the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 08 Feb 2021
| 08 Feb 2021
Low-NO atmospheric oxidation pathways in a polluted megacity
Mike J. Newland
Daniel J. Bryant
Rachel E. Dunmore
Thomas J. Bannan
W. Joe F. Acton
Ben Langford
James R. Hopkins
Freya A. Squires
William Dixon
William S. Drysdale
Peter D. Ivatt
Mathew J. Evans
Peter M. Edwards
Lisa K. Whalley
Dwayne E. Heard
Eloise J. Slater
Robert Woodward-Massey
Chunxiang Ye
Archit Mehra
Stephen D. Worrall
Asan Bacak
Carl J. Percival
C. Nicholas Hewitt
James D. Lee
Tianqu Cui
Jason D. Surratt
Xinming Wang
Alastair C. Lewis
Andrew R. Rickard
Jacqueline F. Hamilton
The impact of emissions of volatile organic compounds (VOCs) to the atmosphere on the production of secondary pollutants, such as ozone and secondary organic aerosol (SOA), is mediated by the concentration of nitric oxide (NO). Polluted urban atmospheres are typically considered to be “high-NO” environments, while remote regions such as rainforests, with minimal anthropogenic influences, are considered to be “low NO”. However, our observations from central Beijing show that this simplistic separation of regimes is flawed. Despite being in one of the largest megacities in the world, we observe formation of gas- and aerosol-phase oxidation products usually associated with low-NO “rainforest-like” atmospheric oxidation pathways during the afternoon, caused by extreme suppression of NO concentrations at this time. Box model calculations suggest that during the morning high-NO chemistry predominates (95 %) but in the afternoon low-NO chemistry plays a greater role (30 %). Current emissions inventories are applied in the GEOS-Chem model which shows that such models, when run at the regional scale, fail to accurately predict such an extreme diurnal cycle in the NO concentration. With increasing global emphasis on reducing air pollution, it is crucial for the modelling tools used to develop urban air quality policy to be able to accurately represent such extreme diurnal variations in NO to accurately predict the formation of pollutants such as SOA and ozone.
- Article
(2887 KB) - Full-text XML
-
Supplement
(2633 KB) - BibTeX
- EndNote





The atmosphere in polluted urban areas has a markedly different chemical composition to that in remote regions (e.g. rainforests). This can lead to changes in the chemical oxidation pathways for volatile organic compounds (VOCs), giving rise to the formation of different secondary pollutants. Oxidation by hydroxyl radicals (OH) is the dominant daytime sink for VOCs, leading to the formation of highly reactive peroxy radicals (RO2). In atmospheres with high concentrations of nitric oxide (NO), emitted by combustion sources such as vehicles, cooking, and energy generation, RO2 radicals react predominantly with NO (Orlando and Tyndall, 2012). This tends to break the initial VOC down to smaller, more oxidised VOCs and can also produce organic nitrates (RONO2). This pathway also produces NO2, the photolysis of which leads to ozone production. In contrast, in low-NO atmospheres RO2 react with other RO2, including hydroperoxyl radicals (HO2), or can isomerise or auto-oxidise to form different multi-functionalised oxygenated RO2 (Crounse et al., 2013). These low-NO pathways tend to maintain the original carbon skeleton. The large highly oxidised molecules formed can efficiently partition to the aerosol phase to yield secondary organic aerosol (SOA) (Bianchi et al., 2019), which often comprises a large fraction of submicron atmospheric particulate matter (PM) in many regions (Jimenez et al., 2009).
In the past 20 years, emissions, and hence atmospheric concentrations, of nitrogen oxides (NOx) have decreased in urban areas throughout the majority of the developed world. In urban areas this has been due to the following: improvements in vehicle emissions technologies; changes to residential heating; and, in many major European cities, restrictions on the types of vehicles that are allowed in certain areas at certain times of the day. In China, through the introduction of the Air Pollution Prevention and Control Action Plan in 2013 (Zhang et al., 2019) there has been a concerted effort to reduce pollutant emissions. Numerous pollution control measures have been introduced, including improved industrial emissions standards, the promotion of clean fuels instead of coal within the residential sector, improving vehicle emissions standards, and taking older vehicles off the road. In Beijing, 900 000 households have converted from using coal to cleaner technologies such as gas or electricity since 2013. These actions have led to a 32 % decrease in NO2 emissions since 2012 (Liu et al., 2016; Krotkov et al., 2016; Miyazaki et al., 2017). Most significant for NOx emissions however is the stringent vehicle control measures introduced within the last decade, accounting for 47 % of the total reduction in emissions for the city (Cheng et al., 2019). Such reductions in NOx emissions are expected to lead to an increased importance of low-NO oxidation pathways for VOCs in urban and suburban areas (e.g. Praske et al., 2018). This will lead to the production of a range of low-volatility multi-functionalised products, efficient at producing SOA, which have previously been found only in remote environments removed from anthropogenic influence.
Surface ozone in Beijing has increased through the 1990s and 2000s (Tang et al., 2009). The city regularly experiences daily peaks in the summertime of over 100 ppb (e.g. Wang et al., 2015). Such high-ozone episodes are a function of both chemistry and meteorology, with air masses coming from the mountainous regions to the northwest tending to bring in clean air low in ozone, while air masses coming from the densely populated regions to the south and west bring processed polluted air high in ozone (Wang et al., 2017). A number of modelling studies have concluded that the sources of the ozone during high-ozone episodes are a combination of both local production and regional transport (Wang et al., 2017; Liu et al., 2019).
Biogenic sources dominate global emissions of VOCs to the atmosphere, with the highly reactive VOC isoprene (2-methyl-1,3-butadiene) contributing ∼70 % by mass (Sindelarova et al., 2014). The gas- and aerosol-phase products of isoprene oxidation have been extensively characterised in the laboratory (Wennberg et al., 2018, and references therein). For isoprene, the low-NO oxidation pathway leads to low-volatility products, such as isoprene hydroperoxides (ISOPOOH), that can go on to form significant quantities of SOA via formation of isoprene epoxides (IEPOX) (Fig. 1) (Paulot et al., 2009; Surratt et al., 2010; Lin et al., 2012). The high-NO pathway can also form SOA via the formation of methacrolein (MACR), which can react further to form SOA constituents such as 2-methylglyceric acid (2-MGA) and corresponding oligomers (Kroll et al., 2006; Surratt et al., 2006, 2010; Nguyen et al., 2015a) (Fig. 1). Other significant contributors to isoprene SOA formed via the high-NO pathway include nitrates (e.g. ISOPONO2) and dinitrates (Schwantes et al., 2019). In this work, a suite of isoprene oxidation products, in both the gas and particle phases, are used as tracers of the changing atmospheric chemical environment throughout the daytime in Beijing.

Figure 1Formation pathways of isoprene oxidation products used as tracers of high- and low-NO chemistry in this work. Following reaction of the primary VOC, isoprene, with OH, a peroxy radical intermediate (ISOPOO) is formed. At low NO concentrations, ISOPOO reacts with HO2 (or other RO2), to yield hydroperoxide (ISOPOOH) isomers ((4,3)-ISOPOOH isomer is shown), which can be rapidly oxidised to isoprene epoxydiol (IEPOX) isomers. At high NO concentrations, ISOPOO reacts with NO, a minor product of which is an isoprene nitrate (ISOPONO2). One of the major products of the ISOPOO reaction with NO is methacrolein (MACR), the subsequent oxidation of which, in the presence of NO2, can lead to 2-methylglyceric acid (2-MGA) and its corresponding oligomers and organosulfates in the aerosol phase. Measurements of these products in the gas or aerosol phase can be used as tracers for the chemical environment in which they were formed.
The site was located at the Institute of Atmospheric Physics (IAP), between the 3rd Ring Road and 4th Ring Road. Measurements took place between 17 May and 24 June 2017. The site is typical of central Beijing, surrounded by residential and commercial properties and is near several busy roads. It is also close to several green spaces, including a tree-lined canal to the south and the Olympic Forest Park to the northeast. Isoprene mixing ratios were measured by dual-channel gas chromatography with flame ionisation detection (DC-GC-FID). IEPOX+ISOPOOH were observed using iodide chemical ionisation mass spectrometry. The sum of MACR + methyl vinyl ketone (MVK) ( 71.05) was measured using proton transfer mass spectrometry. Particle samples were collected onto filter papers at either 3-hourly or 1-hourly time periods, depending on pollution levels. Filters were extracted and analysed with a high-throughput method using ultrahigh-pressure liquid chromatography (LC) coupled to a Q Exactive Orbitrap mass spectrometer. Nitric oxide, NO, was measured by chemiluminescence with a Thermo Scientific Model 42i NOx analyser. Nitrogen dioxide, NO2, was measured using a Teledyne Model T500U cavity attenuated phase shift (CAPS) spectrometer. Ozone, O3, was measured using a Thermo Scientific Model 49i UV photometer.
2.1 Instrumentation
2.1.1 DC-GC-FID
Observations of VOCs were made using dual-channel gas chromatography with flame ionisation detection (DC-GC-FID). Air was sampled at 30 L min−1 at a height of 5 m, through a stainless-steel manifold (0.5 in. internal diameter). Subsamples of 500 mL were taken, dried using a glass condensation finger held at −40 ∘C, and then pre-concentrated using a Markes Unity 2 pre-concentrator on a multi-bed ozone precursor adsorbent trap (Markes International Ltd). These samples were then transferred to the gas chromatography (GC) oven for analysis following methods described by Hopkins et al. (2011).
2.1.2 CIMS
A time-of-flight chemical ionisation mass spectrometer (ToF-CIMS) (Lee et al., 2014; Priestley et al., 2018) using an iodide ionisation system was deployed. The experimental set-up of the University of Manchester ToF-CIMS has been previously described in Zhou et al. (2018). During the campaign, gas-phase backgrounds were established by regularly overflowing the inlet with dry N2 for 5 continuous minutes every 45 min and were applied consecutively. The overflowing of dry N2 will have a small effect on the sensitivity of the instrument to those compounds whose detection is water dependent. Here we find that due to the very low instrumental background for C5H10O3 and C5H9NO4, the absolute error remains small from this effect (<10 ppt in both reported measurements).
Field calibrations were regularly carried out using known-concentration formic acid gas mixtures made in a custom-made gas-phase manifold. A range of other species were calibrated for after the campaign, and relative calibration factors were derived using the measured formic acid sensitivity during these calibrations, as has been performed previously (Le Breton et al., 2018; Bannan et al., 2015). In addition to this, offline calibrations, prior to and after the fieldwork project, of a wide range of organic acids, HNO3, and Cl2 were performed to assess possible large-scale sensitivity changes over the measurement period. No significant changes were observed. Offline calibrations after the fieldwork campaign were performed specific to the isoprene oxidation species observed here. IEPOX (C5H10O3) synthesised by the Department of Environmental Sciences and Engineering, University of North Carolina, was specifically calibrated for. Aliquots of known concentrations of IEPOX (C5H10O3) were thermally desorbed into a known continuous flow of nitrogen. For C5H9NO4 there was no direct calibration source available and concentrations using the calibration factor of C5H10O3 are presented here. Absolute measurement uncertainties are estimated at 50 % for the presented IEPOX+ISOPOOH and ISOPONO2 (C5H9NO4) signals.
2.1.3 PTR-MS
A proton transfer reaction time-of-flight mass spectrometer (PTR-ToF-MS 2000, Ionicon Analytik GmBH, Innsbruck) was deployed at the base of the 325 m meteorological tower at the IAP field site. This instrument has been described in detail by Jordan et al. (2009). The PTR-ToF-MS was operated at a measurement frequency of 5 Hz and an ratio (where E represents the electric field strength and N the buffer gas density) in the drift tube of 130 Td. To enable accurate calibration of the mass scale, trichlorobenzene was introduced by diffusion into the inlet stream.
The instrument was switched between two inlet systems in an hourly cycle. For the first 20 min of each hour the proton transfer reaction mass spectrometer (PTR-MS) sampled from a gradient-switching manifold, and for the next 40 min the instrument subsampled a common flux inlet line running from the 102 m platform on the tower to the container in which the PTR-ToF-MS was housed. Gradient measurements were made from 3, 15, 32, 64, and 102 m with air sampled down 0.25 in. o.d. PFA lines and split between a 3 L min−1 bypass and 300 mL min−1 sample drawn to a 10 L stainless-steel container. During the gradient sampling period, the PTR-ToF-MS subsampled for 2 min from each container giving an hourly average concentration at each height. In this work, only data from the 3 m gradient height are discussed.
Zero air was generated using a platinum catalyst heated to 260 ∘C and was sampled hourly in the gradient-switching cycle. During the field campaign, the instrument was calibrated twice weekly using a 15-component 1 ppmv VOC standard (National Physical Laboratory, Teddington). The calibration gas flow was dynamically diluted into zero air to give a six-point calibration. The sensitivity for each mass was then calculated using a transmission curve. The maximum relative error for PTR-MS calibration using a relative transmission curve has been estimated to be 21 % (Taipale et al., 2008). Data were analysed using PTR-MS Viewer 3.
2.1.4 PM2.5 filter sampling and analysis
PM2.5 filter samples were collected using an Ecotech HiVol 3000 (Ecotech, Australia) high-volume air sampler with a selective PM2.5 inlet, with a flow rate of 1.33 m3 min−1. Filters were baked at 500 ∘C for 5 h before use. After collection, samples were wrapped in foil and then stored at −20 ∘C and shipped to the laboratory. Samples were collected at a height of 8 m, on top of a building in the IAP complex. Hourly samples were taken on 11 June 2017 between 08:00 and 17:00 local time (LT), with one further sample taken overnight. The extraction of the organic aerosol from the filter samples was based on the method of Hamilton et al. (2008). Initially, roughly an eighth of the filter was cut up into 1 cm2 pieces. Then added to the sample was 4 mL of LC–MS-grade H2O, and it was left for 2 h. The samples were then sonicated for 30 min. Using a 2 mL syringe, the water extract was then pushed through a 0.22 µm filter (Millipore) into another sample vial. An additional 1 mL of water was added to the filter sample and then extracted through the filter, to give a combined aqueous extract. This extract was then reduced to dryness using a vacuum solvent evaporator (Biotage, Sweden). The dry sample was then reconstituted in 1 mL 50:50 MeOH:H2O solution, ready for analysis.
The extracted filter samples and standards were analysed using ultra performance liquid chromatography–tandem mass spectrometry (UPLC–MS2), using an UltiMate 3000 UPLC instrument (Thermo Scientific, USA) coupled to a Q Exactive Orbitrap MS (Thermo Fisher Scientific, USA) with heated electrospray ionisation (HESI). The UPLC method uses a reverse-phase, 5 µm, 4.6×100 mm, Accucore column (Thermo Scientific, UK) held at 40 ∘C. The mobile phase consists of LC–MS-grade water and 100 % MeOH (Fisher Chemical, USA). The water was acidified using 0.1 % formic acid to improve peak resolution. The injection volume was 2 µL. The solvent gradient was held for a minute at 90:10 H2O:MeOH; the gradient then changed linearly to 10:90 H2O:MeOH over 9 min; it was then held for 2 min at this gradient before returning to 90:10 H2O:MeOH over 2 min and then held at 90:10 for the remaining 2 min, with a flow rate of 300 µL min−1. The mass spectrometer was operated in negative mode using full-scan MS2. The electrospray voltage was 4.00 kV, with capillary and auxiliary gas temperatures of 320 ∘C. The scan range was set between 50–750 . Organosulfates were quantified using an authentic standard of 2-MGA-OS obtained from the co-author Jason D. Surratt using the method described in Bryant et al. (2020).
2.1.5 OH measurements
The OH radical measurements were made from the roof of the University of Leeds Fluorescence Assay by Gas Expansion (FAGE) instrument container at the IAP field site. Two FAGE detection cells were housed in a weatherproof enclosure at a sampling height of approximately 4 m. OH and HO2 radicals were detected sequentially in the first cell (the HOx cell), whilst and total RO2 radical observations were made using the second FAGE cell (the ROx cell), which was coupled with a flow reactor to facilitate RO2 detection (Whalley et al., 2018). A Nd:YAG pumped Ti:Sapphire laser was used to generate 5 kHz pulsed tunable UV light at 308 nm and to excite OH via the Q1(1) transition of the A2Σ+, , band. On-resonance fluorescence was detected using a gated-microchannel-plate photomultiplier and photon counting. A background signal from laser and solar scatter and detector noise was determined by scanning the laser wavelength away from the OH transition (OHWAVE−BKD). For the entire campaign the HOx cell was equipped with an inlet pre-injector (IPI) which chemically scavenged ambient OH by periodically injecting propane into the airstream just above the FAGE inlet. The removal of ambient OH by chemical reaction provided an alternative means to determine the background signal (OHCHEM−BKD) without the need to tune the laser wavelength. By comparison with OHWAVE−BKD, OHCHEM−BKD was used to identify if any OH was generated internally within the FAGE cell, acting as an interference signal. In general, good agreement between OHCHEM−BKD and OHWAVE−BKD was observed, with a ratio of 1.07 for the whole campaign (Woodward-Massey, 2018). In this paper, the OHCHEM observations are used. The instrument was calibrated every few days by overflowing the detection cell inlet with a turbulent flow of high-purity humid air containing a known concentration of OH (and HO2) radicals generated by photolysing a known concentration of H2O vapour at 185 nm. The product of the photon flux at 185 nm and the time spent in the photolysis region were measured before and after the campaign using N2O actinometry (Commane et al., 2010).
2.1.6 OH reactivity measurements
OH reactivity measurements were made using a laser flash photolysis pump-probe technique (Stone et al., 2016). Ambient air, sampled from the roof of the FAGE container, was drawn into a reaction cell at a flow rate of 15 SLM. A 1 SLM flow of high-purity, humidified air which had passed by a Hg lamp, generating ∼50 ppbv of ozone, was mixed with the ambient air at the entrance to the reaction cell. The ozone present was photolysed by 266 nm laser light at a pulse repetition frequency of 1 Hz along the central axis of the reaction cell, leading to the generation of a uniform profile of OH radicals following the reaction of O(1D) with H2O vapour. The decay in the OH radical concentration by reaction with species present in the ambient air was monitored by sampling a portion of the air in to a FAGE cell positioned at the end of the reaction cell. A fraction of the 5 kHz, 308 nm radiation generated by the Ti:Sapphire laser passed through the OH reactivity FAGE cell perpendicular to the airstream, electronically exciting the OH radicals, and the subsequent laser-induced fluorescence signal was detected with a gated-channel photomultiplier tube. The 1 Hz OH decay profiles were integrated for 5 min and fitted to a first-order rate equation to determine the observed loss rate of OH (kobs). The total OH reactivity, k(OH), was calculated by subtracting the rate coefficient associated with physical losses of OH (kphys) from kobs. kphys was determined by monitoring the decay of OH when the ambient air was replaced with a flow of 15 SLM high-purity air. A small correction to account for dilution of the ambient air by the 1 SLM flow of ozone-containing synthetic air was also applied.
2.2 Box modelling
The box modelling that feeds into Fig. 3 was performed using the Dynamically Simple Model of Atmospheric Chemical Complexity (DSMACC), a zero-dimensional box model (Emmerson and Evans, 2009), together with the isoprene scheme and the relevant inorganic chemistry, from the near-explicit chemical mechanism the Master Chemical Mechanism (MCM) v3.3.1 (Jenkin et al., 1997, 2015). The complete isoprene degradation mechanism in MCM v3.3.1 consists of 1926 reactions of 602 closed-shell and free-radical species, which treat the chemistry initiated by reaction with OH radicals, NO3 radicals, and ozone. It contains much of the isoprene HOx recycling chemistry identified as important in recent years under “low-NO” conditions, including the peroxy radical 1,4 and 1,6 H-shift chemistry described in the LIM1 mechanism (Peeters et al., 2009, 2014), as summarised in Wennberg et al. (2018). Model photolysis rates were calculated using the Tropospheric Ultraviolet and Visible Radiation Model (TUV v5.2) (Madronich, 1993).
The box model was initialised with a range of different concentrations of isoprene (1.7, 3.4, 5.0, 6.7 ppb), and OH (0.25×106, 0.5×106, 1.0×106, 3.0×106, 10×106, 20×106 cm−3). [CH4] was fixed at 1.85 ppmv and [CO] at 110 ppbv; T=298 K, and cm−3. Entrainment loss rates for all model species were set to cm−3 s−1. For the box model, a column average value for deposition velocity, Vd, was calculated according to the functionalities of each species (Table S2 in the Supplement). These terms prevent the build-up of secondary products. The values are based on reported deposition rates in Nguyen et al. (2015b). A boundary layer height (BLH) of 1.5 km was assumed. Loss rates (Ld) for each species to dry deposition are then . Photolysis rates were fixed to mean rates for the daytime period 09:00–17:00 LT calculated for 1 July 2017. The model was then run to steady state for a range of fixed NO mixing ratios from 0–16 000 pptv.
2.3 GEOS-Chem modelling
GEOS-Chem version 11-01 (http://wiki.seas.harvard.edu/geos-chem/index.php/GEOS-Chem_v11-01, last access: 15 January 2021) with the inclusion of the aromatic component of RACM2 (regional atmospheric chemistry mechanism 2) was run nested at 0.25×0.3125∘ resolution, with 4×5∘ boundary conditions using GEOS-FP meteorology. The NO emissions were added via the default MIX emissions inventory, which required a 0.9 multiplier applied to the total daily emissions to match observations from the Atmospheric Pollution and Human Health (APHH) summer campaign. The diurnal scale factor was considerably steeper than the default GEOS-Chem NO diurnal one, with a daytime scale factor on the order of a 1.7× and a 0.25 nighttime multiplier. Isoprene emissions calculated by the MEGAN v2.1 (http://wiki.seas.harvard.edu/geos-chem/index.php/MEGAN_v2.1_plus_Guenther_2012_biogenic_emissions, last access: 15 January 2015; Guenther et al., 2012) biogenic emissions extension were scaled by 2.5 in the Beijing metropolitan region (Jing–Jin–Ji).
Beijing is a megacity (population of 21.4 million) with an atmospheric reactive VOC mix with both biogenic and anthropogenic influences (e.g. Li et al., 2020). Mean diurnal cycles of ozone, NO, and isoprene and a range of gas- and aerosol-phase isoprene oxidation products measured at a city-centre site in summer 2017 (Shi et al., 2019) are shown in Fig. 2. Data are filtered to only include “typical” chemistry days, which are considered to be when the ozone mixing ratio increases through the morning to an afternoon peak of >70 ppb. Such typical days account for 25 of the total of 34 measurement days. Further details of the data filtering is given in Sect. S1 of the Supplement. Ozone increases throughout the day to a mid-afternoon peak (Fig. 2a), driven by the photolysis of NO2, which is rapidly regenerated through the reactions of ozone, RO2, and HO2 with NO. The high level of ozone acts to suppress NO concentrations. Such a diurnal cycle is typical of urban environments (Ren et al., 2003; Whalley et al., 2018). However, ozone is so high in Beijing, with mixing ratios regularly >100 ppbv in the afternoon, that on many days NO concentrations fall to <0.5 ppbv in the afternoon and on some days to <0.1 ppbv (see Figs. S4 and S5 in the Supplement).
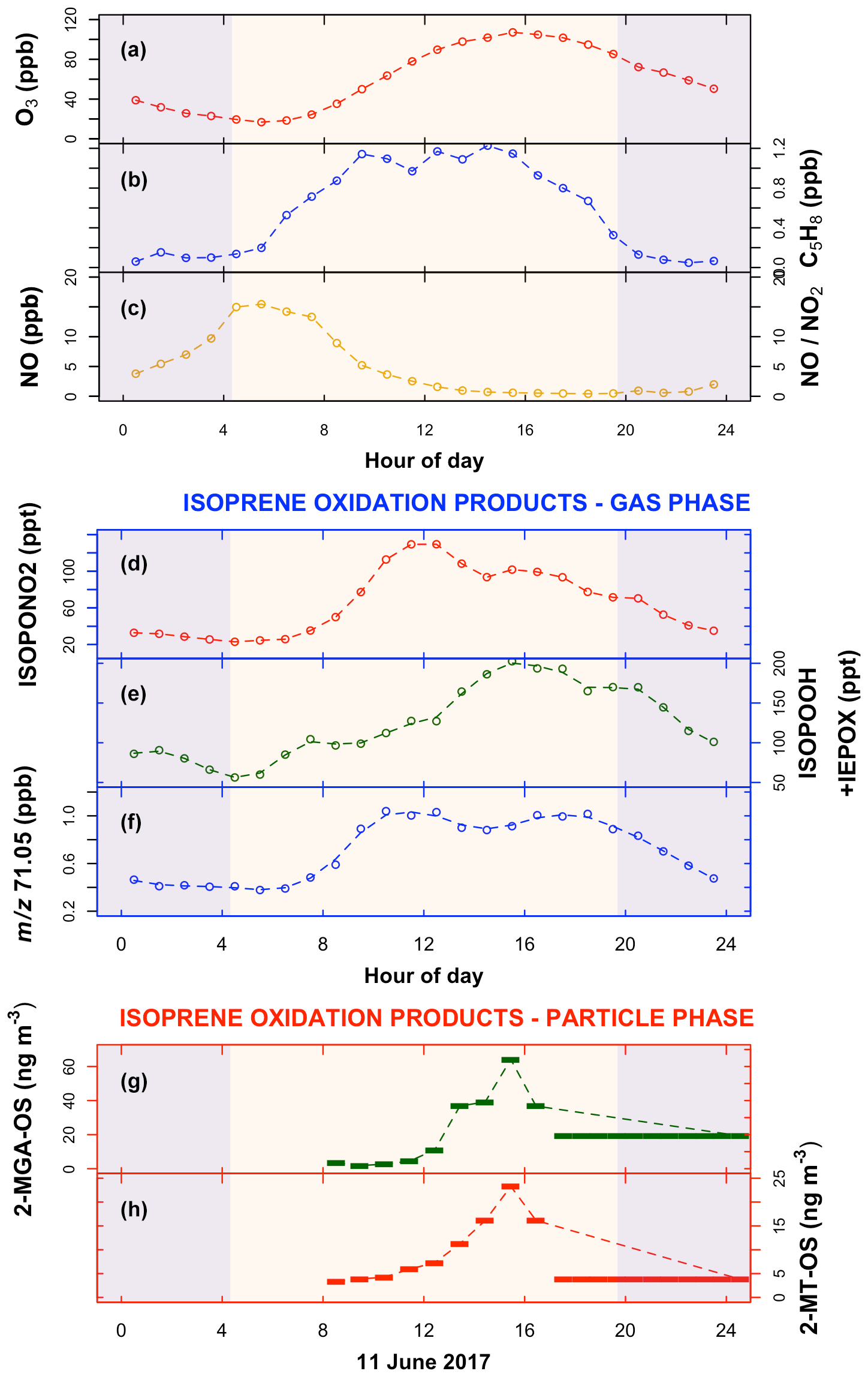
Figure 2Mean diurnal variation in measured organic and inorganic species in the gas and aerosol phase during the Beijing summer observations (data are filtered to only include typical chemistry days – see text for details; the standard deviation of the mean is shown in Fig. S3). Blue-shaded areas are nighttime. (a) Ozone (O3); (b) isoprene (C5H8); (c) nitric oxide (NO) and the ratio ; (d) the gas-phase isoprene high-NO oxidation product, isoprene nitrate (ISOPONO2); (e) the isoprene low-NO oxidation products ISOPOOH+IEPOX; (f) 71.05, assumed to be predominantly the gas-phase isoprene high-NO oxidation products methacrolein (MACR) (precursor to 2-MGA) + methyl vinyl ketone (MVK), signal calibrated with MACR and MVK – see text for further details. (g, h) SOA components: 2-methyltetrol-organosulfate (2-MT-OS) and 2-methylglyceric acid-organosulfate (2-MGA-OS), both measured on 11 and 12 June 2017; the last filter sample was taken from 17:30 LT on 11 June–08:30 LT on 12 June 2017.
The observed diurnal cycles of low-NO and high-NO isoprene oxidation products (Fig. 1) in both the gas and the aerosol phases can be explained by the observed diurnal cycle of NO (Fig. 2c). The high-NO product isoprene nitrate (ISOPONO2), measured using a chemical ionisation mass spectrometer (CIMS) – see Sect. 2.1.2 for further details, is produced throughout the morning from reaction of isoprene peroxy radicals (ISOPOO) with NO (Fig. 2d). During the afternoon, an increasing fraction of ISOPOO begins to react with HO2 or RO2 as the NO concentration drops. This leads to the observed decrease in ISOPONO2 and an increase in the low-NO products IEPOX+ISOPOOH (also measured by CIMS) through the afternoon (Fig. 2e). The profile of the signal at 71.05 is assumed to be dominated by the high-NO products MACR+MVK, measured by a proton transfer reaction time-of-flight mass spectrometer (PTR-ToF-MS) – see Sect. 2.1.2 for further details. This signal is very similar to that of ISOPONO2 until about 15:00 LT, when it begins to increase, with a second peak observed at around 17:00 LT (Fig. 2f). This latter peak may be an observational artefact as a result of the conversion of ISOPOOH to either MVK (via 1,2-ISOPOOH – the dominant isomer; Reeves et al., 2020) or MACR (via 4,3-ISOPOOH) on metal surfaces within the inlet of the PTR instrument (Rivera-Rios et al., 2014). Isoprene oxidation products can also partition into the particle phase and undergo heterogeneous reactions to form organosulfates, with concentrations driven by a number of additional factors such as particulate sulfate and water vapour concentrations. Isoprene organosulfate tracers, 2-MGA-OS (Fig. 2g) and 2-methyltetrol-OS (Fig. 2h), were measured on 11 June 2017, with low concentrations through the morning, increasing during the afternoon to a peak at around 15:00–16:00 LT. Both are tracers for non-NO-driven chemistry. While 2-methyltetrol-OS is formed via the ISOPOO+HO2 IEPOX pathway (Paulot et al., 2009; Surratt et al., 2010; Lin et al., 2012), 2-MGA-OS (Lin et al., 2013) is formed from the OH-initiated oxidation of MPAN (peroxymethacrylic nitric anhydride) (Kjaergaard et al., 2012; Nguyen et al., 2015a), with further oxidation leading to 2-MGA (Surratt et al., 2010; Chan et al., 2010; Nguyen et al., 2015a). MPAN is a product of the OH-initiated oxidation of MACR in an environment with a high ratio. So the observation of 2-MGA-OS formation reflects the observed diurnal NO cycle in Beijing. MACR is formed in the morning through the OH oxidation of isoprene in a high-NO environment, followed by OH oxidation of MACR in a high- environment in the afternoon to form MPAN, which reacts further with OH to yield 2-MGA.
The observed temporal profiles of the isoprene tracer products suggest a chemical cycle switching from a high-NO chemical regime in the morning to a regime with a significant contribution from low-NO chemistry in the afternoon in Beijing. First, isoprene nitrates, formed predominantly during the morning (Fig. 2d), are characteristic of high-NO chemistry. Second, isoprene hydroperoxides (ISOPOOH) and epoxydiols (IEPOX) (Fig. 2e), formed predominantly during the afternoon, are characteristic of low-NO chemistry, where the reaction of ISOPOO with HO2 dominates over the reaction with NO. The formation of highly oxygenated molecules (HOMs), characteristic of RO2 isomerisation and auto-oxidation in low-NO environments, has also been observed during the afternoon at this site (Brean et al., 2019). Third, the observation of large amounts of 2-methylglyceric acid (2-MGA-OS) (Fig. 2g) in the aerosol is suggestive of both high- and low-NO chemistry having occurred.
The chemical box model DSMACC (Emmerson and Evans, 2009), coupled with the near-explicit oxidation mechanism for isoprene from the Master Chemical Mechanism (MCM v3.3.1) (Jenkin et al., 1997, 2015), was used to assess the sensitivity of the fraction of ISOPOO reacting with NO (fNO) to varying NO concentrations and OH reactivities (Eq. 1)
The model was run to steady state at a range of different fixed concentrations of [OH], [NO], and [isoprene], using fixed photolysis rates typical of the Beijing daytime (see Sects. 2 and S5). Figure 3 plots against [NO], where k(OH)* is the OH reactivity less the contribution from OH reactions with NOx and NOy species (Eq. 2), since these reactions do not produce RO2.
As expected, fNO increases with increasing NO concentration. It also shows that fNO is not a fixed value for a given concentration of NO but decreases with the increasing reactivity of the system (the x axis in Fig. 3). The reactivity varies as a function of the VOC mixing ratios, the reactivity of the VOCs, and the OH concentration, i.e. (Eq. 1). Higher reactivity and higher OH concentrations both lead to a higher concentration of peroxy radicals (]), reducing fNO. Average measurements of and [NO] for the afternoon (12:00–20:00 LT) from a range of different environments are shown in Fig. 3 (see also Table S1 in the Supplement). The RO2 chemistry in the rural southeastern USA and the Borneo rainforest lies in the low-NO regime (i.e. fNO<0.5) for the whole afternoon. In the urban areas of London and New York the chemistry remains in the high-NO regime throughout the whole afternoon. However, in Beijing, the extreme suppression of NO concentrations in the afternoon drives the chemistry from a regime in which >95 % of the RO2 is reacting with NO during the morning to one in which less than 70 % is reacting with NO by the mid-afternoon. HO2 concentrations were measured by FAGE during the campaign (Whalley et al., 2020). Concentrations peaked in the mid-afternoon (i.e. when NO is at its lowest), regularly exceeding 5×108 cm−3 and reaching up to 1×109 cm−3 on some days. Based on the relative reaction rates of RO2 with NO and HO2, for cm−3 the contribution of low-NO pathways to RO2 removal would be expected to be roughly 50 % at [NO]=100 ppt and 10 % at [NO]=1 ppb.
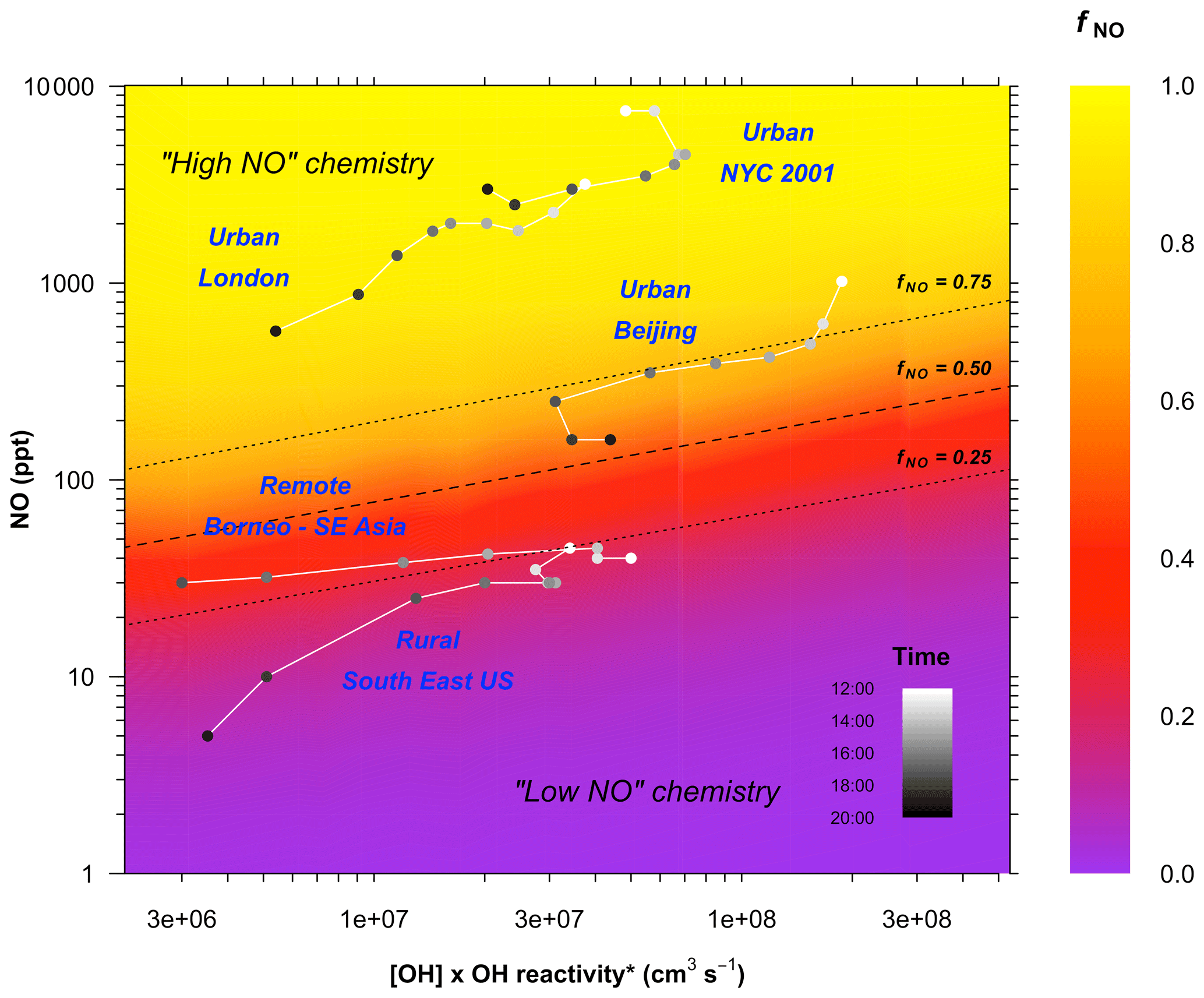
Figure 3Variation in the fraction of ISOPOO reacting with NO as a function of NO concentration and the reactivity of the system. The plot is derived from a series of zero-dimensional box model runs performed as a function of fixed concentrations of [NO], [OH], and [isoprene]. Photolysis is fixed to an average of 09:00-17:00 LT conditions. k(OH)* is the total OH reactivity less the contribution from OH reactions with NOx and NOy species (Eq. 2), since these reactions do not produce RO2. The dashed line shows the fraction of ISOPOO reacting with NOfNO=0.50; dotted lines show fNO=0.25 and 0.75. Points are average diurnal hourly measurements of NO, OH, and k(OH)* for the period 12:00–20:00 LT from a range of different environments: the rural sites, Borneo (Whalley et al., 2011) (only shown for 12:00–18:00 LT) and the southeastern USA (Sanchez et al., 2018) and the urban sites London (Whalley et al., 2016), New York City (Ren et al., 2003), and Beijing (this work). See the Supplement for full details.
Modelling was performed with the global chemical transport model GEOS-Chem, with a nested grid at a 0.25×0.3125∘ resolution over China (see Sect. 2.3 – “GEOS-Chem modelling”), to investigate the modelled diurnal cycle of NO for Beijing. The results are compared to the measurements for the entire campaign (note not the filtered measurements presented in Fig. 2) (Fig. 4). These show that while the model does a good job of recreating the measured ozone and NO2 diurnal profiles for the campaign period, it cannot match the observed diurnal profiles of [NO] or the NO-to-NO2 ratio, particularly at sub-ppb levels typically observed during the afternoon. Thus the model will not capture the formation of low-NO products from isoprene and other VOCs in Beijing. The major driver of the low-NO concentrations in the model is the high level of ozone, which titrates out the NO. The mean afternoon ozone peak is underpredicted by the model by about 10 %. However, this has little impact on the modelled NO concentrations which the model overpredicts by a factor of 3–5 throughout the afternoon. As such, with the chemistry currently in the model there is very little flexibility available to appreciably change this ratio; i.e. changes to other NO sinks in the model, such as RO2, through changes to VOC emissions, will have little effect on [NO]. The fact that the GEOS-Chem modelling cannot recreate the extremely low NO levels observed in the afternoon suggests that there may be additional sinks for NO beyond those currently included in the chemical mechanism. One explanation may be the occurrence of RO2+NO oxidation pathways that lead to the formation of a second RO2 species before forming a stable species, effectively increasing the efficiency of NO-to-NO2 conversion per initial oxidation step (e.g. Whalley et al., 2020). Such reactions are expected to be particularly important for larger and more complex VOCs, for which the detailed oxidation processes have been less studied and which are heavily parameterised in global models. Auto-oxidation processes that regenerate OH, leading to the formation of further RO2, have also been proposed previously for the high-VOC–low-NO (<1 ppbv) conditions seen in Beijing and other cities (Hofzumahaus et al., 2009; Whalley et al., 2018; Tan et al., 2019). Inclusion of such RO2 oxidation processes is one of the main foci of the next generation of atmospheric chemical mechanisms. Another explanation may be the presence of high concentrations of other species (not currently included in the chemical scheme) that can rapidly convert NO to NO2, e.g. halogen oxides.
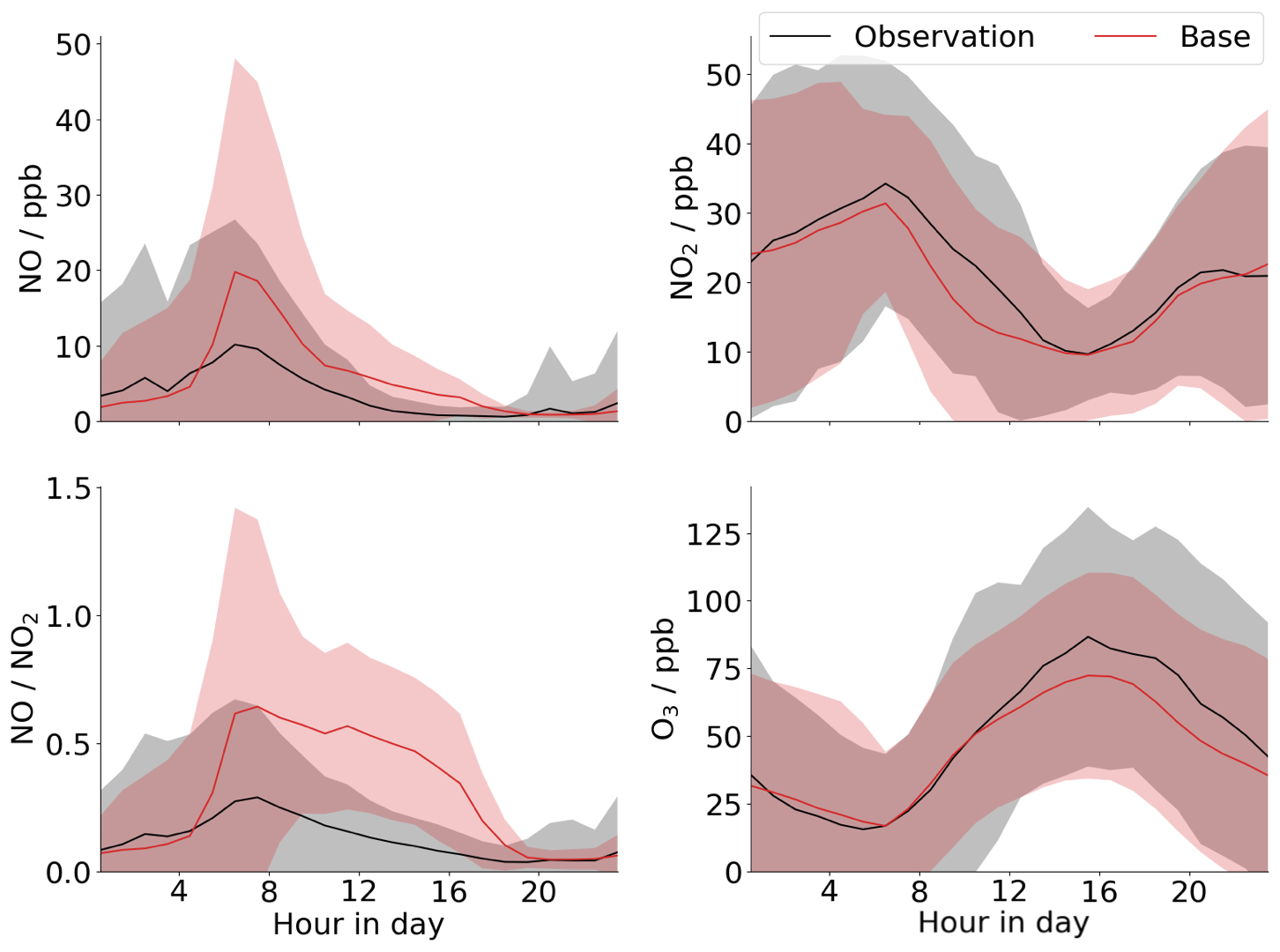
Figure 4Comparison of GEOS-Chem model output (red) to mean diurnal measurements for the entire campaign (black) for NO, NO2, O3, and the ratio. Shaded regions are 2 standard deviations of the mean.
Similar mixed NO regimes to those observed here for Beijing have been observed previously at a suburban site in the Pearl River Delta (Tan et al., 2019), and in the semi-rural southeastern USA (Xiong et al., 2015), albeit with lower morning NO peaks. Such a mixed regime will lead to a range of low-volatility multi-functional products (Xiong et al., 2015; Lee et al., 2016), some of which are only accessible through this regime, which can efficiently partition to the particle phase to contribute to SOA. With the rates of RO2+NO and RO2+HO2 similar for most peroxy radicals (Orlando and Tyndall, 2012), the chemical regime reported herein is not just relevant to isoprene but to all VOCs (see a comparison for butane and toluene in Fig. S11).
Our observations from Beijing challenge the commonly accepted view of polluted urban areas as high-NO atmospheric environments in two ways. First, very high ozone (and other sinks) regularly reduces afternoon NO to <1 ppbv and on some days to <0.1 ppbv. This leads to the formation of low-NO products in the gas and aerosol phase. Second, the level of NO that is required for low-NO chemistry to occur is not a fixed value but is dependent on the concentration and reactivity of the VOCs present and the concentration of OH. Hence NO concentrations that represent low-NO conditions in a tropical rainforest, for example, are different to those that represent low-NO conditions in a highly polluted urban environment with elevated OH and OH reactivity.
Under the conditions observed in Beijing, the production of low-NO SOA and the associated increase in PM is shown to be closely linked to photochemical ozone production. Policies that reduce the afternoon ozone peak might also be expected to reduce the production of these aerosol-phase products. However, such policies must also take account of the complex interactions between NOx, VOCs, ozone, and PM. For example, reducing NOx emissions can counter-intuitively lead to increases in ozone, as has occurred in other major cities (Air Quality Expert Group, 2009), and a recent modelling study (Li et al., 2019) has suggested that reducing PM has led to increases in ozone in China, although a recent experimental study (Tan et al., 2020) in the North China Plain could not observe this effect. With many existing and developing megacities being located in subtropical regions with high emissions of reactive biogenic VOCs, the control of which is very difficult, and with continuing reductions in NOx emissions, such extreme chemical environments as that observed in Beijing can be expected to proliferate. The failure of regional and global models to accurately replicate this chemical regime has wider implications for the prediction of secondary pollutants and hence for determining policies to control air pollution episodes.
The GEOS-Chem model code is available from https://github.com/geoschem/geos-chem (last access: 2 February 2021, International GEOS-Chem User Community, 2017). The DSMACC model (Emmerson and Evans, 2009, https://doi.org/10.5194/acp-9-1831-2009) is available at http://wiki.seas.harvard.edu/geos-chem/index.php/DSMACC_chemical_box_model (last access: 28 January 2021), and the MCMv3.3.1 chemical mechanism (Jenkin et al., 1997: https://doi.org/10.1016/S1352-2310(96)00105-7, Jenkin et al., 2015: https://doi.org/10.5194/acp-15-11433-2015), is available at http://mcm.york.ac.uk (last access: 28 January 2021).
Data are available at http://catalogue.ceda.ac.uk/uuid/7ed9d8a288814b8b85433b0d3fec0300 (last access: 19 January 2021): Isoprene (Nelson et al., 2020, https://catalogue.ceda.ac.uk/uuid/fae65a63910a44b28a3d91268417b7c3); NO, NO2, O3 (Drysdale, 2020, https://catalogue.ceda.ac.uk/uuid/33ba929e3bcd4c08b69871e45a8660aa); 71.05 (Acton et al., 2018, https://catalogue.ceda.ac.uk/uuid/de37c54e59a548ccb9f168ee724f3769; Acton et al., 2020, https://catalogue.ceda.ac.uk/uuid/9e11d5cb819a45068921db5ae296fb57); OH concentrations (Slater and Whalley, 2018, https://catalogue.ceda.ac.uk/uuid/bb339ff791814fc6a8b9a93d339f5bc1); OH reactivity (Whalley and Slater, 2018, https://catalogue.ceda.ac.uk/uuid/8ed84f3c770544c49329df9b068ab662). Specific data are available from the authors on request (jacqui.hamilton@york.ac.uk).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-1613-2021-supplement.
JRH, RED, JFH, WJFA, CNH, BL, and XW provided the VOC measurements. FAS, WSD, and JDL provided the NOx and O3 measurements. TJB, AM, SDW, AB, CJP, and HC collected and analysed the CIMS data. TQ and JDS provided the organosulfate standards. DB, WD, and JFH provided the organosulfate aerosol measurements. LKW, DEH, EJS, RW-M, and CY provided the OH and HO2 data. MN, PME, and ARR provided the MCM box modelling. PDI and MJE provided the GEOS-Chem model run. ACL is the principal investigator of the APHH-Beijing project. MJN, JFH, and ARR conceived and wrote the manuscript with input and discussion from all co-authors.
The authors declare that they have no conflict of interest.
This article is part of the special issue “In-depth study of air pollution sources and processes within Beijing and its surrounding region (APHH-Beijing) (ACP/AMT inter-journal SI)”. It is not associated with a conference.
We acknowledge the support from Pingqing Fu, Zifa Wang, Jie Li, and Yele Sun from the IAP for hosting the APHH-Beijing campaign at the IAP. We thank Zongbo Shi, Roy Harrison, Tuan Vu, and Bill Bloss from the University of Birmingham; Siyao Yue, Liangfang Wei, Hong Ren, Qiaorong Xie, Wanyu Zhao, Linjie Li, Ping Li, Shengjie Hou, and Qingqing Wang from the IAP; Kebin He and Xiaoting Cheng from Tsinghua University; and James Allan from the University of Manchester for providing logistic and scientific support for the field campaigns. Peter Ivatt acknowledges funding from NCAS through one of its Air Pollution and Human Health studentships. Daniel Bryant, William Dixon, William Drysdale, Freya Squires, and Eloise Slater acknowledge the NERC SPHERES Doctoral Training Partnership (DTP) for studentships.
This research has been supported by the Natural Environment Research Council and the Newton Fund and Medical Research Council in the UK and by the National Natural Science Foundation of China (grant nos. NE/N007190/1 and NE/N006917/1).
This paper was edited by Frank Keutsch and reviewed by two anonymous referees.
Acton, J., Hewitt, N., Huang, Z., and Wang, X.: APHH: Volatile organic compound (VOC) mixing ratios made at the IAP-Beijing site during the summer and winter campaigns, Centre for Environmental Data Analysis, available at: https://catalogue.ceda.ac.uk/uuid/de37c54e59a548ccb9f168ee724f3769 (last access: 19 January 2021), 2018.
Acton, J., Hewitt, N., Huang, Z., and Wang, X.: APHH: Volatile organic compound (VOC) flux measurements made during the APHH-Beijing field campaigns 11-12/2016 and 05-06/2017, Centre for Environmental Data Analysis, available at: https://catalogue.ceda.ac.uk/uuid/9e11d5cb819a45068921db5ae296fb57 (last access: 19 January 2021), 2020.
Air Quality Expert Group: Ozone in the United Kingdom, Department for the Environment, Food and Rural Affairs, UK, available at: https://uk-air.defra.gov.uk/assets/documents/reports/aqeg/aqeg-ozone-report.pdf (last access: 15 January 2021), 2009.
Bannan, T. J., Booth, A. M., Bacak, A., Muller, J. B. A., Leather, K. E., Le Breton, M., Jones, B., Young, D., Coe, H., Allan, J., Visser, S., Slowik, J. G., Furger, M., Prevot, A. S. H., Lee, J. Dunmore, R. E., Hopkins, J. R., Hamilton, J. F., Lewis, A. C., Whalley, L. K., Sharp, T., Stone, D., Heard, D. E., Fleming, Z. L., Leigh, R., Shallcross, D. E., and Percival, C. J.: The first U.K. measurements of nitryl chloride using a chemical ionisation mass spectrometer in London, ClearfLo Summer, 2012, and an investigation of the role of Cl atom oxidation, J. Geophys. Res., 120, 5638–5657, 2015.
Bianchi F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, 2019.
Brean, J., Harrison, R. M., Shi, Z., Beddows, D. C. S., Acton, W. J. F., Hewitt, C. N., Squires, F. A., and Lee, J.: Observations of highly oxidized molecules and particle nucleation in the atmosphere of Beijing, Atmos. Chem. Phys., 19, 14933–14947, https://doi.org/10.5194/acp-19-14933-2019, 2019.
Bryant, D. J., Dixon, W. J., Hopkins, J. R., Dunmore, R. E., Pereira, K. L., Shaw, M., Squires, F. A., Bannan, T. J., Mehra, A., Worrall, S. D., Bacak, A., Coe, H., Percival, C. J., Whalley, L. K., Heard, D. E., Slater, E. J., Ouyang, B., Cui, T., Surratt, J. D., Liu, D., Shi, Z., Harrison, R., Sun, Y., Xu, W., Lewis, A. C., Lee, J. D., Rickard, A. R., and Hamilton, J. F.: Strong anthropogenic control of secondary organic aerosol formation from isoprene in Beijing, Atmos. Chem. Phys., 20, 7531–7552, https://doi.org/10.5194/acp-20-7531-2020, 2020.
Chan, A. W. H., Chan, M. N., Surratt, J. D., Chhabra, P. S., Loza, C. L., Crounse, J. D., Yee, L. D., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Role of aldehyde chemistry and NOx concentrations in secondary organic aerosol formation, Atmos. Chem. Phys., 10, 7169–7188, https://doi.org/10.5194/acp-10-7169-2010, 2010.
Cheng, J., Su, J., Cui, T., Li, X., Dong, X., Sun, F., Yang, Y., Tong, D., Zheng, Y., Li, Y., Li, J., Zhang, Q., and He, K.: Dominant role of emission reduction in PM2.5 air quality improvement in Beijing during 2013–2017: a model-based decomposition analysis, Atmos. Chem. Phys., 19, 6125–6146, https://doi.org/10.5194/acp-19-6125-2019, 2019.
Commane, R., Floquet, C. F. A., Ingham, T., Stone, D., Evans, M. J., and Heard, D. E.: Observations of OH and HO2 radicals over West Africa, Atmos. Chem. Phys., 10, 8783–8801, https://doi.org/10.5194/acp-10-8783-2010, 2010.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P.: Autooxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, 2013.
Drysdale, W.: APHH: O3, CO, NO, NO2, NOy and SO2 measurements made at the Indira Gandhi Delhi Technical University for Women (IGDTUW) site during the pre and post monsoon periods for the DelhiFlux field campaign 2018, Centre for Environmental Data Analysis, available at: https://catalogue.ceda.ac.uk/uuid/33ba929e3bcd4c08b69871e45a8660aa (last access: 19 January 2021), 2020.
Emmerson, K. M. and Evans, M. J.: Comparison of tropospheric gas-phase chemistry schemes for use within global models, Atmos. Chem. Phys., 9, 1831–1845, https://doi.org/10.5194/acp-9-1831-2009, 2009 (data available at: http://wiki.seas.harvard.edu/geos-chem/index.php/DSMACC_chemical_box_model, last access: 28 January 2021).
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Hamilton, J. F., Lewis, A.C., Carey, T. J., and Wenger, J. C.: Characterization of Polar Compounds and Oligomers in Secondary Organic Aerosol Using Liquid Chromatography Coupled to Mass Spectrometry, Anal. Chem., 80, 474–480, 2008.
Hofzumahaus, A., Rohrer, F., Lu, K., Bohn, B., Brauers, T., Chang, C.-C., Fuchs, H., Holland, F., Kita, K., Kondo, Y., Li, X., Lou, S., Shao, M., Zeng, L., Wahner, A., and Zhang, Y.: Amplified Trace Gas Removal in the Troposphere, Science, 324, 1702–1704, 2009.
Hopkins, J. R., Jones, C. E., and Lewis, A. C.: A dual channel gas chromatograph for atmospheric analysis of volatile organic compounds including oxygenated and monoterpene compounds, J. Environ. Monit., 13, 2268–2276, 2011.
International GEOS-Chem User Community: geoschem/geos-chem: GEOS-Chem 11.1, Version 11.1.0, GitHub, available at: https://github.com/geoschem/geos-chem (last access: 2 February 2021), 2017.
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997 (data available at: http://mcm.york.ac.uk, last access: 28 January 2021).
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015 (data available at: http://mcm.york.ac.uk, last access: 28 January 2021).
Jimenez, J. L., Canagaratna, M. R., Donahue, Prevot, A. S. H., Zhang, Q., Kroll, J. H. DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C.. Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of Organic Aerosols in the Atmosphere, Science, 326, 1525–1529, 2009.
Jordan, A., Haidacher, S., Hanel, G., Hartungen, E., Märk, L., Seehauser, H., Schottkowsky, R., Sulzer, P., and Märk, T. D.: A high resolution and high sensitivity proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF-MS), Int. J. Mass Spectrom., 286, 122–128, 2009.
Kjaergaard, H. G., Knap, H. C., Ørnsø, K. B., Jørgensen, S., Crounse, J. D., Paulot, F., and Wennberg, P. O.: Atmospheric Fate of Methacrolein. 2. Formation of Lactone and Implications for Organic Aerosol Production, J. Phys. Chem. A, 116, 5763–5768, 2012.
Kroll, J. H., Ng, N. L., Murphy, S. M., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from isoprene photooxidation, Environ. Sci. Technol., 40, 1869–1877, 2006.
Krotkov, N. A., McLinden, C. A., Li, C., Lamsal, L. N., Celarier, E. A., Marchenko, S. V., Swartz, W. H., Bucsela, E. J., Joiner, J., Duncan, B. N., Boersma, K. F., Veefkind, J. P., Levelt, P. F., Fioletov, V. E., Dickerson, R. R., He, H., Lu, Z., and Streets, D. G.: Aura OMI observations of regional SO2 and NO2 pollution changes from 2005 to 2015, Atmos. Chem. Phys., 16, 4605–4629, https://doi.org/10.5194/acp-16-4605-2016, 2016.
Le Breton, M., Wang, Y., Hallquist, Å. M., Pathak, R. K., Zheng, J., Yang, Y., Shang, D., Glasius, M., Bannan, T. J., Liu, Q., Chan, C. K., Percival, C. J., Zhu, W., Lou, S., Topping, D., Wang, Y., Yu, J., Lu, K., Guo, S., Hu, M., and Hallquist, M.: Online gas- and particle-phase measurements of organosulfates, organosulfonates and nitrooxy organosulfates in Beijing utilizing a FIGAERO ToF-CIMS, Atmos. Chem. Phys., 18, 10355–10371, https://doi.org/10.5194/acp-18-10355-2018, 2018.
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurtén, T., Worsnop, D. R., and Thornton, J. A.: An iodide-adduct high-resolution time-of-flight chemical-ionization mass spectrometer: Application to atmospheric inorganic and organic compounds, Environ. Sci. Technol., 48, 6309–6317, 2014.
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurten, T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvery, K., Shepson, P. B., Baumann, K., Edgerton, E., Liu, J., Shilling, J. E., Miller, D. O., Brune, W. H., D'Ambro, E. L., and Thornton, J. A.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci., 113, 1516–1521, 2016.
Li, K., Jacob, D. J., Liao, H., Shen, L., Zhang, Q., and Bates, K. H.: Anthropogenic drivers of 2013–2017 trends in summer surface ozone in China, P. Natl. Acad. Sci. USA, 116, 422–427, 2019.
Li, Q., Su, G., Li, C., Liu, P., Zhao, X., Zhang, C., Sun, X., Mu, Y., Wu, M., Wang, Q., and Sun, B.: An investigation into the role of VOCs in SOA and ozone production in Beijing, China, Sci. Total Environ., 720, 137536, https://doi.org/10.1016/j.scitotenv.2020.137536, 2020.
Lin, Y.-H., Zhang, Z., Docherty, K. S., Zhang, H., Budisulistiorini, S. H., Rubitschin, C. L., Shaw, S. L., Knipping, E. M., Edgerton, E. S., Kleindienst, T. E., Gold, A., and Surratt, J. D.: Isoprene Epoxydiols as Precursors to Secondary Organic Aerosol Formation: Acid-Catalyzed Reactive Uptake Studies with Authentic Compounds, Environ. Sci. Technol., 46, 250–258, 2012.
Lin, Y.-H., Zhang, H., Pye, H. O. T., Zhang, Z., Marth, W. J., Park, S., Arashiro, M., Cui, T., Budisulistiorini, S. H., Sexton, K. G., Vizuete, W., Xie, Y., Luecken, D. J., Piletic, I. R., Edney, E. O., Bartolotti, L.J., Gold, A., and Surratt, J. D.: Epoxide as a precursor to secondary organic aerosol formation from isoprene photooxidation in the presence of nitrogen oxides, P. Natl. Acad. Sci. USA, 110, 6718–6723, https://doi.org/10.1073/pnas.1221150110, 2013.
Liu, F., Zhang, Q., van der A, R. J., Zheng, B., Tong, D., Yan, L., Zheng, Y., and He, K.: Recent reduction in NOx emissions over China: synthesis of satellite observations and emission inventories, Environ. Res. Lett., 11, 114002, https://doi.org/10.1088/1748-9326/11/11/114002, 2016.
Liu, H., Zhang, M., Han, X., Li, J., and Chen, L.: Episode analysis of regional contributions to tropospheric ozone in Beijing using a regional air quality model, Atmos. Environ., 199, 299–312, 2019.
Madronich, S.: The Atmosphere and UV-B Radiation at Ground Level, in: Environmental UV Photobiology, edited by: Young, A. R., Moan, J., Björn, L. O., and Nultsch, W., Boston, MA, Springer US, 1–39, 1993.
Miyazaki, K., Eskes, H., Sudo, K., Boersma, K. F., Bowman, K., and Kanaya, Y.: Decadal changes in global surface NOx emissions from multi-constituent satellite data assimilation, Atmos. Chem. Phys., 17, 807–837, https://doi.org/10.5194/acp-17-807-2017, 2017.
Nelson, B., Hopkins, J. R., Stewart, G., Dunmore, R., and Hamilton, J. F.: APHH: Online measurements of VOC mixing ratios using Gas Chromatography with Flame Ionisation Detector (GC-FID) at Indira Gandhi Delhi Technical University for Women (IGDTUW) field site during the DelhiFlux field campaign, Centre for Environmental Data Analysis, available at: https://catalogue.ceda.ac.uk/uuid/fae65a63910a44b28a3d91268417b7c3 (last access: 19 January 2021), 2020.
Nguyen, T. B., Bates, K. H., Crounse, J. D., Schwantes, R. H., Zhang, X., Kjaergaard, H. G., Surratt, J. D., Lin, P., Laskin, A., Seinfeld, J. H., and Wennberg, P. O.: Mechanism of the hydroxyl radical oxidation of methacryloyl peroxynitrate (MPAN) and its pathway toward secondary organic aerosol formation in the atmosphere, Phys. Chem. Chem. Phys., 17, 17914–17926, https://doi.org/10.1039/C5CP02001H, 2015a.
Nguyen, T. B., Crounse, J. D., Teng, A. P., Clair, J. M. S., Paulot, F., Wolfe, G. M., and Wennberg, P. O.: Rapid deposition of oxidized biogenic compounds to a temperate forest, P. Natl. Acad. Sci. USA, 112, E392–E401, 2015b.
Orlando, J. J. and Tyndall, G. S.: Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 41, 6294–6317, 2012.
Paulot, F., Crounse, J. D., Kjaergaard, H. G., Kürten, A., St. Clair, J. M., Seinfeld, J. H., and Wennberg, P. O.: Unexpected epoxide formation in the gas-phase photooxidation of isoprene, Science, 325, 730–733, 2009.
Peeters, J., Nguyen, T. L., and Vereecken, L.: HOx radical regeneration in the oxidation of isoprene, Phys. Chem. Chem. Phys., 11, 5935–5939, 2009.
Peeters, J., Müller, J.-F., Stavrakou, T., and Nguyen, V. S.: Hydroxyl Radical Recycling in Isoprene Oxidation Driven by Hydrogen Bonding and Hydrogen Tunneling: The Upgraded LIM1 Mechanism, J. Phys. Chem. A., 118, 8625–8643, 2014.
Praske, E., Otkjaer, R. V., Crounse, J. D., Hethcox, J. C., Stoltz, B. M., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric autoxidation is increasingly important in urban and suburban North America, P. Natl. Acad. Sci. USA, 115, 64–69, 2018.
Priestley, M., Le Breton, M., Bannan, T. J., Leather, K. E., Bacak, A., Reyes-Villegas, E., de Vocht, F., Shallcross, B. M. A., Brazier, T., Khan, M. A., Allan, J., Shallcross, D. E., Coe, H., and Percival, C. J.: Observations of isocyanate, amide, nitrate and nitro compounds from an anthropogenic biomass burning event using a ToF-CIMS, J Geophys. Res.-Atmos., 123, 7687–7704, 2018.
Reeves, C. E., Mills, G. P., Whalley, L. K., Acton, W. J. F., Bloss, W. J., Crilley, L. R., Grimmond, S., Heard, D. E., Hewitt, C. N., Hopkins, J. R., Kotthaus, S., Kramer, L. J., Jones, R. L., Lee, J. D., Liu, Y., Ouyang, B., Slater, E., Squires, F., Wang, X., Woodward-Massey, R., and Ye, C.: Observations of speciated isoprene nitrates in Beijing: implications for isoprene chemistry, Atmos. Chem. Phys. Discuss. [preprint], https://doi.org/10.5194/acp-2019-964, in review, 2020.
Ren, X., Harder, H., Martinez, M., Lesher, R. L., Oliger, A., Simpas, J. B., Brune, W. H., Schwab, J. J., Demerjian, K. L., He, Y., Zhou, X., and Gao, H.: OH and HO2 chemistry in the urban atmosphere of New York City, Atmos. Environ., 37, 3639–3651, 2003.
Rivera-Rios, J. C., Nguyen, T. B., Crounse, J. D., Jud, W., St. Clair, J. M., Mikoviny, T., Gilman, J. B., Lerner, B. M., Kaiser, J. B., de Gouw, J., Wisthaler, A., Hansel, A., Wennberg, P. O., Seinfeld, J. H., and Keutsch, F. N.: Conversion of hydroperoxides to carbonyls in field and laboratory instrumentation: Observational bias in diagnosing pristine versus anthropogenically controlled atmospheric chemistry, Geophys. Res. Lett., 41, 8645–8651, https://doi.org/10.1002/2014GL061919, 2014.
Sanchez, D., Jeong, D., Seco, R., Wrangham, I., Park, J. H., Brune, W. H., Koss, A., Gilman, J., de Gouw, J., Misztal, P., Goldstein, A., Baumann, K., Wennberg, P. O., Keutsch, F. N., Guenther, A., and Kim, S.: Intercomparison of OH and OH reactivity measurements in a high isoprene and low NO environment during the Southern Oxidant and Aerosol Study (SOAS), Atmos. Environ., 174, 227–236, 2018.
Schwantes, R. H., Charan, S. M., Bates, K. H., Huang, Y., Nguyen, T. B., Mai, H., Kong, W., Flagan, R. C., and Seinfeld, J. H.: Low-volatility compounds contribute significantly to isoprene secondary organic aerosol (SOA) under high-NOx conditions, Atmos. Chem. Phys., 19, 7255–7278, https://doi.org/10.5194/acp-19-7255-2019, 2019.
Shi, Z., Vu, T., Kotthaus, S., Harrison, R. M., Grimmond, S., Yue, S., Zhu, T., Lee, J., Han, Y., Demuzere, M., Dunmore, R. E., Ren, L., Liu, D., Wang, Y., Wild, O., Allan, J., Acton, W. J., Barlow, J., Barratt, B., Beddows, D., Bloss, W. J., Calzolai, G., Carruthers, D., Carslaw, D. C., Chan, Q., Chatzidiakou, L., Chen, Y., Crilley, L., Coe, H., Dai, T., Doherty, R., Duan, F., Fu, P., Ge, B., Ge, M., Guan, D., Hamilton, J. F., He, K., Heal, M., Heard, D., Hewitt, C. N., Hollaway, M., Hu, M., Ji, D., Jiang, X., Jones, R., Kalberer, M., Kelly, F. J., Kramer, L., Langford, B., Lin, C., Lewis, A. C., Li, J., Li, W., Liu, H., Liu, J., Loh, M., Lu, K., Lucarelli, F., Mann, G., McFiggans, G., Miller, M. R., Mills, G., Monk, P., Nemitz, E., O'Connor, F., Ouyang, B., Palmer, P. I., Percival, C., Popoola, O., Reeves, C., Rickard, A. R., Shao, L., Shi, G., Spracklen, D., Stevenson, D., Sun, Y., Sun, Z., Tao, S., Tong, S., Wang, Q., Wang, W., Wang, X., Wang, X., Wang, Z., Wei, L., Whalley, L., Wu, X., Wu, Z., Xie, P., Yang, F., Zhang, Q., Zhang, Y., Zhang, Y., and Zheng, M.: Introduction to the special issue “In-depth study of air pollution sources and processes within Beijing and its surrounding region (APHH-Beijing)”, Atmos. Chem. Phys., 19, 7519–7546, https://doi.org/10.5194/acp-19-7519-2019, 2019.
Sindelarova, K., Granier, C., Bouarar, I., Guenther, A., Tilmes, S., Stavrakou, T., Müller, J.-F., Kuhn, U., Stefani, P., and Knorr, W.: Global data set of biogenic VOC emissions calculated by the MEGAN model over the last 30 years, Atmos. Chem. Phys., 14, 9317–9341, https://doi.org/10.5194/acp-14-9317-2014, 2014.
Slater, E. and Whalley, L.: APHH: Fluorescence Assay Gas Expansion measurements of OH, HO2 and RO2 made at the IAP-Beijing site during the winter and summer campaigns, Centre for Environmental Data Analysis, available at: https://catalogue.ceda.ac.uk/uuid/bb339ff791814fc6a8b9a93d339f5bc1 (last access: 19 January 2021), 2018.
Stone, D., Whalley, L. K., Ingham, T., Edwards, P. M., Cryer, D. R., Brumby, C. A., Seakins, P. W., and Heard, D. E.: Measurement of OH reactivity by laser flash photolysis coupled with laser-induced fluorescence spectroscopy, Atmos. Meas. Tech., 9, 2827–2844, https://doi.org/10.5194/amt-9-2827-2016, 2016.
Surratt, J. D., Murphy, S. M., Kroll, J. H., Ng, N. L., Hildebrandt, L., Sorooshian, A., Szmigielski, R., Vermeylen, R., Maenhaut, W., Claeys, M., Flagan, R. C., and Seinfeld, J. H.: Chemical composition of secondary organic aerosol formed from the photooxidation of isoprene, J. Phys. Chem. A., 110, 9665–9690, 2006.
Surratt, J. D., Chan, A. W. H., Eddingsaas, N. C., Chan, M. N., Loza, C. L., Kwan, A. J., Hersey, S. P., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Reactive intermediates revealed in secondary organic aerosol formation from isoprene, P. Natl. Acad. Sci. USA, 107, 6640–6645, 2010.
Taipale, R., Ruuskanen, T. M., Rinne, J., Kajos, M. K., Hakola, H., Pohja, T., and Kulmala, M.: Technical Note: Quantitative long-term measurements of VOC concentrations by PTR-MS – measurement, calibration, and volume mixing ratio calculation methods, Atmos. Chem. Phys., 8, 6681–6698, https://doi.org/10.5194/acp-8-6681-2008, 2008.
Tan, Z., Lu, K., Hofzumahaus, A., Fuchs, H., Bohn, B., Holland, F., Liu, Y., Rohrer, F., Shao, M., Sun, K., Wu, Y., Zeng, L., Zhang, Y., Zou, Q., Kiendler-Scharr, A., Wahner, A., and Zhang, Y.: Experimental budgets of OH, HO2, and RO2 radicals and implications for ozone formation in the Pearl River Delta in China 2014, Atmos. Chem. Phys., 19, 7129–7150, https://doi.org/10.5194/acp-19-7129-2019, 2019.
Tan, Z., Hofzumahaus, A., Lu, K., Brown, S. S., Holland, F., Huey, L. G., Kiendler-Scharr, A., Li, X., Liu, X., and Ma, N.: No Evidence for a Significant Impact of Heterogeneous Chemistry on Radical Concentrations in the North China Plain in Summer 2014, Environ. Sci. Technol., 54, 5973–5979, https://doi.org/10.1021/acs.est.0c00525, 2020.
Tang, G., Li, X., Wang, Y., Xin, J., and Ren, X.: Surface ozone trend details and interpretations in Beijing, 2001–2006, Atmos. Chem. Phys., 9, 8813–8823, https://doi.org/10.5194/acp-9-8813-2009, 2009.
Wang, T., Xue, L., Brimblecombe, P., Lam, Y. F., Li, L., and Zhang, L.: Ozone pollution in China: A review of concentrations, meteorological influences, chemical precursors, and effects, Sci. Total. Environ., 575, 1582–1596, 2017.
Wang, Z., Li, Y., Chen, T., Zhang, D., Sun, F., Wei, Q., Dong, X., Sun, R., Huan, N., and Pan, L.: Ground-level ozone in urban Beijing over a 1-year period: Temporal variations and relationship to atmospheric oxidation, Atmos. Res., 164–165, 110–117, 2015.
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., St. Clair, J. M., Teng, A. P., Zhang, X., and Seinfeld J. H.: Gas-phase reactions of isoprene and its major oxidation products, Chem. Rev., 118, 3337–3390, 2018.
Whalley, L. and Slater, E.: APHH: Laser induced fluorescence (LIF) OH reactivity measurements made at the IAP-Beijing site during the winter and summer campaigns, Centre for Environmental Data Analysis, available at: https://catalogue.ceda.ac.uk/uuid/8ed84f3c770544c49329df9b068ab662 (last access: 19 January 2021), 2018.
Whalley, L. K., Edwards, P. M., Furneaux, K. L., Goddard, A., Ingham, T., Evans, M. J., Stone, D., Hopkins, J. R., Jones, C. E., Karunaharan, A., Lee, J. D., Lewis, A. C., Monks, P. S., Moller, S. J., and Heard, D. E.: Quantifying the magnitude of a missing hydroxyl radical source in a tropical rainforest, Atmos. Chem. Phys., 11, 7223–7233, https://doi.org/10.5194/acp-11-7223-2011, 2011.
Whalley, L. K., Stone, D., Bandy, B., Dunmore, R., Hamilton, J. F., Hopkins, J., Lee, J. D., Lewis, A. C., and Heard, D. E.: Atmospheric OH reactivity in central London: observations, model predictions and estimates of in situ ozone production, Atmos. Chem. Phys., 16, 2109–2122, https://doi.org/10.5194/acp-16-2109-2016, 2016.
Whalley, L. K., Stone, D., Dunmore, R., Hamilton, J., Hopkins, J. R., Lee, J. D., Lewis, A. C., Williams, P., Kleffmann, J., Laufs, S., Woodward-Massey, R., and Heard, D. E.: Understanding in situ ozone production in the summertime through radical observations and modelling studies during the Clean air for London project (ClearfLo), Atmos. Chem. Phys., 18, 2547–2571, https://doi.org/10.5194/acp-18-2547-2018, 2018.
Whalley, L. K., Slater, E. J., Woodward-Massey, R., Ye, C., Lee, J. D., Squires, F., Hopkins, J. R., Dunmore, R. E., Shaw, M., Hamilton, J. F., Lewis, A. C., Mehra, A., Worrall, S. D., Bacak, A., Bannan, T. J., Coe, H., Ouyang, B., Jones, R. L., Crilley, L. R., Kramer, L. J., Bloss, W. J., Vu, T., Kotthaus, S., Grimmond, S., Sun, Y., Xu, W., Yue, S., Ren, L., Acton, W. J. F., Hewitt, C. N., Wang, X., Fu, P., and Heard, D. E.: Evaluating the sensitivity of radical chemistry and ozone formation to ambient VOCs and NOx in Beijing, Atmos. Chem. Phys. Discuss. [preprint], https://doi.org/10.5194/acp-2020-785, in review, 2020.
Woodward-Massey, R.: Observations of radicals in the atmosphere: measurement validation and model comparisons, PhD thesis, University of Leeds, UK, 2018.
Xiong, F., McAvey, K. M., Pratt, K. A., Groff, C. J., Hostetler, M. A., Lipton, M. A., Starn, T. K., Seeley, J. V., Bertman, S. B., Teng, A. P., Crounse, J. D., Nguyen, T. B., Wennberg, P. O., Misztal, P. K., Goldstein, A. H., Guenther, A. B., Koss, A. R., Olson, K. F., de Gouw, J. A., Baumann, K., Edgerton, E. S., Feiner, P. A., Zhang, L., Miller, D. O., Brune, W. H., and Shepson, P. B.: Observation of isoprene hydroxynitrates in the southeastern United States and implications for the fate of NOx, Atmos. Chem. Phys., 15, 11257–11272, https://doi.org/10.5194/acp-15-11257-2015, 2015.
Zhang, Q., Zheng, Y. X., Tong, D., Shao, M., Wang, S. X., Zhang, Y. H., Xu, X. D., Wang, J. N., He, H., Liu, W. Q., Ding, Y. H., Lei, Y., Li, J. H., Wang, Z. F., Zhang, X. Y., Wang, Y. S., Cheng, J., Liu, Y., Shi, Q. R., Yan, L., Geng, G. N., Hong, C. P., Li, M., Liu, F., Zheng, B., Cao, J. J., Ding, A. J., Gao, J., Fu, Q. Y., Huo, J. T., Liu, B. X., Liu, Z. R., Yang, F. M., He, K. B., and Hao, J. M.: Drivers of improved PM2.5 air quality in China from 2013 to 2017, P. Natl. Acad. Sci. USA, 116, 24463–24469, 2019.
Zhou, W., Zhao, J., Ouyang, B., Mehra, A., Xu, W., Wang, Y., Bannan, T. J., Worrall, S. D., Priestley, M., Bacak, A., Chen, Q., Xie, C., Wang, Q., Wang, J., Du, W., Zhang, Y., Ge, X., Ye, P., Lee, J. D., Fu, P., Wang, Z., Worsnop, D., Jones, R., Percival, C. J., Coe, H., and Sun, Y.: Production of N2O5 and ClNO2 in summer in urban Beijing, China, Atmos. Chem. Phys., 18, 11581–11597, https://doi.org/10.5194/acp-18-11581-2018, 2018.