the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 08 Dec 2020
| 08 Dec 2020
A comparative and experimental study of the reactivity with nitrate radical of two terpenes: α-terpinene and γ-terpinene
Axel Fouqueau
Manuela Cirtog
Mathieu Cazaunau
Edouard Pangui
Jean-François Doussin
Bénédicte Picquet-Varrault
Biogenic volatile organic compounds (BVOCs) are intensely emitted by forests and crops into the atmosphere. During the night, they react very rapidly with the nitrate radical (NO3), leading to the formation of a variety of functionalized products including organic nitrates and to large amounts of secondary organic aerosols (SOAs). Organic nitrates (ONs) have been shown not only to play a key role in the transport of reactive nitrogen and consequently in the ozone budget but also to be important components of the total organic-aerosol mass, while SOAs are known to play a direct and indirect role in the climate. However, the reactivity of BVOCs with NO3 remains poorly studied. The aim of this work is to provide new kinetic and mechanistic data for two monoterpenes (C10H16), α- and γ-terpinene, through experiments in simulation chambers. These two compounds, which have very similar chemical structures, have been chosen in order not only to overcome the lack of experimental data but also to highlight the influence of the chemical structure on the reactivity.
Rate constants have been measured using both relative and absolute methods. They were found to be and ( cm3 molecule−1 s−1 for α- and γ-terpinene respectively. Mechanistic studies have also been conducted in order to identify and quantify the main reaction products. Total organic nitrate and SOA yields have been determined. While organic nitrate formation yields appear to be similar, SOA yields exhibit large differences with γ-terpinene being a much more efficient precursor of aerosols. In order to provide explanations for this difference, chemical analysis of the gas-phase products was performed at the molecular scale. Detected products allowed for proposing chemical mechanisms and providing explanations through peroxy and alkoxy reaction pathways.
- Article
(2560 KB) - Full-text XML
- Companion paper
-
Supplement
(492 KB) - BibTeX
- EndNote





Since the early 1980s and the discovery of the nitrate radical (NO3) in the nocturnal troposphere (Noxon et al., 1980; Platt et al., 1980) and stratosphere (Naudet et al., 1981; Noxon et al., 1978), nighttime chemistry has been known to be active. NO3 is mainly formed by the reaction of nitrogen dioxide (NO2) with ozone and has two very efficient sinks during the day, its photolysis and its reaction with NO (Brown and Stutz, 2012). Not only during the night but also during the day under low-sunlight conditions (e.g., in forest areas), NO3 has been shown to be an efficient oxidant, reacting with a large variety of volatile organic compounds (VOCs) including alkenes, aromatics and oxygenated VOCs (Atkinson and Arey, 2003). For nighttime conditions, reactions of NO3 with biogenic volatile organic compounds (BVOCs) are particularly rapid. BVOCs, which include not only isoprene (C5H8), monoterpenes (C10H16) and sesquiterpenes (C15H24) but also oxygenated compounds, represent almost 90 % of global emissions of VOCs (Guenther et al., 1995). Most BVOCs have one or several C=C bonds and thus react particularly rapidly with NO3 by addition of the oxidant onto the unsaturation(s) leading to lifetimes below a minute for the most reactive ones. This reaction leads to the formation of nitrooxy alkyl radicals which can then evolve into organic nitrates (ONs). Organic nitrates have been shown to act as reservoirs for reactive nitrogen by undergoing long-range transport in the free troposphere before decomposing and releasing NOx in remote regions. They therefore significantly influence the nitrogenous species (NOy) and ozone budgets in these regions (Ito et al., 2007). Furthermore, some (multifunctional) organic nitrates are low-volatile and highly soluble in both the aqueous phase and organic aerosols (Picquet-Varrault et al., 2020) and are thus capable of strongly partitioning into the atmospheric condensed phases (droplets, aerosols). Recent field observations of the aerosol chemical composition have shown that organic nitrates range from 10 % to 75 % of total organic aerosol (OA) mass (Kiendler-Scharr et al., 2016; Lee et al., 2016; Xu et al., 2015), suggesting that these species are important components of OAs. They can thus significantly affect the aerosols' physical and chemical properties, particularly their optical and hygroscopic properties, controlling their direct and indirect impacts on climate. A good understanding of the reactions of BVOCs + NO3 is thus necessary. Nevertheless this chemistry remains poorly studied and except for with regard to isoprene and α- and β-pinene, which have been largely studied, it is thinly understood so far.
Among the most occurring terpenes (Geron et al., 2000), α-terpinene and γ-terpinene have been detected in many tree emissions. For example, α-terpinene has been shown to represent 12 % of subalpine fir monoterpene emissions and γ-terpinene up to 19 % of Sequoia sempervirens emissions (Geron et al., 2000). These two molecules have very similar structures, the only difference being the position of the double bonds (see Fig. 1), which are conjugated for α-terpinene and not for γ-terpinene. Their reactions with NO3 have been subject to few studies. For both compounds, rate constants have been measured in two studies: γ-terpinene has been the object of absolute- and relative-rate determinations (Martínez et al., 1999, and Atkinson et al., 1985, respectively) which are in good agreement within uncertainties. For α-terpinene, the rate constant was measured only by relative-rate studies using the same reference compound, and the two values differ by 80 % (Atkinson et al., 1985, and Berndt et al., 1998). This compound was shown to be very reactive (with a rate constant around 10−10 cm3 molecule−1 s−1) making an absolute determination technically difficult. The mechanism for the oxidation of γ-terpinene by NO3 has been investigated by only one study (Slade et al., 2017) and no mechanistic study has ever been published for α-terpinene to our knowledge. In the study of Slade et al. (2017), total organic nitrate and SOA yields were determined and some products were detected at the molecular scale allowing for the proposal of a mechanism. New kinetic and mechanistic studies are therefore necessary to better assess the impact of these processes on air quality and radiative forcing.
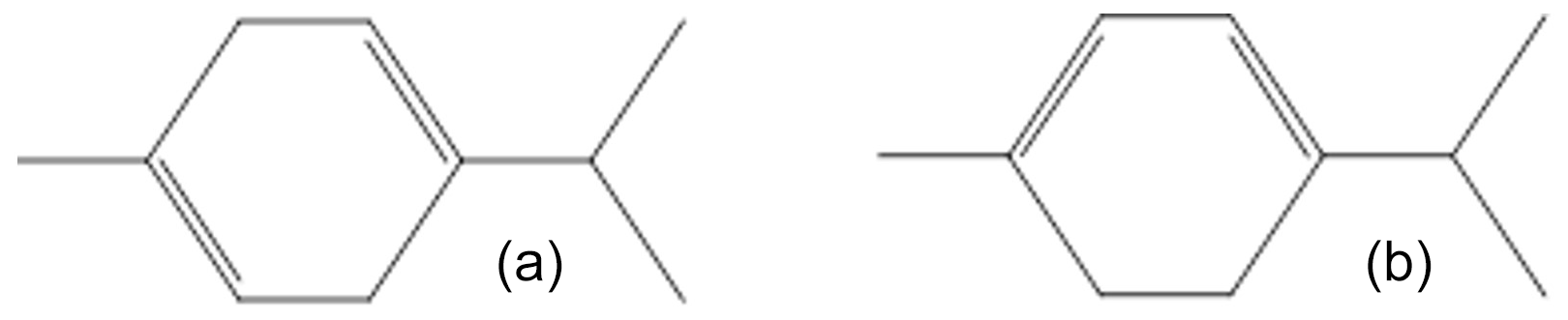
The aim of this work is to provide new kinetic and mechanistic data for the reactions of α- and γ-terpinene with NO3 using atmospheric simulation chambers. To provide precise and accurate rate constants, absolute-rate determinations are conducted for both compounds, using a technique of incoherent broadband cavity-enhanced absorption spectroscopy (IBBCEAS) recently interfaced with the CSA atmospheric simulation chamber at LISA (Fouqueau et al., 2020a). This technique allows in situ monitoring of NO3 with a very good time resolution (10 s) and at very low concentrations (parts-per-trillion level), these two features being mandatory for the kinetic study of fast reactions. For γ-terpinene, a relative-rate determination is also performed. In addition, mechanistic studies have been performed for both compounds by providing total organic nitrate and SOA yields and identification of individual gas-phase products. It allows for proposing reaction mechanisms for the two compounds. For γ-terpinene, these results are compared to those obtained by the previous study. Finally, differences observed for α- and γ-terpinene in kinetic and mechanistic data are discussed in regard to the chemical structures of the two compounds.
2.1 Chemicals
α-Terpinene and γ-terpinene were purchased from Sigma-Aldrich at purities of 85 % and 97 % respectively. For both terpenes, a purification stage has been conducted in a vacuum line prior to their injection into the chamber. This purification is expected to remove high-volatility impurities. For low-volatility impurities, it is expected that they will remain in the sample (condensed phase). However, as the impurities remain unknown, we cannot state with certainty that this purification is 100 % efficient, and it should be considered that it may generate additional uncertainty in the product yields, in particular for α-terpinene. NO3 was generated in situ, by thermal dissociation of N2O5 (Eq. 1), previously synthesized in a vacuum line by the reaction between O3 and NO2 (Eqs. 2 and 3) adapted from Atkinson et al. (1984a) and Schott and Davidson (1958). The detailed protocol is presented in Picquet-Varrault et al. (2009).
2.2 Chamber facilities and analytical devices
Experiments were conducted in two different simulation chambers: the CSA and CESAM (Chamber for Experimental Multiphase Atmospheric Simulation) chambers. The CSA chamber was used for kinetic experiments. It is made of a 6 m long Pyrex® reactor which has a volume of 977 L (Doussin et al., 1997) and is equipped with a homogenization system that allows for a mixing time below 1 min. This chamber is dedicated to gas-phase studies and is hence equipped with several analytical devices for gas-phase monitoring. An in situ multiple reflection optical system coupled to a Fourier-transform infrared (FTIR; Bruker VERTEX 80) spectrometer allows for monitoring organic species in the chamber. Infrared spectra were recorded with a resolution of 0.5 cm−1, an optical path length of 204 m and a spectral range of 700–4000 cm−1. Integrated band intensities (IBIs; cm molecule−1, logarithm base e) used to quantify the species are IBIα-terpinene (790–850 cm−1) , IBIγ-terpinene (920–990 cm−1) , IBI (1530–1680 cm−1) , IBI (840–930 cm−1) and IBI (1205–1275 cm−1) . This technique also allowed for measuring the total organic nitrate concentration by considering that all these species absorb at 1250 and 850 cm−1 which correspond to absorptions of the −ONO2 function and by assuming that their absorption cross sections are similar whatever the organic nitrate considered. This hypothesis was verified in our research group with the analysis of standards. In this study, we used IBION (900–820 cm−1) cm molecule−1. Uncertainties in concentrations of species measured by FTIR spectroscopy include the uncertainty in the IBIs and the uncertainty in the spectra analysis.
For absolute-rate determination, NO3 was monitored from its visible absorption at 662 nm with an in situ IBBCEAS technique which has recently been coupled to the CSA. It is described in detail in Fouqueau et al. (2020a). This technique also allows for NO2 monitoring. Having a precise knowledge of the wavelength-dependent mirror reflectivity, R(λ), is one of the most critical points of the IBBCEAS technique. It was therefore determined prior to each experiment by introducing a known amount of NO2 (several hundred parts per billion) into the chamber. The cross sections used to quantify NO2 are from Vandaele et al. (1997), and those used for NO3 quantification are from Orphal et al. (2003). At 662.1 nm, which corresponds to the NO3 maximum absorption, the cross section is cm2 molecule−1. Thanks to the very high reflectivity of mirrors (99.974±0.002 %), the optimum optical path length was found to be 2.5 km, leading to a detection limit of 6 ppt for 10 s of integration time. The uncertainty in NO3 concentrations by IBBCEAS was estimated to be 9 %, with a minimum absolute value of 3 ppt (Fouqueau et al., 2020a). This uncertainty includes the uncertainties in the reflectivity of the mirrors, the NO3 absorption cross sections and the data treatment.
Finally, in order to monitor organic reactants and products, a high-resolution proton transfer reactor time-of-flight mass spectrometer (PTR-ToF-MS; Kore Series 2e, mass resolution of 4000) was used in two ionization modes, H3O+ and NO+. When used in standard operational conditions, i.e., with H3O+ ionization, it has been shown that organic nitrates are subject to important fragmentation (Müller et al., 2012; Aoki et al., 2007). In order to reduce this fragmentation, Duncianu et al. (2017) have adapted the instrument operating procedure for organic nitrate detection by reducing the electric field in the reactor. The same study has also developed a NO+ ionization mode, by replacing the ionization gas (water vapor) with dry air and also by applying a reduced electric field. These two modes allow for cross-checking for the identification of the products. This method was characterized and validated thanks to experiments with various standards of organic nitrates (alkyl nitrates, carbonyl nitrates and hydroxynitrates), allowing the authors to propose ionization patterns for each type of organic nitrate and for both ionization modes. Hence, in the NO+ ionization mode, organic nitrates were shown to be ionized by charge transfer or by an NO+ adduct formation and therefore to be detected at their own mass (M) or at M+30. Hydroxynitrates have been detected at M−1, suggesting that the ionization proceeds mainly by a hydrogen loss.
The second simulation chamber is the CESAM chamber (Wang et al., 2011). It has been specifically designed to study multiphase processes. In this work, it was used to investigate the mechanisms and the SOA formation. CESAM is a 4177 L stainless-steel evacuable reactor and is equipped with a fan that allows for a mixing time of approximately 1 min. Aerosol lifetimes in CESAM being very long (up to 4 d), it is particularly suited for SOA studies. This chamber is equipped with dedicated analytical instruments for gas and aerosol phases. It is coupled with an in situ long path FTIR spectrometer (Bruker Tensor 37) allowing for acquiring spectra in the 700–4000 cm−1 spectral range with a resolution of 0.5 cm−1 and an optical path of 174.5 m. It is also equipped with a PTR-ToF-MS which was operated in both NO+ and H3O+ ionization modes. The size distribution of the particle phase was measured with a scanning mobility particle sizer (SMPS) composed of a TSI classifier model 3080 and differential mobility analyzer (DMA) model 3081 coupled to a condensation particle counter (CPC) TSI model 3772 which allowed for measurements in the range of 20–880 nm. To convert size distribution into mass distribution, a particle density of 1.4 g cm−3 was used, estimated to be the density of SOAs formed by BVOC+NO3 reactions (Fry et al., 2014; Draper et al., 2015; Boyd et al., 2015). Some experiments conducted on α-terpinene lead to the formation of large particles (diameters >800 nm). In this case, a Palas welas (welas digital 2000) was used in addition to the SMPS, to measure the particle size distribution. This instrument is based on an optical measurement and allows for covering a wider size range (0.2–17 µm).
During experiments, filter sampling proceeded, allowing for the measurement of total ON yield in the aerosol phase. The filter analysis was performed by FTIR spectroscopy after extraction of particles in the liquid phase, following a protocol described by Rindelaub et al. (2015): SOAs are extracted in 5 mL of CCl4. Organic nitrates were quantified from a standard solutions of two types of organic nitrates (nitrooxypropanol and tert-butyl nitrate). IBIs for the two standards were 510 and 580 L mol−1 cm−2 respectively. The difference between the two of them is small, so the integrated absorption cross section of organic nitrates in the liquid phase was considered to be IBIONs (1264–1310 cm−1) L mol−1 cm−2.
The pressure into the chamber was maintained constant by introducing synthetic air in order to compensate for the decrease in pressure due to instrument sampling. This leads to a weak dilution of the mixture, here less than 20 % for an experiment length of 3 h. All data presented in the following sections were corrected by dilution and, for the SOA measurements, by the particles' wall losses, which were characterized in CESAM: wall loss rates were determined as a function of the diameter of the particles and interpolated using the Lai and Nazaroff (2000) model (friction velocity cm s−1 – see Lamkaddam et al., 2017). Due to the material used for the walls (stainless steel), this correction has been found to be small in the present case.
2.3 Kinetic study
Kinetic experiments were performed in the CSA chamber at room temperature and atmospheric pressure, in a mixture of N2∕O2 (generated using 80 % of N2 from liquid nitrogen evaporation, purity >99.995 %, H2O<5 ppm, Messer, and 20 % of O2, quality N5.0, purity >99.995 %, H2O<5 ppm, Air Liquide). Rate constants were determined by using both relative- and absolute-rate methods in order to provide accurate kinetic data. During a typical experiment, organic reactants were introduced into the chamber and left in the dark for a period of approximately 1 h in order to check that there was no significant loss of the compounds. Then, NO3 was generated in situ by thermal decomposition of N2O5 (see Sect. 2.1) which was introduced into the chamber using several stepwise injections for a complete consumption of the BVOC.
For absolute-rate determinations, concentrations of BVOC and NO3 were monitored by the PTR-ToF-MS and IBBCEAS respectively. Low mixing ratios of BVOCs (between 15 and 50 ppb) were used in order to reduce the SOA formation which led to a significant decrease in the IBBCEAS signal due to light absorption or scattering and mirror soiling by particles. In order to allow for monitoring fast decay of reactants, a low integration time (10 s) was used for both techniques. In addition, prior to each experiment, several hundred parts per billion of NO2 (between 450 and 650 ppb) were introduced to (i) measure the reflectivity of the IBBCEAS mirrors and (ii) shift the equilibrium between N2O5, NO3 and NO2, hence slowing down the N2O5 decomposition and the BVOC+NO3 reaction.
Then, the second-order kinetic equation is
This equation can be approximated for small time intervals:
where −Δ[BVOC] corresponds to the consumption of BVOC during the time interval Δt and [BVOC] and [NO3] are averaged concentrations during this interval. By plotting −Δ[BVOC] vs. , a straight line with a slope corresponding to kBVOC is obtained. It should be mentioned that the determination of the rate constant is thus not affected by losses of NO3 due to reaction with other species (products, RO2 radicals, etc.). Uncertainty in the rate constant was calculated by considering twice the standard deviation on the slope.
For relative-rate determination, the decay of the BVOC was monitored relatively to a reference compound using PTR-ToF-MS and FTIR techniques. If it is assumed that reaction with NO3 is the only fate of both the studied compound (BVOC) and the reference compound (Ref.) and that neither of these compounds is reformed at any stage during the experiment, it can be shown that (Atkinson, 1986)
where and are the concentrations of BVOC and Ref. at time t0 (before the beginning of the oxidation), [BVOC]t and [Ref.]t are the concentrations at time t, and kBVOC and kRef. are the rate constants with NO3.
In this work, two different reference compounds with well-known rate constants were used: 2,3-dimethyl-2-butene and 2-methyl-2-butene. Due to the lack of recommendation by the IUPAC for the reaction between these compounds and NO3, rate constants were calculated as the mean values of the determinations available in the literature (Atkinson, 1988; Atkinson et al., 1984a, b; Benter et al., 1992; Berndt et al., 1998). The uncertainties in reference rate constants were calculated as twice the standard deviation of all the values. The obtained rate constants are cm3 molecule−1 s−1 and cm3 molecule−1 s−1. Finally, the uncertainty in kBVOC was calculated by considering the relative uncertainty corresponding to the statistical error in the linear regression (2σ) and the error in the Ref. rate constant.
2.4 Mechanistic study
Mechanistic experiments were performed in the CESAM chamber at room temperature and atmospheric pressure, in the same mixture of N2∕O2 (80∕20) as the kinetic experiments. During a typical experiment, the BVOC is introduced into the chamber and left in the dark to estimate potential wall losses. No significant wall loss was observed ( s−1). Then N2O5 is introduced. Two methods were used to inject N2O5 in order to optimize the decay rate of the BVOC: by stepwise injections and by slow continuous injections. The second method was observed to be more efficient to slow down the oxidation and thus to better control the SOA formation. BVOC and gas-phase products were monitored by both the PTR-ToF-MS and the FTIR spectrometer. Some experiments were conducted with two PTR-ToF-MSs, allowing for analyzing the gas phase in both the NO+ and the H3O+ ionization mode simultaneously. When both instruments were not available at the same time, experiments were duplicated to allow for the product detection with the two ionization modes. Production of SOAs was monitored by the SMPS. No seed particles were introduced in order to determine SOA yields under low aerosol content. Sampling on filters was performed for high-concentration experiments (>150 ppb) in order to determine the total organic nitrate concentration in the particle phase (see Sect. 2.2). The sampling on filters started at the end of the oxidation (when the precursor had completely reacted) and lasted between 3 and 6 h. A charcoal denuder was used to remove organic compounds from the gas phase.
Total organic nitrate yields in the gas phase were determined by plotting the molecular concentration of organic nitrates as a function of the molecular concentration of the BVOC reacted and by calculating the slope of the straight line. Organic nitrate yields in the SOA phase were calculated by measuring the final concentration of organic nitrates and by dividing it by the total reacted BVOC concentration for each experiment. Uncertainty in the yield was calculated as the sum of the relative uncertainties in organic nitrates and BVOC concentrations.
The SOA yield is defined as the ratio of the mass concentration of the SOAs produced, M0, divided by the mass concentration of the BVOC reacted, ΔBVOC. For all experiments, the SOA yield was calculated not only for each data point but also once the BVOC had been totally consumed, hence providing both time-dependent and overall SOA yields. These yields were plotted as a function of the organic aerosol mass and fitted by a two-product model described by Odum et al. (1996):
where α1, α2 and are stoichiometric factors and partitioning coefficients (m3 µg−1) respectively of the two hypothetical products. Due to the slow injections of N2O5, SOA equilibrium was expected to be reached at small time steps and time-dependent yields have been used. This also allowed for obtaining yields for a small aerosol content in the chamber.
As described in Sect. 2.2, oxidation products were detected thanks to PTR-ToF-MS measurements in two ionization modes (H3O+ and NO+). However, quantification of these products was not performed due to the lack of standards. Finally, vapor pressures Pvap have been estimated using the SIMPOL.1 method of Pankow and Asher (2008) in order to evaluate their contribution to SOA formation via the GECKO-A website (http://geckoa.lisa.u-pec.fr/generateur_form.php, last access: 12 May 2020). Raoult's law (Valorso et al., 2011) has also been used to estimate the fraction of a product i in the condensed phase :
where Ni, gas and Ni, aer are the concentrations (molecules cm−3) of the product i in the gas and particle phases respectively, is the mean molecular weight of SOA species (g mol−1), Caer is the total SOA mass concentration (µg m−3), R is the gas constant (atm m3 K−1 mol−1), T the temperature (K), is the vapor pressure, and γi is the activity coefficient of product i (in this study, γi=1). The mean molecular weight has been estimated to be the mean value for low-volatility products which were detected.
The calculation of is highly dependent on the estimated vapor pressure. Pankow and Asher (2008) showed that the SIMPOL.1 technique allows for predicting Pvap with an error of between 50 % and 60 % for atm. is therefore associated with a high uncertainty and can only be used as an indicator. can also be compared to partitioning coefficients Kp in Eq. (8), using the following equation:
3.1 Kinetic results
A list of kinetic experiments and their corresponding experimental conditions are presented in Table 1. Absolute-rate determinations were conducted for α- and γ-terpinene, while relative-rate ones were performed only for γ-terpinene. For each method, between three and five experiments were conducted.
Table 1Experimental conditions of kinetic experiments. [BVOC] is the initial concentration of BVOC; Ref. is the reference compound, and [Ref.] is the initial concentration of the reference compound. For [N2O5] the number of punctual injections is indicated in brackets.
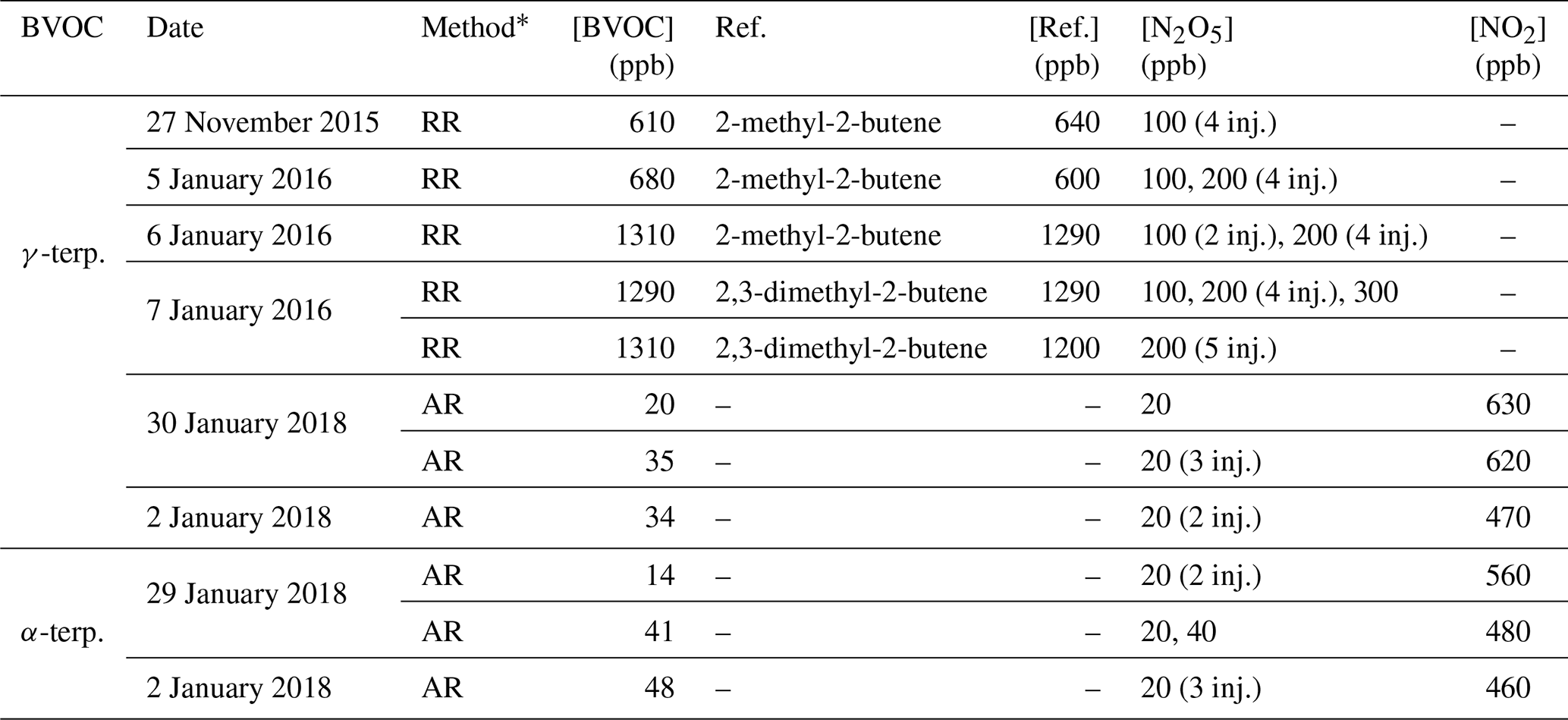
* RR: relative-rate determination; AR: absolute-rate determination.
Kinetic results obtained for γ-terpinene by the relative-rate method are presented in Fig. 2. Good linear tendencies are observed for the two reference compounds, and data obtained using PTR-Tof-MS and FTIR measurements are in good agreement. Linear regressions were first performed for both the individual data sets (PTR-ToF-MS and FTIR). Because the results were in good agreement for both of the measurement techniques, linear regression was applied to all the values (by mixing data sets), leading to cm3 molecule−1 s−1 with 2,3-dimethyl-2-butene and cm3 molecule−1 s−1 with 2-methyl-2-butene. It can be concluded that rate constants obtained with the two references are in very good agreement.
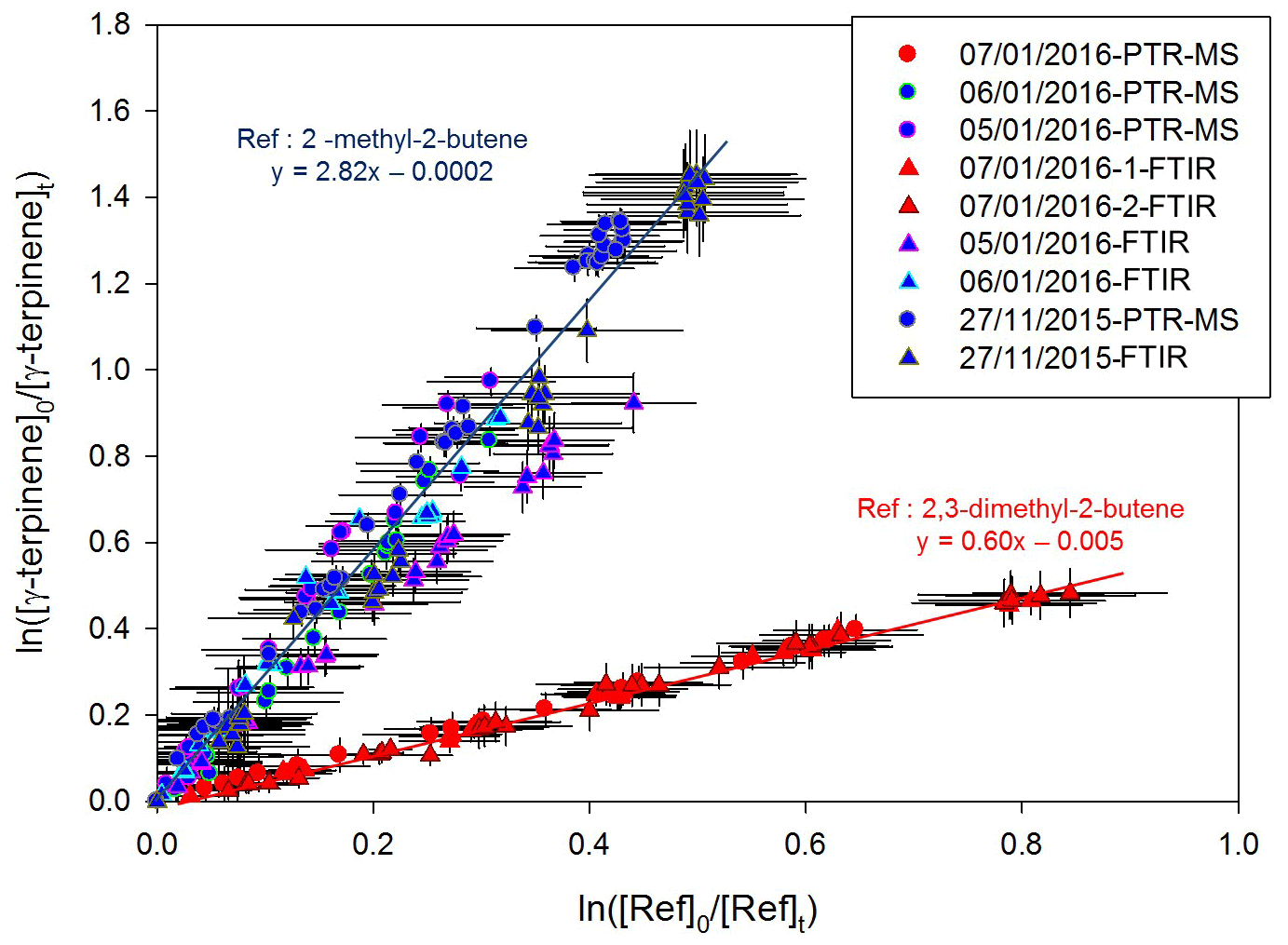
Figure 2Relative kinetic plots measured by FTIR spectrometer (triangle marks) and PTR-ToF-MS (round marks), with 2-methyl-2-butene (blue) and 2,3-dimethyl-2-butene (red) as reference compounds.
For absolute-rate determinations, typical time profiles of reactants measured with the PTR-ToF-MS, FTIR spectroscopy and IBBCEAS are presented in Fig. S1 in the Supplement for the experiment of 30 January 2017 on γ-terpinene. Good agreement is observed for γ-terpinene and between FTIR and PTR-ToF-MS data. Good agreement is also observed for NO2 between FTIR and IBBCEAS data, with an exception for the first experimental point for NO2 following the injection of N2O5, for which a good mixing had probably not been fully achieved yet. These agreements are particularly satisfying considering the fact that the two instruments do not sample in the same volume of the chamber: FTIR spectroscopy provides an integrated measurement of the absorbing species over the whole length of the chamber; IBBCEAS provides an integrated measurement in the width of the chamber, and the PTR-ToF-MS samples the mixture in one point. This comparison demonstrates that the mixing of the chamber is efficient enough to allow for combining data from different instruments for absolute-rate determination.
Kinetic plots for absolute kinetic determinations gathering results from all experiments are presented in Fig. 3 for both BVOCs. As explained above, the first experimental point following the injection of the reactants was not taken into account. Due to the low integration time used for both measurement techniques, a relatively high noise has been observed for BVOC and NO3 concentrations. Kinetic results are thus subject to relatively high uncertainties. Rate constants measured by the absolute-rate method are cm3 molecule−1 s−1 for γ-terpinene and cm3 molecule−1 s−1 for α-terpinene.
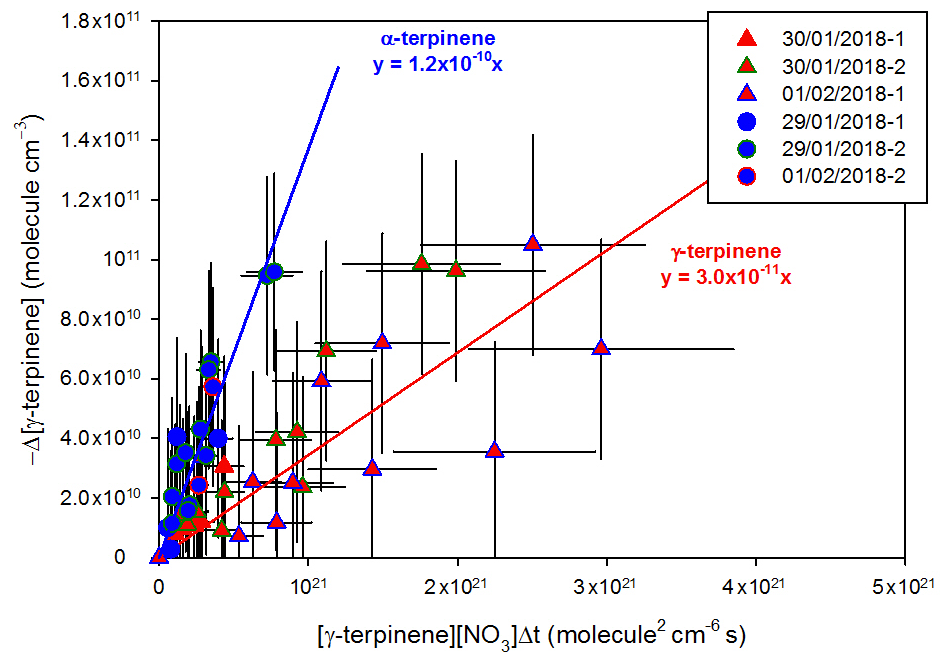
Figure 3Absolute kinetic plots for γ-terpinene (triangle marks) and for α-terpinene (round marks).
The absolute values are compared to those obtained by the relative method (for γ-terpinene) and to those already published in the literature in Table 2. It should be noticed that relative-rate determinations from Atkinson et al. (1985) and Berndt et al. (1996) have been updated by using the same reference rate constants as the ones used for this study (see Sect. 2.3). Uncertainties in the reference rate constants have also been added to the statistical errors provided by the authors.
Table 2Rate constants for the NO3-initiated oxidation of γ-terpinene and α-terpinene: results from this study and comparison with the literature. The bold values are the values measured in our study.
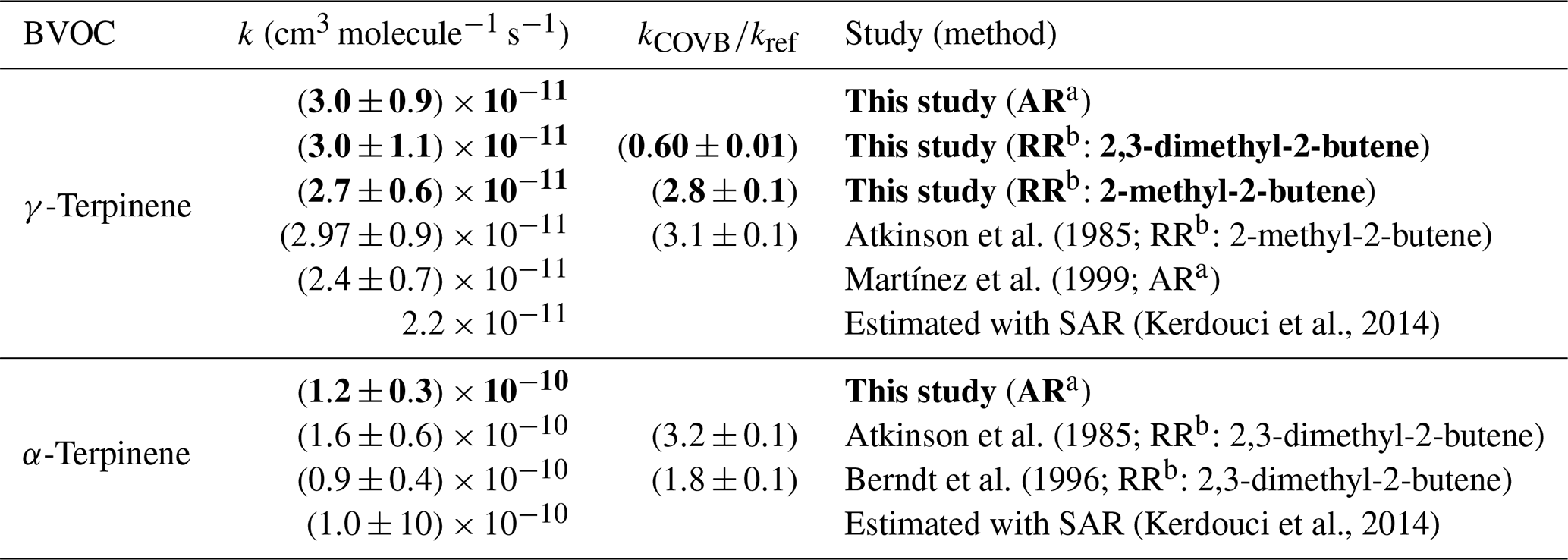
a Absolute-rate determination. b Relative-rate determination.
For γ-terpinene, the three rate constants obtained by this study, i.e., absolute and relative determinations with two reference compounds, are in very good agreement. They are also in good agreement with the relative kinetic study of Atkinson et al. (1985) and with the absolute study of Martínez et al. (1999), even if the second one appears to be 20 % lower. For α-terpinene, the absolute-rate determination provided by this study has been compared to the previous relative determinations provided by Atkinson et al. (1985) and by Berndt et al. (1996). These two relative-rate studies were performed with the same reference compound but using two different experimental setups: a flow reactor (Berndt et al., 1996) and a simulation chamber (Atkinson et al., 1985). When considering the overall uncertainties in these rate constants (approx. 40 %) which include the uncertainty in the reference rate constant, the data seem to be in agreement, but when comparing the ratio kBVOC∕kref, it appears that they are, in fact, not congruent. No explanation was provided by the authors to explain this disagreement. However, for comparison with absolute-rate determination, the overall uncertainty had to be considered. Within uncertainties, the value provided here is in agreement with these two previous values. In conclusion, this study allows for providing new kinetic data for α- and γ-terpinene and for confirming the values obtained by the few previous studies. It also provides the first absolute-rate determination for α-terpinene.
When comparing the reactivity of the two terpenes, it can be seen that α-terpinene is much more reactive than γ-terpinene (by a factor of approximately 4), and this can easily be explained by the conjugation of the double bonds for α-terpinene. Indeed, after the addition of NO3 onto one of the double bonds, the alkyl radical formed is stabilized by the delocalization of the single electron. Experimental data have also been compared to rate constants estimated by the structure–activity relationship (SAR) developed by Kerdouci et al. (2014) for the reaction between BVOCs and NO3 (see Table 2). This SAR has been shown to estimate rate constants within a factor of 2. By taking into account these uncertainties, it can be considered that experimental and estimated rate constants are in good agreement. In particular, the significant increase in the rate constant due to the conjugation of the double bonds is well reproduced by the SAR.
In the CESAM chamber, 11 mechanistic experiments were conducted for γ-terpinene and 8 for α-terpinene, during which the formation of gas-phase products and SOAs was monitored. Experimental conditions as well as organic nitrate and SOA yields obtained for all experiments are presented in Table 3. Figure 4 presents, as an example, time profiles of reactants and products (after correction from dilution) for the experiment of 25 April 2017 on γ-terpinene. During this experiment, N2O5 was introduced into the chamber by slow continuous injection (shown by the red hatched area) in order to ensure a progressive consumption of the BVOC. This injection leads to the formation of large amounts of NO2 and HNO3 which can be explained by N2O5 decomposition and hydrolysis on surfaces (lines, chamber walls) respectively. It should also be noticed that because N2O5 was introduced continuously in small amounts, its concentration remains below the detection limit as long as the concentration of BVOC remains high. γ-Terpinene is totally consumed within approximately 30 min, and this reaction leads to the formation of large amounts of organic nitrates and SOAs. Starting from approximately 500 ppb of γ-terpinene, more than 200 ppb of organic nitrates and 800 µg m3 of aerosols is formed. The aerosol size distribution presented in Fig. 4 shows that particles have mean diameters of around 300–400 nm. PTR-ToF-MS signals (m∕z) are presented in Fig. S2. Several masses corresponding to oxidation products have been detected with the most intense signals being for , 169 and 153. Time profiles and identification of these signals are discussed later.
Table 3Experimental conditions, ONs and SOA yields for mechanistic experiments conducted in the CESAM chamber. The use of an instrument is shown by an “x”, and the non-use is shown by a dash.
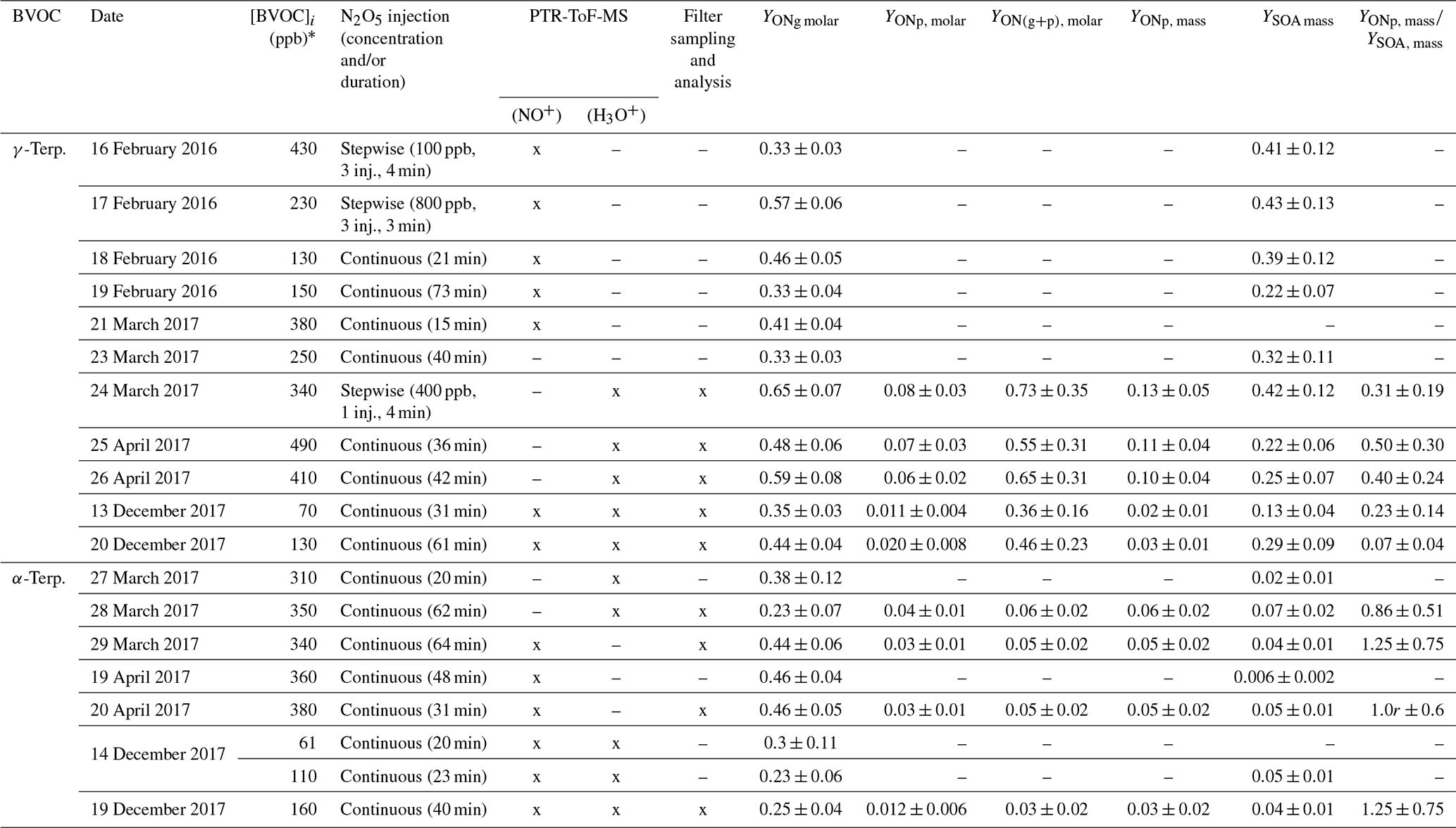
* For all experiments, the BVOC was totally consumed.
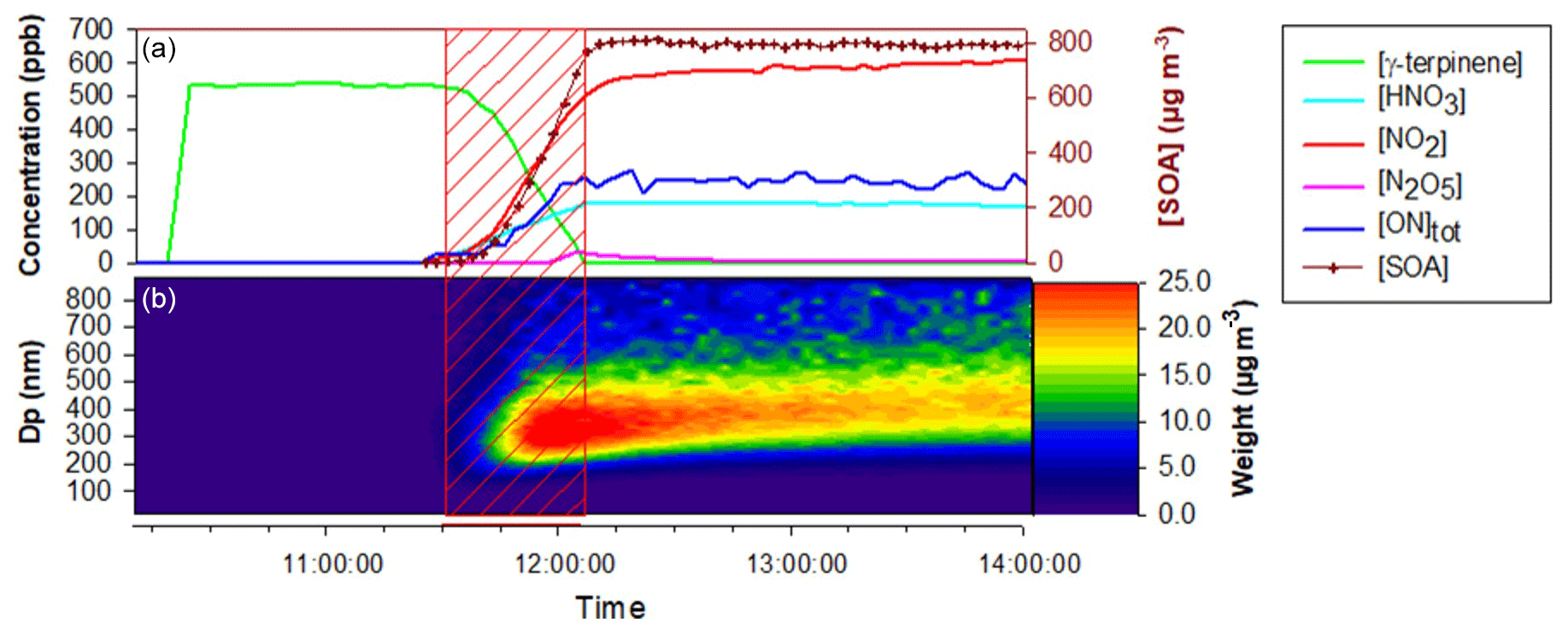
Figure 4Time-dependent concentration of gaseous species, aerosol mass (corrected from dilution), SOA size distribution and PTR-ToF-MS signals during a typical experiment of NO3-initiated oxidation of γ-terpinene (25 April 2017). Red hatched area corresponds to N2O5 injection period. (a) γ-terpinene, N2O5, NO2, HNO3 and total ONs from FTIR spectroscopy and SOA mass concentration from SMPS; b SOA size distribution in mass concentration from SMPS.
4.1 SOA yields
Time-dependent and overall SOA yields (YSOA) for both compounds have been plotted as a function of the aerosol mass (M0) in Fig. 5. A two-product model, defined by Odum et al. (1996) – see Sect. 2.4 – has been applied for the two curves. For each compound, experimental points obtained from different experiments are in fairly good agreement and show similar tendencies. Yields obtained for γ-terpinene can reach 40 %, whereas they are below 2 % for α-terpinene. In the case of α-terpinene this value is subject to possible slight underestimation due to the low purity of the α-terpinene sample (see Sect. 2.1). These results demonstrate that γ-terpinene is a very efficient SOA precursor which is not the case for α-terpinene.
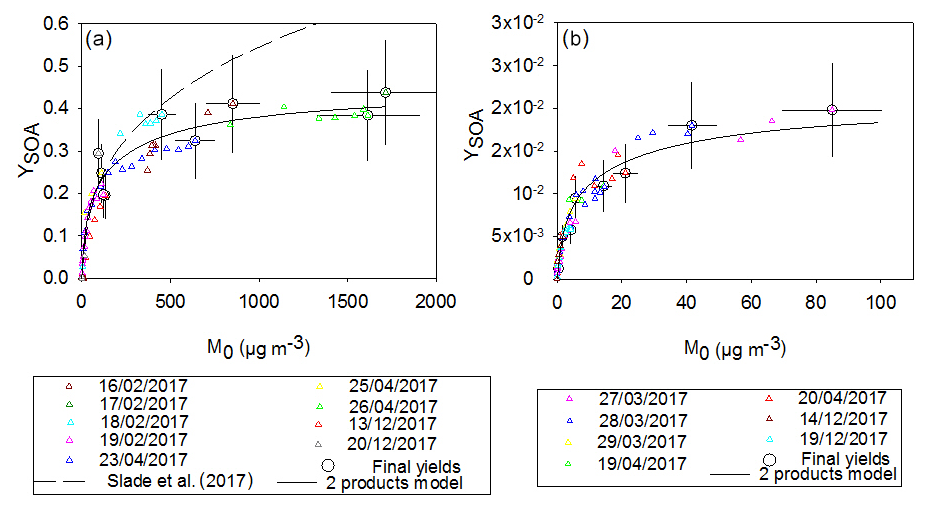
Figure 5SOA yield as a function of the organic aerosol mass concentration measured for γ-terpinene (a) and for α-terpinene (b). Final yields (circle marks) are shown with uncertainties. Data were fitted with a two-product model (solid black curve) and compared with the study of Slade et al. (2017; dashed curve).
As shown in Fig. 5, fitted plots appear to be well constrained for small aerosol content (below 50 µg m−3) due to the many points measured in this area. This is a consequence of the slow injection of N2O5 which allows for a progressive BVOC oxidation. Fitted parameters have been found to be α1=0.22, m3 µg−1 and α2=0.22, m3 µg−1 for γ-terpinene and α1=0.01, m3 µg−1 and α2=0.01, m3 µg−1 for α-terpinene. For both terpenes, SOA production can be successfully modeled by two classes of products having similar stoichiometric factors but different volatilities (Kp, 1 and Kp, 2 differ by more than a factor of 10). However, for γ-terpinene, stoichiometric factors are 2 orders of magnitude higher than those obtained for α-terpinene, leading to higher SOA yields. For α-terpinene, one class of product appears to have very low volatility ( m3 µg−1) but is formed with a very low yield. One can estimate the uncertainties in these parameters by looking at the fit sensitivity. It appears to be very sensitive to α (with an associated error estimated to be 5 %) and less so to Kp (with an error estimated to be 50 %).
For α-terpinene, our study provides the first determination of SOA yields. For γ-terpinene, SOA yields have been compared with those provided by Slade et al. (2017). This study used seeds for half of the experiments and did not observe significantly different yields for experiments conducted with and without seeds. In the study of Slade et al. (2017), a density of 1.7 g cm−3 (which corresponds to the density of seed particles) was used to convert aerosol volume into mass. Data obtained by Slade et al. (2017) have been corrected in order to allow for comparison and to use the same density as the one used here, i.e., 1.4 g cm−3 (Fry et al., 2014). Odum curves have been plotted and compared to our results: up to 200 µg m−3, both Odum curves follow a similar tendency. At higher concentrations, yields measured by Slade et al. (2017) are significantly higher than those obtained here. Despite the use of a progressive N2O5 injection by Slade et al. (2017), reactions were fast and the BVOC was totally consumed within approximately 15 min. In our experiments the oxidation time ranged between 15 min (punctual injection of N2O5) and 70 min (longest continuous injection of N2O5), and we observed that faster oxidations result in higher SOA yields obtained by the local generation of high concentrations of semivolatile species. Hence, the experiment of 15 February 2017 which is one of the fastest continuous injection experiments (21 min) appears to be more congruent with the results from Slade et al. (2017).
In conclusion, both compounds appear to have very different behavior towards SOA production: for an ambient aerosol mass loading of 10 µg m−3, which is typical of biogenic SOA-impacted environments (Slade et al., 2017), yields of 10 % and 1 % have been found for γ-terpinene and α-terpinene respectively. For higher aerosol mass loading observed in polluted atmospheres (between 500 and 1000 µg m−3), yields can reach 30 %–40 % for γ-terpinene and only 2 % for α-terpinene.
4.2 Organic nitrate yields
The formation yields of total organic nitrates in the gas phase (YONg) have been investigated by plotting their concentration as a function of the BVOC consumption for both α- and γ-terpinene (see Fig. 6). The linearity of these plots and the fact that the slopes at the origin are different from zero indicate that (i) organic nitrates are primary products and (ii) if primary organic nitrates are subject to loss processes, e.g., through reaction with NO3, they may produce secondary organic nitrates, so the ON yield is constant. It is also expected that organic nitrates adsorb on the stainless-steel walls. Indeed, the loss rates of several multifunctional organic nitrates (in particular carbonyl nitrates) have been observed in previous studies (Suarez-Bertoa et al., 2012; Picquet-Varrault et al., 2020) and were found to range between 0.5 and s−1. However, as yields of organic nitrates in the gas phase were calculated during a relatively short period (less than 1 h), these wall losses are expected to be low (less than 10 %), and this is confirmed by the good linearity of the plots. Molar YONg values obtained for both BVOCs are very similar: 47±10 % for γ-terpinene and 43±10 % for α-terpinene. These results confirm that organic nitrates are major products of BVOC+NO3 reactions. For γ-terpinene, the yield obtained here has been compared to the only value previously reported in the literature, by Slade et al. (2017): 11±1 %. Despite the fact that experimental conditions are very similar, YONg values differ by a factor of four. Loss reactions of organic nitrates in the particle phase that shift the partitioning equilibrium are invoked by the authors to explain the surprisingly low yield obtained. No influence of relative humidity (RH) on organic nitrate yields has been noticed. Another suggested hypothesis advanced by the authors is an epoxidation of hydroxynitrates in the particle phase, followed by a loss of the NO2 group. However, it is expected that these reactions also occur in our experiments and this hypothesis cannot explain the differences observed between the two studies. Concerning α-terpinene, our study provides the first organic nitrate yield.
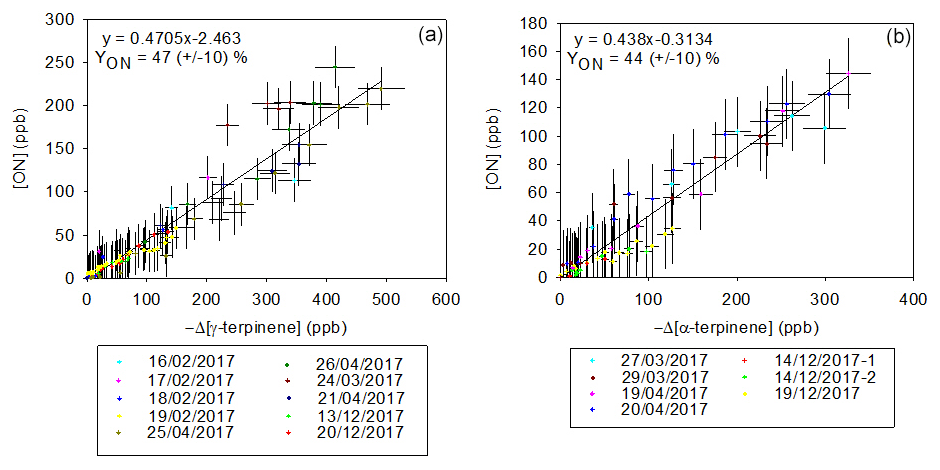
Figure 6Gas-phase organic nitrate production vs. loss of γ-terpinene (a) and α-terpinene (b).
YONg values obtained in this study have also been compared to those obtained for other terpenes available in the literature. Except for α-pinene, for which yields vary between 10 % and 30 % (Fry et al., 2014; Hallquist et al., 1999; Spittler et al., 2006), those obtained for isoprene and monoterpenes are close to our study, with yields higher than 30 %. For β-pinene for example, they vary between 40 % and 74 % (Hallquist et al., 1999; Fry et al., 2014; Boyd et al., 2015), and for limonene, they vary between 30 % and 72 % (Fry et al., 2014, 2011; Hallquist et al., 1999; Spittler et al., 2006).
Considering the fact that organic nitrates may partition into or onto aerosols, yields of total organic nitrates in the particle phase (YONp) have also been measured by collecting particles on filters and analyzing them by FTIR spectroscopy. Values are presented in Table 3. For γ-terpinene, molar yields range between 1 % and 8 %, presenting a high dispersion, and for α-terpinene, they range between 1 % and 3 %. This dispersion can be explained by the fact that (i) YONp directly depends on the total SOA mass concentration and consequently on the concentration of reacted BVOC and (ii) the concentrations of ON are low (minimum is mol L−1) and thus subject to high variability. The first correlation appears to be clear in Table 3: for high-concentration experiments (∼400 ppb), yields reach 10 %, and for low-concentration ones, yields appear to be between 2 % and 3 %. It should also be mentioned that these yields may be affected by wall losses of organic nitrates. Considering that these have been measured by collecting particles several hours after the beginning of the experiment, this may lead to a non-negligible loss of organic nitrates (up to 25 %) and therefore to an underestimation of the yields. Because wall loss rates can vary from one experiment to another, this can also explain the variability in YONp. Also, in the case of α-terpinene the low purity of the sample may lead to a slight underestimation of the calculated yields in both the particle and the gas phase (see Sect. 2.1).
The molar YONp values for γ-terpinene are in good agreement with the one provided by Slade et al. (2017; %) using the same analytical method. The values presented by Slade et al. (2017) also present a high dispersion similar to the one presented here.
In order to estimate the fraction of organic nitrates in SOAs, ON yields in the aerosol phase have been compared to SOA yields, both expressed in terms of mass. Thus, molar YONp values have been converted into mass yields by considering a unique molecular weight which is representative of the expected oxidation products. Here, a hydroxynitrate which has the molecular formula C10H17O4N and the molecular weight of 215 g mol−1 has been used. As discussed later, this compound has been detected as a product by the PTR-ToF-MS. It is clear that this assumption generates large uncertainty in the ON mass yield, especially if products with much higher molecular weight are formed, for example by polymerization in the condensed phase. However, since organic nitrates were not quantified individually, this method allows for estimating the contribution of organic nitrates to SOAs. The values obtained are presented in Table 3. For γ-terpinene, the mass yields have been found to range between 3 % and 13 %, and for α-terpinene, they range between 3 % and 6 %. By comparison with SOA yields, it is estimated that organic nitrates represent ∼50 % of the SOA for γ-terpinene and ∼100 % for α-terpinene. Organic nitrates are therefore major components of the SOA produced by the NO3 oxidation of these two BVOCs.
This conclusion can be compared with some field study results (Kiendler-Scharr et al., 2016; Ng et al., 2017) that found that organic nitrates are a major component of organic aerosols, with a proportion that can reach almost 80 %. Even if organic nitrates can be formed by other chemistries, studies have shown that an enhancement of organic nitrates in SOAs has been observed in NO3-impacted regions: during the night (Gómez-González et al., 2008; Hao et al., 2014; Iinuma et al., 2007) and in forest regions affected by urban air mass (Hao et al., 2014). This result thus confirms the major contribution of organic nitrates to SOA formation and also the importance of NO3 chemistry in this process.
4.3 Products at molecular scale and mechanisms
In order to provide mechanisms and to propose explanations for the different SOA yields between α- and γ-terpinene, an identification of gas-phase products at the molecular scale has been performed by the PTR-ToF-MS. The combination of two different ionization modes, with H3O+ and NO+, for the detection of products allowed for an accurate identification of the molecules. Detected signals in both ionization modes and corresponding raw formulas are summarized in Table 4. Products with molecular weights of 114, 152 and 168 g mol−1 for γ-terpinene and 58, 152, 168 and 171 g mol−1 for α-terpinene have been detected with high intensities. For γ-terpinene the molecular weights of 184, 215 and 229 have also been detected with lower intensities. Many of the detected products are nitrogenous species which is coherent with high production yields of organic nitrates. To explain these observations, mechanisms have been proposed in Fig. 7 for γ-terpinene and in Fig. 8 for α-terpinene. All detected products are framed. Time profiles of PTR-ToF-MS signals were also used to determine whether the products were primary or secondary ones. Typical PTR-ToF-MS profiles are shown in Fig. S2. First-generation products are in blue, and second-generation ones are in red.
Table 4Products detected for γ-terpinene (a) and α-terpinene (b) with PTR-ToF-MS H3O+ and NO+ ionization modes: formula and molar masses, detected masses, ionization processes (H+ is proton adduct; NO+ is NO+ adduct; CT is charge transfer; and PL is proton loss), peak intensity, and comportments.


Figure 7Proposed mechanism for γ-terpinene. First-generation products are in blue, and second-generation ones are in red. Alkoxy fragmentation products are framed according to the location of the fragmentation. Molecular weight, vapor pressures and the gas–particle partition are shown next to the molecules.
4.3.1 γ-Terpinene oxidation scheme
NO3 can react by addition onto one of the two double bonds (H-atom abstraction is considered to be negligible), each addition leading to the formation of two possible nitrooxy alkyl radicals. Kerdouci et al. (2014), who developed a structure–activity relationship for VOC+NO3 reactions, suggest that the additions on the two double bonds of γ-terpinene have the same branching ratios. Thus, the four possible nitrooxy alkyl radicals were considered here. However, to facilitate the reading of the mechanism, only two radicals are shown in Fig. 7, considering that in most cases, products obtained are isomers and cannot be distinguished by the analytical techniques used in this work. The mechanism of the two other alkyl radicals is presented in Fig. S4. Nitrooxy alkyl radicals react with O2 to form a peroxy radical (RO2; Reaction 2). However, the formation of an epoxide (Reaction 3) with a molecular weight of 152 g mol−1 has also been detected in both NO+ (m∕z 152) and H3O+ (m∕z 153) modes. RO2 can then evolve following different pathways: it can react with another RO2 radical to form a hydroxynitrate and a ketonitrate (Reaction 4). The hydroxynitrate (M=215 g mol−1) has been detected at m∕z 216 and m∕z 214 in the H3O+ and NO+ ionization modes respectively. This product is characteristic of the RO2+RO2 pathway. The ketonitrate (M=213 g mol−1) has been detected at m∕z 214 in the H3O+ mode and at m∕z 243 in the NO+ mode (M+30). It should be noticed that this pathway involves an H-atom transfer and so is not possible for tertiary peroxy radicals. RO2 radicals can also react with another RO2 or with NO3 to form alkoxy radicals (RO; Reactions 5 and 5'). Evolution pathways of RO radicals are described by Reactions (6), (7) and (8): (i) alkoxy radicals can react with O2 to form a ketonitrate with M=213 g mol−1 (Reaction 6), which can also be produced by the pathway 4 (RO2+RO2). This ketonitrate is thus not characteristic of a single pathway, contrary to the hydroxynitrate. (ii) They can also decompose by a scission of the C(ONO2)−CH(O•) bond (Reaction 7, framed in orange), leading to the ring opening and to the formation of a dicarbonyl product with M=168 g mol−1 (detected at m∕z 169 in H3O+ mode and at m∕z 168 in NO+ mode). (iii) Finally, they can decompose by a ring opening on the other side of the alkoxy group (Reaction 8), leading to the formation of an alkyl radical which reacts to form a trifunctional compound (two carbonyl groups and one nitrate group) of molecular weight MW = 229 g mol−1 (framed in green in Fig. 7). This product has been detected with a weak signal at m∕z 230 in H3O+ ionization mode but was not observed in the NO+ mode. Because quantification of these products was not possible, branching ratios between these different pathways could not be determined.
The SAR developed by Vereecken and Peeters (2009) which is based on density-functional theory (DFT) calculations, has been used to estimate the energy barriers of the various reaction pathways of the alkoxy radicals. Energy barriers for Reactions (7) and (8) appear to be similar ( kcal mol−1 and kcal mol−1, with an error estimated by the authors of 0.5 kcal mol−1), leading to similar branching ratios for the two possible ring openings. The loss of methyl or isopropyl group presents a significantly higher energy barrier (10.2 kcal mol−1) whatever the alkoxy radical considered. These pathways appear minor compared to the ring opening, and this is in good agreement with the fact that acetone was not detected during the experiments. It should also be noticed that peroxynitrates (RO2NO2), which have a characteristic absorption in the IR region, were not detected in our experiments, either in the gaseous phase or in the aerosol one. This suggests that RO2+NO2 reactions are minor pathways.
Primary products that still have a double bond can also react with the nitrate radical to form second-generation products, in red in the mechanism. This is confirmed by time profiles of primary products MW =152 and 168 g mol−1 which are decreasing with time (see Fig. S2) and by the detection of secondary products with MW =114, 86, 184, 247 and 245 g mol−1. The product with MW =184 g mol−1 corresponds to a second-generation epoxide. Other products can be explained by the reaction of dicarbonyl compounds with NO3. The compounds with MW =114 and 86 g mol−1 correspond to second-generation dicarbonyl products formed by the decomposition of RO radicals (Reaction 7-2). Finally the RO2+RO2 reaction (Reaction 4-2) and the RO+O2 reaction (Reaction 6-2) can lead to the formation of highly functionalized products with MW =247 g mol−1 and MW =245 g mol−1.
Vapor pressures and percentage of partitioning into the aerosol phase were calculated as described in Sect. 2.4 for aerosol mass loading of 800 µg m−3, typical of an experiment end, and are also presented next to the molecules in Fig. 7. Among the first-generation products, two types of products have low vapor pressures and can thus participate in SOA formation: first, the hydroxynitrate (e.g., MW =215 g mol−1) which is characteristic of the RO2+RO2 pathway (Fig. 7 – pathway 4). It was estimated to partition at 40 % in the particle phase. Secondly, trifunctional molecules (Fig. 7 – pathway 8) are expected to have very low volatility and to be present mainly in the aerosol phase (close to 100 %). For these two products, the associate partitioning coefficient, Kp, following Eq. (10) has been calculated. Considering the uncertainty in due to the vapor pressure estimation, it can vary from to m3 µg−1 for the hydroxynitrate (MW =215 g mol−1) and from to m3 µg−1 for the trifunctional compound (MW =229 g mol−1). This appears to be consistent with the partitioning coefficients found with the two product model from Eq. (8) ( m3 µg−1 and m3 µg−1), especially by considering the associated uncertainty estimated in Kp. One can then consider that the two groups of products used by the two products model can be constituted by the hydroxynitrate and the trifunctional compounds or by similar products.
Other first-generation products are estimated to play a minor role in SOA formation. For secondary products, multifunctional products (with four chemical groups) are estimated to be between 80 % and 100 % in the particle phase. Other secondary products which are formed by fragmentation processes (dicarbonyl compounds) are expected to be volatile.
To conclude, the oxidation of γ-terpinene by NO3 leads to the formation of several functionalized products and particularly to multifunctional organic nitrates which were detected in both phases. Most detected products can be formed by different pathways; thus no preferential pathway could clearly be identified. Nevertheless, products with up to four chemical groups (nitrate, carbonyl and alcohol) have been identified and explain the high SOA formation. In particular, the reaction seems to play a significant role in the SOA formation as hydroxynitrates formed have low vapor pressures (both first- and second-generation products) with a large probability of partitioning into the particle phase.
Results obtained in this study have been compared to those furnished by Slade et al. (2017), who used the chemical ionization mass spectrometry (CIMS) technique for the detection of products. Mechanisms appear to be very similar: hydroxynitrates, ketonitrates and dicarbonyl compounds (coming from ring opening) have been observed. No quantification of products has been provided except for hydroxynitrates with an estimated yield in the gas phase of 4 % by using a standard derived from α-pinene. This low yield can be compared with the low total organic nitrate yield found in the same experiments (10 %). In this study, hydroxynitrates were thus found to represent 40 % of total organic nitrates. While detected in our experiments, epoxides were not detected in this previous study. In addition, Slade et al. (2017) have detected hydroperoxides formed by RO2+HO2 reaction which were not observed in our study. Two hypotheses can explain this difference: (i) in our experiments HO2 concentrations are too small for making this reaction significant compared to those with RO2+RO2 or RO2+NO3 or (ii) if formed, hydroperoxides decompose on the stainless-steel walls of the CESAM chamber. The second hypothesis is expected to be more likely as the loss of hydroperoxides on CESAM stainless-steel walls (particularly H2O2) has already been observed.
4.3.2 α-Terpinene oxidation scheme
As described for γ-terpinene, addition of NO3 onto the double bonds of α-terpinene can lead to the formation of four nitrooxy alkyl radicals. Nevertheless, in this case, the conjugation of the two double bonds allows for a delocalization of the single electron and, hence, a stabilization of the two corresponding nitrooxy alkyl radicals (see Fig. S3). Thus, nitrooxy alkyl radicals expected to be the most favorable are those which can undergo an electron delocalization. Note that this delocalization leads to the formation of two tertiary nitrooxy alkyl radicals. A mechanism has been established by considering all possible radicals, but for clarity in Fig. 8, only two radicals are presented. The mechanism of the other two alkyl radicals is presented in Fig. S5.
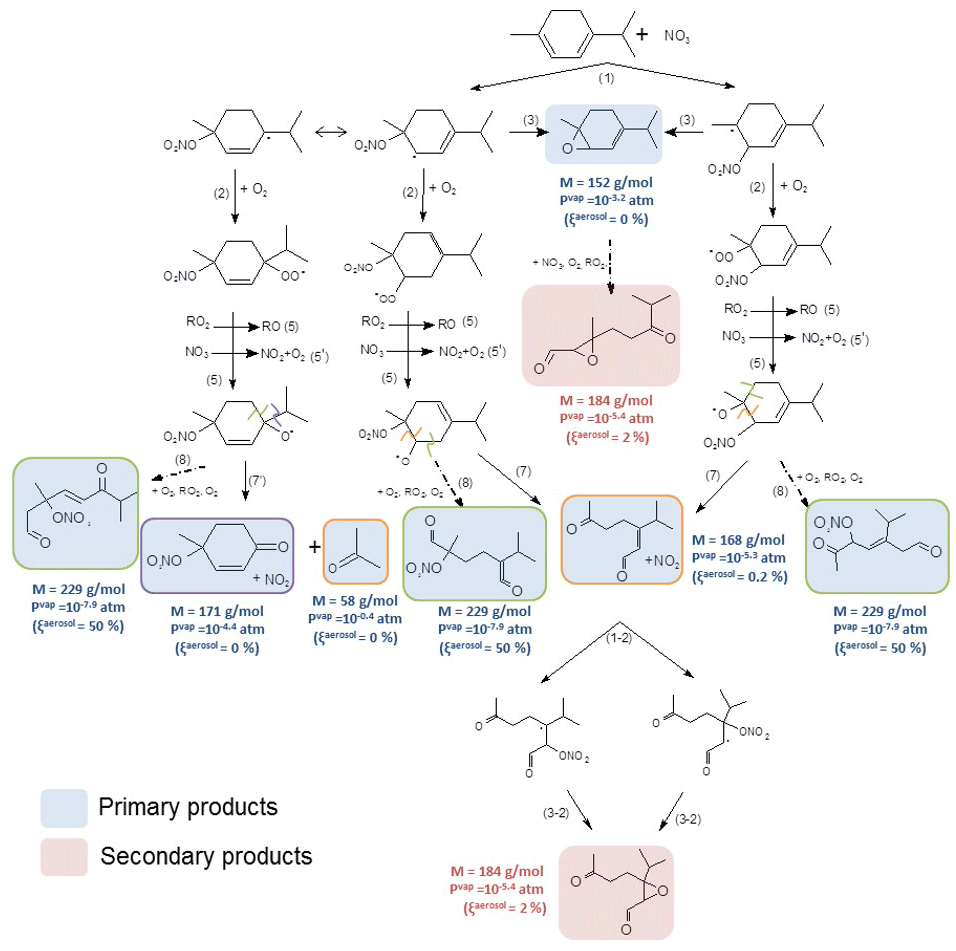
Figure 8Proposed mechanism for α-terpinene. First-generation products are in blue, and second-generation ones are in red. Alkoxy fragmentation products are framed according the location of the fragmentation. Molecular weight, vapor pressures and the gas–particle partition are shown next to the molecules. The reaction of the primary epoxide product is shown in the purple frame.
Like for γ-terpinene, nitrooxy alkyl radicals can lose the NO2 group to form epoxides (Reaction 3) which were detected by the PTR-ToF-MS (m∕z 153 in H3O+ mode and m∕z 152 in NO+ mode). They also react with O2 to form RO2 radicals which then evolve through the RO2+NO3 reaction (Reaction 5) and/or through the RO2+RO2 reaction (Reaction 5') to form alkoxy radicals. Here, the formation of the hydroxynitrate (MW =215 g mol−1), was not observed suggesting that the reaction does not occur. This can be explained by the fact that most favored radicals are tertiary ones and cannot undergo H shift. From product identification we propose the following decomposition pathways for RO radicals: (i) alkoxy radicals can lose the isopropyl group leading to the formation of a cyclic ketonitrate and an isopropyl radical which then evolve to form acetone. The cyclic ketonitrate and the acetone have been detected by the PTR-ToF-MS with high-intensity signals in both ionization modes. Acetone was also detected by FTIR spectroscopy, but its concentration was close to the detection limit of 10 ppb and formation yield could not be precisely measured. Nevertheless, by considering the 10 ppb detection limit, the acetone formation yield is expected to be below 3 %. (ii) They can also decompose by a scission of the C(ONO2)−CH(O•) bond (Reaction 7, framed in orange) leading to a ring opening and to the formation of a dicarbonyl product with MW =168 g mol−1. This compound was detected with high signals by the PTR-ToF-MS in the H3O+ and NO+ modes at m∕z 169 and m∕z 168 respectively. (iii) The formation of trifunctional species (one nitrate group and two carbonyl groups) with MW =229 g mol−1, coming from the ring opening on the other side of alkoxy group (Reaction 8), has also been observed, but it needs confirmation. Indeed, mass m∕z 230 has been detected in the H3O+ mode, but neither m∕z 229 nor 259 was detected in the NO+ mode. As for γ-terpinene, the trifunctional species can be formed with low concentrations and/or be mostly in the particle phase leading to weak signals with the PTR-ToF-MS.
The SAR proposed by Vereecken and Peeters (2009) allowed for estimating the energy barriers of the different evolution pathways of alkoxy radicals. It suggests that ring openings (Reactions 7 and 8) are the two most likely pathways with similar branching ratios (energy barriers are 6.0 and 6.5 kcal mol−1 respectively). As for γ-terpinene, Reaction (7') corresponding to the loss of the isopropyl group appears to be less favorable with an energy barrier of 10.2 kcal mol−1. This is in agreement with the low acetone formation yield. This is also in agreement with the detection of the trifunctional species for which the weak signal observed can be explained by its low volatility. This product is the only primary product expected to contribute to SOA formation (with %). As a reminder, the associated partitioning coefficient Kp for this trifunctional compound (MW =229 g mol−1) was estimated to range between and m3 µg−1. It can correspond to the Kp, 2 estimated by the two-product model in Eq. (8) ( m3 µg−1). The second partitioning coefficient in the two-product model ( m3 µg−1) cannot be attributed, and it can be explained by (i) the fact that this product can be totally in the particle phase and thus not detectable in the gas phase and (ii) the very low production of SOAs, leading to a low precision on the fit.
The only secondary products identified are epoxides that can come not only from the reaction of the primary epoxide with NO3 but also from the oxidation of the unsaturated dicarbonyl compounds. In the last case, they are epoxides coming from the loss of −NO2 from alkyl radicals (Reaction 3-2). Other secondary products were expected but not detected. Signal m∕z 243 has been detected in the NO+ mode, but this signal could not be attributed to a product.
In conclusion, products detected with the highest signals are cyclic ketonitrates, dicarbonyl compounds and epoxides. These three families of products have high vapor pressures and are thus not expected to significantly contribute to SOA formation. Trifunctional products with low vapor pressures have also been detected with low signals, suggesting low formation yields or strong partition in the aerosol phase. Low SOA yields measured for α-terpinene suggest that the first explanation is more favorable.
Even though α- and γ-terpinene have similar chemical structures only differing by the position of the double bonds, their reactions with the nitrate radical have different consequences especially regarding SOA formation. Mean SOA and organic nitrate yields obtained for both compounds are presented in Table 5. Several previous studies on BVOC+NO3 reactions suggest a correlation between organic nitrate yield and SOA formation (Fry et al., 2014; Hallquist et al., 1999). α-Pinene indeed presents a low organic nitrate yield, in good agreement with an SOA yield close to zero, when limonene and Δ-carene both present high SOA and organic nitrate yields. In our study, YONg and YONp for α- and γ-terpinene are similar regarding the uncertainty. Thus α-terpinene does not follow this correlation, as it produces a high amount of organic nitrates but almost no SOAs.
Table 5Mean SOA and organic nitrate yields obtained in this study for γ-terpinene and α-terpinene.

To interpret the difference in SOA formation, the mechanisms have to be compared: on the one hand, for γ-terpinene, the major SOA production can be explained by the formation of (i) a primary hydroxynitrate coming from the RO2+RO2 pathway, (ii) a primary trifunctional nitrate formed by the decomposition of the alkoxy radical and (iii) detection of secondary products, all presenting very low vapor pressures. On the other hand, for α-terpinene (i) no hydroxynitrate and (ii) no secondary products with low vapor pressure were detected in the oxidation products. The absence of hydroxynitrate may be explained by the fact that, due to relocation of the free electron by the mesomeric effect for alkyl radicals, the most stable radicals are tertiary and cannot undergo H shift to produce the hydroxynitrate. Hence, two aspects of the mechanism appear to be critical for the SOA formation:
The peroxy radical reaction pathways. When the carbon that bears the radical group, has a hydrogen available, the reaction can occur, leading here to the formation of an hydroxynitrate. Due to hydrogen bonds, this product has very low vapor pressure. This has already been reported by Ng et al. (2008) who compared isoprene SOA formation for reactions RO2+RO2 and RO2+NO3. The study reported higher formation of SOAs for the pathway RO2+RO2 due to the formation of the specific hydroxynitrate and its secondary reaction. In our study, this product was detected for γ-terpinene but not for α-terpinene for which we expect that most stable radicals are tertiary ones and cannot undergo this reaction pathway.
The alkoxy radical reaction pathways. Several decomposition pathways can occur, leading to products which have very different volatilities. If they decompose by a scission of the C(ONO2)−CH(O•) bond, dicarbonyl products, which are volatile, are formed. If the decomposition occurs by a ring opening on the other side of the alkoxy group, keto-nitrooxy-alkyl radicals are formed, which then evolve towards the formation of low-vapor trifunctional species. This point has already been raised by Kurten et al. (2017), who performed computational calculations on the alkoxy reaction pathways to explain the low SOA yield observed for α-pinene, in comparison to Δ-carene. The authors suggest that for Δ-carene, the decomposition of alkoxy radicals can lead to the formation of keto-nitrooxy-alkyl radicals, whereas for α-pinene, the alkoxy radicals decompose almost exclusively to form the dicarbonyl compound.
The study of Claflin and Ziemann (2018) showed that the hydroxynitrates formed by the RO2+RO2 pathway and the carbonyl compounds, via an acid-catalyzed particle-phase reaction, lead to the formation of acetal dimers and trimers. No molecular analysis of the particle phase, except for the organic nitrates, was conducted. If polymers are formed in the particle phase, for example acetal dimers and trimers, which have a nitrate group, they cannot be distinguished from the monomers. For γ-terpinene, hydroxynitrates and carbonyl nitrates were detected in the gas phase, which have low enough volatility to enter the particle phase and contribute the most to SOA formation. They can then react to form acetal dimers or trimers in the particle phase but with no possible detection. For α-terpinene, no hydroxynitrate formation was detected; the formation of these dimers is expected to be negligible. This is in good agreement with the low SOA yields for this compound.
This study also showed the importance of the RO2+RO2 reaction and alkoxy decomposition, which are the key points in α- and γ-terpinene chemistry. The importance of isomerization and the acid-catalyzed particle-phase reaction has not been proved but is coherent with the results.
It is also interesting to compare the reactivity of γ-terpinene and α-terpinene to those of other monoterpenes. On the one hand, γ-terpinene produces large amounts of SOAs (with yields ranging between 20 % and 40 %), similarly to limonene, β-pinene, Δ-3-carene and sabinene (Fry et al., 2009, 2014; Griffin et al., 1999; Hallquist et al., 1999; Moldanova and Ljungström, 2000; Spittler et al., 2006). On the other hand, α-terpinene produces small amounts of SOAs with yields of around 1 %. It can be compared to α-pinene, with yields between 0 % and 16 % (Hallquist et al., 1999; Spittler et al., 2006; Nah et al., 2016; Fry et al., 2014; Perraud et al., 2010). An explanation for this low SOA yield regarding α-pinene has been given by Kurten et al., (2017).
The two studied compounds have organic nitrate yields of around 50 % which appear similar to those measured for other BVOCs, such as limonene, with yields between 30 % and 72 % (Hallquist et al., 1999; Spittler et al., 2006), or β-pinene, between 22 % and 74 % (Boyd et al., 2015; Fry et al., 2014; Hallquist et al., 1999). Within the uncertainties, they also appear similar to those obtained for Δ-carene (68 %–77 %; Fry et al., 2014; Hallquist et al., 1999) and isoprene (62 %–78 %; Rollins et al., 2009). For most BVOCs, NO3 chemistry is a major organic nitrate precursor. Only α-pinene has been shown to produce less organic nitrate, between 10 % and 25 % (Berndt and Böge, 1997; Fry et al., 2014; Hallquist et al., 1999; Spittler et al., 2006; Wangberg et al., 1997).
One interesting fact is that for both compounds, epoxides have been detected, whereas it is usually admitted that their formation is favored only at low oxygen concentration (Berndt and Böge, 1995). Many studies did not detect these compounds (Jaoui et al., 2013; Slade et al., 2017; Spittler et al., 2006), but their formation has already been observed by Skov et al. (1994), who studied NO3 oxidation of several alkenes and isoprene. Wangberg et al. (1997) also showed low epoxide yields (3 %) for α-pinene, and Ng et al. (2008) showed low epoxide yields (>1 %) for isoprene. In our study, epoxides were not quantified, but based on previous studies, their formation yields are expected to be low.
In summary, this work provides kinetic and mechanistic data on the oxidation by NO3 of α- and γ-terpinene, using simulation chambers. The two compounds present very similar chemical structures (the same carbon skeleton and two double bonds which are conjugated in the case of α-terpinene and not for γ-terpinene), and this work aimed at highlighting the influence of the structure on the reactivity, in particular on the SOA formation.
Absolute and relative kinetic determinations have been performed. This study provides the first absolute determination for α-terpinene. Both α- and γ-terpinene appear to be very reactive towards the nitrate radical due to the presence of two double bonds. α-Terpinene is particularly reactive with NO3 due to the conjugation of the double bonds while γ-terpinene is 4 times less reactive.
As far as we know, this study is the first mechanistic study for the oxidation of α-terpinene by NO3. Our study has confirmed that the NO3 oxidation of α- and γ-terpinene produces large amounts of organic nitrates (with overall yields ∼50 %) which have been shown to be present in both gas and aerosol phases. Nevertheless, major differences in the SOA formation for the two compounds have been pointed out despite their similar structure. γ-Terpinene has been shown to be an efficient SOA precursor, whereas α-terpinene is a poorly efficient SOA precursor. To explain these differences, a molecular-scale study has been conducted, and two reaction pathways have been shown to play a key role in the SOA formation: (i) the peroxy radical reaction pathways leading to the formation of low-volatility hydroxynitrates, which were detected for γ-terpinene but not for α-terpinene and (ii) the alkoxy radical scission pathways which can form either high-volatility dicarbonyl compounds or low-volatility trifunctional products, depending on where the scission occurs.
The atmospheric lifetimes of the two compounds have been estimated by using typical nighttime NO3 concentration (10 ppt) and low insolation diurnal concentration (0.1 ppt; Corchnoy and Atkinson, 1990). These lifetimes are presented and compared to those estimated for oxidation by OH and ozone in Table 6. Prior to the discussion, it is important to remember that monoterpenes are intensively emitted during both the day and the night (Lindwall et al., 2015). From Table 6, it can be observed that the two monoterpenes exhibit very short lifetimes towards NO3 for nighttime conditions (40 s for α-terpinene and 2 min for γ-terpinene) confirming that NO3 oxidation is a major sink for these compounds. As expected, lifetimes estimated for low-sunlight diurnal conditions are longer (a few hours) but are still fairly short. By comparison with lifetimes estimated for other oxidants, it is concluded that all three oxidants are very efficient sinks. For low-insolation diurnal conditions, even though diurnal chemistry is clearly led by OH and O3, NO3 oxidation is not negligible. This result can be compared to the modeling study of Forkel et al. (2006), which has shown that NO3 oxidation is an important sink of BVOCs even during the day under specific conditions (under canopy, with low luminosity and considering a calculated mixing ratio for NO3 of 3 ppt).
Table 6Atmospheric lifetimes of α- and γ-terpinene with respect to their oxidation by NO3 and OH radicals and by ozone.

a Calculated with molecules cm−3 (10 ppt).
b Calculated with molecules cm−3 (0.1 ppt).
c Calculated with molecules cm−3 and
molecules cm−3.
d Calculated with rate constant recommended by the IUPAC.
e Calculated with rate constant from Atkinson et al. (1986).
The short lifetimes indicate that oxidation products are formed close to the BVOCs emission areas. If all three oxidants are major sinks, the products formed by the different processes are very different. Organic nitrates can also be formed by OH oxidation through RO2+NO reactions, but yields are much lower. Lee et al. (2006) have investigated the products formed by the OH oxidation of 16 terpenoids, including α- and γ-terpinene, in the presence of NOx. Organic nitrate yields were shown to be less than 1 %. So a major impact of BVOC oxidation by NO3 is the formation of organic nitrates which are known to act as NOx reservoirs. In our study, dicarbonyl compounds have also been shown to be formed, but the same compounds have been detected as major products of the OH chemistry of α- and γ-terpinene by Lee et al. (2006). So this is not a special feature of the nighttime chemistry.
SOA yields produced by NO3 oxidation can be compared to those formed by ozonolysis and OH oxidation. Friedman and Farmer (2018), Griffin et al. (1999) and Lee et al. (2006) have measured the SOA yields for the OH oxidation of several terpenes, including α- and γ-terpinene. In the experiments performed by Griffin et al. (1999), mixing of terpenes was introduced leading to overall SOA yields which were found to be 4 % at 10 µg m−3 and 20 % at 400 µg m−3. Lee et al. (2006) have provided final SOA yields for α- and γ-terpinene and shown that they are below 20 %. Finally, the study of Friedman and Farmer (2018) was performed with low-NOx conditions, and SOA yields were found to be very low (≤1 %) at M0=10 µg m−3. Regarding these results, the oxidation by NO3 appears to be a much more efficient SOA source than the OH oxidation. This observation has already been made by several previous studies (Hallquist et al., 1997; Griffin et al., 1999; Spittler et al., 2006; Ng et al., 2008; Fry et al., 2014; Boyd et al., 2015; Slade et al., 2017). Regarding the ozonolysis of α- and γ-terpinene, there are, to our knowledge, no data on SOA yields in the literature. However, more generally, the ozonolysis of BVOCs is known to be an important source of SOAs.
In conclusion, the most important impacts of this chemistry rely on the formation of large amounts of organic nitrates (present in both gas and aerosol phases) and SOAs. On the one hand, organic nitrates play a key role in tropospheric chemistry because they behave as NOx reservoirs, carrying reactive nitrogen in remote areas. Their chemistry in gas and aerosol phases is nevertheless still not well documented. Considering that our study shows a large production of multifunctional organic nitrates, it is necessary to better understand their reactivity in order to better evaluate their impacts. Formation of SOAs seems, on the other hand, strongly dependent on the structure of the BVOC. Studies at a molecular scale are thus required to better evaluate the impact of this chemistry on the SOA formation.
The rate constants for the NO3 oxidation of α-and γ-terpinene are available in Table 2. They are also available through the Library of Advanced Data Products (LADP) of the EUROCHAMP data center (https://data.eurochamp.org/data-access/gas-phase-rate-constants/, last access: 1 May 2020; Fouqueau et al., 2020b). Kinetic and mechanistic simulation chamber experiments are available through the Database of Atmospheric Simulation Chamber Studies (DASCS) of the EUROCHAMP data center (https://data.eurochamp.org/data-access/chamber-experiments/, last access: 1 May 2020; Fouqueau et al., 2020c).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-15167-2020-supplement.
BPV and MCi coordinated the research project. AF, BPV, MCi and JFD designed the experiments in the simulation chambers. AF performed the experiments with the technical support of MC and EP and performed the data treatment and interpretation with MCi and BPV. AF, BPV and MCi wrote the paper, and AF was responsible for the final version of the paper. All coauthors revised the content of the original manuscript and approved the final version of the paper.
The authors declare that they have no conflict of interest.
This article is part of the special issue “Simulation chambers as tools in atmospheric research (AMT/ACP/GMD inter-journal SI)”. It is not associated with a conference.
The authors thank Marie Camredon (LISA, Créteil, France) for helping with the GECKO-A website and Marie-Thérèse and Jean-Claude Rayez (ISM, Bordeaux, France) for helping in understanding the reactivity with theoretical calculation.
This research has been supported by the French national program INSU LEFE and the European Commission, Horizon 2020 Research Infrastructures (EUROCHAMP-2020 (grant no. 730997)).
This paper was edited by Nga Lee Ng and reviewed by two anonymous referees.
Aoki, N., Inomata, S., and Tanimoto, H.: Detection of C1–C5 alkyl nitrates by proton transfer reaction time-of-flight mass spectrometry, Int. J. Mass Spectrom., 263, 12–21, 2007.
Atkinson, R.: Kinetics and mechanisms of the gas-phase reactions of the hydroxyl radical with organic compounds under atmospheric conditions, Chem. Rev., 86, 69–201, 1986.
Atkinson, R.: Estimation of gas-phase hydroxyl radical rate constants for organic chemicals, Environ. Toxicol. Chem., 7, 435–442, 1988.
Atkinson, R. and Arey, J.: Gas-phase tropospheric chemistry of biogenic volatile organic compounds: a review, Atmos. Environ., 37, 197–219, https://doi.org/10.1016/S1352-2310(03)00391-1, 2003.
Atkinson, R., Plum, C. N., Carter, W. P. L., Winer, A. M., and Pitts, J. N.: Rate constants for the gas-phase reactions of nitrate radicals with a series of organics in air at 298 1 K, J. Phys. Chem., 88, 1210–1215, https://doi.org/10.1021/j150650a039, 1984a.
Atkinson, R., Aschmann, S. M., Winer, A. M., and Pitts, J. N.: Kinetics of the gas-phase reactions of nitrate radicals with a series of dialkenes, cycloalkenes, and monoterpenes at 295 1 K, Environ. Sci. Technol., 18, 370–375, https://doi.org/10.1021/es00123a016, 1984b.
Atkinson, R., Aschmann, S. M., Winer, A. M., and Pitts, J. N.: Kinetics and atmospheric implications of the gas-phase reactions of nitrate radicals with a series of monoterpenes and related organics at 294 2 K, Environ. Sci. Technol., 19, 159–163, https://doi.org/10.1021/es00132a009, 1985.
Atkinson, R., Aschmann, S. M., and Pitts, J. N.: Rate constants for the gas-phase reactions of the OH radical with a series of monoterpenes at 294±1 K, Int. J. Chem. Kinet., 18, 287–299, https://doi.org/10.1002/kin.550180303, 1986.
Benter, Th., Becker, E., Wille, U., Rahman, M. M., and Schindler, R. N.: The Determination of Rate Constants for the Reactions of Some Alkenes with the NO3 Radical, Berichte der Bunsengesellschaft für physikalische Chemie, 96, 769–775, https://doi.org/10.1002/bbpc.19920960607, 1992.
Berndt, T. and Böge, O.: Products and Mechanism of the Reaction of NO3 with Selected Acyclic Monoalkenes, J. Atmos. Chem., 21, 275–291, 1995.
Berndt, T. and Böge, O.: Products and mechanismof the gas-phase reaction of NO3 radicals with α-pinene, J. Chem. Soc. Faraday T., 93, 3021–3027, 1997.
Berndt, T., Böge, O., Kind, I., and Rolle, W.: Reaction of NO3 radicals with 1,3-cyclohexadiene α-terpinene, and α-phellandrene: Kinetics and products, Berichte der Bunsengesellschaft für physikalische Chemie, 100, 462–469, 1996.
Berndt, T., Kind, I., and Karbach, H.-J.: Kinetics of the Gas-Phase Reaction of NO3 Radicals with 1-Butene, trans-Butene, 2-Methyl-2-butene and 2,3-Dimethyl-2-butene Using LIF Detection, Berichte Bunsengesellschaft für physikalische Chem., 102, 1486–1491, https://doi.org/10.1002/bbpc.199800017, 1998.
Boyd, C. M., Sanchez, J., Xu, L., Eugene, A. J., Nah, T., Tuet, W. Y., Guzman, M. I., and Ng, N. L.: Secondary organic aerosol formation from the β-pinene+NO3 system: effect of humidity and peroxy radical fate, Atmos. Chem. Phys., 15, 7497–7522, https://doi.org/10.5194/acp-15-7497-2015, 2015.
Brown, S. S. and Stutz, J.: Nighttime radical observations and chemistry, Chem. Soc. Rev., 41, 6405–6447, https://doi.org/10.1039/C2CS35181A, 2012.
Claflin M. S. and Ziemann P. J.: Identification and Quantitation of Aerosol Products of the Reaction of β-Pinene with NO3 Radicals and Implications for Gas- and Particle-Phase Reaction Mechanisms J. Phys. Chem. A, 122, 3640–3652, 2018.
Corchnoy, S. B. and Atkinson, R.: Kinetics of the gas-phase reactions of hydroxyl and nitrogen oxide (NO3) radicals with 2-carene, 1,8-cineole, p-cymene, and terpinolene, Environ. Sci. Technol., 24, 1497–1502, 1990.
Draper, D. C., Farmer, D. K., Desyaterik, Y., and Fry, J. L.: A qualitative comparison of secondary organic aerosol yields and composition from ozonolysis of monoterpenes at varying concentrations of NO2, Atmos. Chem. Phys., 15, 12267–12281, https://doi.org/10.5194/acp-15-12267-2015, 2015.
Duncianu, M., David, M., Kartigueyane, S., Cirtog, M., Doussin, J.-F., and Picquet-Varrault, B.: Measurement of alkyl and multifunctional organic nitrates by proton-transfer-reaction mass spectrometry, Atmos. Meas. Tech., 10, 1445–1463, https://doi.org/10.5194/amt-10-1445-2017, 2017.
Doussin, J.-F., Durand-Jolibois, R., Ritz, D., Monod, A., and Carlier, P.: Design of an environmental chamber for the study of atmospheric chemistry: New developments in the analytical device, Analusis, 25, 236–242, 1997.
Forkel, R., Klemm, O., Graus, M., Rappenglück, B., Stockwell, W. R., Grabmer, W., Held, A., Hansel, A., and Steinbrecher, R.: Trace gas exchange and gas phase chemistry in a Norway spruce forest: A study with a coupled 1-dimensional canopy atmospheric chemistry emission model, Atmos. Environ., 40, 28–42, 2006.
Fouqueau, A., Cirtog, M., Cazaunau, M., Pangui, E., Zapf, P., Siour, G., Landsheere, X., Méjean, G., Romanini, D., and Picquet-Varrault, B.: Implementation of an incoherent broadband cavity-enhanced absorption spectroscopy technique in an atmospheric simulation chamber for in situ NO3 monitoring: characterization and validation for kinetic studies, Atmos. Meas. Tech., 13, 6311–6323, https://doi.org/10.5194/amt-13-6311-2020, 2020a.
Fouqueau, A., Cirtog, M., Cazaunau, M., Pangui, E., Doussin, J.-F., and Picquet-Varrault, B.: Library of Advanced Data Products: Gas Phase Rate Constants, EUROCHAMP Data Center, available at: https://data.eurochamp.org/data-access/gas-phase-rate-constants/, last access: 1 May 2020b.
Fouqueau, A., Cirtog, M., Cazaunau, M., Pangui, E., Doussin, J.-F., and Picquet-Varrault, B.: Database of Atmospheric Simulation Chamber Studies, available at: https://data.eurochamp.org/data-access/chamber-experiments/, last access: 1 May 2020c.
Friedman, B. and Farmer, D. K.: SOA and gas phase organic acid yields from the sequential photooxidation ofseven monoterpenes, Atmos. Environ., 187, 335–345, 2018.
Fry, J. L., Kiendler-Scharr, A., Rollins, A. W., Wooldridge, P. J., Brown, S. S., Fuchs, H., Dubé, W., Mensah, A., dal Maso, M., Tillmann, R., Dorn, H.-P., Brauers, T., and Cohen, R. C.: Organic nitrate and secondary organic aerosol yield from NO3 oxidation of β-pinene evaluated using a gas-phase kinetics/aerosol partitioning model, Atmos. Chem. Phys., 9, 1431–1449, https://doi.org/10.5194/acp-9-1431-2009, 2009.
Fry, J. L., Kiendler-Scharr, A., Rollins, A. W., Brauers, T., Brown, S. S., Dorn, H.-P., Dubé, W. P., Fuchs, H., Mensah, A., Rohrer, F., Tillmann, R., Wahner, A., Wooldridge, P. J., and Cohen, R. C.: SOA from limonene: role of NO3 in its generation and degradation, Atmos. Chem. Phys., 11, 3879–3894, https://doi.org/10.5194/acp-11-3879-2011, 2011.
Fry, J. L., Draper, D. C., Barsanti, K. C., Smith, J. N., Ortega, J., Winkler, P. M., Lawler, M. J., Brown, S. S., Edwards, P. M., Cohen, R. C., and Lee, L.: Secondary Organic Aerosol Formation and Organic Nitrate Yield from NO3 Oxidation of Biogenic Hydrocarbons, Environ. Sci. Technol., 48, 11944–11953, https://doi.org/10.1021/es502204x, 2014.
Geron, C., Rasmussen, R., R. Arnts, R., and Guenther, A.: A review and synthesis of monoterpene speciation from forests in the United States, Atmos. Environ., 34, 1761–1781, https://doi.org/10.1016/S1352-2310(99)00364-7, 2000.
Gómez-González, Y., Surratt, J. D., Cuyckens, F., Szmigielski, R., Vermeylen, R., Jaoui, M., Lewandowski, M., Offenberg, J. H., Kleindienst, T. E., Edney, E. O., Blockhuys, F., Alsenoy, C. V., Maenhaut, W., and Claeys, M.: Characterization of organosulfates from the photooxidation of isoprene and unsaturated fatty acids in ambient aerosol using liquid chromatography/(-) electrospray ionization mass spectrometry, J. Mass Spectrom., 43, 371–382, 2008.
Griffin, R. J., Cocker, D. R., Flagan, R. C., and Seinfeld, J. H.: Organic aerosol formation from the oxidation of biogenic hydrocarbons, J. Geophys. Res., 104, 3555–3567, 1999.
Guenther, A., Hewitt, C. N., Erickson, D., Fall, R., Geron, C., Graedel, T., Harley, P., Klinger, L., Lerdau, M., McKay, W. A., Pierce, T., Scholes, B., Steinbrecher, R., Tallamraju, R., Taylor, J., and Zimmerman, P.: A global model of natural volatile organic compound emissions, J. Geophys. Res., 100, 8873–8892, 1995.
Hallquist, M., Wängberg, I., and Ljungström, E.: Atmospheric Fate of Carbonyl Oxidation Products Originating from α-Pinene and Δ3-Carene: Determination of Rate of Reaction with OH and NO3 Radicals, UV Absorption Cross Sections, and Vapor Pressures, Environ. Sci. Technol., 31, 3166–3172, 1997.
Hallquist, M., Wängberg, I., Ljungström, E., Barnes, I., and Becker, K.-H.: Aerosol and Product Yields from NO3 Radical-Initiated Oxidation of Selected Monoterpenes, Environ. Sci. Technol., 33, 553–559, 1999.
Hao, L. Q., Kortelainen, A., Romakkaniemi, S., Portin, H., Jaatinen, A., Leskinen, A., Komppula, M., Miettinen, P., Sueper, D., Pajunoja, A., Smith, J. N., Lehtinen, K. E. J., Worsnop, D. R., Laaksonen, A., and Virtanen, A.: Atmospheric submicron aerosol composition and particulate organic nitrate formation in a boreal forestland–urban mixed region, Atmos. Chem. Phys., 14, 13483–13495, https://doi.org/10.5194/acp-14-13483-2014, 2014.
Iinuma, Y., Müller, C., Berndt, T., Böge, O., Claeys, M., and Herrmann, H.: Evidence for the Existence of Organosulfates from β-Pinene Ozonolysis in Ambient Secondary Organic Aerosol, Environ. Sci. Technol., 41, 6678–6683, https://doi.org/10.1021/es070938t, 2007.
Ito, A., Sillman, S., and Penner, J. E.: Effects of additional nonmethane volatile organic compounds, organic nitrates, and direct emissions of oxygenated organic species on global tropospheric chemistry, J. Geophys. Res., 112, D06309, https://doi.org/10.1029/2005JD006556, 2007.
Jaoui, M., Kleindienst, T. E., Docherty, K. S., Lewandowski, M., and Offenberg, J. H.: Secondary organic aerosol formation from the oxidation of a series of sesquiterpenes: α-cedrene, β-caryophyllene, α-humulene and α-farnesene with O3, OH and NO3 radicals, Environ. Chem., 10, 178–193, 2013.
Kerdouci, J., Picquet-Varrault, B., and Doussin, J. F.: Structure–activity relationship for the gas-phase reactions of NO3 radical with organic compounds: Update and extension to aldehydes, Atmos. Environ., 84, 363–372, https://doi.org/10.1016/j.atmosenv.2013.11.024, 2014.
Kiendler-Scharr, A., Mensah, A., Friese, E., Topping, D., Nemitz, E., Prevot, A. S. H., Äijälä, M., Allan, J., Canonaco, F., Canagaratna, M., Carbone, S., Crippa, M., Dail Osto, M., Day, D. A., Di Marco, C. F., Eibern, H., Eriksson, A., Freney, E., Hao, L., Herrmann, H., Hildebrandt, L., Hillamo, R., Jimenez, J. L., Laaksonen, A., McFiggans, G., Mohr, C., O'Dowd, C., Otjes, R., Ovadnevaite, J., Pandis, S. N., Poulain, L., Schlag, P., Sellegri, K., Swietlicki, E., Tiitta, P., Vermeulen, A., Wahner, A., Wornsnop, D., and Wu, H.-C.: Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol, Geophys. Res. Lett., 43, 7735–7744, https://doi.org/10.1002/2016GL069239, 2016.
Kurten, T., Moller, K. H., Nguyen, T. B., Schwantes, R. H., Misztal, P. K., Su, L., Wennberg, P. O., Fry, J. L., and Kjaergaard, H. G.: Alkoxy Radical Bond Scissions Explain the Anomalously Low Secondary Organic Aerosol and Organonitrate Yields From α-Pinene + NO3, J. Phys. Chem. Lett., 8, 2826–2834, https://doi.org/10.1021/acs.jpclett.7b01038, 2017.
Lai, A. C. K. and Nazaroff, W. W.: Modeling indoor particle deposition from turbulent flow onto smooth surfaces, J. Aerosol. Sci., 31, 463–476, https://doi.org/10.1016/S0021-8502(99)00536-4, 2000.
Lamkaddam, H., Gratien, A., Pangui, E., Cazaunau, M., Picquet-Varrault, B., and Doussin, J.-F.: High-NOx Photooxidation of n-Dodecane: Temperature Dependence of SOA Formation, Env. Sci Technol, 51, 192–201, https://doi.org/10.1021/acs.est.6b03821, 2017.
Lee, A., Goldstein, A. H., Kroll, J. H., Ng, N. L., Varutbangkul, V., Flagan, R. C., and Seinfeld, J. H.: Gas-phase products and secondary aerosol yields from the photooxidation of 16 different terpenes, J. Geophys. Res., 111, D17305, https://doi.org/10.1029/2006JD007050, 2006.
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurten, T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J. A., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., D'Ambro, E. L., and Thornton, J. A.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci. USA, 113, 1516–1521, https://doi.org/10.1073/pnas.1508108113, 2016.
Lindwall, F., Faubert, P., and Rinnan, R.: Diel Variation of Biogenic Volatile Organic Compound Emissions – A field Study in the Sub, Low and High Arctic on the Effect of Temperature and Light, PLosOne, 10, e0123610. https://doi.org/10.1371/journal.pone.0123610, 2015.
Martínez, E., Cabañas, B., Aranda, A., Martín, P., and Salgado, S.: Absolute Rate Coefficients for the Gas-Phase Reactions of NO3 Radical with a Series of Monoterpenes at T = 298 to 433 K, J. Atmos. Chem., 33, 265–282, https://doi.org/10.1023/A:1006178530211, 1999.
Martínez, E., Cabañas, B., Aranda, A., Martín, P., Notario, A., and Salgado, S.: Study on the NO3 Radical Reactivity: Reactions with Cyclic Alkenes, J. Phys. Chem. A, 103, 5321–5327, 1999.
Moldanova, J. and Ljungström, E.: Modelling of particle formation from NO3 oxidation of selected monterpenes, J. Aerosol. Sci., 31, 1317–1333, 2000.
Müller, M., Graus, M., Wisthaler, A., Hansel, A., Metzger, A., Dommen, J., and Baltensperger, U.: Analysis of high mass resolution PTR-TOF mass spectra from 1,3,5-trimethylbenzene (TMB) environmental chamber experiments, Atmos. Chem. Phys., 12, 829–843, https://doi.org/10.5194/acp-12-829-2012, 2012.
Nah, T., Sanchez, J., Boyd, C. M., and Ng, N. L.: Photochemical aging of alpha-pinene and beta-pinene Secondary Organic Aerosol formed from Nitrate Radical Oxidation, Env. Sci. Technol., 50, 222–231, 2016.
Naudet, J. P., Huguenin, D., Rigaud, P., and Cariolle, D.: Stratospheric observations of NO3 and its experimental and theoretical distribution between 20 and 40 km, Planet. Space Sci., 29, 707–712, https://doi.org/10.1016/0032-0633(81)90118-5, 1981.
Ng, N. L., Kwan, A. J., Surratt, J. D., Chan, A. W. H., Chhabra, P. S., Sorooshian, A., Pye, H. O. T., Crounse, J. D., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol (SOA) formation from reaction of isoprene with nitrate radicals (NO3), Atmos. Chem. Phys., 8, 4117–4140, https://doi.org/10.5194/acp-8-4117-2008, 2008.
Ng, N. L., Brown, S. S., Archibald, A. T., Atlas, E., Cohen, R. C., Crowley, J. N., Day, D. A., Donahue, N. M., Fry, J. L., Fuchs, H., Griffin, R. J., Guzman, M. I., Herrmann, H., Hodzic, A., Iinuma, Y., Jimenez, J. L., Kiendler-Scharr, A., Lee, B. H., Luecken, D. J., Mao, J., McLaren, R., Mutzel, A., Osthoff, H. D., Ouyang, B., Picquet-Varrault, B., Platt, U., Pye, H. O. T., Rudich, Y., Schwantes, R. H., Shiraiwa, M., Stutz, J., Thornton, J. A., Tilgner, A., Williams, B. J., and Zaveri, R. A.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 17, 2103–2162, https://doi.org/10.5194/acp-17-2103-2017, 2017.
Noxon, J. F., Norton, R. B., and Henderson, W. R.: Observation of atmospheric NO3, Geophys. Res. Lett., 5, 675–678, 1978.
Noxon, J. F., Norton, R. B., and Marovich, E.: NO3 in the troposphere, Geophys. Res. Lett., 7, 125–128, https://doi.org/10.1029/GL007i002p00125, 1980.
Odum, J. R., Hoffmann, T., Bowman, F., Collins, D., Flagan, R. C., and Seinfeld, J. H.: Gas/Particle Partitioning and Secondary Organic Aerosol Yields, Environ. Sci. Technol., 30, 2580–2585, 1996.
Orphal, J., Fellows, C. E., and Flaud, P.-M.: The visible absorption spectrum of NO3 measured by high-resolution Fourier transform spectroscopy, J. Geophys. Res., 108, 4077, https://doi.org/10.1029/2002JD002489, 2003.
Pankow, J. F. and Asher, W. E.: SIMPOL.1: a simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds, Atmos. Chem. Phys., 8, 2773–2796, https://doi.org/10.5194/acp-8-2773-2008, 2008.
Perraud, V., Bruns, E. A., Ezell, M. J., Johnson, S. N., Greaves, J., and Finlayson-Pitts, B. J.: Identification of organic nitrates in the NO3 radical initiated oxidation of alpha-pinene by atmospheric pressure chemical ionization mass spectrometry, Environ. Sci. Technol., 44, 5887–5893, 2010.
Picquet-Varrault, B., Scarfogliero, M., Helal, W. A., and Doussin, J.-F.: Reevaluation of the rate constant for the reaction propene + NO3 by absolute rate determination, Int. J. Chem. Kinet., 41, 73–81, 2009.
Picquet-Varrault, B., Suarez-Bertoa, R., Duncianu, M., Cazaunau, M., Pangui, E., David, M., and Doussin, J.-F.: Photolysis and oxidation by OH radicals of two carbonyl nitrates: 4-nitrooxy-2-butanone and 5-nitrooxy-2-pentanone, Atmos. Chem. Phys., 20, 487–498, https://doi.org/10.5194/acp-20-487-2020, 2020.
Platt, U., Perner, D., Harris, G. W., Winer, A. M., and Pitts, J. N.: Observations of nitrous acid in an urban atmosphere by differential optical absorption, Nature, 285, 312–314, https://doi.org/10.1038/285312a0, 1980.
Rindelaub, J. D., McAvey, K. M., and Shepson, P. B.: The photochemical production of organic nitrates from a-pinene and loss via acid-dependent particle phase hydrolysis, Atmos. Environ., 100, 193–201, 2015.
Rollins, A. W., Kiendler-Scharr, A., Fry, J. L., Brauers, T., Brown, S. S., Dorn, H.-P., Dubé, W. P., Fuchs, H., Mensah, A., Mentel, T. F., Rohrer, F., Tillmann, R., Wegener, R., Wooldridge, P. J., and Cohen, R. C.: Isoprene oxidation by nitrate radical: alkyl nitrate and secondary organic aerosol yields, Atmos. Chem. Phys., 9, 6685–6703, https://doi.org/10.5194/acp-9-6685-2009, 2009.
Schott, G. and Davidson, N.: Shock waves in chemical kinetics: The decomposition of N2O5 at high temperatures, J. Am. Chem. Soc., 80, 1841–1853, 1958.
Skov, H., Benter, T., Schindler, R. N., Hjorth, J., and Restelli, G.: Epoxide formation in the reactions of the nitrate radical with 2,3-dimethyl-2-butene, cis- and trans-2-butene and isoprene, Atmos. Environ., 28, 1583–1592, 1994.
Slade, J. H., de Perre, C., Lee, L., and Shepson, P. B.: Nitrate radical oxidation of γ-terpinene: hydroxy nitrate, total organic nitrate, and secondary organic aerosol yields, Atmos. Chem. Phys., 17, 8635–8650, https://doi.org/10.5194/acp-17-8635-2017, 2017.
Spittler, M., Barnes, I., Bejan, I., Brockmann, K. J., Benter, Th., and Wirtz, K.: Reactions of NO3 radicals with limonene and α-pinene: Product and SOA formation, Atmos. Environ., 40, 116–127, https://doi.org/10.1016/j.atmosenv.2005.09.093, 2006.
Suarez-Bertoa, R., Picquet-Varrault, B., Tamas, W., Pangui, E., and Doussin, J.-F.: Atmospheric Fate of a Series of Carbonyl Nitrates: Photolysis Frequencies and OH-Oxidation Rate Constants, Environ. Sci. Technol., 46, 12502–12509, 2012.
Valorso, R., Aumont, B., Camredon, M., Raventos-Duran, T., Mouchel-Vallon, C., Ng, N. L., Seinfeld, J. H., Lee-Taylor, J., and Madronich, S.: Explicit modelling of SOA formation from α-pinene photooxidation: sensitivity to vapour pressure estimation, Atmos. Chem. Phys., 11, 6895–6910, https://doi.org/10.5194/acp-11-6895-2011, 2011.
Vandaele, A. C., Hermans, C., Simon, P. C., Carleer, M., Colin, R., Fally, S., Mérienne, M. F., Jenouvrier, A., and Coquart, B.: Measurements of the NO2 absorption cross-section from 42 000 cm−1 to 10 000−1 (238–1000 nm) at 220 K and 294 K, J. Quant. Spectrosc. Ra., 59, 171–184, 1997.
Vereecken, L. and Peeters, J.: Decomposition of substituted alkoxy radicals–part I: a generalized structure–activity relationship for reaction barrier heights, Phys. Chem. Chem. Phys., 11, 9062–9074, https://doi.org/10.1039/b909712k, 2009.
Wang, J., Doussin, J. F., Perrier, S., Perraudin, E., Katrib, Y., Pangui, E., and Picquet-Varrault, B.: Design of a new multi-phase experimental simulation chamber for atmospheric photosmog, aerosol and cloud chemistry research, Atmos. Meas. Tech., 4, 2465–2494, https://doi.org/10.5194/amt-4-2465-2011, 2011.
Wangberg, I., Barnes, I., and Becker, K. H.: Product and mechanistic study of the reaction of NO3 radicals with alpha-pinene, Environ. Sci. Technol., 31, 2130–2135, 1997.
Xu, L., Suresh, S., Guo, H., Weber, R. J., and Ng, N. L.: Aerosol characterization over the southeastern United States using high-resolution aerosol mass spectrometry: spatial and seasonal variation of aerosol composition and sources with a focus on organic nitrates, Atmos. Chem. Phys., 15, 7307–7336, https://doi.org/10.5194/acp-15-7307-2015, 2015.