the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 13 Oct 2022
| 13 Oct 2022
Particle-phase processing of α-pinene NO3 secondary organic aerosol in the dark
Amelie Bertrand
Emelie Graham
Janne Schoonbaert
Stamatios Giannoukos
Urs Baltensperger
Andre S. H. Prevot
Ilona Riipinen
Imad El Haddad
The NO3 radical represents a significant nighttime oxidant which is present downstream of polluted environments. Existing studies have investigated the formation of secondary organic aerosol (SOA) from NO3 radicals, focusing on the yields, general composition, and hydrolysis of organonitrates; however, there is limited knowledge about how the composition of NO3-derived SOA evolves as a result of particle-phase reactions. Here, SOA was formed from the reaction of α-pinene with NO3 radicals generated from N2O5, and the resulting SOA was aged in the dark. The initial composition of NO3-derived α-pinene SOA was slightly dependent upon the concentration of N2O5 injected (excess of NO3 or excess of α-pinene) but was largely dominated by dimer dinitrates (C20H32N2O8−13). Oxidation reactions (e.g., C20H32N2O8→ C20H32N2O9→ C20H32N2O10) accounted for 60 %–70 % of the particle-phase reactions observed. Fragmentation reactions and dimer degradation pathways made up the remainder of the particle-phase processes occurring. The exact oxidant is not known, although suggestions are offered (e.g., N2O5, organic peroxides, or peroxynitrates). Hydrolysis of –ONO2 functional groups was not an important loss term during dark aging under the relative humidity conditions of our experiments (58 %–62 %), and changes in the bulk organonitrate composition were likely driven by evaporation of highly nitrogenated molecules. Overall, 25 %–30 % of the particle-phase composition changes as a function of particle-phase reactions during dark aging, representing an important atmospheric aging pathway.
- Article
(2402 KB) - Full-text XML
- Companion paper
-
Supplement
(711 KB) - BibTeX
- EndNote




Organic aerosol (OA) in the atmosphere can have important impacts on human health and climate (Jimenez et al., 2009). A substantial fraction of the organic aerosol is secondary aerosol, which is formed from reactions that lower the volatility of a molecule (Ziemann and Atkinson, 2012). One of the most prevalent families of molecules undergoing these oxidation reactions to form secondary organic aerosol (SOA) is monoterpenes. Unlike isoprene, monoterpenes are emitted both during the day and night (Pye et al., 2010). Because of the importance of monoterpenes at night, nocturnal oxidants (O3 and NO3) play an important role in their transformation in the atmosphere (Brown and Stutz, 2012). A field study in the southeastern USA found that ∼ 50 % of the total aerosol burden at night comes from NO3-radical-derived chemistry (Xu et al., 2015, 2014; Ayres et al., 2015; Lee et al., 2016). Other studies have shown the importance of nitrate chemistry in both urban and pristine areas with monoterpenes (Yan et al., 2016; Stefenelli et al., 2019; Huang et al., 2019; Kiendler-Scharr et al., 2016).
Although α-pinene is globally the most abundant monoterpene (Guenther et al., 2000), β-pinene has been the most studied of the monoterpenes interacting with NO3 radicals because of the resulting high SOA yields (Takeuchi and Ng, 2019; Nah et al., 2016; Boyd et al., 2015) relative to α-pinene. β-Pinene was estimated to be the precursor of up to ∼ 20 % of the total nighttime OA in the southeastern USA (Ayres et al., 2015). However, recent studies have shown that SOA formation from α-pinene + NO3 is also possible (Romonosky et al., 2017; Takeuchi and Ng, 2019; Nah et al., 2016). In chamber studies, SOA from both α-pinene and β-pinene + NO3 contains a substantial fraction of dimers when the radical balance is dominated by RO2 + NO3 or RO2 + RO2 reactions (Takeuchi and Ng, 2019; Claflin and Ziemann, 2018), whereas monomers are the dominant products when RO2 + HO2 is the dominant pathway (Nah et al., 2016). When experiments were conducted in the dark, monoterpene + NO3 products were shown to steadily evolve, with a steady change in the O:C and N:C ratios reported by Nah et al. (2016). In Takeuchi and Ng (2019), ∼ 9 %–17 % of organonitrates from either α-pinene or β-pinene SOA hydrolyzed at moderate relative humidities (∼ 50 % RH). The work of Nah et al. (2016) relied on acidic seeds, which could impact the particle-phase aging (Riva et al., 2019), when compared with neutral seeds or homogeneously nucleated SOA. Alternatively, Claflin and Ziemann (2018) speculated about the importance of dimer formation within the particle phase itself for β-pinene + NO3 SOA. However, quantifying the absolute magnitude of the importance of the particle-phase processes was not possible in any of these studies. These contrasting studies demonstrate that the processes occurring during the dark aging of NO3-derived SOA are still not well understood and warrant further studies to improve our knowledge.
The ability to observe the chemical changes during aging in real time has been limited to instruments with significant fragmentation, low time resolution, or limited to offline analysis. The advent of new soft-ionization techniques makes it possible to follow these changes in real time with high molecular resolution (Pospisilova et al., 2020). We will employ an extractive electrospray ionization time-of-flight mass spectrometer (EESI-ToF) along with a chemical ionization mass spectrometer with a filter inlet for gases and aerosols (FIGAERO-CIMS) to elucidate the particle-phase composition with high chemical and temporal resolution and uncover the changes occurring therein. Here, we investigate the composition of SOA formed from α-pinene + NO3 under dark conditions, determine the processes occurring in the particle phase, separate them from products formed in the gas phase, and determine the magnitude of the effect dark aging has on the composition of NO3-derived α-pinene SOA.
2.1 Chamber conditions
A series of batch-mode chamber experiments (Table 1) investigating the formation and the aging of SOA formed from NO3-initiated chemistry was performed in the 8 m3 Teflon chamber at the Paul Scherrer Institute, Switzerland, which has been described in existing studies (Platt et al., 2013). Measurements were performed with a proton transfer reaction quadrupole mass spectrometer (PTR-MS, IONICON), an extractive electrospray inlet coupled to a long time-of-flight mass spectrometer (EESI-ToF, TOFWERK), a scanning mobility particle sizer (SMPS, TSI model 3938), a chemical ionization mass spectrometer with a filter inlet for gases and aerosols (FIGAERO-CIMS, Aerodyne), a thermodenuder coupled with an SMPS, an ozone gas monitor (Thermo 49C), and a NOx monitor (Thermo 42C). A zero-air generator (AADCO) was used to supply clean air to the chamber and instrumentation. Isothermal evaporation chambers were used for the investigation of particle volatility. The FIGAERO-CIMS sampled on its own dedicated line from the atmospheric simulation chamber with a flow rate of 5 L min−1. Gas monitors and the PTR-MS operated behind a heated (80 ∘C) 3 m stainless-steel sampling line with a 5 s residence time. The particle instruments sampled from a 3 m stainless-steel line with ∼ 3 s residence time. The chamber was cleaned after every experiment and overnight by purging it with zero air (50 L min−1 input and 50 L min−1 output) and heating it to 30 ∘C. In the morning, the chamber was expanded to its full volume with zero air. The chamber was deemed clean if the particulate concentrations overnight were less than 50 cm−3 and the NOx levels were below 30 ppb. A persistent contamination of cresol (∼ 1 ppb) was present in the chamber from experiments that took place prior to those with α-pinene.
Five experiments generated α-pinene SOA from reactions with NO3 radicals at 58 ± 5 % relative humidity (RH) and 21 ± 3 ∘C. No seed particles were used in this study, in contrast with other studies that have been performed on similar systems (Takeuchi and Ng, 2019). NO3 radicals were formed from the decomposition of N2O5, which was injected by passing dry, clean air over a solid sample of N2O5. For blank experiments, α-pinene was injected ∼ 30 min after N2O5. In these experiments, no new particles were formed after the N2O5 injection and prior to α-pinene injection; moreover, the gaseous cresol contamination did not lead to particle formation during blank experiments. However, in each experiment, contamination was observed by both the FIGAERO-CIMS and EESI-ToF after enough organic mass had formed to allow the contaminants to partition into the particle phase; the cresol contamination constituted ∼ 1 %–2 % of the total EESI-ToF signal and ∼ 5 % of the total FIGAERO signal. In all experiments (Exp.), new particle formation occurred promptly after the injection of N2O5 when a volatile organic compound (VOC) was present in the chamber. α-Pinene was injected volumetrically with the PTR-MS following the absolute concentration; in Exp. 1 and 3, ∼ 100 ppb of α-pinene was injected, whereas 20 ppb of α-pinene was injected in Exp. 4 and 5. It was intended that Exp. 1 and 3 would be identical repeats, as the α-pinene and N2O5 additions were effectively identical; however, there was large differences in the mass loading observed. The reason for this difference in yield was not clear, and it may have resulted from inhomogeneities in the chamber during the N2O5 injection. Therefore, even though Exp. 1 and 3 were intended to be conducted under similar conditions, we cannot state unequivocally that this was true. The FIGAERO-CIMS and EESI-ToF were only present for Exp. 1–3, which will be the focus of the discussion below. Although the FIGAERO-CIMS and EESI-ToF were not present for Exp. 4 and 5, these experiments are included to confirm that the changes in particle mass concentration with dark aging at lower mass concentrations are consistent with the measurements at higher mass concentration.
The Framework for 0-D Atmospheric Modeling (F0AM) platform (Wolfe et al., 2016) with the Master Chemical Mechanism (MCM) (Saunders et al., 2003; Jenkin et al., 1997) was used to model the gas-phase and radical chemistry in the chamber. F0AM was used to estimate the amount of N2O5 injected into the chamber, and the concentration of N2O5 was between 80 and 300 ppb in all cases. During dark aging (3–4 h) the volume of the chamber was reduced by instruments sampling from the chamber, and no additional air was added to the chamber during this time.
Table 1Experimental parameters.

a Measured by the SMPS. b Modeled N2O5 concentration based on VOC decay assuming s−1 as the NO3+α-pinene rate constant. c Volumetric addition of α-pinene.
In all experiments, the mass measured by the SMPS was corrected for particle wall loss. The wall loss rate (kwall) was calculated from the exponential decay of the total particle number concentration (cm−3) measured by the SMPS, corrected for coagulation. The particulate wall loss is defined as follows:
where N is the particle number concentration, and kcoag corresponds to the coagulation coefficient ( s−1). The number of particles lost during the measurement time was scaled according to the mean mass of the entire particle population, which is based on the geometric mean mobility diameter (dm) from the measurement time, assuming a density of 1.2 g cm−3. The wall-loss-corrected mass was divided by the uncorrected mass to obtain the wall loss correction factor that was applied to the EESI-ToF and FIGAERO-CIMS data to correct for particle wall loss (during Exp. 1–3).
2.2 EESI-ToF – extractive electrospray ionization time-of-flight mass spectrometer
The EESI-ToF measured the molecular constituents of the SOA formed in the chamber, sampling the aerosols at 1 L min−1. The aerosol sample passed through a multichannel denuder that strips the gas-phase species from the particles, with minimal evaporation of the particles (Lopez-Hilfiker et al., 2019). The aerosol sample intersected with a spray of droplets (50:50, H2O : acetonitrile) doped with 100 ppm NaI which emanated from an electrospray probe. The soluble portion of the aerosol was extracted into the liquid, and the molecules were observed in the mass spectrometer as Na+ adducts after the droplet evaporated. The adducts were guided through a series of ion guides and are separated based on their mass-to-charge ratio in a time-of-flight mass spectrometer. In the experiments presented here, the resolution of the mass spectrometer was 5500–7000. Background measurements were continually and repeatedly performed by sampling the contents of the atmospheric simulation chamber via a particulate filter (e.g., 4 min chamber air without filter and 1 min with filter). A noise filter was used to remove data points impacted by severe electronic noise, which was periodically observed throughout the campaign. The reported signal was a result of subtracting the background filter periods from the adjacent sampling periods. A filtering threshold was applied to the EESI-ToF data, where signals that were 1σ (standard deviation) greater than background data were considered statistically significant and included in the analysis. The EESI-ToF time series were averaged to a 5 min resolution, including one sample and filter period cycle. The EESI-ToF signal was scaled according to the molecular weight of each respective molecule i and converted to a mass flow rate using
where EESI(Hz) is the raw data from the ToF, and MWi is the molecular weight, normalized using Avogadro's number, and a conversion factor from grams to attograms. Within the EESI-ToF, the detection efficiency varied depending on a number of factors including particle size (Lee et al., 2021), extraction efficiency in the EESI-ToF droplets, and mass transmission of an ion from the inlet to the mass spectrometer. In spite of all of these potential impacts, if all ions were measured equally, the overall agreement between the measured EESI-ToF signal (attograms per second, ag s−1) followed a linear trend with the measured organic mass (SMPS) despite day-to-day variations in absolute intensity (Fig. S2). Experiments 2 and 3 fell along the same slope, as the EESI-ToF capillary and ToF settings were not adjusted between experiments. In contrast, Exp. 1 had a much lower slope because the EESI-ToF was still being optimized (capillary position and ToF settings were being altered) and had low sensitivity during this experiment. Consequently, bulk information was obtained by long averaging times for Exp. 1, but time series of individual molecular formulae were not used due to the noisy signal.
The majority of the ion signal (> 95 %) was from Na+ adducts. However, a consequence of using acetonitrile (C2H3N) as a solvent was that there was also a small fraction of the signal that was associated with Na+-adducts+acetonitrile. For instance, Na+- C20H32N2O8 was one of the main ions observed for α-pinene SOA, and there was an associated peak for Na+-C22H35N3O8 (C20H32N2O8 + C2H3N) which resulted from the interaction between the spray solution and the constituent of SOA. For all C20H32N2Ox molecules, the acetonitrile cluster represented 1 %–3 % of the parent ion. For the cases where unambiguous clustering occurred (e.g., C20 + C2H3N), C2H3N was subtracted from the molecular formula. For some compounds, the molecular formulae of the clusters overlapped with C20 oxidation products (e.g., C18H28−32N0−2Ox). As the C18 molecules represented less than 1 % of the total signal, the possible contribution of the acetonitrile cluster was not corrected for. Because there was not a large variety in the carbon distribution of the SOA formed, as shown in the results below, the majority of the mass was corrected for acetonitrile clustering.
One of the most prevalent peaks in the mass spectra of the EESI-ToF was the C10H16O2 (∼ 50 %–65 % total signal), which resulted from an artifact of specific dimer degradation in the electrospray droplet. A follow-up paper will detail the corrections for the artifact and our confidence in identifying it. In brief, the time series of the C10H16O2 molecule was only strongly correlated (R2 > 0.96) with C20H32N2O8, as shown in Fig. S1 in the Supplement (inset), even during evaporation experiments. Based on this correlation, C20H32N2O8 likely fragments in the electrospray droplet to form two C10H16O2 species, which were observed, while the –ONO2 groups were lost and not detected. If we presume that two C10H16O2 molecules come from a single C20H32N2O8 and correct the mass loss for the nitrate groups lost, then there is good agreement with the measured mass. Therefore, the total contribution of C10H16O2 was converted to C20H32N2O8. Additional potential measurement artifacts included loss of HNO3 (Liu et al., 2019), although this was a minor pathway making up < 1 % of the parent ion signal for any molecular formula (e.g., C20H32N2Ox→ C20H31NO HNO3).
2.3 FIGAERO-CIMS – chemical ionization mass spectrometer with a filter inlet for gases and aerosols
The molecular composition of organic compounds in the gas and particle phases was measured using a FIGAERO-CIMS. The FIGAERO inlet was coupled with a high-resolution time-of-flight chemical ionization mass spectrometer (HR-ToF-CIMS) using I− as reagent ion and an X-ray generator as the ion source. The resolution of the mass spectrometer was 5000–6000. The design and operation of the FIGAERO-CIMS were similar to those described in previous studies (Huang et al., 2019; Lopez-Hilfiker et al., 2014). Briefly, particles were collected on a 25 mm Zefluor® PTFE (polytetrafluorethylene) filter (Pall Corp.) via a sampling port (flow rate 5 L min−1). The duration of particle-phase sampling depended on the mass loadings and was 10–20 min for most of the experiments. During the particle-phase sampling, gases were directly measured by the CIMS from the chamber via a Teflon line at 5 L min−1. When the particle-phase sampling was done, the gas-phase measurement was switched off and particles on the filter were desorbed by a flow of gradually heated ultrahigh-purity (99.999 %) nitrogen before being transported into the CIMS. A FIGAERO-CIMS desorption round lasted about 40 min: 20 min of ramping the temperature of the nitrogen flow up to 200 ∘C followed by a 20 min “soak period” at a constant temperature of 200 ∘C. It was then cooled down to room temperature. The resulting mass spectral signal evolutions during a desorption round as a function of desorption temperature are called thermograms (Lopez-Hilfiker et al., 2014). The raw FIGAERO-CIMS data were analyzed with Tofware (Aerodyne Research, Inc. and TOFWERK AG), and codes were written with MATLAB. The integration of thermograms of individual compounds yields their total signal in counts per deposition. For the first filter in Exp. 2, due to a software failure, the filter was stored wrapped in aluminum foil for ∼ 7 h before being desorbed. However, comparing the first filter with others, the changes in the , , and monomer / dimer ratios were similar to other experiments. For further details about the data analysis, see Wu et al. (2021).
3.1 Initial particle molecular composition and instrumental comparison
In the experiments performed here, the α-pinene rapidly reacts with NO3, with corresponding prompt formation of organic aerosol mass measured by the SMPS (shown in Fig. 1a). After α-pinene consumption and particle formation, the SOA mass steadily decays by evaporation (Fig. 1b), which is consistent with previous studies (Nah et al., 2016). In all experiments, except Exp. 2, the α-pinene is fully consumed; in contrast, there is ∼ 20 ppb of unreacted α-pinene left over in the chamber after the N2O5 is fully consumed in Exp. 2, as the injection of N2O5 was less than the other experiments. The incomplete consumption of α-pinene comes from an injection of N2O5 that was less than the total concentration of α-pinene. The relatively small concentrations of N2O5 injected also change the radical balance in the chamber, which will be discussed in the next section. In the other experiments, with an excess of N2O5 injected, hydrolysis on the walls likely represents a significant sink for any unreacted N2O5. During the experiments presented here, the analysis will focus on the higher loading experiments (Exp. 1–3), as both the FIGAERO-CIMS and EESI-ToF were not present for Exp. 4 and 5 (see Table 1).

Figure 1The evolution of precursor and SOA mass. Panel (a) presents an example from Exp. 2, showing prompt SOA formation and consumption of α-pinene. Panel (b) is an overview of all experiments performed, showing the evaporation occurring during dark aging, as measured by the SMPS, and the organic aerosol mass concentration associated with each experiment, all of which have been wall loss corrected.
In this section, we compare the chemical composition measured by both the EESI-ToF and FIGAERO-CIMS. Detailed mass spectra averaged from Exp. 1–3 for both the EESI-ToF and FIGAERO-CIMS are presented in Fig. 2a and b over the first 10–46 min after particle formation. Figure 2a also colors the composition according to the number of carbon atoms in the molecular formulae. Dinitrates dominate the dimer fraction of α-pinene SOA, with molecular formulae of C20H32N2O8−13 making up ∼ 60 %–85 % of the total composition for the EESI-ToF and 45 %–56 % for the FIGAERO-CIMS (the range of values comes from the variation across Exp. 1–3 for the beginning of each experiment), consistent with previous observations (Takeuchi and Ng, 2019), and with dominant RO2 + RO2 reactions shown for other systems (Zhao et al., 2018; Molteni et al., 2019). There are slight differences when comparing the carbon number distribution observed by both the FIGAERO-CIMS and the EESI-ToF, shown in Fig. 2c and d, reflected in the EESI-ToF detecting more dimers over monomers, when compared with the FIGAERO-CIMS. Additionally, singly nitrated monomers (C10H15NO5−10) are observed by both instruments (5 %–7 % EESI-ToF and 7 %–10 % FIGAERO-CIMS), whereas monomer dinitrates (C10H14,16N2O7−11) are mostly only observed by the FIGAERO-CIMS and are slightly above background levels in the EESI-ToF for Exp. 3. The monomer dinitrates have the highest concentrations in Exp. 1 (10 %) and 3 (12 %), while they are substantially lower in Exp. 2 (2 %). Monomer dinitrates likely form via an RO2 + NO2 reaction to create peroxynitrate functional groups (Chan et al., 2010), which is speculated to form monomer trinitrates in the isoprene + NO3 system (Zhao et al., 2021). The lack of their formation in Exp. 2 agrees with the smaller concentrations of N2O5 injected and the corresponding smaller amount of NO2 present. Overall, the similarity in the measured composition in Exp. 1 and 3 suggests that these conditions were relatively similar, although they should not be considered exact replicates because of the difference in the SOA yield between the experiments (Table 1).

Figure 2The average mass spectrum presented for (a) the EESI-ToF for the first 20 min of the experiments and for (b) the FIGAERO-CIMS from the first desorption from experiments (the first 10–46 min of the experiment) for Exp. 1. The colors denote the number of carbon present in the molecules. The main set of molecules correspond to C20H32N2Ox (x = 8–13) for both instruments. (c, d) The binned carbon distribution for experiments Exp. 1–3 for the same time period for the EESI-ToF and FIGAERO, where Exp. 1 (blue), Exp. 2 (green), and Exp. 3 (orange), respectively, take place between the first 10 and 46 min of the experiment.
The main differences between the instruments come from the oxygen distribution of the dinitrate dimers (C20H32N2O8−13), where the most prevalent molecular formulae are with C20H32N2O8 for the FIGAERO-CIMS, as shown in Fig. 3a and b, respectively. This is similar to differences observed between offline ESI and online FIGAERO-CIMS measurements for β-pinene SOA (Takeuchi and Ng, 2019; Claflin and Ziemann, 2018). As mentioned above, there is slight contamination from cresol (C7H8O) in the chamber, which shows itself in a series of C7 and C17 molecules present in the mass spectra (in Exp. 2, there is 6 % in the FIGAERO-CIMS and 1 % in the EESI-ToF). The contaminant molecule with the largest signal in both instruments is C17H26N2O11, which originates from a dimer formed between a cresol monomer and an α-pinene monomer. The changes in the contaminants with dark aging time is relatively minor with respect to all other particle-phase components;therefore, they are spectators during aging.
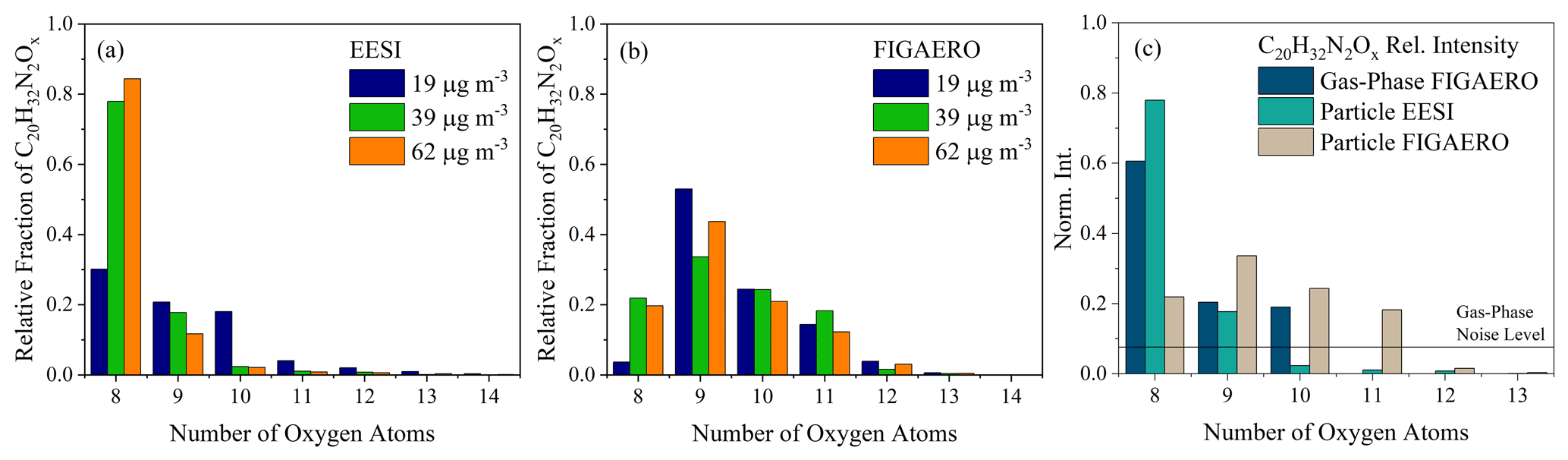
Figure 3The oxygen atom distribution for C20H32N2O8−14 molecules observed by (a) the EESI-ToF and (b) the FIGAERO-CIMS for Exp. 1 (blue), 2 (green), and 3 (orange), respectively, normalized to the total C20H32N2Ox signal. The EESI-ToF data are averaged over the first 10 min of the experiments, whereas the FIGAERO-CIMS data represent the first filter desorption. (c) The oxygen atom distribution for the FIGAERO-CIMS (gas phase) averaged over the first ∼ 5 min from Exp. 2. The solid line denotes the limit of detection for the FIGAERO-CIMS.
Overall, the composition generally agrees between both instruments very well, demonstrating relatively good overlap, with the main molecular classes observed (dimer dinitrates and singly nitrated monomers) making up ∼ 65 %–90 % of the total SOA composition in both instruments. Observed differences do not affect the general scope of the paper focused on the intraparticle reactions of organonitrate species, as will be shown in the following sections.
3.2 Gas-phase radical chemistry and the impact on particle composition
Because particle-phase reactions are not well understood, it is necessary to comprehend which radical pathways control the initial composition of SOA. The radical chemistry of NO3 is important to consider, as Ng et al. (2008) demonstrated that the balance of NO3 and RO2 radical chemistry plays an important role in the yields of formation of SOA from isoprene + NO3. In their study, larger SOA yields were observed under an RO2 + RO2 dominant regime compared with an RO2 + NO3 dominant regime. The RO2 chemistry regime promotes the formation of dimers with lower volatility, increasing yields of SOA, and is consistent with the large prevalence of ROOR dimers observed in isoprene + NO3 SOA (Ng et al., 2008). For monoterpenes, only minor differences were found in the yields of β-pinene + NO3 between the RO2 + NO3 and RO2 + HO2 regimes; however, the RO2 + RO2 dominant regime was not investigated (Boyd et al., 2015).
F0AM was used here to model the fate of RO2 radicals, assuming generalized rate constants for the known RO2 radicals formed via the reaction of α-pinene + NO3. The MCM does not include intramolecular hydrogen shifts, autooxidation reactions, or ROOR formation; however, despite these limitations, the MCM provides general insight into the predicted radical chemistry regimes. Based on the modeling shown in Fig. S3, RO2 + NO3 reactions are predicted to be the dominant pathway when there is an excess of N2O5 (Exp. 1 and 3), whereas RO2 + RO2 reactions will dominate when there is an excess of α-pinene (Exp. 2). HO2 is not an important reaction partner, as there are limited formation mechanisms under our conditions (Boyd et al., 2015).
The first-generation RO2 radical from the reaction of α-pinene + NO3 is C10H16NO5, which can react with itself via the ROOR pathway and form C20H32N2O8 + O2. Based on this reaction pathway, the C20H32N2O8 likely contains two –ONO2 functional groups and a peroxy linkage. This molecule is also the dominant dimer that is observed in the gas phase at the beginning of the experiment (shown in Fig. 3c) and is observed by both instruments in the particle phase. More oxygenated dimers (C20H32N2O9,10) are also observed in the gas phase (Fig. 3c), presumably arising from a similar dimer formation mechanism (RO2 + RO2), but with more highly oxygenated RO2 radicals that can form from the initial RO2 through autooxidation (Bianchi et al., 2019) or the alkoxy pathway (Molteni et al., 2019). Highly oxygenated dinitrate C20 molecules detected here have also been observed in the gas phase in pristine environments (Yan et al., 2016). Gas-phase measurements of C20H32N2O8−10 in Fig. 3c importantly show the presence of dimers in the gas phase, demonstrating they are not exclusively formed in the particle phase, as hypothesized for β-pinene + NO3 (Claflin and Ziemann, 2018).
As we are operating in different predicted radical regimes, RO2 + NO3 dominated (Exp. 1 and 3) and RO2 + RO2 dominated (Exp. 2), there should be differences in the molecules that are observed in the respective experiments. In Exp. 2 (RO2 + RO2 dominant), there is a larger fraction of dimers measured by both instruments (Fig. 2c, d), compared with the other experiments (Exp. 1 and 3) in which RO2 + NO3 reactions are predicted to be the dominant pathway. Consistent with this observation, when dimer formation is suppressed (Exp. 1 and 3), the monomer fraction has a contribution of 17 %–30 % compared with 5 %–7 % in Exp. 2. Given the dominant fraction of dimers in all experiments, the importance of RO2 +RO2 reactions is underestimated, and the RO2 + NO3 reactivity is overestimated by the MCM, especially in Exp. 1 and 3 (see Fig. S3). Despite the potential differences in the predicted radical regime, the formation of dimers dominates the particle-phase composition, meaning that RO2 + RO2 reactions are always very important, even in experiments where RO2 + NO3 is predicted to be the dominant pathway. These findings are consistent with the importance of dimers formed from RO2 + RO2 reactions in other systems (Berndt et al., 2018a, b; Zhao et al., 2018; Ng et al., 2008; Molteni et al., 2019; Rissanen et al., 2015; Simon et al., 2020; Heinritzi et al., 2020) as well as being consistent with higher SOA yields from NO3-initiated oxidation during dominant RO2 + RO2 chemistry (Ng et al., 2008; Bates et al., 2022).
3.3 Evolution in the particle-phase composition
Figure 1b shows that particles steadily evaporate in the chamber after the formation of SOA, suggesting that subsequent changes in composition occur via particle evaporation and/or other particle-phase processes. The high time resolution and detailed molecular information determined by the EESI-ToF, and the FIGAERO-CIMS, were used to determine the processes occurring and their timescales.
A comparison of the temporal evolution of the elemental N:C and O:C ratios for Exp. 1–3 from EESI-ToF data is illustrated in Fig. 4a, colored according to experimental time. Results from Exp. 1 are noisy due to the low sensitivity (see Fig. S2), but the data are included because of the similar trend to the other experiments. The FIGAERO-CIMS is not included because of the low time resolution, which would include only two or three data points. In Fig. 4a, there is a trend toward an increasing O:C during the experiment, coupled with a decreasing N:C which correlates with mass concentration.
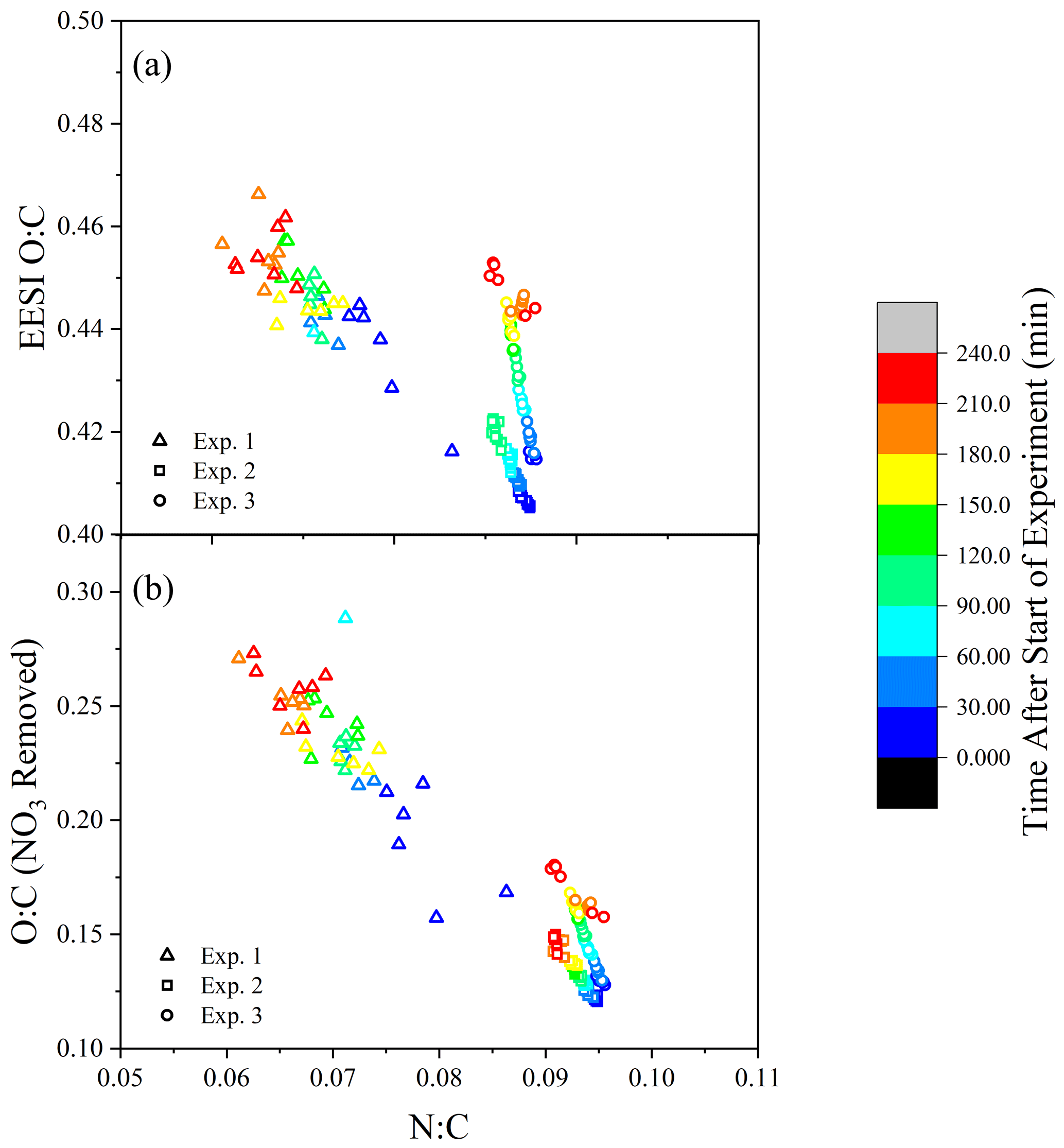
Figure 4(a) The N:C and O:C ratios measured by the EESI-ToF over the course of the experiment (color scale). (b) The O:C ratio is altered by the removal of –ONO2 groups where #O #Ototal – 3⋅ #N.
Considering that each nitrogen atom is likely part of an –ONO2 functional group (Takeuchi and Ng, 2019), the decrease in N:C suggests that the increase in oxidation state (O:C) as a function of aging is larger than the apparent change shown in Fig. 4a. Accounting for the substantial fraction of oxygen in the –ONO2 functional groups, the O:C ratio for the non-nitrate functional group portion is determined by Ono-nitrogen= Omeasured – 3⋅ Nmeasured. With the contribution of the nitrate functional groups removed, the O:C ratio (shown in Fig. 4b) increases by 25 %–60 % during the course of dark aging, demonstrating that the components of SOA either undergo further oxidation during dark aging or that the particles' O:C increases as a function of evaporation of compounds with lower O:C ratios from the particles.
If evaporation is the sole reason for changes in the oxidation state of the SOA, there should be limited changes in the chemical composition as a function of aging, except for those associated with evaporation (i.e., there should be nothing forming in the particle phase). Dimer dinitrates (C20H32N2O8−13) make up between 45 % and 85 % of the total ion signal through the whole dark aging time period, and they will be the focus of the initial discussion on particle-phase processing. The wall-loss-corrected time series of all C20H32N2Ox molecules over the first 180 min of the experiment is shown in Fig. 5a for Exp. 2 (a similar plot is shown for Exp. 3 in Fig. S4; Exp. 1 is not included because of the high relative noise). All molecules exhibit prompt incorporation in SOA, followed by steady changes, with molecules both increasing and decreasing in intensity with time. The C20H32N2O8 molecule steadily decays after particle formation with a decay rate of 0.01 min−1, whereas the C20H32N2O9 molecule steadily increases by 80 % over 3 h. To illustrate the changes more clearly, Fig. 5b shows the changes in the composition relative to t = 10 min after particle formation, when the maximum in particle mass is reached. The C20H32N2O10−11 dinitrates increase by 150 %–450 % during 3 h of dark aging: the contribution of these molecular formulae is ∼ 1 % at the beginning of the experiment and increases to ∼ 2 %–5 % of the total EESI-ToF signal. The FIGAERO-CIMS observes qualitatively similar changes in the composition as a function of dark aging with the C20H32N2O8−13 molecules, although the magnitude of the changes is smaller than those shown in Fig. 5a and b. Overall, Fig. 5a and b demonstrate the production of more highly oxygenated dinitrate dimers in the particle phase. The production observed in Exp. 2 is also observed in Exp. 3, meaning that it occurs regardless of the presence of excess NO3. Therefore, the particle-phase production is not due to gas-phase oxidation reactions with excess NO3 and subsequent condensation to the particle phase; instead, it must be occurring in the particle phase.
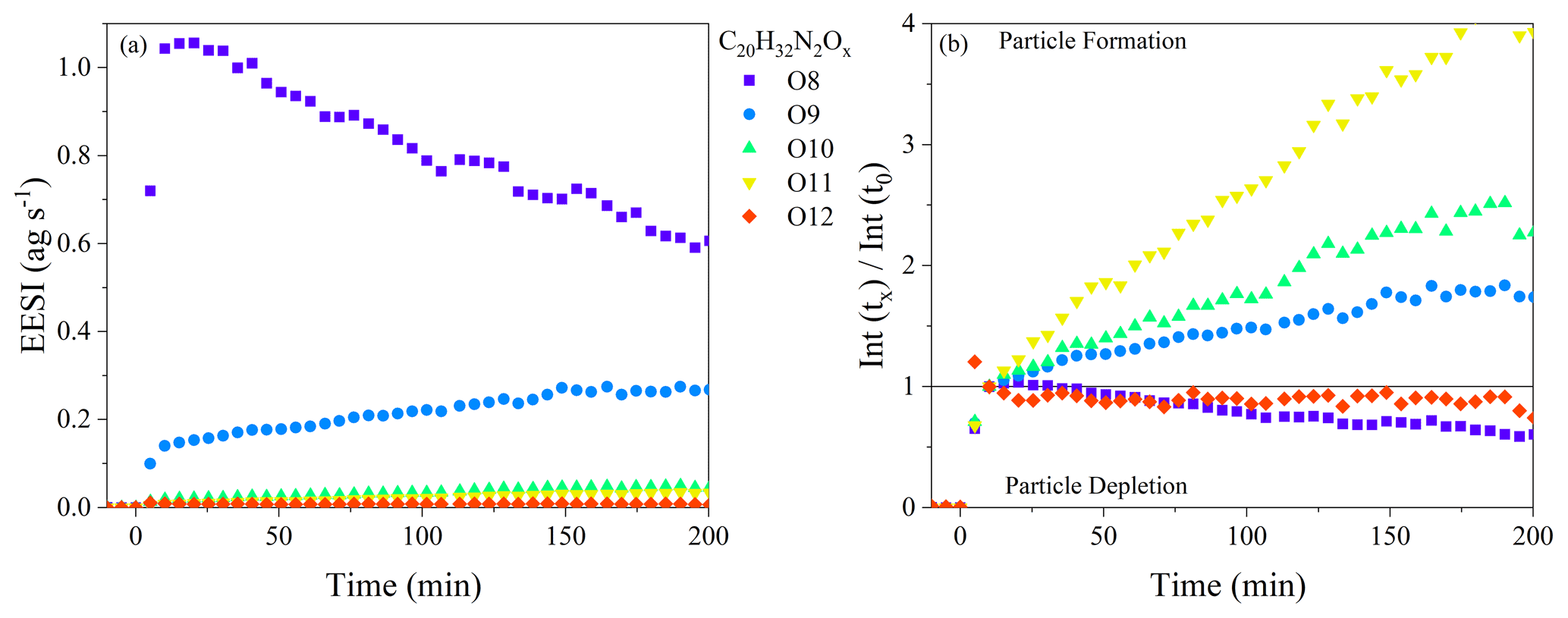
Figure 5(a) Wall-loss-corrected time series of the C20H32N2O8−13 molecules observed by the EESI-ToF over the course of dark aging in the chamber from Exp. 2. (b) All signals are normalized to their intensity at t=10 min.
As we ruled out gas-phase reactions, the production of more highly oxygenated dimers could result from either monomer conversion to dimers or further oxidation in the particle phase. Figure 6a and b compare the carbon distribution of both the EESI-ToF and FIGAERO-CIMS as well as how they change as a function of dark aging for Exp. 3. This experiment is used because it has the best overlap between the instruments (Exp. 1 is also provided in Fig. S5). In both the EESI-ToF and FIGAERO-CIMS, the main species decaying in the particle phase are C20 dimers with an additional contribution from the decay of C10 molecules. Formation is dominated by other C20 molecules and some C10 molecules, with both instruments showing the formation of more highly oxygenated C20 molecules (Fig. 6c, d). The steady evaporation and lack of oxidant in the gas phase likely precludes heterogeneous dimer formation from further gas–particle conversion. We cannot rule out the possibility that dimers decay to monomers and then undergo dimerization reactions to reform as more highly oxygenated dimers. However, if the pathway to more highly oxygenated dimers occurred via the degradation of dimers to monomers, with subsequent dimer formation, there would be a shift to less-oxygenated monomers to compensate for this effect. The oxygen distribution of the C10 molecules for the EESI-ToF is shown as a function of formation and depletion in the Supplement, and they do not present a coherent change with dark aging (Fig. S6).
The argument from the preceding paragraph assumes that the sensitivity of the EESI-ToF holds equally for all molecules. On a molecule-by-molecule basis, there is roughly a spread of 1 order of magnitude in the sensitivity of the EESI-ToF toward different ions (Wang et al., 2021; Lopez-Hilfiker et al., 2019); however, when comparing bulk SOA composition, there is good agreement with the measured mass here (Fig. S2) and during measurements performed in Zurich, Switzerland (Qi et al., 2019; Stefenelli et al., 2019). Additionally, the SOA composition measured by the EESI-ToF compares well to the predicted particle composition based on gas–particle partitioning, using gas-phase measurements performed by the PTR3 (Surdu et al., 2021). This provides confidence that the EESI-ToF is not missing specific molecules here. However, even if the EESI-ToF was not sensitive to the species that are forming the more highly oxygenated molecules (i.e., monomer exchange reactions), the formation of more highly oxygenated dimers (C20H32N2O9−11) would have to be nearly equally balanced by the consumption/disappearance of C20H32N2O8 (in order for the SMPS mass and EESI to agree in Fig. S2). Further, the FIGAERO-CIMS would have to be biased in the exact same way as the EESI-ToF. Considering the agreement presented here and in previous studies, it is more likely that the combination of the EESI and FIGAERO is capturing the change in chemical composition.
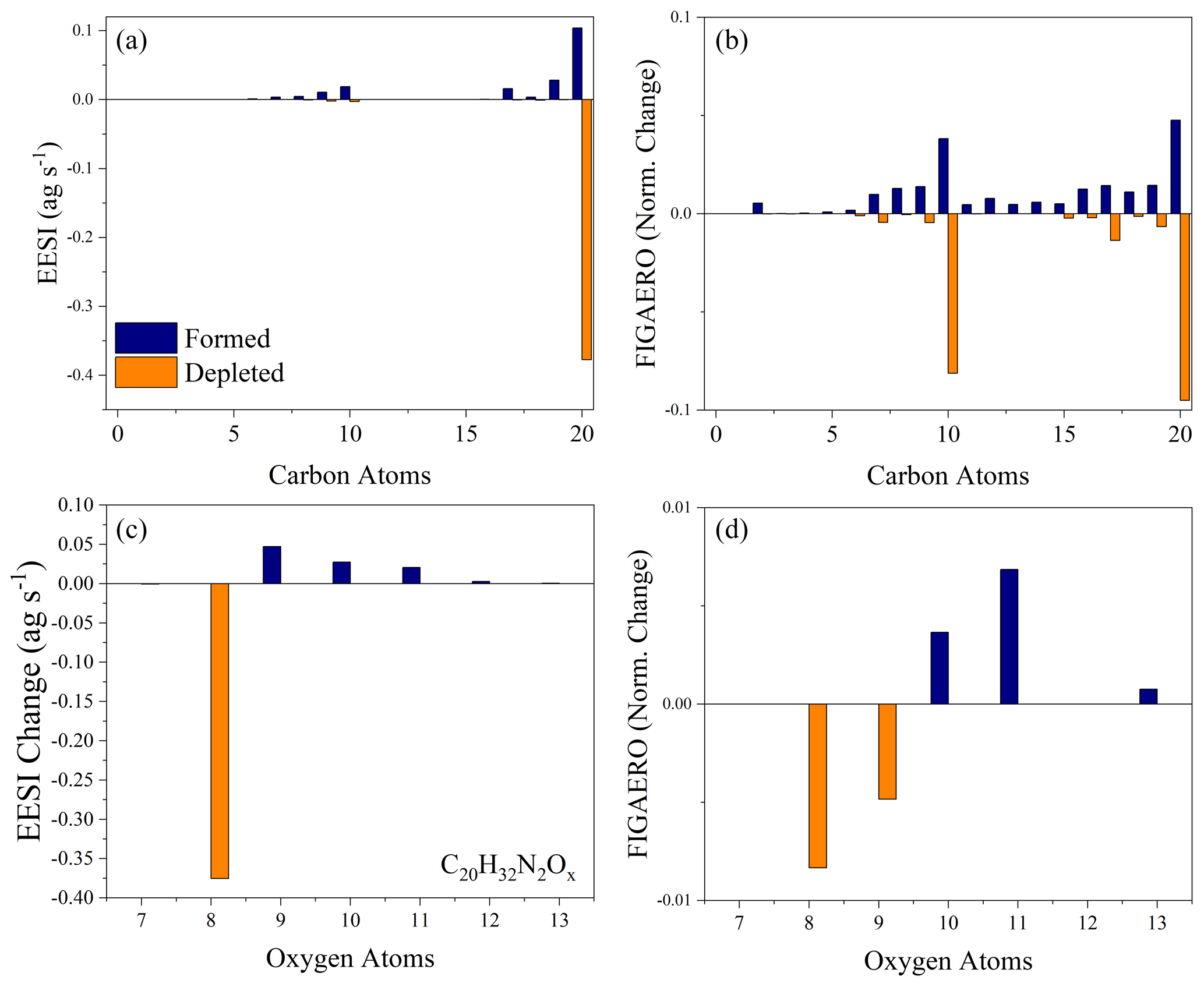
Figure 6The carbon distribution of the molecules formed (blue) and depleted (orange) in the particle phase during aging for the EESI-ToF (a) and FIGAERO-CIMS (b) for Exp. 3. The change in the oxygen distribution for the C20H32N2O8−13 molecules during dark aging for the EESI-ToF (c) and FIGAERO-CIMS (d). For panels (a) and (c), the difference is obtained by taking the difference from t=15 min and t=150 min from the EESI-ToF. Panels (b) and (d) are obtained by taking the difference in the relative sensitivity from the first and third filter. For all figures, the formation is shifted negative on the x axis relative to the nominal carbon number, and the depletion is shifted positive on the x axis comparatively.
Therefore, the species initially formed in the gas phase appear to undergo particle-phase oxidation to form more highly oxygenated molecules, as observed in Figs. 5 and 6. The other important particle-phase reaction pathways shown in Fig. 6 include fragmentation reactions (e.g., C20→ C19) and dimer degradation to monomers, which will be discussed further in terms of their absolute magnitude in the following section.
3.4 Absolute magnitude of particle-phase reactions
Using Figs. 5 and 6, it is possible to assess the magnitude of the processes taking place in the particle phase by taking all molecules observed by the EESI-ToF that are formed relative to t=10 min and creating a mass balance during the whole dark aging period. Figure 7 shows the absolute intensity of all molecules detected by the EESI-ToF from Exp. 2, highlighting the fraction of molecules increasing after the maximum in the organic mass concentration (shown in light blue), the signal of those molecular formulae that decrease (in orange, of which C20H32N2O8 makes up the great majority), and the initial contribution of the molecules that increase (dark blue). The molecules formed during the dark aging period make up ∼ 30 % of the total aerosol composition after 3 h, representing a significant shift in the composition of SOA from the initially formed species. The formation found in the particle phase for Exp. 3 is ∼ 25 % (shown in Fig. S7) and is similar to that reported in Fig. 7. If there was the formation of volatile species, this would not be captured by the current treatment, and the magnitude of the particle-phase processes would be even larger. Therefore, the reported 25 %–30 % formation via particle-phase processes would be closer to 50 % if evaporation resulted from particle-phase reactions and not solely from volatile components repartitioning. Additionally, this treatment also does not account for potential stepwise oxidation reactions (e.g., C20H32N2O8 → C20H32N2O9→ C20H32N2O10), which would again result in an underestimation of the importance of particle-phase reactions. Therefore, a lower estimate of 25 %–30 % of the total composition of α-pinene SOA is altered as a function of particle-phase processes over the 3 h experimental time.
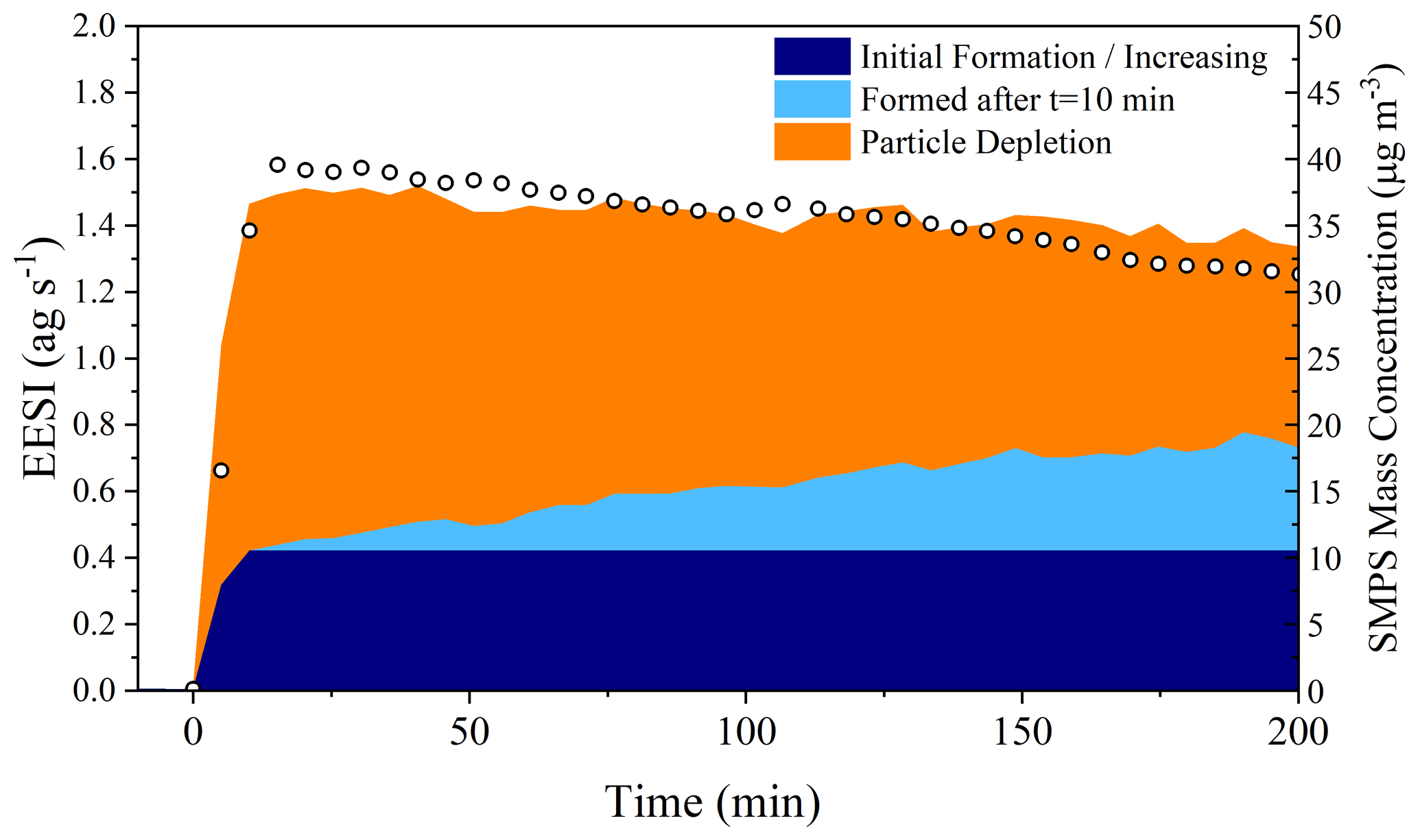
Figure 7The left axis shows the total EESI-ToF intensity plotted as a function of the contribution from different sources, particle-phase formation products determined from those molecular formulae that increase during dark aging after t=10 min from Exp. 2. The right axis shows the measured SMPS mass concentration. Both the EESI and SMPS have been for wall losses.
Figures 7 and S7 also show that particle-phase processes appear to be the most important over the first 2–2.5 h of dark aging. Nearly all of the changes in the particle composition observed by the EESI-ToF come at the expense of C20H32N2O8, which diminishes from 70 % of the total composition to ∼ 35 % after 3 h of aging. Assuming that the steady formation of higher oxidation products is the result of a reaction with C20H32N2O8, the summation of signal forming after t=10 min in Fig. 5a represents the fraction of C20H32N2O8 consumed via particle-phase processes. In Exp. 2, the C20H32N2O8 has a mass flux of 1.05 ag s−1 and decreases to 0.61 ag s−1 after 3 h, with a corresponding increase in oxidation products from 0.17 to 0.38 ag s−1 over that same time frame (shown in Fig. 5a). Approximately half of the total depletion observed arises from an increase in oxidation, with the remainder coming from evaporative losses.
Although oxidation reactions account for a large majority of the particle-phase processes occurring, Fig. 6a–d shows that it is not the only process occurring. The increases in the oxidation state of the C20 dimers make up 60 %–70 % (range for Exp. 2 and 3) of the total fraction of the species formed in the particle phase. Using analysis similar to the results shown in Fig. 6a–d, we can separate the other processes according to how the carbon distribution changes. The other two minor pathways observed by the EESI-ToF are (1) increases in the monomer fraction (10 %–15 % to the total changes) and (2) the formation of C8,9 or C18,19 via fragmentation reactions from C10 or C20 molecules (10 %–20 %). Fragmentation reactions will also lead to the formation of smaller molecules containing one to two carbon atoms, which should rapidly evaporate from the particle phase. Assuming that the molecules leaving the particle phase have a molecular formula of CH2O, as a simple example, this would represent ∼ 10 % loss of mass relative to C20H32N2O8. The amount of mass evaporating from the particle from fragmentation reactions would be on the order of 0.2 %–0.3 % of the total mass (or 0.1 µg m−3 in the experiment shown in Fig. 8). Therefore, fragmentation reactions are not responsible for a significant loss of mass during dark aging. We should note, however, that we do observe a few molecules increasing more than 25 % in the gas phase, which is consistent with fragmentation reactions, including C2H4O3, C3H6O3, C4H8O3, C3H5NO5, C4H7NO5, and C5H9NO5.
In Fig. 4a, we show that the N:C ratio decreases during dark aging. The removal of dimer dinitrates results in a significant mass change during the dark aging period (10 %–30 % of the total signal). Due to the substantial nitrogen content of these dinitrate dimers, they will considerably impact the N:C ratio during aging. Figure 1b shows that the SOA evaporates during aging in the chamber. Evaporative loss of dinitrate dimers is possible, although not common considering that dimers typically have very low volatility. However, when performing isothermal evaporation measurements in the atmospheric simulation chamber, both the C20H32N2O8,9 are susceptible to repartitioning (Fig. S8). As these dimers are low-volatility to semi-volatile molecules, they are in equilibrium between the particles, the gas phase, and the walls (Bertrand et al., 2018; Krechmer et al., 2020). Therefore, part of the loss of C20H32N2O8,9 is due to repartitioning and accounts for a significant fraction of the organonitrate loss from the particle phase observed by the EESI-ToF as well as a significant fraction of the decreasing signal in the FIGAERO-CIMS. Previously, the loss of NO3 from the particle phase has been assumed to come from the hydrolysis of –ONO2 functional groups due to the loss of NO3 measured by the Aerosol Mass Spectrometer (AMS) (Takeuchi and Ng, 2019; Nah et al., 2016). Here, the hydrolysis products (C20H33NO6−12) make up less than 1 % of their corresponding dinitrate species C20H32N2O8−14, demonstrating that hydrolysis of the main dinitrate dimers is not a significant loss term for this system. The lack of hydrolysis could come from the lack of water in the particles, which could differ from other experiments that have used seed aerosols (Takeuchi and Ng, 2019; Nah et al., 2016).
3.5 Possible oxidation pathways
In order to have oxidation reactions occurring in the particle phase, such as the observed C20H32N2O8 to C20H32N2O9 conversion, there needs to be an oxidant or radical present with which to react. Radicals may act as oxidants through hydrogen abstraction reactions, with subsequent O2 addition to the molecule that has lost a hydrogen. This would be a chemical pathway to form more highly oxygenated molecules, and it is analogous to similar reactions occurring in the gas phase (Molteni et al., 2019; Bianchi et al., 2019; Molteni et al., 2018). However, these reactions are not well established in the particle phase. Below, we speculate about some potential pathways and sources of radicals in the particle phase.
Radical chemistry could be initiated from organic peroxides or peroxy nitrates incorporated in the particle phase, initiated from the scission of O–O bonds in organic peroxides. Considering that the peroxy nitrate content is controlled by the concentration of NO2, we would expect that this would only be important for Exp. 1 and 3, not for Exp. 2. Because the magnitude of oxidation is similar between Exp. 2 and 3, peroxy nitrates are likely not an important source of radicals. The degradation of dimers with a peroxy linkage could be prone to this effect, which would likely affect those species formed in the gas phase, C20H32N2O8−10, and these molecules are also abundant in all experiments. For the higher oxygenated molecules (C20H32N2O9,10), this effect would be obscured by the formation that dominates their time series, and disentangling this effect from evaporation for C20H32N2O8 becomes difficult.
The FIGAERO-CIMS also observes a small fraction (< 0.1 µg m−3) of N2O5 in the particle phase, which could act as an oxidant. The fraction of N2O5 in the particle phase decreases with time during the experiment (∼ 50 % over ∼ 3 h) (shown in Fig. S7), indicating that it is being consumed or is evaporating from the particle phase. However, N2O5 is present in the particle phase in experiments both with and without an excess of N2O5, which is surprising considering its high volatility.
Overall, either the presence of N2O5 in the particle phase or organic peroxides could be responsible for the continued oxidation in the particle phase. However, in the system presented here, we are unable to assess which potential oxidant or reaction is more important because each oxidant will likely result in a similar reaction scheme and similar products.
The composition of NO3-derived α-pinene SOA is dominated by dimers formed through RO2–RO2 reactions under the experiment conditions in this study. Pye et al. (2015) modeled the atmospheric conditions during nighttime chemistry in the southeastern USA and showed that RO2 radicals in the atmosphere will react with either other RO2 radicals (∼ 40 %) or HO2 radicals (60 %), with little reactivity with NO3 radicals. In pristine areas, the termination of RO2 radicals can be dominated by reactions with other RO2 species (Yan et al., 2016). Given the prevalence of dimers formed from NO3 chemistry (regardless of the concentration of NO3 radicals), as shown in our study, they will be atmospherically important and form in most environments due to the relatively fast reaction rates between RO2 radicals. These results illustrate that, after formation in the gas phase, dimers will condense to the particle phase where further reactions will proceed. Oxidation reactions are the dominant reaction pathway, accounting for 60 %–70 % of the change in composition, whereas fragmentation (∼ 10 %–20 %) and dimer decay to monomers (10 %–15 %) are the minor pathways. Although there are changes in the nitrogen content, they do not appear to be associated with the hydrolysis of nitrate functional groups but are rather due to the repartitioning of low-volatility/semi-volatile highly nitrated molecules. An open question remains regarding the source of oxidation in the particle phase, as it could come from the degradation of organic peroxides or from the small amount of N2O5 present. Despite not knowing the source of oxidation, the results presented here, along with those shown for α-pinene-derived SOA (from O3) (Pospisilova et al., 2020; D'Ambro et al., 2018), demonstrate that SOA for both NO3 or O3 (two of the major oxidants in the atmosphere) steadily evolves in the dark without external stimuli.
Performing the experiments in a way that balances the radical chemistry, via the addition of HO2, would be ideal to replicate atmospheric chemistry accurately, depending on the region. The results presented here are clearly tilted toward RO2–RO2 reactions or RO2–NO3 reactions. The presence of HO2 radicals will promote the formation of monomer hydroperoxide-containing molecules over dimers, which will affect the ability to form lower-volatility dimers and increase the prevalence of hydroperoxide functional groups. The presence of hydroperoxide functional groups could promote particle-phase reactions if the peroxide groups are able to degrade and act as a source of radicals in the particle phase. Overall, the particle-phase processing of NO3-derived SOA is likely important regardless of the radical regime, as RO2 + RO2 dimer formation is always an important sink of RO2 radicals.
The overall effect of chemical aging of NO3-derived SOA in the dark will be towards less-volatile particles as a function of residence time in the atmosphere. The extent of the oxidation and aging in the particle phase could continue as long as there is a source of oxidant present. The reactions taking place in the particle phase result in changes of 25 %–30 % (lower estimate) of the composition of NO3-derived SOA. The particle-phase reactions slow/cease after 3 h. In the atmosphere, there will be continual production and partitioning of oxidation products, which could continue this process throughout the night.
The atmospheric consequence of these results is that we will typically overpredict the volatility of NO3-derived SOA when aging reactions are not included, underpredict its impact on the lifetime of SOA in the atmosphere, and underpredict the lifetime of organonitrates in the atmosphere. Investigating the detailed chemistry of NO3-derived SOA as a function of relative humidity would provide further insight to assess the potential role of the hydrolysis of nitrate functional groups as well as its impact on these particle-phase processes.
The datasets are available upon request from the corresponding authors. Chamber data are available at https://data.eurochamp.org/ (last access: 11 October 2022) via https://doi.org/10.25326/T8K0-6754 (Bell, 2019a), https://doi.org/10.25326/N0SR-GM67 (Bell, 2019b), and https://doi.org/10.25326/63RB-RT25 (Bell, 2019c).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-22-13167-2022-supplement.
DMB, CW, and CM designed the study. Chamber experiments were carried out by DMB, CW, ELG, AB, SG, and CM. Data analysis and interpretation were performed by DMB, CW, ELG, IR, JS, and CM. DMB wrote the manuscript with input from all the co-authors. All the co-authors read and commented on the manuscript.
At least one of the (co-)authors is a member of the editorial board of Atmospheric Chemistry and Physics. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the special issue “Simulation chambers as tools in atmospheric research (AMT/ACP/GMD inter-journal SI)”. It is not associated with a conference.
We would also like to thank Rene Richter for his assistance with installing and assembling the experimental setup.
This research has been supported by the the Swiss National Science Foundation (SNSF, grant nos. 200021_169787, 200020_172602, and 09802.01.03) and the Horizon 2020 EUROCHAMP-2020 Research and Innovation program through the Infrastructure Activity (grant no. 730997). Financial support from the European Union's Horizon 2020 research and innovation programme (project FORCeS under grant agreement no. 821205), European Research Council (Consolidator grant INTEGRATE no. 865799), and Knut and Alice Wallenberg foundation (Wallenberg Academy Fellowship projects AtmoRemove no. 2015.0162, AtmoCLOUD no. 2021.0169, and CLOUDFORM no. 2017.0165) is gratefully acknowledged.
The article processing charges for this open-access publication were covered by Stockholm University.
This paper was edited by Nga Lee Ng and reviewed by two anonymous referees.
Ayres, B. R., Allen, H. M., Draper, D. C., Brown, S. S., Wild, R. J., Jimenez, J. L., Day, D. A., Campuzano-Jost, P., Hu, W., de Gouw, J., Koss, A., Cohen, R. C., Duffey, K. C., Romer, P., Baumann, K., Edgerton, E., Takahama, S., Thornton, J. A., Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Wennberg, P. O., Nguyen, T. B., Teng, A., Goldstein, A. H., Olson, K., and Fry, J. L.: Organic nitrate aerosol formation via NO3 + biogenic volatile organic compounds in the southeastern United States, Atmos. Chem. Phys., 15, 13377–13392, https://doi.org/10.5194/acp-15-13377-2015, 2015.
Bates, K. H., Burke, G. J. P., Cope, J. D., and Nguyen, T. B.: Secondary organic aerosol and organic nitrogen yields from the nitrate radical (NO3) oxidation of alpha-pinene from various RO2 fates, Atmos. Chem. Phys., 22, 1467–1482, https://doi.org/10.5194/acp-22-1467-2022, 2022.
Bell, D.: Atmospheric simulation chamber study: alpha-pinene + NO3 − Aerosol study – particle formation, AERIS [data set], https://doi.org/10.25326/T8K0-6754, 2019a.
Bell, D.: Atmospheric simulation chamber study: alpha-pinene + NO3 − Aerosol study – particle formation, AERIS [data set], https://doi.org/10.25326/N0SR-GM67, 2019b.
Bell, D.: Atmospheric simulation chamber study: alpha-pinene + NO3 − Aerosol study – particle formation, AERIS [data set], https://doi.org/10.25326/63RB-RT25, 2019c.
Berndt, T., Mentler, B., Scholz, W., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Ozonolysis and OH Radical Reaction of α-Pinene: Mechanistic Insight and the Influence of Isoprene and Ethylene, Environ. Sci. Technol., 52, 11069–11077, https://doi.org/10.1021/acs.est.8b02210, 2018a.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Self- and Cross-Reactions of RO2 Radicals in the Atmosphere, Angew. Chem. Int. Edit., 57, 3820–3824, https://doi.org/10.1002/anie.201710989, 2018b.
Bertrand, A., Stefenelli, G., Pieber, S. M., Bruns, E. A., Temime-Roussel, B., Slowik, J. G., Wortham, H., Prévôt, A. S. H., El Haddad, I., and Marchand, N.: Influence of the vapor wall loss on the degradation rate constants in chamber experiments of levoglucosan and other biomass burning markers, Atmos. Chem. Phys., 18, 10915–10930, https://doi.org/10.5194/acp-18-10915-2018, 2018.
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Boyd, C. M., Sanchez, J., Xu, L., Eugene, A. J., Nah, T., Tuet, W. Y., Guzman, M. I., and Ng, N. L.: Secondary organic aerosol formation from the β-pinene+NO3 system: effect of humidity and peroxy radical fate, Atmos. Chem. Phys., 15, 7497–7522, https://doi.org/10.5194/acp-15-7497-2015, 2015.
Brown, S. S. and Stutz, J.: Nighttime radical observations and chemistry, Chem. Soc. Rev., 41, 6405–6447, https://doi.org/10.1039/C2CS35181A, 2012.
Chan, A. W. H., Chan, M. N., Surratt, J. D., Chhabra, P. S., Loza, C. L., Crounse, J. D., Yee, L. D., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Role of aldehyde chemistry and NOx concentrations in secondary organic aerosol formation, Atmos. Chem. Phys., 10, 7169–7188, https://doi.org/10.5194/acp-10-7169-2010, 2010.
Claflin, M. S. and Ziemann, P. J.: Identification and Quantitation of Aerosol Products of the Reaction of β-Pinene with NO3 Radicals and Implications for Gas- and Particle-Phase Reaction Mechanisms, J. Phys. Chem. A, 122, 3640–3652, https://doi.org/10.1021/acs.jpca.8b00692, 2018.
D'Ambro, E. L., Schobesberger, S., Zaveri, R. A., Shilling, J. E., Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., and Thornton, J. A.: Isothermal Evaporation of α-Pinene Ozonolysis SOA: Volatility, Phase State, and Oligomeric Composition, ACS Earth Space Chem., 2, 1058–1067, https://doi.org/10.1021/acsearthspacechem.8b00084, 2018.
Guenther, A., Geron, C., Pierce, T., Lamb, B., Harley, P., and Fall, R.: Natural emissions of non-methane volatile organic compounds, carbon monoxide, and oxides of nitrogen from North America, Atmos. Environ., 34, 2205–2230, https://doi.org/10.1016/S1352-2310(99)00465-3, 2000.
Heinritzi, M., Dada, L., Simon, M., Stolzenburg, D., Wagner, A. C., Fischer, L., Ahonen, L. R., Amanatidis, S., Baalbaki, R., Baccarini, A., Bauer, P. S., Baumgartner, B., Bianchi, F., Brilke, S., Chen, D., Chiu, R., Dias, A., Dommen, J., Duplissy, J., Finkenzeller, H., Frege, C., Fuchs, C., Garmash, O., Gordon, H., Granzin, M., El Haddad, I., He, X., Helm, J., Hofbauer, V., Hoyle, C. R., Kangasluoma, J., Keber, T., Kim, C., Kürten, A., Lamkaddam, H., Laurila, T. M., Lampilahti, J., Lee, C. P., Lehtipalo, K., Leiminger, M., Mai, H., Makhmutov, V., Manninen, H. E., Marten, R., Mathot, S., Mauldin, R. L., Mentler, B., Molteni, U., Müller, T., Nie, W., Nieminen, T., Onnela, A., Partoll, E., Passananti, M., Petäjä, T., Pfeifer, J., Pospisilova, V., Quéléver, L. L. J., Rissanen, M. P., Rose, C., Schobesberger, S., Scholz, W., Scholze, K., Sipilä, M., Steiner, G., Stozhkov, Y., Tauber, C., Tham, Y. J., Vazquez-Pufleau, M., Virtanen, A., Vogel, A. L., Volkamer, R., Wagner, R., Wang, M., Weitz, L., Wimmer, D., Xiao, M., Yan, C., Ye, P., Zha, Q., Zhou, X., Amorim, A., Baltensperger, U., Hansel, A., Kulmala, M., Tomé, A., Winkler, P. M., Worsnop, D. R., Donahue, N. M., Kirkby, J., and Curtius, J.: Molecular understanding of the suppression of new-particle formation by isoprene, Atmos. Chem. Phys., 20, 11809–11821, https://doi.org/10.5194/acp-20-11809-2020, 2020.
Huang, W., Saathoff, H., Shen, X., Ramisetty, R., Leisner, T., and Mohr, C.: Chemical Characterization of Highly Functionalized Organonitrates Contributing to Night-Time Organic Aerosol Mass Loadings and Particle Growth, Environ. Sci. Technol., 53, 1165–1174, https://doi.org/10.1021/acs.est.8b05826, 2019.
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of organic aerosols in the atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Kiendler-Scharr, A., Mensah, A. A., Friese, E., Topping, D., Nemitz, E., Prevot, A. S. H., Äijälä, M., Allan, J., Canonaco, F., Canagaratna, M., Carbone, S., Crippa, M., Dall Osto, M., Day, D. A., De Carlo, P., Di Marco, C. F., Elbern, H., Eriksson, A., Freney, E., Hao, L., Herrmann, H., Hildebrandt, L., Hillamo, R., Jimenez, J. L., Laaksonen, A., McFiggans, G., Mohr, C., O'Dowd, C., Otjes, R., Ovadnevaite, J., Pandis, S. N., Poulain, L., Schlag, P., Sellegri, K., Swietlicki, E., Tiitta, P., Vermeulen, A., Wahner, A., Worsnop, D., and Wu, H. C.: Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol, Geophys. Res. Lett., 43, 7735–7744, https://doi.org/10.1002/2016GL069239, 2016.
Krechmer, J. E., Day, D. A., and Jimenez, J. L.: Always Lost but Never Forgotten: Gas-Phase Wall Losses Are Important in All Teflon Environmental Chambers, Environ. Sci. Technol., 54, 12890–12897, https://doi.org/10.1021/acs.est.0c03381, 2020.
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurtén, T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., Ambro, E. L., and Thornton, J. A.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci. USA, 113, 1516, https://doi.org/10.1073/pnas.1508108113, 2016.
Lee, C. P., Surdu, M., Bell, D. M., Lamkaddam, H., Wang, M., Ataei, F., Hofbauer, V., Lopez, B., Donahue, N. M., Dommen, J., Prevot, A. S. H., Slowik, J. G., Wang, D., Baltensperger, U., and El Haddad, I.: Effects of aerosol size and coating thickness on the molecular detection using extractive electrospray ionization, Atmos. Meas. Tech., 14, 5913–5923, https://doi.org/10.5194/amt-14-5913-2021, 2021.
Liu, X., Day, D. A., Krechmer, J. E., Brown, W., Peng, Z., Ziemann, P. J., and Jimenez, J. L.: Direct measurements of semi-volatile organic compound dynamics show near-unity mass accommodation coefficients for diverse aerosols, Commun. Chem., 2, 98, https://doi.org/10.1038/s42004-019-0200-x, 2019.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Lutz, A., Hallquist, M., Worsnop, D., and Thornton, J. A.: A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 7, 983–1001, https://doi.org/10.5194/amt-7-983-2014, 2014.
Lopez-Hilfiker, F. D., Pospisilova, V., Huang, W., Kalberer, M., Mohr, C., Stefenelli, G., Thornton, J. A., Baltensperger, U., Prevot, A. S. H., and Slowik, J. G.: An extractive electrospray ionization time-of-flight mass spectrometer (EESI-TOF) for online measurement of atmospheric aerosol particles, Atmos. Meas. Tech., 12, 4867–4886, https://doi.org/10.5194/amt-12-4867-2019, 2019.
Molteni, U., Bianchi, F., Klein, F., El Haddad, I., Frege, C., Rossi, M. J., Dommen, J., and Baltensperger, U.: Formation of highly oxygenated organic molecules from aromatic compounds, Atmos. Chem. Phys., 18, 1909–1921, https://doi.org/10.5194/acp-18-1909-2018, 2018.
Molteni, U., Simon, M., Heinritzi, M., Hoyle, C. R., Bernhammer, A.-K., Bianchi, F., Breitenlechner, M., Brilke, S., Dias, A., Duplissy, J., Frege, C., Gordon, H., Heyn, C., Jokinen, T., Kürten, A., Lehtipalo, K., Makhmutov, V., Petäjä, T., Pieber, S. M., Praplan, A. P., Schobesberger, S., Steiner, G., Stozhkov, Y., Tomé, A., Tröstl, J., Wagner, A. C., Wagner, R., Williamson, C., Yan, C., Baltensperger, U., Curtius, J., Donahue, N. M., Hansel, A., Kirkby, J., Kulmala, M., Worsnop, D. R., and Dommen, J.: Formation of highly oxygenated organic molecules from α-pinene ozonolysis: chemical characteristics, mechanism, and kinetic model development, ACS Earth Space Chem., 3, 873–883, https://doi.org/10.1021/acsearthspacechem.9b00035, 2019.
Nah, T., Sanchez, J., Boyd, C. M., and Ng, N. L.: Photochemical Aging of α-pinene and β-pinene Secondary Organic Aerosol formed from Nitrate Radical Oxidation, Environ. Sci. Technol., 50, 222–231, https://doi.org/10.1021/acs.est.5b04594, 2016.
Ng, N. L., Kwan, A. J., Surratt, J. D., Chan, A. W. H., Chhabra, P. S., Sorooshian, A., Pye, H. O. T., Crounse, J. D., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol (SOA) formation from reaction of isoprene with nitrate radicals (NO3), Atmos. Chem. Phys., 8, 4117–4140, https://doi.org/10.5194/acp-8-4117-2008, 2008.
Platt, S. M., El Haddad, I., Zardini, A. A., Clairotte, M., Astorga, C., Wolf, R., Slowik, J. G., Temime-Roussel, B., Marchand, N., Ježek, I., Drinovec, L., Močnik, G., Möhler, O., Richter, R., Barmet, P., Bianchi, F., Baltensperger, U., and Prévôt, A. S. H.: Secondary organic aerosol formation from gasoline vehicle emissions in a new mobile environmental reaction chamber, Atmos. Chem. Phys., 13, 9141–9158, https://doi.org/10.5194/acp-13-9141-2013, 2013.
Pospisilova, V., Lopez-Hilfiker, F. D., Bell, D. M., El Haddad, I., Mohr, C., Huang, W., Heikkinen, L., Xiao, M., Dommen, J., Prevot, A. S. H., Baltensperger, U., and Slowik, J. G.: On the fate of oxygenated organic molecules in atmospheric aerosol particles, Sci. Adv., 6, eaax8922, https://doi.org/10.1126/sciadv.aax8922, 2020.
Pye, H. O. T., Chan, A. W. H., Barkley, M. P., and Seinfeld, J. H.: Global modeling of organic aerosol: the importance of reactive nitrogen (NOx and NO3), Atmos. Chem. Phys., 10, 11261–11276, https://doi.org/10.5194/acp-10-11261-2010, 2010.
Pye, H. O. T., Luecken, D. J., Xu, L., Boyd, C. M., Ng, N. L., Baker, K. R., Ayres, B. R., Bash, J. O., Baumann, K., Carter, W. P. L., Edgerton, E., Fry, J. L., Hutzell, W. T., Schwede, D. B., and Shepson, P. B.: Modeling the Current and Future Roles of Particulate Organic Nitrates in the Southeastern United States, Environ. Sci. Technol., 49, 14195–14203, https://doi.org/10.1021/acs.est.5b03738, 2015.
Qi, L., Chen, M., Stefenelli, G., Pospisilova, V., Tong, Y., Bertrand, A., Hueglin, C., Ge, X., Baltensperger, U., Prévôt, A. S. H., and Slowik, J. G.: Organic aerosol source apportionment in Zurich using an extractive electrospray ionization time-of-flight mass spectrometer (EESI-TOF-MS) – Part 2: Biomass burning influences in winter, Atmos. Chem. Phys., 19, 8037–8062, https://doi.org/10.5194/acp-19-8037-2019, 2019.
Rissanen, M. P., Kurtén, T., Sipilä, M., Thornton, J. A., Kausiala, O., Garmash, O., Kjaergaard, H. G., Petäjä, T., Worsnop, D. R., Ehn, M., and Kulmala, M.: Effects of Chemical Complexity on the Autoxidation Mechanisms of Endocyclic Alkene Ozonolysis Products: From Methylcyclohexenes toward Understanding α-Pinene, J. Phys. Chem. A, 119, 4633–4650, https://doi.org/10.1021/jp510966g, 2015.
Riva, M., Heikkinen, L., Bell, D. M., Peräkylä, O., Zha, Q., Schallhart, S., Rissanen, M. P., Imre, D., Petäjä, T., Thornton, J. A., Zelenyuk, A., and Ehn, M.: Chemical transformations in monoterpene-derived organic aerosol enhanced by inorganic composition, npj Clim. Atmos. Sci., 2, 2, https://doi.org/10.1038/s41612-018-0058-0, 2019.
Romonosky, D. E., Li, Y., Shiraiwa, M., Laskin, A., Laskin, J., and Nizkorodov, S. A.: Aqueous Photochemistry of Secondary Organic Aerosol of α-Pinene and α-Humulene Oxidized with Ozone, Hydroxyl Radical, and Nitrate Radical, J. Phys. Chem. A, 121, 1298–1309, https://doi.org/10.1021/acs.jpca.6b10900, 2017.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Simon, M., Dada, L., Heinritzi, M., Scholz, W., Stolzenburg, D., Fischer, L., Wagner, A. C., Kürten, A., Rörup, B., He, X.-C., Almeida, J., Baalbaki, R., Baccarini, A., Bauer, P. S., Beck, L., Bergen, A., Bianchi, F., Bräkling, S., Brilke, S., Caudillo, L., Chen, D., Chu, B., Dias, A., Draper, D. C., Duplissy, J., El-Haddad, I., Finkenzeller, H., Frege, C., Gonzalez-Carracedo, L., Gordon, H., Granzin, M., Hakala, J., Hofbauer, V., Hoyle, C. R., Kim, C., Kong, W., Lamkaddam, H., Lee, C. P., Lehtipalo, K., Leiminger, M., Mai, H., Manninen, H. E., Marie, G., Marten, R., Mentler, B., Molteni, U., Nichman, L., Nie, W., Ojdanic, A., Onnela, A., Partoll, E., Petäjä, T., Pfeifer, J., Philippov, M., Quéléver, L. L. J., Ranjithkumar, A., Rissanen, M. P., Schallhart, S., Schobesberger, S., Schuchmann, S., Shen, J., Sipilä, M., Steiner, G., Stozhkov, Y., Tauber, C., Tham, Y. J., Tomé, A. R., Vazquez-Pufleau, M., Vogel, A. L., Wagner, R., Wang, M., Wang, D. S., Wang, Y., Weber, S. K., Wu, Y., Xiao, M., Yan, C., Ye, P., Ye, Q., Zauner-Wieczorek, M., Zhou, X., Baltensperger, U., Dommen, J., Flagan, R. C., Hansel, A., Kulmala, M., Volkamer, R., Winkler, P. M., Worsnop, D. R., Donahue, N. M., Kirkby, J., and Curtius, J.: Molecular understanding of new-particle formation from α-pinene between −50 and +25 °C, Atmos. Chem. Phys., 20, 9183–9207, https://doi.org/10.5194/acp-20-9183-2020, 2020.
Stefenelli, G., Pospisilova, V., Lopez-Hilfiker, F. D., Daellenbach, K. R., Hüglin, C., Tong, Y., Baltensperger, U., Prévôt, A. S. H., and Slowik, J. G.: Organic aerosol source apportionment in Zurich using an extractive electrospray ionization time-of-flight mass spectrometer (EESI-TOF-MS) – Part 1: Biogenic influences and day–night chemistry in summer, Atmos. Chem. Phys., 19, 14825–14848, https://doi.org/10.5194/acp-19-14825-2019, 2019.
Surdu, M., Pospisilova, V., Xiao, M., Wang, M., Mentler, B., Simon, M., Stolzenburg, D., Hoyle, C. R., Bell, D. M., Lee, C. P., Lamkaddam, H., Lopez-Hilfiker, F., Ahonen, L. R., Amorim, A., Baccarini, A., Chen, D., Dada, L., Duplissy, J., Finkenzeller, H., He, X.-C., Hofbauer, V., Kim, C., Kürten, A., Kvashnin, A., Lehtipalo, K., Makhmutov, V., Molteni, U., Nie, W., Onnela, A., Petäjä, T., Quéléver, L. L. J., Tauber, C., Tomé, A., Wagner, R., Yan, C., Prevot, A. S. H., Dommen, J., Donahue, N. M., Hansel, A., Curtius, J., Winkler, P. M., Kulmala, M., Volkamer, R., Flagan, R. C., Kirkby, J., Worsnop, D. R., Slowik, J. G., Wang, D. S., Baltensperger, U., and Haddad, I. E.: Molecular characterization of ultrafine particles using extractive electrospray time-of-flight mass spectrometry, Environ. Sci. Atmos., 1, 434–448, https://doi.org/10.1039/D1EA00050K, 2021.
Takeuchi, M. and Ng, N. L.: Chemical composition and hydrolysis of organic nitrate aerosol formed from hydroxyl and nitrate radical oxidation of α-pinene and β-pinene, Atmos. Chem. Phys., 19, 12749–12766, https://doi.org/10.5194/acp-19-12749-2019, 2019.
Wang, D. S., Lee, C. P., Krechmer, J. E., Majluf, F., Tong, Y., Canagaratna, M. R., Schmale, J., Prévôt, A. S. H., Baltensperger, U., Dommen, J., El Haddad, I., Slowik, J. G., and Bell, D. M.: Constraining the response factors of an extractive electrospray ionization mass spectrometer for near-molecular aerosol speciation, Atmos. Meas. Tech., 14, 6955–6972, https://doi.org/10.5194/amt-14-6955-2021, 2021.
Wolfe, G. M., Marvin, M. R., Roberts, S. J., Travis, K. R., and Liao, J.: The Framework for 0-D Atmospheric Modeling (F0AM) v3.1, Geosci. Model Dev., 9, 3309–3319, https://doi.org/10.5194/gmd-9-3309-2016, 2016.
Wu, C., Bell, D. M., Graham, E. L., Haslett, S., Riipinen, I., Baltensperger, U., Bertrand, A., Giannoukos, S., Schoonbaert, J., El Haddad, I., Prevot, A. S. H., Huang, W., and Mohr, C.: Photolytically induced changes in composition and volatility of biogenic secondary organic aerosol from nitrate radical oxidation during night-to-day transition, Atmos. Chem. Phys., 21, 14907–14925, https://doi.org/10.5194/acp-21-14907-2021, 2021.
Xu, L., Kollman, M. S., Song, C., Shilling, J. E., and Ng, N. L.: Effects of NOx on the Volatility of Secondary Organic Aerosol from Isoprene Photooxidation, Environ. Sci. Technol., 48, 2253–2262, https://doi.org/10.1021/es404842g, 2014.
Xu, L., Suresh, S., Guo, H., Weber, R. J., and Ng, N. L.: Aerosol characterization over the southeastern United States using high-resolution aerosol mass spectrometry: spatial and seasonal variation of aerosol composition and sources with a focus on organic nitrates, Atmos. Chem. Phys., 15, 7307–7336, https://doi.org/10.5194/acp-15-7307-2015, 2015.
Yan, C., Nie, W., Äijälä, M., Rissanen, M. P., Canagaratna, M. R., Massoli, P., Junninen, H., Jokinen, T., Sarnela, N., Häme, S. A. K., Schobesberger, S., Canonaco, F., Yao, L., Prévôt, A. S. H., Petäjä, T., Kulmala, M., Sipilä, M., Worsnop, D. R., and Ehn, M.: Source characterization of highly oxidized multifunctional compounds in a boreal forest environment using positive matrix factorization, Atmos. Chem. Phys., 16, 12715–12731, https://doi.org/10.5194/acp-16-12715-2016, 2016.
Zhao, D., Pullinen, I., Fuchs, H., Schrade, S., Wu, R., Acir, I.-H., Tillmann, R., Rohrer, F., Wildt, J., Guo, Y., Kiendler-Scharr, A., Wahner, A., Kang, S., Vereecken, L., and Mentel, T. F.: Highly oxygenated organic molecule (HOM) formation in the isoprene oxidation by NO3 radical, Atmos. Chem. Phys., 21, 9681–9704, https://doi.org/10.5194/acp-21-9681-2021, 2021.
Zhao, Y., Thornton, J. A., and Pye, H. O. T.: Quantitative constraints on autoxidation and dimer formation from direct probing of monoterpene-derived peroxy radical chemistry, P. Natl. Acad. Sci. USA, 115, 12142, https://doi.org/10.1073/pnas.1812147115, 2018.
Ziemann, P. J. and Atkinson, R.: Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 41, 6582–6605, https://doi.org/10.1039/C2CS35122F, 2012.