the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 26 Aug 2021
| 26 Aug 2021
Atmospheric photooxidation and ozonolysis of Δ3-carene and 3-caronaldehyde: rate constants and product yields
Luisa Hantschke
Anna Novelli
Birger Bohn
Changmin Cho
David Reimer
Franz Rohrer
Ralf Tillmann
Marvin Glowania
Andreas Hofzumahaus
Astrid Kiendler-Scharr
Andreas Wahner
The oxidation of Δ3-carene and one of its main oxidation products, caronaldehyde, by the OH radical and O3 was investigated in the atmospheric simulation chamber SAPHIR under atmospheric conditions for NOx mixing ratios below 2 ppbv. Within this study, the rate constants of the reaction of Δ3-carene with OH and O3 and of the reaction of caronaldehyde with OH were determined to be cm3 s−1 at 304 K, cm3 s−1 at 300 K and cm3 s−1 at 300 K, in agreement with previously published values. The yields of caronaldehyde from the reaction of OH and ozone with Δ3-carene were determined to be 0.30±0.05 and 0.06±0.02, respectively. Both values are in reasonably good agreement with reported literature values. An organic nitrate (RONO2) yield from the reaction of NO with RO2 derived from Δ3-carene of 0.25±0.04 was determined from the analysis of the reactive nitrogen species (NOy) in the SAPHIR chamber. The RONO2 yield of the reaction of NO with RO2 derived from the reaction of caronaldehyde with OH was found to be 0.10±0.02. The organic nitrate yields of Δ3-carene and caronaldehyde oxidation with OH are reported here for the first time in the gas phase. An OH yield of 0.65±0.10 was determined from the ozonolysis of Δ3-carene. Calculations of production and destruction rates of the sum of hydroxyl and peroxy radicals () demonstrated that there were no unaccounted production or loss processes of radicals in the oxidation of Δ3-carene for conditions of the chamber experiments. In an OH-free experiment with added OH scavenger, the photolysis frequency of caronaldehyde was obtained from its photolytical decay. The experimental photolysis frequency was a factor of 7 higher than the value calculated from the measured solar actinic flux density, an absorption cross section from the literature and an assumed effective quantum yield of unity for photodissociation.
- Article
(5272 KB) - Full-text XML
-
Supplement
(3658 KB) - BibTeX
- EndNote





On a global scale, the emission of carbon from biogenic volatile organic compounds (BVOCs) exceeds 1000 Tg per year (Guenther et al., 2012). Among all BVOC emissions, monoterpenes are the second most important class of species, contributing up to 16 % to the total emissions. As they are unsaturated and highly reactive, knowledge of their atmospheric chemistry is crucial to understand the formation of secondary pollutants such as ozone (O3) and particles (e.g. Seinfeld and Pandis, 2006; Zhang et al., 2018).
Of the total global annual monoterpene emissions, Δ3-carene contributes 4.5 %, making it the seventh most abundant monoterpene species (Geron et al., 2000). Δ3-Carene is primarily emitted by pine trees. Measured emission rates are up to (85±17) (nanograms monoterpenes per gram dry weight (dw) of needles and hour) from Scots pine trees (Komenda and Koppmann, 2002) and 57 from maritime pine. Therefore, Δ3-carene regionally gains in importance, for example, in boreal forests and the Mediterranean region. Hakola et al. (2012) measured the mixing ratios of monoterpenes over a boreal forest in Hyytiälä, Finland, and found Δ3-carene to be the second most abundant monoterpene after α-pinene. However, while the atmospheric chemistry of some monoterpenes such as α-pinene or β-pinene has been investigated in a number of experimental and theoretical studies (e.g. Peeters et al., 2001; Aschmann et al., 2002b; Eddingsaas et al., 2012; Rolletter et al., 2019, and references therein), only few studies exist investigating the photooxidation of Δ3-carene.
The similarity of its structure and type of products, e.g. caronaldehyde and pinonaldehyde that have been reported in literature (e.g. Arey et al., 1990; Hakola et al., 1994; Yu et al., 1999), suggest that Δ3-carene behaves similarly to α-pinene (Colville and Griffin, 2004) regarding its oxidation mechanism. The structural relations between Δ3-carene and α-pinene as well as their oxidation products caronaldehyde and pinonaldehyde are shown in Fig. 1. However, recent theoretical and experimental studies show that while the structural differences between the two molecules may be small, they may have an impact on the oxidation mechanism, e.g. due to the impact of unimolecular peroxy radical reactions (Draper et al., 2019; Moller et al., 2020).
In the atmosphere, monoterpenes mainly react with the hydroxyl radical (OH) and ozone (O3) during daytime and O3 and the nitrate radical (NO3) during nighttime. From the reaction of Δ3-carene with OH, organic peroxy radicals (RO2) are formed that can subsequently react with NO regenerating either eventually OH or forming organic nitrates (RONO2). Organic nitrates terminate the radical chain. They are typically less volatile and more hydrophilic than their parent organic compound, making them important for secondary organic aerosol (SOA) formation. Recent measurements suggest that 17 % to 23 % of organic aerosols contain molecules with nitrate functional groups (Rollins et al., 2013). RONO2 species serve also as NOx () reservoirs, hereby influencing, for example, O3 formation (Farmer et al., 2011).

Figure 1Structures of Δ3-carene and its oxidation product caronaldehyde, as well as α-pinene and its oxidation product pinonaldehyde.
Table 1Experimental conditions for all discussed oxidation experiments. Concentrations are given for the conditions in the SAPHIR chamber at the time of the first VOC injection; VOC concentrations refer to the first injection.

a ∼100 ppmv CO added as OH scavenger
In oxidation product studies, caronaldehyde was found to be the main daytime organic oxidation product from the oxidation of Δ3-carene (Arey et al., 1990; Reissell et al., 1999). The vapor pressure of this compound is lower than the vapor pressure of Δ3-carene, possibly making it a contributor to particle formation in the atmosphere (Yokouchi and Ambe, 1985; Hallquist et al., 1997).
In the present study, the following reactions of Δ3-carene were investigated in a series of experiments in the atmospheric simulation chamber SAPHIR:
The reaction rate constants of Reactions (R1)–(R3) and the photolysis frequency of Reaction (R4) are determined from the experimental decay of the respective VOC and compared to previously published values. Furthermore, the OH yield of Reaction (R2) and the caronaldehyde yield of Reactions (R1) and (R2) are determined under atmospheric conditions. The organic nitrate yields of the reactions of NO with RO2 radicals formed in Reactions (R1) and (R3) are obtained for the first time in the gas phase. Radical and trace gas measurements are used for analysis if unaccounted radical reactions are of importance in the oxidation mechanism of Δ3-carene.
2.1 Experiments in the SAPHIR chamber
SAPHIR is a double-walled atmospheric simulation chamber made of Teflon (FEP) film. It is of cylindrical shape (5 m diameter, 18 m length) with an inner volume of 270 m3. A shutter system allows for experiments to be conducted both in the dark (shutters closed) and the sunlit (shutters opened) chamber. With the shutters opened, the chamber is exposed to sunlight, therefore allowing for natural photooxidation conditions. The pressure of the chamber is kept 35 Pa higher than the outside pressure to avoid leakage from ambient air into the chamber. Ultrapure nitrogen and oxygen (Linde, purity 99.99990 %) are used to mix the synthetic air for the experiments. The space between the two FEP films is permanently flushed with ultrapure nitrogen. In order to compensate for the air lost to small leakages and sampling from the instruments, a flow of synthetic air is continuously introduced into the chamber leading to a dilution of trace gases of about 4 % per hour. OH radicals are produced mainly from nitrous acid (HONO) photolysis. HONO is formed on the chamber walls upon illumination of the chamber. The photolysis of HONO also leads to the formation of nitric oxide (NO), so the NOx concentration increases continuously over the course of an experiment. A more detailed description of the SAPHIR chamber and its features can be found elsewhere (e.g. Rohrer et al., 2005).
In total, six experiments were conducted to study the OH-induced photochemical degradation and ozonolysis of Δ3-carene as well as the photolysis and photooxidation of caronaldehyde under different conditions. Experimental conditions for each experiment are summarized in Table 1.
Two experiments, experiment E1 and experiment E2, were conducted to investigate the OH reaction of Δ3-carene with NO mixing ratios of less than 0.8 ppbv. Additionally, the ozonolysis of Δ3-carene was studied under dark conditions (chamber roof closed) in two experiments, one without (experiment E3) and one with (experiment E4) the addition of an OH scavenger. Two experiments focused on the photolysis (experiment E5) and reaction with OH (experiment E6) of caronaldehyde with NO mixing ratios below 2 ppbv.
Before each experiment, the chamber was flushed with synthetic air until the concentrations of trace gases were below the detection limit of the instruments. Experiments E3 and E4 as well as Experiments E5 and E6 were conducted on the same day, and flushing of the chamber was only done overnight prior to experiments E3 and E5, respectively. At the beginning of the experiments, the chamber air was humidified by boiling Milli-Q water and adding the water vapor together with a high flow of synthetic air into the chamber. Relative humidity (RH) for the photooxidation experiments was around 80 % at the beginning of the experiment (water vapor mixing ratio ∼2 %) but decreased during the day mostly due to a rise in temperature inside the chamber. RH values at the end of the experiment were typically 35 % (water vapor mixing ratio ∼0.7 %). For the ozonolysis experiment, relative humidity was 20 % at the beginning of the experiment. For some of the photooxidation experiments, ozone produced by a discharge ozonizer was injected into the chamber to reach mixing ratios of up to 60 ppbv to suppress NO in its reaction with O3. Up to 100 ppmv of CO was injected into the chamber as an OH scavenger for experiments E4 and E5 to study the ozonolysis of Δ3-carene and the photolysis of caronaldehyde under OH-free conditions.
During the first 2 h after opening the roof, the “zero-air” phase, no further reactive species were added, allowing for characterizing chamber sources of, for example, nitrous acid (HONO), formaldehyde (HCHO) and acetone, and to quantify the background OH reactivity. The sum of OH reactant concentrations ([X]) weighted by their reaction rate coefficient with OH (kOH+X) is called OH reactivity (kOH). OH reactivity observed in the SAPHIR chamber in the zero-air phase cannot be attributed to a specific species and is therefore seen as background reactivity. Background OH reactivity in the range of 0.5 s−1 is commonly observed in the SAPHIR chamber due to contaminants coming from the chamber wall upon humidification or irradiation. For the experiments in this study, a higher background reactivity of up to 5 s−1 was observed possibly due to a higher contamination of the chamber caused by previous experiments. Specific contaminants can remain in the chamber even after flushing the chamber with a high flow of ultrapure synthetic air, because they are adsorbed on the chamber's Teflon film. Upon humidification or illumination of the chamber, these contaminants can be desorbed resulting in a higher than usual background OH reactivity. After the injections of the VOCs, OH reactivity was dominated by their reaction with OH, and the background reactivity could be well described from measurements in the zero-air phase, so its influence is negligible for the analysis in this work, which concentrates on the times right after the injection of the VOC, when the total OH reactivity was dominated by the VOC.
Following the zero-air phase, Δ3-carene (Fluka, purity 99 %) was injected two to three times as a liquid into a heated volume. The vapor was transported into the SAPHIR chamber with the abovementioned replenishment flow of synthetic air. Δ3-Carene mixing ratios ranged from 3.1 to 6.0 ppbv in the chamber right after the injections.
Caronaldehyde was synthesized by ChiroBlock GmbH (Wolfen, Germany) as an enantiomeric mixture of (+)- and (−)-caronaldehyde with a purity of >95 % determined by 1H-NMR measurements. The liquid was dissolved in dichloromethane (DCM) for stability reasons. The concentration of caronaldehyde in DCM was 0.5 mol L−1. To exclude influences of the solvent on the chemistry observed in the experiments, additional test experiments were conducted in which only DCM was injected into the chamber. It was found that DCM did not interfere with the measurements and that DCM chemistry did not play a role for the analysis here. Caronaldehyde was injected as a liquid dissolved in DCM and evaporated as done for liquid Δ3-carene. Caronaldehyde mixing ratios in the chamber were 5 to 9 ppbv right after the injections. Potential wall loss of caronaldehyde was determined in a test experiment and was found to be negligible on the timescale of the experiments. Therefore, wall loss of caronaldehyde was not considered in the analysis of experiments.
2.2 Instrumentation
Δ3-Carene and caronaldehyde were detected by proton-transfer-reaction time-of-flight mass spectrometer instruments (VOCUS-PTR-MS, Aerodyne). Both instruments were calibrated for Δ3-carene. The measured concentrations were compared to the rise of the OH reactivity measurements when Δ3-carene was injected. It was found that the Δ3-carene concentration was underestimated by the VOCUS by a factor of 3 throughout all experiments. Therefore, the concentrations were scaled, so the measured rise of Δ3-carene by these mass spectrometric methods matched the increase in the measured OH reactivity at the time of the injections using a Δ3-carene + OH reaction rate constant of cm3 s−1. The uncertainty of this calibration correction is given by the uncertainty in the Δ3-carene + OH reaction rate constant and the uncertainty in the OH reactivity measurements that is 10 % (Fuchs et al., 2017a). Pinonaldehyde was used as a substituent to calibrate the instrument for caronaldehyde, as there is no calibration standard for caronaldehyde. The concentrations obtained from this calibration were in good agreement with the rise of the OH reactivity during caronaldehyde photooxidation experiments using a reaction rate constant of cm3 s−1.
Formaldehyde (HCHO) was measured by a Hantzsch monitor (AL4021, AeroLaser GmbH) and with a cavity ring-down instrument (G2307, Picarro). On average, the concentrations measured by both instruments agreed within 15 % (Glowania et al., 2021). HONO concentrations were measured by a custom-built long-path absorption photometer (LOPAP) (Kleffmann et al., 2002; Li et al., 2014). NO and NO2 were measured using a chemiluminescence instrument (Eco Physics) equipped with a blue-light photolytic converter for the conversion of NO2 to NO. CO and water vapor were measured with a cavity ring-down instrument (G2401, Picarro) and O3 with an UV absorption instrument (Ansyco). Total and diffuse spectral actinic flux densities measured by a spectral radiometer outside of the chamber were used to calculate photolysis frequencies (j) following Eq. (1):
with σ being the absorption cross section, ϕ the quantum yield and Fλ the mean spectral actinic flux density inside the chamber. Absorption cross sections and quantum yields were taken from recommendations in literature. The actinic flux spectra within the chamber were calculated in a model using the spectral radiometer measurements as input. As explained in more detail in Bohn et al. (2005) and Bohn and Zilken (2005), this model takes into account chamber-specific parameters such as the time-dependent effects of shadings of the chamber steel frame and the transmittance of the Teflon film. ROx radicals (OH, HO2, RO2) were measured by laser-induced fluorescence (LIF), in which OH is excited at 308 nm (Holland et al., 1995; Fuchs et al., 2011). HO2 and RO2 are chemically converted to OH so that the sum of radicals could be detected as OH in separate measurement cells (Fuchs et al., 2008, 2011). In the experiments in 2019, HO2 was additionally measured by bromide chemical ionization mass spectrometry (Br-CIMS) as described by Albrecht et al. (2019). The measurements usually agreed within 15 %. The detection sensitivity for RO2 from Δ3-carene was found to be in the range of the detection sensitivity for methyl peroxy radicals. For some experiments in this work, the fraction of RO2 formed from Δ3-carene (RO2,carene) is used and calculated as described in the following section. In all experiments, OH reactivity (kOH) was measured using a pump–probe instrument (Lou et al., 2010; Fuchs et al., 2017b). An overview of the instrumentation including their accuracies is given in the Supplement (Table S1).
2.3 OH reactivity and peroxy radical distribution
The OH reactivity measured in the SAPHIR chamber represents the sum of all species that react with OH. It can be separated into a fraction attributed to inorganic species (NO, CO, NO2) and formaldehyde (here named kOHinorg), as well as a fraction contributed to by VOC species (kOHVOC). This allows us to distinguish between reactions forming RO2 and those that do not. Equation (2) allows us to calculate the fraction contributed to by VOC species by subtracting kOHinorg from the total measured reactivity. k(OH+X) represents the reaction rate constant of the respective compound X with OH.
Included in kOHVOC is the reactivity from Δ3-carene but also from reaction products like oxygenated VOCs (OVOCs). kOHcarene, the fraction of OH reactivity from Δ3-carene, can be calculated using its OH reaction rate constant and measured concentrations (Eq. 3).
Since the RO2 measurement is the sum of all RO2 produced in the chamber, it can be assumed that the fraction of RO2 radicals produced by Δ3-carene to the total RO2 concentration is equal to the ratio of OH reactivity from the Δ3-carene + OH reaction to the total measured OH reactivity, assuming that every VOC + OH reaction leads to the formation of an RO2 radical and that all RO2 species have similar chemical lifetimes. The concentration of RO2 formed by Δ3-carene oxidation can therefore be estimated using Eq. (4).
2.4 Determination of reaction rate constants and OH yield from ozonolysis
Reaction rate constants for the reaction of OH with Δ3-carene as well as its oxidation product caronaldehyde and for the ozonolysis of Δ3-carene are determined by minimizing the root-mean-square error (RMSE) between measured VOC concentration time series and results from a simplified box model while the reaction rate constant is varied. The box model includes a minimum number of reactions required to describe the loss of the VOC. For the ozonoylsis and OH oxidation of Δ3-carene, the model consists of Reactions (R1), (R2) and the dilution of the trace gases from the chamber replenishment flow. The model for the reaction of caronaldehyde with OH consists of Reactions (R3) and (R4) and the dilution of trace gases. The model is constrained to measured oxidant concentrations (OH and O3), temperature, pressure and dilution rates. For the determination of the caronaldehyde + OH reaction rate constant, the model is additionally constrained to the measured photolysis frequency.
To determine the OH yield of the ozonolysis of Δ3-carene, the same method is applied, but the model is constrained to the determined reaction rate constants while the OH yield is varied. When ozonolysis experiments are conducted in the dark chamber, it is assumed that OH production only occurs through the ozonolysis of Δ3-carene and that there are no other photolytic sources (see Sect. 3.1). For this analysis, it is of no importance whether the correct absolute concentration of Δ3-carene or caronaldehyde is used in the box model, as the relative decay of modeled and measured Δ3-carene or caronaldehyde are compared.
2.5 Determination of product yields – organic nitrate RONO2
The yield of nitrates (RONO2) from the reaction of can be determined from the analysis of the concentrations of reactive nitrogen species in the chamber. NO, NO2 and HONO were directly measured in the experiments, and their sum is called for the analysis in this work (Eq. 5):
The source of all reactive nitrogen species in the experiment is the chamber source of HONO in the sunlit chamber. Its variable source strength Q(HONO) depends on temperature, relative humidity and solar ultraviolet radiation (Rohrer et al., 2005). HONO photolysis leads to the production of NO that is further oxidized to higher nitrogen oxides over the course of the experiment.
As HONO can be reformed by the reaction of NO with OH, a photostationary state between HONO, NO and OH is usually reached within several minutes. Therefore, measurements of NO, OH, jHONO and HONO can be used to calculate the source strength of HONO (Eq. 7).
For the experimental conditions in this work ( s−1) photostationarity is reached within 20 min. Chemical loss of in the chamber occurs due to the formation of RONO2 (R6, Table 2) and HNO3 (R2, Table 2). Additionally, species are lost due to replenishment flow that compensates for chamber leakage and gas sampling of analytical instruments. The difference between the time-integrated production and loss terms can then be used to determine the concentration of at a given time t.
where Ldil is the loss due to the replenishment flow, diluting the chamber air with the first-order rate coefficient s−1. Ldil is calculated by Eq. (9).
With respect to analysis of total nitrogen oxide concentration in the chamber, Eq. (8) assumes that HNO3 and RONO2 formation are effective sinks for NOx and that it does not play a role, if nitrates remain as HNO3 or RONO2 in the gas-phase or if they are, for example, deposited on the chamber wall as long as neither NO nor NO2 are reformed. Possible decomposition pathways could be, for example, due to photolysis, which would lead to a reformation of NO2. However, reported atmospheric lifetimes of RONO2 species are in the range of several days due to their small absorption cross sections (Roberts and Fajer, 1989), much longer than the duration of the chamber experiment. NO2 loss due to the formation of nitrate radicals (NO3) from the reaction of NO2 with ozone is also neglected in Eq. (6). For the experimental conditions in this work, the photolytic back-reactions, reforming NO2 from NO3, are fast enough such that the NO3 concentration in the sunlit chamber remains negligibly small. The formation of other oxidized nitrogen species such as acetyl peroxy nitrate (PAN) is also assumed to be negligible. The thermally unstable PAN species are formed from reaction of acyl peroxy radicals with NO2. For experiments in the SAPHIR chamber with comparable temperature conditions, the mixing ratios of PAN formed in the oxidation of acetaldehyde emitted by chamber sources are typically less than 100 pptv. Combining Eqs. (5) and (8), the amount of RONO2 formed can be calculated as follows.
Applying Eq. (10), the reaction yield of organic nitrates ( can be derived from the calculated Δ[RONO2] and measured RO2,carene concentrations.
To prove this concept of calculating organic nitrate yields, reference experiments with CH4 were performed. An upper limit of 0.001 was found for the nitrate yield. Within the uncertainty of the measurements, this value is in good agreement with literature (Scholtens et al., 1999; Butkovskaya et al., 2012). To exclude possible errors in the analysis for larger molecules for which nitrate formation is significant, such as unknown chamber sources of reactive nitrate species, a similar analysis was performed for an α-pinene experiment conducted in the SAPHIR chamber. For this experiment, an organic nitrate yield of (32±6) % was found, which is in reasonable agreement with the reported literature values. A detailed description of the reference experiments with CH4 and α-pinene will be given in a publication currently in preparation.
2.6 Determination of product yields
The experiments conducted in SAPHIR allow us to determine the product yields of caronaldehyde from the reaction of Δ3-carene with OH and O3. The product yield determination for caronaldehyde is done using measured caronaldehyde concentrations and relating them to the concentration of Δ3-carene consumed by OH or O3. The concentrations of Δ3-carene and caronaldehyde were measured by VOCUS or PTR-TOF-MS. A correction was applied to the Δ3-carene and caronaldehyde concentrations similar to corrections described by Galloway et al. (2011) and Kaminski et al. (2017). To derive the concentration of Δ3-carene that reacted with OH or O3, measured Δ3-carene concentrations were corrected for dilution in the chamber and the reaction with O3 or OH, respectively. The measured product concentrations of caronaldehyde were corrected for loss due to photolysis and dilution.
2.7 Determination of radical sources and sinks
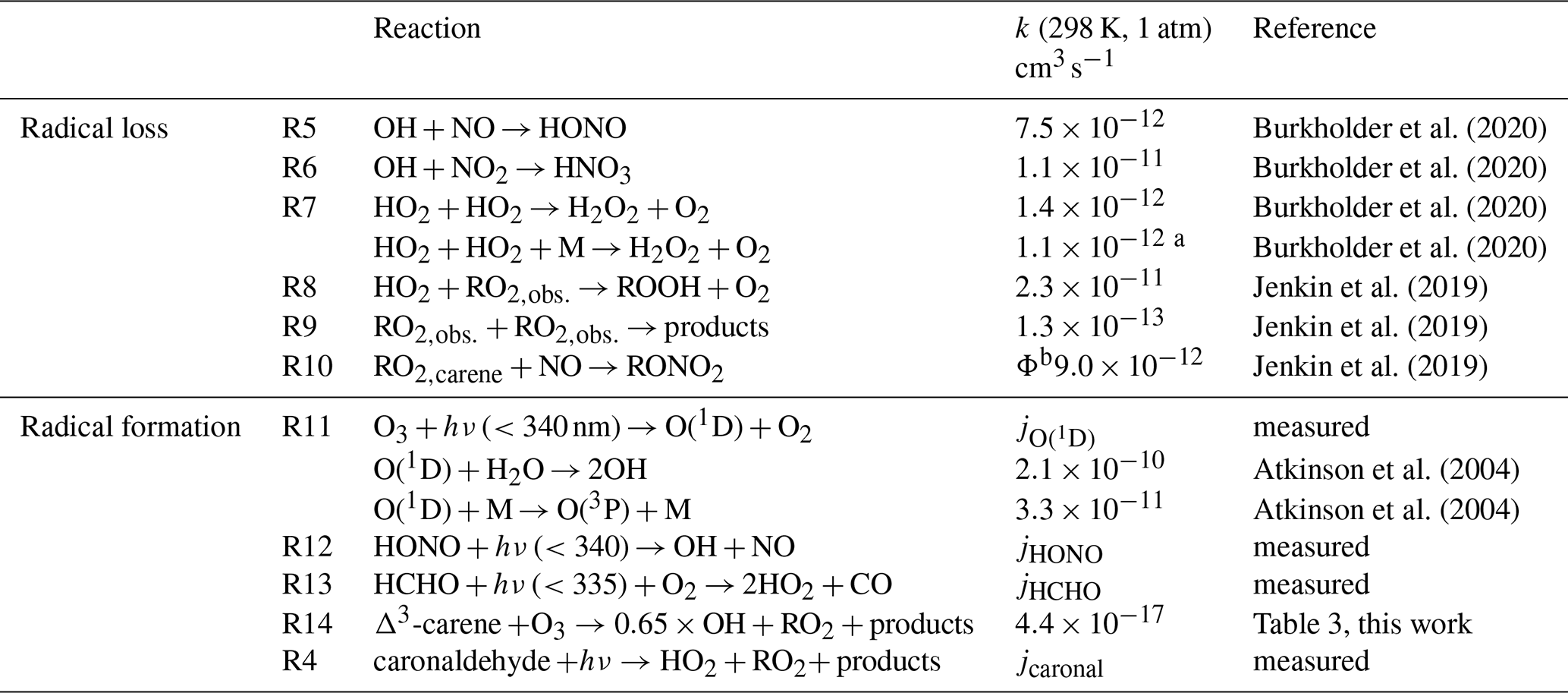
The experiments performed in the SAPHIR chamber that investigated the OH oxidation of Δ3-carene allow us to calculate production and destruction rates of the total ROx concentration. The atmospheric lifetimes of the ROx radicals range from only a few seconds for the OH radical to minutes for HO2 and RO2 radicals. Therefore, steady-state conditions of ROx concentrations can be assumed, so radical production and destruction rates are always balanced for the timescale of the chamber experiments. If there are imbalances between the calculated production and destruction rates, chemical reactions leading to the formation or destruction of radicals must be missing in the calculations. Table 2 gives an overview of the formation and loss reactions considered in the analysis including the respective reaction rate constants used. The reaction rate constants of the individual reactions were either taken from recent experimental studies, from measurements in this study or from calculations applying structure–activity relationship (SAR) as described in Jenkin et al. (2019).
The main ROx formation processes include the photolysis of ozone (Reaction R11), HONO (Reaction R12) and HCHO (Reaction R13) as well as the ozonolysis of Δ3-carene (Reaction R14). The formation of ROx radicals from the photolysis of caronaldehyde (R4) was also considered. The formation rate of ROx radicals P(ROx) can be calculated by Eq. (11).
ΦX indicates the yield of the respective radical X from the given reaction. Loss processes include the reactions of radicals with NOx that lead to the formation of HONO from the reaction of OH with NO (Reaction R5), nitric acid (HNO3) from the reaction of OH with NO2 (Reaction R6) or organic nitrates (RONO2) from the reaction of RO2,carene and NO (Reaction R10). Depending on the experimental conditions, radical loss through radical self-reactions becomes more important, leading to the formation of hydrogen peroxide (H2O2) from the reaction of two HO2 radicals (Reaction R7), the formation of peroxides (ROOH) from the reaction of HO2 with RO2 (Reaction R8) and the self-reaction of RO2 (Reaction R9). The loss rate of ROx radicals L(ROx) is calculated by Eq. (12).
Direct measurements of all relevant species allow us to calculate the total formation and loss rates for the ROx radicals. The error of the loss and production rates of the ROx radicals is determined by error propagation, taking uncertainties in the measurements and kinetic parameters into account.
Burkholder et al. (2020)Burkholder et al. (2020)Burkholder et al. (2020)Burkholder et al. (2020)Jenkin et al. (2019)Jenkin et al. (2019)Jenkin et al. (2019)Atkinson et al. (2004)Atkinson et al. (2004)Table 2Formation and loss reactions of the ROx radicals considered in the budget analysis (Fig. 11). Reaction rate constants are given for 298 K and 1 atm. The reaction rate constants in the actual analysis are calculated using temperature and pressure data measured during the experiments in SAPHIR.
a pressure dependent value given as in Burkholder et al. (2020); b the yield Φ of this reaction is determined in Sect. 3.2.3.
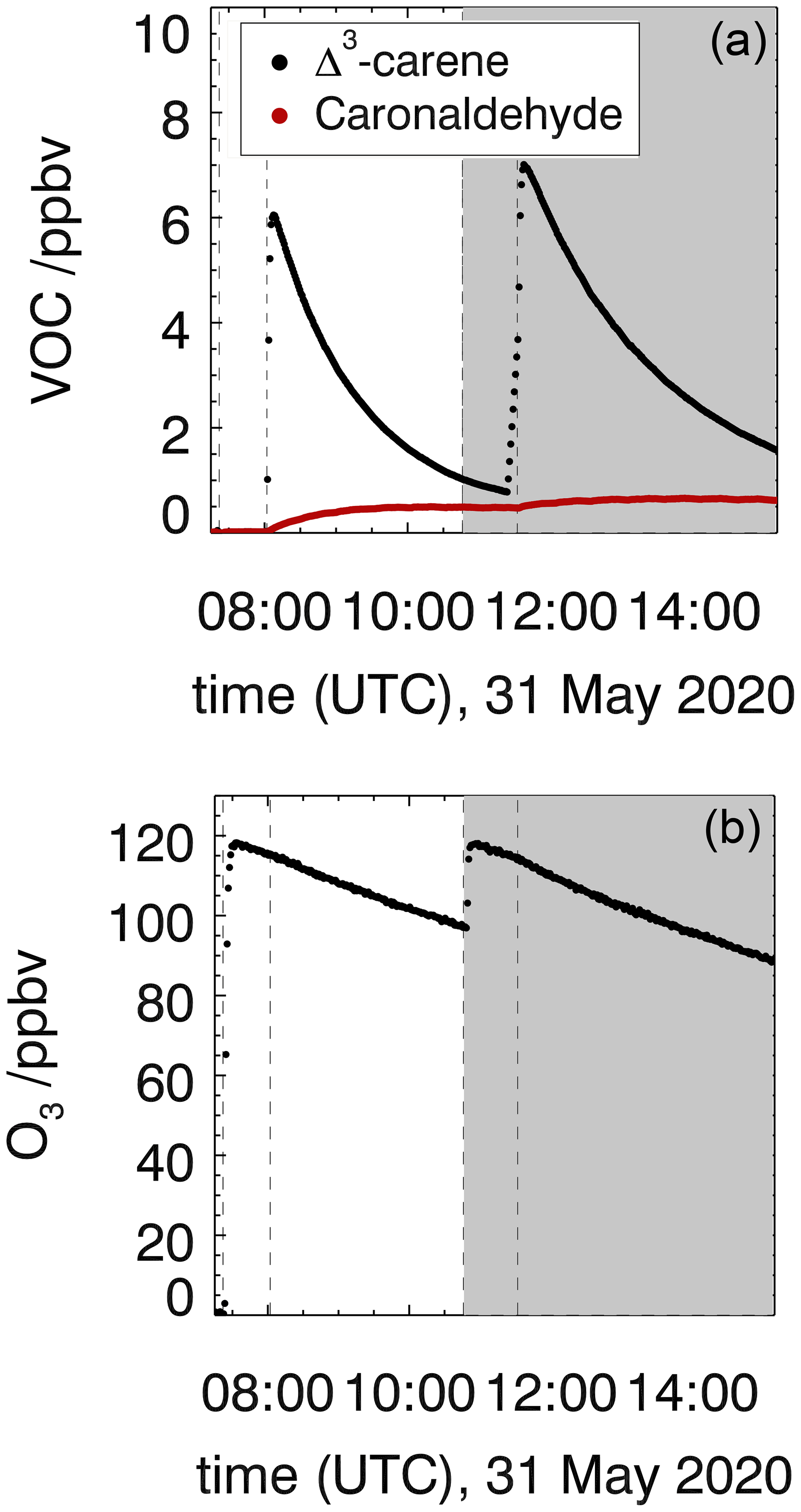
Figure 2Measured Δ3-carene, caronaldehyde (a) and O3 (b) mixing ratios in the experiment investigating the ozonolysis of Δ3-carene (experiment E3). The shaded area indicates the time where CO was injected as an OH scavenger (experiment E4). Vertical lines give the times when trace gases were injected.
Table 3Rate constants of the reaction of Δ3-carene with O3 and OH as well as of the reaction of caronaldehyde with OH. Experimental values obtained in this study are compared to reported literature values.

a temperature-dependent reaction rate coefficient given by Dillon et al. (2017): cm3 s−1;
b measured in a flow reactor;
c measured in a 7.3 m3 Teflon chamber;
d measured in a 90 m3 Teflon chamber;
e structure–activity relationship

Figure 3Measured and modeled Δ3-carene concentrations for the ozonolysis experiment E4. During the second part of the experiment (a), 90 ppmv of an OH scavenger (CO) was injected to suppress OH so that Δ3-carene reacted only with ozone. The modeled decay is fitted with a rate constant of cm3 s−1. During the first part of the experiment (b), when no CO was present, the measured decay of Δ3-carene is significantly faster than expected from ozonolysis alone. The measured time series of Δ3-carene can be best matched if an OH yield of 0.65±0.10 from its ozonolysis reaction is assumed. Red colored areas indicate the accuracy of the measured Δ3-carene concentrations.
Table 4OH yield of the reaction of Δ3-carene with O3. Experimental values obtained in this study are compared to reported literature values.

a with revisited yields of c-hexanone and c-hexanol by Berndt et al. (2003) as explained in the text

Figure 4Yield of caronaldehyde for Reactions (R1) (OH + Δ3-carene) and (R2) (O3+ Δ3-carene) determined from the slope of the relation between consumed Δ3-carene and measured caronaldehyde concentrations. For Reaction (R1), the result from experiments E1 and E2 are shown. Concentrations were corrected for dilution and chemical loss (see text for details). Caronaldehyde yields from the oxidation of Δ3-carene are determined as the slopes of the shown solid linear regression lines (black: OH oxidation R1, green: ozonolysis R2). Dashed lines indicate the error of the regression analysis.
Table 5Caronaldehyde yields for the reaction of Δ3-carene with OH and OH as well as caronaldehyde yields for the reaction of Δ3-carene with O3 obtained from this study compared to literature values. Given Δ3-carene concentrations are initial concentrations.
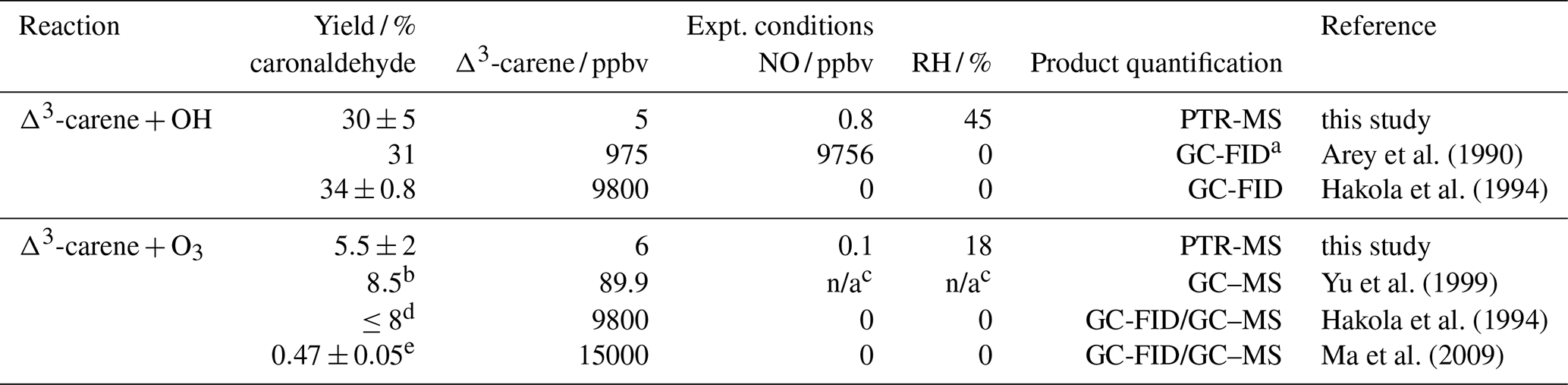
a GC-FID: gas chromatograph with flame ionization detector, GC–MS: gas chromatograph with mass spectrometer; b combined yield for gas and aerosol phase; c no values given in the publication; d yield obtained in the absence of an OH scavenger; e obtained from filter samples.
3.1 Ozonolysis of Δ3-carene
The ozonolysis of Δ3-carene was investigated in the dark SAPHIR chamber in two experiments in order to determine the rate constant (experiment E3) and OH yield (experiment E4) of the ozonolysis reaction, as well as the yield of caronaldehyde. Measured time series of O3, Δ3-carene and caronaldehyde are shown in Fig. 2. In total, 6.5 ppbv of Δ3-carene was consumed in experiment E3, and 7 ppbv was consumed in experiment E4. The roof of the chamber was closed for the whole duration of both experiments, to eliminate photolytical OH production. Since there was no OH scavenger present in experiment E3, the reaction system was also influenced by OH that is formed from ozonolysis (Sect. 3.1.2). OH concentrations were in the range of 1.0 to 2.0×106 cm−3. CO was injected into the chamber as an OH scavenger prior to the beginning of experiment E4.
3.1.1 Rate constant of the ozonolysis reaction of Δ3-carene
Optimization of the ozonolysis reaction rate constants as described in Sect. 2.4 results in a value of cm3 s−1. The stated error arises from the accuracy of the O3 and Δ3-carene measurements. The average temperature inside the SAPHIR chamber during the experiment was 300 K. The value reported in this study is slightly higher than values reported by Atkinson et al. (1990) and Chen et al. (2015) but still agrees within the stated errors (Table 3).
Reaction rate constants determined by Atkinson et al. (1990) and Chen et al. (2015) were and cm3 s−1, respectively. Both values were obtained from relative reaction rate measurements by comparing the rate of decay of Δ3-carene from ozonolysis to the rate of decay of a well-known reference compound (α-pinene in Atkinson et al., 1990, and cyclohexene in Chen et al., 2015). Additionally both studies also determined the reaction rate constant with an absolute rate technique using a pseudo-first-order approach. In Atkinson et al. (1990), reactive impurities in the used Δ3-carene sample were reported, whose presence would reduce the initially measured reaction rate constant from the absolute technique from to cm3 s−1. Chen et al. (2015) used three different set-ups to determine the reaction rate constant with an absolute rate technique: two simulation chambers with volumes of 90 m3 (HELIOS) and 7.3 m3 and a laminar flow reactor. The values determined in the simulation chambers were cm3 s−1 in the smaller simulation chamber and cm3 s−1 in HELIOS. A rate constant of cm3 s−1 was determined in the flow reactor. The 30 % discrepancy between the determined reaction rate constant is explained by interferences in the O3 measurements and increased uncertainties due to higher wall losses in the flow reactor. Witter et al. (2002) reported a faster reaction rate constant of cm3 s−1, obtained with a relative rate technique using 2-methyl-2-butene as reference compound. The value reported by Witter et al. (2002) is higher than the other reported values. Although the reason for this discrepancy is not clear, in their study Witter et al. (2002) also determined the ozonolysis reaction rate constants for other monoterpenes, e.g. limonene, α-terpinene and α-pinene, and the determined values were mostly on the upper end of reported literature values. The rate constants were on average 20 % higher than comparable values, which may point to a general overestimation of monoterpene reaction rate constants. The IUPAC Task Group on Atmospheric Chemical Kinetic Data Evaluation recommend to use a value of cm3 s−1 (Atkinson et al., 2004), which is an average of the relative rate constants determined by Atkinson et al. (1990) and Witter et al. (2002) and is in good agreement with the value determined in this work. Using the SAR published by Jenkin et al. (2020), a reaction rate constant of cm3 s−1 can be calculated. The value reported in this study is in relatively good agreement with this theory-derived value.
3.1.2 Determination of the OH yield from the ozonolysis of Δ3-carene
During experiment E3, when no OH scavenger was present, Δ3-carene was not only consumed by O3 but also by OH that is produced from the ozonolysis reaction. Because OH production from the ozonolysis was the only OH source, the OH yield from ozonolysis of Δ3-carene can be determined from this experiment. The chamber roof was closed for the whole experiment, and there are no photolytic sources for OH formation. The reaction rate constant determined in the previous section for the period, when an OH scavenger was present in the SAPHIR chamber, is used in the box model to determine the OH yield of the ozonolysis as explained in Sect. 2.4. The OH yield from ozonolysis is optimized until the measured decay of Δ3-carene matches the modeled decay (Fig. 3). A comparison of the experimental decay to model runs with different OH yields is shown in Fig. S1.
The OH yield is found to be 0.65±0.10 in the experiment in this work. The error is mainly due to the uncertainties in the reaction rate constants and the accuracies of the O3 measurement. Reported literature values for the OH yield from the ozonolysis of Δ3-carene determined in experiments are 1.1±1.0 (Atkinson et al., 1992) and 0.86±0.11 (Aschmann et al., 2002a). Wang et al. (2019) theoretically calculated the OH yield to be 0.56 to 0.59. Table 4 shows the value determined here compared with reported literature values. The value reported here is closer to the theoretical than to the other measured values, but it agrees with the yield reported by Aschmann et al. (2002a) within the reported uncertainties. As discussed in Wang et al. (2019), the high OH yield reported in Atkinson et al. (1992) could have been overestimated because of the high uncertainty in the determination of the OH yield. Atkinson et al. (1992) derived the OH yield by measuring the concentration of cyclohexanol and cyclohexanone produced in the reaction of OH and cyclohexane that was used as OH scavenger in their experiments. More recent investigations of the yield of cyclohexanol and cyclohexanone from the reaction of cyclohexane and OH indicate a larger yield of 0.88 (Berndt et al., 2003), as compared to the value of 0.5 used by Atkinson et al. (1992). This higher value would reduce the OH yield in the ozonolysis of Δ3-carene in the experiments by Atkinson et al. (1992) to 0.6, which would agree well with the OH yield determined in this work and the value calculated by Wang et al. (2019).
The ozonolysis of Δ3-carene is initiated by O3 attacking the C=C double bond, forming an energy-rich primary ozonide (POZ). Mostly, the energy retained in the POZ leads to a decomposition into Criegee intermediates, retaining one structure bearing a carbonyl functionality on one side of the molecule and the Criegee functionality on the other. These Criegee intermediates then isomerize to form dioxiranes, secondary ozonides (SOZs) or vinyl hydroperoxides. A theoretical study of the ozonolysis of Δ3-carene by Wang et al. (2019) found the formation yields dioxiranes, SOZ and vinyl hydroperoxides to be 0.16, 0.24 and 0.56, respectively. Collisional stabilization of the Criegee intermediates was found to be of minor importance with a yield of only 0.04 in the same study. OH (and an alkoxy radical) is formed from the vinyl hydroperoxide (ROOH) by breakage of the bond (Aschmann et al., 2002a; Ma et al., 2009). The fate of the alkoxy radical formed from this reaction has yet to be investigated, but it might result in the formation of highly oxidized molecules (HOMs) (Wang et al., 2019).
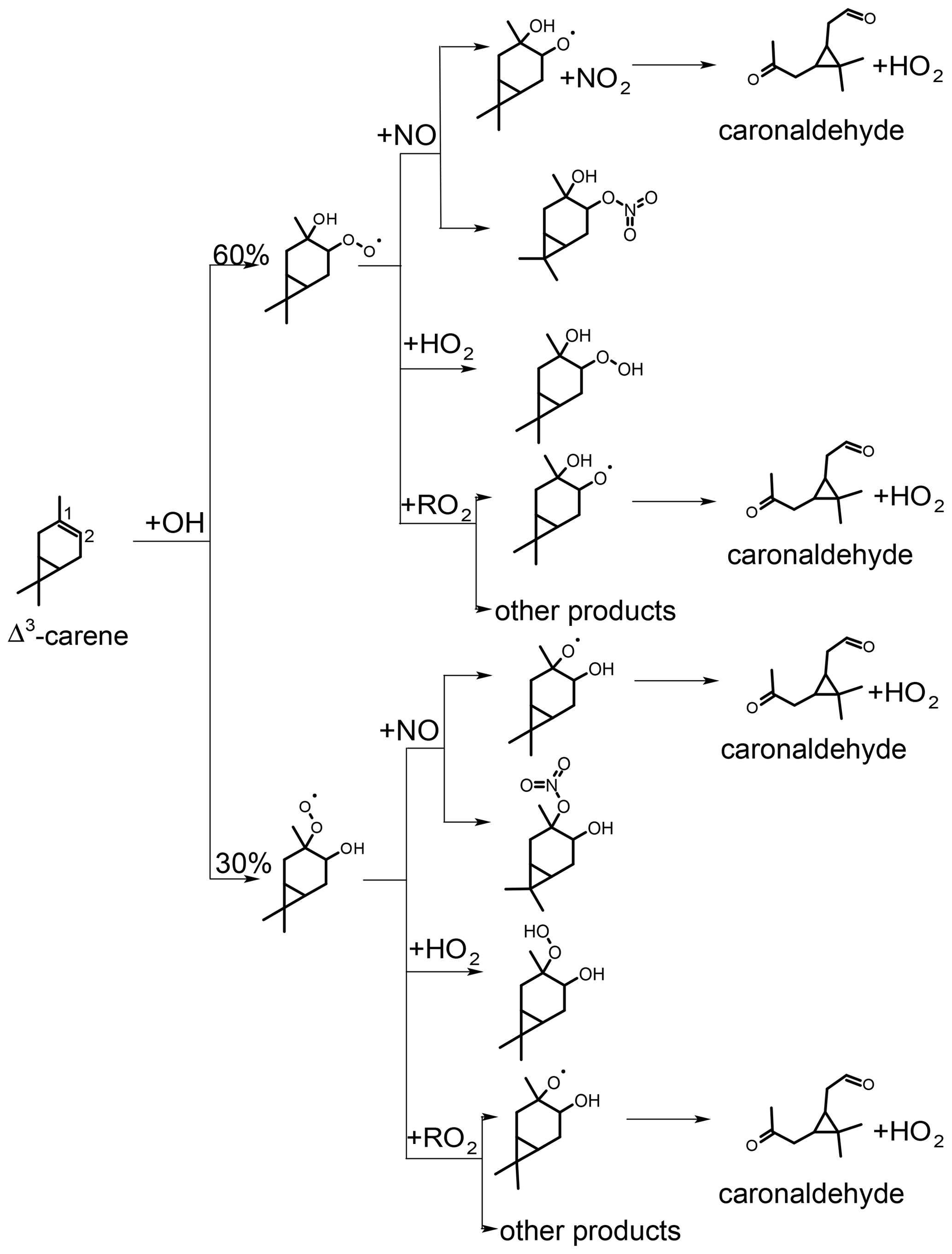
Figure 5Simplified scheme of the first reaction steps of the OH photooxidation of Δ3-carene (adapted from Colville and Griffin, 2004). Yields shown in black are from SAR by Jenkin et al. (2018). H abstraction has only little influence on the presented product yields. RO2+RO2 reactions could lead to other products than the formation of the alkoxy radical that produces caronaldehyde + HO2, so the yield of caronaldehyde from RO2+RO2 reactions is expected to be less than 1.
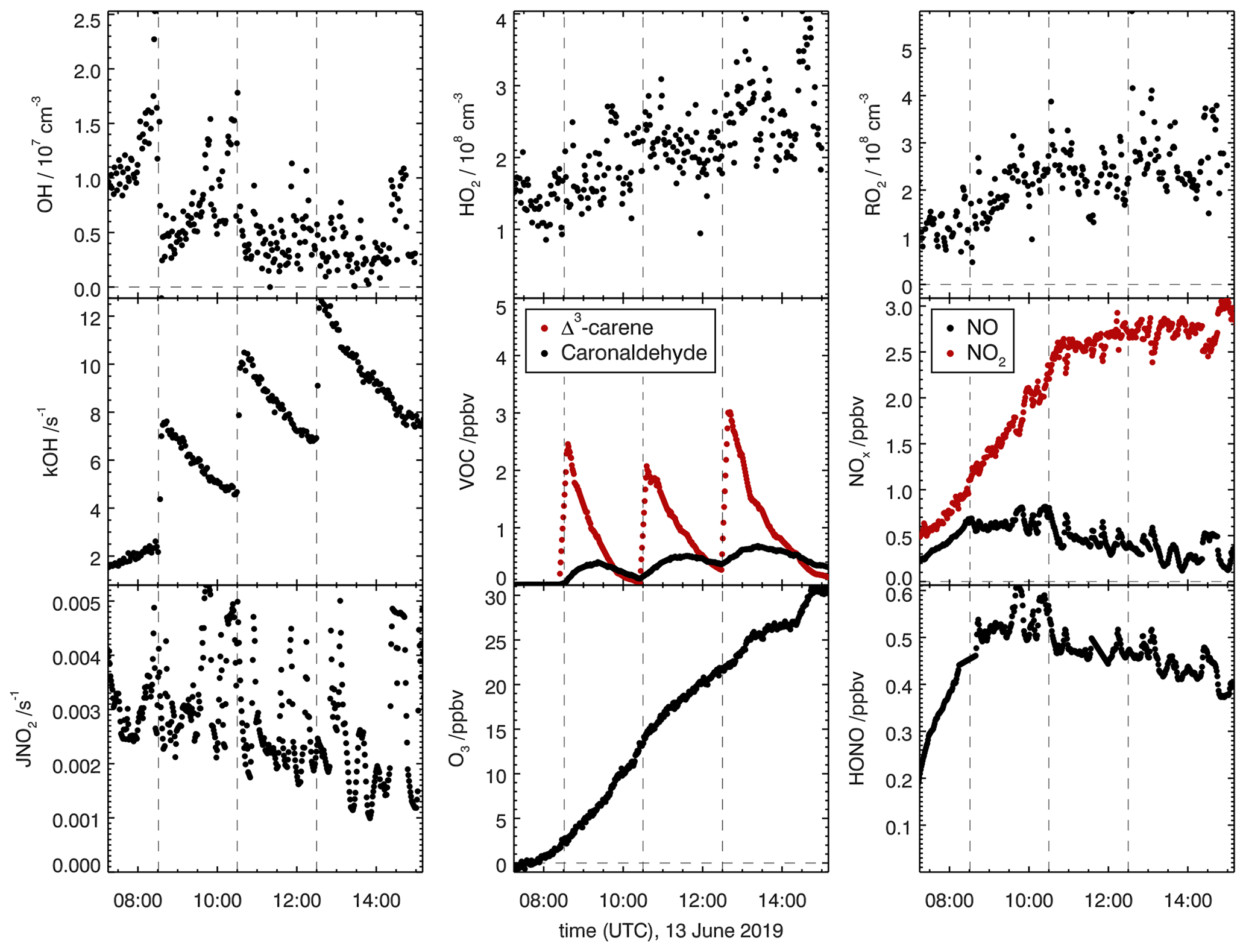
Figure 6Overview of measured concentrations for selected species in the SAPHIR chamber for the Δ3-carene + OH oxidation experiment E1. Dashed lines indicate times when Δ3-carene was injected into the chamber. Measured concentrations are shown in the illuminated chamber only starting with the opening of the roof (zero-air phase).
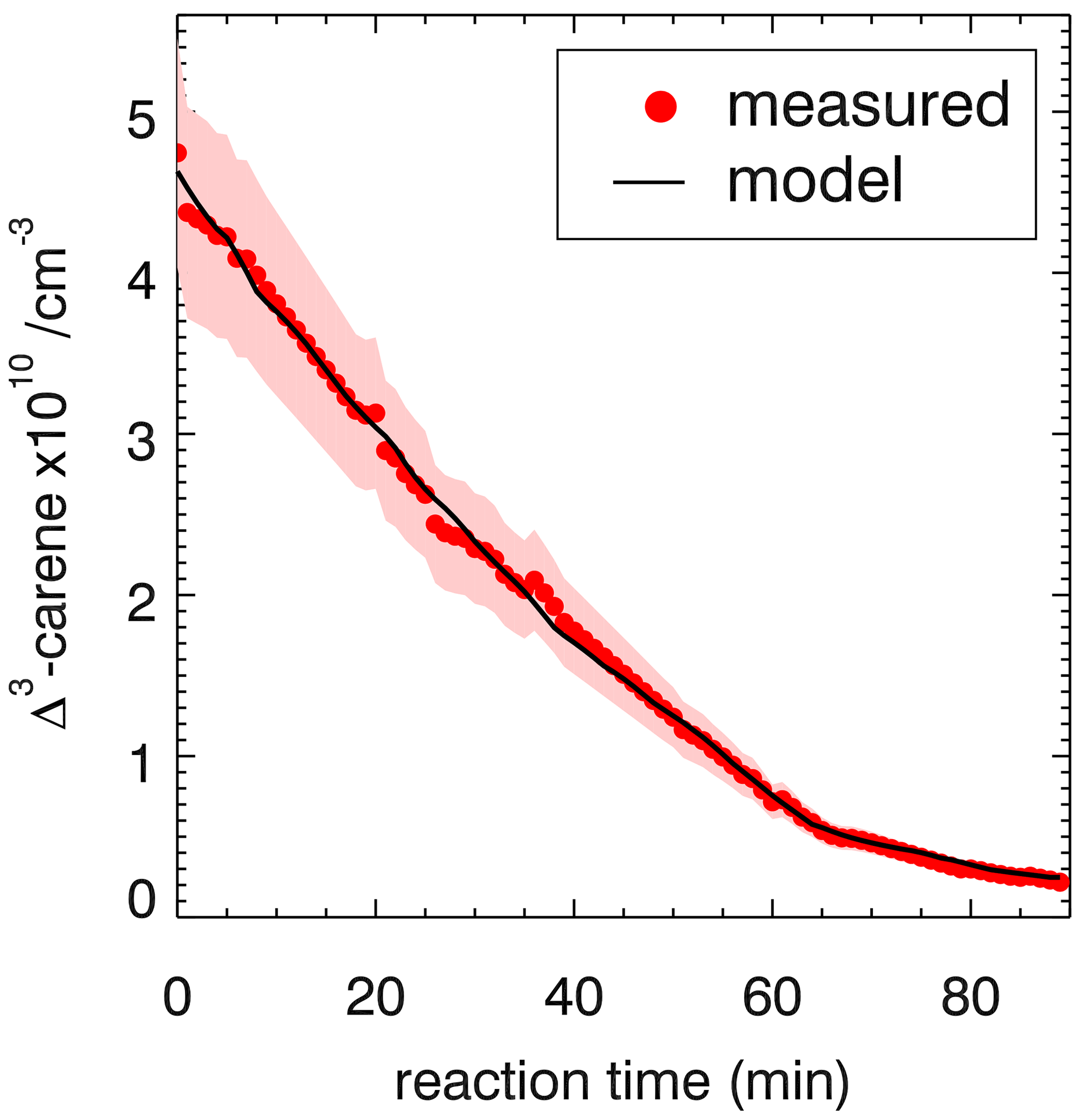
Figure 7Comparison of the modeled and measured Δ3-carene decay for the OH-oxidation experiment E2. Shown is the result of optimization of the rate constant of the OH reaction with Δ3-carene in the model calculations resulting in a reaction rate constant of cm3 s−1.
3.1.3 Caronaldehyde yield from ozonolysis
The caronaldehyde yield for the Δ3-carene + O3 reaction was determined from experiment E4, where an OH scavenger was injected into the chamber so that caronaldehyde is exclusively formed form the ozonolysis reaction. The corrections described in Sect. 2.5 were applied. Caronaldehyde was formed from the ozonolysis of Δ3-carene with a yield of (5.5±2) % as shown in Fig. 4. The uncertainty is derived from measurements and errors of the applied corrections (Sect. 2.6) for the respective experiment.
The determined caronaldehyde yields from this study and reported literature values are given in Table 5. The obtained value is in reasonably good agreement with most of the reported literature values. Yu et al. (1999) reported a caronaldehyde yield of 8 %, using 2-butanol to scavenge OH radicals. Hakola et al. (1994) determined the caronaldehyde yield to be ≤8 %. In the presence of cyclohexane as an OH scavenger, caronaldehyde was not detected in the ozonolysis of Δ3-carene in the experiments conducted by Hakola et al. (1994). Ma et al. (2009) reported a caronaldehyde yield of (0.47±0.05) % from filter samples, using cyclohexane, methanol or ethanol to scavenge OH radicals. However, due to its relatively high vapor pressure, it is likely that caronaldehyde was mainly present in the gas phase, so only a small fraction of the formed caronaldehyde was collected on the filters.
The formation of caronaldehyde from Δ3-carene ozonolysis most likely results from the stabilization and subsequent reaction with water of one of the Criegee intermediates; the formation mechanism of caronaldehyde in the absence of water has yet to be clarified (Ma et al., 2009). The stabilization of the Criegee intermediate is found to be a minor pathway by Wang et al. (2019), possibly explaining the small caronaldehyde yield found for the ozonolysis of Δ3-carene. Further reaction products that have not been measured in this study include a range of multifunctional organic acids according to studies conducted by, for example, Ma et al. (2009).
3.2 OH reaction of Δ3-carene
The first oxidation steps of the OH-induced photochemical oxidation of Δ3-carene relevant for this study are shown in Fig. 5 (Colville and Griffin, 2004). The OH oxidation is initiated by addition of OH to the C=C double bond or by the abstraction of an H atom rapidly followed by addition of O2. From the OH addition, two peroxy radical isomers are formed. The branching ratios of the specific attack on the C=C double bond (Fig. 5) and the H abstraction are estimated using the structure–activity relationship (SAR) method described in Jenkin et al. (2018). Branching ratios are 10 % for H abstraction, 30 % for OH addition to C1 and 60 % for OH addition to C2 of the double bond. Due to the small fraction of H abstraction, a relatively small influence of these reactions on the results of the following analysis can be expected.
Peroxy radicals (RO2) can react with NO, HO2 or undergo radical self-reactions with RO2. The reaction of Δ3-carene-derived RO2,carene with NO and possible reaction products are discussed in Sect. 3.2.2 and 3.2.3. RO2+RO2 and RO2+HO2 reactions are discussed in Sect. 3.2.4.
OH-induced photooxidation of Δ3-carene was investigated in the SAPHIR chamber during two experiments with NOx mixing ratios below 1 ppbv (Table 1). Figure 6 shows an overview of all measured species in experiment E1 that are representative for experiments at low NOx mixing ratios. Time series of concentration measurements for the other experiment are shown in the Supplement (Figs. S2 and S3). For all experiments, Δ3-carene was injected three times into the chamber, increasing the mixing ratio by approximately 5 ppbv each injection.
3.2.1 Rate constant of the OH + Δ3-carene reaction
In order to determine the rate constant of the reaction of Δ3-carene with OH, measured time series were compared to model results from a box model, in which OH and O3 concentrations were constrained to the measurements. A reaction rate constant was determined from all experiments. The determined value represents the mean value from all experiments. The given error is the standard error of the mean. The optimization error was much smaller than the variability of the results in different experiments. On average, the temperature inside the SAPHIR chamber was 304 K during the experiments. OH concentrations usually ranged from 5 to 8×106 cm−3. Ozonolysis only played a minor role in the experiments, contributing to a maximum of 5 % to the Δ3-carene consumption. The reaction rate constant for the ozonolysis reaction was taken from this work (Sect. 3.1.1).
The optimized rate constant of the OH reaction with Δ3-carene is cm3 s−1 as shown in Fig. 7.
Table 3 shows a comparison of reported literature values to the reaction rate constants obtained in this study. The determined value for the OH + Δ3-carene reaction rate constant from this study agrees well with both experimentally and theoretically derived literature values. Dillon et al. (2017) determined a temperature-dependent rate coefficient; for comparability with data in this work, only the rate coefficient determined at 304 K is used. Dillon et al. (2017) determined a reaction rate constant of cm3 s−1 using an absolute rate approach, and Atkinson et al. (1986) reported a value of cm3 s−1 at 294 K using a relative rate determination approach. From a site-specific structure–activity relationship (SAR), Peeters et al. (2007) predict the reaction rate constant to be cm3 s−1 at 298 K. In this reaction rate constant, the contribution of H abstraction is not considered.
3.2.2 Caronaldehyde yield from the reaction of Δ3-carene with OH
Table 5 compares the product yields of the Δ3-carene + OH reaction obtained in this study to reported literature values. The caronaldehyde yield of (30±5) % determined in this study as shown in Fig. 4 in good agreement with reported literature values within the range of the uncertainty of the measurements. The given error reflects the range of values determined in the considered experiments. Arey et al. (1990) reported a yield of 31 % and Hakola et al. (1994) a yield of (34±0.8) %. In both studies, caronaldehyde was quantified using GC-FID. Hakola et al. (1994) additionally used GC–MS and 1H NMR to verify the structure and purity of the measured compound.
As shown in Fig. 5, caronaldehyde is mainly formed from the decomposition of alkoxy radicals (RO). These alkoxy radicals are mainly formed from the reaction of RO2 with NO. A similar RO radical is also formed from the RO2+RO2 reaction, as well as from the photolysis of hydroperoxides (ROOH) that result from the RO2+HO2 reaction. Since the RO2+NO reaction mainly leads to the formation of RO (the branching ratio of an alternative pathway is discussed in Sect. 3.2.3), yields of caronaldehyde as found in this and previous studies can be expected. Other reaction products of the Δ3-carene + OH reaction determined in previous studies include formaldehyde with a yield of 20 % (Orlando et al., 2000) and acetone with a yield of 15 % (Reissell et al., 1999; Orlando et al., 2000). Caronic acid, hydroxy-caronic acid isomers and hydroxy-caronaldehyde isomers have additionally been found in the aerosol phase of smog chamber experiments investigating the Δ3-carene + OH reaction by Larsen et al. (2001).
3.2.3 Determination of alkyl nitrate yield for the reaction of Δ3-carene + OH
Organic nitrates are formed from the reaction of RO2 radicals with NO as shown in Fig. 5 for the reaction of the RO2 formed in the first oxidation step of Δ3-carene. An alternative pathway for the NO+RO2 reaction is the formation of an alkoxy radical and NO2 that ultimately leads to the formation of caronaldehyde (Sect. 3.2.2). The nitrate yield for the reaction of Δ3-carene + OH is determined following the procedure described in Sect. 2.4 using RO2,carene. Figure 8 shows the accumulation of reactive NOy species over the course of experiment E2. In total, 8.1 ppbv of reactive nitrogen species were formed over the course of the experiment. The contributions of NO, NO2 and HONO are almost constant with 2.2 ppbv, while the contributions of RONO2 and HNO3 increase continuously over the course of the experiments due to their continuous production from the reaction of RO2,carene with NO and OH with NO2. The formation of HNO3 and RONO2 before the injection of Δ3-carene is not relevant for the analysis and therefore not shown in Fig. 8.
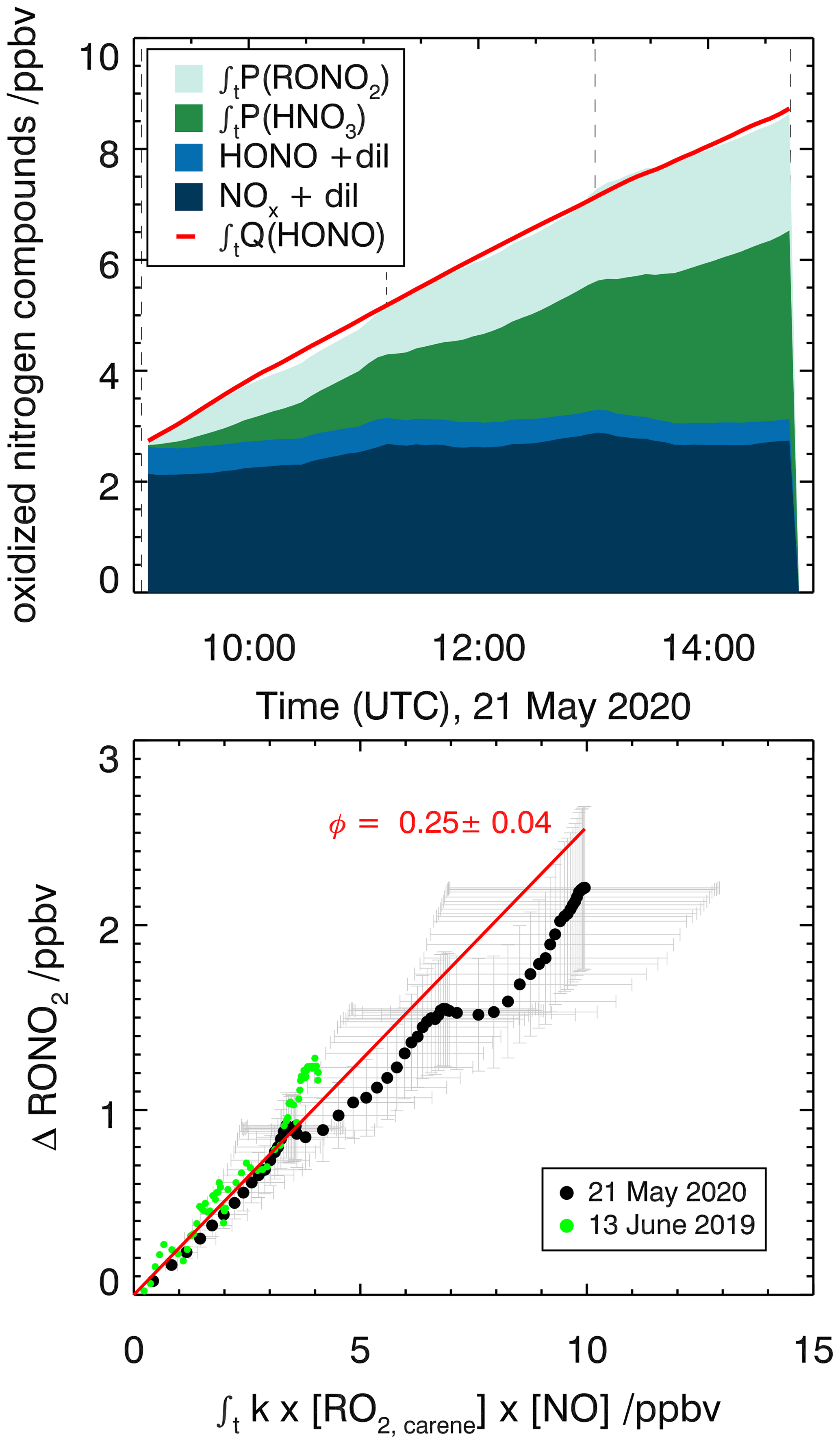
Figure 8Determination of the organic nitrate yield for the Δ3-carene + OH oxidation experiment E2. The red line shows the time-integrated HONO emission calculated as described in the text. NOx and HONO concentrations were measured, while the time-integrated NO2 loss by HNO3 formation and time-integrated NO loss by RONO2 formation are calculated as described in the text. The yield of (25±4) % for alkyl nitrates is optimized such that the total NOy produced over the course of both experiments, E1 and E2, is accounted for.
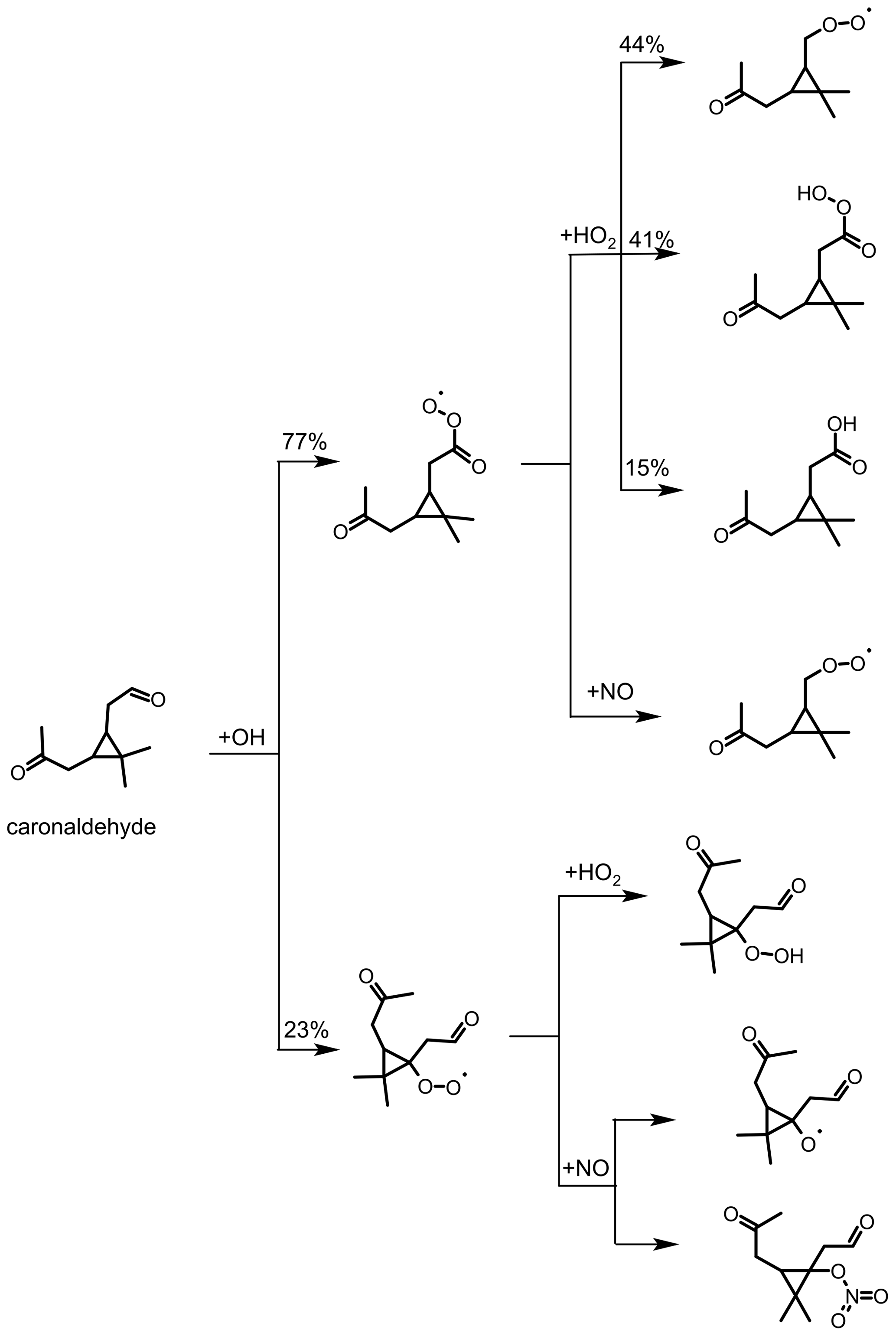
Figure 9Simplified scheme of the first reaction steps of the OH photooxidation of caronaldehyde, adapted from the oxidation mechanism of pinonaldehyde. Yields shown in black are from SAR by Jenkin et al. (2018). Additional reactions like, for example, the formation of carbonic acids from RO2 with HO2 are not shown in the scheme.
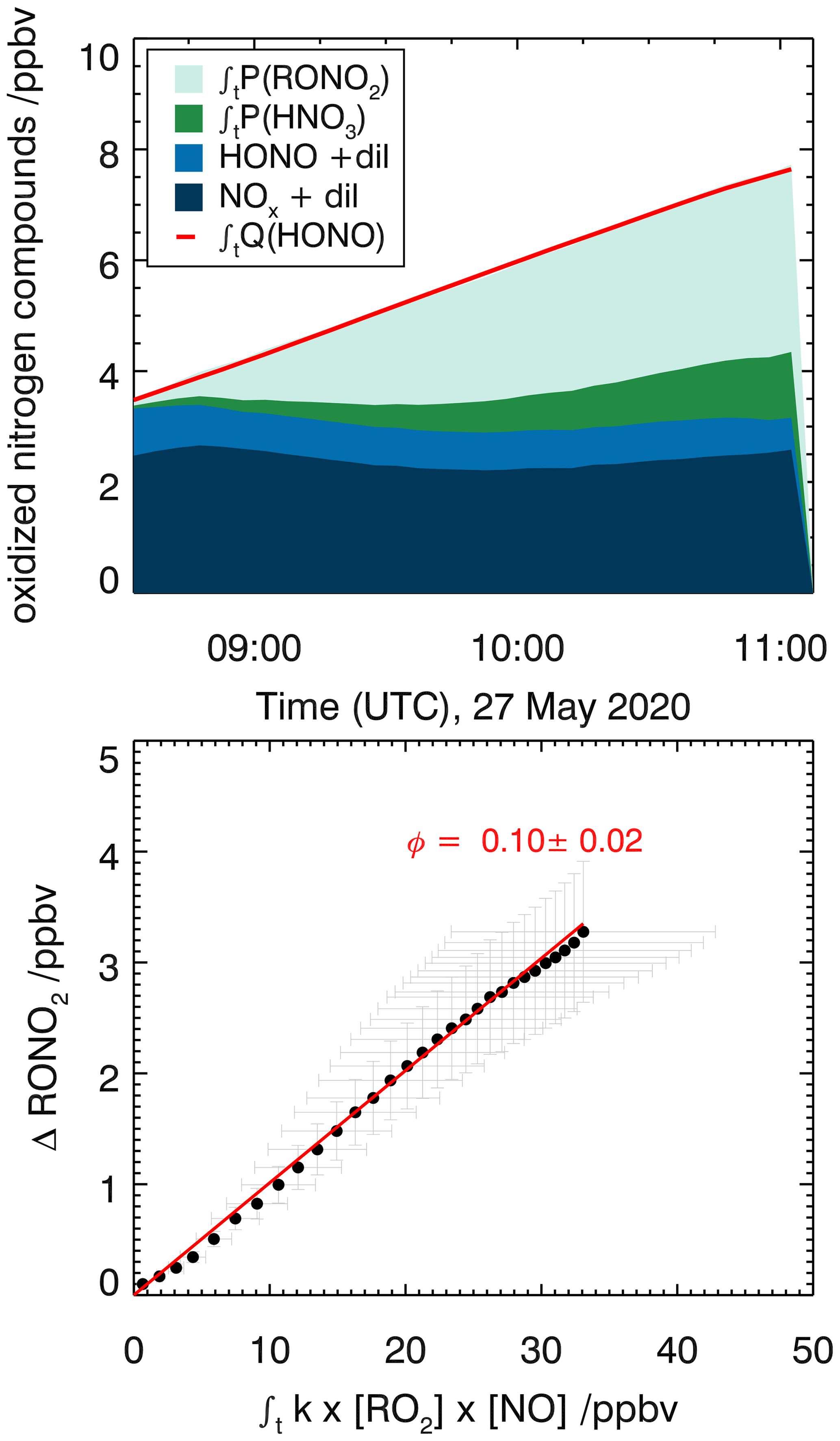
Figure 10Determination of the organic nitrate yield for the reaction of caronaldehyde with OH for experiment E6. HONO and NOx concentrations are measured, while RONO2 and HNO3 concentrations are calculated as described in the text.
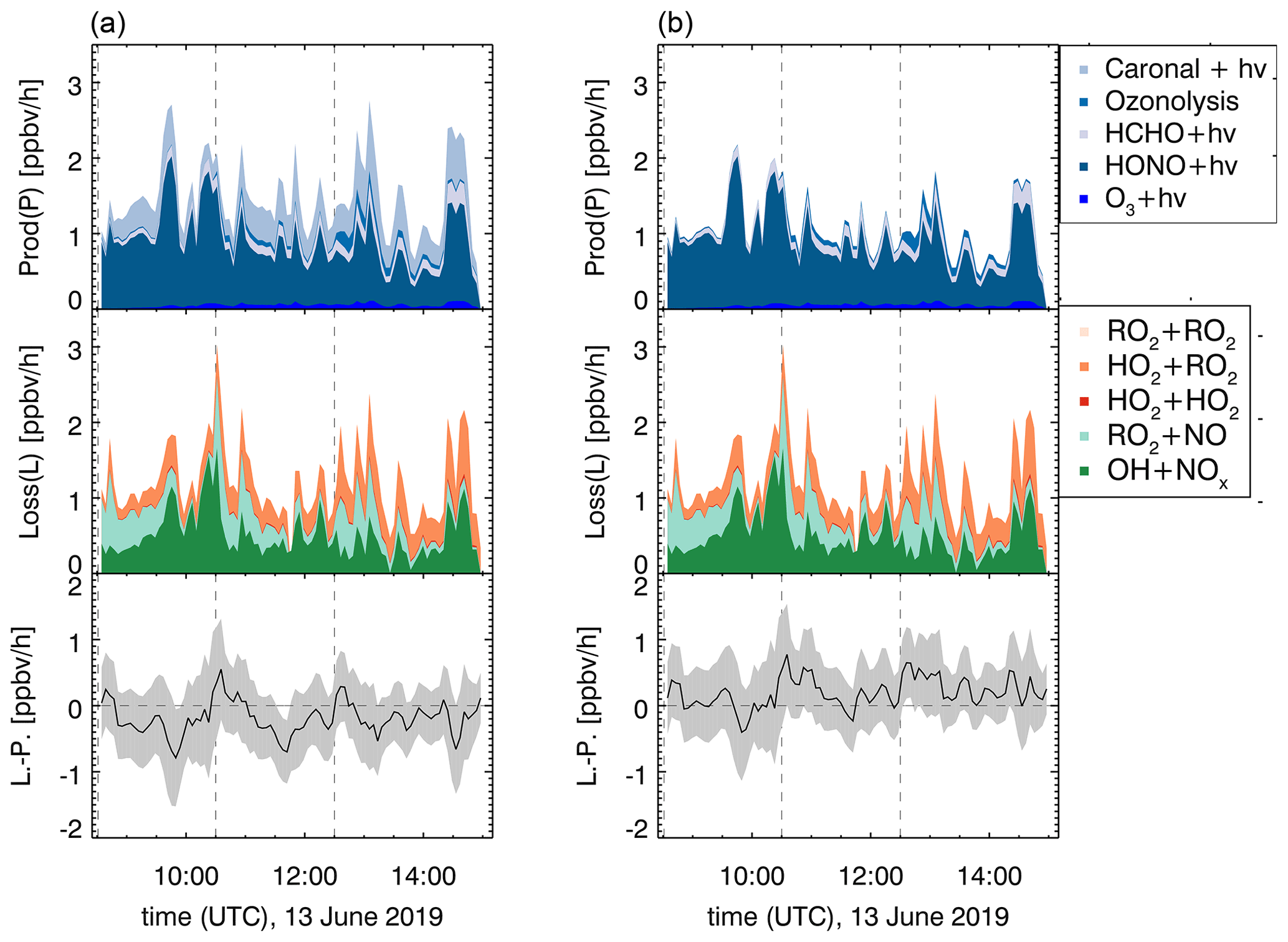
Figure 11ROx production and destruction rates for the OH-induced photooxidation in experiment E1, excluding (a) and including (b) the higher photolysis frequency determined in this work. Dashed lines indicate times when trace gases were injected.
An organic nitrate yield of (25±4) % was found from experiments E1 and E2. To our knowledge, only one other study investigated the organic nitrate yield of Δ3-carene. Based on RONO2 measurements in the aerosol phase using a TD-LIF instrument (thermal dissociation laser-induced fluorescence), Rollins et al. (2010) calculated an organic nitrate yield of 25 % for the photooxidation of Δ3-carene in experiments with high NOx conditions (100 ppbv of Δ3-carene and 500 ppbv of NO). This yield represents the fraction of SOA molecules that are hydroxynitrates, and Rollins et al. (2010) suggest that the organic nitrate fraction of SOA molecules produced in photooxidation are similar to the total yield of RONO2 from RO2+NO. The SAR by Jenkin et al. (2019) predicts an organic nitrate yield of 19 %. This calculation is based on the number of C atoms in the peroxy radical and therefore possibly has a relatively high uncertainty. The value obtained in this study for experiments with NOx mixing ratios below 1 ppbv is in good agreement with both the experimental and the SAR-derived values. Due to their structural similarities, it can be assumed that organic nitrate yields for Δ3-carene could be comparable to reported determined organic nitrate yields of α-pinene. Noziere et al. (1999) report an organic nitrate yield of (18±9) % and Rindelaub et al. (2015) (26±7) % for the reaction of α-pinene with OH. The organic nitrate yield of 1 % by Aschmann et al. (2002b) was not measured directly but approximated from API-MS measurements. This leads to a high uncertainty and likely explains the difference with the values by Noziere et al. (1999) and Rindelaub et al. (2015). The nitrate yield obtained in this study for Δ3-carene is in reasonable agreement with the values reported for α-pinene, as can be expected due to their structural similarities.
3.3 Photooxidation of caronaldehyde
A simplified reaction scheme for caronaldehyde degradation chemistry is shown in Fig. 9. Caronaldehyde can be oxidized by the reaction with OH, forming RO2 radicals through H abstraction and fast subsequent addition of O2. The formed peroxy radicals can then undergo reactions with NO, HO2 and RO2, resulting in similar product species such as explained before. Photolysis of caronaldehyde is an additional loss path. Due to their structural similarities, it can be assumed that caronaldehyde photolyzes in a similar way like pinonaldehyde. Photolysis leads to the C−C bond scission next to the aldehydic functional unit of the molecule, leading to the formation of HCO and an alkyl radical. Both species subsequently react with O2, forming CO, HO2 and a peroxy radical.
The reaction of caronaldehyde with OH was investigated in experiment E6. Caronaldehyde was injected twice into the chamber to reach mixing ratios of 5 ppbv directly after the first and 6 ppbv directly after the second injection. The OH concentration was 7×106 cm−3 for the period after the first injection. Prior to the second injection, 50 ppmv of CO was injected into the chamber as an OH scavenger to study the photolysis of caronaldehyde separately. The temperature in the chamber ranged from 295 to 305 K during the experiment.
3.3.1 Photolysis frequency of caronaldehyde
As CO was injected into the chamber as an OH scavenger and as wall loss was found to be negligible in the timeframe of the experiment, the decay of caronaldehyde measured can only be due to photolysis and dilution. The absorption spectrum of caronaldehyde was measured by Hallquist et al. (1997) in the range from 275 to 340 nm at 300 K. This spectrum and an assumed quantum yield for dissociation of 1 was used to calculate the photolysis frequency for the conditions of the experiment in the sunlit chamber using Eq. (1) (Sect. 2.2), resulting in a mean loss of caronaldehyde due to photolysis of s−1. The calculated photolysis frequency was included in a model to compare the measured to the modeled decay. The modeled decay was found to be too slow and requires a photolysis frequency that is larger by a factor of 7 to match the experimental decay. The absorption cross section by Hallquist et al. (1997) used in this work is the only reported measurement. It was measured in a 0.48 m3 borosilicate reactor at low pressure. During their experiments, dichloromethane and methanol solvents used in the synthesis of caronaldehyde were present in the reactor. Even though the authors corrected for possible interferences caused by these two species regarding the caronaldehyde concentration measured by IR spectroscopy, potential errors in the measurement of caronaldehyde concentrations might explain the discrepancy. Although there is no mechanistic explanation, it cannot be fully excluded that the faster decay observed in the experiments in this work compared to that by Hallquist et al. (1997) is the result of an alternative OH independent loss process occurring in the illuminated chamber. The absorption cross sections that have to be applied to explain the decay of caronaldehyde in photolysis experiments in SAPHIR suggest further investigation of this reaction and the absorption cross section of caronaldehyde.
3.3.2 Rate constant of the caronaldehyde + OH reaction
The measured decay of caronaldehyde was compared to the results from the box model constraining the photolysis frequency to the value with a correction by a factor of 7 calculated as explained above. The temperature inside the SAPHIR chamber was 300 K, and the OH concentration was 8×106 cm−3. The optimum OH reaction rate constant is cm3 s−1. The reaction rate constant was also optimized with the model constrained to the photolysis frequency as calculated with the absorption spectrum measured by Hallquist and a quantum yield of 1. This yields an OH reaction rate constant of cm3 s−1. Both values agree reasonably well with the reaction rate constant of cm3 s−1 measured by Alvarado et al. (1998) within the error. The structure–activity relationship by Jenkin et al. (2018) predicts a total reaction rate constant of cm3 s−1 which is consistent with the two measured values within the uncertainty of SAR. Hallquist et al. (1997) also determined the reaction rate constant of caronaldehyde with OH resulting in a value of cm3 s−1. This value is significantly higher than both values reported here and by Alvarado et al. (1998). Both Alvarado et al. (1998) and Hallquist et al. (1997) determined the reaction rate constant using a relative rate technique, and both studies were performed in smaller reaction volumes than the 270 m3 SAPHIR chamber. The measurements by Hallquist et al. (1997) were performed in a 0.153 m3 borosilicate glass reactor, while the studies by Alvarado et al. (1998) were performed in an 7.9 m3 Teflon chamber. Wall losses of caronaldehyde in the range of (4– s−1 were observed in the borosilicate chamber in the dark and further increased when the chamber was irradiated. Even though the determined reaction rate constant is corrected for the measured wall loss, it causes further uncertainty. Additionally, the initial caronaldehyde mixing ratio in the experiments in this work and the study performed by Alvarado et al. (1998) were significantly lower than those used by Hallquist et al. (1997) (20 ppbv, this study; 113 ppbv, Alvarado et al., 1998; 3252 ppbv, Hallquist et al., 1997). The reason for the high reaction rate constant reported by Hallquist et al. (1997) is not entirely clear, but it may arise from enhanced wall losses in the irradiated chamber unaccounted for in the experiments by Hallquist et al. (1997).
3.3.3 Determination of alkyl nitrate yields for the reaction of caronaldehyde + OH
The nitrate yield from the reaction of caronaldehyde with OH is determined for experiment E6. The organic nitrate yield is found to be (10±2) %, following the procedure described in Sect. 2.5. As shown in Fig. 10, 3.5 ppbv of RONO2 was formed in the chamber during experiment E6 along with 1.2 ppbv of HNO3. NOx and HONO concentrations were 2 and 1 ppbv, respectively. The error is derived from the accuracies of measured concentrations. To our knowledge, this is the first report of a measured organic nitrate yield for the caronaldehyde + OH reaction.
Generally, the nitrate yields from aldehydes are typically smaller than those of monoterpenes. Organic nitrate yields for other aldehydes used in current chemical models like the MCM3.3.1 are 5 % for pinonaldehyde, the main daytime oxidation product of α-pinene, and 12 % for pentanal. Calculation of the organic nitrate yield from the caronaldehyde oxidation according to Jenkin et al. (2019) is 5.5 %. The value determined in this study is within the range of nitrate yields reported in the literature but somewhat higher than values reported for similar aldehydes and calculations from SAR.
3.4 Production and destruction rates of ROx radicals in the oxidation of Δ3-carene
Field studies in forested environments, where Δ3-carene was one of the main monoterpenes emitted, found that both measured OH and HO2 concentrations cannot be reproduced by models (Kim et al., 2013; Hens et al., 2014). The analysis of radical formation and loss reactions for those environments revealed that a photolytic HO2 source missing in the model calculations is one possible explanation for the observed discrepancies. In the following, radical formation and loss rates of the sum of radicals (ROx) and their differences are determined according to Eqs. (6) and (7). The results of this analysis for experiment E1 with NOx mixing ratios below 1 ppbv are shown in Fig. 11.
The turnover rates in this experiment reach maximum values between 2 and 3 ppbv h−1. The main radical formation process is HONO photolysis, contributing more than 70 % to the average formation rate. Assuming that the photolysis frequency is higher by a factor of 7 (see Sect. 3.3.1), the formation of one HO2 and one RO2 radical per photolyzed caronaldehyde molecule would have a higher influence on the radical budgets of the Δ3-carene + OH reaction than previously assumed. This may therefore help to reduce the discrepancies between measured and modeled HO2 concentrations in field studies. The photolysis of caronaldehyde contributes on average 0.3 ppbv h−1; 60 % of the average loss rate is due to the reaction of ROx with NOx species. The remaining ROx losses can be explained by radical self-reactions, as can be expected for the experimental conditions because the reaction of the peroxy radical with HO2 (35 %) or RO2 radicals becomes competitive.
For experiments with NOx mixing ratios below 1 ppbv in the experiments in this work, the ROx budgets are closed within the uncertainty. It can therefore be assumed that there are no primary radical production or loss processes that are unaccounted for in this analysis. The contribution of the photolysis of caronaldehyde to the radical formation is in the range of the error of the analysis, and the loss and formation of radicals would also be closed if this process was not considered. In the scope of this work, it is therefore not possible to distinguish whether the caronaldehyde photolysis will contribute significantly to reduce the previously observed discrepancies between measured and modeled HO2 concentration in field studies. Possible reaction pathways leading to the formation and loss of radicals in the Δ3-carene oxidation are shown in Fig. 5. The reaction of RO2 with HO2 proceeds with a reaction rate constant of cm3 s−1 (Jenkin et al., 2019), mainly leading the formation of hydroperoxide species ROOH. The reaction of RO2 with RO2 results in the formation of either a diol or an alkoxy radical RO, which again most likely decomposes forming caronaldehyde. The reaction rate constant of this reaction is calculated to be cm3 s−1 (Jenkin et al., 2019). In the experiments presented here, this reaction only contributes <1 % to the loss of total ROx radicals. Isomerization reactions like intramolecular H shifts as investigated by Vereecken and Noziere (2020) can form a new pathway to product formation from RO2. For the RO2 described here, an intramolecular 1,5-H-shift from the OH functional group to the peroxy radical unit −OO would be the fastest of the possible isomerization reactions with a reaction rate constant of s−1 expected from the SAR in (Vereecken and Noziere, 2020). Compared to the other described RO2 loss rates, especially the loss in the reaction with NO (0.07 s−1 at 0.3 ppbv NO) and HO2 ( s−1 at 1.0×108 cm−3 HO2), this isomerization reaction is too slow to be significant for atmospheric conditions like in the experiments here.
The photooxidation of Δ3-carene was investigated for NOx mixing ratios below 1.5 ppbv in the atmospheric simulation chamber SAPHIR. Photooxidation experiments were performed under atmospheric conditions with Δ3-carene mixing ratios in the range of 5 to 7 ppbv. In this study, the gas-phase organic nitrate yield of the Δ3-carene + OH reaction was determined for the first time and found to be (25±4) %. The comparison of the obtained organic nitrate yield to yields obtained in the aerosol phase (Rollins et al., 2010) and to the organic nitrate yield of the structurally similar monoterpene α-pinene shows that the determined value is in accordance with the reported values. The reaction rate constant of the reaction of Δ3-carene with OH was determined to be cm3 s−1 using an absolute rate technique approach. The obtained value was found to be in good agreement with reaction rate constants reported in the literature within the stated error. Additionally, the ozonolysis of Δ3-carene was studied. A reaction rate constant of cm3 s−1 was found, with an OH yield of 0.65±0.10. The yield of the oxidation products caronaldehyde was determined for the Δ3-carene + OH and Δ3-carene + O3 reactions and found to be (0.33±0.03) and (0.055±0.02), respectively, in good agreement with reported literature values. The photolysis and OH-induced photooxidation of caronaldehyde were also studied. The photolysis frequency was calculated using the absorption spectrum measured by Hallquist et al. (1997), but it was found that to explain the observed caronaldehyde decay the absorption cross section would need to be higher by a factor of 7 assuming a maximum quantum yield of 1. The caronaldehyde + OH reaction rate constant was found to be cm3 s−1, in relatively good agreement with reported literature values. The experimental budget analysis of the loss and production processes of ROx radicals for the Δ3-carene + OH reaction shows that primary loss and production reactions are balanced within the uncertainty of the experiment of ±0.5 ppbv h−1. The formation of HO2 and RO2 radicals from the photolysis of caronaldehyde was considered an additional radical source with the photolysis frequency determined in this work. The contribution of the photolysis reaction to the radical formation was about 10 %. The fact that radical formation and loss reactions are well balanced indicates that there are no primary formation or loss processes unaccounted for in this analysis.
Data from the experiments in the SAPHIR chamber used in this work are available on the EUROCHAMP data home page. 13 June 2019: https://doi.org/10.25326/BQNH-P286 (Hantschke et al., 2021a), 21 May 2020: https://doi.org/10.25326/AGPD-MP70 (Hantschke et al., 2021b), 27 May 2020: https://doi.org/10.25326/8K59-JC53 (Hantschke et al., 2021c), 30 May 2020: https://doi.org/10.25326/PXFM-3967 (Hantschke et al., 2021d), 31 May 2020: https://doi.org/10.25326/QACC-KJ83 (Hantschke et al., 2021e).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-12665-2021-supplement.
LH and HF designed the experiments. AN and CC conducted the ROx radical measurements and the OH reactivity measurements. MG conducted the HONO measurements. RT, DR and SW were responsible for the PTR-TOF-MS and VOCUS measurements. BB conducted the radiation measurements. FR was responsible for the NOx and O3 data. LH analyzed the data and wrote the paper with the help of HF. All co-authors commented and discussed the article.
Astrid Kiendler-Scharr and Andreas Hofzumahaus are editors of ACP.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the special issue “Simulation chambers as tools in atmospheric research (AMT/ACP/GMD inter-journal SI)”. It is not associated with a conference.
This research has been supported by the European Research
Council, H2020 European Research Council (SARLEP (grant no. 681529)) and the
European Commission, Horizon 2020 Framework Programme (EUROCHAMP-2020 (grant
no. 730997)).
The article processing charges for this open-access publication were covered by the Forschungszentrum Jülich.
This paper was edited by Hang Su and reviewed by two anonymous referees.
Albrecht, S. R., Novelli, A., Hofzumahaus, A., Kang, S., Baker, Y., Mentel, T., Wahner, A., and Fuchs, H.: Measurements of hydroperoxy radicals (HO2) at atmospheric concentrations using bromide chemical ionisation mass spectrometry, Atmos. Meas. Tech., 12, 891–902, https://doi.org/10.5194/amt-12-891-2019, 2019. a
Alvarado, A., Arey, J., and Atkinson, R.: Kinetics of the gas-phase reactions of OH and NO3 radicals and O3 with the monoterpene reaction products pinonaldehyde, caronaldehyde, and sabinaketone, J. Atmos. Chem., 31, 281–297, https://doi.org/10.1023/a:1006010915971, 1998. a, b, c, d, e, f, g
Arey, J., Atkinson, R., and Aschmann, S. M.: Product study of the gas-phase reaction of monoterpenes with the OH radical in the presence of NOx, J. Geophys. Res.-Atmos., 95, 18539–18546, https://doi.org/10.1029/JD095iD11p18539, 1990. a, b, c, d
Aschmann, S. M., Arey, J., and Atkinson, R.: OH radical formation from the gas-phase reactions of O3 with a series of terpenes, Atmos. Environ., 36, 4347–4355, https://doi.org/10.1016/s1352-2310(02)00355-2, 2002a. a, b, c
Aschmann, S. M., Atkinson, R., and Arey, J.: Products of reaction of OH radicals with α-pinene, J. Geophys. Res.-Atmos., 107, 7, https://doi.org/10.1029/2001jd001098, 2002b. a, b, c
Atkinson, R., Aschmann, S. M., and Pitts, J. N.: Rate constants for the gas-phase reactions of the OH – radical with a series of monoterpenes (294±1) K, Int. J. Chem. Kinet., 18, 287–299, https://doi.org/10.1002/kin.550180303, 1986. a, b
Atkinson, R., Hasegawa, D., and Aschmann, S. M.: Rate constants for the gas-phase reactions of O3 with a series of monoterpenes and related compounds at 296±2 K, Int. J. Chem. Kinet., 22, 871–887, https://doi.org/10.1002/kin.550220807, 1990. a, b, c, d, e, f, g
Atkinson, R., Aschmann, S. M., Arey, J., and Shorees, B.: Formation of OH radicals in the gas-phase reaction of O3 with a series of terpenes, J. Geophys. Res.-Atmos., 97, 6065–6073, https://doi.org/10.1029/92jd00062, 1992. a, b, c, d, e, f, g
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I - gas phase reactions of Ox, HOx, NOx and SOx species, Atmos. Chem. Phys., 4, 1461–1738, https://doi.org/10.5194/acp-4-1461-2004, 2004. a, b, c
Berndt, T., Boge, O., and Stratmann, F.: Gas-phase ozonolysis of α-pinene: gaseous products and particle formation, Atmos. Environ., 37, 3933–3945, https://doi.org/10.1016/s1352-2310(03)00501-6, 2003. a, b
Bohn, B. and Zilken, H.: Model-aided radiometric determination of photolysis frequencies in a sunlit atmosphere simulation chamber, Atmos. Chem. Phys., 5, 191–206, https://doi.org/10.5194/acp-5-191-2005, 2005. a
Bohn, B., Rohrer, F., Brauers, T., and Wahner, A.: Actinometric measurements of NO2 photolysis frequencies in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 5, 493–503, https://doi.org/10.5194/acp-5-493-2005, 2005. a
Burkholder, J. B., Sander, S. P., Abbatt, J., Barker, J. R., Cappa, C., Crounse, J. D., Dibble, T. S., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Percival, C. J., Wilmouth, D. M., and Wine, P. H.: Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 19, JPL Publication 19-5, Jet Propulsion Laboratory, Pasadena, available at: http://jpldataeval.jpl.nasa.gov/ (last access: 16 April 2021), 2020. a, b, c, d, e
Butkovskaya, N., Kukui, A., and Le Bras, G.: Pressure and Temperature Dependence of Methyl Nitrate Formation in the CH3O2+NO Reaction, J. Phys. Chem. A, 116, 5972–5980, https://doi.org/10.1021/jp210710d, 2012. a
Chen, H., Ren, Y., Cazaunau, M., Daële, V., Hua, Y., Chena, J., and Mellouki, A.: Rate coefficients for the reaction of ozone with 2- and 3-carene, Chem. Phys. Lett., 621, 71–77, https://doi.org/10.1016/j.cplett.2014.12.056, 2015. a, b, c, d, e, f, g, h
Colville, C. J. and Griffin, R. J.: The roles of individual oxidants in secondary organic aerosol formation from Δ-3-carene: 1. gas-phase chemical mechanism, Atmos. Environ., 38, 4001–4012, https://doi.org/10.1016/j.atmosenv.2004.03.064, 2004. a, b, c
Dillon, T. J., Dulitz, K., Groß, C. B. M., and Crowley, J. N.: Temperature-dependent rate coefficients for the reactions of the hydroxyl radical with the atmospheric biogenics isoprene, alpha-pinene and delta-3-carene, Atmos. Chem. Phys., 17, 15137–15150, https://doi.org/10.5194/acp-17-15137-2017, 2017. a, b, c, d
Draper, D. C., Myllys, N., Hyttinen, N., Moller, K. H., Kjaergaard, H. G., Fry, J. L., Smith, J. N., and Kurten, T.: Formation of Highly Oxidized Molecules from NO3 Radical Initiated Oxidation of Δ-3-Carene: A Mechanistic Study, ACS Earth Space Chem., 3, 1460–1470, https://doi.org/10.1021/acsearthspacechem.9b00143, 2019. a
Eddingsaas, N. C., Loza, C. L., Yee, L. D., Seinfeld, J. H., and Wennberg, P. O.: α-pinene photooxidation under controlled chemical conditions – Part 1: Gas-phase composition in low- and high−NOx environments, Atmos. Chem. Phys., 12, 6489–6504, https://doi.org/10.5194/acp-12-6489-2012, 2012. a
Farmer, D. K., Perring, A. E., Wooldridge, P. J., Blake, D. R., Baker, A., Meinardi, S., Huey, L. G., Tanner, D., Vargas, O., and Cohen, R. C.: Impact of organic nitrates on urban ozone production, Atmos. Chem. Phys., 11, 4085–4094, https://doi.org/10.5194/acp-11-4085-2011, 2011. a
Fuchs, H., Holland, F., and Hofzumahaus, A.: Measurement of tropospheric RO2 and HO2 radicals by a laser-induced fluorescence instrument, Rev. Sci. Instrum., 79, 12, https://doi.org/10.1063/1.2968712, 2008. a
Fuchs, H., Bohn, B., Hofzumahaus, A., Holland, F., Lu, K. D., Nehr, S., Rohrer, F., and Wahner, A.: Detection of HO2 by laser-induced fluorescence: calibration and interferences from RO2 radicals, Atmos. Meas. Tech., 4, 1209–1225, https://doi.org/10.5194/amt-4-1209-2011, 2011. a, b
Fuchs, H., Novelli, A., Rolletter, M., Hofzumahaus, A., Pfannerstill, E. Y., Kessel, S., Edtbauer, A., Williams, J., Michoud, V., Dusanter, S., Locoge, N., Zannoni, N., Gros, V., Truong, F., Sarda-Esteve, R., Cryer, D. R., Brumby, C. A., Whalley, L. K., Stone, D., Seakins, P. W., Heard, D. E., Schoemaecker, C., Blocquet, M., Coudert, S., Batut, S., Fittschen, C., Thames, A. B., Brune, W. H., Ernest, C., Harder, H., Muller, J. B. A., Elste, T., Kubistin, D., Andres, S., Bohn, B., Hohaus, T., Holland, F., Li, X., Rohrer, F., Kiendler-Scharr, A., Tillmann, R., Wegener, R., Yu, Z., Zou, Q., and Wahner, A.: Comparison of OH reactivity measurements in the atmospheric simulation chamber SAPHIR, Atmos. Meas. Tech., 10, 4023–4053, https://doi.org/10.5194/amt-10-4023-2017, 2017a. a
Fuchs, H., Tan, Z., Lu, K., Bohn, B., Broch, S., Brown, S. S., Dong, H., Gomm, S., Häseler, R., He, L., Hofzumahaus, A., Holland, F., Li, X., Liu, Y., Lu, S., Min, K.-E., Rohrer, F., Shao, M., Wang, B., Wang, M., Wu, Y., Zeng, L., Zhang, Y., Wahner, A., and Zhang, Y.: OH reactivity at a rural site (Wangdu) in the North China Plain: contributions from OH reactants and experimental OH budget, Atmos. Chem. Phys., 17, 645–661, https://doi.org/10.5194/acp-17-645-2017, 2017b. a
Galloway, M. M., Huisman, A. J., Yee, L. D., Chan, A. W. H., Loza, C. L., Seinfeld, J. H., and Keutsch, F. N.: Yields of oxidized volatile organic compounds during the OH radical initiated oxidation of isoprene, methyl vinyl ketone, and methacrolein under high−NOx conditions, Atmos. Chem. Phys., 11, 10779–10790, https://doi.org/10.5194/acp-11-10779-2011, 2011. a
Geron, C., Rasmussen, R., Arnts, R. R., and Guenther, A.: A review and synthesis of monoterpene speciation from forests in the United States, Atmos. Environ., 34, 1761–1781, https://doi.org/10.1016/s1352-2310(99)00364-7, 2000. a
Glowania, M., Rohrer, F., Dorn, H.-P., Hofzumahaus, A., Holland, F., Kiendler-Scharr, A., Wahner, A., and Fuchs, H.: Comparison of formaldehyde measurements by Hantzsch, CRDS and DOAS in the SAPHIR chamber, Atmos. Meas. Tech., 14, 4239–4253, https://doi.org/10.5194/amt-14-4239-2021, 2021. a
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012. a
Hakola, H., Arey, J., Aschmann, S. M., and Atkinson, R.: Product formation from the gas-phase reactions of OH radicals and O3 with a series of monoterpenes, J. Atmos. Chem., 18, 75–102, https://doi.org/10.1007/bf00694375, 1994. a, b, c, d, e, f, g
Hakola, H., Hellén, H., Hemmilä, M., Rinne, J., and Kulmala, M.: In situ measurements of volatile organic compounds in a boreal forest, Atmos. Chem. Phys., 12, 11665–11678, https://doi.org/10.5194/acp-12-11665-2012, 2012. a
Hallquist, M., Wangberg, I., and Ljungstrom, E.: Atmospheric fate of carbonyl oxidation products originating from α-pinene and Δ-(3)-carene: Determination of rate of reaction with OH and NO3radicals, UV absorption cross sections, and vapor pressures, Environ. Sci. Technol., 31, 3166–3172, https://doi.org/10.1021/es970151a, 1997. a, b, c, d, e, f, g, h, i, j, k, l, m
Hantschke, L., Fuchs, H., Novelli, A., Bohn, B., Cho, C., Reimer, D., Rohrer, F., Tillmann, R., and Glowania, M.: Atmospheric simulation chamber study: delta-carene + OH – Gas-phase oxidation – product study, AERIS [data set], https://doi.org/10.25326/BQNH-P286, 2021a. a
Hantschke, L., Fuchs, H., Novelli, A., Bohn, B., Cho, C., Reimer, D., Rohrer, F., Tillmann, R., and Glowania, M.: Atmospheric simulation chamber study: delta-carene + OH – Gas-phase oxidation – product study, AERIS [data set], https://doi.org/10.25326/AGPD-MP70, 2021b. a
Hantschke, L., Fuchs, H., Novelli, A., Bohn, B., Cho, C., Reimer, D., Rohrer, F., Tillmann, R., and Glowania, M.: Atmospheric simulation chamber study: caronaldehyde + OH – Gas-phase oxidation – product study, AERIS [data set], https://doi.org/10.25326/8K59-JC53, 2021c. a
Hantschke, L., Fuchs, H., Novelli, A., Bohn, B., Cho, C., Reimer, D., Rohrer, F., Tillmann, R., and Glowania, M.: Atmospheric simulation chamber study: caronaldehyde + OH – Gas-phase oxidation – product study, AERIS [data set], https://doi.org/10.25326/PXFM-3967, 2021d. a
Hantschke, L., Fuchs, H., Novelli, A., Bohn, B., Cho, C., Reimer, D., Rohrer, F., Tillmann, R., and Glowania, M.: Atmospheric simulation chamber study: delta-carene + OH – Gas-phase oxidation – product study, AERIS [data set], https://doi.org/10.25326/QACC-KJ83, 2021e. a
Hens, K., Novelli, A., Martinez, M., Auld, J., Axinte, R., Bohn, B., Fischer, H., Keronen, P., Kubistin, D., Nölscher, A. C., Oswald, R., Paasonen, P., Petäjä, T., Regelin, E., Sander, R., Sinha, V., Sipilä, M., Taraborrelli, D., Tatum Ernest, C., Williams, J., Lelieveld, J., and Harder, H.: Observation and modelling of HOx radicals in a boreal forest, Atmos. Chem. Phys., 14, 8723–8747, https://doi.org/10.5194/acp-14-8723-2014, 2014. a
Holland, F., Hessling, M., and Hofzumahaus, A.: In-situ measurement of tropospheric OH radicals by laser-induced fluorescence – a description of the KFA instrument, J. Atmos. Sci., 52, 3393–3401, https://doi.org/10.1175/1520-0469(1995)052<3393:Ismoto>2.0.Co;2, 1995. a
Jenkin, M. E., Valorso, R., Aumont, B., Rickard, A. R., and Wallington, T. J.: Estimation of rate coefficients and branching ratios for gas-phase reactions of OH with aliphatic organic compounds for use in automated mechanism construction, Atmos. Chem. Phys., 18, 9297–9328, https://doi.org/10.5194/acp-18-9297-2018, 2018. a, b, c, d, e
Jenkin, M. E., Valorso, R., Aumont, B., and Rickard, A. R.: Estimation of rate coefficients and branching ratios for reactions of organic peroxy radicals for use in automated mechanism construction, Atmos. Chem. Phys., 19, 7691–7717, https://doi.org/10.5194/acp-19-7691-2019, 2019. a, b, c, d, e, f, g, h
Jenkin, M. E., Valorso, R., Aumont, B., Newland, M. J., and Rickard, A. R.: Estimation of rate coefficients for the reactions of O3 with unsaturated organic compounds for use in automated mechanism construction, Atmos. Chem. Phys., 20, 12921–12937, https://doi.org/10.5194/acp-20-12921-2020, 2020. a, b
Kaminski, M., Fuchs, H., Acir, I.-H., Bohn, B., Brauers, T., Dorn, H.-P., Häseler, R., Hofzumahaus, A., Li, X., Lutz, A., Nehr, S., Rohrer, F., Tillmann, R., Vereecken, L., Wegener, R., and Wahner, A.: Investigation of the β-pinene photooxidation by OH in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 17, 6631–6650, https://doi.org/10.5194/acp-17-6631-2017, 2017. a
Kim, S., Wolfe, G. M., Mauldin, L., Cantrell, C., Guenther, A., Karl, T., Turnipseed, A., Greenberg, J., Hall, S. R., Ullmann, K., Apel, E., Hornbrook, R., Kajii, Y., Nakashima, Y., Keutsch, F. N., DiGangi, J. P., Henry, S. B., Kaser, L., Schnitzhofer, R., Graus, M., Hansel, A., Zheng, W., and Flocke, F. F.: Evaluation of HOx sources and cycling using measurement-constrained model calculations in a 2-methyl-3-butene-2-ol (MBO) and monoterpene (MT) dominated ecosystem, Atmos. Chem. Phys., 13, 2031–2044, https://doi.org/10.5194/acp-13-2031-2013, 2013. a
Kleffmann, J., Heland, J., Kurtenbach, R., Lorzer, J., and Wiesen, P.: A new instrument (LOPAP) for the detection of nitrous acid (HONO), Environ. Sci. Pollut. R., 4, 48–54, 2002. a
Komenda, M. and Koppmann, R.: Monoterpene emissions from Scots pine (Pinus sylvestris): Field studies of emission rate variabilities, J. Geophys. Res.-Atmos., 107, 13, https://doi.org/10.1029/2001jd000691, 2002. a
Larsen, B. R., Di Bella, D., Glasius, M., Winterhalter, R., Jensen, N. R., and Hjorth, J.: Gas-phase OH oxidation of monoterpenes: Gaseous and particulate products, J. Atmos. Chem., 38, 231–276, https://doi.org/10.1023/a:1006487530903, 2001. a
Li, X., Rohrer, F., Hofzumahaus, A., Brauers, T., Haseler, R., Bohn, B., Broch, S., Fuchs, H., Gomm, S., Holland, F., Jager, J., Kaiser, J., Keutsch, F. N., Lohse, I., Lu, K. D., Tillmann, R., Wegener, R., Wolfe, G. M., Mentel, T. F., Kiendler-Scharr, A., and Wahner, A.: Missing Gas-Phase Source of HONO Inferred from Zeppelin Measurements in the Troposphere, Science, 344, 292–296, https://doi.org/10.1126/science.1248999, 2014. a
Lou, S., Holland, F., Rohrer, F., Lu, K., Bohn, B., Brauers, T., Chang, C. C., Fuchs, H., Häseler, R., Kita, K., Kondo, Y., Li, X., Shao, M., Zeng, L., Wahner, A., Zhang, Y., Wang, W., and Hofzumahaus, A.: Atmospheric OH reactivities in the Pearl River Delta – China in summer 2006: measurement and model results, Atmos. Chem. Phys., 10, 11243–11260, https://doi.org/10.5194/acp-10-11243-2010, 2010. a
Ma, Y., Porter, R. A., Chappell, D., Russell, A. T., and Marston, G.: Mechanisms for the formation of organic acids in the gas-phase ozonolysis of 3-carene, Phys. Chem. Chem. Phys., 11, 4184–4197, 2009. a, b, c, d, e
Moller, K. H., Otkjaer, R. V., Chen, J., and Kjaergaard, H. G.: Double Bonds Are Key to Fast Unimolecular Reactivity in First-Generation Monoterpene Hydroxy Peroxy Radicals, J. Phys. Chem. A, 124, 2885–2896, https://doi.org/10.1021/acs.jpca.0c01079, 2020. a
Noziere, B., Barnes, I., and Becker, K. H.: Product study and mechanisms of the reactions of α-pinene and of pinonaldehyde with OH radicals, J. Geophys. Res.-Atmos., 104, 23645–23656, https://doi.org/10.1029/1999jd900778, 1999. a, b
Orlando, J. J., Noziere, B., Tyndall, G. S., Orzechowska, G. E., Paulson, S. E., and Rudich, Y.: Product studies of the OH- and ozone-initiated oxidation of some monoterpenes, J. Geophys. Res.-Atmos., 105, 11561–11572, https://doi.org/10.1029/2000jd900005, 2000. a, b
Peeters, J., Vereecken, L., and Fantechi, G.: The detailed mechanism of the OH-initiated atmospheric oxidation of α-pinene: a theoretical study, Phys. Chem. Chem. Phys., 3, 5489–5504, https://doi.org/10.1039/b106555f, 2001. a
Peeters, J., Boullart, W., Pultau, V., Vandenberk, S., and Vereecken, L.: Structure-activity relationship for the addition of OH to (poly)alkenes: Site-specific and total rate constants, J. Phys. Chem. A, 111, 1618–1631, https://doi.org/10.1021/jp066973o, 2007. a, b
Reissell, A., Harry, C., Aschmann, S. M., Atkinson, R., and Arey, J.: Formation of acetone from the OH radical- and O3-initiated reactions of a series of monoterpenes, J. Geophys. Res.-Atmos., 104, 13869–13879, https://doi.org/10.1029/1999jd900198, 1999. a, b
Rindelaub, J. D., McAvey, K. M., and Shepson, P. B.: The photochemical production of organic nitrates from α-pinene and loss via acid-dependent particle phase hydrolysis, Atmos. Environ., 100, 193–201, https://doi.org/10.1016/j.atmosenv.2014.11.010, 2015. a, b
Roberts, J. M. and Fajer, R. W.: UV absorption cross sections of organic nitrates of potential atmospheric importance and estimation of atmospheric lifetimes, Environ. Sci. Technol., 23, 945–951, https://doi.org/10.1021/es00066a003, 1989. a
Rohrer, F., Bohn, B., Brauers, T., Brüning, D., Johnen, F.-J., Wahner, A., and Kleffmann, J.: Characterisation of the photolytic HONO-source in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 5, 2189–2201, https://doi.org/10.5194/acp-5-2189-2005, 2005. a, b
Rolletter, M., Kaminski, M., Acir, I.-H., Bohn, B., Dorn, H.-P., Li, X., Lutz, A., Nehr, S., Rohrer, F., Tillmann, R., Wegener, R., Hofzumahaus, A., Kiendler-Scharr, A., Wahner, A., and Fuchs, H.: Investigation of the α-pinene photooxidation by OH in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 19, 11635–11649, https://doi.org/10.5194/acp-19-11635-2019, 2019. a
Rollins, A. W., Smith, J. D., Wilson, K. R., and Cohen, R. C.: Real Time In Situ Detection of Organic Nitrates in Atmospheric Aerosols, Environ. Sci. Technol., 44, 5540–5545, https://doi.org/10.1021/es100926x, 2010. a, b, c
Rollins, A. W., Pusede, S., Wooldridge, P., Min, K. E., Gentner, D. R., Goldstein, A. H., Liu, S., Day, D. A., Russell, L. M., Rubitschun, C. L., Surratt, J. D., and Cohen, R. C.: Gas/particle partitioning of total alkyl nitrates observed with TD-LIF in Bakersfield, J. Geophys. Res.-Atmos., 118, 6651–6662, https://doi.org/10.1002/jgrd.50522, 2013. a
Scholtens, K. W., Messer, B. M., Cappa, C. D., and Elrod, M. J.: Kinetics of the CH3O2+NO reaction: Temperature dependence of the overall rate constant and an improved upper limit for the CH3ONO2 branching channel, J. Phys. Chem. A, 103, 4378–4384, https://doi.org/10.1021/jp990469k, 1999. a
Seinfeld, J. H. and Pandis, S. N.: Atmospheric Chemistry and Physics From Air Pollution to Climate Change, John Wiley and Sons, Inc., Hoboken, New Jersey, 2nd edn., 2006. a
Vereecken, L. and Nozière, B.: H migration in peroxy radicals under atmospheric conditions, Atmos. Chem. Phys., 20, 7429–7458, https://doi.org/10.5194/acp-20-7429-2020, 2020. a, b
Wang, L. Y., Liu, Y. H., and Wang, L. M.: Ozonolysis of 3-carene in the atmosphere. Formation mechanism of hydroxyl radical and secondary ozonides, Phys. Chem. Chem. Phys., 21, 8081–8091, https://doi.org/10.1039/c8cp07195k, 2019. a, b, c, d, e, f, g
Witter, M., Berndt, T., Boge, O., Stratmann, F., and Heintzenberg, J.: Gas-phase ozonolysis: Rate coefficients for a series of terpenes and rate coefficients and OH yields for 2-methyl-2-butene and 2,3-dimethyl-2-butene, Int. J. Chem. Kinet., 34, 394–403, https://doi.org/10.1002/kin.10063, 2002. a, b, c, d, e
Yokouchi, Y. and Ambe, Y.: Aersols formed from the chemical reaction of monoterpenes and ozone, Atmos. Environ., 19, 1271–1276, https://doi.org/10.1016/0004-6981(85)90257-4, 1985. a
Yu, J. Z., Cocker, D. R., Griffin, R. J., Flagan, R. C., and Seinfeld, J. H.: Gas-phase ozone oxidation of monoterpenes: Gaseous and particulate products, J. Atmos. Chem., 34, 207–258, https://doi.org/10.1023/a:1006254930583, 1999. a, b, c
Zhang, H. F., Yee, L. D., Lee, B. H., Curtis, M. P., Worton, D. R., Isaacman-VanWertz, G., Offenberg, J. H., Lewandowski, M., Kleindienst, T. E., Beaver, M. R., Holder, A. L., Lonneman, W. A., Docherty, K. S., Jaoui, M., Pye, H. O. T., Hu, W. W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Guo, H. Y., Weber, R. J., de Gouw, J., Koss, A. R., Edgerton, E. S., Brune, W., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Kreisberg, N. M., Spielman, S. R., Hering, S. V., Wilson, K. R., Thornton, J. A., and Goldstein, A. H.: Monoterpenes are the largest source of summertime organic aerosol in the southeastern United States, P. Natl. Acad. Sci. USA, 115, 2038–2043, https://doi.org/10.1073/pnas.1717513115, 2018. a