the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 22 Jul 2021
| 22 Jul 2021
Impact of stratospheric air and surface emissions on tropospheric nitrous oxide during ATom
Yenny Gonzalez
Ethan Manninen
Bruce C. Daube
Luke D. Schiferl
J. Barry McManus
Kathryn McKain
Eric J. Hintsa
James W. Elkins
Stephen A. Montzka
Colm Sweeney
Fred Moore
Jose L. Jimenez
Pedro Campuzano Jost
Thomas B. Ryerson
Ilann Bourgeois
Jeff Peischl
Chelsea R. Thompson
Paul O. Wennberg
John Crounse
Michelle Kim
Hannah M. Allen
Paul A. Newman
Britton B. Stephens
Eric C. Apel
Rebecca S. Hornbrook
Benjamin A. Nault
Eric Morgan
Steven C. Wofsy
We measured the global distribution of tropospheric N2O mixing ratios during the NASA airborne Atmospheric Tomography (ATom) mission. ATom measured concentrations of ∼ 300 gas species and aerosol properties in 647 vertical profiles spanning the Pacific, Atlantic, Arctic, and much of the Southern Ocean basins, nearly from pole to pole, over four seasons (2016–2018). We measured N2O concentrations at 1 Hz using a quantum cascade laser spectrometer (QCLS). We introduced a new spectral retrieval method to account for the pressure and temperature sensitivity of the instrument when deployed on aircraft. This retrieval strategy improved the precision of our ATom QCLS N2O measurements by a factor of three (based on the standard deviation of calibration measurements). Our measurements show that most of the variance of N2O mixing ratios in the troposphere is driven by the influence of N2O-depleted stratospheric air, especially at mid- and high latitudes. We observe the downward propagation of lower N2O mixing ratios (compared to surface stations) that tracks the influence of stratosphere–troposphere exchange through the tropospheric column down to the surface. The highest N2O mixing ratios occur close to the Equator, extending through the boundary layer and free troposphere. We observed influences from a complex and diverse mixture of N2O sources, with emission source types identified using the rich suite of chemical species measured on ATom and the geographical origin calculated using an atmospheric transport model. Although ATom flights were mostly over the oceans, the most prominent N2O enhancements were associated with anthropogenic emissions, including from industry (e.g., oil and gas), urban sources, and biomass burning, especially in the tropical Atlantic outflow from Africa. Enhanced N2O mixing ratios are mostly associated with pollution-related tracers arriving from the coastal area of Nigeria. Peaks of N2O are often associated with indicators of photochemical processing, suggesting possible unexpected source processes. In most cases, the results show how difficult it is to separate the mixture of different sources in the atmosphere, which may contribute to uncertainties in the N2O global budget. The extensive data set from ATom will help improve the understanding of N2O emission processes and their representation in global models.
- Article
(6864 KB) - Full-text XML
-
Supplement
(6285 KB) - BibTeX
- EndNote




Nitrous oxide (N2O) is a powerful greenhouse gas and, due to its oxidation to NOx, a major contributor to both stratospheric ozone loss and to the passivation of stratospheric oxy-halogen radicals (Forster et al., 2007; Ravishankara et al., 2009). The rate of increase in atmospheric N2O since the Industrial Revolution, 0.93 ppb yr−1, implies a significant (∼ 30 %) imbalance between emission rates and destruction in the stratosphere. Seasonal cycles in tropospheric N2O are driven by both stratosphere-to-troposphere exchange and surface emissions (Nevison et al., 2011; Assonov et al., 2013; Thompson et al., 2014a). Most N2O emissions are attributed to microbial nitrification and denitrification in natural and cultivated soils, freshwaters, and oceans plus emissions related to human activities, such as biomass burning and industrial emissions (Butterbach-Bahl et al., 2013; Saikawa et al., 2014; Thompson et al., 2014a; Upstill-Goddard et al., 2017; WMO, 2018).
Much effort has been made to reduce the uncertainties in the individual components of the N2O global budget (e.g., Tian et al., 2012, 2020; Xiang et al., 2013; Thompson et al., 2014a, b; Ganesan et al., 2020; Yang et al., 2020). Recent estimates of global total N2O emissions to the atmosphere from bottom-up and top-down methods average 17 Tg N yr−1 (12.2–23.5 from bottom-up analysis and 15.9–17.7 Tg N yr−1 from top-down approaches, Tian et al., 2020). The most recent estimates of the global ocean emissions of N2O range between 2.5 and 4.3 Tg N yr−1 (∼ 20 % of total emissions), with the tropics, upwelling coastal areas, and subpolar regions the major contributors to these fluxes (Yang et al., 2020; Tian et al., 2020). However, the magnitude of marine N2O emissions is subject to large uncertainty due to spatial and temporal heterogeneity (Nevison et al., 1995, 2005; Ganesan et al., 2020; Yang et al., 2020). According to Tian et al. (2020), anthropogenic sources account for ∼ 43 % of global N2O emissions (7.3 Tg N yr−1), with industry and biomass burning emissions estimated to be 1.6–1.9 Tg N yr−1, respectively (Syakila and Kroeze, 2011; Tian et al., 2020) and the rest originating from agriculture. N2O emissions from biogenic sources and fires in Africa are estimated at 3.3 ± 1.3 Tg N2O yr−1 (Valentini et al., 2014). Agricultural N2O emission estimates (up to ∼ 37 %) range between 2.5 and 5.8 Tg N yr−1, and between 4.9 and 6.5 Tg N yr−1 in the case of natural soils (Kort et al., 2008, 2010; Syakila and Kroeze, 2011; Tian et al., 2020). Recent estimates of N2O emissions from fertilized tropical and subtropical agricultural systems are 3 ± 5 kg N ha−1 yr−1 (Albanito et al., 2017). Most of these estimates are derived from short-term local-scale in-situ measurements and are difficult to extrapolate with confidence to large regions or to the globe.
In the atmosphere, N2O is destroyed by oxidation (10 %, O(1D) reaction) and photolysis (90 %, 190–230 nm photolysis) in the upper stratosphere (> 20 km altitude; SPARC, 2013), which makes it a good candidate for tracing the air exchange between the stratosphere and the troposphere (Hintsa et al., 1998; Nevison et al., 2011; Assonov et al., 2013; Krause et al., 2018). Atmospheric models tend to underestimate the interhemispheric N2O gradient, which Thompson et al. (2014a) attribute to an overestimation of N2O emissions in the Southern Ocean, an underestimate of Northern Hemisphere emissions, and/or an overestimate of stratosphere-to-troposphere exchange in the Northern Hemisphere. Overall, the largest uncertainties in modeled N2O emissions are found in tropical South America and South Asia (Thompson et al., 2014b).
We present atmospheric N2O altitude profiles at high temporal resolution collected during the NASA Atmospheric Tomography (ATom) mission. ATom was a global-scale airborne deployment conducted over a 3 year period (2016–2018) using the NASA DC-8 aircraft. In ATom, the DC-8 flew vertical profiles (0.2–13 km) nearly continuously almost from pole to pole while measuring mixing ratios of ∼ 300 trace gases and aerosol physical and chemical properties over the Pacific and Atlantic basins and during each of the four seasons. Each deployment (1–4) started and ended in Palmdale (California, USA) and generally consisted of a loop southward from the Arctic through the central Pacific, across the Southern Ocean to South America, northward through the Atlantic, and across Greenland and the Arctic Ocean. During ATom-3 and -4, two additional flights from Punta Arenas (Chile) sampled the Antarctic troposphere and upper troposphere/lower stratosphere (UT/LS) to 80∘ S.
In this work, we focus on the measurements taken during January–February 2017 (ATom-2), September–October 2017 (ATom-3), and April–May 2018 (ATom-4) (no quantum cascade laser spectrometer (QCLS) N2O data are available for ATom-1 in August 2016). The motivation for this paper is twofold. Firstly, we present a new retrieval strategy to account for the pressure and temperature dependence of laser-based instruments, and specifically for the use of quantum cascade laser spectrometers on aircraft. Secondly, we report on the global distribution of N2O from the surface to 13 km and examine the processes contributing to the variability of tropospheric N2O based on the vertical profiles of N2O and a broad variety of covariate chemical species and aerosol properties.
2.1 Specifications of QCLS
We measured N2O mixing ratios with the Harvard/NCAR/Aerodyne Research Inc. Quantum Cascade Laser Spectrometer (QCLS). This instrument was previously deployed on the NCAR/NSF Gulfstream V for the HIAPER Pole-to-Pole Observations mission (HIPPO, Wofsy et al., 2011; https://www.eol.ucar.edu/field_projects/hippo, last access: 14 October 2020) and the O2 N2 Ratio and CO2 Southern Ocean Study (ORCAS, Stephens et al., 2018; https://www.eol.ucar.edu/field_projects/orcas, last access: 14 October 2020). Detailed information about the spectrometer configuration can be found in Jiménez et al. (2005, 2006) and Santoni et al. (2014). A brief description follows.
QCLS provides continuous (1 Hz) measurements of N2O, methane (CH4), and carbon monoxide (CO) using two thermoelectrically cooled pulsed quantum cascade lasers, a 76 m pathlength multiple-pass absorption cell (∼ 0.5 L volume), and two liquid-nitrogen-cooled solid-state HgCdTe detectors. All these components are mounted on a temperature-stabilized, vibrationally isolated optical bench. The temperature in QCLS is controlled by Peltier elements coupled with a closed-circuit recirculating fluid kept at 288.0 ± 0.1 K. QCLS measures CH4 and N2O by scanning the spectral interval of 1275.45 ± 0.15 cm−1. A second laser is used to scan CO at 2169.15 ± 0.15 cm−1. The supply currents to QCLS are ramped at a rate of 3.8 kHz to scan the laser frequency for 200 channels (steps in frequency) in laser 1 and 50 channels in laser 2; an extra 10 channels are used to measure the laser shut off (zero-light level). The spectra and fit residuals for CH4, N2O, and CO are shown in Fig. S1 of the Supplement. Mixing ratios are derived at a rate of 1 Hz by a least-squares spectral fit assuming a Voigt line profile at the pressure and temperature measured inside the sample cell and using molecular line parameters from the HIgh-resolution TRANsmission molecular absorption database (HITRAN, Rothman et al., 2005). The temperature and pressure inside the cell are monitored with a 30 kΩ thermistor and a capacitance manometer (133 hPa full scale), respectively.
During sampling, the air passes through a 50-tube Nafion drier to remove the bulk water vapor. A Teflon diaphragm pump downstream of the cell reduces the air pressure to ∼ 60 hPa. Both ambient air and calibration gases pass through a Teflon dry-ice trap to reduce the dew point to −70 ∘C. After ATom-1, we added a bypass between the inlet and the instrument to increase the flushing rate of the inlet and inlet tubing. The calibration sequence includes 2 min of ultra-high-purity zero air followed by 1 min each of low- and high-mixing ratio gases every 30 min (see Fig. S2). We measured zero air every 15 min during ATom-1 and -2, and every 30 min during ATom-3 and -4. A data logger (CR10X, Campbell Scientific) was used to automate the sampling sequence. The CR10X controlled the pressure controller on the cell and managed the data transfer.
We use gas cylinders traceable to the National Oceanic and Atmospheric Administration World Meteorological Organization scales for calibration (NOAA-WMO-X2004A scale for CH4, WMO-X2014A for CO, and NOAA-2006A for N2O). These gas standards were recalibrated before, during and after the deployments to maintain traceability. The low mixing ratio gas cylinder contained 298.5 ± 0.3 ppb of N2O, 1692.4 ± 0.2 ppb of CH4, and 119.1 ± 0.3 ppb of CO. The high mixing ratio gas cylinder contained 399.1 ± 0.3 ppb of N2O, 2182.5 ± 0.3 ppb of CH4, and 192.8 ± 0.5 ppb of CO. Detailed information on calibrations of the gas cylinders used during ATom is given in Table S1 of the Supplement.
QCLS also measures carbon dioxide (CO2) in a separate unit. Detailed information about QCLS CO2 measurements can be found in Santoni et al. (2014).
2.2 Spectral analysis and calibration
QCLS was damaged during shipping to the deployment site before the start of ATom-1, and the resulting alteration in the optical alignment modified the sensitivity of the instrument to temperature and pressure changes during aircraft maneuvers. This increased sensitivity was observed in all ATom deployments. At constant altitude, instrumental precision was similar to the precision measured during HIPPO (see the Allan–Werle variance analysis in Fig. 2 in Santoni et al., 2014 for HIPPO and Fig. S3 for ATom), but drifts were observed during altitude changes due to the effects of changes in cabin pressure and temperature on the spectral location of interference fringes that arise in the optical path outside the sample cell. In addition, flight altitude changes mechanically stressed the optical elements surrounding the cell, further modulating fringes or changing the shape of the detected laser intensity profile. These spectral artifacts ultimately reduced the accuracy of mixing ratios retrieved from spectral fitting. The spectral artifacts most strongly affected the measurements of CH4 and N2O. Several post-processing methods using the TDL-Wintel software were explored to improve the precision and accuracy of ATom QCLS N2O data, most with little success. Since the measured spectra were all saved, it is possible to refit the data with different fit parameters. A limited number of interference fringes may be included in the set of fitting functions. However, none of the previously used full refitting strategies significantly improved the data accuracy.
We have achieved significant improvements in the precision and accuracy of the ATom QCLS N2O data using a new method dubbed the “Neptune algorithm,” developed by Aerodyne Research, Inc., and that method has been further developed and applied to the data sets described here. Using this algorithm, the precision of the retrieved N2O data measured with the damaged QCLS was similar to that reported in HIPPO. The Neptune algorithm generates corrections to the mixing ratios retrieved from the original fits by associating specific spectral features with anomalies in retrieved mixing ratios observed during calibrations, i.e., during intervals when the mixing ratios are held constant. The spectral baseline is defined as the spectral channels outside the boundaries of the spectral lines of the target gas. Fluctuations in the spectral baselines are quantified for the entire data set by means of principal component analysis (PCA). PCA provides an efficient description of the spectral fluctuations, naturally producing an ordered set from the strongest to the weakest orthogonal vectors (spectral forms), each with an amplitude history spanning the data set. The PCAs are defined by an optimization procedure during calibrations, when mixing ratio fluctuations are designed to be ∼ 0. The finite fluctuations in retrieved mixing ratios during calibrations are fitted in the spectral space of the baseline as linear combinations of the leading PCA vector amplitudes, creating a linear combination of amplitudes of spectral fluctuations that predict errors in the mixing ratios for each gas for an entire flight. The error-producing linear combination of amplitudes of PCA spectral fluctuations produces a full set of anomaly estimates that are subtracted from the retrieved mixing ratios during the flight. The computational time for a 10 h long data set is only seconds, so variations in the algorithm's parameters (i.e., how many PCAs are retained) can be optimized rapidly.
The Neptune–PCA analysis improved the overall precision by a factor of four for CH4 and a factor of three in the case of N2O with respect to the precision of the original retrievals, as measured by the standard deviation of the retrieved mixing ratios during calibrations. The repeatability of the retrieved calibrations was 0.2 ppb for N2O and 1 ppb for CH4 (Fig. S4). The laser path of the CH4 N2O laser was realigned between ATom-1 and -2, and the Neptune retrieval was applied to CH4 and N2O measurements corresponding to the ATom-2, -3, and -4 deployments. Mixing ratios of CH4 and N2O could not be retrieved during ATom-1 because light levels were too low for the CH4 N2O laser due to the damage-induced misalignment.
The steps involved in the Neptune correction process were as follows:
-
We paired the mixing ratio records with the corresponding spectra (1 s resolution) for each species (CH4 and N2O).
-
We grouped the mixing ratios and spectra by type – into calibrations (zeros, low span, and high span) and air samples – and by time. The spectral data were thus arranged in an array with point number in the spectrum as x and spectrum number as y. We calculated an average spectrum for each group type and subtracted these from each individual spectrum within a group.
-
We zeroed out the spectral arrays at the positions of the absorption lines to concentrate on the fluctuations observed in the baseline and to prevent the PCA from finding line-depth fluctuations as relevant vectors during the calibrations. Some degree of smoothing (in x) was applied to the subtracted spectra so that high-frequency fluctuations, which have little influence on the mixing ratio determination, are not represented. An example of such a processed spectral array is shown in Fig. 1a.
-
We applied PCA to the whole line-zeroed spectral array to evaluate the fluctuations. PCA was applied in two steps: multiply the spectral array by its transpose to generate an autocovariance array and then perform singular value decomposition on the autocovariance array. The PCA generated an efficient description of how the baseline of the spectrum changed with cabin pressure and temperature. The description of spectral fluctuations consisted of a set of products of vectors and amplitudes.
-
We fitted the spectra to the PCAs to express mixing ratio fluctuations during the set of calibrations and zeros as a linear combination of PCA vector histories. The number of vector histories included in the fit is typically limited to less than 30 because the weaker PCA amplitudes tend to just describe random noise.
The linear combination of amplitudes that links spectral fluctuations in the baseline to mixing ratio fluctuations during calibrations was then applied to the full data set. That generated the retrieval errors for uncalibrated mixing ratios for the whole time series. We subtracted the errors from the initial retrievals from the TDLWintel-QCLS software and computed calibrated mixing ratios using the corrected retrievals for both calibrations and samples. An example of the result of applying the Neptune algorithm to the N2O samples and calibrations for the ATom-4 flight on 12 May 2018 is shown in Fig. 1b. The approach used here to minimize the effect of changes in pressure and temperature in optical instruments was based on the observation of fluctuations of the baseline during calibrations. Hence, this methodology does not provide any improvement in cases where altitude changes occurred during sampling but not during any of the calibrations for an individual flight. Due to frequent calibrations, we did not observe this rare scenario in the whole mission. To evaluate the ultimate accuracy of our measurements, we compared the QCLS N2O measurements with other onboard N2O measurements as well as with the surface N2O measurements of stations located along the flight tracks.
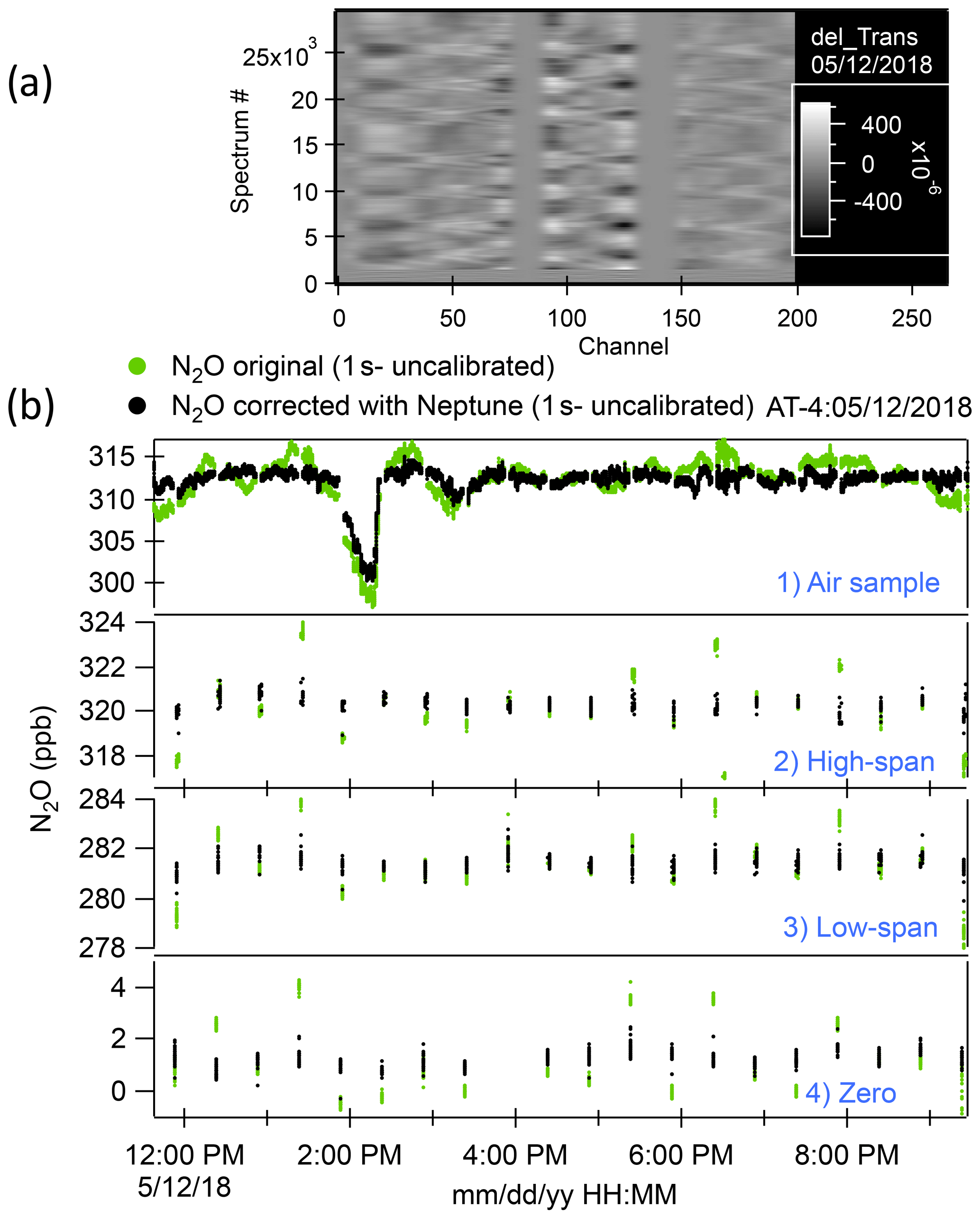
Figure 1(a) A processed spectral array from the ATom-4 flight on 12 May 2018. “Channel” represents a point number in the spectra. Spectra have been grouped by type (i.e., calibration or ambient), with averages subtracted, absorption lines zeroed out (near channels 75, 140, and 225). This residual spectral array is then smoothed with a binomial filter where the filter width corresponds to the linewidth of the original spectra. Shifts in fringe phases during altitude changes are apparent. (b) Time series of ambient air samples and high-span, low-span, and zero calibrations for the same flight as (a). Green dots are the original N2O data record. Black dots are the N2O data corrected with Neptune (no calibration applied at this point).

Figure 2(a) Comparisons between Neptune-corrected QCLS N2O and (1) UCATS N2O, (2) PANTHER N2O, and (3) PFP N2O for ATom-2 (orange circles), ATom-3 (green stars), and ATom-4 (blue squares). We used the 10 s averaged merged file to compare QCLS, UCATS, and PANTHER data. The PFP flask samples had longer sampling times (30 s to a few minutes). The 1:1 line is shown as a dashed line. (b) Comparisons between NOAA N2O surface flask measurements and Neptune-corrected and airborne data from (1) QCLS N2O, (2) UCATS N2O, (3) PANTHER N2O, (4) and PFP N2O for ATom-2, -3, and -4, similar to (a1)–(a3). The solid line shows the 1:1 relationship + offset. For plots (b1)–(b4), the airborne data are the mean N2O values within ± 5∘ of latitude of each surface station and between 1 and 4 km.
We evaluated N2O mixing ratios measured by QCLS against three other instruments that measured N2O on the NASA DC-8 aircraft during ATom. In addition, we compared the set of four airborne measurements to data from the flask sampling network at ground stations from the NOAA Global Monitoring Laboratory (GML, https://gml.noaa.gov/, last access: 10 December 2020) to evaluate the differences between the airborne data and the ground-based measurements in the NOAA reference network.
3.1 Comparison between airborne N2O measurements
Measurements of N2O on the DC-8 during ATom were obtained by four instruments: (i) the Unmanned Aircraft Systems Chromatograph for Atmospheric Trace Species (UCATS, Hintsa et al., 2021), (ii) the PAN and other Trace Hydrohalocarbon ExpeRiment (PANTHER; Moore et al., 2006; Wofsy et al., 2011), (iii) the Programmable Flask Package Whole Air Sampler (PFP; Montzka et al., 2019), and (iv) our 1 Hz QCLS.
We compared QCLS, PANTHER, and UCATS at 10 s intervals, as provided in the ATom merged file MER10_DC8_ATom-1.nc available at the Oak Ridge National Laboratory Distributed Active Archive Center (ORNL-DAAC, Wofsy et al., 2018, https://doi.org/10.3334/ORNLDAAC/1581, last access: 28 February 2021). The ATom file MER-PFP merged with the PFP sampling interval (also available in the above repository) was used to compare QCLS and PFP data. A one-to-one comparison between these instruments showed an approximately 1 ppb positive bias in N2O mixing ratios from QCLS (see Fig. 2a1–a3). The 95 % confidence interval of the mean difference for each pair (95 % C.I.) was 0.75 ± 0.04 ppb between QCLS and PANTHER, 1.13 ± 0.03 ppb between QCLS and UCATS, and 1.18 ± 0.09 ppb between QCLS and PFP, respectively, for the full data set (ATom-2, -3, and -4). Information about the coefficients of the linear fit for each instrument comparison and the 95 % C.I. of the difference for each pair are shown in Table S2. The offset that QCLS N2O shows against PFP N2O coincides with the offset already reported by Santoni et al. (2014) during HIPPO in 2009–2011, which may be attributed to our calibration procedure. PFP flasks are considered the reference measurement on board as the flasks are analyzed with excellent precision and accuracy.
3.2 Comparison between airborne and surface measurements of N2O
We evaluated the traceability of lower-troposphere N2O mixing ratios by ATom by comparing the four airborne instruments with the surface measurements of N2O from the NOAA flask sampling network. If a surface station was encountered within a latitude range of 5∘ north and south with respect to the flight track during a flight, that surface station was used in the study.
The mean value of N2O within that latitude grid of ± 5∘ and at instrument altitudes of 1–4 km was compared with the mean N2O observed at the surface station during the period ± 5 d relative to the flight (due to the non-daily frequency of flask samples). We chose the altitude range between 1 to 4 km to agree with the low free tropospheric conditions that characterized most of the selected ground stations. Information about the surface stations used here is shown in Table S3 of the Supplement.
The comparison of the whole data set (ATom-2, -3, -4) shows that, overall, QCLS and PANTHER overestimated N2O mixing ratios with respect to the surface data by 1.37 ± 0.35 and 0.44 ± 0.51 ppb (95 % C.I.), respectively. In contrast, UCATS and PFP showed low bias with respect to the surface data: 0.27 ± 0.37 and 0.008 ± 0.34 ppb (95 % C.I.), respectively (Fig. 2b1–b4). Due to the excellent agreement between PFP and the surface stations and the consistent offset that QCLS showed against PFP and the stations, the QCLS N2O data presented in the following sections of this publication were corrected by subtracting the offset with respect to the PFP data onboard in each deployment: 1.03 ± 0.13 ppb in AT-2, 1.49 ± 0.19 ppb in AT-3, and 1.18 ± 0.17 ppb in AT-4. The final official archive data file includes a new column where these corrections have been applied (N2O_QCLS_ad).
These results show the very close comparability of the ATom airborne N2O instruments (differences were < 0.5 ppb for UCATS and PANTHER instruments and 0 ppb in the case of PFP) relative to the surface stations and demonstrate the feasibility of using ATom N2O measurements to evaluate the impact of stratospheric air and meridional transport of N2O emissions on N2O tropospheric column measurements over the ocean basins. In the following section, we define the boundary conditions that were used to evaluate that impact, which were based on the NOAA Greenhouse Gas Marine Boundary Layer Reference from the NOAA GML Carbon Cycle Group (NOAA/ESRL GML CCGG, https://gml.noaa.gov/, last access: 10 December 2020). The NOAA-MBL N2O product is a synthetic latitude profile generated at 0.05 sine latitude and weekly resolution from individual flask measurements of marine boundary-layer surface stations distributed along the two ocean basins, and provides the scenario needed to evaluate the traceability of aircraft measurements relative to ground measurements at remote sites (https://gml.noaa.gov/ccgg/arc/?id=13, last access: 10 December 2020).
The vertical profiles of N2O from ATom provide a global overview of the N2O distribution in the troposphere, with observations performed over the Pacific and Atlantic basins. For this study, we do not include data collected over and close to land. In ATom, N2O ranged between 280 and 335 ppb over the oceans. In each season, the lowest N2O mixing ratios are observed at high latitudes (HL, > 60∘) in the UT/LS (8–12.5 km), in air transported downward from the stratosphere. The highest N2O mixing ratios are found close to the Equator (30∘ S–30∘ N, 326 to 335 ppb), and extend along the tropospheric column up to 6 km. They are influenced by convective activity over the tropical regions (Kort et al., 2011; Santoni et al., 2014). At mid-latitudes (ML, 30–60∘ N), tropospheric N2O values range between 322 and 333 ppb. Tropospheric N2O tends to increase towards northern latitudes as a result of higher anthropogenic emissions in the Northern Hemisphere relative to the Southern Hemisphere. More details on the variability of N2O mixing ratios along the tropospheric column are described in Sect. S1.
We study the impact of N2O sources and stratospheric air on the N2O column based on the anomalies (enhancements and depletions) we observed in the airborne N2O mixing ratios relative to the N2O “background,” defined here as the NOAA-MBL product. We use the NOAA-MBL product to constrain a latitudinal gradient of N2O mixing ratios at the surface for each deployment. These data have been widely used to estimate the N2O background (Assonov et al., 2013; Nevison et al., 2011). More information about the NOAA-MBL product and the latitudinal gradient of their measurements is discussed in Sect. S2. This approach highlights the extra information that aircraft profiles can provide. Cross-sections of N2O anomalies are shown in Fig. 3. The data describe the overall homogeneity of N2O in the troposphere (30 % of the anomalies ranged between ± 0.5 ppb). We suppose that the ± 0.5 ppb interval accounts for the day-to-day and seasonal variability of N2O. Episodes of N2O depletion (< −0.5 ppb) that relate to the influence of stratospheric air are observed in 53.5 % of the aircraft samples during ATom-2 to -4, whereas episodes of N2O enhancement (> 0.5 ppb) that relate to the contribution of N2O sources account for 16.5 % of the calculated anomalies.
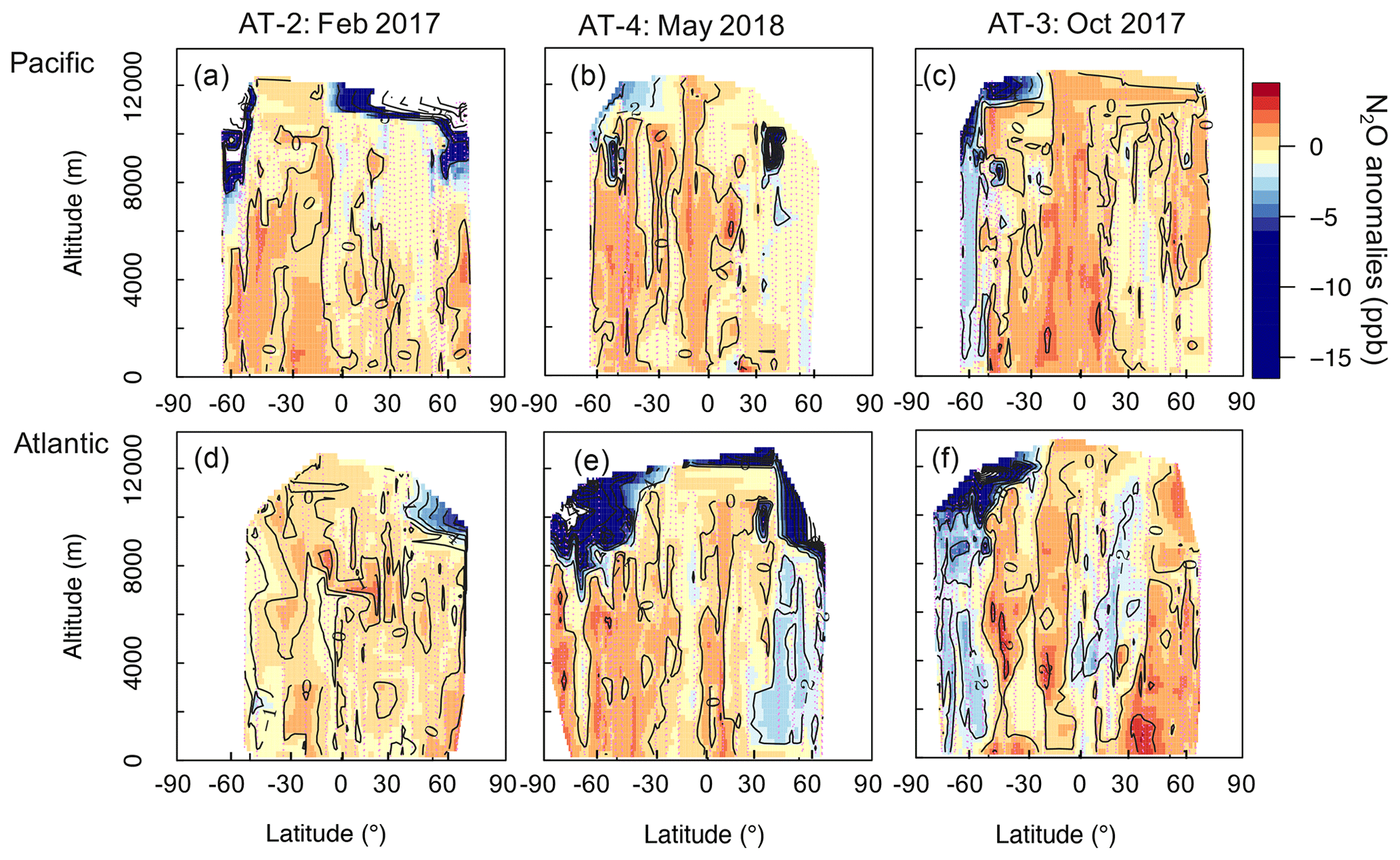
Figure 3Cross-sections of N2O anomalies (ppb) representing the differences between the airborne N2O (10 s resolution) and the surface N2O mixing ratios, interpolated to 0.25∘ latitude and 250 m altitude for each deployment. Shown are the N2O anomalies over (a)–(c) the Pacific and (d)–(f) the Atlantic, with each column representing a deployment (ordered by season: ATom-2, -4, and -3). The color scale ranges from −15 to 5 ppb. Values between −50 and −15 ppb, observed at the highest altitudes (> 10 km), are shown in white to allow better visualization of small changes in positive anomalies. Lilac dashed lines represent the flight tracks. Black contours are areas of N2O anomalies.
Trajectories and associated surface influence functions were computed using the Traj3D model (Bowman, 1993) and wind fields from the National Center for Environmental Prediction Global Forecast System (NCEP GFS). Model trajectories were initialized at receptors spaced 1 min apart along the ATom flight tracks, followed backwards for 30 d, and reported at 3 h resolution. From these trajectories, we calculated the surface influence for each receptor point (footprints in units of concentration mixing ratio per emission flux; ppt nmol−1 m2 s). The footprint can be convolved with a known flux inventory of a nonreactive gas to calculate the expected enhancement/depletion of that gas for each receptor point.
4.1 Impact of stratospheric air on tropospheric N2O mixing ratios during ATom
We observe the strongest depletions (> 5 ppb) in N2O mixing ratios at high latitudes and altitudes, consistent with stratospherically influenced air (Fig. 3). Stratosphere–troposphere exchange processes allow stratospheric-depleted N2O to be distributed throughout the troposphere. The NOAA surface network shows a seasonal minimum of N2O 2–4 months later than the stratospheric polar vortex break-up season. This seasonal minimum is observed at the surface around May in the Southern Hemisphere and around July in the Northern Hemisphere (see Figs. S8 and S9) (see Nevison et al., 2011 and references therein). The enhanced downwelling of the Brewer–Dobson circulation (BDC) in late winter–spring reinforces the downward transport of stratospheric air depleted in N2O throughout the free troposphere (1–8 km), as observed in October in the Southern Hemisphere (ATom-3, Fig. 3c and f) and in May in the North Atlantic (ATom-4, Fig. 3e). The N2O depletion is likely the result of stratospheric air being moved downwards by the BDC and trapped by the polar vortex, with a more pronounced effect in the Southern Hemisphere, where the polar vortex is stronger. These results support previous work suggesting that downward transport of stratospheric air with low N2O exerts a strong influence on the variance of tropospheric N2O mixing ratios (Nevison et al., 2011; Assonov et al., 2013).
The impact of stratosphere-to-troposphere transport can be studied by combining information on tracers of stratospheric air such as ozone (O3 from the NOAA – NOyO3; Bourgeois et al., 2020), sulfur hexafluoride (SF6 from PANTHER), CFC12 (from PANTHER), and carbon monoxide (CO from QCLS). These tracers are usually used either because they are strongly produced in the stratosphere (e.g., O3) or because they are tracers of anthropogenic emissions in the troposphere with a strong stratospheric sink (e.g., CO, SF6, and CFC12). In addition, meteorological parameters such as potential vorticity (PV), the product of absolute vorticity and thermodynamic stability (PV was generated by GEOS5-FP for ATom), can be used to trace the stratosphere-to-troposphere transport.
Overall, the interhemispheric gradient of N2O is much smaller than those of CO and SF6 (Fig. 4), but the difference for each species is driven by larger anthropogenic emissions in the Northern Hemisphere. The tracer–tracer correlations shown in Fig. 4 show different patterns. The linear trend between N2O and O3 or CFC-12 highlights the role of depletion (N2O and CFC-12) and production (O3) in the stratosphere (Fig. 4a1, a4). When N2O is plotted against the anthropogenic tracers CO and SF6, two distinct trends are observed. Tropospheric N2O can be identified as the horizontal band containing high N2O (> 328 ppb) and variable CO and SF6, whereas the vertical band with variable N2O and small changes in CO and SF6 is due to the mixing between tropospheric air and stratospheric air depleted in N2O (Fig. 4a1–a3). The N2O versus CO plot shows an L-shaped (bimodal) curve similar to those typically observed in O3–CO correlations during stratosphere-to-troposphere airmass mixing events (Fig. 4a2, Krause et al., 2018). A quasi-vertical line in the N2O–CO plot (i.e., constant CO) is indicative of a strong impact of stratospheric air, as the stratospheric equilibrium mixing ratio of CO is observed (Krause et al., 2018). The lower the CO background, the greater the influence of the stratospheric air during the airmass mixing (North Atlantic high latitudes in Fig. 4a2) and vice versa. A strong correlation is also indicative of rapid mixing between the two air masses. During ATom, the strongest impact of stratospheric air was observed in the Pacific mid- and high latitudes in February (ATom-2) and in the Atlantic in May (ATom-4, Fig. S11). At the North Pacific mid- and high latitudes (NMHL > 30∘ N), we find a consistent linear relationship between N2O and O3, with a relatively constant N2O O3 slope (−0.05 to −0.04) during all seasons. Linear correlations between N2O and CFC-12 highlight the dominant influence of stratospheric air that was depleted in these two substances in the range of mixing ratios observed at mid- and high latitudes (Fig. S11).
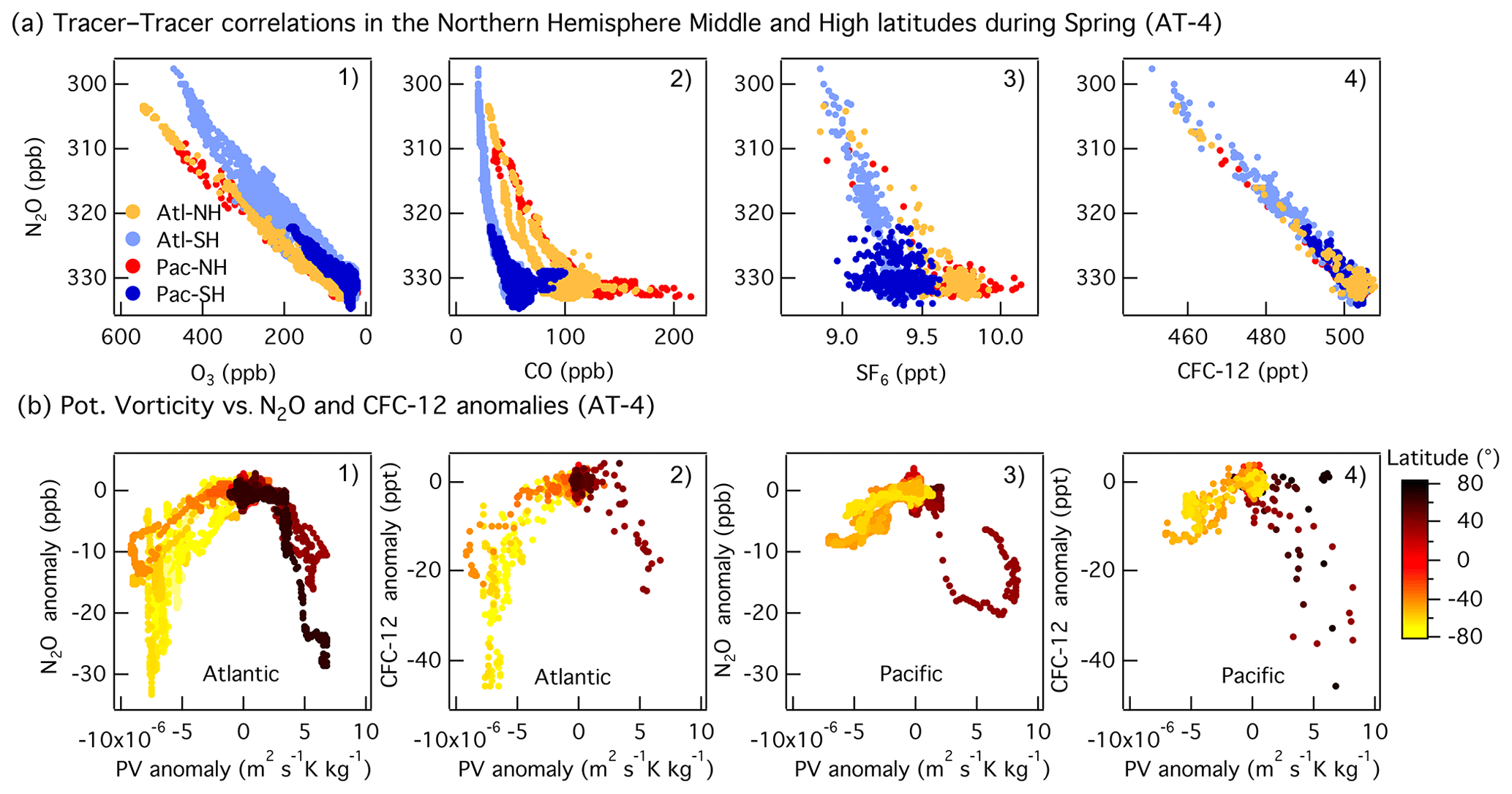
Figure 4(a) Correlations between N2O and O3 (a1), CO (a2), SF6 (a3), and CFC-12 (a4) at mid- and high latitudes (30–85∘ N) during Northern Hemisphere spring (ATom-4). The data are colored as a function of the ocean basin and hemisphere: North Pacific mid–high latitudes (Pac-NH, > 30∘ N) in red, South Pacific mid–high latitudes (Pac-SH, < 30∘ S) in dark blue, South Atlantic mid–high latitudes (Atl-SH, < 30∘ S) in light blue, and North Atlantic mid–high latitudes (Atl-NH, > 30∘ N) in orange. Note that the N2O and O3 axes are reversed. (b) Correlations between anomalies in potential vorticity relative to its mean latitudinal distribution in the free troposphere (2–8 km) and anomalies in N2O (b1, b3) and CFC-12 (b2, b4) as a function of latitude during spring (ATom-4) over the Pacific and Atlantic basins. Mid-latitudes are shown in orange in the SH and in light brown in the NH.
During spring, the mid-latitudes are strongly impacted by stratospheric air due to the occurrence of tropopause folds and cutoff lows to the south of the westerly subtropical jets (Hu et al., 2010 and references therein). The stronger depletion of N2O mixing ratios observed over the Atlantic relative to the Pacific during spring is due to a greater number of deep stratosphere-to-troposphere transport events at mid-latitudes in the region between May and July (Fig. 3e; Cuevas et al., 2013 and references therein). Anomalies in PV relative to its mean latitudinal distribution in the free troposphere (2–8 km) highlight events involving the strong downward transport of stratospheric air. Negative PV, N2O, CO, and CFC-12 anomalies (positive for O3) describe the transport of stratospheric air into the troposphere in the SH, whereas positive PV and negative N2O, CO, and SF6 anomalies (positive for O3) describe the downward transport of stratospheric air in the NH (Fig. 4b1–b4). The correlations between N2O and PV and the similarities with CFC-12 indicate that stratosphere-to-troposphere exchange leads to variations in tropospheric N2O of up to 10 ppb at the higher latitudes for the altitudes covered during the flights. This influence is notably larger than the 2–4 ppb enhancements associated with regional emissions (see below).
4.2 Impact of emissions on tropospheric N2O mixing ratios during ATom
During ATom, episodes of positive N2O anomalies relative to the surface station MBL reference occurred close to the equator (Fig. 3a–c) and in a few locations at mid-latitudes in both ocean basins across all seasons. We used the information from the vertical profiles, including back trajectories and correlated chemical tracers, to trace the origins of these enhancements. We investigated data on CO, CH4, and CO2 measured by QCLS and NOAA Picarro 2401 m; hydrogen cyanide (HCN), sulfur dioxide (SO2), hydrogen peroxide (H2O2), and peroxyacetic acid (PAA) measured with the California Institute of Technology Chemical Ionization Mass Spectrometer (CIT-CIMS, Crounse et al., 2006; St. Clair et al., 2010); ammonium (NH), sulfate (SO), nitrate (NO), and organic aerosols (OA) from the Colorado University Aircraft High-Resolution Time-of-Flight Aerosol Mass Spectrometer (HR-AMS, DeCarlo et al., 2006; Canagaratna et al., 2007; Jimenez et al., 2019; Guo et al., 2021; Hodzic et al., 2020); NOy from the NOAA NOyO3 four-channel chemiluminescence instrument (CL, Ryerson et al., 2019); CH2Br2, CH3CN, benzene, and propane from the NCAR Trace Organic Gas Analyzer (TOGA, Apel et al., 2019); and atmospheric potential oxygen (APO ≈ O2+1.1 × CO2) from the NCAR Airborne Oxygen Instrument (AO2, Stephens et al., 2020).
We calculated the correlations between N2O and the mentioned species in three layers (0–2000, 2000–4000, and 4000–6000 m). Correlation coefficients in each layer for a given profile were calculated using a minimum threshold of 15 data points per layer. These profiles show that many of the most prominent enhancements of N2O are closely associated with pollutants such as HCN, CH3CN, H2O2,, and other pollutants associated with combustion and photochemical air pollution. Some profiles show peaks that are closely correlated with SO2 and enhanced PM1 particles, and vertical gradients were sometimes correlated with gradients of APO and HCN.
Several N2O peaks are observed together with enhancements of H2O2 and PAA, which are primarily formed in chemical processes that occur in the atmosphere. For the altitude range 2–4 km, regressions produced r2 > 0.7 for 16 profiles of N2O vs. H2O2 and 15 profiles of N2O vs. HCN (a tracer for combustion of biomass), but only three such profiles produced these strong associations for both H2O2 and HCN in common. Some of these profiles also showed correlated enhancements of SO2 and NO (nine profiles with r2 > 0.6). This result raises the question of whether globally significant production of N2O may be occurring in heterogeneous reactions involving SO2, NO redox chemistry, and HONO near to strong sources of reactive pollutants, which have been observed in heavily polluted atmospheres (Wang et al., 2020) and have been theorized to occur in the plumes of refineries or power plants (e.g., Pires and Rossi, 1997).
In most cases, because we were sampling in the middle of each ocean and not over the source regions, it was not possible to distinguish between the different sources that contributed to the observed N2O enhancements. We also observed that the impacts of the different sources on N2O mixing ratios were region dependent. Here, we describe, with some examples, the sources that contribute to the major N2O enhancements observed during ATom by oceanic region, although we cannot precisely pinpoint the source processes.
4.2.1 N2O enhancements over the Pacific
Episodes of N2O enhancement were frequently observed at mid-latitudes in the southern Pacific Ocean, and these were linked by the associated footprints to emissions over the continents. In this region, N2O enhancements are predominantly associated with air masses with enhanced H2O2, PAA, and CO. For example, consider Fig. 5, which shows data from profile 12, obtained at 49.5–50∘ S and near the Date Line on 3 May 2018. A distinct peak in N2O of amplitude 1 ppb at 1700 m altitude is significantly correlated with enhancements in CH3CN. These associations and the footprints suggest a regional contribution from fuel types from the industrial zone of Australia (Fig. 5c), which is also supported by the aerosol characterization from PALMS (not shown for brevity). In this profile, close to the surface, the lowest QCLS N2O mixing ratios agree with the NOAA MBL N2O (dashed line in Fig. 5b). At higher altitudes (2.5–6 km), strong correlations between N2O, H2O2, PAA, CO, and HCN but not SO2 suggest the influence of biomass burning from central Australia (3–5 km) and South America (6 km) (Fig. 5b, middle and right-hand panels in Fig. 5c, and Fig. S11f). The relatively low mixing ratios of short-lived trace gases (PAA, H2O2, and PM1 aerosols with lifetimes ranging from hours to a few days) and the surface influence based on the back trajectories (Fig. S13a) indicate that most of these profiles sampled significantly aged air masses that were transported for extended periods over the South Pacific.
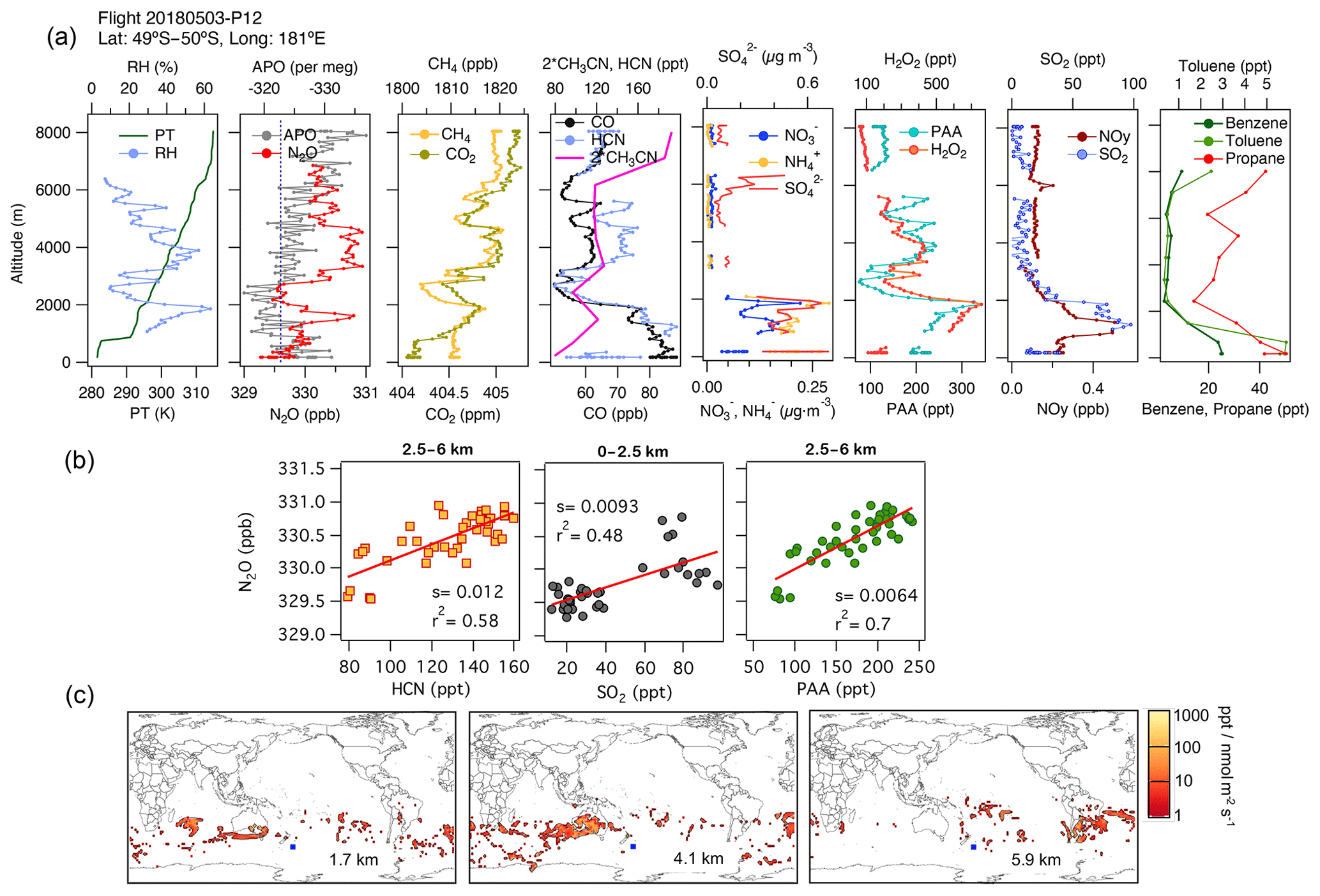
Figure 5(a) Vertical profiles of potential temperature (PT), relative humidity (RH), N2O, APO, CH4, CO2, CO, HCN, CH3CN, NO, NH, SO, H2O2, PAA (CH3C(O)OOH), SO2, NOy, benzene, toluene, and propane from profile 12, obtained on 3 May 2018. The dotted blue line in the plot of APO and N2O represents the NOAA-MBL reference (N2O-MBL) at the latitude of the flight. (b) Correlations between N2O and HCN and PAA for altitudes between 2.5 and 6 km, and between N2O and SO2 for altitudes between 0 and 2.5 km, indicate an admixture of marine, biomass-burning, urban, and oil and gas industry contributions to N2O mixing ratios (s represents the slope of the linear fit). (c) Footprint maps tracing surface regions that influence mixing ratios measured at the altitude ranges of 1–2, 2.5–5, and 5–7 km, respectively. Blue squares show sampling locations. Values below 3 ppt nmol−1 m2 s are not included. Note that the APO axes are reversed.
In the equatorial Pacific, episodes of N2O enhancement were frequently associated with a mixture of potential marine, industrial, and biomass burning emissions. Atmospheric potential oxygen (APO) is primarily a tracer of oxygen exchange with the oceans, defined as deviations in the oxygen-to-nitrogen ratio (δ(O2 N2)) corrected for changes in O2 due to terrestrial photosynthesis and respiration and for influences from combustion (Stephens et al., 1998),
Here, δ(O2 N2) is the deviation in the O2 N2 ratio (per meg), 1.1 is an approximation to the O2 CO2 ratio for photosynthesis and respiration, is the mole fraction of O2 in dry air, and is the mole fraction of CO2 in the air sample (dry, µmol mol−1). Since APO primarily tracks oxygen exchange between the ocean and the atmosphere, APO depletions can indicate marine N2O emissions from areas with strong upwelling (Lueker et al., 2003; Ganesan et al., 2020). However, APO is also sensitive to pollution such as biomass burning and fossil fuel combustion (Lueker et al., 2001) and, because both N2O and APO have meridional gradients resulting from many influences, correlations can result simply from sampling air transported from different latitudes. In ATom, nine profiles showed significant correlations (r2 > 0.7) between N2O and APO (or δ(O2 N2), which has lower measurement noise) for altitude bins 0–2 (8) and 2–4 km (1), with back trajectories indicating that they originated close to the west coast of North America and the Mauritanian coast as well as in the equatorial Pacific. The median slope of regressions of APO vs. N2O for these profiles in ATom is −0.04 ppb per meg, and the mean is −0.05 (± 0.04, 1σ) ppb per meg – very similar to the range found by Ganesan et al. (2020) and Lueker et al. (2003) in coastal areas.
An example is shown in Fig. 6 for 1 May 2018. We observe a high correlation between N2O and APO (r2=0.66) between 0 and 4 km altitude. At these altitudes we also see enhancements in dibromomethane (CH2Br2), a tracer of phytoplankton biomass (Liu et al., 2013 and references therein), consistent with a marine biological flux of halogenated VOCs (Asher et al., 2019), dimethyl sulfide (DMS), and methanesulfonic acid (MSA), the main particulate product of DMS oxidation in the MBL. However, on this flight, the footprints and the influence of the surface ocean (Fig. S12b) indicate that this N2O gradient represents a difference between sampling a near-surface marine air mass from the south and a more continental air mass from the east at 4 km (Fig. 6a–c). Close to the surface, the lowest QCLS N2O mixing ratios agree with the NOAA MBL N2O at the origin of the air masses suggested by the footprints (25∘ S, dashed red line in Fig. 6b), whereas the lowest QCLS N2O mixing ratios at 4 km agree with the NOAA MBL N2O (dotted blue line in Fig. 6b). Thus, the N2O to APO correlation most likely represents the latitudinal and ocean–land gradients established for a combination of reasons, with higher APO and lower N2O originating from higher southern latitudes away from continents. During this flight, there were particularly noticeable N2O variations between 4 and 6 km height that appear to be related to biomass-burning plumes from fires occurring in Venezuela and the Caribbean, in agreement with simultaneous enhancements in CO and HCN mixing ratios (Fig. 6a, c and Fig. S12), and there was increasing SO2 in the first 2 km, which was linked to oil and gas pollution sources near coastlines. The nature of these emissions was also confirmed by aerosol characterization using the PALMS instrument (figure not shown).
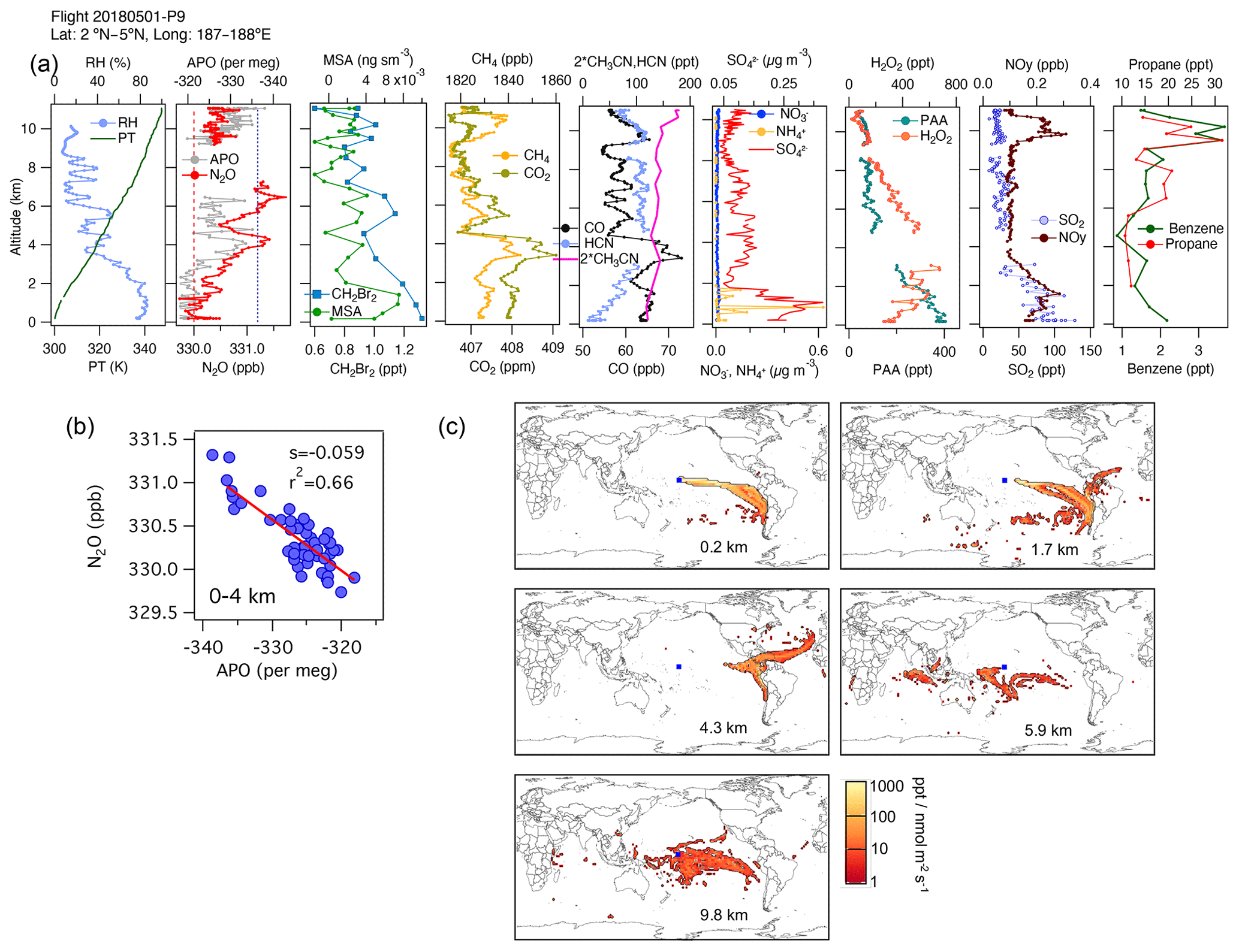
Figure 6(a) Vertical profiles of PT, RH, and the tracers N2O, APO, MSA, CH2Br2, CH4, CO2, CO, HCN, CH3CN, NO, NH, SO, H2O2, PAA (CH3C(O)OOH), SO2, NOy, benzene, toluene, and propane from profile 9 on 1 May 2018. The dotted blue line in the plot of APO and N2O represents the NOAA-MBL reference (N2O-MBL) at the latitude of the flight; the dashed red line shows the N2O-MBL at the origin of the air masses suggested by the footprints (25∘ S). (b) N2O–APO correlations between 0 and 4 km that possibly describe the latitudinal gradient of N2O (s represents the slope of the linear fit). (c) Footprint maps tracing surface regions that influence mixing ratios measured in the altitude ranges 0–2, 2–4, 3–5, 5–7, and 9–11 km, respectively. Blue squares show sampling locations. Values below 3 ppt nmol−1 m2 s are not included. Note that the APO axes are reversed to illustrate the negative correlation with N2O.
4.2.2 N2O enhancements over the Atlantic
Much more of a continental influence was observed during the Atlantic Basin ATom flights than during the Pacific flights. In the North Atlantic at around 30∘ N during winter, we observe small enhancements of N2O that contrast with the overall influence of stratospheric air on the tropospheric column (AT-2, Fig. 3d). The contribution is much higher during the fall season (AT-3, Fig. 3f). Several episodes of N2O enhancement are associated with enhancements of CH4, CO, and HCN. We also observe some episodes where N2O increases while CO2 decreases (figure not shown), which could reflect the accumulation of agricultural emissions over the summer or just greater sampling of Northern Hemisphere summer air masses, whereas increases of N2O with CO are indicators of urban pollution and are, together with HCN, associated with a few episodes of biomass burning.
The influences of different regions on the N2O mixing ratios over the Atlantic on 14 May 2018 are shown in Fig. 7. This profile shows the contributions to tropospheric N2O from pollution transported down over the Mauritanian coast from Western Europe, biomass-burning emissions, urban and industrial emissions from southern Africa and the Middle East (between 1.5 and 3 km), and polluted air masses from South America and the west coast of Africa, which are mixed with the oceanic contribution to N2O (∼ 10 km, Fig. 7a–c and Fig. S13). The aerosol characterization (from PALMS, not shown) indicates that mineral dust and biomass-burning emissions influence the atmospheric layer between 1 and 6 km in altitude, while oil combustion influences the layer below 4 km. At high altitudes, N2O enhancements are caused by the interception of polluted air masses from South America and the west coast of Africa mixed with the oceanic contribution to N2O (∼ 10 km). The N2O:APO correlations for the feature between 1.5 and 3 km most likely represent APO depletion through industrial combustion, which is stoichiometrically consistent with the observed increases in CO2 and CH4 for this feature.
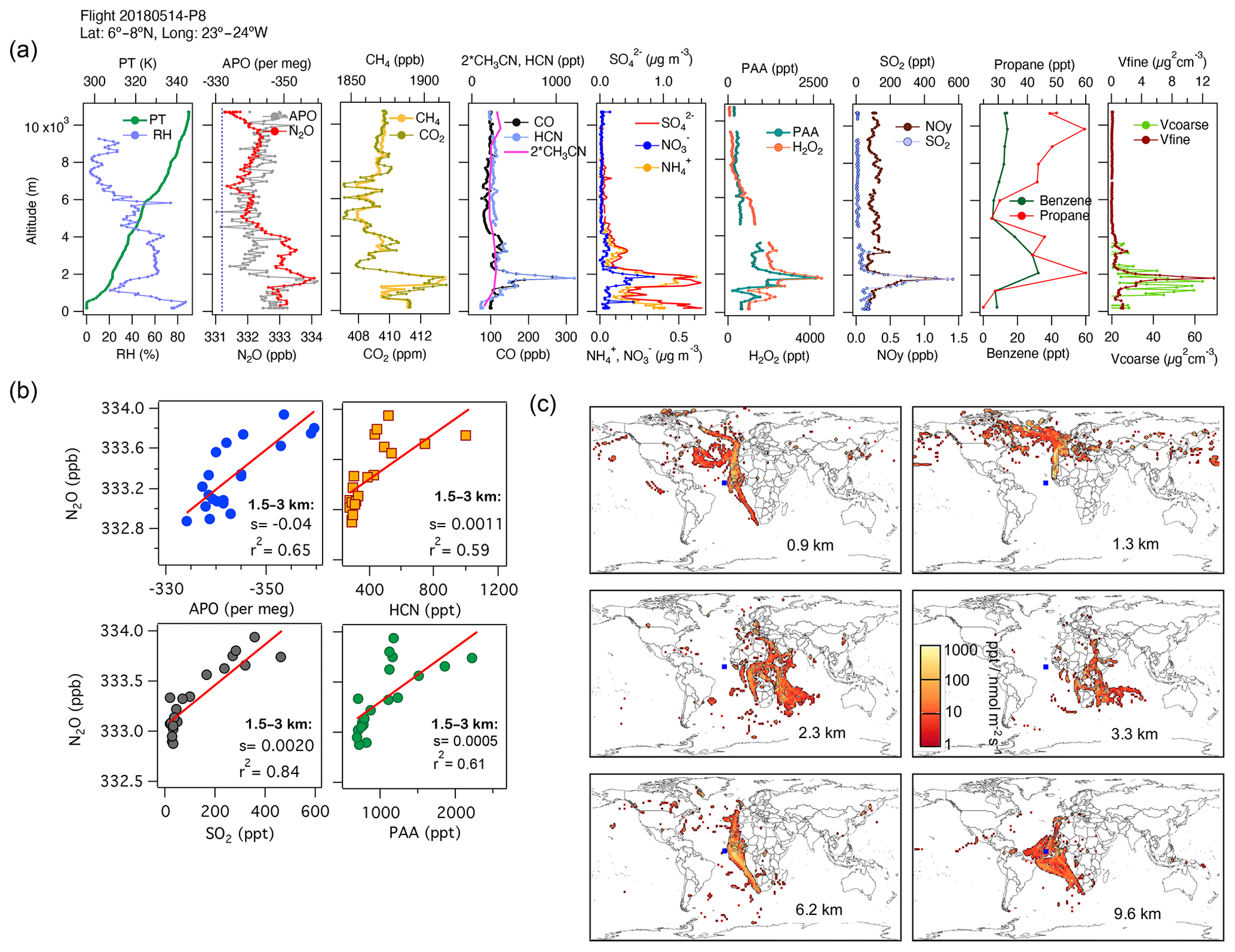
Figure 7(a) Vertical profiles of PT, RH, the tracers N2O, APO, CH4, CO2, CO, HCN, CH3CN, NO, NH, SO, H2O2, PAA, SO2, NOy, benzene, and propane, and the volumes of coarse and fine particles from profile 8 on 14 May 2018. The dotted blue line in the plot of APO and N2O represents the NOAA-MBL reference (N2O-MBL) at the latitude of the flight. (b) Correlations between N2O and APO, HCN, SO2, and propane at altitudes of 1–3 km show possible contributions from marine upwelling, biomass burning, and the oil and gas industry, as supported by the footprints (s represents the slope of the linear fit). (c) Footprint maps tracing surface regions that influence mixing ratios measured in the altitude ranges 0–1, 2–4, 4–5, 5–7, and 7–10 km, respectively. The blue square shows the sampling location. Values below 3 ppt nmol−1 m2 s are not included. Note that the APO axes are reversed.
During ATom, we observed large contributions to the tropospheric N2O over the Atlantic Ocean from Africa, along with some contributions from Europe and South America. During AT-2, we found strong correlations in the subtropical and tropical regions over the Atlantic between N2O, H2O2, PAA, HCN, CO, CO2, SO2, OA, NH, and SO at altitudes between 0 and 2.5 km, representing the combined influence of photochemistry (), biomass-burning events from the Congo region (), and the industrial production of N2O from oil and gas emissions from the Niger River Delta in Africa (). An example is shown in Fig. 8 for 15 February 2017 (see also Fig. S12 and the land contribution in Fig. S13).
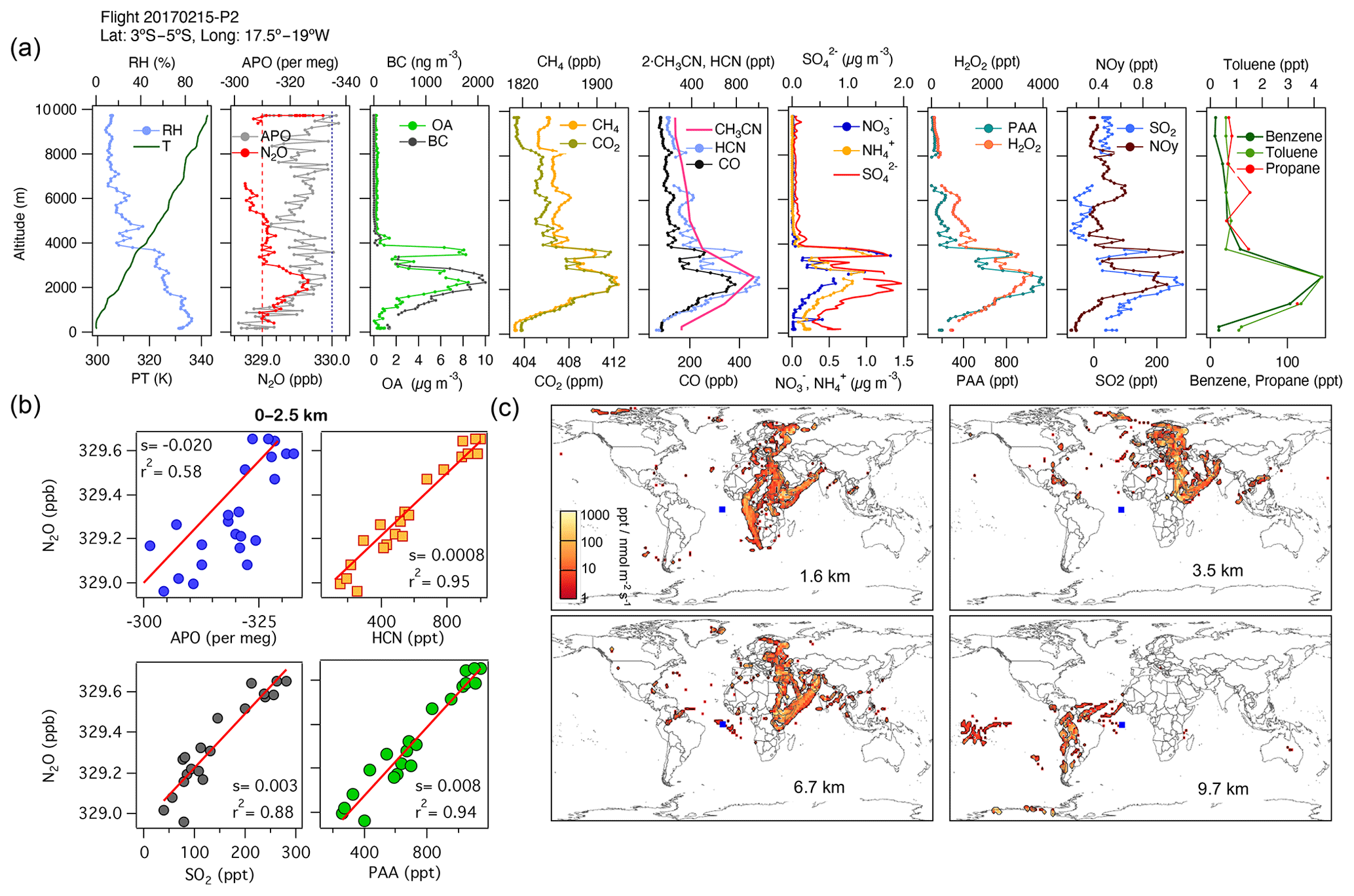
Figure 8(a) Vertical profiles of PT, RH, and the tracers N2O, APO, organic aerosols (OA), black carbon (BC), CH4, CO2, CO, HCN, CH3CN, NO, NH, SO, H2O2, PAA, SO2, NOy, benzene, toluene, and propane from profile 2 on 15 February 2017. The dotted blue line in the plot of APO and N2O represents the NOAA-MBL reference (N2O-MBL) at the latitude of the flight, and the red dashed line shows the NOAA-MBL at the origin of the southern air masses shown by the footprints below 2 km (20∘ S). (b) Correlations between N2O and APO, HCN, and SO2 for data observed below 2.5 km indicate an admixture of marine, biomass-burning, urban, and oil and gas industry contributions to N2O mixing ratios (s represents the slope of the linear fit). (c) Footprint maps tracing surface regions that influence mixing ratios measured in the altitude ranges 0–2, 2–3, 3–4, and 4–7 km, respectively. Blue squares show sampling locations. Values below 3 ppt nmol−1 m2 s are not included in the footprint plot. Note that the APO axes are reversed.
To understand the origin of the enhancements in N2O, we calculated the enhancement expected in the atmosphere based on monthly mean estimates of anthropogenic emissions from the Emissions Database for Global Atmospheric Research (EDGAR, http://edgar.jrc.ec.europa.eu/, last access: 5 February 2021). We convolved the calculated surface influence (footprint) with the inventory to calculate the N2O enhancement expected for each receptor. We also calculated the contribution of each region and source sector to the overall enhancement. This allowed us to quantify the dominant sources for various layers within each profile. Each of the calculated enhancements was then compared to the enhancement in N2O observed for the profiles. The observed N2O enhancements were calculated relative to the NOAA MBL reference (Fig. 8a, dashed red line) for each 10 s observation, with background concentrations selected from locations close to the origin of the air mass as indicated by the surface influence (shown as dashed and dotted lines in the N2O altitude profiles in Figs. 5–8). We also included 0.4 ppb of uncertainty for the observed enhancements based on our measurement precision.

Figure 9N2O enhancements estimated by EDGAR for the entire globe (gray polygons) and for the African region (blue polygons), and the observed QCLS-N2O enhancement relative to the NOAA-MBL N2O reference at the origin of the southern air masses shown by the footprints below 2 km for the profile 2017/02/15-P2 (20∘ S, 329 ppb, shown in Fig. 8). QCLS-N2O was observed every 10 s and receptors were calculated every 60 s.
The largest N2O enhancement (peaking at 2 ppb at 2 km) observed over the Atlantic during ATom-2 (February 2017; Fig. 9) can be attributed to African agriculture, along with smaller but significant influences from Asia and Europe (0.5 ppb each at 2–4 km, Fig. S14). The observed and modeled N2O enhancements agree within an order of magnitude for the profile, but the model underestimates the high-altitude (4–7 km) N2O enhancement by < 1 ppb and overestimates the lower-altitude enhancement (2–4 km) by ∼ 1 ppb. This difference in N2O enhancement could be due to a strong latitudinal gradient in N2O across this profile or the timing of N2O emissions sampled along this single profile compared to a monthly mean estimate from the inventory. Strong correlations between N2O and HCN (r2=0.95), CO, and CH3CN suggest that N2O from burning emissions also contributes to the N2O enhancement (Figs. 8 and S12). However, when we convolved the monthly mean fire contributions from the Global Fire Emissions Database (GFED, https://www.globalfiredata.org, last access: 5 February 2021) with the surface influence footprints (as described above), we found that the wildfire-produced N2O is minimal for this profile (∼ 0.2 ppb), suggesting that fires of anthropogenic or urban origin might be the source of that contribution (Figs. 8a–c, 9, S12, and S13).
N2O mixing ratios at 1 Hz were obtained during the NASA ATom airborne program by applying a new spectral retrieval method to account for the pressure and temperature sensitivity of quantum cascade laser spectrometers deployed on aircraft. This method improved the precision of our QCLS N2O measurements by a factor of three (based on the standard deviation of calibration measurements), allowing us to provide N2O measurements to the level of precision shown in previous aircraft missions.
The N2O altitude profiles observed during ATom show that tropospheric N2O variability is strongly driven by the influence of stratospheric air depleted in N2O, especially at mid- and high latitudes. At high latitudes, our profiles showed a strongly depleted N2O signal around the time of the vortex break-up season, persisting for several months. Combining the information from N2O profiles and other chemical tracers such as CO, SF6, O3, and CFC-12, we traced the propagation of stratospheric air along the tropospheric column down to the surface. This transport dominates the N2O seasonal cycle and creates the seasonal surface minima 2–3 months after the peak stratosphere–troposphere exchange in spring.
The high resolution of this data set (10 s) allowed us to study the factors influencing the enhancements in the N2O tropospheric mixing ratios, which are associated with biomass burning and human activities such as urban and industrial emissions. The highest N2O mixing ratios occur close to the Equator, where they extend throughout the tropospheric column. The strongest N2O enhancements were observed close to the Equator and at a number of mid-latitude locations. We used the information given by the vertical profiles of N2O and a variety of chemical tracers together with footprints computed every 60 s along the flight track to identify and trace the sources of these N2O enhancements. N2O enhancement events were more frequent in the Atlantic than in the Pacific.
Over the Atlantic, the co-occurrence of excess N2O together with other pollutants suggested that industrial and urban N2O emissions originating from distant locations such as western and southern Africa, the Middle East, Europe, and South America may be significantly greater than the emissions from biomass burning in Africa. This view is supported by our observations of a strong contribution to N2O from oil and gas emissions from the Niger River Delta in Africa. The correlations observed between N2O and SO2 (r2=0.90) could possibly be used to estimate N2O emissions from oil and gas.
Over the southern Pacific Ocean and the tropical Atlantic Ocean, we observed a significant number (> 12) of profiles where enhancements in N2O were associated with increased H2O2 and PAA and notably less well correlated with HCN or CO. Since H2O2 and PAA are products of photochemical pollution, this observation raised the question of whether significant N2O may be produced by heterogeneous processes involving HONO or NOx reactions in acidic aerosols close to sources or in very heavily polluted areas. It is hard to draw a definite conclusion based on measurements obtained so far from the most active regions. Studies performed to address this question would have to be carried out directly in the polluted areas. Because agricultural activities do not have unique tracer signatures, we were not able to distinguish contributions from cultivated and natural soils to N2O emissions from the ATom data. Previous airborne studies have observed these inputs using flights in agricultural areas (Kort et al., 2008) and at towers in these regions (e.g., Nevison et al., 2017; Miller et al., 2008).
Our study shows that airborne campaigns such as ATom can help trace the origins of biomass-burning and industrial emissions and investigate their impact on the variability of tropospheric N2O, providing unique signatures in vertical profiles and with covariate tracers. We hope that the information provided by the global tropospheric N2O profiles from the ATom mission will help better diagnose and reduce uncertainties in atmospheric chemical transport models for constraining the N2O global budget.
| Description | Acronym |
|---|---|
| Atmospheric Potential Oxygen | APO |
| Atmospheric Tomography | ATom |
| California Institute of Technology – Chemical Ionization Mass Spectrometer | CIT-CIMS |
| CU Aircraft High-Resolution Time-of-Flight Aerosol Mass Spectrometer | HR-AMS |
| Global Monitoring Laboratory | GML |
| HIAPER Pole-to-Pole Observations | HIPPO |
| High Latitudes | HL |
| HIgh-resolution TRANsmission molecular absorption database | HITRAN |
| Marine Boundary Layer | MBL |
| Middle Latitudes | ML |
| Modern-Era Retrospective analysis for Research and Applications 2 model | MERRA2 |
| National Center for Environmental Prediction Global Forecast System model | NCEP GFS |
| National Oceanic and Atmospheric Administration | NOAA |
| NCAR Airborne Oxygen Instrument | AO2 |
| NOAA Halocarbons and other Atmospheric Trace Species Flask Sampling Program | NOAA-HATS |
| NOAA NOyO3 4-channel chemiluminescence | CL |
| Northern Hemisphere | NH |
| PAN and other Trace Hydrohalo-carbon ExpeRiment | PANTHER |
| Particle Analysis by Laser Mass Spectrometry instrument | PALMS |
| Potential Vorticity | PV |
| Principal Component Analysis | PCA |
| Programmable Flask Package Whole Air Sampler | PFP |
| Quantum Cascade Laser Spectrometer | QCLS |
| Southern Hemisphere | SH |
| Stochastic Time-Inverted Lagrangian Transport Model | STILT |
| Trace Organic Gas Analyzer | TOGA |
| Unmanned Aircraft Systems Chromatograph for Atmospheric Trace Species | UCATS |
| Upper Troposphere/Lower Stratosphere | UT/LS |
| World Meteorological Organization | WMO |
Data from the ATom mission can be found in the NASA ESPO archive (https://espoarchive.nasa.gov/archive/browse/atom, last access: 10 February 2021), and in the ATom data repository at the NASA/ORNL DAAC (https://doi.org/10.3334/ORNLDAAC/1581, Wofsy et al., 2018). The QCLS N2O data is available at https://doi.org/10.3334/ORNLDAAC/1747 (Commane et al., 2020).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-21-11113-2021-supplement.
YG did the data analysis and wrote and revised the paper. SCW and RC actively contributed to the design of the study and data analysis. JBM designed the Neptune software for spectral re-analysis and contributed to the writing. RC and BCD performed and analyzed QCLS measurements of CH4, N2O, and CO and contributed to the discussions. EM and LDS contributed to the data analysis. KM performed and analyzed NOAA Picarro measurements of CH4, CO, and CO2. JWE, EJH, and FM performed and analyzed N2O, SF6, and CFC-12 measurements from PANTHER and UCATS instruments. FM, SM, and CS performed and analyzed N2O measurements with the Programmable Flask Package Whole Air Sampler (PFP). POW, JC, MK, and HMA performed and analyzed the CIT-CIMS measurements of HCN and SO2 shown here. KF performed and analyzed PALMS measurements. JLJ, PCJ, and BAN performed and analyzed HR-AMS measurements for a variety of aerosols. ER provided back trajectories for each minute during the flight tracks, and PN provided the GEOS5 FP meteorological products. TBR, IB, JP, and CRT performed and analyzed NOyO3 measurements of NOy and O3. BBS and EJM performed and analyzed AO2 and Medusa Whole Air Sampler measurements of O2 N2 and CO2 and assisted with the interpretation. ECA and RSH performed and analyzed TOGA measurements of volatile organic compounds. All coauthors provided comments on the paper.
The authors declare that they have no conflict of interest.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
We would like to thank the ATom leadership team, the science team, and the NASA DC-8 pilot, technicians, and mechanics for their contribution and support during the mission. We thank Karl Froyd for the aerosol products during ATom that support this study. We also thank the National Aeronautics and Space Administration and the National Science Foundation for providing the financial support that made possible this study.
This research has been supported by the National Aeronautics and Space Administration (grant nos. NNX15AJ23G, NNX17AF54G, NNX15AG58A, NNX15AH33A, and 80NSSC19K0124) and the National Science Foundation (grant nos. 1852977, AGS-1547626, and AGS-1623745).
This paper was edited by Andreas Engel and reviewed by three anonymous referees.
Albanito, F., Lebender, U., Cornulier, T., Sapkota, T. B., Brentrup, F., Stirling, C., and Hillier, J.: Direct Nitrous Oxide Emissions from Tropical and Sub-Tropical Agricultural Systems – A Review and Modelling Of Emission Factors, Sci. Rep., 7, 44235, https://doi.org/10.1038/srep44235, 2017.
Apel, E., Asher, E. C., Hills, A. J., and Hornbrook, R. S.: ATom: L2 Volatile Organic Compounds (VOCs) from the Trace Organic Gas Analyzer (TOGA), ORNL DAAC, , Oak Ridge, Tennessee, USA, https://doi.org/10.3334/ORNLDAAC/1749, 2019.
Asher, E., Hornbrook, R. S., Stephens, B. B., Kinnison, D., Morgan, E. J., Keeling, R. F., Atlas, E. L., Schauffler, S. M., Tilmes, S., Kort, E. A., Hoecker-Martínez, M. S., Long, M. C., Lamarque, J.-F., Saiz-Lopez, A., McKain, K., Sweeney, C., Hills, A. J., and Apel, E. C.: Novel approaches to improve estimates of short-lived halocarbon emissions during summer from the Southern Ocean using airborne observations, Atmos. Chem. Phys., 19, 14071–14090, https://doi.org/10.5194/acp-19-14071-2019, 2019.
Assonov, S. S., Brenninkmeijer, C. A. M., Schuck, T., and Umezawa, T.: N2O as a tracer of mixing stratospheric and tropospheric air based on CARIBIC data with applications for CO2, Atmos. Environ., 79, 769–779, https://doi.org/10.1016/j.atmosenv.2013.07.035, 2013.
Bourgeois I., Peischl, J., Thompson, C. R., Aikin, K. C., Campos, T., Clark, H., Commane, R., Daube, B., Diskin, G. W., Elkins, J. W., Gao, R.-S., Gaudel1, A., Hintsa, E. J., Johnson, B. J., Kivi, R., McKain, K., Moore, F. L., Parrish, D. D., Querel, R., Ray, E., Saìnchez, R., Sweeney, C., Tarasick, D. W., Thompson, A. M., Thouret, V., Witte, J. C., Wofsy, S. C., and Ryerson, T. B.: Global-scale distribution of ozone in the remote troposphere from ATom and HIPPO airborne field missions, Atmos. Chem. Phys., 20, 10611–10635, https://doi.org/10.5194/acp-20-10611-2020, 2020.
Bowman, K. P.: Large-scale isentropic mixing properties of the Antarctic polar vortex from analyzed winds, J. Geophys. Res.-Atmos., 98, 23013–23027, https://doi.org/10.1029/93JD02599, 1993.
Butterbach-Bahl, K., Baggs, E. M., Dannenmann, M., Kiese, R., and Zechmeister-Boltenstern, S.: Nitrous oxide emissions from soils: how well do we understand the processes and their controls?, Philos. T. R. Soc. B, 368, 20130122, https:// doi.org/10.1098/rstb.2013.0122, 2013.
Canagaratna, M. R., Jayne, J. T., Jimenez, J. L., Allan, J. D., Alfarra, M. R., Zhang, Q., Onasch, T. B., Drewnick, F., Coe, H., Middlebrook, A., Delia, A., Williams, L. R., Trimborn, A. M., Northway, M. J., Decarlo, P. F., Kolb, C. E., Davidovits, P., and Worsnop, D. R.: Chemical and microphysical characterization of ambient aerosols with the Aerodyne Aerosol Mass Spectrometer, Mass Spectrom. Rev., 26, 185–222, 2007.
Commane, R., Budney, J. W., Gonzalez Ramos, Y., Sargent, M., Wofsy, S. C., and Daube, B. C.: ATom: Measurements from the Quantum Cascade Laser System (QCLS). ORNL DAAC, Oak Ridge, Tennessee, USA, https://doi.org/10.3334/ORNLDAAC/1747, 2020.
Crounse, J. D., McKinney, K. A., Kwan, A. J., and Wennberg, P. O.: Measurement of Gas-Phase Hydroperoxides by Chemical Ionization Mass Spectrometry, Anal. Chem., 78, 6726–6732, https://doi.org/10.1021/ac0604235, 2006.
Cuevas, E., González, Y., Rodríguez, S., Guerra, J. C., Gomez-Peláez, A. J., Alonso-Pérez, S., Bustos, J., and Milford, C.: Assessment of atmospheric processes driving ozone variations in the subtropical North Atlantic free troposphere, Atmos. Chem. Phys., 13, 1973–1998 , https://doi.org/10.5194/acp-13-1973-2013, 2013.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer, Anal. Chem., 78, 8281–8289, 2006.
Forster, P., Ramaswamy, V., Artaxo, P., Berntsen, T., Betts, R., Fa- hey, D. W., Haywood, J., Lean, J., Lowe, D. C., Myhre, G., Nganga, J., Prinn, R., Raga, G., Schulz, M., and Van Dorland, R.: Changes in Atmospheric Constituents and in Radiative Forcing, in: Climate Change 2007: The Physical Science Basis, Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press Cambridge, UK and New York, NY, USA, 2007.
Ganesan, A. L., Manizza, M., Morgan, E. J., Harth, C. M., Kozlova, E., Lueker, T., Manning, A. J., Lunt, M.F., Mühle, J., Lavric, J. V., Heimann, M., Weiss, R. F., and Rigby, M.: Marine Nitrous Oxide Emissions From Three Eastern Boundary Upwelling Systems Inferred From Atmospheric Observations, Geophys. Res. Lett., 47, e2020GL087822, https://doi.org/10.1029/2020GL087822, 2020.
Guo, H., Campuzano-Jost, P., Nault, B. A., Day, D. A., Schroder, J. C., Kim, D., Dibb, J. E., Dollner, M., Weinzierl, B., and Jimenez, J. L.: The importance of size ranges in aerosol instrument intercomparisons: a case study for the Atmospheric Tomography Mission, Atmos. Meas. Tech., 14, 3631–3655, https://doi.org/10.5194/amt-14-3631-2021, 2021.
Hintsa, E., Boering, K. A., Weinstock, E. M., Anderson, J. G., Gary, B. L., Pfister, L., Daube, B. C., Wofsy, S. C., Loewenstein, M., Podolske, J. R., Margitan, J. J., and Bu, T. T.: Troposphere-to-stratosphere transport in the lowermost stratosphere from measurements of H2O, CO, N2O and O3, Geosphys. Res. Lett., 25, 14, 2655–2658, https://doi.org/10.1029/98GL01797, 1998.
Hintsa, E. J., Moore, F. L., Hurst, D. F., Dutton, G. S., Hall, B. D., Nance, J. D., Miller, B. R., Montzka, S. A., Wolton, L. P., McClure-Begley, A., Elkins, J. W., Hall, E. G., Jordan, A. F., Rollins, A. W., Thornberry, T. D., Watts, L. A., Thompson, C. R., Peischl, J., Bourgeois, I., Ryerson, T. B., Daube, B. C., Pittman, J. V., Wofsy, S. C., Kort, E., Diskin, G. S., and Bui, T. P.: UAS Chromatograph for Atmospheric Trace Species (UCATS) – a versatile instrument for trace gas measurements on airborne platforms, Atmos. Meas. Tech. Discuss. [preprint], https://doi.org/10.5194/amt-2020-496, in review, 2021.
Hodzic, A., Campuzano-Jost, P., Bian, H., Chin, M., Colarco, P. R., Day, D. A., Froyd, K. D., Heinold, B., Jo, D. S., Katich, J. M., Kodros, J. K., Nault, B. A., Pierce, J. R., Ray, E., Schacht, J., Schill, G. P., Schroder, J. C., Schwarz, J. P., Sueper, D. T., Tegen, I., Tilmes, S., Tsigaridis, K., Yu, P., and Jimenez, J. L.: Characterization of organic aerosol across the global remote troposphere: a comparison of ATom measurements and global chemistry models, Atmos. Chem. Phys., 20, 4607–4635, https://doi.org/10.5194/acp-20-4607-2020, 2020.
Hu, K., Lu, R., and Wang, D.: Seasonal climatology of cut-off lows and associated precipitation patterns over Northeast China, Meteorol. Atmos. Phys., 106, 37–48, https://doi.org/10.1007/s00703-009-0049-0, 2010.
Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Nault, B. A., Price, D. J., and Schroder, J. C.: ATom: L2 Measurements from CU High-Resolution Aerosol Mass Spectrometer (HR-AMS), ORNL DAAC, Oak Ridge, Tennessee, USA, https://doi.org/10.3334/ORNLDAAC/1716, 2019.
Jiménez, R., Herndon, S., Shorter, J. H., Nelson, D. D., McManus, J. B., and Zahniser, M. S.: Atmospheric trace gas measurements using a dual quantum-cascade laser mid-infrared absorption spectrometer, Proc. SPIE, 5738, 318–331, https://doi.org/10.1117/12.597130, 2005.
Jiménez, R., Park, S., Daube, B. C., McManus, J. B., Nelson, D. D., Zahniser, M. S., and Wofsy, S. C.: A new quantum-cascade laser-based spectrometer for high-precision airborne CO2 measurements, 13th WMO/IAEA Meeting of Experts on Carbon Dioxide Concentration and Related Tracers Measurement Techniques, WMO/TD-No. 1359; GAW Report No. 168, 100–105, 2006.
Kort, E. A., Eluszkiewicz, J., Stephens, B. B., Miller, J. B., Gerbig, C., Nehrkorn, T., Daube, B. C., Kaplan, J. O., Houweling, S., and Wofsy, S. C.: Emissions of CH4 and N2O over the United States and Canada based on a receptor-oriented modeling framework and COBRA-NA atmospheric observations, Geophys. Res. Lett., 35, L18808, https://doi.org/10.1029/2008GL034031, 2008.
Kort, E. A., Andrews, A. E., Dlugokencky, E., Sweeney, C., Hirsch, A., Eluszkiewicz, J., Nehrkorn, T., Michalak, A., Stephens, B., Gerbig, C., Miller, J. B., Kaplan, J., Houweling, S., Daube, B. C., Tans, P., and Wofsy, S. C.: Atmospheric constraints on 2004 emissions of methane and nitrous oxide in North America from atmospheric measurements and a receptor-oriented modeling framework, J. Int. Environ. Sci., 7, 125–133, https://doi.org/10.1080/19438151003767483, 2010.
Kort, E. A., Patra, P. K., Ishijima, K., Daube, B. C., Jimeìnez, R., Elkins, J., Hurst, D., Moore, F. L., Sweeney, C., and Wofsy, S. C.: Tropospheric distribution and variability of N2O: Evidence for strong tropical emissions, Geophys. Res. Lett., 38, L15806, https://doi.org/10.1029/2011GL047612, 2011.
Krause, J., Hoor, P., Engel, A., Plöger, F., Grooß, J.-U., Bönisch, H., Keber, T., Sinnhuber, B.-M., Woiwode, W., and Oelhaf, H.: Mixing and ageing in the polar lower stratosphere in winter 2015–2016, Atmos. Chem. Phys., 18, 6057–6073, https://doi.org/10.5194/acp-18-6057-2018, 2018.
Liu, Y., Yvon-Lewis, S. A., Thornton, D. C. O., Butler, J. H., Bianchi, T. S., Campbell, L., Hu, L., and Smith, R. W.: Spatial and temporal distributions of bromoform and dibromomethane in the Atlantic Ocean and their relationship with photosynthetic biomass, J. Geophys. Res.-Ocean., 118, 3950–3965, https://doi.org/10.1002/jgrc.20299, 2013.
Lueker, T. J., Keeling, R. F., and Dubey, M. K.: The oxygen to Carbon Dioxide Ratios observed in Emissions from a Wildfire in the Northern California, Geophys. Res. Lett., 28, 2413–2416, https://doi.org/10.1029/2000GL011860, 2001.
Lueker, T. J., Walker, S. J., Vollmer, M. K., Keeling, R. F., Nevison, C. D., Weiss, R. F., and Garcia, H. E.: Coastal upwelling air-sea fluxes revealed in atmospheric observations of O2 N2, CO2 and N2O, Geophys. Res. Lett., 30, 1292, https://doi.org/10.1029/2002GL016615, 2003.
Miller, S. M., Matross, D. M., Andrews, A. E., Millet, D. B., Longo, M., Gottlieb, E. W., Hirsch, A. I., Gerbig, C., Lin, J. C., Daube, B. C., Hudman, R. C., Dias, P. L. S., Chow, V. Y., and Wofsy, S. C.: Sources of carbon monoxide and formaldehyde in North America determined from high-resolution atmospheric data, Atmos. Chem. Phys., 8, 7673–7696, https://doi.org/10.5194/acp-8-7673-2008, 2008.
Montzka, S., Moore, F., and Sweeney, C.: ATom: L2 Measurements from the Programmable Flask Package (PFP) Whole Air Sampler, ORNL DAAC, https://doi.org/10.3334/ORNLDAAC/1746, 2019.
Moore, F., Dutton, G., Elkins, J. W., Hall, B., Hurst, D., Nance, J. D., and Thompson, T.: PANTHER Data from SOLVE-II Through CR-AVE: A Contrast Between Long- and Short-Lived Compounds, American Geophysical Union, Fall Meeting 2006, abstract no. A41A-0025, 2006.
Nevison, C. D., Weiss, R. F., and Erickson III, D. J.: Global oceanic emissions of nitrous oxide, J. Geophys. Res.-Ocean., 100, 5809–15820, https://doi.org/10.1029/95JC00684, 1995.
Nevison, C. D., Keeling, R. F., Weiss, R. F., Popp, B. N., Jin, X., Fraser, P. J., Porter, L. W., and Hess, P. G.: Southern Ocean ventilation inferred from seasonal cycles of atmospheric N2O and O2 N2 at Cape Grim, Tasmania, Tellus B, 57, 218–229, 2005.
Nevison, C. D., Dlugokencky, E., Dutton, G., Elkins, J. W., Fraser, P., Hall, B., Krummel, P. B., Langenfelds, R. L., O'Doherty, S, Prinn, R. G., Steele, L. P., and Weiss, R. F.: Exploring causes of interannual variability in the seasonal cycles of tropospheric nitrous oxide, Atmos. Chem. Phys., 11, 3713–3730, https://doi.org/10.5194/acp-11-3713-2011, 2011.
Pires M. and Rossi, M. J.: The Heterogeneous Formation of N2O in the Presence of Acidic Solutions: Experiments and Modeling, Int. J. Chem. Kin., 29, 869–891, 1997.
Ravishankara, A. R., Daniel, J. S., and Portmann, R. W.: Nitrous oxide (N2O): the dominant ozone-depleting substance emitted in the 21st century, Science, 326, 123–5, https://doi.org/10.1126/science.1176985, 2009.
Rothman, L. S., Jacquemart, D., Barbe, A., Benner, D. C., Birk, M., Brown, L. R., Carleer, M.R., Chackerian Jr., C., Chance, K., Couderth, L. H., Dana, V., Devi, V. M., Flaud, J.-M., Gamache, R.R., Goldman, A., Hartmann, J.-M., Jucks, K. W., Maki, A. G., Mandin, J.-Y., Massie, S. T., Orphal, J., Perrin, A., Rinsland, C. P., Smith, M. A. H., Tennyson, J., Tolchenov, R. N., Toth, R. A., Vander Auwera, J., Varanasi, P., and Wagner, G.: The HITRAN 2004 molecular spectroscopic database, J. Quant. Spectrosc. Ra., 96, 139–204, 2005.
Ryerson, T. B., Thompson, C., Peischl, J., and Bourgeois, I.: ATom: L2 In Situ Measurements from NOAA Nitrogen Oxides and Ozone (NOyO3) Instrument, ORNL DAAC, 2019, https://doi.org/10.3334/ORNLDAAC/1734, 2019.
Saikawa, E., Prinn, R. G., Dlugokencky, E., Ishijima, K., Dutton, G. S., Hall, B. D., Langenfelds, R., Tohjima, Y., Machida, T., Manizza, M., Rigby, M. , O'Doherty, S., Patra, P. K., Harth, C. M., Weiss, R. F., Krummel, P. B., van der Schoot, M., Fraser, P. J., Steele, L. P., Aoki, S., Nakazawa, T., and Elkins, J. W.: Global and regional emissions estimates for N2O, Atmos. Chem. Phys., 14, 4617–4641, https://doi.org/10.5194/acp-14-4617-2014, 2014.
Santoni, G. W., Daube, B. C., Kort, E. A., Jimeìnez, R., Park, S., Pittman, J. V., Gottlieb, E., Xiang, B., Zahniser, M. S., Nelson, D. D., McManus, J. B., Peischl, J., Ryerson, T. B., Holloway, J. S., Andrews, A. E., Sweeney, C., Hall, B., Hintsa, E. J., Moore, F. L., Elkins, J. W., Hurst, D. F., Stephens, B. B., Bent, J., and Wofsy, S. C.: Evaluation of the airborne quantum cascade laser spectrometer (QCLS) measurements of the carbon and greenhouse gas suite – CO2, CH4, N2O, and CO – during the CalNex and HIPPO campaigns, Atmos. Meas. Tech., 7, 1509–1526, https://doi.org/10.5194/amt-7-1509-2014, 2014.
SPARC: Report on the Lifetimes of Stratospheric Ozone-Depleting Substances: Their Replacements, and Related Species, edited by: Ko, M., Newman, P., Reimann, S., and Strahan, S., SPARC Report No. 6, WCRP-15/2013, 2013.
St. Clair, J. M., McCabe, D. C., Crounse, J. D., Steiner, U., and Wennberg, P. O.: Chemical ionization tandem mass spectrometer for the in situ measurement of methyl hydrogen peroxide, Rev. Sci. Instrum., 81, 094102, https://doi.org/10.1063/1.3480552, 2010.
Stephens, B. B., Keeling, R. F., Heimann, M., Six, K. D., Murnane, R., and Caldeira, K.: Testing global ocean carbon cycle models using measurements of atmospheric O2 and CO2 concentration, Global Biochem. Cy., 12, 213–230, 1998.
Syakila, A. and Kroeze, C.: The global nitrous oxide budget revisited, Greenhouse Gas Meas. Manag., 1, 17–26, https://doi.org/10.3763/ghgmm.2010.0007, 2011.
Thompson, R. L., Patra, P. K., Ishijima, K., Saikawa, E., Corazza, M., Karstens, U., Wilson, C., Bergamaschi, P., Dlugokencky, E., Sweeney, C., Prinn, R. G., Weiss, R. F., O'Doherty, S., Fraser, P. J., Steele, L. P., Krummel, P. B., Saunois, M., Chipperfield, M., and Bousquet, P.: TransCom N2O model inter-comparison – Part 1: Assessing the influence of transport and surface fluxes on tropospheric N2O variability, Atmos. Chem. Phys., 14, 4349–4368, https://doi.org/10.5194/acp-14-4349-2014, 2014a.
Thompson, R. L., Ishijima, K., Saikawa, E., Corazza, M., Karstens, U., Patra, P. K., Bergamaschi, P., Chevallier, F., Dlugokencky, E., Prinn, R. G., Weiss, R. F., O'Doherty, S., Fraser, P. J., Steele, L. P., Krummel, Vermeulen, A., Tohjima, Y., Jordan, A., Haszpra, L., Steinbacher, M., Van der Laan, S., Aalto, T., Meinhardt, F., Popa, M. E., Moncrieff, J., and Bousquet, P.: TransCom N2O model inter-comparison – Part 2: Atmospheric inversion estimates of N2O emissions, Atmos. Chem. Phys., 14, 6177–6194, https://doi.org/10.5194/acp-14-6177-2014, 2014b.
Tian, H., Lu, C., Chen G., Tao, B., Pan, S., Del Grosso, S. J., Xu, X., Bruhwiler, L., Wofsy, S. C., Kort, E. A., and Prior, S. A.: Contemporary and projected biogenic fluxes of methane and nitrous oxide in North American terrestrial ecosystems, Front. Ecol. Environ., 10, 528–536, https://doi.org/10.1890/120057, 2012.
Tian, H., Xu, R., Canadell, J. G., Thompson, R. L., Winiwater, W., Suntharalingam, P., Davidson, E. A., Ciais, P., Jackson, R. B., Janssens-Maenhout, G., Prather, M. J., Regnier, P., Pan, N., Pan, S., Peters, G. P., Shi, H., Tubiello, F. N., Zaehle, S., Zhou, F., Arneth, A., Battaglia, G., Berthet, S., Bopp, L., Bouwman, A. F., Buitenhuis, E. T., Chang, J., Chipperfield, M. P., Dangal, S. R. S., Dlugokencky, E., Elkins, J. W., Eyre, B. D., Fu, B., Hall, B., Ito, A., Joos, F., Krummel, P. B., Landolfi, A., Laruelle, G. G., Lauerwald, R., Li, W., lienert, S., Maavara, T., MacLeod, M., Millet, D. B., Olin, S., Patra, P. K., Prinn, R. G., Raymond, P. A., Ruiz, D. J., van der Werf, G. R., Vuichard, N., Wang, J., Weiss, R. F., Wells, K. C., Wilson, C., Yang, J., and Yao, Y.: A comprehensive quantification of global nitrous oxide sources and sinks, Nature, 586, 248–256, https://doi.org/10.1038/s41586-020-2780-0, 2020.
Upstill-Goddard, R. C., Salter, M. E., Mann, P. J., Barnes, J., Poulsen, J., Dinga, B., Fiske, G. J., and Holmes, R. M.: The riverine source of tropospheric CH4 and N2O from the Republic of Congo, Western Congo Basin, Biogeosciences, 14, 2267–2281, https://doi.org/10.5194/bg-14-2267-2017, 2017.
Valentini, R., Arneth, A., Bombelli, A., Castaldi, S., Cazzolla Gatti, R., Chevallier, F., Ciais, P., Grieco, E., Hartmann, J., Henry, M., Houghton, R. A., Jung, M., Kutsch, W. L., Malhi, Y., Mayorga, E., Merbold, L., Murray-Tortarolo, G., Papale, D., Peylin, P., Poulter, B., Raymond, P. A., Santini, M., Sitch, S., Vaglio Laurin, G., van der Werf, G. R., Williams, C. A., and Scholes, R. J.: A full greenhouse gases budget of Africa: synthesis, uncertainties, and vulnerabilities, Biogeosciences, 11, 381–407, https://doi.org/10.5194/bg-11-381-2014, 2014.
Wang, J., Li, J., Ye, J., Zhao, J., Wu, Y., Hu, J., Liu, D., Nie, D., Shen, F., Huang, X., Huang, D. D., Ji, D., Sun, X., Xu, W., Guo, J., Song, S., Qin, Y., Liu, P., Turner, J. R., Lee, H. C., Hwang, S., Liao, H., Martin, S. T., Zhang, Q., Chen, M., Sun, Y., Ge, X., and Jacob, D. J.: Fast sulfate formation from oxidation of SO2 by NO2 and HONO observed in Beijing haze, Nat. Commun., 11, 2844, https://doi.org/10.1038/s41467-020-16683-x, 2020.
WMO: WMO Greenhouse Gas Bulletin, No. 14, ISSN 2078-0796, available at: https://library.wmo.int/doc_num.php?explnum_id=5455 (last access: 15 December 2020), 2018.
Wofsy, S. C., the HIPPO Science Team, and Cooperating Modelers and Satellite Teams: HIAPER Pole-to-Pole Observations (HIPPO): Fine grained, global scale measurements for determining rates for transport, surface emissions, and removal of climatically important atmospheric gases and aerosols, Philos. T. R. Soc. A, 369, 2073–2086, https://doi.org/10.1098/rsta.2010.0313, 2011.
Wofsy, S. C., Afshar, S., Allen, H. M., Apel, E., Asher, E. C., Barletta, B., Bent, J., Bian, H., Biggs, B. C., Blake, D. R., Blake, N., Bourgeois, I., Brock, C. A., Brune, W. H., Budney, J. W., Bui, T. P., Butler, A., Campuzano-Jost, P., Chang, C. S., Chin, M., Commane, R., Correa, G., Crounse, J. D., Cullis, P. D., Daube, B. C., Day, D. A., Dean-Day, J. M., Dibb, J. E., DiGangi, J. P., Diskin, G. S., Dollner, M., Elkins, J. W., Erdesz, F., Fiore, A. M., Flynn, C. M., Froyd, K., Gesler, D. W., Hall, S. R., Hanisco, T. F., Hannun, R. A., Hills, A. J., Hintsa, E. J., Hoffman, A., Hornbrook, R. S., Huey, L. G., Hughes, S., Jimenez, J. L., Johnson, B. J., Katich, J. M., Keeling, R. F., Kim, M. J., Kupc, A., Lait, L. R., Lamarque, J.-F., Liu, J., McKain, K., Mclaughlin, R. J., Meinardi, S., Miller, D. O., Montzka, S. A., Moore, F. L., Morgan, E. J., Murphy, D. M., Murray, L. T., Nault, B. A., Neuman, J. A., Newman, P. A., Nicely, J. M., Pan, X., Paplawsky, W., Peischl, J., Prather, M. J., Price, D. J., Ray, E., Reeves, J. M., Richardson, M., Rollins, A. W., Rosenlof, K. H., Ryerson, T. B., Scheuer, E., Schill, G. P., Schroder, J. C., Schwarz, J. P., St. Clair, J. M., Steenrod, S. D., Stephens, B. B., Strode, S. A., Sweeney, C., Tanner, D., Teng, A. P., Thames, A. B., Thompson, C. R., Ullmann, K., Veres, P. R., Vieznor, N., Wagner, N. L., Watt, A., Weber, R., Weinzierl, B., Wennberg, P., Williamson, C. J., Wilson, J. C., Wolfe, G. M., Woods, C. T., and Zeng, L. H.: ATom: Merged Atmospheric Chemistry, Trace Gases, and Aerosols, ORNL DAAC, Oak Ridge, Tennessee, USA, https://doi.org/10.3334/ORNLDAAC/1581, 2018.
Xiang, B., Miller, S. M., Kort, E. A., Santoni, G. W., Daube, B. C., Commane, R., Angevine, W. M., Ryerson, T. B. , Trainer, M. K. , Andrews, A. E., Nehrkorn, T., Tian, H., and Wofsy, S. C.: Nitrous oxide (N2O) emissions from California based on 2010 CalNex airborne measurements, J. Geophys. Res.-Atmos., 118, 2809–2820, https://doi.org/10.1002/jgrd.50189, 2013.
Yang, Simon, Chang, B. X., Warner, M. J., Weber, T. S., Bourbonnais, A. M., Santoro, A. E., Kock, A., Sonnerup, R. E., Bullister, J. L. Wilson, S. T., and Bianchi, D.: Global reconstruction reduces the uncertainty of oceanic nitrous oxide emissions and reveals a vigorous seasonal cycle, P. Natl. Acad. Sci. USA, 117, 11954–11960; https://doi.org/10.1073/pnas.1921914117, 2020.
- Abstract
- Introduction
- Instrument specifications, spectral analysis, and calibration
- Accuracy of N2O measurements from QCLS
- Results and discussion
- Conclusions
- Appendix A: List of frequently used symbols and acronyms
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement
- Abstract
- Introduction
- Instrument specifications, spectral analysis, and calibration
- Accuracy of N2O measurements from QCLS
- Results and discussion
- Conclusions
- Appendix A: List of frequently used symbols and acronyms
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement