the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 30 Apr 2020
| 30 Apr 2020
Formation of highly oxygenated organic molecules from chlorine-atom-initiated oxidation of alpha-pinene
Matthieu Riva
Hongbin Xie
Liine Heikkinen
Simon Schallhart
Qiaozhi Zha
Xu-Cheng He
Otso Peräkylä
Mikael Ehn
Highly oxygenated organic molecules (HOMs) from atmospheric oxidation of alpha-pinene can irreversibly condense to particles and contribute to secondary organic aerosol (SOA) formation. Recently, the formation of nitryl chloride (ClNO2) from heterogeneous reactions, followed by its subsequent photolysis, is suggested to be an important source of chlorine atoms in many parts of the atmosphere. However, the oxidation of monoterpenes such as alpha-pinene by chlorine atoms has received very little attention, and the ability of this reaction to form HOMs is completely unstudied. Here, chamber experiments were conducted with alpha-pinene and chlorine under low- and high-nitrogen-oxide (NOx, ) conditions. A nitrate-based CI-APi-ToF (chemical ionization–atmospheric pressure interface–time of flight) mass spectrometer was used to measure HOM products. Clear distributions of monomers with 9–10 carbon atoms and dimers with 18–20 carbon atoms were observed under low-NOx conditions. With increased concentration of NOx within the chamber, the formation of dimers was suppressed due to the reactions of peroxy radicals with NO. We estimated the HOM yields from chlorine-initiated oxidation of alpha-pinene under low-NOx conditions to be around 1.8 %, though with a substantial uncertainty range (0.8 %–4 %) due to lack of suitable calibration methods. Corresponding yields at high NOx could not be determined because of concurrent ozonolysis reactions. Our study demonstrates that also the oxidation of alpha-pinene by chlorine atoms and yield low-volatility organic compounds.
- Article
(1801 KB) - Full-text XML
- BibTeX
- EndNote





Highly oxygenated organic molecules (HOMs) have been identified as key species in the formation of new atmospheric aerosol particles and secondary organic aerosol (SOA; Ehn et al., 2014, 2017; Kulmala et al., 2013; Bianchi et al., 2019). Recently, the formation of HOMs in the gas phase was described as an autoxidation process of peroxy radicals (RO2) via multiple intramolecular H atom shifts (Crounse et al., 2013; Jokinen et al., 2014; Mentel et al., 2015; Rissanen et al., 2014). Oxygen-containing moieties such as carbonyl, carboxylic acid and hydroxyl groups can weaken nearby C−H bonds, making H abstraction and autoxidation competitive with bimolecular RO2 reactions, e.g. with NO (Crounse et al., 2013; Praske et al., 2018). Until now, all studies on the formation of HOMs have focused on reactions initiated by oxygen-containing oxidants (O3 and OH; Berndt et al., 2016; Ehn et al., 2014; Jokinen et al., 2015).
Increasing evidence indicates that the chlorine atom (Cl) may also play an important role in transforming atmospheric organics (Tham et al., 2016; Thornton et al., 2010). Chlorine atoms have the greatest reactivity toward volatile organic compounds (VOCs), with rate constants that are, with some exceptions, an order of magnitude higher than those of hydroxyl radicals (OH; Riva et al., 2015). Historically, chlorine atoms were thought to be formed primarily from heterogeneous reaction cycles involving sea salt, and their concentrations estimated to be around 1 %–10 % of that of OH (Thornton et al., 2010). Therefore, the role of chlorine atoms in atmospheric oxidation processes has traditionally been thought to be limited to the marine boundary layer only. In recent years, ClNO2, as a significant chlorine atom source, was found in continental regions of America, Canada and Germany, and high concentrations of ClNO2 were also detected in the urban atmosphere in China (Reyes-Villegas et al., 2018; Tham et al., 2016; Thornton et al., 2010; Wang et al., 2017). The new findings have expanded the potential importance of chlorine atoms from coastal areas to continental urban areas. A recent study also reported that chlorine atoms can be more important than OH radicals for the oxidation of alkanes in the North China Plain (Liu et al., 2017). Therefore, it is desirable to probe the role of chlorine radicals in the degradation of VOCs and related SOA formation.
Emission of biogenic volatile organic compounds (BVOCs) to the atmosphere dominates total hydrocarbon emissions on a global scale, with methane, isoprene and terpenes having the highest source strengths (Guenther et al., 1995). Alpha-pinene is the most abundant monoterpene in the atmosphere, and its oxidation products from ozonolysis and photooxidation contribute to a substantial fraction of SOA mass (Riccobono et al., 2014; Zhang et al., 2018). Many of the regions listed earlier, where ClNO2 was identified as a chlorine atom source, may also have substantial monoterpene emissions, making Cl a relevant oxidant also for BVOC. Chlorine-atom-initiated reactions of alpha-pinene have also been shown to contribute to the formation of SOA, which implies that low-volatility compounds are efficiently produced also in this process (Cai and Griffin, 2006; Ofner et al., 2013).
Similar to the reaction with OH radicals, the reaction of VOCs with chlorine atoms may proceed either via addition of Cl to unsaturated bonds or via H abstraction. Wang et al. (2017) found that the Cl addition to isoprene can lead to the formation of low-volatility organic compounds. In principle, Cl-initiated reactions could form HOMs in a similar manner to OH-initiated reactions (Berndt et al., 2016), as the initial addition or abstraction step is comparable for both oxidants. In view of the increased understanding of the importance of chlorine atoms in atmospheric chemistry, it is desirable to investigate the formation of HOMs from reactions of common atmospheric VOC with Cl.
Here, laboratory chamber experiments were performed to investigate the ability of the chlorine atom to form HOMs from the oxidation of alpha-pinene. HOMs were characterized using a nitrate-based chemical ionization mass spectrometer, under both low- and high-NOx conditions. The yields of these HOMs were determined under the low-NOx conditions, and the atmospheric implications of this study are discussed.
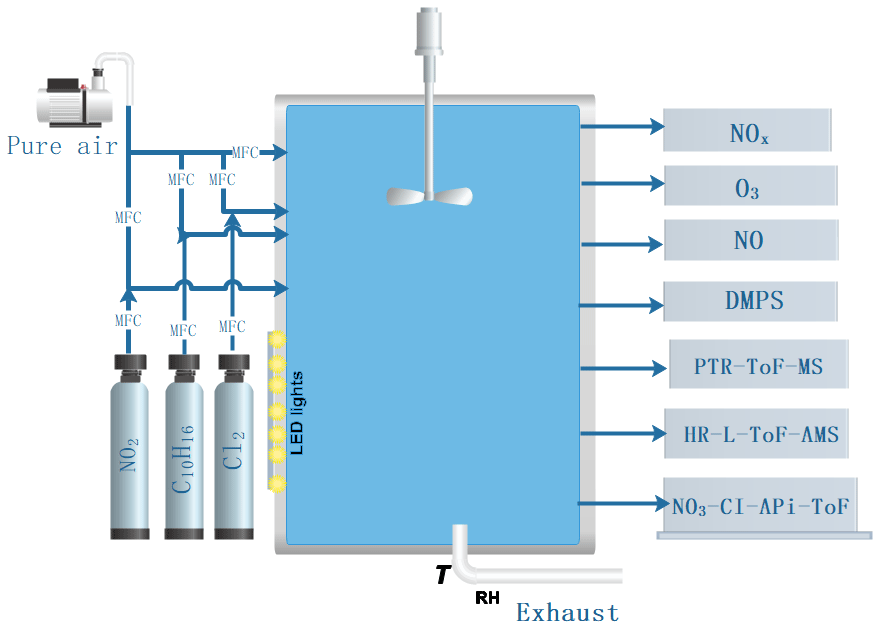
2.1 Experimental set-up
The experiments were conducted in the “COALA” chamber at the University of Helsinki (Peräkylä et al., 2020; Riva et al., 2019b). This is a 2 m3 Teflon chamber, run as a continuously stirred tank reactor, used with a flow of 45 L min−1 (L min−1), resulting in an average residence time of about 45 min. The chamber is surrounded by housing to provide dark conditions. No water vapour was added to the chamber, resulting in an RH <1 %, and the temperature was the same as the temperature of the room, around 25 ∘C. The HOM formation targeted in this work is not expected to change markedly as a function of RH, as also indicated in a previous study on both ozone and OH-initiated oxidation of monoterpenes (Li et al., 2019). Although SOA mass yields were not studied in this work, these may be affected by RH (Jonsson et al., 2006, 2008). A general schematic of the chamber facility is shown in Fig. 1. Our experiment was aimed to probe chlorine-atom-initiated formation of HOMs under low- and high-NOx conditions. We use 400 nm LED lights to photolyse chlorine and NO2 and produce chlorine atoms and NO as follows:
The concentration of Cl atoms was varied by changing the amount of 400 nm light. In practice this was done by turning on different numbers of the available lights, with the maximum corresponding to seven. We will refer below to the number of lights that were turned on, although each “light” corresponds to a group of LED strips.
2.2 Instrumentation and data analysis
A nitrate-ion-based (-based) chemical ionization–atmospheric pressure interface–time-of-flight (CI-APi-ToF) mass spectrometer was used for measuring HOMs. The instrument has been shown to be sensitive towards this group of compounds, detecting them as adducts with the nitrate ion. Due to a lack of suitable calibration methods, the CI-APi-ToF was not calibrated for HOMs during this study. In order to estimate rough HOM concentrations ([HOM]), we directly use the calibration coefficient molec. cm−3 utilized by Ehn et al. (2014) to convert the measured HOM ion signals according to the equation below (Jokinen et al., 2014):
This value of C is very close to values utilized in several other studies using a CI-APi-ToF (Jokinen et al., 2014, 2015; Riva et al., 2019a). Ehn et al. (2014) obtained their calibration factor from a permeation source using a perfluorinated acid and also showed through calculations that there were theoretical limitations for having a much larger or a much smaller value for C.
This approach obviously brings large uncertainties to the HOM concentrations, which we estimate to be at least −50 % ± 100 % according to previous calibration results (Jokinen et al., 2014, 2015; Riva et al., 2019). More detailed information about the instrument can be found in Jokinen et al. (2012), noting that compared with the CI-APi-ToF used before, the long time-of-flight mass spectrometer used here has a doubled mass-resolving power enabling a more accurate assignment of molecular formulas. Simultaneously, we also used a high-resolution long time-of-flight aerosol mass spectrometer (HR-L-ToF-AMS) to measure bulk aerosol chemical properties (Decarlo et al., 2006). As no seed aerosol particles were added to the chamber, the VOC oxidation products lead to new particle formation and growth to large enough sizes to be measured by the AMS. We also periodically used a filter in front of the AMS inlet to see the influence of the background signal on measured aerosol mass concentration. There were 10 min of filter measurements per hour during our experiments. A PTR-ToF-MS (TOF-8000, Ionicon) was used to measure the concentration of alpha-pinene in the chamber. The instrument background was determined every day for 20 min by guiding the chamber air through a catalytic converter, which removes the VOCs. Then, the background-corrected signals were used to obtain alpha-pinene mixing ratios by using the calibration coefficient determined before the experiments. A description of the used set-up employed for the calibration and zero-air measurements has been introduced earlier (Schallhart et al., 2018). A custom-built DMPS system was used to measure the particle number size distribution from 10 to 400 nm in the chamber. The NO concentration was measured with an ECO-PHYSICS CLD 780 TR instrument with a detection limit of 3 ppt. ) concentrations were determined by using a Thermo Fisher 42i analyser. O3 concentration was measured with a Thermo Fisher 49i analyser.
2.3 Estimation of chlorine atom concentrations
During steady state in the chamber, average concentrations of the chlorine atom were calculated using the rate coefficients cm3 molecule−1 s−1 of Cl atoms with alpha-pinene (Finlayson-Pitts et al., 1999), as follows:
where Qin is the flow rate of alpha-pinene continuously injected into the chamber, and Qout is the flow rate that exited the chamber. The term-Qin-related concentration was 13.3 ppb, while the term Qout varied depending on the conditions and is calculated as [AP] ∕ 45 min. During steady state, d[AP]∕dt is zero, and then [Cl] concentration is calculated accordingly. As shown in Fig. 2, the concentration of HOMs decreased and alpha-pinene increased as the number of lights switched on changed from seven to four, two and one. We use the variation in alpha-pinene and HOM concentrations during this run to calculate both chlorine atom concentrations and HOM yields. Each change in alpha-pinene concentration was due to the change in Cl atom concentration, and with knowledge of the reaction rate, the concentration of Cl atoms as a function of the number of lights turned on was determined (Fig. 3). The calculated [Cl] concentrations are in the range of molecules cm−3, which is within atmospherically relevant concentration ranges (Tham et al., 2016). Raw data from the CI-APi-ToF were recorded in 10 s resolution in HDF format. We used tofTools for data analysis, and detailed protocols of the software have been introduced by Junninen et al. (2010).
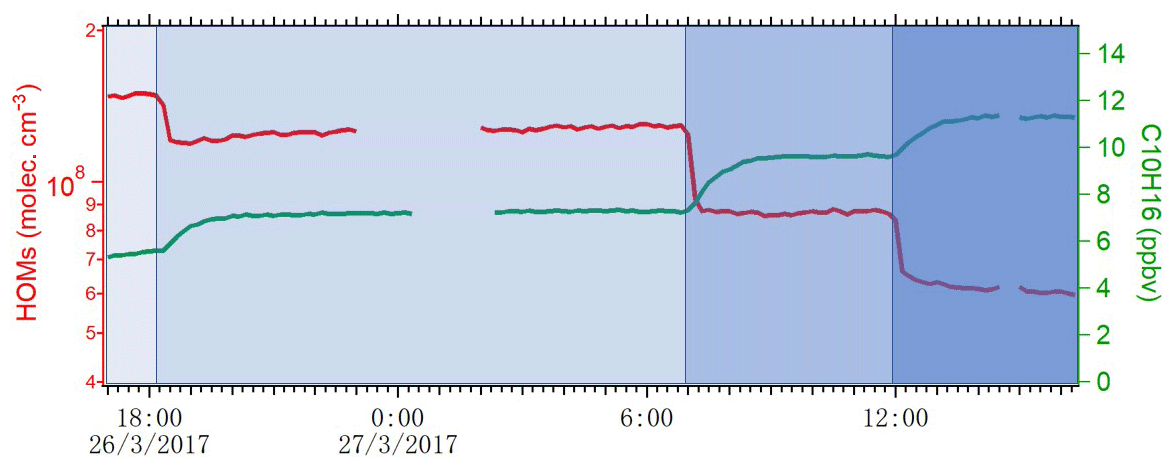
Figure 2The variation in total HOM concentration and alpha-pinene during four experiments where the 400 nm lights were decreased stepwise from seven lights to four lights, two lights and one light.
2.4 HOM molar yield
The change of HOM concentration with time can be described as follows, analogous to Ehn et al. (2014):
Here, k1 is the reaction rate coefficient of alpha-pinene with chlorine atoms and γ is the molar yield of HOMs, i.e., the fraction of alpha-pinene + Cl reactions that produced HOMs. kloss is the loss rate of HOMs to the chamber walls and particles, though the latter was negligible in this study due to the low aerosol loadings. We used 300 s as a lifetime of HOMs, i.e. s−1, in our previous study in the COALA chamber (Riva et al., 2019).
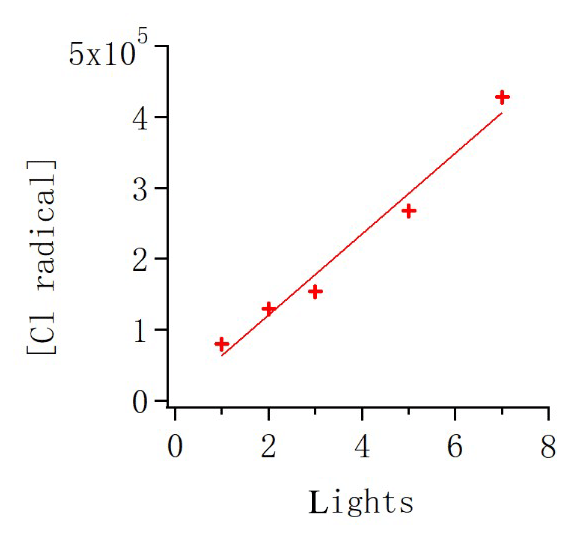
Figure 3The variation in chlorine radical concentration as a function of lights. The input alpha-pinene concentration was kept constant throughout the experiments.
3.1 Formation of HOMs under low-NO condition
Figure 4a–d show mass spectra measured by the NO3-CI-APi-ToF during steady-state alpha-pinene oxidation with different numbers of lights switched on. The x axis represents mass-to-charge ratio, in units of thomson (Th). The y axis represents signals in units of counts per second. As we can see, both monomers (280–400 Th) and dimers (440–580 Th) showed increased signals with an increased number of lights and, consequently, increased [Cl]. The most abundant peaks are labelled in Fig. 4d, with some of the largest signals in the monomer range attributed to C9H12O7,8 and . The formation of both groups could correspond to the oxidation being initiated by H-atom abstraction by Cl and final termination (from uni- or bimolecular reactions) leading to loss of OH or HO2. For the C9 compounds, an additional loss of formaldehyde (CH2O) during the oxidation process would explain the number of observed C and H atoms. During the oxidation of C10H16, in the absence of NO, the fate of RO2 radicals depends on the concentrations of HO2 and RO2. Autoxidation competes with bimolecular reactions, becoming more likely at lower RO2 and HO2 concentrations.
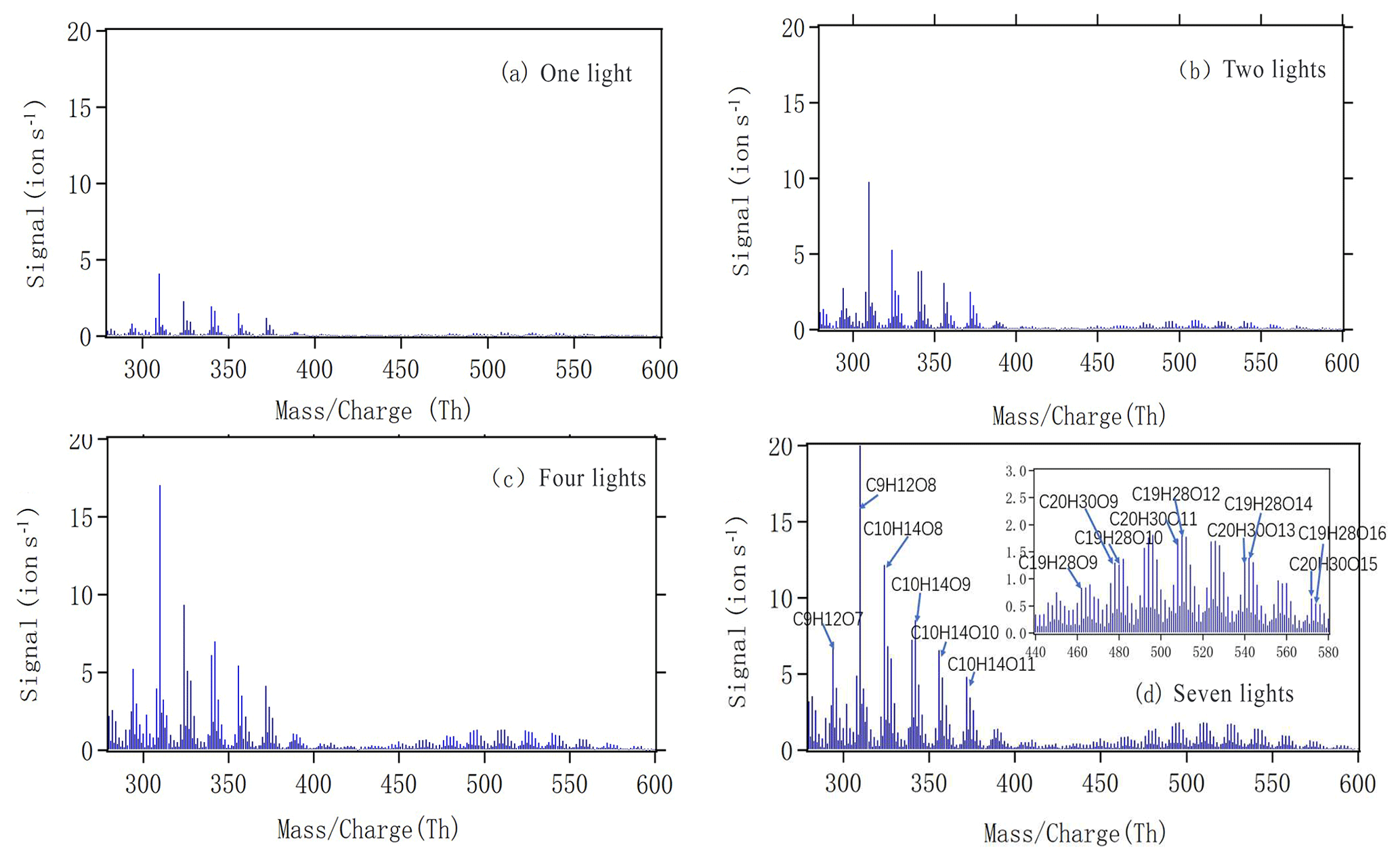
Figure 4The mass spectra obtained by NO3 CI-APi-ToF during steady state with one (a), two (b), four (c) and seven (d) lights. All peaks are detected as clusters with . The spectra are plotted as unit mass resolution, with background signals removed, but the peak identifications (labelled in panel d) are based on high-resolution analyses. The spectra correspond to the same four steady-state conditions depicted in Fig. 2.
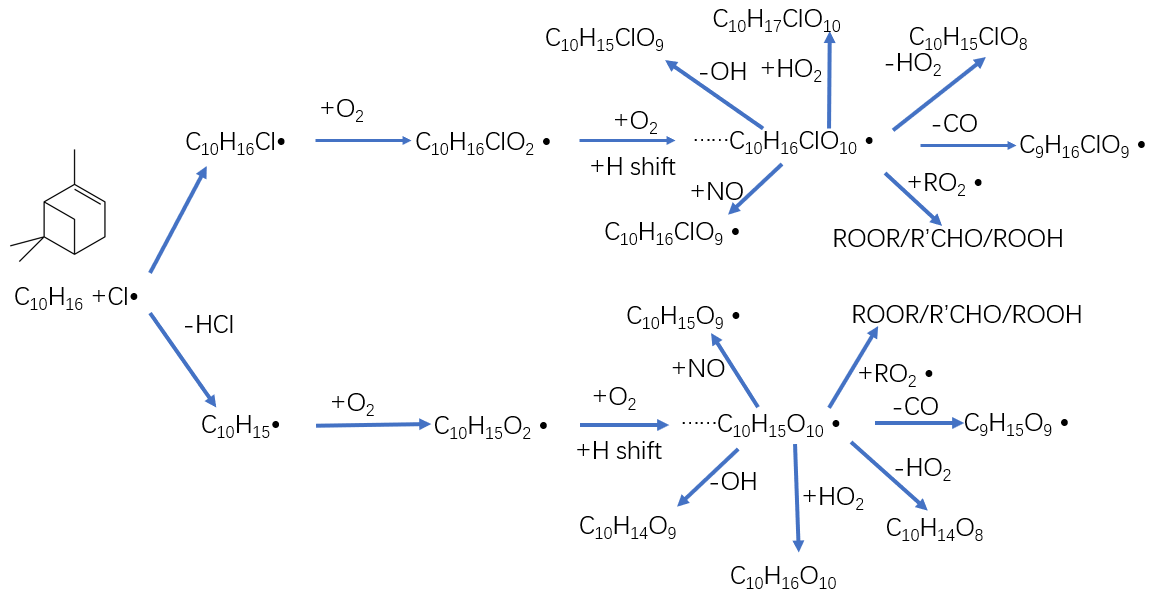
Figure 5Proposed chemical pathways for chlorine radical oxidation of alpha-pinene and subsequent autoxidation and HOM formation. The upper path shows the chlorine radical addition pathway, while the lower chain shows hydrogen atom abstraction pathway. In both cases, initially a C-centred radical forms (C10H16Cl or C10H15) to which O2 is added to form an initial peroxy radical. This peroxy radical may then undergo multi-step autoxidation to reach the example molecules C10H16ClO10 or C10H15O10 before termination.
As we show in the Fig. 4d, C10H14O8−12 compounds are large peaks in the monomer range observed with the NO3 CI-APi-ToF. These compounds with 14 hydrogens may come from decomposition of C10H15On peroxy radicals via loss of OH or HO2 or following reactions with other RO2, as depicted schematically in Fig. 5. Another abundant group is C10H16O6−12, which may result from RO2 terminated by HO2. In the dimer range, the most abundant compounds are C19H28O8−14 and C20H30O11−14. These compounds come from RO2 cross reactions, as has been shown in multiple earlier studies (Ehn et al., 2014; Jokinen et al., 2015; Mentel et al., 2015). The C20H30On dimers are most likely formed from reactions of two C10H15Ox radicals, as were many abundant monomers. As noted earlier, Cl oxidation of alkenes may occur via a Cl addition (forming an initial radical containing 16 H atoms and 1 Cl atom) or via an H-abstraction reaction (forming a radical with 15 H atoms and no Cl; Fig. 5). The abstraction pathway either leads to HOM formation or the Cl atom is lost during the subsequent reaction in the oxidation processes. With our data, we cannot rule out either of these explanations for this result. Loss of HCl from alpha-pinene products from Cl oxidation has, to our knowledge, only been reported to take place in the aerosol phase (Ofner et al., 2013).
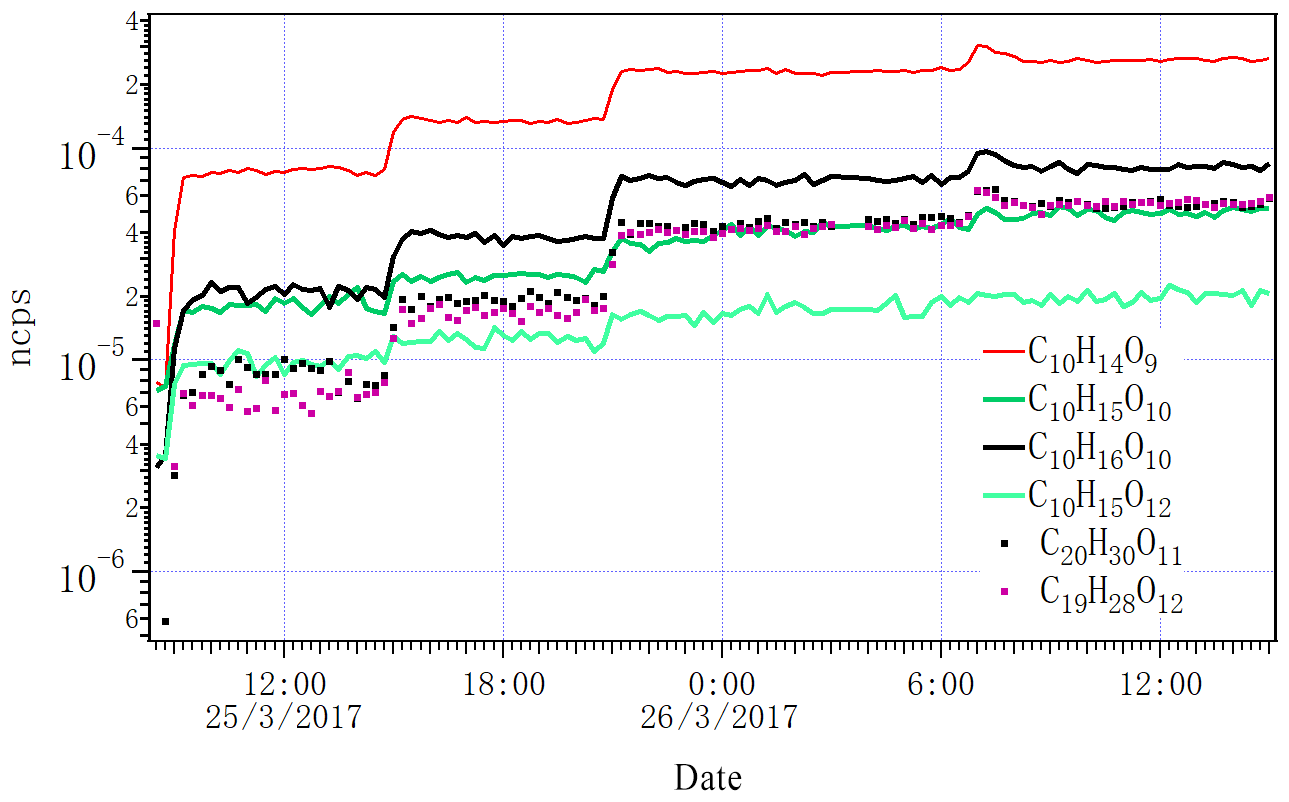
Figure 6Time series of selected closed-shell HOM monomers, dimers and two RO2 radicals (C10H15O10 and C10H15O12) detected by NO3-CI-APi-ToF as the lights increased from zero to one, two, four and seven. ncps stands for normalized counts per second.
Figure 6 shows the variation in several close-shelled HOM products and the peroxy radical C10H15O10 measured by NO3 CI-APi-ToF when we changed the lights from dark conditions to one, two, four and seven lights switched on. Given the low Cl atom concentration, it is expected that no multi-generation oxidation by Cl can take place, and the behaviour of all closed-shell oxidation products should follow similar patterns. As seen in Fig. 6, this was the case both for monomers and dimers. The less steep increase in the radical is also according to expectations, as the formation of RO2 is linear with the alpha-pinene oxidation rate, but the loss rate (when dominated by RO2 cross reactions) is proportional to (RO2)2. For closed-shell species, the wall loss-driven loss rate stays constant throughout the experiment, and therefore they increase linearly with the alpha-pinene oxidation rate while the RO2 radicals increase as the square root of the oxidation rate. For more detailed discussion on RO2 dynamics in a steady-state chamber, see Ehn et al. (2014).
In Fig. 7, we plotted time series of the particle number size distribution and the total number concentration together with mass concentrations of particulate chloride and organics as we changed the number of lights. Particle formation was detected even at the lowest Cl atom concentration, as indicated by the increases in aerosol number concentration. An increase in aerosol mass concentration as detected by the AMS only took place at the two highest Cl atom concentrations, when the particles were able to grow into a size range measurable by the AMS. Particulate chloride mass concentrations also increased relatively linearly with the concentration of organics as we increased the number of lights. The Chl / Org ratio was only around 3 %, suggesting that the majority of condensed OVOCs (oxygenated volatile organic compounds) did not contain Cl atoms. However, the exact Chl quantification from organochlorides using the AMS may contain uncertainties (Wang and Ruiz, 2017), and we avoid drawing conclusions that are too far-reaching from this value, keeping also in mind that some fraction of the size distribution was below the lowest detectable size of our AMS. In addition, some part of the chloride signal may also result from adsorption of HCl to particles.
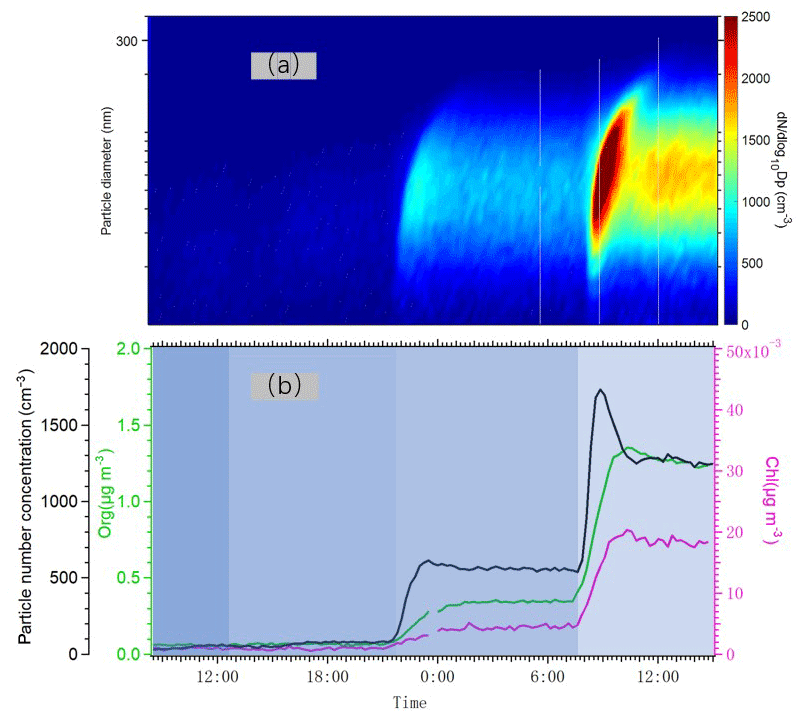
Figure 7(a) Particle number size distribution measured by DMPS from 10 to 400 nm, when the lights varied from one to two, four and seven. (b) Time series of total number concentration (black) measured by DMPS and organic aerosol concentration (green) and particulate chloride concentration (pink) measured by AMS.
3.2 Formation of HOMs at high NOx
Anthropogenic emissions have a significant influence on the formation of SOA, in large part due to the influence of NOx on the atmospheric oxidation chemistry (Lee et al., 2016). In general, the fate of peroxy radicals in chamber experiments can be dominated by reactions with other RO2, HO2 or NO, depending on the exact conditions. In our experiments without NOx addition, RO2 was expected to be the main terminator, as also supported by the high number of detected ROOR dimers. In the atmosphere, all of the three mentioned reaction partners may be relevant at the same time. However, with increased anthropogenic influence, the reaction of RO2 with NO will often become dominant. Therefore, we added NOx to the chamber, as it allowed for the isolation of the formation pathways leading to HOMs in cases where NO was the main terminator for RO2 radicals. Figure 8 depicts a HOM mass spectrum at steady state during alpha-pinene oxidation by chlorine radicals in the presence of ∼ 10 ppb NOx, with the maximum seven lights turned on. As anticipated, the dimers above 440 Th were greatly reduced compared to the runs without NOx. As more lights were turned on, both the Cl atoms and NO formation increased, as the 400 nm lights photolyse both Cl2 and NO2. This coupling, together with the fact that the NO2 photolysis leads to ozone formation, which subsequently can react with alpha-pinene to form HOMs, limits our quantitative analysis of these experiments. However, we conclude that efficient HOM formation took place also under these high-NOx conditions, and thus the autoxidation occurs rapidly enough to still compete with RO2 termination reactions. The NOx addition also formed an abundance of organonitrate compounds like , as shown in Fig. 8. This family of compounds may also form from H abstraction by the chlorine radical, followed by autoxidation and finally radical termination by NO. We calculated reacted alpha-pinene from the ozone and chlorine atom as follows: , where the result is around 0.2. With the results, we conclude that alpha-pinene reaction with the chlorine atom is the main reaction in the system.
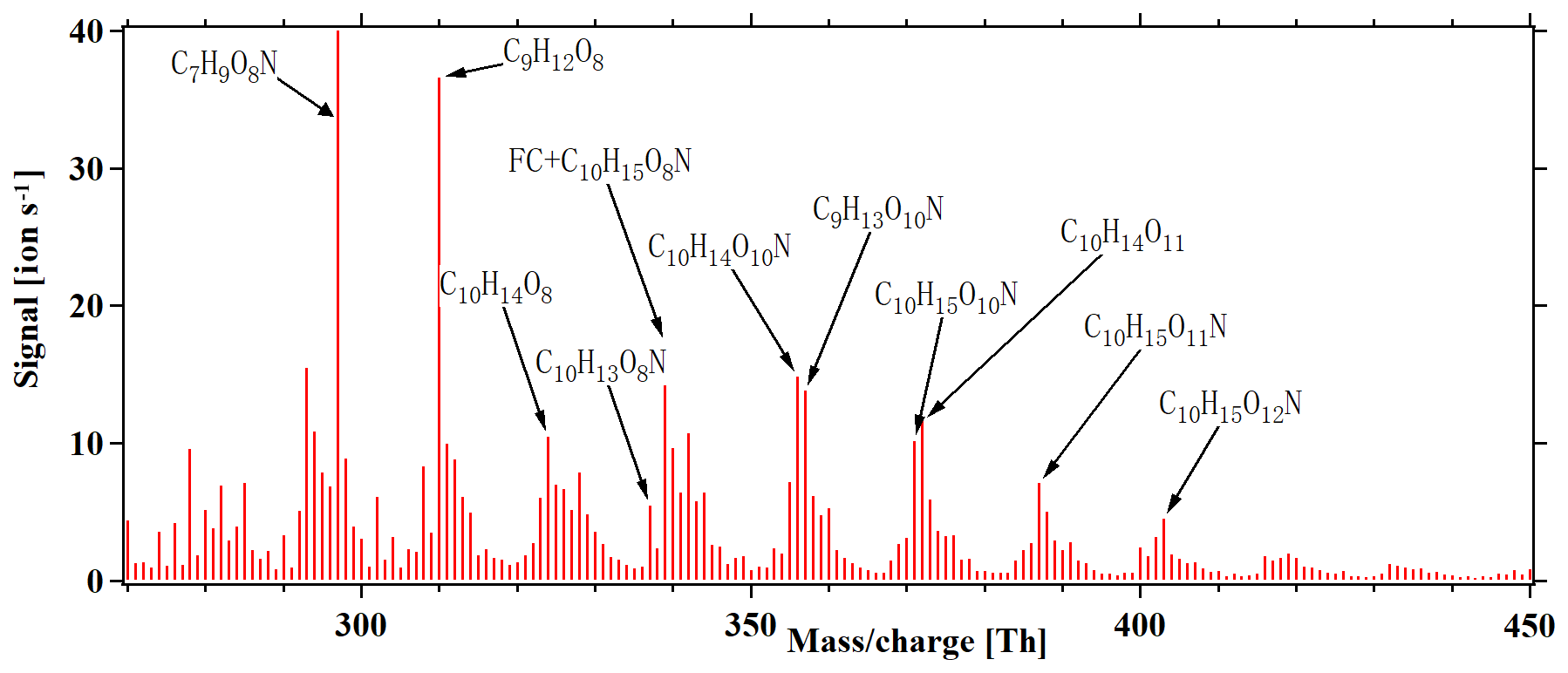
Figure 8HOM mass spectrum during steady-state alpha-pinene oxidation in the presence of 10 ppb NOx, with seven lights switched on. In addition to molecules detected also in the experiments without NOx, several abundant organic nitrate peaks are formed. Note that a fluorinated compound (FC) overlaps with the organic nitrate C10H15O8N at 339 Th. All peaks are detected as clusters with .
The concurrent formation of ozone means that also some alpha-pinene ozonolysis reaction will take place, though oxidation by Cl atoms was still the main loss for alpha-pinene also under these conditions.
Figure 9 shows variation in some nitrogen-containing HOMs and variation in alpha-pinene, ozone, NO and NOx, as we changed the lights from dark conditions to one, two, four or seven lights switched on. The concentrations of alpha-pinene and NO2 decreased because of the consumption by chlorine radicals and photolysis of NO2 into NO. Importantly, we did not observe any SOA when we had NO in the chamber. NO may have suppressed the particle formation by suppressing the dimer formation, as these have been shown to be important for initial particle formation (Tröstl et al., 2016).

Figure 9Time series of (a) selected HOMs measured by NO3 CI-APi-ToF and (b) NOx, alpha-pinene, ozone and NO as the lights switched on from zero to two, four and seven.

Figure 10HOM loss rate ([HOM] × Kloss) as a function of the alpha-pinene oxidation rate. In steady state the loss rate equals the formation rate, and thus the slope of the points gives the molar yield of HOM from the alpha-pinene + Cl reaction. The data correspond to the conditions with one, two, four and seven lights switched on in the chamber. The slope indicates an average molar yield of HOMs of 1.8 %.
3.3 Estimated HOM production yields
Quantifying the molar yields of HOMs is essential to knowing their potential importance from a specific system. We attempt to estimate the molar yield in the case of Cl oxidation of alpha-pinene in the absence of NOx. The initial C10H16 concentration is around 13.3 ppb without any UV lights switched on in the chamber. As we changed the lights, alpha-pinene and HOM concentrations varied, as shown in Fig. 3. In addition, we calculated the concentration of Cl radicals as introduced in the “Experiment and method” section. With this information, we can calculate the formation rate of HOM, which in the steady state equals the HOM loss rate [HOM]×kloss. We can also calculate the oxidation rate of alpha-pinene as . The ratio of these two numbers corresponds to the HOM molar yield. We selected the same runs as in Fig. 3, used also for calculating the chlorine radical concentration, and calculated the ratio as a linear fit to these four conditions (Fig. 10). We get a slope of 0.018, meaning a HOM yield of 1.8 %. Considering the uncertainty in estimating absolute HOM concentrations, we conservatively estimate that the molar HOM yield from alpha-pinene + Cl is within the range of 0.8 %–4 %. These values are similar to HOM yields reported for alpha-pinene oxidation by ozone and OH (Berndt et al., 2016; Ehn et al., 2014).
We systematically explored the reactions of alpha-pinene with chlorine atoms in a simulation smog chamber under atmospherically relevant conditions. We measured substantial numbers of highly oxidized organic molecules (HOMs) with a NO3 CI-APi-ToF. With increasing UV lights, and consequently higher chlorine radical concentrations, the concentrations of both HOM and secondary organic aerosol increased. With addition of NOx, HOM monomer formation was still efficient, but the particle formation decreased greatly. We estimated HOM molar yields of around 1.8 % (0.8 %–4 %) from the reaction of alpha-pinene with Cl atoms. Our study thus indicates that in regions where chlorine atom oxidation is of importance, its possible reactions with monoterpenes can be an important source of HOM and, consequently, SOA.
The readers can access the data by contacting Yonghong Wang (yonghong.wang@helisnkifi).
YW, HX and ME came up with the original idea of the study. YW, MR and HX conducted the chamber experiments. YW, MR, HX, LH and ME interpreted the data. YW plotted the figures and wrote the paper, with comments and suggestions from all co-authors.
The authors declare that they have no conflict of interest.
This work is supported by the European Research Council project and Academy of Finland, via the Centre of Excellence in Atmospheric Science. We acknowledge the tofTools team for providing the software.
This work is supported by a European Research Council (grant no. 638703-COALA) project and Academy of Finland, via the Center of Excellence in Atmospheric Sciences and project numbers 317380 and 320094.
This paper was edited by Timothy Bertram and reviewed by two anonymous referees.
Berndt, T., Richters, S., Jokinen, T., Hyttinen, N., Kurtén, T., Otkjaer, R. V, Kjaergaard, H. G., Stratmann, F., Herrmann, H., Sipilä, M., Kulmala, M., and Ehn, M.: ARTICLE Hydroxyl radical-induced formation of highly oxidized organic compounds, Nat. Commun., 7, 13677, https://doi.org/10.1038/ncomms13677, 2016.
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Cai, X. and Griffin, R. J.: Secondary aerosol formation from the oxidation of biogenic hydrocarbons by chlorine atoms, J. Geophys. Res.-Atmos., 111, 1–14, https://doi.org/10.1029/2005JD006857, 2006.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of organic compounds in the atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013.
Decarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Ehn, M., Berndt, T., Wildt, U., and Mentel, T.: Highly Oxygenated Molecules from Atmospheric Autoxidation of Hydrocarbons: A Prominent Challenge for Chemical Kinetics Studies, Int. J. Chem. Kinet., 49, 821–831, https://doi.org/10.1002/kin.21130, 2017.
Finlayson-Pitts, B. J., Keoshian, C. J., Buehler, B., and Ezell, A. A.: Kinetics of Reaction of Chlorine Atoms with Some, Int. J. Chem. Kinet., 31, 491–499, 1999.
Guenther, A., Hewitt, C. N., Erickson, D., Fall, R., Geron, C., Graedel, T., Harley, P., Klinger, L., Lerdau, M., Mckay, W. A., Pierce, T., Scholes, B., Steinbrecher, R., Tallamraju, R., Taylor, J., and Zimmerman, P.: A global model of natural volatile organic compound emissions, J. Geophys. Res.-Atmos., 100, 8873–8892, https://doi.org/10.1029/94JD02950, 1995.
Jokinen, T., Sipilä, M., Junninen, H., Ehn, M., Lönn, G., Hakala, J., Petäjä, T., Mauldin III, R. L., Kulmala, M., and Worsnop, D. R.: Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF, Atmos. Chem. Phys., 12, 4117–4125, https://doi.org/10.5194/acp-12-4117-2012, 2012.
Jokinen, T., Sipilä, M., Richters, S., Kerminen, V., Paasonen, P., Stratmann, F., Worsnop, D., Kulmala, M., Ehn, M., Herrmann, H., and Berndt, T.: Rapid Autoxidation Forms Highly Oxidized RO2 Radicals in the Atmosphere, Angew. Chem. Int. Ed., 53, 14596–14600, https://doi.org/10.1002/anie.201408566, 2014.
Jokinen, T., Berndt, T., Makkonen, R., Kerminen, V.-M., Junninen, H., Paasonen, P., Stratmann, F., Herrmann, H., Guenther, A. B., Worsnop, D. R., Kulmala, M., Ehn, M., and Sipilä, M.: Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications, P. Natl. Acad. Sci. USA, 112, 7123–7128, https://doi.org/10.1073/pnas.1423977112, 2015.
Jonsson, Å. M., Hallquist, M., and Ljungström, E.: Impact of humidity on the ozone initiated oxidation of limonene, Δ3-carene, and α-pinene, Environ. Sci. Technol., 40, 188–194, https://doi.org/10.1021/es051163w, 2006.
Jonsson, Å. M., Hallquist, M., and Ljungström, E.: Influence of OH scavenger on the water effect on secondary organic aerosol formation from ozonolysis of limonene, Δ3-carene, and α-pinene, Environ. Sci. Technol., 42, 5938–5944, https://doi.org/10.1021/es702508y, 2008.
Kulmala, M., Kontkanen, J., Junninen, H., Lehtipalo, K., Manninen, H. E., Nieminen, T., Petäjä, T., Sipilä, M., Schobesberger, S., Rantala, P., Franchin, A., Jokinen, T., Järvinen, E., Äijälä, M., Kangasluoma, J., Hakala, J., Aalto, P. P., Paasonen, P., Mikkilä, J., Vanhanen, J., Aalto, J., Hakola, H., Makkonen, U., Ruuskanen, T., Mauldin, R. L., Duplissy, J., Vehkamäki, H., Bäck, J., Kortelainen, A., Riipinen, I., Kurtén, T., Johnston, M. V., Smith, J. N., Ehn, M., Mentel, T. F., Lehtinen, K. E. J., Laaksonen, A., Kerminen, V. M., and Worsnop, D. R.: Direct observations of atmospheric aerosol nucleation, Science, 339, 943–946, https://doi.org/10.1126/science.1227385, 2013.
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurtén, T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., D'Ambro, E. L., and Thornton, J. A.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci. USA, 113, 1516–1521, https://doi.org/10.1073/pnas.1508108113, 2016.
Li, X., Chee, S., Hao, J., Abbatt, J. P. D., Jiang, J., and Smith, J. N.: Relative humidity effect on the formation of highly oxidized molecules and new particles during monoterpene oxidation, Atmos. Chem. Phys., 19, 1555–1570, https://doi.org/10.5194/acp-19-1555-2019, 2019.
Liu, X., Qu, H., Huey, L. G., Wang, Y., Sjostedt, S., Zeng, L., Lu, K., Wu, Y., Hu, M., Shao, M., Zhu, T., and Zhang, Y.: High Levels of Daytime Molecular Chlorine and Nitryl Chloride at a Rural Site on the North China Plain, Environ. Sci. Technol., 51, 9588–9595, https://doi.org/10.1021/acs.est.7b03039, 2017.
Mentel, T. F., Springer, M., Ehn, M., Kleist, E., Pullinen, I., Kurtén, T., Rissanen, M., Wahner, A., and Wildt, J.: Formation of highly oxidized multifunctional compounds: autoxidation of peroxy radicals formed in the ozonolysis of alkenes – deduced from structure–product relationships, Atmos. Chem. Phys., 15, 6745–6765, https://doi.org/10.5194/acp-15-6745-2015, 2015.
Ofner, J., Kamilli, K. A., Held, A., Lendl, B., and Zetzsch, C.: Halogen-induced organic aerosol (XOA): a study on ultra-fine particle formation and time-resolved chemical characterization, Faraday Discuss., 165, 135–149, https://doi.org/10.1039/c3fd00093a, 2013.
Peräkylä, O., Riva, M., Heikkinen, L., Quéléver, L., Roldin, P., and Ehn, M.: Experimental investigation into the volatilities of highly oxygenated organic molecules (HOMs), Atmos. Chem. Phys., 20, 649–669, https://doi.org/10.5194/acp-20-649-2020, 2020.
Praske, E., Otkjær, R. V., Crounse, J. D., Hethcox, J. C., Stoltz, B. M., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric autoxidation is increasingly important in urban and suburban North America, P. Natl. Acad. Sci. USA, 115, 64–69, https://doi.org/10.1073/pnas.1715540115, 2018.
Reyes-Villegas, E., Priestley, M., Ting, Y.-C., Haslett, S., Bannan, T., Le Breton, M., Williams, P. I., Bacak, A., Flynn, M. J., Coe, H., Percival, C., and Allan, J. D.: Simultaneous aerosol mass spectrometry and chemical ionisation mass spectrometry measurements during a biomass burning event in the UK: insights into nitrate chemistry, Atmos. Chem. Phys., 18, 4093–4111, https://doi.org/10.5194/acp-18-4093-2018, 2018.
Riccobono, F., Schobesberger, S., Scott, C. E., Dommen, J., Ortega, I. K., Rondo, L., Almeida, J., Amorim, A., Bianchi, F., Breitenlechner, M., David, A., Downard, A., Dunne, E. M., Duplissy, J., Ehrhart, S., Flagan, R. C., Franchin, A., Hansel, A., Junninen, H., Kajos, M., Keskinen, H., Kupc, A., Kürten, A., Kvashin, A. N., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Mathot, S., Nieminen, T., Onnela, A., Petäjä, T., Praplan, A. P., Santos, F. D., Schallhart, S., Seinfeld, J. H., Sipilä, M., Spracklen, D. V, Stozhkov, Y., Stratmann, F., Tomé, A., Tsagkogeorgas, G., Vaattovaara, P., Viisanen, Y., Vrtala, A., Wagner, P. E., Weingartner, E., Wex, H., Wimmer, D., Carslaw, K. S., Curtius, J., Donahue, N. M., Kirkby, J., Kulmala, M., Worsnop, D. R., and Baltensperger, U.: Oxidation Products of Biogenic Emissions Contribute to Nucleation of Atmospheric Particles, Science, 344, 717–721, https://doi.org/10.1126/science.1243527, 2014.
Rissanen, M. P., Kurtén, T., Sipilä, M., Thornton, J. A., Kangasluoma, J., Sarnela, N., Junninen, H., Jørgensen, S., Schallhart, S., Kajos, M. K., Taipale, R., Springer, M., Mentel, T. F., Ruuskanen, T., Petäjä, T., Worsnop, D. R., Kjaergaard, H. G., and Ehn, M.: The formation of highly oxidized multifunctional products in the ozonolysis of cyclohexene, J. Am. Chem. Soc., 136, 15596–15606, https://doi.org/10.1021/ja507146s, 2014.
Riva, M., Healy, R. M., Flaud, P. M., Perraudin, E., Wenger, J. C., and Villenave, E.: Gas- and Particle-Phase Products from the Chlorine-Initiated Oxidation of Polycyclic Aromatic Hydrocarbons, J. Phys. Chem. A, 119, 11170–11181, https://doi.org/10.1021/acs.jpca.5b04610, 2015.
Riva, M., Heikkinen, L., Bell, D. M., Peräkylä, O., Zelenyuk, A., and Ehn, M.: Chemical transformations in monoterpene-derived organic aerosol enhanced by inorganic composition, npj Clim. Atmos. Sci., 2, 2, https://doi.org/10.1038/s41612-018-0058-0, 2019a.
Riva, M., Rantala, P., Krechmer, J. E., Peräkylä, O., Zhang, Y., Heikkinen, L., Garmash, O., Yan, C., Kulmala, M., Worsnop, D., and Ehn, M.: Evaluating the performance of five different chemical ionization techniques for detecting gaseous oxygenated organic species, Atmos. Meas. Tech., 12, 2403–2421, https://doi.org/10.5194/amt-12-2403-2019, 2019b.
Schallhart, S., Rantala, P., Kajos, M. K., Aalto, J., Mammarella, I., Ruuskanen, T. M., and Kulmala, M.: Temporal variation of VOC fluxes measured with PTR-TOF above a boreal forest, Atmos. Chem. Phys., 18, 815–832, https://doi.org/10.5194/acp-18-815-2018, 2018.
Tham, Y. J., Wang, Z., Li, Q., Yun, H., Wang, W., Wang, X., Xue, L., Lu, K., Ma, N., Bohn, B., Li, X., Kecorius, S., Größ, J., Shao, M., Wiedensohler, A., Zhang, Y., and Wang, T.: Significant concentrations of nitryl chloride sustained in the morning: investigations of the causes and impacts on ozone production in a polluted region of northern China, Atmos. Chem. Phys., 16, 14959–14977, https://doi.org/10.5194/acp-16-14959-2016, 2016.
Thornton, J. A., Kercher, J. P., Riedel, T. P., Wagner, N. L., Cozic, J., Holloway, J. S., Dubé, W. P., Wolfe, G. M., Quinn, P. K., Middlebrook, A. M., Alexander, B., and Brown, S. S.: A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry, Nature, 464, 271–274, https://doi.org/10.1038/nature08905, 2010.
Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., Wagner, R., Simon, M., Lehtipalo, K., Williamson, C., Craven, J. S., Duplissy, J., Adamov, A., Almeida, J., Bernhammer, A.-K., Breitenlechner, M., Brilke, S., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Gysel, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Keskinen, H., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lawler, M., Leiminger, M., Mathot, S., Möhler, O., Nieminen, T., Onnela, A., Petäjä, T., Piel, F. M., Miettinen, P., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Sipilä, M., Smith, J. N., Steiner, G., Tomè, A., Virtanen, A., Wagner, A. C., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Carslaw, K. S., Curtius, J., Dommen, J., Kirkby, J., Kulmala, M., Riipinen, I., Worsnop, D. R., Donahue, N. M., and Baltensperger, U.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 533, 527–531, https://doi.org/10.1038/nature18271, 2016.
Wang, D. S. and Ruiz, L. H.: Secondary organic aerosol from chlorine-initiated oxidation of isoprene, Atmos. Chem. Phys., 17, 13491–13508, https://doi.org/10.5194/acp-17-13491-2017, 2017.
Wang, Z., Wang, W., Tham, Y. J., Li, Q., Wang, H., Wen, L., Wang, X., and Wang, T.: Fast heterogeneous N2O5- uptake and ClNO2 production in power plant and industrial plumes observed in the nocturnal residual layer over the North China Plain, Atmos. Chem. Phys., 17, 12361–12378, https://doi.org/10.5194/acp-17-12361-2017, 2017.
Zhang, H., Yee, L. D., Lee, B. H., Curtis, M. P., Worton, D. R., Isaacman-VanWertz, G., Offenberg, J. H., Lewandowski, M., Kleindienst, T. E., Beaver, M. R., Holder, A. L., Lonneman, W. A., Docherty, K. S., Jaoui, M., Pye, H. O. T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Guo, H., Weber, R. J., de Gouw, J., Koss, A. R., Edgerton, E. S., Brune, W., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Kreisberg, N. M., Spielman, S. R., Hering, S. V., Wilson, K. R., Thornton, J. A., and Goldstein, A. H.: Monoterpenes are the largest source of summertime organic aerosol in the southeastern United States, P. Natl. Acad. Sci. USA, 115, 2038–2043, https://doi.org/10.1073/pnas.1717513115, 2018.