the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 15 Jan 2020
| 15 Jan 2020
Multi-generation OH oxidation as a source for highly oxygenated organic molecules from aromatics
Matti P. Rissanen
Iida Pullinen
Sebastian Schmitt
Oskari Kausiala
Ralf Tillmann
Defeng Zhao
Carl Percival
Thomas J. Bannan
Michael Priestley
Åsa M. Hallquist
Einhard Kleist
Astrid Kiendler-Scharr
Mattias Hallquist
Torsten Berndt
Gordon McFiggans
Jürgen Wildt
Thomas F. Mentel
Recent studies have recognised highly oxygenated organic molecules (HOMs) in the atmosphere as important in the formation of secondary organic aerosol (SOA). A large number of studies have focused on HOM formation from oxidation of biogenically emitted monoterpenes. However, HOM formation from anthropogenic vapours has so far received much less attention. Previous studies have identified the importance of aromatic volatile organic compounds (VOCs) for SOA formation. In this study, we investigated several aromatic compounds, benzene (C6H6), toluene (C7H8), and naphthalene (C10H8), for their potential to form HOMs upon reaction with hydroxyl radicals (OH). We performed flow tube experiments with all three VOCs and focused in detail on benzene HOM formation in the Jülich Plant Atmosphere Chamber (JPAC). In JPAC, we also investigated the response of HOMs to NOx and seed aerosol. Using a nitrate-based chemical ionisation mass spectrometer (CI-APi-TOF), we observed the formation of HOMs in the flow reactor oxidation of benzene from the first OH attack. However, in the oxidation of toluene and naphthalene, which were injected at lower concentrations, multi-generation OH oxidation seemed to impact the HOM composition. We tested this in more detail for the benzene system in the JPAC, which allowed for studying longer residence times. The results showed that the apparent molar benzene HOM yield under our experimental conditions varied from 4.1 % to 14.0 %, with a strong dependence on the OH concentration, indicating that the majority of observed HOMs formed through multiple OH-oxidation steps. The composition of the identified HOMs in the mass spectrum also supported this hypothesis. By injecting only phenol into the chamber, we found that phenol oxidation cannot be solely responsible for the observed HOMs in benzene experiments. When NOx was added to the chamber, HOM composition changed and many oxygenated nitrogen-containing products were observed in CI-APi-TOF. Upon seed aerosol injection, the HOM loss rate was higher than predicted by irreversible condensation, suggesting that some undetected oxygenated intermediates also condensed onto seed aerosol, which is in line with the hypothesis that some of the HOMs were formed in multi-generation OH oxidation. Based on our results, we conclude that HOM yield and composition in aromatic systems strongly depend on OH and VOC concentration and more studies are needed to fully understand this effect on the formation of HOMs and, consequently, SOA. We also suggest that the dependence of HOM yield on chamber conditions may explain part of the variability in SOA yields reported in the literature and strongly advise monitoring HOMs in future SOA studies.
- Article
(3666 KB) - Full-text XML
-
Supplement
(92 KB) - BibTeX
- EndNote





Highly oxygenated organic molecules (HOMs) have been identified as large contributors to atmospheric secondary organic aerosol (SOA) in forested environments (Ehn et al., 2014; Öström et al., 2017; Bianchi et al., 2019). HOMs form through a process called autoxidation, where intramolecular hydrogen shifts in organic peroxy radicals are followed by addition of molecular oxygen (Crounse et al., 2013), causing a rapid increase in the oxygen content of the molecules. The product is a new peroxy radical, with an additional hydroperoxide functionality, that may be able to experience additional H shifts. A wealth of studies have shown that this process is especially efficient in the oxidation of molecules with endocyclic double bonds (e.g. Rissanen et al., 2014; Jokinen et al., 2014; Mentel et al., 2015; Berndt et al., 2015; Rissanen et al., 2015; Berndt et al., 2016), a feature typical of biogenically emitted volatile organic compounds (VOCs) such as monoterpenes.
While the formation pathways of HOMs from biogenic VOCs as well as their impact on atmospheric aerosol formation has been studied extensively over the past years, the potential of anthropogenic VOCs to form HOMs has received much less attention. Wang et al. (2017) showed both computationally and experimentally that the yield of HOMs from the hydroxyl radical (OH) initiated oxidation of alkyl benzenes increased with the size of the alkyl group. A second study investigated HOM formation from the OH oxidation of seven different aromatics, finding HOM yields mainly within 0.1 %–1 % for single-ring aromatics, and a few percent for two polycyclic aromatics, naphthalene and biphenyl (Molteni et al., 2018). These yields are comparable in magnitude with those reported from ozone and OH oxidation of monoterpenes (e.g. Jokinen et al., 2015; Berndt et al., 2016). As aromatics are thought to be the most efficient precursors of SOA in urban areas (Kroll and Seinfeld, 2008), further studies of HOM formation, as well as their contribution to SOA, are necessary.
The most abundant aromatics in the atmosphere are benzene and alkylated benzenes, i.e. toluene, xylenes, and trimethylbenzenes. Their primary sources are traffic, fuel handling, and industrial processes. Aromatic compounds can constitute up to 20 % of urban VOCs (Calvert et al., 2002) and in extremely polluted environments, such as next to a road with heavy traffic, their total concentrations can reach up to tens of parts per billion (Liu et al., 2008). In addition, vegetation also emits a wide range of aromatic compounds, often in oxygenated form, and the total amount of the potential emissions may even match the anthropogenic sources (Misztal et al., 2015).
The major sink of aromatics in the atmosphere is the reaction with OH (Atkinson and Arey, 2003), which in most cases involves OH addition to the aromatic ring and the formation of a carbon-centred radical. In the case of benzene, more than half of these radicals will end up forming phenol (Volkamer et al., 2002; Berndt and Böge, 2006). The remainder of the products can undergo O2 additions and isomerisation, forming bicyclic peroxy radicals (BPRs), or result in epoxides (Bloss et al., 2005; Glowacki et al., 2009; Wang et al., 2013). As suggested by Molteni et al. (2018), the BPR may undergo further autoxidation to form HOMs. However, the produced phenol will be abundant, which upon reaction with OH can also produce a BPR with low yield, about 10 % (Master Chemical Mechanism, MCMv3.3.1; Bloss et al., 2005). The reaction rate coefficient of phenol with OH is about 20 times higher than that of benzene, meaning that we cannot ignore its role in the total HOM formation following further oxidation steps. For instance, Schwantes et al. (2017) showed that methylphenol (cresol) formed in toluene oxidation was a much more important SOA precursor than its branching ratio (20 %) would suggest. In their study, only a minor fraction of the identified compounds would classify as HOMs, according to the definition suggested by Bianchi et al. (2019) that six or more O atoms are required for a molecule to be classified as HOMs. However, the authors demonstrated the importance of multiple OH oxidation steps for SOA formation.
Several studies over the last decades examined the SOA yields from oxidation of aromatics, with disparate results that remain largely unexplained. The suggested causes are the differences in the exact experimental conditions (Ng et al., 2007; Hildebrandt et al., 2009; Emanuelsson et al., 2013). These include differences in VOC loading, UV light intensity, and the concentration of NOx (NO+NO2). Being a by-product of combustion, NOx is on a large scale co-emitted with aromatic VOCs. NOx, and especially NO, will decrease the lifetime of RO2 radicals in the atmosphere, in direct competition with autoxidation (Praske et al., 2018; Rissanen, 2018). Additionally, highly oxygenated RO2 radicals can combine efficiently to form ROOR' dimers (Berndt et al., 2018a, b). These dimers are often the least volatile oxidation products, with a particularly large influence on the formation of new particles (e.g. Tröstl et al., 2016; Lehtipalo et al., 2018), but under high-NOx conditions their formation becomes suppressed (e.g. Ehn et al., 2014; Rissanen, 2018).
The measurement of HOMs relies mainly on the use of the chemical ionisation atmospheric pressure interface time-of-flight mass spectrometer (CI-APi-TOF; Jokinen et al., 2012). In combination with a wall-less CI inlet, nitrate ion ionisation is typically used due to its selectivity towards molecules with several H-bond donors, such as the multi-hydroperoxides typically formed in autoxidation (Hyttinen et al., 2015). Until now, the application of the CI-APi-TOF to measuring HOMs from aromatics has been limited to a few studies (Wang et al., 2017; Molteni et al., 2018), and these have been performed in flow reactors with residence times of 20 s or less. To understand the importance of aromatic-derived HOMs in the atmosphere, systematic studies, including experiments at varying conditions and longer timescales, are needed.
In this study, we investigated the OH-initiated oxidation of aromatics, with a strong focus on benzene. We conducted experiments in a flow reactor and a continuously stirred tank reactor (Jülich Plant Atmosphere Chamber, JPAC) in order to determine HOM composition and yield over a wide range of conditions. In the JPAC runs, we varied both VOC and OH concentrations and tested the influence of NOx on the HOM distribution. Benzene was also substituted by phenol in order to test different oxidation pathways. Finally, we explored the contribution of HOMs to SOA formation by adding seed aerosol.
In this section, we outline the relevant oxidation steps of aromatic compounds with a focus on benzene. In oxidation reactions initiated by OH, the oxidation propagation and termination will determine the chemical composition of the product molecules. These reactions will change the amount of hydrogen (H), carbon (C), oxygen (O), and nitrogen (N) atoms in the detected oxidised species and are therefore central to our discussion. In this section, we do not attempt to review all of the existing studies. Instead, we present an overview of relevant products and radicals formed in benzene oxidation by OH. We also discuss the relevant chain propagating and terminating reactions of organic peroxy radicals (RO2) as the main intermediates of gas-phase oxidation. Detailed mechanistic descriptions of benzene oxidation can be found in the literature (Calvert et al., 2002; Volkamer et al., 2002; Bloss et al., 2005; Glowacki et al., 2009; Wang et al., 2013; Vereecken, 2019, and references therein).
2.1 Oxidation by OH
Benzene (C6H6) oxidation by OH almost exclusively initiates via addition of OH to the aromatic ring (Glowacki and Pilling, 2010; Bloss et al., 2005), while abstraction of H atoms from the ring is a minor pathway. The addition of OH creates a carbon-centred radical C6H7O•. According to previous studies, about 53 %–61 % of these radicals will form phenol, where the aromatic ring is retained and the C6H6O molecule has one OH group (one more O atom) (Volkamer et al., 2002; Berndt and Böge, 2006). The remaining fraction of C6H7O• will add molecular oxygen (O2), forming a peroxy radical (Lay et al., 1996). This RO2 can undergo endo-cyclization, where RO2 attacks its own double bond to form an oxygen bridge, resulting in an alkyl radical. This radical then reacts again with O2 and forms a BPR (Glowacki et al., 2009; Birdsall et al., 2010; Wang et al., 2013). In this pathway OH attachment and addition of two O2 molecules increases the molecular composition of parent benzene by five O atoms (and one H), and subsequent reactions generally lead to radical termination and potential molecular fragmentation (Jenkin et al., 2003; Wang et al., 2013). Studies have also reported a minor channel in which the bicyclic alkyl radical isomerises and forms an epoxide functionality, though the importance of this pathway under atmospheric conditions is yet unclear (Bloss et al., 2005; Glowacki et al., 2009; Wang et al., 2013).
For substituted aromatics, the set of reactions is similar to that described above, though branching ratios are different (Birdsall and Elrod, 2011). For instance, in toluene oxidation by OH, the BPR forms with about 65 % yield, which is about twice that formed in the case of benzene (MCMv3.3.1, Bloss et al., 2005). In addition, the presence of methyl groups increases the chances of H abstraction by OH radicals and also increases the OH-VOC reaction rate coefficient (kOH) (Bloss et al., 2005; Atkinson, 1994a, b; Atkinson and Aschmann, 1989).
A distinct feature of aromatic oxidation is the faster oxidation rates of first-generation products as compared to the parent molecule. For instance, benzene has a kOH of 1.22×10−12 cm3 s−1 at 298 K, while kOH of phenol is about 20 times larger (2.82×10−11 cm3 s−1), and kOH of catechol (a primary product of phenol oxidation) is about 100 times higher (1.0×10−10 cm3 s−1) (Atkinson et al., 2006). In the case of less thoroughly investigated oxidation products, kOH is likely to increase in comparison to benzene itself, as the pi-electron structure of benzene makes it less susceptible towards OH oxidation compared to most organic molecules. The process of sequential oxidation is commonly known as ageing and in general should lead to eventual fragmentation of the products retained in the gas phase (Chacon-Madrid and Donahue, 2011).
2.2 RO2 radical reactions
2.2.1 Chain propagation
Chain propagation refers to the reactions that result in another radical (i.e. a molecule that still has an unpaired electron). These reactions can be bimolecular, happening upon collision with another molecule, or unimolecular, occurring within the molecule. The reaction rates depend on the structure of the compound as well as the concentration of potential bimolecular reaction partners.
A bimolecular propagation reaction proceeds through formation of alkoxy radicals (RO) when an RO2 radical reacts with another R′O2 (forming ) or NO (forming RO+NO2). This reaction decreases the oxygen content per molecule by one and is one of the most common reactions for peroxy radicals (Finlayson-Pitts and Pitts, 2000). The fate of the RO radicals depends on their structure. They can decompose, undergo H shifts, or react with O2. In the case of benzene, decomposition of alkoxy radicals may lead to ring scission and potentially further autoxidation. However, in the case of a first-generation BPR from benzene, the MCMv3.3.1 predicts that the BPR will react with HO2 or RO2, forming RO radicals with branching ratios of 23 % and 60 %, respectively, which eventually decompose into smaller molecules (Jenkin et al., 2003; Bloss et al., 2005).
Autoxidation of RO2 radicals is one important reaction chain recently shown in the oxidation of monoterpenes and other alkenes (Crounse et al., 2013; Ehn et al., 2014). It involves intramolecular hydrogen shifts to the peroxide group from other carbon atoms and subsequent addition of oxygen to the produced carbon-centred radicals. While autoxidation involves both uni- and bimolecular reactions, the high abundance of oxygen in the air allows autoxidation to be pseudo-unimolecular (Rissanen et al., 2015). Autoxidation is more likely to happen at lower RO2 concentrations and for RO2 with a larger amount of functional groups (Crounse et al., 2013). It may also occur in aromatic molecules following the initial bicyclic peroxy radical, i.e. in the case of benzene (Wang et al., 2017). The H shift itself does not modify the molecular composition, but O2 additions increase the oxygen content by an even number. Autoxidation can proceed until the H shift potential is exhausted and, at least in monoterpene systems, can often be competitive with bimolecular termination reactions under atmospheric conditions (see next section). However, autoxidation for aromatic compounds is not yet well understood, and until recently, the bicyclic peroxy radical was considered the most oxygenated first-generation product.
Other propagation reactions mostly include fragmentation. During autoxidation, H atoms may be abstracted from a terminal carbonyl group, creating an acyl radical (RC•=O), which may eliminate a CO from the molecule and leave an alkyl radical to which further O2 can attach itself. In this reaction, one C atom and one O atom are lost (Crounse et al., 2012; Rissanen et al., 2014). If CO is not eliminated, O2 will add and, upon a reaction with another RO2 (or NO), an RO radical will split CO2, losing one C and two O atoms instead (Orlando et al., 2003; Atkinson and Arey, 2003; Vereecken and Peeters, 2009).
2.2.2 Chain termination
Termination reactions proceed in competition with the chain propagation reactions described above. Termination reactions result in “closed-shell” molecules containing only paired electrons. An example of a unimolecular termination process is the ejection of OH following an H abstraction from a carbon with a hydroperoxide group, forming a carbonyl (C=O), meaning a loss of one H and one O atom (Rissanen et al., 2014).
A number of bimolecular termination reactions can take place. First, RO2 can react with HO2 and form ROOH hydroperoxides, which adds one H atom (as O2 is ejected). Alternatively, RO2 can react with another R′O2 and form a dimer ROOR' and O2, where the number of C and H atoms of RO2 and R′O2 in sum are conserved, while two O atoms are lost. RO2 can also upon collision with R′O2 form an alcohol (ROH) or an aldehyde (RCHO) and O2 in which the molecule will have lost an oxygen and either gained or lost a hydrogen, compared to the initial RO2 (Finlayson-Pitts and Pitts, 2000). In addition, RO radicals, mentioned in the previous section, may terminate upon reaction with O2, forming a carbonyl compound with one less H atom.
In the atmosphere, NO and NO2 can be effective in terminating RO2, although the major reaction between RO2 and NO is chain-propagating to form NO2 and RO. NO can add to RO2 to form organonitrates while NO2 upon reaction with RO2 can form thermally unstable peroxynitrates (RO2NO2) or more stable peroxyacylnitrates RO2(O)NO2 (Zabel, 1995; Atkinson, 2000; Orlando and Tyndall, 2012; Rissanen, 2018). In the case of aromatics, RO can also be long-lived enough to react with NO2 to form nitrophenol-type compounds (Olariu et al., 2002; Jenkin et al., 2003; Bloss et al., 2005). NO and NO2 addition to the molecule consequently changes its composition and is easy to identify based on the added N atom. However, distinguishing between nitrogen-containing HOMs with nitro-, nitrate-, or peroxynitrate functionalities is impossible with our instrumentation, and can only be speculated based on experimental conditions like the NO∕NO2 ratio.
3.1 Experimental set-up
The gas-phase oxidation experiments were conducted in two different laboratory settings. Initial experiments were performed in a flow reactor at the University of Helsinki, focusing on determination of HOM distributions during the oxidation of benzene, toluene, and naphthalene. The flow reactor allowed fast oxidation experiments at high VOC concentrations providing a possibility for comparison with previous studies. Motivated by the findings in the flow reactor, we performed subsequent quantitative studies on HOM formation from benzene oxidation at the Jülich Plant Atmosphere Chamber facility at Research Centre Jülich (Mentel et al., 2009). Using JPAC allowed us to do experiments at longer timescales and under more varied experimental conditions. In the following sub-sections, the two facilities are described in more detail, as are the types of experiments conducted in each of them.
3.1.1 University of Helsinki flow reactor
The flow reactor utilised in this work was made of quartz and had a volume of 2 L. At the inlet of the flow reactor, reactant gases were mixed in a Swagelok steel cross. We operated the system at room temperature (∼22 ∘C) with high VOC concentrations. Synthetic air (79 % N2 ∕ 21 % O2; AGA purity 5.0, 99.999 %) was used as the main carrier gas, while a VOC and water were added via separate lines by bubbling nitrogen (cryogenic N2, AGA) through vials containing the respective liquids (see Fig. 1 for a schematic depiction of the set-up). In the case of the solid naphthalene, nitrogen flow was passed over granules of the compound. An ultraviolet (UV) lamp was attached on the top of the reactor, irradiating a small part of it through a light shield. Hydroxyl radicals were produced via photolysis of water at 184.9 nm, a reaction that also caused the co-production of HO2. The total flow through the flow reactor was 12 standard litres per minute (slpm), leading to a 10 s residence time inside the reactor.
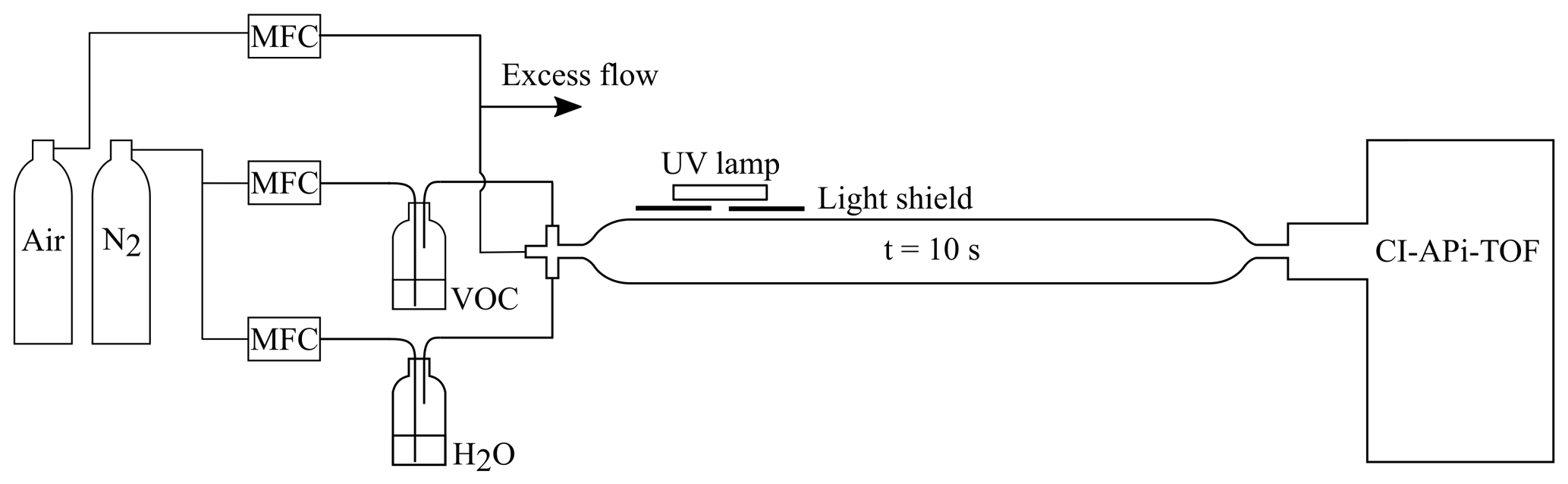
Figure 1Flow reactor set-up used at the University of Helsinki. The aromatic VOC and water vapour were mixed with synthetic air at the inlet of the flow reactor. A shielded UV lamp irradiated a small part of the reactor, forming OH radicals by photolysing the water vapour. The inlet flow to the CI-APi-TOF mass spectrometer (see Sect. 3.2.1) defined the residence time (∼10 s) in the reactor.
In the flow reactor experiments, uncertainties in the VOC and OH concentrations were large enough that no quantitative analysis was attempted, but instead we focused on the chemical composition of the HOM products. We could only roughly approximate the VOC concentrations in the flow reactor. Assuming the flow over the VOC in the vial (0.01–0.05 slpm) was saturated, we got the following estimated concentrations in the flow reactor for different experiments: benzene ∼400 ppm, toluene ∼25 ppm, and naphthalene ∼0.4 ppm. The photolysis rate could not be determined in the present set-up, and thus no attempt was made to calculate OH concentrations in the flow reactor. We also conducted direct VOC photolysis experiment in the absence of water to determine the effect of this process on the product spectrum.
3.1.2 Jülich Plant Atmosphere Chamber (JPAC)
In this study, we used the larger chamber of the JPAC facility (1450 L), made from borosilicate glass (Mentel et al., 2009). It was operated as a continuously stirred tank reactor with modifications as described in Mentel et al. (2015). The chamber was positioned in temperature-controlled housing and the temperature throughout the experiments was kept at 14.2±0.3 ∘C. Purified air was fed into the system at a flow rate of ∼30 L min−1, allowing a ∼48 min residence time. A slight overpressure of 5 mbar was maintained to reduce the leaking of ambient air into the chamber. Inflow to the chamber was from two separate lines, one used to feed ozone and humidified air, the other to introduce a VOC and NOx mixed into dry air. The RH in the chamber was maintained at 65±3 %.
Benzene was fed into the chamber from a diffusion source with a constant flow, and the concentration in the chamber could be varied according to what fraction of this flow was diverted into the chamber. The procedure was identical in the experiments where phenol was used instead of benzene. OH radicals were produced by ozone photolysis in the presence of water vapour. The UV lamp (Philips, TUV 40 W, nm) was located inside the chamber and was shielded from both ends with UV-absorbing glass tubes. OH production could be varied by adjusting either the concentration of ozone or the light intensity by changing the size of the gap between the UV-absorbing tubes on the UV lamp. Starting ozone concentration was varied between 15 and 100 ppb, resulting in OH concentrations of 1.2–45×107 cm−3. These parameters, together with the concentration of benzene, determined the final concentration of OH inside the chamber. The OH concentration integrated over the residence time would define an OH dose, which could be used to compare the results between different systems or to ambient atmosphere. By definition, the OH dose would recognise that a 48 min experiment with OH concentration of 108 cm−3 is equivalent to a 480 min experiment with OH concentration of 107 cm−3. Since in our JPAC experiments the residence time is kept constant, we use the OH concentration to describe our system.
The influence of NOx on the benzene oxidation system was studied by injecting NO into the chamber. The injected NO resulted in 4.3 ppb of NOx. 12 UV-A lamps (Philips, TL 60W/10-R, nm) placed around the chamber were used to photolyse NO2 to NO and O, the latter reacting with O2 to form ozone. Ozone consequently reacts with NO to reform NO2. A photostationary state with a constant NO2:NO ratio of roughly 3 : 1 was achieved at a given ozone concentration (∼58 ppb) and photolysis rate (, s−1).
In certain experiments, monodisperse 100 nm seed aerosol particles consisting of dry ammonium sulfate ((NH4)2SO4, AS) were introduced into the chamber. The AS particles were formed by atomising an ammonium sulfate water solution and were then dried using silica gel and size-selected using a differential mobility classifier (TSI Inc, 3071). Before particles were added, pure water was nebulised to ensure a constant flow into the chamber. The achieved aerosol had a bimodal distribution, as ∼25 % of the particles were doubly charged particles of larger size, which, having the same electrical mobility, entered the chamber. This was considered when calculating the condensation sink (CS) in the chamber. The seed addition experiments helped to assess the amount of SOA that was formed from low-volatile compounds, as the increased CS shifted their main sink from the chamber walls to the aerosol. The method is described in more detail by Ehn et al. (2014).
Table 1Summary of the experimental parameters from the JPAC chamber. QMS/TOF refers to the quadrupole/time-of-flight detector in the proton transfer reaction (PTR) mass spectrometer used for measuring VOCs.
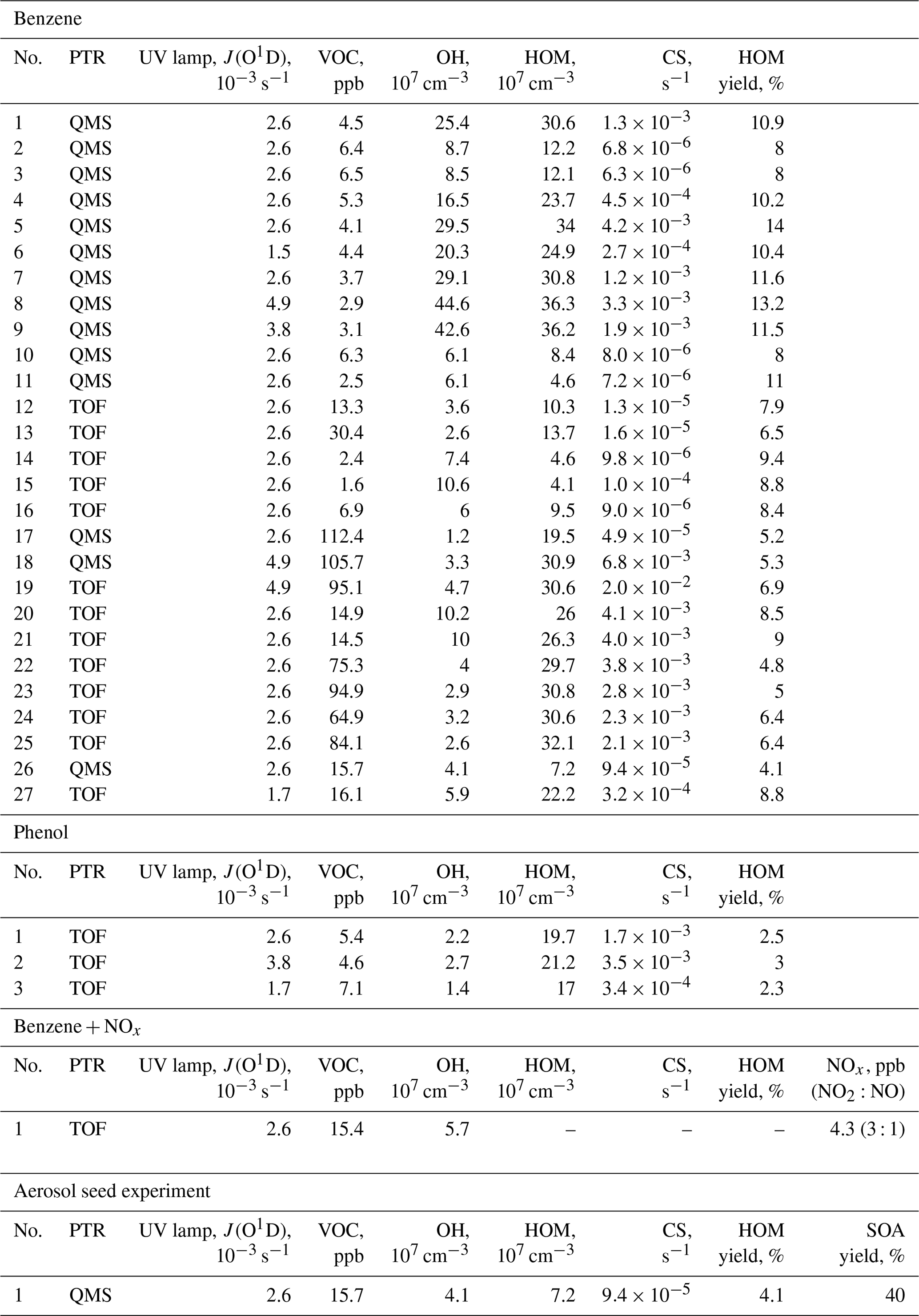
In this work, we utilise a total of 27 benzene + OH experiments, 3 phenol + OH experiments, 1 benzene + OH + NOx experiment, and 1 seed-addition experiment (Table 1). The reaction of benzene and ozone under dark conditions as well as photolysis in the absence of ozone were also tested. In these tests, no HOMs were detected, and we thus assume the VOC + OH reaction to be the initiator of all measured HOMs in this work. The parameters for each experiment were determined when the chamber had reached steady state. Typically, each experiment started by adjusting VOC and O3 concentrations, after which the UV lamp was switched on. Especially at high VOC concentrations, this initiated a strong particle formation event, and it took several hours to reach a steady state. An example experiment is presented in Fig. 2.
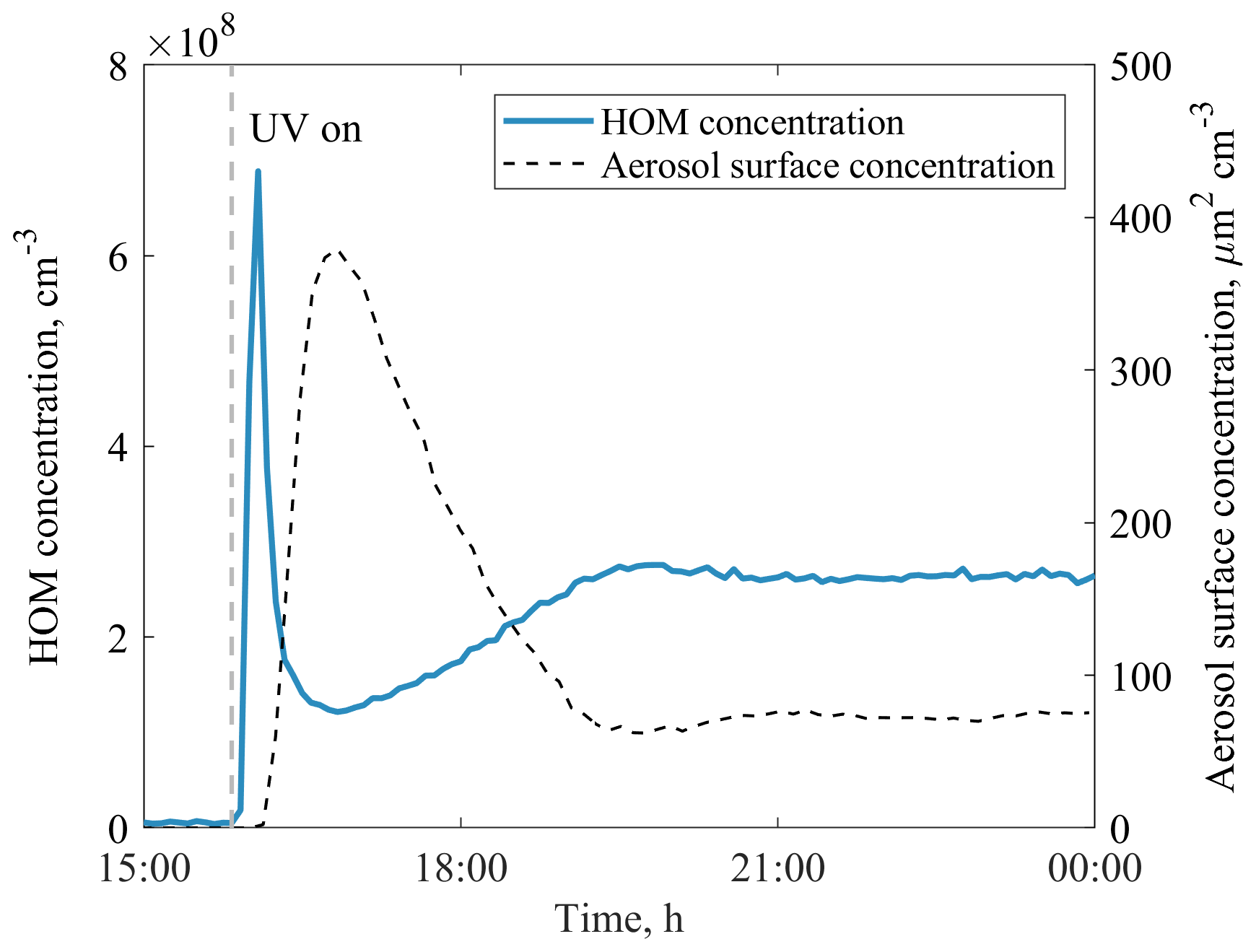
Figure 2An example of JPAC reaching steady state after the UV lamp was switched on in experiment no. 20. Within a few minutes, the concentration of HOMs increased significantly, triggering particle formation in the chamber, which acted as a sink for HOMs. After both the gas- and particle-phase signals reached steady state (in ∼ 5 h) the HOM yield was calculated.
3.2 Instrumentation
3.2.1 CI-APi-TOF
A chemical ionisation atmospheric pressure interface time-of-flight mass spectrometer (Jokinen et al., 2012) was used to measure HOMs in the Helsinki flow reactor and at JPAC. It consists of a chemical ionisation inlet (CI, Airmodus Oy) and an APi-TOF online high-resolution mass spectrometer (Junninen et al., 2010, Tofwerk AG/Aerodyne Research Inc.). The CI inlet is designed to minimise wall contact during sampling and utilises a high sample flow rate of around 10 slpm. Inside the CI inlet, the sample air is co-axially merged with a sheath flow (∼20 slpm) of filtered compressed air that contains nitric acid and nitrate ions. The ions were produced by exposure to either a radioactive source (241Am α-emitter in JPAC) or soft X-rays (<9.5 keV, Hamamatsu L9490 photoioniser in a Helsinki flow reactor). Upon collisions with neutral nitric acid, the nitrate ions can form adducts, which are referred to as reagent ions. Using an electric field, the reagent ions are pushed into the sample flow and, after ∼200 ms of interaction between sample molecules and reagent ions, guided into the APi-TOF through a critical orifice admitting 0.8 L min−1.
If energetically favourable, molecules in the sample air can be ionised via proton transfer or adduct formation. In most cases, the ionisation of a molecule M happens as
In cases where M is a strong acid, such as sulfuric acid, it may transfer a proton to and be detected in its deprotonated form, but most molecules are detected in clusters with .
Nitrate is strongly bound to neutral nitric acid, and, therefore, ionisation through adduct formation will only happen with molecules that can compete for nitrate against neutral nitric acid (Hyttinen et al., 2015). As a result, the nitrate CI-APi-TOF is selective towards HOMs, as they often contain two or more hydrogen bond donors in the form of –OOH (or –OH) groups and are able to form stable adducts with the nitrate ion (Hyttinen et al., 2015).
Inside the atmospheric pressure interface (APi), sampled ions are guided through two differentially pumped quadrupoles and an ion lens assembly, in which the pressure is gradually decreased. After this, the ions enter the time-of-flight (TOF) chamber, where they are orthogonally extracted, and their flight time to the micro-channel plate (MCP) detector is measured. This flight time is converted to mass-to-charge ratio (Th) for each ion in data post-processing. All data processing, including averaging, mass axis calibration, and peak integration, was done using the tofTools software package for MATLAB (Junninen, 2013).
The molecular formulas of sampled ions could be resolved owing to the high resolution (∼4000 Th∕Th at 125 Th) of the TOF analyser. If an ion was identified to be a HOM, defined here simply as a molecule with 6 or more O atoms (Bianchi et al., 2019), and was the dominant ion (>80 % of the signal) at its integer mass, the intensity was determined by integrating over the whole integer mass where this HOM was observed. This approach was concluded to be the most robust method, as an accurate mass axis calibration was at times problematic to achieve, and at the 5 min time resolution used, peak fitting uncertainties increased due to a limited signal-to-noise ratio. By using this type of unit mass resolution (UMR) analysis, we avoided having small variations leading to signals “leaking” into closely lying ions that were also being fitted. While our approach does add uncertainty to the quantification, it is believed to be on the order of 10 % (as we limited ourselves to masses where the HOM was the dominant ion). This is much smaller than the uncertainty in the absolute sensitivity calibration of the CI-APi-TOF (see below). In addition, when determining the average HOM intensity for a particular experiment, the background signal, determined before the UV lights were switched on, was subtracted.
The HOM ion count rate was converted to concentration (molecules cm−3) using the following equation (Jokinen et al., 2012):
where HOM is the count rate of individual HOM clusters with and the denominator describes the count rate of the reagent ions. Cf is a calibration coefficient, which in the JPAC experiments in this work was approximated as 1.6×1010 molecules cm−3 following Ehn et al. (2014), who used gravimetric calibration with perfluoroheptanoic acid (PFHA) for the same set-up as used in this study. Ehn et al. (2014) reported the uncertainty of this method as ±50 %, and we estimate a slightly larger uncertainty here due to the lack of calibrations during our measurement campaign. We estimate an uncertainty in determination of the absolute concentration of ±70 % with the precision in relative changes of less than 10 %.
3.2.2 PTR-QMS and PTR-TOF
VOCs and their oxidation products in JPAC were measured by a high-sensitivity Proton-Transfer-Reaction Mass Spectrometer (PTR-QMS, Ionicon Analytik GmbH). The technique is described by Lindinger et al. (1998). Calibrations of the VOCs were performed using diffusion sources (Gautrois and Koppmann, 1999). The PTR-QMS operated at 2 min time resolution and the sampling switched every 20 min between the inlet and the outlet of JPAC. The sampling lines consisted of ∼10 m long PFA tubing of 4 mm inner diameter and were heated to 60 ∘C. The sampling flow rate was 0.5 L min−1. During part of the campaign, a high-resolution PTR-TOF, equipped with a time-of-flight mass spectrometer, was deployed (Graus et al., 2010). The PTR-TOF was calibrated using an advanced Liquid Calibration Unit (LCUa, Ionicon Analytik GmbH) for phenol. Benzene calibrations were performed using a self-made compressed gas standard also containing, among other VOCs, 670 ppb of benzene, further diluted using the LCUa. Sampling from the outlet of JPAC was performed via a 2 m long, 1 mm inner diameter PEEK-sampling line heated to 60 ∘C. In order to cover the VOC measurements during all experiments, the data from both instruments were used, giving preference to the PTR-TOF data when they were available.
3.2.3 Aerosol instrumentation
To measure the particle number size distribution in JPAC a scanning mobility particle sizer (SMPS, electrostatic classifier, TSI 3071, and condensation particle counter, TSI 3025, TSI Inc.) was deployed. The SMPS measured particle concentrations in the size range from 14 to 820 nm in diameter, which were used to calculate the condensation sink (see Sect. 3.4). A high-resolution time-of-flight aerosol mass spectrometer (AMS; DeCarlo et al., 2006; Rubach, 2013, Aerodyne Research Inc.) was used to measure the composition of particles from ∼40 nm to 1 µm in diameter in the JPAC. In the AMS, aerosol particles were vaporised at 600 ∘C and ionised by electron impact ionisation at 70 eV, after which the ions were guided via an ion lens into the time-of-flight mass detector. The AMS was calibrated using ammonium nitrate particles, and the concentration of ammonium sulfate and organic aerosol was determined by summing the corresponding fragment ions from the mass spectra. SOA yield was estimated from the AMS as the ratio of produced organic aerosol mass to the consumed VOC.
3.3 Determination of OH concentration
In JPAC, the concentration of OH radicals in the experiment was calculated based on the amount of reacted VOC molecules for which the reaction rate coefficient with OH is known. In the chamber, the concentration of a VOC unreactive to O3 is represented by the following equation:
where the V is the chamber volume, F is the flow rate through the chamber, and k1 is the reaction rate coefficient for OH with the VOC. [VOC]in indicates the average concentration of the precursor compound in the total flow entering the chamber, and [VOC] and [OH] describe the actual concentrations in the chamber, whereby [VOC] is measured at the outflow of the chamber. During steady-state conditions, OH concentration in the chamber can be calculated as follows:
where is the residence time in the chamber. t was approximately constant throughout the campaign (2900 s) and k1 for 14 ∘C was taken as 1.19×10−12 cm3 s−1 for benzene and 3.30×10−11 cm3 s−1 for phenol (Bloss et al., 2005; Atkinson and Aschmann, 1989). [VOC]in and [VOC] were both determined by PTR-QMS or PTR-TOF. This method is independent of the instrumental calibration; however, it assumes that benzene is lost solely through the reaction with OH. The determination of [OH] was verified in some experiments by introducing 1,8-cineole in addition to benzene, which confirmed the determined OH concentrations within 6 %–12 %.
3.4 Determination of HOM yield in JPAC
We were able to calculate HOM molar yields from JPAC experiments. For HOM yields in this work, we take a slightly different approach than earlier studies in which the yield has directly been equated with a branching ratio of a certain VOC-oxidant reaction. We define the molar yield γ of HOMs as the fraction of the reacted VOC that produced HOMs during the residence time in our chamber. This definition also includes HOM formation from molecules reacting multiple times with OH, i.e. multi-generation OH oxidation. We take this approach since the oxidation products will react with OH much more rapidly than the parent VOC benzene, which subsequently means that the primary fate of the first-generation oxidation products of benzene will be to undergo further OH reactions. In other words, the more atmospherically relevant quantity, for instance relating to SOA formation, is the ultimate amount of HOMs formed, rather than only the HOM branching ratio in the initial OH reaction. The change in HOM concentration in time is defined as HOM production rate minus HOM loss rate:
where k1 is the VOC-OH reaction rate coefficient as described in Sect. 3.3, γ is a HOM molar yield, and kloss is the total loss coefficient of HOMs to the chamber walls (kwall) and to aerosol particles present in the chamber (i.e. the condensation sink):
Here we assume that HOMs are of low enough volatility that these are the dominant loss pathways, and that flush-out from the chamber, at a rate of 1∕48 min−1, can be ignored. We stress again that γ is not only the branching ratio for the initial VOC+OH reaction, but the fraction of reacted VOC molecules that become converted into HOMs in the chamber, irrespective of detailed formation pathways. In steady state in JPAC, the concentration of HOMs is constant, so
and therefore
Then, the molar HOM yield can be calculated as
For kloss, we needed to assume that HOMs condense irreversibly, which is a valid assumption based on earlier studies (e.g. Ehn et al., 2014). In accordance with Ehn et al. (2014), and verified in our experiments (not shown), kwall of 0.011 s−1 was used. Average HOM concentrations for runs were calculated as a sum of all identified peaks with an oxygen content more than or equal to six atoms. In the case of phenol experiments, the peaks of the same composition were used as those in benzene experiments for better comparison. The condensation sink was calculated using the following equation (Kulmala et al., 2012).
where D is the diffusion coefficient for condensing vapour and is the correction factor for the transition regime calculated based on the Fuchs–Sutugin approximation. D was approximated as 0.06 cm2 s−1 based on the mean molar mass 237 g mol−1 and approximated diffusion volume 170 of the observed HOMs, according to the approach described by Fuller et al. (1966). dp is the diameter of particle size bins, and is the concentration of particles in the chamber in the size bin dp.
Finally, we stress that the HOM yield depends on our ability to determine the HOM concentrations and is thus associated with at least the same ±70 % uncertainty. Additional uncertainty will arise from the other parameters in Eq. (5), but these are likely to be much smaller than the uncertainty arising from HOM quantification. As stated earlier, only clearly identifiable peaks were utilised for HOM concentration calculations, in order to make the quantification as robust as possible. These peaks constituted approximately 50 % of the total signal in mass range from m∕z 200 to 550. Although isotopes account for some of this unexplained fraction, our approach may cause an underestimation of the HOM yields by up to 50 %.
3.5 Chamber kinetic model
In order to model HOM condensation during the seed addition in JPAC, we have constructed a simple kinetic model. The HOM mass concentration was modelled with 0.1 s resolution and the model assumed that the chamber was perfectly mixed for every time point. HOM molecular concentration for each point in time j for each HOM i was calculated by adding the HOMs produced in a cubic centimetre in 0.1 s and subtracting the HOMs lost from the HOM concentration in the previous time point (j−1) as follows:
The molar yield of total HOMs was set to 5 % to match the measured HOM concentration before seed addition. For an individual HOMi, the relative abundance in the spectra determined its yield. [VOC] was set to constant 15.7 ppb (as measured by PTR-QMS), while [OH] concentration was scaled to match start and end measured HOM concentration (see Sect. 4.2.4). In Eq. (7), kloss took into account both wall loss and CS. The loss of HOMs due to the flush-out from the chamber was excluded as it is negligible compared to wall loss and CS in JPAC. Then, the total HOM mass concentration at point in time j equaled
where Mi is the molar mass of HOM i and NA is Avogadro's constant. If a HOM was detected as a cluster with , Mi was calculated as the m∕z value of the peak minus the mass of . For this model, we used the peaks corresponding to the same HOMs as in the HOM yield calculation, 69 peaks in total. The list of the m∕z values and corresponding compositions can be found in Table S4 in the Supplement.
4.1 Flow reactor study
In the first part of this work, we studied the OH oxidation of benzene, toluene, and naphthalene in the Helsinki flow reactor using a nitrate-based CI-APi-TOF. In all three systems, we observed the formation of HOMs. Product distributions are shown in Fig. 3 and include both HOMs (products with six or more O atoms) and less oxidised species, which were detected as adducts with . The following discussion focuses on peaks detected by adduct formation. We omit the reagent ion when presenting the molecular formulas. However, the mass of the molecules refers to the correct mass, including the nitrate ion.
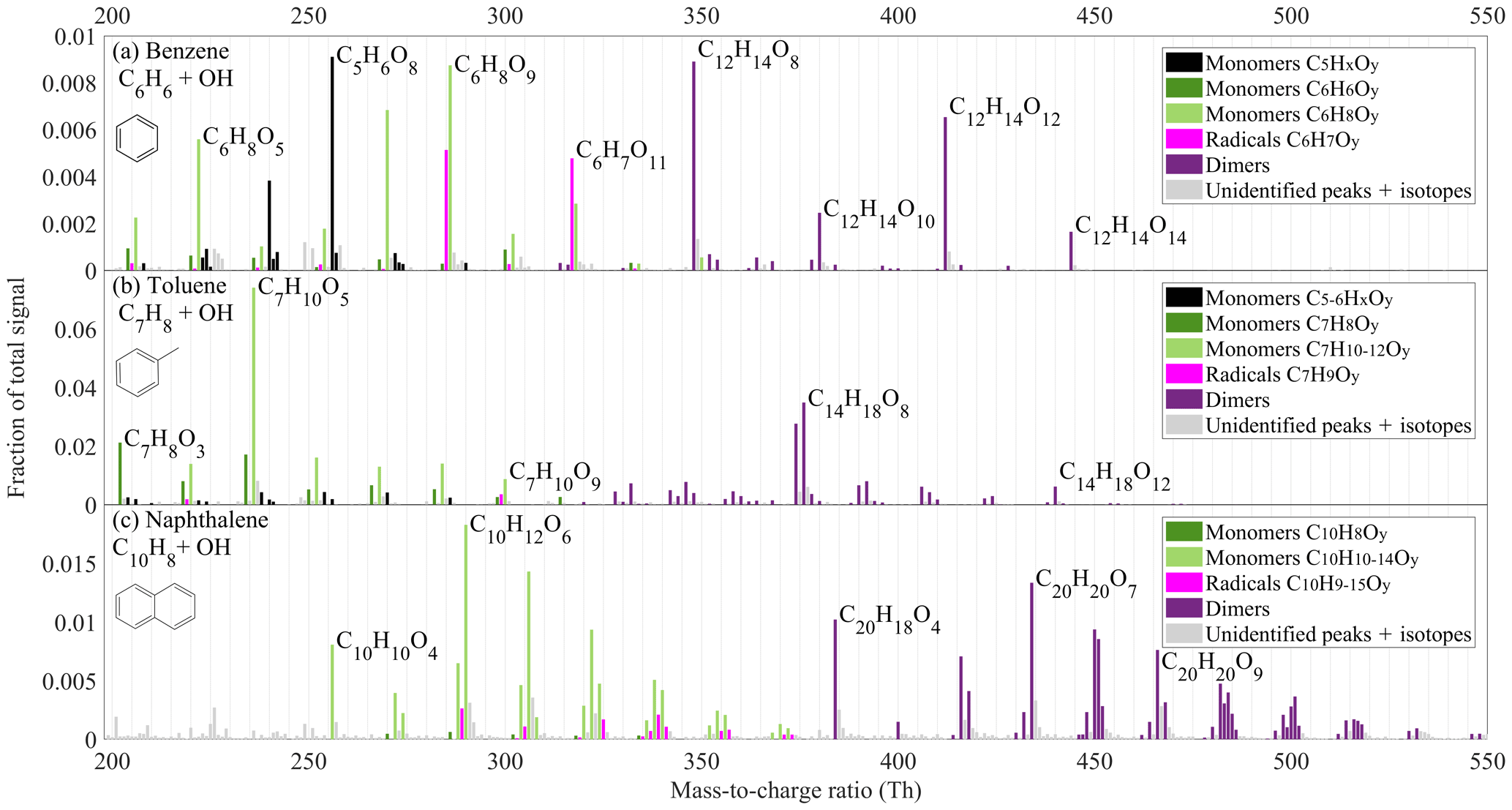
Figure 3Spectra of organic oxidation products observed in oxidation of (a) benzene, (b) toluene and (c) naphthalene. The y axis shows the signal normalised by the total ion count of the instrument. The colours indicate different compound groups, as described in the legend of each subplot. The unit masses with more than one peak are marked with the colour of the most abundant peak. All the peaks above 200 Th are detected as adducts with , which is excluded from the labels. The full list of peaks can be found in the Supplement (Tables S1–S3). Unidentified masses, isotopes, and contaminant peaks are marked in light grey.
A few prominent peaks clearly dominated the benzene spectrum (Fig. 3a), with oxygen content of the products ranging from 4 to 14 atoms. Among the closed-shell HOMs, C5H6O7, C5H6O8, C6H8O8, C6H8O9, and C6H8O11 monomers and C12H14O8, C12H14O10, C12H14O12, and C12H14O14 dimers dominated the signal. Two radicals, and , were also detected.
The BPR, in the case of benzene, is potentially an intermediate in the formation of many HOMs in the oxidation of aromatics. It was proposed in earlier studies that BPRs from substituted aromatics can undergo further autoxidation (Molteni et al., 2018; Wang et al., 2017). In the case of benzene, it would form radicals with chemical composition , where x is an odd number larger than five. This hypothesis is supported by the presence of and radicals, while the BPR itself and the radical are detected only as very small signals. This is most likely due to the reduced detection efficiency for smaller radicals. While the BPR is expected to have only one OH group and its detection is unlikely, the reason for the low abundance of in the spectrum is unclear. It would be explained, however, if would contain two endoperoxides and a peroxy group, as also proposed by Molteni et al. (2018), thus still having only one OH group to supply a hydrogen bond to a nitrate ion.
While (BPR) is weakly detected as such, we can observe products consistent with its termination reactions. For instance, C6H8O5 at 222 Th can be formed through a BPR reacting with HO2. In the photo-oxidation of any VOC, HO2 production from RO2 is efficient. HO2 will also be co-produced from water photolysis in our reactor, making a reaction with HO2 an important bimolecular termination pathway. This is also supported by the observation of C6H8O7, C6H8O9, C6H8O11, and C6H8O13. However, the high oxygen content of HOMs as well as the existence of dimeric species shows that the termination of RO2 by HO2 was not a dominant process in our system
The other important termination agents in our system are RO2 radicals. The dominance of the C12H14O8 dimer in the spectrum, likely formed from BPR self-reaction, strongly indicates the importance of ROOR' dimer formation. The prominence of dimers with even oxygen numbers (also C12H14O10, C12H14O12, and C12H14O14) is consistent with primarily odd-oxygen RO2 being formed in the benzene system. If even-oxygen RO2 were also abundant, odd-oxygen dimers, from cross-reactions of odd- and even-oxygen RO2, should be more prominent in the spectrum.
Monomer HOMs with an even number of O atoms are also abundant in Fig. 3a, and these can be formed from RO2 cross-reactions forming a carbonyl and an alcohol, or via alkoxy (RO) radical pathways. We are not able to separate formation pathways in such detail based on our data. However, the importance of RO radicals is suggested by some C5 radicals that we observed, namely , , and , with one carbon less than benzene. Since benzene is a plain aromatic ring, loss of carbon from this molecule is only possible after a ring opening, potentially due to RO decomposition or another reaction causing the break of a bond between carbon atoms. After the ring is broken, CO or CO2 could be lost and, after a reaction with another O2 molecule, radicals are formed. These RO2 radicals would terminate by reacting with HO2 or another RO2. Indeed, C10 and C11 dimers as well as closed-shell C5 products are observed, of which C5H6O8 (256 Th) is one of the dominant peaks in Fig. 3a. We cannot rule out other pathways for loss of carbon atoms from the molecules and only conclude that it is a non-negligible pathway for HOM formation in benzene oxidation under our conditions.
The product spectrum from our flow reactor study of benzene oxidation shown in Fig. 3a was similar to the previous study by Molteni et al. (2018). For instance, the three largest signals in their study above 200 Th were C6H8O5, C5H6O8, and C12H14O8, which are also prominent signals in our spectrum. Overall, almost the same molecules are present, with some variations in relative abundance. Specific differences worth noting are the larger fractions of RO2 radicals visible in our spectrum, with two radicals ( and ) being among the highest peaks. In contrast to our experiment, Molteni et al. (2018) observed dimers with odd and even amounts of oxygen at comparable concentrations, suggesting the presence of both even- and odd-oxygen radicals in their system. While the specific experimental conditions between the studies of Molteni et al. (2018) and ours were not identical (benzene concentration ∼100 times higher, residence time 50 % shorter, UV lamp irradiating part of the flow reactor in our study), some differences in the spectra are expected. However, based on the good agreement in product composition between the two studies, we conclude that direct photolysis of the VOC or its oxidation products (whether radicals or closed-shell species) were not affecting our results to a large extent. In addition, in our flow reactor, a direct photolysis experiment in the absence of water showed no HOM formation. Nevertheless, future studies are needed to determine the exact role of photolysis in comparison to OH oxidation in initiating HOM formation in such systems.
In our flow reactor, we also tested the oxidation of toluene (C7H8, Fig. 3b). While the composition of toluene oxidation products is generally consistent with the reactions described for the benzene system above, some notable differences are observed. For instance, compared to the benzene experiment, the signal spreads out more evenly over many ions in the monomer product mass range, except for the dominant C7H10O5 peak. Analogously to the benzene system, this is likely a termination product of a BPR in reaction with HO2. Another difference is that even-oxygen dimers do not dominate the dimer spectrum. Instead, only two peaks, C14H18O8 and C14H16O8, are dominant. While the former dimer could originate from toluene-BPR self-reaction, the origin of the latter is unclear. We also observed some monomers with five or six carbon atoms, though at much lower contribution to the total than the contribution of C5 monomers in the benzene experiment. Overall, in comparison to the study by Molteni et al. (2018), where the toluene concentration was about 25 times smaller than in our experiments, many of the peaks are similar. Specifically, it is interesting that in the toluene system, we also observed a few C7H12O4–8 products with four hydrogen atoms more than in toluene itself, indicating the potential secondary OH oxidation (addition) step (Molteni et al., 2018). These products overlapped with C6H8Ox compounds in the spectrum, so while they can be separated in high-resolution analysis, they are not recognisable in Fig. 3b.
In our naphthalene (C10H8) experiment, which is presented in Fig. 3c, the signal was distributed among an even larger amount of product peaks than in the toluene experiment. Interestingly, the largest monomer (C10H12O6) contained 4 H atoms more than the naphthalene precursor (C10H8). This suggests that an oxidation pathway including two OH attacks in combination with two HO2-termination reactions was important in the naphthalene system. Evidence of RO2 radicals formed through two OH attacks is also seen in H20 dimers, which likely formed through cross-reaction of H9 and H11 RO2. In the benzene spectrum we did not observe any monomers containing four hydrogens more than the parent VOC, while in the toluene spectrum we observed only a minor fraction of such peaks. We attribute this to the higher VOC concentration used in our benzene experiment (400 ppm), in comparison to naphthalene (0.4 ppm) and toluene (25 ppm). The OH production from H2O photolysis stayed constant in our experiments, but the VOC acted as a sink for the OH radicals, which means that higher VOC concentrations resulted in lower OH concentrations. This, in turn, decreases the likelihood of oxidation products reacting with OH a second time in our flow reactor. Therefore, not only the competition between autoxidation and bimolecular RO2 termination reactions will govern the exact concentration and distribution of HOMs, but also the amount of secondary (or higher) OH attacks. For determining the importance of multi-generation OH oxidation as a source of HOMs, longer timescales and lower VOC concentrations than reachable in our flow reactor were needed. Our further investigations in the JPAC chamber facility at the Research Centre Jülich were well suited for such a task.
4.2 JPAC chamber studies
4.2.1 HOM yields
To continue our study on the formation of HOMs from aromatics, we performed systematic studies of benzene oxidation in the JPAC chamber (see Table 1). In JPAC, we were able to control the experimental conditions in more detail than in our flow reactor. In experiments without NOx, the main parameters determining the oxidation process were the concentrations of OH and the VOC. As described in Sect. 3.1.2, these two parameters could be adjusted by changing the inflow of the VOC or ozone, or by adjusting the photolysis rate by changing the gap width of the UV filter. Out of these, the input of the VOC had the largest range, spanning around 2 orders of magnitude (1.6–112 ppb). An increase in the VOC also meant a larger sink for OH, and thus the VOC and OH concentrations in the chamber were codependent.
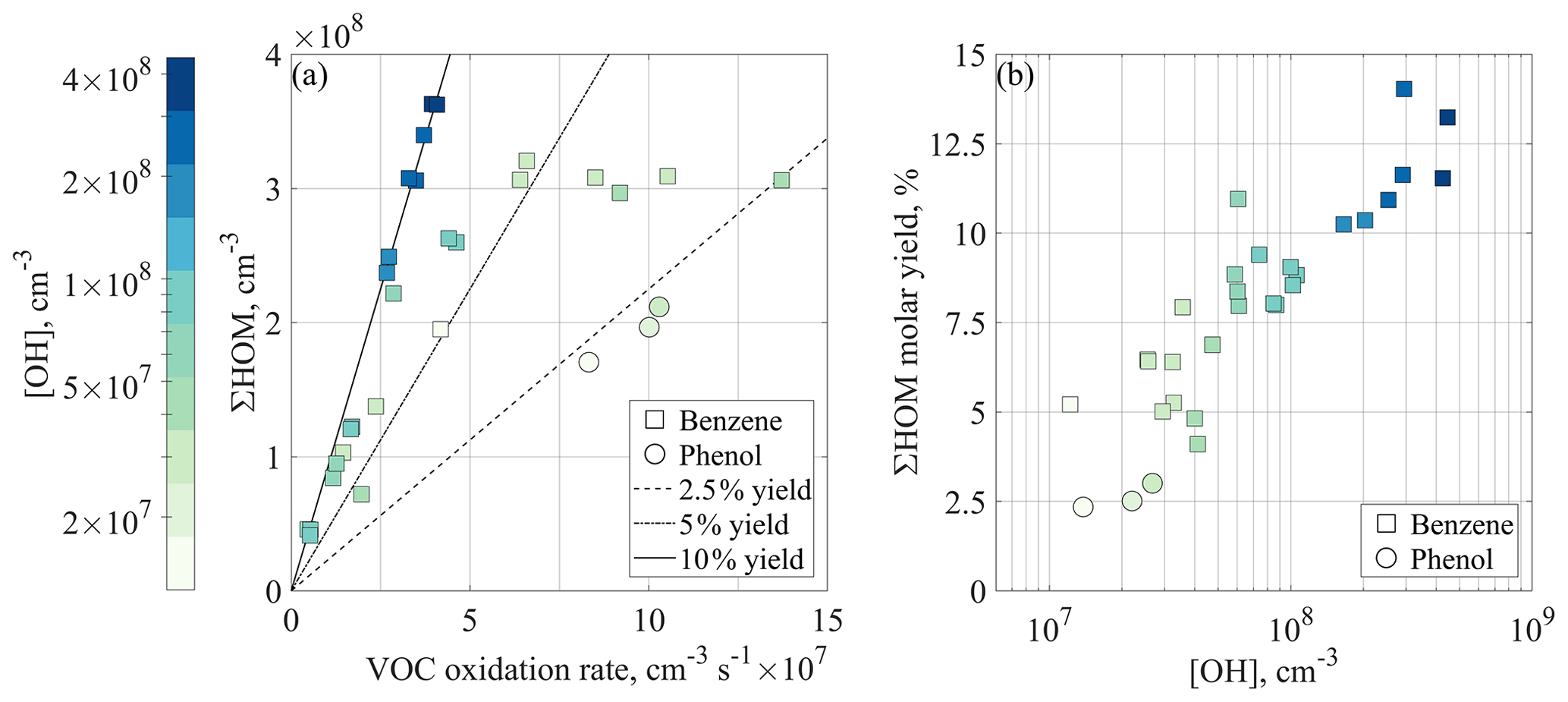
Figure 4HOM concentrations and yields observed in the JPAC experiments. (a) Total HOM concentration plotted as a function of VOC oxidation rate. If the HOM loss rate is constant between the experiments, conditions with the same molar HOM yields should fall on the same lines. The included lines (for 2.5 %, 5 %, and 10 % yields, respectively) neglect the condensation sink (CS) and depict the yields in the case where loss to walls is the only sink for HOMs. The colour represents the concentration of hydroxyl radicals, squares depict benzene runs, while circles show phenol runs. (b) Calculated HOM molar yields as a function of OH concentration in the chamber corrected for CS. Markers are the same as in panel (a), as is the colour code for easier comparison.
Figure 4a shows the measured HOM concentration as a function of the VOC oxidation rate (k1×[VOC]×[OH]), including primarily benzene experiments (square markers) but also three phenol experiments (circles). In benzene experiments at small oxidation rates, the total HOM concentration increased linearly, but a plateau at around 3×108 cm−3 was visible at higher oxidation rate. If the loss coefficient (kloss) of HOMs were constant throughout all runs, experiments with the same HOM yield would fall on the same line. Assuming the loss of HOMs is only determined by wall loss (kloss=kwall), the plotted lines in Fig. 4a would correspond to 2.5 %, 5 %, and 10 % yields. However, especially in the high [VOC] experiments (markers on the right-hand side of Fig. 4a), the CS due to particles formed in the chamber was of the same order as the wall loss and thus the approximation that kloss equals kwall is not valid anymore. In addition, the high-OH experiments (dark blue points) seem to result in the highest HOM yields.
In order to identify the role of OH concentration for HOM yields, we calculated the molar yields, i.e. the number of HOMs formed per reacted precursor VOC molecule, according to Eq. (5), properly accounting also for the CS. The results are shown in Fig. 4b. It is clear that under the conditions probed in JPAC, the main determining factor for the HOM yield was the OH concentration. It should be noted that the specific dependency of HOM yield on OH may vary if other gases and loss mechanisms would be present. Since the observed HOM molar yield increased, we can conclude that the undetected lower oxygenated products reacted again with OH to form more of the detectable HOMs. These intermediate precursors could also be higher oxygenated compounds that were detected in our instrument with ionisation efficiency below the collision limit (Hyttinen et al., 2015).
Our estimated HOM yields from benzene oxidation were 4.1 %–14.0 %, which can be compared to a value of 0.2 % provided by Molteni et al. (2018). The difference in the results is expected due to the substantial difference in the studied timescales (20 s in their study). In addition, in their flow reactor, an air parcel was exposed to an initial OH concentration that decreased as OH reacted away, while in JPAC, the OH was produced continuously. This resulted in different OH doses in the systems. Considering these differences, fewer oxidation steps would be expected in a flow reactor. As a result, the yield in Molteni et al. (2018) likely corresponds to the HOM yield of the first OH oxidation step, potentially also impacted by a second step. This suggests that more than 90 % of the “HOM-forming potential” of benzene comes from multi-generation OH oxidation in combination with slower isomerisation reactions that may not be observed on shorter timescales.
To test secondary OH oxidation, we conducted three similar experiments starting with phenol as the precursor, a known first-generation oxidation product of benzene. Examining Fig. 4b, the phenol experiments show the lowest HOM yields (2.3 %–3 %), suggesting that the phenol oxidation pathway is not the major route to form HOMs from benzene. However, the phenol experiments do not fall far from the trend produced by the benzene experiments, and thus phenol is likely to contribute to the total HOM formation from benzene. The OH concentration in the chamber was clearly higher than in the atmosphere, but the average reaction time in the chamber was limited to approximately 48 min. If utilising the concept of an equivalent OH dose, a 48 min residence time with [OH]=107–5×108 cm−3 is equivalent to atmospheric oxidation times of roughly 10 h–15 d at OH concentration of 106 cm−3. In other words, our experiments span a reasonable range of atmospheric lifetimes.
In comparison to biogenic VOCs, our results were closest to the HOM yields observed in ozonolysis of α-pinene and limonene, 3.4 %–7 % and 17 % respectively (Bianchi et al., 2019). In the biogenic systems, especially if a VOC contains an endocyclic double bond, the first oxidation step by O3 is known to form HOMs at large yields. On the other hand, the observed yields in first-step OH oxidation are reported to be low (∼1 %, Bianchi et al., 2019). To our knowledge, no studies exist that explore HOM yields of biogenic VOC oxidation as a function of OH concentration. However, McFiggans et al. (2019) indicated a non-linear increase in HOM concentration with increasing α-pinene oxidation rate. We would therefore expect that in biogenic systems, an increase in HOM yield due to multi-generation OH oxidation could also be observed.
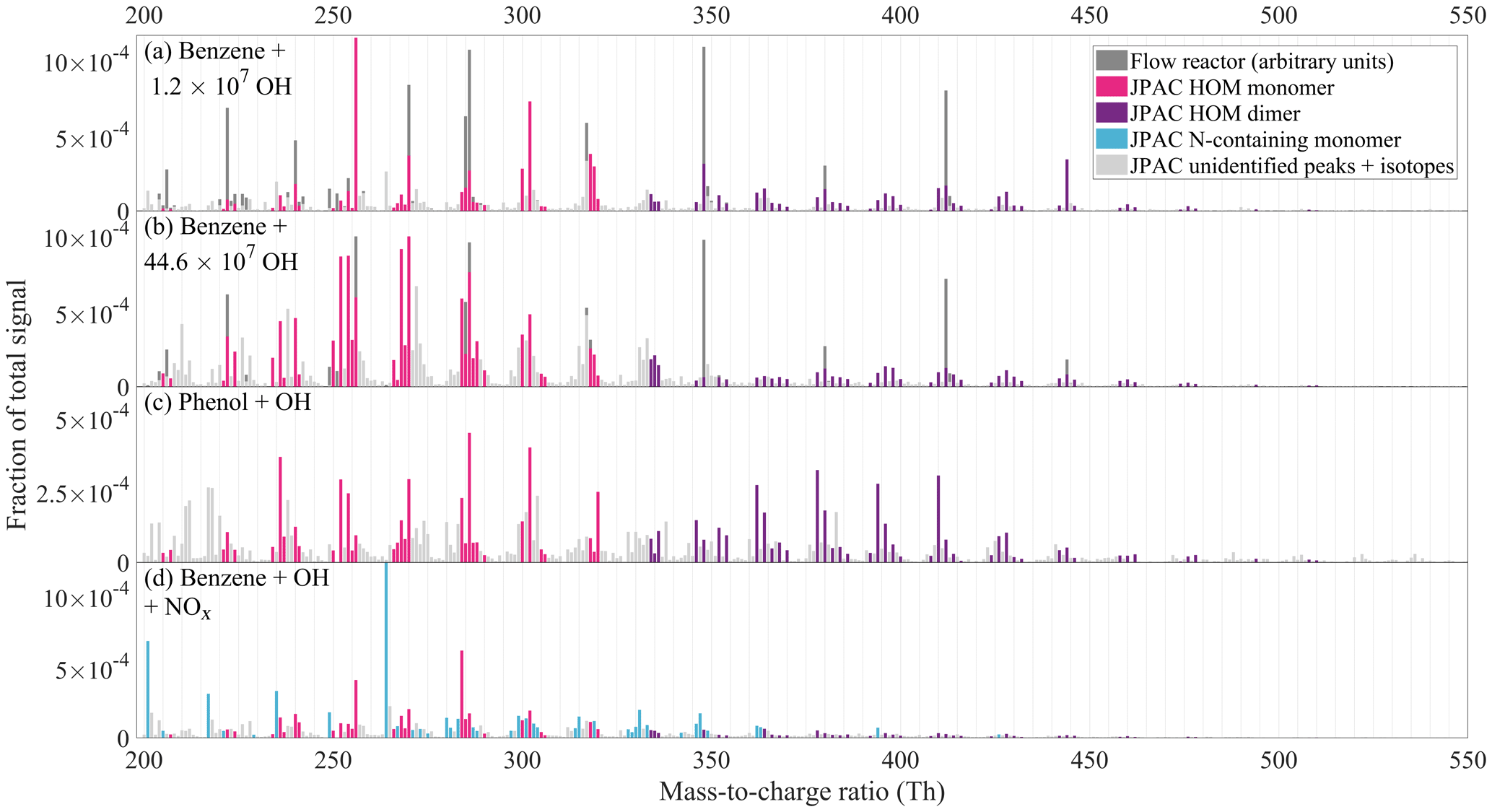
Figure 5CI-APi-TOF spectra observed during experiments at JPAC. The y axis shows the signal normalised to total ion counts. Panel (a) shows the mass spectrum of benzene oxidation at the lowest OH concentration among our experiments, while panel (b) corresponds to the highest OH concentration. In panels (a) and (b), the scaled flow reactor mass spectrum is also included for comparison (dark grey bars, in arbitrary units). Panel (c) shows the oxidation products of a phenol oxidation experiment. In panels (a), (b), and (c), the colour schemes are identical and non-grey peaks represent those that were included in HOM yield calculation, with the exception of a few peaks. The full list of peaks is presented in the Supplement (Table S4). Panel (d) shows the benzene + OH experiment in presence of NOx, where N-containing ions (“N-containing monomers”) dominate the spectrum.
4.2.2 Variability in HOM spectra
In addition to total HOM yield, OH concentration also affected the distribution of monomers and dimers in the benzene HOM spectrum, which is seen in Fig. 5a–b. Higher OH concentration produced a spectrum with more peaks than did lower [OH], indeed pointing at multiple oxidation steps. At lower [OH], the monomers somewhat resembled the benzene flow reactor experiment. In Fig. 5a and b, the VOC oxidation rate is very similar (4.2×107 versus 3.9×107 cm−3 s−1), while OH concentration is 35 times larger in Fig. 5b (4.5×108 versus 1.2×107 cm−3). The HOM yields corresponding to Fig. 5a and b are 5.2 % and 13.2 %, respectively.
As OH concentration increased in benzene experiments, an increase in the abundance of products with more H atoms than the parent molecule due to secondary OH addition was expected; however, we observed an increase in products with lower H content, H=4–6. This means that OH oxidation through H abstraction started to play a role. Oxidation of benzoquinone (formed in OH oxidation of phenol, MCM3.3.1; Bloss et al., 2005) could also potentially explain H4–6 monomer HOMs. After ring-opening, the BPR will contain one double bond, and if the products retain this bond, one more OH addition is possible. However, after this, OH oxidation can only proceed via H abstraction, and if the subsequent termination reactions occur by loss of OH or HO2, a decrease in H atoms will take place. In other words, it is to be expected, that multi-generation OH oxidation will also produce molecules with fewer H atoms than the parent VOC.
The dimers detected in JPAC experiments had up to 18 oxygen atoms, which was a higher number than seen in flow reactor study. Dimers in JPAC had larger variability in the H atom content, from 10 to 16. As in monomers, the dimer distribution also varied with OH concentration. At higher OH concentrations, a larger fraction of dimers was C11 dimers, suggesting more efficient formation of C5 radicals. At lower OH concentrations, the dimer distribution was more similar to the distribution seen in the flow reactor. In addition, in JPAC, the dimer-to-monomer ratio was observed to decrease with increased OH concentration. This may be explained by higher HO2 concentrations at higher [OH]. Another possible explanation is that RO2 formed at higher [OH] would have less favourable structures for dimer formation. The dimer formation rate has been shown to be highly dependent on the structure of the reacting RO2 (Berndt et al., 2018a).
In the phenol experiments (Fig. 5c), most elemental compositions were similar to those starting with benzene, as could be expected given that phenol has the same amount of C and H atoms as benzene. However, the relative distribution of peaks in the phenol spectrum did not directly resemble either the low or the high OH concentration benzene spectrum, again suggesting that a considerable fraction of HOMs were produced from non-phenol pathways. In Fig. 5c, the peaks in colour are the same peaks as were observed in the benzene experiments and were used for HOM yield calculations. Compared to the benzene experiments, phenol produced more dimers, of which H12 dimers were a significant fraction, suggesting that H5 radical production in phenol oxidation was somewhat more important than in benzene oxidation (H5 and H7 radicals would react to form H12 dimers).
4.2.3 NOx experiment
While not being the main focus of this study, we also added NOx to the chamber in order to see its effect on HOM formation from benzene. Aromatic VOCs and NOx are often co-emitted, and thus our no-NOx experiments are mainly relevant in places where the emissions were sufficiently diluted following transport from the vicinity of the sources. As seen in Fig. 5d, we observed many nitrogen-containing HOMs as well as less oxidised compounds likely relating to nitrophenol-type compounds (i.e. nitrophenol with additional -OH or -NO2 groups). The list of observed products is presented in the Supplement (Table S4). In addition, we observed HOMs without nitrogen, presumably from the reaction pathways involving alkoxy radicals (formed from RO2+NO).
The nitrophenol-type compounds reacted much slower to changes in the chamber compared to HOMs, likely indicating condensation and re-evaporation from chamber walls (i.e. semi-volatile compounds). As such, they can likely be transported long distances in the atmosphere, as shown in a recent study, which found a large nitrophenol signal in a CI-APi-TOF in the boreal forest (Yan et al., 2016). This study also showed that nitrophenol, despite only having one OH group, is readily detected by the CI-APi-TOF. Hyttinen et al. (2017) confirmed the stability of nitrophenol clusters with a nitrate ion using quantum chemical calculations.
4.2.4 HOM contribution to SOA
To examine the role of the observed HOMs on SOA formation, we added ammonium sulfate seed aerosol to the chamber during one experiment. Aerosol addition increased the condensation sink for low-volatility species in the chamber. Many of these compounds would otherwise condense onto the chamber walls. Upon addition of the seed aerosol, HOM concentrations decreased and SOA concentrations increased (Fig. 6). Ehn et al. (2014) observed a similar behaviour in JPAC experiments during ozonolysis of α-pinene. In the Ehn et al. (2014) experiments, the total aerosol mass loading was 30 µg m−3, out of which 7 µg m−3 was the SOA mass formed during the experiment. The condensed HOMs explained more than 50 % of that SOA mass. In our benzene case, at total aerosol mass loading of 22 µg m−3, the removed HOMs explained around 30 % of the 2.7 µg m−3 SOA mass. The chamber did not fully reach steady state before the seed addition, which adds some uncertainty to the estimates, as discussed in the next paragraph. We expect that addition of seed aerosol will not affect the VOC oxidation rate, therefore the amount of reacted VOC should be the same over the whole experiment. Since the SOA yield is defined as SOA mass formed over VOC mass reacted, the observed increase in SOA directly indicates an increased SOA yield. In our case, the calculated SOA yield was negligible before seed addition, and increased to about 40 % at the peak aerosol concentration of 22 µg m−3. This clearly suggests that aerosol loadings can greatly influence SOA yield estimates from chamber studies as long as wall loss can compete with CS (Ehn et al., 2014; Kokkola et al., 2014; Zhang et al., 2014). We acknowledge that most SOA yield studies have been performed in larger chambers where the wall loss rate can be much smaller than in our chamber, and thus the effect is unlikely to be quite as large as observed here. The final SOA yield of 40 % in our study is similar to 37 % yield in a low-NOx regime reported in previous work (Ng et al., 2007).
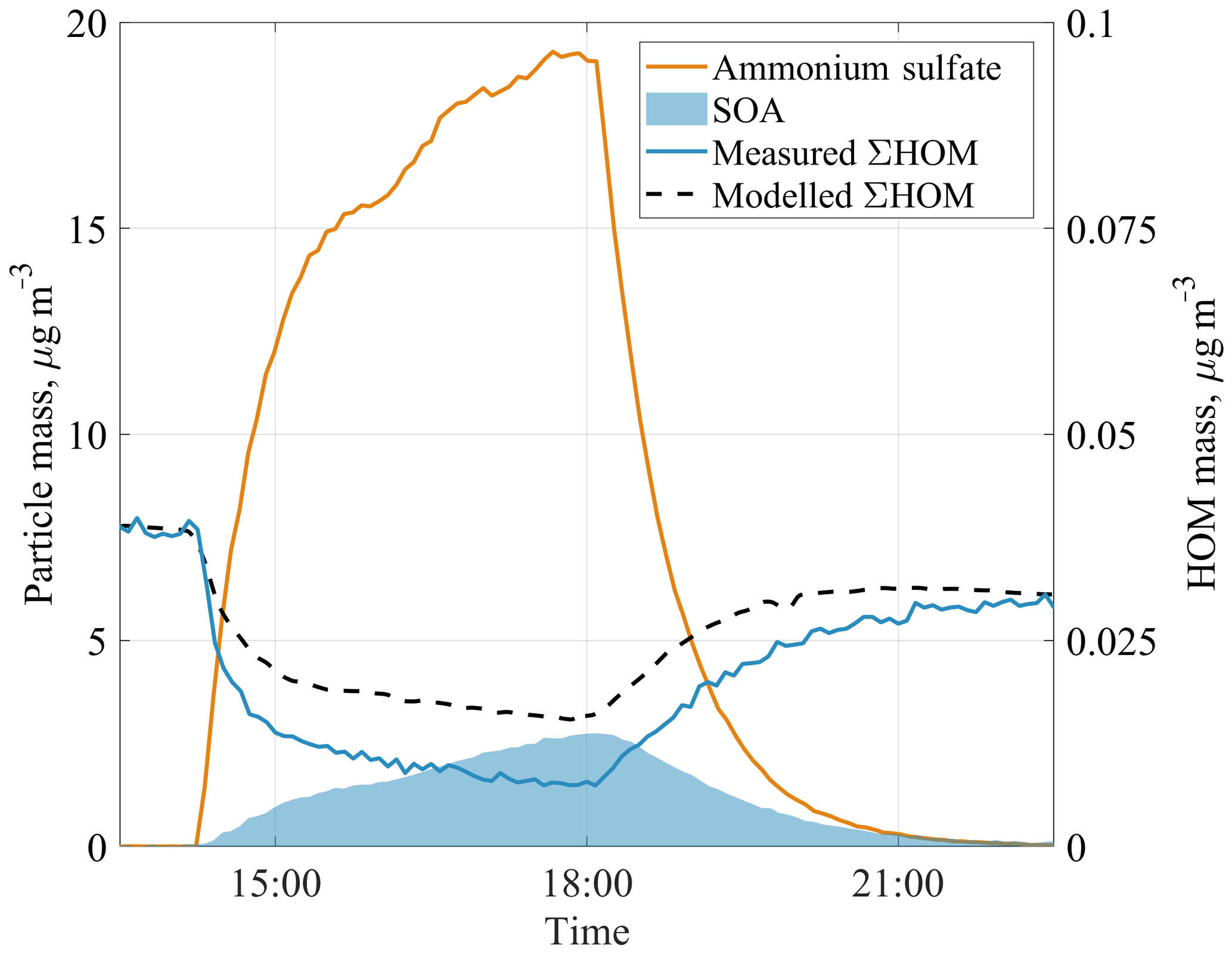
Figure 6The evolution of components of aerosol mass and HOM mass concentrations during a seed aerosol experiment. The modelled total HOM concentration (dashed line) underestimates the removal of HOMs from the chamber when aerosol seed is added, suggesting that not only HOMs but also some HOM precursors decrease during the seed addition.
We constructed a simple chamber model to test the expected loss of HOMs at different seed loadings, matching HOM concentrations to the periods before and after seed addition. HOM loss rates are a sum of wall loss rate (estimated as 0.011 s−1) and condensation sink, which is calculated for every point in time according to Eq. (6), using the measured aerosol number size distribution. It should be noted that condensation sink assumes that the vapour is non-volatile. The reason behind the HOM concentration not returning to the same level as before seed addition (∼25 % lower at the end of the experiment) is unclear. VOC in- and outflows were stable, as were O3 concentration and RH. As a consequence, [OH] was also constant within the error ranges, with a tendency to drop by about 10 % over the time when seed aerosol was present. In our model we included a linear decrease in the OH concentration over the experiments to match the start and end HOM concentrations.
Using our model, we capture the shape of the HOM decrease very well but find that our model underpredicts the loss of HOMs to the particles (Fig. 6). A possible explanation is that we underestimate the condensation sink or overestimate the wall loss rate (kwall) in our model. For kwall, the value would need to be ∼2.5 times lower, corresponding to an inverse lifetime of 220 s, which is not supported by earlier experiments (Ehn et al., 2014) and observed lifetimes of individual HOMs in our experiments. A similar correction factor of ∼2.5 would be required for the CS in order to match the measurements, and this is a much greater value than the uncertainty in the aerosol loading data used for the CS calculation. In addition, the discrepancy is larger for some of the detected HOMs, while for others the model matches the observed loss (Figs. A1, A2 in Appendix A).
The most likely explanation for the mismatch in Figs. 6, A1, and A2 is that by introducing the seed aerosol we introduce a sink not only for the HOMs detected by the CI-APi-TOF, but also for some of the undetected oxidation products (or detected at low sensitivity) that could have formed detectable HOMs upon further OH oxidation steps. This explanation is plausible and is in support of our hypothesis that some of the HOMs were formed in multi-generation OH oxidation. It also suggests that both the detected benzene-derived HOMs and some of the HOM precursors are of sufficiently low volatility to condense on 100 nm seed aerosol. If a HOM were not to condense irreversibly onto the aerosol surface, it would lead to the opposite effect, i.e. that our model would overpredict the loss of the HOM due to seed addition. Based on the explanation above, we note that our earlier estimate of HOM contribution to benzene SOA of 30 % is a slight overestimation. Based on our current understanding of HOMs and the results from our SOA experiment, we expect that the change of HOM yield with OH would affect in turn the formed SOA yield. It is likely that this effect will be mainly pronounced in SOA studies conducted without seed aerosol or in studies where seed aerosol is added at low concentrations (Ehn et al., 2014).
In this study, we confirmed the production of highly oxygenated organic molecules (HOMs) in the OH-induced oxidation of aromatic compounds. We tested this chemical system in a flow reactor (10 s residence time) and in the Jülich Plant Atmosphere Chamber (JPAC; 48 min residence time).
In benzene oxidation experiments in the flow reactor, we most likely observed first-generation HOMs formed after a single OH attack. In experiments of toluene and naphthalene, we observed a broader distribution of HOMs, within which no particular compound clearly dominated the signal. We attributed this difference to lower VOC concentrations in the toluene and naphthalene systems compared to the benzene system, resulting in higher OH concentrations and consequent multiple OH reactions.
Complementary to the flow reactor study, we further investigated the multi-generation OH oxidation as a source for HOMs in JPAC, specifically focusing on quantifying the benzene-derived HOM yield. The HOM molar yield, which in our definition also included multi-generation oxidation, in JPAC varied from 4.1 % to 14.0 % and strongly depended on the OH concentration. This dependence suggested that multi-generation oxidation produced a major portion of HOMs. When examining the HOM composition, higher OH concentrations caused a larger variety in HOM products, with H-abstraction oxidation becoming more significant. We also noted a decrease in the dimer-to-monomer ratio as [OH] increased.
In a phenol oxidation experiment (a first-generation product of the benzene reaction with OH), we observed a lower HOM molar yield in comparison to the benzene oxidation at a comparable VOC oxidation rate and OH concentration. The lower HOM yield in phenol oxidation suggests that the non-phenolic pathway must be significant for HOM formation from benzene. This was further supported by the difference of the spectral distribution of HOM products between phenol and benzene.
Upon addition of about 4 ppb NOx to the benzene system in JPAC, we observed a production of N-containing HOMs. These likely contained both nitrate and nitro functionalities. While termination reactions by NOx were significant, many HOMs without nitrogen were still observed. The HOM spectrum observed in this experiment is likely more representative of the ambient urban air, where NOx concentrations are high. On the other hand, experiments without NOx are representative of the emissions after considerable dilution.
We also tested the ability of HOMs from benzene oxidation to form secondary organic aerosol (SOA). We introduced seed aerosol to JPAC and investigated the rate of condensation of HOMs. The loss of HOMs was faster than the simple kinetic model predicted, which likely means that precursors for the detected HOMs, which were not observed by our instrument, were also condensing. This further supported our hypothesis that a large fraction of HOMs in the benzene system was produced via multiple OH oxidation steps.
Our study confirmed the formation of HOMs from aromatic compounds on both short and long timescales. We have determined the HOM yield from benzene oxidation at relevant atmospheric lifetimes. In addition, we examined the phenol branching pathway and confirmed the production of nitrogen-containing HOMs upon NOx addition. Based on our findings, we conclude that HOM yield and composition is very sensitive to the reaction conditions. This sensitivity of HOM yield may partly explain the variability of SOA yield and time dependency observed in previous studies. Based on current understanding of HOMs as well as our SOA experiment result, we can suggest that HOMs observed in this study may play an important role in initial particle growth in the ambient atmosphere, where aromatic VOCs are abundant.
In addition, we also conclude that more studies are required to fully understand how HOM yield and composition in aromatic systems depends on OH concentration and how the differences in HOMs will affect the rate and magnitude of SOA formation. It would be valuable to sample different timescales and low and high reactant concentrations as well as the effect of other important parameters, such as lights and NOx. We also propose that future studies on aerosol formation from aromatic precursors would greatly benefit from including measurements of HOMs in order to elucidate the detailed influence of experimental conditions on aromatic-derived highly oxygenated organic molecules and SOA, in the laboratory and the atmosphere.
Data from Tables 1 and S1–S4 and Figs. 6, A1, and A2 are available at https://doi.org/10.5281/zenodo.3543607 (Garmash, 2019). Raw and high-resolution mass spectrometry data are available upon request from the corresponding authors.
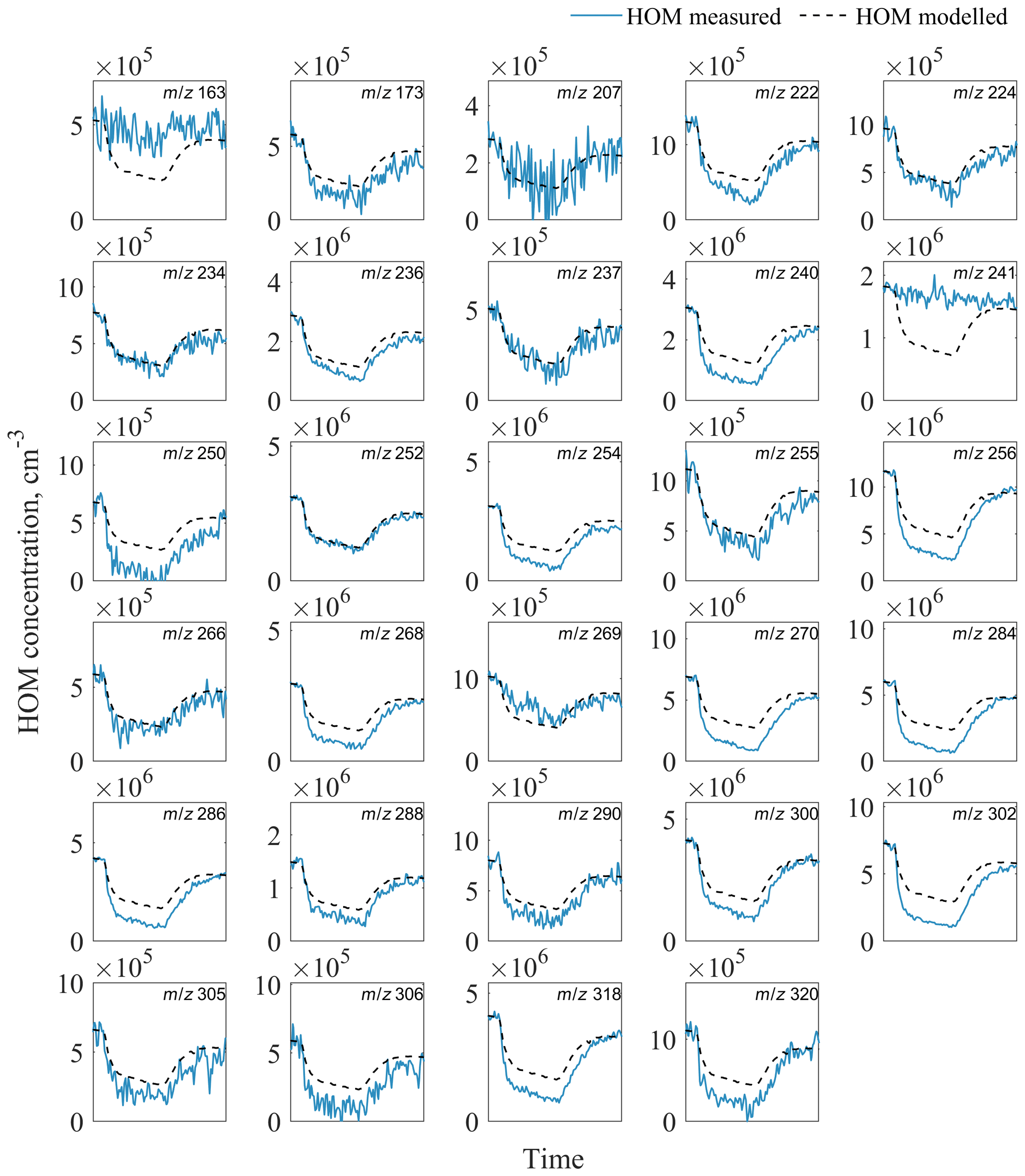
Figure A1Evolution of measured and modelled HOM monomers during the seed addition experiment. The list of HOM compositions for each peak at corresponding m∕z is presented in Table S4 in the Supplement.
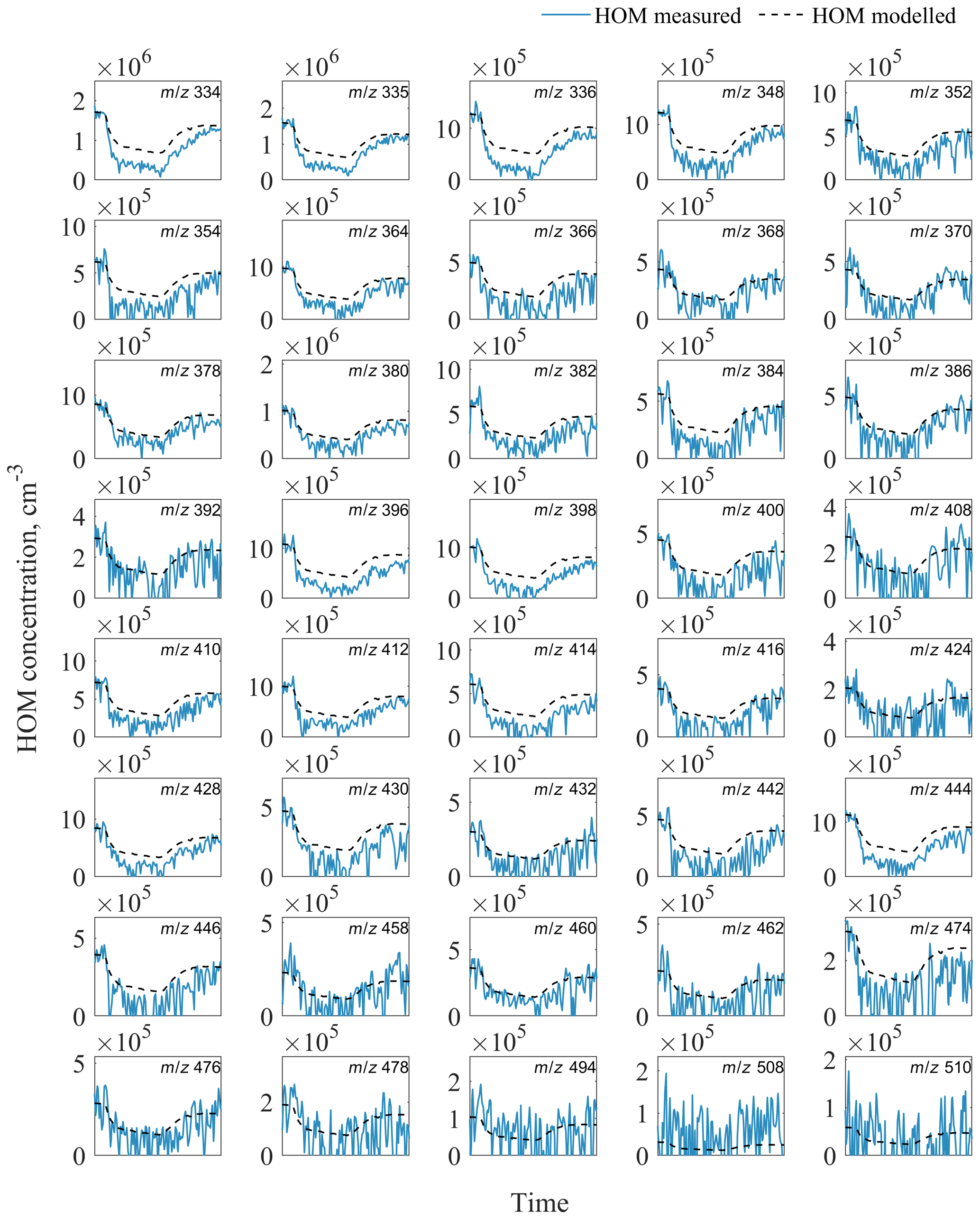
Figure A2Evolution of measured and modelled HOM dimers during the seed addition experiment. The list of HOM compositions for each peak at corresponding m∕z is presented in Table S4 in the Supplement.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-515-2020-supplement.
ME, MPR, TFM, JW, EK, and IP designed the experiments. Instrument deployment and operation were carried out by OG, MPR, IP, SS, OK, RT, DZ, TJB, MP, ÅMH, EK, JW, and TFM. Data analysis was done by OG, TFM, SS, and RT. OG interpreted the compiled data set. OG, ME, and MPR wrote the paper. All co-authors discussed the results and commented on the paper.
Oskari Kausiala works for Kärsa Oy and Sebastian Schmitt works for TSI GmbH. The authors declare that they have no conflict of interest.
We thank the tofTools team for providing tools for mass spectrometry data analysis.
This research has been supported by the European Research Council (grant no. 638703), the Academy of Finland (grant nos. 299574, 317380, 320094, 326948, 272041, and 307331), the Svenska Forskningsrådet Formas (grant no. 214-2013-1430), the VINNOVA (grant no. 2013-03058), and the Doctoral School in Atmospheric Sciences at the University of Helsinki (ATM-DP).
Open access funding provided by Helsinki University Library.
This paper was edited by Nga Lee Ng and reviewed by two anonymous referees.
Atkinson, R.: Kinetics and mechanisms of the gas-phase reactions of the hydroxyl radical with organic compounds, J. Phys. Chem. Ref. Data, 1, 1–246, 1994a. a
Atkinson, R.: Gas-Phase Tropospheric Chemistry of Organic Compounds, J. Phys. Chem. Ref. Data, 2, 1–216, 1994b. a
Atkinson, R.: Atmospheric chemistry of VOCs and NOx, Atmos. Environ., 34, 2063–2101, https://doi.org/10.1016/s1352-2310(99)00460-4, 2000. a
Atkinson, R. and Arey, J.: Atmospheric Degradation of Volatile Organic Compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003. a, b
Atkinson, R. and Aschmann, S. M.: Rate constants for the gas-phase reactions of the OH radical with a series of aromatic hydrocarbons at 296±2 K, Int. J. Chem. Kinet., 21, 355–365, https://doi.org/10.1002/kin.550210506, 1989. a, b
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006. a
Berndt, T. and Böge, O.: Formation of phenol and carbonyls from the atmospheric reaction of OH radicals with benzene, Phys. Chem. Chem. Phys., 8, 1205–1214, https://doi.org/10.1039/b514148f, 2006. a, b
Berndt, T., Richters, S., Kaethner, R., Voigtländer, J., Stratmann, F., Sipilä, M., Kulmala, M., and Herrmann, H.: Gas-Phase Ozonolysis of Cycloalkenes: Formation of Highly Oxidized RO2 Radicals and Their Reactions with NO, NO2, SO2, and Other RO2 Radicals, J. Phys. Chem. A, 119, 10336–10348, https://doi.org/10.1021/acs.jpca.5b07295, 2015. a
Berndt, T., Richters, S., Jokinen, T., Hyttinen, N., Kurtén, T., Otkjær, R. V., Kjaergaard, H. G., Stratmann, F., Herrmann, H., Sipilä, M., Kulmala, M., and Ehn, M.: Hydroxyl radical-induced formation of highly oxidized organic compounds, Nat. Commun., 7, 13677, https://doi.org/10.1038/ncomms13677, 2016. a, b
Berndt, T., Mentler, B., Scholz, W., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Ozonolysis and OH Radical Reaction of α-Pinene: Mechanistic Insight and the Influence of Isoprene and Ethylene, Environ. Sci. Technol., 52, 11069–11077, https://doi.org/10.1021/acs.est.8b02210, 2018a. a, b
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Self- and Cross-Reactions of RO2 Radicals in the Atmosphere, Angew. Chem. Int. Edit., 57, 3820–3824, https://doi.org/10.1002/anie.201710989, 2018b. a
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019. a, b, c, d, e
Birdsall, A. W. and Elrod, M. J.: Comprehensive NO-Dependent Study of the Products of the Oxidation of Atmospherically Relevant Aromatic Compounds, J. Phys. Chem. A, 115, 5397–5407, https://doi.org/10.1021/jp2010327, 2011. a
Birdsall, A. W., Andreoni, J. F., and Elrod, M. J.: Investigation of the Role of Bicyclic Peroxy Radicals in the Oxidation Mechanism of Toluene, J. Phys. Chem. A, 114, 10655–10663, https://doi.org/10.1021/jp105467e, 2010. a
Bloss, C., Wagner, V., Jenkin, M. E., Volkamer, R., Bloss, W. J., Lee, J. D., Heard, D. E., Wirtz, K., Martin-Reviejo, M., Rea, G., Wenger, J. C., and Pilling, M. J.: Development of a detailed chemical mechanism (MCMv3.1) for the atmospheric oxidation of aromatic hydrocarbons, Atmos. Chem. Phys., 5, 641–664, https://doi.org/10.5194/acp-5-641-2005, 2005. a, b, c, d, e, f, g, h, i, j, k
Calvert, J. G., Atkinson, R., Becker, K. H., Kamens, R. M., Seinfeld, J. H., Wallington, T. J., and Yarwood, G.: The Mechanisms of Atmospheric Oxidation of the Aromatic Hydrocarbons, Oxford University Press, New York, USA, 2002. a, b
Chacon-Madrid, H. J. and Donahue, N. M.: Fragmentation vs. functionalization: chemical aging and organic aerosol formation, Atmos. Chem. Phys., 11, 10553–10563, https://doi.org/10.5194/acp-11-10553-2011, 2011. a
Crounse, J. D., Knap, H. C., Ørnsø, K. B., Jørgensen, S., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric Fate of Methacrolein. 1. Peroxy Radical Isomerization Following Addition of OH and O2, J. Phys. Chem. A, 116, 5756–5762, https://doi.org/10.1021/jp211560u, 2012. a
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013. a, b, c
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006. a
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014. a, b, c, d, e, f, g, h, i, j, k, l, m
Emanuelsson, E. U., Hallquist, M., Kristensen, K., Glasius, M., Bohn, B., Fuchs, H., Kammer, B., Kiendler-Scharr, A., Nehr, S., Rubach, F., Tillmann, R., Wahner, A., Wu, H.-C., and Mentel, Th. F.: Formation of anthropogenic secondary organic aerosol (SOA) and its influence on biogenic SOA properties, Atmos. Chem. Phys., 13, 2837–2855, https://doi.org/10.5194/acp-13-2837-2013, 2013. a
Finlayson-Pitts, B. J. and Pitts, J. N. J.: Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications, Academic Press, San Diego, USA, 2000. a, b
Fuller, E. N., Schettler, P. D., and Giddings, J. C.: New method for prediction of binary gas-phase diffusion coefficients, Ind. Eng. Chem., 58, 18–27, https://doi.org/10.1021/ie50677a007, 1966. a
Garmash, O.: Laboratory simulations of benzene oxidation and formation of highly oxygenated organic molecules (HOM), Zenodo, https://doi.org/10.5281/zenodo.3543607, 2019. a
Gautrois, M. and Koppmann, R.: Diffusion technique for the production of gas standards for atmospheric measurements, J. Chromatogr. A, 848, 239–249, https://doi.org/10.1016/s0021-9673(99)00424-0, 1999. a
Glowacki, D. R. and Pilling, M. J.: Unimolecular Reactions of Peroxy Radicals in Atmospheric Chemistry and Combustion, ChemPhysChem, 11, 3836–3843, https://doi.org/10.1002/cphc.201000469, 2010. a
Glowacki, D. R., Wang, L., and Pilling, M. J.: Evidence of Formation of Bicyclic Species in the Early Stages of Atmospheric Benzene Oxidation, J. Phys. Chem. A, 113, 5385–5396, https://doi.org/10.1021/jp9001466, 2009. a, b, c, d
Graus, M., Müller, M., and Hansel, A.: High resolution PTR-TOF: Quantification and formula confirmation of VOC in real time, J. Am. Soc. Mass Spectr., 21, 1037–1044, https://doi.org/10.1016/j.jasms.2010.02.006, 2010. a
Hildebrandt, L., Donahue, N. M., and Pandis, S. N.: High formation of secondary organic aerosol from the photo-oxidation of toluene, Atmos. Chem. Phys., 9, 2973–2986, https://doi.org/10.5194/acp-9-2973-2009, 2009. a
Hyttinen, N., Kupiainen-Määttä, O., Rissanen, M. P., Muuronen, M., Ehn, M., and Kurtén, T.: Modeling the Charging of Highly Oxidized Cyclohexene Ozonolysis Products Using Nitrate-Based Chemical Ionization, J. Phys. Chem. A, 119, 6339–6345, https://doi.org/10.1021/acs.jpca.5b01818, 2015. a, b, c, d
Hyttinen, N., Otkjær, R. V., Iyer, S., Kjaergaard, H. G., Rissanen, M. P., Wennberg, P. O., and Kurtén, T.: Computational Comparison of Different Reagent Ions in the Chemical Ionization of Oxidized Multifunctional Compounds, J. Phys. Chem. A, 122, 269–279, https://doi.org/10.1021/acs.jpca.7b10015, 2017. a
Jenkin, M. E., Saunders, S. M., Wagner, V., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part B): tropospheric degradation of aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 181–193, https://doi.org/10.5194/acp-3-181-2003, 2003. a, b, c
Jokinen, T., Sipilä, M., Junninen, H., Ehn, M., Lönn, G., Hakala, J., Petäjä, T., Mauldin III, R. L., Kulmala, M., and Worsnop, D. R.: Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF, Atmos. Chem. Phys., 12, 4117–4125, https://doi.org/10.5194/acp-12-4117-2012, 2012. a, b, c
Jokinen, T., Sipilä, M., Richters, S., Kerminen, V.-M., Paasonen, P., Stratmann, F., Worsnop, D., Kulmala, M., Ehn, M., Herrmann, H., and Berndt, T.: Rapid Autoxidation Forms Highly Oxidized RO2 Radicals in the Atmosphere, Angew. Chem. Int. Edit., 53, 14596–14600, https://doi.org/10.1002/anie.201408566, 2014. a
Jokinen, T., Berndt, T., Makkonen, R., Kerminen, V.-M., Junninen, H., Paasonen, P., Stratmann, F., Herrmann, H., Guenther, A. B., Worsnop, D. R., Kulmala, M., Ehn, M., and Sipilä, M.: Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications, P. Natl. Acad. Sci. USA, 112, 7123–7128, https://doi.org/10.1073/pnas.1423977112, 2015. a
Junninen, H.: Data cycle in atmospheric physics: From detected millivolts to understanding the atmosphere, Thesis, Report Series in Aerosol Science, 145, Helsinki, available at: http://urn.fi/URN:ISBN:978-952-5822-81-6 (last access: 4 November 2019), 2013. a
Junninen, H., Ehn, M., Petäjä, T., Luosujärvi, L., Kotiaho, T., Kostiainen, R., Rohner, U., Gonin, M., Fuhrer, K., Kulmala, M., and Worsnop, D. R.: A high-resolution mass spectrometer to measure atmospheric ion composition, Atmos. Meas. Tech., 3, 1039–1053, https://doi.org/10.5194/amt-3-1039-2010, 2010. a
Kokkola, H., Yli-Pirilä, P., Vesterinen, M., Korhonen, H., Keskinen, H., Romakkaniemi, S., Hao, L., Kortelainen, A., Joutsensaari, J., Worsnop, D. R., Virtanen, A., and Lehtinen, K. E. J.: The role of low volatile organics on secondary organic aerosol formation, Atmos. Chem. Phys., 14, 1689–1700, https://doi.org/10.5194/acp-14-1689-2014, 2014. a
Kroll, J. H. and Seinfeld, J. H.: Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 42, 3593–3624, https://doi.org/10.1016/j.atmosenv.2008.01.003, 2008. a
Kulmala, M., Petäjä, T., Nieminen, T., Sipilä, M., Manninen, H. E., Lehtipalo, K., Maso, M. D., Aalto, P. P., Junninen, H., Paasonen, P., Riipinen, I., Lehtinen, K. E. J., Laaksonen, A., and Kerminen, V.-M.: Measurement of the nucleation of atmospheric aerosol particles, Nat. Protoc., 7, 1651–1667, https://doi.org/10.1038/nprot.2012.091, 2012. a
Lay, T. H., Bozzelli, J. W., and Seinfeld, J. H.: Atmospheric Photochemical Oxidation of Benzene: Benzene + OH and the Benzene-OH Adduct (Hydroxyl-2, 4-cyclohexadienyl) + O2, J. Phys. Chem., 100, 6543–6554, https://doi.org/10.1021/jp951726y, 1996. a
Lehtipalo, K., Yan, C., Dada, L., Bianchi, F., Xiao, M., Wagner, R., Stolzenburg, D., Ahonen, L. R., Amorim, A., Baccarini, A., Bauer, P. S., Baumgartner, B., Bergen, A., Bernhammer, A.-K., Breitenlechner, M., Brilke, S., Buchholz, A., Mazon, S. B., Chen, D., Chen, X., Dias, A., Dommen, J., Draper, D. C., Duplissy, J., Ehn, M., Finkenzeller, H., Fischer, L., Frege, C., Fuchs, C., Garmash, O., Gordon, H., Hakala, J., He, X., Heikkinen, L., Heinritzi, M., Helm, J. C., Hofbauer, V., Hoyle, C. R., Jokinen, T., Kangasluoma, J., Kerminen, V.-M., Kim, C., Kirkby, J., Kontkanen, J., Kürten, A., Lawler, M. J., Mai, H., Mathot, S., Mauldin, R. L., Molteni, U., Nichman, L., Nie, W., Nieminen, T., Ojdanic, A., Onnela, A., Passananti, M., Petäjä, T., Piel, F., Pospisilova, V., Quéléver, L. L. J., Rissanen, M. P., Rose, C., Sarnela, N., Schallhart, S., Schuchmann, S., Sengupta, K., Simon, M., Sipilä, M., Tauber, C., Tomé, A., Tröstl, J., Väisänen, O., Vogel, A. L., Volkamer, R., Wagner, A. C., Wang, M., Weitz, L., Wimmer, D., Ye, P., Ylisirniö, A., Zha, Q., Carslaw, K. S., Curtius, J., Donahue, N. M., Flagan, R. C., Hansel, A., Riipinen, I., Virtanen, A., Winkler, P. M., Baltensperger, U., Kulmala, M., and Worsnop, D. R.: Multicomponent new particle formation from sulfuric acid, ammonia, and biogenic vapors, Sci. Adv., 4, eaau5363, https://doi.org/10.1126/sciadv.aau5363, 2018. a
Lindinger, W., Hansel, A., and Jordan, A.: On-line monitoring of volatile organic compounds at pptv levels by means of proton-transfer-reaction mass spectrometry (PTR-MS) medical applications, food control and environmental research, Int. J. Mass Spectrom., 173, 191–241, https://doi.org/10.1016/s0168-1176(97)00281-4, 1998. a
Liu, P.-W. G., Yao, Y.-C., Tsai, J.-H., Hsu, Y.-C., Chang, L.-P., and Chang, K.-H.: Source impacts by volatile organic compounds in an industrial city of southern Taiwan, Sci. Total Environ., 398, 154–163, https://doi.org/10.1016/j.scitotenv.2008.02.053, 2008. a
McFiggans, G., Mentel, T. F., Wildt, J., Pullinen, I., Kang, S., Kleist, E., Schmitt, S., Springer, M., Tillmann, R., Wu, C., Zhao, D., Hallquist, M., Faxon, C., Le Breton, M., Hallquist, Å. M., Simpson, D., Bergström, R., Jenkin, M. E., Ehn, M., Thornton, J. A., Alfarra, M. R., Bannan, T. J., Percival, C. J., Priestley, M., Topping, D., and Kiendler-Scharr, A.: Secondary organic aerosol reduced by mixture of atmospheric vapours, Nature, 565, 587–593, https://doi.org/10.1038/s41586-018-0871-y, 2019. a
Mentel, T. F., Wildt, J., Kiendler-Scharr, A., Kleist, E., Tillmann, R., Dal Maso, M., Fisseha, R., Hohaus, Th., Spahn, H., Uerlings, R., Wegener, R., Griffiths, P. T., Dinar, E., Rudich, Y., and Wahner, A.: Photochemical production of aerosols from real plant emissions, Atmos. Chem. Phys., 9, 4387–4406, https://doi.org/10.5194/acp-9-4387-2009, 2009. a, b
Mentel, T. F., Springer, M., Ehn, M., Kleist, E., Pullinen, I., Kurtén, T., Rissanen, M., Wahner, A., and Wildt, J.: Formation of highly oxidized multifunctional compounds: autoxidation of peroxy radicals formed in the ozonolysis of alkenes – deduced from structure–product relationships, Atmos. Chem. Phys., 15, 6745–6765, https://doi.org/10.5194/acp-15-6745-2015, 2015. a, b
Misztal, P., Hewitt, C., Wildt, J., Blande, J., Eller, A., Fares, S., Gentner, D., Gilman, J., Graus, M., Greenberg, J., Guenther, A., Hansel, A., Harley, P., Huang, M., Jardine, K., Karl, T., Kaser, L., Keutsch, F., Kiendler-Scharr, A., Kleist, E., Lerner, B., Li, T., Mak, J., Nölscher, A., Schnitzhofer, R., Sinha, V., Thornton, B., Warneke, C., Wegener, F., Werner, C., Williams, J., Worton, D., Yassaa, N., and Goldstein, A.: Atmospheric benzenoid emissions from plants rival those from fossil fuels, Sci. Rep., 5, 12064, https://doi.org/10.1038/srep12064, 2015. a
Molteni, U., Bianchi, F., Klein, F., El Haddad, I., Frege, C., Rossi, M. J., Dommen, J., and Baltensperger, U.: Formation of highly oxygenated organic molecules from aromatic compounds, Atmos. Chem. Phys., 18, 1909–1921, https://doi.org/10.5194/acp-18-1909-2018, 2018. a, b, c, d, e, f, g, h, i, j, k, l
Ng, N. L., Kroll, J. H., Chan, A. W. H., Chhabra, P. S., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from m-xylene, toluene, and benzene, Atmos. Chem. Phys., 7, 3909–3922, https://doi.org/10.5194/acp-7-3909-2007, 2007. a, b
Olariu, R. I., Klotz, B., Barnes, I., Becker, K. H., and Mocanu, R.: FT–IR study of the ring-retaining products from the reaction of OH radicals with phenol, o-, m-, and p-cresol, Atmos. Environ., 36, 3685–3697, https://doi.org/10.1016/s1352-2310(02)00202-9, 2002. a
Orlando, J. J. and Tyndall, G. S.: Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 41, 6294–6317, https://doi.org/10.1039/c2cs35166h, 2012. a
Orlando, J. J., Tyndall, G. S., and Wallington, T. J.: The Atmospheric Chemistry of Alkoxy Radicals, Chem. Rev., 103, 4657–4690, https://doi.org/10.1021/cr020527p, 2003. a
Öström, E., Putian, Z., Schurgers, G., Mishurov, M., Kivekäs, N., Lihavainen, H., Ehn, M., Rissanen, M. P., Kurtén, T., Boy, M., Swietlicki, E., and Roldin, P.: Modeling the role of highly oxidized multifunctional organic molecules for the growth of new particles over the boreal forest region, Atmos. Chem. Phys., 17, 8887–8901, https://doi.org/10.5194/acp-17-8887-2017, 2017. a
Praske, E., Otkjær, R. V., Crounse, J. D., Hethcox, J. C., Stoltz, B. M., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric autoxidation is increasingly important in urban and suburban North America, P. Natl. Acad. Sci. USA, 115, 64–69, https://doi.org/10.1073/pnas.1715540115, 2018. a
Rissanen, M. P.: NO2 Suppression of Autoxidation–Inhibition of Gas-Phase Highly Oxidized Dimer Product Formation, ACS Earth and Space Chemistry, 2, 1211–1219, https://doi.org/10.1021/acsearthspacechem.8b00123, 2018. a, b, c
Rissanen, M. P., Kurtén, T., Sipilä, M., Thornton, J. A., Kangasluoma, J., Sarnela, N., Junninen, H., Jørgensen, S., Schallhart, S., Kajos, M. K., Taipale, R., Springer, M., Mentel, T. F., Ruuskanen, T., Petäjä, T., Worsnop, D. R., Kjaergaard, H. G., and Ehn, M.: The Formation of Highly Oxidized Multifunctional Products in the Ozonolysis of Cyclohexene, J. Am. Chem. Soc., 136, 15596–15606, https://doi.org/10.1021/ja507146s, 2014. a, b, c
Rissanen, M. P., Kurtén, T., Sipilä, M., Thornton, J. A., Kausiala, O., Garmash, O., Kjaergaard, H. G., Petäjä, T., Worsnop, D. R., Ehn, M., and Kulmala, M.: Effects of Chemical Complexity on the Autoxidation Mechanisms of Endocyclic Alkene Ozonolysis Products: From Methylcyclohexenes toward Understanding α-Pinene, J. Phys. Chem. A, 119, 4633–4650, https://doi.org/10.1021/jp510966g, 2015. a, b
Rubach, F.: Aerosol processes in the Planetary Boundary Layer: High resolution Aerosol Mass Spectrometry on a Zeppelin NT Airship, Forschungszentrum Jülich, Reihe Energie & Umwelt/Energy & Environment, 196. iii, 141, available at: http://hdl.handle.net/2128/5160 (last access: 4 November 2019), 2013. a
Schwantes, R. H., Schilling, K. A., McVay, R. C., Lignell, H., Coggon, M. M., Zhang, X., Wennberg, P. O., and Seinfeld, J. H.: Formation of highly oxygenated low-volatility products from cresol oxidation, Atmos. Chem. Phys., 17, 3453–3474, https://doi.org/10.5194/acp-17-3453-2017, 2017. a
Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., Wagner, R., Simon, M., Lehtipalo, K., Williamson, C., Craven, J. S., Duplissy, J., Adamov, A., Almeida, J., Bernhammer, A.-K., Breitenlechner, M., Brilke, S., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Gysel, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Keskinen, H., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lawler, M., Leiminger, M., Mathot, S., Möhler, O., Nieminen, T., Onnela, A., Petäjä, T., Piel, F. M., Miettinen, P., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Sipilä, M., Smith, J. N., Steiner, G., Tomè, A., Virtanen, A., Wagner, A. C., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Carslaw, K. S., Curtius, J., Dommen, J., Kirkby, J., Kulmala, M., Riipinen, I., Worsnop, D. R., Donahue, N. M., and Baltensperger, U.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 533, 527–531, https://doi.org/10.1038/nature18271, 2016. a
Vereecken, L.: Reaction Mechanisms for the Atmospheric Oxidation of Monocyclic Aromatic Compounds, in: Advances in Atmospheric Chemistry, 377–527, https://doi.org/10.1142/9789813271838_0006, 2019. a
Vereecken, L. and Peeters, J.: Decomposition of substituted alkoxy radicals – part I: a generalized structure–activity relationship for reaction barrier heights, Phys. Chem. Chem. Phys., 11, 9062, https://doi.org/10.1039/b909712k, 2009. a
Volkamer, R., Klotz, B., Barnes, I., Imamura, T., Wirtz, K., Washida, N., Becker, K. H., and Platt, U.: OH-initiated oxidation of benzene, Phys. Chem. Chem. Phys., 4, 1598–1610, https://doi.org/10.1039/b108747a, 2002. a, b, c
Wang, L., Wu, R., and Xu, C.: Atmospheric Oxidation Mechanism of Benzene. Fates of Alkoxy Radical Intermediates and Revised Mechanism, J. Phys. Chem. A, 117, 14163–14168, https://doi.org/10.1021/jp4101762, 2013. a, b, c, d, e
Wang, S., Wu, R., Berndt, T., Ehn, M., and Wang, L.: Formation of Highly Oxidized Radicals and Multifunctional Products from the Atmospheric Oxidation of Alkylbenzenes, Environ. Sci. Technol., 51, 8442–8449, https://doi.org/10.1021/acs.est.7b02374, 2017. a, b, c, d
Yan, C., Nie, W., Äijälä, M., Rissanen, M. P., Canagaratna, M. R., Massoli, P., Junninen, H., Jokinen, T., Sarnela, N., Häme, S. A. K., Schobesberger, S., Canonaco, F., Yao, L., Prévôt, A. S. H., Petäjä, T., Kulmala, M., Sipilä, M., Worsnop, D. R., and Ehn, M.: Source characterization of highly oxidized multifunctional compounds in a boreal forest environment using positive matrix factorization, Atmos. Chem. Phys., 16, 12715–12731, https://doi.org/10.5194/acp-16-12715-2016, 2016. a
Zabel, F.: Unimolecular Decomposition of Peroxynitrates, Z. Phys. Chem., 188, 119–142, https://doi.org/10.1524/zpch.1995.188.part_1_2.119, 1995. a
Zhang, X., Cappa, C. D., Jathar, S. H., McVay, R. C., Ensberg, J. J., Kleeman, M. J., and Seinfeld, J. H.: Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol, P. Natl. Acad. Sci. USA, 111, 5802–5807, https://doi.org/10.1073/pnas.1404727111, 2014. a