the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 21 Jan 2022
| 21 Jan 2022
Atmospheric measurements at Mt. Tai – Part II: HONO budget and radical (ROx + NO3) chemistry in the lower boundary layer
Chaoyang Xue
Jörg Kleffmann
Wenjin Zhang
Xiaowei He
Pengfei Liu
Chenglong Zhang
Xiaoxi Zhao
Chengtang Liu
Zhuobiao Ma
Junfeng Liu
Jinhe Wang
Keding Lu
Valéry Catoire
Abdelwahid Mellouki
In the summer of 2018, a comprehensive field campaign, with measurements on HONO and related parameters, was conducted at the foot (150 m a.s.l.) and the summit of Mt. Tai (1534 m a.s.l.) in the central North China Plain (NCP). With the implementation of a 0-D box model, the HONO budget with six additional sources and its role in radical chemistry at the foot station were explored. We found that the model default source, NO + OH, could only reproduce 13 % of the observed HONO, leading to a strong unknown source strength of up to 3 ppbv h−1. Among the additional sources, the NO2 uptake on the ground surface dominated (∼ 70 %) nighttime HONO formation, and its photo-enhanced reaction dominated (∼ 80 %) daytime HONO formation. Their contributions were sensitive to the mixing layer height (MLH) used for the parameterizations, highlighting the importance of a reasonable MLH for exploring ground-level HONO formation in 0-D models and the necessity of gradient measurements. A ΔHONONOx ratio of 0.7 % for direct emissions from vehicle exhaust was inferred, and a new method to quantify its contribution to the observations was proposed and discussed. Aerosol-derived sources, including the NO2 uptake on the aerosol surface and the particulate nitrate photolysis, did not lead to significant HONO formation, with their contributions lower than NO + OH.
HONO photolysis in the early morning initialized the daytime photochemistry at the foot station. It was also a substantial radical source throughout the daytime, with contributions higher than O3 photolysis to OH initiation. Moreover, we found that OH dominated the atmospheric oxidizing capacity in the daytime, while modeled NO3 appeared to be significant at night. Peaks of modeled NO3 time series and average diurnal variation reached 22 and 9 pptv, respectively. NO3-induced reactions contribute 18 % of nitrate formation potential (P(HNO3)) and 11 % of the isoprene (C5H8) oxidation throughout the whole day. At night, NO3 chemistry led to 51 % and 44 % of P(HNO3) or the C5H8 oxidation, respectively, implying that NO3 chemistry could significantly affect nighttime secondary organic and inorganic aerosol formation in this high-O3 region. Considering the severe O3 pollution in the NCP and the very limited NO3 measurements, we suggest that besides direct measurements of HOx and primary HOx precursors (O3, HONO, alkenes, etc.), NO3 measurements should be conducted to understand the atmospheric oxidizing capacity and air pollution formation in this and similar regions.
- Article
(6653 KB) - Full-text XML
- Companion paper
-
Supplement
(1142 KB) - BibTeX
- EndNote




Numerous field campaigns coupled with model simulations have been conducted worldwide to understand summertime atmospheric chemistry as it is linked to the regional air quality and global climate (Alicke et al., 2003; Elshorbany et al., 2012; Heard et al., 2004; Kanaya et al., 2009, 2013; Michoud et al., 2012; Ren et al., 2003; Rohrer and Berresheim, 2006; Tan et al., 2017; Travis et al., 2020). One of the key issues is the level and the production–loss paths of the atmospheric oxidizing capacity governed by radicals (OH, NO3, etc.). This is also essential in converting primary to secondary pollutants and the removal of greenhouse gases (Lu et al., 2019; Seinfeld and Pandis, 2016).
On a global scale, OH controls atmospheric oxidation. As the detergent in the troposphere, OH can oxidize most trace gases, including inorganic (SO2, NO2, etc.) and organic compounds (VOCs, etc.), and determines the lifetime of greenhouse gases (e.g., CH4). In addition, the fast conversion of HO2 to OH (Reaction R1) as part of the radical propagation cycle, primary OH (radical initiation) mainly originates from photolysis reactions, including O3 (Reactions R2 to R4), HONO (Reaction R5), HCHO (Reactions R6 to R9, and R1), and H2O2 (Reaction R10), and the ozonolysis of alkenes (not shown in detail here). In particular, HONO photolysis is reported to be an important or even the major OH source in the lower atmosphere of polluted regions, with a contribution of 20 %–90 % (Alicke et al., 2003; Elshorbany et al., 2009; Kleffmann et al., 2005; Platt et al., 1980; Slater et al., 2020; Whalley et al., 2021; Xue et al., 2020). However, this process still needs more global quantification due to the incomplete understanding of HONO formation and its vertical distribution in the atmosphere (Kleffmann, 2007). A state-of-art summary of the reported HONO sources can be found in our recent study (Xue et al., 2020).
In addition, other oxidants can also be of importance on a regional scale. For example, the NO3 radical could be a major oxidant in forests (vegetation shadows slow down its photolysis) or in the nocturnal boundary layer at high-O3 regions (Brown and Stutz, 2012). Formed by the reaction of NO2 + O3 (Reaction R11), high NO3 levels usually occur at night, due to its very rapid photolysis during the daytime (Reactions R12 and R13). Moreover, high NO3 concentrations are only observed for high O3 and medium NOx concentrations in the absence of significant levels of NO caused by Reaction (R15). Like OH, NO3 also has high reactivity with various trace gases (Brown and Stutz, 2012; Mellouki et al., 2021). For example, NO3 reacts with NO2 to form N2O5 (Reaction R14), which can undergo hydrolysis on wet surfaces or clouds to produce HNO3 (or NO) (Reaction R16) or decomposition back to NO3 + NO2 (Reaction R17). NO3 can also react with various organic compounds to form secondary organic aerosol (SOA). For instance, NO3 reacts with isoprene (C5H8), leading to significant organic nitrate (e.g., alkyl nitrate) production (Rollins et al., 2009).
In the past decade, particle pollution, such as PM2.5, has been going down, while O3 pollution is increasing in many cities of China (Han et al., 2020; Li et al., 2019; Sun et al., 2016, 2019). Especially in the North China Plain (NCP), air pollution has become a major environmental risk for the public health of >330 million people living in this region. This raises efforts in exploring the NOx–VOC–O3 chemistry. Meanwhile, high O3 indicates an enhanced atmospheric oxidizing capacity; that is, elevated OH and NO3 levels are expected. However, very few observations have been reported of OH and NO3 levels, as well as their production (e.g., HONO photolysis or NO2 + O3) or loss (e.g., to oxidize primary pollutants) in the high-O3 region of the NCP so far (Lu et al., 2019; Suhail et al., 2019). Herein, we provide the first HONO measurements at the foot of Mt. Tai (in Tai'an, a typical urban site), followed by measurements at the summit of Mt. Tai in the summer of 2018. Data from the summit station are presented in the companion paper, in which daytime HONO formation and its role in the atmospheric oxidizing capacity at the summit level are studied. In this paper, coupled with the box model, the HONO budget and the radical chemistry at the ground level are explored and discussed.
2.1 Field campaign
2.1.1 Measurement sites
In the summer (from late May to July) of 2018, a comprehensive field campaign was conducted to understand the atmospheric oxidizing capacity and O3 pollution in Tai'an, a city in the middle of the NCP. Tai'an is located nearly in the middle between Beijing and Shanghai. The city has a population of about 5.6 million and is about 60 km south of Jinan (the capital city of Shandong province, population: ∼ 8.7 million). Measurements were conducted both at the ground level (the foot of Mt. Tai, 150 m a.s.l.) and the summit level (the summit of Mt. Tai, 1534 m a.s.l.; 36.23∘ N, 117.11∘ E). The foot station was inside Shandong College of Electric Power (36.18∘ N, 117.11∘ E) in the Taishan district of Tai'an. There are no industrial activities around this site, which is surrounded by the campus, a residential area, and a business district. The 801st province road is in the northeast of this typical urban site. Inside the campus (about 50 ha), frequent traffic was not observed, but it sometimes occurred on the urban roads nearby. Mt. Tai is located in the north part of Tai'an. Locations of these two stations on the map can be found in the companion ACP paper (entitled “Atmospheric measurements at Mt. Tai – Part I: HONO formation and its role in the oxidizing capacity of the upper boundary layer”; Xue et al., 2021a).
Measurements at these two stations allow us to study HONO formation and its role in the atmospheric oxidizing capacity of the lower (the foot study) and the upper boundary layer (the summit study). Briefly, in the summit study, we found remarkably high daytime HONO levels as well as high unknown HONO source strength, which was mainly caused by rapid vertical transport from the ground to the summit levels driven by mountain winds.
2.1.2 Instrumentation
HONO mixing ratios were continuously measured by the LOPAP technique (LOPAP-03, QUMA GmbH, Germany) (Heland et al., 2001; Kleffmann et al., 2006) from 29 May to 8 July 2018 at the foot station, followed by measurements at the summit station from 9 to 31 July 2017. At the foot station, NO–NO2–NOx, O3, CO, and SO2 were measured online by a series of Thermo Fisher Scientific instruments (42i, 49i, 48i, and 43i, respectively). Because chemiluminescence techniques using molybdenum converters were reported to overestimate the NO2 level caused by interferences of other NOy species, the measured NO2 was corrected with a family constraint in a model run (see Sect. 3.1.2). VOCs (56 species) and OVOCs (15 species) were measured by a homemade GC-FID (gas chromatography–flame ionization detector) instrument (Liu et al., 2016) and the USEPA DNPH-HPLC (high-performance liquid chromatography) method (Wang et al., 2020), respectively. Gas-phase H2O2 was measured by a monitor based on the wet chemical method (AL2021, Aerolaser GmbH, Germany), details of which can be found in Ye et al. (2018a). Water-soluble inorganic particle species (i.e., NO, SO, Cl−, Na+, K+, Ca2+, etc.) of PM2.5 were collected on Teflon filters every 2 h at a sampling flow of 100 L min−1 and analyzed by an ion chromatograph (Liu et al., 2020).
Meteorological parameters, including atmospheric temperature (T), pressure (p), relative humidity (RH), wind direction (WD), wind speed (WS), and solar irradiance (Ra), were continuously measured by an automated meteorological station. J(NO2) was measured by a 4π filter radiometer (Metcon GmbH, Germany). For the analysis of time series and statistic descriptions of the data, 10 min average data (except for PM2.5) were used. In contrast, hourly data were used for model simulations. PM2.5 measurements were obtained from the Tai'an monitoring station (200 m east of the foot station), and only hourly average data were available. Other J values used in this study, including J(HONO), J(O(1D)), J(H2O2), J(HCHO)rad, and J(HNO3), are calculated by the box model based on trigonometric SZA (solar zenith angle) functions (MCM default photolysis frequency calculation; see Jenkin et al., 1997) and scaled by the measured J(NO2). For instance, J(HONO) = J(HONO)model×J(NO2)(NO2)model.
2.2 Model description
2.2.1 Box model and constraints
The Framework for 0-D Atmospheric Modeling, F0AM v4.0 (available at https://github.com/AirChem/F0AM, last access: 1 May 2021) developed by Wolfe et al. (2016) was used to explore the HONO budget and the radical chemistry. The MCM v3.3.1 Chemical Mechanism was obtained from http://mcm.york.ac.uk/home.htt (last access: 1 May 2021). Note that the present F0AM model could also be run with family constraints (see details in Wolfe et al., 2016), such as the NOy and Cly families. Hence, it allows us to correct for interferences in the NO2 measurements using the chemiluminescence method (see Sect. 3.1.2).
The model was constrained by the measured J(NO2), T, RH, P, VOCs, OVOCs, and all the other measured inorganic species, including the corrected NO2 by the family constraint. Continuous VOC measurements were available from 12 June to 6 July, and hence box model simulations were performed during this period. While J(NO2) measurement was available from 16 June, J(NO2) from 12 to 16 June was estimated through the high quadratic correlation (R2=0.96, Fig. S1) between J(NO2) and solar irradiance.
2.2.2 Model scenarios
Table 1 shows the description of different model scenarios. A base case (Sce-0) with all the measured parameters as constraints was run to simulate radical concentrations and their production–loss rates. The family constraint was used in this scenario to correct for interferences of NO2 measurements (Sect. 3.1.2). Meanwhile, the role of HONO in radical chemistry was also explored by several model sensitivity tests that involved reducing or increasing the constrained HONO.
With the simulated OH and the corrected NO2 from the base case, we could further explore the HONO budget. Three model scenarios were conducted to assess the potential contributions of different HONO sources, including one with only the default model source (Sce-1) and one with all the six additional sources, including direct emission, dark and photo-enhanced NO2 uptake on aerosol and ground surfaces and nitrate photolysis (Sce-2). In Sce-3, photo-enhanced NO2 uptake on the ground surface was reduced by a factor of 10, and aerosol-derived sources (NO2 uptake on the aerosol surface or particulate nitrate photolysis) were significantly enhanced (see Sect. 3.2.3) to test whether the aerosol-derived sources could explain the observations well.
3.1 Overview of the measurements and potential interferences
3.1.1 Overview of the measurements
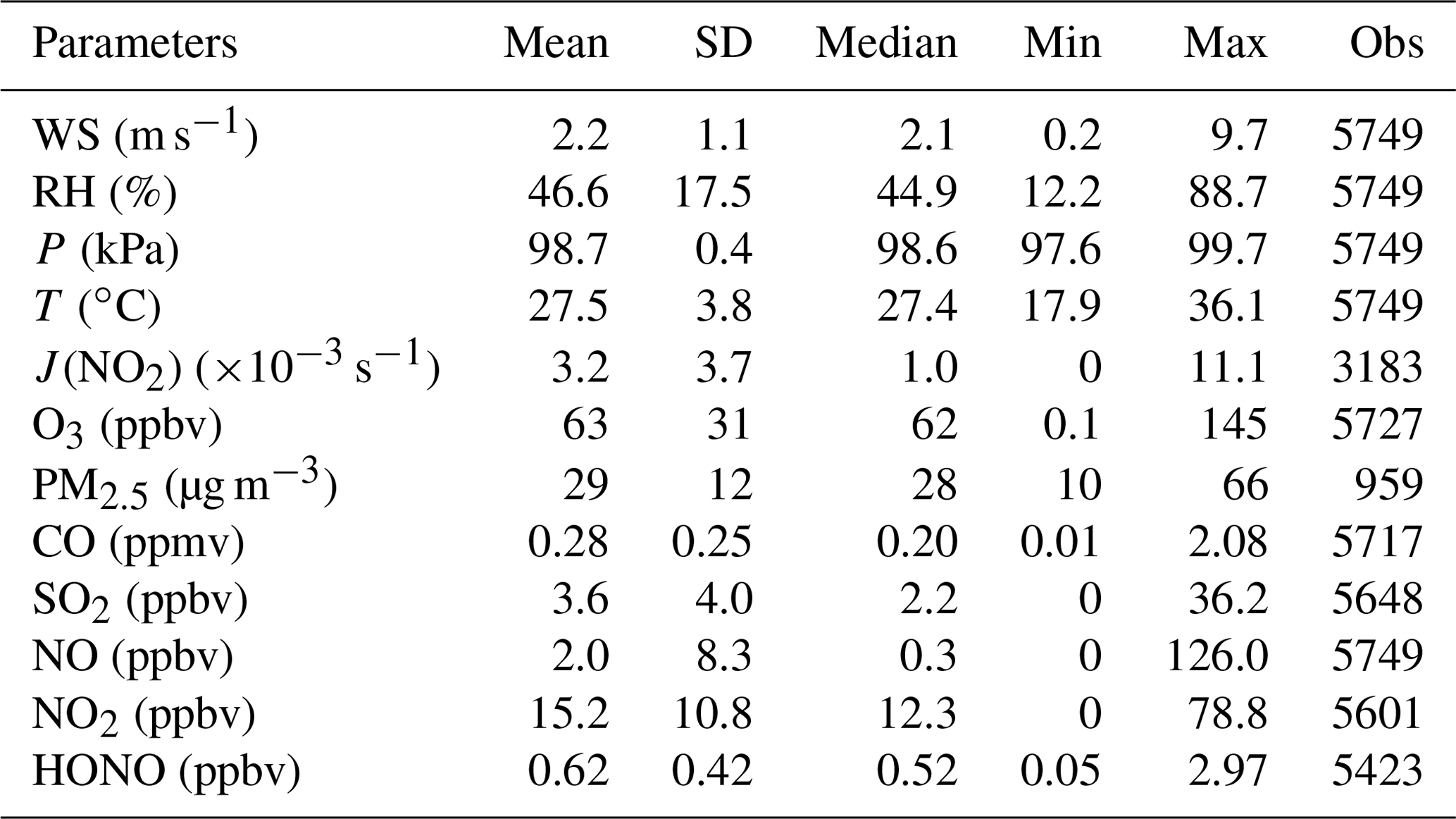
Figure 1 shows the meteorological parameters measured at the foot station. During the campaign, it was generally sunny except for slight rain (<10 mm) on 9, 10, 13, and 28 June and heavy rain (∼ 100 mm) in the night of 25/26 June. Ambient temperature was normally around 25 ∘C at night and around 30 ∘C during the daytime, except for rainy days when the temperature was relatively low. The relative humidity (RH) was high (up to 80 %) on those rainy or cloudy days and low (around 40 %) on other days. Campaign-averaged temperature and RH were 27.5 ∘C and 46.6 %, respectively (Table 2). Air masses observed at this site originated from multiple directions, including west, south, and east, which are shown in the wind rose plot in Fig. S2a. Wind speed was generally low, with an average of about 2 m s−1. In addition, the wind rose results generally agree well with HYSPLIT trajectory results in Fig. S2b and c.

Table 2Statistic summary of meteorological parameters and the measured species. Note that the observation data point number (Obs) of hourly PM2.5 is about of others (10 min time resolution). The measured rather than the corrected NO2 was used here. SD: standard deviation. Min: minimum. Max: maximum.
In Figs. 2 and 3 the measured HONO and related species at the foot station and their diurnal variation profiles are presented, respectively. The measured HONO showed a typical diurnal variation with accumulation after sunset and decay after sunrise. Mixing ratios of the measured HONO varied from 0.05 to about 3 ppbv, with an average of 0.62 ± 0.42 ppbv. The measured NO2 showed a very similar variation to HONO, and their correlation was high (R=0.73), indicating a potential role of NO2 in HONO formation. Severe O3 pollution (maximum: 145 ppbv) was observed at this site, with O3 levels frequently exceeding the Class-1 limit value (1 h 160 µg m−3, equivalent to 82 ppbv at 298 K and 101 kPa) of the National Ambient Air Quality Standard of China (GB3095-2012), while the NOx level was typically lower than O3. Consequently, a relatively low NO was frequently found, whose concentration was generally lower than 1 ppbv, except for some fresh plumes that contained higher NO concentrations. The two primary pollutants, CO and SO2, were generally lower than 0.5 ppmv and 5 ppbv, respectively, except for several polluted events, within which CO and SO2 reached around 2 ppmv and around 35 ppbv, respectively. However, all the primary pollutants, including NO, CO, and SO2, showed poor correlations with HONO (R=0.49, 0.44, and 0.13, respectively), implying the minor role of direct emission in HONO formation. The measured hourly PM2.5 varied from 10 to 66 µg m−3, with an average of 29 µg m−3. The correlation of PM2.5 and HONO was very low (R=0.06), suggesting a minor role of aerosol-derived sources in HONO formation. More discussion on the HONO budget is presented in Sect. 3.2.
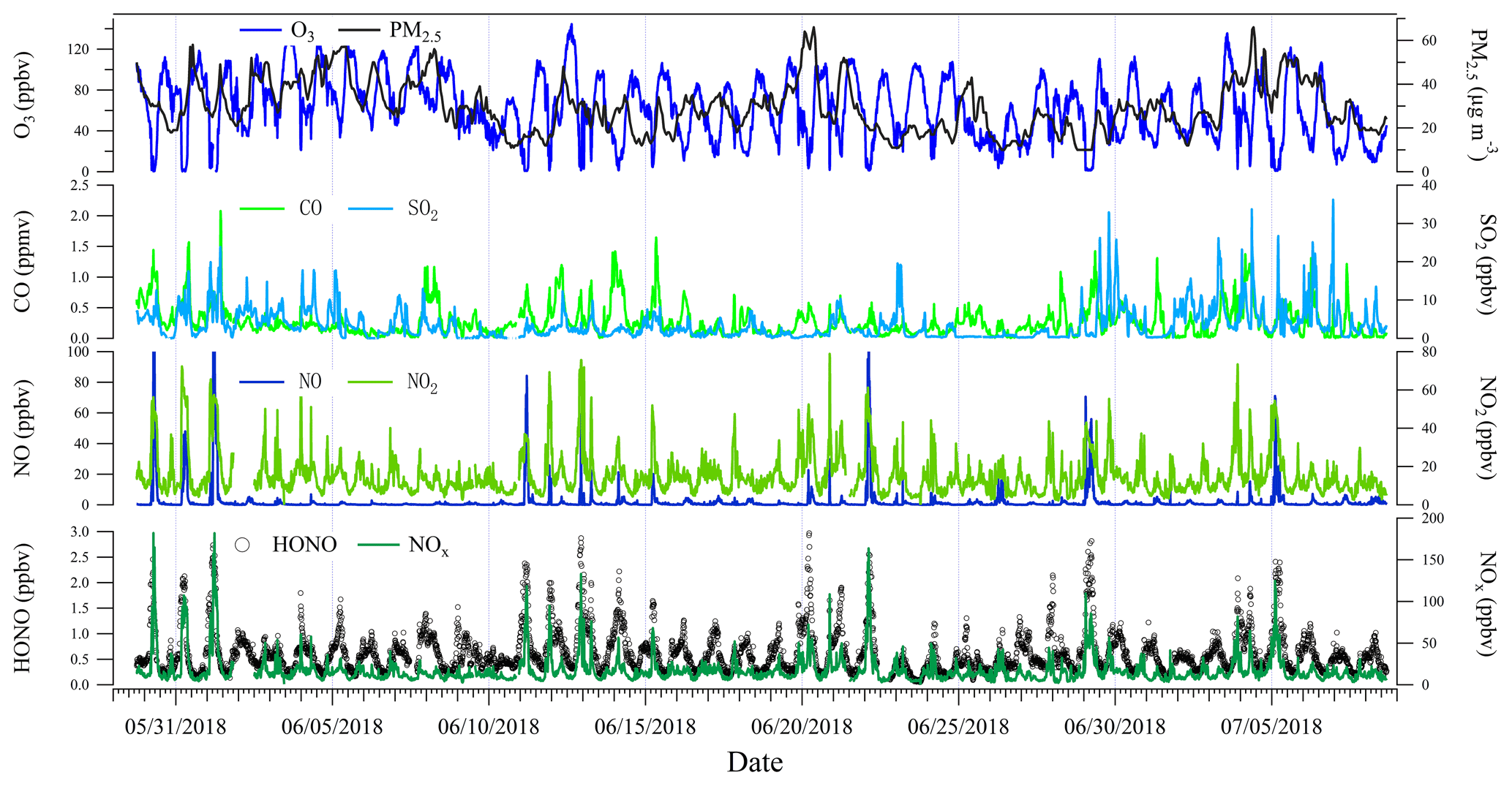
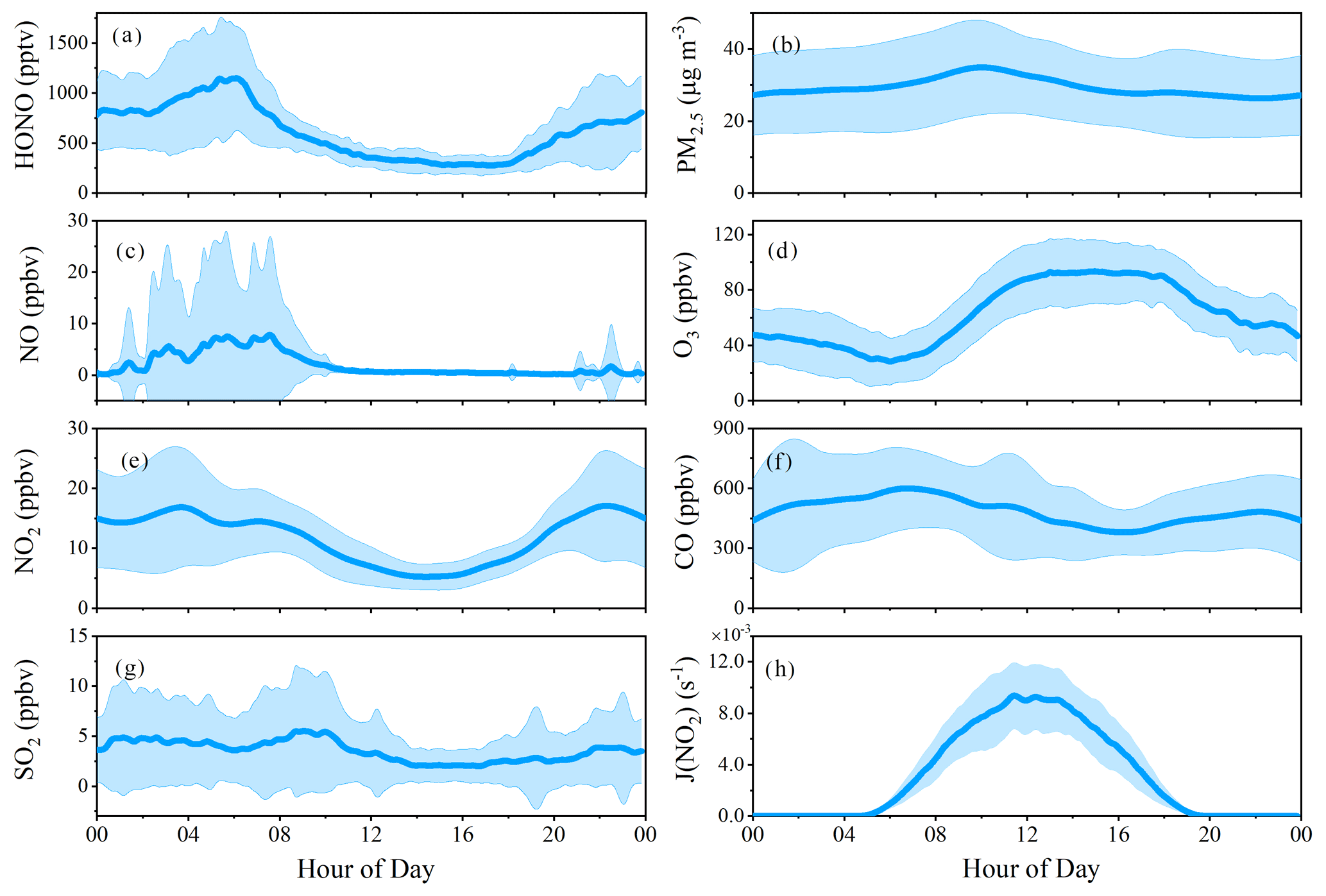
Figure 3Average diurnal profiles of HONO and related species measured during the campaign. Note that some of these data were also shown in the companion ACP paper for comparison between measurements at the foot and the summit stations.
Compared to other summertime measurements worldwide (Table 3), the measured HONO level at this site is similar to some measurements in China, such as Beijing in 2007 (Hendrick et al., 2014), Beijing in 2008 and 2017 (Crilley et al., 2019; Hendrick et al., 2014), and Guangzhou in 2006 (Yang et al., 2014); in Europe, such as Milan in 1998 (Alicke et al., 2002) and Rome in 2001 (Acker et al., 2006); and in North America, such as New York in 2001 (Ren et al., 2003) and Colorado in 2011 (Vandenboer et al., 2013). In addition, it is lower than measurements in cities during polluted periods, such as Jinan in 2016 (Li et al., 2018), Santiago de Chile in 2005 (Elshorbany et al., 2009), Santiago de Chile in 2009 (Villena et al., 2011), and Mexico in 2003 (Volkamer et al., 2010) but higher than recent measurements near European cities, including Forschungszentrum Karlsruhe (Kleffmann et al., 2003), Forschungszentrum Jülich (Elshorbany et al., 2012), suburban Paris (Michoud et al., 2014), and Cyprus (Meusel et al., 2016). It is noteworthy that the measured HONO at the foot station is significantly higher than that observed at the summit station in the same summer, indicating possibly different roles and formation paths of HONO at these two stations.
Table 3Examples of worldwide HONO measurements at the ground level.
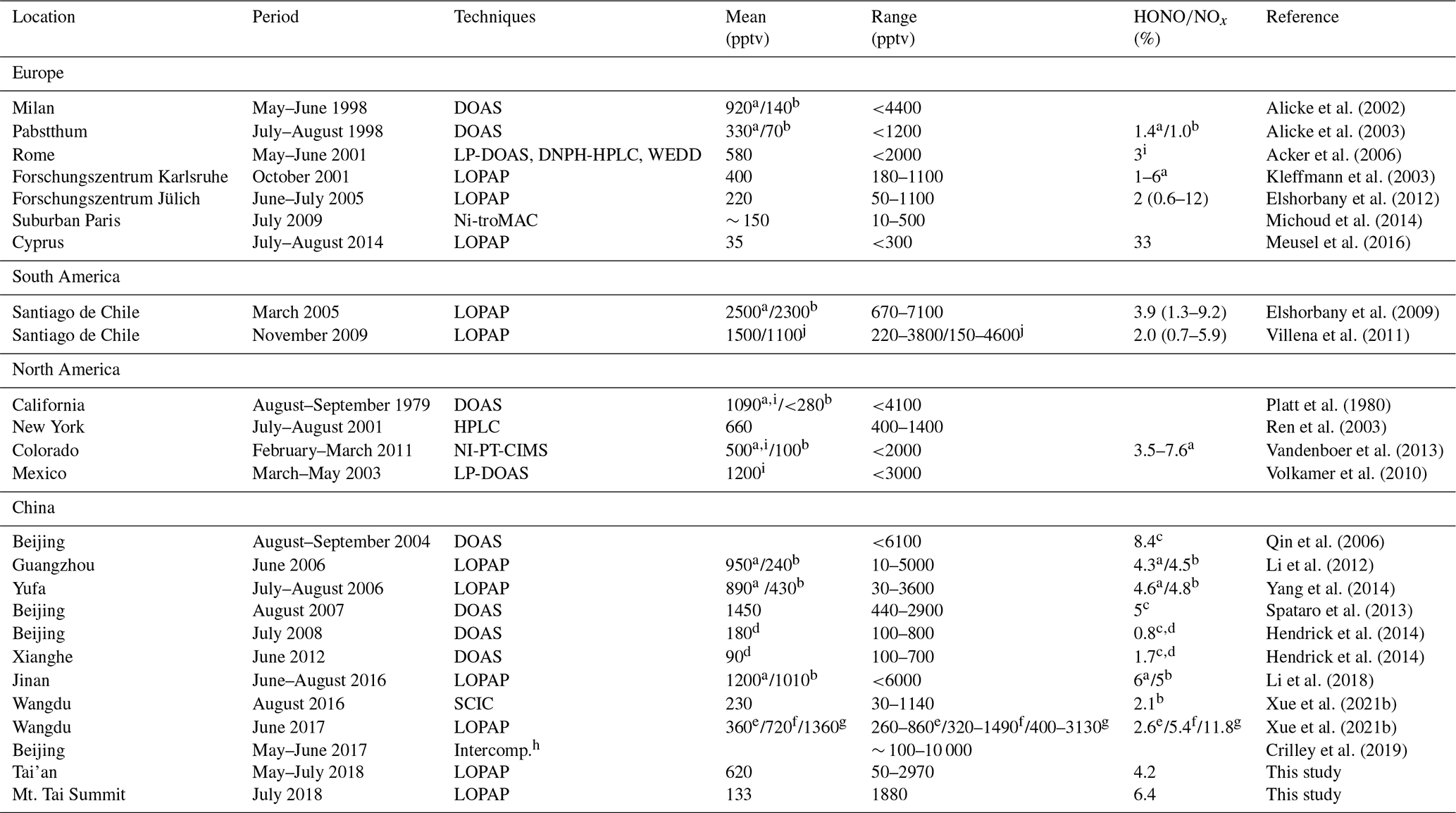
a Nighttime. b Daytime. c HONONO2. d Noontime. e Non-fertilization period. f Pre-fertilization period. g Intensive fertilization period. Intercomph: intercomparison of five instruments. i Half of the diurnal maximum. j 3rd and 21st floors, respectively.
3.1.2 NO2 interference and correction
As the most important HONO precursor, accurate measurement of NO2 plays a key role in analyzing HONO formation. The NOx monitor used in this study could specifically detect NO. To measure NO2, a molybdenum converter is used to convert NO2 to NO. However, this chemical conversion process suffers from the interference of other NOy species (Villena et al., 2012), primarily including inorganic species such as (measured) HONO, (non-measured) HNO3, HNO4, N2O5, and NO3, peroxyacyl nitrates (PANs, RC(O)OONO2), organic nitrates (RONO2), and peroxy nitrates (ROONO2). In this study, we defined the sum of RONO2 and ROONO2 as organic nitrates*. Hence, the measured NO2 is the sum of real NO2 and those interfering species. HONO was measured and subtracted from the measured NO2, and we defined NO = the measured NO2 − HONO. As NO2 is the most important HONO precursor, we used the family constraint (NO = NO2 + HNO4 + 2N2O5 + NO3 + PANs + organic nitrates*) in the base case (Sce-0) to separate each species from NO. To describe the PAN class, PAN, PPN, and MPAN (MCM names; see their structures at http://mcm.york.ac.uk/home.htt, last access: 1 May 2021) were considered. In the organic nitrates* class, CH3NO3, C2H5NO3, NC3H7NO3, IC3H7NO3, TC4H9NO3, NOA, ISOP34NO3, ISOPANO3, ISOPDNO3, ISOPCNO3, and ISOPBNO3 (MCM names) were included. Considering that HNO3 is very sticky, we expect HNO3 was mostly absorbed by the filter and/or sampling tubes before the converter rather than being converted to NO by the converter. Therefore, HNO3 was generally not included in the family constraint and only considered for the uncertainty analysis.
Figure 4 shows the model results of the relative contribution of each NO species to NO. At night with the absence of photochemistry, the real NO2 dominated NO components, with a contribution of >95 %, suggesting a small interference on the NO2 measurement. However, the contribution of real NO2 was found to decrease during the daytime due to the increasing interference. For example, at 11:00, the real NO2 contributed 82 % of the NO, indicating that the interferences could be as high as +22 % (calculated from 18 %82 %). In particular, at 13:00, PANs alone caused the highest interference by +21 % (calculated from 17 %81 %).
The variations of the simulated PANs and NO3 and their ratios to NO2 were similar to previous observations (Brown and Stutz, 2012; Roberts et al., 1998; Su et al., 2008; Villena et al., 2012; Xue et al., 2011), indicating that the uncertainty of the method is small. For the following model simulations and analysis, only the corrected NO2 was used. In addition, Fig. S3 exhibits the parallel test results in which HNO3 was included in the family constraint. It can be found that the interference became more significant; for instance, the interference could be as high as +75 % (calculated from 43 %57 %, at 11:00). This represents the upper limit of the interferences if the sampling tubes and the inlet filter are heated so that HNO3 could have reached the converter.
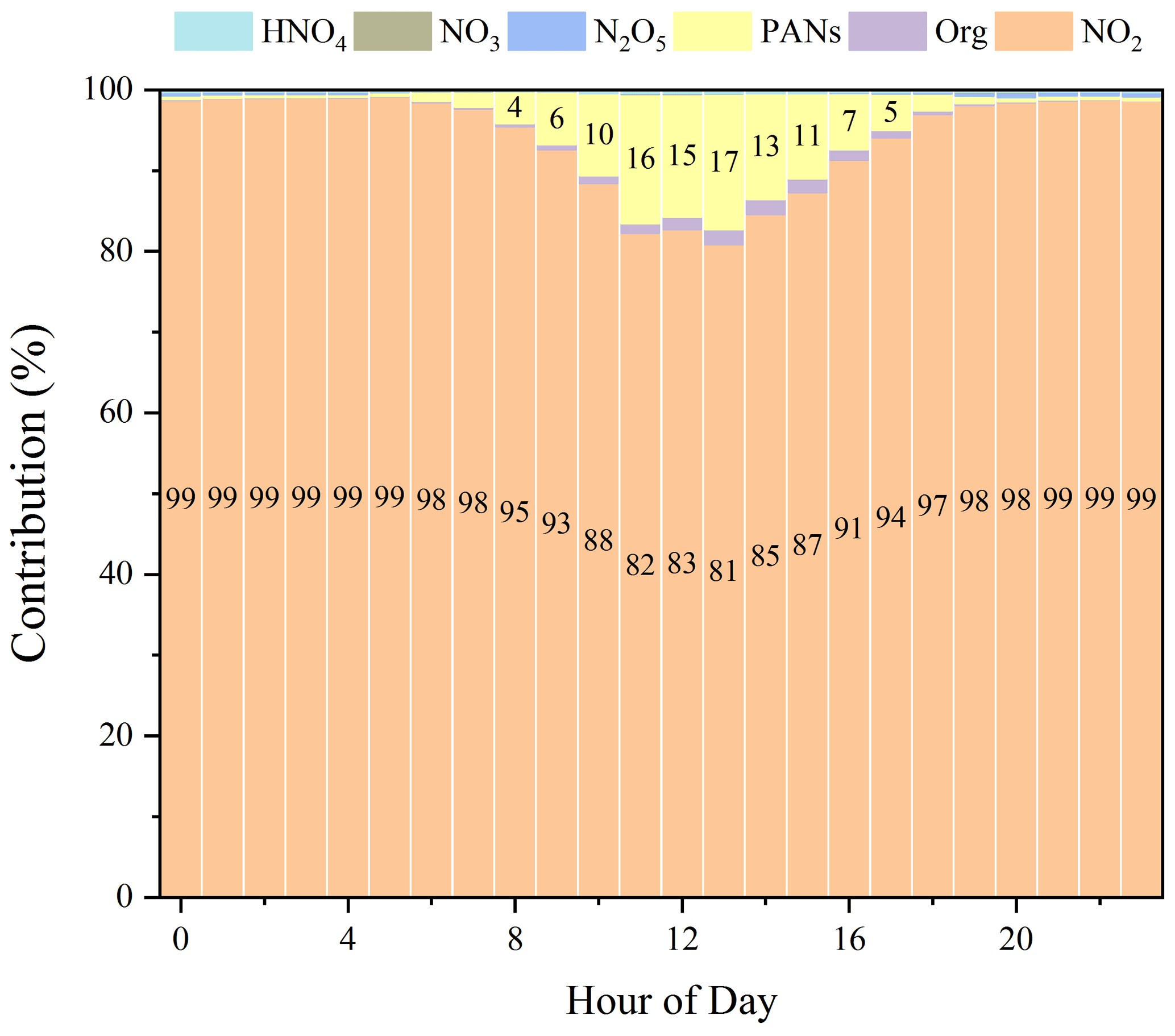
Figure 4Relative contribution of each NO species. PANs = PAN + PPN + MPAN. Org represents organic nitrates* (RONO2 + ROONO2).
Additionally, as shown in Fig. S4, the simulated HNO4 showed (1) a different diurnal variation from the observed HONO, (2) concentrations generally 1–2 orders of magnitude lower than the observed HONO, and (3) a very poor correlation (R2=0.06) with the observed HONO, indicating its negligible interference on the HONO measurement by the LOPAP technique (Legrand et al., 2014). It is worth noting that for the description of O3 formation in the polluted atmosphere, accurate measurements of NOx and HONO are necessary.
3.2 HONO sources and budget
3.2.1 Model default source (NO + OH) and unknown source strength
The homogeneous reaction of NO and OH has been adopted as the default HONO source in atmospheric chemistry models, including MCM. Model results from Sce-1 that only contains the homogeneous source with the modeled OH from Sce-0 are shown in Fig. 5. Apparently, the source of NO + OH is too small to explain the observed HONO as the simulated one is almost 1 order of magnitude lower than observations. Its contributions to the measured daytime and nighttime HONO are 15 % and 12 %, respectively.
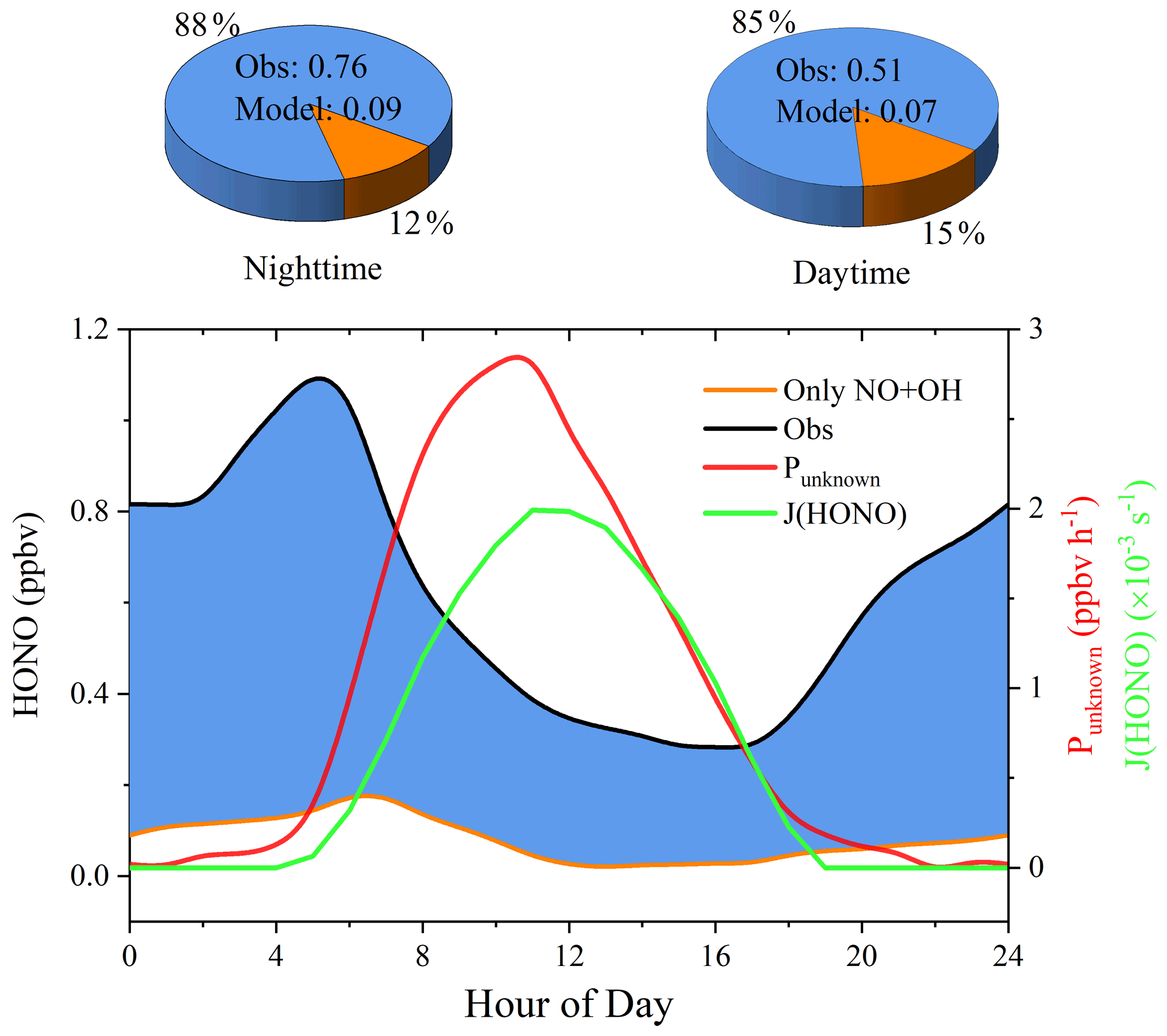
Figure 5Simulated HONO by the default mechanism (Sce-1, left axis) compared with the observations (Obs, left axis), unknown source strength (Punknown, right axis), HONO photolysis frequency (J(HONO), right axis). The blue shaded area in the plot and the pie charts represents the difference between the observation and modeled values. The relative contributions of NO + OH to the observations at night (19:00–04:50) and in the day (05:00–18:50) are shown in the left and the right pie charts, respectively.
Then we calculated the unknown source strength (Punknown) based on the following equation (Sörgel et al., 2011; Su et al., 2008).
The HONO loss rates through photolysis (L(HONO)pho) and reaction with OH (L(HONO)HONO+OH) and production rate from NO + OH were obtained from the base model scenario (Sce-0 with a constraint of the measured HONO). HONO mixing ratio differences within a 1 h interval, , were calculated from the measurements and are compared with Punknown in Fig. S5. As shown in Fig. 5, Punknown rapidly increased in the morning and peaked at nearly 3 ppbv h−1 at 11:00, followed by a decrease, revealing a photo-enhanced source. Note that the profile of Punknown is asymmetric around 12:00 solar noon, indicating the unknown source is not simply photolytic but also includes its precursors. For instance, higher NO2 levels were observed in the morning than in the afternoon (Fig. 3e), which preliminarily implies the importance of NO2-to-HONO conversion. The possible additional HONO sources responsible for Punknown are discussed in the following section.
3.2.2 Additional sources vs. Punknown
Direct emission: ΔHONONOx ratio
The ΔHONONOx ratio for the direct emission was determined from fresh plumes, which reached the following requirements: (1) measured at night when photolysis was absent and (2) a rapid NO increase by >10 ppbv within 10 min. Only 17 cases were obtained throughout the campaign due to the persistent high O3 and the fast NO-to-NO2 conversion. If NO is completely titrated by O3, there is a risk that the plume may not be fresh and that the inferred ΔHONONOx might be overestimated due to heterogeneous secondary HONO sources. In Table 4 the obtained ΔHONONOx was shown, varying from 0.18 % to 1.86 %, with an average of 0.98 % and a median of 0.90 %. The inferred value might be larger than the real one as potential NO2-to-HONO conversion leads to an overestimation. This is consistent with the observation that the inferred ΔHONONOx is generally higher in high RH conditions due to the potential NO2-to-HONO conversion (Fig. 6). More importantly, we found that the observed HONONOx is convergent as NONO2 increases (Fig. 6), which allows for a further correction of ΔHONONOx. The reported NONO2 ratios from combustion processes show significant variations, e.g., 6.7 in Wuppertal (Kurtenbach et al., 2012), ∼ 18 in Denver (Wild et al., 2017), 5–30 in London (Carslaw and Beevers, 2005), and 13–43 from China IV/V vehicles (He et al., 2020). Furthermore, in the emission inventory, the NONO2 emission ratio in the NCP is about 9 (Zhang et al., 2009). However, the measured nighttime NONO2 ratios were less than 3 (Fig. 6), much lower compared to direct emission measurements, indicating that the observed plumes were not fresh enough to directly obtain the primary ΔHONONOx emission ratio. By using a typical NONO2 ratio of 10 from car exhaust, the calculated ΔHONONOx through the convergent function (Fig. 6) is 0.7 %, similar to that obtained from laboratory or tunnel experiments (Kirchstetter et al., 1996; Kramer et al., 2020; Kurtenbach et al., 2001; Liu et al., 2017a).
Caused by its fast daytime photolysis, direct HONO emissions (HONOemi) are likely significantly overestimated when a constant ΔHONONOx ratio is considered, as done in former studies (Kramer et al., 2020; Liu et al., 2017b). Due to the much shorter lifetime of HONO (τ(HONO)) compared to NOx (τ(NOx)), a much smaller fraction of the emitted HONO compared to NOx will survive in the daytime atmosphere. Accordingly, for the first time, we calculated τ(HONO) and τ(NOx) and considered them in the parameterization of HONOemi (see the method in Sect. S1 of the Supplement). As shown in Fig. S6a, daytime τ(NOx) was typically 1 order of magnitude higher than τ(HONO) (Fig. S6a), indicating a remarkable overestimation of the contribution of HONOemi to the measured HONO when using a constant ΔHONONOx emission ratio (Fig. S6b). Hence, HONOemi was quantified by the following equations:
In summary, direct emission contributed about 1 %–26 % of the measured HONO, with an average of 13 %. Moreover, the new method developed here may still have some uncertainties but largely reduces the significant overestimation of the contribution of HONOemi to the observations during daytime compared to using a constant ΔHONONOx (Fig. S6b).
Table 4ΔHONONOx ratios determined from nighttime (19:00–04:50) data of the campaign. Concentrations (ppbv) of HONO, NO, and NOx at the start and the end of each plume of fresh emissions are also shown.
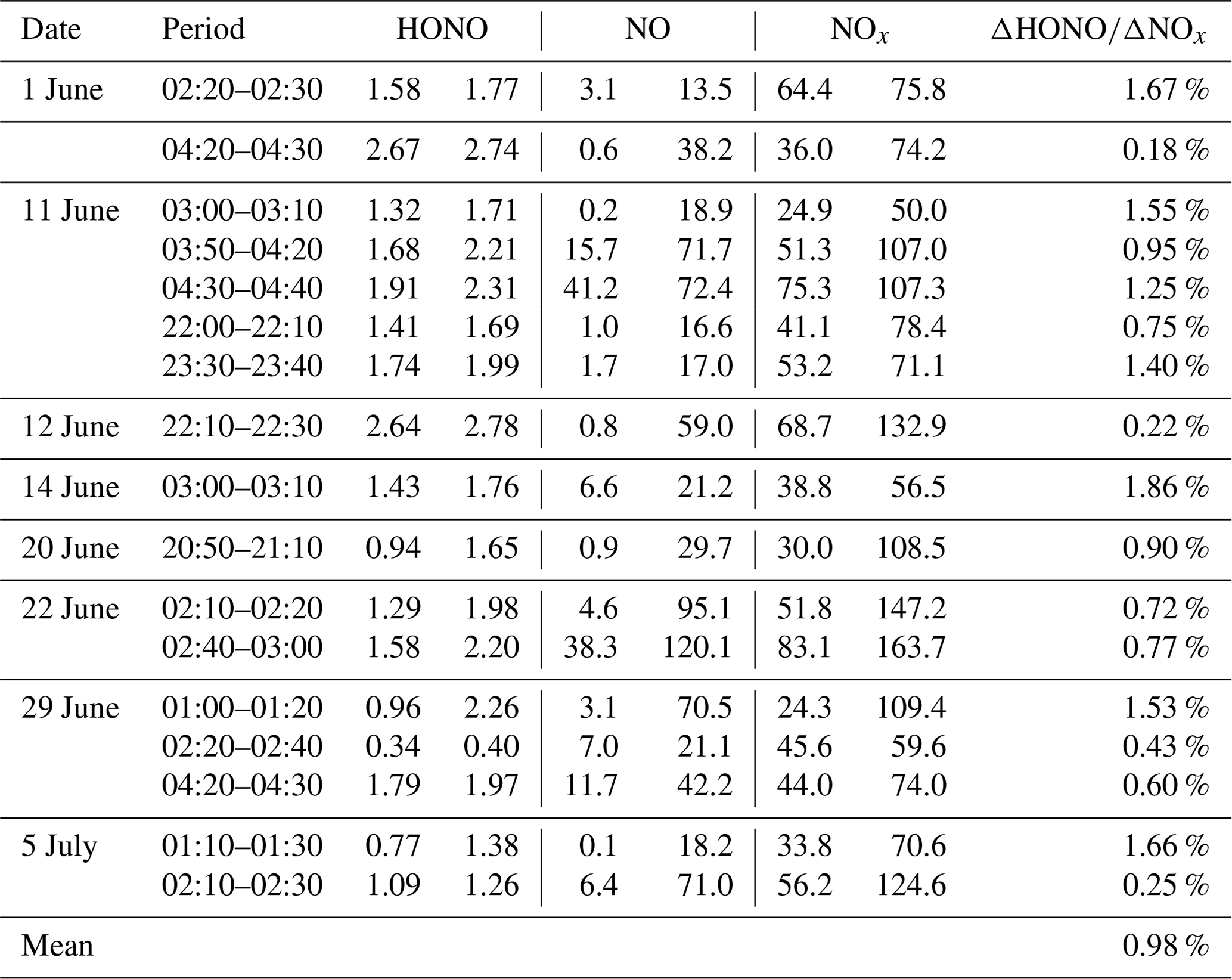
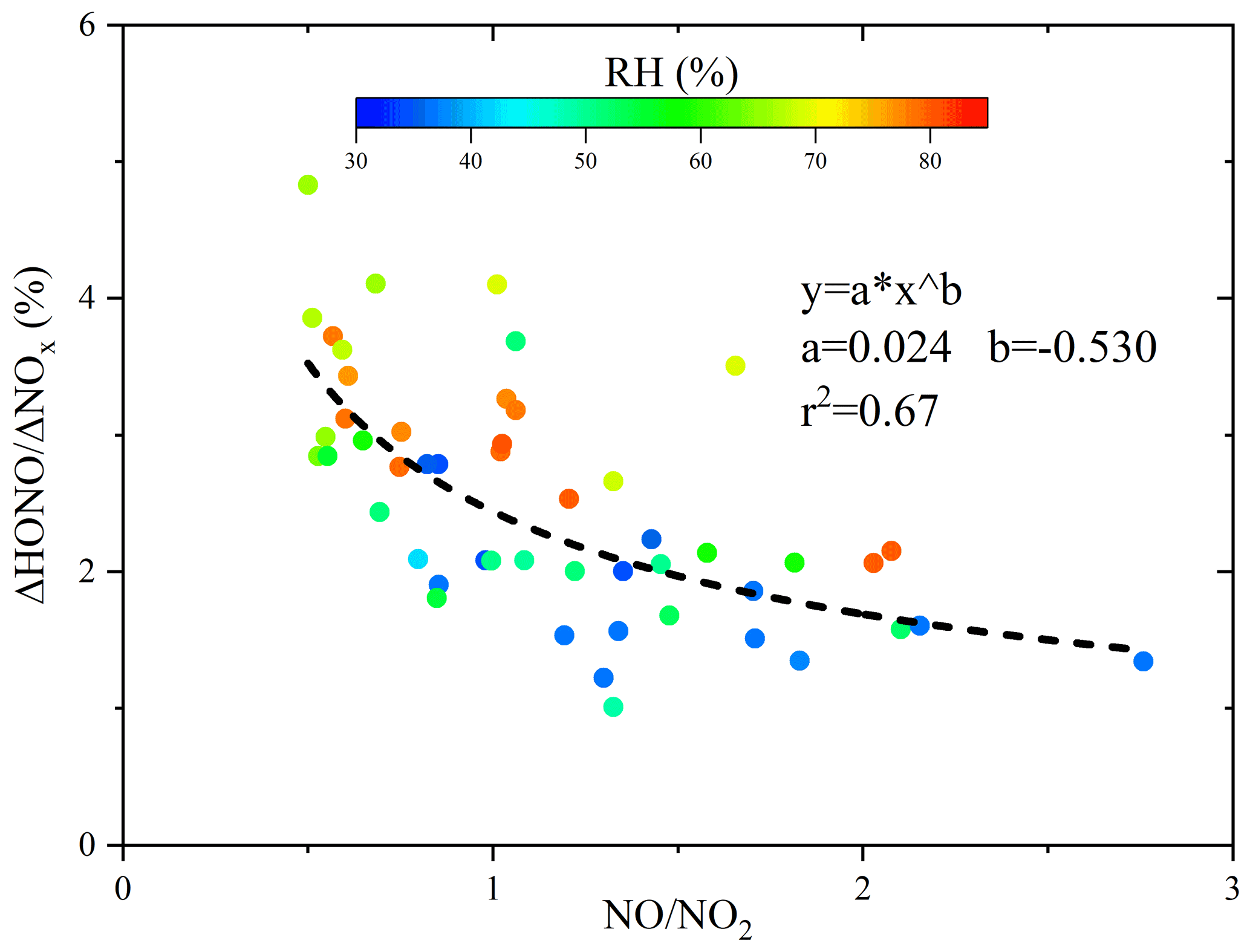
Figure 6The inferred direct emission ratio (ΔHONONOx) and NONO2 colored by RH. Only data with NONO2 >0.5 were shown as lower ratios indicate more aged rather than fresh plumes.
NO2 uptake on the aerosol surface
Parameterizations of HONO formation from the NO2 uptake on the aerosol surface without and with photo-enhanced effects (P(HONO)a_dark and P(HONO)a) are described by Eqs. (4) and (5), respectively. In Eqs. (4) and (5), HONO yields of 50 % and 100 % were considered for the dark and the photo-enhanced NO2 conversion, respectively (Finlayson-Pitts et al., 2003; George et al., 2005). A relatively large NO2 uptake coefficient γa_dark of 1 × 10−5 was used here to represent its upper limit. Its overestimation should not cause significant uncertainties as P(HONO)a_dark was negligible in HONO formation (see the following discussion). For the photo-enhanced NO2 uptake coefficient γa values of 1.3 × 10−4 (upper limit derived from the summit measurements in our companion paper; see Xue et al., 2021a) and 2 × 10−5 (more realistic value derived from laboratory experiments; see Stemmler et al., 2006 and 2007) were used in Eq. (5) to constrain the upper limit and a more realistic value of P(HONO)a.
where v(NO2), Sa, [NO2], and J(NO2)measured denote the average NO2 molecular speed (m s−1), aerosol surface density (m−1), NO2 concentration (ppbv), and the measured NO2 photolysis frequency (s−1), respectively. As aerosol size distribution measurements were not available at the foot station, we estimated Sa based on the measured PM2.5 concentrations as they were highly correlated. For instance, measurements at the summit station during this campaign and other sites in the NCP found high correlations between PM2.5 and Sa (derived from particle size distribution measurement), with a PM2.5 ratio of about 8 × 10−6–1.3 × 10−5 m2 µg−1 (Wu et al., 2008; Xue et al., 2020). Here a PM2.5 ratio of 1.0 × 10−5 m2 µg−1 was used, and its uncertainty will not cause significant changes in the HONO simulations because of its small contribution (see the following discussion).
Diurnal variations of P(HONO)a_dark and P(HONO)a, in comparison with Punknown and P(HONO)NO+OH, are shown in Fig. 7a. Clearly, both P(HONO)a_dark and P(HONO)a were negligible compared to daytime Punknown. P(HONO)a increased with γa, but even when using the upper limit of γa=1.3 × 10−4, it was still too small to be comparable to P(HONO)NO+OH and far from explaining Punknown. This observation reveals minor impacts of heterogeneous HONO formation on particle surfaces to the missing HONO source, particularly during daytime.
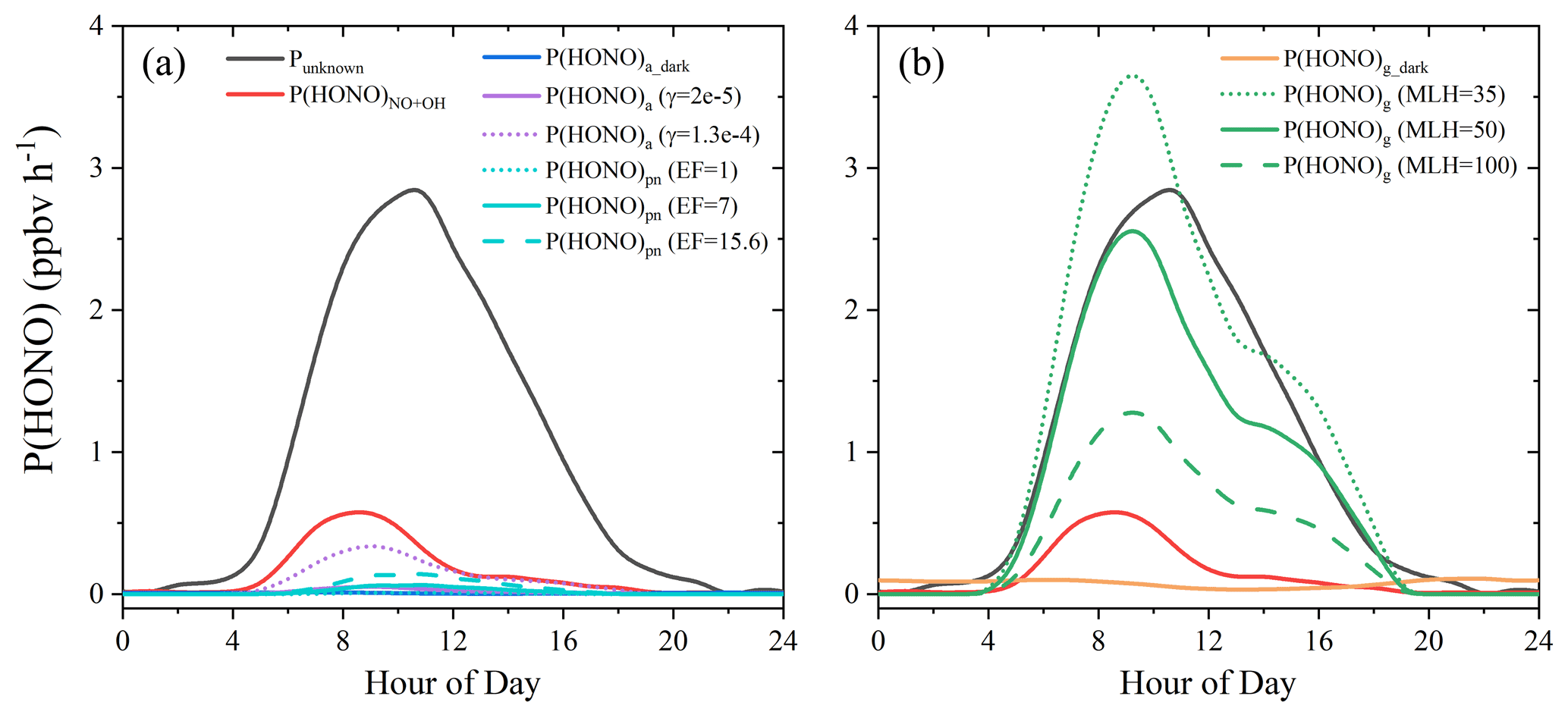
Figure 7HONO production rates from different (a: aerosol-derived, b: ground-derived) sources and unknown HONO source strength.
pNO3 photolysis
Parameterization of HONO formation from particulate nitrate photolysis (P(HONO)pn) is presented in Eq. (6). Recent studies found that EF (enhancement factor) values were generally lower than 10; for instance, a value of 7 was reported from a recent field study (Romer et al., 2018). In laboratory studies, even lower values of ∼ 1 were observed (Laufs and Kleffmann, 2016; Shi et al., 2021; Wang et al., 2021). Hence an EF value of 7 was used in the P(HONO)pn calculation, and values of 1 and 15.6 were also used to test the sensitivities (the upper limit value was derived from the summit measurement; see Xue et al., 2021a).
where pNO3 and J(HNO3) represent the measured particulate nitrate (with units converted from µg m−3 to ppbv) and the photolysis frequency of gas-phase HNO3 (s−1), respectively.
Diurnal variations of P(HONO)pn with different EF values are shown in Fig. 7a. With EF varying from 1 to 7, P(HONO)pn was 1–2 orders of magnitude lower than Punknown. Even using a high EF = 15.6, P(HONO)pn was still significantly less than half of P(HONO)NO+OH. Therefore, model results constrained by field measurements and recent kinetics suggested that the two aerosol-derived sources (NO2 conversion and nitrate photolysis) may not have significant impacts on daytime HONO formation, with their contributions significantly lower than half of P(HONO)NO+OH.
NO2 uptake on the ground surface
Parameterizations of HONO production from the NO2 uptake on the ground surface without and with photo-enhanced effects (P(HONO)g_dark and P(HONO)g) are demonstrated in Eqs. (7) and (8), respectively. NO2 uptake coefficients of γg_dark and γg were set to 1.6 × 10−6 and 2 × 10−5 (Han et al., 2016; Stemmler et al., 2006, 2007), respectively. The photo-enhancement effect was described by (Vogel et al., 2003; Wong et al., 2013; Xue et al., 2020).
As shown above, one of the most important parameters for calculating ground HONO formation in a box model is the mixing layer height (MLH) as it is part of the denominators in both Eqs. (7) and (8). MLH for HONO should be significantly lower than the boundary layer height (BLH) due to its formation on the ground level and its short lifetime, which could be confirmed by gradient measurements (Kleffmann et al., 2003; Meng et al., 2020; Vandenboer et al., 2013; Vogel et al., 2003; Wang et al., 2019; Wong et al., 2012; Xing et al., 2021; Ye et al., 2018b). For instance, a recent gradient HONO study by the MAX-DOAS technique in southwest China found a steep HONO gradient from 0 to 4 km (Xing et al., 2021). When considering their measurement at 17:00 (UTC + 8) as an example, HONO levels rapidly decreased from 4.8 ppbv at the ground level (∼ 4 m above the ground surface) to 1.6, 0.7, 0.3, 0.2, and 0.1 ppbv averaged in the layers of 0–100, 100–200, 200–300, 300–400, and 400–500 m above the ground level, respectively. In contrast, both NO2 and aerosol extinction remarkably increased from the ground level to about 200 m and then decreased with altitude (>200 m). These observations indicate that (1) ground-derived sources dominated daytime HONO formation; (2) the MLH for HONO was much less than 100 m; and (3) significant overestimation, i.e., by a factor of >3 in Xing et al. (2021), could be expected if using measurements on the ground surface to represent the average HONO within an MLH higher than 100 m. A similar phenomenon could also be found in tower-based vertical measurements in Germany and the USA. For instance, from the ground level (4–10 m) to 100 m above the ground surface, Vogel et al. (2003) and Vandenboer et al. (2013) observed similarly declining HONO levels from ∼ 0.6 to 0.3 (representative cases from Fig. 4 in Vogel et al., 2003, and Fig. 8 in Vandenboer et al., 2013), respectively. All of these cases suggest that near-ground surface measurements were more weighted by ground-derived sources. Moreover, this phenomenon was observed during their whole campaigns including daytime and nighttime, suggesting a similar level of MLH. Hence, the maximum of MLH could be reasonably assumed to be 100 m for near-ground surface measurements. A minimum MLH of 35 m was derived based on the assumption that all the Punknown could be wholly explained by photosensitized heterogeneous NO2 reaction on the ground surface in our recent study (Xue et al., 2021b). Therefore, in the present study, the MLH for HONO was set at a constant height of 50 m, with sensitivity tests performed with the MLH set at 35 and 100 m. In general, a ∼ 50 m level could represent a general MLH for 0D models to study HONO measurements near the ground surface. Similar values (25–100 m) were also used in previous box model studies (Harrison et al., 1996; Lee et al., 2016; Xue et al., 2020, 2021b). Nevertheless, it should be highlighted that a box model as used in the present study is not an ideal tool for studying a ground source when comparing with near-ground surface measurements in the atmosphere. For the future, gradient measurements are recommended, which should be compared with 1-D model simulations.
Diurnal variations of P(HONO)g_dark and P(HONO)g, in comparison with Punknown and P(HONO)NO+OH, are shown in Fig. 7b. P(HONO)g_dark is the largest HONO source during the nighttime, while it was negligible during the daytime, which is consistent with many previous studies (Li et al., 2010; Liu et al., 2019; Vogel et al., 2003; Xue et al., 2020; Zhang et al., 2019b, a, 2016). The photo-enhanced formation, P(HONO)g, shows a similar shape and a similar level to daytime Punknown, indicating the potential dominance of P(HONO)g in the daytime HONO formation. When changing MLH to 100 (or 35) m, the level of P(HONO)g becomes much lower (or higher) than Punknown. Due to this, sensitivity tests on MLH were conducted, but in Sce-2 a constant MLH of 50 m was used. Small differences in the shapes of measured and modeled results may also be caused by the variable MLH induced by variable vertical mixing in the atmosphere and the variable photolytic lifetime of HONO during the daytime.
3.2.3 HONO budget
Along with the previous discussion, we conducted a model run (Sce-2) with all the discussed HONO sources. As shown in Fig. 8, this model performed well in predicting HONO, indicating reasonable parameterizations of the HONO sources. Here, the time series of the modeled HONO were very consistent with those of the observations, both for the variations and the absolute levels. The only major exception was a period of heavy rain from 25 to 28 June (see the next section). In particular, the model exhibited very high performance in predicting noontime (10:00–16:00) HONO as the modeled HONO was very close to the observed HONO (Fig. 8b). Moreover, in Sce-3 we reduced γg by a factor of 10 and enlarged γa from 2 × 10−5 to 1.2 × 10−3 or EF from 7 to 400. While the modified model could also generally predict the observed HONO levels (Figs. S7a and S8a), it largely failed to reproduce the noontime observations in levels and variations (Figs. S7b and S8b), including its asymmetry, as mentioned in Sect. 3.2.1. This observation reinforces our conclusion that aerosol-derived sources only play a minor role in daytime HONO formation.

Figure 8Modeled HONO mixing ratios (model, in blue) in comparison with observations (Obs, in black). (a) Time series. (b) Average diurnal variations. Model + HONOemi represents the sum of the modeled HONO and HONOemi.
Figure 9 displays the diurnal relative contributions of different HONO sources. It clearly shows that dark NO2 uptake on the ground surface dominated (∼ 70 %) nighttime HONO formation, while photo-enhanced NO2 uptake on the ground surface dominated (∼ 80 %) daytime HONO formation. P(HONO)NO+OH played a moderate role throughout the whole day, with a contribution of 5 %–15 %, except for a relatively larger contribution (∼ 20 %) in the early morning due to high NO levels. Direct emissions made moderate contributions of 15 %–25 % at night but negligible ones during the daytime. Contributions of P(HONO)a_dark, P(HONO)a, and P(HONO)pn were always lower than 10 %, and their contributions could be even smaller when using realistic kinetic parameters derived in recent studies. Therefore, aerosol-derived HONO sources may not significantly contribute to HONO formation for near-ground surface measurements (Chen et al., 2019; Neuman et al., 2016; Sarwar et al., 2008; Vogel et al., 2003; Wong et al., 2013; Xue et al., 2020; Zhang et al., 2016, 2019b).
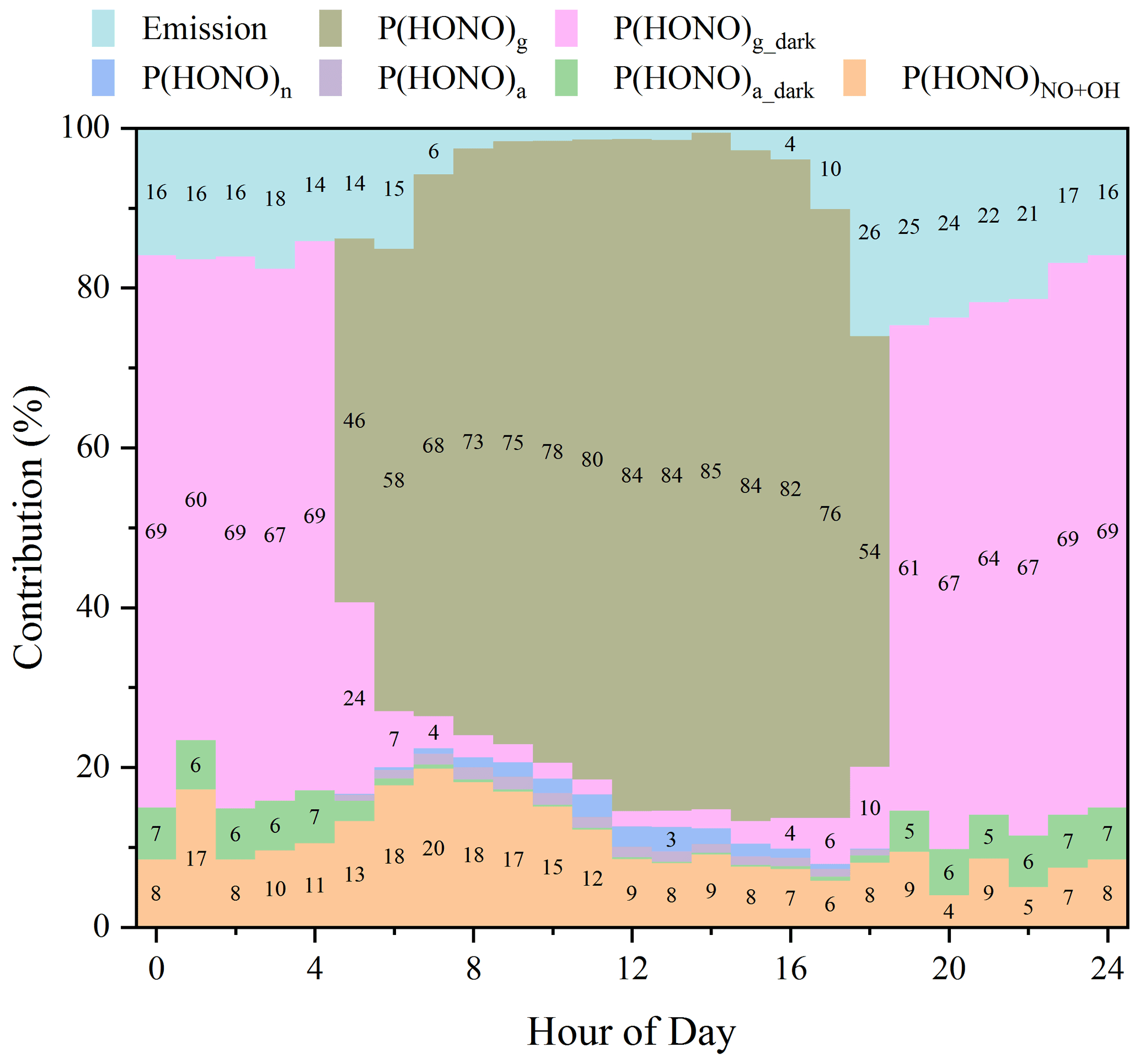
Figure 9Relative contributions of different HONO sources. Note that the contribution from direct emission was calculated based on the ratio of HONOemi to the observed HONO.
3.2.4 Other potential sources
As discussed before, the model (Sce-2) could generally well reproduce most observations, except for the period from 25 to 27 June. A significant overestimation occurred from midday of 25 June to the morning of 26 June, which was caused by the enhanced wet/dry deposition due to the heavy rain (>100 mm, Fig. S9) during the night of 24/25 June. In contrast, from midday on 26 June to the night of 27/28 June, a significant underestimation by the model was obtained. Additionally, an elevation of HONONOx was found during this period (Fig. S9). This might be caused by (1) the enhanced HONO emission from urban soil or (2) the enhanced NO2 uptake on the ground surface. HONO emissions from soils may occur through biological processes observed in the laboratory experiments or field measurements over the agricultural fields (Oswald et al., 2013; Scharko et al., 2015; Tang et al., 2019; Xue et al., 2019), while evidence for its occurrence on urban soil surfaces after the rain is still uncertain. At 13:00 on 26 and 27 June, the model predicted lower HONO by almost a factor of 2–4 (observation: 0.45 ppbv on both days; model: 0.13 and 0.21 ppbv), which needs an enhancement of at least 2–4 in γg if using NO2 uptake on the ground surface to explain the underestimation. Current laboratory studies have studied the enhancement effect of atmospheric RH (in the range of 10 %–70 %) on the NO2 uptake coefficient on the humic acid surface with enhancement factors of less than 3 (Han et al., 2016; Stemmler et al., 2006, 2007). Campaign averages of the measured NO2 and RH at 13:00 were 7.4 ppbv and 35.5 %, respectively. At 13:00 on 26 (27) June, the measured NO2 of 7.9 (4.3) ppbv was similar to (or lower than) the campaign average, but the RH of 67.6 % (53.1 %) was significantly higher than the campaign average. But the RH was still in the range (10 %–70 %) for which RH showed a small enhancement effect on γg (less than 3). Hence, after rain, an enhanced NO2 uptake may be responsible for the underestimation. In addition, soil HONO emission may co-exist, but more evidence for urban regions was needed. However, the impact of highly humid surfaces (e.g., rainwater on the ground surface) on different HONO sources and sinks was still unclear. Further studies on the impact of rain on urban soil surface processes are necessary, such as field studies of soil HONO emission fluxes and laboratory studies of NO2 uptake kinetics on relevant surfaces.
3.3 Radical chemistry
Comprehensive field measurements in comparison to model studies allow the role of HONO in the radical chemistry of the atmosphere to be studied. HONO is expected to strongly impact OH levels in the lower atmosphere due to strong daytime HONO sources and due to its fast photolysis. In addition, considering high O3 levels at the present field site, NO3 chemistry could also be important, particularly during nighttime, which will also be discussed in this section.
3.3.1 Role of HONO in radical concentrations
Figure 10 shows the simulated radical concentrations in different model scenarios, in which their sensitivities to the constrained HONO were tested. It can be obtained that ROx radicals (OH, HO2, and RO2) were significantly affected by the constrained HONO, implying the vital role of HONO in the ROx budget. For instance, the peak OH concentration in the base case was 0.42 pptv (equivalent to 1.0 × 107 molecules cm−3). It decreased to 0.37 (or 0.32) pptv when HONO was reduced by 50 % (or 100 %) and increased to 0.46 (or 0.51) pptv when HONO was enlarged by 50 % (or 100 %). In contrast, modeled NO3 concentrations showed very small variations whether HONO was reduced or enlarged. NO3 concentrations are mainly governed by the levels of O3 + NO2 during nighttime, when HONO has no impact on radical levels, due to its missing photolysis. The radical concentrations that are almost the same in the model cases NO + OH and case −100 % indicate the minor role of NO + OH in the radical budget as this OH sink is exactly compensated for by the OH production through Reaction (R5).

Figure 10Simulated concentrations of (a) OH, (b) HO2, (c) RO2, and (d) NO3. Different colored lines show results in different scenarios. NO + OH: only with the homogeneous source. Base: constrained by the observed HONO. +50 %: constrained by the observed HONO × 1.5. +100 %: constrained by the observed HONO × 2. −50 %: constrained by the observed HONO × 0.5. −100 %: constrained by HONO = 0.
3.3.2 Radical production–loss rates and reactivity
Figure 11a and b illustrate the production–loss rates of OH and NO3, respectively. The total production rates of these radicals were similar to their loss rates due to their short lifetimes and high reactivities. For OH (Fig. 11a), its largest source was the reaction of HO2 + NO (average contribution: 70 %), which is part of the propagation cycle and which is not a radical initiation source (Elshorbany et al., 2010). HONO photolysis was the second largest OH source (average contribution: 11 %), and it is expected to be the largest primary OH source after subtracting OH loss through HONO + OH and NO + OH (see Sect. 3.3.4). Reactions with NO2, CO, and C5H8 acted as the top three OH sinks but did not dominate OH loss due to high OH reactivity caused by various other reactions, particularly those with other VOCs (see below).
Figures 11c and S10a show the OH reactivity with different classes of pollutants and their relative contributions, respectively. Total OH reactivity showed a small peak of 20 s−1 in the morning and then stayed almost constant around 17 s−1. Among different classes of pollutants, the measured inorganics (ordered by OH reactivity contribution: NO2 > CO > NO > O3 > HONO > SO2 > H2O2) contributed the largest to the OH reactivity with values in the range of 2.6–8.4 s−1. Their total contribution was larger in the morning (43 %) due to high NO, NO2, and CO levels (Fig. 2) and decreased to 15 % at noontime. Reactivities with the measured alkanes, alkenes, aromatics, and OVOCs were 0.95–1.2, 3.3–3.9, 2.2–2.9, and 1.65–1.9 s−1, leading to relative contributions of around 5 %–7 %, 18 %–22 %, 11 %–17 %, and 9 %–12 % throughout the whole day, respectively. Likewise, C5H8 alone contributed 4 % of the OH reactivity in the early morning (0.85 s−1), and its contribution increased to 12 % at noontime (2.1 s−1) as a result of high levels of C5H8 at noontime.
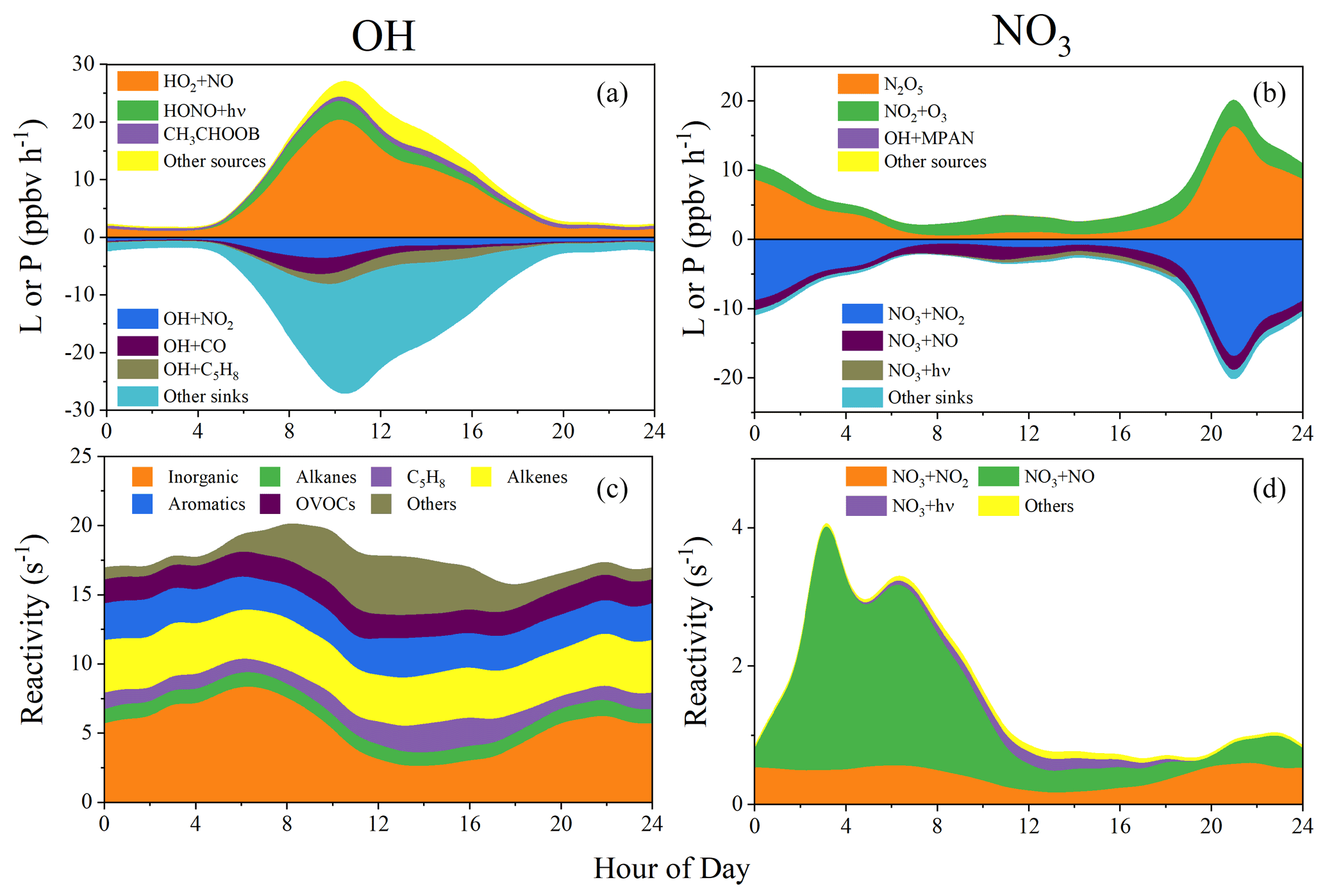
Figure 11Production rates (P), loss rates (L), and reactivities of radicals. (a) L and P of OH. (b) L and P of NO3. (c) Reactivities of OH. (d) Reactivities of NO3. In (a) and (b), the top three sources or sinks are shown, and all the others are summarized as “Other sources” or “Other sinks”. In (c), OH reactivities with different families of the measured species are shown, and reactivities with all the unmeasured species are summarized as “Others”. In (d), NO3 reactivities from the top three reactions are shown, and all the others are summarized as “Others”.
Figures 11d and S10b show NO3 reactivity with different pollutant classes and their relative contributions, respectively. Compared with the total OH reactivity, the total NO3 reactivity exhibited lower values and a different variation profile. It showed a minimum of 1 s−1 at noontime and increased to around 4 s−1 at 02:00. The reaction NO2 + O3 (Reaction R11) is the most important net NO3 source. In contrast, NO3 formation by the N2O5 decomposition (Reaction R17) is almost compensated for by the same amount of NO3 loss during N2O5 production (Reaction R14). NO3 loss was dominated by its photolysis and its reactions with NO during the daytime and reactions with NO2 at night. More discussion on the NO3 chemistry is presented in the following section.
3.3.3 NO3 chemistry
As shown in Fig. 10d, high NO3 levels (diurnal peak: 9.3 pptv, time-series peak: 22 pptv) were simulated by the model. High NO3 concentrations, as well as high NO3 reactivities (Fig. 10d), generally appeared at night (18:00 to 04:00 in the next day) when OH was very low and NO3 was not lost by photolysis, indicating that the NO3-initialized chemistry may play an important role in nighttime chemistry at this site. To verify this assumption, we compared the C5H8 oxidation and nitrate formation through NO3-induced reactions with other reactions.
C5H8 oxidation
Figure 12 shows (a) the C5H8 loss rates (L(C5H8)) through different oxidation paths and (b) their relative contributions. L(C5H8) through O3 was generally in the range of 0.04–0.12 ppbv h−1. L(C5H8) through OH showed high values in the daytime and low ones during the night. Opposite to OH, loss rates L(C5H8) through NO3 were low in the daytime and high during the night. On average, L(C5H8) values through OH, O3, and NO3 oxidation were 1.3, 0.07, and 0.16 ppbv h−1, with relative contributions of 84 %, 5 %, and 11 %, respectively. During the daytime, L(C5H8) through OH oxidation was generally 1 order of magnitude higher than those through NO3 or O3 oxidation, leading to a dominant C5H8 loss contribution of generally >90 % through OH oxidation (Fig. 12b). However, at night, OH was much lower and NO3 was higher due to the absence of photochemistry, resulting in an increasing contribution of L(C5H8) through NO3 oxidation (Fig. 12b). Average L(C5H8) through nighttime NO3 oxidation increased to 0.30 ppbv h−1, but L(C5H8) through OH oxidation decreased to 0.33 ppbv h−1, resulting in a relatively high contribution of NO3 oxidation (32 %–57 %). NO3 oxidation contributed to 44 % of the nighttime C5H8 loss, which is comparable to OH oxidation (48 %) and much higher than O3 oxidation (8 %). C5H8 is an important common hemiterpene emitted by many species of vegetation, and its oxidation plays a key role in secondary organic aerosol (SOA) formation. The results of the present study show that daytime OH-induced C5H8 oxidation is the most important, while NO3-induced oxidation of C5H8 may also significantly affect the SOA formation during the nighttime (Brown and Stutz, 2012; Mellouki et al., 2021).
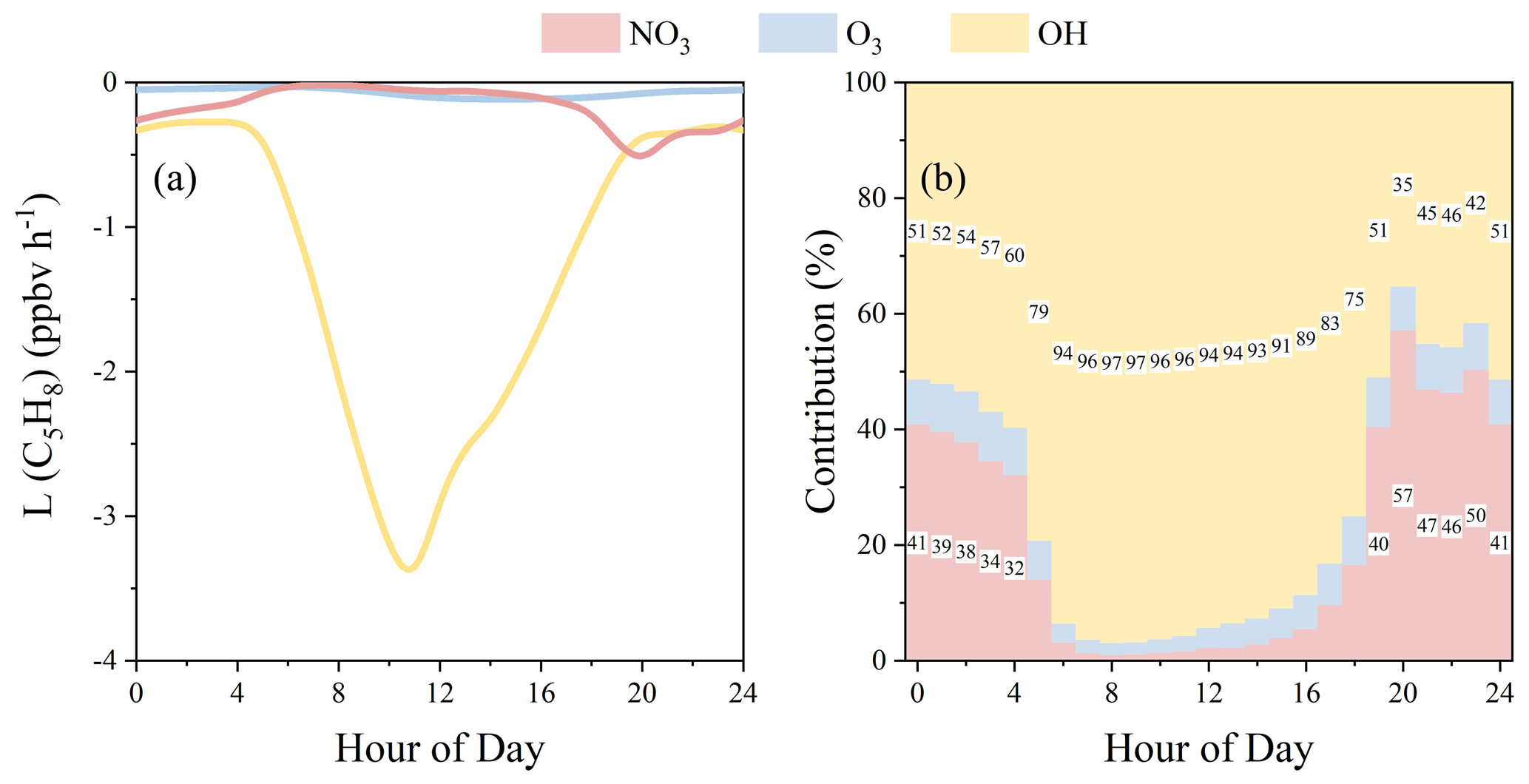
Figure 12(a) Diurnal loss rate of C5H8 by NO3, O3, and OH oxidation and (b) their diurnal relative contributions.
HNO3 formation
As an important component of particulate matter, inorganic nitrate (pNO3) is produced by the partitioning of gas-phase HNO3 and by heterogeneous uptake of N2O5. HNO3 formation from OH + NO2 and from N2O5 hydrolysis on the aqueous aerosol surface is also the major daytime and nighttime sinks for removing NOx from the atmosphere. Hence, the production of HNO3, defined as P(HNO3) = P(HNO3)OH + P(HNO3), represents the upper limits of pNO3 production. P(HNO3)OH denotes the HNO3 production through Reaction (R18) in the model (Sce-0). For calculation of P(HNO3), both HNO3 formation through N2O5 heterogeneous uptake on the aerosol surface (Reaction R16) and other NO3-induced reactions were considered (the former is the dominant one).
In the model, parameterization for the heterogeneous N2O5 uptake is described by Eq. (9).
where v(N2O5), [N2O5], and represent the molecular speed, concentration, and heterogeneous uptake coefficient of N2O5, respectively. was typically set as 0.1 reported in previous studies (Brown and Stutz, 2012; Wang et al., 2017).
As shown in Fig. 13, the overall P(HNO3) was high during the daytime and low during the night. During the daytime, P(HNO3) was generally much lower than P(HNO3)OH, leading to high contributions of P(HNO3)OH (>90 %). However, during the night, P(HNO3)OH and its relative contribution to P(HNO3) remarkably increased. On average throughout all day, P(HNO3) contributed 18 %, significantly lower than P(HNO3)OH (82 %). However, at night, P(HNO3) contribution increased to 51 %, slightly higher than P(HNO3)OH (49 %). Model results may have uncertainties but shed light on the atmospheric chemistry in this polluted region. So far, very few NO3 measurements are available in China (Lu et al., 2019; Suhail et al., 2019), while its high concentration and important role in chemical oxidation presented in this study indicate the necessity of direct NO3 (as well as related species such as N2O5 and ClNO2) measurements in the NCP, where summertime O3 levels are substantially increasing (Han et al., 2020; Li et al., 2019; Sun et al., 2016, 2019).
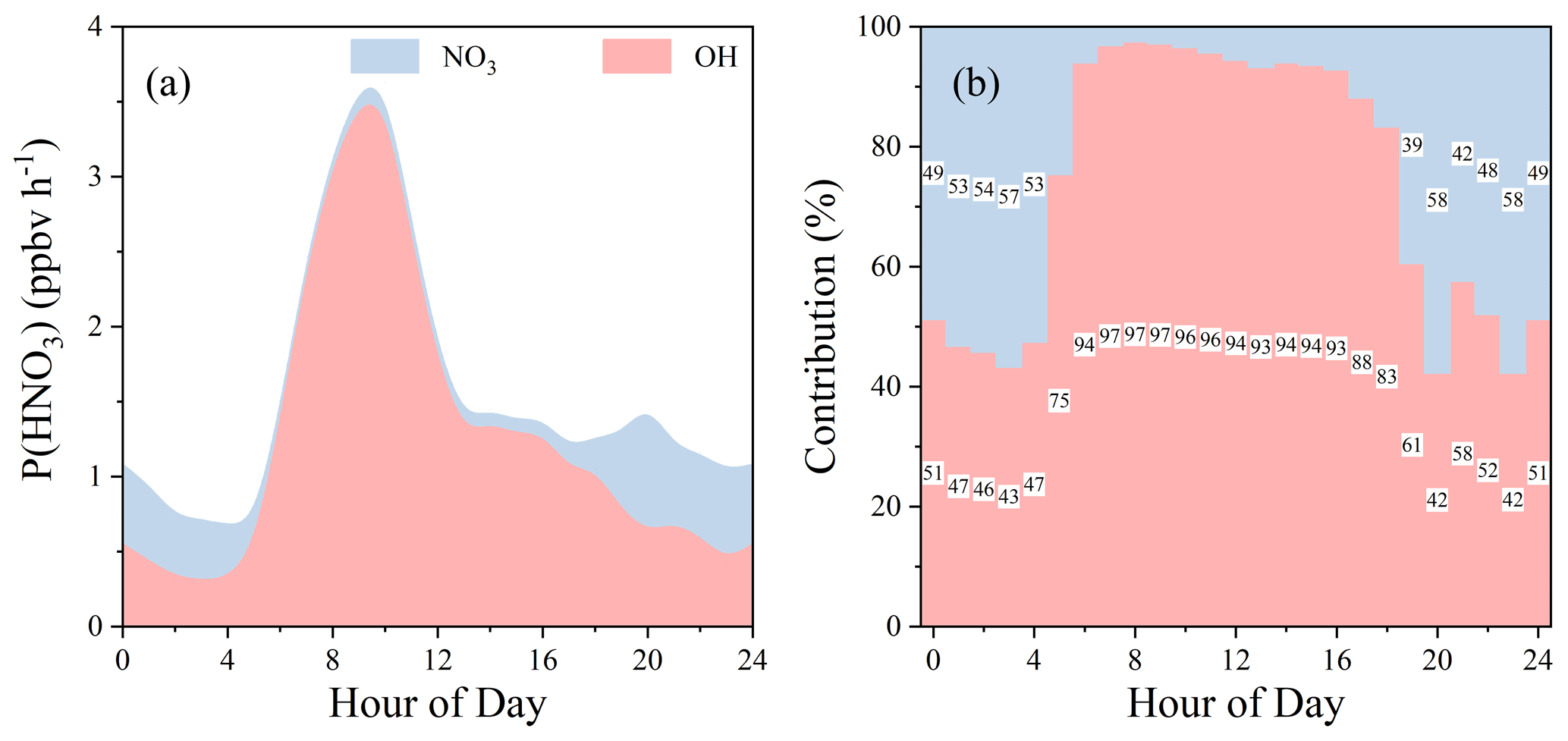
Figure 13(a) HNO3 production (P(HNO3)) from NO3- or OH-induced reactions and (b) their relative contribution at each hour of the day. NO3-induced reactions include heterogeneous uptake of N2O5 on the aerosol surface and all the other NO3 reactions that produce HNO3.
3.3.4 Radical initiation vs. termination
Measurements of other radical precursors, such as H2O2 (through photolysis to produce OH), HCHO (through photolysis to produce HO2), and alkenes (through ozonolysis via Criegee intermediate to produce OH, HO2, and RO2), were available, which allows for a comparison of radical initiation (primary production) and termination (T(ROx)). As shown in Fig. 14, the overall radical initiation and termination showed similar variations and levels. Both the sum of the ROx initiation and termination showed peaks of about 7.6 ppbv h−1 at noon, which is in the range of 2.5–12.2 ppbv h−1 reported in previous studies (Elshorbany et al., 2010, 2012; Hofzumahaus et al., 2009; Kukui et al., 2014; Liu et al., 2012; Ren et al., 2003). During the daytime, it is evident that HONO photolysis (P(ROx)HONO_net) made the largest contribution (20 %–70 %, Fig. 14b) to ROx initiation, with an average of 37 % (or 32 % for all day; Fig. S11), followed by ozonolysis (29 %), O3 photolysis (21 %), HCHO photolysis (13 %), and H2O2 photolysis (1 %). In particular, ROx production from the ozonolysis of alkenes was significantly lower than that from HONO during 06:00–14:00 until later after 17:00 when it started to dominate ROx production. At night with the absence of photochemistry, ozonolysis was the major source for primary ROx. Due also to the high production of radicals during the daytime, ozonolysis played an important role in primary ROx production (39 % for all day). The termination of radicals T(ROx) was dominated by NO2 + OH → HNO3, NO2 + CH3COO2 → PAN, and HO2 + HO2 → H2O2 (Elshorbany et al., 2010, 2012; Hofzumahaus et al., 2009; Kukui et al., 2014; Liu et al., 2012; Stone et al., 2012).
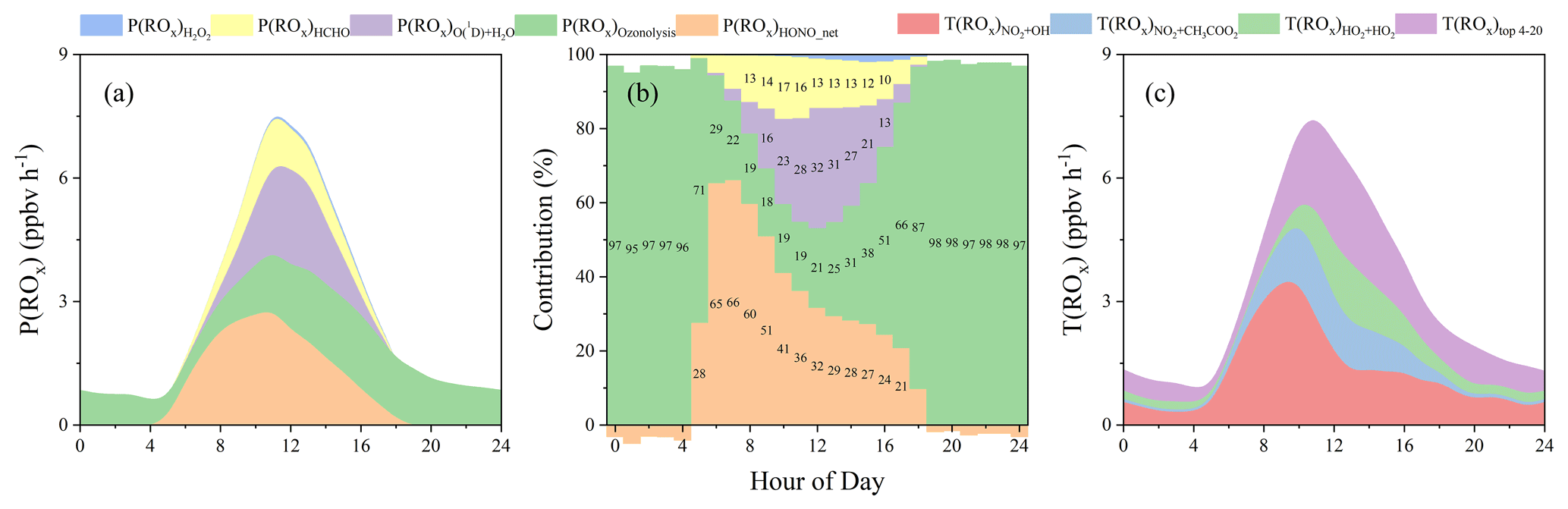
Figure 14Primary ROx production and net ROx loss. (a) Production from different sources and (b) their relative contributions at different hours of the day. (c) The top 20 ROx loss rates. Note that the top 20 net radical loss paths are summarized here. It could represent the majority of total T(ROx) as others (<0.03 ppbv h−1) were at least 2 orders of magnitude lower than the sum of the top 20. Nighttime P(ROx)HONO_net was negative (a net sink for OH), so that its contribution was also negative at night. The same amounts of radical loss or production from equilibrium reactions (e.g., HO2 + NO2 ↔ HNO4; CH3COO2 + NO2 ↔ PAN) were excluded from radical initiation or termination. T(ROx) represents the net PAN formation.
3.4 Implications for further studies
For the first time, we considered HONO and NOx lifetimes to quantify the contribution of direct emission to daytime HONO formation. The method developed here remarkably reduced the overestimation of contribution from direct emission. It is universal and should be used for all ground measurements to quantify the contribution of direct emission to daytime HONO formation.
In the present study, we also conclude that heterogeneous NO2 reaction on the ground surfaces is the major HONO source. Constraints on aerosol-derived sources, including NO2 uptake on the aerosol surfaces and particulate nitrate photolysis, are conducted by our measurements at the summit of Mt. Tai and recent laboratory studies. Therefore, it could be expected that similar conclusions can be found in other studies when considering ground measurements. Additionally, parameterizations of HONO sources used for box model simulations are applicable for other studies. The values of some parameters, such as NO2 uptake coefficients, MLH, and particulate nitrate photolysis frequency, are obtained or derived from laboratory or field studies. Further studies may improve the understanding of the variation of those parameters; for instance, NO2 uptake coefficients may vary with locations that have different landscapes. In particular, based on three vertical measurements in Germany, the USA, and China, similar levels of MLH (<100 m) were derived, indicating the potential application of this method to ground measurements worldwide. This could significantly reduce the underestimation of HONO formation from ground-derived sources compared to models in which the BLH was used. Meanwhile, sensitivity tests should be conducted, and uncertainties should be discussed accordingly.
It has been recognized that HONO photolysis could initiate daytime atmospheric chemistry in the early morning and also act as a substantial OH source during the daytime in polluted regions. The significant contribution and impact of HONO on radical levels could motivate studies by chemistry-transport models, most of which currently do not include HONO chemistry.
Furthermore, O3 pollution is becoming a key environmental issue in China. These high levels of O3 are often accompanied by moderate levels of NOx. In this case, NO3 chemistry could make a considerable contribution to atmospheric oxidization capacity, especially during the nighttime. Its follow-up impacts on atmospheric composition, such as the formation of nitrate and SOA, need further field measurements and model quantifications.
Atmospheric HONO and related parameters (VOCs, NOx, PM2.5, J(NO2), etc.) were measured at the foot and the summit of Mt. Tai in the summer of 2018. The present study was conducted mainly based on measurements at the foot station. The observed HONO varied from 0.05 to about 3 ppbv, with an average of 0.62 ± 0.42 ppbv. With the implementation of a 0-D box model (F0AM) coupled with the Master Chemical Mechanism (MCM v3.3.1), the HONO budget and the radical (ROx + NO3) chemistry were explored.
The main conclusions are summarized as follows:
-
The default HONO source, NO + OH, significantly underestimated the observed HONO concentrations. This reaction could only account for 13 % of the observed HONO, revealing a strong unknown source (Punknown). The diurnal profile of Punknown rapidly increased in the morning and peaked nearly 3 ppbv h−1 at ca. 1 h before noon, suggesting additional photo-enhanced HONO formation processes.
-
A HONONOx ratio of 0.7 % was derived for direct emission, and its contribution (15 %–25 % at night but negligible during the daytime) was quantified by a new method developed in this study. Based on the constraints on the aerosol-derived HONO sources (NO2 uptake on the aerosol surface and nitrate photolysis) obtained from the summit measurement (see the companion paper) and from recent laboratory studies, we found that the aerosol-derived HONO sources may not significantly contribute to HONO formation at the ground level. Their contributions to HONO formation were always smaller than NO + OH. Heterogeneous NO2 conversion on the ground surface made the largest contribution to Punknown, but it was sensitive to the MLH used for its parameterization. This addressed the importance of a reasonable MLH for exploring ground-level HONO formation in 0-D models and the necessity of vertical measurements.
-
HONO played an important role in ROx but a negligible role in NO3 concentrations. OH dominated the atmospheric oxidizing capacity in the daytime, while NO3 appeared to be significant at night. Peaks of NO3 time series and diurnal variation reached 22 and 9 pptv, respectively. NO3-induced reactions contribute 18 % of nitrate formation potential and 11 % of the C5H8 oxidation throughout the whole day, while at night, NO3 chemistry led to 51 % and 44 % of the nitrate formation potential and the C5H8 oxidation, respectively. NO3 chemistry may significantly affect nighttime secondary organic and inorganic aerosol formation in this high-O3 region. Hence, the direct measurement of NO3 (along with HOx, N2O5, ClNO2, etc.) in this region should be conducted.
All the data used in this study are available upon request from the corresponding authors.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-22-1035-2022-supplement.
CX, CY, WZ, XH, PL, CZ, XZ, CL, ZM, JL, and JW performed the field measurements. CX analyzed the observation data, performed model simulations, and wrote the paper with input from all co-authors. CY and JK also contributed with fruitful discussions and comments on model simulations and writing. JK, CY, KL, VC, AM, and YM revised the manuscript.
The contact author has declared that neither they nor their co-authors have any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
We are grateful to Shuyu Sun for her help with OVOC measurements. We thank all researchers involved in this campaign from the Research Centre for Eco-Environmental Sciences, Chinese Academy of Sciences, Fudan University, Shandong Jianzhu University, Shandong University, and the Municipal Environmental Protection Bureau of Tai'an. Chaoyang Xue thanks the University of Leeds and the University of York for providing the MCM v3.3.1 and Glenn M. Wolfe for providing the F0AM platform. We appreciate the three anonymous reviewers and the editor, John Orlando, for their careful reading of our paper and many insightful comments, suggestions, and discussion.
This work was supported by the National Natural Science Foundation of China (grant nos. 91544211, 41727805, 21976190, and 41975164), and the PIVOTS project provided by the Region Centre-Val de Loire (ARD 2020 program and CPER 2015–2020).
This paper was edited by John Orlando and reviewed by three anonymous referees.
Acker, K., Febo, A., Trick, S., Perrino, C., Bruno, P., Wiesen, P., Möller, D., Wieprecht, W., Auel, R., Giusto, M., Geyer, A., Platt, U., and Allegrini, I.: Nitrous acid in the urban area of Rome, Atmos. Environ., 40, 3123–3133, https://doi.org/10.1016/j.atmosenv.2006.01.028, 2006.
Alicke, B., Platt, U., and Stutz, J.: Impact of nitrous acid photolysis on the total hydroxyl radical budget during the Limitation of Oxidant Production/Pianura Padana Produzione di Ozono study in Milan, J. Geophys. Res.-Atmos., 107, 8196, https://doi.org/10.1029/2000JD000075, 2002.
Alicke, B., Geyer, A., Hofzumahaus, A., Holland, F., Konrad, S., Pätz, H.-W., Schäfer, J., Stutz, J., Volz-Thomas, A., and Platt, U.: OH formation by HONO photolysis during the BERLIOZ experiment, J. Geophys. Res.-Atmos., 108, 8247, https://doi.org/10.1029/2001JD000579, 2003.
Brown, S. S. and Stutz, J.: Nighttime radical observations and chemistry, Chem. Soc. Rev., 41, 6405–6447, https://doi.org/10.1039/c2cs35181a, 2012.
Carslaw, D. C. and Beevers, S. D.: Estimations of road vehicle primary NO2 exhaust emission fractions using monitoring data in London, Atmos. Environ., 39, 167–177, https://doi.org/10.1016/j.atmosenv.2004.08.053, 2005.
Chen, Q., Edebeli, J., McNamara, S. M., Kulju, K. D., May, N. W., Bertman, S. B., Thanekar, S., Fuentes, J. D., and Pratt, K. A.: HONO, Particulate Nitrite, and Snow Nitrite at a Midlatitude Urban Site during Wintertime, ACS Earth Sp. Chem., 3, 811–822, https://doi.org/10.1021/acsearthspacechem.9b00023, 2019.
Crilley, L. R., Kramer, L. J., Ouyang, B., Duan, J., Zhang, W., Tong, S., Ge, M., Tang, K., Qin, M., Xie, P., Shaw, M. D., Lewis, A. C., Mehra, A., Bannan, T. J., Worrall, S. D., Priestley, M., Bacak, A., Coe, H., Allan, J., Percival, C. J., Popoola, O. A. M., Jones, R. L., and Bloss, W. J.: Intercomparison of nitrous acid (HONO) measurement techniques in a megacity (Beijing), Atmos. Meas. Tech., 12, 6449–6463, https://doi.org/10.5194/amt-12-6449-2019, 2019.
Elshorbany, Y. F., Kurtenbach, R., Wiesen, P., Lissi, E., Rubio, M., Villena, G., Gramsch, E., Rickard, A. R., Pilling, M. J., and Kleffmann, J.: Oxidation capacity of the city air of Santiago, Chile, Atmos. Chem. Phys., 9, 2257–2273, https://doi.org/10.5194/acp-9-2257-2009, 2009.
Elshorbany, Y. F., Kleffmann, J., Kurtenbach, R., Lissi, E., Rubio, M., Villena, G., Gramsch, E., Rickard, A. R., Pilling, M. J., and Wiesen, P.: Seasonal dependence of the oxidation capacity of the city of Santiago de Chile, Atmos. Environ., 44, 5383–5394, https://doi.org/10.1016/j.atmosenv.2009.08.036, 2010.
Elshorbany, Y. F., Kleffmann, J., Hofzumahaus, A., Kurtenbach, R., Wiesen, P., Brauers, T., Bohn, B., Dorn, H.-P., Fuchs, H., Holland, F., Rohrer, F., Tillmann, R., Wegener, R., Wahner, A., Kanaya, Y., Yoshino, A., Nishida, S., Kajii, Y., Martinez, M., Kubistin, D., Harder, H., Lelieveld, J., Elste, T., Plass-Dümer, C., Stange, G., Berresheim, H., and Schurath, U.: HOx budgets during HOxComp: A case study of HOx chemistry under NOx-limited conditions, J. Geophys. Res.-Atmos., 117, D03307, https://doi.org/10.1029/2011JD017008, 2012.
Finlayson-Pitts, B. J., Wingen, L. M., Sumner, A. L., Syomin, D., and Ramazan, K. A.: The heterogeneous hydrolysis of NO2 in laboratory systems and in outdoor and indoor atmospheres: An integrated mechanism, Phys. Chem. Chem. Phys., 5, 223–242, https://doi.org/10.1039/b208564j, 2003.
George, C., Strekowski, R. S., Kleffmann, J., Stemmler, K., and Ammann, M.: Photoenhanced uptake of gaseous NO2 on solid organic compounds: a photochemical source of HONO?, Faraday Discuss., 130, 195–210, https://doi.org/10.1039/b417888m, 2005.
Han, C., Yang, W., Wu, Q., Yang, H., and Xue, X.: Heterogeneous Photochemical Conversion of NO2 to HONO on the Humic Acid Surface under Simulated Sunlight, Environ. Sci. Technol., 50, 5017–5023, https://doi.org/10.1021/acs.est.5b05101, 2016.
Han, S., Yao, Q., Tie, X., Zhang, Y., Zhang, M., Li, P., and Cai, Z.: Analysis of surface and vertical measurements of O3 and its chemical production in the NCP region, China, Atmos. Environ., 241, 117759, https://doi.org/10.1016/j.atmosenv.2020.117759, 2020.
Harrison, R. M., Peak, J. D., and Collins, G. M.: Tropospheric cycle of nitrous acid, J. Geophys. Res.-Atmos., 101, 14429–14439, https://doi.org/10.1029/96JD00341, 1996.
He, L., Zhang, S., Hu, J., Li, Z., Zheng, X., Cao, Y., Xu, G., Yan, M., and Wu, Y.: On-road emission measurements of reactive nitrogen compounds from heavy-duty diesel trucks in China, Environ. Pollut., 262, 114280, https://doi.org/10.1016/j.envpol.2020.114280, 2020.
Heard, D. E., Carpenter, L. J., Creasey, D. J., Hopkins, J. R., Lee, J. D., Lewis, A. C., Pilling, M. J., Seakins, P. W., Carslaw, N., and Emmerson, K. M.: High levels of the hydroxyl radical in the winter urban troposphere, Geophys. Res. Lett., 31, L18112, https://doi.org/10.1029/2004GL020544, 2004.
Heland, J., Kleffmann, J., Kurtenbach, R., and Wiesen, P.: A New Instrument To Measure Gaseous Nitrous Acid (HONO) in the Atmosphere, Environ. Sci. Technol., 35, 3207–3212, https://doi.org/10.1021/es000303t, 2001.
Hendrick, F., Müller, J.-F., Clémer, K., Wang, P., De Mazière, M., Fayt, C., Gielen, C., Hermans, C., Ma, J. Z., Pinardi, G., Stavrakou, T., Vlemmix, T., and Van Roozendael, M.: Four years of ground-based MAX-DOAS observations of HONO and NO2 in the Beijing area, Atmos. Chem. Phys., 14, 765–781, https://doi.org/10.5194/acp-14-765-2014, 2014.
Hofzumahaus, A., Rohrer, F., Lu, K., Bohn, B., Brauers, T., Chang, C.-C., Fuchs, H., Holland, F., Kita, K., Kondo, Y., Li, X., Lou, S., Shao, M., Zeng, L., Wahner, A., and Zhang, Y.: Amplified trace gas removal in the troposphere, Science, 324, 1702–1704, https://doi.org/10.1126/science.1164566, 2009.
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997.
Kanaya, Y., Pochanart, P., Liu, Y., Li, J., Tanimoto, H., Kato, S., Suthawaree, J., Inomata, S., Taketani, F., Okuzawa, K., Kawamura, K., Akimoto, H., and Wang, Z. F.: Rates and regimes of photochemical ozone production over Central East China in June 2006: a box model analysis using comprehensive measurements of ozone precursors, Atmos. Chem. Phys., 9, 7711–7723, https://doi.org/10.5194/acp-9-7711-2009, 2009.
Kanaya, Y., Akimoto, H., Wang, Z.-F., Pochanart, P., Kawamura, K., Liu, Y., Li, J., Komazaki, Y., Irie, H., Pan, X.-L., Taketani, F., Yamaji, K., Tanimoto, H., Inomata, S., Kato, S., Suthawaree, J., Okuzawa, K., Wang, G., Aggarwal, S. G., Fu, P. Q., Wang, T., Gao, J., Wang, Y., and Zhuang, G.: Overview of the Mount Tai Experiment (MTX2006) in central East China in June 2006: studies of significant regional air pollution, Atmos. Chem. Phys., 13, 8265–8283, https://doi.org/10.5194/acp-13-8265-2013, 2013.
Kirchstetter, T. W., Harley, R. A., and Littlejohn, D.: Measurement of nitrous acid in motor vehicle exhaust, Environ. Sci. Technol., 30, 2843–2849, https://doi.org/10.1021/es960135y, 1996.
Kleffmann, J.: Daytime sources of nitrous acid (HONO) in the atmospheric boundary layer, ChemPhysChem, 8, 1137–1144, https://doi.org/10.1002/cphc.200700016, 2007.
Kleffmann, J., Kurtenbach, R., Lörzer, J., Wiesen, P., Kalthoff, N., Vogel, B., and Vogel, H.: Measured and simulated vertical profiles of nitrous acid – Part I: Field measurements, Atmos. Environ., 37, 2949–2955, https://doi.org/10.1016/S1352-2310(03)00242-5, 2003.
Kleffmann, J., Gavriloaiei, T., Hofzumahaus, A., Holland, F., Koppmann, R., Rupp, L., Schlosser, E., Siese, M., and Wahner, A.: Daytime formation of nitrous acid: A major source of OH radicals in a forest, Geophys. Res. Lett., 32, L05818, https://doi.org/10.1029/2005GL022524, 2005.
Kleffmann, J., Lörzer, J. C., Wiesen, P., Kern, C., Trick, S., Volkamer, R., Rodenas, M., and Wirtz, K.: Intercomparison of the DOAS and LOPAP techniques for the detection of nitrous acid (HONO), Atmos. Environ., 40, 3640–3652, https://doi.org/10.1016/j.atmosenv.2006.03.027, 2006.
Kramer, L. J., Crilley, L. R., Adams, T. J., Ball, S. M., Pope, F. D., and Bloss, W. J.: Nitrous acid (HONO) emissions under real-world driving conditions from vehicles in a UK road tunnel, Atmos. Chem. Phys., 20, 5231–5248, https://doi.org/10.5194/acp-20-5231-2020, 2020.
Kukui, A., Legrand, M., Preunkert, S., Frey, M. M., Loisil, R., Gil Roca, J., Jourdain, B., King, M. D., France, J. L., and Ancellet, G.: Measurements of OH and RO2 radicals at Dome C, East Antarctica, Atmos. Chem. Phys., 14, 12373–12392, https://doi.org/10.5194/acp-14-12373-2014, 2014.
Kurtenbach, R., Becker, K. H., Gomes, J. A. G., Kleffmann, J., Lörzer, J. C., Spittler, M., Wiesen, P., Ackermann, R., Geyer, A., and Platt, U.: Investigations of emissions and heterogeneous formation of HONO in a road traffic tunnel, Atmos. Environ., 35, 3385–3394, https://doi.org/10.1016/S1352-2310(01)00138-8, 2001.
Kurtenbach, R., Kleffmann, J., Niedojadlo, A., and Wiesen, P.: Primary NO2 emissions and their impact on air quality in traffic environments in Germany, Environ. Sci. Eur., 24, 21, https://doi.org/10.1186/2190-4715-24-21, 2012.
Laufs, S. and Kleffmann, J.: Investigations on HONO formation from photolysis of adsorbed HNO3 on quartz glass surfaces, Phys. Chem. Chem. Phys., 18, 9616–9625, https://doi.org/10.1039/c6cp00436a, 2016.
Lee, J. D., Whalley, L. K., Heard, D. E., Stone, D., Dunmore, R. E., Hamilton, J. F., Young, D. E., Allan, J. D., Laufs, S., and Kleffmann, J.: Detailed budget analysis of HONO in central London reveals a missing daytime source, Atmos. Chem. Phys., 16, 2747–2764, https://doi.org/10.5194/acp-16-2747-2016, 2016.
Legrand, M., Preunkert, S., Frey, M., Bartels-Rausch, Th., Kukui, A., King, M. D., Savarino, J., Kerbrat, M., and Jourdain, B.: Large mixing ratios of atmospheric nitrous acid (HONO) at Concordia (East Antarctic Plateau) in summer: a strong source from surface snow?, Atmos. Chem. Phys., 14, 9963–9976, https://doi.org/10.5194/acp-14-9963-2014, 2014.
Li, D., Xue, L., Wen, L., Wang, X., Chen, T., Mellouki, A., Chen, J., and Wang, W.: Characteristics and sources of nitrous acid in an urban atmosphere of northern China: Results from 1-yr continuous observations, Atmos. Environ., 182, 296–306, https://doi.org/10.1016/j.atmosenv.2018.03.033, 2018.
Li, G., Lei, W., Zavala, M., Volkamer, R., Dusanter, S., Stevens, P., and Molina, L. T.: Impacts of HONO sources on the photochemistry in Mexico City during the MCMA-2006/MILAGO Campaign, Atmos. Chem. Phys., 10, 6551–6567, https://doi.org/10.5194/acp-10-6551-2010, 2010.
Li, K., Jacob, D. J., Liao, H., Shen, L., Zhang, Q., and Bates, K. H.: Anthropogenic drivers of 2013–2017 trends in summer surface ozone in China, P. Natl. Acad. Sci. USA, 116, 422–427, https://doi.org/10.1073/pnas.1812168116, 2019.
Li, X., Brauers, T., Häseler, R., Bohn, B., Fuchs, H., Hofzumahaus, A., Holland, F., Lou, S., Lu, K. D., Rohrer, F., Hu, M., Zeng, L. M., Zhang, Y. H., Garland, R. M., Su, H., Nowak, A., Wiedensohler, A., Takegawa, N., Shao, M., and Wahner, A.: Exploring the atmospheric chemistry of nitrous acid (HONO) at a rural site in Southern China, Atmos. Chem. Phys., 12, 1497–1513, https://doi.org/10.5194/acp-12-1497-2012, 2012.
Liu, C., Mu, Y., Zhang, C., Zhang, Z., Zhang, Y., Liu, J., Sheng, J., and Quan, J.: Development of gas chromatography-flame ionization detection system with a single column and liquid nitrogen-free for measuring atmospheric C2–C12 hydrocarbons, J. Chromatogr. A, 1427, 134–141, https://doi.org/10.1016/j.chroma.2015.11.060, 2016.
Liu, P., Ye, C., Xue, C., Zhang, C., Mu, Y., and Sun, X.: Formation mechanisms of atmospheric nitrate and sulfate during the winter haze pollution periods in Beijing: gas-phase, heterogeneous and aqueous-phase chemistry, Atmos. Chem. Phys., 20, 4153–4165, https://doi.org/10.5194/acp-20-4153-2020, 2020.
Liu, Y., Lu, K., Ma, Y., Yang, X., Zhang, W., Wu, Y., Peng, J., Shuai, S., Hu, M., and Zhang, Y.: Direct emission of nitrous acid (HONO) from gasoline cars in China determined by vehicle chassis dynamometer experiments, Atmos. Environ., 169, 89–96, https://doi.org/10.1016/j.atmosenv.2017.07.019, 2017a.
Liu, Y., Lu, K., Ma, Y., Yang, X., Zhang, W., Wu, Y., Peng, J., Shuai, S., Hu, M., and Zhang, Y.: Direct emission of nitrous acid (HONO) from gasoline cars in China determined by vehicle chassis dynamometer experiments, Atmos. Environ., 169, 89–96, https://doi.org/10.1016/j.atmosenv.2017.07.019, 2017b.
Liu, Y., Lu, K., Li, X., Dong, H., Tan, Z., Wang, H., Zou, Q., Wu, Y., Zeng, L., Hu, M., Min, K.-E., Kecorius, S., Wiedensohler, A., and Zhang, Y.: A Comprehensive Model Test of the HONO Sources Constrained to Field Measurements at Rural North China Plain, Environ. Sci. Technol., 53, 3517–3525, https://doi.org/10.1021/acs.est.8b06367, 2019.
Liu, Z., Wang, Y., Gu, D., Zhao, C., Huey, L. G., Stickel, R., Liao, J., Shao, M., Zhu, T., Zeng, L., Amoroso, A., Costabile, F., Chang, C.-C., and Liu, S.-C.: Summertime photochemistry during CAREBeijing-2007: ROx budgets and O3 formation, Atmos. Chem. Phys., 12, 7737–7752, https://doi.org/10.5194/acp-12-7737-2012, 2012.
Lu, K., Guo, S., Tan, Z., Wang, H., Shang, D., Liu, Y., Li, X., Wu, Z., Hu, M., and Zhang, Y.: Exploring atmospheric free-radical chemistry in China: the self-cleansing capacity and the formation of secondary air pollution, Natl. Sci. Rev., 6, 579–594, https://doi.org/10.1093/nsr/nwy073, 2019.
Mellouki, A., Ammann, M., Cox, R. A., Crowley, J. N., Herrmann, H., Jenkin, M. E., McNeill, V. F., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: volume VIII – gas-phase reactions of organic species with four, or more, carbon atoms (≥ C4), Atmos. Chem. Phys., 21, 4797–4808, https://doi.org/10.5194/acp-21-4797-2021, 2021.
Meng, F., Qin, M., Tang, K., Duan, J., Fang, W., Liang, S., Ye, K., Xie, P., Sun, Y., Xie, C., Ye, C., Fu, P., Liu, J., and Liu, W.: High-resolution vertical distribution and sources of HONO and NO2 in the nocturnal boundary layer in urban Beijing, China, Atmos. Chem. Phys., 20, 5071–5092, https://doi.org/10.5194/acp-20-5071-2020, 2020.
Meusel, H., Kuhn, U., Reiffs, A., Mallik, C., Harder, H., Martinez, M., Schuladen, J., Bohn, B., Parchatka, U., Crowley, J. N., Fischer, H., Tomsche, L., Novelli, A., Hoffmann, T., Janssen, R. H. H., Hartogensis, O., Pikridas, M., Vrekoussis, M., Bourtsoukidis, E., Weber, B., Lelieveld, J., Williams, J., Pöschl, U., Cheng, Y., and Su, H.: Daytime formation of nitrous acid at a coastal remote site in Cyprus indicating a common ground source of atmospheric HONO and NO, Atmos. Chem. Phys., 16, 14475–14493, https://doi.org/10.5194/acp-16-14475-2016, 2016.
Michoud, V., Kukui, A., Camredon, M., Colomb, A., Borbon, A., Miet, K., Aumont, B., Beekmann, M., Durand-Jolibois, R., Perrier, S., Zapf, P., Siour, G., Ait-Helal, W., Locoge, N., Sauvage, S., Afif, C., Gros, V., Furger, M., Ancellet, G., and Doussin, J. F.: Radical budget analysis in a suburban European site during the MEGAPOLI summer field campaign, Atmos. Chem. Phys., 12, 11951–11974, https://doi.org/10.5194/acp-12-11951-2012, 2012.
Michoud, V., Colomb, A., Borbon, A., Miet, K., Beekmann, M., Camredon, M., Aumont, B., Perrier, S., Zapf, P., Siour, G., Ait-Helal, W., Afif, C., Kukui, A., Furger, M., Dupont, J. C., Haeffelin, M., and Doussin, J. F.: Study of the unknown HONO daytime source at a European suburban site during the MEGAPOLI summer and winter field campaigns, Atmos. Chem. Phys., 14, 2805–2822, https://doi.org/10.5194/acp-14-2805-2014, 2014.
Neuman, J. A., Trainer, M., Brown, S. S., Min, K.-E., Nowak, J. B., Parrish, D. D., Peischl, J., Pollack, I. B., Roberts, J. M., Ryerson, T. B., and Veres, P. R.: HONO emission and production determined from airborne measurements over the Southeast U.S., J. Geophys. Res.-Atmos., 121, 9237–9250, https://doi.org/10.1002/2016JD025197, 2016.
Oswald, R., Behrendt, T., Ermel, M., Wu, D., Su, H., Cheng, Y., Breuninger, C., Moravek, A., Mougin, E., Delon, C., Loubet, B., Pommerening-Röser, A., Sörgel, M., Pöschl, U., Hoffmann, T., Andreae, M. O., Meixner, F. X., and Trebs, I.: HONO emissions from soil bacteria as a major source of atmospheric reactive nitrogen, Science, 341, 1233–1235, https://doi.org/10.1126/science.1242266, 2013.
Platt, U., Perner, D., Harris, G. W., Winer, A. M., and Pitts Jr., J. N.: Observations of nitrous acid in an urban atmosphere by differential optical absorption, Nature, 285, 312–314, https://doi.org/10.1038/285312a0, 1980.
Qin, M., Xie, P.-H., Liu, W.-Q., Li, A., Dou, K., Fang, W., Liu, J.-G., and Zhang, W.-J.: Observation of atmospheric nitrous acid with DOAS in Beijing, China, J. Environ. Sci., 18, 69–75, 2006.
Ren, X., Harder, H., Martinez, M., Lesher, R. L., Oliger, A., Simpas, J. B., Brune, W. H., Schwab, J. J., Demerjian, K. L., He, Y., Zhou, X., and Gao, H.: OH and HO2 chemistry in the urban atmosphere of New York City, Atmos. Environ., 37, 3639–3651, https://doi.org/10.1016/S1352-2310(03)00459-X, 2003.
Roberts, J. M., Williams, J., Baumann, K., Buhr, M. P., Goldan, P. D., Holloway, J., Hübler, G., Kuster, W. C., McKeen, S. A., Ryerson, T. B., Trainer, M., Williams, E. J., Fehsenfeld, F. C., Bertman, S. B., Nouaime, G., Seaver, C., Grodzinsky, G., Rodgers, M., and Young, V. L.: Measurements of PAN, PPN, and MPAN made during the 1994 and 1995 Nashville Intensives of the Southern Oxidant Study: Implications for regional ozone production from biogenic hydrocarbons, J. Geophys. Res.-Atmos., 103, 22473–22490, https://doi.org/10.1029/98JD01637, 1998.
Rohrer, F. and Berresheim, H.: Strong correlation between levels of tropospheric hydroxyl radicals and solar ultraviolet radiation, Nature, 442, 184–187, https://doi.org/10.1038/nature04924, 2006.
Rollins, A. W., Kiendler-Scharr, A., Fry, J. L., Brauers, T., Brown, S. S., Dorn, H.-P., Dubé, W. P., Fuchs, H., Mensah, A., Mentel, T. F., Rohrer, F., Tillmann, R., Wegener, R., Wooldridge, P. J., and Cohen, R. C.: Isoprene oxidation by nitrate radical: alkyl nitrate and secondary organic aerosol yields, Atmos. Chem. Phys., 9, 6685–6703, https://doi.org/10.5194/acp-9-6685-2009, 2009.
Romer, P. S., Wooldridge, P. J., Crounse, J. D., Kim, M. J., Wennberg, P. O., Dibb, J. E., Scheuer, E., Blake, D. R., Meinardi, S., Brosius, A. L., Thames, A. B., Miller, D. O., Brune, W. H., Hall, S. R., Ryerson, T. B., and Cohen, R. C.: Constraints on Aerosol Nitrate Photolysis as a Potential Source of HONO and NOx, Environ. Sci. Technol., 52, 13738–13746, https://doi.org/10.1021/acs.est.8b03861, 2018.
Sarwar, G., Roselle, S. J., Mathur, R., Appel, W., Dennis, R. L., and Vogel, B.: A comparison of CMAQ HONO predictions with observations from the Northeast Oxidant and Particle Study, Atmos. Environ., 42, 5760–5770, https://doi.org/10.1016/j.atmosenv.2007.12.065, 2008.
Scharko, N. K., Schütte, U. M. E., Berke, A. E., Banina, L., Peel, H. R., Donaldson, M. A., Hemmerich, C., White, J. R., and Raff, J. D.: Combined Flux Chamber and Genomics Approach Links Nitrous Acid Emissions to Ammonia Oxidizing Bacteria and Archaea in Urban and Agricultural Soil, Environ. Sci. Technol., 49, 13825–13834, https://doi.org/10.1021/acs.est.5b00838, 2015.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, 3rd edn., John Wiley & Sons, ISBN 978-1-118-94740-1, 2016.
Shi, Q., Tao, Y., Krechmer, J. E., Heald, C. L., Murphy, J. G., Kroll, J. H., and Ye, Q.: Laboratory Investigation of Renoxification from the Photolysis of Inorganic Particulate Nitrate, Environ. Sci. Technol., 55, 854–861, https://doi.org/10.1021/acs.est.0c06049, 2021.
Slater, E. J., Whalley, L. K., Woodward-Massey, R., Ye, C., Lee, J. D., Squires, F., Hopkins, J. R., Dunmore, R. E., Shaw, M., Hamilton, J. F., Lewis, A. C., Crilley, L. R., Kramer, L., Bloss, W., Vu, T., Sun, Y., Xu, W., Yue, S., Ren, L., Acton, W. J. F., Hewitt, C. N., Wang, X., Fu, P., and Heard, D. E.: Elevated levels of OH observed in haze events during wintertime in central Beijing, Atmos. Chem. Phys., 20, 14847–14871, https://doi.org/10.5194/acp-20-14847-2020, 2020.
Sörgel, M., Regelin, E., Bozem, H., Diesch, J.-M., Drewnick, F., Fischer, H., Harder, H., Held, A., Hosaynali-Beygi, Z., Martinez, M., and Zetzsch, C.: Quantification of the unknown HONO daytime source and its relation to NO2, Atmos. Chem. Phys., 11, 10433–10447, https://doi.org/10.5194/acp-11-10433-2011, 2011.
Spataro, F., Ianniello, A., Esposito, G., Allegrini, I., Zhu, T., and Hu, M.: Occurrence of atmospheric nitrous acid in the urban area of Beijing (China), Sci. Total Environ., 447, 210–224, https://doi.org/10.1016/j.scitotenv.2012.12.065, 2013.
Stemmler, K., Ammann, M., Donders, C., Kleffmann, J., and George, C.: Photosensitized reduction of nitrogen dioxide on humic acid as a source of nitrous acid, Nature, 440, 195–198, https://doi.org/10.1038/nature04603, 2006.
Stemmler, K., Ndour, M., Elshorbany, Y., Kleffmann, J., D'Anna, B., George, C., Bohn, B., and Ammann, M.: Light induced conversion of nitrogen dioxide into nitrous acid on submicron humic acid aerosol, Atmos. Chem. Phys., 7, 4237–4248, https://doi.org/10.5194/acp-7-4237-2007, 2007.
Stone, D., Whalley, L. K., and Heard, D. E.: Tropospheric OH and HO2 radicals: Field measurements and model comparisons, Chem. Soc. Rev., 41, 6348–6404, https://doi.org/10.1039/c2cs35140d, 2012.
Su, H., Cheng, Y. F., Shao, M., Gao, D. F., Yu, Z. Y., Zeng, L. M., Slanina, J., Zhang, Y. H., and Wiedensohler, A.: Nitrous acid (HONO) and its daytime sources at a rural site during the 2004 PRIDE-PRD experiment in China, J. Geophys. Res.-Atmos., 113, D14312, https://doi.org/10.1029/2007JD009060, 2008.
Suhail, K., George, M., Chandran, S., Varma, R., Venables, D. S., Wang, M., and Chen, J.: Open path incoherent broadband cavity-enhanced measurements of NO3 radical and aerosol extinction in the North China Plain, Spectrochim. Acta A, 208, 24–31, https://doi.org/10.1016/j.saa.2018.09.023, 2019.
Sun, L., Xue, L., Wang, T., Gao, J., Ding, A., Cooper, O. R., Lin, M., Xu, P., Wang, Z., Wang, X., Wen, L., Zhu, Y., Chen, T., Yang, L., Wang, Y., Chen, J., and Wang, W.: Significant increase of summertime ozone at Mount Tai in Central Eastern China, Atmos. Chem. Phys., 16, 10637–10650, https://doi.org/10.5194/acp-16-10637-2016, 2016.
Sun, L., Xue, L., Wang, Y., Li, L., Lin, J., Ni, R., Yan, Y., Chen, L., Li, J., Zhang, Q., and Wang, W.: Impacts of meteorology and emissions on summertime surface ozone increases over central eastern China between 2003 and 2015, Atmos. Chem. Phys., 19, 1455–1469, https://doi.org/10.5194/acp-19-1455-2019, 2019.
Tan, Z., Fuchs, H., Lu, K., Hofzumahaus, A., Bohn, B., Broch, S., Dong, H., Gomm, S., Häseler, R., He, L., Holland, F., Li, X., Liu, Y., Lu, S., Rohrer, F., Shao, M., Wang, B., Wang, M., Wu, Y., Zeng, L., Zhang, Y., Wahner, A., and Zhang, Y.: Radical chemistry at a rural site (Wangdu) in the North China Plain: observation and model calculations of OH, HO2 and RO2 radicals, Atmos. Chem. Phys., 17, 663–690, https://doi.org/10.5194/acp-17-663-2017, 2017.
Tang, K., Qin, M., Duan, J., Fang, W., Meng, F., Liang, S., Xie, P., Liu, J., Liu, W., Xue, C., and Mu, Y.: A dual dynamic chamber system based on IBBCEAS for measuring fluxes of nitrous acid in agricultural fields in the North China Plain, Atmos. Environ., 196, 10–19, https://doi.org/10.1016/j.atmosenv.2018.09.059, 2019.
Travis, K. R., Heald, C. L., Allen, H. M., Apel, E. C., Arnold, S. R., Blake, D. R., Brune, W. H., Chen, X., Commane, R., Crounse, J. D., Daube, B. C., Diskin, G. S., Elkins, J. W., Evans, M. J., Hall, S. R., Hintsa, E. J., Hornbrook, R. S., Kasibhatla, P. S., Kim, M. J., Luo, G., McKain, K., Millet, D. B., Moore, F. L., Peischl, J., Ryerson, T. B., Sherwen, T., Thames, A. B., Ullmann, K., Wang, X., Wennberg, P. O., Wolfe, G. M., and Yu, F.: Constraining remote oxidation capacity with ATom observations, Atmos. Chem. Phys., 20, 7753–7781, https://doi.org/10.5194/acp-20-7753-2020, 2020.
Vandenboer, T. C., Brown, S. S., Murphy, J. G., Keene, W. C., Young, C. J., Pszenny, A. A. P., Kim, S., Warneke, C., De Gouw, J. A., Maben, J. R., Wagner, N. L., Riedel, T. P., Thornton, J. A., Wolfe, D. E., Dubé, W. P., Öztürk, F., Brock, C. A., Grossberg, N., Lefer, B., Lerner, B., Middlebrook, A. M., and Roberts, J. M.: Understanding the role of the ground surface in HONO vertical structure: High resolution vertical profiles during NACHTT-11, J. Geophys. Res.-Atmos., 118, 10155–10171, https://doi.org/10.1002/jgrd.50721, 2013.
Villena, G., Kleffmann, J., Kurtenbach, R., Wiesen, P., Lissi, E., Rubio, M. A., Croxatto, G., and Rappenglück, B.: Vertical gradients of HONO, NOx and O3 in Santiago de Chile, Atmos. Environ., 45, 3867–3873, https://doi.org/10.1016/j.atmosenv.2011.01.073, 2011.
Villena, G., Bejan, I., Kurtenbach, R., Wiesen, P., and Kleffmann, J.: Interferences of commercial NO2 instruments in the urban atmosphere and in a smog chamber, Atmos. Meas. Tech., 5, 149–159, https://doi.org/10.5194/amt-5-149-2012, 2012.
Vogel, B., Vogel, H., Kleffmann, J., and Kurtenbach, R.: Measured and simulated vertical profiles of nitrous acid – Part II. Model simulations and indications for a photolytic source, Atmos. Environ., 37, 2957–2966, https://doi.org/10.1016/S1352-2310(03)00243-7, 2003.
Volkamer, R., Sheehy, P., Molina, L. T., and Molina, M. J.: Oxidative capacity of the Mexico City atmosphere – Part 1: A radical source perspective, Atmos. Chem. Phys., 10, 6969–6991, https://doi.org/10.5194/acp-10-6969-2010, 2010.
Wang, J., Sun, S., Zhang, C., Xue, C., Liu, P., Zhang, C., Mu, Y., Wu, H., Wang, D., Chen, H., and Chen, J.: The pollution levels, variation characteristics, sources and implications of atmospheric carbonyls in a typical rural area of North China Plain during winter, J. Environ. Sci., 95, 256–265, https://doi.org/10.1016/j.jes.2020.05.003, 2020.
Wang, X., Wang, H., Xue, L., Wang, T., Wang, L., Gu, R., Wang, W., Tham, Y. J., Wang, Z., Yang, L., Chen, J., and Wang, W.: Observations of N2O5 and ClNO2 at a polluted urban surface site in North China: High N2O5 uptake coefficients and low ClNO2 product yields, Atmos. Environ., 156, 125–134, https://doi.org/10.1016/j.atmosenv.2017.02.035, 2017.
Wang, X., Dalton, E. Z., Payne, Z. C., Perrier, S., Riva, M., Raff, J. D., and George, C.: Superoxide and Nitrous Acid Production from Nitrate Photolysis Is Enhanced by Dissolved Aliphatic Organic Matter, Environ. Sci. Technol. Lett., 8, 53–58, https://doi.org/10.1021/acs.estlett.0c00806, 2021.
Wang, Y., Dörner, S., Donner, S., Böhnke, S., De Smedt, I., Dickerson, R. R., Dong, Z., He, H., Li, Z., Li, Z., Li, D., Liu, D., Ren, X., Theys, N., Wang, Y., Wang, Y., Wang, Z., Xu, H., Xu, J., and Wagner, T.: Vertical profiles of NO2, SO2, HONO, HCHO, CHOCHO and aerosols derived from MAX-DOAS measurements at a rural site in the central western North China Plain and their relation to emission sources and effects of regional transport, Atmos. Chem. Phys., 19, 5417–5449, https://doi.org/10.5194/acp-19-5417-2019, 2019.
Whalley, L. K., Slater, E. J., Woodward-Massey, R., Ye, C., Lee, J. D., Squires, F., Hopkins, J. R., Dunmore, R. E., Shaw, M., Hamilton, J. F., Lewis, A. C., Mehra, A., Worrall, S. D., Bacak, A., Bannan, T. J., Coe, H., Percival, C. J., Ouyang, B., Jones, R. L., Crilley, L. R., Kramer, L. J., Bloss, W. J., Vu, T., Kotthaus, S., Grimmond, S., Sun, Y., Xu, W., Yue, S., Ren, L., Acton, W. J. F., Hewitt, C. N., Wang, X., Fu, P., and Heard, D. E.: Evaluating the sensitivity of radical chemistry and ozone formation to ambient VOCs and NOx in Beijing, Atmos. Chem. Phys., 21, 2125–2147, https://doi.org/10.5194/acp-21-2125-2021, 2021.
Wild, R. J., Dubé, W. P., Aikin, K. C., Eilerman, S. J., Neuman, J. A., Peischl, J., Ryerson, T. B., and Brown, S. S.: On-road measurements of vehicle NONOx emission ratios in Denver, Colorado, USA, Atmos. Environ., 148, 182–189, https://doi.org/10.1016/j.atmosenv.2016.10.039, 2017.
Wolfe, G. M., Marvin, M. R., Roberts, S. J., Travis, K. R., and Liao, J.: The Framework for 0-D Atmospheric Modeling (F0AM) v3.1, Geosci. Model Dev., 9, 3309–3319, https://doi.org/10.5194/gmd-9-3309-2016, 2016.
Wong, K. W., Tsai, C., Lefer, B., Haman, C., Grossberg, N., Brune, W. H., Ren, X., Luke, W., and Stutz, J.: Daytime HONO vertical gradients during SHARP 2009 in Houston, TX, Atmos. Chem. Phys., 12, 635–652, https://doi.org/10.5194/acp-12-635-2012, 2012.
Wong, K. W., Tsai, C., Lefer, B., Grossberg, N., and Stutz, J.: Modeling of daytime HONO vertical gradients during SHARP 2009, Atmos. Chem. Phys., 13, 3587–3601, https://doi.org/10.5194/acp-13-3587-2013, 2013.
Wu, Z., Hu, M., Lin, P., Liu, S., Wehner, B., and Wiedensohler, A.: Particle number size distribution in the urban atmosphere of Beijing, China, Atmos. Environ., 42, 7967–7980, https://doi.org/10.1016/j.atmosenv.2008.06.022, 2008.
Xing, C., Liu, C., Hu, Q., Fu, Q., Wang, S., Lin, H., Zhu, Y., Wang, S., Wang, W., Javed, Z., Ji, X., and Liu, J.: Vertical distributions of wintertime atmospheric nitrogenous compounds and the corresponding OH radicals production in Leshan, southwest China, J. Environ. Sci., 105, 44–55, https://doi.org/10.1016/j.jes.2020.11.019, 2021.
Xue, C., Ye, C., Zhang, Y., Ma, Z., Liu, P., Zhang, C., Zhao, X., Liu, J., and Mu, Y.: Development and application of a twin open-top chambers method to measure soil HONO emission in the North China Plain, Sci. Total Environ., 659, 621–631, https://doi.org/10.1016/j.scitotenv.2018.12.245, 2019.
Xue, C., Zhang, C., Ye, C., Liu, P., Catoire, V., Krysztofiak, G., Chen, H., Ren, Y., Zhao, X., Wang, J., Zhang, F., Zhang, C., Zhang, J., An, J., Wang, T., Chen, J., Kleffmann, J., Mellouki, A., and Mu, Y.: HONO Budget and Its Role in Nitrate Formation in the Rural North China Plain, Environ. Sci. Technol., 54, 11048–11057, https://doi.org/10.1021/acs.est.0c01832, 2020.
Xue, C., Ye, C., Kleffmann, J., Zhang, C., Catoire, V., Bao, F., Mellouki, A., Xue, L., Chen, J., Lu, K., Zhao, Y., Liu, H., Guo, Z., and Mu, Y.: Atmospheric Measurements at the Foot and the Summit of Mt. Tai – Part I: HONO Formation and Its Role in the Oxidizing Capacity of the Upper Boundary Layer, Atmos. Chem. Phys. Discuss. [preprint], https://doi.org/10.5194/acp-2021-529, in review, 2021a.
Xue, C., Ye, C., Zhang, C., Catoire, V., Liu, P., Gu, R., Zhang, J., Ma, Z., Zhao, X., Zhang, W., Ren, Y., Krysztofiak, G., Tong, S., Xue, L., An, J., Ge, M., Mellouki, A., and Mu, Y.: Evidence for Strong HONO Emission from Fertilized Agricultural Fields and its Remarkable Impact on Regional O3 Pollution in the Summer North China Plain, ACS Earth Sp. Chem., 5, 340–347, https://doi.org/10.1021/acsearthspacechem.0c00314, 2021b.
Xue, L. K., Wang, T., Zhang, J. M., Zhang, X. C., Deliger, Poon, C. N., Ding, A. J., Zhou, X. H., Wu, W. S., Tang, J., Zhang, Q. Z., and Wang, W. X.: Source of surface ozone and reactive nitrogen speciation at Mount Waliguan in western China: New insights from the 2006 summer study, J. Geophys. Res.-Atmos., 116, D07306, https://doi.org/10.1029/2010JD014735, 2011.
Yang, Q., Su, H., Li, X., Cheng, Y., Lu, K., Cheng, P., Gu, J., Guo, S., Hu, M., Zeng, L., Zhu, T., and Zhang, Y.: Daytime HONO formation in the suburban area of the megacity Beijing, China, Sci. China Chem., 57, 1032–1042, https://doi.org/10.1007/s11426-013-5044-0, 2014.
Ye, C., Liu, P., Ma, Z., Xue, C., Zhang, C., Zhang, Y., Liu, J., Liu, C., Sun, X., and Mu, Y.: High H2O2 Concentrations Observed during Haze Periods during the Winter in Beijing: Importance of H2O2 Oxidation in Sulfate Formation, Environ. Sci. Technol. Lett., 5, 757–763, https://doi.org/10.1021/acs.estlett.8b00579, 2018a.
Ye, C., Zhou, X., Pu, D., Stutz, J., Festa, J., Spolaor, M., Tsai, C., Cantrell, C., Mauldin III, R. L., Weinheimer, A., Hornbrook, R. S., Apel, E. C., Guenther, A., Kaser, L., Yuan, B., Karl, T., Haggerty, J., Hall, S., Ullmann, K., Smith, J., and Ortega, J.: Tropospheric HONO distribution and chemistry in the southeastern US, Atmos. Chem. Phys., 18, 9107–9120, https://doi.org/10.5194/acp-18-9107-2018, 2018b.
Zhang, J., An, J., Qu, Y., Liu, X., and Chen, Y.: Impacts of potential HONO sources on the concentrations of oxidants and secondary organic aerosols in the Beijing-Tianjin-Hebei region of China, Sci. Total Environ., 647, 836–852, https://doi.org/10.1016/j.scitotenv.2018.08.030, 2019a.
Zhang, J., Chen, J., Xue, C., Chen, H., Zhang, Q., Liu, X., Mu, Y., Guo, Y., Wang, D., Chen, Y., Li, J., Qu, Y., and An, J.: Impacts of six potential HONO sources on HOx budgets and SOA formation during a wintertime heavy haze period in the North China Plain, Sci. Total Environ., 681, 110–123, https://doi.org/10.1016/j.scitotenv.2019.05.100, 2019b.
Zhang, L., Wang, T., Zhang, Q., Zheng, J., Xu, Z., and Lv, M.: Potential sources of nitrous acid (HONO) and their impacts on ozone: A WRF-Chem study in a polluted subtropical region, J. Geophys. Res.-Atmos., 121, 3645–3662, https://doi.org/10.1002/2015JD024468, 2016.
Zhang, Q., Streets, D. G., Carmichael, G. R., He, K. B., Huo, H., Kannari, A., Klimont, Z., Park, I. S., Reddy, S., Fu, J. S., Chen, D., Duan, L., Lei, Y., Wang, L. T., and Yao, Z. L.: Asian emissions in 2006 for the NASA INTEX-B mission, Atmos. Chem. Phys., 9, 5131–5153, https://doi.org/10.5194/acp-9-5131-2009, 2009.