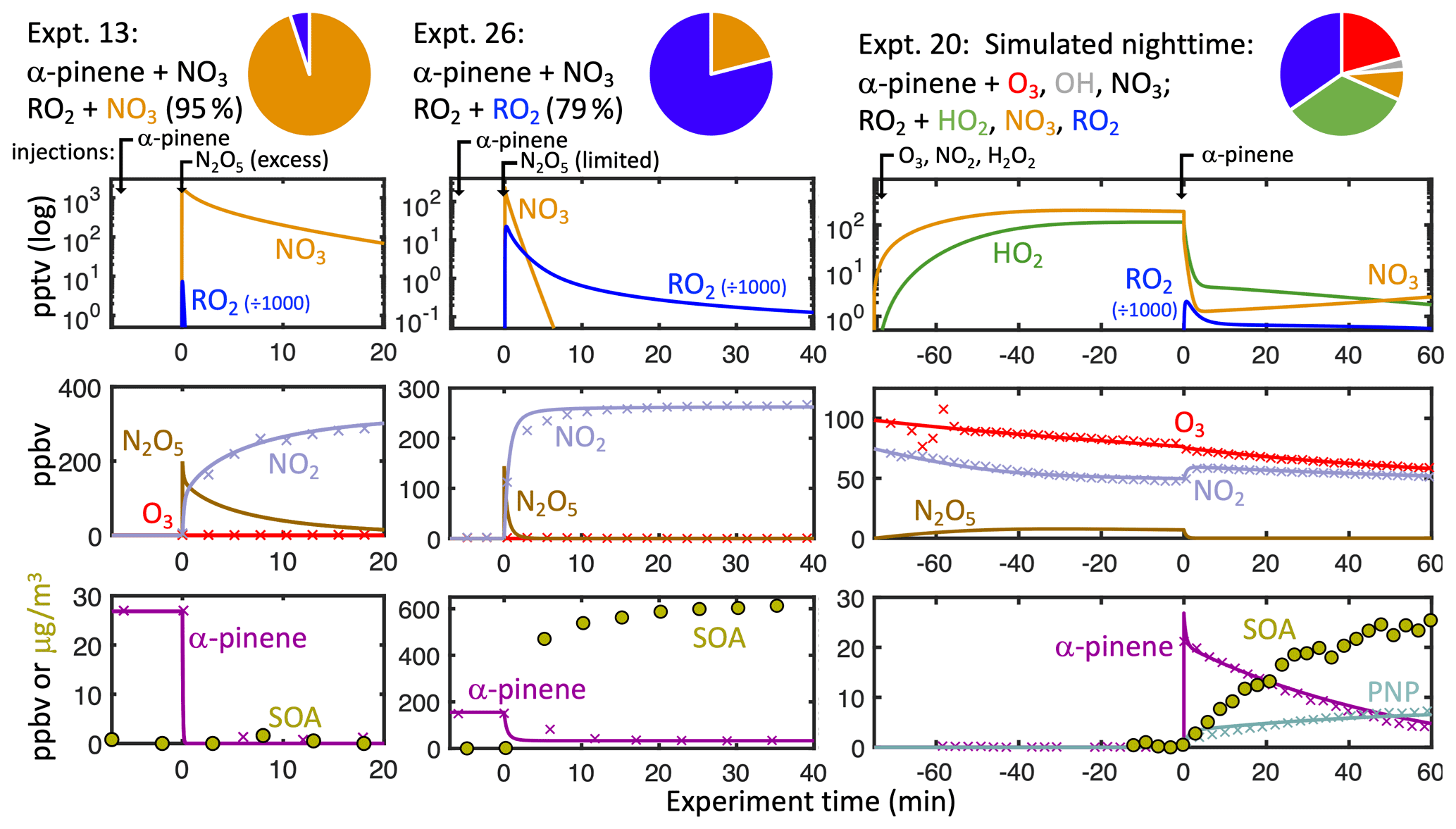
Figure 2Modeled (solid lines) and measured (points) concentrations of key reactive species and products in three example chamber experiments. Timing of chamber injections is demarcated at the top. Experiment time of 0 corresponds to the beginning of α-pinene oxidation. Pie charts show the percent contribution of each α-pinene oxidative pathway (α-pinene + O3 in red, α-pinene + OH in grey and α-pinene + NO3 speciated by subsequent RO2 reaction partner: orange for NO3, blue for RO2, green for HO2).
source. Using excess N2O5 as a source causes a rapid initial spike of NO3, making the nRO2+ NO3 pathway dominant in experiment 13 (Fig. 1, left). The nRO2+ NO pathway is similarly easy to isolate with excess NO added before N2O5 injection, as in experiment 15 (Fig. S1). nRO2+nRO2 chemistry can be maximized by injecting excess α-pinene prior to N2O5 addition; the NO3 is thus dominantly consumed by reaction with α-pinene, leaving the subsequently produced nRO2 to react with each other, as in experiment 26 (Fig. 1, middle), for which we calculate that 79 % of nRO2 reacted with other nRO2.
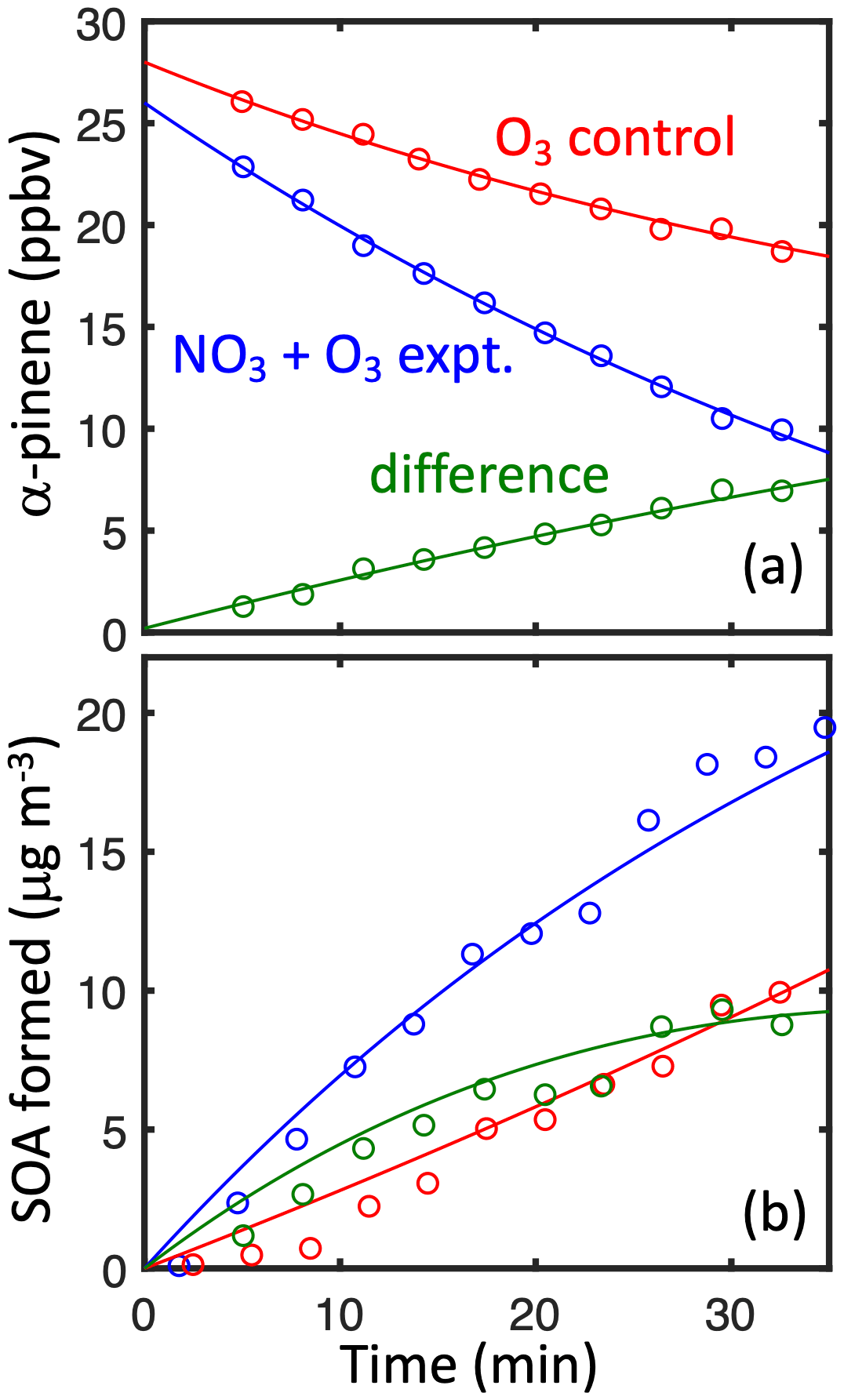
Figure 3(a) α-pinene loss and (b) SOA formation in simulated nighttime experiment 22 (blue) and the ozonolysis control experiment (red). The difference (green) shows the contribution from α-pinene + NO3.
nRO2+ HO2 chemistry is more difficult to isolate due to the scarcity of clean HOx sources in the dark. Here, we describe a method whereby reaction with HO2 represents a majority of the nRO2 fate without additional carbon inputs, initiated by the slow production of NO3 in situ via NO2+ O3, which will introduce three reaction partners for α-pinene (NO3, O3, OH). Alkene ozonolysis has been used previously to produce dark OH in chamber experiments (Xu et al., 2019). Leveraging the ozonolysis of α-pinene already occurring in the chamber, we can amplify HO2 production by injecting excess H2O2 prior to α-pinene to scavenge OH. Thus, the OH + H2O2→ HO2+ H2O reaction simultaneously produces HO2 and suppresses the side chemistry of α-pinene + OH. This HO2 source is relevant to the nighttime atmosphere, as ozonolysis always occurs with NO3 reaction in the ambient atmosphere due to the major source chemistry from NO2+ O3.
The added H2O2 provides an additional benefit – its reaction with NO3 is a source of HO2 prior to α-pinene injection (Fig. S2). The H2O2+ NO3→ HO2+ HNO3 reaction has been estimated by Burrows et al. (1985) to have an upper limit of < cm3 molec.−1 s−1 – too slow for atmospheric relevance but sufficient to produce significant HO2 in chambers when H2O2 is in excess. The rate coefficient of this reaction was further constrained from its upper limit based on the ratio of (Burkholder et al., 2019), resulting in a rate coefficient of 1.1 × 10−16 cm3 molec.−1 s−1 used in our simulations. The formation of PNP was not adequately reproduced if HO2 is assumed to originate from H2O2+ OH alone; i.e., omitting the H2O2+ NO3 reaction in the kinetic model, the yield of PNP would need to be unphysically high to reconcile the difference.
The α-pinene + NO3 reaction is modeled to be the major α-pinene sink in experiments initiated by NO2+ O3 (e.g., 73 % in experiment 22, Fig. 3, Table 2). Even so, the background chemistry from α-pinene + O3 and α-pinene + OH (from the ozone control experiments without NO2) are substantial sources of SOA and require careful subtraction (Fig. 3). Caveats to this approach include the following.
-
In the controls, ozone and OH are larger sinks for α-pinene due to a lack of competition from NO3; thus, a larger fraction of α-pinene produces SOA from ozonolysis in the control compared to the experiment, and the subsequent subtraction obtains a lower-limit SOA yield.
-
Synergistic reactions between RO2 intermediates from the different oxidation pathways are not possible to isolate and contributed roughly 20 % to the analytical signal from the SOA composition analysis (Sect. 3.3); however, it is now appreciated that these synergies also occur in the ambient atmosphere and are not realistic to ignore in laboratory and modeling studies (Kenseth et al., 2018; Inomata, 2021).
These “simulated nighttime” experiments (e.g., experiment 20, Fig. 2, right) are termed as such because they provide an atmospherically relevant balance of reactive pathways, in which fractional contributions are comparable to those on summer nights in the southeast United States (60 %–80 % α-pinene + NO3, 20 %–40 % α-pinene + O3; 30 %–50 % nRO2+ HO2, 30 %–50 % nRO2+ RO2; Ayres et al., 2015; Romer et al., 2018). This is corroborated by gas phase data from the CIMS (Fig. 4), which show that NO3-initiated products are the major volatile products formed in the simulated nighttime experiments. Furthermore, these experiments are able to reproduce gaseous organonitrates observed during the Southern Oxidant and Aerosol Study (SOAS; Carlton et al., 2018), including PNP – a major monoterpene semivolatile at the SOAS site in rural Alabama – and an unknown compound at 314 that has also been observed in the ambient atmosphere with similar time profiles to PNP (Fig. S3).
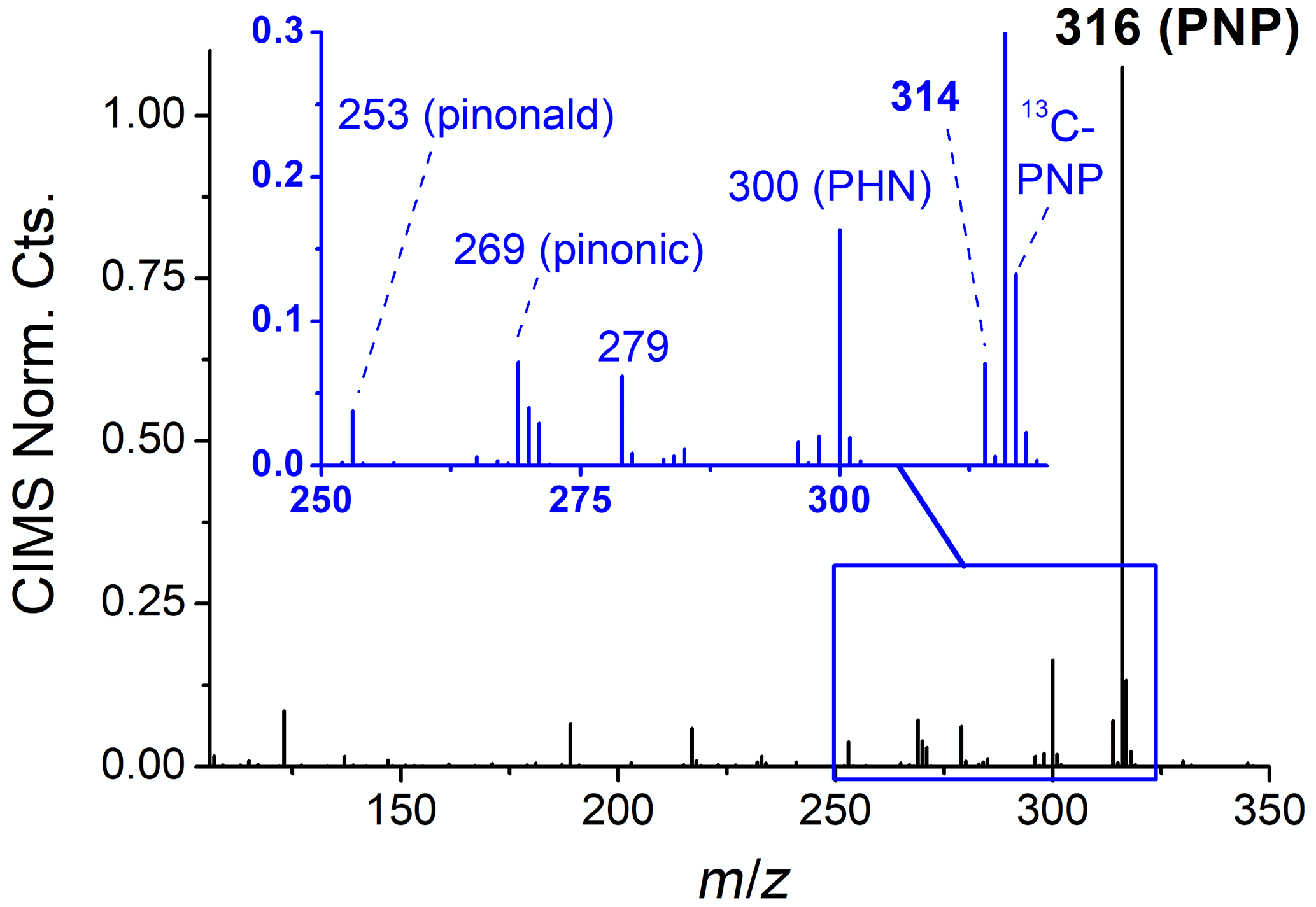
Figure 4Negative ion CIMS mass spectrum after 1 h of oxidation under simulated nighttime conditions, showing PNP, PHN, and another organic nitrogen product at 314. Pinonaldehyde is observed at 253 and pinonic acid at 269.
Product yield results from different nRO2 reaction pathways are shown in Table 2, along with the modeled contributions of each nRO2 reaction pathway. We observe low SOA formation in experiments maximizing nRO2+ NO3 (3 (±3) % mass yield, experiment 13) and nRO2+ NO (12 (±7) %, experiment 15). This is consistent with previous observations of 0 %–16 % SOA mass yields from N2O5-initiated experiments (in which the nRO2+ NO3 pathway tends to dominate), where the larger yields were obtained only with slow continuous introductions of N2O5 (Bell et al., 2021; Fry et al., 2014; Hallquist et al., 1999; Moldanova et al., 2000; Mutzel et al., 2021; Spittler et al., 2006). We also observe no correlation between SOA yields in simulated nighttime experiments and the fractional contribution of the nRO2+ HO2 pathway (Fig. S4), suggesting no SOA production from this pathway, consistent with Kurtén et al. (2017) and Nah et al. (2016). This result conforms to expectations as nRO2+ HO2 produces either the RO radical (Iyer et al., 2018) leading to the volatile pinonaldehyde (Kurtén et al., 2017) or the hydroperoxide, which has high enough volatility to be observed by the CIMS. We do, however, observe high yields of SOA in experiments targeting nRO2+nRO2 chemistry (up to 82 % mass yield, experiment 26). This includes both N2O5-initiated experiments with excess α-pinene and O3+ NO2+ H2O2-initiated simulated nighttime experiments, during which we measured SOA mass yields of 19 %–55 %.
Table 2Peroxy radical pathway contributions and measured experimental outcomes for experiments performed in this work.
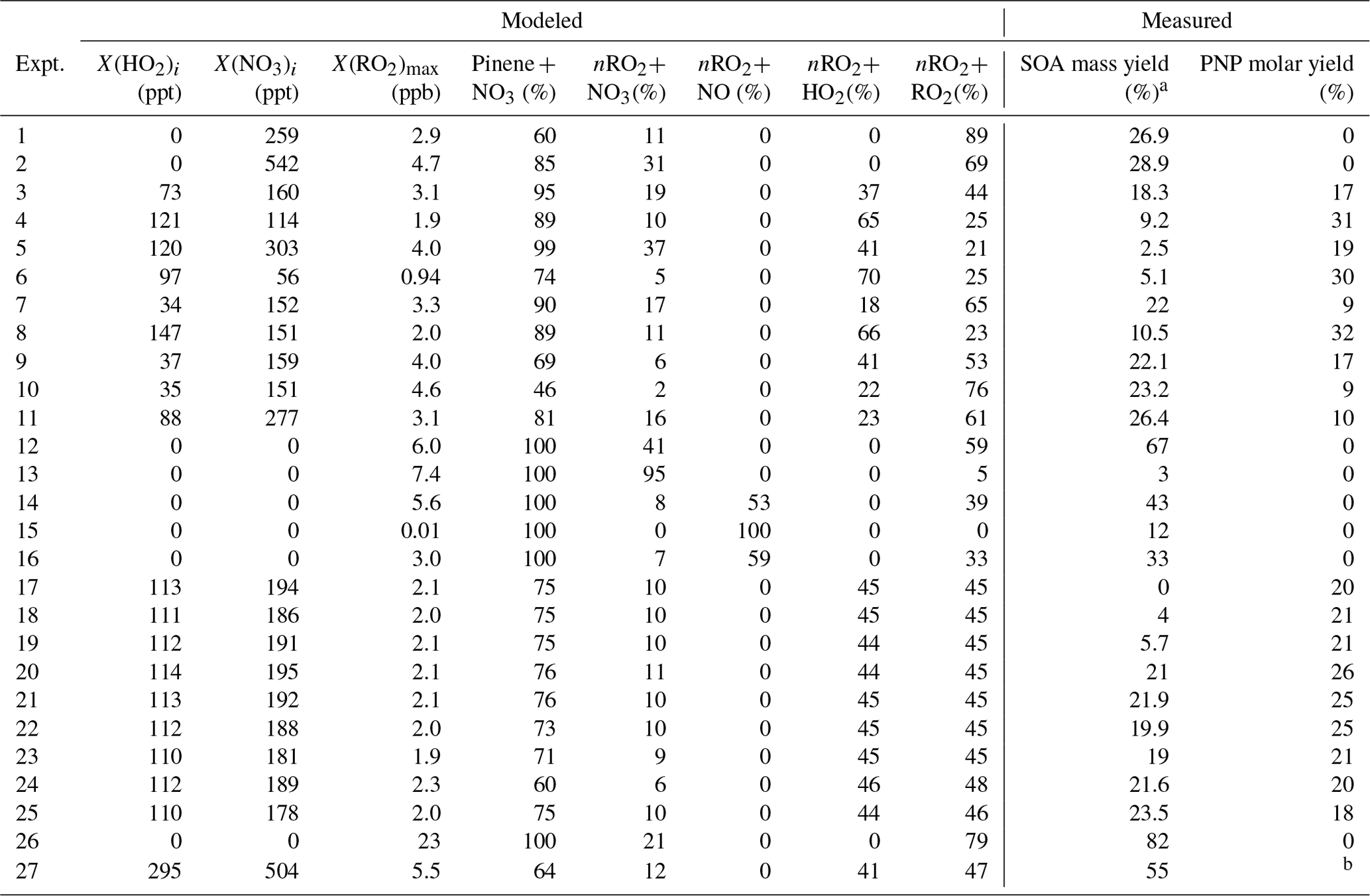
a Ozonolysis-corrected (see Table S1). b Not measured.
Across all experiments, the modeled fractional contribution of the nRO2+ RO2 pathway correlates strongly with SOA yield (Fig. 5). An error-weighted linear regression (York et al., 2004) of ozonolysis-corrected SOA yield against the modeled nRO2+ RO2 contribution for seeded experiments suggests a 58 (±6) % mass yield (R2= 0.54) from this pathway alone. With such a large number of experiments spanning a range of different pathway contributions, however, we can improve upon this
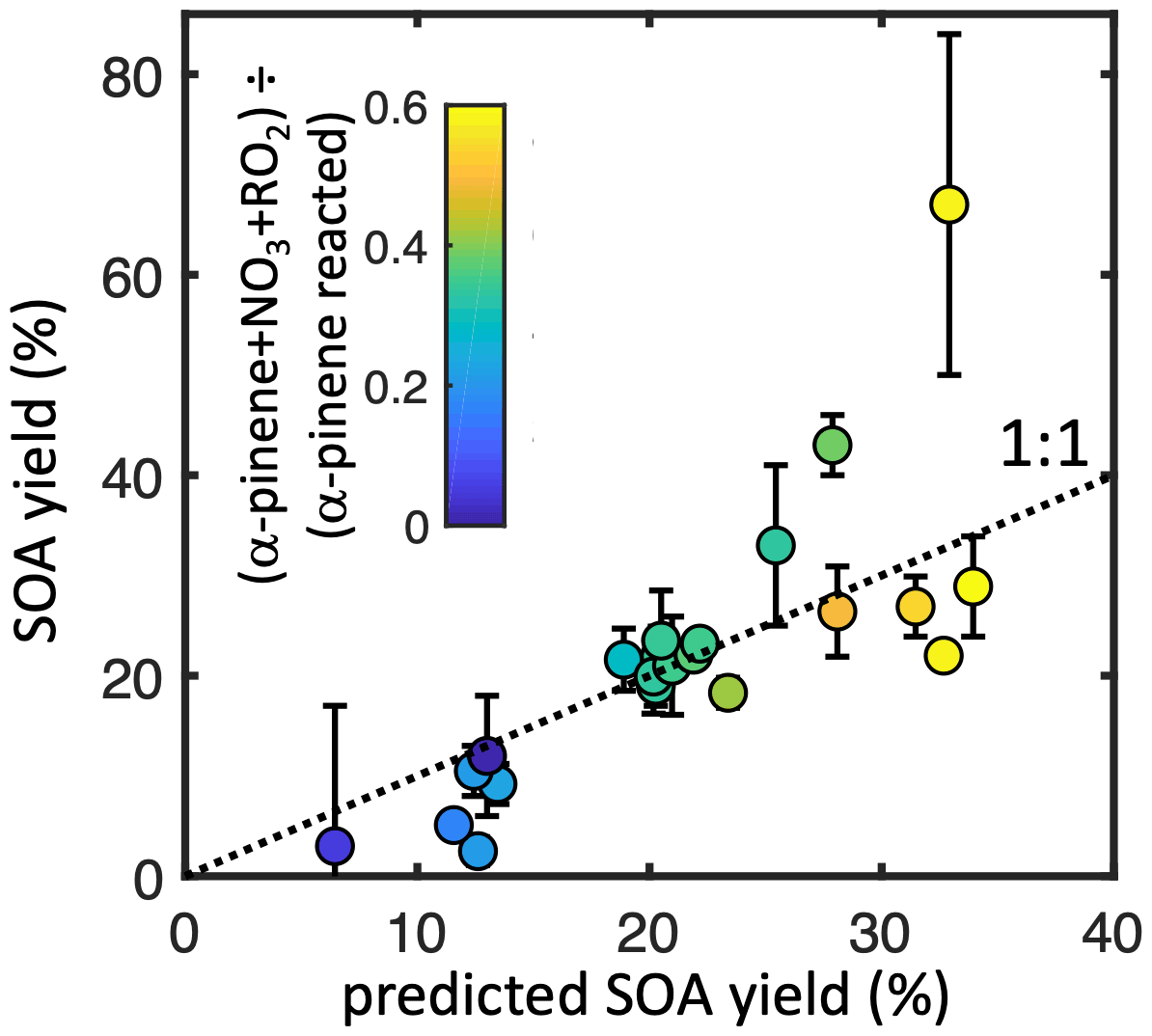
Figure 5Measured (non-ozonolysis-corrected) SOA mass yields from each experiment as a function of the modeled contribution of nRO2+ nRO2 (color) and of the predicted SOA yield from the sum of each pathway contribution (x axis). Error bars denote uncertainty from SMPS measurements.
simple fit by performing a multivariate linear regression to estimate SOA yields from all pathways, with coefficients limited to be ≥ 0. This results in a predicted SOA mass yield from the nRO2+ RO2 pathway identical to results from the simple linear estimate within error: 56 (±7) %. SOA mass yields from the other pathways are not significantly different from zero: 13 (±11) % for nRO2+ NO, 4 (±8) % from nRO2+ NO3, and 0 (±5) % from nRO2+ HO2. Results from this regression analysis, which gives an R2 of 0.66 against measured SOA yields, are shown in Fig. 5.
Repeating the regression analysis for all α-pinene sinks in the simulated nighttime experiments, including the ozonolysis and OH pathways as additional independent variables, also gives coefficients indistinguishable within error (0 % for α-pinene + OH, 15 % for α-pinene + O3). Furthermore, neither the SOA yields themselves nor the residual regression-measurement differences are significantly correlated with reactive pathways other than nRO2+ RO2 or with other experimental parameters (e.g., initial reactant concentrations; see Fig. S5).
By inherently treating individual reactive pathways as independent variables, these regression analyses cannot separate the possible contributions of interactions between multiple pathways, e.g., from synergistic dimerization between nRO2 and ozonolysis-derived RO2 (see Sect. 3.3). The reported coefficients may therefore misrepresent what each pathway on its own would contribute to SOA formation without such synergy. However, because the analysis was performed on experiments predominantly designed to replicate the reactive pathways experienced by α-pinene under ambient nighttime conditions, we expect the coefficients reported above to represent reasonable estimates of each pathway's contribution to SOA formation in the nighttime atmosphere, including any synergistic reactive pathways that occur in ambient conditions. Conditions which deviate substantially from the nighttime atmosphere and therefore lack the same synergistic reactions (e.g., experiments isolating single reactive pathways) may measure different SOA yields. Among the experiments performed here, those with high nRO2+ RO2 contributions without any ozonolysis (experiments 12, 14, and 26) exhibited some of the highest measured SOA yields – higher than the regression model would predict – suggesting perhaps that the nRO2+ nRO2 pathway on its own results in even higher SOA yields while nRO2+ other RO2 pathways have lower yields. Without knowing the relative rates of various nRO2+ RO2 reactions, though, we cannot sufficiently constrain these differences, and additional regression analyses including interaction terms between the reactive pathways did not yield statistically robust results.
These regression analyses also intrinsically depend on kinetic model parameters such as bimolecular RO2 reaction rates, some of which are uncertain. While we are unable to fully quantify these rates, we find that certain ratios between rates are constrained by our experimental outcomes. For example, the negligible SOA yield in the high-NO3 experiment 13 suggests that nRO2+ RO2 chemistry cannot play a major role in that experiment. This can only be achieved if the bulk rate coefficient for nRO2+ RO2 reactions () is approximately 2 orders of magnitude slower than that of nRO2+ NO3 reactions (). We therefore set the bulk to cm3 molec.−1 s−1 with the corresponding bulk equal to cm3 molec.−1 s−1(a value higher than most reported although few measurements exist for these reactions). The PNP yields in simulated nighttime experiments also provide a constraint on the relative rates of the nRO2+ RO2 and nRO2+ HO2 reactions. RO2 + HO2 rate coefficients are, in general, better constrained than those of RO2+ RO2 or RO2+ NO3 reactions; we use the parameterization in Wennberg et al. (2018), which gives cm3 molec.−1 s−1, similar to the measured value for pinene-derived radicals of cm3 molec.−1 s−1 from Boyd et al. (2003). Increasing in our simulations causes a decrease in the contribution of RO2+ HO2 chemistry, but this decrease is only plausible until the measured PNP yield is 100 %, which occurs at a value slightly above cm3 molec.−1 s−1. The substantial yield of pinonaldehyde instead of PNP from nRO2+ HO2 calculated by Kurtén et al. (2017) suggests that must be well below cm3 molec.−1 s−1 to keep the PNP yield well below 100 %.
Based on these constraints, a value of cm3 molec.−1 s−1 represents our optimal fit, and a value of cm3 molec.−1 s−1 represents an upper limit for the bulk of α-pinene nRO2. A higher value would require implausibly high values in order to maintain dominance of the nRO2+ NO3 pathway in experiment 13 and would require implausibly high values in order to maintain a measured PNP branching ratio below the maximum of 100 %. Our best estimate is reasonably within the broad range of measured for other peroxy radicals, particularly given that rate coefficients tend to decrease with size and degree of substitution at the radical site (Orlando and Tyndall, 2012). It is smaller, however, than some recent measurements of the of peroxy radicals derived from α-pinene ozonolysis ( to cm3 molec.−1 s−1), highlighting the uncertainty in reaction rates of this type (Zhao et al., 2018; Berndt et al., 2018b). A slower bulk of cm3 molec.−1 s−1, which would still be consistent with our constraints on relative rate ratios, would scale the contribution of nRO2+ RO2 chemistry in the simulated nighttime experiments by a factor of 0.67, which in turn would require scaling SOA yields from this pathway up by 50 %. A faster bulk of cm3 molec.−1 s−1 would scale the nRO2+ RO2 contribution in the same experiments by a factor of 1.16, which in turn would require scaling the nRO2+ RO2 SOA yield down by 14 %. Despite our constraints, the bulk remains highly uncertain and is also likely to differ between nRO2 isomers for self- and cross-reactions (Orlando and Tyndall, 2012).
To constrain the sequestration of reactive nitrogen under ambient conditions from the α-pinene + NO3 reaction, we also quantify PNP yields in our simulated nighttime experiments. Figure 6 (top) shows measured PNP molar yields as a function of the nRO2+ HO2 fate contribution. Regression analysis (York et al., 2004) suggests a branching ratio for PNP formation of 58 (±2) % from nRO2+ HO2 (R2= 0.88), which we implement in our kinetic model (Fig. 6, bottom). Including the uncertainty in the CIMS calibration increases the uncertainty bounds to 58 (±20) %. The bulk yield likely represents a combination of different branching ratios to PNP formation from the different nRO2 isomers. We hypothesize that the secondary (minor) nRO2 produces exclusively PNP in its reaction with HO2, which would imply a branching ratio of 37 % for PNP formation from the tertiary (major) nRO2+ HO2, assuming an initial branching of 65 : 35 major : minor isomers from α-pinene + NO3 (Jenkin et al., 1997; Saunders et al., 2003). Our measured bulk PNP yield is somewhat higher than the 30 % estimated in the FIXCIT studies (Kurtén et al., 2017), which used similar instrumentation but slightly different chamber conditions (formaldehyde instead of H2O2 and slow addition of α-pinene to minimize RO2+ RO2 chemistry). Some of this discrepancy might be explained by an overestimate of the contribution of nRO2+ HO2 chemistry in the FIXCIT experiment, where they assumed that all NO3 produced by O3+ NO2 reacted with α-pinene and that all nRO2 reacted with HO2 (Kurtén et al., 2017).
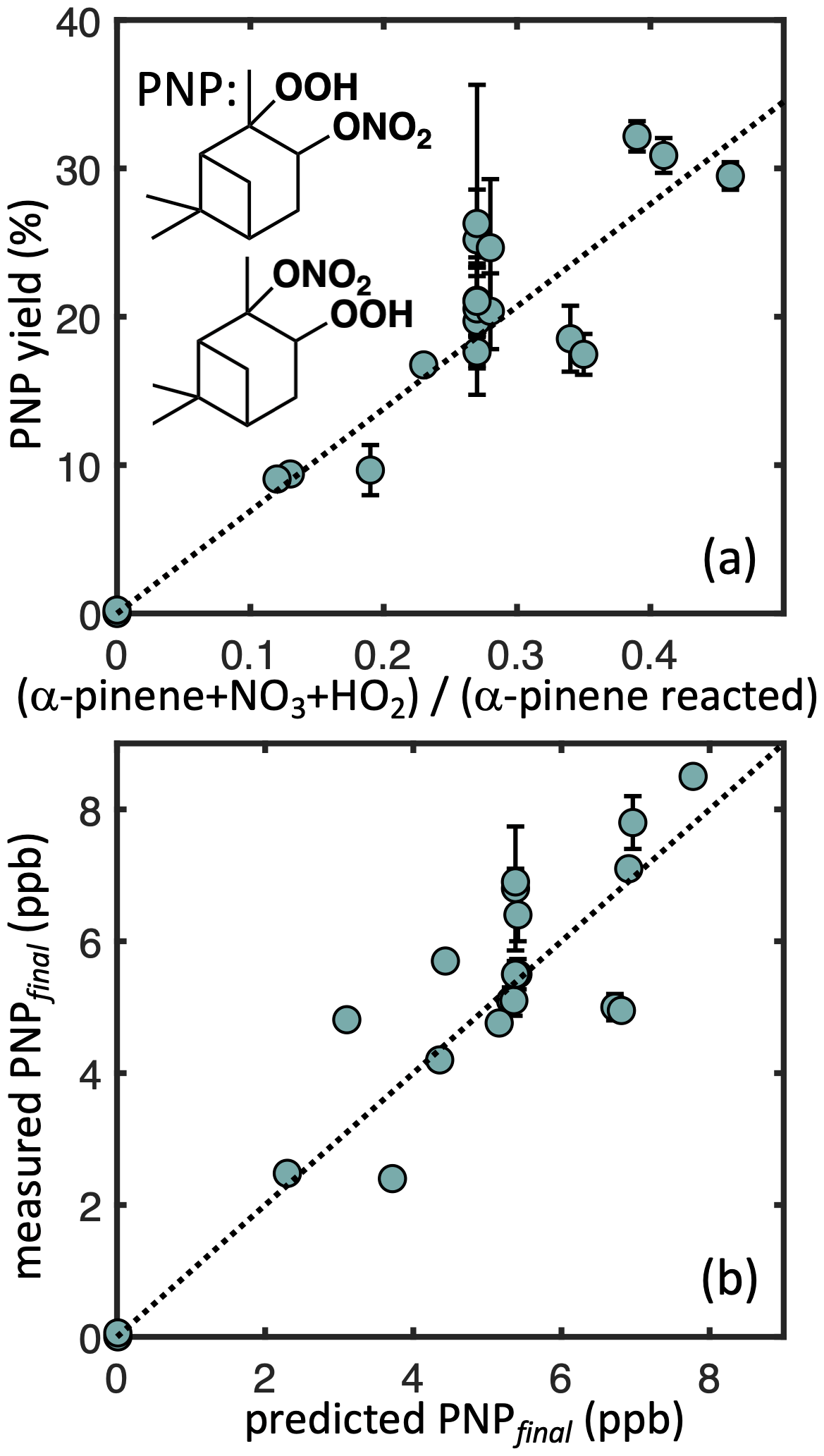
Figure 6Measured α-pinene nitrooxy-hydroperoxide (PNP) yields as a function of the modeled contribution of the nRO2+ HO2 pathway (a) and measured PNP at the end of each experiment as a function of the predicted PNP based on the simple kinetic model. Dotted lines denote error-weighted ordinary least squares fits with intercepts constrained to zero (R2= 0.88 a, 0.74 b; slope = 0.58 a, 1.0 b). Error bars denote uncertainty from CIMS measurements.
We also detect α-pinene hydroxy nitrate (PHN), a product of nRO2+nRO2 chemistry, and α-pinene dinitrate (PDN), a product of nRO2+ NO or nRO2+ NO3 chemistry, by CF3O− CIMS. Based on similar regression analyses of observations and modeling results (Fig. S6), we estimate a bulk PHN yield from nRO2+nRO2 of 11.7 (±3.3) %. However, because the major nRO2 isomer (with the tertiary peroxy radical) is unable to donate an α-hydrogen for PHN formation, a bulk yield of 11.8 % represents a 34 % branching ratio from the minor (secondary) nRO2 isomer (assuming an initial ratio of 65 : 35 major : minor isomers from α-pinene + NO3; Jenkin et al., 1997; Saunders et al., 2003). While observed PDN correlates with nRO2+ NO3 chemistry, its low signal and uncertain CIMS sensitivity make quantification difficult.
3.2 SOA yield dependence on seed surface area
SOA from gas–particle partitioning tends to exhibit a strong dependence on seed aerosol surface area, because of competition between particle surfaces and chamber walls (Schwantes et al., 2019; Zhang et al., 2014, 2015). This effect can cause an underestimation of SOA yields in atmospheric chambers if insufficient seed aerosol is used. To quantify this effect and ensure that RO2 fate experiments (1–16) were initiated with sufficient seed aerosol, we performed an additional set of experiments (17–25) under simulated nighttime conditions to investigate the dependence of SOA yield on seed surface area (Fig. 7). No SOA formation was observed in the absence of seed aerosol, indicating that α-pinene + NO3 does not nucleate. This is in contrast to our ozonolysis control experiments where nucleation was observed, in agreement with other accounts (Burkholder et al., 2007; Hoppel et al., 2001; Inomata, 2021; Takeuchi et al., 2019). The lack of nucleation is consistent with the expectation that the α-pinene + NO3 reaction dominates the nighttime experiments over ozonolysis.
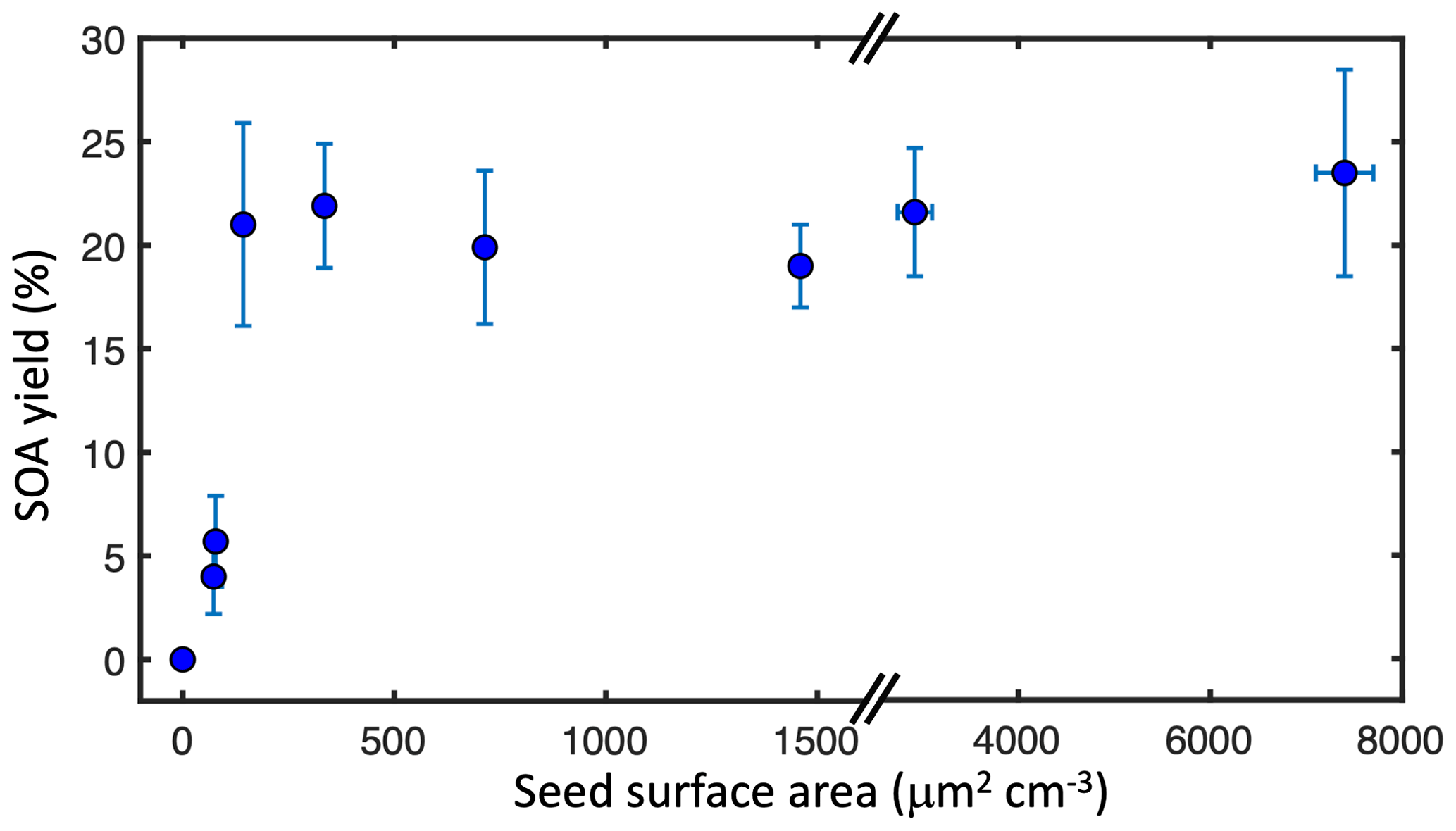
Figure 7Measured ozonolysis-corrected SOA yields from seed area experiments (17–25) plotted against initial seed surface area. Error bars denote experimental uncertainty from SMPS measurements.
SOA yields reached a maximum when initial seed surface area reached 143 µm2 cm−3 (equivalent under experimental conditions to 5.3 µg m−3). This threshold is at least an order of magnitude lower than those measured by Schwantes et al. (2019) for SOA from isoprene OH oxidation under high-NO conditions and by Zhang et al. (2014) for SOA from toluene oxidation. Although vapor wall losses may still cause a decrease in measured SOA yields regardless of seed surface area (Krechmer et al., 2020), we assume here that these effects are minimal at the high seed surface areas (> 600 µm2 cm−3) used in our experiments to investigate RO2 fate. However, true SOA yields may be higher than reported if vapor wall losses are considered.
3.3 SOA molecular composition
Filters collected following SOA formation in experiments 26 (high nRO2+nRO2) and 27 (simulated nighttime) and the ozonolysis control (without NO2) for experiment 27 were analyzed by HRMS to determine the SOA molecular composition (Fig. 8). As expected, the mass spectrum from the simulated nighttime experiment (27, Fig. 8b) exhibits substantial overlap with those from the N2O5-initiated experiment in which the nRO2+nRO2 pathway dominated (26, Fig. 8d), corroborating the results in Sect. 3.1 that suggested the nRO2+nRO2 pathway is responsible for most SOA formation from α-pinene + NO3 chemistry. The SOA from nRO2+nRO2 chemistry is dominated by compounds containing 17–20 carbon atoms, suggesting that dimerization is the primary mechanism by which nRO2+nRO2 reactions lead to SOA formation.
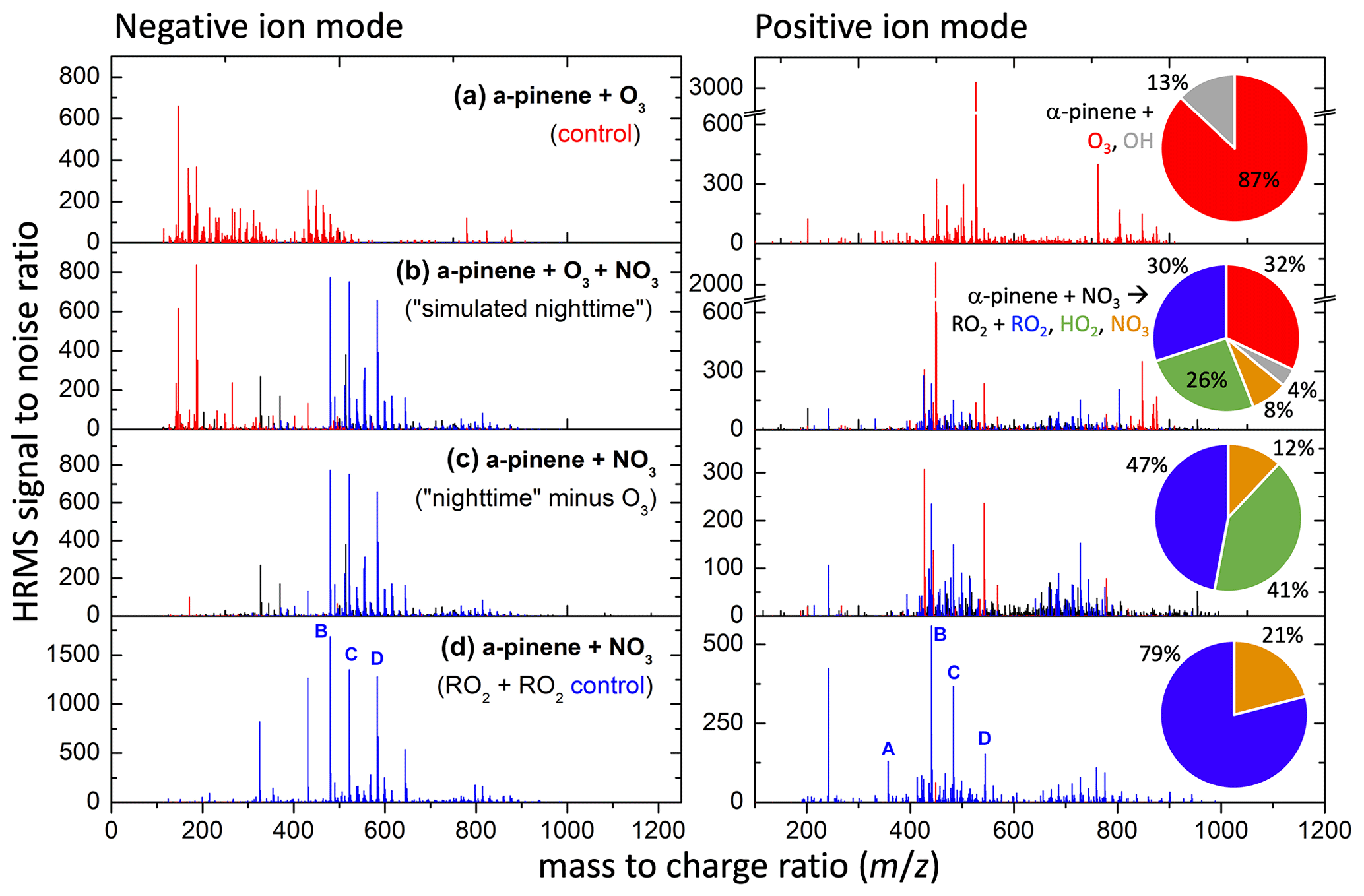
Figure 8Mass spectra of filter samples from experiments 26 (d) and 27 (b), along with the ozonolysis-only control for experiment 27 (a) and the signal difference (c) between (b) and (a). Peaks uniquely attributable to α-pinene ozonolysis alone are shown in red, while those uniquely attributable to α-pinene + NO3 followed by RO2 + RO2 chemistry are shown in blue, with all other peaks in black. Pie charts denote the fraction of α-pinene reacting via each pathway for a given experiment. Key dimer species (A–D) identified in the bottom panels and their peroxy radical precursors are shown in Figs. 9–10.
Masses in the simulated nighttime SOA that are also observed in the nRO2+nRO2 control experiment (26) account for 39 % of the negative mode peak signal and 29 % of the positive mode peak signal in the SOA from the simulated nighttime experiment. Negative mode may overestimate the contributions from dinitrated dimers from nRO2+ nRO2 chemistry, whereas positive mode may underestimate them. In the positive mode, it was found that 34 % of the overall peak signal in the simulated nighttime experiment can also be observed from ozonolysis chemistry (including OH reactions), 14 % of peak signal is found in both the ozonolysis and nRO2+nRO2 control experiments, and 24 % of overall peak signal is completely unique to the nighttime chemistry. SOA from α-pinene ozonolysis is dominated by oxygenated monomers in the negative mode and lacks organonitrate functionalities, making it easy to distinguish from the mass spectrum of NO3-oxidized SOA. The peaks from α-pinene ozonolysis can thus be subtracted off from the simulated nighttime SOA mass spectrum, leaving a spectrum attributable to α-pinene + NO3 under simulated nighttime conditions (Fig. 8c).
The signal-weighted composition of SOA from the simulated nighttime reaction (Fig. 8b), which includes background ozonolysis, is 60 %–70 % nitrogen-containing organics with one to three nitrate groups, as determined in both negative and positive modes (where organic nitrogen species are not enhanced). Of the organic nitrogen, it appears that 2N species are the most abundant and comprise ∼ 30 % of the SOA on average. This suggests dimerization from nRO2+nRO2 to form C14–20 compounds is highly important to SOA formation. Previous analyses of the molecular composition of SOA from α-pinene + NO3 have also found a dominant contribution of dimers in the particle phase (Bell et al., 2021; Takeuchi et al., 2019). The 0N, 1N, and 3N species may also be dimers (4N were not highly observed), as each of the expected RO2s from all channels can have 0–2 N (Fig. 10). There is also a clear population of trimers from α-pinene SOA that is observed here (Fig. 8, centered around 700, with associated carbons C21–C30) and elsewhere (Claflin and Ziemann, 2018; Romonosky et al., 2017). This may suggest RO2 can react with neutral compounds to propagate RO2 radicals that then terminate with another RO2 or may alternatively be attributable to second-generation accretion reactions of dimers (especially those still containing a double bond, which would react rapidly with NO3) with RO2 monomers.

Figure 9Proposed mechanisms for the formation of stable products (red) from α-pinene + NO3 oxidation, including nitrooxy-hydroperoxide (PNP), α-pinene hydroxynitrate (PHN), pinene dinitrate (PDN), the oxidation product observed by CIMS at 314, and pinonaldehyde, as well as α-pinene-derived peroxy radicals (nRO2α-φ, blue) that may contribute to particle-phase dimers (Fig. 10) following the dominant secondary addition of NO3 to α-pinene. “X” stands in for any of RO2, HO2, NO, or NO3 as bimolecular reaction partners.
The data show that SOA formation from α-pinene + NO3 originates from a varied cohort of distinct nRO2 isomers. In Fig. 9 we suggest multiple pathways involving bimolecular reactions, β-scission, and intramolecular isomerization that can produce different C7-C10nRO2 isomers following the initial reaction of α-pinene with NO3. These pathways are consistent with RO2 chemistry previously suggested from theoretical (Kurtén et al., 2017) or lab studies (Xu et al., 2019) of similar systems.
Some reactions of nRO2 with NO, NO3, or HO2 shown in Fig. 9 propagate nRO2 radicals due to unimolecular decomposition rather than forming stable products; this implies that dimer-containing SOA from nRO2 can originate from all nRO2 fate pathways. Figure 10c shows the most abundantly observed dimers in the SOA from nRO2+nRO2 chemistry in experiment 26 (labeled peaks a–d in Fig. 8d), which include C20H30O4 (α-α, Fig. 8d, bar A; presumably following the loss of 2 HONO from dimer C20H32N2O8 in the ESI source), C17H26N2O10 (δ-α, Fig. 8d, bar B), C20H32N2O10 (β-β, Fig. 8d, bar C), and C20H31N3O13 (ε-α, Fig. 8d, bar D). Bell et al. (2021) also observed C20H32N2O8 and C20H32N2O10 among the most abundant compounds in SOA from α-pinene + NO3 chemistry in which nRO2+ nRO2 reactions were thought to dominate.
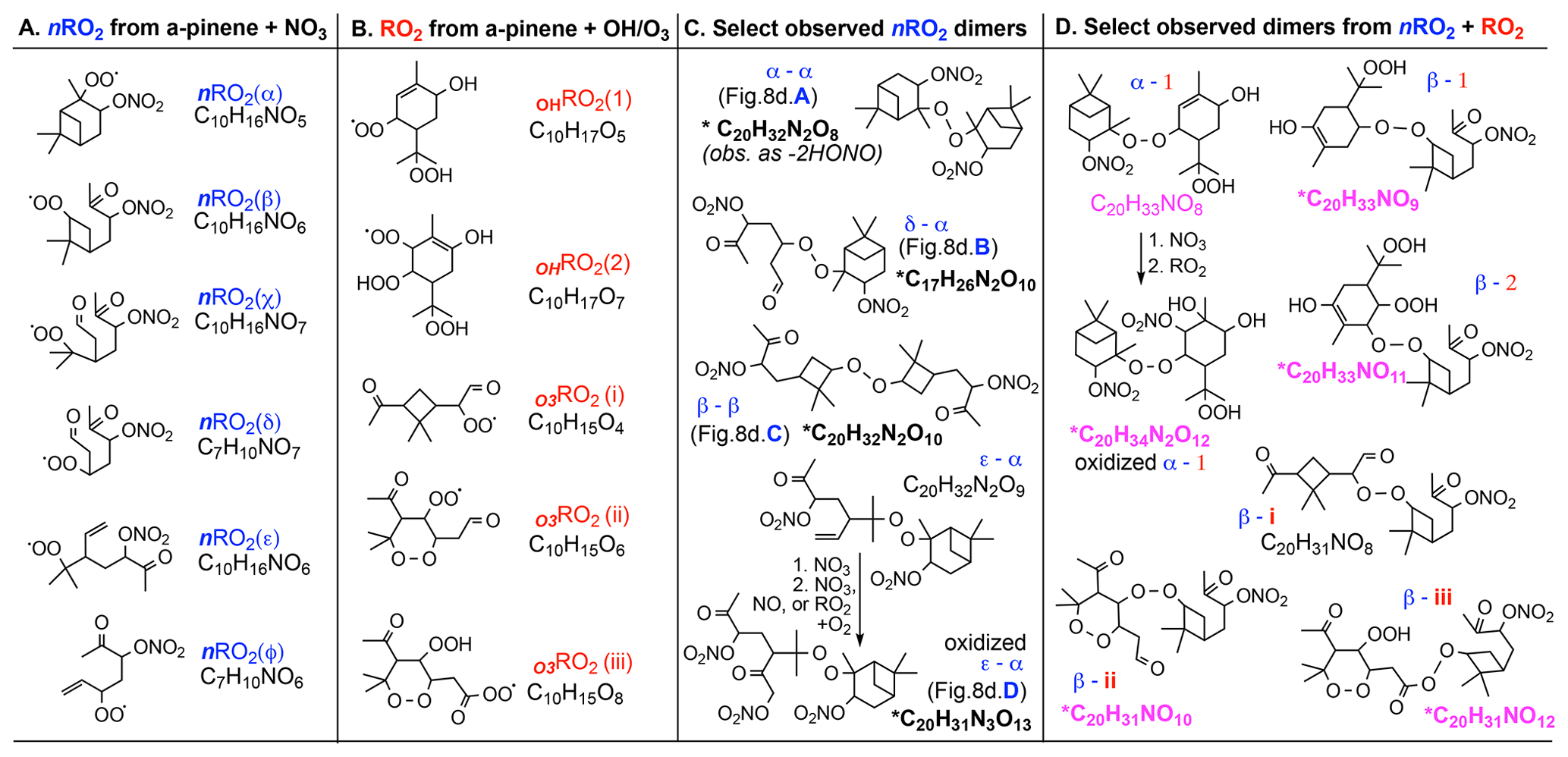
Figure 10Structures and molecular formulas for (a) nRO2 from NO3-initiated chemistry (Fig. 9), (b) RO2 from OH or O3 chemistry (Xu et al., 2019; Iyer et al., 2021), (c) prominent dimers observed from nRO2 self- or cross-reactions, and (d) dimers from synergistic nRO2+ RO2 couplings observed in the simulated nighttime reactions. Dimers with chemical formulas written in pink are uniquely observed in the simulated nighttime experiments; those marked with an asterisk have moderate to high signal (40 > S/N).
Dimers from the ozonolysis control experiments are notably absent from the simulated nighttime experiment. This is likely because the peroxy radicals from α-pinene ozonolysis, which would have reacted with each other to dimerize in the ozonolysis-only experiment, instead react with the more abundant nRO2 radicals from α-pinene + NO3 in the simulated nighttime experiment. This dimerization from cross-reactions of peroxy radicals from both oxidation pathways is responsible for the ∼ 24 % additional HRMS signals in the α-pinene + NO3 SOA spectrum (Fig. 8c, black peaks) but not in the nRO2+nRO2 SOA spectrum (Fig. 8d). Figure 10d shows how RO2 formed from ozonolysis (i–iii; Iyer et al., 2021), initially via the vinylhydroperoxide channel, and OH reactions (1–2; Xu et al., 2019) can couple with nRO2 to produce synergistic dimers during nighttime oxidation of α-pinene. This type of synergy is missing when lab studies cleanly isolate reaction pathways but may be an important part of SOA formation in the ambient atmosphere as reactions occur simultaneously (Berndt et al., 2018a, b; Kenseth et al., 2018; Zhao et al., 2018; McFiggans et al., 2019; Inomata, 2021). These results underscore the importance of conducting chamber experiments under atmospherically relevant conditions.
While we do not seek to represent each individual nRO2 and dimerization pathway in our kinetic model, we adjust the product yields from the first-generation nRO2+nRO2 reactions to provide a reasonable estimate of dimer formation. The observed combined positive and negative mode peak signal attributable to C14–20 compounds (presumed dimers) in nRO2+nRO2 SOA (experiment 27, Fig. 8d) constitutes 40 %–60 % of the total signal. To produce this 40 %–60 % of the 56 % SOA mass yield estimated from nRO2+nRO2 chemistry in Sect. 3.1 using dimers, assuming a mean dimer molar mass of 485 g mol−1 (the average of the positive mode and negative mode peak-signal-weighted mean), a molar yield of 0.16 dimer molecules from the nRO2+nRO2 reaction is required (0.09 per nRO2). This bulk effective yield represents an average across the various nRO2+nRO2 isomer permutations; branching ratios from individual nRO2+nRO2 isomer reactions may differ substantially, but in our kinetic model we simply apply the bulk effective yield to all peroxy radical self- and cross-reactions and scale down the pinonaldehyde-forming pathway accordingly. This 16 % dimer yield is somewhat higher than has previously been measured in other systems (Orlando and Tyndall, 2012), including from α-pinene ozonolysis (Zhao et al., 2018), but fits with recent evidence suggesting that increased molecular size and functionalization can significantly increase dimer branching ratios (Molteni et al., 2019; Berndt et al., 2018a). However, our estimated dimer yield is highly uncertain for a wide range of reasons, including (a) variations in compound-specific sensitivities within the ESI HRMS, which may accentuate the dimer contribution as ESI efficiency has been shown to increase with molecular size (Kenseth et al., 2020); (b) sensitivity to the uncertain estimates of RO2 fate, which depend on poorly constrained RO2+ RO2 and RO2+ NO3 rates (see Sect. 3.1); (c) the possibility of particle-phase reactions, or reactions during filter extraction, which may form or break dimers; and (d) lack of consideration of trimers, which account for 14 % of overall peak intensity and may form from dimers, but are also subject to even greater uncertainty from ESI sensitivity. For all these reasons, we caution that the dimer yield estimated here is only a best guess meant to fit our kinetic model and that further investigation is needed with more quantitative methodology.
Contrary to previous chamber experimental results, we have shown here that the reaction of α-pinene with NO3 can form high mass yields of SOA (> 21 %, at ∼ 30 ppbv of α-pinene) under RO2 fate branching ratios designed to mimic those of summer nights in the southeast United States. Much of this SOA originates from the self- and cross-reactions of nitrated peroxy radicals (nRO2+ nRO2); we estimate that this pathway alone has an SOA mass yield of 56 %, with an associated dimer branching ratio of ∼ 16 %, while SOA formation from other nRO2 pathways is negligible within uncertainty. This hypothesis is also supported by the dominance of dimers in the ozonolysis-subtracted mass spectra of SOA collected from simulated nighttime experiments, the diversity of which suggests that many different peroxy radical rearrangements and dimerization channels contribute to SOA formation from nRO2+ RO2 chemistry.
The magnitude of α-pinene concentrations in the southeast United States will be lower than that used in our chamber; thus, we model the α-pinene + NO3 chemistry at nighttime concentrations measured during the SOAS campaign in rural Alabama in the summer (1 ppb α-pinene, 1 ppb other terpenes, 20 ppb ozone, 1 ppt NO3, and 2 ppt HO2; Ayres et al., 2015; Romer et al., 2018) and assuming steady state for nRO2. At atmospheric concentrations, our kinetic model predicts that 80 % of α-pinene reacts with NO3. Of the nRO2 formed, approximately 20 % is predicted to react with α-pinene-derived nRO2 or RO2 at to cm3 molec.−1 s−1. Combined with our measured 56 % SOA mass yield from nRO2+ RO2, this suggests an overall SOA mass yield of 11 % from α-pinene + NO3 chemistry under the ambient conditions of SOAS. Combined with our measured 60 %–70 % contribution of organonitrates to the SOA composition, this SOA yield corresponds to a 7 % particulate nitrate yield. The mass of the -ONO2 group alone represents 13 % of the SOA mass. A rough calculation predicts that the nighttime reaction of 1 ppbv (∼ 5.5 µg m−3) α-pinene may produce ∼ 0.56 µg m−3 SOA (∼ 0.38 µg m−3 of which is particulate nitrates) through its reaction with NO3. These calculations neglect contributions from cross reactions between nRO2 and other terpene RO2, estimated to comprise another 20 % of the nRO2 fate, which may also lead to SOA formation. Our model excludes the contribution of other non-terpene RO2 from precursors such as isoprene, which, while minor at night, may still contribute to RO2 reactivity and will tend to depress SOA yields due to the smaller dimers formed in those reactions (McFiggans et al., 2019). Although the yields in this work would predict α-pinene + NO3 represents a significant fraction of aerosol and particulate nitrate mass observations during SOAS, previous modeling efforts did not track the SOA or organic nitrates from α-pinene + NO3 due to belief that this reaction does not produce aerosol (Pye et al., 2015; Ayres et al., 2015). Atmospheric models should therefore include SOA formation from α-pinene + NO3, ideally with a dependence on nRO2 fate.
Since HRMS analysis suggests that most compounds in α-pinene + NO3 SOA retain their organonitrate functionality, they will become a permanent sink of NOx following particle deposition. The α-pinene + NO3 chemistry can also have important implications for reactive nitrogen budgets in the gas phase. The high yield of PNP (58 %) from nRO2+ HO2 will temporarily sequester NOy, which may become a permanent sink when the PNP undergoes deposition (Nguyen et al., 2015) or retains its nitrate functionality following further oxidation. Approximately 45 % of nRO2 is expected to react with HO2 during the conditions of SOAS; thus, the nighttime reaction of 1 ppbv α-pinene is expected to produce ∼ 260 pptv of volatile PNP. During SOAS, PNP is observed with maximum concentrations of ∼ 30 pptv, possibly limited by its atmospheric deposition and transport, among other fates (Nguyen et al., 2015). Together, both particle and gas-phase organonitrate formation and deposition from α-pinene + NO3 chemistry may amount to a substantial removal pathway for atmospheric NOx.
Our results also highlight the necessity of performing chamber experiments under conditions that more closely match those in the ambient atmosphere. Measured SOA yields depend heavily on the reactive fate of RO2 in chamber experiments. Previous experiments using N2O5 as a source of NO3 to study SOA formation from α-pinene + NO3 likely observed low mass yields due to the dominance of nRO2+ NO3 reactions, from which we observe little to no SOA formation. Similarly, experiments that maximize nRO2+ HO2 may observe primarily volatile products. Only in experiments designed to allow sufficient nRO2+ RO2 reactions was SOA formation observed. Furthermore, HRMS analysis shows that a large fraction of the compounds in SOA formed under simulated nighttime conditions are not observed in either ozonolysis-only or nRO2+nRO2-only SOA, suggesting that synergistic reaction pathways can enhance SOA formation when multiple oxidation channels occur simultaneously. Because ozonolysis and NO3 oxidation occur together at night in the atmosphere, chamber experiments isolating one or the other will necessarily be biased in their representation of nighttime SOA from α-pinene and potentially other volatile hydrocarbons.
Chamber experiment data are available online at the Index of Chamber Atmospheric Research in the United States (ICARUS; https://icarus.ucdavis.edu/experimentset/214, Bates, 2022). The chemical mechanism for kinetic modeling can be found in the Supplement (Mechanism S1).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-22-1467-2022-supplement.
TBN, KHB, and GJPB designed the chamber experiments. KHB and GJPB performed the chamber experiments. KHB performed the kinetic modeling. JDC assisted with chemical syntheses and experiments. TBN and KHB prepared the manuscript.
The contact author has declared that neither they nor their co-authors have any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This research has been supported by the US National Oceanic and Atmospheric Administration (NOAA) Atmospheric Chemistry, Carbon Cycle, & Climate (AC4) program (grant no. NA18OAR4310121).
This paper was edited by Eleanor Browne and reviewed by two anonymous referees.
Ayres, B. R., Allen, H. M., Draper, D. C., Brown, S. S., Wild, R. J., Jimenez, J. L., Day, D. A., Campuzano-Jost, P., Hu, W., de Gouw, J., Koss, A., Cohen, R. C., Duffey, K. C., Romer, P., Baumann, K., Edgerton, E., Takahama, S., Thornton, J. A., Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Wennberg, P. O., Nguyen, T. B., Teng, A., Goldstein, A. H., Olson, K., and Fry, J. L.: Organic nitrate aerosol formation via NO3 + biogenic volatile organic compounds in the southeastern United States, Atmos. Chem. Phys., 15, 13377–13392, https://doi.org/10.5194/acp-15-13377-2015, 2015.
Bates, K. H.: Experiment Set: a-pinene nighttime chemistry, ICARUS [data set], available at: https://icarus.ucdavis.edu/experimentset/214 (last accessed: 24 January 2022), 2021.
Bell, D. M., Wu, C., Bertrand, A., Graham, E., Schoonbaert, J., Giannoukos, S., Baltensperger, U., Prevot, A. S. H., Riipinen, I., El Haddad, I., and Mohr, C.: Particle-phase processing of α-pinene NO3 secondary organic aerosol in the dark, Atmos. Chem. Phys. Discuss. [preprint], https://doi.org/10.5194/acp-2021-379, in review, 2021.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion product formation from self- and cross-reactions of RO2 radicals in the atmosphere, Angew. Chem. Int. Ed., 57, 3820–3824, https://doi.org/10.1002/anie.201710989, 2018a.
Berndt, T., Mentler, B., Scholz, W., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Ozonolysis and OH Radical Reaction of α-Pinene: Mechanistic Insight and the Influence of Isoprene and Ethylene, Environ. Sci. Technol., 52, 11069–11077, https://doi.org/10.1021/acs.est.8b02210, 2018b.
Boyd, A. A., Flaud, P.-M., Daugey, N., and Lesclaux, R.: Rate constants for RO2 + HO2 reactions measured under a large excess of HO2, J. Phys. Chem. A, 107, 818–821, 2003.
Boyd, C. M., Sanchez, J., Xu, L., Eugene, A. J., Nah, T., Tuet, W. Y., Guzman, M. I., and Ng, N. L.: Secondary organic aerosol formation from the β-pinene + NO3 system: effect of humidity and peroxy radical fate, Atmos. Chem. Phys., 15, 7497–7522, https://doi.org/10.5194/acp-15-7497-2015, 2015.
Burkholder, J. B., Baynard, T., Ravishankara, A. R., and Lovejoy, E. R.: Particle nucleation following the O3 and OH initiated oxidation of α-pinene and β-pinene between 278 and 320 K, J. Geophys. Res., 112, D10216, https://doi.org/10.1029/2006JD007783, 2007.
Burkholder, J. B., Sander, S. P., Abbatt, J., Barker, J. R., Cappa, C., Crounse, J. D., Dibble, T. S., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Percival, C. J., Wilmouth, D. M., and Wine, P. H.: Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 19, JPL Publication 19-5, Jet Propulsion Laboratory, Pasadena, available at: http://jpldataeval.jpl.nasa.gov (last access: 24 January 2022), 2019.
Burrows, J. P., Tyndall, G. S., and Moortgat, G. K.: Absorption spectrum of NO3 and kinetics of the reactions of NO3 with NO2, Cl, and several stable atmospheric species at 298 K, J. Phys. Chem., 89, 4848–4856, https://doi.org/10.1021/j100268a038, 1985.
Carlton, A. G., de Gouw, J., Jimenez, J. L., Ambrose, J. L., Attwood, A. R., Brown, S., Baker, K. R., Brock, C., Cohen, R. C., Edgerton, S., Farkas, C. M., Farmer, D., Goldstein, A. H., Gratz, L., Guenther, A., Hunt, S., Jaeglé, L., Jaffe, D. A., Mak, J., McClure, C., Nenes, A., Nguyen, T. K., Pierce, J. R., de Sa, S., Selin, N. E., Shah, V., Shaw, S., Shepson, P. B., Song, S., Stutz, J., Surratt, J. D., Turpin, B. J., Warneke, C., Washenfelder, R. A., Wennberg, P. O., and Zhou, X. Synthesis of the Southeast Atmosphere Studies: Investigating Fundamental Atmospheric Chemistry Questions, B. Am. Meteorol. Soc. 99, 547–567, https://doi.org/10.1175/BAMS-D-16-0048.1, 2018.
Chan, L. K., Nguyen, K. Q., Karim, N., Yang, Y., Rice, R. H., He, G., Denison, M. S., and Nguyen, T. B.: Relationship between the molecular composition, visible light absorption, and health-related properties of smoldering woodsmoke aerosols, Atmos. Chem. Phys., 20, 539–559, https://doi.org/10.5194/acp-20-539-2020, 2020.
Claflin, M. S. and Ziemann, P. J.: Identification and quantitation of aerosol products of the reaction of β-pinene with NO3 radicals and implications for gas-and particle-phase reaction mechanisms, J. Phys. Chem. A, 122, 3640–3652, 2018.
Crounse, J. D., McKinney, K. A., Kwan, A. J., and Wennberg, P. O.: Measurement of Gas-Phase Hydroperoxides by Chemical Ionization Mass Spectrometry, Anal. Chem., 78, 6726–6732, https://doi.org/10.1021/ac0604235, 2006.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of organic compounds in the atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, 2013.
Fry, J. L., Draper, D. C., Barsanti, K. C., Smith, J. N., Ortega, J., Winkler, P. M., Lawler, M. J., Brown, S. S., Edwards, P. M., and Cohen, R. C.: Secondary Organic Aerosol Formation and Organic Nitrate Yield from NO3 Oxidation of Biogenic Hydrocarbons, Environ. Sci. Technol., 48, 11944–11953, 2014.
Hakola, H., Tarvainen, V., Laurila, T., Hiltunen, V., Hellén, H., and Keronen, P.: Seasonal variation of VOC concentrations above a boreal coniferous forest, Atmos. Environ., 37, 1623–1634, 2003.
Hallquist, M., Waengberg, I., Ljungstroem, E., Barnes, I., and Becker, K.-H.: Aerosol and Product Yields from NO3 Radical-Initiated Oxidation of Selected Monoterpenes, Environ. Sci. Technol., 33, 553–559, 1999.
Hidy, G. M., Blanchard, C. L., Baumann, K., Edgerton, E., Tanenbaum, S., Shaw, S., Knipping, E., Tombach, I., Jansen, J., and Walters, J.: Chemical climatology of the southeastern United States, 1999–2013, Atmos. Chem. Phys., 14, 11893–11914, https://doi.org/10.5194/acp-14-11893-2014, 2014.
Hoppel, W., Fitzgerald, J., Frick, G., Caffrey, P., Pasternack, L., Hegg, D., Gao, S., Leaitch, R., Shantz, N., Cantrell, C., Albrechcinski, T., Ambrusko, J., and Sullivan, W.: Particle formation and growth from ozonolysis of α-pinene, J. Geophys. Res., 106, 27603–27618, https://doi.org/10.1029/2001JD900018, 2001.
Inomata, S.: New particle formation promoted by OH reactions during α-pinene ozonolysis, ACS Earth Space Chem, 5, 8, 1929–1933, https://doi.org/10.1021/acsearthspacechem.1c00142, 2021.
Iyer, S., Reiman, H., Møller, K. H., Rissanen, M. P., Kjaergaard, H. G., and Kurtén, T.: Computational Investigation of RO2 + HO2 and RO2 + RO2 Reactions of Monoterpene Derived First-Generation Peroxy Radicals Leading to Radical Recycling, J. Phys. Chem. A, 122, 9542–9552, https://doi.org/10.1021/acs.jpca.8b09241, 2018.
Iyer, S., Rissanen, M. P., Valiev, R., Barua, S., Krechmer, J. E., Thornton, J., Ehn, M., and Kurtén, T.: Molecular mechanism for rapid autoxidation in α-pinene ozonolysis, Nat. Comm., 12, 878, https://doi.org/10.1038/s41467-021-21172-w, 2021.
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The Tropospheric Degradation of Volatile Organic Compounds: A Protocol for Mechanism Development, Atmos. Environ., 31, 81–104, 1997.
Kenseth, C. M., Huang, Y., Zhao, R., Dalleska, N. F., Hethcox, J. C., Stoltz, B. M., and Seinfeld, J. H.: Synergistic O3 + OH oxidation pathway to extremely low-volatility dimers revealed in β-pinene secondary organic aerosol, P. Natl. Acad. Sci. USA, 115, 8301–8306, 2018.
Kenseth, C. M., Hafeman, N. J., Huang, Y., Dalleska, N. F., Stoltz, B. M., and Seinfeld, J. H.: Synthesis of Carboxylic Acid and Dimer Ester Surrogates to Constrain the Abundance and Distribution of Molecular Products in α-Pinene and β-Pinene Secondary Organic Aerosol, Environ. Sci. Technol., 54, 12829–12839, https://doi.org/10.1021/acs.est.0c01566, 2020.
Kind, T. and Fiehn, O.: Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry, BMC Bioinformatics, 8, 105, https://doi.org/10.1186/1471-2105-8-105, 2007.
Krechmer, J. E., Day, D. A., and Jimenez, J. L.: Always Lost but Never Forgotten: Gas-Phase Wall Losses Are Important in All Teflon Environmental Chambers, Environ. Sci. Technol., 54, 12890–12897, https://doi.org/10.1021/acs.est.0c03381, 2020.
Kruve, A., Kaupmees, K., Liigand, J., Oss, M., and Leito, I.: Sodium adduct formation efficiency in ESI source, J. Mass Spectrom., 48, 695–702, 2013.
Kurtén, T., Møller, K. H., Nguyen, T. B., Schwantes, R. H., Misztal, P. K., Su, L., Wennberg, P. O., Fry, J. L., and Kjærgaard, H. G.: Alkoxy Radical Bond Scissions Explain the Anomalously Low Secondary Organic Aerosol and Organonitrate Yields from α-Pinene + NO3, J. Phys. Chem. Lett., 8, 2826–2834, 2017.
Lee, L., Teng, A. P., Wennberg, P. O., Crounse, J. D., and Cohen, R. C.: On rates and mechanism of OH and O3 reactions with isoprene-derived hydroxy nitrates, J. Phys. Chem. A, 118, 1622–1637, 2014.
Mathis, J. A. and McCord, B. R.: The analysis of high explosives by liquid chromatography/electrospray ionization mass spectrometry: multiplexed detection of negative ion adducts, Rapid Commun. Mass Sp., 19, 99–104, 2005.
McFiggans, G., Mentel, T. F., Wildt, J., Pullinen, I., Kang, S., Kleist, E., Schmitt, S., Springer, M., Tillmann, R., Wu, C., Zhao, D., Hallquist, M., Faxon, C., Le Breton, M., Hallquist, Å. M., Simpson, D., Bergström, R., Jenkin, M. E., Ehn, M., Thornton, J. A., Alfarra, M. R., Bannan, T. J., Percival, C. J., Priestley, M., Topping, D., Kiendler-Scharr, A.: Secondary organic aerosol reduced by mixture of atmospheric vapours, Nature, 565, 587–593, https://doi.org/10.1038/s41586-018-0871-y, 2019.
Moldanova, J. and Ljungström, E.: Modelling of particle formation from NO3 oxidation of selected monoterpenes, J. Aerosol Sci., 31, 1317–1333, https://doi.org/10.1016/S0021-8502(00)00041-0, 2000.
Molteni, U., Simon, M., Heinritz, M., Hoyle, C. R., Bernhammer, A.-K., Bianchi, F., Breitenlechner, M., Brilke, S., Dias, A., Duplissy, J., Frege, C., Gordon, H., Heyn, C., Jokinen, T., Kurten, A., Lehtipalo, K., Makhmutov, V., Petäjä, T., Pieber, S. M., Praplan, A. P., Schobesberger, S., Steiner, G., Stozhkov, Y., Tomé, A., Tröstl, J., Wagner, A. C., Wagner, R., Williamson, C., Yan, C., Baltensperger, U., Curtius, J., Donahue, N., Hansel, A., Kirkby, J., Kulmala, M., Worsnop, D. R., and Dommen, J. Formation of highly oxygenated organic molecules from α-pinene ozonolysis: chemical characteristics, mechanism, and kinetic model development, ACS Earth Space Chem, 3, 873–883, https://doi.org/10.1021/acsearthspacechem.9b00035, 2019.
Mutzel, A., Zhang, Y., Böge, O., Rodigast, M., Kolodziejczyk, A., Wang, X., and Herrmann, H.: Importance of secondary organic aerosol formation of α-pinene, limonene, and m-cresol comparing day- and nighttime radical chemistry, Atmos. Chem. Phys., 21, 8479–8498, https://doi.org/10.5194/acp-21-8479-2021, 2021.
Nah, T., Sanchez, J., Boyd, C. M., and Ng, N. L.: Photochemical Aging of α-pinene and β-pinene Secondary Organic Aerosol formed from Nitrate Radical Oxidation, Environ. Sci. Technol., 50, 222–231, 2016.
Nguyen, T. B., Bateman, A. P., Bones, D. L., Nizkorodov, S. A., Laskin, J., and Laskin, A.: High-resolution mass spectrometry analysis of secondary organic aerosol generated by ozonolysis of isoprene, Atmos. Environ., 44, 1032–1042, 2010.
Nguyen, T. B., Laskin, J., Laskin, A., and Nizkorodov, S. A.: Nitrogen containing organic compounds and oligomers in secondary organic aerosol formed by photooxidation of isoprene, Environ. Sci. Technol., 45, 6908–6918, 2011.
Nguyen, T. B., Crounse, J. D., Schwantes, R. H., Teng, A. P., Bates, K. H., Zhang, X., St. Clair, J. M., Brune, W. H., Tyndall, G. S., Keutsch, F. N., Seinfeld, J. H., and Wennberg, P. O.: Overview of the Focused Isoprene eXperiment at the California Institute of Technology (FIXCIT): mechanistic chamber studies on the oxidation of biogenic compounds, Atmos. Chem. Phys., 14, 13531–13549, https://doi.org/10.5194/acp-14-13531-2014, 2014a.
Nguyen, T. B., Coggon, M. M., Bates, K. H., Zhang, X., Schwantes, R. H., Schilling, K. A., Loza, C. L., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Organic aerosol formation from the reactive uptake of isoprene epoxydiols (IEPOX) onto non-acidified inorganic seeds, Atmos. Chem. Phys., 14, 3497–3510, https://doi.org/10.5194/acp-14-3497-2014, 2014b.
Nguyen, T. B., Crounse, J. D., Teng, A. P., St. Clair, J. M., Paulot, F., Wolfe, G. M., and Wennberg, P. O.: Rapid deposition of oxidized biogenic compounds to a temperate forest, P. Natl. Acad. Sci. USA, 112, E392–E401, 2015.
Orlando, J. J. and Tyndall, G. S.: Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 41, 6294–6317, https://doi.org/10.1039/c2cs35166h, 2012.
Praske, E., Crounse, J. D., Bates, K. H., Kurtén, T., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric fate of methyl vinyl ketone: peroxy radical reactions with NO and HO2, J. Phys. Chem. A, 119, 4562–4572, https://doi.org/10.1021/jp5107058, 2015.
Pye, H. O., Luecken, D. J., Xu, L., Boyd, C. M., Ng, N. L., Baker, K. R., Ayres, B. R., Bash, J. O., Baumann, K., and Carter, W. P.: Modeling the current and future roles of particulate organic nitrates in the southeastern United States, Environ. Sci. Technol., 49, 14195–14203, 2015.
Roach, P. J., Laskin, J., and Laskin, A.: Higher-Order Mass Defect Analysis for Mass Spectra of Complex Organic Mixtures, Anal. Chem., 83, 4924–4929, 2011.
Romer, P. S., Duffey, K. C., Wooldridge, P. J., Edgerton, E., Baumann, K., Feiner, P. A., Miller, D. O., Brune, W. H., Koss, A. R., de Gouw, J. A., Misztal, P. K., Goldstein, A. H., and Cohen, R. C.: Effects of temperature-dependent NOx emissions on continental ozone production, Atmos. Chem. Phys., 18, 2601–2614, https://doi.org/10.5194/acp-18-2601-2018, 2018.
Romonosky, D. E., Li, Y., Shiraiwa, M., Laskin, A., Laskin, J., and Nizkorodov, S. A.: Aqueous photochemistry of secondary organic aerosol of α-pinene and α-humulene oxidized with ozone, hydroxyl radical, and nitrate radical, J. Phys. Chem. A, 121, 1298–1309, https://doi.org/10.1021/acs.jpca.6b10900, 2017.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Schwantes, R. H., Charan, S. M., Bates, K. H., Huang, Y., Nguyen, T. B., Mai, H., Kong, W., Flagan, R. C., and Seinfeld, J. H.: Low-volatility compounds contribute significantly to isoprene secondary organic aerosol (SOA) under high-NOx conditions, Atmos. Chem. Phys., 19, 7255–7278, https://doi.org/10.5194/acp-19-7255-2019, 2019.
Sindelarova, K., Granier, C., Bouarar, I., Guenther, A., Tilmes, S., Stavrakou, T., Müller, J.-F., Kuhn, U., Stefani, P., and Knorr, W.: Global data set of biogenic VOC emissions calculated by the MEGAN model over the last 30 years, Atmos. Chem. Phys., 14, 9317–9341, https://doi.org/10.5194/acp-14-9317-2014, 2014.
Sisco, E. and Forbes, T. P.: Rapid detection of sugar alcohol precursors and corresponding nitrate ester explosives using direct analysis in real time mass spectrometry, Analyst, 140, 2785–2796, 2015.
Spittler, M., Barnes, I., Bejan, I., Brockmann, K. J., Benter, T., and Wirtz, K.: Reactions of NO3 radicals with limonene and α-pinene: Product and SOA formation, Atmos. Environ., 40, S116–S127, https://doi.org/10.1016/j.atmosenv.2005.09.093, 2006.
St. Clair, J. M., McCabe, D. C., Crounse, J. D., Steiner, U., and Wennberg, P. O.: Chemical Ionization Tandem Mass Spectrometer for the In Situ Measurement of Methyl Hydrogen Peroxide, Rev. Sci. Instrum., 81, 094102, https://doi.org/10.1063/1.3480552, 2010.
Takeuchi, M. and Ng, N. L.: Chemical composition and hydrolysis of organic nitrate aerosol formed from hydroxyl and nitrate radical oxidation of α-pinene and β-pinene, Atmos. Chem. Phys., 19, 12749–12766, https://doi.org/10.5194/acp-19-12749-2019, 2019.
Teng, A. P., Crounse, J. D., and Wennberg, P. O.: Isoprene peroxy radical dynamics, J. Am. Chem. Soc., 139, 5367–5377, 2017.
Vasquez, K. T., Crounse, J. D., Schulze, B. C., Bates, K. H., Teng, A. P., Xu, L., and Wennberg, P. O. Rapid hydrolysis of tertiary isoprene nitrate efficiently removes NOx from the atmosphere, P. Natl Acad. Sci. USA, 117, 33011–33016, https://doi.org/10.1073/pnas.2017442117, 2020.
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., St. Clair, J. M., Teng, A. P., Zhang, X., and Seinfeld, J.: The gas-phase oxidation of isoprene and its first-generation products, Chem. Rev., 118, 3337–3390, https://doi.org/10.1021/acs.chemrev.7b00439, 2018.
Xu, L., Møller, K. H., Crounse, J. D., Otkjær, R. V., Kjaergaard, H. G., and Wennberg, P. O.: Unimolecular reactions of peroxy radicals formed in the oxidation of α-pinene and β-pinene by hydroxyl radicals, J. Phys. Chem. A, 123, 1661–1674, 2019.
York, D., Evensen, N., Martinez, M., and Delgado, J.: Unified Equations for the Slope, Intercept, and Standard Errors of the Best Straight Line, Am. J. Phys., 72, 367–375, https://doi.org/10.1119/1.1632486, 2004.
Zhang, H., Yee, L. D., Lee, B. H., Curtis, M. P., Worton, D. R., Isaacman-VanWertz, G., Offenberg, J. H., Lewandowski, M., Kleindienst, T. E., and Beaver, M. R.: Monoterpenes are the largest source of summertime organic aerosol in the southeastern United States, P. Natl. Acad. Sci. USA, 115, 2038–2043, 2018.
Zhang, X., Cappa, C. D., Jathar, S. H., McVay, R. C., Ensberg, J. J., Kleeman, M. J., and Seinfeld, J. H.: Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol, P. Natl. Acad. Sci. USA, 111, 5802–5807, 2014.
Zhang, X., Schwantes, R. H., McVay, R. C., Lignell, H., Coggon, M. M., Flagan, R. C., and Seinfeld, J. H.: Vapor wall deposition in Teflon chambers, Atmos. Chem. Phys., 15, 4197–4214, https://doi.org/10.5194/acp-15-4197-2015, 2015.
Zhao, Y., Thornton, J. A., and Pye, H. O. T.: Quantitative constraints on autoxidation and dimer formation from direct probing of monoterpene-derived peroxy radical chemistry, P. Natl. Acad. Sci. USA, 115, 12142–12147, https://doi.org/10.1073/pnas.1812147115, 2018.