the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 29 Jul 2021
| 29 Jul 2021
Atmospheric gaseous hydrochloric and hydrobromic acid in urban Beijing, China: detection, source identification and potential atmospheric impacts
Xiaolong Fan
Jing Cai
Jian Zhao
Yishuo Guo
Chang Li
Kaspar R. Dällenbach
Feixue Zheng
Zhuohui Lin
Yonghong Wang
Lubna Dada
Qiaozhi Zha
Jenni Kontkanen
Theo Kurtén
Siddhart Iyer
Joni T. Kujansuu
Tuukka Petäjä
Douglas R. Worsnop
Veli-Matti Kerminen
Yongchun Liu
Federico Bianchi
Markku Kulmala
Gaseous hydrochloric (HCl) and hydrobromic acid (HBr) are vital halogen species that play essential roles in tropospheric physicochemical processes. Yet, the majority of the current studies on these halogen species were conducted in marine or coastal areas. Detection and source identification of HCl and HBr in inland urban areas remain scarce, thus limiting the full understanding of halogen chemistry and potential atmospheric impacts in the environments with limited influence from the marine sources. Here, both gaseous HCl and HBr were concurrently measured in urban Beijing, China, during winter and early spring of 2019. We observed significant HCl and HBr concentrations ranging from a minimum value at 1 × 108 molecules cm−3 (4 ppt) and 4 × 107 molecules cm−3 (1 ppt) up to 6 × 109 molecules cm−3 (222 ppt) and 1 × 109 molecules cm−3 (37 ppt), respectively. The HCl and HBr concentrations are enhanced along with the increase of atmospheric temperature, UVB and levels of gaseous HNO3. Based on the air mass analysis and high correlations of HCl and HBr with the burning indicators (HCN and HCNO), gaseous HCl and HBr are found to be related to anthropogenic burning aerosols. The gas–particle partitioning may also play a dominant role in the elevated daytime HCl and HBr. During the daytime, the reactions of HCl and HBr with OH radicals lead to significant production of atomic Cl and Br, up to 2 × 104 molecules cm−3 s−1 and 8 × 104 molecules cm−3 s−1, respectively. The production rate of atomic Br (via HBr + OH) is 2–3 times higher than that of atomic Cl (via HCl + OH), highlighting the potential importance of bromine chemistry in the urban area. On polluted days, the production rates of atomic Cl and Br are faster than those on clean days. Furthermore, our observations of elevated HCl and HBr may suggest an important recycling pathway of halogen species in inland megacities and may provide a plausible explanation for the widespread halogen chemistry, which could affect the atmospheric oxidation in China.
- Article
(5786 KB) - Full-text XML
-
Supplement
(1491 KB) - BibTeX
- EndNote





Tropospheric halogen chemistry plays a variety of roles in perturbing the fate of chemical compositions, including ozone (O3) and volatile organic compounds (VOCs) in the troposphere (Saiz-Lopez and von Glasow, 2012; Simpson et al., 2015; Artiglia et al., 2017). Halogen radicals, in particular atomic chlorine (Cl•) and bromine (Br•), can deplete the O3, react rapidly with VOCs with reaction rates of up to 2 orders of magnitude faster than those of the hydroxyl radical (OH) reaction with VOCs and accelerate the depletion of gaseous elemental mercury (Atkinson et al., 2007; Calvert and Lindberg, 2004). Significant halogen-induced O3 reduction of about 10 % of the annually averaged tropospheric ozone column was reported over the tropical marine boundary layer (Saiz-Lopez et al., 2012). However, in polluted coastal regions with high NOx, the coupling between halogen chemistry and NOx chemistry contributes to a significant enhancement of ozone production of up to 7 ppb (parts per billion by volume) (Li et al., 2020; Sherwen et al., 2017; Sarwar et al., 2014). Besides affecting the ozone chemistry, the oxidation processes of VOCs by halogen radicals can potentially lead to secondary aerosol production. Wang and Ruiz (2017) demonstrated that the chlorine-initiated oxidation of isoprene contributed to the formation of particulate organochloride and the yield of secondary organic aerosol (SOA) ranged from 7 % to 36 %. A recent study also found that the oxidation of α-pinene by chlorine atoms yields low-volatility organic compounds, which are essential precursors for secondary particle formation and growth (Wang et al., 2020).
It is known that sea salt particles are a major source of atomic halogens in the marine environment. Chloride (Cl−) and bromide (Br−) in sea salt particles can be displaced by strong acids (i.e., nitric acid (HNO3) and sulfuric acid (H2SO4)) to release gas-phase hydrogen halides HX (Reaction R1; X = Cl or Br) into the atmosphere (Gard et al., 1998; Thornton et al., 2010). The HX then can react with an OH radical to form a X• via Reaction (R2).
On the other hand, the heterogeneous uptake of dinitrogen pentoxide (N2O5) onto sea salt particles can form nitryl halides XNO2 via Reaction (R3) (Finlayson-Pitts et al., 1989; Osthoff et al., 2008; Tham et al., 2014), which is a reservoir of halogen during the nighttime. At sunrise, the XNO2 undergoes rapid photolysis to liberate highly reactive halogen atom (X•), which subsequently reacts with VOCs to produce HX and peroxy radicals (RO2; Reactions R4 and R5). In addition, the heterogeneous oxidation of Br− by O3 at the aqueous phase–vapor interface can lead to the formation of a pre-complex intermediate (Br•OOO−), which contributes to the formation of atmospheric HOBr (Artiglia et al., 2017).
The atmospheric lifetimes of hydrochloric (HCl) and hydrobromic acid (HBr) due to Reaction (R2) are approximately 35.6 and 2.5 h (when OH = 1 × 107 molecules cm−3), respectively, making them a significant daytime recycling source of atomic halogen in the marine atmosphere. Riedel et al. (2012) showed that the reaction of HCl with OH accounts for about 45 % of the integrated Cl atom production over the entire day along the Santa Monica Bay of Los Angeles (Riedel et al., 2012). Another shipborne study reported that the Cl atom production rate peaks at 3 × 105 molecules cm−3 s−1 during the noontime in southern coastal California (Crisp et al., 2014). The produced HCl and HBr can also end up in particle phase during the nighttime (Chen et al., 2016; Roberts et al., 2019; Crisp et al., 2014), further promoting the heterogeneous reaction of N2O5 (Reaction R3).
The discovery of Thornton et al. (2010) has changed the paradigm of halogen chemistry, where it was thought to be restricted to the marine environment (Thornton et al., 2010). A significant source of atomic chlorine from the heterogeneous reaction of N2O5 onto chloride aerosol (Reaction R3) was observed in Boulder, United States, which is 1400 km from the nearest coastline, indicating that active chlorine chemistry also occurs in regions far from the ocean (Thornton et al., 2010). Follow-up studies have confirmed the presence of halogen activation spreading over the continental regions of North America, Canada, Europe and Asia (Mielke et al., 2011; Phillips et al., 2012; Riedel et al., 2013; Tham et al., 2016; Wang et al., 2017; Tham et al., 2018; Liu et al., 2017; Xia et al., 2020; Zhou et al., 2018; McNamara et al., 2020). These findings suggest the crucial role of HCl gas–particle partitioning in sustaining the aerosol chloride concentrations in continental regions for Reaction (R3) to take place (Brown and Stutz, 2012).
On the global scale, sea salt sprays were estimated to be the dominant source of halogens such as Cl and Br (X. Wang et al., 2019; Keene et al., 1999). Through acid displacement and other heterogeneous processes, 64 and 6.2 Tg a−1 gas-phase inorganic Cl and Br from sea salt were emitted to the troposphere, while anthropogenic emissions such as biomass burning, fossil combustion and incineration were supposed to be minor on a global scale (X. Wang et al., 2019; Keene et al., 1999). For the emissions of Cl, anthropogenic emissions were quite crucial for both gaseous and particulate Cl in the urban environment and heavily polluted areas. For example, the anthropogenic emissions for gaseous HCl and particulate Cl were 458 and 486 Gg in 2014 in China, of which biomass burning is the largest contributor (Fu et al., 2018). Many recent field studies reported elevated ClNO2 and particulate chloride concentrations in the plumes influenced by biomass burning and coal-fired power plants, suggesting they could be the driving force for the Cl activation process in continental areas (Riedel et al., 2013; Tham et al., 2016; Wang et al., 2017; Liu et al., 2017; Yang et al., 2018). Furthermore, Bannan et al. (2019) showed that ClNO2 is consistently formed at a landfill site in London, highlighting the potential contribution from landfill emissions of Cl in promoting Reactions (R3) and (R4) (Bannan et al., 2019). Other possible anthropogenic Cl sources include the emissions from industry and water and sewage treatment plants (Hara et al., 1989; Graedel and Keene, 1995; Thornton et al., 2010). During the wintertime, the use of road salt could also be a dominant source of atmospheric Cl in city areas (McNamara et al., 2020).
Atmospheric bromine is much less abundant than chlorine in the stratosphere, with concentrations of around 25 ppt (parts per trillion by volume) compared to 3.7 ppb of chlorine (Bedjanian and Poulet, 2003; Rotermund et al., 2021). HBr is known as a principal bromine sink species for ozone loss chemistry in the stratosphere, showing an average concentration of 1.3 ± 0.39 ppt between 20.0 and 36.5 km altitude (Bedjanian and Poulet, 2003; Nolt et al., 1997; Yang et al., 2005), and also one of the dominant inorganic bromine species in the marine boundary layer, free troposphere and tropical tropopause layer as well (Fernandez et al., 2014; Glasow and Crutzen, 2014; Nolt et al., 1997; Bedjanian and Poulet, 2003). In the urban environment, atmospheric Br was previously known to be strongly affected by traffic emissions since ethylene dibromide (C2H4Br2) used to be used as an anti-knock compound in leaded gasoline (Glasow and Crutzen, 2014). Yet, since the phasing out of leaded gasoline, long-term atmospheric Br has exhibited a continuous decreasing trend for 2 to 3 decades in Germany (Lammel et al., 2002), and a similar situation is expected in Beijing as the usage of leaded gasoline was banned from the years around the 2000s in China (Cai et al., 2017).
Despite the advances in the understanding of concentrations and sources of global halogen species, atmospheric gaseous HCl and HBr in the continental and especially urban environments are much less studied. Some limited studies focused on atmospheric HCl; for example, Crisp et al. (2014) summarized that the concentration of HCl is typically less than 1 ppb over the continental regions, and McNamara et al. (2020) measured the concentration of HCl to be around 100 ppt from inland sources, while an airborne measurement showed HCl concentrations of around 100 ppt to be typically observed over the land area of the northeast United States, except near power plant plumes with concentrations over 1 ppb (Crisp et al., 2014; McNamara et al., 2020; Haskins et al., 2018). Furthermore, much less information is available on the presence of HBr in the continental environment. Until very recently, an airborne measurement detected significant levels of gas-phase reactive bromine species in the exhaust of coal-fired power plants (Lee et al., 2018). Therefore, the measurement of gas-phase HCl and HBr in inland urban environments is necessary to fully assess their effects on the tropospheric chemistry, such as gas–particle partitioning effects on the particulate halide concentrations that can undergo rapid activation via Reaction (R3). These would be more important in polluted regions such as the North China Plain, where Beijing is located and a large amount of chloride is emitted to the atmosphere (Tham et al., 2016; Zhou et al., 2018; Fu et al., 2018).
In this study, we deployed a chemical ionization–atmospheric pressure interface–long-time-of-flight mass spectrometer (CI-APi-LTOF) to measure the atmospheric gas-phase HCl and HBr from 1 February to 31 March 2019, in urban Beijing, China. To the best of our knowledge, it is the first time a simultaneous measurement of HCl and HBr is presented with high time resolution in urban Beijing. In addition, we identify the potential source that contributed to the high levels of gaseous HCl and HBr during wintertime and early springtime. In addition, we estimate the contribution of gaseous HCl and HBr to the production rates of atomic Cl and Br in urban Beijing.
2.1 Sampling site
The field measurements were conducted at Beijing University of Chemical Technology (BUCT) monitoring station (39.94∘ N, 116.30∘ E), located in an urban area of Beijing, China (Fig. 1); the nearest coastline is located about 150 km away in the southeast. The sampling site is about 130 m north of the Zizhuyuan Road and 550 m west of the West Third Ring Road, which is one of the main roads in Beijing. Besides the effect of traffic, this site is also surrounded by local commercial properties and residential dwellings. Thus, the BUCT sampling site can be regarded as a typical urban site. More information about this sampling site can be found in previous studies (Cai et al., 2020; Kontkanen et al., 2020; Zhou et al., 2020; Chu et al., 2021). The instruments were deployed on the roof of a teaching building, which is approximately 15 m above ground level.
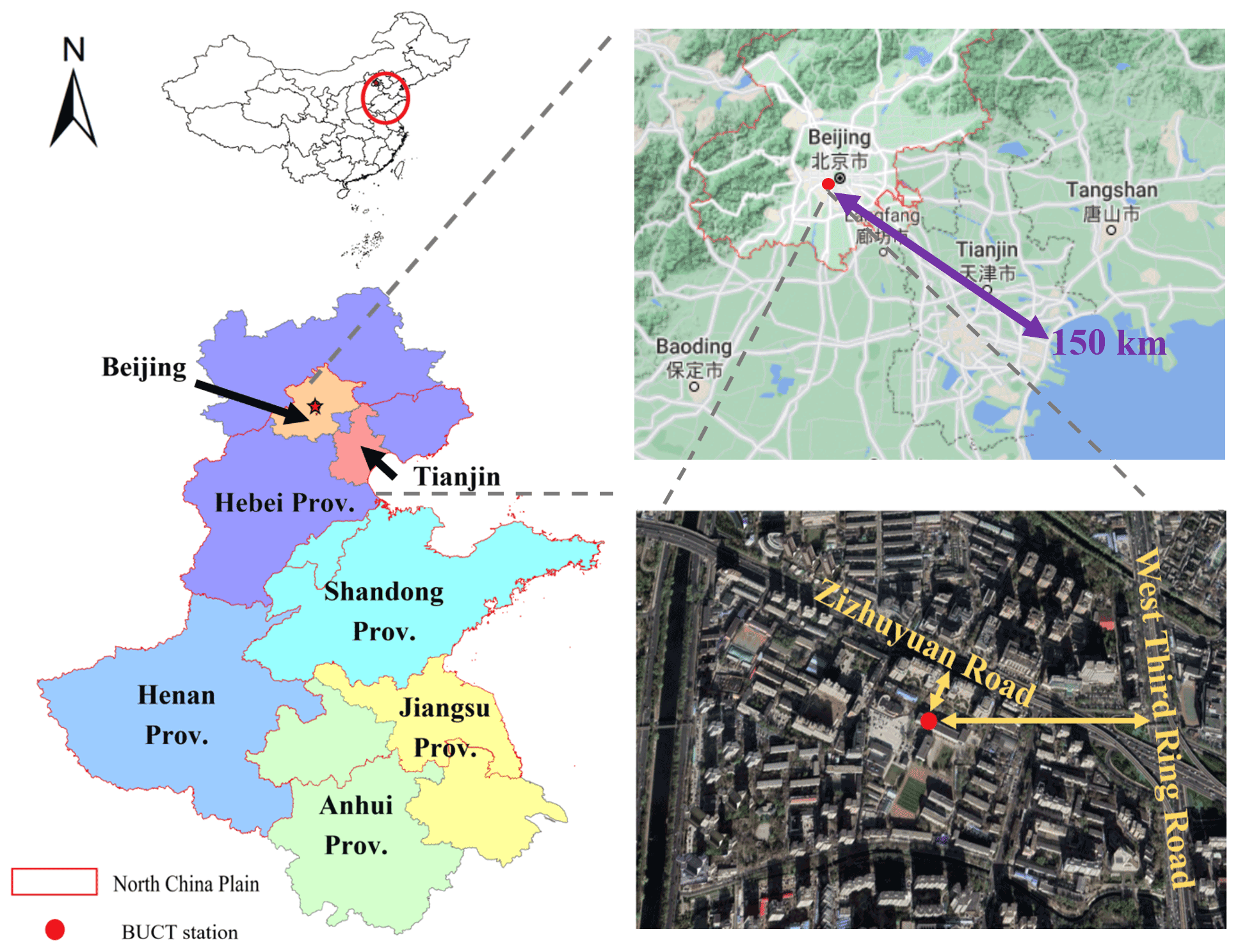
Figure 1The location of BUCT measurement station. The terrain and satellite maps were revised from © Google Maps.
2.2 CI-APi-LTOF mass spectrometer
The working principle of CI-APi-LTOF (Aerodyne Research Inc. and Tofwerk AG) has been described elsewhere (Yao et al., 2020; Eisele and Tanner, 1993; Yao et al., 2018); therefore only details relevant to this present work are discussed here. A typical mass spectrum during our field measurement is depicted in Fig. S1. The dominant reagent ions were nitrate ions (NO, and HNO3•NO) and nitrite ions (NO). Among them, nitrate ions were generated by exposure of sheath flow (pure air with RH ∼5 %), which carried gaseous HNO3. Besides the nitrate ions that acted as dominate reagent ions, nitrite ions were formed from the reaction of a small amount of NO2 (∼1 ppb) in the sheath flow with O and OH−, which were generated from the exposure of sheath flow (pure air with RH ∼5 %) to an X-ray source (Hamamatsu L9491) (Fig. S5) (Arnold et al., 1995; Skalny et al., 2004). Considering nitrate ions were still the dominant reagent ions (Fig. S1), the CI-APi-LTOF was actually operated as a typical nitrate-CI-APi-LTOF.
Ambient air was drawn into the CI inlet through a 0.75 in. stainless steel tube with a flow of ∼8 L min−1. A small mixed flow (∼0.8 L min−1 controlled by a critical orifice with 300 µm diameter) entered the APi-LTOF and was analyzed. The CI-APi-LTOF was operated in the negative V-mode with a mass resolving power of ∼10 000 Th/Th and a mass accuracy better than 5 ppm. Data of CI-APi-LTOF were acquired with 5 s time resolution, and the recorded data were further analyzed with the MATLAB tofTools package (Junninen et al., 2010).
2.3 Detection and quantification of HCl and HBr
From Table 1, it can be seen that the gas-phase acidity (−ΔG) of HCl is 1354 kJ mol−1, which is larger than that of HNO3 (1329 kJ mol−1). In addition, the enthalpy (ΔH) of HNO3 and Cl− is 32.8 kcal mol−1, which is higher than that of HCl and NO (22.9 kcal mol−1), hinting that the reaction of HCl and NO was unlikely to occur (Fig. S4a). Additionally, a previous study showed that the reaction rate (<10−12 molecules cm−3 s−1) between NO and HCl was significantly less than that (1.4 × 10−9 molecules cm−3 s−1) of NO with HCl (Ferguson et al., 1972). Therefore, the HCl is likely mainly charged by NO instead of NO to result in Cl− formation. The ion–molecule reaction between nitrite ions and HCl can be written as follows (Ferguson et al., 1972):
In addition to NO, HCl can also react with O, leading to Cl− formation via Reaction (R7).
Therefore, HCl can be quantified according to
where CHCl (in units of molecules cm−3) is a calibration coefficient of HCl. (Cl−), (NO) and (O) represent the signals of Cl−, NO and O from CI-APi-LTOF, respectively. Based on ambient data, a very small fraction (less than 5 %) of Cl− (or HCl) would react with HNO3 (or NO) in the sheath flow to form Cl−•HNO3 (or HCl•NO). Thus, the signals of Cl−•HNO3 (or HCl•NO) were not taken into account for HCl quantification. The background measurement was carried out by sampling zero air. Figure S7 shows that the background signals were significantly lower than those of ambient air and injected HCl and HBr. The limits of detection (LODs, 3σ) were 1 × 108 and 1 × 107 molecules cm−3 (i.e., 4 and 0.5 ppt) for HCl and HBr, respectively. Using 4 d synchronous gaseous HCl concentrations measured by the Monitor for AeRosols and Gases in Ambient air (MARGA; Metrohm Inc., Switzerland), an indirect calibration was adopted to quantify the HCl measured by the CI-APi-LTOF (Sect. S5 in the Supplement). The obtained calibration factor CHCl for HCl is 3 ± 0.1 × 1012 molecules cm−3 (Fig. S8b), and an uncertainty of ±30 % (Sect. S5) was applied to the reported HCl concentrations. Similar to HCl, the same uncertainty was also adopted for HBr mixing ratios. It should be noted that our assumptions lead towards a semi-quantitative estimation of HBr concentrations, due to other potential uncertainties (e.g., different sensitivities of HCl and HBr) not being taken into account.
Table 1Gas-phase acidities and deprotonated anion of a few compounds of interest.

* Gas-phase acidity is defined as −ΔG for the protonation reaction (H HA). Data are obtained from the NIST Chemistry WebBook.
On the basis of −ΔG of HBr, HNO3, HNO2 and HO2 and the enthalpy (ΔH) calculations (Table 1, Figs. 2 and S4), besides the reaction with NO and O, similar to HCl, some HBr could also react with NO to form Br− via Reaction (R8) (Ferguson et al., 1972).
Hence, HBr should be quantified according to
where CHBr (in units of molecules cm−3) is a calibration coefficient of HBr. (Br−), (NO), (O) and (NO) represent the signals of Br−, NO, O and NO from CI-APi-LTOF, respectively. However, as direct calibration for HBr was not available, the calibration coefficient of HCl (CHCl) was utilized to semi-quantify HBr based on the following equation:
Since the enthalpies (ΔH) of HBr•NO formed by HBr with NO (27.3 kcal mol−1) and Br− with HNO3 (27.9 kcal mol−1) were very close to each other (Fig. S4b), it was difficult to quantify the specific contribution to Br− from the reaction of HBr with NO. Also, the ratios of Br−•HNO3 (or HBr•NO) to Br− were less than 4 %. Therefore, in Eq. (3), the reaction pathway of HBr with NO was not considered. The presented HBr concentrations should be treated as semi-quantitative ones.
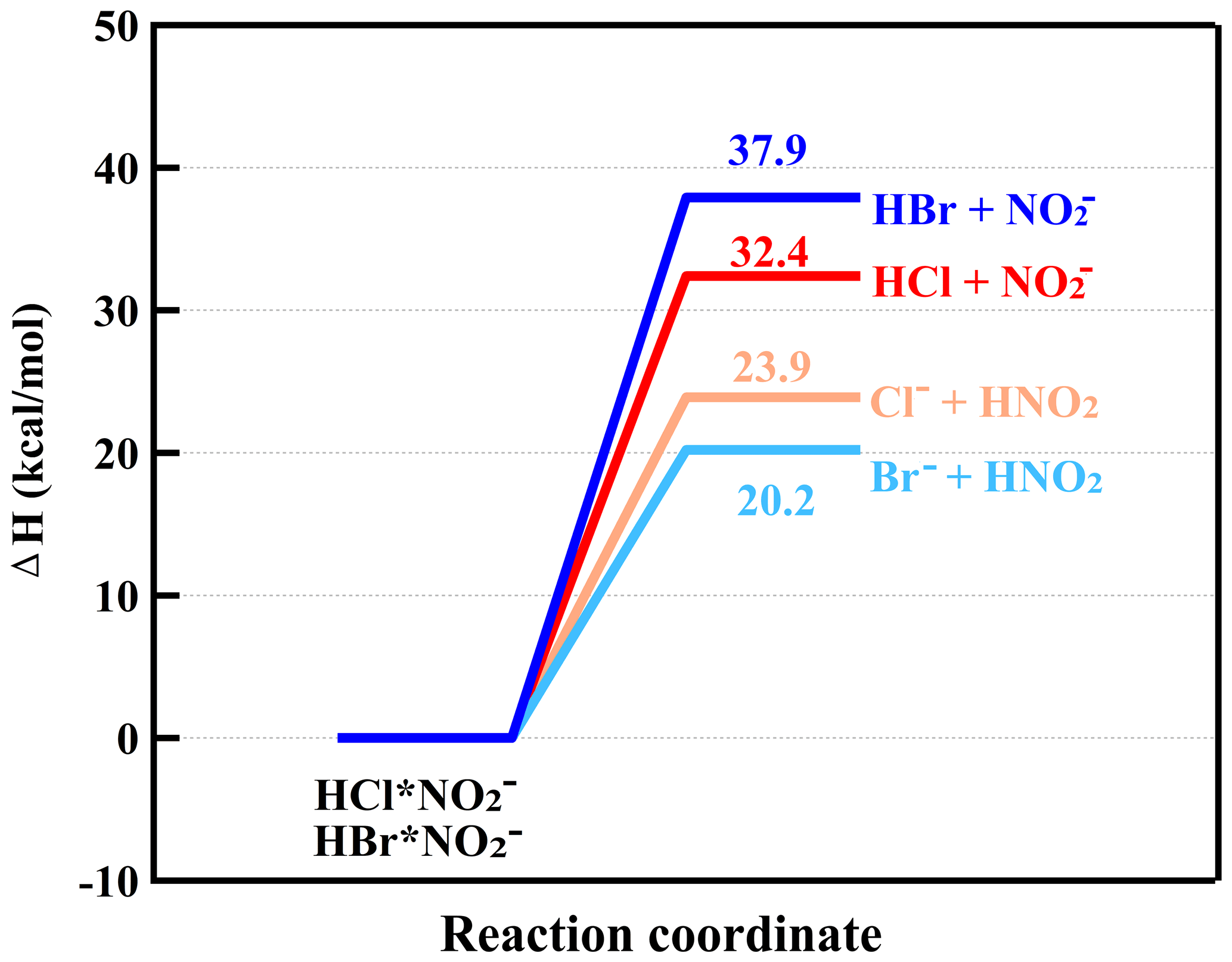
Figure 2The calculated enthalpies of HCl•NO formed by HCl with NO and Cl− with HNO2 and enthalpies of HBr•NO formed by HBr with NO and Br− with HNO2 at the DLPNO-CCSD(T)/def2-QZVPP// ωB97X-D/aug-cc-pVTZ-PP level of theory.
To confirm these ion–molecule reactions, high concentrations (undetermined) of gaseous HCl and HBr were mixed with zero air generated from a zero-air generator (Aadco 737) and then measured by CI-APi-LTOF (Sect. S4). After the injection of HCl and HBr, the signals of Cl−, Br−, Cl−•HNO3 (or HCl•NO) and Br−•HNO3 (or HBr•NO) started to increase (Fig. S7), confirming that HCl and HBr can be detected as Cl−, Br−, Cl− •HNO3 and Br−•HNO3 by CI-APi-LTOF.
2.4 Other auxiliary measurements
Gaseous HCN and HCNO can also be detected by O through the ion–molecule reactions as follows:
The −ΔG values of HCN and HCNO are 1433 and 1415 kJ mol−1, respectively, which are higher than the value of NO (1393 kJ mol−1) (Table 1) and lower than that of O (1450 kJ mol−1). Therefore, HCN and HCNO are able to be charged by O (but not NO) via a deprotonation reaction to lead to CN− and CNO− formation. In this study, direct calibrations for HCN and HCNO were not available. Instead, the normalized signals of CN− and CNO− by O were tentatively utilized to indicate the abundance and trend of HCN and HCNO.
The meteorological parameters, including temperature and UVB intensities, were recorded by a weather station (Vaisala Inc., Finland). NO2 was measured with a Thermo 42i NO–NO2–NOx analyzer (Thermal Environment Instruments Inc. USA). The mass concentrations of particulate chlorine and black carbon (BC) in PM2.5 were measured by a time-of-flight aerosol chemical speciation monitor (ToF-ACSM, Aerodyne Research Inc., USA) and an aethalometer (AE33, Magee Inc., USA), respectively (Sect. S1 in the Supplement).
Meanwhile, we applied 24 h air mass back trajectory and potential source contribution function (PSCF) analyses to help elucidate the potential source regions (i.e., air masses) of high levels of HCl and HBr. The detailed descriptions of PSCF and air mass trajectory analysis are described in the Supplement (Sect. S6) and previous literature (Wang, 2014; Y. Q. Wang, 2019). It is noted that the lifetime of gaseous HCl and HBr could be shorter than the length of the air mass trajectories. These analyses mainly aimed to point out the source regions of pollutant air masses that brought high levels of Cl and Br rather than the real-time origins of air parcels.
3.1 HCl and HBr measurement
Figure 3 shows the time series of gaseous HCl and HBr, temperature (T) and ultraviolet radiation b (UVB, 280–315 nm) intensities for the entire measurement period in winter and early spring of 2019 (February to April). High concentrations of HCl and HBr were observed for the whole measurement period, with a clear diurnal variation (Fig. 3g). The mean concentrations of HCl and HBr are 1 × 109 molecules cm−3 (37 ppt) and 2 × 108 molecules cm−3 (7 ppt), respectively. The maximum concentrations reach up to 6 × 109 molecules cm−3 (222 ppt) for HCl and 1 × 109 molecules cm−3 (37 ppt) for HBr during the daytime. The concentrations of HCl and HBr showed a similar change in atmospheric temperature and UVB. For the first period of measurement (from 1 to 15 February), HCl and HBr concentrations are lower when the atmospheric temperature is close to 0∘ and the UVB intensities are relatively low. Yet, for the later period of March, the HCl and HBr concentrations begin to increase, along with the rising of temperature and UVB. In late March, even with higher temperature, due to the lower abundance of HNO3 and particulate chloride, the HCl and HBr concentrations remain at a relatively low level (Fig. 3).
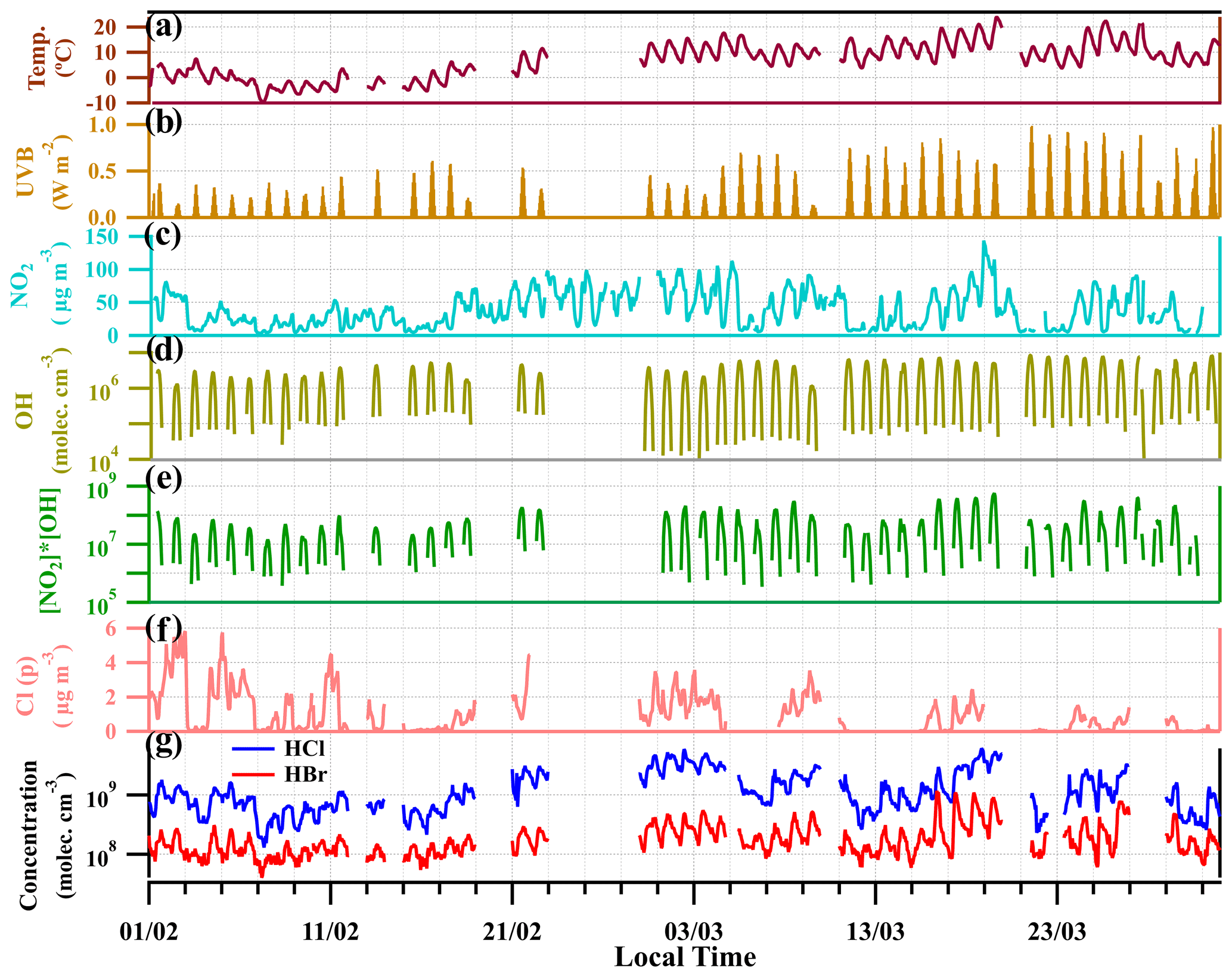
Figure 3Time profiles of temperature (a), UVB intensities (b), NO2 concentration (c), OH concentration from calculation (d), [NO2] × [OH] (µg m−3 × molecules cm−3) (e), particulate chloride concentration (Cl(p)) (f) and the mixing ratios of HCl and HBr (g). The data points are in hourly average intervals.
The diurnal cycles of HCl and HBr are depicted in Fig. 4a and b, respectively. The HCl concentrations are typically higher than HBr by approximately an order of magnitude; nevertheless, the diel patterns showed by these two species are quite similar to each other. It is noticed that both HCl and HBr began to increase after sunrise, and a relatively high concentration was observed during the daytime (08:00 to 17:00). From Fig. 4d, it also can be found that elevated HCl is associated with high temperature and [NO2] × [OH] value during the daytime. The reaction of NO2 with the OH radical being one of the dominant formation pathways of gaseous HNO3 during the daytime (Stavrakou et al., 2013) implies that strong photochemical reactions and the following potential elevated HNO3 could intensify the HCl release from particulate chloride in the daytime from 08:00 to 17:00. The OH radical concentrations were calculated using JO1D (Sect. S8). This phenomenon is consistent with our observation results above where the increase of temperature and UVB could reinforce the formation of chemicals (e.g., HNO3) that promote the gas–particle partitioning or directly increase the gas-phase formation rate of HCl and HBr (Crisp et al., 2014; Riedel et al., 2012), thus further enhancing the HCl and HBr (Fig. 3). Although there is no direct measurement of particulate bromide (Br), considering the similarity in diurnal patterns and good correlation (r=0.70) between HBr and HCl (Fig. 4c) and HBr tracking well with the temperature and [NO2] × [OH] (see Fig. 3), it is rational to suppose HBr also predominantly derived from the gas–particle partitioning process. The contribution by the reaction of bromine atoms with hydrocarbons to form HBr is likely not the dominant pathway as the bromine atom is less reactive to hydrocarbons compared to the chlorine atom and most often reacts with ozone (Simpson et al., 2015).
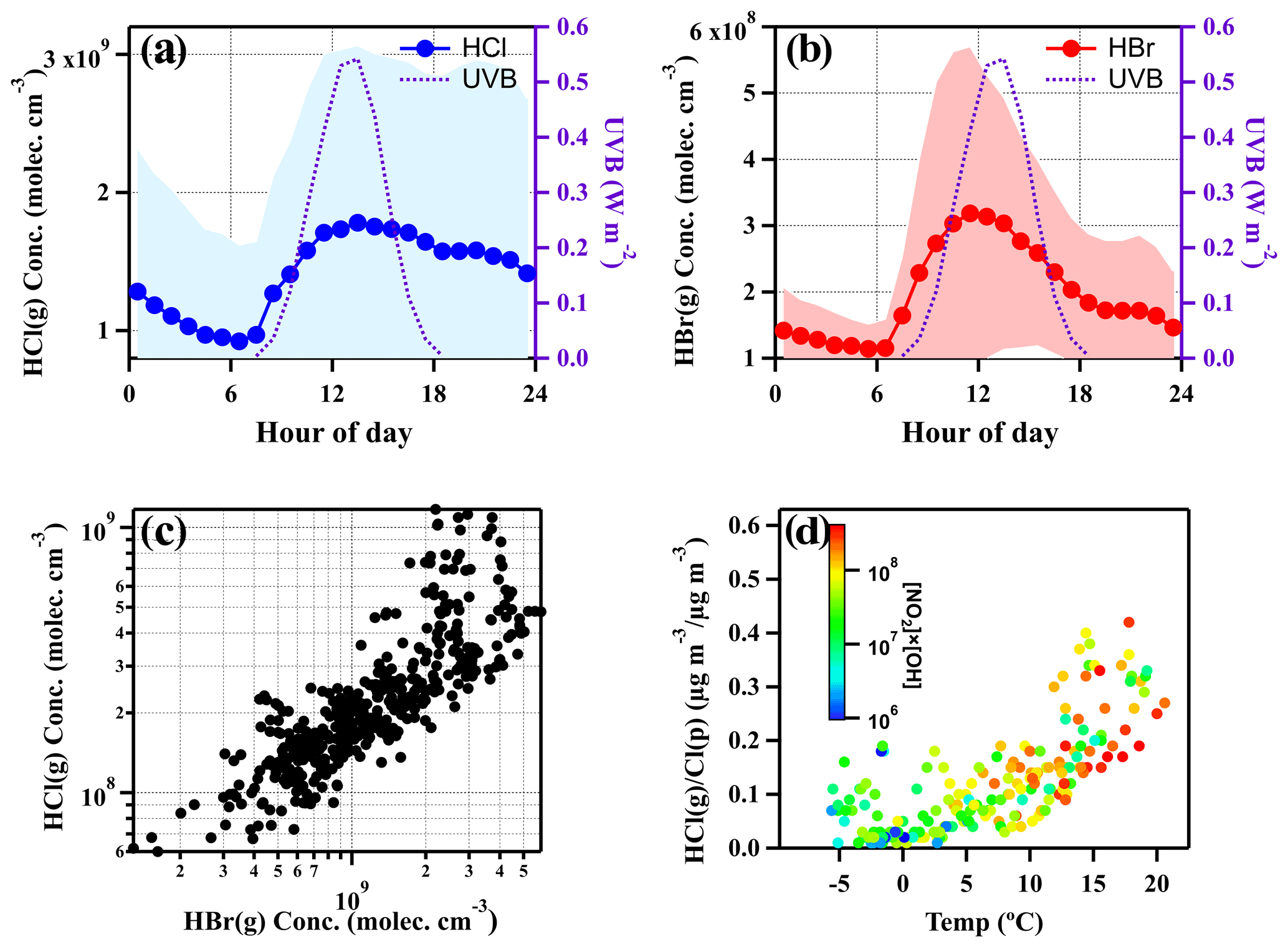
Figure 4Diurnal variations of UVB intensities, HCl and HBr concentrations (averaged values ± one standard deviation) (a, b) and the correlation between HCl and HBr (c). Temperature dependence of gas–particle partitioning ratios of mass concentration of chloride, color-coded by [NO2] × [OH], which indicates the abundance of HNO3 in the daytime (d). In panel (c) and (d), the data points are hourly averaged ones during daytime (08:00–17:00). All snowy and rainy days during the sampling period were excluded.
Our observation of daily averaged mass concentrations of particulate chloride (Cl (p)) in PM2.5 showed a similar trend with daily averaged mixing ratios of gaseous HCl (Fig. 5a). The difference from the ratios of HCl(g) to Cl(p) in February and March is likely due to the higher temperature in March (Figs. 3 and 5a). In contrast, the diurnal variations of HCl and particulate Cl showed the opposite trend at daytime from 08:00 to 17:00 (Fig. 5b). The mole ratios of HCl(g) to Cl(p) ranged from <0.1 at nighttime and early morning to >0.3 in the afternoon (Fig. 5b). The enhancement of HCl(g)Cl(p) during the noontime is due to the large increase of gaseous HCl. It also suggests that the higher temperature and stronger photochemical reactions during the daytime would strongly influence HCl release from particulate chloride in Beijing, which will be further discussed in the following sections. During the period between the late afternoon and midnight, the increase of Cl(p) and HCl(g) could be explained by the higher nighttime emissions of residential combustion such as wood and coal burning in Beijing (Hu et al., 2017; Sun et al., 2016), and the high abundance of gaseous HNO3 is attributed to efficient nocturnal N2O5 chemistry (Tham et al., 2018).
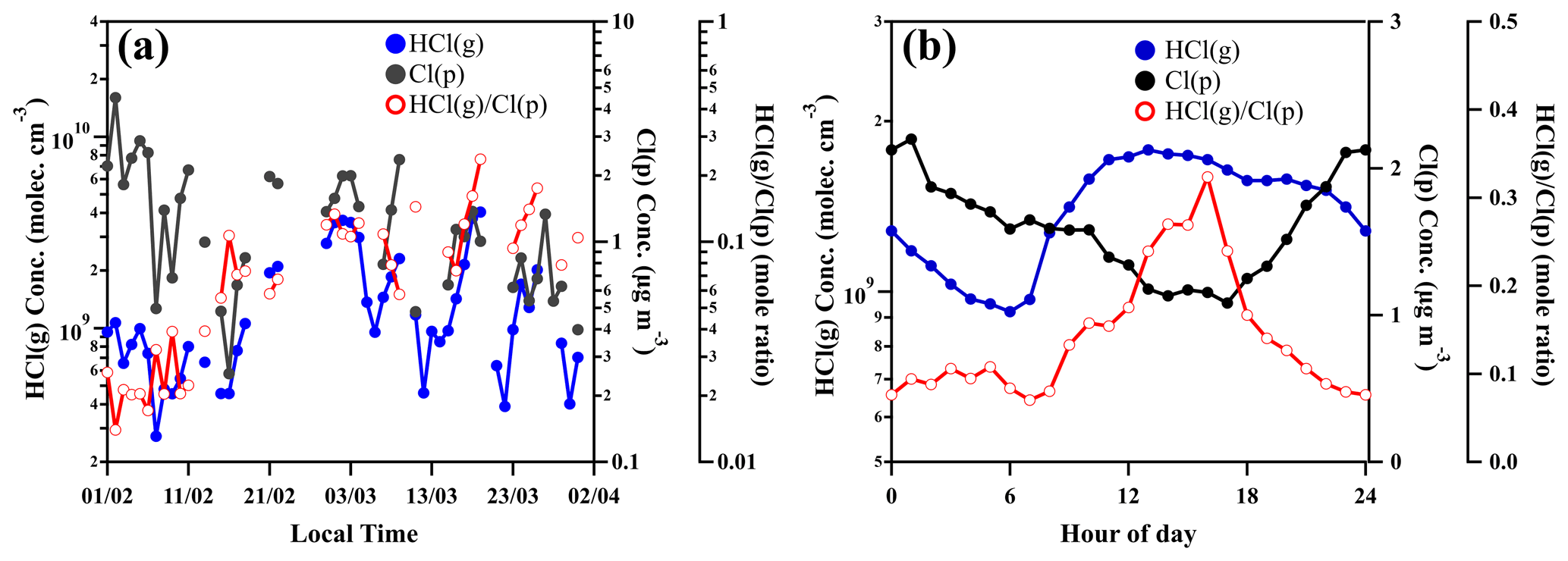
Figure 5Time variation of daily averaged concentration of particulate chloride (Cl(p)) measured by ACSM, gaseous HCl (HCl(g)) measured by CI-APi-LTOF and mole ratios of HCl(g)Cl(p) (a) and diurnal variation of HCl(g), Cl(p) and mole ratios of HCl(g)Cl(p) (b).
These observations showed that there is an abundance of gaseous HCl and HBr in the polluted urban environment. To our best of knowledge, this is the first concurrent observation of gaseous HCl and HBr in a polluted inland urban atmosphere. Although it is well known that the HCl is abundant in the polluted coastal and inland regions, previous studies show that the typical HCl mixing ratios over the continental urban areas are less than 1 ppb (Crisp et al., 2014; Faxon and Allen, 2013; Le Breton et al., 2018; McNamara et al., 2020), which are similar to our observations at Beijing. In contrast, the presence of gaseous HBr in the urban regions is unknown prior to our observation. The significant concentration of HBr in the urban atmosphere of Beijing is even comparable to the simulated concentrations in the marine environment, where concentration up to 2 ppt was reported (Fernandez et al., 2014). These elevated HCl and HBr in urban Beijing may point to the existence of Cl and Br sources in this region.
3.2 Source identification
The natural sources of atmospheric Cl and Br include sea salt spray, wildfires and volcano emissions, while the anthropogenic emissions include coal combustion and traffic emissions, as well as other industries such as pesticides, battery industry and waste incineration (Simpson et al., 2015). Compared with the sources of particulate Cl and Br that are widely studied and identified in previous literature, the origins of gaseous HCl and HBr are much less studied, due to their much shorter lifetime in the troposphere (Simpson et al., 2015).
According to air mass analysis (24 h back-trajectory) for HCl and HBr during February and March (Fig. 6a and b), the potential source regions of the selected periods with high-level concentrations of HCl (above 75 % percentile) were located in the south of the North China Plain, such as the south of Hebei province, where heavy residential coal, biomass burning and industry emissions occurred (Fu et al., 2018). These figures further suggest that the high concentrations of HCl seemed not to be strongly affected by marine regions during our sampling period. Instead, the good correlation (r=0.67) between hourly particulate Cl and BC together with the similar trend between particulate Cl and HCl suggested that HCl is likely to have the same original sources with particulate Cl and black carbon (BC) in PM2.5 rather than marine sources (Figs. 5a and S10a). Hydrocyanic acid (HCN) and isocyanic acid (HCNO) were typically regarded as tracers for burning emissions, especially in the biomass burning process (Vigouroux et al., 2012; Adachi et al., 2019; Leslie et al., 2019; Wren et al., 2018; Priestley et al., 2018). Although a recent study showed that HCNO came from both primary emissions and secondary formation in the scale of the North China Plain (NCP) during the daytime (Wang et al., 2020), the high correlations between HCN and HCNO (daytime, 08:00–17:00, r=0.94 and nighttime, 18:00–07:00, r=0.96) indicated that in urban Beijing, HCN and HCNO are mainly from primary emission (Fig. 7c and d) and can be regarded as the tracers of combustion emissions. Thus, high correlations of measured gaseous HCl with HCN (r=0.83) and HCNO (r=0.90) further suggested that the HCl during our sampling period was more likely coming from combustion origins rather than marine sources in urban Beijing (Fig. 7a and b). Since gaseous HCl could be affected by both emissions and gas–particle partitioning (shown in Fig. 4d), we compared the daily concentrations of gaseous HCl and particulate Cl to minimize the influence of temperature and partitioning. The daily averaged HCl concentration had a high correlation with daily averaged particulate Cl (r=0.84 and 0.70 for winter and early spring periods, respectively) and BC concentration (r=0.82), which is consistent with previous studies that particulate Cl, coal combustion organic aerosol (CCOA) and BC were highly correlated and likely to be from the same source in winter and early spring in Beijing (Hu et al., 2017, 2016).

Figure 6The results of PSCF analysis for HCl (a) and HBr (b). Darker colors indicate greater potential source distribution. The terrain base map was made from © Yahoo Maps. Black stars mark the location of the sampling site.
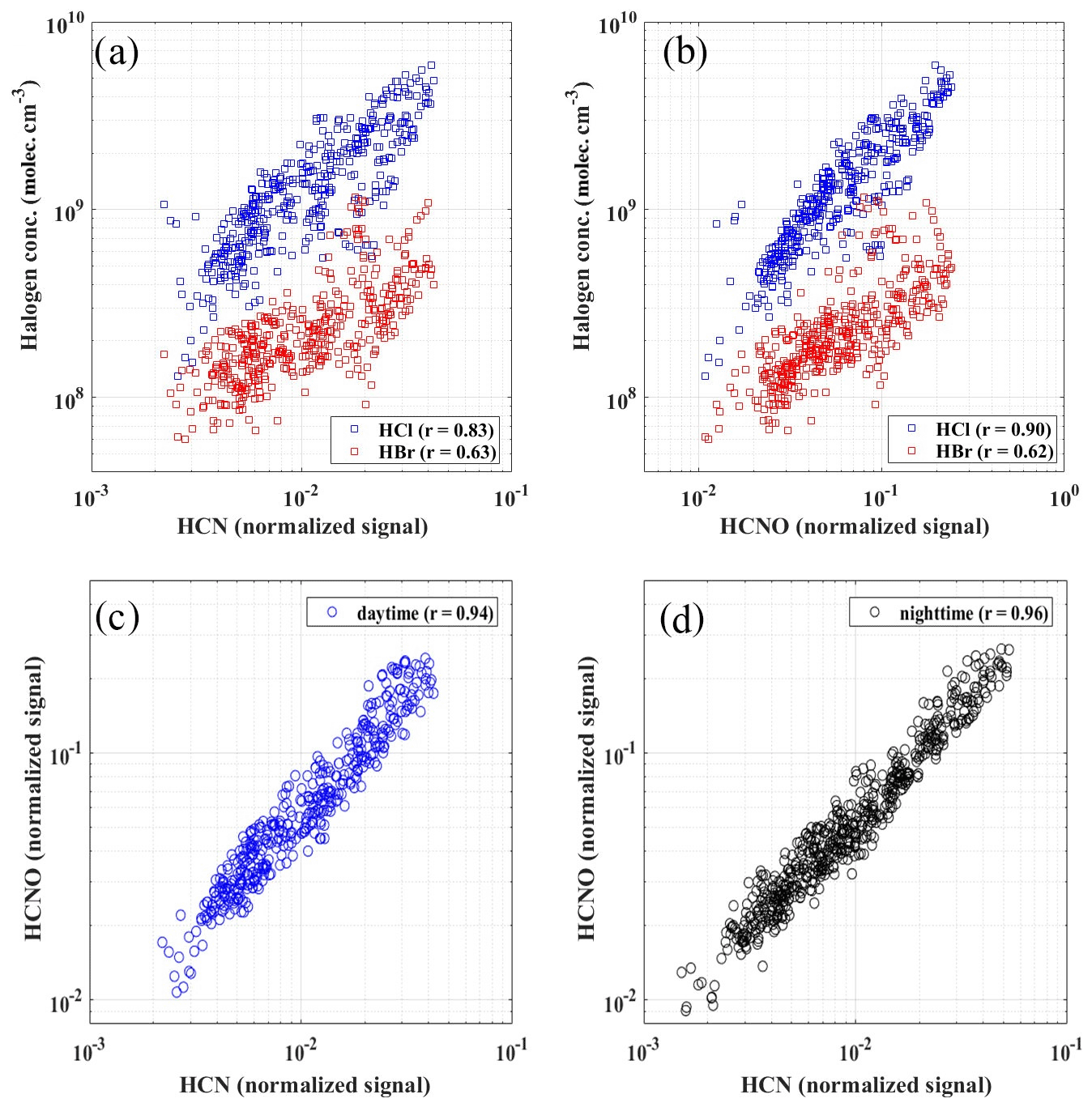
Figure 7The relationship of HCl and HBr concentrations with HCN and HCNO during the daytime (08:00–17:00) (a, b) and the correlations between HCN and HCNO during both daytime (08:00–17:00) (c) and nighttime (18:00–07:00 the next day) (d).
Similar to HCl, the potential source regions for high Br concentrations were also located in the inland, demonstrating marine sources might not be the dominant source for gaseous HBr in winter of Beijing (Fig. 6b). The ratio of particulate Br / Na from previous literature in Beijing was 0.04 (He et al., 2001), which was much higher than the ratios from seawater (0.018) and crustal dust (0.0006 to 0.0008) but much closer to the ratios of biomass burning aerosols (0.01 to 0.06) (Sander et al., 2003). As discussed before, the good correlation (r=0.70) between gaseous HCl and HBr also implied their similar origins. In our study, moderate correlation coefficients were also observed between gaseous HBr and combustion tracers such as HCN, HCNO (0.63 and 0.62, respectively) and daily BC (r=0.60) (Figs. 7a, b and S10b). Multiple gaseous organic and inorganic Br compounds such as CH3Br, Br2, BrNO2, BrCl, CH3Br and CH2Br2 were also observed in different combustion processes such as biomass burning, coal combustions and waste incineration in previous studies, further supporting the possibilities of combustion origins of the gaseous HBr in this study (Lee et al., 2018; Keene et al., 1999; Manö and Andreae, 1994). A recent airborne observation conducted in the United States found that high levels of reactive inorganic Br species in the plume from a coal power plant, likely due to the application of calcium bromide as additives in coal fuel (Lee et al., 2018). Taking these observations together, in urban Beijing, the measured HBr was more likely coming from combustion sources such as biomass burning and coal combustion in the south of Beijing rather than marine sources. It is also interesting to note that in a previous marine study conducted in Oahu, Hawaii, gaseous Br was found to be 4 to 10 times higher than particulate Br (Moyers and Duce, 1972). On the other hand, from a previous observation conducted in urban Beijing, high levels of both gaseous (7 ng m−3) and particulate (in total suspended particles (TSP), 18 ng m−3) bromine were measured by an offline sampling–organic solvent extraction and instrumental neutron activation analysis (INAA) method (Tian et al., 2005). Considering the high concentration and reactivity of Br, gaseous Br from anthropogenic sources may play a more critical role in the urban atmosphere.
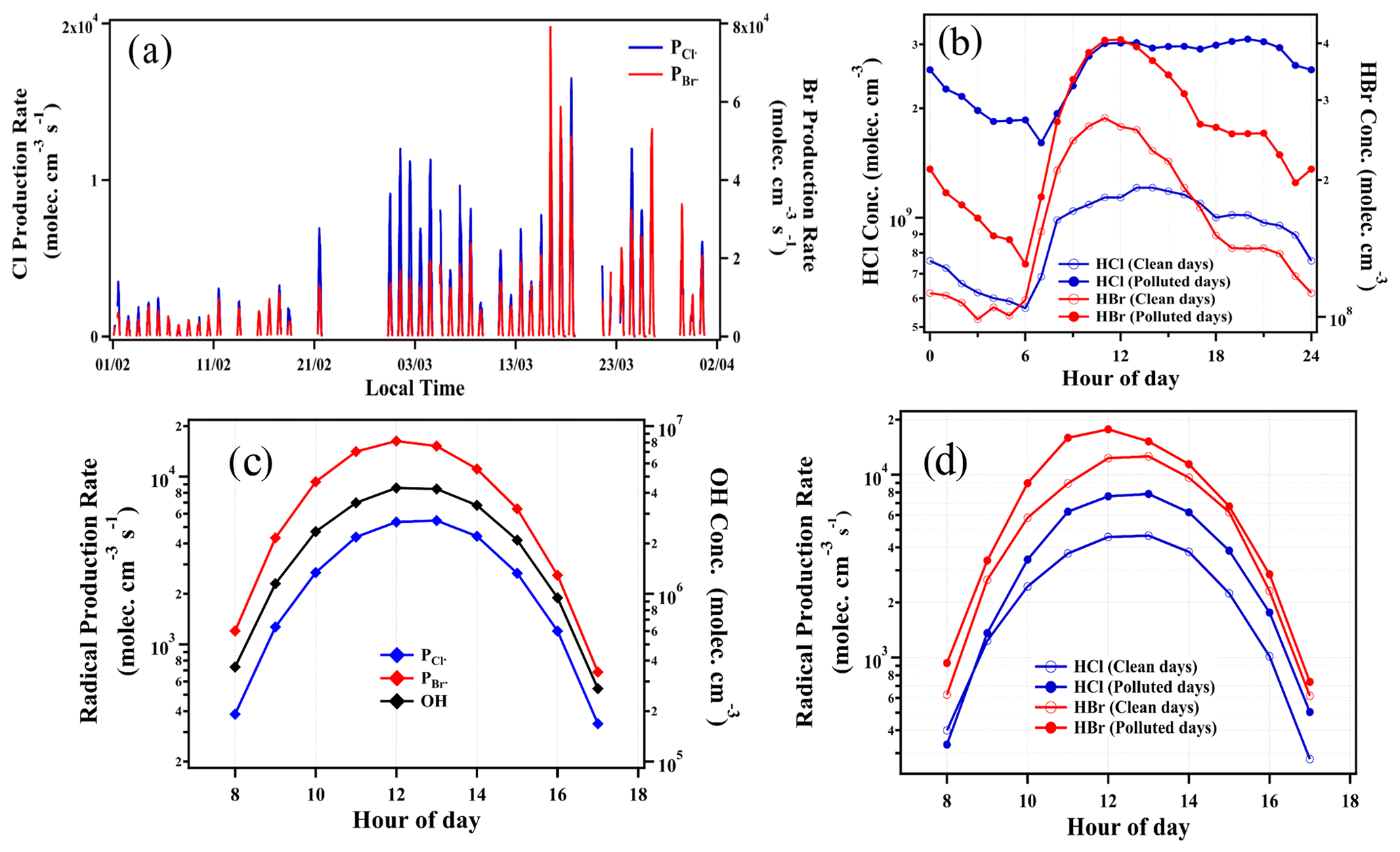
Figure 8Time series of calculated production rates of Cl and Br radicals during the observation period (a); diurnal variations of HCl and HBr concentrations on clean and polluted days (b); diurnal variations of production rates of Cl and Br radicals from 08:00 to 17:00, together with calculated OH radical concentrations (c); and production rates of Cl and Br radicals on clean and polluted days (d). The clean and polluted days were classified as daily PM2.5 < 75 µg m−3 and PM2.5≥75 µg m−3, respectively. The data points are hourly average intervals and measured during observation periods from 1 February to 31 March 2019.
3.3 Halogen-atom production
To investigate the potential atmospheric implications of HCl and HBr on atmospheric oxidation capacity, we calculated the production rate of atomic Cl (PCl•) and Br (PBr•) via the reactions of HCl and HBr with OH radicals. Figure 8 shows the time series of PCl• and PBr• and the estimated diel concentration of OH calculated from the photolysis rate (JO1D) (Sect. S8). Note that the estimated peak concentrations of OH radicals varied between ∼3 × 105 and ∼4 × 106 molecules cm−3 during noontime. The reaction of HCl with OH radicals led to a mean Cl atom production rate of 3 × 103 molecules cm−3 s−1 during daytime from 08:00 to 17:00 (Fig. 8c). These rates fall within the range of Cl atom production rates (∼103 to 106 molecules cm−3 s−1) reported in polluted environments (Crisp et al., 2014; Hoffmann et al., 2018; McNamara et al., 2020). The reaction of HBr with OH is estimated to produce a daytime mean of 8 × 103 molecules cm−3 s−1 of the Br atom (Fig. 8c). This result shows that in addition to the Cl atom, the Br atom could also be present in urban Beijing and may be as important as the Cl atom in terms of reaction with OH, since the PBr• is about 2–3 times faster than the PCl• (Fig. 8c). The average HCl and HBr concentrations were observed to be higher during the polluted days (daily mean PM2.5≥75 µg m−3), about 2–3 times higher than the clean days (daily mean PM2.5 < 75 µg m−3), as shown in Fig. 8b. Consequently, the radical production rate also showed a difference between clean and polluted days (Fig. 8d). The mean diurnal pattern shows that the values of PCl (up to 8 × 103 molecules cm−3 s−1) and PBr (up to 2 × 104 molecules cm−3 s−1) on polluted days were both higher than those on clean days by up to 2 times (Fig. 8d). This hints that the roles of HCl and HBr may be more significant in polluted environments. Recent studies in several polluted sites of China suggested that the photolysis of ClNO2 and Cl2 is the dominant daytime Cl atom source, leading to a Cl atom production rate of up to 8 × 106 molecules cm−3 s−1 (Tham et al., 2016; Liu et al., 2017; Xia et al., 2020), while our observation of the Cl atom production rate from HCl + OH could reach up to 2 × 104 molecules cm−3 s−1 in the daytime. Despite the lower production rate, the reaction of HCl with OH may also act as an important recycling process of the Cl atom, which ultimately enhanced the atmospheric oxidation capacity (Riedel et al., 2012). Analogous to chlorine chemistry, the reaction of HBr with OH could be a significant source of the Br atom in the daytime, although rapid photolysis of Br2 and BrNO2 is believed to be the major Br atom source in a polluted urban environment as ubiquitous bromine species (e.g., Br2, BrCl and BrNO2) have been previously observed in residential coal burning and coal-fired power plant plumes (Lee et al., 2018; Peng et al., 2020).
In conclusion, we present the first concurrent measurement of both gaseous HCl and HBr in urban Beijing, a megacity with strong anthropogenic emissions in the North China Plain. Our observation surprisingly shows significant concentrations of HBr in urban Beijing, together with the elevated levels of HCl, throughout the winter and spring during our sampling period. Gaseous HCl and HBr are most likely originated from anthropogenic emissions such as burning activities (e.g., biomass burning and fossil fuel combustion) in the inland region rather than marine sources. In addition, the gas–particle partitioning may play a crucial role in contributing to elevated levels of HCl and HBr in urban Beijing. On polluted days, the concentrations of HCl and HBr are higher than those on clean days. The abundance of HCl and HBr in the polluted urban troposphere may further influence the photochemistry of the atmosphere through the following two aspects: (1) direct contributions to the production of highly reactive halogen atom (e.g., Cl•and Br•), which can rapidly oxidize VOCs (Reaction R5); and (2) replenishment of the halide ion (Cl− and Br−) in the aerosols, supporting the nocturnal heterogeneous production of ClNO2 and BrNO2, which are major sources of highly reactive halogen atom at sunrise (Reactions R3 and R4). Our observation of elevated HCl and HBr may indicate an important recycling pathway of Cl and Br species and may provide a plausible explanation for the recent observations of widespread halogen activation in polluted areas of China (e.g., Tham et al., 2016; Zhou et al., 2018; Xia et al., 2020; Peng et al., 2020), which could have a significant influence on the atmospheric oxidation capacity and secondary aerosol formation. The atomic Cl and Br on polluted days might contribute to oxidation capacity to a greater extent than on clean days. Furthermore, additional insight into the HBr levels in Beijing shows that bromine chemistry, a previously neglected chemistry, may be important in inland megacities of China. Our results also suggest that understanding of gaseous HCl and HBr would be of much importance to photochemistry studies, as well as air quality improvement in urban areas of China.
All data related to this study can be obtained from the corresponding authors (Lei Yao and Yee Jun Tham) via email.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-11437-2021-supplement.
LY and YJT designed the research. XF, LY, YJT, JC, CY, YG, CL, KRD, FZ, ZL, BC, YW, LD, WD, JK, JTK, JZ, QZ, TK, SI, TP, DRW, VMK, YL, FB and MK carried out the observation, analyzed the data and interpreted the results. SI and TK provided quantum calculation results. XF, LY, YJT and JC prepared the manuscript with contributions from all co-authors.
The authors declare that they have no conflict of interest.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the special issue “Pan-Eurasian Experiment (PEEX) – Part II”. It is not associated with a conference.
The work is supported by the Academy of Finland (ACCC Flagship, grant no. 337549, Center of Excellence in Atmospheric Sciences, project no. 307331, and PROFI3 funding, 311932), the European Research Council via ATM-GTP (grant no. 742206), CHAPAs (grant no. 850614) and the EMME-CARE project, which has received funding from the European Union's Horizon 2020 Research and Innovation.
This research has been supported by the Academy of Finland (ACCC Flagship, grant no. 337549, Center of Excellence in Atmospheric Sciences, project no. 307331, and PROFI3 funding, 311932) and the European Research Council via ATM-GTP (grant no. 742206) and CHAPAs (grant no. 850614).
Open-access funding was provided by the Helsinki University Library.
This paper was edited by Thorsten Bartels-Rausch and reviewed by three anonymous referees.
Adachi, K., Sedlacek, A. J., Kleinman, L., Springston, S. R., and Buseck, P. R.: Spherical tarball particles form through rapid chemical and physical changes of organic matter in biomass-burning smoke, P. Natl. Acad. Sci. USA, 116, 201900129, https://doi.org/10.1073/pnas.1900129116, 2019.
Arnold, S., Morris, R., Viggiano, A., and Jayne, J.: Ion chemistry relevant for chemical ionization detection of SO3, J. Geophys. Res-Atmos., 100, 14141–14146, https://doi.org/10.1029/95JD01004, 1995.
Artiglia, L., Edebeli, J., Orlando, F., Chen, S., Lee, M.-T., Corral Arroyo, P., Gilgen, A., Bartels-Rausch, T., Kleibert, A., Vazdar, M., Carignano, M. A., Francisco, J. S., Shepson, P. B., Gladich, I., and Ammann, M.: A surface-stabilized ozonide triggers bromide oxidation at the aqueous solution-vapour interface, Nat. Commun., 8, 700, https://doi.org/10.1038/s41467-017-00823-x, 2017.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume III – gas phase reactions of inorganic halogens, Atmos. Chem. Phys., 7, 981–1191, https://doi.org/10.5194/acp-7-981-2007, 2007.
Bannan, T. J., Khan, M. A. H., Le Breton, M., Priestley, M., Worrall, S. D., Bacak, A., Marsden, N. A., Lowe, D., Pitt, J., Allen, G., Topping, D., Coe, H., McFiggans, G., Shallcross, D. E., and Percival, C. J.: A Large Source of Atomic Chlorine From ClNO2 Photolysis at a U.K. Landfill Site, Geophys. Res. Lett., 46, 8508–8516, https://doi.org/10.1029/2019GL083764, 2019.
Bedjanian, Y. and Poulet, G.: Kinetics of halogen oxide radicals in the stratosphere, Chem. Rev., 103, 4639, https://doi.org/10.1021/cr0205210, 2003.
Brown, S. S. and Stutz, J.: Nighttime radical observations and chemistry, Chem. Soc. Rev., 41, 6405–6447, https://doi.org/10.1039/C2CS35181A, 2012.
Cai, J., Wang, J., Zhang, Y., Tian, H., Zhu, C., Gross, D. S., Hu, M., Hao, J., He, K., and Wang, S.: Source apportionment of Pb-containing particles in Beijing during January 2013, Environ. Pollut., 226, 30–40, https://doi.org/10.1016/j.envpol.2017.04.004, 2017.
Cai, J., Chu, B., Yao, L., Yan, C., Heikkinen, L. M., Zheng, F., Li, C., Fan, X., Zhang, S., Yang, D., Wang, Y., Kokkonen, T. V., Chan, T., Zhou, Y., Dada, L., Liu, Y., He, H., Paasonen, P., Kujansuu, J. T., Petäjä, T., Mohr, C., Kangasluoma, J., Bianchi, F., Sun, Y., Croteau, P. L., Worsnop, D. R., Kerminen, V.-M., Du, W., Kulmala, M., and Daellenbach, K. R.: Size-segregated particle number and mass concentrations from different emission sources in urban Beijing, Atmos. Chem. Phys., 20, 12721–12740, https://doi.org/10.5194/acp-20-12721-2020, 2020.
Calvert, J. and Lindberg, S.: The potential influence of iodine-containing compounds on the chemistry of the troposphere in the polar spring. II. Mercury depletion, Atmos. Environ., 38, 5105–5116, 2004.
Chen, D., Huey, L. G., Tanner, D. J., Salawitch, R. J., Anderson, D. C., Wales, P. A., Pan, L. L., Atlas, E. L., Hornbrook, R. S., Apel, E. C., Blake, N. J., Campos, T. L., Donets, V., Flocke, F. M., Hall, S. R., Hanisco, T. F., Hills, A. J., Honomichl, S. B., Jensen, J. B., Kaser, L., Montzka, D. D., Nicely, J. M., Reeves, J. M., Riemer, D. D., Schauffler, S. M., Ullmann, K., Weinheimer, A. J., and Wolfe, G. M.: Airborne measurements of BrO and the sum of HOBr and Br2 over the Tropical West Pacific from 1 to 15 km during the CONvective TRansport of Active Species in the Tropics (CONTRAST) experiment, J. Geophys. Res-Atmos., 121, 12560–12578, https://doi.org/10.1002/2016JD025561, 2016.
Chu, B. W., Dada, L., Liu, Y. C., Yao, L., Wang, Y. H., Du, W., Cai, J., Dallenbach, K. R., Chen, X. M., Simonen, P., Zhou, Y., Deng, C. J., Fu, Y. Y., Yin, R. J., Li, H. Y., He, X. C., Feng, Z. M., Yan, C., Kangasluoma, J., Bianchi, F., Jiang, J. K., Kujansuu, J., Kerminen, V. M., Petaja, T., He, H., and Kulmala, M.: Particle growth with photochemical age from new particle formation to haze in the winter of Beijing, China, Sci. Total Environ., 753, 142207, https://doi.org/10.1016/j.scitotenv.2020.142207, 2021.
Crisp, T., Lerner, B., Williams, E., Quinn, P., Bates, T., and Bertram, T.: Observations of gas-phase hydrochloric acid in the polluted marine boundary layer, J. Geophys. Res.-Atmos., 119, 6897–6915, https://doi.org/10.1002/2013JD020992, 2014.
Eisele, F. L. and Tanner, D. J.: Measurement of the Gas-Phase Concentration of H2SO4 and Methane Sulfonic-Acid and Estimates of H2SO4 Production and Loss in the Atmosphere, J. Geophys. Res.-Atmos., 98, 9001–9010, 1993.
Faxon, C. B. and Allen, D. T.: Chlorine chemistry in urban atmospheres: a review, Environ. Chem., 10, 221–233, https://doi.org/10.1071/EN13026, 2013.
Ferguson, E. E., Dunkin, D. B., and Fehsenfeld, F. C.: Reactions of NO and NO with HCl and HBr, J. Chem. Phys., 57, 1459–1463, 1972.
Fernandez, R. P., Salawitch, R. J., Kinnison, D. E., Lamarque, J.-F., and Saiz-Lopez, A.: Bromine partitioning in the tropical tropopause layer: implications for stratospheric injection, Atmos. Chem. Phys., 14, 13391–13410, https://doi.org/10.5194/acp-14-13391-2014, 2014.
Finlayson-Pitts, B. J., Ezell, M. J., and Pitts, J. N.: Formation of chemically active chlorine compounds by reactions of atmospheric NaCl particles with gaseous N2O5 and ClONO2, Nature, 337, 241–244, https://doi.org/10.1038/337241a0, 1989.
Fu, X., Wang, T., Wang, S., Zhang, L., Cai, S., Xing, J., and Hao, J.: Anthropogenic Emissions of Hydrogen Chloride and Fine Particulate Chloride in China, Environ. Sci. Technol., 52, 1644–1654, 2018.
Gard, E. E., Kleeman, M. J., Gross, D. S., Hughes, L. S., Allen, J. O., Morrical, B. D., Fergenson, D. P., Dienes, T., E. Gälli, M., Johnson, R. J., Cass, G. R., and Prather, K. A.: Direct Observation of Heterogeneous Chemistry in the Atmosphere, Science, 279, 1184, https://doi.org/10.1126/science.279.5354.1184, 1998.
Glasow, R. V. and Crutzen, P. J.: Tropospheric Halogen Chemistry, Treatise on Geochemistry, 5, 19–69, 2014.
Graedel, T. E. and Keene, W. C.: Tropospheric budget of reactive chlorine, Global Biogeochem. Cy., 9, 47–77, 1995.
Hara, H., Kato, T., and Matsushita, H.: The Mechanism of Seasonal Variation in the Size Distributions of Atmospheric Chloride and Nitrate Aerosol in Tokyo, B. Chem. Soc. Jpn., 62, 2643–2649, https://doi.org/10.1246/bcsj.62.2643, 1989.
Haskins, J. D., Jaeglé, L., Shah, V., Lee, B. H., Lopez-Hilfiker, F. D., Campuzano-Jost, P., Schroder, J. C., Day, D. A., Guo, H., Sullivan, A. P., Weber, R., Dibb, J., Campos, T., Jimenez, J. L., Brown, S. S., and Thornton, J. A.: Wintertime Gas-Particle Partitioning and Speciation of Inorganic Chlorine in the Lower Troposphere Over the Northeast United States and Coastal Ocean, J. Geophys. Res.-Atmos., 123, 12897–12916, https://doi.org/10.1029/2018JD028786, 2018.
He, K. B., Yang, F. M., Ma, Y. L., Zhang, Q., Yao, X. H., Chan, C. K., Cadle, S., Chan, T., and Mulawa, P.: The characteristics of PM2.5 in Beijing, China, Atmos. Environ., 35, 4959–4970, https://doi.org/10.1016/s1352-2310(01)00301-6, 2001.
Hoffmann, E., Tilgner, A., Wolke, R., and Herrmann, H.: Enhanced Chlorine and Bromine Atom Activation by Hydrolysis of Halogen Nitrates from Marine Aerosols at Polluted Coastal Areas, Environ. Sci. Technol., 53, 771–778, https://doi.org/10.1021/acs.est.8b05165, 2018.
Hu, W., Hu, M., Hu, W., Jimenez, J., Yuan, B., Chen, W., Wang, M., Wu, Y., Chen, C., Wang, Z., Peng, J., Zeng, L., and Shao, M.: Chemical composition, sources, and aging process of submicron aerosols in Beijing: Contrast between summer and winter, J. Geophys. Res.-Atmos., 121, 1955–1977, https://doi.org/10.1002/2015JD024020, 2016.
Hu, W., Hu, M., Hu, W.-W., Zheng, J., Chen, C., Wu, Y., and Guo, S.: Seasonal variations in high time-resolved chemical compositions, sources, and evolution of atmospheric submicron aerosols in the megacity Beijing, Atmos. Chem. Phys., 17, 9979–10000, https://doi.org/10.5194/acp-17-9979-2017, 2017.
Junninen, H., Ehn, M., Petäjä, T., Luosujärvi, L., Kotiaho, T., Kostiainen, R., Rohner, U., Gonin, M., Fuhrer, K., Kulmala, M., and Worsnop, D. R.: A high-resolution mass spectrometer to measure atmospheric ion composition, Atmos. Meas. Tech., 3, 1039–1053, https://doi.org/10.5194/amt-3-1039-2010, 2010.
Keene, W. C., Khalil, M. A. K., Erickson, D. J., McCulloch, A., Graedel, T. E., Lobert, J. M., Aucott, M. L., Gong, S. L., Harper, D. B., Kleiman, G., Midgley, P., Moore, R. M., Seuzaret, C., Sturges, W. T., Benkovitz, C. M., Koropalov, V., Barrie, L. A., and Li, Y. F.: Composite global emissions of reactive chlorine from anthropogenic and natural sources: Reactive Chlorine Emissions Inventory, J. Geophys. Res.-Atmos., 104, 8429–8440, https://doi.org/10.1029/1998jd100084, 1999.
Kontkanen, J., Deng, C., Fu, Y., Dada, L., Zhou, Y., Cai, J., Daellenbach, K. R., Hakala, S., Kokkonen, T. V., Lin, Z., Liu, Y., Wang, Y., Yan, C., Petäjä, T., Jiang, J., Kulmala, M., and Paasonen, P.: Size-resolved particle number emissions in Beijing determined from measured particle size distributions, Atmos. Chem. Phys., 20, 11329–11348, https://doi.org/10.5194/acp-20-11329-2020, 2020.
Lammel, G., Röhrl, A., and Schreiber, H.: Atmospheric lead and bromine in Germany, Environ. Sci. Pollut. Res., 9, 397, https://doi.org/10.1007/BF02987589, 2002.
Le Breton, M., Hallquist, Å. M., Pathak, R. K., Simpson, D., Wang, Y., Johansson, J., Zheng, J., Yang, Y., Shang, D., Wang, H., Liu, Q., Chan, C., Wang, T., Bannan, T. J., Priestley, M., Percival, C. J., Shallcross, D. E., Lu, K., Guo, S., Hu, M., and Hallquist, M.: Chlorine oxidation of VOCs at a semi-rural site in Beijing: significant chlorine liberation from ClNO2 and subsequent gas- and particle-phase Cl–VOC production, Atmos. Chem. Phys., 18, 13013–13030, https://doi.org/10.5194/acp-18-13013-2018, 2018.
Lee, B. H., Lopez-Hilfiker, F. D., Schroder, J. C., Campuzano-Jost, P., Jimenez, J. L., McDuffie, E. E., Fibiger, D. L., Veres, P. R., Brown, S. S., Campos, T. L., Weinheimer, A. J., Flocke, F. F., Norris, G., O'Mara, K., Green, J. R., Fiddler, M. N., Bililign, S., Shah, V., Jaegle, L., and Thornton, J. A.: Airborne Observations of Reactive Inorganic Chlorine and Bromine Species in the Exhaust of Coal-Fired Power Plants, J. Geophys. Res.-Atmos., 123, 11225–11237, https://doi.org/10.1029/2018JD029284, 2018.
Leslie, M. D., Ridoli, M., Murphy, J. G., and Borduas-Dedekind, N.: Isocyanic acid (HNCO) and its fate in the atmosphere: a review, Environ. Sci.: Processes Impacts, 21, 793–808, https://doi.org/10.1039/C9EM00003H, 2019.
Li, Q., Badia, A., Wang, T., Sarwar, G., Fu, X., Zhang, L., Zhang, Q., Fung, J., Cuevas, C. A., Wang, S., Zhou, B., and Saiz-Lopez, A.: Potential Effect of Halogens on Atmospheric Oxidation and Air Quality in China, J. Geophys. Res.-Atmos., 125, e2019JD032058, https://doi.org/10.1029/2019JD032058, 2020.
Liu, X., Qu, H., Huey, L. G., Wang, Y., Sjostedt, S., Zeng, L., Lu, K., Wu, Y., Hu, M., Shao, M., Zhu, T., and Zhang, Y.: High Levels of Daytime Molecular Chlorine and Nitryl Chloride at a Rural Site on the North China Plain, Environ. Sci. Technol., 51, 9588–9595, https://doi.org/10.1021/acs.est.7b03039, 2017.
Manö, S. and Andreae, M. O.: Emission of Methyl Bromide from Biomass Burning, Science, 263, 1255–1257, 1994.
McNamara, S. M., Kolesar, K. R., Wang, S., Kirpes, R. M., May, N. W., Gunsch, M. J., Cook, R. D., Fuentes, J. D., Hornbrook, R. S., Apel, E. C., China, S., Laskin, A., and Pratt, K. A.: Observation of Road Salt Aerosol Driving Inland Wintertime Atmospheric Chlorine Chemistry, ACS Central Science, 6, 684–694, https://doi.org/10.1021/acscentsci.9b00994, 2020.
Mielke, L. H., Furgeson, A., and Osthoff, H. D.: Observation of ClNO2 in a Mid-Continental Urban Environment, Environ. Sci. Technol., 45, 8889–8896, https://doi.org/10.1021/es201955u, 2011.
Moyers, J. L. and Duce, R. A.: Gaseous and particulate bromine in the marine atmosphere, J. Geophys. Res., 77, 5330–5338, https://doi.org/10.1029/JC077i027p05330, 1972.
Nolt, I. G., Ade, P., Alboni, F., Carli, B., Carlotti, M., Cortesi, U., Epifani, M., Griffin, M. J., Hamilton, P. A., Lee, C., Lepri, G., Mencaraglia, F., Murray, A. G., Park, J. H., Park, K., Raspollini, P., Ridolfi, M., and Vanek, M. D.: Stratospheric HBr concentration profile obtained from far-infrared emission spectroscopy, Geophys. Res. Lett., 24, 281–284, https://doi.org/10.1029/97GL00034, 1997.
Osthoff, H. D., Roberts, J. M., Ravishankara, A. R., Williams, E. J., Lerner, B. M., Sommariva, R., Bates, T. S., Coffman, D., Quinn, P. K., Dibb, J. E., Stark, H., Burkholder, J. B., Talukdar, R. K., Meagher, J., Fehsenfeld, F. C., and Brown, S. S.: High levels of nitryl chloride in the polluted subtropical marine boundary layer, Nat. Geosci., 1, 324–328, https://doi.org/10.1038/ngeo177, 2008.
Peng, X., Wang, W., Xia, M., Chen, H., Ravishankara, A., Li, Q., Alfonso, S.-L., Liu, P., Zhang, F., Zhang, C., Xue, L., Wang, X., George, C., Wang, J., Mu, Y., Chen, J.-M., and Tao, W.: An unexpected large continental source of reactive bromine and chlorine with significant impact on wintertime air quality, Natl. Sci. Rev., nwaa304, https://doi.org/10.1093/nsr/nwaa304, 2020.
Phillips, G. J., Tang, M. J., Thieser, J., Brickwedde, B., Schuster, G., Bohn, B., Lelieveld, J., and Crowley, J. N.: Significant concentrations of nitryl chloride observed in rural continental Europe associated with the influence of sea salt chloride and anthropogenic emissions, Geophys. Res. Lett., 39, L10811, https://doi.org/10.1029/2012GL051912, 2012.
Priestley, M., Breton, M., Bannan, T., Leather, K., Bacak, A., Reyes Villegas, E., Vocht, F., Shallcross, B., Brazier, T., Khan, A., Allan, J., Shallcross, D., Coe, H., and Percival, C.: Observations of Isocyanate, Amide, Nitrate, and Nitro Compounds From an Anthropogenic Biomass Burning Event Using a ToF-CIMS, J. Geophys. Res.-Atmos., 123, 7687–7704, https://doi.org/10.1002/2017JD027316, 2018.
Riedel, T. P., Bertram, T. H., Crisp, T. A., Williams, E. J., Lerner, B. M., Vlasenko, A., Li, S.-M., Gilman, J., de Gouw, J., Bon, D. M., Wagner, N. L., Brown, S. S., and Thornton, J. A.: Nitryl Chloride and Molecular Chlorine in the Coastal Marine Boundary Layer, Environ. Sci. Technol., 46, 10463–10470, https://doi.org/10.1021/es204632r, 2012.
Riedel, T. P., Wagner, N. L., Dubé, W. P., Middlebrook, A. M., Young, C. J., Öztürk, F., Bahreini, R., VandenBoer, T. C., Wolfe, D. E., Williams, E. J., Roberts, J. M., Brown, S. S., and Thornton, J. A.: Chlorine activation within urban or power plant plumes: Vertically resolved ClNO2 and Cl2 measurements from a tall tower in a polluted continental setting, J. Geophys. Res.-Atmos., 118, 8702–8715, https://doi.org/10.1002/jgrd.50637, 2013.
Roberts, T., Dayma, G., and Oppenheimer, C.: Reaction Rates Control High-Temperature Chemistry of Volcanic Gases in Air, Front. Earth Sci., 7, 154, https://doi.org/10.3389/feart.2019.00154, 2019.
Rotermund, M. K., Bense, V., Chipperfield, M. P., Engel, A., Grooß, J.-U., Hoor, P., Hüneke, T., Keber, T., Kluge, F., Schreiner, B., Schuck, T., Vogel, B., Zahn, A., and Pfeilsticker, K.: Organic and inorganic bromine measurements around the extratropical tropopause and lowermost stratosphere: Insights into the transport pathways and total bromine, Atmos. Chem. Phys. Discuss. [preprint], https://doi.org/10.5194/acp-2021-202, in review, 2021.
Saiz-Lopez, A. and von Glasow, R.: Reactive halogen chemistry in the troposphere, Chem. Soc. Rev., 41, 6448–6472, https://doi.org/10.1039/c2cs35208g, 2012.
Saiz-Lopez, A., Lamarque, J.-F., Kinnison, D. E., Tilmes, S., Ordóñez, C., Orlando, J. J., Conley, A. J., Plane, J. M. C., Mahajan, A. S., Sousa Santos, G., Atlas, E. L., Blake, D. R., Sander, S. P., Schauffler, S., Thompson, A. M., and Brasseur, G.: Estimating the climate significance of halogen-driven ozone loss in the tropical marine troposphere, Atmos. Chem. Phys., 12, 3939–3949, https://doi.org/10.5194/acp-12-3939-2012, 2012.
Sander, R., Keene, W. C., Pszenny, A. A. P., Arimoto, R., Ayers, G. P., Baboukas, E., Cainey, J. M., Crutzen, P. J., Duce, R. A., Hönninger, G., Huebert, B. J., Maenhaut, W., Mihalopoulos, N., Turekian, V. C., and Van Dingenen, R.: Inorganic bromine in the marine boundary layer: a critical review, Atmos. Chem. Phys., 3, 1301–1336, https://doi.org/10.5194/acp-3-1301-2003, 2003.
Sarwar, G., Simon, H., Xing, J., and Mathur, R.: Importance of tropospheric ClNO2 chemistry across the Northern Hemisphere, Geophys. Res. Lett., 41, 4050–4058, https://doi.org/10.1002/2014GL059962, 2014.
Sherwen, T., Evans, M. J., Sommariva, R., Hollis, L. D. J., Ball, S. M., Monks, P. S., Reed, C., Carpenter, L. J., Lee, J. D., Forster, G., Bandy, B., Reeves, C. E., and Bloss, W. J.: Effects of halogens on European air-quality, Faraday Discuss., 200, 75–100, https://doi.org/10.1039/C7FD00026J, 2017.
Simpson, W., Brown, S., Alfonso, S. L., Thornton, J., and Glasow, R.: Tropospheric Halogen Chemistry: Sources, Cycling, and Impacts, Chem. Rev., 115, 4035–4062, https://doi.org/10.1021/cr5006638, 2015.
Skalny, J., Mikoviny, T., Matejcik, S., and Mason, N.: An analysis of mass spectrometric study of negative ions extracted from negative corona discharge in air, Int. J. Mass Spectrom., 233, 317–324, https://doi.org/10.1016/j.ijms.2004.01.012, 2004.
Stavrakou, T., Müller, J.-F., Boersma, K. F., van der A, R. J., Kurokawa, J., Ohara, T., and Zhang, Q.: Key chemical NOx sink uncertainties and how they influence top-down emissions of nitrogen oxides, Atmos. Chem. Phys., 13, 9057–9082, https://doi.org/10.5194/acp-13-9057-2013, 2013.
Sun, Y., Du, W., Fu, P., Wang, Q., Li, J., Ge, X., Zhang, Q., Zhu, C., Ren, L., Xu, W., Zhao, J., Han, T., Worsnop, D. R., and Wang, Z.: Primary and secondary aerosols in Beijing in winter: sources, variations and processes, Atmos. Chem. Phys., 16, 8309–8329, https://doi.org/10.5194/acp-16-8309-2016, 2016.
Tham, Y. J., Yan, C., Xue, L., Zha, Q., Wang, X., and Wang, T.: Presence of high nitryl chloride in Asian coastal environment and its impact on atmospheric photochemistry, Chinese Sci. Bull., 59, 356–359, https://doi.org/10.1007/s11434-013-0063-y, 2014.
Tham, Y. J., Wang, Z., Li, Q., Yun, H., Wang, W., Wang, X., Xue, L., Lu, K., Ma, N., Bohn, B., Li, X., Kecorius, S., Größ, J., Shao, M., Wiedensohler, A., Zhang, Y., and Wang, T.: Significant concentrations of nitryl chloride sustained in the morning: investigations of the causes and impacts on ozone production in a polluted region of northern China, Atmos. Chem. Phys., 16, 14959–14977, https://doi.org/10.5194/acp-16-14959-2016, 2016.
Tham, Y. J., Wang, Z., Li, Q., Wang, W., Wang, X., Lu, K., Ma, N., Yan, C., Kecorius, S., Wiedensohler, A., Zhang, Y., and Wang, T.: Heterogeneous N2O5 uptake coefficient and production yield of ClNO2 in polluted northern China: roles of aerosol water content and chemical composition, Atmos. Chem. Phys., 18, 13155–13171, https://doi.org/10.5194/acp-18-13155-2018, 2018.
Thornton, J. A., Kercher, J. P., Riedel, T. P., Wagner, N. L., Cozic, J., Holloway, J. S., Dubé, W. P., Wolfe, G. M., Quinn, P. K., and Middlebrook, A. M.: A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry, Nature, 464, 271–274, 2010.
Tian, Q., Xu, D., Chai, Z., Lu, Y., and Xiong, Y.: Analysis on the organic bromine in the atmosphere in Beijing, Journal of Nuclear and Radiochemistry, 27, 236–238, https://doi.org/10.3969/j.issn.0253-9950.2005.04.010, 2005.
Vigouroux, C., Stavrakou, T., Whaley, C., Dils, B., Duflot, V., Hermans, C., Kumps, N., Metzger, J.-M., Scolas, F., Vanhaelewyn, G., Müller, J.-F., Jones, D. B. A., Li, Q., and De Mazière, M.: FTIR time-series of biomass burning products (HCN, C2H6, C2H2, CH3OH, and HCOOH) at Reunion Island (21∘ S, 55∘ E) and comparisons with model data, Atmos. Chem. Phys., 12, 10367–10385, https://doi.org/10.5194/acp-12-10367-2012, 2012.
Wang, D. S. and Ruiz, L. H.: Secondary organic aerosol from chlorine-initiated oxidation of isoprene, Atmos. Chem. Phys., 17, 13491–13508, https://doi.org/10.5194/acp-17-13491-2017, 2017.
Wang, X., Jacob, D. J., Eastham, S. D., Sulprizio, M. P., Zhu, L., Chen, Q., Alexander, B., Sherwen, T., Evans, M. J., Lee, B. H., Haskins, J. D., Lopez-Hilfiker, F. D., Thornton, J. A., Huey, G. L., and Liao, H.: The role of chlorine in global tropospheric chemistry, Atmos. Chem. Phys., 19, 3981–4003, https://doi.org/10.5194/acp-19-3981-2019, 2019.
Wang, Y. Q.: MeteoInfo: GIS software for meteorological data visualization and analysis, Meteorol. Appl., 21, 360–368, https://doi.org/10.1002/met.1345, 2014.
Wang, Y. Q.: An Open Source Software Suite for Multi-Dimensional Meteorological Data Computation and Visualisation, Journal of Open Research Software, 7, p. 21, https://doi.org/10.5334/jors.267, 2019.
Wang, Z., Wang, W., Tham, Y. J., Li, Q., Wang, H., Wen, L., Wang, X., and Wang, T.: Fast heterogeneous N2O5 uptake and ClNO2 production in power plant and industrial plumes observed in the nocturnal residual layer over the North China Plain, Atmos. Chem. Phys., 17, 12361–12378, https://doi.org/10.5194/acp-17-12361-2017, 2017.
Wang, Z., Yuan, B., Ye, C., Roberts, J., Wisthaler, A., Lin, Y., Li, T., Wu, C., Peng, Y., Wang, C., Wang, S., Yang, S., Wang, B., Qi, J., Wang, C., Song, W., Hu, W., Wang, X., Xu, W., Ma, N., Kuang, Y., Tao, J., Zhang, Z., Su, H., Cheng, Y., Wang, X., and Shao, M.: High Concentrations of Atmospheric Isocyanic Acid (HNCO) Produced from Secondary Sources in China, Environ. Sci. Tech., 54, 11818–11826, https://doi.org/10.1021/acs.est.0c02843, 2020.
Wren, S. N., Liggio, J., Han, Y., Hayden, K., Lu, G., Mihele, C. M., Mittermeier, R. L., Stroud, C., Wentzell, J. J. B., and Brook, J. R.: Elucidating real-world vehicle emission factors from mobile measurements over a large metropolitan region: a focus on isocyanic acid, hydrogen cyanide, and black carbon, Atmos. Chem. Phys., 18, 16979–17001, https://doi.org/10.5194/acp-18-16979-2018, 2018.
Xia, M., Peng, X., Wang, W., Yu, C., Sun, P., Li, Y., Liu, Y., Xu, Z., Wang, Z., Xu, Z., Nie, W., Ding, A., and Wang, T.: Significant production of ClNO2 and possible source of Cl2 from N2O5 uptake at a suburban site in eastern China, Atmos. Chem. Phys., 20, 6147–6158, https://doi.org/10.5194/acp-20-6147-2020, 2020.
Yang, X., Cox, R., Warwick, N., Pyle, J., Carver, G., O'Connor, F., and Savage, N.: Tropospheric bromine chemistry and its impacts on ozone: A model study, J. Geophys. Res., 110, D23311, https://doi.org/10.1029/2005JD006244, 2005.
Yang, X., Wang, T., Xia, M., Gao, X., Li, Q., Zhang, N., Gao, Y., Lee, S., Wang, X., Xue, L., Yang, L., and Wang, W.: Abundance and origin of fine particulate chloride in continental China, Sci. Total Environ., 624, 1041–1051, https://doi.org/10.1016/j.scitotenv.2017.12.205, 2018.
Yao, L., Garmash, O., Bianchi, F., Zheng, J., Yan, C., Kontkanen, J., Junninen, H., Mazon, S. B., Ehn, M., Paasonen, P., Sipila, M., Wang, M., Wang, X., Xiao, S., Chen, H., Lu, Y., Zhang, B., Wang, D., Fu, Q., Geng, F., Li, L., Wang, H., Qiao, L., Yang, X., Chen, J., Kerminen, V. M., Petaja, T., Worsnop, D. R., Kulmala, M., and Wang, L.: Atmospheric new particle formation from sulfuric acid and amines in a Chinese megacity, Science, 361, 278–281, https://doi.org/10.1126/science.aao4839, 2018.
Yao, L., Fan, X., Yan, C., Kurten, T., Daellenbach, K. R., Li, C., Wang, Y., Guo, Y., Dada, L., Rissanen, M. P., Cai, J., Tham, Y. J., Zha, Q., Zhang, S., Du, W., Yu, M., Zheng, F., Zhou, Y., Kontkanen, J., Chan, T., Shen, J., Kujansuu, J. T., Kangasluoma, J., Jiang, J., Wang, L., Worsnop, D. R., Petaja, T., Kerminen, V. M., Liu, Y., Chu, B., He, H., Kulmala, M., and Bianchi, F.: Unprecedented Ambient Sulfur Trioxide (SO3) Detection: Possible Formation Mechanism and Atmospheric Implications, Environ. Sci. Tech. Let., 7, 809–818, https://doi.org/10.1021/acs.estlett.0c00615, 2020.
Zhou, W., Zhao, J., Ouyang, B., Mehra, A., Xu, W., Wang, Y., Bannan, T. J., Worrall, S. D., Priestley, M., Bacak, A., Chen, Q., Xie, C., Wang, Q., Wang, J., Du, W., Zhang, Y., Ge, X., Ye, P., Lee, J. D., Fu, P., Wang, Z., Worsnop, D., Jones, R., Percival, C. J., Coe, H., and Sun, Y.: Production of N2O5 and ClNO2 in summer in urban Beijing, China, Atmos. Chem. Phys., 18, 11581–11597, https://doi.org/10.5194/acp-18-11581-2018, 2018.
Zhou, Y., Dada, L., Liu, Y., Fu, Y., Kangasluoma, J., Chan, T., Yan, C., Chu, B., Daellenbach, K. R., Bianchi, F., Kokkonen, T. V., Liu, Y., Kujansuu, J., Kerminen, V.-M., Petäjä, T., Wang, L., Jiang, J., and Kulmala, M.: Variation of size-segregated particle number concentrations in wintertime Beijing, Atmos. Chem. Phys., 20, 1201–1216, https://doi.org/10.5194/acp-20-1201-2020, 2020.