the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 22 May 2021
| 22 May 2021
On the similarities and differences between the products of oxidation of hydrocarbons under simulated atmospheric conditions and cool flames
Roland Benoit
Nesrine Belhadj
Maxence Lailliau
Philippe Dagaut
Atmospheric oxidation chemistry and, more specifically, photooxidation show that the long-term oxidation of organic aerosol (OA) progressively erases the initial signature of the chemical compounds and can lead to a relatively uniform character of oxygenated organic aerosol (OOA). This uniformity character observed after a long reaction time seems to contrast with the great diversity of reaction mechanisms observed in the early stages of oxidation. The numerous studies carried out on the oxidation of terpenes, and more particularly on limonene for its diversity of reaction sites (endo- and oxocyclic), allow this evolution to be studied. We have selected, for their diversity of experimental conditions, nine studies of limonene oxidation at room temperature over long reaction times to be compared to the present data set obtained at elevated temperature and short reaction time in order to investigate the similarities in terms of reaction mechanisms and chemical species formed. Here, the oxidation of limonene–oxygen–nitrogen mixtures was studied using a jet-stirred reactor at elevated temperature and atmospheric pressure. Samples of the reacting mixtures were collected and analyzed by high-resolution mass spectrometry (Orbitrap) after direct injection or after separation by reverse-phase ultra-high-pressure liquid chromatography and soft ionization, i.e., () HESI and () APCI. Unexpectedly, because of the diversity of experimental conditions in terms of continuous-flow tank reactor, concentration of reactants, temperature, reaction time, mass spectrometry techniques, and analysis conditions, the results indicate that among the 1138 presently detected molecular formulae, many oxygenates found in earlier studies of limonene oxidation by OH and/or ozone are also produced under the present conditions. Among these molecular formulae, highly oxygenated molecules and oligomers were detected in the present work. The results are discussed in terms of reaction pathways involving the initial formation of peroxy radicals (RO2), isomerization reactions yielding keto-hydroperoxides, and other oxygenated intermediates and products up to C25H32O17, products which could derive from RO2 autoxidation via sequential H shift and O2 addition () and products deriving from the oxidation of alkoxy radicals (produced by RO2 self-reaction or reaction with HO2) through multiple H shifts and O2 additions (). The oxidation of RO2, with possible occurrence of the Waddington mechanism and of the Korcek mechanism, involving H shifts is also discussed. The present work demonstrates similitude between the oxidation products and oxidation pathways of limonene under simulated atmospheric conditions and in those encountered during the self-ignition of hydrocarbons at elevated temperatures. These results complement those recently reported by Vereecken and Nozière and confirm for limonene the existence of an oxidative chemistry of the alkylperoxy radical beyond 450 K based on the H shift (Nozière and Vereecken, 2019; Vereecken and Nozière, 2020).
- Article
(3421 KB) - Full-text XML
-
Supplement
(1252 KB) - BibTeX
- EndNote





Air particulates are responsible for increasing death rates and diseases worldwide (Lim et al., 2012). Furthermore they have a negative impact on climate (Myhre et al., 2013). With increasing temperatures, particularly in summer, biogenic emissions of volatile organic compounds (VOCs) are more important than anthropogenic. Among them, terpenes emitted by vegetation represent a large fraction of the volatile organic compounds present in the troposphere (Seinfeld and Pandis, 2006; Llusià and Peñuelas, 2000). In addition, one should note that these cyclic hydrocarbons are also considered to be potential high-density biojet fuels (Pourbafrani et al., 2010; Meylemans et al., 2012; Harvey et al., 2010, 2015). Their use as drop-in ground transportation fuel could also be of interest, considering their cetane number around 20 (Yanowitz et al., 2017). Their use as fuel would likely increase their emission into the troposphere. The atmospheric oxidation kinetics of terpenes has been extensively studied, although we are far from a detailed understanding of the many processes involved (Berndt et al., 2015).
Monoterpenes such as α-pinene, β-pinene, and limonene are among the most abundant terpenes in the troposphere (Witkowski and Gierczak, 2017; Zhang et al., 2018). Their oxidation can yield a large variety of oxygenated organic compounds such as highly oxygenated molecules (HOMs), which are considered to play an important role in secondary organic aerosol (SOA) formation (Bianchi et al., 2019).
Recently, Kourtchev et al. (2016) have shown that the concentration of SOA (directly related to that of VOC) mainly influences the apparition of oligomers, whereas environmental or experimental conditions (relative humidity, RH, ozonolysis vs. OH oxidation/photolysis, long-term atmospheric aging) preferentially influence the evolution of these oligomers (Kourtchev et al., 2016). This initial concentration is presented as one of the determining factors of the chemical nature of SOAs and their evolution into oligomers. It was also shown that, under different conditions of oxidative aging, most of the chemical evolution of SOA was due to the reaction of the OH radical (Kourtchev et al., 2015).
The evolution of these primary compounds during atmospheric oxidation has been widely studied in atmospheric chambers, in potential aerosol mass (PAM), but also in field measurements in order to understand the oxidation processes. In the case of limonene, although the fundamental aspects of the oxidation processes are mostly known, there is a real difficulty to observe and characterize these chemical species in the initial phase of oxidation under the above conditions. This difficulty is notably reinforced, in the first steps, by the isomerization phenomena present during the oxidation of alkylperoxy radicals, RO2, and the instability of these chemical species. These reaction mechanisms are mainly related to the H shift, whose rate constant varies with temperature but may also be dependent on the presence of double bonds (Nozière and Vereecken, 2019). In atmospheric chemistry, hydrogen migration on carbon chains is a critical step in the formation of highly oxygenated molecules. It has been recently shown that this mechanism, which is also responsible for the formation of OH radicals, increases with temperature and requires further modeling over a temperature range between 200 and 450 K (Vereecken and Nozière, 2020). Studies show that this H shift and autoxidation mechanism continues beyond 450 K and increases to 600 K depending on the chemical nature of the compound (Belhadj et al., 2021a, b). Among the numerous studies available in the literature on the oxidation of terpenes, limonene has the particularity of having endocyclic and exocyclic bonds, which favor the formation of SOA.
Whereas in atmospheric chemistry peroxy radicals self- and cross-reactions are very important (Wallington et al., 1992), in combustion (Bailey and Norrish, 1952; Benson et al., 1981; Cox and Cole, 1985; Morley et al., 1987), it is commonly accepted that the low-temperature oxidation of hydrocarbons (RH), also named cool flame, can lead to the formation of oxygenated intermediates, but generally, it is assumed that the autoxidation proceeds through the formation of keto-hydroperoxides (KHPs), which provide chain branching by decomposition: RH + OH⇆R + H2O, R+O2⇆ROO, ROO⇆QOOH, QOOH + O2⇆OOQOOH, + OH, + OH. However, recent studies reported the formation of HOMs during the so-called low-temperature oxidation (500–600 K) of hydrocarbons and other organics, e.g., alcohols, aldehydes, ethers, and esters (Wang et al., 2018; Z. Wang et al., 2017; Belhadj et al., 2020). There, the H atom transfer in the OOQOOH intermediate does not involve the group but another H−C group, opening new oxidation pathways. Such alternative pathways do not yield keto-hydroperoxides, and a third O2 addition to HOOQ'OOH yielding OOQ′(OOH)2 can occur. This sequence of reactions can proceed again, yielding highly oxygenated products (Z. Wang et al., 2017; Belhadj et al., 2020, 2021a, b). Also, QOOH can decompose via cyclic ether, carbonyl + olefin, and olefin. In few studies devoted to the understanding of atmospheric oxidation mechanism of hydrocarbons yielding highly oxidized products, autoxidation was proposed as a pathway to organic aerosols (e.g., Jokinen et al., 2014, 2015; Mutzel et al., 2015; Berndt et al., 2016; Crounse et al., 2013; Ehn et al., 2014). The early H shift, ROO⇆QOOH, is favored by increased temperature, which explains its importance in autoignition, but the presence of substituents such as OH, C=O, and C=C in the ROO radical can significantly increase the rate of H shift, making it of significance at atmospheric temperatures (Bianchi et al., 2019).
Beside these processes, the Waddington mechanism (Ray et al., 1973), involving OH and O2 successive additions on a C=C double bond, followed by H atom transfer from −OH to −OO, can occur, yielding carbonyl compounds: , . The Korcek mechanism (Jensen et al., 1981) through which γ-keto-hydroperoxides are transformed into a carboxylic acid and a carbonyl compound can occur too. The formation of carboxylic acids and carbonyl products via the Korcek mechanism has already been postulated by Mutzel et al. (2015), and it is frequently considered in recent kinetic combustion modeling (Ranzi et al., 2015).
Then, the following questions arise: what are the similarities and differences between the products of oxidation of hydrocarbons under simulated atmospheric conditions and cool flames? Do oxidation routes observed in autoxidation (cool flames) play a significant role under atmospheric conditions? Can atmospheric chemistry benefit from combustion chemistry studies and vice versa?
The aim of this work is to characterize the oxidation products of limonene and more particularly those resulting from chain branching. The identification of the isomers resulting from the oxidation of RO2 will be carried out thanks to an ultra-high-resolution mass spectrometer (UHRMS) Orbitrap coupled with ultra-high-performance liquid chromatography (UHPLC) in tandem mode. The initial oxidation concentrations of hydrocarbons used in the laboratory, of the order of a few parts per million (ppm), only allow for the detection of the presence of HOMs without being able to exploit their fragmentation and their chemical speciation within a mixture of isomers. The confirmation of the reaction mechanisms remains difficult.
To overcome this limitation, and to form these compounds in gas-phase oxidation processes with a concentration compatible with the UHPLC separation, the ionization mode, the transfer function, the fragmentation, and low residence time, we chose to increase the initial concentration of limonene. Given these conditions, the study focused only on the mechanistic reaction and qualitative aspects of chemical speciation.
The impact of this initial concentration and the experimental conditions on the range of chemical formulae was investigated by comparing our results to other limonene oxidation studies chosen for their experimental diversity (i.e., oxidation mode, concentration, type of characterization, aging time).
To this end, we studied the oxidation of limonene–oxygen–nitrogen mixtures in a jet-stirred reactor (JSR) at atmospheric pressure, large excess of oxygen, and elevated temperature. Our results are compared to literature data obtained under tropospheric relevant conditions where terpenes are oxidized by OH and/or ozone. Inventory and chemical speciation of oxidation products, as well as the comparison with products of other modes of oxidation (ozonolysis, OH• and photolysis), should lead to a better comprehension of the specificities of each oxidation mode and provide new target data for field experiments. For the sake of clarity, the present oxidation experiments will be called “autoxidation” in the following sections.
The present experiments were carried out in a fused silica jet-stirred reactor (JSR) setup presented earlier (Dagaut et al., 1986) and used in previous studies (Dayma et al., 2011; Dagaut and Lecomte, 2003; Dagaut et al., 1998). As in earlier works (Thion et al., 2017; Dayma et al., 2011), limonene (R)-(+) (>97 % pure from Sigma-Aldrich) was pumped by an HPLC pump (high-performance liquid chromatography pump, Shimadzu LC10 AD VP) with an online degasser (Shimadzu DGU-20 A3) and sent to a vaporizer assembly where it was diluted by a nitrogen flow. Limonene and oxygen were sent separately to a 42 mL JSR to avoid oxidation before reaching the injectors (four nozzles of 1 mm i.d.) providing stirring. The flow rates of nitrogen and oxygen were controlled by mass flow meters. Good thermal homogeneity along the vertical axis of the JSR was recorded (gradients of <1 K cm−1) by thermocouple measurements (0.1 mm Pt-Pt/Rh-10 % wires located inside a thin-wall silica tube). In order to be able to observe the oxidation of limonene, which is not prompt to strong autoignition (cetane number of 20, similar to that of iso-octane), the oxidation of 1 % limonene (C10H16) under fuel lean conditions (equivalence ratio of 0.25, 56 % O2, 43 % N2) was performed at 590 K, at atmospheric pressure, and at a residence time of 2 s. Under these conditions, the oxidation of limonene is initiated by H atom abstraction by molecular oxygen. The radicals of the fuel rapidly react with O2 to form peroxy radicals, which undergo further oxidation, as presented in the Introduction. The absence of ozone, and no need for the addition of a scavenger, allows reaction mechanisms to be probed and chemical species to be observed that are potentially specific to the oxidation by OH radical.
A low-pressure sonic probe was used to freeze the reactions and take samples. To measure low-temperature oxidation products ranging from hydroperoxides and keto-hydroperoxides (KHPs) to highly oxidized molecules, the sonic probe samples were bubbled into cooled acetonitrile (UHPLC grade ≥99.9, T=0 ∘C, 250 mL) for 90 min. The resulting solution was stored in a freezer at −30 ∘C. Analyses were performed by direct sample instillation (flow injection analyses heated electrospray ionization/atmospheric pressure chemical ionization: FIA HESI/APCI) settings (sheath gas 12 arbitrary units (a.u.), auxiliary gas flow 0, vaporizer temperature 120 ∘C, capillary temperature 350 ∘C, spray voltage 3.8 kV, flow injection of 3 µL min−1 recorded for 1 min for data averaging) in the ionization chamber of a high-resolution mass spectrometer (Orbitrap® Q-Exactive from Thermo Scientific, mass resolution of 140 000 and mass accuracy <0.5 ppm rms). Mass calibrations in positive and negative HESI were performed using Pierce™ calibration mixtures (Thermo Scientific). Ultra-high-pressure liquid chromatography (UHPLC) analyses were performed using an analytical column at a controlled temperature of 40 ∘C (C18 Phenomenex Luna, 1.6 µm, 100 Å, 100×2.1 mm) for products separation after injection of 3 µL of sample eluted by water-acetonitrile (ACN) at a flow rate of 250 µL min−1 (gradient 5 % to 90 % ACN, during 14 min). Both heated electrospray ionization (HESI) and atmospheric chemical ionization (APCI) were used in positive and negative modes for the ionization of products. APCI settings were as follows: spray voltage 3.8 kV, vaporizer temperature of 120 ∘C, capillary temperature of 300 ∘C, sheath gas flow of 55 a.u., auxiliary gas flow of 6 a.u., sweep gas flow of 0 a.u., and corona current of 3 µA. In HESI mode, setting were as follows: spray voltage 3.8 kV, T vaporizer of 120 ∘C, T capillary 300 ∘C, sheath gas flow of 12 a.u., auxiliary gas flow of 6 a.u., sweep gas flow of 0 a.u., and mass range between 50 and 1000 Da. Because oxidation of analytes in HESI has been reported previously (Pasilis et al., 2008; Chen and Cook, 2007), we verified that no significant oxidation occurred in the HESI and APCI ion sources by injecting a limonene–ACN mixture. The optimization of the Orbitrap ionization parameters in HESI and APCI did not show any clustering phenomena on pure limonene. The parameters evaluated were as follows: injection source–capillary distance, vaporization and capillary temperatures, potential difference, injected volume, and flow rate of nitrogen in the ionization source. The APCI source, more versatile on polarities and adapted to low masses, was used to identify, in a second phase, KHPs and HOMs. This source, used in positive mode (minimization of salt adducts), has improved the response of low masses necessary for fragmentation. Nevertheless, it should be considered that some of the molecules presented in this study could result from our experimental conditions (continuous-flow tank reactor, concentration of reagents, temperature, reaction time) and to some extent to our acquisition conditions, different from those in the cited studies (Table 1). Indeed, the use of a continuous-flow tank reactor operating at elevated temperature, as well as a high initial concentration of reactants, can induce the formation of unrealistic atmospheric compounds. With regards to the mass spectrometry (MS) acquisition parameters, the selected mass scan range has an influence on the ion transmission, especially at the higher mass range. Figure S9 in the Supplement compares two spectra of oxidized limonene with different acquisition mass ranges. A decrease in trapping efficiency at higher masses is clearly visible when changing the mass scan range from 150–750 to 50–750. It is also necessary to consider the possible formation of non-covalent artifacts, without excluding an incidence on the double bond equivalent (DBE) number. A more detailed description of these technical aspects is available in a recent review (Hecht et al., 2019).
Table 1Main experimental parameters for studies of limonene oxidation.

To determine the structure of limonene oxidation products, MS-MS analyses were performed at collision cell energy of 10–30 eV. 2,4-Dinitrophenylhydrazine (DNPH) was also used to characterize carbonyl compounds. As in previous work (Z. Wang et al., 2017; Belhadj et al., 2020), the fast exchange was used to prove the presence of hydroxyl or hydroperoxyl functional groups in the products. We added 300 µL of D2O (Sigma-Aldrich) to 1.5 mL of sample. The resulting solution was analyzed by flow injection and HESI/APCI mass spectrometry.
High-resolution mass spectrometry (HR-MS) generates a significant amount of data that are easier to interpret with two- or three-dimensional visualization tools (Nozière et al., 2015; X. Wang et al., 2017; Walser et al., 2008; Tu et al., 2016). In this study, we used Kendrick's mass analysis, double bond equivalent (DBE), van Krevelen diagrams, and carbon oxidation state (OSc).
Kendrick's mass analysis (Sleno, 2012; Hughey et al., 2001; Kune et al., 2019; Kendrick, 1963) allows for representation of a complex mass spectrum of an organic mixture in two dimensions and in a new reference frame. This reference frame is based on a mass defect calculated from structural units (CH2, O, CHO). In a Kendrick representation, the homologous series (constructed by the repeated addition of structural units CH2, O, CHO) are aligned on the same horizontal line. This mass defect is calculated by the difference between the Kendrick mass and the nominal mass. In this study, CH2 was chosen as the structural unit.
In Kendrick's plots, the x axis represents the Kendrick mass:
and the y axis represents the Kendrick mass defect:
The belonging of unknown chemical compounds to an homogeneous series of compounds can be used for their identification.
The number of double bond equivalent (DBE) represents the sum of the number of unsaturation and rings present in a compound (Nozière et al., 2015). The decrease in the number of hydrogen atoms increases its value, but it is independent of the number of oxygen atoms. It can be used to identify certain groups of chemical compounds or reaction mechanisms (Kundu et al., 2012). The decimal values of this number were not taken into account in this study. The van Krevelen diagram (Kim et al., 2003; Van Krevelen, 1950) shows the evolution of the ratio as a function of for a set of identified molecules. In a complex organic mixture, it allows the chemical products to be classified according to their degree of oxidation or their degree of reduction/saturation. This type of representation allows for the identification of classes of compounds such as aliphatics, aromatics, or highly oxidized compounds (Fig. 3).
The oxidation state of carbon allows the degree of oxidation of organic species (alcohols, aldehydes, carboxylic acids, esters, ethers, and ketones but not peroxides) (Kroll et al., 2011) to be measured. It is defined by the simple equation:
These data can be used together with the atomic ratios of van Krevelen's diagrams to identify families of organic compounds (Tu et al., 2016; X. Wang et al., 2017; Bianchi et al., 2019). In the case of HOMs, three families of compounds can be distinguished according to their oxidation state and the and ratios:
-
and OSc≥0 (Region 1, highly oxygenated and highly oxidized)
-
and OSc<0 (Region 2, very oxygenated and moderately oxidized)
-
OSc≥0 and (Region 3, moderately oxygenated and highly oxidized).
The oxidation of 1 % limonene (C10H16) was studied at 590 K, at atmospheric pressure, and at a residence time of 2 s. Under these conditions, the fuel conversion is moderate, but formation of low-temperature oxidation products is maximized.
To study the nature of the chemical products formed and the particularity of autoxidation, we compared our results with those obtained by ozonolysis and OH-initiated photooxidation of limonene. This comparison was carried out using visualization methods adapted to large intrinsic data sets of high resolution and high sensitivity reached with current mass spectrometry. At this scale, these tools allow for the differentiation of families of compounds or chemical processes that are hardly perceptible at the level of a few individuals (chemical species).
The comparison of the oxidation modes of limonene (autoxidation and ozonolysis/photooxidation) was based only on the nature of the chemical formula of products, without considering the quantitative, sensitivity, or ionization aspects that are difficult to exploit given the diversity of chemical products formed in this study (i) of the analytical methods and (ii) the large number of instruments involved in this comparison.
To carry out this comparison, and in order to obtain the greatest representativeness of the oxidation of limonene by ozonolysis and OH-initiated photooxidation, we have selected nine previous studies for their diversity of oxidation and characterization processes. Table 1 presents the main experimental parameters of these studies.
These nine experimental studies performed under diverse initial conditions, as shown in Table 1, yielded a first set of 1233 molecular formulae for an inventory, which, although incomplete, gives a broad representativeness of the chemical products which can result from limonene ozonolysis and OH-initiated photooxidation.The second set was obtained using the chemical formulae observed here during limonene autoxidation. It should be noted that this second set of data is generated from different experimental conditions than the previous one, i.e., with a continuous flow reactor, a high concentration of reactants, a high temperature, and a reaction time of 2 s. As previously mentioned, MS detector type and MS acquisition parameters (e.g., mass scan range) in the present study and those compared in Table 1 can affect the inventory of this second set.
For the identification phase, we favored direct injection for its sensitivity. We used UHPLC for its capacity of isomers separation and advanced isomers identification through MS–MS analysis. For direct injection, we have chosen a HESI source operating in negative mode for its wide range of polar molecule ionization. After verification of the absence of oxidation induced by the ionization source (Pasilis et al., 2008; Chen and Cook, 2007) and elimination of the ions common to the reference, attribution rules were made on the basis of molecules composed solely of carbon, hydrogen and oxygen, respecting a deviation of less than 3 ppm by mass over the range 50–1000 Da. Chemical formulae with relative intensity less than 1 ppm to the highest mass peak in the mass spectrum were not considered. Following these rules, we identified 1138 chemical formulae in our oxidized limonene sample, which we split into three groups centered on the number of monomers, i.e., a limonene molecule with different degrees of oxidation: I (); II (); and III () (Kundu et al., 2012; Leungsakul et al., 2005; Nørgaard et al., 2013).
These groups are identified in Fig. 1, which shows the mass spectrum of oxidized limonene. Generally speaking, these groups are built around one or more monomers and a few families of chemical reactions. Group I corresponds to compounds resulting from multiple oxidation reactions, including fragmentation and condensation. Groups II and III correspond to higher molecular masses, resulting from addition and condensation reactions, including, in the case of ozonolysis, reactions of (i) hemiacetalization, (ii) with radicals (hydroperoxy or Griegee), and (iii) of condensation of aldols and/or esterification (Kundu et al., 2012).
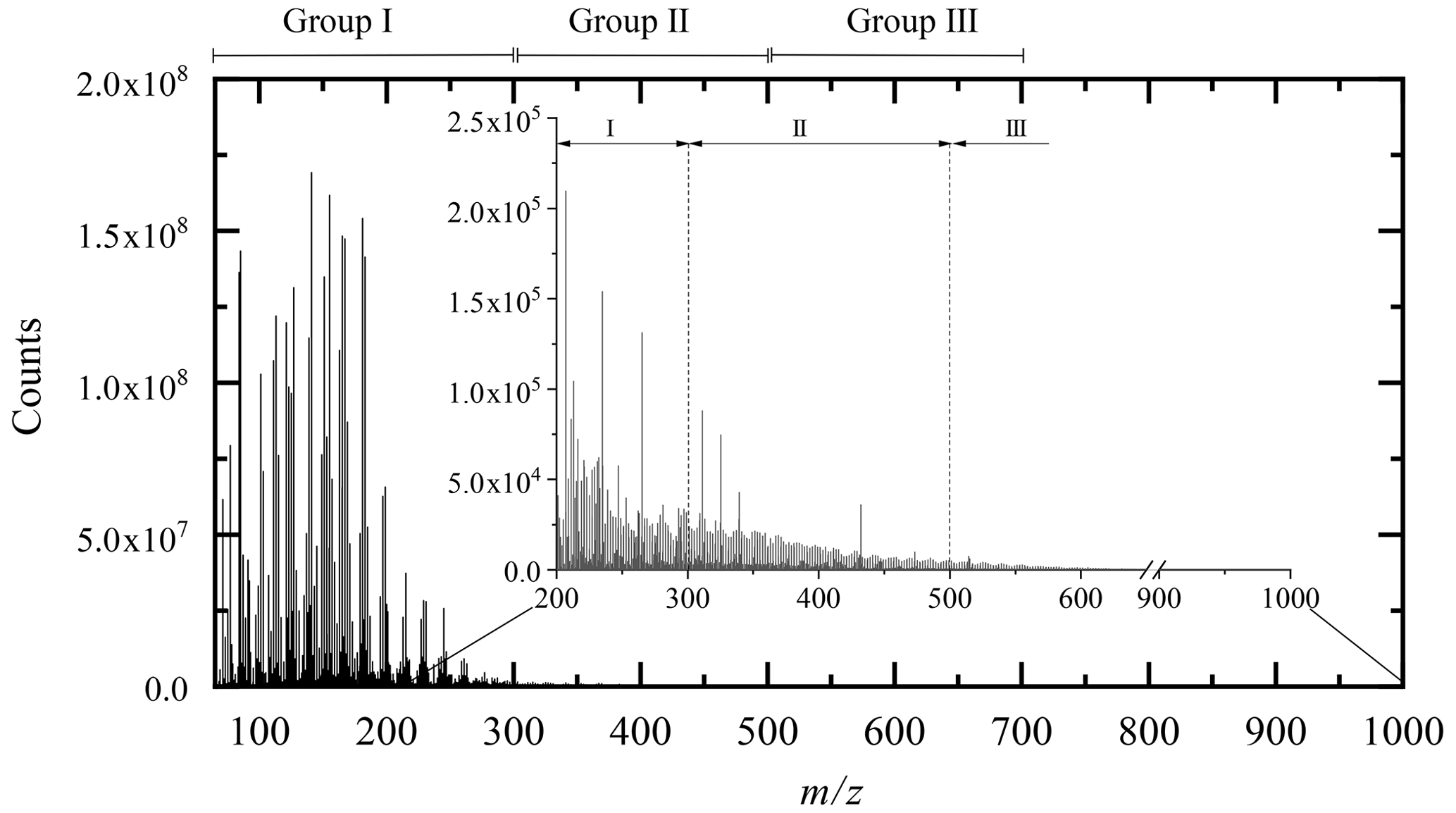
Figure 1Limonene oxidation sample analyzed by FIA and HESI (negative mode, sheath gas flow of 12 a.u., auxiliary gas flow of 6 a.u., sweep gas flow of 0 a.u., voltage 3.8 kV, capillary temperature 300 ∘C, vaporizer temperature 120 ∘C, Hamilton syringe at a flow rate of 3 µL min−1).
The two sets obtained from each oxidation mode were merged, forming a new set of 1600 molecules. In this set, ∼50 % of the chemical formulae (771) are common to both oxidation modes, while 462 molecular formulae are obtained solely by ozonolysis/photooxidation, and 367 are produced in autoxidation experiments only.
All the molecular formulae were represented in a Kendrick diagram (based on a CH2 structural unit) associated with the DBE number (Fig. 2). The representation of Kendrick highlights the families of compounds; the addition of a third dimension makes it possible to study the reaction mechanisms linking these different families (Bateman et al., 2009; Kundu et al., 2012).
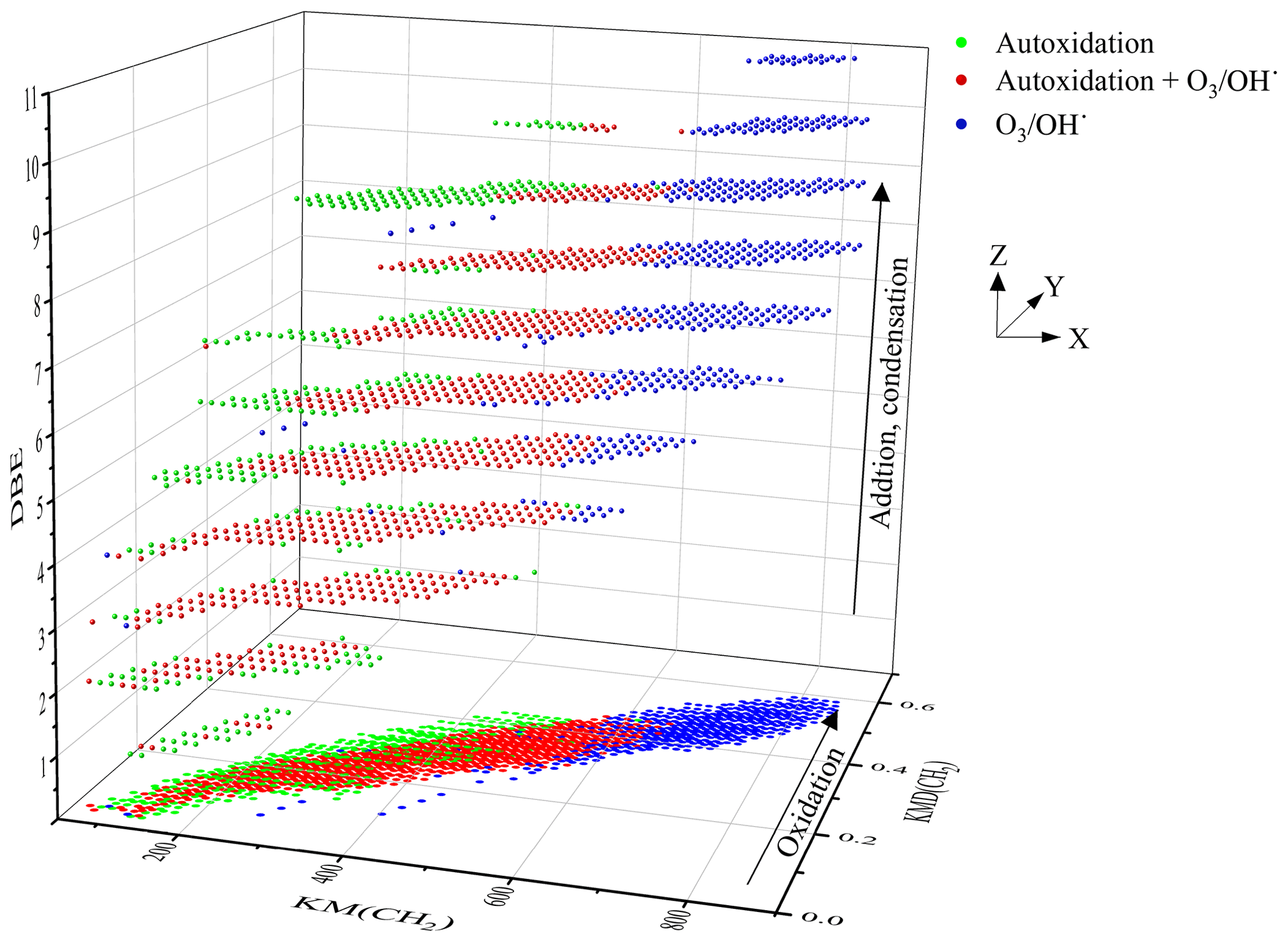
Figure 2All the chemical products, resulting from limonene oxidation by ozonolysis/photooxidation and autoxidation gathered in the form of a Kendrick diagram correlated to the DBE (with projection on the XY plane). Green dots: new chemical products from autoxidation experiments (JSR), red dots: common to the three modes of oxidation, blue dots: chemical products with molecular formulae not observed in autoxidation.
The chemical formulae are distributed into 11 x–y planes according to their DBE number. The representation of each of these x–y planes is given in Fig. S1. This representation immediately highlights the origin of the chemical formulae, i.e., those specific to autoxidation, common to the three modes of oxidation, or specific to ozonolysis and photooxidation. The chemistry of these chemical species can then be read graphically on the axes: the x axis represents the extent of the family, the y axis represents oxidation, and the z axis denotes addition and condensation reactions. This is illustrated in Fig. S1.
Unexpectedly, the distribution of chemical formulae is homogeneous and forms a continuum between oxidation by autoxidation and by ozonolysis/photooxidation.
In autoxidation, the above-mentioned pathways, probably constrained by short oxidation time (residence time of 2 s), seem to extend the molecular weight growth of products up to ∼300 Da, favor splitting (decrease of DBE), additions or condensation (increase of DBE) of unsaturated chemical groups. Ozonolysis and photooxidation experiments, performed over longer periods of time (several seconds few hours), promote, in addition to the previous reactions, the appearance of oligomers and an increase of products' molecular weight (Zhao et al., 2015).
In the Kendrick diagram (Fig. 2), the DBE =3 XY plane containing limonene includes chemical formulae of products deriving from the early oxidation steps. In this case, the breaking of unsaturation, double bond, and ring are counterbalanced by the formation of carbonyls or ozonides, keeping DBE constant. In this XY plane, 83 % of the chemical formulae of products obtained by autoxidation are identical to those formed by ozonolysis/photooxidation, although ozone is most likely absent from JSR autoxidation experiments. Many of the molecular formulae of products described in previous ozonolysis/photooxidation works involve reaction mechanisms based on ozonides and Criegee intermediates. These molecular formulae can be primary products, i.e., weakly oxidized (Bateman et al., 2009) (C9H14O3, C9H14O4, C10H14O3, C10H14O4, C10H16O2, C10H16O3, C10H16O4) but also secondary, with an increasing number of oxygen atoms (C7H10O4, C7H10O5, C8H12O3, C8H12O4, C8H12O5, C9H14O3, C9H14O4, C9H14O5, C10H16O3) (Hammes et al., 2019) and (C7H10O6, C8H12O6, C9H14O6) (Kundu et al., 2012). Additional chemical formulae observed in the current study (also referred to as “new chemical formulae” below) do not form a new group but are in the continuity of the families of chemical molecules found in previous studies (Witkowski and Gierczak, 2017; Jokinen et al., 2015; Walser et al., 2008; Kundu et al., 2012; Fang et al., 2017; Nørgaard et al., 2013; Bateman et al., 2009; Warscheid and Hoffmann, 2001; Hammes et al., 2019). These families are built on the basis of a simple difference in alkyl groups (CH2 basic unit of the Kendrick diagram).
The decrease of DBE from 3 to 2 and then to 1 reflects a greater reactivity on double bonds and on the limonene ring. This reactivity on unsaturated sites is accompanied by a fragmentation of the C−C bonds and a decrease in molecular weight. The new molecular formulae of products, specific to autoxidation, are located at the extremities of the DBE =2 plane or on almost the entire DBE =1 plane. They are characterized by a lower ratio, which can be explained by less advanced oxidation, and certainly by fragmentation. The observation of these new chemical formulae of products, compared to previous studies, can be explained, in addition to the short oxidation time and the elevated temperature in the JSR experiment, by a termination in the radical chain process or by bimolecular reactions (Rissanen et al., 2014; Walser et al., 2008).
The increase in DBE from 3 to 11 characterizes the increase in double bonds and degree of unsaturation obtained by the addition or condensation of limonene oxidized species. In the case of ozonolysis/photooxidation, this increase is usually explained by reactions between Criegee radicals (Criegee intermediate) and acids or alcohols, by hemiacetal formation, aldolization, or esterification (Bateman et al., 2009; Docherty et al., 2005). Each of these reactions is associated with an increase in DBE (hemiacetal: +2; aldolization: +3). For example, in the case of aldol condensation between two aldehydes (limonoaldehyde + 7-OH-limonoaldehyde), the DBE increases from 3 to 6. The emergence of new chemical formulae of products with DBE =9 is mostly observed for molecular weights between 200 and 500 Da. These molecules probably correspond to the addition or condensation of several oxidized limonene compounds (condensation/addition of several cycles) favored, under short time oxidation, by the elevated experimental temperature (590 K).
In general, the new molecular formulae of products observed in autoxidation, compared to ozonolysis (Tu et al., 2016), have a lower molecular weight and are better found in group I (, 52 %) associated with addition or splitting reactions around the limonene skeleton (monomer channel).
In ozonolysis/photooxidation, the products which molecular formulae are rather located in groups II and III can easily oligomerize. This process is highly time-dependent in the presence of ozone (Kundu et al., 2012).
In addition to the identification of chemical families by Kendrick's analysis, it is possible to specify the nature of the chemical products using a van Krevelen diagram. Figure 3 shows a representation of all the chemical formulae of products observed in this work, overlaid on the locations of the different families of chemical compounds described in the literature (Bianco et al., 2018; Nozière et al., 2015). These families are defined by and ratios and shown in Fig. 3.

Figure 3van Krevelen representation of all the chemical products and the different families of chemical compounds described in the literature: (a) aliphatic compounds; (b) unsaturated hydrocarbons; (c) aromatics hydrocarbons; (d) compounds obtained by ozonolysis (including OH-initiated oxidation) (Kundu et al., 2012); (e) HOMs, (e') HOMs formed without .
In the limonene oxidation process, observable from the left to the right on this figure, the first oxidation steps concern both the products resulting from JSR autoxidation and those from ozonolysis/photooxidation.
4.1 Characterization of KHPs
Oxidation, at the initial stage, forms compounds with number of carbon atoms varying from 7 to 11, number of hydrogen atoms ranging from 12 to 16, and up to 9 oxygen atoms. These chemical compounds are mainly in the (d) area. In ozonolysis/photooxidation studies, initial oxidation phase, by ozone and/or the radical OH• have been widely described (Walser et al., 2008; Kundu et al., 2012; Nørgaard et al., 2013; Librando and Tringali, 2005). Although the experimental conditions and the associated reaction mechanisms are different, it is observed, similarly to Fang et al. (2017), many common products of these different oxidation modes. In the case of autoxidation, in the absence of ozone, H abstraction by the radical OH• initiates further oxidation steps yielding ROO• radicals and hydroperoxides.
These reactions will themselves lead to the formation of, among others, KHPs, diketones, or will proceed further (see Sect. 1) and lead to the formation of HOMs (Jokinen et al., 2014; Wang et al., 2019, 2016). In the initial stage of this autoxidation, we studied the formation of the compounds C10H14O3, C10H12O2, C10H16O2, C10H14O5−11 corresponding respectively to the chemical formulae of KHPs, diketones, oxyhydroxides, and HOMs. Experiments were carried out using UHPLC-Orbitrap coupling in tandem mode in order to isolate these molecules and fragment them with a higher energy collisional dissociation (HCD) cell at energy ranging from 10 to 30 eV. Considering a mass range of initial molecules detected in autoxidation lower than 700 Da, we used an APCI source in positive mode, well suited to this mass range.
For the KHPs (C10H14O3), whose mechanisms of formation from limonene are described in Fig. S4 (with example of reaction mechanisms of KHP formation and probable sites of attack of OH radicals), the analyses by high-resolution mass spectrometry confirm the presence of 12 isomers among the compounds formed, compared to a maximum of 18 potentially produced (Fig. S5).
Among these 12 isomers, MS–MS fragmentation allowed for the identification of three groups of compounds presented in Fig. 4. The limits of separation and detection of these chemical compounds made it impossible to specify the position of the functional groups in these isomers.

Figure 4MS analyses of the KHPs isomers together with their fragmentation and number of possible isomers for each group (Fig. S5).
In order to verify the presence of carbonyl groups, 20 µL of a mixture containing DNPH (20 µL of H3PO4 (85 %) in ACN with 20 µL of 2,4-DNPH) were added to 1 mL of sample. This mixture was allowed to react for 4 h before analysis. Characterization, at different reaction times (0.5, 1, and 4 h), was performed by UHPLC-MS APCI (−) in tSIM mode following 361.1153 Da mass of the C16H18O6N4 compound (Fig. S6). One could note an increase in the intensity of the signal (inset Fig. S6). Nearly 12 isomers were observed, with an elution time that is longer and consistent with the initial retention time of the KHPs, thus confirming the presence of carbonyl groups. However, the fragmentation carried out on all these chromatographic peaks did not make it possible to complete the chemical speciation of all the isomers. Nevertheless, for the first time, this study confirms the formation of KHPs initiated by the OH• radical during the oxidation of limonene.
The difficulty for characterizing KHPs lies in the fact that these products are unstable and transform according to different mechanisms. One of the instabilities described in the literature consists in spontaneous dehydration of KHPs to give diketones (Herbinet et al., 2012). Analysis of the data confirms the presence of a diketone (C10H12O2). It shows that diketones are detected both by elution of oxidized limonene (7.79 and 8 min), and systematically in the chromatogram of KHP isomers. This means that this spontaneous transformation can occur in the spectrometer without questioning the presence of diketones in the initial sample. Figure S7 compares the two profiles of diketones resulting from the fragmentation of KHPs and by elution.
Other transformation pathways of KHPs are possible, e.g., via the Korcek mechanism (Jensen et al., 1981; Mutzel et al., 2015) where δ-KHPs decompose into carbonyl compounds and carboxylic acids. Among the 18 proposed KHPs (Fig. S5), 4 isomers (#4, 11, 15, and 18) could react via the Korcek mechanism, but only the #4 isomer is likely to form a cyclic peroxide between a carbonyl group and a hydroperoxide group. The other three isomers will give, after ring opening, isomers of the compound C10H14O3. For the #4 isomer, the Korcek mechanism leads to the formation of a carbonyl compound, C9H12O, and the formic acid CH2O2 (Fig. 5). UHPLC analyses confirm the presence of products with chemical formula C9H12O in the form of two isomers (Fig. 5), but only the peak located at 5.64 min shows a C8H12 fragment consistent with the transformation of the initial KHP.
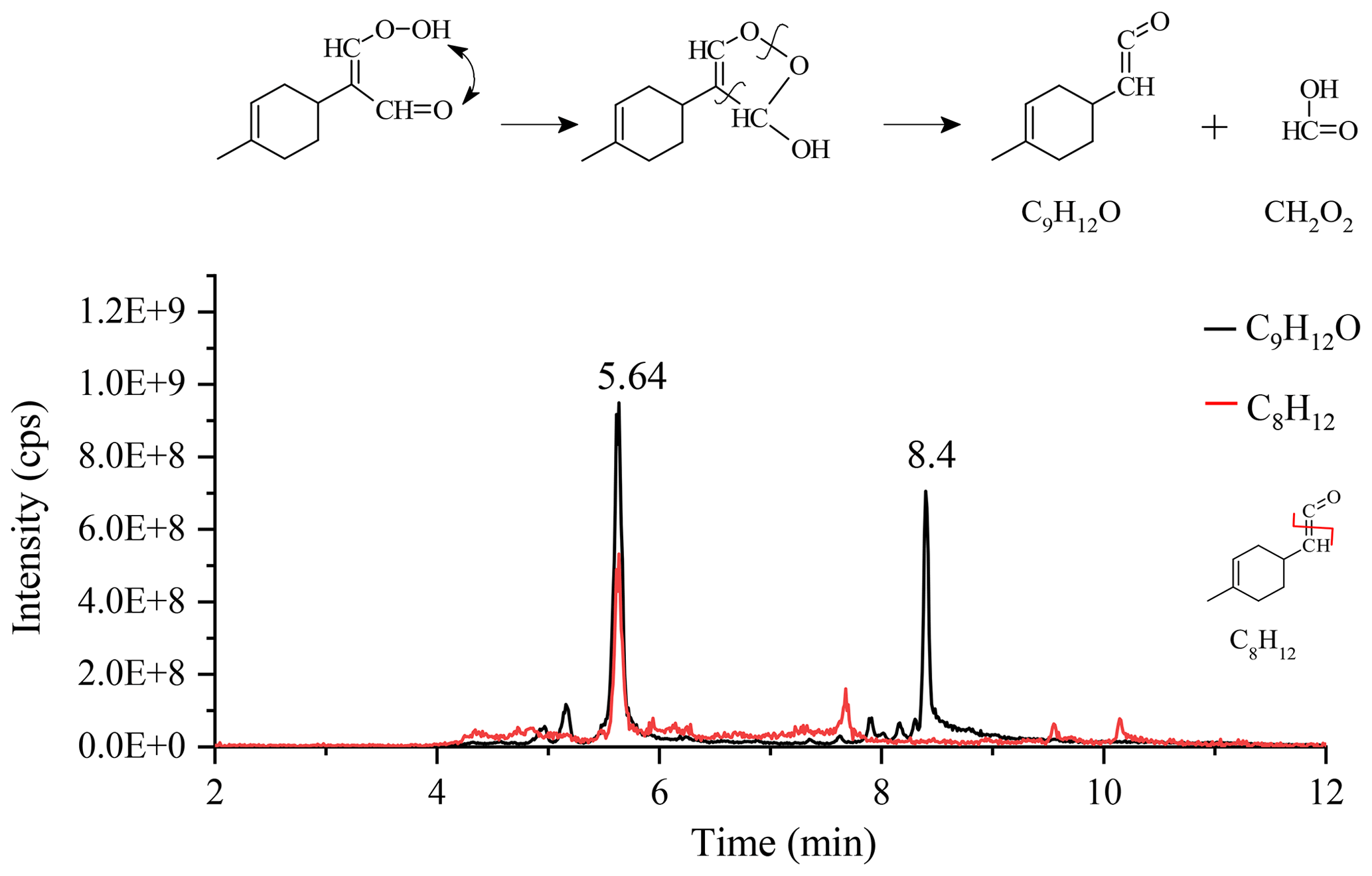
Figure 5Korcek mechanism for the #4 KHP isomer and chromatograms of C9H12O and the fragment C8H12 (APCI(+), vaporizer temperature =120 ∘C, sheath gas flow of 50 a.u., auxiliary gas flow of 0 a.u.; sweep gas flow of 0 a.u., capillary temperature of 300 ∘C, corona current of 3 µA).
KHPs can also give rise to branching reactions that will generate radicals
promoting autoxidation by OH• (Wang et al., 2016).
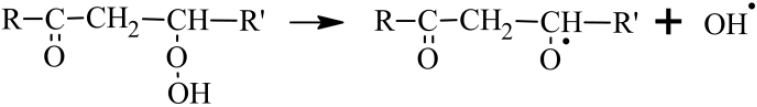
Otherwise, if the H atom transfer in the OOQOOH intermediate does not
involve the group but another H−C group, then no keto-hydroperoxide is formed and a third O2 addition to HOOQ′OOH yielding OOQ′(OOH)2 can occur. If oxidation proceeds further following this pathway, it can lead to the formation of HOMs.
Considering the presence of the radicals OH•, we also searched for chemical compounds resulting from the Waddington mechanism (Li et al., 2020). This mechanism, through which oxidation of alkenes can occur, has two reaction pathways in the case of limonene. The first pathway leads to the formation of a C9H14O ketone and formaldehyde via oxidation of the exocyclic double bond. The second, involving the endocyclic double bond of limonene, gives the compound C10H16O2. For each pathway, we obtained three isomers on the chromatograms (Fig. 6).
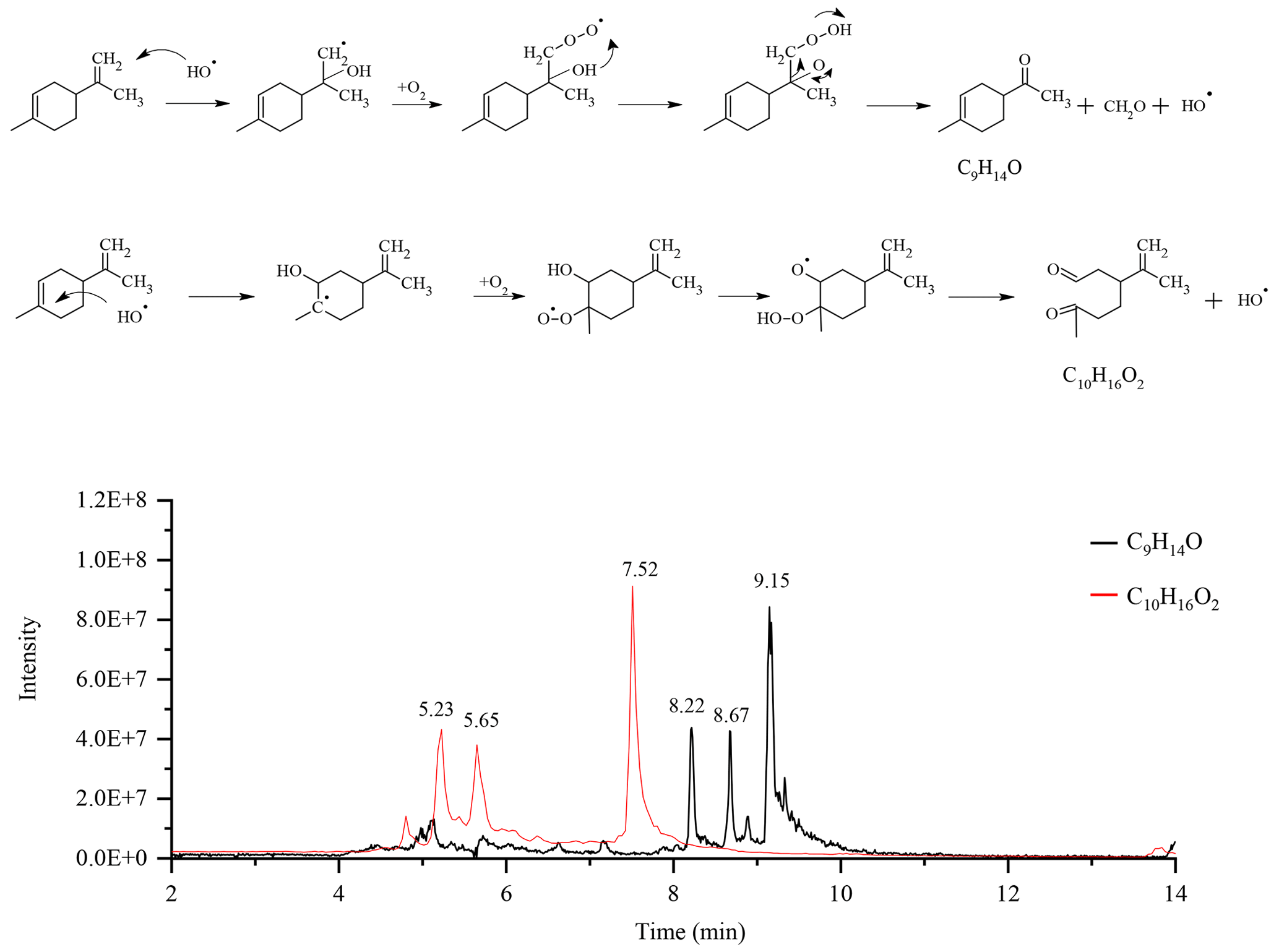
Figure 6Oxidation of limonene according to the Waddington mechanism and chromatograms of the compounds obtained: C9H14O and C10H16O2 (APCI+).
The fragmentation of these isomers did not allow for their identification. However, in order to confirm the presence of carbonyl groups, we added 20 µL of a mixture containing DNPH (Same experimental condition as above) to 1 mL of sample, and let this mixture react for 4 h before analyses. In order to facilitate the detection of isomers and their addition compounds, we used the APCI(-) mode for UHPLC analyses. Only the compound C16H20O5N4 (C10H16O2+DNPH) was detected. The second carbonyl compound (yielding C22H24O8N8) could not be confirmed (Fig. S8). Moreover, the addition of D2O, in order to test the presence of −OH or −OOH groups, gave no results for the two chemical compounds. Recent modeling work on the Waddington mechanism (Lizardo-Huerta et al., 2016) has shown that structural parameters have an impact on the energy barriers associated with the β-scission step. There, a decrease in the activation energy is observed when the substitution of the carbon atom carrying the peroxyl function increases. According to that study, this effect is amplified by the degree of substitution of the carbon atom carrying the hydroxy group. These results are in agreement with the preferential detection of the compound C10H16O2 whose carbon atoms, with the peroxy groups, is the most substituted. Beyond the complete identification of the isomers, this study confirms the presence of chemical compounds which could well result from the Waddington mechanism during limonene autoxidation in JSR.
4.2 Characterization of HOMs
Different strategies were considered for tracking the production of HOMs. Because of higher sensitivity and difficulty in separating isomers, FIA was preferred. Besides KHPs (C10H14O3), the compounds C10H14O5, C10H14O7, C10H14O9, C10H14O11 (Fig. 3, area d) were detected by FIA (APCI +, vaporizer temperature =120 ∘C, sheath gas flow of 12 a.u., auxiliary gas flow of 0 a.u.; sweep gas flow of 0 a.u., capillary temperature of 300 ∘C, corona discharge current of 3 µA, flow of 8 ,µL min−1). We also detected C10H14O2 (keto-hydroxide), C10H14O4, C10H14O6, C10H14O8, and C10H14O10 (Fig. 7a). Whereas the products with odd numbers of oxygen atoms can derive from “combustion” oxidation pathways (Fig. 7b), as presented in the Introduction, those with pair numbers of oxygen atoms can be formed via the classical atmospheric oxidation pathway yielding alkoxy radicals, i.e., , , and followed by alkoxy H shift (Baldwin and Golden, 1978; Atkinson and Carter, 1991) and peroxidation, ; . The reaction can continue with sequential H shift and oxygen addition, yielding HOMs via up to six O2 addition in the present study.

Figure 7Reaction pathways to highly oxygenated products considered in atmospheric chemistry (a, b) (Bianchi et al., 2019). Recently extended reaction pathways in combustion (b) (Z. Wang et al., 2017).
The intensity of ions signal decreases with increasing number of O atoms in the (by 5 orders of magnitude) and (by 6 orders of magnitude) products. Nevertheless, the diversity of reaction pathways, associated with the increasing number of chemical compounds, makes it difficult within a population of several hundred chemical compounds to identify all HOMs. Therefore, we have used the van Krevelen diagram again, which allows us to follow the evolution of the oxidation of the first HOMs and to identify them according to definitions that seem to be consensus (Walser et al., 2008; Tu et al., 2016; Nozière et al., 2015; X. Wang et al., 2017). To this end, we used the average carbon oxidation state OSc which allows three regions to be distinguished according to the nature of the functional groups: Region 1 ( ≥0.6 and OSc≥0) consists of highly oxygenated and highly oxidized compounds (acids and carbonyls), Region 2 ( ≥0.6 and OSc<0), consists of highly oxygenated and moderately oxidized compounds (alcohols, esters and peroxides), finally, Region 3 (OSc≥0 and ≤1.2) includes compounds with a moderate level of oxygen but strongly oxidized (Tu et al., 2016).
It can be seen from Fig. 3 that autoxidation enhances the development of HOMs, compared to ozonolysis/photooxidation, and that the majority of these new products are found in Regions 1 and 3 of the inset of Fig. 3. Thus, further oxidation can go on. We observed products of addition of up to 17 oxygen atoms yielding C25H32O17.
4.3 Complementary method of screening
Further study of the oxidation of chemical compounds and their reaction mechanisms is limited by the complexity of the exponential increase of chemical reactions, chemical species, and their isomers. Nevertheless, monitoring the evolution of these complex mixtures is possible by correlating the OSc to the number of carbon atoms (nc). We have reported in Fig. 8 our measurements and have associated to these results the different biogenic VOCs families defined in the literature (low-volatility oxygenated organic aerosol, LV-OOA; semi-volatile oxygenated organic aerosol, SV-OOA; hydrocarbon-like organic aerosol, HOA; and biomass burning organic aerosol, BBOA) corresponding to particulates (Kroll et al., 2011; An et al., 2019). The development of advanced oxidation, specific to autoxidation, is confirmed with an OSc close to 1. As it stands, it is difficult to make hypotheses on the evolution of these new chemical products and, in the absence of speciation, to assess their environmental impact.
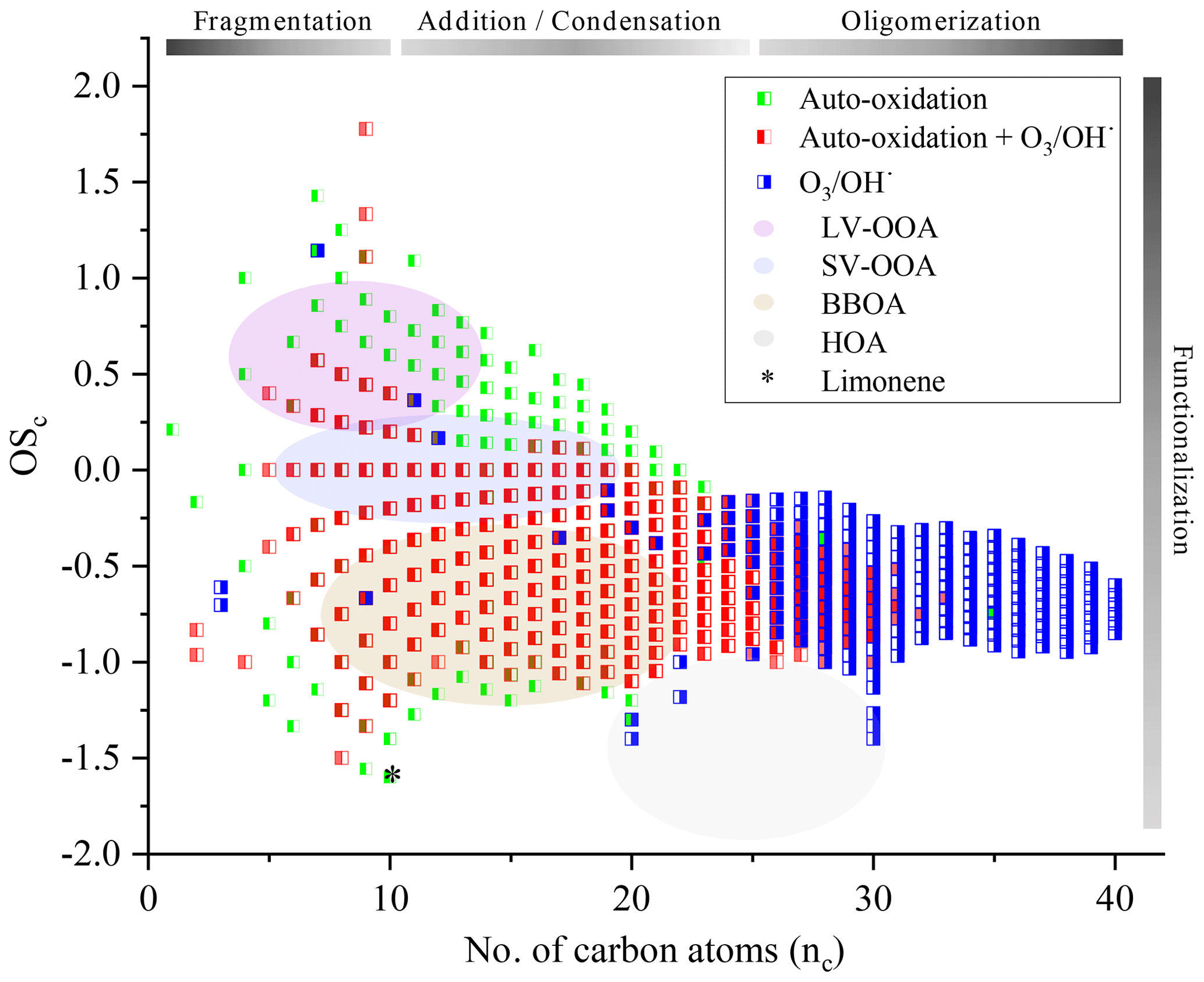
Figure 8Representation in the OSc–nc space of all the chemical formulae considered in this study and analysis of the degree of oxidation.
Finally, it is also possible to further exploit the van Krevelen diagram by introducing reaction mechanisms. Until now, this diagram has been used to identify reaction pathways or families of compounds (Kim et al., 2003; Wu et al., 2004). One can refine this identification by associating with a reaction mechanism a vector whose amplitude and direction will allow reagents and products to be linked. By applying this method to all the experimental data points, one scans the space of the possibilities of formation of a compound or its isomers.
If this method is applied to the formation of C10H14O3 (KHPs and isomers), the vector is defined by the loss of two hydrogen atoms and the gain of three oxygen atoms. By focusing only on molecules composed of 10 carbon atoms, 31 C10H14O3 isomers were identified, 17 of which are new chemical formulae detected in autoxidation only. This is an exhaustive inventory of the possibilities of formation of these compounds based on the experimental data points and not on thermodynamic and kinetic considerations. Applying the same method to the search for keto-di-hydroperoxides (−2H; +5O) and di-keto-hydroperoxide (−2H; +7O), we observed the formation of 24 and 23 compounds, respectively. All these results are presented in Fig. 9.
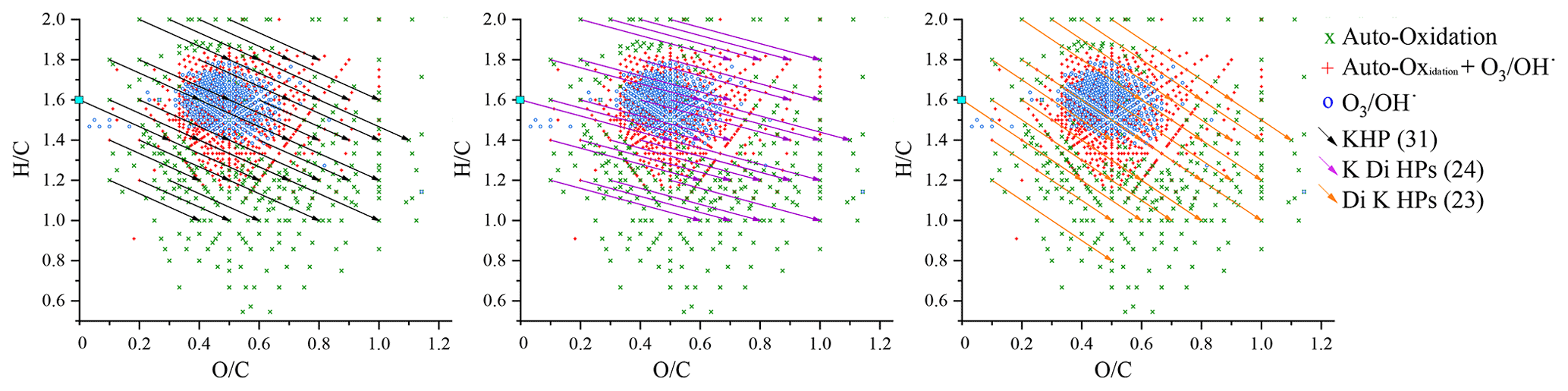
Figure 9Representation in the van Krevelen diagram of the vectors associated with reaction mechanisms for the formation of KHPs, keto-di-hydroperoxides (K Di HPs), and di-keto-hydroperoxides (Di K HPs).
Numerous studies on the ozonolysis of limonene have allowed the reaction mechanisms of its oxidation to be characterized by describing a large fraction of chemical products. In these mechanisms, the formation of a Criegee intermediate has often been described as the major pathway to oxidized compounds, associated with the more restricted formation of the OH• radical.
Among these studies, some have shown that despite different oxidation conditions being used, implying differences in oxidation mechanisms in ozonolysis and OH-oxidation/photolysis, many of the products were similar.
Our study suggests that in the absence of ozone, the oxidation by the OH• radical, common to ozonolysis, gives similar results in terms of observed chemical formulae under the present conditions and those of the studies considered for comparison (Table 1). These results are in agreement with the previous study by Kourtchev et al. (2015), which shows that the evolution of chemical species is mainly dominated by the concentration of OH radical. The present study has allowed us to highlight autoxidation-specific processes, such as formation of KHPs and diketones and occurrence of the Korcek and Waddington reaction mechanisms.
The present results indicate that one should pay more attention to the Korcek and Waddington mechanisms yielding specific products observed here and in previous smog chamber experiments and field measurements.
Extensive oxidation of peroxy radicals yielding HOMs has been considered in atmospheric chemistry, but only recently was a third-O2 addition added to combustion models, showing some influence on ignition modeling (Wang and Sarathy, 2016). Here, limonene oxidation was initiated by reaction with molecular oxygen, yielding alkyl radicals which form peroxy radicals by reaction with O2. The oxidation proceeds further by sequential H shift and O2 addition, yielding a wide range of products with odd numbers of O atoms (). In addition, products with even numbers of O atoms were measured in this work (). They are expected to come from the oxidation of limonene via the commonly accepted tropospheric oxidation mechanism forming alkoxy radicals, i.e., . The following sequential H shift and O2 addition on the alkoxy radicals yielded products of up to six O2 addition in the present work (C10H14O10). Such products have been reported in the previous studies considered here for comparison (Table 1). Although diverse experimental conditions were used here, in terms of concentration of reactants, temperature, reaction time, and analysis conditions, we observed strong similitude in terms of molecular formulae detected in atmospheric and “combustion” chemistry experiments. Additionally, these two routes can produce a pool of OH radicals via decomposition of intermediates, e.g., •OOQOOH→•OH + HOOQ′O (KHP) and (HOO)2Q′OO•→•OH + (HOO)2Q′′O (keto-di-hydroperoxide) for the “combustion” route and •OOQOH→ •OH + OQ′OH (keto alcohol) and HOOQ′(OH)OO•→ •OH + HOOQ′′(OH)O (keto-hydroxy-hydroperoxide) for the “tropospheric” oxidation route. Furthermore, similarly to what has been reported in atmospheric chemistry studies (Witkowski and Gierczak, 2017; Jokinen et al., 2015; Walser et al., 2008; Kundu et al., 2012; Fang et al., 2017; Nørgaard et al., 2013; Bateman et al., 2009; Warscheid and Hoffmann, 2001; Hammes et al., 2019), a wide range of highly oxygenated products were detected, with molecular formulae up to C25H32O7 in the present work.
Analysis at the molecular level was complemented by observation at chemical family scale using Kendrick and van Krevelen visualization tools, necessary to compare and identify features in large data sets. Indeed, the formation of new HOMs and the development of combustion-related autoxidation are perfectly perceptible using these tools. The same is true for the oligomerization, which is not very important in autoxidation, in favor of addition and/or condensation reactions on limonene that are prompt to increase the DBE. As it stands, the meshing within these chemical families according to reaction, thermodynamic, or kinetic criteria remains sketchy but will certainly develop in the light of all the available experimental and theoretical inputs. The observed similarity in terms of chemical formulae obtained by different reaction mechanisms remains qualitative. It does not cover the aspects of quantification and chemical speciation specific to a reaction mechanism. Nevertheless, we noticed that products' similarity is predominant in ozonolysis and photolysis, whereas it is only close to 50 % for limonene low-temperature combustion. Indeed, lower similarity for the production of the lighter species, mostly observed in low-temperature combustion, was noticed. Further studies are needed to clarify the reasons for this difference and assess the impact of residence time and/or aging on the observed degree of similarity. Visualization tools (e.g., VK diagrams, DBE plots) allowed us to differentiate a number of the molecules that are likely related to the experimental conditions used in the current study (e.g., low-temperature combustion). Among the chemical formulae observed in this work, some have not been reported in the studies considered here for comparison. It should be noted that other factors including experimental conditions (e.g., the use of flow tube reactor vs. smog chambers) and/or MS instrument acquisition parameters (e.g., as demonstrated in Fig. S9) can be responsible for the observed differences with the compared studies. It would be interesting to perform additional experiments under conditions relevant to the atmosphere to verify that these chemical formulae are absent. Additional experiments in a JSR at lower initial temperatures and concentrations could also be undertaken to clarify the variation in product formation as temperature and reactant concentration change.
Further studies involving others terpenes are underway. They should confirm the results presented here.
The data used in this study are available from the first author upon request: please contact Roland Benoit (roland.benoit@cnrs-orleans.fr).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-7845-2021-supplement.
RB and PD designed the research; RB, NB, and ML performed the research; RB, NB, and PD analyzed the data; RB and PD wrote the paper; and NB and ML proofread the paper.
The authors declare that they have no conflict of interest.
The authors gratefully acknowledge funding from Labex Caprysses, the Labex Voltaire, and CPER and EFRD (PROMESTOCK and APPROPOR-e projects).
This research has been supported by Labex Caprysses (grant no. ANR-11-LABX-0006-01), the Labex Voltaire (grant no. ANR-10-LABX-100-01), and CPER and EFRD (PROMESTOCK and APPROPOR-e projects).
This paper was edited by Ivan Kourtchev and reviewed by Andrew Wozniak and two anonymous referees.
An, Y., Xu, J., Feng, L., Zhang, X., Liu, Y., Kang, S., Jiang, B., and Liao, Y.: Molecular characterization of organic aerosol in the Himalayas: insight from ultra-high-resolution mass spectrometry, Atmos. Chem. Phys., 19, 1115–1128, https://doi.org/10.5194/acp-19-1115-2019, 2019.
Atkinson, R. and Carter, W. P. L.: Reactions of alkoxy radicals under atmospheric conditions: The relative importance of decomposition versus reaction with O2, J. Atmos. Chem., 13, 195–210, https://doi.org/10.1007/BF00115973, 1991.
Bailey, H. C. and Norrish, R. G. W.: The oxidation of hexane in the cool-flame region, P. R. Soc. London, 212, 311–330, https://doi.org/10.1098/rspa.1952.0084, 1952.
Baldwin, A. C. and Golden, D. M.: Alkoxy radical reactions: the isomerization of n-butoxy radicals generated from the pyrolysis of n-butyl nitrite, Chem. Phys. Lett., 60, 108–111, https://doi.org/10.1016/0009-2614(78)85722-4, 1978.
Bateman, A. P., Nizkorodov, S. A., Laskin, J., and Laskin, A.: Time-resolved molecular characterization of limonene/ozone aerosol using high-resolution electrospray ionization mass spectrometry, Phys. Chem. Chem. Phys., 11, 7931–7942, https://doi.org/10.1039/B905288G, 2009.
Belhadj, N., Benoit, R., Dagaut, P., Lailliau, M., Serinyel, Z., Dayma, G., Khaled, F., Moreau, B., and Foucher, F.: Oxidation of di-n-butyl ether: Experimental characterization of low-temperature products in JSR and RCM, Combust. Flame, 222, 133–144, https://doi.org/10.1016/j.combustflame.2020.08.037, 2020.
Belhadj, N., Benoit, R., Dagaut, P., Lailliau, M., Serinyel, Z., and Dayma, G.: Oxidation of di-n-propyl ether: Characterization of low-temperature products, P. Combust. Inst., 38, 337–344, https://doi.org/10.1016/j.proci.2020.06.350, 2021a.
Belhadj, N., Benoit, R., Dagaut, P., and Lailliau, M.: Experimental characterization of n-heptane low-temperature oxidation products including keto-hydroperoxides and highly oxygenated organic molecules (HOMs), Combust. Flame, 224, 83–93, https://doi.org/10.1016/j.combustflame.2020.10.021, 2021b.
Benson, S. W.: The kinetics and thermochemistry of chemical oxidation with application to combustion and flames, Prog. Energ. Combust., 7, 125–134, https://doi.org/10.1016/0360-1285(81)90007-1, 1981.
Berndt, T., Richters, S., Kaethner, R., Voigtlaender, J., Stratmann, F., Sipilae, M., Kulmala, M., and Herrmann, H.: Gas-phase ozonolysis of cycloalkenes: Formation of highly oxidized RO2 radicals and their reactions with NO, NO2, SO2, and other RO2 adicals, J. Phys. Chem. A, 119, 10336–10348, https://doi.org/10.1021/acs.jpca.5b07295, 2015.
Berndt, T., Richters, S., Jokinen, T., Hyttinen, N., Kurtén, T., Otkjær, R. V., Kjaergaard, H. G., Stratmann, F., Herrmann, H., Sipilä, M., Kulmala, M., and Ehn, M.: Hydroxyl radical-induced formation of highly oxidized organic compounds, Nat. Commun., 7, 13677, https://doi.org/10.1038/ncomms13677, 2016.
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Bianco, A., Deguillaume, L., Vaïtilingom, M., Nicol, E., Baray, J.-L., Chaumerliac, N., and Bridoux, M.: Molecular Characterization of Cloud Water Samples Collected at the Puy de Dôme (France) by Fourier Transform Ion Cyclotron Resonance Mass Spectrometry, Environ. Sci. Technol., 52, 10275–10285, https://doi.org/10.1021/acs.est.8b01964, 2018.
Chen, M. and Cook, K. D.: Oxidation artifacts in the electrospray mass spectrometry of A beta peptide, Anal. Chem., 79, 2031–2036, https://doi.org/10.1021/ac061743r, 2007.
Cox, R. A. and Cole, J. A.: Chemical aspects of the autoignition of hydrocarbon-air mixtures, Combust. Flame, 60, 109–123, https://doi.org/10.1016/0010-2180(85)90001-X, 1985.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013.
Dagaut, P. and Lecomte, F.: Experiments and kinetic modeling study of NO-reburning by gases from biomass pyrolysis in a JSR, Energy Fuels, 17, 608–613, https://doi.org/10.1021/ef020256l, 2003.
Dagaut, P., Cathonnet, M., Rouan, J. P., Foulatier, R., Quilgars, A., Boettner, J. C., Gaillard, F., and James, H.: A Jet-Stirred Reactor for Kinetic-Studies of Homogeneous Gas-Phase Reactions at Pressures up to 10-Atmospheres (∼ 1 MPa), J. Phys. E Sci. Instrum., 19, 207–209, https://doi.org/10.1088/0022-3735/19/3/009, 1986.
Dagaut, P., Daly, C., Simmie, J. M., and Cathonnet, M.: The oxidation and ignition of dimethylether from low to high temperature (500–1600 K): Experiments and kinetic modeling, Symposium (International) on Combustion, 27, 361–369, https://doi.org/10.1016/S0082-0784(98)80424-4, 1998.
Dayma, G., Togbe, C., and Dagaut, P.: Experimental and Detailed Kinetic Modeling Study of Isoamyl Alcohol (Isopentanol) Oxidation in a Jet-Stirred Reactor at Elevated Pressure, Energy Fuels, 25, 4986–4998, https://doi.org/10.1021/ef2012112, 2011.
Docherty, K. S., Wu, W., Lim, Y. B., and Ziemann, P. J.: Contributions of organic peroxides to secondary aerosol formed from reactions of monoterpenes with O3, Environ. Sci. Technol., 39, 4049–4059, https://doi.org/10.1021/es050228s, 2005.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Fang, W., Gong, L., and Sheng, L.: Online analysis of secondary organic aerosols from OH-initiated photooxidation and ozonolysis of α-pinene, β-pinene, Δ3-carene and d-limonene by thermal desorption-photoionisation aerosol mass spectrometry, Environ. Chem., 14, 75–90, https://doi.org/10.1071/EN16128, 2017.
Hammes, J., Lutz, A., Mentel, T., Faxon, C., and Hallquist, M.: Carboxylic acids from limonene oxidation by ozone and hydroxyl radicals: insights into mechanisms derived using a FIGAERO-CIMS, Atmos. Chem. Phys., 19, 13037–13052, https://doi.org/10.5194/acp-19-13037-2019, 2019.
Harvey, B. G., Wright, M. E., and Quintana, R. L.: High-Density Renewable Fuels Based on the Selective Dimerization of pinenes, Energy Fuels, 24, 267–273, https://doi.org/10.1021/ef900799c, 2010.
Harvey, B. G., Merriman, W. W., and Koontz, T. A.: High-Density Renewable Diesel and Jet Fuels Prepared from Multicyclic Sesquiterpanes and a 1-Hexene-Derived Synthetic Paraffinic Kerosene, Energy Fuels, 29, 2431–2436, https://doi.org/10.1021/ef5027746, 2015.
Hecht, E. S., Scigelova, M., Eliuk, S., and Makarov, A.: Fundamentals and Advances of Orbitrap Mass Spectrometry, in: Encyclopedia of Analytical Chemistry, edited by: Meyers, R. A., John Wiley & Sons, Ltd., Chichester, UK, 1–40, https://doi.org/10.1002/9780470027318.a9309.pub2, 2019.
Herbinet, O., Husson, B., Serinyel, Z., Cord, M., Warth, V., Fournet, R., Glaude, P.-A., Sirjean, B., Battin-Leclerc, F., Wang, Z., Xie, M., Cheng, Z., and Qi, F.: Experimental and modeling investigation of the low-temperature oxidation of n-heptane, Combust. Flame, 159, 3455–3471, https://doi.org/10.1016/j.combustflame.2012.07.008, 2012.
Hughey, C. A., Hendrickson, C. L., Rodgers, R. P., Marshall, A. G., and Qian, K.: Kendrick Mass Defect Spectrum: A Compact Visual Analysis for Ultrahigh-Resolution Broadband Mass Spectra, Anal. Chem., 73, 4676–4681, https://doi.org/10.1021/ac010560w, 2001.
Jensen, R. K., Korcek, S., Mahoney, L. R., and Zinbo, M.: Liquid-phase autoxidation of organic-compounds at elevated-temperatures, 2. Kinetics and mechanisms of the formation of cleavage products in normal-hexadecane autoxidation, J. Am. Chem. Soc., 103, 1742–1749, https://doi.org/10.1021/ja00397a026, 1981.
Jokinen, T., Sipilä, M., Richters, S., Kerminen, V.-M., Paasonen, P., Stratmann, F., Worsnop, D., Kulmala, M., Ehn, M., Herrmann, H., and Berndt, T.: Rapid Autoxidation Forms Highly Oxidized RO2 Radicals in the Atmosphere, Angew. Chem. Int. Edit., 53, 14596–14600, https://doi.org/10.1002/anie.201408566, 2014.
Jokinen, T., Berndt, T., Makkonen, R., Kerminen, V.-M., Junninen, H., Paasonen, P., Stratmann, F., Herrmann, H., Guenther, A. B., Worsnop, D. R., Kulmala, M., Ehn, M., and Sipilä, M.: Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications, P. Natl. Acad. Sci. USA, 112, 7123–7128, https://doi.org/10.1073/pnas.1423977112, 2015.
Kendrick, E.: Mass Scale Based on CH2 = 14 0000 for High-Resolution Mass Spectrometry of Organic Compounds, Anal. Chem., 35, 2146–2154, https://doi.org/10.1021/ac60206a048, 1963.
Kim, S., Kramer, R. W., and Hatcher, P. G.: Graphical Method for Analysis of Ultrahigh-Resolution Broadband Mass Spectra of Natural Organic Matter, the Van Krevelen Diagram, Anal. Chem., 75, 5336–5344, https://doi.org/10.1021/ac034415p, 2003.
Kourtchev, I., Doussin, J.-F., Giorio, C., Mahon, B., Wilson, E. M., Maurin, N., Pangui, E., Venables, D. S., Wenger, J. C., and Kalberer, M.: Molecular composition of fresh and aged secondary organic aerosol from a mixture of biogenic volatile compounds: a high-resolution mass spectrometry study, Atmos. Chem. Phys., 15, 5683–5695, https://doi.org/10.5194/acp-15-5683-2015, 2015.
Kourtchev, I., Giorio, C., Manninen, A., Wilson, E., Mahon, B., Aalto, J., Kajos, M., Venables, D., Ruuskanen, T., Levula, J., Loponen, M., Connors, S., Harris, N., Zhao, D., Kiendler-Scharr, A., Mentel, T., Rudich, Y., Hallquist, M., Doussin, J.-F., Maenhaut, W., Bäck, J., Petäjä, T., Wenger, J., Kulmala, M., and Kalberer, M.: Enhanced Volatile Organic Compounds emissions and organic aerosol mass increase the oligomer content of atmospheric aerosols, Sci. Rep.-UK, 6, 35038, https://doi.org/10.1038/srep35038, 2016.
Kroll, J. H., Donahue, N. M., Jimenez, J. L., Kessler, S. H., Canagaratna, M. R., Wilson, K. R., Altieri, K. E., Mazzoleni, L. R., Wozniak, A. S., Bluhm, H., Mysak, E. R., Smith, J. D., Kolb, C. E., and Worsnop, D. R.: Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol, Nat. Chem., 3, 133–139, https://doi.org/10.1038/nchem.948, 2011.
Kundu, S., Fisseha, R., Putman, A. L., Rahn, T. A., and Mazzoleni, L. R.: High molecular weight SOA formation during limonene ozonolysis: insights from ultrahigh-resolution FT-ICR mass spectrometry characterization, Atmos. Chem. Phys., 12, 5523–5536, https://doi.org/10.5194/acp-12-5523-2012, 2012.
Kune, C., McCann, A., Raphaël, L. R., Arias, A. A., Tiquet, M., Van Kruining, D., Martinez, P. M., Ongena, M., Eppe, G., Quinton, L., Far, J., and De Pauw, E.: Rapid Visualization of Chemically Related Compounds Using Kendrick Mass Defect As a Filter in Mass Spectrometry Imaging, Anal. Chem., 91, 13112–13118, https://doi.org/10.1021/acs.analchem.9b03333, 2019.
Leungsakul, S., Jeffries, H. E., and Kamens, R. M.: A kinetic mechanism for predicting secondary aerosol formation from the reactions of d-limonene in the presence of oxides of nitrogen and natural sunlight, Atmos. Environ., 39, 7063–7082, https://doi.org/10.1016/j.atmosenv.2005.08.024, 2005.
Li, Y., Zhao, Q., Zhang, Y., Huang, Z., and Sarathy, S. M.: A Systematic Theoretical Kinetics Analysis for the Waddington Mechanism in the Low-Temperature Oxidation of Butene and Butanol Isomers, J. Phys. Chem. A, 124, 5646–5656, https://doi.org/10.1021/acs.jpca.0c03515, 2020.
Librando, V. and Tringali, G.: Atmospheric fate of OH initiated oxidation of terpenes: Reaction mechanism of α-pinene degradation and secondary organic aerosol formation, J. Environ. Manage., 75, 275–282, https://doi.org/10.1016/j.jenvman.2005.01.001, 2005.
Lim, S. S., Vos, T., Flaxman, A. D., Danaei, G., Shibuya, K., Adair-Rohani, H., AlMazroa, M. A., Amann, M., Anderson, H. R., Andrews, K. G., Aryee, M., Atkinson, C., Bacchus, L. J., Bahalim, A. N., Balakrishnan, K., Balmes, J., Barker-Collo, S., Baxter, A., Bell, M. L., Blore, J. D., Blyth, F., Bonner, C., Borges, G., Bourne, R., Boussinesq, M., Brauer, M., Brooks, P., Bruce, N. G., Brunekreef, B., Bryan-Hancock, C., Bucello, C., Buchbinder, R., Bull, F., Burnett, R. T., Byers, T. E., Calabria, B., Carapetis, J., Carnahan, E., Chafe, Z., Charlson, F., Chen, H., Chen, J. S., Cheng, A. T.-A., Child, J. C., Cohen, A., Colson, K. E., Cowie, B. C., Darby, S., Darling, S., Davis, A., Degenhardt, L., Dentener, F., Des Jarlais, D. C., Devries, K., Dherani, M., Ding, E. L., Dorsey, E. R., Driscoll, T., Edmond, K., Ali, S. E., Engell, R. E., Erwin, P. J., Fahimi, S., Falder, G., Farzadfar, F., Ferrari, A., Finucane, M. M., Flaxman, S., Fowkes, F. G. R., Freedman, G., Freeman, M. K., Gakidou, E., Ghosh, S., Giovannucci, E., Gmel, G., Graham, K., Grainger, R., Grant, B., Gunnell, D., Gutierrez, H. R., Hall, W., Hoek, H. W., Hogan, A., Hosgood III, H. D., Hoy, D., Hu, H., Hubbell, B. J., Hutchings, S. J., Ibeanusi, S. E., Jacklyn, G. L., Jasrasaria, R., Jonas, J. B., Kan, H., Kanis, J. A., Kassebaum, N., Kawakami, N., Khang, Y.-H., Khatibzadeh, S., Khoo, J.-P., Kok, C., Laden, F., Lalloo, R., Lan, Q., Lathlean, T., Leasher, J. L., Leigh, J., Li, Y., Lin, J. K., Lipshultz, S. E., London, S., Lozano, R., Lu, Y., Mak, J., Malekzadeh, R., Mallinger, L., Marcenes, W., March, L., Marks, R., Martin, R., McGale, P., McGrath, J., Mehta, S., Memish, Z. A., Mensah, G. A., Merriman, T. R., Micha, R., Michaud, C., Mishra, V., Hanafiah, K. M., Mokdad, A. A., Morawska, L., Mozaffarian, D., Murphy, T., Naghavi, M., Neal, B., Nelson, P. K., Nolla, J. M., Norman, R., Olives, C., Omer, S. B., Orchard, J., Osborne, R., Ostro, B., Page, A., Pandey, K. D., Parry, C. D. H., Passmore, E., Patra, J., Pearce, N., Pelizzari, P. M., Petzold, M., Phillips, M. R., Pope, D., Pope III, C. A., Powles, J., Rao, M., Razavi, H., Rehfuess, E. A., Rehm, J. T., Ritz, B., Rivara, F. P., Roberts, T., Robinson, C., Rodriguez-Portales, J. A., Romieu, I., Room, R., Rosenfeld, L. C., Roy, A., Rushton, L., Salomon, J. A., Sampson, U., Sanchez-Riera, L., Sanman, E., Sapkota, A., Seedat, S., Shi, P., Shield, K., Shivakoti, R., Singh, G. M., Sleet, D. A., Smith, E., Smith, K. R., Stapelberg, N. J. C., Steenland, K., Stöckl, H., Stovner, L. J., Straif, K., Straney, L., Thurston, G. D., Tran, J. H., Van Dingenen, R., van Donkelaar, A., Veerman, J. L., Vijayakumar, L., Weintraub, R., Weissman, M. M., White, R. A., Whiteford, H., Wiersma, S. T., Wilkinson, J. D., Williams, H. C., Williams, W., Wilson, N., Woolf, A. D., Yip, P., Zielinski, J. M., Lopez, A. D., Murray, C. J. L., and Ezzati, M.: A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010, Lancet, 380, 2224–2260, https://doi.org/10.1016/S0140-6736(12)61766-8, 2012.
Lizardo-Huerta, J. C., Sirjean, B., Bounaceur, R., and Fournet, R.: Intramolecular effects on the kinetics of unimolecular reactions of β-HOROO• and HOQ•OOH radicals, Phys. Chem. Chem. Phys., 18, 12231–12251, https://doi.org/10.1039/C6CP00111D, 2016.
Llusià, J. and Peñuelas, J.: Seasonal patterns of terpene content and emission from seven Mediterranean woody species in field conditions, Am. J. Bot., 87, 133–140, https://doi.org/10.2307/2656691, 2000.
Meylemans, H. A., Quintana, R. L., and Harvey, B. G.: Efficient conversion of pure and mixed terpene feedstocks to high density fuels, Fuel, 97, 560–568, https://doi.org/10.1016/j.fuel.2012.01.062, 2012.
Morley, C.: A Fundamentally Based Correlation Between Alkane Structure and Octane Number, Combust. Sci. Technol., 55, 115–123, https://doi.org/10.1080/00102208708947074, 1987.
Mutzel, A., Poulain, L., Berndt, T., Iinuma, Y., Rodigast, M., Böge, O., Richters, S., Spindler, G., Sipilä, M., Jokinen, T., Kulmala, M., and Herrmann, H.: Highly Oxidized Multifunctional Organic Compounds Observed in Tropospheric Particles: A Field and Laboratory Study, Environ. Sci. Technol., 49, 7754–7761, https://doi.org/10.1021/acs.est.5b00885, 2015.
Myhre, G., Samset, B. H., Schulz, M., Balkanski, Y., Bauer, S., Berntsen, T. K., Bian, H., Bellouin, N., Chin, M., Diehl, T., Easter, R. C., Feichter, J., Ghan, S. J., Hauglustaine, D., Iversen, T., Kinne, S., Kirkevåg, A., Lamarque, J.-F., Lin, G., Liu, X., Lund, M. T., Luo, G., Ma, X., van Noije, T., Penner, J. E., Rasch, P. J., Ruiz, A., Seland, Ø., Skeie, R. B., Stier, P., Takemura, T., Tsigaridis, K., Wang, P., Wang, Z., Xu, L., Yu, H., Yu, F., Yoon, J.-H., Zhang, K., Zhang, H., and Zhou, C.: Radiative forcing of the direct aerosol effect from AeroCom Phase II simulations, Atmos. Chem. Phys., 13, 1853–1877, https://doi.org/10.5194/acp-13-1853-2013, 2013.
Nørgaard, A. W., Vibenholt, A., Benassi, M., Clausen, P. A., and Wolkoff, P.: Study of Ozone-Initiated Limonene Reaction Products by Low Temperature Plasma Ionization Mass Spectrometry, J. Am. Soc. Mass Spectr., 24, 1090–1096, https://doi.org/10.1007/s13361-013-0648-3, 2013.
Nozière, B. and Vereecken, L.: Direct Observation of Aliphatic Peroxy Radical Autoxidation and Water Effects: An Experimental and Theoretical Study, Angew. Chem. Int. Edit., 39, 13976–13982, https://doi.org/10.1002/anie.201907981, 2019.
Nozière, B., Kalberer, M., Claeys, M., Allan, J., D'Anna, B., Decesari, S., Finessi, E., Glasius, M., Grgić, I., Hamilton, J. F., Hoffmann, T., Iinuma, Y., Jaoui, M., Kahnt, A., Kampf, C. J., Kourtchev, I., Maenhaut, W., Marsden, N., Saarikoski, S., Schnelle-Kreis, J., Surratt, J. D., Szidat, S., Szmigielski, R., and Wisthaler, A.: The Molecular Identification of Organic Compounds in the Atmosphere: State of the Art and Challenges, Chem. Rev., 115, 3919–3983, https://doi.org/10.1021/cr5003485, 2015.
Pasilis, S. P., Kertesz, V., and Van Berkel, G. J.: Unexpected analyte oxidation during desorption electrospray ionization-mass spectrometry, Anal. Chem., 80, 1208–1214, https://doi.org/10.1021/ac701791w, 2008.
Pourbafrani, M., Forgács, G., Horváth, I. S., Niklasson, C., and Taherzadeh, M. J.: Production of biofuels, limonene and pectin from citrus wastes, Bioresource Technol., 101, 4246–4250, https://doi.org/10.1016/j.biortech.2010.01.077, 2010.
Ranzi, E., Cavallotti, C., Cuoci, A., Frassoldati, A., Pelucchi, M., and Faravelli, T.: New reaction classes in the kinetic modeling of low temperature oxidation of n-alkanes, Combust. Flame, 162, 1679–1691, https://doi.org/10.1016/j.combustflame.2014.11.030, 2015.
Ray, D. J. M., Redfearn, A., and Waddington, D. J.: Gas-phase oxidation of alkenes: decomposition of hydroxy-substituted peroxyl radicals, J. Chem. Soc. Perk. T. 2, Issue 5, 540–543, https://doi.org/10.1039/p29730000540, 1973.
Rissanen, M. P., Kurtén, T., Sipilä, M., Thornton, J. A., Kangasluoma, J., Sarnela, N., Junninen, H., Jørgensen, S., Schallhart, S., Kajos, M. K., Taipale, R., Springer, M., Mentel, T. F., Ruuskanen, T., Petäjä, T., Worsnop, D. R., Kjaergaard, H. G., and Ehn, M.: The Formation of Highly Oxidized Multifunctional Products in the Ozonolysis of Cyclohexene, J. Am. Chem. Soc., 136, 15596–15606, https://doi.org/10.1021/ja507146s, 2014.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, 2nd edn., Wiley-Interscience, Hoboken, New Jersey, USA, 1232 pp., ISBN: 978-1-118-94740-1, 2006.
Sleno, L.: The use of mass defect in modern mass spectrometry, J. Mass Spectrom., 47, i–i, https://doi.org/10.1002/jms.2978, 2012.
Thion, S., Togbe, C., Serinyel, Z., Dayma, G., and Dagaut, P.: A chemical kinetic study of the oxidation of dibutyl-ether in a jet-stirred reactor, Combust. Flame, 185, 4–15, https://doi.org/10.1016/j.combustflame.2017.06.019, 2017.
Tu, P., Hall, W. A., and Johnston, M. V.: Characterization of Highly Oxidized Molecules in Fresh and Aged Biogenic Secondary Organic Aerosol, Anal. Chem., 88, 4495–4501, https://doi.org/10.1021/acs.analchem.6b00378, 2016.
Van Krevelen, D. W.: Graphical-statistical method for the study of structure and reaction processes of coal, Fuel, 29, 269–284, 1950.
Vereecken, L. and Nozière, B.: H migration in peroxy radicals under atmospheric conditions, Atmos. Chem. Phys., 20, 7429–7458, https://doi.org/10.5194/acp-20-7429-2020, 2020.
Wallington, T. J., Dagaut, P., and Kurylo, M. J.: UV absorption cross sections and reaction kinetics and mechanisms for peroxy radicals in the gas phase, Chem. Rev., 92, 667–710, https://doi.org/10.1021/cr00012a008, 1992.
Walser, M. L., Desyaterik, Y., Laskin, J., Laskin, A., and Nizkorodov, S. A.: High-resolution mass spectrometric analysis of secondary organic aerosol produced by ozonation of limonene, Phys. Chem. Chem. Phys., 10, 1009–1022, https://doi.org/10.1039/B712620D, 2008.
Wang, X., Hayeck, N., Brüggemann, M., Yao, L., Chen, H., Zhang, C., Emmelin, C., Chen, J., George, C., and Wang, L.: Chemical Characteristics of Organic Aerosols in Shanghai: A Study by Ultrahigh-Performance Liquid Chromatography Coupled With Orbitrap Mass Spectrometry, J. Geophys. Res.-Atmos., 122, 11703–11722, https://doi.org/10.1002/2017jd026930, 2017.
Wang, Z. and Sarathy, S. M.: Third O2 addition reactions promote the low-temperature auto-ignition of n-alkanes, Combust. Flame, 165, 364–372, https://doi.org/10.1016/j.combustflame.2015.12.020, 2016.
Wang, Z., Zhang, L., Moshammer, K., Shankar, V. S. B., Lucassen, A., Hemken, C., Taatjes, C. A., Leone, S. R., Kohse-Höinghaus, K., Hansen, N., Dagaut, P., and Sarathy, S. M.: Additional chain-branching pathways in the low-temperature oxidation of branched alkanes, Combust. Flame, 164, 386–396, https://doi.org/10.1016/j.combustflame.2015.11.035, 2016.
Wang, Z., Popolan-Vaida, D. M., Chen, B., Moshammer, K., Mohamed, S. Y., Wang, H., Sioud, S., Raji, M. A., Kohse-Höinghaus, K., Hansen, N., Dagaut, P., Leone, S. R., and Sarathy, S. M.: Unraveling the structure and chemical mechanisms of highly oxygenated intermediates in oxidation of organic compounds, P. Natl. Acad. Sci. USA, 114, 13102–13107, https://doi.org/10.1073/pnas.1707564114, 2017.
Wang, Z., Herbinet, O., Hansen, N., and Battin-Leclerc, F.: Exploring hydroperoxides in combustion: History, recent advances and perspectives, Prog. Energ. Combust., 73, 132–181, https://doi.org/10.1016/j.pecs.2019.02.003, 2019.
Wang, Z. D., Chen, B. J., Moshammer, K., Popolan-Vaida, D. M., Sioud, S., Shankar, V. S. B., Vuilleumier, D., Tao, T., Ruwe, L., Brauer, E., Hansen, N., Dagaut, P., Kohse-Hoinghaus, K., Raji, M. A., and Sarathy, S. M.: n-Heptane cool flame chemistry: Unraveling intermediate species measured in a stirred reactor and motored engine, Combust. Flame, 187, 199–216, https://doi.org/10.1016/j.combustflame.2017.09.003, 2018.
Warscheid, B. and Hoffmann, T.: Structural elucidation of monoterpene oxidation products by ion trap fragmentation using on-line atmospheric pressure chemical ionisation mass spectrometry in the negative ion mode, Rapid Commun. Mass Sp., 15, 2259–2272, https://doi.org/10.1002/rcm.504, 2001.
Witkowski, B. and Gierczak, T.: Characterization of the limonene oxidation products with liquid chromatography coupled to the tandem mass spectrometry, Atmos. Environ., 154, 297–307, https://doi.org/10.1016/j.atmosenv.2017.02.005, 2017.
Wu, Z., Rodgers, R. P., and Marshall, A. G.: Two- and Three-Dimensional van Krevelen Diagrams: A Graphical Analysis Complementary to the Kendrick Mass Plot for Sorting Elemental Compositions of Complex Organic Mixtures Based on Ultrahigh-Resolution Broadband Fourier Transform Ion Cyclotron Resonance Mass Measurements, Anal. Chem., 76, 2511–2516, https://doi.org/10.1021/ac0355449, 2004.
Yanowitz, J., Ratcliff, M. A., McCormick, R. L., Taylor, J. D., and Murphy, M. J.: Compendium of Experimental Cetane Numbers, Tech. Rep., National Renewable Energy Lab. (NREL), Golden, Colorado, USA, NREL/TP-5400-67585, https://doi.org/10.2172/1345058, 2017.
Zhang, H., Yee, L. D., Lee, B. H., Curtis, M. P., Worton, D. R., Isaacman-VanWertz, G., Offenberg, J. H., Lewandowski, M., Kleindienst, T. E., Beaver, M. R., Holder, A. L., Lonneman, W. A., Docherty, K. S., Jaoui, M., Pye, H. O. T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Guo, H., Weber, R. J., de Gouw, J., Koss, A. R., Edgerton, E. S., Brune, W., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Kreisberg, N. M., Spielman, S. R., Hering, S. V., Wilson, K. R., Thornton, J. A., and Goldstein, A. H.: Monoterpenes are the largest source of summertime organic aerosol in the southeastern United States, P. Natl. Acad. Sci. USA, 115, 2038–2043, https://doi.org/10.1073/pnas.1717513115, 2018.
Zhao, D. F., Kaminski, M., Schlag, P., Fuchs, H., Acir, I.-H., Bohn, B., Häseler, R., Kiendler-Scharr, A., Rohrer, F., Tillmann, R., Wang, M. J., Wegener, R., Wildt, J., Wahner, A., and Mentel, Th. F.: Secondary organic aerosol formation from hydroxyl radical oxidation and ozonolysis of monoterpenes, Atmos. Chem. Phys., 15, 991–1012, https://doi.org/10.5194/acp-15-991-2015, 2015.