the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 22 Jul 2020
| 22 Jul 2020
Biomass-burning-derived particles from a wide variety of fuels – Part 2: Effects of photochemical aging on particle optical and chemical properties
Christopher Y. Lim
David H. Hagan
Matthew Coggon
Abigail Koss
Kanako Sekimoto
Joost de Gouw
Timothy B. Onasch
Carsten Warneke
Jesse H. Kroll
Particles in smoke emitted from biomass combustion have a large impact on global climate and urban air quality. There is limited understanding of how particle optical properties – especially the contributions of black carbon (BC) and brown carbon (BrC) – evolve with photochemical aging of smoke. We analyze the evolution of the optical properties and chemical composition of particles produced from combustion of a wide variety of biomass fuels, largely from the western United States. The smoke is photochemically aged in a reaction chamber over atmospheric-equivalent timescales ranging from 0.25 to 8 d. Various aerosol optical properties (e.g., the single-scatter albedo, the wavelength dependence of absorption, and the BC mass absorption coefficient, MACBC) evolved with photochemical aging, with the specific evolution dependent on the initial particle properties and conditions. The impact of coatings on BC absorption (the so-called lensing effect) was small, even after photochemical aging. The initial evolution of the BrC absorptivity (MACBrC) varied between individual burns but decreased consistently at longer aging times; the wavelength dependence of the BrC absorption generally increased with aging. The observed changes to BrC properties result from a combination of secondary organic aerosol (SOA) production and heterogeneous oxidation of primary and secondary OA mass, with SOA production being the major driver of the changes. The SOA properties varied with time, reflecting both formation from precursors having a range of lifetimes with respect to OH and the evolving photochemical environment within the chamber. Although the absorptivity of BrC generally decreases with aging, the dilution-corrected absorption may actually increase from the production of SOA. These experimental results provide context for the interpretation of ambient observations of the evolution of particle optical properties in biomass-combustion-derived smoke plumes.
- Article
(3710 KB) - Full-text XML
- Companion paper
-
Supplement
(1420 KB) - BibTeX
- EndNote





Open and contained biomass combustion contributes substantial amounts of particulate matter to the atmosphere (Bond et al., 2004). The emitted particles have a strong influence on global climate by scattering and absorbing solar radiation and impacting cloud properties and stratospheric water content (Penner et al., 1992; Sherwood, 2002; Jacobson, 2014). Biomass-burning-derived particles (BB particles, for short) also have substantial negative impacts on human health globally (Lelieveld et al., 2015), especially when produced as a result of indoor combustion associated with residential cooking and heating. In some parts of the world the frequency and severity of uncontrolled fires is rising and projected to become worse in the future as a consequence of climate change (Dale et al., 2001; Stephens et al., 2013), with effects already being seen in some regions (McClure and Jaffe, 2018).
Particles produced from biomass burning are primarily composed of organic material and black carbon (Andreae and Merlet, 2001). The relative contribution of organic aerosol (OA) and black carbon (BC) depends on the burn conditions, which are strongly related to the fuel type and other environmental factors (McMeeking et al., 2009; McClure et al., 2020). The chemical, optical, and physical properties of freshly emitted BB particles produced from burning of various biomass fuel types under various conditions are reasonably well studied (e.g., Lewis et al., 2008; McMeeking et al., 2009; Levin et al., 2010; Cheng et al., 2016; Fortner et al., 2018; McClure et al., 2020). Such measurements have established that some fraction of the emitted OA is light absorbing, with the absorptivity dependent upon the burn conditions (Saleh et al., 2014). Light-absorbing OA is commonly referred to as brown carbon (BrC). Compared to BC, brown carbon is generally less absorbing and exhibits a much stronger wavelength dependence (Kirchstetter et al., 2004; Andreae and Gelencsér, 2006). As such, the importance of BrC to light absorption tends to increase as wavelength decreases.
The influence of atmospheric processing on the properties of biomass combustion smoke has received less attention, especially in the context of how aging influences BB particle optical properties. (Here, we use “smoke” to indicate the mixture of particles and gases emitted from biomass combustion.) Understanding how BB particles evolve over time is key to establishing their atmospheric impacts (Wang et al., 2018). Field observations of the effects of photochemical aging on BB particle optical properties, and in particular light absorption, are sparse. Forrister et al. (2015) observed that absolute absorption by water-soluble BrC decreased over time within a biomass-burning plume, with a decay time constant of about a day. Wang et al. (2016) observed that the BrC absorptivity (as opposed to the absolute absorption) decreased with photochemical age, with a similar time constant as reported by Forrister et al. (2015). However, both studies suggest that there is some fraction of the BrC that is more persistent and less subject to photochemical degradation, and Zhang et al. (2017) found no decrease in BrC absorption with age for particles in convective outflow. Laboratory measurements can help provide the mechanistic understanding necessary to comprehensively interpret the field observations. Most laboratory studies investigating photochemical aging effects on optical properties, and in particular light absorption, of BB particles have done so for only a small number of individual fuel types or burn conditions and often for particles alone rather than smoke (e.g., Saleh et al., 2013; Sumlin et al., 2017; Tasoglou et al., 2017; Wong et al., 2017; Kumar et al., 2018). Thus, while these studies have proven insightful, the limited number of fuels and conditions considered makes extending the observations to the atmosphere thus far challenging.
In this study, we examine how the optical properties of BB particles evolve as a result of OH-radical-dominated photochemical aging for smoke derived from combustion of a multitude of fuel types and spanning a wide range of burn conditions. We characterize the influence of photochemical aging on optical properties for a substantially greater number of fuel types than have been reported previously in the literature for burn conditions ranging from mostly flaming to entirely smoldering. We access aging timescales ranging from a fraction of a day to many days and characterize the continuous evolution of the particle properties. We show that the BB particle optical properties evolve with photochemical oxidation, linked to both chemical evolution of the primary particles and production of secondary organic aerosol. We characterize and quantify this behavior, developing a generalizable model for the evolution of brown carbon properties in wildfire plumes.
2.1 Campaign overview and sampling strategy
The Fire Influence on Regional to Global Environments and Air Quality Experiment (FIREX-AQ) lab campaign took place at the Missoula Fire Sciences Laboratory in November 2016 (NOAA, 2013). A full description of the sampling strategy and methods, including descriptions of instruments used, for our study is provided in Lim et al. (2019) and McClure et al. (2020). Only a short description is provided here. A wide variety of fuels (Table S1 in the Supplement) were combusted under realistic conditions in a large combustion chamber (ca. 12 m × 12 m × 19 m). Fuels included bear grass, rice straw, chaparral (chamise and manzanita), juniper, sagebrush, canopy, litter and mixed components from soft woods (fir, pine, spruce), rotten logs, peat, and dung. Data from the FIREX lab study are available via the NOAA website (https://www.esrl.noaa.gov/csd/projects/firex/firelab/; last access: 24 January 2020), with data specific to this work also archived as Cappa et al. (2019a).
The particle and gas emissions from individual stack burns were injected into a 0.15 m3 photochemical reaction chamber (the mini chamber). A burn typically lasted about 10–20 min. Smoke was transferred from the burn room to the adjacent room housing the mini chamber and associated instrumentation through a 30 m long high-velocity community inlet. The residence time in the community inlet was determined as <2 s. Smoke from the community inlet was subsampled into the mini chamber through a PM1 cyclone using an ejector diluter. There were some losses of particles and gases during transfer. However, comparison between the volatile organic compound (VOC) concentrations measured prior to sampling through the community inlet and from the mini chamber prior to photooxidation demonstrates losses were relatively minor (<8 % per volatility bin, across the entire measured VOC distribution) and should not substantially impact the results here (Coggon et al., 2019; Lim et al., 2019).
2.2 The mini chamber
Prior to each burn, the chamber was flushed with clean air with RH ranging from 25 % to 40 %. Smoke was sampled into the mini chamber across an entire burn or until shortly after the concentration in the mini chamber reached maximum. An instrument suite continuously sampled air from the mini chamber (see below). Clean makeup air added from a zero-air generator (Teledyne 701H) ensured the total inflow (sample + makeup air) equaled the air being sampled out of the chamber. The actual flow rate out of the chamber varied slightly between experiments dependent upon the exact instrument suite. Given the mini chamber volume and the typical flow rates, the net dilution from the community inlet was about a factor of 7.
Details of the mini chamber and its operation are provided in Lim et al. (2019). The FIREX mini chamber operated in semibatch mode, where unoxidized smoke-laden air is first sampled into the mini chamber. After the sampling period, a clean airflow replaces the sample flow, maintaining the size of the bag over time. Oxidation is initiated by turning on one 254 nm UV light. Externally generated ozone added at 50–100 ppb serves to initiate production of OH radicals via generation of O(1D). The concentrations of gases and particles continuously decreased owing to dilution by clean air and wall losses. The observed decay rate of acetonitrile (ACN) provides the dilution rate (see Sect. 2.3); gases that are substantially more sticky than ACN may be lost at a faster rate. The ACN loss rate, characterized as , was around 0.024 min−1, consistent with the gas flow rates. The particle loss rate was greater owing to additional losses of particles to the walls of the chamber. The general particle mass loss rate was min−1. However, for BC-rich particles the loss rate was initially enhanced when the lights were turned on (discussed further below). OH exposures were determined from the dilution-corrected decay of deuterated n-butanol (D9). These are converted to an equivalent time-evolving photochemical age (tOH) assuming [OH] molecules cm−3. ThetOH ranged from 0.25 to about 8 d of equivalent aging. OH concentrations determined for real-world biomass burning plumes may be higher than assumed here (Akagi et al., 2012), and thus aging may happen on shorter timescales. For the model calculations in Sect. 3.2.1, the time-evolving OH exposures were converted to equivalent OH concentrations based on the time since the lights were turned on. The n-butanol (D9) measurements were available for only about half of the individual burn experiments owing to data availability limitations of the VOC measurements. While there is some experiment-to-experiment variability in thetOH values accessed, the relationship between experiment time andtOH is reasonably consistent. Thus, we use the average behavior to estimate the photochemical ages for experiments lacking direct measurement.
As discussed in Coggon et al. (2019), the conditions of the mini chamber do not perfectly represent the photochemical conditions of the atmosphere. The use of the 254 nm UV lights enhances photolysis of a small number of select nonmethane organic gases (NMOGs), especially furfural. In contrast, photolysis of NO2 is likely substantially slower than in the atmosphere. This impacts the time evolution of the NO∕NO2 ratio and consequently radical reactions (e.g., RO2+NO) and NOx loss processes. The influence of photobleaching of BrC is likely underestimated relative to the atmosphere as the actual exposure time (tens of minutes) is much shorter than the equivalent photochemical age (many days). Overall, the photochemical environment in the mini chamber emphasizes OH-driven oxidation under initially high NOx conditions that rapidly shift towards low-NOx conditions (Coggon et al., 2019).
2.3 Instrumentation
The suite of instruments sampling from the mini chamber are listed in Table S2 and further details of instrument operation and uncertainties are provided in McClure et al. (2020), Lim et al. (2019), and Coggon et al. (2019). In brief, particle-phase instruments sampled alternatingly through a two-stage thermodenuder that operated at 150 and 250 ∘C with a residence time of ∼5 s. The cycle rate between ambient and thermodenuder sampling was 2 min. The ambient (bypass) line on the thermodenuder was lined with charcoal cloth to remove excess NMOG, O3, and NO2 that might otherwise interfere with the measurements. Light absorption and extinction coefficients (babs and bext, respectively) were measured at 405 and 532 nm using the UC Davis cavity ring-down photoacoustic spectrometer (CRD-PAS) and at 781 nm using a DMT photoacoustic soot spectrometer (PASS-3). Estimated uncertainties for the absorption measurements were 8 %, 5 %, and 10 % at 405, 532, and 781 nm, respectively. Particle size distributions were measured using a scanning electrical mobility spectrometer (SEMS; model 2002, Brechtel Mfg., Inc.). The overall concentration and composition of submicron nonrefractory particulate matter (NR-PM1) were measured using a high-resolution time-of-flight aerosol mass spectrometer (HR-AMS; Aerodyne Research, Inc.). The concentration and composition of refractory-BC-containing particles were characterized using a soot particle aerosol mass spectrometer (SP-AMS; Aerodyne Research, Inc.); the SP-AMS was operated with only laser vaporization of particles such that it was sensitive to only those particles containing refractory black carbon (rBC). Refractory black carbon concentrations and size distributions for volume-equivalent diameters from 90 to 350 nm were quantitatively measured using a single-particle soot photometer (SP2; DMT). Refractory BC outside of this size window was accounted for through multimodal fitting of the observed mass-weighted size distributions. The estimated uncertainty in the rBC concentration is 30 %. Gas-phase instruments sampled directly from the mini chamber through Teflon sampling lines. These included a proton-transfer-reaction time-of-flight mass spectrometer (PTR-ToF-MS) to characterize primary organic gases (Yuan et al., 2017), an I− chemical ionization mass spectrometer to characterize various gas-phase organic oxidation products, an ozone monitor (2B Technologies, model 202), a CO monitor (Teledyne, model T300), and a CO2 monitor (LI-COR, LI-840A).
2.4 Brown carbon, coatings, and particle classification
Mass absorption coefficients, referenced to BC, are calculated as
where babs is the absorption coefficient (Mm−1) and [BC] is the concentration of refractory black carbon (µg m−3). The observed MACBC may include contributions to absorption from both the BrC (the BrC enhancement, Eabs,BrC) and from the so-called lensing effect that can occur when nonabsorbing coatings mix with BC (the coating-induced enhancement, Eabs,coat) (Cappa et al., 2012; Lack et al., 2012). The observable absorption enhancement is characterized as
where MACBC,obs is the observed value and MACBC,pure is a reference value for pure, uncoated BC. The reference MACBC,pure can be established from the literature or from the observations; we take the latter approach here, using wavelength-specific MACBC,pure values determined by McClure et al. (2020) for this data set by extrapolation of the observed MACBC,obs versus [OA]∕[BC] relationship to zero [OA]∕[BC]. The MACBC,pure equals 11.8±1.2 m2 g−1 (405 nm), 8.8±0.9 m2 g−1 (532 nm), and 5.5±0.6 m2 g−1 (781 nm). We assume these reference values are applicable to all burns, consistent with the negligible dependence of the primary particle MACBC,781 nm on the coating-to-BC mass ratio (Rcoat-rBC) (McClure et al., 2020) and the lack of size dependence of the MACBC,pure measured for uncoated BC particles produced from a laboratory flame (Forestieri et al., 2018). The use of a campaign-specific value helps to mitigate instrumental uncertainties in determining Eabs (Cappa et al., 2019b).
We observe limited change in the Eabs,781 nm with photochemical aging here, with Eabs,781 nm for aged particles averaging 1.19 for Rcoat-rBC<9 (Fig. S2). The Eabs,781 nm (and MACBC,781 nm) only increases notably (by more than 10 %) when Rcoat-rBC>9, while theory predicts the MACBC,781 nm should increase by this amount for individual particles when Rcoat-rBC>1 (Fuller et al., 1999). Other laboratory experiments, often performed using monodisperse BC seed particles, have observed substantial coating-induced enhancements and a dependence on the Rcoat-rBC (e.g., Cross et al., 2010; Shiraiwa et al., 2010; Cappa et al., 2012; Metcalf et al., 2013; J. Peng et al., 2016; You et al., 2016). We suggest that the small Eabs,coat for photochemically aged biomass combustion particles results from three phenomena: (i) condensation occurring onto a polydisperse rBC distribution, leading to some particles having very thick coatings and some quite thin, yielding a large average Rcoat-rBC yet small enhancement (Fierce et al., 2016, 2020); (ii) the coated biomass-burning-derived rBC particles not having a core–shell morphology and reduced Eabs,coat (Helgestad, 2016; Liu et al., 2017); and (iii) weak absorption by BrC at 781 nm by both internally and externally mixed BrC that becomes notable only when the total [OA] greatly exceeds [BC] (McClure et al., 2020). This is supported by the comparably much stronger relationship for photochemically aged particles between MACBC,781 nm and the [OA]∕[rBC] ratio than with the Rcoat-rBC (Fig. S2). This suggests that the majority of the variability in the MACBC,781 nm derives from varying contributions of BrC, rather than in Eabs,coat, and that Eabs,coat is near unity.
The wavelength-specific BrC absorption (babs,BrC) is estimated as
Given the above discussion and the complementary discussion in McClure et al. (2020), we assume Eabs,coat is unity, with the derived babs,BrC an upper limit. A lower limit for babs,BrC can be obtained using an empirical Eabs,coat versus RBC-coat relationship. For the aged particles, the average upper-limit babs,BrC values exceed the lower limit babs,BrC values by 11 (±9) % at 405 nm and 29 (±16) % at 532 nm. The brown-carbon-specific mass absorption coefficient (MACBrC) is
The MACBrC values are bulk-average values and do not account for different molecules and classes of molecules likely having different absorptivities. Uncertainty in the estimated MACBC,pure and measured babs, as well as [rBC], also contributes to uncertainty in the estimated babs,BrC and MACBrC. The uncertainties in babs,BrC and the derived MACBrC are estimated by error propagation and scale inversely with the [OA]∕[rBC] ratio as the OA contribution to the total absorption decreases (McClure et al., 2020).
The Ångström absorption exponent (AAE) characterizes the wavelength dependence of absorption:
where λ1 and λ2 indicate two different measurement wavelengths. The AAE can be calculated based on the total absorption or just for BrC, with the latter designated as AAEBrC. The single-scatter albedo (SSA) characterizes the fraction of light extinction attributable to scattering:
McClure et al. (2020) classified individual experiments into six classes dependent on the primary particles (SSA405 nm); the SSA405 nm exhibited strong relationships with a variety of other intensive optical properties and with the [OA]∕[rBC] mass ratio. We use these same classifications, ranging from class 1 – primary particles having low SSA405 nm values (0.22–0.38) and small [OA]∕[rBC] (0.3–2.6) – to class 6 – primary particles having high SSA405 nm values (0.94 to 0.96) and very large [OA]∕[rBC] (400 to 1×105) – to interpret the influence of photochemical aging. Table S3 provides the general relationship between fuel type, SSA, and log ([OA]∕[BC]). As noted by McClure et al. (2020), the classifications relate somewhat to the burn-average modified combustion efficiency (MCE), which characterizes the burn efficiency, with class 1 generally deriving from more efficient, flaming burns having higher MCE and class 6 from more less efficient, smoldering burns having lower MCE. The average dependence of the various aerosol properties on photochemical age is determined for each SSA class. Grouping experiments by SSA classification are justified given the substantial variability in the primary particle properties between individual burns. The focus on average behavior increases robustness of the resulting interpretation as sensitivity to experiment-by-experiment differences (e.g., length of experiment, initial concentration, amount of fuel consumed) is reduced.
3.1 Observations of the influence of photochemical aging on optical properties
3.1.1 Total particles (black + brown carbon)
The suite of intensive optical (e.g., SSA, MAC, AAE) and chemical and physical properties (e.g., [OA]∕[rBC], Rcoat-rBC) vary with photochemical aging; the average behavior for each particle class is shown in Fig. 1 and the experiment-specific behavior within each class in Figs. S3 through S8. The majority of the mass formed is OA, with minor contributions from inorganic species. A detailed assessment of secondary organic aerosol (SOA) formation is provided by Lim et al. (2019).
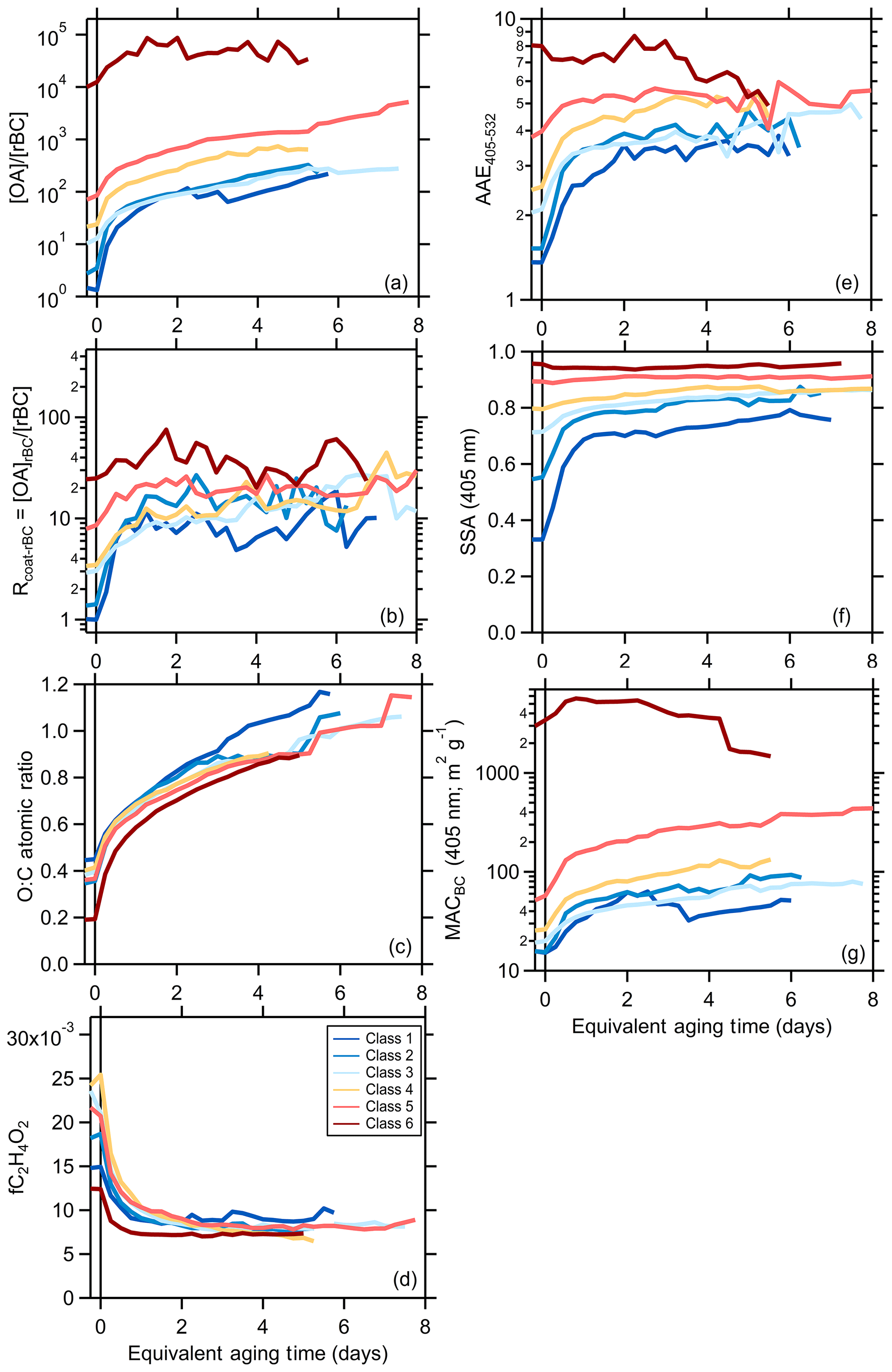
Figure 1Relationship between (a) the [OA]∕[rBC] ratio, (b) Rcoat-rBC, (c) AAE405–532, (d) the SSA at 405 nm, (e) the O : C atomic ratio, (f) the AMS f60, and (g) the MACBC at 405 nm as a function of equivalent photochemical aging time (assuming [OH] molecules cm−3). The different lines are colored according to the SSA classification and are averages for all burns within a class (see panel d). Figures showing results for each burn are available in the Supplement. Some of the apparent discontinuities for individual classes with certain properties results from a change in the number of individual experiments contributing to the average above a given aging time.
The changes observed for the lower-number (more BC-rich) particle classes are typically more pronounced than for the higher-number (more OA-rich) particle classes. For example, the class 1 SSA405 nm increases from 0.3 to 0.7 and the AAE405–532 from 1.4 to 3.5 with aging, while the class 6 SSA405 nm remains near constant at 0.94 and the AAE405–532 decreases slightly from ca. 7 to 5. Notably, the AAE405–532 converges towards a common value, around 4, with aging. While there remain some class-specific differences, this indicates that the SOA that is forming has similar optical properties independent of the initial burn conditions.
Also, the [OA]∕[BC] for class 1 increases by a factor of nearly 70 while the increase for class 6 is 10 times smaller, and the particle class 1 Rcoat-rBC increases by a factor of 11 while there is negligible change for particle class 6. Generally, the most rapid changes occur when the equivalent aging time is less than a day, with slower changes after this. Some of the observed changes, in particular for class 1 and class 2 particles, result from enhanced loss of BC-rich particles when the lights are turned on, although this alone cannot explain the differing extents of change and would not directly impact the Rcoat-rBC (Sect. 3.2.1). These observations indicate substantial formation of secondary OA and a net increase in the overall (dilution-corrected) OA, consistent with Lim et al. (2019). If secondary OA formation were offset by evaporation of primary OA then the SSA would not have increased, nor would the [OA]∕[rBC] or Rcoat-rBC ratios have increased.
Additionally, the OA O : C atomic ratio increases and the AMS f60 (a marker for primary biomass burning OA attributed to levoglucosan and similar species) decreases rapidly with photochemical aging (Fig. 1). However, unlike the above properties, the O : C and f60 evolution are generally similar between SSA classes, despite some variability in the primary particle properties (McClure et al., 2020). The changes in both O : C and f60 occur more rapidly than can be explained by heterogeneous oxidation, implying rapid photochemical production of secondary OA (Lim et al., 2019). The O : C increases rapidly at tOH<1 d with continual increases with further aging. The f60 values decline rapidly at tOH<1 d to around f60=0.008, changing negligibly with continued aging. It is noteworthy that the f60 plateaus around 0.008 upon aging rather than at 0.003, the value denoted by Cubison et al. (2011) as the nominal background outside of biomass-burning-influenced areas. This suggests that observations of f60 values much smaller than 0.008 in ambient measurements indicate an influence of OA from non-biomass-combustion sources (both POA + SOA).
3.1.2 Brown carbon
Observations
The class-average MACBrC,405 and the AAEBrC,405–532 evolve with photochemical aging (Fig. 2). Results for individual experiments are shown in Figs. S10 and S11. Overall, the MACBrC,405 values decrease from the primary values with aging. However, for the initially less absorbing BrC, corresponding to the higher SSA classes (i.e., class 5 and 6), the MACBrC,405 values first increase at tOH<1 d before decreasing. This behavior suggests rapid formation of SOA more absorbing than the primary OA for these classes, with production of less-absorbing SOA or conversion of POA or SOA via heterogeneous oxidation into less-absorbing forms at later times (discussed further in Sect. 3.2.2). The MACBrC,532 values behave similarly (Fig. S12). The class-average MACBrC,405 values all converge around 0.2 m2 g−1 after many days (∼5) of equivalent aging. The class-average AAEBrC,405–532 values converge towards AAEBrC,405–532 ∼5 after many days of equivalent aging, with the exact behavior dependent on the initial AAEBrC,405–532.
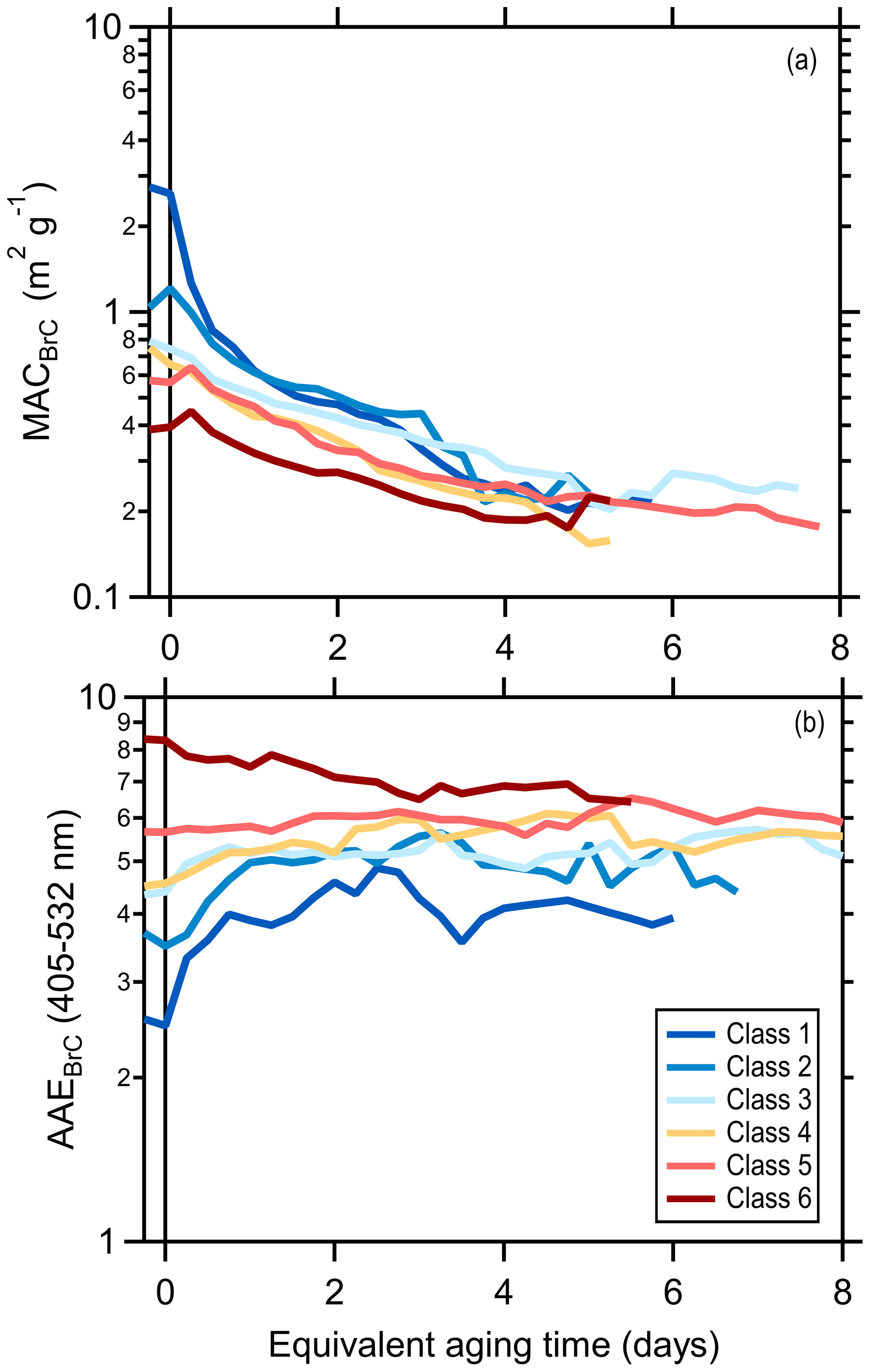
Figure 2Relationship between the brown carbon (a) MACBrC and (b) AAEBrC as a function of equivalent photochemical aging time (assuming [OH] molecules cm−3). The different lines are colored according to the SSA classification and are averages for all burns within a class. Class 1 corresponds to particles having low SSA405 and class 6 to particles having high SSA405 nm. Figures showing results for each burn are available in the Supplement.
Comparison with literature
Our observations can be compared with various literature observations, where we use reported SSA or AAE values to infer the equivalent particle class (Table S4). Martinsson et al. (2015) observed that the 370 nm OA absorptivity of class 1 particles decreased after rapidly aging for tOH=8.3 d. Saleh et al. (2013) reported the AAE for class 1 particles produced from oak combustion changed negligibly (1.38 to 1.42) while the AAE for particles from pine combustion increased slightly (1.48 to 1.73) after photochemically aging smoke for tOH of a few hours; the extent of aging was likely greater for the pine than oak. The AAE for class 1 particles during FIREX increased to a greater extent (1.4 to 2.2) after similar aging (tOH∼6 h). Saleh et al. (2013) also report smaller SOA absorptivity compared to POA at 550 nm, qualitatively consistent with our observations. Zhong and Jang (2014) observed the 550 nm absorptivity of class 5 particles initially increased over ∼2 h but then gradually declined upon photochemical aging of smoke from smoldering combustion. This general behavior is consistent with the behavior of the class 5 and 6 particles here, for which the MACBrC increased slightly in the first few hours of equivalent aging followed by a continuous decline.
Similar to our observations, Kumar et al. (2018) observed AAE370–660 values for class 1 particles increased from around 1.4 to 2 for photochemically aged beechwood smoke over tOH∼1 d. They report that the primary OA is more absorbing than the SOA formed. We interpolate their results to estimate MAC405 values for BC (12.4 m2 g), primary BrC (4.0 m2 g), and secondary BrC (1.6 m2 g). The MACBC,405 value agrees well with our estimate of 11.8 m2 g−1. However, their primary MACBrC,405 substantially exceeds our class 1 average (2.25 m2 g) as does their primary BrC AAE (4.6 versus 2.2±1.1, respectively), while their AAE for SOA is similar to that observed here at longer aging times (5.5 versus ∼5, respectively). Over their ∼1 d of equivalent aging they find little variability in the MACBrC for SOA. Cappa et al. (2019b) reported MACBrC,405 and AAE405–532 for relatively fresh biomass-burning-derived organic aerosol in the ambient atmosphere, reporting values (0.84 m2 g−1 and 3.02, respectively) similar to those for class 2 particles here.
Sumlin et al. (2017) heterogeneously aged class 6 particles with OH and observed negligible changes in the absorptivity and SSA405 at 1 and 3.5 d of equivalent aging but then a sudden change at 4.5 d of equivalent aging. This differs substantially from our observations, where changes occurred continuously; the reasons for this difference are unclear, although a discontinuous change with increasing exposure is unexpected. Browne et al. (2019) also heterogeneously aged class 6 particles with OH but observed a continuous decrease in the absorptivity with OH oxidation, consistent with the long-term behavior observed here. Wong et al. (2017) photolytically aged (at 300–400 nm) water-soluble and water-insoluble solution extracts of particles from smoldering combustion and observed that the particle absorptivity initially increased over the course of about a day, peaked, and then declined over longer times to below the initial value. While this general behavior is consistent with the evolution of our class 5 and 6 particles, the photon flux here was likely too low for direct photolytic aging to contribute substantially (Z. Peng et al., 2016), especially given the short absolute aging timescale in the mini chamber (tens of minutes). Lee et al. (2014) photolytically aged water-soluble extracts of secondary BrC produced from photooxidation of naphthalene and observed a continuous decrease in the absorptivity, but again with a time constant that was likely too long to have influenced our experiments. Fleming et al. (2020) photolytically aged particles from 12 of the FIREX burns that were collected on Teflon filters. They observed a wide diversity of equivalent lifetimes (referenced to the time-averaged photon flux in Los Angeles, CA) for different BrC chromophores, some of which were quite short (<1 d). However, the equivalent lifetime of the total absorption was >10 d for all samples, with a recalcitrant component making up ca. 30 % of the total absorption.
Overall, our results combined with the literature indicate that differences in starting conditions (i.e., particle class) are important to consider when comparing between different photochemical aging experiments on biomass-combustion-derived particles. Additionally, it is important consider whether a given experiment is likely to favor one process over another, e.g., SOA formation, heterogeneous oxidation, or photolysis.
3.2 Understanding and modeling the photochemical evolution of smoke
3.2.1 Chamber photochemical model description
Overview
We observe that a wide range of particle properties evolve as the smoke from all SSA classes is photochemically aged. To facilitate interpretation, we have developed a simplified model of photooxidation that accounts for SOA formation from oxidation of gas-phase species, heterogeneous oxidation, differential losses of particle types, and dilution in the mini chamber. We aim to simulate the evolution of the brown carbon absorptivity (i.e., MACBrC), specifically, using the initial primary particle properties, gas-phase species, and evolution of the [OA]∕[rBC], O : C, and AMS f60 as constraints or guides as to the reasonableness of the general model formulation. Model parameters are constrained as possible by the observations or tuned to provide good agreement with the observations for all particle classes (Tables 1 and 2). Our modeling approach complements the analysis of Lim et al. (2019), who show that the amount of SOA formed is directly related to the total amount of precursor nonmethane organic gases (NMOGs) in the chamber and that the effective SOA mass yield increases with aging. Here, we take a more mechanistic approach to understanding SOA formation in these experiments and focus on the influence of SOA formation on the observed optical properties. The model framework, constraints, and assumptions are described below.
Table 1Model parameters for NMOG.
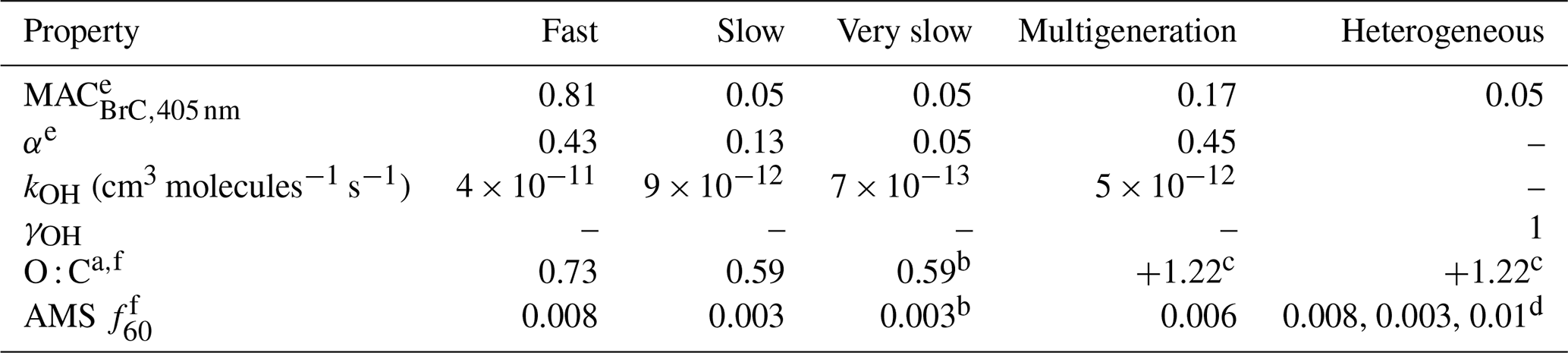
– means not applicable. a It is assumed that the carbon backbone has eight carbon atoms. The assumed O : C values therefore correspond to 5.8, 4.7, and 4.7 oxygen atoms per molecule for the fast, slow, and very slow SOA. b Assumed to equal the slow SOA. c It is assumed that every reaction leads to the addition of n oxygen atoms, where n is the indicated value. Consequently, the average O : C for multigeneration products increases over time as the multigeneration species accumulate oxygen. d Values for oxidation products from internally mixed POA, externally mixed POA, and SOA. e Determined from a global fit to the MACBrC,405 nm and [OA]∕[rBC] observations. f Determined from a global fit to the O : C or f60 observations after determining the α values.
Table 2Model parameters by particle class.

a Directly constrained from observations. b Mass ratio (unitless). c Units are m2 g−1. d Determined from a global fit to the MACBrC,405 nm and [OA]∕[rBC]. e Specified as a constant.
Reactive gases
Biomass burning releases a wide variety of NMOGs (Koss et al., 2018; Sekimoto et al., 2018; and references therein). Upon oxidation, these NMOGs can form SOA. In general, higher-molecular-weight NMOGs have a higher propensity to form SOA (Cappa and Wilson, 2012). We consider the suite of gas-phase compounds measured by PTR-ToF-MS (Koss et al., 2018) having molecular weights greater than 50 amu as potential SOA precursor compounds. (Consideration of all species measured, including those with MW <50 amu does not change the results, consistent with Lim et al., 2019.) These 107 identified gas-phase compounds have been binned according to their OH reaction rate coefficients reported by Koss et al. (2018) (Figs. S13 and S14). The OH rate coefficients range from cm3 molecules−1 s−1 (for sesquiterpenes) to (for nitrobenzene); most compounds have kOH values greater than cm3 molecules−1 s−1. Based on these observations, we make a simplifying assumption that there are three general classes of SOA precursor molecules: those that react fast (), those that react slowly (), and those that react very slowly (). The corresponding SOA-type-specific kOH values we use in the model are cm3 molecules−1 s−1 ( d), cm3 molecules−1 s−1 ( d), and cm3 molecules−1 s−1 ( d) (Table 1 and Fig. S13). (The time constants given correspond to [OH] molecules cm−3.) The dividing lines between the NMOG classes are somewhat arbitrary but capture the general overall behavior and are consistent with variable observed formation timescales of secondary NMOG (Coggon et al., 2019). Loss of primary species due to photolysis is not considered; Coggon et al. (2019) show that rapid photolysis dominates loss for a small number of primary species, notably furfural. These species tend to be fast reacting and thus are incorporated in the fast NMOG group, although their actual loss rate is likely underestimated.
The initial total NMOG concentration is specified relative to the initial [OA], consistent with Lim et al. (2019). The observed average ratio between the total carbon mass concentration measured by the PTR-ToF-MS and the initial [OA] carbon mass is 7±3. SSA-class-specific initial [NMOG]∕[OA] values are specified in the model, constrained to fall around this range (Table 2). This initial gas-phase mass is apportioned between the fast, slow, and very slow NMOGs. The PTR-ToF-MS measurements indicate that the relative abundances of fast-reacting, slow-reacting, and very slow reacting NMOG average approximately 0.5 : 0.4 : 0.1 on a mass basis. We specify the very slow NMOG fraction as a constant and allow the relative abundances of fast- and slow-reacting NMOGs to vary between the particle classes, with specific values determined from fitting to the observations; the derived values are in general agreement with the observed range (Table 2). Allowing for some variability is reasonable, given that the NMOG composition varied between experiments and with burn type (Sekimoto et al., 2018).
The influence of multigeneration chemistry is accounted for by assuming all gas-phase products from reaction of primary precursors are reactive towards OH radicals. This multigenerational (MG) chemistry is simplified by assuming that all MG species react identically, independent of the actual generation number (i.e., number of times reacted) or precursor identity. This allows for a substantial reduction in the number of species tracked relative to more explicit methods (Aumont et al., 2005; Cappa and Wilson, 2012). The assumed multigenerational oxidation OH reaction rate coefficient, kOH,MG, is set to cm−3 molecules−1 s−1 ( d). The kOH,MG falls between the fast and slow species. MG species are assumed to fragment upon reaction, with a fragmentation probability (f) of 15 %, serving to limit the maximum SOA formation; assuming a constant f is a simplification over more detailed approaches (Cappa and Wilson, 2012).
SOA formation and processing
SOA forms upon reaction of the primary NMOG types and MG species, referred to here as SOAfast, SOAslow, SOAVS, and SOAMG. SOA formation occurs here with fixed yields (α) assuming both instantaneous and irreversible (i.e., nonvolatile) condensation, which are simplifications over more detailed approaches. The influence of vapor wall losses (Zhang et al., 2014) is not explicitly considered and thus reflected in the derived α values. SOA and POA also react heterogeneously with OH, producing oxidized OA that can also react with OH. We assume an OH reactive uptake coefficient (γOH) of unity for all OA types. Upon oxidation, 15 % of the POA and SOA mass is assumed lost owing to fragmentation and volatilization of the products (i.e., ). Heterogeneous oxidation of oxidized OA leads only to fragmentation reflecting the greater propensity for more oxidized species to fragment upon reaction (Kroll et al., 2015). The kOH,het values are calculated using the [OH] with the observed surface-weighted particle diameters and appropriate mass-transfer corrections (Smith et al., 2009).
The overall reaction scheme is summarized below:
Reactions (R1)–(R4) represent oxidation of gas-phase species and SOA formation, where and for a given reaction ; the subscript i indicates which NMOG type reacted. Also,
Reaction (R5) represents the heterogeneous oxidation of fast, slow, very slow, or multigeneration SOA and conversion to an oxidized SOA product, while Reaction (R6) represents heterogeneous oxidation of POA to an oxidized product. Reaction (R7) indicates that reaction of oxidized OA only produces volatile products.
Dilution and particle wall losses
Given the semibatch operation method for the mini chamber, the concentration of particles and gases decreased over time owing to dilution and losses to the chamber walls. The non-OH loss rate of gases was assumed constant in the model, consistent with observations during an experiment where the lights were left off. However, we observed that the overall decay of BC particles containing little coating material (e.g., class 1 and class 2) was enhanced when the lights were turned on, while it was not for particles having thicker initial coating amounts (Figs. S15 and S16). This enhanced loss was unique to BB particles, as no such behavior was observed for either atomized fullerene soot (highly absorbing) or ammonium sulfate (nonabsorbing). That enhanced decay occurs for the BC-rich particles suggests that this phenomenon is related to the amount of coating material, since the class 1 and 2 particles have substantially smaller initial Rcoat,rBC values compared to the other classes (Fig. 1 and McClure et al., 2020). We speculate that the enhanced loss of BC-rich particles results from their having a greater propensity towards photoelectric charging with the 254 nm light, as photoelectric charging is highly sensitive to the state of the particle surface (Burtscher, 1992). We determined an empirical relationship between the observed time-varying rBC decay rate and the coating amount for use in the model (Fig. S17). The decay rate slows as particles become more coated (i.e., have larger Rcoat,rBC), eventually reaching a plateau. We also account for the mixing state of the particles when determining particle loss rates, as McClure et al. (2020) showed that the fraction of total OA that is internally mixed with BC decreases as [OA]∕[rBC] increases. We assume that the population of particles containing BC exhibits faster loss than the population that does not. Independent of the enhanced decay for BC-rich particles, particle decay is faster than for dilution-driven decay of gases because the particles are more subject to wall losses. The reference decay rates for particles and gases were determined from experiments conducted using atomized ammonium sulfate, atomized fullerene soot, and a “dark” smoke experiment. It is assumed that POA does not evaporate upon dilution in the chamber and thus does not contribute further to the available NMOGs (discussed further in Sect. 3.2.6).
SOA properties and model fitting
The different types of SOA in the model, which form from different NMOGs, can have different properties. The properties that are allowed to differ between SOA types and heterogeneous oxidation products, besides kOH, are the MACBrC, SOA yields, O : C, and the f60 (Tables 1 and 2). While allowed to vary between SOA type, these parameters are assumed to be independent of the SSA class. In this manner, we aim to find an overall solution that allows for one set of SOA-type-specific parameters that gives good model–measurement agreement for all of the SSA classes. The choice of SOA yields influences all model outputs, while the choice of the other parameters influences only that parameter output. The type-specific MACBrC and SOA yields (α) are determined by fitting the model to the observations, specifically to the evolution of [OA]∕[rBC] and MACBrC with the OH exposure (Table 1). The Global Fit package in Igor v.8.03 (WaveMetrics) allowed for simultaneous fitting of the observed [OA]∕[rBC] and MACBrC for all particle classes. Thus, the retrieved MACBrC and α values are those that minimize the model–observation difference across all of the particle classes. After determining the best-fit, type-specific MACBrC and SOA yields, separate global fits were performed to the observed O : C and f60 to determine the type-specific O : C and f60 (Table 1). The model time step was 2 min, as this matched the averaging time for the observations.
Our analysis focuses on the evolution of the particle optical properties and specifically the BrC absorptivity. Sect. 3.2.2 provides a detailed discussion of the evolution of the MACBrC with oxidation and the associated determination of SOA-type-specific MACBrC values. Values for the other parameters (SOA yields, O : C and the f60) are briefly discussed here. The tuned SOA yields were αfast=0.43, αslow=0.15, αVS=0.05, and αMG=0.45. These are effective yields averaged across all NMOGs of a given type. The derived average SOA yields are within the range of SOA yields expected for the individual precursor species (Bruns et al., 2016).
Tuned values for the O : C for the SOA types are consistent with expected physical behavior, guided by single-component studies (Chhabra et al., 2011). The fit O : C atomic ratios of the first-generation SOA formed are 0.73 (fast), 0.59 (slow), and 0.59 (very slow, assumed to be the same as slow). The O : C of multigeneration SOA and heterogeneously oxidized POA and SOA vary with time, with the model fit indicating 1.22 oxygen atoms added per oxidation reaction, generally consistent with expectations (Kroll et al., 2015). The model agrees well with the observations (i) when the f60 values for the various SOA types are , , and and (ii) when the f60 for the heterogeneous oxidation products are assumed to depend on the identity of the oxidized component, with for SOA oxidation, for externally mixed POA oxidation, and for internally mixed POA oxidation. We note that similar results are obtained if values are used instead of f60. ( is the high-resolution ion at that most corresponds to levoglucosan.)
3.2.2 Model–measurement comparison
The optimized model does a good job (reduced chi-square =0.4) of describing the evolution of MACBrC, [OA]∕[rBC], O : C atomic ratio, and AMS f60 (Fig. 3). The reasonable agreement for [OA]∕[rBC] indicates the appropriateness of the empirical correction for enhanced decay of rBC-containing particles, the specified SOA yields and initial [NMOG]∕[OA] ratio, and the apportionment between the different NMOG types. The model–measurement agreement improves if the unknown model parameters are allowed to vary somewhat between particle classes; again, some variability is expected given the burn-to-burn variability in the mix of NMOG species (Sekimoto et al., 2018). However, that use of class-independent parameters provides for a generally good description across classes demonstrates an overall general nature of the photochemical evolution.
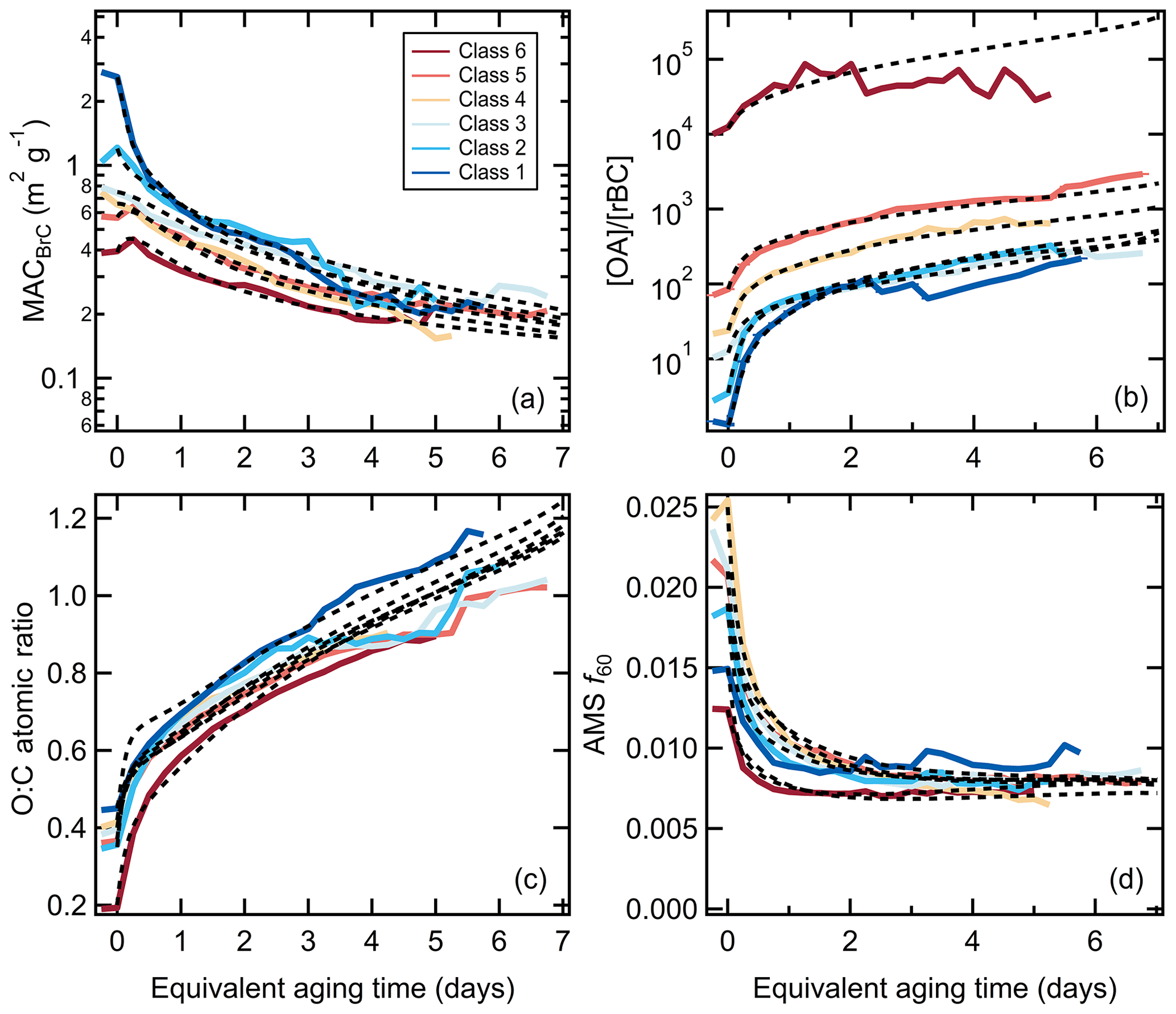
Figure 3Comparison between observed (solid lines) and modeled (dashed lines) values of the (a) MACBrC, (b) the [OA]∕[rBC] ratio, (c) the O : C atomic ratio, and (d) the AMS f60 and the equivalent photochemical aging time (assuming [OH] molecules cm−3), with results shown for each SSA class.
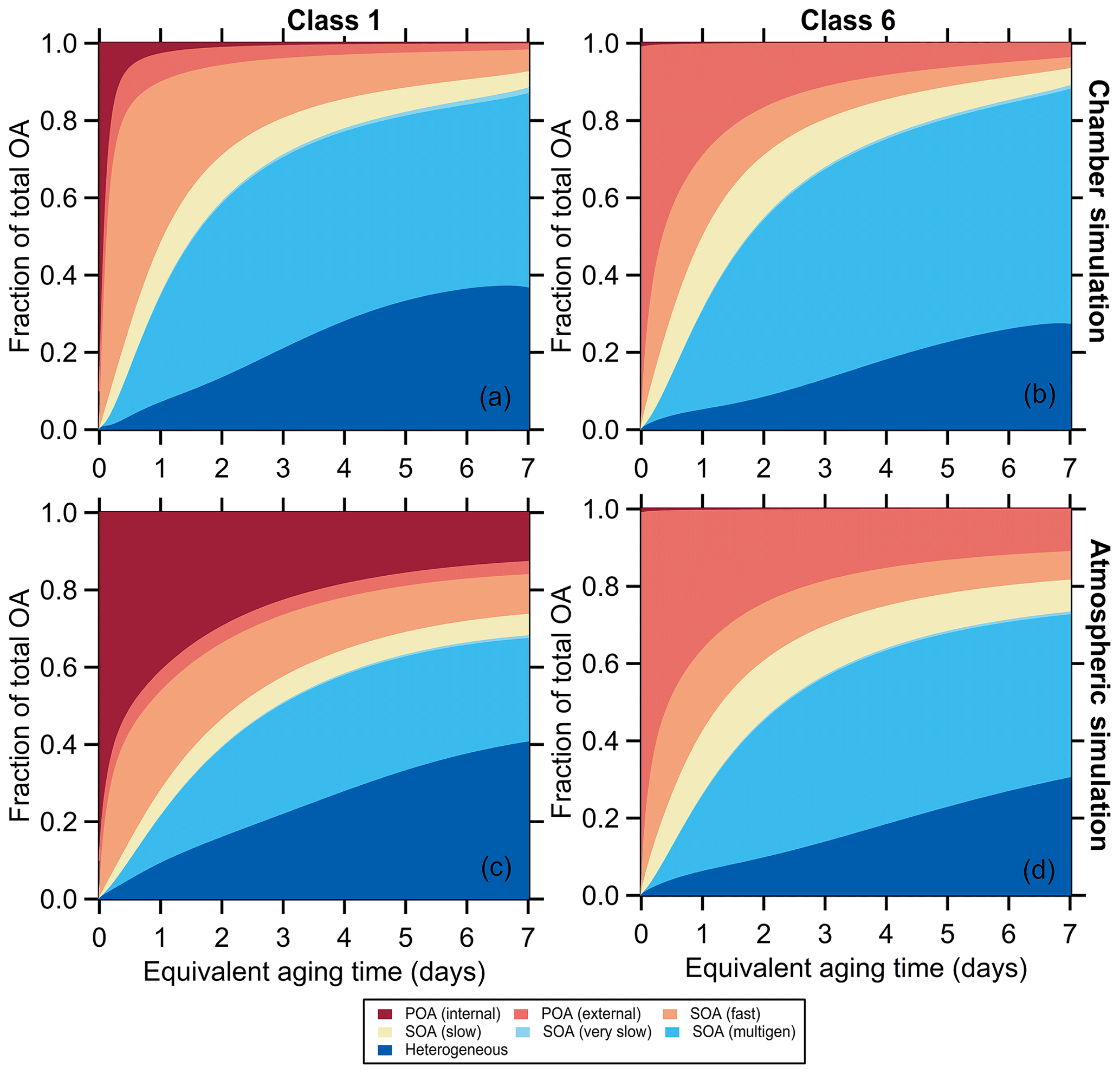
Figure 4Variation in the simulated OA fractional composition with equivalent aging time for (a, c) SSA class 1 (BC-rich) and (b, d) SSA class 6 (OA-rich) for the mini chamber experiments. For the chamber simulations in panels (a) and (b) the differences in decay rates between particle types and gases are accounted for. For the atmospheric simulations in panels (c) and (d) it is assumed that gases and particles all decay with the same rate.
The evolution of the MACBrC, [OA]∕[rBC], O : C atomic ratio, and AMS f60 derives from changes in the OA composition. Examples of the simulated time-dependent variation in OA composition for particle class 1 and class 6 are shown in Fig. 4. The observations demonstrate the importance of SOA formation, with the predicted fractional contribution of POA decreasing over time. For class 1, the MACBrC decays rapidly owing to a large fraction of the POA being internally mixed with BC and therefore subject to enhanced decay in our experiments. Importantly, simulations with heterogeneous oxidation only (i.e., no SOA formation is allowed) show much too small increases in [OA]∕[rBC] values and too slow a decay of the AMS f60 and rise of the O : C (Fig. S18, discussed further in Sect. 3.2.4).
SOA-type-specific 405 nm MACBrC values determined from the model were MACfast=0.81 (±0.2) m2 g−1, MACslow= MACVS=0.05 () m2 g−1, MAC2G=0.17 (±0.05) m2 g−1, and MAChet=0.05 (±0.025) m2 g−1. The SOAfast is substantially more absorbing than the other SOA types. It is difficult to estimate a comprehensive uncertainty on these values; the above uncertainties were qualitatively estimated based on the model sensitivity to changing the parameter values (see Supplement). The strong absorptivity of the fast-forming SOA and the substantial difference between MACfast and MACslow and MAC2G are unexpected and somewhat surprising. The identities of the likely precursor NMOG characterized (Koss et al., 2018) can be interrogated to understand this difference along with the evolving chemical conditions in the chamber.
The measurements indicate precursor NMOGs most likely to contribute to fast SOA formation include monoterpenes, guaiacol, benzenediol, methyl furfural, methyl guaiacol, vanillin, vinyl guaiacol, creosol, isoeugenol, syringol, and styrene (Fig. S13). The precursor NMOGs most likely to contribute to slow SOA formation include toluene, 2-furanmethanol, phenol, o-cresol, highly oxygenated levoglucosan dehydration products, naphthalene, tolualdehyde, 5-hydroxymethyl tetrahydro 2-furanone, and C9 aromatic species. Benzene is the most likely very slow SOA contributor, although it contributes little here. These species are identified based on their (i) measured emission factors, (ii) reaction rates with OH radicals (Koss et al., 2018), and (iii) estimated SOA yields (Bruns et al., 2016).
SOA absorptivity varies between precursors (Lambe et al., 2013; Romonosky et al., 2016; Xie et al., 2017), suggesting different precursors contribute differentially to the SOA absorption here. Monoterpenes form SOA that is non-to-weakly absorbing. In contrast, SOA from aromatic precursors can be quite absorbing, more so in the presence of NOx. Formation of nitroaromatics is often linked to the enhanced absorptivity when NOx is present, and condensed-phase reactions producing highly conjugated, potentially N containing compounds also contribute to light absorption (Laskin et al., 2015). Garmash et al. (2020) recently reported ROOR′ dimer formation in the gas phase from OH oxidation of aromatics, which we speculate could also contribute to light absorption.
Given the potential importance of nitroaromatic formation to SOA light absorption, the organonitrogen (ON) aerosol contribution here is characterized from the HR-AMS using the method of Kiendler-Scharr et al. (2016; see Supplement in McClure et al., 2020). (We use the term “organonitrogen” quite generally here, as the measurements do not directly provide the particular chemical nature of the N-containing organic species, which could be nitroaromatics or organic nitrates. Further work is required to quantitatively differentiate ON functionalities in the AMS.) The [ON]∕[OA] ratio for primary particles varies by SSA class and inversely with [OA]∕[rBC], from 5.7 % (class 1) to 0.25 % (class 6; McClure et al., 2020). Upon oxidation, the [ON]∕[OA] ratio exhibits an initial rapid increase for all SSA classes (Figs. 5 and S19), demonstrating rapid formation of ON linked to oxidation of the fast-reacting NMOG. However, over time the [ON]∕[OA] ratio decreases for all SSA classes, with the exception of class 6, which remains approximately constant. The slow and very slow NMOG and the MG species are, apparently, comparably less likely to form ON in our experiments, leading to a decline in their relative contribution to OA.
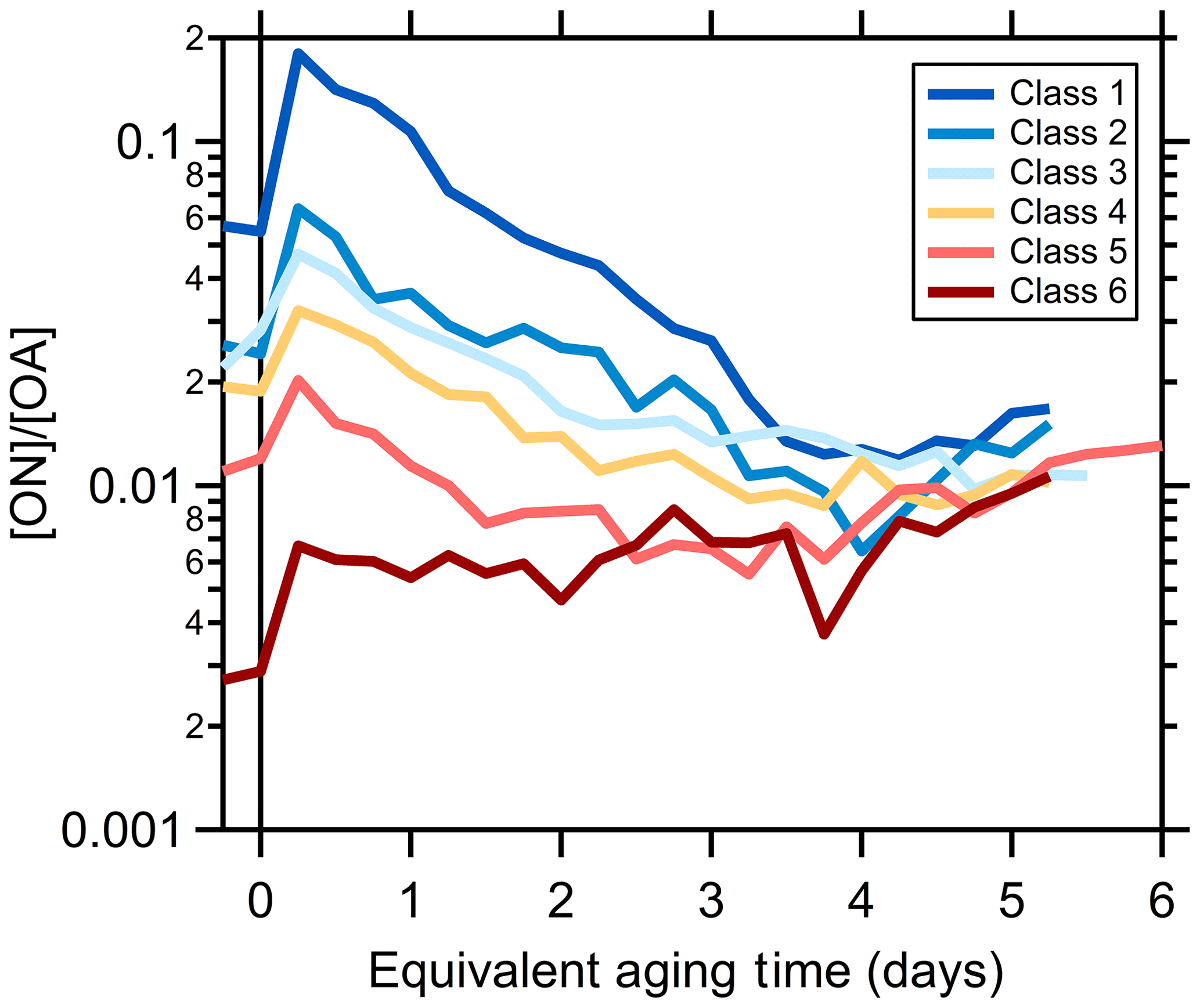
Figure 5Dependence of the observed organic-nitrate-to-total-OA ratio for each SSA class on the equivalent aging time (assuming [OH] molecules cm−3).
That the observed [ON]∕[OA] ratio first increases then decreases with aging while the average MACBrC derived for the SOA decreases with aging (owing to the MACfast> MAC2G> MACslow= MACVS) suggests a relationship between the processes driving these behaviors as well as a shift with aging. While the [ON]∕[OA] ratio would depend on the formation of any organonitrogen species, the SOA absorption likely depends, at least in part, on nitroaromatic formation. Organic nitrates form from reaction of NO with peroxy radicals (RO2), which competes with RO2+HO2, RO2+RO2, and RO2 autoxidation and is thus sensitive to variations in the availability of NO (Orlando and Tyndall, 2012; Bianchi et al., 2019). However, nitroaromatics typically form in the gas phase from reaction of NO2 with phenoxy radicals or with OH–aromatic adducts (Grosjean, 1984; Berndt and Böge, 2003; Vereecken, 2019). Phenoxy radicals generally derive from OH reaction with phenolic molecules and both NO and O3 compete with NO2 for reaction with phenoxy radicals; products from reaction with NO and O3 are not well established (Vereecken, 2019). The importance of the OH–aromatic adduct +NO2 pathway to nitroaromatic formation is suggested as small for species such as toluene and phenol owing to efficient reaction with O2 (Atkinson et al., 1992; Vereecken, 2019), although it has been proposed as the major pathway for nitroaromatic formation from guaiacol and similar species (Lauraguais et al., 2014; Sun et al., 2019).
The [NO]∕[NO2] ratio in the primary smoke varied from 4.88 to 0.84 (Selimovic et al., 2018). Modeling by Coggon et al. (2019) for a few of the experiments indicates a rapid decrease in the [NO]∕[NO2] ratio towards zero with time owing to inefficient photolysis of NO2 in the mini chamber. Consequently, organic nitrate formation should be fastest early on, becoming very small as time progresses. This can help explain the observed dependence of [ON]∕[OA] on aging. However, as nitroaromatic formation involves NO2, rather than NO, the evolving [NO]∕[NO2] would not explain the evolution of the MACBrC. The fast-reacting aromatics (e.g., guaiacol, syringol, eugenol, styrene, vanillin, vinyl guaiacol, creosol, catechol) tend to be more substituted than the slow-reacting aromatics (e.g., phenol, o-cresol, toluene, benzaldehyde, tolualdehyde, naphthalene). SOA formed from guaiacol with NOx present is substantially more absorbing than SOA from either toluene or m-xylene (Romonosky et al., 2016). Additionally, the nitroaromatic yield from reaction of guaiacol exceeds that of phenol, reflecting, in part, faster H abstraction from the phenol group in more substituted aromatics (Harrison et al., 2005; Lauraguais et al., 2014), and production of nitroaromatic species from reaction of nonphenolic molecules, such as toluene, requires multiple reactions with OH. Thus, it may be that the fast-reacting aromatic species generally produce more-absorbing nitroaromatic species with higher SOA yields compared to the slow-reacting aromatic species, contributing to the decrease in both the [ON]∕[OA] and the MACBrC with aging in the mini chamber.
Analogous to the treatment of the MACBrC for SOA, we have attempted to model the ON formation by assigning a NMOG-type-specific, yet particle-class-independent, ON yield. Unlike the MACBrC, no model parameters allow for good model–measurement agreement using this approach. This primarily results from the different particle classes having very different [ON]∕[OA]. For example, for the [ON]∕[OA] ratio for class 1 particles to increase from the initial value (5.7 %) to the peak value (18 %) requires a much larger model ON species SOA yield than for the class 6 particles, for which the [ON]∕[OA] increases from 0.3 % to only 0.6 %. The reason for these substantial differences likely relates to the factors discussed above.
The reasonableness of the derived MACBrC values for SOA is assessed by estimating approximate upper (high NOx) and lower (low NOx) MACBrC values for the SOA types based on literature MAC values for SOA from individual precursors (Romonosky et al., 2016), the NMOG emission factors (Koss et al., 2018), and estimated SOA yields (Bruns et al., 2016):
where x is the SOA type and i is for an individual NMOG. We note the highly uncertain nature of these estimates and therefore only consider them qualitatively. The estimated MACBrC,fast values fall in the range 0.3 m2 g−1 (low NOx) to 2.4 m2 g−1 (high NOx). This encompasses contributions from highly absorbing and abundant guaiacol SOA, moderately absorbing benzene diol (and related species), and effectively nonabsorbing monoterpene SOA. For slow SOA, the estimated MACBrC,slow ranges from 0.1 to 0.6 m2 g−1, encompassing major contributions from moderately absorbing phenol and cresol SOA. The estimated MACBrC,VS depends almost entirely on the benzene SOA, ranging from 0.21 m2 g−1 (low NOx) to 0.88 m2 g−1 (high NOx). Our derived MACBrC,fast falls between the upper and lower estimates, while the MACBrC,slow, MACBrC,VS, and MACBrC,MG are more similar to the lower estimates.
3.2.3 Extending the mini chamber results to the atmosphere
In the experiments, particles and gases experience different decay rates, with particle loss rates exceeding NMOG precursor loss rates and the loss rates of BC-rich particles enhanced. The model allows for simulation of the evolution of the overall system in the absence of such experimental differences by setting the dilution of all components to the same value and wall losses to zero. This provides insights into the likely influence of photochemical oxidation on the evolution of smoke in the atmosphere, although it likely does not fully account for the influence of NOx and the evolving NO∕NO2 ratio. For simplicity, we use a dilution rate of zero for these atmospheric simulations, but any value could be used so long as the focus is on variability in intensive properties.
Simulated OA composition profiles are compared for particle classes 1 and 6, as examples, for chamber (“chm”) and atmospheric (“atm”) simulations in Fig. 4. In the atmospheric simulations, the POA influence persists to much longer times than in the chamber simulations. For example, for class 1 the POA fraction after 2 d of equivalent aging is 0.05 for the chamber simulation but 0.35 for the atmospheric simulations; for class 6 the increase in the POA fraction, from 0.2 (chamber) to 0.3 (atmospheric), is smaller but still notable. Some of the increase in the POA contribution for the atmospheric versus chamber simulations results from the POA and NMOG having the same loss rates in the atmospheric simulations. This leads to a decreased relative contribution of SOA. The larger difference between the atmospheric and chamber simulations for particle class 1 also reflects the comparably greater influence of enhanced decay of BC-rich particles.
The fractional contribution of the second-generation SOA decreases in the atmospheric simulations for all classes. This too results from the particle decay rate equaling the gas decay rate in the atmospheric simulations. In the chamber simulations, the OA is lost at a faster rate than the gases, and thus the second-generation SOA, which forms later than the fast and slow SOA, is effectively enhanced; this enhancement does not influence the atmospheric simulations. There is little difference in the fractional contribution of heterogeneous oxidation products between simulations, a consequence of the ensemble treatment of heterogeneous oxidation products here.
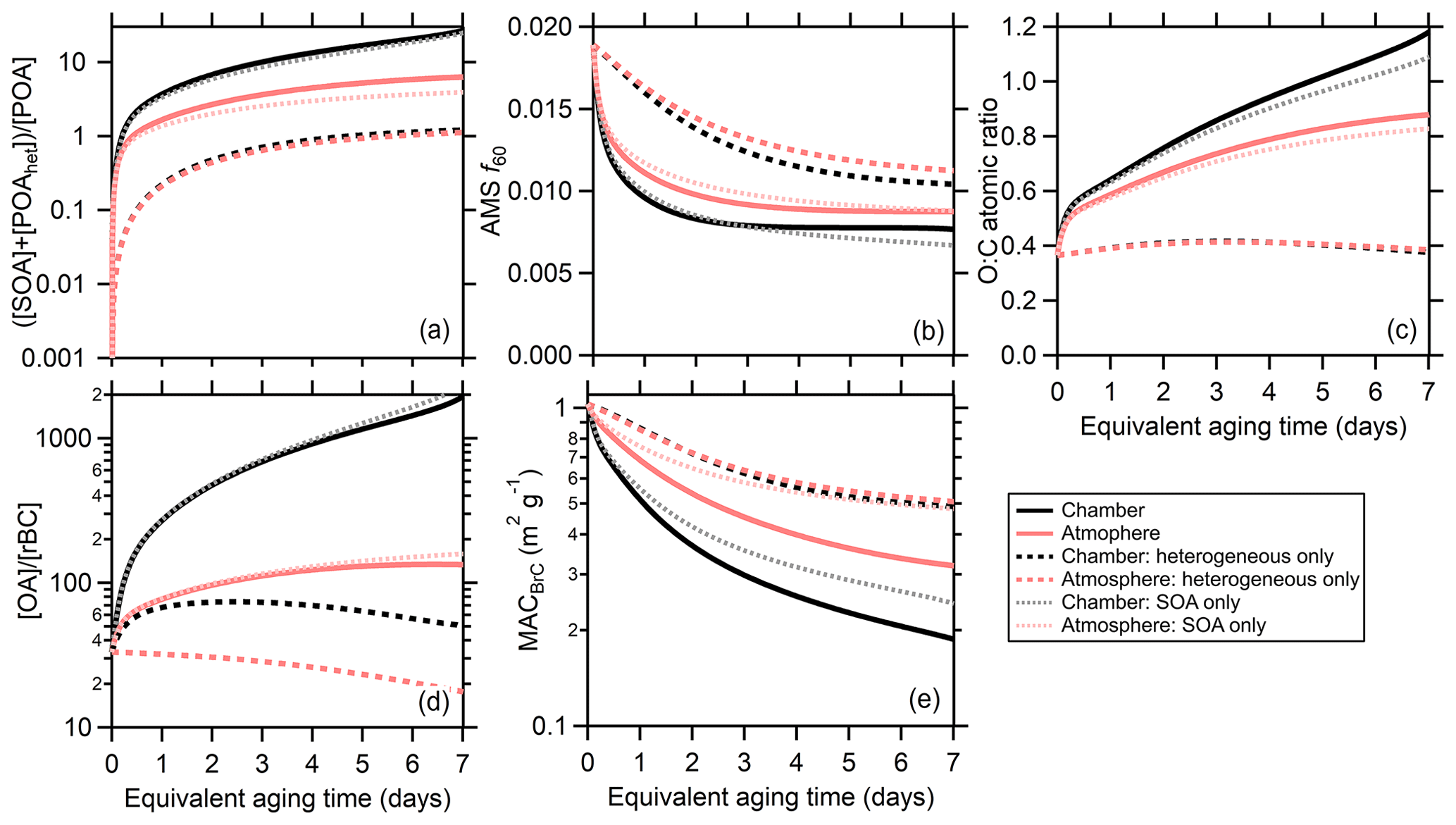
Figure 6Comparison between the model results when dilution and wall losses are included (the chamber simulation, black lines) or turned off (the atmospheric simulation, red lines). The model has been run with both SOA formation and heterogeneous oxidation (solid lines), with heterogeneous oxidation only (dashed lines), or with SOA formation only (dotted lines). Averages (either arithmetic or geometric) across all particle classes are shown for (a) the SOA-plus-oxidized-POA-to-unoxidized-POA ratio, (b) the AMS f60, (c) the organic O : C, (d) the OA-to-rBC ratio, and (e) the MACBrC,405.
Differences between the chamber and atmospheric simulations are shown in Fig. 6 for the MACBrC,405, [OA]∕[rBC], O : C, AMS f60, and the [SOA]∕[POA] ratio. Averages across all particle classes are shown to facilitate comparison; atmospheric simulation results for individual particle classes are compared with the observations in Fig. S20. Given the relatively large (albeit particle class dependent) absorptivity of the POA, the MACBrC,atm decays more slowly than the MACBrC,chm. This is accentuated by the fraction of fast SOA being somewhat greater at longer aging times for the atmospheric simulations, since the MACfast> MACsec> MACslow. The evolution of the MACBrC for the atmospheric simulations is empirically well described by a biexponential decay. A fit to the model prediction for the average behavior across all classes (Fig. 6) yields time constants of 0.4 and 2.8 d. (The overall fit equation is MAC, where tOH is in days and the MACBrC in m2 g−1.)
Additionally, the increase in the [OA]∕[rBC] is much smaller in the atmospheric simulations and the O : C values somewhat smaller. The AMS f60 values at longer aging times remain similar to the observations but are somewhat larger at shorter times for the atmospheric simulations. The is substantially smaller for the atmospheric simulations, with the difference growing over time. After tOH=1 d the geometric average atmospheric equals 1.7 while the chamber equals 3.7. After tOH=6 d the atmospheric equals 5.8 while the chamber equals 20.8.
3.2.4 Secondary OA versus heterogeneous oxidation
The particle properties evolve from both SOA formation and heterogeneous oxidation. To separate these processes, two additional sets of model simulations are run: (i) with the gas-phase rate coefficients set to zero such that only heterogeneous oxidation occurs, and (ii) with the OH uptake coefficient set to zero, and thus only SOA formation occurs (Fig. 6). It is evident that the evolution of all intensive properties occurs much more slowly with only heterogeneous oxidation. With heterogeneous oxidation only, the class-average MACBrC,405 decays to a much lesser extent, even for the atmospheric simulations. Similarly, the increase in O : C is much too small and the AMS f60 decays to a much lesser extent. The OA-to-BC ratio for the heterogeneous-only simulations differs notably from the full simulations. For the heterogeneous-only chamber simulations, this ratio increases initially until about 1.5 d of aging, at which point it decreases. The decrease results from loss of mass over time from fragmentation, while the increase results from faster loss of BC-rich particles in the chamber. For the atmospheric simulations, the OA-to-BC ratio decreases continuously when only heterogeneous oxidation is included. In contrast, for most properties the SOA-only simulations differ by only small amounts from the full simulations. The exception is the MACBrC, for which the decrease in the class-average value with time is notably smaller in the SOA-only simulation compared to the full simulation for both chamber and atmospheric simulations. Our simulations assumed an OH reactive uptake coefficient of unity. We note that if a substantially larger value is assumed (γOH∼10), the predicted changes due to heterogeneous oxidation are much larger and reasonably in line with the observations. Values substantially greater than unity have been found for some chemical systems (Richards-Henderson et al., 2016; Schnitzler and Abbatt, 2018), but for compounds such as levoglucosan – a notable component of BB particles – values less than unity are common (Kroll et al., 2015).
Under the assumption that γOH≤1 for BB particles, these observations point to an important conclusion that is in line with various ambient observations. Large changes in key intensive properties of BB particles, such as O : C and AMS f60, result primarily from secondary OA formation, with heterogeneous oxidation having a smaller influence. While the BrC absorptivity is comparably more sensitive to heterogeneous oxidation, SOA formation remains the major driver of the observed changes. Thus, in ambient observations of biomass burning plumes (Vakkari et al., 2014; Forrister et al., 2015; Garofalo et al., 2019), a notable increase in the O : C (or related measures) and a decrease in the f60 can be taken as a clear indication of SOA formation.
3.2.5 Absolute absorption
The observations indicate that the observable (i.e., bulk average) MACBrC decreases with photochemical aging. However, this results primarily from mixing of absorbing POA with various types of absorbing SOA, with some contribution from heterogeneous oxidation. As such, the absolute absorption in the atmospheric simulations typically increases over time, at least initially, due to the production of new, absorbing particle mass. The exact behavior is particle-class-specific (Fig. 7). The initial increase slows over time as the pool of NMOG precursors depletes. At even longer times the absolute absorption for some classes decreases as heterogeneous oxidation converts more-absorbing BrC (specifically, POA and SOAfast) into less-absorbing BrC and engenders some mass loss over time due to fragmentation. However, this is only true for some of the particle classes. The simulations indicate that, on average, after 1 d of equivalent aging the absolute absorption increases by a factor of 1.6 owing to production of new, absorbing OA mass.
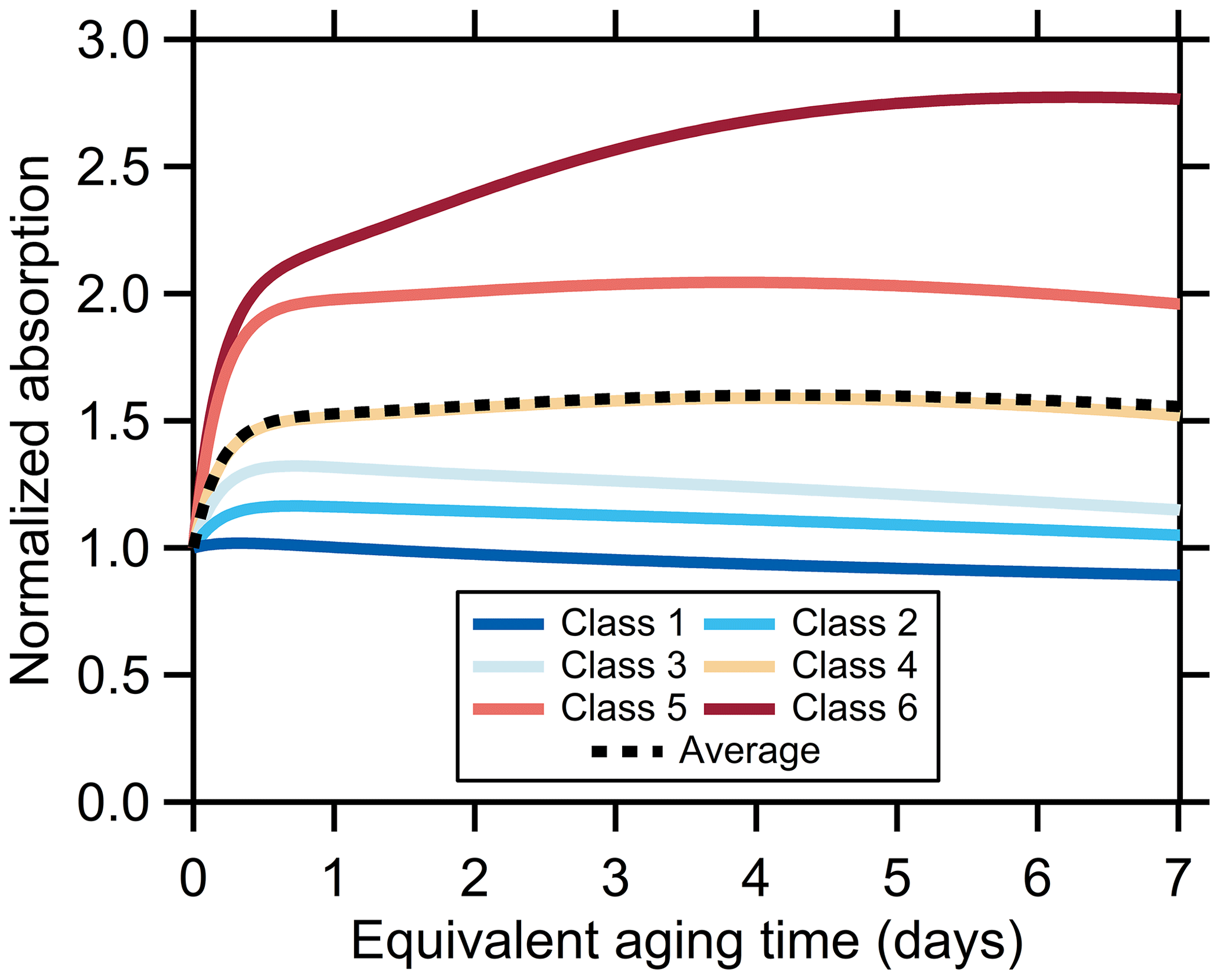
Figure 7Modeled change in the absolute absorption with aging, assuming no dilution, normalized to the value at t=0. Results are shown for each particle class (colors) and the average (black).
3.2.6 POA volatility and the impact of dilution
Upon dilution, semivolatile particle components may evaporate, which should lead to a decrease in the OA-to-BC ratio and, potentially, changes in other particle properties; the MACBrC could change if the absorptivity of the evaporating versus low-volatility (or nonvolatile) components differs. Given the semibatch operation (Sect. 2.2), substantial, continual dilution occurs throughout an experiment. Rapid dilution also occurs upon sampling of smoke into the clean mini chamber air. Dilution-driven evaporation has been suggested as an important factor in the chemical evolution of BB in ambient biomass burning plumes (Hodshire et al., 2019).
We assessed the impact of continual dilution on our observations by conducting one experiment during which the chamber was left dark, although ozone was present at about 50 ppb (Fig. 8). The primary particles corresponded to class 3 particles (SSA405 nm=0.73). After sampling, the OA and rBC concentrations decreased by about a factor of 33 over 45 min owing to dilution and other losses. For comparison, the [ACN] dilution tracer decreased by a factor of 21.
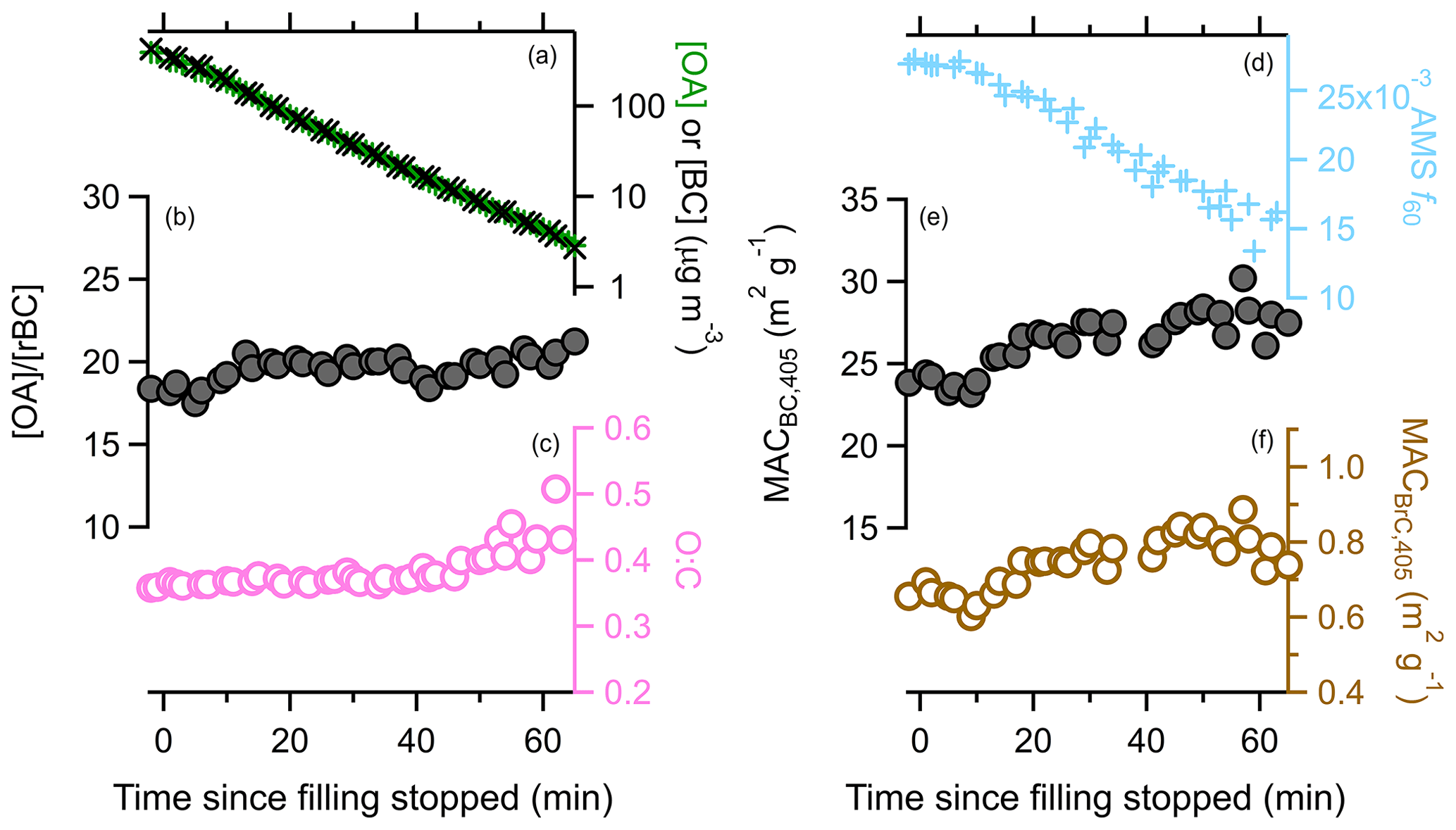
Figure 8Variation in various particle properties with time during the dark experiment. The change in (a) either the [OA] (green) or [rBC] (black), with [rBC] multiplied by 20; (b) the [OA]∕[rBC] ratio; (c) the O : C ratio (circles) or OA mass fraction remaining (triangles); (d) the AMS f60 biomass burning marker; (e) the MACBC,405 nm; and (f) the MACBrC,405 nm.
During this experiment the [OA]∕[rBC] ratio was nearly constant (19.6±0.7, 1σ), even slightly increasing over time. Also, the O : C remained constant (0.39±0.03, 1σ), as did the H : C (1.74±0.01, 1σ). These observations together indicate that there was little, if any, evaporative loss of OA. However, the AMS f60 declined continually, by about 30 %. The signal at is only a small fraction of the total OA, and thus changes in f60 can occur even if only small changes in total mass occur. The AMS f60 could also evolve over time from in-particle reactions that occur even in the absence of oxidants. The mass fraction of OA remaining (MFROA) after heating in the thermodenuder increased slightly over time, from 0.05 to 0.07. This could indicate evaporation of more volatile components, although it could result from “ripening” of the particles over time (Tritscher et al., 2011).
SOA from reaction of some NMOGs with O3 may have offset some evaporative OA mass loss, keeping the total OA mass constant while the f60 and MFRorg changed. Monoterpenes are the most important SOA precursor class here that reacts readily with O3. The measured initial monoterpene concentration (8.6 ppb =50 µg m−3) constrains the potential SOA formed. Accounting for differential losses of particles and gases in the mini chamber, with 50 ppb O3 and an assumed SOA yield of 0.3, we estimate that SOA formation might have increased the [OA]∕[rBC] ratio by about 4 %. Given the constancy of the [OA]∕[rBC], we estimate no more than 8 % of the OA evaporated despite the substantial dilution over the 45 min experiment. Much greater POA evaporation is expected based on the volatility distribution of May et al. (2013) for biomass burning OA, assuming evaporation is facile. Using their distribution, we estimate a factor of 33 decrease in the OA should have resulted in a decrease in the [OA]∕[rBC] from 19.6 to 11.2 from evaporation; a factor of 21 dilution (the observed value for ACN) should have caused a decrease to . Thus, we conclude that evaporation due to dilution had minimal influence on our observations and, more broadly, suggests minimal influence of dilution in general.
The reason for the insensitivity to dilution of our experiments may result from the concentration range considered. May et al. (2013) established the volatility distribution for biomass burning OA from thermodenuder measurements. They compared predictions from their derived volatility distribution to observations of OA emission factors for different fuel types at varying levels of dilution and initial concentrations. At the lowest initial [OA] (∼100 µg m−3), active dilution had little influence and in some cases the OA emission factor actually increased after dilution in their experiments. In contrast, when the initial [OA] was much larger, on the order of 1000 µg m−3, there was a clearer relationship between the OA emission factor and the OA concentration and better agreement with their predictions. For comparison, in our experiments, the initial [OA] in the mini chamber had a geometric average of 76 µg m−3, ranging from 8 to 384 µg m−3; notably, the dark experiment was at the highest initial [OA]. Dilution into the mini chamber was a factor of 7. Most likely, evaporation resulting from dilution occurred upon initial injection of the particles into the mini chamber with little additional evaporation as experiments progressed, to the extent that the particles evaporated much at all.
3.3 Comparison with ambient observations
There are very few ambient assessments of how photochemical oxidation influences brown carbon absorptivity. Forrister et al. (2015) measured dilution-corrected water-soluble plus methanol-soluble BrC absorption in aboveground ambient wildfire plumes at various distances downwind from the fires. The measured O : C and AMS f60 changed with time as the BrC absorption decreased, indicating that chemical changes occurred as the plume aged. They observed that the absolute BrC absorption decreased over time, with a timescale of only 10 h; this was associated with a decay in the AAE determined from in situ measurements. Surface measurements of aged smoke plumes by Selimovic et al. (2019) are also suggestive of a decay in the AAE with aging. The BrC decay time from Forrister et al. (2015) is substantially faster than we observe for any of the particle classes. Interestingly, we predict that over 10 h the absorption by BrC in a wildfire plume should have increased, and only at much later times should it have decreased (Fig. 7). In the case where only heterogeneous oxidation is included (with γOH=1), the predicted decay in BrC absorption based on our observations has a timescale of ca. 4 d, substantially slower than observed by Forrister et al. (2015).
It is possible that the smoke sampled by Forrister et al. (2015) differed significantly from any of the burns sampled here. However, this seems unlikely given the wide range of fuels and burn conditions considered here and since the plumes sampled by Forrister et al. (2015) likely derived from combustion of similar fuels as used here. Also unlikely is that greater dilution in the atmosphere played an important role, as it is thought that more-absorbing primary OA has lower volatility than less-absorbing primary OA (Saleh et al., 2014). Thus, it is possible that the difference between Forrister et al. (2015) and our results indicates that direct photobleaching rapidly degrades BrC absorptivity, as direct photolysis in our experiments was likely of minimal importance owing to the relatively low light intensity, single initiation wavelength (254 nm), and short absolute timescale (<1 h) compared to the atmosphere.
However, an additional difference is that we measured absorption by suspended particles at low-to-moderate relative humidity, whereas Forrister et al. (2015) characterized BrC absorption after extracting material collected onto filters into water and then methanol. It is possible that solvent extraction alters the BrC absorptivity in some OA-composition-dependent manner, leading to an apparent time-dependent decay in the BrC absorption. Consistent with this suggestion, Fleming et al. (2020) reported that (i) the total absorptivity lifetime of BB particles photolytically aged on a filter greatly exceeded that of individual chromophores (>10 d versus <2 d) and (ii) the apparent changes in total absorption depended on whether the absorption was measured for particles on the filter versus for solution extracts. There is also evidence that the larger molecules comprising OA from biomass burning, which are often more absorbing yet less soluble than small molecules (Di Lorenzo et al., 2017; Saleh et al., 2018), are less susceptible to photobleaching (Wong et al., 2017, 2019). This could lead to absorption measurements from solvent extracts overestimating the effects of photobleaching. Nonetheless, the suggestion that the measurement method contributes to the lab–field difference remains speculative as direct, quantitative comparisons between BrC absorption measured for suspended particles versus from solution extracts are limited; we suggest that targeted comparisons between absorption measurement methods would be informative.
Wang et al. (2016) also characterized changes in BrC absorptivity of BB particles in the Amazon. They found that the BrC absorptivity decreased initially over an estimated ca. 20 h of photochemical aging, after which the absorptivity remained constant. As above, this timescale seems too short for heterogeneous oxidation to have a major impact, thus implicating direct photolysis as the reason for the photobleaching. However, there are two important considerations. First, Wang et al. (2016) note that they cannot rule out production of less-absorbing SOA as the reason for the absorptivity decrease. While the OA concentration was constant over time and therefore suggestive of little SOA formation, it may be that dilution of the plume offset SOA formation and consequently that SOA formation precipitated the decrease in absorptivity. Second, their estimated lifetimes are determined from NOx losses and thus sensitive to assumptions about the average OH; as such, longer lifetimes cannot be ruled out.
Finally, Zhang et al. (2017) characterized water-plus-methanol-extracted BrC in the outflow of storm clouds and, in one case, were able to measure BrC absorption for freshly expelled particles versus particles sampled 1 d later. The BrC absorptions for the fresh and aged particles were nearly identical, with no indication of bleaching despite the high altitude, 11 km, and thus high UV photon flux. Thus, this study indicates that photobleaching of BrC may have little influence on at least some types of BrC. Further, Zhang et al. (2017) report absolute absorption values that, it seems, are not dilution corrected. Thus, the constancy of the absolute absorption between the fresh and aged particles implies potential BrC production, as dilution is expected. Ultimately, further experiments investigating direct photolysis of biomass-derived BrC and additional field observations of the evolution of BrC absorptivity will be necessary to reconcile our findings and the limited number of field observations.
We characterized the photochemical evolution of smoke (particles + gases) produced from combustion of a wide variety of biomass fuels. Particle properties were characterized as a function of equivalent photochemical aging, with a focus on the particle optical properties. Photochemical aging led to dramatic changes in the particle chemical composition, as evidenced by a large and rapid increase in the O : C atomic ratio of the organic aerosol and a decrease in the marker ion f60 in the OA mass spectrum. These chemical changes occurred concurrent with large changes in the [OA]∕[rBC] ratio, indicating substantial production of secondary organic aerosol mass. The particle optical properties also changed substantially upon aging. Experiments were classified into six classes according to the SSA of the primary particles. The average evolution of the optical properties of particles in each class was characterized; the observed behavior was generally class specific. The total particle absorptivity normalized to black carbon (the MACBC) generally increased with aging due to production of new, somewhat absorbing OA. The single-scatter albedo also generally increased with aging, although to a much lesser extent for experiments in which the initial particles already had a large SSA. The absorptivity of the organic aerosol components, i.e., of the brown carbon mass absorption coefficient (MACBrC), exhibited class-specific behavior. For particles having an initially large SSA, the MACBrC exhibited an initial increase at short times (<0.5 d of equivalent aging) but then decreased monotonically with continued aging. For particles having an initially small SSA, there was a rapid decline in the MACBrC at short times and then a continued slower decline at longer times. At long times the MACBrC for the high-SSA and low-SSA initial particles behaved similarly.
The evolution of the BrC absorptivity was shown via application of a mechanistic model to be consistent with a combination of production of strongly absorbing and much more weakly absorbing secondary OA, along with heterogeneous conversion of absorbing POA and SOA into a less-absorbing oxidized OA. This was the case for all particle classes. The SOA formed could be distinguished into four general types: (i) a fast-forming, relatively highly absorbing type; (ii) a more slowly forming, weakly absorbing type; (iii) a very slowly forming, weakly absorbing type; and (iv) a weakly absorbing, second-generation type. The relative abundances of these different types were similar between the different particle classes. The combination of SOA formation and heterogeneous oxidation causes the MACBrC to evolve in time on three timescales: (i) an initial rapid increase, occurring at aging times <0.25 d; (ii) a moderately fast decrease, occurring with a timescale of ∼1 d; and (iii) a longer decay, occurring with a timescale of about ∼1 week. Our results demonstrate that, while primary particle properties derived from biomass combustion can vary dramatically in terms of their properties, the overall evolution upon photochemical aging is reasonably independent of the initial conditions.
All data used to create this paper are available at https://www.esrl.noaa.gov/csl/projects/firex/firelab/ (National Oceanic and Atmospheric Administration, 2020) and in Cappa et al. (2019a).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-8511-2020-supplement.
CDC and JHK designed the experiments. CDC, CYL, DHH, MC, AK, TBO, and KS carried out the measurements and data processing, with additional assistance from CW and JdG. CDC and CL analyzed data. CDC wrote the manuscript, with contributions from all coauthors.
The authors declare that they have no conflict of interest.
The entire FIREX team, especially Bob Yokelson and Jim Roberts and the staff of the Missoula Fire Sciences Laboratory, are acknowledged for their assistance. Putting together the community inlet was a community effort, and thank you to all who contributed. Shuka Schwarz and Gavin McMeeking are also thanked for their assistance with the SP2.
This research has been supported by the National Oceanic and Atmospheric Administration (Atmospheric Chemistry, Carbon Cycle, and Climate Program, Climate Program Office, grant nos. NA16OAR4310111, NA16OAR4310112, and NA16OAR4310104), the National Science Foundation (Division of Atmospheric and Geospace Sciences, grant no. 1748266), and the National Science Foundation Graduate Research Fellowship Program.
This paper was edited by Ryan Sullivan and reviewed by two anonymous referees.
Akagi, S. K., Craven, J. S., Taylor, J. W., McMeeking, G. R., Yokelson, R. J., Burling, I. R., Urbanski, S. P., Wold, C. E., Seinfeld, J. H., Coe, H., Alvarado, M. J., and Weise, D. R.: Evolution of trace gases and particles emitted by a chaparral fire in California, Atmos. Chem. Phys., 12, 1397–1421, https://doi.org/10.5194/acp-12-1397-2012, 2012.
Andreae, M. O. and Gelencsér, A.: Black carbon or brown carbon? The nature of light-absorbing carbonaceous aerosols, Atmos. Chem. Phys., 6, 3131–3148, https://doi.org/10.5194/acp-6-3131-2006, 2006.
Andreae, M. O. and Merlet, P.: Emission of trace gases and aerosols from biomass burning, Global Biogeochem. Cy., 15, 955–966, https://doi.org/10.1029/2000GB001382, 2001.
Atkinson, R., Aschmann, S. M., and Arey, J.: Reactions of OH and NO3 radicals with phenol, cresols, and 2-nitrophenol at K, Environ. Sci. Technol., 26, 1397–1403, https://doi.org/10.1021/es00031a018, 1992.
Aumont, B., Szopa, S., and Madronich, S.: Modelling the evolution of organic carbon during its gas-phase tropospheric oxidation: development of an explicit model based on a self generating approach, Atmos. Chem. Phys., 5, 2497–2517, https://doi.org/10.5194/acp-5-2497-2005, 2005.
Berndt, T. and Böge, O.: Gas-phase reaction of OH radicals with phenol, Phys. Chem. Chem. Phys., 5, 342–350, https://doi.org/10.1039/B208187C, 2003.
Bianchi, F., Kurten, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Bond, T. C., Streets, D. G., Yarber, K. F., Nelson, S. M., Woo, J.-H., and Klimont, Z.: A technology-based global inventory of black and organic carbon emissions from combustion, J. Geophys. Res., 109, D14203, https://doi.org/10.1029/2003jd003697, 2004.
Browne, E. C., Zhang, X., Franklin, J. P., Ridley, K. J., Kirchstetter, T. W., Wilson, K. R., Cappa, C. D., and Kroll, J. H.: Effect of heterogeneous oxidative aging on light absorption by biomass burning organic aerosol, Aerosol Sci. Tech., 53, 663–674, https://doi.org/10.1080/02786826.2019.1599321, 2019.
Bruns, E. A., El Haddad, I., Slowik, J. G., Kilic, D., Klein, F., Baltensperger, U., and Prévôt, A. S. H.: Identification of significant precursor gases of secondary organic aerosols from residential wood combustion, Sci. Rep., 6, 27881, https://doi.org/10.1038/srep27881, 2016.
Burtscher, H.: Measurement and characteristics of combustion aerosols with special consideration of photoelectric charging and charging by flame ions, J. Aerosol Sci., 23, 549–595, https://doi.org/10.1016/0021-8502(92)90026-R, 1992.
Cappa, C. D. and Wilson, K. R.: Multi-generation gas-phase oxidation, equilibrium partitioning, and the formation and evolution of secondary organic aerosol, Atmos. Chem. Phys., 12, 9505–9528, https://doi.org/10.5194/acp-12-9505-2012, 2012.
Cappa, C. D., Onasch, T. B., Massoli, P., Worsnop, D., Bates, T. S., Cross, E., Davidovits, P., Hakala, J., Hayden, K., Jobson, B. T., Kolesar, K. R., Lack, D. A., Lerner, B., Li, S. M., Mellon, D., Nuaanman, I., Olfert, J., Petaja, T., Quinn, P. K., Song, C., Subramanian, R., Williams, E. J., and Zaveri, R. A.: Radiative absorption enhancements due to the mixing state of atmospheric black carbon, Science, 337, 1078–1081, https://doi.org/10.1126/science.1223447, 2012.
Cappa, C. D., Lim, C. Y., Hagan, D. H., and Kroll, J. H.: Measurements from the Fire Influence on Regional and Global Environments Experiment (FIREX) Fire Lab Mini Chamber Experiment, UC Davis DASH, Dataset, version 1, https://doi.org/10.25338/B8CK5N, 2019a.
Cappa, C. D., Zhang, X., Russell, L. M., Collier, S., Lee, A. K. Y., Chen, C.-L., Betha, R., Chen, S., Liu, J., Price, D. J., Sanchez, K. J., McMeeking, G., Williams, L. R., Onasch, T. B., Worsnop, D. R., Abbatt, J., and Zhang, Q.: Light absorption by ambient black and brown carbon and its dependence on black carbon coating state for two California, USA cities in winter and summer, J. Geophys. Res.-Atmos., 124, 1550–1577, https://doi.org/10.1029/2018JD029501, 2019b.
Cheng, Y., Engling, G., Moosmaller, H., Arnott, W. P., Chen, L. W. A., Wold, C. E., Hao, W. M., and He, K. B.: Light absorption by biomass burning source emissions, Atmos. Environ., 127, 347–354, https://doi.org/10.1016/j.atmosenv.2015.12.045, 2016.
Chhabra, P. S., Ng, N. L., Canagaratna, M. R., Corrigan, A. L., Russell, L. M., Worsnop, D. R., Flagan, R. C., and Seinfeld, J. H.: Elemental composition and oxidation of chamber organic aerosol, Atmos. Chem. Phys., 11, 8827–8845, https://doi.org/10.5194/acp-11-8827-2011, 2011.
Coggon, M. M., Lim, C. Y., Koss, A. R., Sekimoto, K., Yuan, B., Gilman, J. B., Hagan, D. H., Selimovic, V., Zarzana, K. J., Brown, S. S., Roberts, J. M., Müller, M., Yokelson, R., Wisthaler, A., Krechmer, J. E., Jimenez, J. L., Cappa, C., Kroll, J. H., de Gouw, J., and Warneke, C.: OH chemistry of non-methane organic gases (NMOGs) emitted from laboratory and ambient biomass burning smoke: evaluating the influence of furans and oxygenated aromatics on ozone and secondary NMOG formation, Atmos. Chem. Phys., 19, 14875–14899, https://doi.org/10.5194/acp-19-14875-2019, 2019.
Cross, E. S., Onasch, T. B., Ahern, A., Wrobel, W., Slowik, J. G., Olfert, J., Lack, D. A., Massoli, P., Cappa, C. D., Schwarz, J. P., Spackman, J. R., Fahey, D. W., Sedlacek, A., Trimborn, A., Jayne, J. T., Freedman, A., Williams, L. R., Ng, N. L., Mazzoleni, C., Dubey, M., Brem, B., Kok, G., Subramanian, R., Freitag, S., Clarke, A., Thornhill, D., Marr, L. C., Kolb, C. E., Worsnop, D. R., and Davidovits, P.: Soot Particle Studies – Instrument Inter-Comparison – Project Overview, Aerosol Sci. Tech., 44, 592–611, https://doi.org/10.1080/02786826.2010.482113, 2010.
Cubison, M. J., Ortega, A. M., Hayes, P. L., Farmer, D. K., Day, D., Lechner, M. J., Brune, W. H., Apel, E., Diskin, G. S., Fisher, J. A., Fuelberg, H. E., Hecobian, A., Knapp, D. J., Mikoviny, T., Riemer, D., Sachse, G. W., Sessions, W., Weber, R. J., Weinheimer, A. J., Wisthaler, A., and Jimenez, J. L.: Effects of aging on organic aerosol from open biomass burning smoke in aircraft and laboratory studies, Atmos. Chem. Phys., 11, 12049–12064, https://doi.org/10.5194/acp-11-12049-2011, 2011.
Dale, V. H., Joyce, L. A., McNulty, S., Neilson, R. P., Ayres, M. P., Flannigan, M. D., Hanson, P. J., Irland, L. C., Lugo, A. E., Peterson, C. J., Simberloff, D., Swanson, F. J., Stocks, B. J., and Wotton, B. M.: Climate Change and Forest Disturbances: Climate change can affect forests by altering the frequency, intensity, duration, and timing of fire, drought, introduced species, insect and pathogen outbreaks, hurricanes, windstorms, ice storms, or landslides, BioScience, 51, 723–734, https://doi.org/10.1641/0006-3568(2001)051[0723:CCAFD]2.0.CO;2, 2001.
Di Lorenzo, R. A., Washenfelder, R. A., Attwood, A. R., Guo, H., Xu, L., Ng, N. L., Weber, R. J., Baumann, K., Edgerton, E., and Young, C. J.: Molecular-Size-Separated Brown Carbon Absorption for Biomass-Burning Aerosol at Multiple Field Sites, Environ. Sci. Technol., 51, 3128–3137, https://doi.org/10.1021/acs.est.6b06160, 2017.
Fierce, L., Bond, T. C., Bauer, S. E., Mena, F., and Riemer, N.: Black carbon absorption at the global scale is affected by particle-scale diversity in composition, Nat. Comm., 7, 12361, https://doi.org/10.1038/ncomms12361, 2016.
Fierce, L., Onasch, T. B., Cappa, C. D., Mazzoleni, C., China, S., Bhandari, J., Davidovits, P., Fischer, D. A., Helgestad, T. M., Lambe, A., Sedlacek, A. J., Smith, G. D., and Wolff, L.: Absorption enhancements by black carbon controlled by particle-to-particle heterogeneity in composition, P. Nat. Acad. Sci. USA, 117, 5196–5203, https://doi.org/10.1073/pnas.1919723117, 2020.
Fleming, L. T., Lin, P., Roberts, J. M., Selimovic, V., Yokelson, R., Laskin, J., Laskin, A., and Nizkorodov, S. A.: Molecular composition and photochemical lifetimes of brown carbon chromophores in biomass burning organic aerosol, Atmos. Chem. Phys., 20, 1105–1129, https://doi.org/10.5194/acp-20-1105-2020, 2020.
Forestieri, S. D., Helgestad, T. M., Lambe, A. T., Renbaum-Wolff, L., Lack, D. A., Massoli, P., Cross, E. S., Dubey, M. K., Mazzoleni, C., Olfert, J. S., Sedlacek III, A. J., Freedman, A., Davidovits, P., Onasch, T. B., and Cappa, C. D.: Measurement and modeling of the multiwavelength optical properties of uncoated flame-generated soot, Atmos. Chem. Phys., 18, 12141–12159, https://doi.org/10.5194/acp-18-12141-2018, 2018.
Forrister, H., Liu, J., Scheuer, E., Dibb, J., Ziemba, L., Thornhill, K. L., Anderson, B., Diskin, G., Perring, A. E., Schwarz, J. P., Campuzano-Jost, P., Day, D. A., Palm, B. B., Jimenez, J. L., Nenes, A., and Weber, R. J.: Evolution of brown carbon in wildfire plumes, Geophys. Res. Lett., 42, 4623–4630, https://doi.org/10.1002/2015GL063897, 2015.
Fortner, E., Onasch, T., Canagaratna, M., Williams, L. R., Lee, T., Jayne, J., and Worsnop, D.: Examining the chemical composition of black carbon particles from biomass burning with SP-AMS, J. Aerosol Sci., 120, 12–21, https://doi.org/10.1016/j.jaerosci.2018.03.001, 2018.
Fuller, K. A., Malm, W. C., and Kreidenweis, S. M.: Effects of mixing on extinction by carbonaceous particles, J. Geophys. Res.-Atmos., 104, 15941–15954, https://doi.org/10.1029/1998jd100069, 1999.
Garmash, O., Rissanen, M. P., Pullinen, I., Schmitt, S., Kausiala, O., Tillmann, R., Zhao, D., Percival, C., Bannan, T. J., Priestley, M., Hallquist, Å. M., Kleist, E., Kiendler-Scharr, A., Hallquist, M., Berndt, T., McFiggans, G., Wildt, J., Mentel, T. F., and Ehn, M.: Multi-generation OH oxidation as a source for highly oxygenated organic molecules from aromatics, Atmos. Chem. Phys., 20, 515–537, https://doi.org/10.5194/acp-20-515-2020, 2020.
Garofalo, L. A., Pothier, M. A., Levin, E. J. T., Campos, T., Kreidenweis, S. M., and Farmer, D. K.: Emission and Evolution of Submicron Organic Aerosol in Smoke from Wildfires in the Western United States, ACS Earth Space Chem., 3, 1237–1247, https://doi.org/10.1021/acsearthspacechem.9b00125, 2019.
Grosjean, D.: Atmospheric reactions of ortho cresol: Gas phase and aerosol products, Atmos. Environ., 18, 1641–1652, https://doi.org/10.1016/0004-6981(84)90386-X, 1984.
Harrison, M. A. J., Barra, S., Borghesi, D., Vione, D., Arsene, C., and Iulian Olariu, R.: Nitrated phenols in the atmosphere: a review, Atmos. Environ., 39, 231–248, https://doi.org/10.1016/j.atmosenv.2004.09.044, 2005.
Helgestad, T. M.: Characterizing the optical properties of coated black carbon particles, MS Thesis, Civil and Environmental Engineering, University of California, Davis, USA, 2016.
Hodshire, A. L., Akherati, A., Alvarado, M. J., Brown-Steiner, B., Jathar, S. H., Jimenez, J. L., Kreidenweis, S. M., Lonsdale, C. R., Onasch, T. B., Ortega, A. M., and Pierce, J. R.: Aging Effects on Biomass Burning Aerosol Mass and Composition: A Critical Review of Field and Laboratory Studies, Environ. Sci. Technol., 53, 10007–10022, https://doi.org/10.1021/acs.est.9b02588, 2019.
Jacobson, M. Z.: Effects of biomass burning on climate, accounting for heat and moisture fluxes, black and brown carbon, and cloud absorption effects, J. Geophys. Res.-Atmos., 119, 8980–9002, https://doi.org/10.1002/2014JD021861, 2014.
Kiendler-Scharr, A., Mensah, A. A., Friese, E., Topping, D., Nemitz, E., Prevot, A. S. H., Äijälä, M., Allan, J., Canonaco, F., Canagaratna, M., Carbone, S., Crippa, M., Dall Osto, M., Day, D. A., De Carlo, P., Di Marco, C. F., Elbern, H., Eriksson, A., Freney, E., Hao, L., Herrmann, H., Hildebrandt, L., Hillamo, R., Jimenez, J. L., Laaksonen, A., McFiggans, G., Mohr, C., O'Dowd, C., Otjes, R., Ovadnevaite, J., Pandis, S. N., Poulain, L., Schlag, P., Sellegri, K., Swietlicki, E., Tiitta, P., Vermeulen, A., Wahner, A., Worsnop, D., and Wu, H.-C.: Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol, Geophys. Res. Lett., 43, 7735–7744, https://doi.org/10.1002/2016GL069239, 2016.
Kirchstetter, T. W., Novakov, T., and Hobbs, P. V.: Evidence that the spectral dependence of light absorption by aerosols is affected by organic carbon, J. Geophys. Res.-Atmos., 109, D21208, https://doi.org/10.1029/2004JD004999, 2004.
Koss, A. R., Sekimoto, K., Gilman, J. B., Selimovic, V., Coggon, M. M., Zarzana, K. J., Yuan, B., Lerner, B. M., Brown, S. S., Jimenez, J. L., Krechmer, J., Roberts, J. M., Warneke, C., Yokelson, R. J., and de Gouw, J.: Non-methane organic gas emissions from biomass burning: identification, quantification, and emission factors from PTR-ToF during the FIREX 2016 laboratory experiment, Atmos. Chem. Phys., 18, 3299–3319, https://doi.org/10.5194/acp-18-3299-2018, 2018.
Kroll, J. H., Lim, C. Y., Kessler, S. H., and Wilson, K. R.: Heterogeneous Oxidation of Atmospheric Organic Aerosol: Kinetics of Changes to the Amount and Oxidation State of Particle-Phase Organic Carbon, J. Phys. Chem. A, 119, 10767–10783, https://doi.org/10.1021/acs.jpca.5b06946, 2015.
Kumar, N. K., Corbin, J. C., Bruns, E. A., Massabó, D., Slowik, J. G., Drinovec, L., Močnik, G., Prati, P., Vlachou, A., Baltensperger, U., Gysel, M., El-Haddad, I., and Prévôt, A. S. H.: Production of particulate brown carbon during atmospheric aging of residential wood-burning emissions, Atmos. Chem. Phys., 18, 17843–17861, https://doi.org/10.5194/acp-18-17843-2018, 2018.
Lack, D. A., Langridge, J., Bahreni, R., Cappa, C. D., Middlebrook, A., and Schwarz, J. P.: Brown Carbon and Internal Mixing in Biomass Burning Particles, P. Natl. Acad. Sci. USA, 10, 14802–14807, https://doi.org/10.1073/pnas.1206575109, 2012.
Lambe, A. T., Cappa, C. D., Massoli, P., Onasch, T. B., Forestieri, S. D., Martin, A. T., Cummings, M. J., Croasdale, D. R., Brune, W. H., Worsnop, D. R., and Davidovits, P.: Relationship between Oxidation Level and Optical Properties of Secondary Organic Aerosol, Environ. Sci. Technol., 47, 6349–6357, https://doi.org/10.1021/es401043j, 2013.
Laskin, A., Laskin, J., and Nizkorodov, S. A.: Chemistry of Atmospheric Brown Carbon, Chem. Rev., 115, 4335–4382, https://doi.org/10.1021/cr5006167, 2015.
Lauraguais, A., Coeur-Tourneur, C., Cassez, A., Deboudt, K., Fourmentin, M., and Choël, M.: Atmospheric reactivity of hydroxyl radicals with guaiacol (2-methoxyphenol), a biomass burning emitted compound: Secondary organic aerosol formation and gas-phase oxidation products, Atmos. Environ., 86, 155–163, https://doi.org/10.1016/j.atmosenv.2013.11.074, 2014.
Lee, H. J., Aiona, P. K., Laskin, A., Laskin, J., and Nizkorodov, S. A.: Effect of Solar Radiation on the Optical Properties and Molecular Composition of Laboratory Proxies of Atmospheric Brown Carbon, Environ. Sci. Technol., 48, 10217–10226, https://doi.org/10.1021/es502515r, 2014.
Lelieveld, J., Evans, J. S., Fnais, M., Giannadaki, D., and Pozzer, A.: The contribution of outdoor air pollution sources to premature mortality on a global scale, Nature, 525, 367–371, https://doi.org/10.1038/nature15371, 2015.
Levin, E. J. T., McMeeking, G. R., Carrico, C. M., Mack, L. E., Kreidenweis, S. M., Wold, C. E., Moosmüller, H., Arnott, W. P., Hao, W. M., Collett, J. L., and Malm, W. C.: Biomass burning smoke aerosol properties measured during Fire Laboratory at Missoula Experiments (FLAME), J. Geophys. Res.-Atmos., 115, D18210, https://doi.org/10.1029/2009JD013601, 2010.
Lewis, K., Arnott, W. P., Moosmüller, H., and Wold, C. E.: Strong spectral variation of biomass smoke light absorption and single scattering albedo observed with a novel dual-wavelength photoacoustic instrument, J. Geophys. Res.-Atmos., 113, D16203, https://doi.org/10.1029/2007JD009699, 2008.
Lim, C. Y., Hagan, D. H., Coggon, M. M., Koss, A. R., Sekimoto, K., de Gouw, J., Warneke, C., Cappa, C. D., and Kroll, J. H.: Secondary organic aerosol formation from the laboratory oxidation of biomass burning emissions, Atmos. Chem. Phys., 19, 12797–12809, https://doi.org/10.5194/acp-19-12797-2019, 2019.
Liu, D. T., Whitehead, J., Alfarra, M. R., Reyes-Villegas, E., Spracklen, D. V., Reddington, C. L., Kong, S. F., Williams, P. I., Ting, Y. C., Haslett, S., Taylor, J. W., Flynn, M. J., Morgan, W. T., McFiggans, G., Coe, H., and Allan, J. D.: Black-carbon absorption enhancement in the atmosphere determined by particle mixing state, Nat. Geosci., 10, 184–188, https://doi.org/10.1038/ngeo2901, 2017.
Martinsson, J., Eriksson, A. C., Nielsen, I. E., Malmborg, V. B., Ahlberg, E., Andersen, C., Lindgren, R., Nyström, R., Nordin, E. Z., Brune, W. H., Svenningsson, B., Swietlicki, E., Boman, C., and Pagels, J. H.: Impacts of Combustion Conditions and Photochemical Processing on the Light Absorption of Biomass Combustion Aerosol, Environ. Sci. Technol., 49, 14663–14671, https://doi.org/10.1021/acs.est.5b03205, 2015.
May, A. A., Levin, E. J. T., Hennigan, C. J., Riipinen, I., Lee, T., Collett, J. L., Jimenez, J. L., Kreidenweis, S. M., and Robinson, A. L.: Gas-particle partitioning of primary organic aerosol emissions: 3. Biomass burning, J. Geophys. Res.-Atmos., 118, 11327–11338, https://doi.org/10.1002/jgrd.50828, 2013.
McClure, C. D. and Jaffe, D. A.: US particulate matter air quality improves except in wildfire-prone areas, P. Natl. Acad. Sci. USA, 115, 7901–7906, https://doi.org/10.1073/pnas.1804353115, 2018.
McClure, C. D., Lim, C. Y., Hagan, D. H., Kroll, J. H., and Cappa, C. D.: Biomass-burning-derived particles from a wide variety of fuels – Part 1: Properties of primary particles, Atmos. Chem. Phys., 20, 1531–1547, https://doi.org/10.5194/acp-20-1531-2020, 2020.
McMeeking, G. R., Kreidenweis, S. M., Baker, S., Carrico, C. M., Chow, J. C., Collett, J. L., Hao, W. M., Holden, A. S., Kirchstetter, T. W., Malm, W. C., Moosmüller, H., Sullivan, A. P., and Wold, C. E.: Emissions of trace gases and aerosols during the open combustion of biomass in the laboratory, J. Geophys. Res.-Atmos., 114, D19210, https://doi.org/10.1029/2009JD011836, 2009.
Metcalf, A. R., Loza, C. L., Coggon, M. M., Craven, J. S., Jonsson, H. H., Flagan, R. C., and Seinfeld, J. H.: Secondary Organic Aerosol Coating Formation and Evaporation: Chamber Studies Using Black Carbon Seed Aerosol and the Single-Particle Soot Photometer, Aerosol Sci. Technol., 47, 326–347, https://doi.org/10.1080/02786826.2012.750712, 2013.
National Oceanic and Atmospheric Administration, Chemical Sciences Laboratory: Fire Lab 2016, available at: https://www.esrl.noaa.gov/csl/projects/firex/firelab/, last access: 19 July 2020.
NOAA: Fire Influence on Regional to Global Environments and Air Quality (FIREX-AQ), available at: https://www.esrl.noaa.gov/csl/projects/firex-aq/whitepaper.pdf (last access: 19 July 2020), 2013.
Orlando, J. J. and Tyndall, G. S.: Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 41, 6294–6317, https://doi.org/10.1039/c2cs35166h, 2012.
Peng, J., Hu, M., Guo, S., Du, Z., Zheng, J., Shang, D., Zamora, M. L., Zeng, L., Shao, M., Wu, Y.-S., Zheng, J., Wang, Y., Glen, C. R., Collins, D. R., Molina, M. J., and Zhang, R.: Markedly enhanced absorption and direct radiative forcing of black carbon under polluted urban environments, P. Natl. Acad. Sci. USA, 113, 4266–4271, https://doi.org/10.1073/pnas.1602310113, 2016.
Peng, Z., Day, D. A., Ortega, A. M., Palm, B. B., Hu, W., Stark, H., Li, R., Tsigaridis, K., Brune, W. H., and Jimenez, J. L.: Non-OH chemistry in oxidation flow reactors for the study of atmospheric chemistry systematically examined by modeling, Atmos. Chem. Phys., 16, 4283–4305, https://doi.org/10.5194/acp-16-4283-2016, 2016.
Penner, J. E., Dickinson, R. E., and O'Neill, C. A.: Effects of Aerosol from Biomass Burning on the Global Radiation Budget, Science, 256, 1432–1434, https://doi.org/10.1126/science.256.5062.1432, 1992.
Richards-Henderson, N. K., Goldstein, A. H., and Wilson, K. R.: Sulfur Dioxide Accelerates the Heterogeneous Oxidation Rate of Organic Aerosol by Hydroxyl Radicals, Environ. Sci. Technol., 50, 3554–3561, https://doi.org/10.1021/acs.est.5b05369, 2016.
Romonosky, D. E., Ali, N. N., Saiduddin, M. N., Wu, M., Lee, H. J., Aiona, P. K., and Nizkorodov, S. A.: Effective absorption cross sections and photolysis rates of anthropogenic and biogenic secondary organic aerosols, Atmos. Environ., 130, 172–179, https://doi.org/10.1016/j.atmosenv.2015.10.019, 2016.
Saleh, R., Hennigan, C. J., McMeeking, G. R., Chuang, W. K., Robinson, E. S., Coe, H., Donahue, N. M., and Robinson, A. L.: Absorptivity of brown carbon in fresh and photo-chemically aged biomass-burning emissions, Atmos. Chem. Phys., 13, 7683–7693, https://doi.org/10.5194/acp-13-7683-2013, 2013.
Saleh, R., Robinson, E. S., Tkacik, D. S., Ahern, A. T., Liu, S., Aiken, A. C., Sullivan, R. C., Presto, A. A., Dubey, M. K., Yokelson, R. J., Donahue, N. M., and Robinson, A. L.: Brownness of organics in aerosols from biomass burning linked to their black carbon content, Nat. Geosci., 7, 647–650, https://doi.org/10.1038/ngeo2220, 2014.
Saleh, R., Cheng, Z., and Atwi, K.: The Brown–Black Continuum of Light-Absorbing Combustion Aerosols, Environ. Sci. Technol. Lett., 5, 508–513, https://doi.org/10.1021/acs.estlett.8b00305, 2018.
Schnitzler, E. G. and Abbatt, J. P. D.: Heterogeneous OH oxidation of secondary brown carbon aerosol, Atmos. Chem. Phys., 18, 14539–14553, https://doi.org/10.5194/acp-18-14539-2018, 2018.
Sekimoto, K., Koss, A. R., Gilman, J. B., Selimovic, V., Coggon, M. M., Zarzana, K. J., Yuan, B., Lerner, B. M., Brown, S. S., Warneke, C., Yokelson, R. J., Roberts, J. M., and de Gouw, J.: High- and low-temperature pyrolysis profiles describe volatile organic compound emissions from western US wildfire fuels, Atmos. Chem. Phys., 18, 9263–9281, https://doi.org/10.5194/acp-18-9263-2018, 2018.
Selimovic, V., Yokelson, R. J., Warneke, C., Roberts, J. M., de Gouw, J., Reardon, J., and Griffith, D. W. T.: Aerosol optical properties and trace gas emissions by PAX and OP-FTIR for laboratory-simulated western US wildfires during FIREX, Atmos. Chem. Phys., 18, 2929–2948, https://doi.org/10.5194/acp-18-2929-2018, 2018.
Selimovic, V., Yokelson, R. J., McMeeking, G. R., and Coefield, S.: In situ measurements of trace gases, PM, and aerosol optical properties during the 2017 NW US wildfire smoke event, Atmos. Chem. Phys., 19, 3905–3926, https://doi.org/10.5194/acp-19-3905-2019, 2019.
Sherwood, S.: A Microphysical Connection Among Biomass Burning, Cumulus Clouds, and Stratospheric Moisture, Science, 295, 1272–1275, https://doi.org/10.1126/science.1065080, 2002.
Shiraiwa, M., Kondo, Y., Iwamoto, T., and Kita, K.: Amplification of Light Absorption of Black Carbon by Organic Coating, Aerosol Sci. Tech., 44, 46–54, https://doi.org/10.1080/02786820903357686, 2010.
Smith, J. D., Kroll, J. H., Cappa, C. D., Che, D. L., Liu, C. L., Ahmed, M., Leone, S. R., Worsnop, D. R., and Wilson, K. R.: The heterogeneous reaction of hydroxyl radicals with sub-micron squalane particles: a model system for understanding the oxidative aging of ambient aerosols, Atmos. Chem. Phys., 9, 3209–3222, https://doi.org/10.5194/acp-9-3209-2009, 2009.
Stephens, S. L., Agee, J. K., Fulé, P. Z., North, M. P., Romme, W. H., Swetnam, T. W., and Turner, M. G.: Managing Forests and Fire in Changing Climates, Science, 342, 41–42, https://doi.org/10.1126/science.1240294, 2013.
Sumlin, B. J., Pandey, A., Walker, M. J., Pattison, R. S., Williams, B. J., and Chakrabarty, R. K.: Atmospheric Photooxidation Diminishes Light Absorption by Primary Brown Carbon Aerosol from Biomass Burning, Environ. Sci. Technol. Lett., 4, 540–545, https://doi.org/10.1021/acs.estlett.7b00393, 2017.
Sun, Y. H., Xu, F., Li, X. F., Zhang, Q. Z., and Gu, Y. X.: Mechanisms and kinetic studies of OH-initiated atmospheric oxidation of methoxyphenols in the presence of O2 and NOx, Phys. Chem. Chem. Phys., 21, 21856–21866, https://doi.org/10.1039/c9cp03246k, 2019.
Tasoglou, A., Saliba, G., Subramanian, R., and Pandis, S. N.: Absorption of chemically aged biomass burning carbonaceous aerosol, J. Aerosol Sci., 113, 141–152, https://doi.org/10.1016/j.jaerosci.2017.07.011, 2017.
Tritscher, T., Dommen, J., DeCarlo, P. F., Gysel, M., Barmet, P. B., Praplan, A. P., Weingartner, E., Prévôt, A. S. H., Riipinen, I., Donahue, N. M., and Baltensperger, U.: Volatility and hygroscopicity of aging secondary organic aerosol in a smog chamber, Atmos. Chem. Phys., 11, 11477–11496, https://doi.org/10.5194/acp-11-11477-2011, 2011.
Vakkari, V., Kerminen, V.-M., Beukes, J. P., Tiitta, P., Zyl, P. G., Josipovic, M., Venter, A. D., Jaars, K., Worsnop, D. R., Kulmala, M., and Laakso, L.: Rapid changes in biomass burning aerosols by atmospheric oxidation, Geophys. Res. Lett., 41, 2644–2651, https://doi.org/10.1002/2014GL059396, 2014.
Vereecken, L.: Reaction Mechanisms for the Atmospheric Oxidation of Monocyclic Aromatic Compounds, in: Advances in Atmospheric Chemistry, edited by: Barker, J. R., Steiner, A. L., and Wallington, T. J., World Scientific, Hackensack, USA, 377–527, 2019.
Wang, X., Heald, C. L., Sedlacek, A. J., de Sá, S. S., Martin, S. T., Alexander, M. L., Watson, T. B., Aiken, A. C., Springston, S. R., and Artaxo, P.: Deriving brown carbon from multiwavelength absorption measurements: method and application to AERONET and Aethalometer observations, Atmos. Chem. Phys., 16, 12733–12752, https://doi.org/10.5194/acp-16-12733-2016, 2016.
Wang, X., Heald, C. L., Liu, J., Weber, R. J., Campuzano-Jost, P., Jimenez, J. L., Schwarz, J. P., and Perring, A. E.: Exploring the observational constraints on the simulation of brown carbon, Atmos. Chem. Phys., 18, 635–653, https://doi.org/10.5194/acp-18-635-2018, 2018.
Wong, J. P. S., Nenes, A., and Weber, R. J.: Changes in Light Absorptivity of Molecular Weight Separated Brown Carbon Due to Photolytic Aging, Environ. Sci. Technol., 51, 8414–8421, https://doi.org/10.1021/acs.est.7b01739, 2017.
Wong, J. P. S., Tsagkaraki, M., Tsiodra, I., Mihalopoulos, N., Violaki, K., Kanakidou, M., Sciare, J., Nenes, A., and Weber, R. J.: Atmospheric evolution of molecular-weight-separated brown carbon from biomass burning, Atmos. Chem. Phys., 19, 7319–7334, https://doi.org/10.5194/acp-19-7319-2019, 2019.
Xie, M., Chen, X., Hays, M. D., Lewandowski, M., Offenberg, J., Kleindienst, T. E., and Holder, A. L.: Light Absorption of Secondary Organic Aerosol: Composition and Contribution of Nitroaromatic Compounds, Environ. Sci. Technol., 51, 11607–11616, https://doi.org/10.1021/acs.est.7b03263, 2017.
You, R., Radney, J. G., Zachariah, M. R., and Zangmeister, C. D.: Measured Wavelength-Dependent Absorption Enhancement of Internally Mixed Black Carbon with Absorbing and Nonabsorbing Materials, Environ. Sci. Technol., 50, 7982–7990, https://doi.org/10.1021/acs.est.6b01473, 2016.
Yuan, B., Koss, A. R., Warneke, C., Coggon, M., Sekimoto, K., and de Gouw, J. A.: Proton-Transfer-Reaction Mass Spectrometry: Applications in Atmospheric Sciences, Chem. Rev., 117, 13187–13229, https://doi.org/10.1021/acs.chemrev.7b00325, 2017.
Zhang, X., Cappa, C. D., Jathar, S. H., McVay, R. C., Ensberg, J. J., Kleeman, M. J., and Seinfeld, J. H.: Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol, P. Natl. Acad. Sci. USA, 111, 5802–5807, https://doi.org/10.1073/pnas.1404727111, 2014.
Zhang, Y., Forrister, H., Liu, J., Dibb, J., Anderson, B., Schwarz, J. P., Perring, A. E., Jimenez, J. L., Campuzano-Jost, P., Wang, Y., Nenes, A., and Weber, R. J.: Top-of-atmosphere radiative forcing affected by brown carbon in the upper troposphere, Nat. Geosci., 10, 486–489, https://doi.org/10.1038/ngeo2960, 2017.
Zhong, M. and Jang, M.: Dynamic light absorption of biomass-burning organic carbon photochemically aged under natural sunlight, Atmos. Chem. Phys., 14, 1517–1525, https://doi.org/10.5194/acp-14-1517-2014, 2014.