the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 27 Apr 2021
| 27 Apr 2021
Observations of speciated isoprene nitrates in Beijing: implications for isoprene chemistry
Graham P. Mills
Lisa K. Whalley
W. Joe F. Acton
William J. Bloss
Leigh R. Crilley
Sue Grimmond
Dwayne E. Heard
C. Nicholas Hewitt
James R. Hopkins
Simone Kotthaus
Louisa J. Kramer
Roderic L. Jones
James D. Lee
Yanhui Liu
Bin Ouyang
Eloise Slater
Freya Squires
Xinming Wang
Robert Woodward-Massey
Chunxiang Ye
Isoprene is the most important biogenic volatile organic compound in the atmosphere. Its calculated impact on ozone (O3) is critically dependent on the model isoprene oxidation chemical scheme, in particular the way the isoprene-derived organic nitrates (IN) are treated. By combining gas chromatography with mass spectrometry, we have developed a system capable of separating and unambiguously measuring individual IN isomers. In this paper we use measurements from its first field deployment, which took place in Beijing as part of the Atmospheric Pollution and Human Health in a Chinese Megacity programme, to test understanding of the isoprene chemistry as simulated in the Master Chemical Mechanism (MCM) (v.3.3.1). Seven individual isoprene nitrates were identified and quantified during the campaign: two β-hydroxy nitrates (IHN), four δ-carbonyl nitrates (ICN), and propanone nitrate.
Our measurements show that in the summertime conditions experienced in Beijing the ratio of (1-OH, 2-ONO2)-IHN to (4-OH, 3-ONO2)-IHN (the numbers indicate the carbon atom in the isoprene chain to which the radical is added) increases at NO mixing ratios below 2 ppb. This provides observational field evidence of the redistribution of the peroxy radicals derived from OH oxidation of isoprene away from the kinetic ratio towards a new thermodynamic equilibrium consistent with box model calculations. The observed amounts of δ-ICN demonstrate the importance of daytime addition of NO3 to isoprene in Beijing but suggest that the predominant source of the δ-ICN in the model (reaction of NO with δ-nitrooxy peroxy radicals) may be too large. Our speciated measurements of the four δ-ICN exhibit a mean C1 : C4 isomer ratio of 1.4 and a mean trans : cis isomer ratio of 7 and provide insight into the isomeric distribution of the δ-nitrooxy peroxy radicals. Together our measurements and model results indicate that propanone nitrate was formed from the OH oxidation of δ-ICN both during the day and night, as well as from NO3 addition to propene at night.
This study demonstrates the value of speciated IN measurements in testing understanding of the isoprene degradation chemistry and shows how more extensive measurements would provide greater constraints. It highlights areas of the isoprene chemistry that warrant further study, in particular the impact of NO on the formation of the IHN and the NO3-initiated isoprene degradation chemistry, as well as the need for further laboratory studies on the formation and the losses of IN, in particular via photolysis of δ-ICN and hydrolysis.
- Article
(3192 KB) - Full-text XML
-
Supplement
(1523 KB) - BibTeX
- EndNote





Isoprene is the most important biogenic volatile organic compound (BVOC) in the atmosphere, with its emissions accounting for around 500 Tg yr−1, about half of the global biogenic non-methane VOC emissions (Guenther et al., 2012). It is emitted by vegetation primarily during the daytime as a function of temperature and solar radiation and is readily oxidised by the hydroxyl (OH) and nitrate (NO3) radicals and ozone (O3). Through its degradation chemistry, isoprene impacts O3 and the formation of secondary organic aerosols (SOAs), which together impact the oxidising capacity of the atmosphere and radiative forcing. Global and regional model studies have shown that the calculated impact of isoprene on O3 is critically dependent on the model isoprene oxidation chemical scheme, in particular the way the isoprene-derived nitrates (IN) are treated (e.g. Emmerson and Evans, 2009; Fiore et al., 2005; Squire et al., 2015; von Kuhlman et al., 2004; Wu et al., 2007; Bates and Jacob, 2019; Schwantes et al., 2020). Much of the uncertainty in this chemistry is related to the yield and fate of IN, in particular whether NOx (nitrogen oxides) and radicals, which are tied up in the nitrates, are later recycled or lost from the atmosphere.
First-generation IN are formed following oxidation of isoprene by either OH or NO3 (Wennberg et al., 2018) (Fig. 1). On oxidation by OH, peroxy radicals are formed, which when they react with nitric oxide (NO) can lead to the formation of hydroxy nitrates (IHN). These are dominated by β-IHN, but some δ-IHN are also formed. Depending on the fate of the peroxy radicals formed following NO3 addition, a variety of IN can be produced: isoprene hydroperoxy nitrates (IPN), isoprene dinitrates (IDN), isoprene carbonyl nitrates (ICN), and IHN. While the fate of first-generation IN is poorly understood, much advancement in recent years has been made through new laboratory studies following the synthesis of some of the IN (Jacobs et al., 2014; Lee et al., 2014; Lockwood, et al., 2010; Teng et al., 2017; Xiong et al., 2016), but these are still limited to specific IN isomers (six IHN and one ICN), and reaction rate constants for others are based on extrapolation and structural activity relationships. The IN are lost via reaction with OH, O3, and NO3 (Wennberg et al., 2018) and by photolysis (Xiong et al., 2016; Müller et al., 2014) and deposition (Nguyen et al., 2015) and have lifetimes of the order of a few hours. Reactions of the IHN and ICN with OH can lead to the formation of carbonyls and release of NO2, as well as the formation of shorter-chained nitrates such as methyl vinyl ketone nitrate, methacrolein nitrate, propanone nitrate (acetone nitrate), and ethanal nitrate, with the ratio between these two pathways differing for specific IN isomers (Wennberg et al., 2018, hereafter referred to as W2018). More details of the chemistry of IN are given in Sect. S1 of the Supplement.
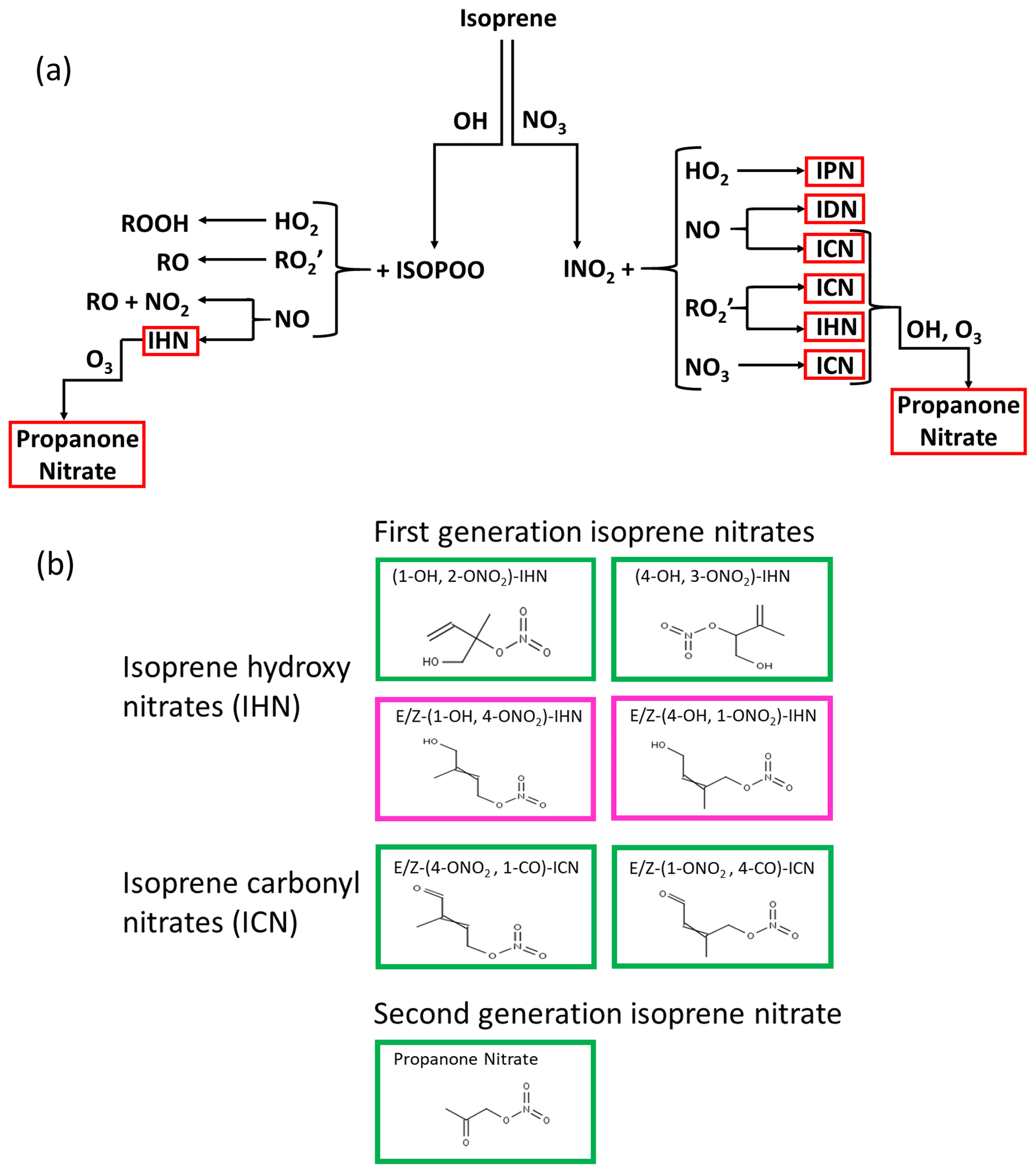
Figure 1(a) Formation of IN (red boxes) from isoprene oxidation by OH and NO3: isoprene hydroxy nitrates (IHN), isoprene hydroperoxy nitrates (IPN), isoprene dinitrates (IDN), isoprene carbonyl nitrates (ICN), and propanone nitrate. (b) The skeletal formula of the specific IN discussed in this paper. Box colours are as follows: green represents measured in Beijing, and pink represents measured by the analytical system previously in the laboratory but not discernible in Beijing.
In this study we deploy a new gas chromatography (GC) negative ionisation (NI) mass spectrometry (MS) system (Bew et al., 2016; Mills et al., 2016) in the field for the first time. By separating and unambiguously measuring individual IHN and ICN isomers along with propanone nitrate during a field campaign in Beijing, we are then able to test the isoprene chemistry of the MCM using a box model. We examine how the ratios of the IHN, primarily the β-IHN, can provide insight into the peroxy radicals (ISOPOO) derived from the OH oxidation of isoprene and in particular their relationship with NO (left-hand side of Fig. 1a), and we use data on ICN, IHN, and propanone nitrate to provide insight into the chemistry of the δ-nitrooxy peroxy radicals (INO2) formed from NO3 addition to isoprene and the (right-hand side of Fig. 1a).
In this paper, when naming the IN, we have followed the nomenclature described by W2018. We assign numbers to the carbons of isoprene based on the conjugated butadiene backbone being comprised of carbons 1–4, with the methyl substituent (carbon 5) connected to carbon 2. We refer to these carbons as “C#” without subscripts (e.g. “C2”). For functionalised isoprene oxidation products, we drop the “C” when describing substituent positions; for example, (1-OH, 2-ONO2)-isoprene hydroxy nitrate (IHN) has a hydroxy group at C1 and a nitrooxy group at C2. This is different to the way we named the IN in Mills et al. (2016), in which the IHN naming followed that of Lockwood et al. (2010) and the ICN were named similarly to the equivalent IHN, except they have “–al” as a suffix. We referred to acetone nitrate as NOA in Mills et al. (2016), whereas here we refer to it as propanone nitrate.
3.1 Field campaign overview
The GC-NI-MS system was deployed at the Institute of Atmospheric Physics (IAP, Chinese Academy of Sciences) in the summer of 2017 (21 May to 22 June) as part of the Atmospheric Pollution and Human Health in a Chinese Megacity (APHH-Beijing) programme (Shi et al., 2019). The IAP is located at 39.97∘ N, 116.38∘ E in a residential area between the 3rd and 4th north ring roads of Beijing. The site contained small trees and grass, with roads 150 m away. During the campaign air quality was poor (Fig. S1) with average concentrations of PM2.5 of ∼ 30 µg m−3 and average mixing ratios of NO2 of 15 ppb, CO of 450 ppb and O3 of ∼ 45 ppb (Shi et al., 2019). Details of the isoprene nitrate measurement technique are provided below, whilst details of instrumentation used for the supporting data are provided in the Supplement.
3.2 Isoprene nitrate measurement method
Seven individual isoprene nitrates were identified and quantified during the campaign (Figs. 1 and S2): two β-IHN ((1-OH, 2-ONO2)-IHN, (4-OH, 3-ONO2)-IHN), four ICN (E-(1-ONO2, 4-CO)-ICN, Z-(1-ONO2, 4-CO)-ICN, E-(4-ONO2, 1-CO)-ICN, Z-(4-ONO2, 1-CO)-ICN), and propanone nitrate.
Measurements were made approximately hourly. Air was drawn at 10 L min−1 down a 2.5 m heated inlet ( PFA and 45 ∘C) mounted on the roof (a height of approximately 3 m above the ground) of a mobile laboratory. Three different instrument setups were employed. (1) From the start of the measurements to 31 May, samples of 500 mL were taken off the inlet line down a 0.3 m length of 0.53 mm ID MxT-200 transfer line held at 50 ∘C, preconcentrated on a Tenax adsorption trap at 35 ∘C and 50 mL min−1, and injected onto the column via a metal six-port Valco valve by heating to 150 ∘C (Mills et al., 2016). A 30.5 m, 0.32 mm (internal diameter, ID) combination column was used, which was comprised of 28 m of Rtx-200 followed by 2.5 m of Rtx-1701 column. The GC oven was temperature profiled from 40 to 200 ∘C, with a constant column flow of 4.5 mL min−1 of helium. (2) Between 10 and 16 June, the system was operated without a trap but instead consisted of direct injection of a 3 mL sample through a plastic Valco Cheminert valve connected to a short 0.32 mm ID combination column (2.5 m of Rtx-200 joined to 0.5 m of Rtx-1701). The GC oven was temperature-programmed from 10 to 200 ∘C and cooled with carbon dioxide (CO2). A constant flow of 6.5 mL min−1 of helium was used as the carrier gas. (3) From 18 June to 22 June, the system again used the 30 m column and Tenax trapping as described above, but the metal valve was replaced with the Cheminert valve that was used for the direct injections.
Of the compounds reported here, all but those of (1-OH, 2-ONO2)-IHN and E-(1-ONO2, 4-CO)-ICN were confirmed by injection of known isomers (Mills et al., 2016) after the campaign. (1-OH, 2-ONO2)-IHN was identified based on its expected elution just before (4-OH, 3-ONO2)-IHN (Nguyen et al., 2014) and the similarity of the observed ions to those of (4-OH, 3-ONO2)-IHN. The E-(1-ONO2, 4-CO)-ICN peak was identified by its relative elution position compared to the other ICN (Schwantes et al., 2015), its expected retention time estimated from the relative retention times of known δ-IHN on this system and their aldehydic equivalents, and the similarity of observed ions to the other ICN.
During several comparisons of samples measured immediately before and after the valve was changed from metal to plastic and vice versa (1 h between samples), it was evident that the (4-OH, 3-ONO2)-IHN and the ICN were lost to varying degrees on the metal valve as suggested by Crounse (John D. Crounse, personal communication, 2016), while simple alkyl nitrates were not. To account for this, all data obtained with the metal valve were scaled by the ratio of peak areas from the samples on either side of the valve changes to give results equivalent to those obtained when using the Cheminert valve.
Calibrations for (4-OH, 3-ONO2)-IHN and propanone nitrate were derived from the relative sensitivity of the compound to that of n-butyl nitrate (Mills et al., 2016) corrected for the relative ion abundances of the specific measurement ions used for each compound ( 71 and 73, respectively). 73 is a relatively minor ion for propanone nitrate, but we were unable to use a more major ion due to interferences from other compounds. N-butyl nitrate calibrations were performed every few days by attaching the transfer line to the standard in place of the inlet. We were unable to measure the relative sensitivities of (1-OH, 2-ONO2)-IHN and the ICN to n-butyl nitrate directly. To obtain an estimate, we have assumed that the ICN and propanone nitrate all have the same total ion yields compared to those of n-butyl nitrate and scaled this relative total ion yield by the fraction of the ion yield that the measurement ion represents. Similarly, we have assumed that the total ion yields of (1-OH, 2-ONO2)-IHN and (4-OH, 3-ONO2)-IHN are the same (and thus the n-butyl nitrate 71: total IN ion ratio) and scaled this to reflect the proportion of the total ions that 101 represents for (1-OH, 2-ONO2)-IHN.
3.3 Isoprene nitrate measurement uncertainties
We had previously determined the uncertainty for the measurement of the IN in the laboratory (including the GC-MS precision and calibration uncertainties) to be ±14 % (Mills et al., 2016), which includes an uncertainty of 5 % for the GC-MS precision. For determination of propanone nitrate in the field, we had to use a minor ion, and, with much smaller peaks, the precision was worse than it had been in the laboratory using more abundant ions. Based on the signal to noise on a peak, we estimate that the precision was 10 % rather than the 5 %. We obtained ion counts in parts per trillion of NOy for three isoprene nitrates: (4-OH, 3-ONO2)-IHN (2030), propanone nitrate (2202), and Z-(4-OH, 1-ONO2)-IHN (2365). Using this range, we assume an additional uncertainty of 17 % for the electron capture–ionisation efficiency of (1-OH, 2-ONO2)-IHN and the ICN. During the campaign, we swapped between a metal and plastic valve twice. Using the peak areas for the last sample with the old valve and first sample with the new valve we calculated loss correction factors and the uncertainties in these correction factors of (4-OH, 3-ONO2)-IHN (±5.2 %), propanone nitrate (±6.4 %), E-(1-ONO2, 4-CO)-ICN (±14.8 %), E-(4-ONO2, 1-CO)-ICN (±9.0 %), Z-(1-ONO2, 4-CO)-ICN (±7.5 %), and Z-(4-ONO2, 1-CO)-ICN (±9.2 %). Loss corrections were applied to the data collected with the metal valve, and these additional uncertainties were included in the overall uncertainties calculated for the periods when the metal valve was used. Based on Mills et al. (2016), the detection limit (DL) of our system with the column and trap is 0.1 ppt, but this increased to 1 ppt when run with direct injection. Combining these uncertainties, we get overall uncertainties for the measurements of the IN as shown in Table 1.
Table 1Uncertainties in the measurements of the isoprene nitrates.
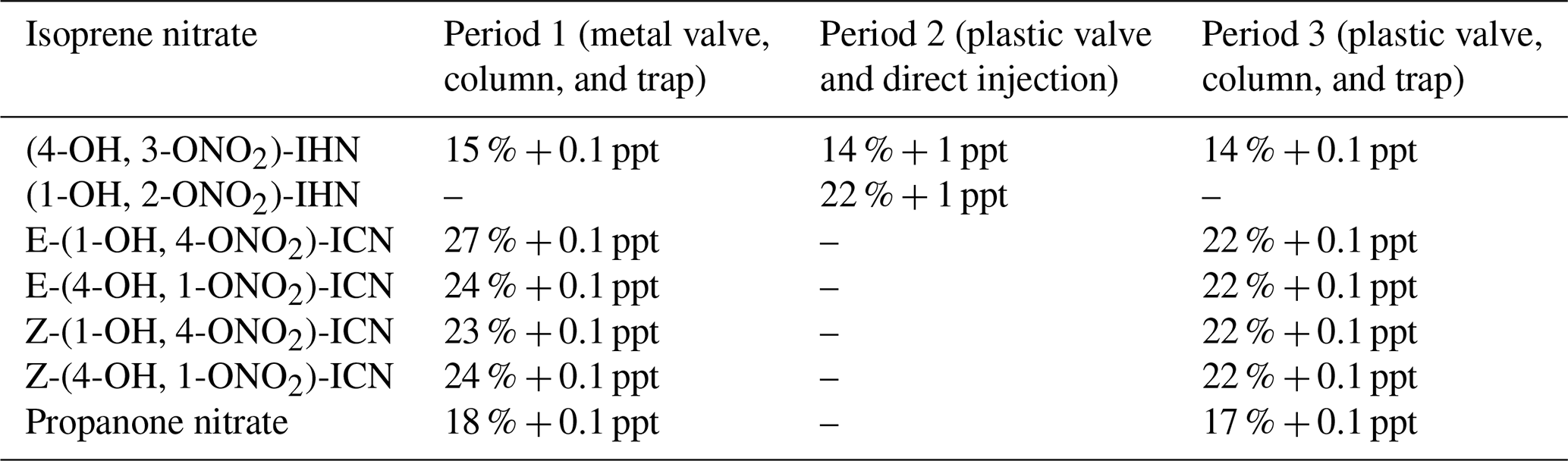
When determining the uncertainties in the ratios between IN, we first calculated the uncertainties for each individual IN measurement excluding the calibration uncertainties that were common to both. We then combined the uncertainties in these to derive overall uncertainties in the ratios. We only assessed the ratios of (4-OH, 3-ONO2)-IHN : (1-OH, 2-ONO2)-IHN in period 2, when we the used the plastic valve and direct injection; i.e. for the ratio (4-OH, 3-ONO2)-IHN : (1-OH, 2-ONO2)-IHN, we considered the GC-MS precision of 5 % for each β-IHN and the additional 17 % uncertainty for the electron capture–ionisation efficiency of (1-OH, 2-ONO2)-IHN, plus the 1 ppt for the DL. We only assessed the ICN ratio in period 3 when we the used the plastic valve, along with column and trap; i.e. we considered the GC-MS precision of 5 % and the additional 17 % uncertainty for the electron capture–ionisation efficiency for each of the ICN and 0.1 ppt for the DL.
A zero-dimensional box model, utilising a subset of the chemistry described within the Master Chemical Mechanism, MCMv3.3.1 (Jenkin et al., 2015), was used to calculate the concentrations of the various isoprene nitrates for the campaign. The MCMv3.3.1 includes an update of the isoprene degradation chemistry to reflect findings of recent laboratory and theoretical studies.
The model was constrained by measured values of water vapour, temperature, pressure, NO, NO2, NO3, O3, CO, SO2, HONO, and HCHO. Speciated VOC measurements of alcohols, alkanes, alkenes, dialkenes (including isoprene), multi-functional aromatics, carbonyls, and monoterpenes were included as further model constraints. The concentrations of H2 and CH4 were held constant at 500 ppb and 1.8 ppm, respectively. The photolysis rates for j(O1D), j(NO2), and j(HONO), calculated from the measured actinic flux and published absorption cross sections and quantum yields, were included as model inputs. Other photolysis frequencies used in the model were calculated. For UV-active species, such as HCHO and CH3CHO, photolysis rates were calculated by scaling the ratio of clear-sky j(O1D) to observed j(O1D) to account for clouds. For species able to photolyse further into the visible, the ratio of clear-sky j(NO2) to observed j(NO2) was used. The variation of the clear-sky photolysis rates (j) with solar zenith angle (χ) was calculated within the model using the following expression:
with the parameters l, m, and n optimised for each photolysis frequency (see Table 2 in Saunders et al., 2003).
The model was run for the entirety of the campaign (21 May 2017–25 June 2017) in overlapping 7 d segments, with the model constraints updated every 15 min. By this method, a model time series was produced that could be directly compared with observations and from which diel averages were generated. There was a spike of very high concentrations of isoprene in the early hours of the morning of 16 June 2017, which led to extremely high concentrations of modelled ICN, propanone nitrate, and (4-OH, 1-ONO2)-IHN. These have been removed from the diel averages presented in this paper. Fluxes through each reaction were calculated for every 15 min period to allow an analysis of the production and loss terms of the chemical species.
The loss due to mixing of all non-constrained model-generated species, including the speciated isoprene nitrates, was parameterised and evaluated by comparing the model-predicted glyoxal concentration with the observed glyoxal concentration. Applying a loss rate proportional to the observationally derived mixed-layer height (Fig. S4), the model was able to reproduce glyoxal observations reasonably well (Fig. S4). As a result of this first-order loss process, the partial lifetime of the model-generated species was ∼ 2 h at night and then decreased rapidly to a lifetime of < 30 min in the morning as the mixed layer grew, effectively simulating ventilation of the model box. With the collapse of the mixed layer in the late afternoon, the model lifetime with respect to ventilation of glyoxal (and other model-generated species) increased. However, the model has a tendency to underestimate glyoxal concentrations between 16:00 and 00:00 LT. This underestimation suggests that either the lifetime with respect to ventilation should be even longer or that the model is underestimating oxidation processes that lead to glyoxal production at these times. We do not consider uptake of glyoxal onto aerosol, the rate of which is highly uncertain (Volkamer et al., 2007; Washenfelder et al., 2011; Li et al., 2016). Including one would have led to use of a slower ventilation rate. The same first-order ventilation loss was applied to all species and no specific assumptions were made about the background concentrations. Consequently, the rate of mixing may be overestimated for longer-lived species with significant background concentrations and conversely underestimated for shorter-lived species.
5.1 β-IHN
Figures 2 and S3 compare the measured and modelled β-IHN. The shaded areas in Fig. 2 represent ±1 SD in the data for each hour of the day and illustrate the large day-to-day variability in the mixing ratios of β-IHN. Note that for (1-OH, 2-ONO2)-IHN there are only 6 d worth of data, and thus the average diel patterns are strongly affected by the day-to-day variability. This is particularly the case for the measurements where three of the hourly bins contain just one measurement, while the rest have between three and eight measurements.
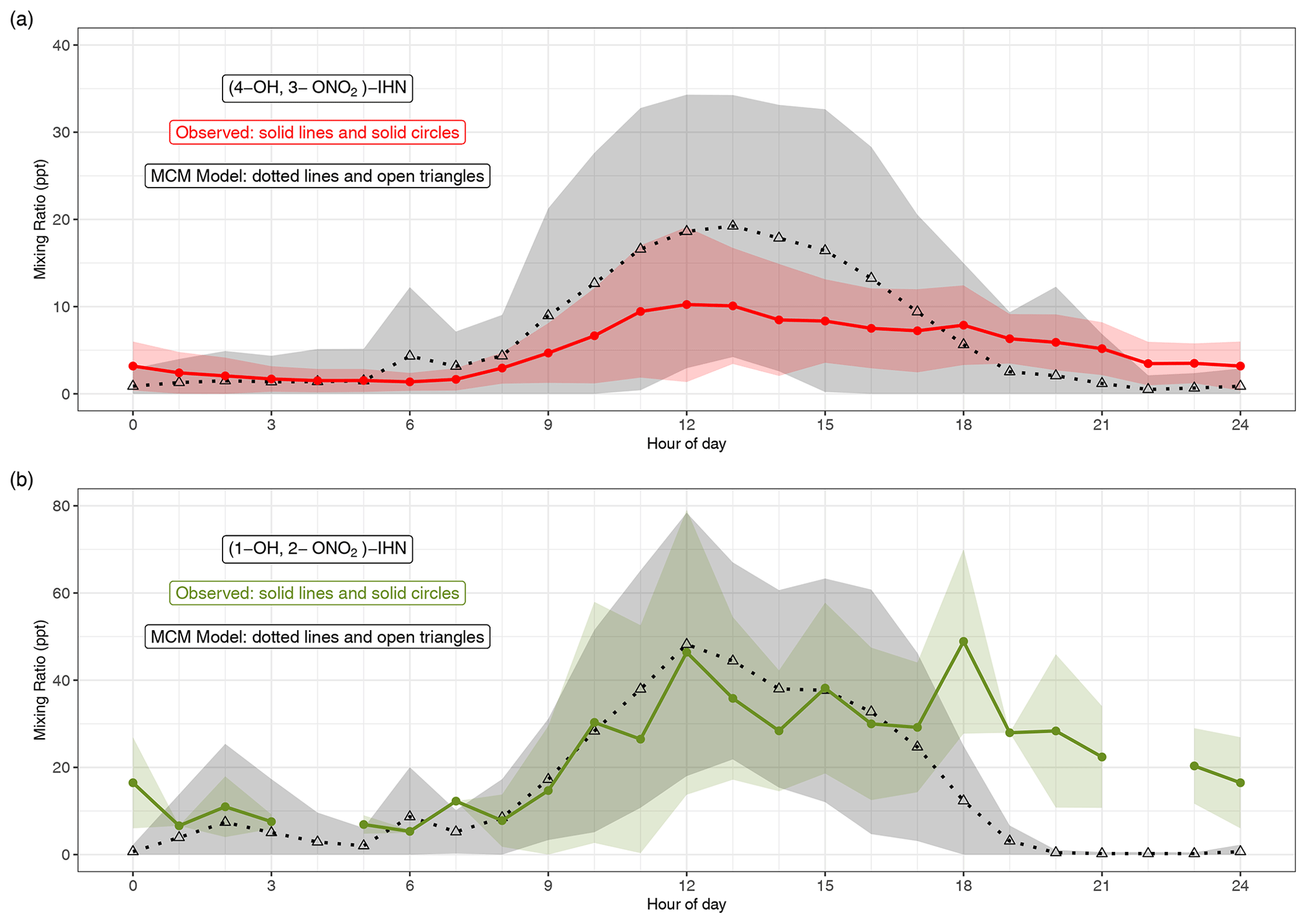
Figure 2Modelled and observed mixing ratios of (a) (4-OH, 3-ONO2)-IHN and (b) (1-OH, 2-ONO2)-IHN. Data points are the mean, and the shaded areas represent ±1 SD in the variability of values for each hour of the day.
The observed β-IHN exhibit diel patterns that are broadly in agreement with those modelled and consistent with daytime formation from OH oxidation of isoprene and a shift in competition from the reactions of isoprene-derived peroxy radicals (ISOPOO) with NO to reaction with HO2 as mixing ratios of NO decline from an early morning peak (Fig. S5) and those of the peroxy radicals maximise in the mid-afternoon (Whalley et al., 2021). The observed β-IHN peak around midday, and these levels are mostly maintained until around sunset when they decline to reach minimum values just after sunrise. This pattern is also broadly similar to that of total IHN observed during the Southern Oxidant and Aerosol Study (SOAS) (Xiong et al., 2015).
The absolute values of the observed (1-OH, 2-ONO2)-IHN daytime mixing ratios are very similar to those modelled (Figs. 2 and S3). On some days, the observed and modelled (4-OH, 3-ONO2)-IHN mixing ratios are reasonably similar, but on a few days the model simulates considerably larger mixing ratios than observed (Fig. S3) such that the mean daytime observed mixing ratios tend to be lower than modelled and exhibit far less day-to-day variability (Fig. 2). The observed evening mixing ratios of both β-IHN are higher than modelled, suggesting that, like glyoxal, the model overestimates their loss with respect to ventilation. Alternatively, there may be greater production or slower chemical loss than simulated. It is worth noting that the MCM assumes 8 % of the OH addition to isoprene occurs at the C2 and C3 positions instead of the C1 and C4 positions, thus reducing the potential to form (1-OH, 2-ONO2)-IHN or (4-OH, 3-ONO2)-IHN, whereas W2018 recommend that the C2 and C3 additions are negligible.
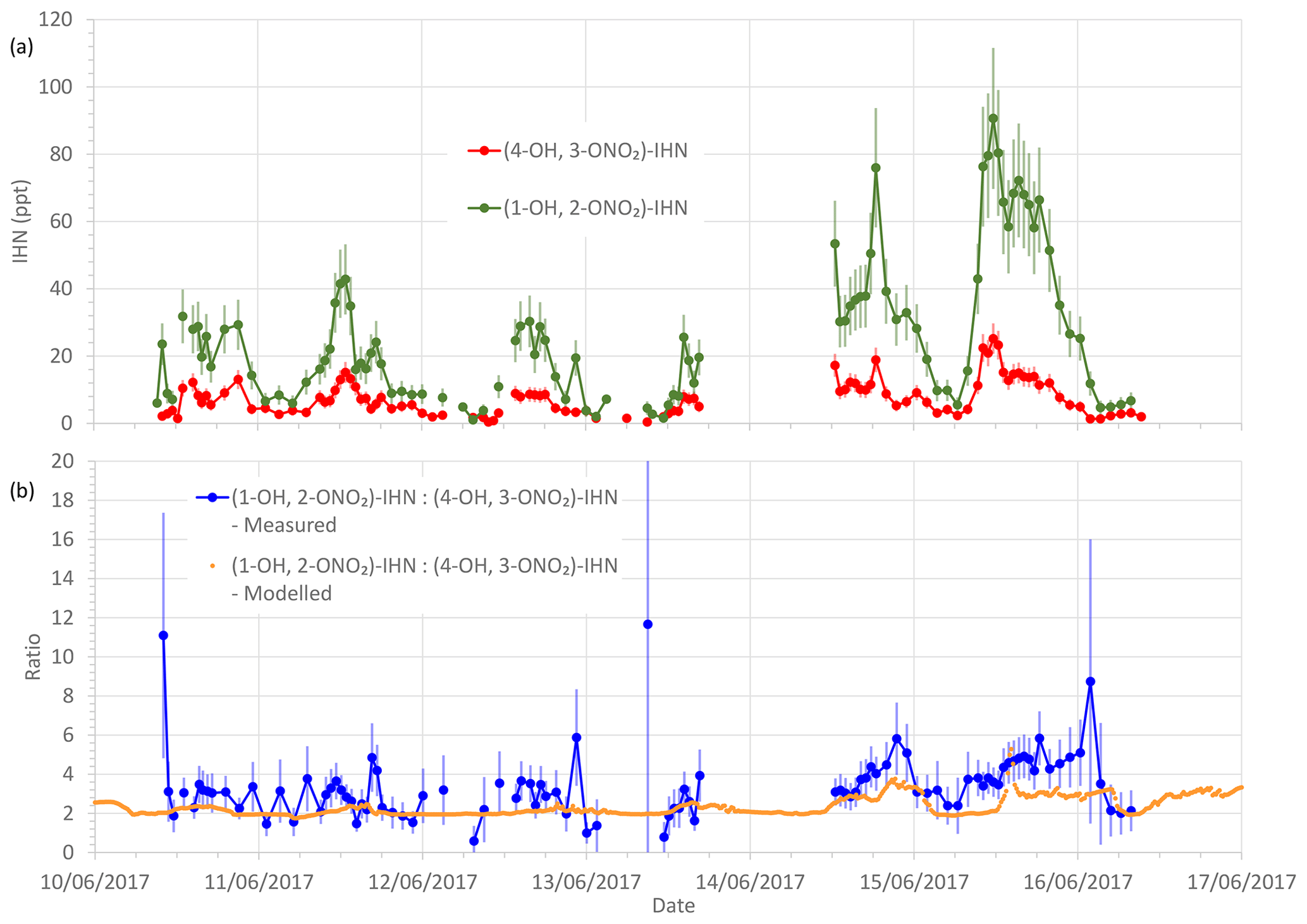
Figure 3(a) Measured β-IHN mixing ratios. (b) Measured and modelled ratio (1-OH, 2-ONO2)-IHN:(4-OH, 3-ONO2)-IHN. Error bars are the measurement uncertainties (see Sect. 3.3 for details).
To limit the impact of ventilation on the comparison between the model and observations we compare the ratio of (1-OH, 2-ONO2)-IHN to (4-OH, 3-ONO2)-IHN (Figs. 3 and 4). When looking at the time series (Fig. 3) of this ratio, the model and measurements often agree within the measurement uncertainties, although there are times when the observed values are greater than modelled. The shaded areas in Fig. 4 represent ±1 SD in the data for each hour of the day. The large variability in the observed data is caused by some hours having very few data points, sometimes affected by a single high value (Fig. 3). The observed mean ratios are generally higher than the modelled mean, although there is often agreement within the day-to-day variability.
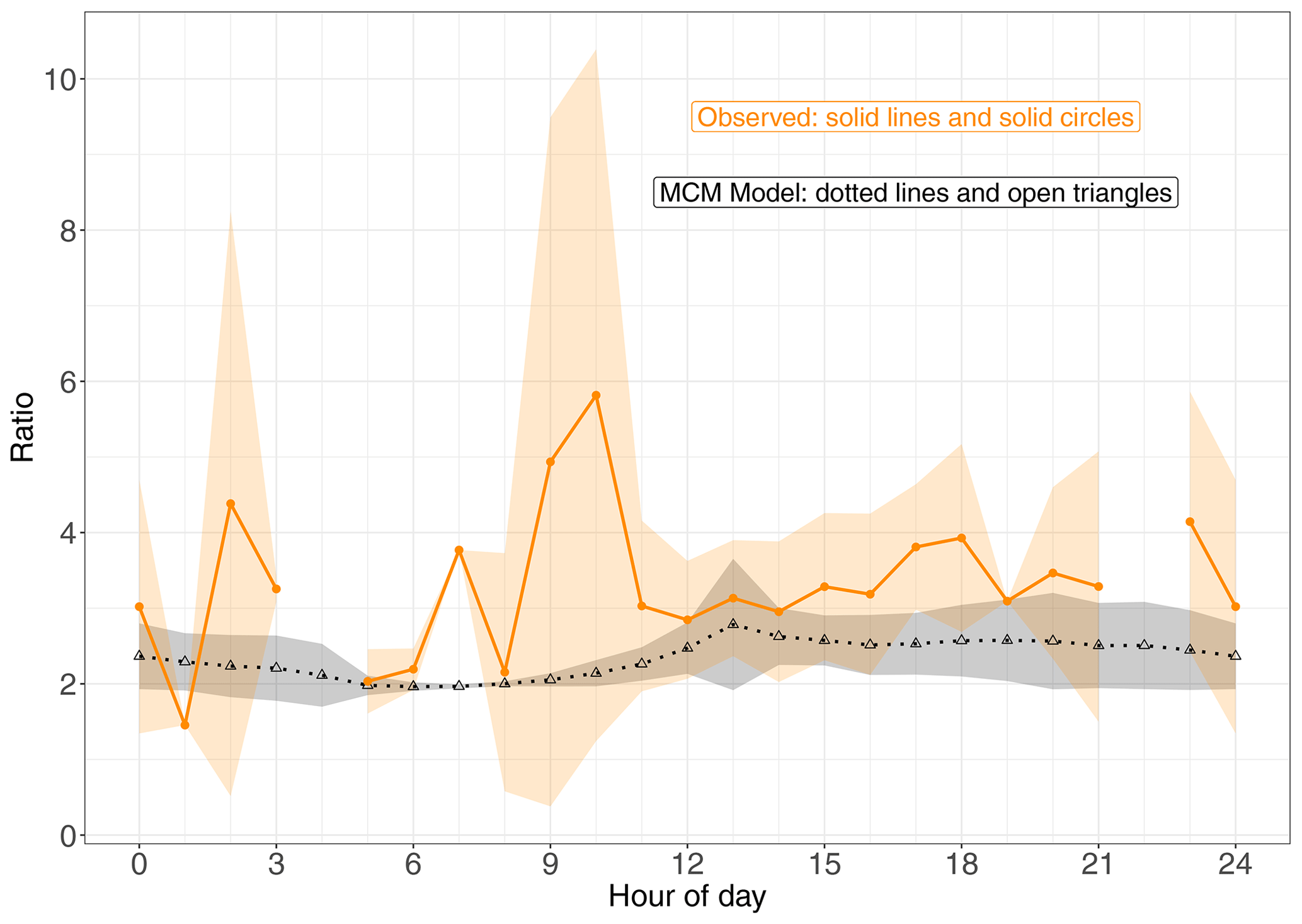
Figure 4Modelled and observed (1-OH, 2-ONO2)-IHN (4-OH, 3-ONO2)-IHN ratio. Data points are the means and the shaded areas represent ±1 SD in the variability of values for each hour of the day.
It should be noted that the ratio we obtain from our measurements is not based on an independent calibration for (1-OH, 2-ONO2)-IHN but is instead based on the assumption that the analytical system has the same sensitivity to (1-OH, 2-ONO2)-IHN as it does to (4-OH, 3-ONO2)-IHN. We have tried to account for this in the uncertainty calculations by assuming that the error in this sensitivity is equal to the percentage range of sensitivities that we observed for the other IN (see Sect. 3.3). It is possible that this is an underestimate.
There are four main factors that determine the ratio of the β-IHN: (1) the yields of their respective peroxy radicals (ISOPOO) following oxidation of isoprene by OH addition (φ), (2) the fraction of the respective ISOPOO that reacts with NO (γ), (3) the branching ratios for the formation of the IHN from the reaction of NO with the ISOPOO (α), and (4) the relative loss rates of the β-IHN, including via deposition.
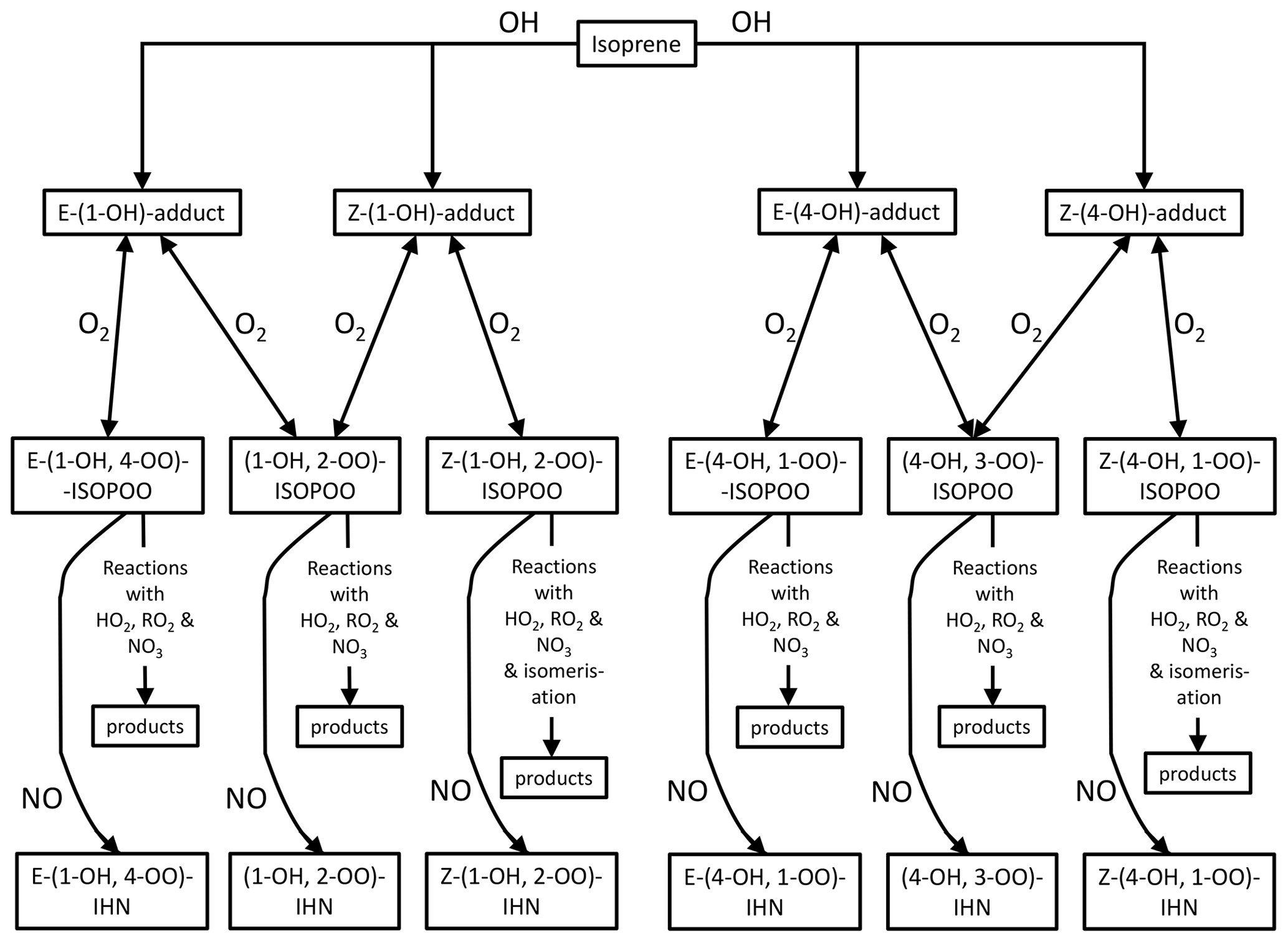
Figure 5Schematic of the formation of the IHN following addition of OH to the C1 and C4 positions.
For the first two factors, the concentration of NO is largely the determining influence. The adducts formed from OH addition to a specific C in isoprene can form a β-ISOPOO and either an E or Z δ-ISOPOO upon reaction with O2 (Fig. 5). These reactions are reversible and, since the lifetimes of these peroxy radicals differ, they inter-convert within two subgroups defined by the position of the OH (i.e. C1 on the left-hand side of Fig. 5 and C4 on the right-hand side). NO is often present in large amounts (Figs. S2 and S5) and thus reaction with it is the dominant loss process for the ISOPOO. However, at low NO mixing ratios other losses of the ISOPOO become relatively more important, and the different rates of 1,6 H atom shift isomerisation of the Z-(1-OH, 4-OO)-ISOPOO and Z-(4-OH, 1-OO)-ISOPOO in particular mean that redistribution of the ISOPOO within each sub-group differs, such that (1-OH, 2-OO)-ISOPOO increases relative to (4-OH, 3-OO)-ISOPOO. Consequently, at lower NO mixing ratios the modelled ratio of φ-(1-OH, 2-OO)-ISOPOO to φ-(4-OH, 3-OO)-ISOPOO becomes larger (Fig. 6a). For mixing ratios of NO greater than ∼ 2 ppb, the ratio of the values of φ decreases approximately linearly from around 2 to about 1.7 at 100 ppb of NO. The ratio of the kinetic yields in the MCM is 1.58, which is the ratio of the values of φ that we get if we switch off the reverse pathway of the O2 reactions. This implies that even at 100 ppb of NO, the ratio of the yields of the (1-OH, 2-OO)-ISOPOO to (4-OH, 3-OO)-ISOPOO is shifted to values slightly greater than the kinetic ratio. At NO mixing ratios less than ∼ 2 ppb, the ratio of the values of φ increase with decreasing NO, such that at ∼ 10–100 ppt NO the ratio is typically between 2.5 and 4.
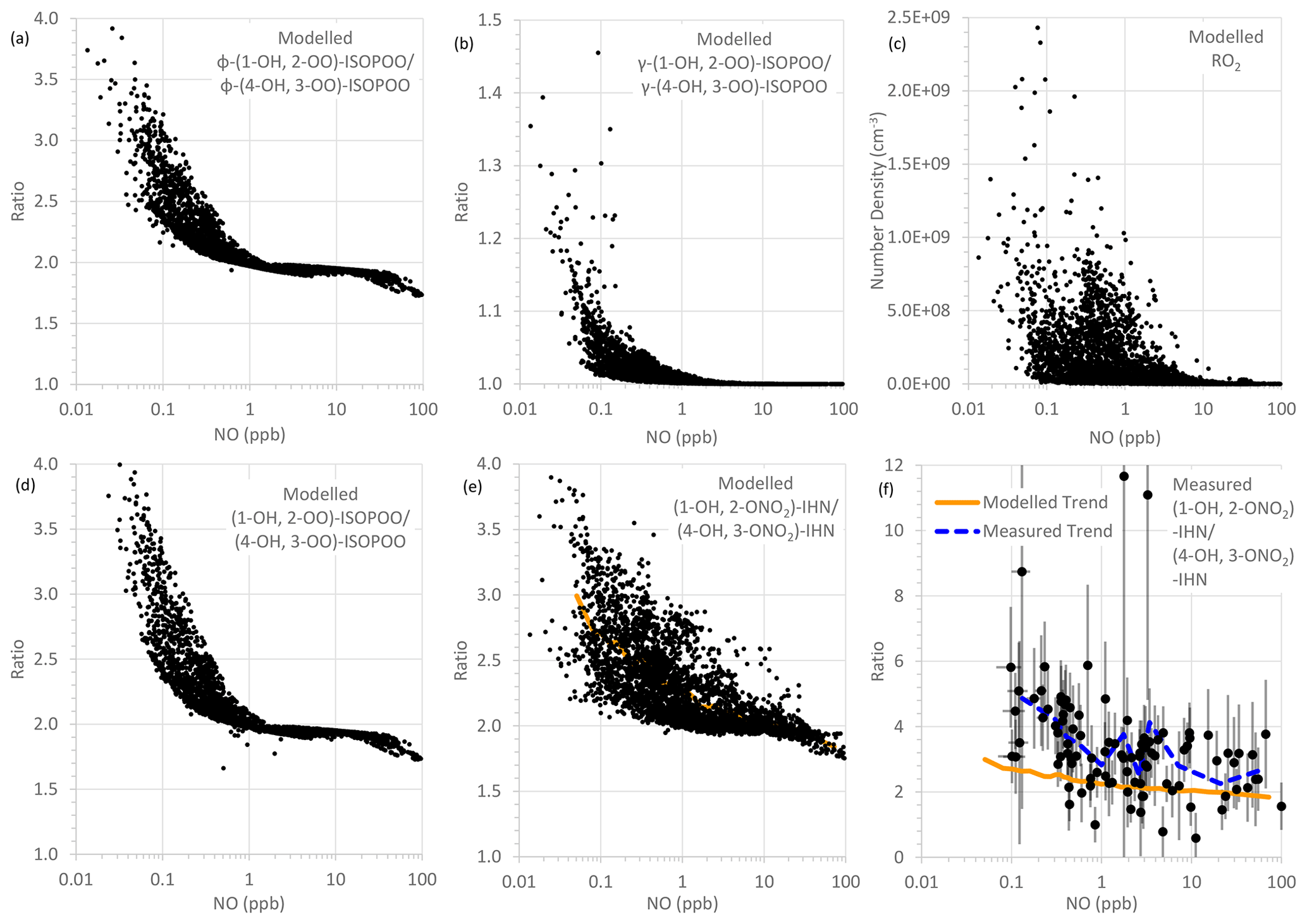
Figure 6Modelled and measured parameters as a function of NO mixing ratio: (a) modelled ratio of φ-(1-OH, 2-OO)-ISOPOO to φ-(4-OH, 3-OO)-ISOPOO, (b) modelled ratio of γ-(1-OH, 2-OO)-ISOPOO to γ-(4-OH, 3-OO)-ISOPOO, (c) modelled RO2 number density, (d) modelled ratio of (1-OH, 2-OO)-ISOPOO to (4-OH, 3-OO)-ISOPOO, (e) modelled ratio of (1-OH, 2-ONO2)-IHN to (4-OH, 3-ONO2)-IHN, and (f) measured ratio of (1-OH, 2-ONO2)-IHN to (4-OH, 3-ONO2)-IHN (error bars are the measurement uncertainties; see Sect. 3.3 for details). In panel (e), the orange line is a trend line produced by plotting the mean modelled (1-OH, 2-ONO2)-IHN to (4-OH, 3-ONO2)-IHN ratio for each bin of 100 NO mixing ratios. This same line is plotted in panel (f) for comparison with the observed data. The dashed blue line in panel (f) is a trend line produced by plotting the mean measured (1-OH, 2-ONO2)-IHN to (4-OH, 3-ONO2)-IHN ratio for each bin of nine NO mixing ratios.
The rates at which the ISOPOO are assumed to be lost via the reactions with NO, HO2, and NO3 are the same for both β-ISOPOO. However, the rate constants for reaction of (1-OH, 2-OO)-ISOPOO with RO2 and for its rate of isomerisation are slower than those of (4-OH, 3-OO)-ISOPOO. At lower NO mixing ratios, these reactions become relatively more important, and thus the modelled value of γ is lower for (4-OH, 3-OO)-ISOPOO than for (1-OH, 2-OO)-ISOPOO, and the ratio of γ-(1-OH, 2-OO)-ISOPOO to γ-(4-OH, 3-OO)-ISOPOO is larger (Fig. 6b). This is further enhanced as the concentrations of RO2 can also be much greater at the lower NO concentrations, particularly below 1 ppb NO (Fig. 6c), which leads to the ratio in the γ values often being considerably greater than 1 at NO concentrations below 100 ppt.
It should be noted that the MCM model underestimates the measured RO2 mixing ratios (Whalley et al., 2021). This will lead to underestimation of the ratio of γ-(1-OH, 2-OO)-ISOPOO to γ-(4-OH, 3-OO)-ISOPOO, primarily at mixing ratios of NO below ∼ 2 ppb. This might explain some of the differences between the modelled and observed β-IHN ratios.
The net effect of these relationships is that the modelled ratio of (1-OH, 2-OO)-ISOPOO to (4-OH, 3-OO)-ISOPOO increases with decreasing NO (Fig. 6d); i.e. for NO mixing ratios greater than 2 ppb the ratio is around 1.7–2.0, but at NO mixing ratios less than 2 ppb the ratio increases up towards a value of around 4. The ratio of the rate of production of (1-OH, 2-ONO2)-IHN to (4-OH, 3-ONO2)-IHN will have the same relationship with NO as the ratio of their precursor ISOPOO since the MCM assumes that the branching ratios for the formation of the two β-IHN from the reaction of NO with the ISOPOO (i.e. α, third factor) are the same. However, there are still considerable uncertainties in these branching ratios (Sect. S1.1).
As for the loss processes of the β-IHN (fourth factor), the dominant loss in the model is the mixing term, which is set at the same rate for both β-IHN. Photolysis is assumed to be faster for (1-OH, 2-ONO2)-IHN than for (4-OH, 3-ONO2)-IHN in the MCM, but it is only a minor loss process. However, (1-OH, 2-ONO2)-IHN reacts with both OH and O3 more slowly than (4-OH, 3-ONO2)-IHN does, and since the dominant chemical loss process for the β-IHN by far is their reactions with OH, the net effect of these loss processes is to increase the ratio of (1-OH, 2-ONO2)-IHN to (4-OH, 3-ONO2)-IHN above their production ratio. The diel pattern in OH (Fig. S5) will tend to increase the ratio of (1-OH, 2-ONO2)-IHN to (4-OH, 3-ONO2)-IHN during the daytime.
Overall, this means that the modelled ratio of (1-OH, 2-ONO2)-IHN to (4-OH, 3-ONO2)-IHN increases with decreasing NO mixing ratios (Fig. 6e) (as also seen by Jenkin et al. (2015) in a box model using the MCM) and generally does not drop below the ratio of the β-ISOPOO (Fig. 6d). In the conditions modelled for Beijing it ranges from between 1.75 and 2.0 at NO mixing ratios above ∼ 30 ppb, typically rising up to between 2 and 3 but sometimes up to 4 at NO mixing ratios below 1 ppb. There are several cases at these low NO mixing ratios when the ratio of the β-IHN is below the ratio of the β-ISOPOO, but these occur at night when the production rates and the mixing ratios of the β-IHN are very small.
In comparison, the observed ratios of (1-OH, 2-ONO2)-IHN to (4-OH, 3-ONO2)-IHN show a similar but weaker relationship with NO (Fig. 6f). The strength of the observed relationship is limited by the number of data points and uncertainties in the measurements but shows a tendency for relatively more (1-OH, 2-ONO2)-IHN at NO mixing ratios of less than 1 ppb.
Newland et al. (2021) point out that during the campaign a high NOx environment existed in the morning but then switched to a low NOx environment in the afternoon. The mean hourly NO mixing ratios were typically above 2 ppb between 06:00 and 12:00 LT but mostly below this value in the afternoon (Fig. S5). The relationship between β-IHN and NO as illustrated in (Fig. 6e) largely explains why the modelled ratio of the β-IHN (Fig. 4) is ∼ 2 and exhibits little variability between about 06:00 and 09:00 LT and then rises up to around 2.5 in the afternoon.
Our mean observed (1-OH, 2-ONO2)-IHN to (4-OH, 3-ONO2)-IHN ratio of ∼ 3.4 is higher than the daytime values reported by Vasquez et al. (2018) in the PROPHET campaign (∼ 2.6) and in Pasadena (∼ 1.4), but their data show a similar pattern to ours in that the ratio is higher in the low NOx environment of PROPHET compared to the high NOx environment in Pasadena. We cannot rule out calibration differences affecting this comparison and, like us, Vasquez et al. (2018) relied on relative calibration estimates. In addition, differences in the observed β-IHN ratios may be due to the amount and reactivity of the peroxy radicals present in the different studies. However, the ratio of 1.4 observed for Pasadena is lower than the kinetic φ ratios of 1.58 and 1.85 based on MCM and W2018 kinetic yields, respectively. Xiong et al. (2015) calculate a ratio ranging from 2.6 to 6.0 based on the conditions experienced in SOAS.
5.2 δ-ICN
The MCM assumes all of the δ-ICN formed can be represented by a single species, (1-ONO2, 4-CO)-ICN, called NC4CHO. We shall therefore compare the sum of the observed δ-ICN with the modelled NC4CHO and then look at the speciation as exhibited in the observations.
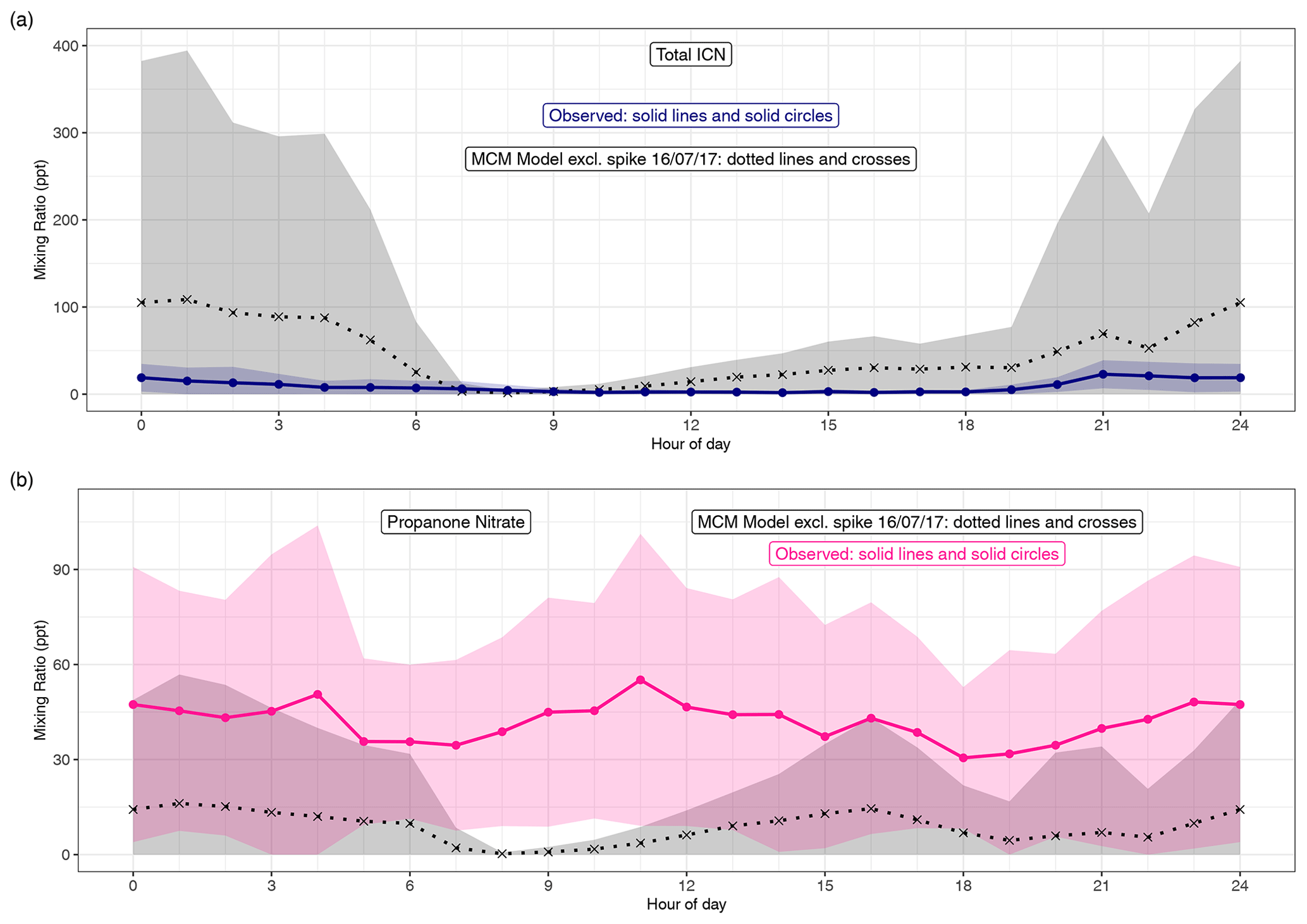
Figure 7(a) Diel pattern of total ICN as modelled using the MCM and measured. For the MCM this is the species NC4CHO, whilst the measurements are the sum of the four δ-ICN (E and Z-(1-ONO2, 4-CO)-ICN and E and Z-(4-ONO2, 1-CO)-ICN). (b) Diel pattern of propanone as modelled using the MCM and measured. Data points are the means and the shaded areas represent ±1 SD in the variability of values for each hour of the day.
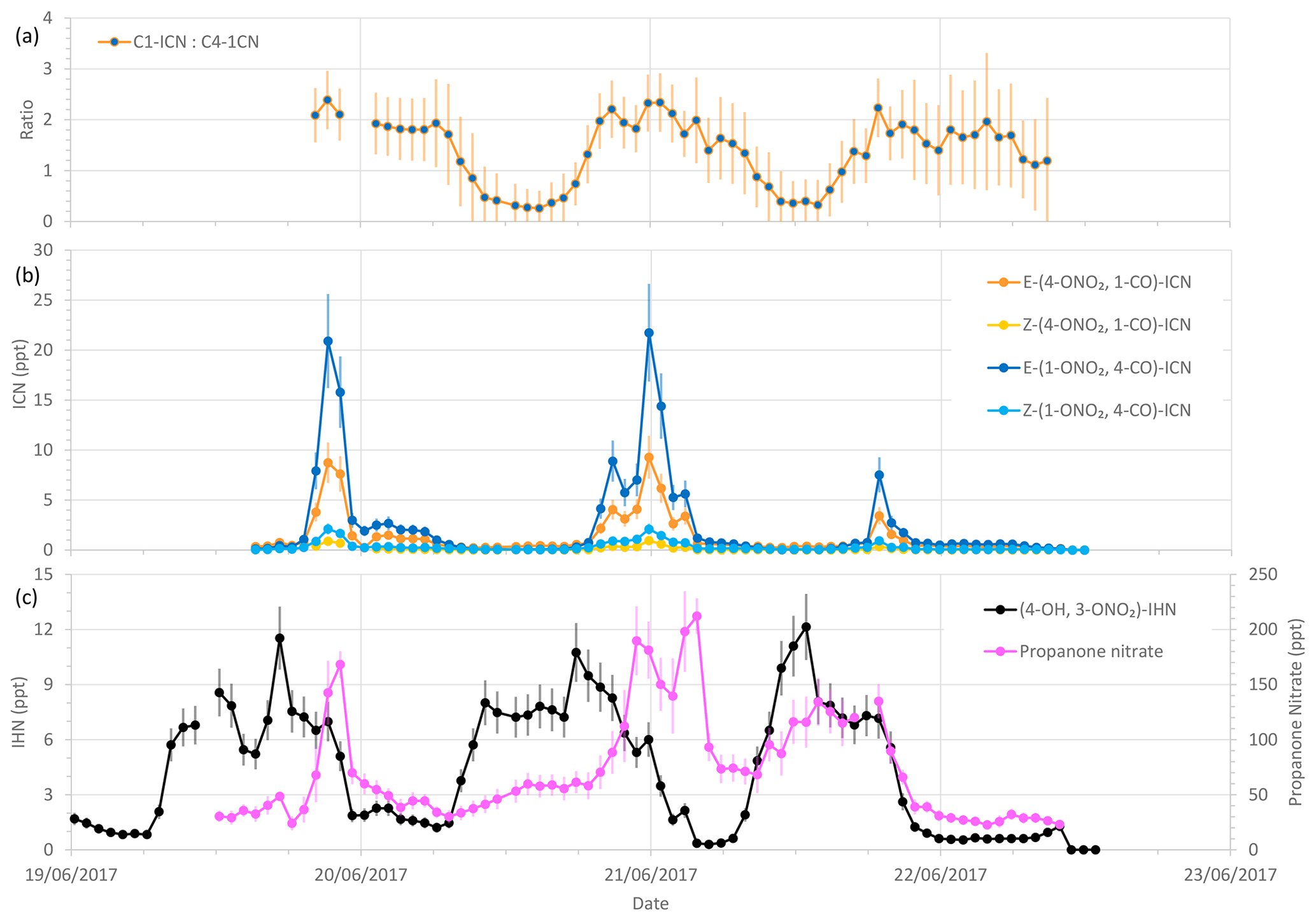
Figure 8Measured δ-ICN mixing ratios and ratios of C1-ICN to C4-ICN, along with (4-OH, 3-ONO2)-IHN and propanone nitrate mixing ratios during the last 4 d of the summer campaign. Error bars are the measurement uncertainties (see Sect. 3.3 for details). The vertical grid lines indicate midnight on each day.
Both the observed and modelled total δ-ICN peak at night, but the observed total was typically less than that modelled (Figs. 7 and S6), particularly on occasions at night (Fig. S6). This is also illustrated by the large day-to-day variability in the modelled diel patterns not seen in the observations (Fig. 7). The observed diel patterns in the ICN can be seen more clearly in Figs. 8 and S5 and show maximum values in the early night and minimum values during the daytime.
The source of δ-ICN is via the addition of NO3 to isoprene followed by addition of O2. This produces δ-nitrooxy peroxy radicals (INO2) (NISOPO2 in the MCM) and, in the conditions simulated for Beijing, the major loss of INO2 is reaction with NO to form NO2 and a δ-nitrooxy alkoxy radical (NISOPO in the MCM), which then reacts rapidly with O2 to form the δ-ICN. Other production pathways for δ-ICN exist in the MCM, but the reaction of INO2 with NO is by far the dominant source of δ-ICN in our simulations. There are some nights when the model simulates large sources of INO2, but typically the modelled production of INO2 maximises in the mid-afternoon. The model is constrained by the observed concentrations of isoprene and NO3, and in the mid-afternoon the observed isoprene mixing ratios are still high and NO3 is around 2 ppt (Fig. S5).
The dominant loss of δ-ICN in the model is the ventilation term, which is greatest during the daytime when the mixed layer is fully developed. The next most important loss processes for δ-ICN are simulated to be photolysis and reaction with OH, which are also both predominantly daytime losses.
The net effect of the production and loss terms is that the modelled δ-ICN increases during the afternoon and maximises during the night-time, and the observed δ-ICN are broadly consistent with this result (Figs. 7, S5 and S6). We observed around 1–2 ppt E-(4-ONO2, 1-CO)-ICN during the daytime, and this requires a significant daytime source assuming a lifetime of around 30 min with respect to photolysis (based on a value of 4.6 × 10−4 s−1 for a solar zenith angle of 0∘ and adjusting for latitude, time of year, and time of day; Xiong et al., 2016). This source is consistent with the observed presence of NO3 during the daytime. However, the considerably larger modelled mixing ratios of δ-ICN compared to the observed suggests that the modelled source via NO3 oxidation of isoprene might be too fast or that the loss processes are too slow.
The same ventilation process has been applied to all model-generated species (Sect. 4). For glyoxal, (1-OH, 2-ONO2)-IHN, and (4-OH, 3-ONO2)-IHN, the model tends to overestimate the decrease in concentrations from the late afternoon onwards, suggesting that the lifetime with respect to ventilation should be longer at these times but increasing the lifetime of δ-ICN would lead to their further overestimation. Applying the same loss term to all model species is of course an approximation, not least because the dilution term depends on the concentration of the species in the diluent air.
The MCM uses a photolysis frequency for δ-ICN based on that measured for propanone nitrate, which is equivalent to 3.16 × 10−4 s−1 for a solar zenith angle of 0∘. Xiong et al. (2016) determined a higher rate of 4.6 × 10−4 s−1 for (4-ONO2, 1-CO)-ICN for a solar zenith angle of 0∘. Increasing rate of photolysis in the model would not only reduce the daytime increase in δ-ICN but would also reduce the amount of modelled δ-ICN that would persist into the night.
Reaction with OH constitutes a similar size loss for δ-ICN as photolysis in the model. The MCM treats all the δ-ICN as (1-ONO2, 4-CO,)-ICN and uses a rate constant for reaction with OH of 4.16 × 10−11 cm3 s−1. W2018 suggests a lower rate constant for reaction of OH with (1-ONO2, 4-CO)-ICN (3.4 × 10−11 cm3 s−1) but a similar rate for (4-ONO2, 1-CO)-ICN (4.1 × 10−11 cm3 s−1). Therefore, treating the two separately in the model and using the W2018 recommended rate constants would overall reduce the loss of δ-ICN with respect to OH, increasing discrepancy with the model.
Night-time losses of δ-ICN are reaction with O3 and NO3. The MCM uses a rate coefficient of 2.4 × 10−17 cm3 s−1 for the reaction of δ-ICN with O3, which is 5 times faster than the value of 4.4 × 10−18 cm3 s−1 recommended by W2018, giving a partial lifetime on the order of 12 h for an O3 mixing ratio of 40 ppb. On the other hand, the MCM uses a rate constant for the reaction of δ-ICN with NO3, which is 10 times slower than that recommended by W2018, but even so the lifetime of δ-ICN with respect to the reaction with NO3 as estimated by W2018 is of the order of 4 d, so this loss pathway would have to be much faster to reduce the modelled night-time δ-ICN close to that which was observed.
Of the observed δ-ICN, the two trans (E) isomers have the highest mixing ratios with E-(1-ONO2, 4-CO)-ICN being the most abundant (Figs. 8 and S2). Focusing on the last 4 d (three nights) of the campaign (Fig. 8), when we have most confidence in the data (i.e. when the plastic valve was used; see Sect. 3.2), we see that the observed ICN C1 : C4 isomer ratio exhibits a diel cycle with higher values at night (mean of 2.0 and SD of 0.3) and an overall mean of 1.4 (SD of 0.6). These values are considerably lower than would be expected based solely on the addition of NO3 to isoprene occurring in the C1 and C4 positions in a ratio of 6 (C1 : C4) (W2018). Our observed ratios are more comparable to the C1 : C4 isomer ratio of 2.8 reported in Schwantes et al. (2015) for their environmental chamber, although in their experiment the ICN mostly came from RO2 + RO2 reactions (see Sect. S1.2) because the NO and NO3 concentrations were low. Turning to the E : Z ratios, we observed the E-ICN isomers as dominating over the Z-ICN isomers. The (1-ONO2, 4-CO)-ICN isomers exhibit a mean night-time E : Z ratio of 8 (SD of 1.4), whilst the (4-ONO2, 1-CO)-ICN isomers exhibit a mean night-time E : Z ratio of 11 (SD of 1.5), giving an overall mean night-time E : Z ratio of 9 (SD of 1.0). These values are far greater than the trans : cis ratio of 1 presumed by W2018 for the reaction of NO3 addition to isoprene based on the OH addition to C1 of isoprene calculated by Peeters et al. (2009). However, it should be noted that the peroxy radicals formed from the reaction of the adducts with O2 may be in a different ratio as these reactions are reversible, similar to those for peroxy radicals formed following OH addition to isoprene.
As noted above, the ratios of C1-ICN to C4-ICN exhibit diel patterns (Fig. 8). The ratios are higher at night and lower in the daytime. The evening ratios are driven by the preferential addition of NO3 to the C1 position as discussed above. The decrease in this ratio during the morning could be explained if the lifetime of C1-ICN were shorter than for the other isomers. However, the rate coefficients for reaction with OH recommended by W2018 are about 20 % slower for C1-ICN than for C4-ICN. Photolysis is expected to be the largest daytime sink, but Xiong et al. (2016) only determined this for E-(4-ONO2, 1-CO)-ICN.
5.3 Propanone nitrate
Figures S6 and 7 show the time series and diel patterns of the measured and modelled propanone nitrate. The observed mixing ratios are generally higher than the modelled values. The chemical lifetime of propanone nitrate is calculated to be around 10 h during the daytime and considerably longer at night, so transport is expected to play an important role in the distribution of propanone nitrate. The mixing term dominates the modelled lifetime and the resulting mixing ratios are highly dependent on the assumptions regarding this term. As discussed in Sect. 4, the same first-order mixing loss rate is used for all species. As significant concentrations of propanone nitrate are expected to remain in the residual layer, this may lead to an overestimation of the mixing, but reducing this would worsen the comparison with the observed propanone nitrate. Despite these issues, the model can still provide insight into the dominant chemical production processes.
The primary source of propanone nitrate in the model is the OH oxidation of δ-ICN, which is formed from NO3 oxidation of isoprene. Consequently, the modelled propanone nitrate and δ-ICN time series share many similarities (Fig. S6). As discussed above, the production of δ-ICN and its loss via OH oxidation occur mostly during the daytime, so this source of propanone nitrate is predominantly during the daytime. On nights when OH is present even at low concentrations it can be a sizable source due to the relatively large amounts of δ-ICN at night. Propanone nitrate is also formed from oxidation of δ-ICN by O3. This is a relatively small source except on nights when O3 was present (Fig. S1). Propanone nitrate can also be produced following the NO3 addition to propene. Overall, the model results suggest this to be a relatively small source, but that it is often calculated to be the dominant source of some of the night-time peaks in propanone nitrate.
Both the modelled and observed propanone nitrate reflect the fact that production of propane nitrate can occur both during the daytime and at night (Figs. S6 and 8). Figure 8 shows the temporal variation of the observed propanone nitrate, along with (4-ONO2, 1-CO)-ICN for the last 4 d of the campaign. Propanone nitrate exhibits three peaks: two on the nights of the 19–20 and 20–21 June 2017 and one during the daytime on the 21 June 2017. These peaks are replicated, but to a lesser extent by the model, which suggests that the peak on the night of the 19–20 June 2017 was OH oxidation of δ-ICN and, to a lesser extent, NO3 addition to propene. The next night NO3 addition to propene was modelled to be the dominant source of the propanone nitrate, with OH oxidation of δ-ICN being the main source during the following day. The consequence of this pattern in sources is that both the observed and modelled mixing ratios show no clear diel cycle, possibly a weak bimodal pattern in the mean, and large day-to-day variability (Fig. 7).
The modelled propanone nitrate is generally less than the δ-ICN. In contrast, the observed propanone nitrate is typically a lot greater than total δ-ICN. This might in part be due to the model being unable to simulate the mixing correctly, but, as discussed in Sect. 5.2, the model simulates considerably larger amounts of δ-ICN than observed, and getting the wrong balance between the various production and loss terms of the δ-ICN will likely impact the modelled propanone nitrate.
5.4 δ-IHN
Four δ-IHN can be formed from the OH addition to isoprene in the C1 and C4 positions: E-(1-OH, 4-ONO2)-IHN, Z-(1-OH, 4-ONO2)-IHN, E-(4-OH, 1-ONO2)-IHN), and Z-(4-OH, 1-ONO2)-IHN) (Fig. 5). The MCM treats the trans and cis δ-IHN isomers as a single species and thus considers two δ-IHN: (1-OH, 4-ONO2)-IHN and (4-OH, 1-ONO2)-IHN. The two modelled δ-IHN are simulated to have very similar mixing ratios during the daytime with peaks values of around 1 ppt (Figs. 9 and S7). Whilst we have previously demonstrated that our system can measure the four δ-IHN (Mills et al., 2016), we found no evidence of them in Beijing despite the modelled daytime mixing ratios being above our detection limit of 0.1 ppt.
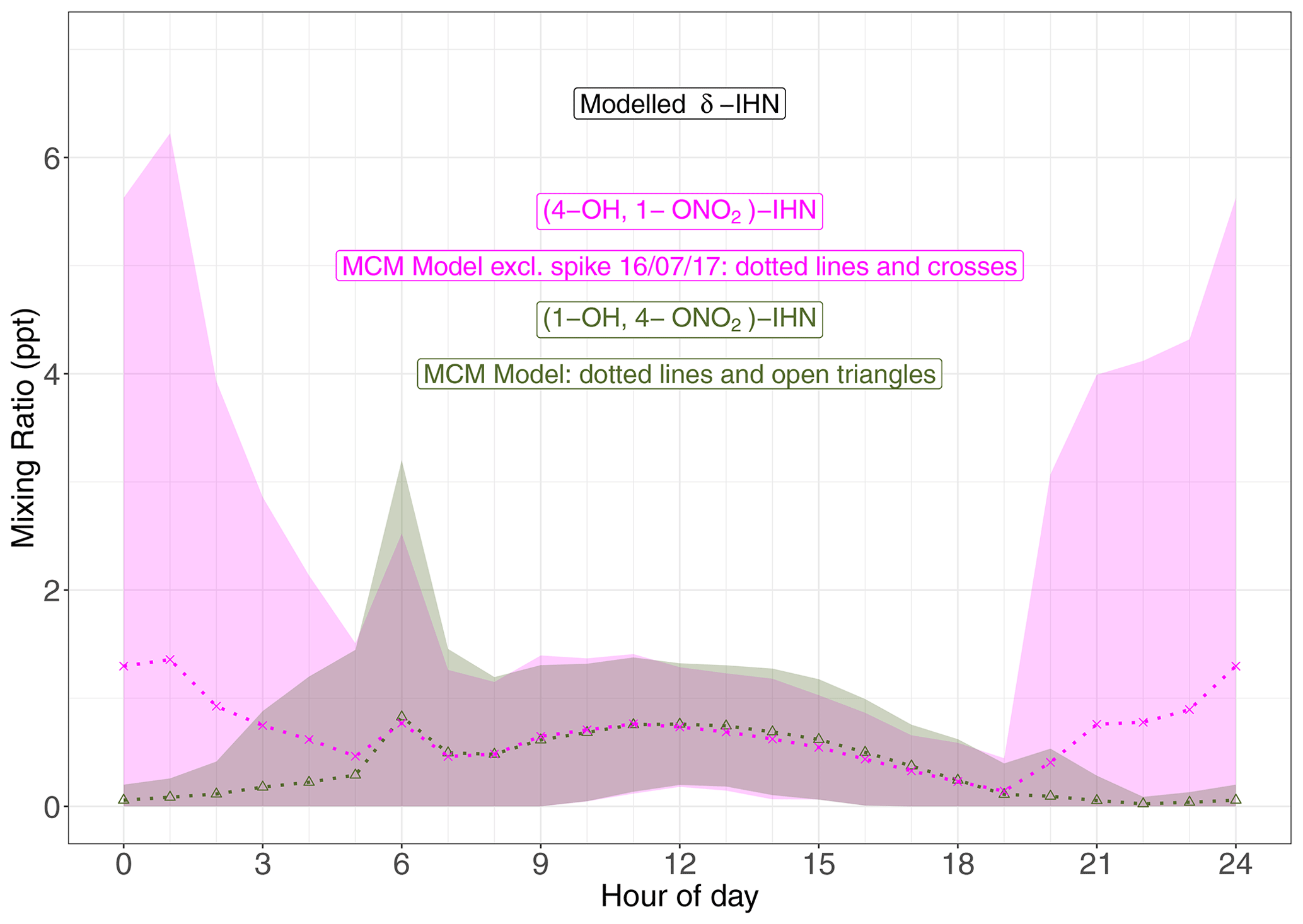
Figure 9Diel pattern of MCM modelled δ-IHN ((1-OH, 4-ONO2)-IHN and (4-OH, 1-ONO2)-IHN). Data points are the means, and the shaded areas represent ±1 SD in the variability of values for each hour of the day.
The model also simulates enhancements of (4-OH, 1-ONO2)-IHN of around 15–30 ppt on several nights coincident with enhanced mixing ratios of the δ-ICN (Figs. 9 and S7) because (4-OH, 1-ONO2)-IHN is also formed when the INO2 radicals react with organic peroxy radicals. As discussed in Sect. 5.2, INO2 is mostly produced during the daytime, but on some nights INO2 mixing ratios were simulated to be high, leading to these elevated mixing ratios of both (4-OH, 1-ONO2)-IHN and δ-ICN. We were only making measurements on a few of the nights when the model simulates these enhancements in (4-OH, 1-ONO2)-IHN, but again we did not detect it, and although we saw small enhancements in δ-ICN, they were far smaller than modelled.
Following OH oxidation of isoprene, the concentration of NO is critical in determining the lifetime, fate, and redistribution of the isoprene-derived peroxy radicals, ISOPOO. Measuring IHN, products of the reactions between NO and ISOPOO, provides observational insight into this chemistry. Our measurements show that in the summertime conditions experienced in Beijing the β-IHN ratio ((1-OH, 2-ONO2)-IHN to (4-OH, 3-ONO2)-IHN) increases at NO mixing ratios below 2 ppb, providing observational field evidence of the redistribution of the ISOPOO away from the kinetic ratio towards a new thermodynamic equilibrium consistent with box model calculations.
There are absolute discrepancies between the modelled and observed ratio of β-IHN, with the observed values being higher. Some of this might be resolved with a more accurate calibration of (1-OH, 2-ONO2)-IHN. However, there may be issues with the chemical scheme, and it is noteworthy that the model underestimates the measured RO2 mixing ratios for both those classed as simple and those as complex, the latter which include ISOPOO (Whalley et al., 2021). Whilst an underestimation of the β-ISOPOO might impact the absolute mixing ratios of modelled β-IHN, an underestimation of the total RO2 can affect the β-IHN ratio as the rate constant for the reaction of RO2 with (1-OH, 2-OO)-ISOPOO is slower than that for reaction of RO2 with (4-OH, 3-OO)-ISOPOO and this contributes to the higher β-IHN ratios at the lower NO mixing ratios.
The diel pattern in observed NO (along with OH and peroxy radicals) in Beijing suggests that the ratio of (1-OH, 2-ONO2)-IHN to (4-OH, 3-ONO2)-IHN should have increased during the afternoon, but unfortunately we were unable to make enough measurements to observe this given the day-to-day variability in atmospheric composition. However, our observations demonstrate that more extensive measurements of individual β-IHN should provide insight into the β-ISOPOO speciation and how it changes under different chemical regimes.
Like the β-IHN, δ-IHN are formed following OH addition to isoprene in the C1 and C4 positions, and their rates of production are dependent on the kinetic yields of their respective ISOPOO from O2 reaction with the C1 and C4 adducts and redistribution amongst the ISOPOO. The rapid isomerisation of the Z-δ-ISOPOO leads to a greater β : δ ISOPOO ratio than the kinetic one. Whilst the model strongly favours production of β-IHN over the δ-IHN, it still suggests that there should be enough δ-IHN present during the daytime for us to detect with our measurement system. However, we found no evidence of their presence, which may suggest that the model underestimates the β : δ ISOPOO ratio or the δ-IHN losses.
The observed amounts of δ-ICN demonstrate the importance of daytime addition of NO3 to isoprene in Beijing. Hamilton et al. (2021) have also shown this source of organic nitrates to be important for the formation secondary organic aerosol in Beijing. We did, however, observe far less δ-ICN than we modelled, which may suggest that the predominant source of the δ-ICN in the model (reaction of NO with δ-nitrooxy peroxy radicals) is too large or that the sink is too small. The main source of propanone nitrate in the model is the OH oxidation of δ-ICN, so the atmospheric budgets of these two nitrates are linked. Observations of propanone nitrate suggest it can be a marker of this chemistry, although other sources, such as the NO3 addition to propene and the transport of propane nitrate, need to be taken into consideration. The model suggests that reaction of the δ-nitrooxy peroxy radicals with organic peroxy radicals is a significant source of (4-OH, 1-ONO2)-IHN at night, but this is not supported by our observations.
Our speciated measurements of the four δ-ICN isomers provide insight into the isomeric distribution of the δ-nitrooxy peroxy radicals. The two trans δ-ICN isomers are observed to have the highest mixing ratios, with E-(1-ONO2, 4-CO)-ICN being the most abundant. However, the mean C1 : C4 isomer ratio is 1.4, which is considerably lower than would be expected based solely on the addition of NO3 to isoprene occurring in the C1 and C4 positions in a 6 : 1 ratio. This raises the question as to whether it is appropriate to represent the δ-ICN by a single C1 nitrated isomer, as has been done in the MCM. We observed the trans-ICN isomers to dominate over the cis-ICN isomers with a mean ratio of 7, far greater than the trans : cis ratio of 1 presumed by W2018 for the reaction of NO3 addition to isoprene. This suggests that thermodynamic redistribution of the δ-nitrooxy peroxy radicals may also be important.
This study demonstrates the value of speciated IN measurements in testing the understanding of isoprene degradation chemistry, and more measurements would provide more robust constraints. One reason for the limited data was the need for a different instrument setup for the measurement of (1-OH, 2-ONO2)-IHN. Resolving this would increase data capture and provide concurrent measurements of a range of speciated IN that can be used to test different aspects of the isoprene degradation system simultaneously, including the balance between OH and NO3 oxidation of isoprene. Observations of speciated IN in a wide range of NO VOC chemical space at different times of day would provide greater constraint on their chemistry, in particular the isomeric distribution and fate of the peroxy radicals. The chemistry of the isoprene degradation chemistry is complex, involving multiple species and reactions. Analysis of field measurements of IN can help constrain aspects of this, but interpretation would be enhanced by simultaneous measurements of other chemical species (e.g. other products of the nitrooxy peroxy radicals), as well as improved quantification of several reaction rate constants. Further laboratory studies are required to improve quantification of the IN lifetimes, in particular with respect to photolysis of the ICN, deposition, and hydrolysis, and to better constrain the peroxy radical reactions, including the branching ratio of the NO reaction that leads to IHN.
The MCM code is available from the authors on request.
The observational data and diel cycles from the MCM in the figures are provided in the Supplement. The 15 min data from the MCM are available from the authors on request.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-6315-2021-supplement.
CER led the data interpretation and writing of the manuscript. GPM made the measurements of the IN with the assistance of YL. LKW did the MCM modelling. CER, WJB, SG, DEH, CNH, RLJ, JDL, XW, and CY were involved in the project planning and leading the measurement groups. WJFA, LRC, JRH, SK, LJK, BO, ES, FS, and RWM provided measurement data. All authors commented on the manuscript.
The authors declare that they have no conflict of interest.
This article is part of the special issue “In-depth study of air pollution sources and processes within Beijing and its surrounding region (APHH-Beijing) (ACP/AMT inter-journal SI)”. It is not associated with a conference.
We are grateful for funding provided by the UK Natural Environment Research Council (NERC), UK Medical Research Council, and the Natural Science Foundation of China (NSFC) under the framework of the Newton Innovation Fund. Eloise Slater and Robert Woodward-Massey are grateful to the NERC SPHERES Doctoral Training Programme for funding their PhD studentships. Claire E. Reeves acknowledges Andrew Rickard (NCAS, University of York) for providing information on the MCM.
This research has been supported by the UK Natural Environment Research Council (NERC; grant nos. NE/N006909/1, NE/N006895/1, NE/N006976/1, and NE/N00700X/1), UK Medical Research Council, the Natural Science Foundation of China (NSFC) under the framework of the Newton Innovation Fund (grant no. 41571130031) and the NERC SPHERES Doctoral Training Programme.
This paper was edited by Frank Keutsch and reviewed by three anonymous referees.
Bates, K. H. and Jacob, D. J.: A new model mechanism for atmospheric oxidation of isoprene: global effects on oxidants, nitrogen oxides, organic products, and secondary organic aerosol, Atmos. Chem. Phys., 19, 9613–9640, https://doi.org/10.5194/acp-19-9613-2019, 2019.
Bew, S. P., Hiatt-Gipson, G. D., Mills, G. P., and Reeves, C. E.: Efficient syntheses of climate impacting isoprene nitrates and (1R,5S)-(-)-myrtenol nitrate, Beilstein J. Org. Chem., 12, 1081–1095, https://doi.org/10.3762/bjoc.12.103, 2016.
Emmerson, K. M. and Evans, M. J.: Comparison of tropospheric gas-phase chemistry schemes for use within global models, Atmos. Chem. Phys., 9, 1831–1845, https://doi.org/10.5194/acp-9-1831-2009, 2009.
Fiore, A. M., Horowitz, L. W., Purves, D. W., Levy II, H., Evans, M. J., Wang, Y., Li, Q., and Yantosca, R. M.: Evaluating the contribution of changes in isoprene emissions to surface ozone trends over the eastern United States, J. Geophys. Res., 110, D12303, https://doi.org/10.1029/2004JD005485, 2005.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Hamilton, J. F., Bryant, D. J., Edwards, P. E., Quyang, B., Bannan, T. J., Mehra, A., Mayhew, A. W., Hopkins, J. R., Dunmore, R. E., Squires, F. A., Lee, J. D., Newland, M. J., Worrall, S. D., Bacak, A., Coe, H., Percival, C., Whalley, L. K., Heard, D. E., Slater, E. J., Jones, R. L., Cui, T., Surratt, J. D., Reeves, C. E., Mills, G. P., Grimmond, S., Sun, Y., Xu, W., Shi, Z., and Rickard, A. R.: Key Role of NO3 Radicals in the Production of Isoprene Nitrates and Nitrooxyorganosulfates in Beijing, Environ. Sci. Technol., 55, 842–853, doi.org/10.1021/acs.est.0c05689, 2021.
Jacobs, M. I., Burke, W. J., and Elrod, M. J.: Kinetics of the reactions of isoprene-derived hydroxynitrates: gas phase epoxide formation and solution phase hydrolysis, Atmos. Chem. Phys., 14, 8933–8946, https://doi.org/10.5194/acp-14-8933-2014, 2014.
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015.
Lee, L., Teng, A. P., Wennberg, P. O., Crounse, J. D., and Cohen, R. C.: On Rates and Mechanisms of OH and O3 Reactions with Isoprene Derived Hydroxy Nitrates, J. Phys. Chem., 118, 1622–1637, https://doi.org/10.1021/jp4107603, 2014.
Li, J., Mao, J., Min, K.-E., Washenfelder, R. A., Brown, S. S., Kaiser, J., Keutsch, F. N., Volkamer, R., Wolfe, G. M., Hanisco, T. F., Pollack, I. B., Ryerson, T. B., Graus, M., Gilman, J. B., Lerner, B. M., Warneke, C., de Gouw, J. A., Middlebrook, A. M., Liao, J., Welt, A., Henderson, B. H., McNeill, V. F., Hall, S. R., Ullmann, K., Donner, L. J., Paulot, F., and Horowitz, L. W.: Observational constraints on glyoxal production from isoprene oxidation and its contribution to organic aerosol over the Southeast United States, J. Geophys. Res.-Atmos., 121, 9849–9861, https://doi.org/10.1002/2016JD025331, 2016.
Lockwood, A. L., Shepson, P. B., Fiddler, M. N., and Alaghmand, M.: Isoprene nitrates: preparation, separation, identification, yields, and atmospheric chemistry, Atmos. Chem. Phys., 10, 6169–6178, https://doi.org/10.5194/acp-10-6169-2010, 2010.
Mills, G. P., Hiatt-Gipson, G. D., Bew, S. P., and Reeves, C. E.: Measurement of isoprene nitrates by GCMS, Atmos. Meas. Tech., 9, 4533–4545, https://doi.org/10.5194/amt-9-4533-2016, 2016.
Müller, J.-F., Peeters, J., and Stavrakou, T.: Fast photolysis of carbonyl nitrates from isoprene, Atmos. Chem. Phys., 14, 2497–2508, https://doi.org/10.5194/acp-14-2497-2014, 2014.
Newland, M. J., Bryant, D. J., Dunmore, R. E., Bannan, T. J., Acton, W. J. F., Langford, B., Hopkins, J. R., Squires, F. A., Dixon, W., Drysdale, W. S., Ivatt, P. D., Evans, M. J., Edwards, P. M., Whalley, L. K., Heard, D. E., Slater, E. J., Woodward-Massey, R., Ye, C., Mehra, A., Worrall, S. D., Bacak, A., Coe, H., Percival, C. J., Hewitt, C. N., Lee, J. D., Cui, T., Surratt, J. D., Wang, X., Lewis, A. C., Rickard, A. R., and Hamilton, J. F.: Low-NO atmospheric oxidation pathways in a polluted megacity, Atmos. Chem. Phys., 21, 1613–1625, https://doi.org/10.5194/acp-21-1613-2021, 2021.
Nguyen, T. B., Crounse, J. D., Schwantes, R. H., Teng, A. P., Bates, K. H., Zhang, X., St. Clair, J. M., Brune, W. H., Tyndall, G. S., Keutsch, F. N., Seinfeld, J. H., and Wennberg, P. O.: Overview of the Focused Isoprene eXperiment at the California Institute of Technology (FIXCIT): mechanistic chamber studies on the oxidation of biogenic compounds, Atmos. Chem. Phys., 14, 13531–13549, https://doi.org/10.5194/acp-14-13531-2014, 2014.
Nguyen, T. B., Crounse, J. D., Teng, A. P., St Clair, J. M., Paulot, F., Wolfe, G. M., and Wennberg, P. O.: Rapid deposition of oxidized biogenic compounds to a temperate forest, P. Natl. Acad. Sci. USA, 112, E392–E401, https://doi.org/10.1073/pnas.1418702112, 2015.
Peeters, J., Nguyen, T. L., and Vereecken, L.: HOx radical regeneration in the oxidation of isoprene, Phys. Chem. Chem. Phys., 28, 5935–5939, 2009.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Schwantes, R. H., Teng, A. P., Nguyen, T. B., Coggon, M. M., Crounse, J. D., St Clair, J. M., Zhang, X., Schilling, K. A., Seinfeld, J. H., and Wennberg, P. O.: Isoprene NO3 Oxidation Products from the RO2 + HO2 Pathway, J. Phys. Chem., 119, 10158–10171, https://doi.org/10.1021/acs.jpca.5b06355, 2015.
Schwantes, R. H., Emmons, L. K., Orlando, J. J., Barth, M. C., Tyndall, G. S., Hall, S. R., Ullmann, K., St. Clair, J. M., Blake, D. R., Wisthaler, A., and Bui, T. P. V.: Comprehensive isoprene and terpene gas-phase chemistry improves simulated surface ozone in the southeastern US, Atmos. Chem. Phys., 20, 3739–3776, https://doi.org/10.5194/acp-20-3739-2020, 2020.
Shi, Z., Vu, T., Kotthaus, S., Harrison, R. M., Grimmond, S., Yue, S., Zhu, T., Lee, J., Han, Y., Demuzere, M., Dunmore, R. E., Ren, L., Liu, D., Wang, Y., Wild, O., Allan, J., Acton, W. J., Barlow, J., Barratt, B., Beddows, D., Bloss, W. J., Calzolai, G., Carruthers, D., Carslaw, D. C., Chan, Q., Chatzidiakou, L., Chen, Y., Crilley, L., Coe, H., Dai, T., Doherty, R., Duan, F., Fu, P., Ge, B., Ge, M., Guan, D., Hamilton, J. F., He, K., Heal, M., Heard, D., Hewitt, C. N., Hollaway, M., Hu, M., Ji, D., Jiang, X., Jones, R., Kalberer, M., Kelly, F. J., Kramer, L., Langford, B., Lin, C., Lewis, A. C., Li, J., Li, W., Liu, H., Liu, J., Loh, M., Lu, K., Lucarelli, F., Mann, G., McFiggans, G., Miller, M. R., Mills, G., Monk, P., Nemitz, E., O'Connor, F., Ouyang, B., Palmer, P. I., Percival, C., Popoola, O., Reeves, C., Rickard, A. R., Shao, L., Shi, G., Spracklen, D., Stevenson, D., Sun, Y., Sun, Z., Tao, S., Tong, S., Wang, Q., Wang, W., Wang, X., Wang, X., Wang, Z., Wei, L., Whalley, L., Wu, X., Wu, Z., Xie, P., Yang, F., Zhang, Q., Zhang, Y., Zhang, Y., and Zheng, M.: Introduction to the special issue “In-depth study of air pollution sources and processes within Beijing and its surrounding region (APHH-Beijing)”, Atmos. Chem. Phys., 19, 7519–7546, https://doi.org/10.5194/acp-19-7519-2019, 2019.
Squire, O. J., Archibald, A. T., Griffiths, P. T., Jenkin, M. E., Smith, D., and Pyle, J. A.: Influence of isoprene chemical mechanism on modelled changes in tropospheric ozone due to climate and land use over the 21st century, Atmos. Chem. Phys., 15, 5123–5143, https://doi.org/10.5194/acp-15-5123-2015, 2015.
Teng, A. P., Crounse, J. D., and Wennberg, P. O.: Isoprene Peroxy Radical Dynamics, J. Am. Che. Soc., 139, 5367–5377, https://doi.org/10.1021/jacs.6b12838, 2017.
Vasquez, K. T., Allen, H. M., Crounse, J. D., Praske, E., Xu, L., Noelscher, A. C., and Wennberg, P. O.: Low-pressure gas chromatography with chemical ionization mass spectrometry for quantification of multifunctional organic compounds in the atmosphere, Atmos. Meas. Tech., 11, 6815–6832, https://doi.org/10.5194/amt-11-6815-2018, 2018.
Volkamer, R., San Martini, F., Molina, L. T., Salcedo, D. Jimenez, J. L., and Molina, M. J.: A missing sink for gas-phase glyoxal in Mexico City: Formation of secondary organic aerosol, Geophys. Res. Letts., 34, L19807, https://doi.org/10.1029/2007GL030752, 2007.
von Kuhlmann, R., Lawrence, M. G., Pöschl, U., and Crutzen, P. J.: Sensitivities in global scale modeling of isoprene, Atmos. Chem. Phys., 4, 1–17, https://doi.org/10.5194/acp-4-1-2004, 2004.
Washenfelder, R. A., Young, C. J., Brown, S. S., Angevine, W. M. Atlas, E. L., Blake, D, R., Bon, D. M., Cubison, M. J., de Gouw, J. A., Dusanter, S., Flynn, J., Gilman, J. B., Graus, M., Griffith, S., Grossberg, N., Hayes, P. L., Jimenez, J. L., Kuster, W. C., Lefer, B. L. Pollack, I. B., Ryerson, T. B., Stark, H., Stevens, P. S., and Trainer, M. K: The glyoxal budget and its contribution to organic aerosol for Los Angeles, California, during CalNex 2010, J. Geophys. Res., 116, D00V02, https://doi.org/10.1029/2011JD016314, 2011.
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., St Clair, J. M., Teng, A. P., Zhang, X., and Seinfeld, J. H.: Gas-Phase Reactions of Isoprene and Its Major Oxidation Products, Chem. Rev., 118, 3337–3390, https://doi.org/10.1021/acs.chemrev.7b00439, 2018.
Whalley, L. K., Slater, E. J., Woodward-Massey, R., Ye, C., Lee, J. D., Squires, F., Hopkins, J. R., Dunmore, R. E., Shaw, M., Hamilton, J. F., Lewis, A. C., Mehra, A., Worrall, S. D., Bacak, A., Bannan, T. J., Coe, H., Percival, C. J., Ouyang, B., Jones, R. L., Crilley, L. R., Kramer, L. J., Bloss, W. J., Vu, T., Kotthaus, S., Grimmond, S., Sun, Y., Xu, W., Yue, S., Ren, L., Acton, W. J. F., Hewitt, C. N., Wang, X., Fu, P., and Heard, D. E.: Evaluating the sensitivity of radical chemistry and ozone formation to ambient VOCs and NOx in Beijing, Atmos. Chem. Phys., 21, 2125–2147, https://doi.org/10.5194/acp-21-2125-2021, 2021.
Wu, S., Mickley, L. J., Jacob, D. J., Logan, J. A. Yantosca, R. M., and Rind, D.: Why are there large differences between models in global budgets of tropospheric ozone?, J. Geophys. Res., 112, D05302, https://doi.org/10.1029/2006JD007801, 2007.
Xiong, F., McAvey, K. M., Pratt, K. A., Groff, C. J., Hostetler, M. A., Lipton, M. A., Starn, T. K., Seeley, J. V., Bertman, S. B., Teng, A. P., Crounse, J. D., Nguyen, T. B., Wennberg, P. O., Misztal, P. K., Goldstein, A. H., Guenther, A. B., Koss, A. R., Olson, K. F., de Gouw, J. A., Baumann, K., Edgerton, E. S., Feiner, P. A., Zhang, L., Miller, D. O., Brune, W. H., and Shepson, P. B.: Observation of isoprene hydroxynitrates in the southeastern United States and implications for the fate of NOx, Atmos. Chem. Phys., 15, 11257–11272, https://doi.org/10.5194/acp-15-11257-2015, 2015.
Xiong, F., Borca, C. H., Slipchenko, L. V., and Shepson, P. B.: Photochemical degradation of isoprene-derived 4,1-nitrooxy enal, Atmos. Chem. Phys., 16, 5595–5610, https://doi.org/10.5194/acp-16-5595-2016, 2016.