the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 08 Jun 2020
| 08 Jun 2020
Atmospheric chemical loss processes of isocyanic acid (HNCO): a combined theoretical kinetic and global modelling study
Simon Rosanka
Giang H. T. Vu
Hue M. T. Nguyen
Tien V. Pham
Umar Javed
Domenico Taraborrelli
Luc Vereecken
Isocyanic acid (HNCO) is a chemical constituent suspected to be harmful to humans if ambient concentrations exceed ∼1 ppbv. HNCO is mainly emitted by combustion processes but is also inadvertently released by NOx mitigation measures in flue gas treatments. With increasing biomass burning and more widespread usage of catalytic converters in car engines, good prediction of HNCO atmospheric levels with global models is desirable. Little is known directly about the chemical loss processes of HNCO, which limits the implementation in global Earth system models. This study aims to close this knowledge gap by combining a theoretical kinetic study on the major oxidants reacting with HNCO with a global modelling study. The potential energy surfaces of the reactions of HNCO with OH and NO3 radicals, Cl atoms, and ozone were studied using high-level CCSD(T)/CBS(DTQ)//M06-2X/aug-cc-pVTZ quantum chemical methodologies, followed by transition state theory (TST) theoretical kinetic predictions of the rate coefficients at temperatures of 200–3000 K. It was found that the reactions are all slow in atmospheric conditions, with , and that product formation occurs predominantly by H abstraction; the predictions are in good agreement with earlier experimental work, where available. The reverse reactions of NCO radicals with H2O, HNO3, and HCl, of importance mostly in combustion, were also examined briefly.
The findings are implemented into the atmospheric model EMAC (ECHAM/MESSy Atmospheric Chemistry) to estimate the importance of each chemical loss process on a global scale. The EMAC predictions confirm that the gas-phase chemical loss of HNCO is a negligible process, contributing less than 1 % and leaving heterogeneous losses as the major sinks. The removal of HNCO by clouds and precipitation contributes about 10 % of the total loss, while globally dry deposition is the main sink, accounting for ∼90 %. The global simulation also shows that due to its long chemical lifetime in the free troposphere, HNCO can be efficiently transported into the UTLS by deep convection events. Daily-average mixing ratios of ground-level HNCO are found to regularly exceed 1 ppbv in regions dominated by biomass burning events, but rarely exceed levels above 10 ppt in other areas of the troposphere, though locally instantaneous toxic levels are expected.
- Article
(2223 KB) - Full-text XML
-
Supplement
(3062 KB) - BibTeX
- EndNote





The existence of isocyanic acid (HNCO) in the atmosphere has been established only recently (Roberts et al., 2011; Wentzell et al., 2013) despite its molecular structure and chemical synthesis being first discovered in the 19th century (Liebig and Wöhler, 1830). HNCO can form H-bonded clusters (Zabardasti et al., 2009, 2010; Zabardasti and Solimannejad, 2007) and in pure form appreciably polymerizes to other species (Belson and Strachan, 1982) but becomes relatively stable in the gaseous phase (ppm level) under ambient temperature conditions (Roberts et al., 2010). It is thus near-exclusively present as a monomer in the gaseous phase under ambient temperature conditions (Fischer et al., 2002; Roberts et al., 2010). The background ambient mixing ratios of HNCO as determined by Young et al. (2012) using a global chemistry transport model vary in the range of a few parts per trillion by volume (pptv) over the ocean and remote Southern Hemisphere to tens of pptv over land. In urban regions, HNCO mixing ratio increases from tens of pptv to hundreds of pptv (Roberts et al., 2014; Wentzell et al., 2013). Peak levels can reach up to a few parts per billion by volume under the conditions impacted by direct emissions (Chandra and Sinha, 2016).
HNCO has been linked to adverse health effects such as cataracts, cardiovascular disease, and rheumatoid arthritis via a process called protein carbamylation (see Leslie et al., 2019; Roberts et al., 2011; Suarez-Bertoa and Astorga, 2016; SUVA, 2016; Wang et al., 2007, and references therein). To our knowledge, no past studies have been performed to provide a direct link between inhalation exposure and related adverse health effects. However, human exposure to HNCO concentrations of 1 ppbv is estimated to be potentially sufficient to start the process of protein carbamylation (Roberts et al., 2011). Unfortunately, an air quality standard for HNCO does not exist in most of the countries, whereas an occupational exposure limit has been established by law in only a few countries, including the Swedish Work Environment Authority (SWEA, 2011) and the Swiss National Accident Insurance Fund (SUVA, 2016). For example, the Swedish Work Environment Authority sets the level limit value (LLV) for HNCO at about 0.018 mg m−3, i.e. 10 ppbv (SWEA, 2011). The potential negative impact on health makes it important to assess the atmospheric sources and sinks of HNCO to determine its fate and lifetime.
HNCO emission into the atmosphere is driven primarily by combustion processes based on both natural and anthropogenic activities (see Leslie et al., 2019, and references therein), where the pyrolysis of nitrogen-containing biomass materials during the events of wildfires and agricultural fires leads to the emission of HNCO into the atmosphere. The presence of HNCO in cigarette smoke has been established via the pyrolysis of urea used as a cigarette additive (Baker and Bishop, 2004), oxidation of nicotine (Borduas et al., 2016a), and oxidation of formamide (Barnes et al., 2010; Borduas et al., 2015; Bunkan et al., 2015). Even the combustion of almost all sorts of common household materials, including fibre glass, rubber, wood, PVC-based carpet, and cables (Blomqvist et al., 2003), and polyurethane-based foam (Blomqvist et al., 2003; Jankowski et al., 2014), leads to HNCO emissions along with other isocyanates (Leslie et al., 2019). HNCO emissions from traffic are originating mainly from usage of recent catalytic converters in the exhaust systems of gasoline-based (Brady et al., 2014) and diesel-based (Heeb et al., 2011) vehicles. These converters are implemented to control the emission of primary pollutants such as hydrocarbons, carbon monoxide, particulate matter, and nitrogen oxides. However, these implementations have promoted (Suarez-Bertoa and Astorga, 2016) the formation and emissions of HNCO via surface-bound chain reactions at different stages of the flue gas exhaust and additionally due to emission of unreacted HNCO in the most commonly used urea-based SCR (selective catalytic reduction) system (Heeb et al., 2011). The usages of catalytic converters in modern vehicles potentially give rise to the emission of HNCO especially in urban regions with a growing density of vehicles. A few studies also reported a direct formation of HNCO in the diesel engines during fuel combustion without any after-treatments (Heeb et al., 2011; Jathar et al., 2017). A tabular overview of past studies for HNCO emissions related to gasoline or diesel exhausts can be found in Wren et al. (2018) and Leslie et al. (2019). HNCO emissions via fossil fuel usage are not limited to on-road activity. Off-road fossil fuel activities (e.g. tar sands) also contribute to significant HNCO emissions on regional scales (Liggio et al., 2017). Finally, secondary HNCO formation in the atmosphere is also known through the oxidation of amines and amides (e.g. Borduas et al., 2016a; Parandaman et al., 2017).
The number of studies examining HNCO gas-phase chemistry is limited and mostly focused on its role in the chemistry in NOx mitigation strategies in combustion systems. The scarce data suggest that HNCO destruction in the atmosphere by typical pathways such as reactions with oxidizing agents or by photolysis is ineffective. We give a short overview here to supplement a recent review (Leslie et al., 2019). The reaction of HNCO with the hydroxyl radical (OH), the most important daytime oxidizing agent, has only been studied experimentally at temperatures between 620 and 2500 K (Baulch et al., 2005; Mertens et al., 1992; Tsang, 1992; Tully et al., 1989; Wooldridge et al., 1996), where the extrapolated rate expressions lead to an estimated rate coefficient of 5– at 298 K, i.e. a HNCO lifetime towards OH of over 25 years when assuming a typical OH concentration of 1×106 molecule cm−3. Early theoretical work by Sengupta and Nguyen (1997) at temperatures ≥500 K showed that the mechanism proceeds predominantly by H abstraction, forming NCO+H2O, with an energy barrier of ∼6 kcal mol−1. Wooldridge et al. (1996) determined an upper limit of ≤0.1 for the fraction of CO2+NH2 formation. To our knowledge, no experimental or theoretical data are available on HNCO reactions with other dominant atmospheric oxidants, including the nitrate radicals (NO3), chlorine atoms (Cl), or ozone (O3). Some data are available for H- and O-atom co-reactants of importance in combustion, as well as estimates for HCO and CN (Baulch et al., 2005; Tsang, 1992), but these are not reviewed here. There is no direct measurement for the dry deposition of HNCO. In a global chemical-transport-model-based study, the deposition velocity was considered to be similar to formic acid, yielding a HNCO lifetime of 1–3 d (over the ocean) to 1–2 weeks (over vegetation) (Young et al., 2012). The UV absorption for HNCO is only reported at wavelengths <262 nm, and photolysis is mostly reported for energies at wavelengths below 240 nm by excitation to the first singlet excited states, forming H+NCO or NH+CO (Keller-Rudek et al., 2013; Okabe, 1970; Spiglanin et al., 1987; Spiglanin and Chandler, 1987; Uno et al., 1990; Vatsa and Volpp, 2001). In the troposphere photolysis occurs only at the UV absorption wavelength band >290 nm due to filtering of shorter-wavelength radiation (Hofzumahaus et al., 2002). DrozGeorget et al. (1997) have reported the photolysis of HNCO forming at 332.4 nm, but the HNCO absorption cross section at this wavelength would lead to a lifetime of months (Roberts et al., 2011). Therefore, HNCO loss due to photo-dissociation appears to be negligible in the lower atmosphere. HNCO has absorption bands in the infrared (Sharpe et al., 2004), but at these wavelengths the photon energy is generally too limited for photo-dissociation (Hofzumahaus et al., 2002). The main atmospheric loss processes are considered to be the transfer to the liquid phase, followed by hydrolysis, and deposition. This process depends on the varying atmospheric liquid water contents, relevant temperatures, and pH of cloud droplets. Therefore, the gas-to-liquid partitioning, in the varying atmospheric properties, i.e. water content, temperature, and pH of cloud droplets, becomes important to determine the atmospheric fate of HNCO (Leslie et al., 2019). The gas-to-liquid partitioning has been described by the Henry's law coefficient KH (ranging from 20 to 26±2 M atm−1) and related parameters by a handful of studies (Borduas et al., 2016b; Roberts et al., 2011; Roberts and Liu, 2019). Based on a recent study (Barth et al., 2013), the lifetime of HNCO due to heterogeneous processes is known to be of the order of a few hours (in-cloud reactions) to weeks (aerosol deposition).
The emissions and sources of HNCO have been focused on by many past studies, but there remain large uncertainties in our understanding of HNCO removal processes, especially in gas-phase chemistry. This missing information on HNCO removal processes limits global models to predict HNCO with confidence. To alleviate the dearth of direct data and therefore improve the representation of HNCO in global models, we first provide a theoretical analysis of the chemical reactions of HNCO with the dominant atmospheric oxidants: OH and NO3 radicals, Cl atoms, and O3 molecules, including the prediction of each rate coefficient at atmospheric conditions. In a second step, these results are included in a global numerical chemistry and climate model to assess the impact of chemical loss of HNCO in competition against hydrolysis within cloud droplets and against deposition to the Earth's surface. Additionally, the model is used to provide an estimate of the relative importance of primary and secondary HNCO sources.
2.1 Theoretical methodologies
The potential energy surfaces of the initiation reactions of all four reaction systems were characterized at the M06-2X/aug-cc-pVTZ level of theory (Dunning, 1989; Zhao and Truhlar, 2008), optimizing the geometries and rovibrational characteristics of all minima and transition states. The relative energy of the critical points was further refined at the CCSD(T) level of theory in a set of single-point energy calculations using a systematic series of basis sets, aug-cc-pVxZ (x=D, T, Q) (Dunning, 1989; Purvis and Bartlett, 1982). These energies were extrapolated to the complete basis set (CBS) limit using the aug-Schwartz6(DTQ) scheme as proposed by Martin (1996). The rate coefficients were then obtained by transition state theory (Truhlar et al., 1996) in a rigid rotor, harmonic oscillator approximation, applying a scaling factor of 0.971 to the vibrational wavenumbers (Alecu et al., 2010; Bao et al., 2017). The spin–orbit splitting of the OH radicals of 27.95 cm−1 was taken into account (Huber and Herzberg, 1979). Tunnelling was incorporated using an asymmetric Eckart correction (Johnston and Heicklen, 1962).
To further complete our knowledge on some of the reactions beyond their initiation steps, the full potential energy surfaces of the HNCO+Cl and HNCO+O3 reactants were characterized at the M06-2X/aug-cc-pVTZ or B3LYP/aug-cc-pVTZ level of theory (Becke, 1993; Dunning, 1989; Lee et al., 1988), combined with CCSD(T)/aug-cc-pVTZ single-point energy calculations. To our knowledge, these are the first characterizations of these surfaces. At atmospheric temperatures, most of the reaction channels are negligible, and a detailed kinetic analysis is not performed at this time.
The expected uncertainty of the rate predictions at room temperature is of a factor of 4, based on an estimated uncertainty on the barrier height of at least 0.5 kcal mol−1, and on the tunnelling correction of a factor of 1.5. Though the level of theory used is robust, there are some aspects that are not treated with the highest possible precision. For example, post-CCSD(T)/CBS calculations could refine the predicted energies but are not expected to change our values by more than a few tenths of a kilocalorie per mole (kcal mol−1). The calculation of the state densities could be improved for internal rotation (especially at temperatures outside the atmospheric range), for the notoriously complex rovibronic structure of the NO3 radical (Stanton, 2007, 2009; Stanton and Okumura, 2009), or by treating the transition states (micro)variationally to better characterize the energy-specific kinetic bottleneck. Another aspect is the effect of redissociation of chemically activated adducts, which decreases the effective rate of HNCO loss. Finally, tunnelling corrections for the H-abstraction reactions could benefit from higher-dimensional (curvature and corner-cutting) corrections. The tunnelling corrections are currently predicted to be smaller than a factor of 15 at room temperature due to the low and broad energy barriers, except for a factor of ∼40 for the HNCO+NO3 H abstraction with a somewhat higher barrier. Incorporating any of the aforementioned improvements in the theoretical predictions, however, has a high to very high computational burden with strongly diminished return, as none are expected to change the rate coefficient by a factor large enough to affect the conclusions of our calculations; i.e. that the reactions are negligibly slow by many orders of magnitude compared to other HNCO loss processes (see further text). This is also illustrated in Fig. 2. We refer to Vereecken and Francisco (2012), Vereecken et al. (2015), and Papajak and Truhlar (2012) for further information on theoretical methodologies in atmospheric chemistry.
2.2 Global modelling
The ECHAM/MESSy Atmospheric Chemistry (EMAC) model is a numerical chemistry and climate simulation system that includes submodels describing tropospheric and middle-atmosphere processes and their interaction with oceans, land, and human influences (Jöckel et al., 2010). It uses the second version of the Modular Earth Submodel System (MESSy2) to link multi-institutional computer codes. The core atmospheric model is the fifth-generation European Centre Hamburg general circulation model (ECHAM5) (Roeckner et al., 2006). A hierarchal diagram of EMAC is given in Jöckel et al. (2005). Additionally, Jöckel et al. (2010) provide an update on all modelling components used. For the present study, we applied EMAC (ECHAM5 version 5.3.02, MESSy version 2.54.0) in the T63L90MA resolution, i.e. with a spherical truncation of T63 (corresponding to a quadratic Gaussian grid of approximately 1.875∘ by 1.875∘ in latitude and longitude) with 90 vertical hybrid pressure levels up to 0.01 hPa. By using this horizontal resolution, assessing the global impact is still feasible while at the same time being of a computationally reasonable cost. The 90 vertical layers used (focusing on the lower and middle atmosphere) represent tropospheric and stratospheric transport processes reasonable well (Jöckel et al., 2010) such that the tropospheric impact and the impact on the UTLS (upper troposphere/lower stratosphere) can be addressed. The applied model setup comprised the submodel MECCA (Module Efficiently Calculating the Chemistry of the Atmosphere) to calculate atmospheric chemistry using parts of the Mainz Organic Mechanism (MOM) (Sander et al., 2011). Within MOM, aromatics and terpenes were excluded to reduce the computational demand of all simulations performed; this chemistry has no relevant impact on HNCO. The mechanism was extended to include the proposed changes of this study: formamide as an additional chemical source of HNCO (Bunkan et al., 2016) and chemical mechanisms for nitromethane (Calvert, 2008; Taylor et al., 1980), methylamine, dimethylamine, and trimethylamine (Nielsen et al., 2012). The reaction rates used for the latter three are average values of the measured values reported in Nielsen et al. (2012). The product yields reported in the same source are simplified to suit a global model application. The submodel SCAV (SCAVenging submodel) was used to simulate the physical and chemical removal of trace gases and aerosol particles by clouds and precipitation (Tost et al., 2006). The aqueous-phase mechanism was extended to include the HNCO and formamide mechanism proposed by Borduas et al. (2016b), Barnes et al. (2010), and Behar (1974). These lead to the formation to ammonia in the aqueous phase, which was before limited to the acid–base equilibrium in cloud droplets. The representation of cyanide was improved based on Buechler et al. (1976). Tables S1 and S2 in the Supplement summarize all additional changes to the chemical mechanism in gas and aqueous phases, respectively. The submodel DDEP (Dry DEPosition) is used to simulate the dry deposition of HNCO using the default scheme, where non-stomatal uptake is effectively disabled by using a large and constant resistance (Kerkweg et al., 2006a). The effective Henry's law coefficient (H*) is used, as proposed by Borduas et al. (2016b), modified to a pH of 7. Differently from Young et al. (2012), the same H* over the ocean is used. This approximation is reasonable since the levels of HNCO in the marine boundary layer are expected to be minor. In a global context, the major sources of HNCO and formamide are biomass burning emissions. From literature, two emission factors are available, which differ substantially: 0.53 g kg−1 (Koss et al., 2018) versus 0.2 g kg−1 (Kumar et al., 2018). Thus two simulations are performed to quantify the uncertainty due to those emission factors. The MESSy submodel BIOBURN is used to calculate biomass burning fluxes based on the selected emission factor and Global Fire Assimilation System (GFAS) data. GFAS data are calculated based on fire radiative power observations from the Moderate Resolution Imaging Spectroradiometer (MODIS) satellite instruments, which are used to calculate the dry-matter combustion rates (Kaiser et al., 2012). The biomass burning emission fluxes are then obtained by combining these dry-matter combustion rates with the defined biomass burning emission factors per unit of dry matter burned. The MESSy submodel OFFEMIS (OFFline EMISsions) then calculates the resulting concentration changes for each tracer due to the biomass burning emissions (Kerkweg et al., 2006b). Anthropogenic HNCO emissions from diesel cars are scaled to ammonia EDGAR (Crippa et al., 2016) road emissions by 15 % (Heeb et al., 2011). Other known sources of HNCO (e.g. cigarette smoke) were not taken into account due to the resolution of the spatial grid used. The model was run for 2 years (2010–2011) in which the first year was used as spin-up and 2011 for analysis. In 2010, the biomass burning emissions were particularly high (Kaiser et al., 2012), providing higher background HNCO concentrations during spin-up and improving the representation of HNCO, which allows for a more representative comparison in 2011.
3.1 HNCO+OH
The reaction of HNCO with OH can proceed by four distinct pathways: H abstraction or OH addition on the carbon, nitrogen, or oxygen atom of HNCO; a potential energy surface is shown in Fig. 1. Formation of the HN=C•OOH and HN(OH)C•=O adducts through OH addition on the oxygen or nitrogen atom is highly endothermic by 20 kcal mol−1 or more, and it is not competitive at any temperature. The two remaining pathways are exothermic, with HN•C(=O)OH being the most stable nascent product, 19.8 kcal mol−1 below the reactants, followed by , at 7.5 kcal mol−1 exoergicity. Despite the higher energy of the products, we predict this last reaction to have a lower barrier, 6.0 kcal mol−1, compared to the addition process, 8.7 kcal mol−1, in agreement with the theoretical predictions of Sengupta and Nguyen (1997). Furthermore, the H-abstraction process allows for faster tunnelling, making this process the fastest reaction channel, while addition contributes less than 0.5 % of product formation at temperatures below 400 K. From these data, we derive the following rate coefficient expressions (see also Fig. 2):
Our predictions are in very good agreement between 624 and 875 K, when compared with experimental data from Tully et al. (1989), which served as the basis for the recommendation of Tsang (1992); our predictions reproduce the rate coefficients within a factor of 1.7, comparable to the experimental uncertainty of a factor of 1.5 (see Fig. 2). Likewise, our predictions agree within a factor of 1.7 with the experimental determination by Wooldridge et al. (1996) over the entire 620–1860 K temperature range. Our predictions overshoot the upper limit estimated by Mertens et al. (1992) by a factor of up to 4 at the upper end of the temperature range (2120 to 2500 K). At these elevated temperatures, it is expected that our theoretical kinetic calculations are less accurate since anharmonicity, internal rotation, and possibly pressure effects are not fully accounted for. At this time, we choose not to invest the computational cost to improve the predictions at these temperatures. The predicted rate at room temperature is within a factor of 2 of the extrapolation of the recommended expression derived by Tsang (1992), and very close to the extrapolation of the expression by Wooldridge et al. (1996), which is . The good agreement of our rate coefficient with the experimental data extrapolated to room temperature is mainly due to the curvature predicted in the temperature dependence (see Fig. 2), as our calculations have a slightly steeper temperature dependence than the experiments in the high-temperature range. Though negligible at low temperature, we find that OH addition on the C atom of HNCO accounts for 7 % to 8 % of the reaction rate between 2000 and 3000 K, with other non-H-abstraction channels remaining negligible (<0.1 %). The addition channel is the likely origin of CO2+NH2 products (Sengupta and Nguyen, 1997), for which Wooldridge et al. (1996) experimentally determined an upper limit of ≤0.1 over the temperature range 1250–1860 K, corroborating our predictions to its low contribution.
Typical concentrations of the OH radical during daytime are measured at ∼106 molecule cm−3 (Stone et al., 2012), leading to a pseudo-first order rate coefficient for HNCO loss by OH radicals of s−1, i.e. suggesting an atmospheric chemical lifetime of decades to several centuries, depending on local temperature and OH concentration, negligible compared to other loss processes like scavenging. Even in extremely dry conditions, where aqueous uptake is slow, heterogeneous loss processes will dominate, or alternatively atmospheric mixing processes will transport HNCO to more humid environments where it will hydrolyze.
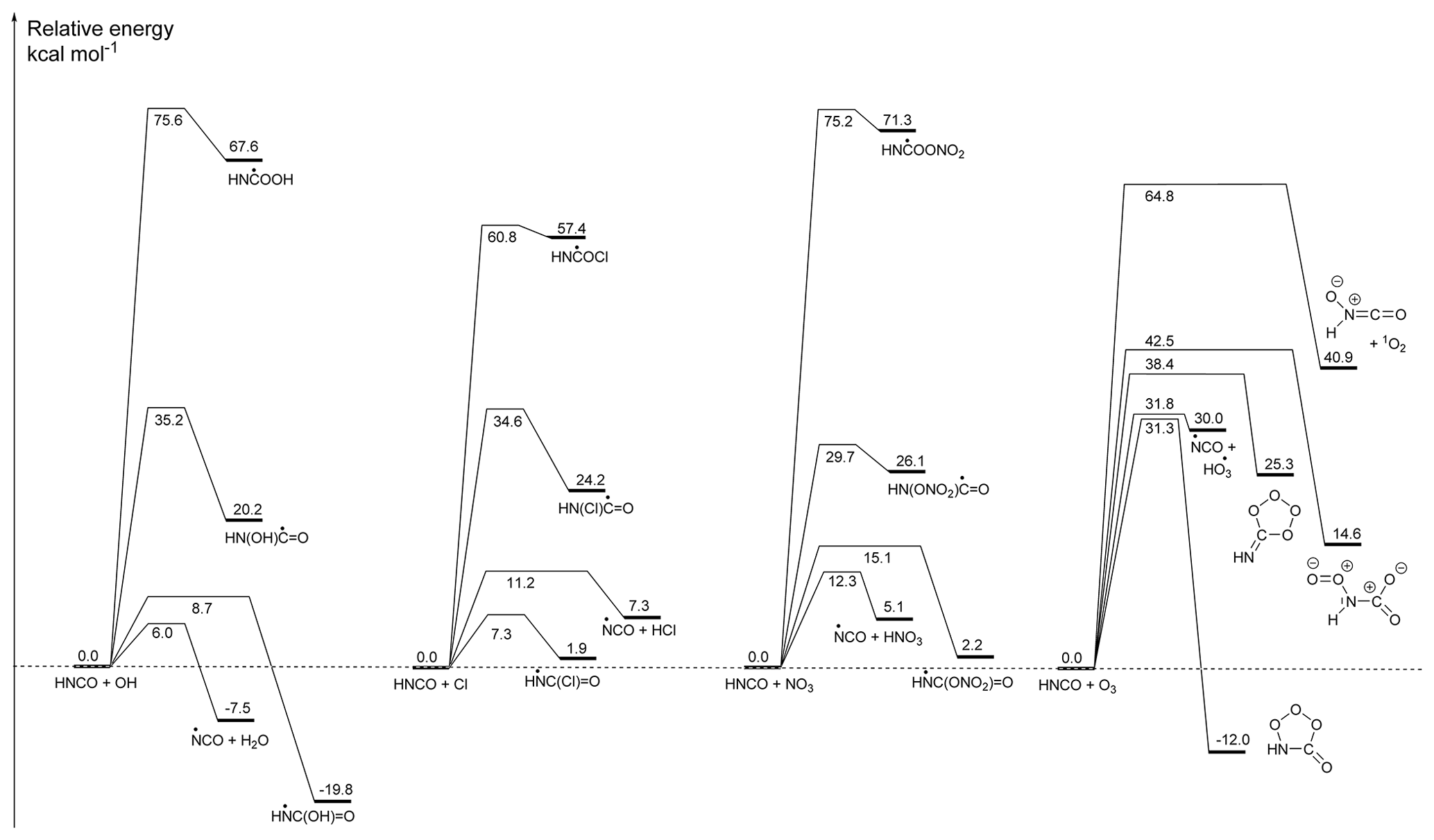
Figure 1Potential energy surfaces for the initiation reactions of HNCO with OH radicals, Cl atoms, NO3 radicals, and ozone, showing CCSD(T)/CBS(DTQ) energies (kcal mol−1) based on M06-2X/aug-cc-pVTZ geometries. The pre-reactive complexes are omitted as they do not influence the kinetics; similarly, the subsequent reactions of the products are not shown. The Supplement has additional energetic and rovibrational data, more complete potential energy surfaces for some of the reactions, and three-dimensional representations of the molecular structures with bond lengths and angles.
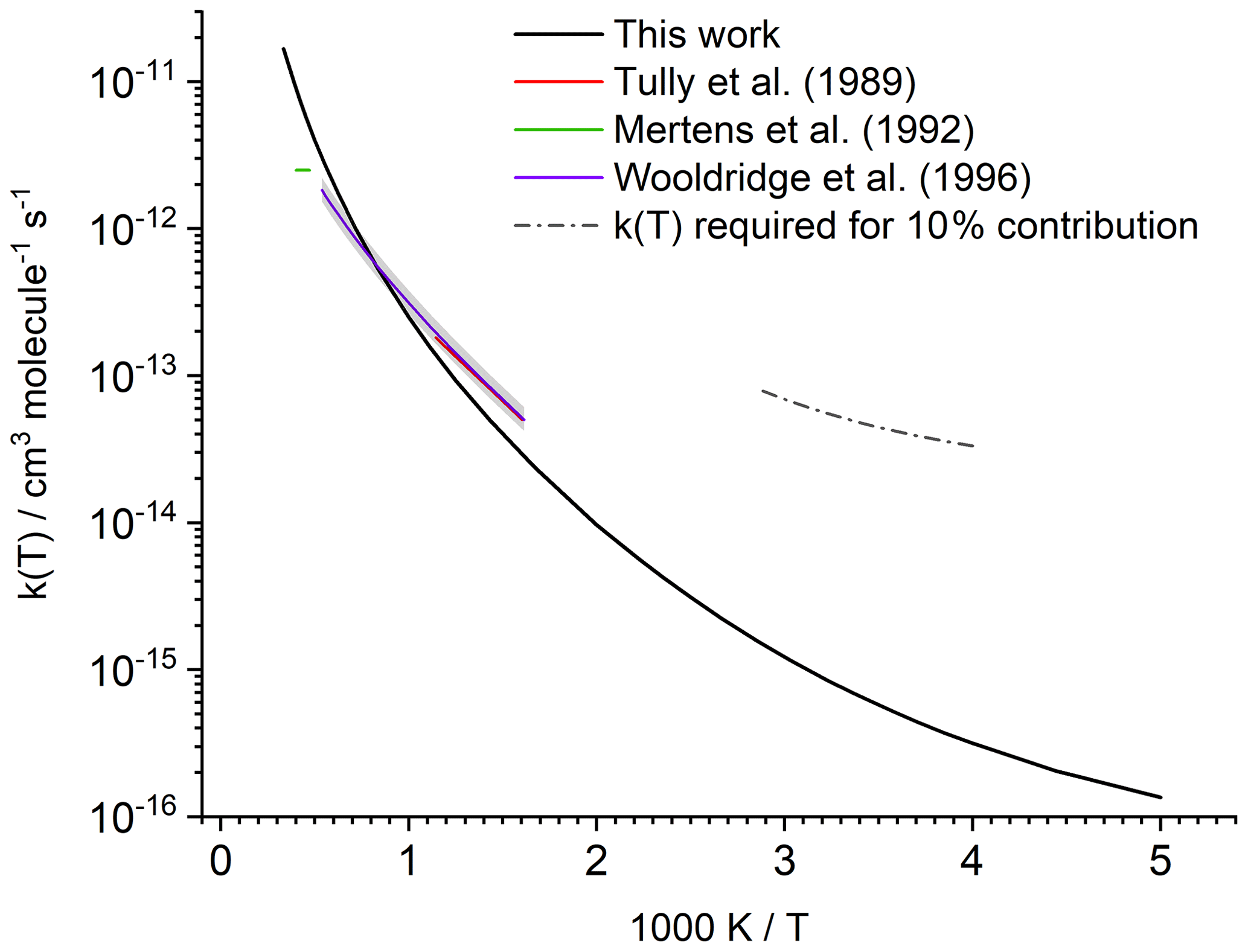
Figure 2Predicted rate coefficient k(T) for the reaction of HNCO+OH compared against experimental data. The shaded area indicates the experimental uncertainty reported by Wooldridge et al. (1996). The dashed line estimates the 298 K rate coefficient that would be needed to remove 10 % of the atmospheric HNCO by reaction with OH (see text).

Figure 3Total rate coefficient predictions for the reaction of HNCO with NO3, Cl, and O3. The addition of Cl atoms on HNCO leads to the formation of a very short-lived adduct, which rapidly redissociates to the reactants; the effective rate coefficient for HNCO loss by Cl atoms, keff (Cl), is thus equal to the H-abstraction rate forming HCl+NCO (see text).
3.2 HNCO+Cl
From the potential energy surface (PES) shown in Fig. 1, we see that the reaction between HNCO and the Cl atom can occur by abstraction of the H atom from HNCO or by addition of the Cl atom on the C, N, or O atoms. Contrary to the OH reaction, all entrance reactions are endothermic, with formation of the HN•C(Cl)=O alkoxy radical nearly energy neutral (see Fig. 1). Formation of this latter product, proceeding by the addition of a Cl atom to the carbon atom of HNCO, also has the lowest energy barrier, which is 7.3 kcal mol−1 above the reactants. The hydrogen abstraction, forming HCl and •NCO, requires passing a higher barrier of 11.2 kcal mol−1, whereas additions on the N and O atoms have very high barriers exceeding 34 kcal mol−1. The product energy difference between addition and H abstraction is much smaller compared to the HNCO+OH reaction. Despite this reduced reaction energy, the addition barrier remains 4 kcal mol−1 below the H-abstraction barrier, making the HNCO+Cl reaction the only reaction studied here where H abstraction is not dominant. For the HNCO+Cl reaction, we then obtain the following rate coefficients (see also Fig. 3):
We find that the overall rate coefficient of the HNCO+Cl reaction is almost 1 order of magnitude below that for the OH radical. The HN•C(Cl)=O radical formed, however, has a weak C−Cl bond requiring only 5.4 kcal mol−1 to redissociate. The rate coefficient of 8×108 s−1 for dissociation at room temperature ( s−1) is over an order of magnitude faster than O2 addition under atmospheric conditions, assuming the latter is equally fast as for H2C•CH=O vinoxy radicals, i.e. k(298 K, 0.2 atm O2)≤107 s−1 (IUPAC Subcommittee on Atmospheric Chemical Kinetic Data Evaluation, 2017). This makes redissociation to the reactants the most likely fate of the HN•C(Cl)=O adduct. Addition is thus an ineffective channel for HNCO removal, and the effective reaction with Cl atoms is dominated by the H-abstraction reaction, forming HCl+•NCO, with the following rate coefficient (see also Fig. 3):
Globally, Cl atoms have a lower concentration, about 5×103 atom cm−3, compared to OH radicals (Finlayson-Pitts and Pitts, 1999). Under such conditions, lifetimes estimated for HNCO towards Cl atoms are about 3×107 years, which is much longer than towards the OH radial. Therefore, HNCO loss by Cl radicals is negligible.
The supporting information provides information on the extended potential energy surface of the HNCO+Cl reaction, with information on nine intermediates, 19 transition states, and 16 products.
3.3 HNCO+NO3
The reaction of NO3 with HNCO shows the same four radical mechanisms found for OH and Cl, i.e. H abstraction and addition on the three heavy atoms. As for Cl atoms, none of the reactions are exothermic, and the energy difference between the two most stable products is reduced to 3 kcal mol−1, indicating that NO3 addition is even less favourable than Cl addition. Formation of HNO3+•NCO is more favourable than HCl+NCO formation by about 2 kcal mol−1. The barrier for H abstraction, however, is larger compared to abstraction by both OH and Cl and exceeds 12 kcal mol−1. The most favourable addition process, forming HN•C(=O)NO3, has a barrier of 15.1 kcal mol−1, but it contributes less than 0.01 % to the reaction rate at room temperature. The overall reaction thus proceeds nearly exclusively by H abstraction forming HNO3+•NCO for which we derived the following rate coefficients (see also Fig. 3):
While this rate coefficient is almost 5 orders of magnitude below that of the OH radical, the nitrate radical is known to be present in higher concentrations during night-time, reaching concentrations as high as 109 molecule cm−3 (Finlayson-Pitts and Pitts, 1999). The effective rate of the NO3 reaction at night-time is similar to the reaction with OH at daytime. The NO3 radical is thus likewise considered to be ineffective for atmospheric removal of HNCO, compared to heterogeneous loss processes.
3.4 HNCO+O3
The chemistry of ozone with organic compounds is drastically different from radicals, where O3 typically reacts by cycloaddition on double bonds in unsaturated compounds. For HNCO, cycloaddition pathways have been characterized for both double bonds (). Only cycloaddition on the N=C bond leads to an exothermic reaction, with the oxo-ozonide product being 12 kcal mol−1 more stable than the reactants (see Fig. 1). In addition to the traditional cycloaddition channels, three further channels were found, corresponding to H abstraction, forming HO3+NCO; oxygen transfer to the N atom, forming ON(H)CO+1O2; and addition on the C and N atoms, forming HN(OO)C(O)O. The HO3 product radical is known to be only weakly bonded by 2.94 kcal mol−1, falling apart to OH+O2 (Bartlett et al., 2019; Le Picard et al., 2010; Varandas, 2014).
The cyclo-addition channels on the hetero-atom double bonds have high-energy barriers, exceeding 30 kcal mol−1, significantly larger than typical barriers for C=C bonds with aliphatic substitutions. Surprisingly, this allows H abstraction to become competitive to cycloaddition, with a comparable barrier of 32 kcal mol−1. For the overall reaction, we obtain the following rate coefficients (see also Fig. 3):
At room temperature, H abstraction contributes 80 % to the total reaction and cycloaddition on the N=C bond the remaining 20 %. All other channels are negligible. The rate coefficient is exceedingly low, , such that even in areas with very high ozone concentrations of 100 ppbv the loss by ozonolysis is expected to be negligible.
The Supplement provides information on the extended potential energy surface of the HNCO+O3 reaction, with information on 10 intermediates, 30 transition states, and 15 products. The lowest-energy unimolecular product channel leads to formation of CO2+HNOO by breaking of the cyclic primary ozonide (see Fig. 1) following the traditional Criegee mechanism (Criegee, 1975).
The radical reactions characterized above proceed by H abstraction, forming the NCO radical with an H2O, HNO3, or HCl co-product. Likewise, the ozonolysis reaction proceeds for a large part by H abstraction, forming NCO with a HO3 co-product that readily dissociates to OH+O2. Though NCO radical formation through these reactions is found to be negligibly slow in atmospheric conditions, this radical remains of interest in other environments. Examples include combustion chemistry, where it can be formed directly from nitrogen-containing fuels and where it is a critical radical intermediate in, for example, the RAPRENOx (RAPid REmoval of nitrogen oxides) NOx mitigation strategy which employs HNCO introduced in the combustion mixture through (HOCN)3 (cyanuric acid) injection (Fenimore, 1971; Gardiner, 2000). The NCO radical has also been observed in space (Marcelino et al., 2018). There is extensive experimental and theoretical information on the reactions of NCO radicals, e.g. tabulated in Tsang (1992), Baulch et al. (2005), and other works. To our knowledge, the rate coefficients of the reactions of NCO radicals with H2O, HNO3, and HCl have not been determined before, but Tsang (1992) has estimated a rate coefficient based on the equilibrium constant and rate coefficient of the HNCO+OH reaction. Since the H–N bond in HNCO is quite strong, with a bond energy of ∼110 kcal mol−1 (Ruscic, 2014; Ruscic and Bross, 2019), it is expected that NCO can readily abstract a hydrogen atom from most hydrogen-bearing species to produce HNCO, and that H abstraction is the main reaction channel. Hence, despite that our potential energy surfaces do not include an exhaustive search of all possible reaction channels in the NCO radical chemistry, we expect that the single-channel H-abstraction rate predictions for NCO from H2O, HNO3, and HCl are sufficiently dominant that these rates are fair estimates of the total rate coefficients including all possible channels for each of these reactions.
The energy barriers for the NCO radical reactions with H2O, HNO3, and HCl, being 14, 7, and 4 kcal mol−1, respectively (see Fig. 1), follow the bond strength trend in these reactants, with kcal mol−1, kcal mol−1, and kcal mol−1 (Luo, 2007; Ruscic et al., 2002). Figure 1 also shows that the NCO+H2O reaction is endothermic by 8 kcal mol−1, while the HNO3 and HCl paths are exothermic by −5 and −7 kcal mol−1, respectively. The predicted rate coefficients are then the following:
The indirect estimate of Tsang (1992) compares well to our prediction for NCO+H2O, reproducing our values within a factor of 15 at 1000 K and a factor of 3 at 2000 K, i.e. within the stated uncertainties. An analysis of the impact of the NCO reactions in combustion or non-terrestrial environments is well outside the scope of this paper, and reactions with other co-reactants not discussed in this paper are likely to be of higher importance, e.g. H abstraction from organic compounds or recombination with other radicals. In atmospheric conditions, the fate of the NCO radical is likely recombination with an O2 molecule, with a rate coefficient of (Manion et al., 2020; Schacke et al., 1974), leaving H2O, HNO3, and HCl as negligible co-reactants. Hence, the NCO radical will not affect the atmospheric fate of any of these compounds to any extent. Subsequent chemistry of the •OONCO radical is assumed to be conversion to an •ONCO alkoxy radical through reactions with NO, HO2, or RO2, followed by dissociation to NO+CO.
Table 1Yearly global HNCO budget in 2011 for both biomass burning emission datasets by Kumar et al. (2018) and Koss et al. (2018). Additionally, the HNCO budget from Young et al. (2012) is given for comparison.
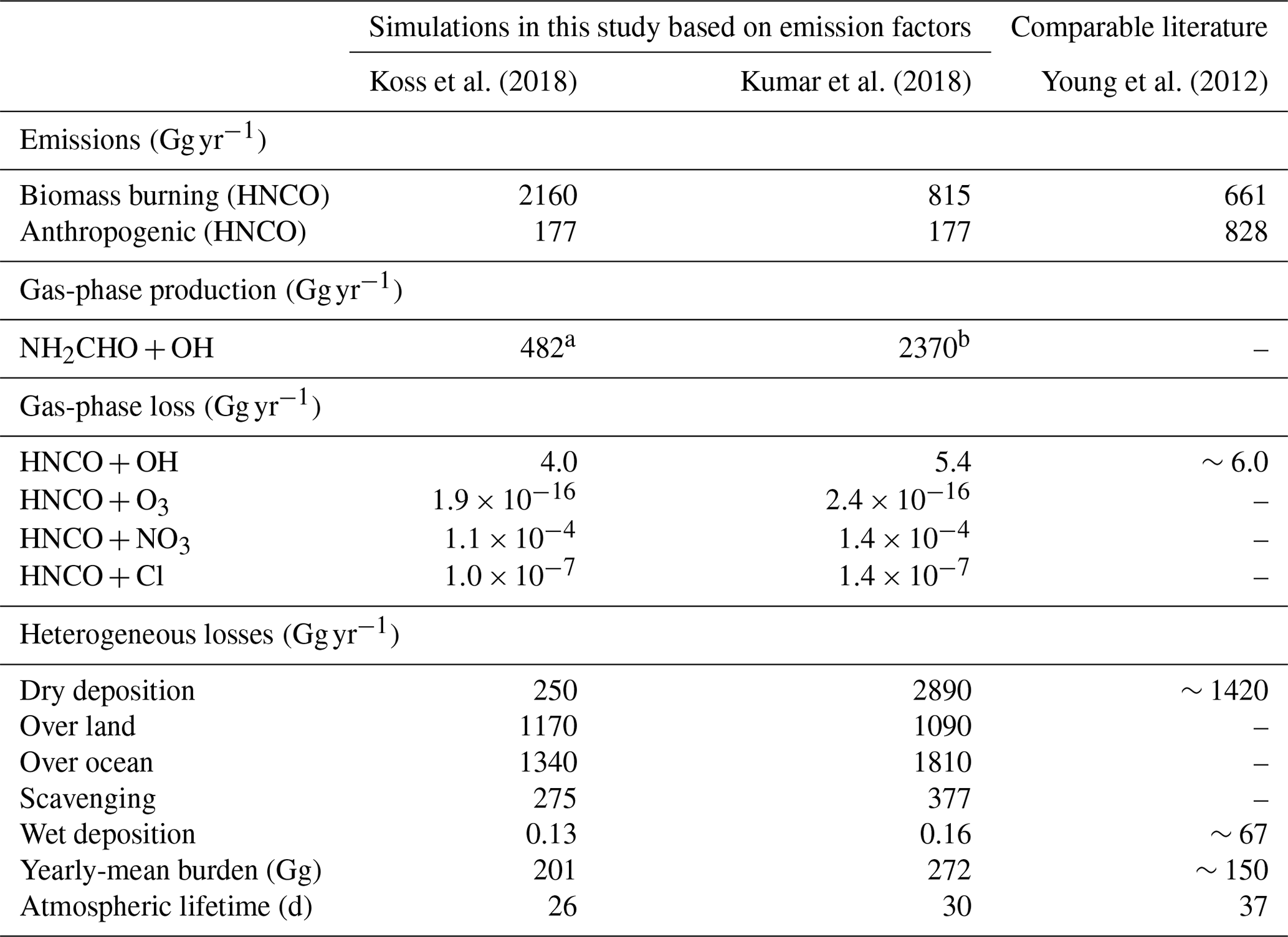
a of which 51 Gg yr−1 is NH2CHO biomass burning emissions
(Koss et al., 2018)
b of which 2340 Gg yr−1 is NH2CHO biomass burning emissions
(Kumar et al., 2018)
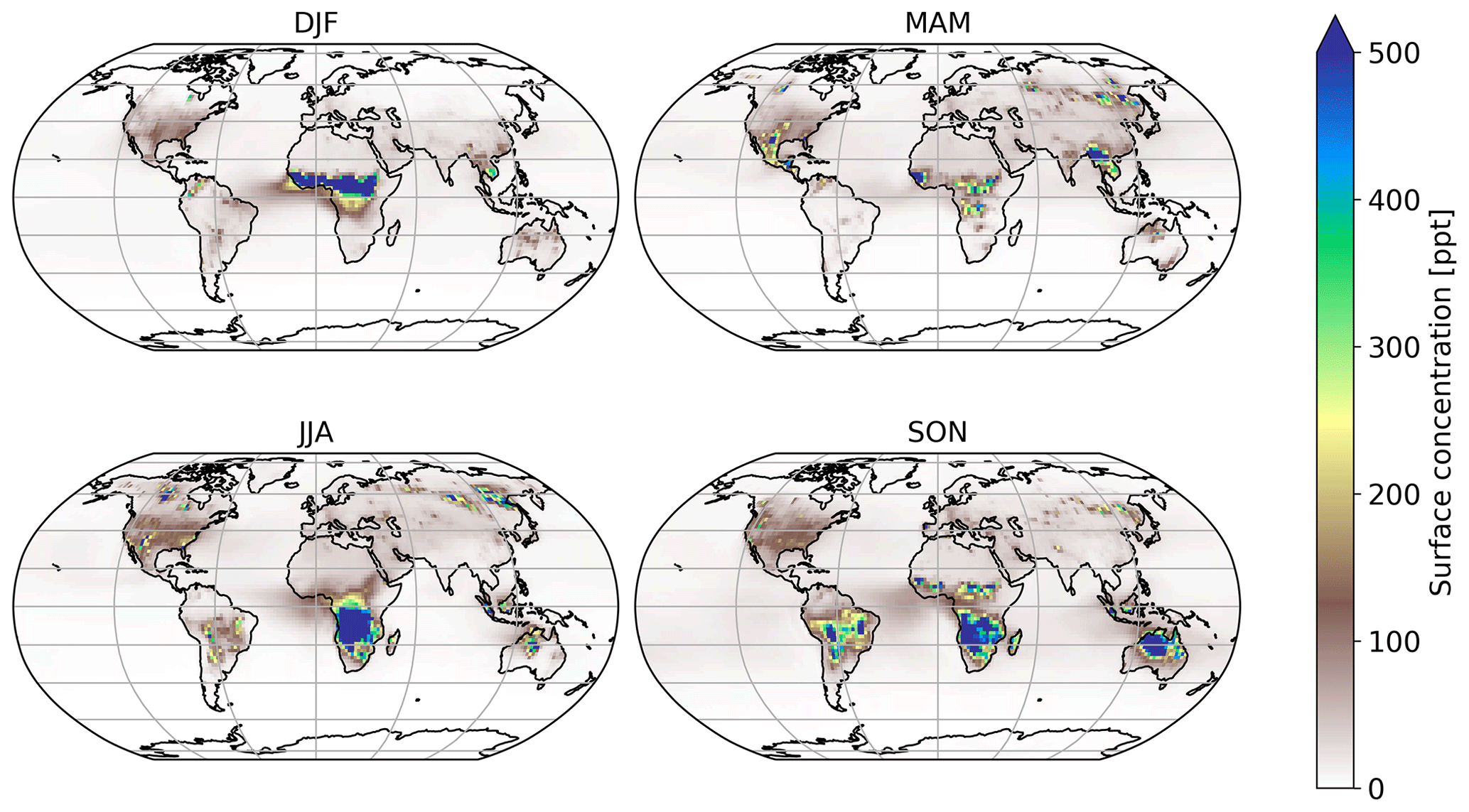
Figure 4Mean seasonal surface concentration of HNCO using Koss et al. (2018) biomass burning emission factors.
Global atmospheric simulations allow us to gain insights into the significance of the chemical loss processes of HNCO and its distribution. Table 1 shows the corresponding HNCO budget for both performed simulations. The full kinetic model including our theoretically predicted gas-phase chemical reactions of HNCO is detailed in Tables S1 and S2 of the Supplement. Figure 4 shows the mean seasonal surface mixing ratio of HNCO using the biomass burning emission factors by Koss et al. (2018). It can be observed that high levels persist in each season. In general, high HNCO levels occur in regions associated with frequent biomass burning activities. Regions with no biomass burning activities have low HNCO concentrations, mainly caused by free tropospheric entrainment from regions with higher concentrations. The global vertical profile of HNCO is well illustrated by that for January as given in Fig. 5, showing that the free troposphere contains about 81 % of the total HNCO mass. The gas-phase production via formamide differs greatly depending on the biomass burning emissions used. In the case of Kumar et al. (2018), significantly more formamide is emitted, leading to a higher production of HNCO in the gas phase. The hydrolysis of HNCO produces ∼120 Tg yr−1 of ammonia and thus contributing little to the global ammonia budget. Our estimate is a factor of 5–6 lower than the upper limit estimated by Leslie et al. (2019).
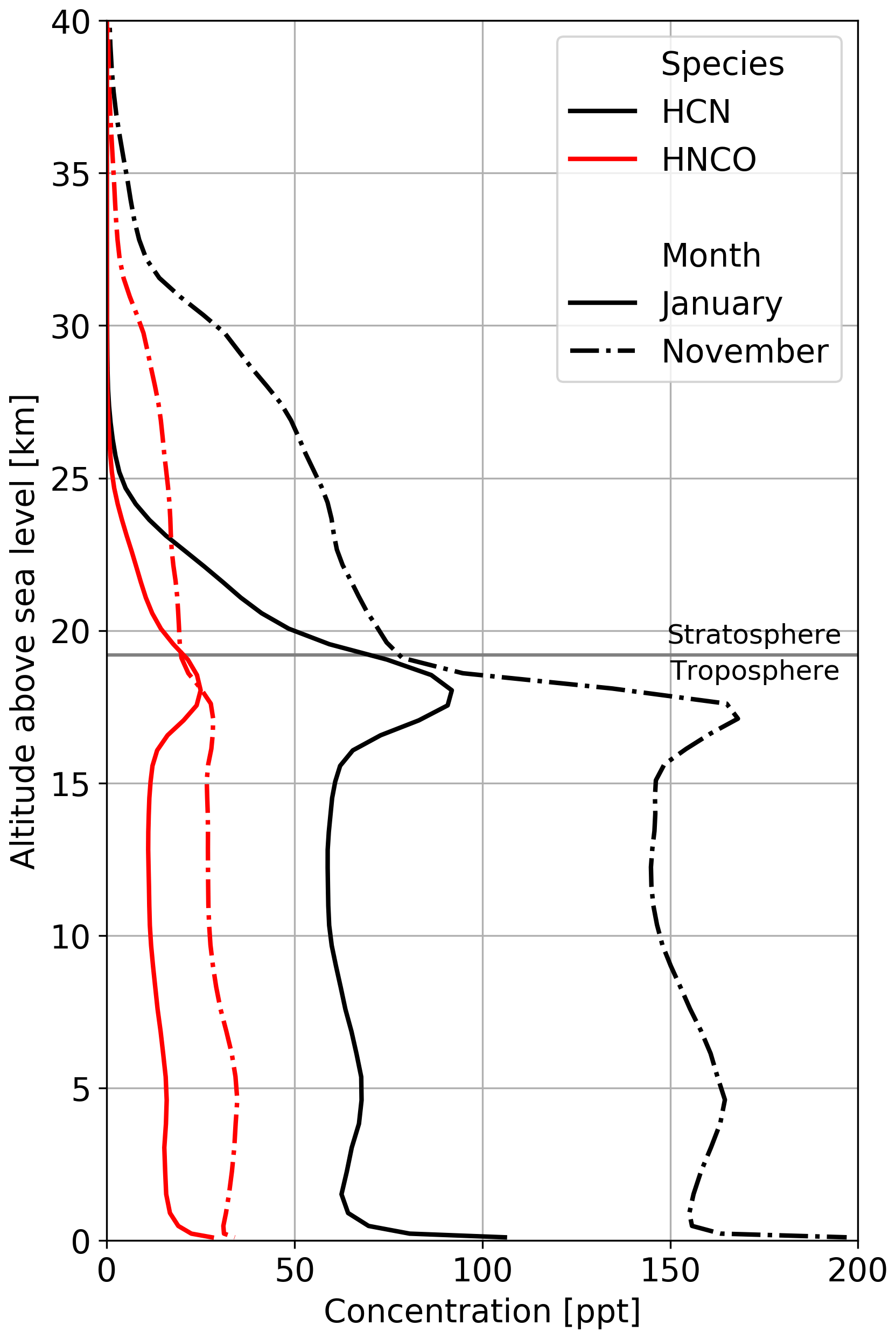
Figure 5Mean vertical profiles of HCN (black) and HNCO (red) for January (solid lines) and November (dashed-dotted lines) over South East Asia. Biomass burning emission factors are based on Koss et al. (2018).
The model predictions for local OH radical concentrations range from 1.15×100 to 1.56×107 molecule cm−3, with a weighted atmospheric global average of 1.14×106 molecule cm−3; in the air parcel where the highest OH concentration is found this leads to a HNCO lifetime towards OH of more than 500 years when accounting for the temperature-dependent rate coefficient (∼276 K). In the planetary boundary layer, the highest OH concentration predicted is 7.6×106 molecule cm−3 at a temperature of 297.8 K, leading to a HNCO lifetime to OH of ∼6 years in that air parcel. The calculated average OH concentration of 1.2×106 molecule cm−3 in the boundary layer leads to lifetimes towards OH of about 40 years near the surface. For O3, Cl, and NO3 – with maximum oxidant concentrations of 1.0×1013, 7.8×105, 1.5×109 molecule cm−3 and atmospheric average concentrations of 1.0×1012, 2.0×103, and 1.1×107 molecule cm−3, respectively – even longer temperature-dependent lifetimes are found, exceeding 5000 years even in the air parcels with the most favourable co-reactant concentration and temperature. The relative contributions of the different co-reactants varies locally and temporally, and shorter lifetimes might occur locally when co-reactant concentration and temperature are at their most favourable, but it is clear that gas-phase chemical losses of HNCO are small. Only the reaction of HNCO with OH leads to some destruction of HNCO, while the other chemical sinks (O3, NO3, and Cl) are negligible. When compared to the major loss processes, however, all these loss processes are negligible on a global scale (see Table 1). Young et al. (2012) have a somewhat higher chemical loss via OH compared to our result, which is due to the higher rate constant used. Figure 2 shows the rate coefficient that would be required to allow for the gas-phase loss of HNCO by reaction with OH radicals to contribute 10 % of the total atmospheric sink, which is well outside the expected uncertainty of the theoretical kinetic rate predictions. It can therefore be robustly concluded that the gas-phase chemical sinks predicted and assessed in this study (OH, Cl, NO3, O3) are insignificant when compared to heterogeneous loss processes, confirming earlier assumptions. This is independent of the high uncertainty in the available biomass burning emission factors or missing road emission datasets.
As seen in Table 1 the major sinks are dry deposition and scavenging (heterogeneous losses), where the former contributes between 2520 and 2890 Gg yr−1 and the latter from 274 to 377 Gg yr−1, when using the emission factors by Koss et al. (2018) and Kumar et al. (2018), respectively. The results in this study are in a similar range as the modelling study by Young et al. (2012). These authors had lower total HNCO emissions and did not include formamide as a secondary source of HNCO. The lower total HNCO emissions could be explained by a different year simulated in that study and different biomass burning emission model approaches used. Young et al. (2012) also scaled their HNCO emissions to the HCN emissions by a factor of 0.3, whereas in this study actual measured emission factors are used. In our study, formamide contributes between 17 % and 70 % of the total HNCO emissions when using the biomass burning emission factors by Koss et al. (2018) and Kumar et al. (2018) respectively. Young et al. (2012) find a higher HNCO lifetime due to generally lower total heterogeneous loss terms (dry and wet deposition). The total dry deposition varies slightly depending on the biomass burning emission factor used (see Table 1). In both scenarios, most HNCO is deposited over the ocean. For biomass burning emission factors from Koss et al. (2018), this contribution 53 %, is significantly lower when compared to the simulation using emission factors from Kumar et al. (2018), where about 62 % of the total HNCO deposition is deposited over the ocean. The larger fraction of computed HNCO deposition over the ocean is a consequence of the much larger secondary HNCO production from formamide far from its source regions (continents). Young et al. (2012) found that the importance of both heterogeneous loss processes depends on the cloud pH. In the SCAV submodel, as used in this work, cloud droplet pH is calculated online and includes an explicit hydrolysis scheme for HNCO, whereas Young et al. (2012) used a simplified approach. The relative importance of dry deposition is higher in the simulation in which Young et al. (2012) calculated pH online, when compared to the findings in this study.
The atmospheric lifetime of HNCO is dominated by its heterogeneous loss processes, leading to an atmospheric lifetime of multiple weeks when accounting for all HNCO losses (chemical and heterogeneous), as opposed to a gas-phase lifetime in the free troposphere of about 50 years when calculated solely based on the chemical losses towards the four chemical oxidants described in this study. This long gas-phase lifetime and the fact that mainly surface sources are relevant indicate that atmospheric HNCO distribution is significantly affected by transport processes. Our simulations even show that HNCO is transported from the surface into the UTLS and that about 10 % of the total atmospheric HNCO mass is located in the stratosphere (see Fig. 5), with modelled concentrations of HNCO in the lower stratosphere of typically tens of parts per trillion by volume but reaching up to hundreds of parts per trillion by volume in tropical regions. In the chemical model, photolysis in the stratosphere was not taken into account. Thus, OH is the only significant stratospheric sink included, resulting in a stratospheric lifetime of more than 330 years. During the monsoon period, the total stratospheric HNCO mass increases from 15 Gg before to 20 Gg at the end of the monsoon season. Pumphrey et al. (2018) demonstrated that in 2015 and 2016, elevated levels of stratospheric hydrogen cyanide (HCN) can be linked to biomass burning emissions from Indonesian fires. Figure 5 shows the vertical profiles of HCN and HNCO over South East Asia well before (January) and after (November) the Indian monsoon. It becomes evident that, similar to HNCO in our simulations, tropospheric and stratospheric concentrations of HCN increase during the Indian monsoon period. In the performed simulations, the ratio between stratospheric HCN and HNCO is very similar throughout the year, indicating that HCN and HNCO are similarly affected by transport processes within this period. The combination of strong biomass burning events and strong vertical transport during the monsoon period leads to high HNCO concentrations in the UTLS, indicating that pollutants from biomass burning events could potentially influence stratospheric chemistry.
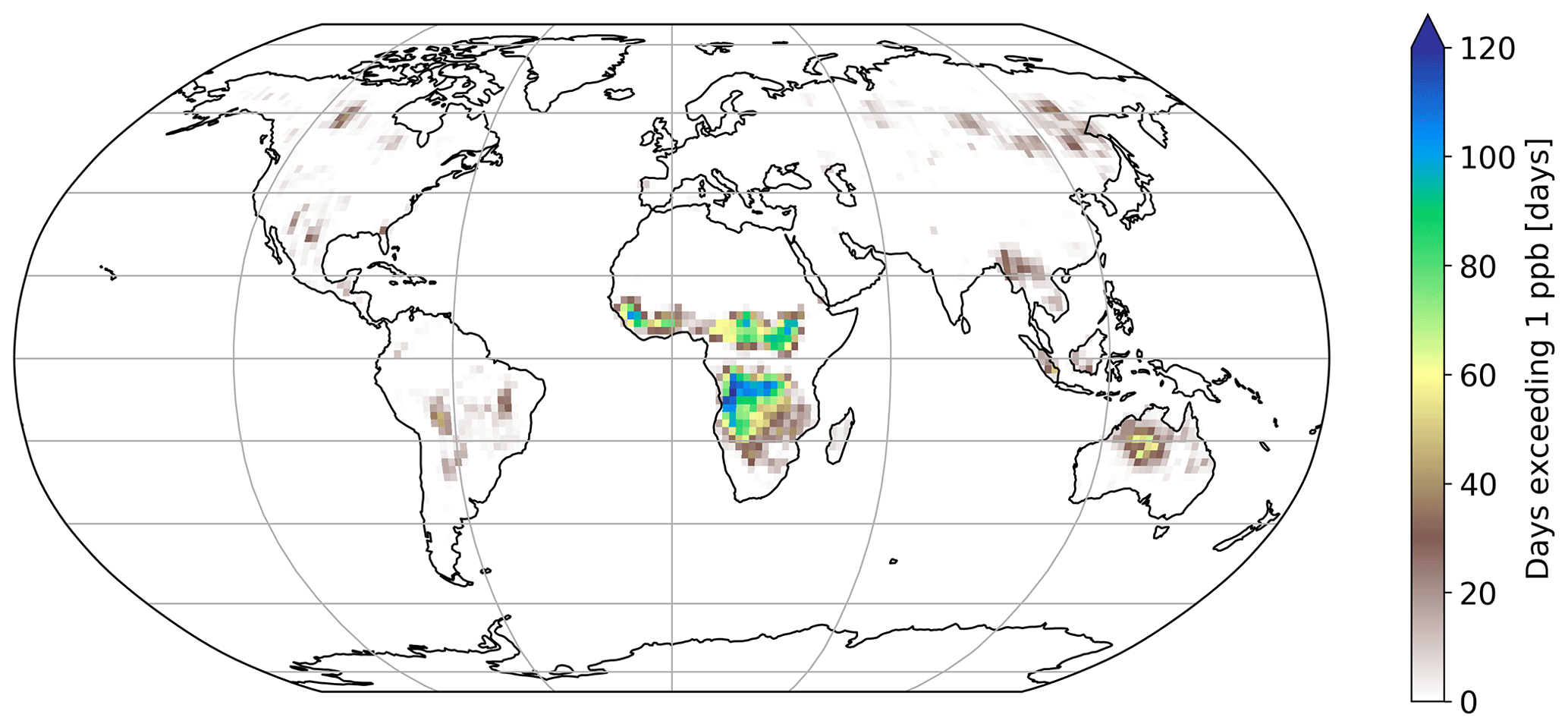
Figure 6Number of days exceeding 1 ppb of HNCO at the surface. Biomass burning emission factors are based on Koss et al. (2018).
Figure 6 shows the number of days exceeding a daily mean HNCO concentration of 1 ppbv. Mainly regions impacted by biomass burning events have frequent concentrations above this threshold. When using 10 ppbv as a limit for toxic concentrations of HNCO, as proposed by the Swedish Work Environment Authority (SWEA, 2011), only a few days can be observed in which this limit is exceeded. The maximum number of days exceeding 10 ppbv is 10 d over Africa, compared to 120 d above 1 ppbv. It is important to take into account that this analysis is limited by the computational output available in this study, which has only daily averages. Therefore, it is expected that areas which frequently exceed daily averages of 1 ppbv are potentially areas in which peak HNCO can be observed above 10 ppbv throughout the day.
No correlation exists between the number of days exceeding 1 or 10 ppbv and road traffic emissions. This becomes evident since typical areas of high road traffic activities (i.e. USA and Europe) do not exceed daily averages of 1 ppbv (see Fig. 6). Road traffic activities occur on a smaller spatial scale than biomass burning events. The EMAC model used is not capable of representing, for example, inner-city road traffic activities, due to the spatial resolution of the model used (1.875∘ by 1.875∘ in latitude and longitude). Therefore, we are not capable of drawing any conclusion if 10 ppbv is exceeded regionally in densely populated areas, impacted by high traffic emissions.
The isocyanic acid molecule, HNCO, is found to be chemically unreactive towards the dominant atmospheric gas-phase oxidants, i.e. OH and NO3 radicals, Cl atoms, and O3 molecules. The reactions all remove HNCO predominantly by H abstraction and have low rates of reactions with , leading to chemical gas-phase lifetimes of decades to centuries. Yearly loss of HNCO towards these reactants is only ∼5 Gg yr−1 out of ∼ 3000 Gg yr−1 total losses. Removal of HNCO by clouds and precipitation (“scavenging”), with hydrolysis to ammonia, is also implemented in the global model and was found to contribute significantly more, ∼300 Gg yr−1, than the gas-phase loss processes. Still, these combined processes are overwhelmed by the loss of HNCO by dry deposition, which is removing ∼2700 Gg yr−1. These conclusions are robust against modifications of the emission scenarios, where two distinct sets of emission factors were used, incorporating HNCO formation from biomass burning, as well as anthropogenic sources such as formamide oxidation and road traffic. The inefficiency of gas-phase chemical loss processes confirms earlier assumptions; inclusion of the gas-phase chemical loss processes in kinetic models appears superfluous except in specific experimental conditions with very high co-reactant concentrations. The long gas-phase chemical lifetime (multiple decades to centuries) allows HNCO to be transported efficiently into the upper troposphere lower stratosphere (UTLS) demonstrating that surface emissions may impact the upper troposphere. Further research is necessary to identify the importance of strong biomass burning events coupled to strong vertical transport processes (i.e. monsoon systems) on the chemical composition of the UTLS.
On a global scale, the daily-average concentrations of HNCO rarely exceed 10 ppbv, which is the threshold assumed here for toxicity; the exceedances are mainly located in regions with strong biomass burning emissions. Average daily concentrations of the order of 1 ppbv are encountered more frequently, with about one-third of the year exceeding this limit. This suggests that local concentrations might peak to much higher values, e.g. in urban environments where road traffic emissions are highest, or in the downwind plume of biomass burning events, and could impact regional air quality. Such regional effects were not studied in the current work, as the resolution of the global model used here is not sufficiently fine grained.
Though not important for the atmosphere, we briefly examined the reactions of the NCO radical formed in the chemical reactions studied. The rate coefficients of the H-abstraction reactions with H2O, HNO3, and HCl suggest that these reactions might contribute in high-temperature environments, such as combustion processes.
The simulation results are archived at the Jülich Supercomputing Centre (JSC) and are available on request from Domenico Taraborrelli (d.taraborrelli@fz-juelich.de).
The supplement related to this article contains extended information on the chemical model and the quantum chemical characterizations (geometric, energetic, and entropic data). The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-6671-2020-supplement.
The quantum chemical calculations were performed by HMTN, GHTV, and TVP, while LV performed the theoretical kinetic calculations. UJ, SR, and DT collected the literature data on HNCO sources and sinks and implemented these in the kinetic model; the model runs were performed by SR and DT. All authors contributed significantly to the writing of the article.
The authors declare that they have no conflict of interest.
Hue M. T. Nguyen, Giang H. T. Vu, and Tien V. Pham thank the National Foundation for Science and Technology Development (Nafosted), Vietnam, for sponsoring this work under project number 104.06-2015.85. Simon Rosanka and Domenico Taraborrelli gratefully acknowledge the Earth System Modelling Project (ESM) for funding this work by providing computing time on the ESM partition of the supercomputer JUWELS at the Jülich Supercomputing Centre (Forschungszentrum Jülich, 2019).
This research has been supported by the National Foundation for Science and Technology Development (Nafosted), Vietnam (grant no. 104.06-2015.85).
The article processing charges for this open-access
publication were covered by a Research
Centre of the Helmholtz Association.
This paper was edited by James Roberts and reviewed by two anonymous referees.
Alecu, I. M., Zheng, J., Zhao, Y., and Truhlar, D. G.: Computational Thermochemistry: Scale Factor Databases and Scale Factors for Vibrational Frequencies Obtained from Electronic Model Chemistries, J. Chem. Theory Comput., 6, 2872–2887, https://doi.org/10.1021/ct100326h, 2010.
Baker, R. R. and Bishop, L. J.: The pyrolysis of tobacco ingredients, J. Anal. Appl. Pyrolysis, 71, 223–311, https://doi.org/10.1016/S0165-2370(03)00090-1, 2004.
Bao, J. L., Zheng, J., Alecu, I. M., Lynch, B. J., Zhao, Y., and Truhlar, D. G.: Database of Frequency Scale Factors for Electronic Model Chemistries (Version 3 Beta 2), available at: http://comp.chem.umn.edu/freqscale/index.html (last access: 29 May 2020), 2017.
Barnes, I., Solignac, G., Mellouki, A., and Becker, K. H.: Aspects of the Atmospheric Chemistry of Amides, Chemphyschem, 11, 3844–3857, https://doi.org/10.1002/cphc.201000374, 2010.
Barth, M. C., Cochran, A. K., Fiddler, M. N., Roberts, J. M., and Bililign, S.: Numerical modeling of cloud chemistry effects on isocyanic acid (HNCO), J. Geophys. Res.-Atmos., 118, 8688–8701, https://doi.org/10.1002/jgrd.50661, 2013.
Bartlett, M. A., Kazez, A. H., Schaefer, H. F., and Allen, W. D.: Riddles of the structure and vibrational dynamics of HO3 resolved near the ab initio limit, J. Chem. Phys., 151, 094304, https://doi.org/10.1063/1.5110291, 2019.
Baulch, D. L., Bowman, C. T., Cobos, C. J., Cox, R. A., Just, Th., Kerr, J. A., Pilling, M. J., Stocker, D., Troe, J., Tsang, W., Walker, R. W., and Warnatz, J.: Evaluated Kinetic Data for Combustion Modeling: Supplement II, J. Phys. Chem. Ref. Data, 34, 757, https://doi.org/10.1063/1.1748524, 2005.
Becke, A. D.: A New Mixing of Hartree-Fock and Local Density-Functional Theories, J. Chem. Phys., 98, 1372–1377, https://doi.org/10.1063/1.464304, 1993.
Behar, D.: Pulse-Radiolysis Study of Aqueous Hydrogen-Cyanide and Cyanide Solutions, J. Phys. Chem., 78, 2660–2663, https://doi.org/10.1021/j100619a005, 1974.
Belson, D. J. and Strachan, A. N.: Preparation and Properties of Isocyanic Acid, Chem. Soc. Rev., 11, 41–56, https://doi.org/10.1039/cs9821100041, 1982.
Blomqvist, P., Hertzberg, T., Dalene, M., and Skarping, G.: Isocyanates, aminoisocyanates and amines from fires – a screening of common materials found in buildings, Fire Mater., 27, 275–294, https://doi.org/10.1002/fam.836, 2003.
Borduas, N., da Silva, G., Murphy, J. G., and Abbatt, J. P. D.: Experimental and Theoretical Understanding of the Gas Phase Oxidation of Atmospheric Amides with OH Radicals: Kinetics, Products, and Mechanisms, J. Phys. Chem. A, 119, 4298–4308, https://doi.org/10.1021/jp503759f, 2015.
Borduas, N., Murphy, J. G., Wang, C., da Silva, G., and Abbatt, J. P. D.: Gas Phase Oxidation of Nicotine by OH Radicals: Kinetics, Mechanisms, and Formation of HNCO, Environ. Sci. Technol. Lett., 3, 327–331, https://doi.org/10.1021/acs.estlett.6b00231, 2016a.
Borduas, N., Place, B., Wentworth, G. R., Abbatt, J. P. D., and Murphy, J. G.: Solubility and reactivity of HNCO in water: insights into HNCO's fate in the atmosphere, Atmos. Chem. Phys., 16, 703–714, https://doi.org/10.5194/acp-16-703-2016, 2016b.
Brady, J. M., Crisp, T. A., Collier, S., Kuwayama, T., Forestieri, S. D., Perraud, V., Zhang, Q., Kleeman, M. J., Cappa, C. D., and Bertram, T. H.: Real-Time Emission Factor Measurements of Isocyanic Acid from Light Duty Gasoline Vehicles, Environ. Sci. Technol., 48, 11405–11412, https://doi.org/10.1021/es504354p, 2014.
Büchler, H., Bühler, R. E., and Cooper, R.: Pulse radiolysis of aqueous cyanide solutions. Kinetics of the transient hydroxyl radical and hydrogen atom adducts and subsequent rearrangements, J. Phys. Chem., 80, 1549–1553, https://doi.org/10.1021/j100555a006, 1976.
Bunkan, A. J. C., Hetzler, J., Mikoviny, T., Wisthaler, A., Nielsen, C. J., and Olzmann, M.: The reactions of N-methylformamide and N,N-dimethylformamide with OH and their photo-oxidation under atmospheric conditions: experimental and theoretical studies, Phys. Chem. Chem. Phys., 17, 7046–7059, https://doi.org/10.1039/c4cp05805d, 2015.
Bunkan, A. J. C., Mikoviny, T., Nielsen, C. J., Wisthaler, A., and Zhu, L.: Experimental and Theoretical Study of the OH-Initiated Photo-oxidation of Formamide, J. Phys. Chem. A, 120, 1222–1230, https://doi.org/10.1021/acs.jpca.6b00032, 2016.
Calvert, J. G.: Mechanisms of atmospheric oxidation of the alkanes, Oxford University Press, Oxford, New York, 2008.
Chandra, B. P. and Sinha, V.: Contribution of post-harvest agricultural paddy residue fires in the NW Indo-Gangetic Plain to ambient carcinogenic benzenoids, toxic isocyanic acid and carbon monoxide, Environ. Int., 88, 187–197, https://doi.org/10.1016/j.envint.2015.12.025, 2016.
Criegee, R.: Mechanism of Ozonolysis, Angew. Chem. Int. Ed. Engl., 14, 745–752, https://doi.org/10.1002/anie.197507451, 1975.
Crippa, M., Janssens-Maenhout, G., Dentener, F., Guizzardi, D., Sindelarova, K., Muntean, M., Van Dingenen, R., and Granier, C.: Forty years of improvements in European air quality: regional policy-industry interactions with global impacts, Atmos. Chem. Phys., 16, 3825–3841, https://doi.org/10.5194/acp-16-3825-2016, 2016.
DrozGeorget, T., Zyrianov, M., Reisler, H., and Chandler, D. W.: Correlated distributions in the photodissociation of HNCO to NH(X-3 Sigma(-),a(1)Delta)+CO(X-1 Sigma(+)) near the barrier on S-1, Chem. Phys. Lett., 276, 316–324, https://doi.org/10.1016/S0009-2614(97)00804-X, 1997.
Dunning, T. H.: Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 90, 1007–1023, https://doi.org/10.1063/1.456153, 1989.
Fenimore, C. P.: Formation of nitric oxide in premixed hydrocarbon flames, Proc. Combust. Inst., 13, 373–380, 1971.
Finlayson-Pitts, B. J. and Pitts, J. N.: Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications, Academic Press, San Diego, 1999.
Fischer, G., Geith, J., Klapotke, T. M., and Krumm, B.: Synthesis, properties and dimerization study of isocyanic acid, Z. Naturforschung Sect. B-J. Chem. Sci., 57, 19–24, 2002.
Forschungszentrum Jülich: JUWELS: Modular Tier-0/1 Supercomputer at Jülich Supercomputing Centre, J. Large-Scale Res. Facil., 5, A135, https://doi.org/10.17815/jlsrf-5-171, 2019.
Gardiner, W. C. (Ed.): Gas-phase combustion chemistry, Springer, New York, 2000.
Heeb, N. V., Zimmerli, Y., Czerwinski, J., Schmid, P., Zennegg, M., Haag, R., Seiler, C., Wichser, A., Ulrich, A., Honegger, P., Zeyer, K., Emmenegger, L., Mosimann, T., Kasper, M., and Mayer, A.: Reactive nitrogen compounds (RNCs) in exhaust of advanced PM-NOx abatement technologies for future diesel applications, Atmos. Environ., 45, 3203–3209, https://doi.org/10.1016/j.atmosenv.2011.02.013, 2011.
Hofzumahaus, A., Kraus, A., Kylling, A., and Zerefos, C. S.: Solar actinic radiation (280–420 nm) in the cloud-free troposphere between ground and 12 km altitude: Measurements and model results, J. Geophys. Res.-Atmos., 107, D18, https://doi.org/10.1029/2001jd900142, 2002.
Huber, K.-P. and Herzberg, G.: Molecular Spectra and Molecular Structure IV. Constants of diatomic molecules, Van Nostrand Reinhold, New York, 1979.
IUPAC Subcommittee on Atmospheric Chemical Kinetic Data Evaluation: Evaluated Kinetic Data, English, IUPAC, available at: http://iupac.pole-ether.fr/index.html (last access: 29 May 2020), 2017.
Jankowski, M. J., Olsen, R., Nielsen, C. J., Thomassen, Y., and Molander, P.: The applicability of proton transfer reaction-mass spectrometry (PTR-MS) for determination of isocyanic acid (ICA) in work room atmospheres, Environ. Sci.-Process. Impacts, 16, 2423–2431, https://doi.org/10.1039/c4em00363b, 2014.
Jathar, S. H., Heppding, C., Link, M. F., Farmer, D. K., Akherati, A., Kleeman, M. J., de Gouw, J. A., Veres, P. R., and Roberts, J. M.: Investigating diesel engines as an atmospheric source of isocyanic acid in urban areas, Atmos. Chem. Phys., 17, 8959–8970, https://doi.org/10.5194/acp-17-8959-2017, 2017.
Jöckel, P., Sander, R., Kerkweg, A., Tost, H., and Lelieveld, J.: Technical Note: The Modular Earth Submodel System (MESSy) – a new approach towards Earth System Modeling, Atmos. Chem. Phys., 5, 433–444, https://doi.org/10.5194/acp-5-433-2005, 2005.
Jöckel, P., Kerkweg, A., Pozzer, A., Sander, R., Tost, H., Riede, H., Baumgaertner, A., Gromov, S., and Kern, B.: Development cycle 2 of the Modular Earth Submodel System (MESSy2), Geosci. Model Dev., 3, 717–752, https://doi.org/10.5194/gmd-3-717-2010, 2010.
Johnston, H. S. and Heicklen, J.: Tunneling corrections for unsymmetrical Eckart potential energy barriers, J. Phys. Chem., 66, 532–533, https://doi.org/10.1021/j100809a040, 1962.
Kaiser, J. W., Heil, A., Andreae, M. O., Benedetti, A., Chubarova, N., Jones, L., Morcrette, J.-J., Razinger, M., Schultz, M. G., Suttie, M., and van der Werf, G. R.: Biomass burning emissions estimated with a global fire assimilation system based on observed fire radiative power, Biogeosciences, 9, 527–554, https://doi.org/10.5194/bg-9-527-2012, 2012.
Keller-Rudek, H., Moortgat, G. K., Sander, R., and Sörensen, R.: The MPI-Mainz UV/VIS Spectral Atlas of Gaseous Molecules of Atmospheric Interest, Earth Syst. Sci. Data, 5, 365–373, https://doi.org/10.5194/essd-5-365-2013, 2013.
Kerkweg, A., Buchholz, J., Ganzeveld, L., Pozzer, A., Tost, H., and Jöckel, P.: Technical Note: An implementation of the dry removal processes DRY DEPosition and SEDImentation in the Modular Earth Submodel System (MESSy), Atmos. Chem. Phys., 6, 4617–632, https://doi.org/10.5194/acp-6-4617-2006, 2006a.
Kerkweg, A., Sander, R., Tost, H., and Jöckel, P.: Technical note: Implementation of prescribed (OFFLEM), calculated (ONLEM), and pseudo-emissions (TNUDGE) of chemical species in the Modular Earth Submodel System (MESSy), Atmos. Chem. Phys., 6, 3603–3609, https://doi.org/10.5194/acp-6-3603-2006, 2006b.
Koss, A. R., Sekimoto, K., Gilman, J. B., Selimovic, V., Coggon, M. M., Zarzana, K. J., Yuan, B., Lerner, B. M., Brown, S. S., Jimenez, J. L., Krechmer, J., Roberts, J. M., Warneke, C., Yokelson, R. J., and de Gouw, J.: Non-methane organic gas emissions from biomass burning: identification, quantification, and emission factors from PTR-ToF during the FIREX 2016 laboratory experiment, Atmos. Chem. Phys., 18, 3299–3319, https://doi.org/10.5194/acp-18-3299-2018, 2018.
Kumar, V., Chandra, B. P., and Sinha, V.: Large unexplained suite of chemically reactive compounds present in ambient air due to biomass fires, Sci. Rep.-UK, 8, 626, https://doi.org/10.1038/s41598-017-19139-3, 2018.
Lee, C., Yang, W., and Parr, R. G.: Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B, 37, 785–789, https://doi.org/10.1103/PhysRevB.37.785, 1988.
Le Picard, S. D., Tizniti, M., Canosa, A., Sims, I. R., and Smith, I. W. M.: The Thermodynamics of the Elusive HO3 Radical, Science, 328, 1258–1262, https://doi.org/10.1126/science.1184459, 2010.
Leslie, M. D., Ridoli, M., Murphy, J. G., and Borduas-Dedekind, N.: Isocyanic acid (HNCO) and its fate in the atmosphere: a review, Environ. Sci.-Process. Impacts, 21, 793–808, https://doi.org/10.1039/c9em00003h, 2019.
Liebig, J. and Wöhler, F.: Untersuchungen über die Cyansäure, Ann. Phys., 96, 369–400, https://doi.org/10.1002/andp.18300961102, 1830.
Liggio, J., Stroud, C. A., Wentzell, J. J. B., Zhang, J. H., Sommers, J., Darlington, A., Liu, P. S. K., Moussa, S. G., Leithead, A., Hayden, K., Mittermeier, R. L., Staebler, R., Wolde, M., and Li, S. M.: Quantifying the Primary Emissions and Photochemical Formation of Isocyanic Acid Downwind of Oil Sands Operations, Environ. Sci. Technol., 51, 14462–14471, https://doi.org/10.1021/acs.est.7b04346, 2017.
Luo, Y.-R.: Comprehensive Handbook of Chemical Bond Energies, 1st edn., CRC Press, Boca Raton, 2007.
Manion, J. A., Huie, R. E., Levin, R. D., Burgess Jr., D. R., Orkin, V. L., Tsang, W., McGivern, W. S., Hudgens, J. W., Knyazev, V. D., Atkinson, D. B., Chai, E., Tereza, A. M., Lin, C.-Y., Allison, T. C., Mallard, W. G., Westley, F., Herron, J. T., Hampson, R. F., and Frizzell, D. H.: NIST Chemical Kinetics Database – Standard Reference Database 17, Version 7.0 (Web Version), Release 1.6.8, Data Version 2017.07, available at: http://kinetics.nist.gov/kinetics/, last access: 29 May 2020.
Marcelino, N., Agundez, M., Cernicharo, J., Roueff, E., and Tafalla, M.: Discovery of the elusive radical NCO and confirmation of H2NCO+ in space, Astron. Astrophys., 612, L10, https://doi.org/10.1051/0004-6361/201833074, 2018.
Martin, J. M. L.: Ab initio total atomization energies of small molecules – towards the basis set limit, Chem. Phys. Lett., 259, 669–678, https://doi.org/10.1016/0009-2614(96)00898-6, 1996.
Mertens, J. D., Chang, A. Y., Hanson, R. K., and Bowman, C. T.: A Shock-Tube Study of Reactions of Atomic Oxygen with Isocyanic Acid, Int. J. Chem. Kinet., 24, 279–295, https://doi.org/10.1002/kin.550240306, 1992.
Nielsen, C. J., Herrmann, H., and Weller, C.: Atmospheric chemistry and environmental impact of the use of amines in carbon capture and storage (CCS), Chem. Soc. Rev., 41, 6684–6704, https://doi.org/10.1039/c2cs35059a, 2012.
Okabe, H.: Photodissociation of HNCO in Vacuum Ultraviolet – Production of NCO A2Σ and NH(A3π, πc1), J. Chem. Phys., 53, 3507–3515, https://doi.org/10.1063/1.1674525, 1970.
Papajak, E. and Truhlar, D. G.: What are the most efficient basis set strategies for correlated wave function calculations of reaction energies and barrier heights?, J. Chem. Phys., 137, 064110, https://doi.org/10.1063/1.4738980, 2012.
Parandaman, A., Tangtartharakul, C. B., Kumar, M., Francisco, J. S., and Sinha, A.: A Computational Study Investigating the Energetics and Kinetics of the HNCO + (CH3)2NH Reaction Catalyzed by a Single Water Molecule, J. Phys. Chem. A, 121, 8465–8473, https://doi.org/10.1021/acs.jpca.7b08657, 2017.
Pumphrey, H. C., Glatthor, N., Bernath, P. F., Boone, C. D., Hannigan, J. W., Ortega, I., Livesey, N. J., and Read, W. G.: MLS measurements of stratospheric hydrogen cyanide during the 2015–2016 El Niño event, Atmos. Chem. Phys., 18, 691–703, https://doi.org/10.5194/acp-18-691-2018, 2018.
Purvis, G. D. and Bartlett, R. J.: A full coupled-cluster singles and doubles model: The inclusion of disconnected triples, J. Chem. Phys., 76, 1910, https://doi.org/10.1063/1.443164, 1982.
Roberts, J. M. and Liu, Y.: Solubility and solution-phase chemistry of isocyanic acid, methyl isocyanate, and cyanogen halides, Atmos. Chem. Phys., 19, 4419–4437, https://doi.org/10.5194/acp-19-4419-2019, 2019.
Roberts, J. M., Veres, P., Warneke, C., Neuman, J. A., Washenfelder, R. A., Brown, S. S., Baasandorj, M., Burkholder, J. B., Burling, I. R., Johnson, T. J., Yokelson, R. J., and de Gouw, J.: Measurement of HONO, HNCO, and other inorganic acids by negative-ion proton-transfer chemical-ionization mass spectrometry (NI-PT-CIMS): application to biomass burning emissions, Atmos. Meas. Tech., 3, 981–990, https://doi.org/10.5194/amt-3-981-2010, 2010.
Roberts, J. M., Veres, P. R., Cochran, A. K., Warneke, C., Burling, I. R., Yokelson, R. J., Lerner, B., Gilman, J. B., Kuster, W. C., Fall, R., and de Gouw, J.: Isocyanic acid in the atmosphere and its possible link to smoke-related health effects, P. Natl. Acad. Sci. USA, 108, 8966–8971, https://doi.org/10.1073/pnas.1103352108, 2011.
Roberts, J. M., Veres, P. R., VandenBoer, T. C., Warneke, C., Graus, M., Williams, E. J., Lefer, B., Brock, C. A., Bahreini, R., Ozturk, F., Middlebrook, A. M., Wagner, N. L., Dube, W. P., and de Gouw, J. A.: New insights into atmospheric sources and sinks of isocyanic acid, HNCO, from recent urban and regional observations, J. Geophys. Res.-Atmos., 119, 1060–1072, https://doi.org/10.1002/2013JD019931, 2014.
Roeckner, E., Brokopf, R., Esch, M., Giorgetta, M., Hagemann, S., Kornblueh, L., Manzini, E., Schlese, U., and Schulzweida, U.: Sensitivity of simulated climate to horizontal and vertical resolution in the ECHAM5 atmosphere model, J. Climate, 19, 3771–3791, https://doi.org/10.1175/JCLI3824.1, 2006.
Ruscic, B.: Uncertainty quantification in thermochemistry, benchmarking electronic structure computations, and Active Thermochemical Tables, Int. J. Quantum Chem., 114, 1097–1101, https://doi.org/10.1002/qua.24605, 2014.
Ruscic, B. and Bross, D. H.: Active Thermochemical Tables (ATcT) values based on ver. 1.122g of the Thermochemical Network (2019), available at: http://atct.anl.gov/ (last access: 29 May 2020), Argonne Natl. Lab. Act. Thermochem, 2019.
Ruscic, B., Wagner, A. F., Harding, L. B., Asher, R. L., Feller, D., Dixon, D. A., Peterson, K. A., Song, Y., Qian, X. M., Ng, C. Y., Liu, J. B., and Chen, W. W.: On the enthalpy of formation of hydroxyl radical and gas-phase bond dissociation energies of water and hydroxyl, J. Phys. Chem. A, 106, 2727–2747, https://doi.org/10.1021/jp013909s, 2002.
Sander, R., Baumgaertner, A., Gromov, S., Harder, H., Jöckel, P., Kerkweg, A., Kubistin, D., Regelin, E., Riede, H., Sandu, A., Taraborrelli, D., Tost, H., and Xie, Z.-Q.: The atmospheric chemistry box model CAABA/MECCA-3.0, Geosci. Model Dev., 4, 373–380, https://doi.org/10.5194/gmd-4-373-2011, 2011.
Schacke, H., Schmatjko, K. J., and Wolfrum, J.: Reaktionen von CN-Radikalen im H-C-N-O-System, Arch. Proces. Spalania, 5, 363, 1974.
Sengupta, D. and Nguyen, M. T.: Mechanism of NH2+CO2 formation in OH + HNCO reaction: Rate constant evaluation via ab initio calculations and statistical theory, J. Chem. Phys., 106, 9703–9707, https://doi.org/10.1063/1.474090, 1997.
Sharpe, S. W., Johnson, T. J., Sams, R. L., Chu, P. M., Rhoderick, G. C., and Johnson, P. A.: Gas-phase databases for quantitative infrared spectroscopy, Appl. Spectrosc., 58, 1452–1461, https://doi.org/10.1366/0003702042641281, 2004.
Spiglanin, T. A. and Chandler, D. W.: Rotational State Distributions of NH(a1Δ) from HNCO Photodissociation, J. Chem. Phys., 87, 1577–1581, https://doi.org/10.1063/1.453216, 1987.
Spiglanin, T. A., Perry, R. A., and Chandler, D. W.: Internal State Distributions of CO from HNCO Photodissociation, J. Chem. Phys., 87, 1568–1576, https://doi.org/10.1063/1.453215, 1987.
Stanton, J. F.: On the vibronic level structure in the NO3 radical. I. The ground electronic state, J. Chem. Phys., 126, 134309, https://doi.org/10.1063/1.2715547, 2007.
Stanton, J. F.: On the vibronic level structure in the NO3 radical: II. Adiabatic calculation of the infrared spectrum, Mol. Phys., 107, 1059–1075, https://doi.org/10.1080/00268970902740530, 2009.
Stanton, J. F. and Okumura, M.: On the vibronic level structure in the NO3 radical?: Part III. Observation of intensity borrowing via ground state mixing, Phys. Chem. Chem. Phys., 11, 4742, https://doi.org/10.1039/b902252j, 2009.
Stone, D., Whalley, L. K., and Heard, D. E.: Tropospheric OH and HO2 radicals: field measurements and model comparisons, Chem. Soc. Rev., 41, 6348–6404, https://doi.org/10.1039/c2cs35140d, 2012.
Suarez-Bertoa, R. and Astorga, C.: Isocyanic acid and ammonia in vehicle emissions, Transp. Res. Part-Transp. Environ., 49, 259–270, https://doi.org/10.1016/j.trd.2016.08.039, 2016.
SUVA: Grenzwerte am Arbeitsplatz 2016, available at: https://www.suva.ch/1903.d (last access: 29 May 2020), 2016.
SWEA: Occupational exposure limit values. Provision (AFS) 2011:18, Swedish Work Environment Authority, Stockholm, Sweden, ISBN 978-91-7930-559-8, 2011.
Taylor, W. D., Allston, T. D., Moscato, M. J., Fazekas, G. B., Kozlowski, R., and Takacs, G. A.: Atmospheric Photo-Dissociation Lifetimes for Nitromethane, Methyl Nitrite, and Methyl Nitrate, Int. J. Chem. Kinet., 12, 231–240, https://doi.org/10.1002/kin.550120404, 1980.
Tost, H., Jöckel, P., Kerkweg, A., Sander, R., and Lelieveld, J.: Technical note: A new comprehensive SCAVenging submodel for global atmospheric chemistry modelling, Atmos. Chem. Phys., 6, 565–574, https://doi.org/10.5194/acp-6-565-2006, 2006.
Truhlar, D. G., Garrett, B. C., and Klippenstein, S. J.: Current Status of Transition-State Theory, J. Phys. Chem., 100, 12771–12800, https://doi.org/10.1021/jp953748q, 1996.
Tsang, W.: Chemical Kinetic Data Base for Propellant Combustion. II. Reactions Involving CN, NCO, and HNCO, J. Phys. Chem. Ref. Data, 21, 753, https://doi.org/10.1063/1.555914, 1992.
Tully, F. P., Perry, R. A., Thorne, L. R., and Allendorf, M. D.: Free-radical oxidation of isocyanic acid, Symp. Int. Combust., 22, 1101–1106, https://doi.org/10.1016/S0082-0784(89)80120-1, 1989.
Uno, K., Hikida, T., Hiraya, A., and Shobatake, K.: Formation of NH(C1Π), NH(A3Π) and NCO(A2Σ) in the VUV Photolysis of HNCO, Chem. Phys. Lett., 166, 475–479, https://doi.org/10.1016/0009-2614(90)87136-F, 1990.
Varandas, A. J. C.: Odd-Hydrogen: An Account on Electronic Structure, Kinetics, and Role of Water in Mediating Reactions with Atmospheric Ozone. Just a Catalyst or Far Beyond?, Int. J. Quantum Chem., 114, 1327–1349, https://doi.org/10.1002/qua.24580, 2014.
Vatsa, R. K. and Volpp, H. R.: Absorption cross-sections for some atmospherically important molecules at the H atom Lyman-alpha wavelength (121.567 nm), Chem. Phys. Lett., 340, 289–295, https://doi.org/10.1016/S0009-2614(01)00373-6, 2001.
Vereecken, L. and Francisco, J. S.: Theoretical studies of atmospheric reaction mechanisms in the troposphere, Chem. Soc. Rev., 41, 6259–6293, https://doi.org/10.1039/c2cs35070j, 2012.
Vereecken, L., Glowacki, D. R., and Pilling, M. J.: Theoretical Chemical Kinetics in Tropospheric Chemistry: Methodologies and Applications, Chem. Rev., 115, 4063–4114, https://doi.org/10.1021/cr500488p, 2015.
Wang, Z., Nicholls, S. J., Rodriguez, E. R., Kummu, O., Horkko, S., Barnard, J., Reynolds, W. F., Topol, E. J., DiDonato, J. A., and Hazen, S. L.: Protein carbamylation links inflammation, smoking, uremia and atherogenesis, Nat. Med., 13, 1176–1184, https://doi.org/10.1038/nm1637, 2007.
Wentzell, J. J. B., Liggio, J., Li, S.-M., Vlasenko, A., Staebler, R., Lu, G., Poitras, M.-J., Chan, T., and Brook, J. R.: Measurements of Gas phase Acids in Diesel Exhaust: A Relevant Source of HNCO?, Environ. Sci. Technol., 47, 7663–7671, https://doi.org/10.1021/es401127j, 2013.
Wooldridge, M. S., Hanson, R. K., and Bowman, C. T.: A shock tube study of and products via simultaneous laser adsorption measurements of OH and CO2, Int. J. Chem. Kinet., 28, 361–372, https://doi.org/10.1002/(SICI)1097-4601(1996)28:5<361::AID-KIN5>3.0.CO;2-T, 1996.
Wren, S. N., Liggio, J., Han, Y., Hayden, K., Lu, G., Mihele, C. M., Mittermeier, R. L., Stroud, C., Wentzell, J. J. B., and Brook, J. R.: Elucidating real-world vehicle emission factors from mobile measurements over a large metropolitan region: a focus on isocyanic acid, hydrogen cyanide, and black carbon, Atmos. Chem. Phys., 18, 16979–17001, https://doi.org/10.5194/acp-18-16979-2018, 2018.
Young, P. J., Emmons, L. K., Roberts, J. M., Lamarque, J.-F., Wiedinmyer, C., Veres, P., and VandenBoer, T. C.: Isocyanic acid in a global chemistry transport model: Tropospheric distribution, budget, and identification of regions with potential health impacts, J. Geophys. Res.-Atmos., 117, D10308, https://doi.org/10.1029/2011JD017393, 2012.
Zabardasti, A. and Solimannejad, M.: Theoretical study and AIM analysis of hydrogen bonded clusters of water and isocyanic acid, J. Mol. Struct.-Theochem, 819, 52–59, https://doi.org/10.1016/j.theochem.2007.05.032, 2007.
Zabardasti, A., Amani, S., Solimannejad, M., and Salehnassaj, M.: Theoretical study and atoms in molecule analysis of hydrogen bonded clusters of ammonia and isocyanic acid, Struct. Chem., 20, 1087–1092, https://doi.org/10.1007/s11224-009-9513-1, 2009.
Zabardasti, A., Kakanejadifard, A., Kikhaei, M., and Solimannejad, M.: Theoretical studies and topological analysis of the electron density of clusters of O3 with HNCO and HCNO, J. Mol. Struct.-Theochem., 961, 1–5, https://doi.org/10.1016/j.theochem.2010.08.015, 2010.
Zhao, Y. and Truhlar, D. G.: The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 120, 215–241, https://doi.org/10.1007/s00214-007-0310-x, 2008.