the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 09 Nov 2021
| 09 Nov 2021
Mapping gaseous dimethylamine, trimethylamine, ammonia, and their particulate counterparts in marine atmospheres of China's marginal seas – Part 1: Differentiating marine emission from continental transport
Dihui Chen
Yanjie Shen
Juntao Wang
Huiwang Gao
Xiaohong Yao
To study sea-derived gaseous amines, ammonia, and primary particulate aminium ions in the marine atmosphere of China's marginal seas, an onboard URG-9000D Ambient Ion Monitor-Ion Chromatograph (AIM-IC, Thermo Fisher) was set up on the front deck of the R/V Dongfanghong-3 to semi-continuously measure the spatiotemporal variations in the concentrations of atmospheric trimethylamine (TMAgas), dimethylamine (DMAgas), and ammonia (NH3gas) along with their particulate matter (PM2.5) counterparts. In this study, we differentiated marine emissions of the gas species from continental transport using data obtained from 9 to 22 December 2019 during the cruise over the Yellow and Bohai seas, facilitated by additional short-term measurements collected at a coastal site near the Yellow Sea during the summer, fall, and winter of 2019. The data obtained from the cruise and coastal sites demonstrated that the observed TMAgas and protonated trimethylamine (TMAH+) in PM2.5 over the Yellow and Bohai seas overwhelmingly originated from marine sources. During the cruise, no significant correlation (P>0.05) was observed between the simultaneously measured TMAH+ and TMAgas concentrations. Additionally, the concentrations of TMAH+ in the marine atmosphere varied around 0.28±0.18 µg m−3 (average ± standard deviation), with several episodic hourly average values exceeding 1 µg m−3, which were approximately 1 order of magnitude larger than those of TMAgas (approximately 0.031±0.009 µg m−3). Moreover, there was a significant negative correlation (P<0.01) between the concentrations of TMAH+ and in PM2.5. Therefore, the observed TMAH+ in PM2.5 was overwhelmingly derived from primary sea-spray aerosols. Using TMAgas and TMAH+ in PM2.5 as tracers for sea-derived basic gases and sea-spray particulate aminium ions, the values of non-sea-derived DMAgas, NH3gas, and non-sea-spray particulate DMAH+ in PM2.5 were estimated. The estimated average values of each species contributed 16 %, 34 %, and 65 % of the observed average concentrations for non-sea-derived DMAgas, NH3gas, and non-sea-spray particulate DMAH+ in PM2.5, respectively. Uncertainties remained in the estimations, as TMAH+ may decompose into smaller molecules in seawater to varying extents. The non-sea-derived gases and non-sea-spray particulate DMAH+ likely originated from long-range transport from the upwind continents based on the recorded offshore winds and increased concentrations of non-sea-salt (nss-) and in PM2.5. The lack of a detectable increase in particulate DMAH+, , and nss- concentrations in several SO2 plumes did not support the secondary formation of particulate DMAH+ in the marine atmosphere.
- Article
(1401 KB) - Full-text XML
-
Supplement
(397 KB) - BibTeX
- EndNote





Gaseous amines and their particulate counterparts are vital for reduced nitrogen compounds in the marine atmosphere (Facchini et al., 2008; Müller et al., 2009; Hu et al., 2015, 2018; van Pinxteren et al., 2015, 2019; Yu et al., 2016; Xie et al., 2018; Zhou et al., 2019) and are primarily derived from seawater, where glycine betaine (GBT), trimethylamine N-oxide (TMAO), and choline are the three major precursors (Burg and Ferraris, 2008; Lidbury et al., 2015a, b; Jameson et al., 2016; Taubert et al., 2017). GBT, TMAO, and choline are critical for maintaining the osmotic pressure in marine organisms. When released into the environment, they can be degraded by bacteria to trimethylamine (TMA), dimethylamine (DMA), or methylamine (MA) (Lidbury et al., 2015a, b). Gaseous DMA, TMA, and MA may be vital in the formation of secondary particles in the atmosphere by nucleation (Almeida et al., 2013; Chen et al., 2016; Yao et al., 2018; Zhu et al., 2019). In addition to biogenic amines, anthropogenic emissions are known as important sources of amines in the continental atmosphere but not in the marine atmosphere (Ge et al., 2011). Modeling studies have shown that the continental amine species in the gas and/or particle phases can be transported regionally, including downwind marine atmospheres (Yu and Luo, 2014; Mao et al., 2018). Simultaneous real-time measurement of gaseous amines and their particulate counterparts in the marine atmosphere over the ocean remains challenging because of artifact signals related to self-vessel emissions and amine-containing dew evaporation; however, this is not the case in the continental atmosphere (VandenBoer et al., 2011). The lack of direct measurements restricts the determination of their sources and the relationship between the reduced nitrogen compounds and acid–base neutralization reactions in the marine atmosphere.
Reduced nitrogen compounds in the ocean can finally decompose into ammonium ions () and other smaller molecules. in surface seawater releases into the marine atmosphere as atmospheric ammonia (NH3gas) under favorable conditions (Johnson et al., 2008; Carpenter et al., 2012; Paulot et al., 2015). The ocean is an important source of NH3gas, contributing approximately 40 % of the natural NH3 emissions on Earth (Carpenter et al., 2012; Paulot et al., 2015). In the literature, large uncertainties remain in estimating NH3 emissions from the ocean; for example, the annual emission flux ranges from 2 to 23 Tg N a−1 (Clarke and Porter, 1993; Dentener and Crutzen, 1994; Sutton et al., 2013; Paulot et al., 2015). These uncertainties are primarily derived from two factors: (1) the major marine sources of NH3gas are still disputed, such as seawater, seabirds, or the photolysis of marine organic nitrogen at the ocean surface or in the atmosphere; (2) direct NH3gas observations in marine atmospheres are restricted as onboard ambient NH3gas measurement techniques sometimes suffer from large artifacts from NH3gas contamination associated with onboard human activities, dew evaporation, and water vapor interference (Quinn et al., 1990; Clarke and Porter, 1993; Johnson et al., 2008; Keene et al., 2009; Wentworth et al., 2016; Teng et al., 2017). Additionally, the long-range transport of atmospheric NH3gas from the continent may also complicate the source analysis of NH3gas in marine atmospheres (McNaughton et al., 2004; Uematsu et al., 2004; Zhao et al., 2015; Lutsch et al., 2016).
To identify and characterize sea-derived gaseous amines, ammonia, and sea-spray particulate aminium ions as well as secondary particulate aminium ions from continental transport in the atmospheres of China's marginal seas, we conducted two cruise campaigns: one over the Yellow and Bohai seas in China from 9 to 22 December 2019 (Campaign A) and another over the Eastern China and Yellow seas from 27 December 2019 to 16 January 2020 (Campaign B). Winter cruise campaigns provide great opportunities for observational studies due to the following: (1) higher concentrations of nutrients in the seas at lower sea surface water temperatures, which may favor higher primary production (Guo et al., 2020) and subsequently increase marine emissions of gaseous amines and/or aminium-containing sea-spray aerosols; (2) periodically enhanced air–sea exchanges driven by the strong winter Asian monsoon every 4–10 d (Zhu et al., 2018); (3) periodically enhanced long-range transport of anthropogenic pollutants from continents to the seas, which may enhance the formation of secondary ammonium and aminium aerosols (Guo et al., 2016; Yu et al., 2016; Xie et al., 2018; Wang et al., 2019).
In this study, an onboard URG-9000D Ambient Ion Monitor-Ion Chromatograph (AIM-IC, Thermo Fisher) instrument was used to simultaneously measure the spatiotemporal variations in the concentrations of gaseous amines and NH3gas along with their counterparts in PM2.5. Semi-continuous measurement data were analyzed to identify the study targets. This study was divided into two parts. In this section, we distinguish the marine sources from the continental transport of reduced nitrogen compounds in marine atmospheres and subsequently quantify each contribution to the observed species during the 9–22 December 2019 campaign. In the companion paper (Gao et al., 2021), we analyzed the spatiotemporal heterogeneity and related causes and subsequently delivered a hypothesis regarding the marine emissions of reduced nitrogen compounds using the data from the two campaigns and an additional cruise campaign previously reported by Hu et al. (2015).
2.1 Sampling periods, locations, and instruments
Campaign A was conducted from 9 to 19 December 2019 on the R/V Dongfanghong-3 with a displacement tonnage of 5000. The research vessel was still within its testing period and used state-of-the-art combustion technology with low-sulfur diesel. Campaign B started from 27 December 2019 to 17 January 2020 and was organized by another research team. During 20–22 December, the vessel was anchored at the port while the sampling continued. The 44 h were referred to as the transition period between campaigns A and B. A standard-sized air-conditioned container was set up on the front deck to house a suite of instruments including the AIM-IC, a fast-mobility particle sizer (FMPS, Tsi), a cloud condensation nuclei counter (CCN-100, Droplet MT), and a single-particle aerosol mass spectrometer (SPAMS 05, Hexin). No human activities occurred on the front deck during cruising, excluding anchoring at the port. Even during the anchoring period, human activity on the front deck was rare. The use of the container on the front deck effectively minimized the self-vessel contamination by NH3gas and gaseous amines. The front deck was approximately 10 m a.s.l., and the container height was 2.8 m.
To ensure that the onboard AIM-IC was operated properly, it was housed in a mobile air-conditioned mini-container, which was further placed in a standard container with a 1 m stainless steel sampling probe connected to the ambient air. The inlet of the sampling probe extended from the top corner of the standard container facing the sea. The AIM-IC consists of two major parts: an ambient air sampling system and an ion chromatography analysis system. For the sampling system, the AIM-IC was equipped with a PM2.5 cyclone and operated at a rate of 3 L min−1. The sampled gases and particles in the water solution were stored in two syringes prior to their injection for analysis. The ion chromatography analysis system measured the semi-continuous concentrations of chemically reactive gases. These included NH3gas, gaseous amines, and acidic gases such as SO2 and HNO3, along with their particulate counterparts, at a temporal resolution of 1 h. This facilitated the identification of possible interference from onboard dew evaporation, which typically occurs with sunrise (Teng et al., 2017).
An automatic weather system providing real-time meteorological data is available on the R/V Dongfanghong-3. The heading wind was corrected to determine the true wind speed and direction. The surface seawater temperature was not measured during this cruise campaign and typically had a delay of a few hours compared to the ambient air temperature (Deng et al., 2014).
On 1–9 August, 12 September to 1 October, and 16 November to 1 December 2019, the AIM-IC was set up at a coastal site in Qingdao (36.34∘ N, 120.67∘ E) to conduct routine measurements (Fig. 1). Coastal measurement data were obtained from 2 weeks to 4 months before the winter cruise campaign. The sampling site was located in a new high-technology zone near the Yellow Sea, with the shortest distance from the sea being approximately 1 km in the south. The AIM-IC was housed in a research lab on the fifth floor of a building, approximately 16 m a.g.l. The sampling probe extended out of the window and was directly connected to the ambient air. Typically, higher biogenic emissions of reduced nitrogen compounds over the continents are expected in the summer than in the winter owing to the temperature effect (Yu et al., 2016; Teng et al., 2017).
2.2 Chemical analysis
The AIM-IC includes an ICS-1100 ion chromatograph, wherein an analytical column (Ion Pac CS17A – 2×250 mm) was used to measure cations, including Na+, , protonated dimethylamine (DMAH+), and protonated trimethylamine (TMAH+), and an AS11-HC (2×50 mm) was used to measure anions, including , , Cl−, and organic ions. Methanesulfonic acid solution (5 mM) was used as the eluent for cation analysis, while potassium hydroxide solution (varying from 3 to 40 mM) was used as the gradient eluent for anion analysis. Each analysis took 26–28 min to obtain a complete ion spectrum. The volume of the injection loop installed on the low-pressure valve was 250 µL, which substantially reduced the limits of detection for all ions. The limits of detection for , DMAH+, and TMAH+ were 0.0004, 0.004, and 0.002 µg m−3, respectively, in ambient air. The limits of detection for and were 0.05 and 0.015 µg m−3, respectively, in ambient air. The ICS-1100 was calibrated onboard prior to obtaining regular measurements, and the second calibration was conducted when the vessel was anchored at the port. The AIM-IC analysis was not affected by ambient water vapor, as the device directly measured the ions. Detailed information regarding the AIM-IC analysis is provided in Teng et al. (2017) and Xie et al. (2018). Notably, strong K+ interference occurred unexpectedly and occasionally and then disappeared during different campaigns. When the interference occurred, DMAH+ and TMAH+ were undetectable because of the increased baseline at the corresponding residence time in the ion chromatograph (Fig. S1 in the Supplement); consequently, some PM2.5 DMAH+ and TMAH+ concentration data are unavailable in Fig. 1. However, the concentrations of gaseous amines were still detected correctly, with a low baseline at the residence. K+ interference remains to be investigated. Additionally, a few surface seawater samples were collected from different sea zones. The and aminium ion concentrations in the samples were not measured, as the analytical methods were still hindered by high sea-salt ion contents.
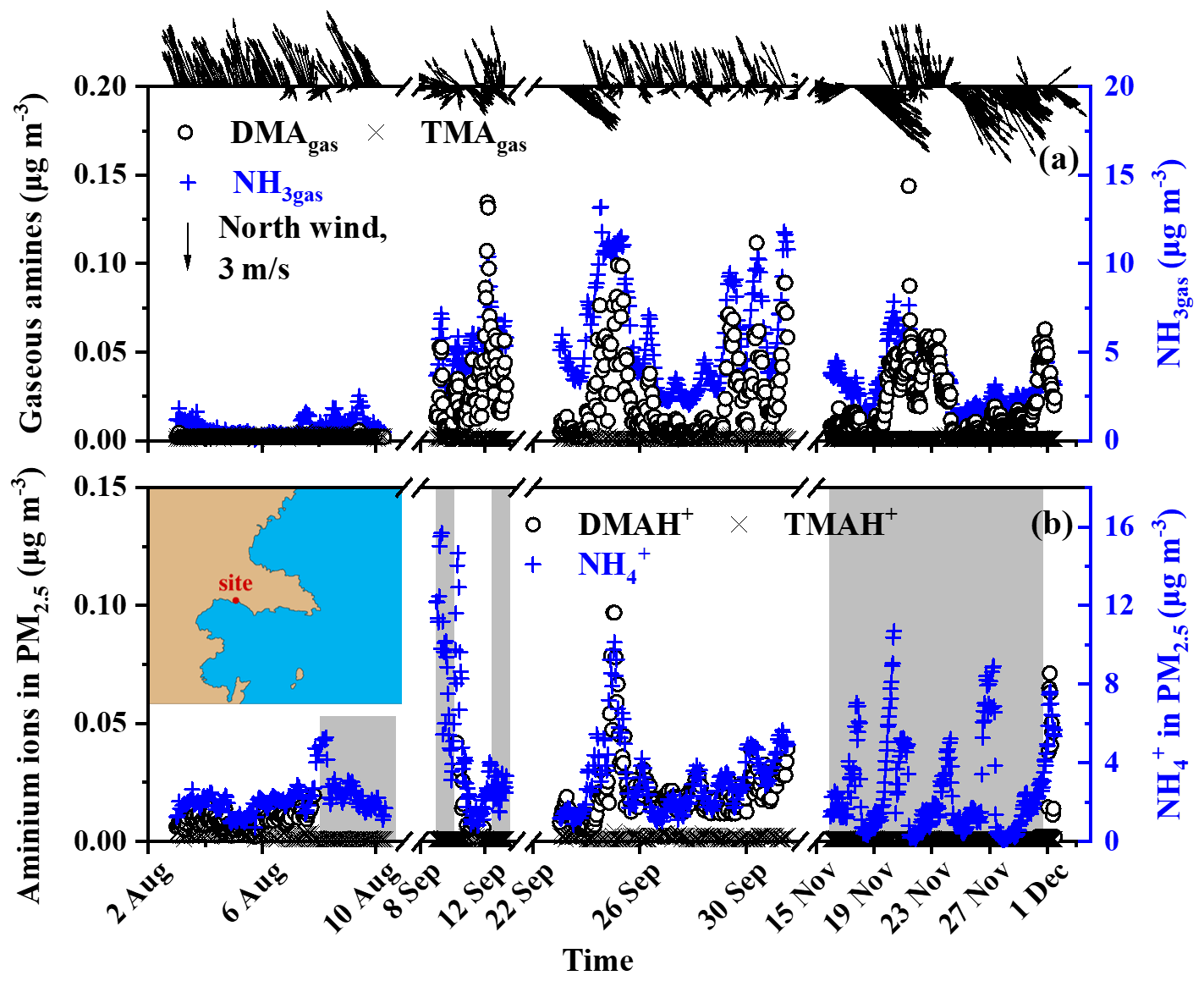
Figure 1Temporal variations in the concentrations of NH3gas, gaseous amines, and their counterparts in PM2.5 at a coastal site during three seasons of 2019. (a) NH3gas and gaseous amines; (b) counterparts in PM2.5; wind speed and direction superimposed on the top of (a); a map of the sampling site superimposed on (b); the missing data regarding aminium ions in the PM2.5 shading in the gray shadow were due to occasional K+ interference (b).
3.1 Temporal variations in the concentrations of basic gases and their PM2.5 counterparts in the coastal atmosphere
Before analyzing the basic gases and their counterparts in the marine atmosphere, we initially presented their continental concentrations at the coastal site facing the Yellow Sea as important evidence to facilitate the analysis of the contributors to these species in the marine atmosphere. Figure 1a and b show that the TMAgas and TMAH+ concentrations in PM2.5 were mostly below the detection limit, varying at approximately (average ± standard deviation), regardless of the presence of offshore or onshore winds during short-term measurements in the three seasons of 2019. The DMAgas and DMAH+ concentrations varied at 0.018±0.021 and , respectively, which were approximately 1 order of magnitude larger than those of TMAgas and TMAH+. TMAgas and TMAH+ concentrations in the upwind continental and coastal atmospheres were substantially lower than those reported in the literature, by up to a few tens of ng m−3 (Ge et al., 2011). However, Gibb et al. (1999) reported a low average TMAgas (0.5 ng m−3) and particulate TMAH+ (0.5 ng m−3) in the marine atmosphere over the Arabian Sea on 16 November to 19 December 1994. Xie et al. (2018) reported that TMAH+ concentrations were comparable to those of DMAH+ in atmospheric particles collected at two other coastal sites located approximately 20 km from the study area, as listed in Table S1 in the Supplement. The cause of this change is beyond the scope of this study but may be due to the large decrease in manure application, based on our recent survey in the Qingdao area.
The DMAgas and DMAH+ concentrations in PM2.5 with offshore winds from the north were substantially higher than those with onshore winds from the south or southeast (top of Fig. 1a), suggesting that their continental emissions and related secondary sources were stronger. Moreover, the concentrations of DMAgas and DMAH+ were moderately correlated with those of NH3gas and , namely, (R2=0.79, P<0.01) and (R2=0.84, P<0.01). Generally, the DMAgas and DMAH+ concentrations were approximately 1/200 of those of the corresponding NH3gas and .
3.2 Spatiotemporal variations in the concentrations of basic gases over the seas
Throughout Campaign A, the TMAgas concentrations varied at approximately 0.031±0.009 µg m−3 (Fig. 2a–c), with three peaks occurring at 4–5 d intervals (gray shadowing in Fig. 2c). Peaks 1 and 2 were generally associated with offshore winds, while peak 3 was mostly associated with onshore winds (Fig. 2b). The peaks lasted from tens to dozens of hours and were not induced by the onboard dew evaporation at sunrise. For example, the highest value (0.060 µg m−3) occurred at 23:00 LT (local time; UTC+08:00) on 16 December. The observed TMAgas concentrations were 1 order of magnitude higher than those measured in the coastal atmosphere during the summer, fall, and winter. This suggested that the TMAgas observed during Campaign A was largely derived from marine sources rather than from long-range continental transport. The same conclusion can be drawn by analyzing the three peaks of TMAgas and its temporal variations during the port-anchoring period. For example, during peak 1 (Fig. 2a), the concentrations of TMAgas increased by approximately 100 % from 20:00 LT on 9 December to 11:00 LT on 10 December, with an approximately 30 % decrease in the non-sea-salt (nss-) concentration (from 22 to 16 µg m−3; Fig. 2b). Moreover, the peaks in the TMAgas concentrations corresponded to troughs in the nss- concentrations during peak 3, as shown in Fig. 2c and d. The self-vessel emissions of nss- in PM2.5 were negligible because of the use of low-sulfur diesel, which is discussed later. The increased nss- concentrations in PM2.5 may be a good indicator of continental transport and vice versa.
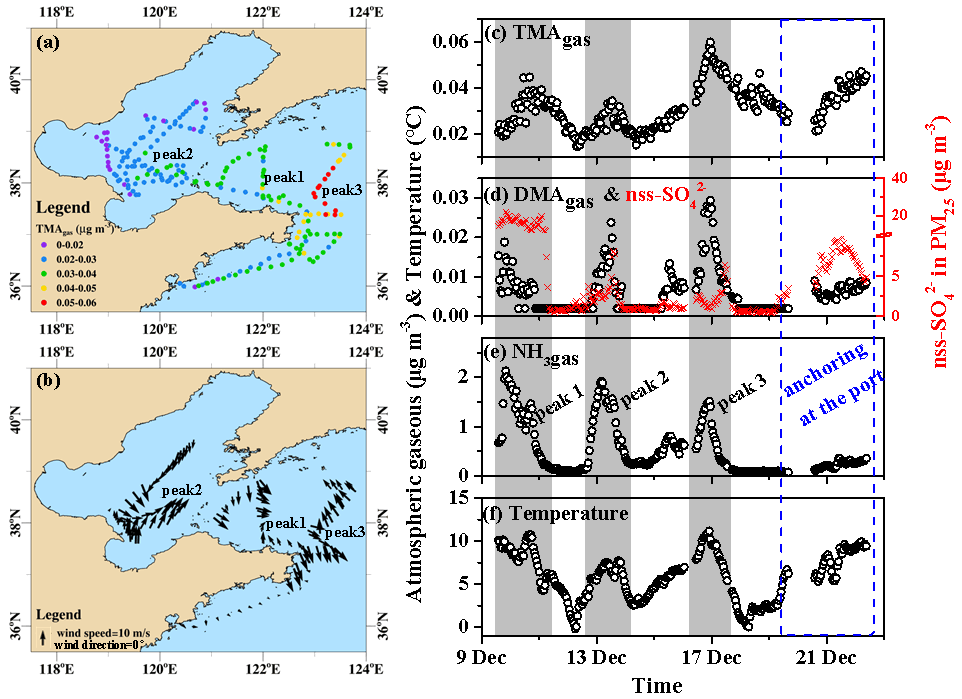
Figure 2Spatiotemporal variations in the concentrations of basic gases and other parameters during cruise campaigns in the Yellow and Bohai seas on 9–22 December 2019. (a) Mapping TMAgas by concentration; (b) mapping onboard recorded wind speeds and directions; time series of (c) TMAgas, (d) DMAgas, (e) NH3gas, and (f) ambient air temperature recorded onboard. The time series of nss- in PM2.5 were shown as indicators of anthropogenic air pollutants in (d); not all data were shown in (b) to avoid clustering.
The concentrations of DMAgas varied at approximately (Fig. 2d) and were significantly higher than those of TMAgas (P<0.01). Unlike TMAgas, continental transport was likely an important contributor to the DMAgas and NH3gas observed in the marine atmosphere, particularly during peak 1, when higher nss- concentrations were observed in PM2.5 (Fig. 2c–e). The DMAgas and NH3gas concentrations were negatively correlated with those of TMAgas during peak 1, namely, R2=0.35 (P<0.01) between TMAgas and DMAgas and R2=0.17 (P<0.01) between TMAgas and NH3gas. This suggested that most of the DMAgas and NH3gas were likely derived from continental transport rather than marine sources. During peak 2, increased TMAgas, DMAgas, and NH3gas concentrations were concurrently observed with increasing nss- concentrations, suggesting that both marine emissions and continental transport simultaneously contributed to the observed DMAgas and NH3gas. During the port-anchoring period from 20 to 22 December, the DMAgas and NH3gas concentrations varied slightly and were moderate and low, respectively. However, the TMAgas concentrations continuously increased by over 100 % as the ambient temperature increased (Fig. 2c and f). Additionally, the nss- concentrations of PM2.5 varied greatly and followed a bell-shaped pattern during the port-anchoring period.
Additionally, the NH3gas concentrations varied at approximately 0.53±0.53 µg m−3 from 9 to 22 December. The variation narrowed to approximately 0.24±0.07 µg m−3 during the port-anchoring period from 20 to 22 December. When the data during Campaign A were used for the analysis, the NH3gas concentrations were significantly correlated with those of DMAgas, namely, (R2=0.71, P<0.01). However, there was no correlation between NH3gas and TMAgas concentrations.
3.3 Spatiotemporal variations in the aminium and ion concentrations of PM2.5 over the seas
Figure 3a–f show the spatiotemporal variations in the TMAH+, DMAH+, and concentrations of PM2.5 throughout Campaign A from 9 to 22 December, during which the TMAH+ concentrations varied greatly at approximately 0.28±0.18 µg m−3. However, they narrowed at approximately 0.21±0.04 µg m−3 during the port-anchoring period. The TMAH+ concentrations generally increased from 0.13±0.05 µg m−3 on 9 December to 0.46±0.05 µg m−3 on 16 December (Fig. 3a) and subsequently decreased to approximately 0.2 µg m−3 thereafter, excluding some strong peaks from 0.62 to 1.24 µg m−3 at 03:00–05:59 LT and from 1.02 to 1.81 µg m−3 at 14:00–16:59 LT on 18 December (gray shadowing representing peak 4 in Fig. 3a–d). The peaks reproduced the episodes observed in the marine atmosphere over the Yellow Sea in May 2012 (Hu et al., 2015) and were repeatedly observed during Campaign B (Gao et al., 2021). However, they were not observed in several other marine cruise campaigns conducted across the marginal seas of China and the northwestern Pacific Ocean (Hu et al., 2018; Xie et al., 2018).
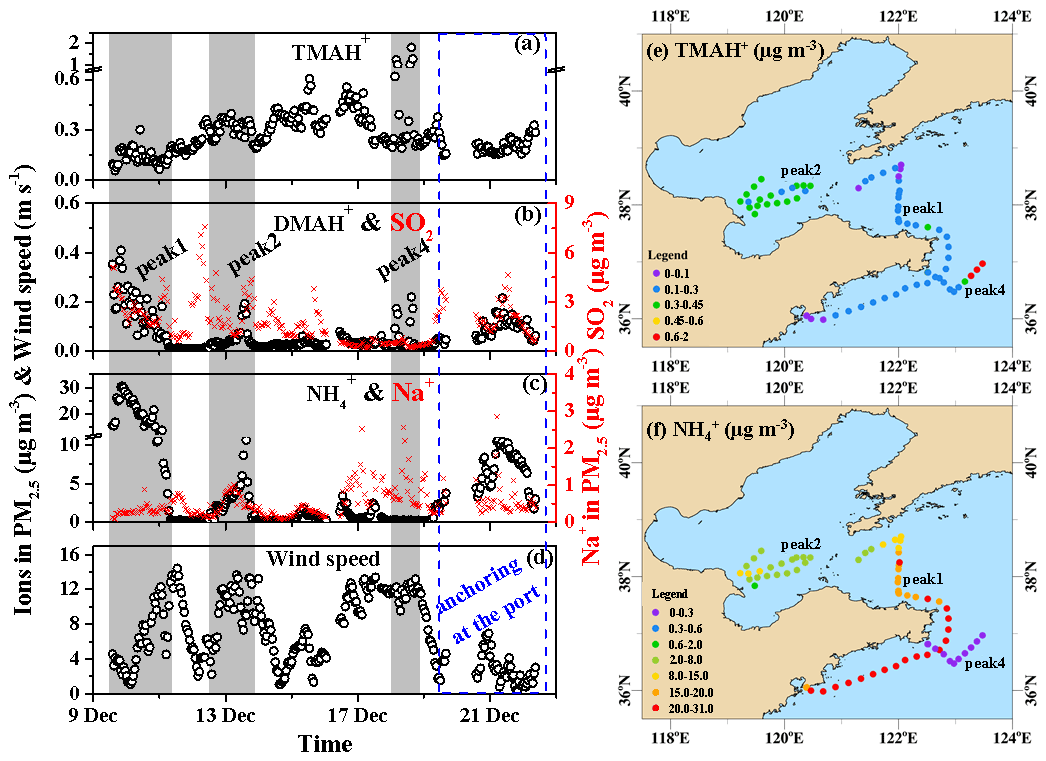
Figure 3Spatiotemporal variations in the aminium ions and concentrations of PM2.5 and other parameters during cruise campaigns over the Yellow and Bohai seas on 9–22 December 2019 (time series of (a) TMAH+, (b) DMAH+, and (c) in PM2.5; (d) wind speeds (WS); (e) mapping of the TMAH+ in concentration; (f) mapping of the concentration. The time series of SO2 is shown as an indicator in (b); that of Na+ in PM2.5 is shown as an indicator of sea-spray aerosols in (c). To better show the spatiotemporal distributions of TMAH+ and during peaks 1, 2, and 4, only the data during periods shaded in (a–d) were used in (e) and (f) to avoid clustering).
As the TMAH+ concentrations were approximately 2 orders of magnitude higher than those observed at the coastal site during the three seasons of 2019, the observed TMAH+ was largely derived from marine sources. The TMAH+ concentrations followed a spatiotemporal pattern that clearly differed from those of DMAH+ and , while the latter two ions exhibited a similar spatiotemporal pattern during most periods of Campaign A (Fig. 3a–c). A significant negative correlation (P<0.01) was observed between the concentrations of TMAH+ and in PM2.5 (not shown). The spatiotemporal pattern of the TMAH+ concentration also significantly differed from those of nss- (Fig. 2d) and SO2 (Fig. 3b), which are regarded as tracers of long-range transported continental pollutants and fresh-vessel plumes. For example, extremely strong TMAH+ peaks occurred concurrently with low nss-, , and SO2 concentrations accompanied by high Na+ concentrations under high wind speeds, which are common indicators of sea-spray aerosols (Feng et al., 2017). Moreover, the TMAH+ concentrations were approximately 1 order of magnitude larger than those of TMAgas, and no significant correlation was observed between them (P>0.05). This suggests that the observed TMAH+ may not be derived from the neutralization reactions of TMAgas with acids in the marine atmosphere and may have been derived from primary sea-spray organic aerosols (Hu et al., 2015, 2018). Primary sea-spray organic aerosols mainly contain primary and degraded biogenic organics (Ault et al., 2013; Prather et al., 2013; Quinn et al., 2015; Dall'Osto et al., 2019).
The DMAH+ concentrations varied at approximately from 9 to 22 December; however, they varied at approximately 0.10±0.04 µg m−3 during the port-anchoring period. The 25th percentile value of DMAH+ during Campaign A was 0.021 µg m−3, suggesting a low background concentration in the marine area. The DMAH+ concentrations were significantly correlated with those of (R2=0.71, P<0.01; data not shown). When the data obtained at 03:00–05:59 LT and 14:00–16:59 LT on 18 December (strong peaks of TMAH+ with a simultaneous increase in DMAH+) were removed for correlation, the R2 value improved to 0.78. Unlike TMAH+, the observed DMAH+ may have been partially derived from acid–base neutralization reactions in the ambient air in addition to the primary sea-spray organic aerosols. For example, a large increase in DMAH+ concentrations occurred concurrently with strong peaks in the TMAH+ concentrations (gray shadowed peak 4 in Fig. 3a and b).
The concentrations of PM2.5 varied greatly at approximately 4.7±7.2 µg m−3 during Campaign A (Fig. 3c). However, the 25th percentile values were as low as 0.21 µg m−3, suggesting low marine background values. The 50th percentile value was also only 1.2 µg m−3, which was considerably smaller than the average owing to the presence of strong peaks in concentrations. The increased concentrations associated with and nss- during Campaign A were likely due to the long-range transport from the upwind continents.

Figure 4Correlations between the concentrations of basic gases and ambient temperature. (a) TMAgas; (b) DMAgas; (c) NH3. The colored bar represents different wind speeds; full symbols represent the data observed throughout the campaign, excluding the period from 15:00 LT on 16 December to 01:00 LT on 19 December 2019.
4.1 Effects of temperature on the observed basic gases in the marine atmosphere
As mentioned above, the observed TMAgas likely originated from marine sources. We plotted the concentrations of TMAgas against the ambient air temperature (T) in Fig. 4a, which generally increased with increasing T. We further separated the average hourly wind speed (WS) into three categories: WS≤5.0, , and . At , the data obtained from 15:00 LT on 16 December to 01:00 LT on 19 December, including peaks 3 and 4, were separately considered as half-full symbols in Fig. 4a. The concentrations of TMAgas (half-full symbols) generally exceeded those of the other gases at the same T, with which they exhibited a moderately good exponent correlation ( with R2=0.72). From 15:00 LT on 16 December to 01:00 LT on 19 December, stronger emission potentials of TMAgas to the marine atmosphere were expected in the corresponding marine zone. However, the measured concentrations of TMAH+ and seawater pH in surface seawater are required to confirm this.
Following the same approach, the DMAgas and NH3gas concentrations were plotted against T, as shown in Fig. 4b and c, respectively. These values generally increased with increasing T. The NH3gas concentrations (half-full symbols) were strongly correlated with T ( with R2=0.96). As lower concentrations of nss-, , and SO2 were generally observed simultaneously, the continental transport of NH3gas was greatly reduced; therefore, the observed NH3gas was mainly derived from the seas. Therefore, the seas were the net source of NH3gas at the time of measurement. However, at the same T, the NH3gas concentrations (half-full symbols) were generally lower than those during other periods in this study. The concentrations of in surface seawater may have been lower at the time of measurement. However, this may not be the case, as higher concentrations of TMAH+ were expected. Alternatively, the continental transport of NH3gas may have made an important contribution to the observed NH3gas during most other periods when the seas were the net NH3gas sink.
DMAgas exhibited an extremely good exponent correlation with T (half-full symbols) at the measurement time ( with R2=0.91). At the same T, the DMAgas concentrations (half-full symbols) were not always higher or lower than the others. We considered these two hypotheses: in hypothesis 1, the observed DMAgas concentrations exceeded those predicted by the regression equation using the ambient T as the input; the seas were the net sinks of DMAgas. In hypothesis 2, including all the others, measurements of DMAH+ in the surface seawater were required to confirm whether the seas were net sources or sinks of DMAgas.
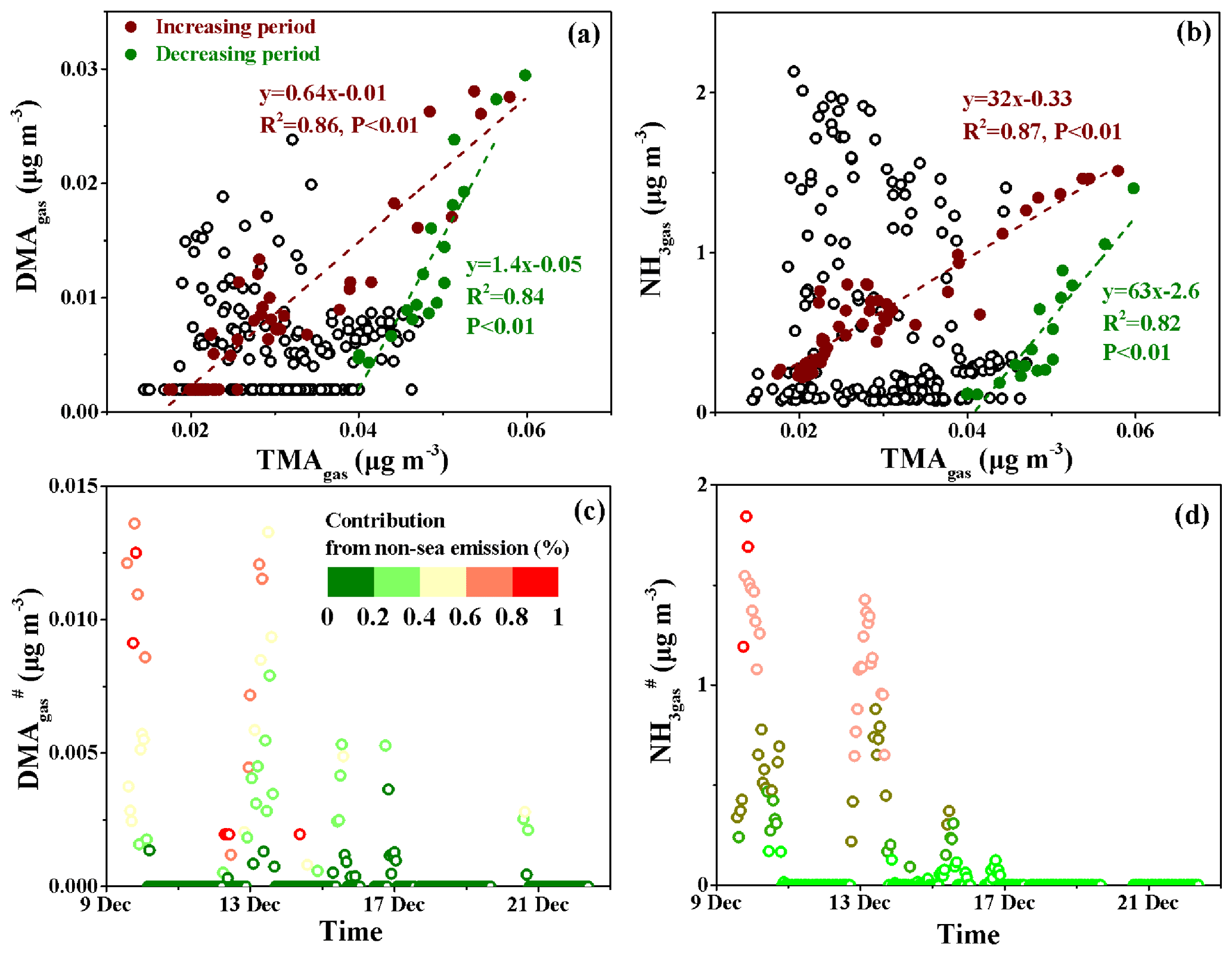
Figure 5Correlations of DMAgas and NH3gas with TMAgas and time series of the calculated DMA and NH. (a) DMAgas vs. TMAgas; (b) NH3gas vs. TMAgas; (c) DMA; (d) NH. The colored bars in (c) and (d) represent the percentages of transported DMA and NH in each corresponding observed value.
4.2 Estimating the sea-derived DMAgas and NH3gas in the marine atmosphere
To estimate the sea-derived DMAgas and NH3gas concentrations in the marine atmosphere, we plotted the DMAgas and NH3gas concentrations against TMAgas, as shown in Fig. 5a and b. The purple-red and dark-green markers represent the data obtained, with increasing concentrations of the three species from 10:00 LT on 14 December to 23:00 LT on 16 December (increasing period) and with decreasing concentrations from 23:00 LT on 16 December to 19:59 LT on 17 December (decreasing period) during peak 3, respectively; these were analyzed separately. A good correlation was obtained between DMAgas and TMAgas during the increasing period (, R2=0.86, and P<0.01). The good correlation suggested that DMAgas was likely released with TMAgas from the seawater and facilitated the estimation of non-sea-derived DMAgas (DMA) concentrations using the regression equation. We assumed that any data beyond the purple-red dashed line reflected the non-sea-derived DMAgas, which can be attributed to continental transport. Therefore, we assumed that the DMA concentrations were equal to the observed values of DMAgas minus the predicted values obtained using ; the calculated DMA values are shown in Fig. 5c. During peak 1, the calculated DMA contributed over 40 % of the observed DMAgas for 12 h. Similar calculated results for DMA were obtained during peak 2.
However, the equation for the decreasing period was as follows: , R2=0.84, and P<0.01. The decreasing R2 value and the increasing slope suggest that the TMAH+ in the surface seawater may decompose into DMAH+ to different extents (Lidbury et al., 2015a, b; Xie et al., 2018). The two regression curves (purple-red and dark-green dashed lines in Fig. 5a and b) created a large triangular zone that likely reflected the different ratios of DMATMAgas in primary marine emissions on the cruise route. Based on the triangular zone in Fig. 5a, the aforementioned calculations should be considered the lower limit of DMA.
The same approach was employed to analyze the NH3gas results, as shown in Fig. 5b and d. During peak 1, the calculated non-sea-derived NH3gas () contributed over 40 % of the observed NH3gas for 17 h. During peak 2, the calculated contributed over 40 % of the observed NH3gas for 24 h.
Overall, the DMA and concentrations varied at approximately 0.001±0.002 and 0.18±0.39 µg m−3, respectively. The calculated average DMA and values accounted for 16 % and 34 % of the observed averages of each species, respectively. The estimations suggested an appreciable continental contribution to the observed DMAgas and NH3gas during Campaign A.
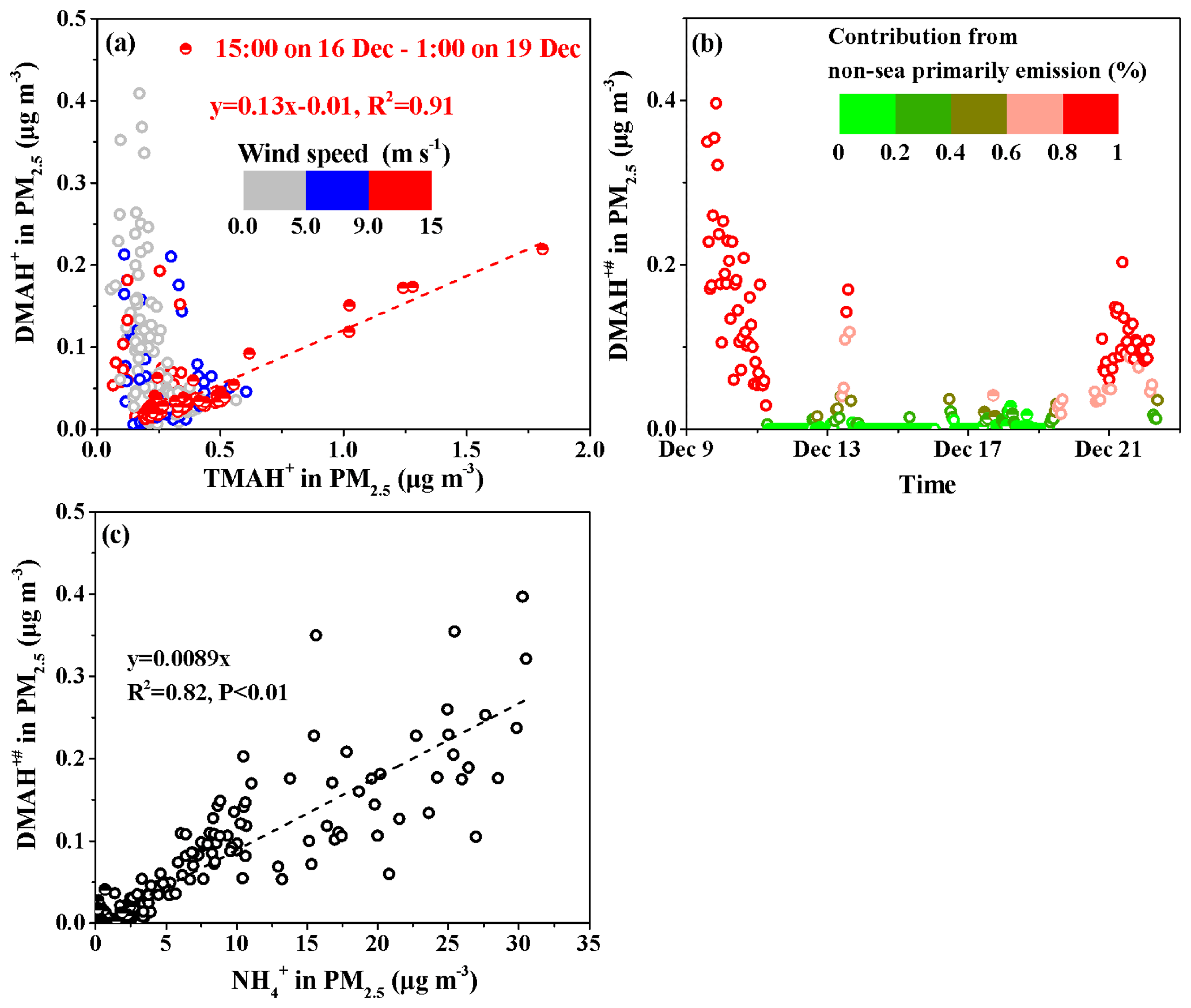
Figure 6Correlation analyses of different variables in PM2.5 and the time series of the calculated DMAH+# in PM2.5. (a) DMAH+ vs. TMAH+; (b) time series of DMAH+#; (c) DMAH+# vs. .
4.3 Estimation of non-sea-spray particulate DMAH+ in the marine atmosphere
We plotted the concentrations of DMAH+ against those of TMAH+ in PM2.5 (Fig. 6a) using the data obtained from 15:00 LT on 16 December to 01:00 LT on 19 December (, R2=0.91, P<0.01). During this period, largely increased concentrations of DMAH+ and TMAH+ were observed under high wind speeds of 9–13 m s−1. The good correlation suggested that the observed DMAH+ was likely released with TMAH+ as amine-containing sea-spray aerosols in the atmosphere and facilitated the calculation of sea-derived DMAH+ using TMAH+ as a tracer of sea-spray aerosols. Thus, the non-sea-derived DMAH+ concentrations in PM2.5, marked as DMAH+#, were assumed to be equal to the observed DMAH+ values minus the predicted values (sea-derived DMAH+) using the regression equation. The calculated DMAH+# values are shown in Fig. 6b. The DMAH+# concentrations varied at approximately 0.042±0.070 µg m−3 throughout Campaign A, during which the calculated average DMAH+# accounted for 65 % of the observed average. Additionally, the calculated DMAH+# values accounted for over 80 % of the observed values in 26 % of the Campaign A period. The estimations suggested that the observed DMAH+ originated predominantly from long-range continental transport and/or secondary formation in the marine atmosphere. The analysis was supported by the good correlation between the concentrations of DMAH+# and those of , namely, (R2=0.82, P<0.01; Fig. 6c). The slope of 0.0089 was approximately 50 % larger than that obtained in the coastal atmosphere (0.0059), suggesting more DMAgas partitioning in PM2.5 in the marine atmosphere than in the coastal atmosphere (Pankow, 2015; Xie et al., 2018).
Moreover, the decomposition of TMAH+ to DMAH+ may have occurred in surface seawater and/or the marine atmosphere, to an extent, and the estimated DMAH+# should be considered the upper limit. Notably, the and TMAH+ concentrations were negatively correlated during Campaign A, and no primary particulate from sea-spray aerosols was identified.
4.4 Formation and chemical conversion of aminium ions in the transported and self-vessel SO2 plumes
When the sea-spray particulate DMAH+ was deducted, the increased concentrations of DMAH+# were generally associated with increased nss- and SO2 concentrations. Combining this with the moderate correlation between DMAH+# and , we inferred that DMAH+# likely originated from concurrent secondary formation with . However, we separated the air pollutant plumes into two groups. Group 1 represented an increase in nss- and together with SO2, while group 2 represented an increase in SO2 without increases in nss- and . Group 1 likely reflected the transport of aged air pollutant plumes from the continents, while group 2 may reflect self-vessel SO2 plumes. As shown in Figs. 6b and 3b–c, the concentrations of DMAH+# and in the self-vessel SO2 plumes did not increase in the intervals between peaks 1 and 2 and between peaks 2 and 3. Therefore, no fresh formation of DMAH+# and in the self-vessel emissions was detected. However, the concentrations of TMAH+ decreased in some self-vessel SO2 plumes. The TMAH+ concentrations were approximately 1 order of magnitude higher than those of TMAgas in the marine atmosphere. Assuming that the decreased TMAH+ was released from PM2.5 to the gas phase, a simultaneous large spike in TMAgas should be observed. However, this was not the case, as shown in Fig. 1c. The decreased TMAH+ may persist in PM2.5 but could not be detected by AIM-IC.
In continental China upwind of the Yellow Sea, the TMAgas and TMAH+ concentrations in PM2.5 were extremely low (0.001±0.001 µg m−3), mostly below the detection limit of the AIM-IC. Considering the observations as a reference, the largely increased TMAgas (0.031±0.009 µg m−3) and particulate TMAH+ (0.28±0.18 µg m−3) concentrations in the marine atmosphere were attributed to marine emissions. Therefore, TMAgas and particulate TMAH+ can be used as unique tracers to quantify the marine emissions of DMAgas, NH3gas, and particulate DMAH+ as well as the long-range transport from upwind continental China.
Through comprehensive comparison and correlation analyses, the high concentrations of TMAH+ in PM2.5 observed over the Yellow and Bohai seas, with episodic hourly averages exceeding 1 µg m−3, were inferred to originate from strong primary sea-spray aerosol emissions. Moreover, the TMAgas concentrations generally increased with increasing ambient temperature and sea surface wind speeds, suggesting that the observed TMAgas was likely released from the surface seawater. However, the TMAgas concentrations were substantially lower than those of particulate TMAH+ and were not significantly correlated. Although different mechanisms have been reported in the literature for the release of TMAgas and particulate TMAH+ from the seas, the lack of a significant correlation between them was surprising and was explored in the companion study.
The DMAgas and NH3gas concentrations varied at approximately 0.006±0.006 and 0.53±0.53 µg m−3 during Campaign A, wherein at least 16 % and 34 % of the observational values were derived from continental transport, respectively. The sea-derived DMAgas and NH3gas were likely released with TMAgas as they peaked simultaneously. The DMAH+ concentrations in PM2.5 varied at approximately 0.065±0.068 µg m−3 during Campaign A, 65 % of which was derived from continental transport.
Our analysis results did not support the occurrence of photolysis of marine organic nitrogen to generate NH3gas in the marine atmosphere during winter, as there was no correlation between the sea-derived NH3gas and particulate TMAH+ concentrations. Additionally, peaks 2 and 3 of NH3gas persisted for dozens of hours under strong winds and were therefore unlikely to be derived from seabird emissions. A good exponent correlation was observed between the observed NH3gas concentrations and T during the period without continental air pollutant transport, suggesting that the observed NH3gas was released from seawater. NH3 emissions from seabirds were unlikely contributors to the observed NH3gas in the marine atmosphere during winter; however, this may not have been the case during other seasons.
Additionally, no formation of particulate and DMAH+ in the self-vessel SO2 plume was observed in the marine atmosphere. However, the particulate TMAH+ concentration clearly decreased in the self-vessel SO2 plume without a simultaneous increase in the TMAgas concentration. Chemical conversion of particulate TMAH+ likely occurred in the plume, while AIM-IC could not detect the products. This requires further investigation.
The datasets related to this work can be accessed via https://doi.org/10.17632/3kpnkkr55c.1 (Chen, 2021).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-16413-2021-supplement.
XY designed the research. DC carried out the field measurements and analyzed the data. XY helped in interpretation of the results. All the authors wrote the paper and contributed toward improving the paper.
The authors declare that they have no conflict of interest.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This research is supported by the Natural Science Foundation of China (grant no. 41776086), the National Key Research and Development Program of China (grant no. 2016YFC0200504), and the Fundamental Research Funds for the Central Universities (202072002).
This research is supported by the Natural Science Foundation of China (grant no. 41776086), the National Key Research and Development Program of China (grant no. 2016YFC0200504), and the Fundamental Research Funds for the Central Universities (grant no. 202072002).
This paper was edited by Maria Kanakidou and reviewed by three anonymous referees.
Almeida, J., Schobesberger, S., Kürten, A., Ortega, I. K., Kupiainen-Määttä, O., Praplan, A. P., Adamov, A., Amorim, A., Bianchi, F., Breitenlechner, M., David, A., Dommen, J., Donahue, N. M., Downard, A., Dunne, E., Duplissy, J., Ehrhart, S., Flagan, R. C., Franchin, A., Guida, R., Hakala, J., Hansel, A., Heinritzi, M., Henschel, H., Jokinen, T., Junninen, H., Kajos, M., Kangasluoma, J., Keskinen, H., Kupc, A., Kurtén, T., Kvashin, A. N., Laaksonen, A., Lehtipalo, K., Leiminger, M., Leppä, J., Loukonen, V., Makhmutov, V., Mathot, S., McGrath, M. J., Nieminen, T., Olenius, T., Onnela, A., Petäjä, T., Riccobono, F., Riipinen, I., Rissanen, M., Rondo, L., Ruuskanen, T., Santos, F. D., Sarnela, N., Schallhart, S., Schnitzhofer, R., Seinfeld, J. H., Simon, M., Sipilä, M., Stozhkov, Y., Stratmann, F., Tomé, A., Tröstl, J., Tsagkogeorgas, G., Vaattovaara, P., Viisanen, Y., Virtanen, A., Vrtala, A., Wagner, P. E., Weingartner, E., Wex, H., Williamson, C., Wimmer, D., Ye, P., Yli-Juuti, T., Carslaw, K. S., Kulmala, M., Curtius, J., Baltensperger, U., Worsnop, D. R., Vehkamäki, H., and Kirkby, J.: Molecular understanding of sulphuric acid–amine particle nucleation in the atmosphere, Nature, 502, 359–363, https://doi.org/10.1038/nature12663, 2013.
Ault, A. P., Moffet, R. C., Baltrusaitis, J., Collins, D. B., Ruppel, M. J., Cuadra-Rodriguez, L. A., Zhao, D., Guasco, T. L., Ebben, C. J., Geiger, F. M., Bertram, T. H., Prather, K. A., and Grassian, V. H.: Size-dependent changes in sea spray aerosol composition and properties with different seawater conditions, Environ. Sci. Technol., 47, 5603–5612, https://doi.org/10.1021/es400416g, 2013.
Burg, M. B. and Ferraris, J. D.: Intracellular organic osmolytes: Function and regulation, J. Biol. Chem., 283, 7309–7313, https://doi.org/10.1074/jbc.R700042200, 2008.
Carpenter, L. J., Archer, S. D., and Beale, R.: Ocean-atmosphere trace gas exchange, Chem. Soc. Rev., 41, 6473–6506, https://doi.org/10.1039/C2CS35121H, 2012.
Chen, D.: Amines and ammonia concentrations in the atmospheres datasets, V1, Mendeley Data [data set], https://doi.org/10.17632/3kpnkkr55c.1, 2021.
Chen, H., Varner, M. E., Gerber, R. B., and Finlayson-Pitts, B. J.: Reactions of methanesulfonic acid with amines and ammonia as a source of new particles in air, J. Phys. Chem. B, 120, 1526–1536, https://doi.org/10.1021/acs.jpcb.5b07433, 2016.
Clarke, A. D. and Porter, J. N.: Pacific marine aerosol: 2. Equatorial gradients in chlorophyll, ammonium, and excess sulfate during SAGA 3, J. Geophys. Res., 98, 16997–17010, https://doi.org/10.1029/92JD02481, 1993.
Dall'Osto, M., Airs, R. L., Beale, R., Cree, C., Fitzsimons, M. F., Beddows, D., Harrison, R. M., Ceburnis, D., O'Dowd, C., Rinaldi, M., Paglione, M., Nenes, A., Decesari, S., and Simó, R.: Simultaneous detection of alkylamines in the surface ocean and atmosphere of the Antarctic sympagic environment, ACS Earth Space Chem., 3, 854–862, https://doi.org/10.1021/acsearthspacechem.9b00028, 2019.
Deng, Y., Gao, T., Gao, H., Yao, X., and Xie, L.: Regional precipitation variability in East Asia related to climate and environmental factors during 1979–2012, Sci. Rep., 4, 5693, https://doi.org/10.1038/srep05693, 2014.
Dentener, F. J. and Crutzen, P. J.: A three-dimensional model of the global ammonia cycle, J. Atmos. Chem., 19, 331–369, https://doi.org/10.1007/BF00694492, 1994.
Facchini, M. C., Decesari, S., Rinaldi, M., Carbone, C., Finessi, E., Mircea, M., Fuzzi, S., Moretti, F., Tagliavini, E., Ceburnis, D., and O'Dowd, C. D.: Important source of marine secondary organic aerosol from biogenic amines, Environ. Sci. Technol., 42, 9116–9121, https://doi.org/10.1021/es8018385, 2008.
Feng, L., Shen, H., Zhu, Y., Gao, H., and Yao, X.: Insight into generation and evolution of sea-salt aerosols from field measurements in diversified marine and coastal atmospheres, Sci. Rep., 7, 41260, https://doi.org/10.1038/srep41260, 2017.
Gao, Y., Chen, D., Shen, Y., Gao, Y., Gao, H., and Yao, X.: Mapping gaseous amines, ammonia, and their particulate counterparts in marine atmospheres of China's marginal seas: Part 2 – spatiotemporal heterogeneity, causes and hypothesis, Atmos. Chem. Phys. Discuss. [preprint], https://doi.org/10.5194/acp-2021-301, in review, 2021.
Ge, X., Wexler, A. S., and Clegg, S. L.: Atmospheric amines – Part I. A review, Atmos. Environ., 45, 524–546, https://doi.org/10.1016/j.atmosenv.2010.10.012, 2011.
Gibb, S. W., Mantoura, R. F. C., and Liss, P. S.: Ocean-atmosphere exchange and atmospheric speciation of ammonia and methylamines in the region of the NW Arabian Sea, Global Biogeochem. Cy., 13, 161–178, https://doi.org/10.1029/98GB00743, 1999.
Guo, C., Zhang, G., Sun, J., Leng, X., Xu, W., Wu, C., Li, X., and Pujari, L.: Seasonal responses of nutrient to hydrology and biology in the southern Yellow Sea, Cont. Shelf Res., 206, 104207, https://doi.org/10.1016/j.csr.2020.104207, 2020.
Guo, T., Li, K., Zhu, Y., Gao, H., and Yao, X.: Concentration and size distribution of particulate oxalate in marine and coastal atmospheres – Implication for the increased importance of oxalate in nanometer atmospheric particles, Atmos. Environ., 142, 19–31, https://doi.org/10.1016/j.atmosenv.2016.07.026, 2016.
Hu, Q., Yu, P., Zhu, Y., Li, K., Gao, H., and Yao, X.: Concentration, size distribution, and formation of trimethylaminium and dimethylaminium ions in atmospheric particles over marginal seas of China, J. Atmos. Sci., 72, 3487–3498, https://doi.org/10.1175/JAS-D-14-0393.1, 2015.
Hu, Q., Qu, K., Gao, H., Cui, Z., Gao, Y., and Yao, X.: Large increases in primary trimethylaminium and secondary dimethylaminium in atmospheric particles associated with cyclonic eddies in the northwest Pacific Ocean, J. Geophys. Res.-Atmos., 123, 12133–12146, https://doi.org/10.1029/2018JD028836, 2018.
Jameson, E., Doxey, A. C., Airs, R., Purdy, K. J., Murrell, J. C., and Chen, Y.: Metagenomic data-mining reveals contrasting microbial populations responsible for trimethylamine formation in human gut and marine ecosystems, Microbial Genomics, 2, e000080, https://doi.org/10.1099/mgen.0.000080, 2016.
Johnson, M. T., Liss, P. S., Bell, T. G., Lesworth, T. J., Baker, A. R., Hind, A. J., Jickells, T. D., Biswas, K. F., Woodward, E. M. S., and Gibb, S. W.: Field observations of the ocean-atmosphere exchange of ammonia: Fundamental importance of temperature as revealed by a comparison of high and low latitudes, Global Biogeochem. Cy., 22, GB1019, https://doi.org/10.1029/2007GB003039, 2008.
Keene, W. C., Long, M. S., Pszenny, A. A. P., Sander, R., Maben, J. R., Wall, A. J., O'Halloran, T. L., Kerkweg, A., Fischer, E. V., and Schrems, O.: Latitudinal variation in the multiphase chemical processing of inorganic halogens and related species over the eastern North and South Atlantic Oceans, Atmos. Chem. Phys., 9, 7361–7385, https://doi.org/10.5194/acp-9-7361-2009, 2009.
Lidbury, I., Kimberley, G., Scanlan, D. J., Murrell, J. C., and Chen, Y.: Comparative genomics and mutagenesis analyses of choline metabolism in the marine Roseobacter clade, Environ. Microbiol., 17, 5048–5062, https://doi.org/10.1111/1462-2920.12943, 2015a.
Lidbury, I. D., Murrell, J. C., and Chen, Y.: Trimethylamine and trimethylamine N-oxide are supplementary energy sources for a marine heterotrophic bacterium: Implications for marine carbon and nitrogen cycling, ISME J., 9, 760–769, https://doi.org/10.1038/ismej.2014.149, 2015b.
Lutsch, E., Dammers, E., Conway, S., and Strong, K.: Long-range transport of NH3, CO, HCN, and C2H6 from the 2014 Canadian wildfires, Geophys. Res. Lett., 43, 8286–8297, https://doi.org/10.1002/2016GL070114, 2016.
Mao, J., Yu, F., Zhang, Y., An, J., Wang, L., Zheng, J., Yao, L., Luo, G., Ma, W., Yu, Q., Huang, C., Li, L., and Chen, L.: High-resolution modeling of gaseous methylamines over a polluted region in China: source-dependent emissions and implications of spatial variations, Atmos. Chem. Phys., 18, 7933–7950, https://doi.org/10.5194/acp-18-7933-2018, 2018.
McNaughton, C. S., Clarke, A. D., Howell, S. G., Moore II, K. G., Brekhovskikh, V., Weber, R. J., Orsini, D. A., Covert, D. S., Buzorius, G., Brechtel, F. J., Carmichael, G. R., Tang, Y., Eisele, F. L., Mauldin, R. L., Bandy, A. R., Thornton, D. C., and Blomquist, B.: Spatial distribution and size evolution of particles in Asian outflow: Significance of primary and secondary aerosols during ACE-Asia and TRACE-P, J. Geophys. Res.-Atmos., 109, D19S06, https://doi.org/10.1029/2003JD003528, 2004.
Müller, C., Iinuma, Y., Karstensen, J., van Pinxteren, D., Lehmann, S., Gnauk, T., and Herrmann, H.: Seasonal variation of aliphatic amines in marine sub-micrometer particles at the Cape Verde islands, Atmos. Chem. Phys., 9, 9587–9597, https://doi.org/10.5194/acp-9-9587-2009, 2009.
Pankow, J. F.: Phase considerations in the gas/particle partitioning of organic amines in the atmosphere, Atmos. Environ., 122, 448–453, https://doi.org/10.1016/j.atmosenv.2015.09.056, 2015.
Paulot, F., Jacob, D. J., Johnson, M. T., Bell, T. G., Baker, A. R., Keene, W. C., Lima, I. D., Doney, S. C., and Stock, C. A.: Global oceanic emission of ammonia: Constraints from seawater and atmospheric observations, Global Biogeochem. Cy., 29, 1165–1178, https://doi.org/10.1002/2015GB005106, 2015.
Prather, K. A., Bertram, T. H., Grassian, V. H., Deane, G. B., Stokes, M. D., Demott, P. J., Aluwihare, L. I., Palenik, B. P., Azam, F., Seinfeld, J. H., Moffet, R. C., Molina, M. J., Cappa, C. D., Geiger, F. M., Roberts, G. C., Russell, L. M., Ault, A. P., Baltrusaitis, J., Collins, D. B., Corrigan, C. E., Cuadra-Rodriguez, L. A., Ebben, C. J., Forestieri, S. D., Guasco, T. L., Hersey, S. P., Kim, M. J., Lambert, W. F., Modini, R. L., Mui, W., Pedler, B. E., Ruppel, M. J., Ryder, O. S., Schoepp, N. G., Sullivan, R. C., and Zhao, D.: Bringing the ocean into the laboratory to probe the chemical complexity of sea spray aerosol, P. Natl. Acad. Sci. USA, 110, 7550–7555, https://doi.org/10.1073/pnas.1300262110, 2013.
Quinn, P. K., Bates, T. S., Johnson, J. E., Covert, D. S., and Charlson, R. J.: Interactions between the sulfur and reduced nitrogen cycles over the central Pacific Ocean, J. Geophys. Res., 95, 16405–16416, https://doi.org/10.1029/JD095iD10p16405, 1990.
Quinn, P. K., Collins, D. B., Grassian, V. H., Prather, K. A., and Bates, T. S.: Chemistry and related properties of freshly emitted sea spray aerosol, Chem. Rev., 115, 4383–4399, https://doi.org/10.1021/cr500713g, 2015.
Sutton, M. A., Reis, S., Riddick, S. N., Dragosits, U., Nemitz, E., Theobald, M. R., Tang, Y. S., Braban, C. F., Vieno, M., Dore, A. J., Mitchell, R. F., Wanless, S., Daunt, F., Fowler, D., Blackall, T. D., Milford, C., Flechard, C. R., Loubet, B., Massad, R., Cellier, P., Personne, E., Coheur, P. F., Clarisse, L., van Damme, M., Ngadi, Y., Clerbaux, C., Skjøth, C. A., Geels, C., Hertel, O., Wichink Kruit, R. J., Pinder, R. W., Bash, J. O., Walker, J. T., Simpson, D., Horváth, L., Misselbrook, T. H., Bleeker, A., Dentener, F., and de Vries, W.: Towards a climate-dependent paradigm of ammonia emission and deposition, Philos. T. Roy. Soc. B, 368, 20130166, https://doi.org/10.1098/rstb.2013.0166, 2013.
Taubert, M., Grob, C., Howat, A. M., Burns, O. J., Pratscher, J., Jehmlich, N., von Bergen, M., Richnow, H. H., Chen, Y., and Murrell, J. C.: Methylamine as a nitrogen source for microorganisms from a coastal marine environment, Environ. Microbiol., 19, 2246–2257, https://doi.org/10.1111/1462-2920.13709, 2017.
Teng, X., Hu, Q., Zhang, L., Qi, J., Shi, J., Xie, H., Gao, H., and Yao, X.: Identification of major sources of atmospheric NH3 in an urban environment in northern China during wintertime, Environ. Sci. Technol., 51, 6839–6848, https://doi.org/10.1021/acs.est.7b00328, 2017.
Uematsu, M., Toratani, M., Kajino, M., Narita, Y., Senga, Y., and Kimoto, T.: Enhancement of primary productivity in the western North Pacific caused by the eruption of the Miyake-jima Volcano, Geophys. Res. Lett., 31, L06106, https://doi.org/10.1029/2003GL018790, 2004.
VandenBoer, T. C., Petroff, A., Markovic, M. Z., and Murphy, J. G.: Size distribution of alkyl amines in continental particulate matter and their online detection in the gas and particle phase, Atmos. Chem. Phys., 11, 4319–4332, https://doi.org/10.5194/acp-11-4319-2011, 2011.
van Pinxteren, M., Fiedler, B., van Pinxteren, D., Iinuma, Y., Körtzinger, A., and Herrmann, H.: Chemical characterization of sub-micrometer aerosol particles in the tropical Atlantic Ocean: Marine and biomass burning influences, J. Atmos. Chem., 72, 105–125, https://doi.org/10.1007/s10874-015-9307-3, 2015.
van Pinxteren, M., Fomba, K. W., van Pinxteren, D., Triesch, N., Hoffmann, E. H., Cree, C. H. L., Fitzsimons, M. F., von Tümpling, W., and Herrmann, H.: Aliphatic amines at the Cape Verde Atmospheric Observatory: Abundance, origins and sea-air fluxes, Atmos. Environ., 203, 183–195, https://doi.org/10.1016/j.atmosenv.2019.02.011, 2019.
Wang, B., Chen, Y., Zhou, S., Li, H., Wang, F., and Yang, T.: The influence of terrestrial transport on visibility and aerosol properties over the coastal East China Sea, Sci. Total Environ., 649, 652–660, https://doi.org/10.1016/j.scitotenv.2018.08.312, 2019.
Wentworth, G. R., Murphy, J. G., Benedict, K. B., Bangs, E. J., and Collett Jr., J. L.: The role of dew as a night-time reservoir and morning source for atmospheric ammonia, Atmos. Chem. Phys., 16, 7435–7449, https://doi.org/10.5194/acp-16-7435-2016, 2016.
Xie, H., Feng, L., Hu, Q., Zhu, Y., Gao, H., Gao, Y., and Yao, X.: Concentration and size distribution of water-extracted dimethylaminium and trimethylaminium in atmospheric particles during nine campaigns – Implications for sources, phase states and formation pathways, Sci. Total Environ., 631–632, 130–141, https://doi.org/10.1016/j.scitotenv.2018.02.303, 2018.
Yao, L., Garmash, O., Bianchi, F., Zheng, J., Yan, C., Kontkanen, J., Junninen, H., Mazon, S. B., Ehn, M., Paasonen, P., Sipilä, M., Wang, M., Wang, X., Xiao, S., Chen, H., Lu, Y., Zhang, B., Wang, D., Fu, Q., Geng, F., Li, L., Wang, H., Qiao, L., Yang, X., Chen, J., Kerminen, V.-M., Petäjä, T., Worsnop, D. R., Kulmala, M., and Wang, L.: Atmospheric new particle formation from sulfuric acid and amines in a Chinese megacity, Science, 361, 278–281, https://doi.org/10.1126/science.aao4839, 2018.
Yu, F. and Luo, G.: Modeling of gaseous methylamines in the global atmosphere: impacts of oxidation and aerosol uptake, Atmos. Chem. Phys., 14, 12455–12464, https://doi.org/10.5194/acp-14-12455-2014, 2014.
Yu, P., Hu, Q., Li, K., Zhu, Y., Liu, X., Gao, H., and Yao, X.: Characteristics of dimethylaminium and trimethylaminium in atmospheric particles ranging from supermicron to nanometer sizes over eutrophic marginal seas of China and oligotrophic open oceans, Sci. Total Environ., 572, 813–824, https://doi.org/10.1016/j.scitotenv.2016.07.114, 2016.
Zhao, Y., Zhang, L., Pan, Y., Wang, Y., Paulot, F., and Henze, D. K.: Atmospheric nitrogen deposition to the northwestern Pacific: seasonal variation and source attribution, Atmos. Chem. Phys., 15, 10905–10924, https://doi.org/10.5194/acp-15-10905-2015, 2015.
Zhou, S., Li, H., Yang, T., Chen, Y., Deng, C., Gao, Y., Chen, C., and Xu, J.: Characteristics and sources of aerosol aminiums over the eastern coast of China: insights from the integrated observations in a coastal city, adjacent island and surrounding marginal seas, Atmos. Chem. Phys., 19, 10447–10467, https://doi.org/10.5194/acp-19-10447-2019, 2019.
Zhu, J., Shi, J., Guo, X., Gao, H., and Yao, X.: Air-sea heat flux control on the Yellow Sea Cold Water Mass intensity and implications for its prediction, Cont. Shelf Res., 152, 14–26, https://doi.org/10.1016/j.csr.2017.10.006, 2018.
Zhu, Y., Li, K., Shen, Y., Gao, Y., Liu, X., Yu, Y., Gao, H., and Yao, X.: New particle formation in the marine atmosphere during seven cruise campaigns, Atmos. Chem. Phys., 19, 89–113, https://doi.org/10.5194/acp-19-89-2019, 2019.