the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 09 Nov 2020
| 09 Nov 2020
Long-term observational constraints of organic aerosol dependence on inorganic species in the southeast US
Joel A. Thornton
Nga Lee Ng
Hansen Cao
Daven K. Henze
Erin E. McDuffie
Weiwei Hu
Jose L. Jimenez
Eloise A. Marais
Eric Edgerton
Organic aerosol (OA), with a large biogenic fraction in the summertime southeast US, adversely impacts air quality and human health. Stringent air quality controls have recently reduced anthropogenic pollutants including sulfate, whose impact on OA remains unclear. Three filter measurement networks provide long-term constraints on the sensitivity of OA to changes in inorganic species, including sulfate and ammonia. The 2000–2013 summertime OA decreases by 1.7 % yr−1–1.9 % yr−1 with little month-to-month variability, while sulfate declines rapidly with significant monthly difference in the early 2000s. In contrast, modeled OA from a chemical-transport model (GEOS-Chem) decreases by 4.9 % yr−1 with much larger monthly variability, largely due to the predominant role of acid-catalyzed reactive uptake of epoxydiols (IEPOX) onto sulfate. The overestimated modeled OA dependence on sulfate can be improved by implementing a coating effect and assuming constant aerosol acidity, suggesting the needs to revisit IEPOX reactive uptake in current models. Our work highlights the importance of secondary OA formation pathways that are weakly dependent on inorganic aerosol in a region that is heavily influenced by both biogenic and anthropogenic emissions.
- Article
(2331 KB) - Full-text XML
-
Supplement
(1069 KB) - BibTeX
- EndNote





Organic aerosol (OA) accounts for a large fraction of ambient fine particulate matter mass, which strongly affects air quality, regional climate and human welfare (Jimenez et al., 2009). Since the implementation of the Clean Air Act Amendments of 1990, there has been a significant decline in ambient aerosol in the United States, mostly due to reductions in inorganic aerosol mass following changes in emissions of sulfur dioxide (SO2) and nitrogen oxides (NOx = NO + NO2), as well as reductions in black carbon (EPA, 2011), leaving OA as the major component of fine particulate matter (50 %–70 %) over the southeast US, especially in summer (Attwood et al., 2014; Kim et al., 2015). OA can be directly emitted by combustion processes (primary organic aerosol, POA) or secondarily formed (secondary organic aerosol, SOA) from the atmospheric oxidation of biogenic volatile organic compounds (BVOCs), mainly isoprene and monoterpenes, and also precursors from anthropogenic sources and biomass burning (Hayes et al., 2015; Hodshire et al., 2019). OA has also been declining across much of the US over the past few decades, primarily due to decreased anthropogenic emissions from vehicle and residential fuel burning, except for the southeast US (Ridley et al., 2018). The southeast US is one of the largest BVOC emission hotspots in the world (Guenther et al., 2006), and at the same time it is heavily populated with large anthropogenic emissions of pollutants. Biogenic SOA (formed from atmospheric oxidation of BVOCs) may account for 60 %–100 % of OA in the summertime southeast US (Kim et al., 2015; Xu et al., 2015a). To what extent biogenic SOA could be mediated through emission control strategies remains an open question (Carlton et al., 2010; Mao et al., 2018).
The oxidation of BVOCs produces hundreds of intermediate products. Some products have low volatility that can partition onto the condensed phase, while some gas-phase products can react in the aqueous phase to form SOA. SOA formed from uptake of isoprene epoxydiols (IEPOX SOA) (Paulot et al., 2009) appears to be the major confirmed aqueous SOA product globally, being important in all high-isoprene and lower-NO regions (Hu et al., 2015), along with glyoxal formed from isoprene and aromatics (Fu et al., 2008). Formation of SOA in clouds was investigated in the southeast US and found to be not statistically significant (Wagner et al., 2015). These pathways have been implemented into three-dimensional global atmospheric chemistry and climate models using two different approaches. First, to simulate the partitioning of organic vapors, the BVOC oxidation products can be grouped based on their volatility (volatility basis set, VBS), and the product yields and vapor pressures are parameterized for each surrogate precursor (Donahue et al., 2006; Pankow, 1994). Such empirical VBS schemes are usually derived using dry laboratory chamber experiments (with relative humidity RH < 10 %) and do not explicitly depend on aerosol water, RH, or inorganic aerosol mass or composition. Therefore, here we refer to the SOA formed through partitioning calculated by VBS as dry SOA. Second, a more explicit representation of aqueous SOA formation from isoprene products has been used recently, which incorporates dependence on inorganic aerosol volume and aerosol acidity (Budisulistiorini et al., 2017; Ervens et al., 2011; Fu et al., 2008; Marais et al., 2016; Pye et al., 2013). The relative contribution of dry vs. aqueous SOA to total OA mass in the atmosphere is uncertain and has limited observational constraints.
Long-term field measurements show a decreasing trend of OA in the southeast US (Attwood et al., 2014; Hidy et al., 2014; Kim et al., 2015), which is likely linked to reductions in anthropogenic POA and SOA (Blanchard et al., 2016; Ridley et al., 2018), sulfate (Blanchard et al., 2016; Malm et al., 2017; Marais et al., 2017; Xu et al., 2015a, 2016), and NOx (Carlton et al., 2010; Pye et al., 2010, 2019; Xu et al., 2015a). The influence of sulfate on OA is thought to be mainly due to its influence on the uptake of isoprene gas-phase oxidation products, which are often small molecules that cannot directly condense due to high vapor pressure but may undergo aqueous-phase reactive uptake onto wet sulfate particles to form aqueous SOA, as suggested by extensive laboratory and field studies (Budisulistiorini et al., 2015; Hu et al., 2015; Li et al., 2016; Liggio et al., 2005; McNeill et al., 2012; Riedel et al., 2016; Shrivastava et al., 2017; Surratt et al., 2010; Tan et al., 2012; Xu et al., 2016, 2015a). NOx plays a complex role in regulating oxidation capacity, different oxidation pathways and aerosol water content through aerosol nitrate (Kiendler-Scharr et al., 2016; Kroll et al., 2005, 2006; Li et al., 2018; Ng et al., 2017; Presto et al., 2005; Shrivastava et al., 2019; Zheng et al., 2015; Ziemann and Atkinson, 2012). Prior 3-D modeling studies with different SOA mechanisms provide different explanations for the long-term OA trend observed in the southeast US. For example, the dry SOA calculated by the VBS framework with NOx-dependent yields implies a small decrease in OA following the reductions of NOx (Pye et al., 2013; Zheng et al., 2015) but has little dependence on changes in inorganic aerosol mass such as sulfate. On the other hand, models using aqueous SOA formation from isoprene attributed the decreasing OA from 1991 to 2013 to reductions in sulfate (Marais et al., 2017) but showed greater interannual variability than was observed. The driving mechanism for the OA trend in the southeast US remains to be elucidated.
Here we use observations from three surface filter-based networks (IMPROVE, SEARCH, CSN), combined with the three-dimensional chemical-transport model GEOS-Chem v12.1.0, to examine the long-term trend and, more importantly, the month-to-month variability of OA in the southeast US during 2000–2013. The results provide new observational constraints on the drivers of OA variability and the SOA formation mechanisms in the southeast US.
2.1 Observational datasets
We use surface filter-based measurement of fine particulate matter mass and composition (including organic carbon, OC) in 2000–2013 from three networks: the Interagency Monitoring of Protected Visual Environments (IMPROVE) (Solomon et al., 2014), the SouthEastern Aerosol Research and Characterization (SEARCH) (Edgerton et al., 2005), and the Environmental Protection Agency's PM2.5 National Chemical Speciation Network (CSN) (Solomon et al., 2014). We select 21 IMPROVE sites, 3 SEARCH rural sites and 36 CSN sites within the southeast US region [29–37∘ N, 74–96∘ W] (Fig. S1 in the Supplement). The SEARCH sites are organized in rural/urban pairs (Edgerton et al., 2005), and only the data from the rural sites are used here to represent background conditions. IMPROVE sites are mostly rural (Solomon et al., 2014). The OC measurement in the CSN network in 2004–2009 gradually shifted to a different protocol and analytical technique than the early 2000s, which led to the discontinuity in the long-term trend (Fig. S2 in the Supplement); therefore, we only use CSN data to examine the monthly variability of OA and focus on IMPROVE and SEARCH for all analysis. The 3 d OC measurement from IMPROVE and daily OC from SEARCH and CSN are averaged to monthly values. A factor of 2.1 is used to convert measured organic carbon (OC) to organic aerosol mass, as suggested by the southeast US field measurements (Pye et al., 2017; Schroder et al., 2018).
We use OA measurements by aerosol mass spectrometer (AMS) from the Southern Oxidant and Aerosol Study campaign (SOAS) at the Centerville, AL, site on 1 June–15 July 2013 (SOAS 2013). The OA measurements and derived IEPOX-SOA factor calculated by positive matrix factorization (PMF) analysis (Hu et al., 2015; Xu et al., 2015b, 2018) are from two independent groups: one group from the Georgia Institute of Technology led by Nga Lee Ng and the other from the University of Colorado Boulder led by Jose L. Jimenez, denoted as Obs_GT and Obs_CU, respectively.
2.2 Modeling framework
2.2.1 GEOS-Chem
In this study we use the three-dimensional global chemical-transport model GEOS-Chem version 12.1.1 (https://doi.org/10.5281/zenodo.2249246, https://github.com/geoschem/, last access: 5 June 2019) with detailed O3–NOx–HOx–CO–VOC–aerosol tropospheric chemistry (Bey et al., 2001; Mao et al., 2013). Isoprene chemistry is described in Fisher et al. (2016) and Travis et al. (2016). GEOS-Chem is driven by offline meteorology from 1999 to 2013 from the NASA Modern-Era Retrospective analysis for Research and Applications, version 2 (MERRA-2 https://gmao.gsfc.nasa.gov/reanalysis/MERRA-2/, last access: 5 June 2019). The global anthropogenic (including agricultural) emissions are from the Community Emissions Data System (CEDS) inventory, with the US region replaced by the EPA's National Emission Inventory for 2011 (NEI11v1). The monthly mean anthropogenic emissions of CO, SO2, NOx, NH3, VOCs, OC and black carbon are scaled to the year 2011 using the ratio of the EPA's national annual emission totals from 2000 to 2013 (Travis et al., 2016). Biomass burning emissions are from the Global Fire Emissions Database version 4 (GFED4) (Randerson et al., 2015). Biogenic emissions of isoprene and terpenes are online calculated by the Model of Emissions of Gases and Aerosols from Nature (MEGAN2.1) (Guenther et al., 2012) that is also driven by MERRA-2 meteorology.
For organic aerosol, we employ the complexSOA scheme for SOA modeling for all simulations in this study (Marais et al., 2016; Pai et al., 2020; Pye et al., 2010). POAs are regarded as nonvolatile. This SOA modeling includes a four-product volatility basis set (VBS) for SOA formation from reversible condensation of oxidation products of biogenic terpenes (including monoterpenes and sesquiterpenes) and anthropogenic VOCs, referred to as terpene SOA and anthropogenic SOA, respectively. The SOA calculated through VBS parameterization is fitted based on dry chamber (RH < 10 %) results independent of inorganic aerosol, aerosol water and RH (Pye et al., 2010). The complexSOA scheme also includes aqueous SOA formed from reactive uptake of isoprene oxidation products, including IEPOX, glyoxal, C4 epoxides, methylglyoxal, non-IEPOX product of the ISOPOOH oxidation and hydroxynitrates from NO3-initiated oxidation (Marais et al., 2016). GEOS-Chem v12.1.1 considers sulfate, nitrate and ammonium from all sectors, as well as fine-mode Na+, Ca2+, Mg2+ and Cl− from anthropogenic and sea salt sources, and employs the ISORROPIA II thermodynamic model (Fountoukis and Nenes, 2007; Pye et al., 2009; Song et al., 2018) to calculate aerosol water content and aerosol acidity (Pye et al., 2020).
We run the default GEOS-Chem model at 4∘ × 5∘ latitude by longitude continuously from 1 October 1999 to 31 December 2013. For each year, the restart file at 1 May from the continuous 4∘ × 5∘ simulation has been regridded to 2∘ × 2.5∘ and is used to initiate 2∘ × 2.5∘ simulations from 1 May to 31 August each year. The May results are discarded as spin-up, and the results of June, July and August are used for analysis.
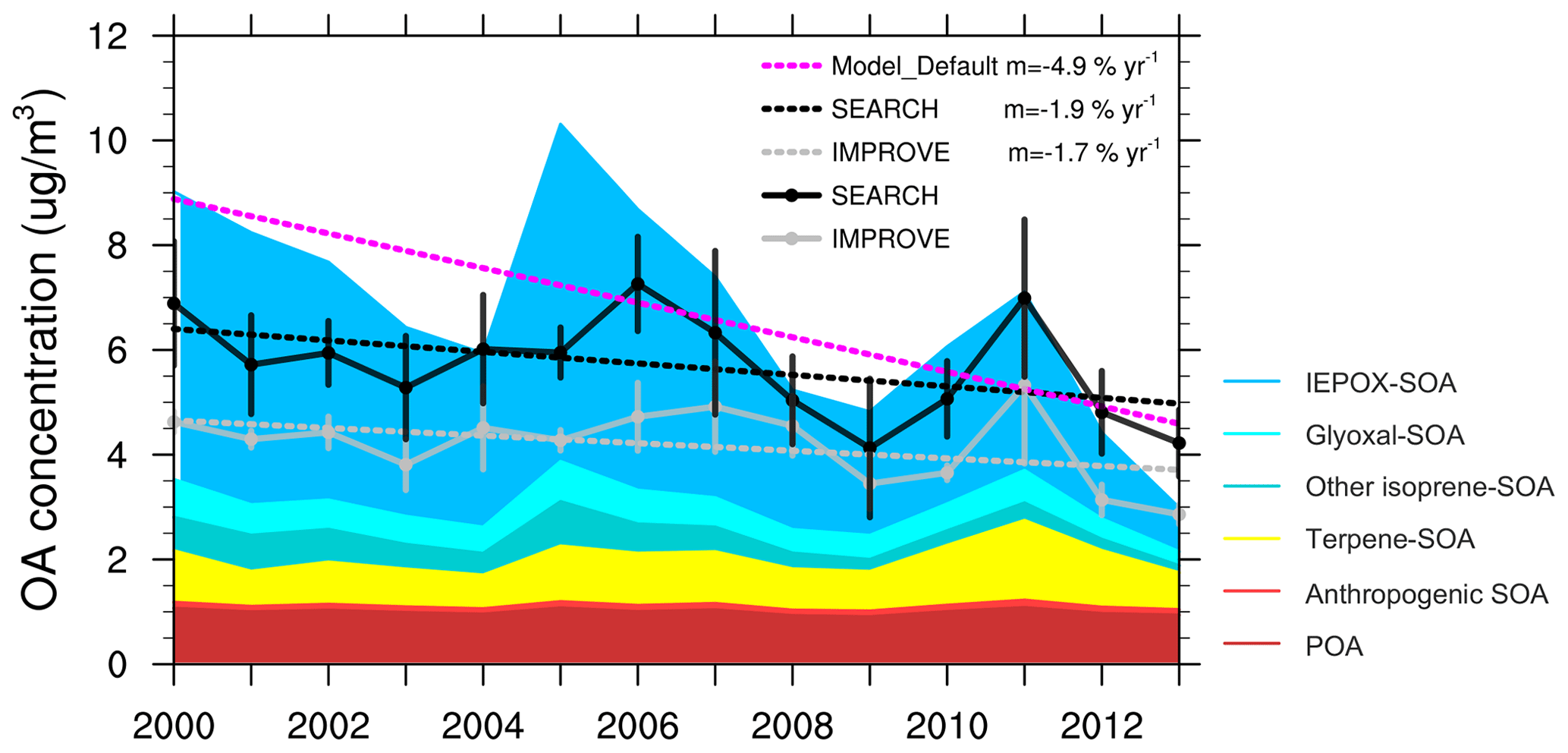
Figure 1Comparison of June–July–August averaged surface OA concentration (µg m−3) over the southeast US between the default model and the observation from the IMPROVE and SEARCH network. Colored shades represent different components of modeled OA. IEPOX SOA, glyoxal SOA, and other isoprene SOA are from aqueous uptake of isoprene oxidation products. Terpene and anthropogenic SOA are dry SOA calculated using the volatility basis set.
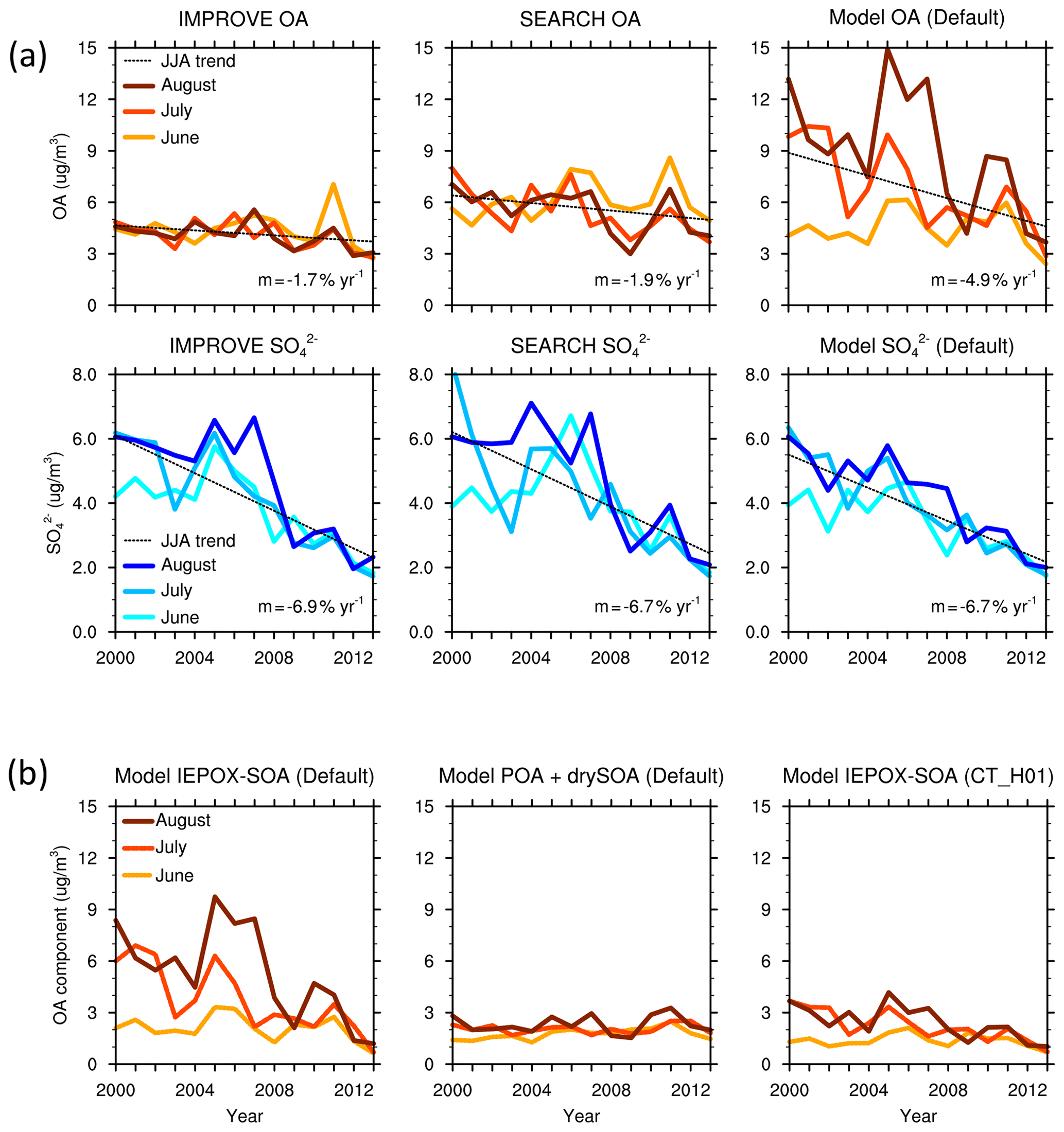
Figure 2(a) Monthly surface OA and sulfate () concentration (µg m−3) averaged over the southeast US from IMPROVE, SEARCH and the default model. (b) Monthly surface concentrations of IEPOX SOA and the sum of POA and dry SOA from the default model, and IEPOX SOA from the CT_H01 simulation.
The default modeled OA shows a stronger decreasing trend from 2000 to 2013 and a large month-to-month variability in the early 2000s, which is different from the observations (more details in Figs. 1 and 2a and Sect. 3.1). To address this model–observation discrepancy, we do four sets of 2∘ × 2.5∘ simulations: default (using default complexSOA scheme); CT (with coating effect for IEPOX reactive uptake); CT_newNH3 (with coating effect and US NH3 emissions replaced by satellite-derived NH3 inventory); CT_H01 (with coating effect and fixing aerosol acidity at 0.1 mol L−1 when calculating IEPOX reactive uptake). The sensitivity simulations are further explained in Sects. 2.2.2, 2.2.3 and 3.3.
2.2.2 Coating
The default IEPOX-SOA mechanism in GEOS-Chem uses aerosol-phase reaction rates from laboratory chamber studies with pure acidic inorganic particles (Gaston et al., 2014; Riedel et al., 2015), as well as a representative effective Henry law constant obtained by matching the model to the observations from the SOAS 2013 campaign (Marais et al., 2016), to estimate the reactive uptake coefficient γIEPOX. In the default scheme, γIEPOX is calculated as follows:
where Rp is the particle radius of the inorganic sulfate–nitrate–ammonium particle (cm), ω is the mean molecular speed (cm s−1), Dg is the gas-phase diffusion coefficient (0.1 cm2 s−1), α is the mass accommodation coefficient (α = 0.1), Sa is the total (wet) particle surface area (cm2 cm−3), V is the total (wet) particle volume (cm3 cm−3), R is the ideal gas constant (), T is temperature (K), Haq is the Henry law coefficient (1.7 × 107 M atm−1) and kaq is the first-order reaction rate constant (s−1):
where (= 0.036 ), knuc (= 2 × 10−4 ) and kga (= 7.3 × 10−4 ) are the reaction rates due to acid-catalyzed ring opening, presence of nucleophiles (including nitrate and sulfate) and presence of bisulfate acids, respectively (Gaston et al., 2014; Marais et al., 2016).
In the real atmosphere, inorganic aerosol is generally internally mixed with other organics. The presence of an organic coating may alter the aerosol properties and suppress the uptake of IEPOX onto acidified sulfate aerosol (Anttila et al., 2006; Gaston et al., 2014). We implement a linear coating effect for the IEPOX-SOA formation. The coating effect is fitted using laboratory-derived values of γIEPOX on particles containing both ammonium bisulfate and ethylene glycol under RH = 50 % conditions (Gaston et al., 2014). In the coating scheme, is calculated as above with Rp, V and Sa updated considering OA coated outside the inorganic core. Then, the fitted function is applied to modify :
where χorg is the mass fraction of OA in the mixed particle including both the inorganic aerosol and OA. When χorg > 0.7, the IEPOX uptake will be terminated, i.e., γIEPOX_modified=0. In the real atmosphere when inorganic cores are coated with more viscous SOA (Y. Zhang et al., 2018), the coating effect may be stronger because ethylene glycol is a low-viscosity material. However, this simplified linear function does not consider the decreased viscosity and reduced coating effect at higher-RH conditions (which is common in the summertime southeast US) (Gaston et al., 2014; Y. Zhang et al., 2018) and prevents further IEPOX uptake when the mass fraction of OA (χorg) is larger than 0.7; therefore, this linear function may mimic a strong coating effect even though ethylene glycol is less viscous than real atmospheric SOA. The uncertainties need to be addressed in further studies with a more realistic coating parameterization (Li et al., 2020; Schmedding et al., 2019; Zhang et al., 2019b). We assume all OA is coated outside the inorganic aerosol core when calculating the IEPOX reactive uptake. The default GEOS-Chem with no organic coating calculates the surface area of inorganic aerosol (Jo et al., 2019). By adding the coating effect, the increased particle radius Rp and surface area Sa of the mixed particle will partially offset (but does not outweigh) the impact of reduced reaction probability γIEPOX_modified.
2.2.3 Satellite-derived NH3 emissions
We use the Cross-track Infrared Sounder (CrIS) satellite-derived NH3 emissions (Cao et al., 2020) in a sensitivity test in this study. The top-down monthly NH3 emissions over the contiguous US at 0.25∘ × 0.3125∘ latitude by longitude are derived from CrIS v1.5 measurements of NH3 profiles (Shephard and Cady-Pereira, 2015) for the year 2014 through a 4D-Var approach using GEOS-Chem and its adjoint model (Henze et al., 2007). The CrIS-derived emissions are then regridded to 0.1∘ × 0.1∘ to replace the default NEI11 emissions for the year 2011 and applied the same annual scaling factors for 2000–2013. The default NEI11 and CrIS-derived NH3 emissions averaged over 2000–2013 are compared in Fig. S3 in the Supplement. There is no significant trend of NH3 emissions from 2000 to 2013 (Fig. S4 in the Supplement), consistent with other studies suggesting nearly constant NH3 emissions from 2001 to 2014 (Butler et al., 2016). The CrIS-derived emissions used the HTAPv2 emissions inventory as the prior emissions, which is based on the 2008 NEI emissions over the US (Janssens-Maenhout et al., 2015). The CrIS-derived NH3 emissions have been validated against surface observations of NH3 concentration from the Ammonia Monitoring Network (AMoN) and wet deposition measurements from the National Atmospheric Deposition Program (NADP). More details can be found in Cao et al. (2020). Using the top-down emissions in GEOS-Chem increases the correlation coefficient (r) between modeled monthly mean NH3 concentration and surface observations from 0.74 to 0.93 and reduces the normalized mean bias of domain-averaged annual mean simulated NH3 by a factor of 1.9. The seasonal cycle of simulated wet deposition is also improved (r increased from 0.70 to 0.86), but the normalized mean bias of domain-averaged annual simulated wet increases from 0.34 to 0.96 due to overly strong wet scavenging in the model. The latter issue was ultimately resolved in Cao et al. (2020), and the final top-down emissions reported therein differ from those reported here; nevertheless, the emissions estimates used here provide a valuable basis for conducting a sensitivity experiment.
2.3 Multivariate linear regression analysis
In this study we did a multivariate regression analysis of modeled monthly IEPOX SOA (µg m−3) against modeled sulfate aerosol (µg m−3), aerosol acidity (mol L−1) and isoprene emissionISOPemis ():
Mean values have been subtracted from all variables, which are then divided by SDs. β1, β2 and β3 are standardized partial regression coefficients associated with sulfate aerosol, and isoprene emission, and they can be directly compared to evaluate the relative importance of the three variables. We apply the regression analysis using monthly data within different time frames (2000–2013, 2000–2004, 2005–2008 and 2009–2013 as in Table S1 in the Supplement) to determine the evolving importance of variables.
3.1 Long-term trend and month-to-month variability (MMV) of OA
In the southeast US, observations from the IMPROVE and SEARCH network both show a reduction in summertime surface OA concentration from 2000 to 2013 (Fig. 1). Observational results are averaged using 21 IMPROVE sites and 3 SEARCH sites within the southeast US. OA concentration averaged over June–July–August (JJA) 2000–2013 is 4.2 µg m−3 from the IMPROVE sites and 5.7 µg m−3 from SEARCH sites. A similar ∼ 30 % summertime low bias on the IMPROVE sites was documented by Kim et al. (2015) compared to the SEARCH sites, which is thought to be due to evaporation of OA from the filters after collection, as the IMPROVE filters stay several days on site after sampling and are shipped without refrigeration, while the SEARCH filters are analyzed in situ. Despite different magnitudes, OAs from the two networks demonstrate similar trends and interannual variability. The 2000–2013 trend of JJA OA mass is −1.7 % yr−1 for IMPROVE and −1.9 % yr−1 for SEARCH. Compared to the slow decrease in OA, a faster declining trend is found for sulfate from IMPROVE (−6.9 % yr−1) and SEARCH (−6.7 % yr−1) for the same period (Fig. 2).
Compared to the observations, the default GEOS-Chem model predicts a steeper decreasing trend of OA mass during 2000–2013 (Fig. 1). Modeling results are averaged over the domain [29–37∘ N, 74–96∘ W] excluding ocean grid cells (Fig. S1). The 2000–2013 JJA-averaged OA from the default model is 6.7 µg m−3, which is higher than OA from IMPROVE and SEARCH. Modeled total OA mass decreases at a rate of 4.9 % yr−1, which is about 1.9 (1.6) times faster than IMPROVE (SEARCH) OA (Student's t test p < 0.001). By sampling the model results at the locations of the IMPROVE and SEARCH sites, the modeled summertime OA has an average of 6.9 µg m−3 and a trend of 5.0 % yr−1, similar to the model results averaged over the whole southeast US domain. For simplicity, we show only the domain-averaged model results in all figures and analyses. The strong reduction in total OA mass is dominated by aqueous SOA, especially through reactive uptake of IEPOX, with no decreasing trend in other components (Fig. 1). The contribution of IEPOX SOA to total OA mass decreases from 61 % in the early 2000s to 28 % in 2013. The simulated IEPOX SOA in 2013 compares well with previous field studies which suggested that IEPOX SOA contributed to 18 % –40 % in southeast US sites in summer 2013 (Budisulistiorini et al., 2016; Xu et al., 2015a).
A main constraint comes from the MMV of OA in the southeast US. IMPROVE and SEARCH OA observations show little variability among June, July and August, despite large MMV of sulfate in the early 2000s (Fig. 2a). We find similar behavior from another observation network, CSN. The discontinuity in the OA trend in the CSN network is due to different protocols applied (Fig. S2). Within sites using the same protocol, there are no systematic monthly differences, which agrees with IMPROVE and SEARCH. In contrast, modeled OA displays large MMV between June, July and August from 2000 to 2008, where OA in July and August is 1–3 times that of June values (Fig. 2a). Such a large MMV is dominated by aqueous SOA, especially from the reactive uptake of IEPOX. Prior to 2008, the simulated IEPOX SOA alone can be up to a factor 2 higher than the observed total OA (Fig. 2). The other components including POA and dry SOA (including terpene SOA and anthropogenic SOA) formed through partitioning together have low concentrations and small MMV. The default model captures the variability of observed sulfate well (Fig. 2a), with an average of 3.8 µg m−3 and a trend of −6.9 % yr−1, as compared to −6.9 % yr−1 (average concentration 4.2 µg m−3) from IMPROVE and −6.7 % yr−1 (average concentration 4.3 µg m−3) from SEARCH.
The large MMV in the model suggests a much stronger modeled OA dependence on sulfate than observations. In 2000–2004, changes in modeled sulfate from June to July and/or August correspond to large MMV of modeled OA mass. In contrast, little MMV is found in observed OA mass during the same months despite large MMV in observed sulfate (Fig. 2a). From a linear regression analysis using all monthly data in 2000–2013, the OA-to-sulfate regression slope is m=0.29 (r2=0.25) from IMPROVE, m=0.51 (r2=0.43) from SEARCH and m=1.87 (r2=0.57) from the default model, even though the default model captures the magnitude, trend and monthly variability of observed sulfate well. In summary, simulated total OA mass in the standard GEOS-Chem model, dominated by IEPOX SOA, has a steeper decreasing trend from 2000 to 2013 than the observations and has a large MMV indicating strong dependence on sulfate.
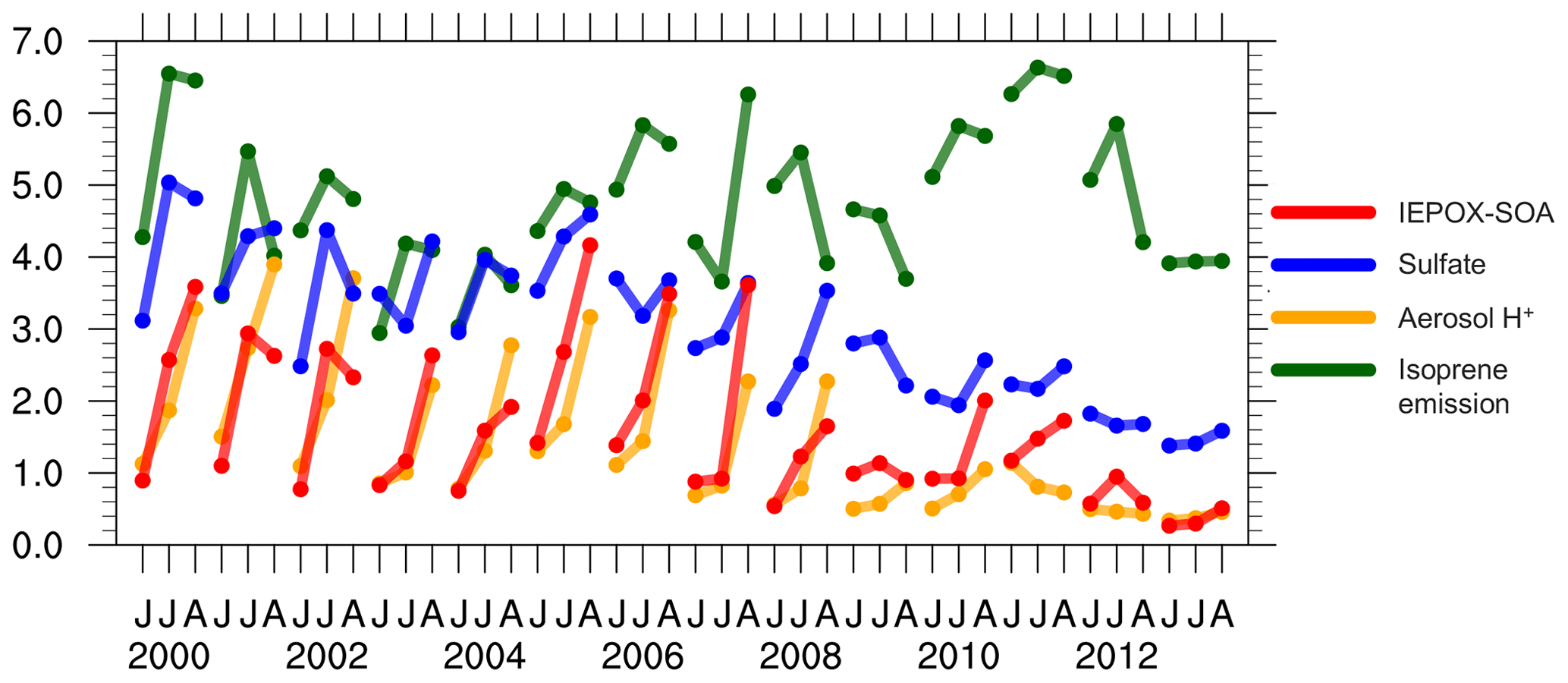
Figure 3Standardized monthly surface IEPOX-SOA concentration, sulfate concentration, aerosol H+ activity and isoprene emission from the default model. All variables are averaged over the southeast US, have been divided by 1 SDs and therefore are unitless.
3.2 What controls the modeled IEPOX-SOA variability?
The strong dependence of IEPOX SOA on sulfate is well established by laboratory and field work: wet sulfate particles provide the surface and volume of liquid media for IEPOX reactive uptake (Budisulistiorini et al., 2017; Eddingsaas et al., 2010; Riva et al., 2016; Xu et al., 2015a, 2016) and serve as nucleophiles for nucleophilic addition to form organosulfates (Nguyen et al., 2014; Surratt et al., 2007a). Sulfate (), together with ammonium (), nitrate () and other ions, regulates proton (H+) activity () that can catalyze the ring opening of epoxide group leading to the formation of IEPOX SOA (Gaston et al., 2014; Pye et al., 2013; Surratt et al., 2007b). However, some recent studies suggest that IEPOX SOA is not well correlated with aerosol acidity estimated from thermodynamic models (Budisulistiorini et al., 2015; Lin et al., 2013; Xu et al., 2015a), although the lack of direct measurements of aerosol acidity may be a limitation. We use the GEOS-Chem model here to examine the simulated IEPOX-SOA dependence on sulfate, aerosol acidity and emissions of isoprene which produce IEPOX at high yields under low-NOx conditions (Paulot et al., 2009). Temperature impacts the formation of IEPOX SOA mainly through regulating isoprene emissions but does not influence partitioning as IEPOX SOA is treated as nonvolatile in GEOS-Chem. Therefore, temperature is not examined as another driver in addition to isoprene emissions. We do not treat aerosol water as an independent driver because the dilution effect of aerosol water is implicitly considered in the inorganic sulfate–ammonium–nitrate aerosol volume and acidity calculation, and studies have shown that particle water is not a limiting factor unless the particle is purely dry (Nguyen et al., 2014; Riva et al., 2016; Xu et al., 2015a), which is rare in summertime in the southeast US.

Figure 4(a) Schematic diagram of IEPOX reactive uptake coefficient (γIEPOX). Colored lines indicate the position of JJA-averaged organic mass fraction and aerosol H+ activity in 2000–2013 from the default, CT and CT_H01 simulations. (b) Simulated aerosol acidity (mol L−1) from the default, CT_newNH3 and CT_H01 simulations. (c) JJA-averaged surface OA (µg m−3) from IMPROVE, SEARCH and all model simulations. (d) Standard deviation of OA (µg m−3) between June, July and August from IMPROVE, SEARCH and all model simulations. All results are averaged over the southeast US.
We find that the large MMV of OA in the model is mainly driven by sulfate concentrations and aerosol acidity. Figure 3 shows the standardized monthly surface IEPOX-SOA concentration, sulfate concentration, aerosol H+ activity and isoprene emission from the default model. For each variable, the monthly gridded data have been first averaged over the southeast US. Then, we calculate the 1 SD of all monthly data (June, July and August data from 2000 to 2013). Finally, the domain-averaged monthly data have been divided by their SD, so the variables are standardized to be unitless and their variability can be compared directly. Prior to 2008, IEPOX-SOA production is largely enhanced by abundant sulfate (Gaston et al., 2014). Due to this high level of sulfate (about > 4 µg m−3), the modeled aerosol acidity becomes particularly sensitive to variations in NH3 emissions. The default NH3 emissions from NEI11v1 suggest no significant long-term trend from 2000 to 2013. In general, ammonium aerosol is strongly correlated with sulfate and has a similar declining trend to sulfate (Silvern et al., 2017). However, the NH3 emissions in August are about 25 % lower than in June and July (Fig. S4). As a result, in August before 2008, the aerosol ratio is smaller (Fig. S4) and is up to 3 times higher than in June (Fig. 4b), leading to high production of IEPOX SOA in August. Both sulfate and aerosol acidity appear to be the dominant contributors to MMV of OA during this period. After 2008, IEPOX-SOA formation is substantially suppressed, due to small SO2 emissions and low modeled aerosol acidity with small monthly variability. Isoprene emissions also contribute to the month-to-month and interannual OA variability in the model.
The multivariate linear regression analysis of IEPOX SOA quantitatively determines the relative importance of its three drivers in the model. Using all monthly data in 2000–2013, the standardized regression coefficients (β) associated with , sulfate aerosol concentration and isoprene emission are β=0.50 (r2=0.71), β=0.39 (r2=0.64) and β=0.34 (r2=0.18), respectively, suggesting that aerosol acidity is the dominant controlling factor. The three variables together explain 88 % of the variability of IEPOX SOA. Their relative importance changes over time (Table S1). Aerosol acidity strongly correlates with IEPOX SOA in 2005–2008 (β=0.57, r2=0.82) but its role becomes much weaker after 2008 (β=0.27, r2=0.56). Sulfate aerosol is always the first or second most important driver, especially in 2000–2004 (β=0.46, r2=0.76). Isoprene emission contributes to the overall interannual variability, for example leading to the relatively low IEPOX SOA in 2003/04 and the peaks in 2000, 2006 and 2011 (Fig. 3). The high IEPOX SOA in 2000/01 and 2005–2007 is a result of high-sulfate aerosol, high aerosol acidity due to low NH3 supply relative to high-sulfate and high-isoprene emissions during these periods (Figs. 3 and 4b).
3.3 Narrowing the gap between model and observation
3.3.1 Coating
Several reasons may lead to the large monthly variations of the modeled OA. The modeled IEPOX SOA shows a much stronger sensitivity to aerosol acidity than suggested by field observations, which found weak or no correlation between observed IEPOX SOA and derived aerosol acidity (Budisulistiorini et al., 2015; Lin et al., 2013; Worton et al., 2013; Xu et al., 2015a). Lack of consideration of organic coating effect may provide one possible explanation. In the real atmosphere, inorganic aerosol is generally internally mixed with other organics (Anttila et al., 2006; Murphy et al., 2006). The presence of an organic coating may alter the solubility and diffusion properties at the surface of inorganic particles and diminish further uptake of IEPOX. We implemented a linear coating effect for the IEPOX uptake in a sensitivity simulation, CT, in which both the magnitude of γIEPOX and its sensitivity to acidity have been reduced. Figure 4a shows a schematic illustrating the dependence of the γIEPOX coating effect on acidity and organic mass fraction (χorg). The original γIEPOX without coating is represented at χorg=0. The orange line in Fig. 4a shows the approximate position of JJA-averaged acidity and organic mass fraction in the CT simulation. Adding a coating reduces γIEPOX by almost half, but the impact on the total reactive uptake rate of IEPOX is partially compensated for by the corresponding increase in particle surface area. The sensitivity of γIEPOX to acidity has also been reduced, especially during the early 2000s (Fig. 4a). The CT simulation reduces the southeast US JJA-averaged IEPOX-SOA concentrations by 0.3–1.8 µg m−3 (Fig. 4c).
3.3.2 NH3 emissions and aerosol acidity
Second, recent studies present contradictory results and explanations on the long-term trend of aerosol acidity in the southeast US (Pye et al., 2020; Silvern et al., 2017; Weber et al., 2016). In this study, we show that the decreasing trend of aerosol acidity from the standard GEOS-Chem model is mainly caused by high acidity in August before 2008, which corresponds to insufficient NH3 emissions in high-sulfate environments. The NEI11v1 inventory is used in the default configuration, in which NH3 emissions in June and July are 30 % higher than in August (Fig. S4), but not all NH3 emission inventories agree with such a pattern (Paulot et al., 2014). We did a sensitivity test (CT_newNH3) replacing the default US NH3 emissions from NEI11v1 by a new NH3 emission product derived from CrIS satellite observations, which has higher emissions and smaller MMV among June, July and August (Fig. S4). In the CT_newNH3 simulation, the resulting simulated aerosol acidity is substantially changed in 2000–2008 (Fig. 4b). The high acidity ( = 0.55–0.9 mol L−1) in August has been reduced to around 0.2 mol L−1 and is much closer to June and July values (Fig. 3). The results suggest that the fine particles in the southeast US are within a regime where the acidity ( in units of mol L−1) is sensitive to NH3 emissions relative to sulfate concentration, though corresponding pH changes are small (pH within 0.5–1.5, Fig. S4). Small changes in NH3 may lead to large changes in especially when sulfate concentrations are high, resulting in high month-to-month variability of the IEPOX uptake. After updating the NH3 emissions using the satellite-based estimates, the model simulates a much more stable trend in aerosol acidity from 2000 to 2013 (Fig. 4b), consistent with recent thermodynamic modeling studies that suggested steady aerosol acidity despite large reductions in observed sulfate (Pye et al., 2020; Weber et al., 2016).
Due to the high uncertainty associated with the derived NH3 emission product and acidity calculation (Guo et al., 2015, 2018; Silvern et al., 2017; Song et al., 2018; Tao and Murphy, 2019), we conducted another simulation, CT_H01, that fixes level at 0.1 mol L−1 when calculating IEPOX uptake rate, corresponding to the predicted value (constrained by observations) during the 2013 SOAS campaign (Weber et al., 2016). The two simulations, CT_newNH3 and CT_H01, yield similar long-term trends of IEPOX SOA in the southeast US (Fig. S5 in the Supplement), and they agree better with the long-term surface OA measurements from IMPROVE and SEARCH than the default model (Fig. 4c and d). For the SOAS 2013 campaign, the CT_H01 scheme simulates an average IEPOX-SOA concentration of 0.74 µg m−3, similar to 0.81 µg m−3 in the default model, and agrees well with the two independent aerosol mass spectrometer measurements (0.97 µg m−3 from obs_GT and 0.68 µg m−3 from obs_CU; see daily time series in Fig. S6 in the Supplement). The CT_newNH3 scheme simulates an average IEPOX-SOA concentration of 0.34 µg m−3, lower than the observation and the other models by a factor of > 2, due to both the simplified coating effect and small aerosol values ( < 0.1 mol L−1, Fig. 4b). In general, the fixed acidity in the CT_H01 simulation captures the measured IEPOX SOA from the SOAS 2013 campaign well (Fig. S6) and improves the modeled total OA mass relative to the observations: the modeled long-term decreasing rate of JJA-average OA from 2000 to 2013 has been reduced from 4.9 to 3.2 % yr−1, which is better compared to the IMPROVE (1.7 % yr−1) and SEARCH (1.9 % yr−1) observations, but is still higher (Fig. 4c). The modeled MMV of OA have also been greatly reduced (Fig. 4d).
3.3.3 Relationships between OA and sulfate
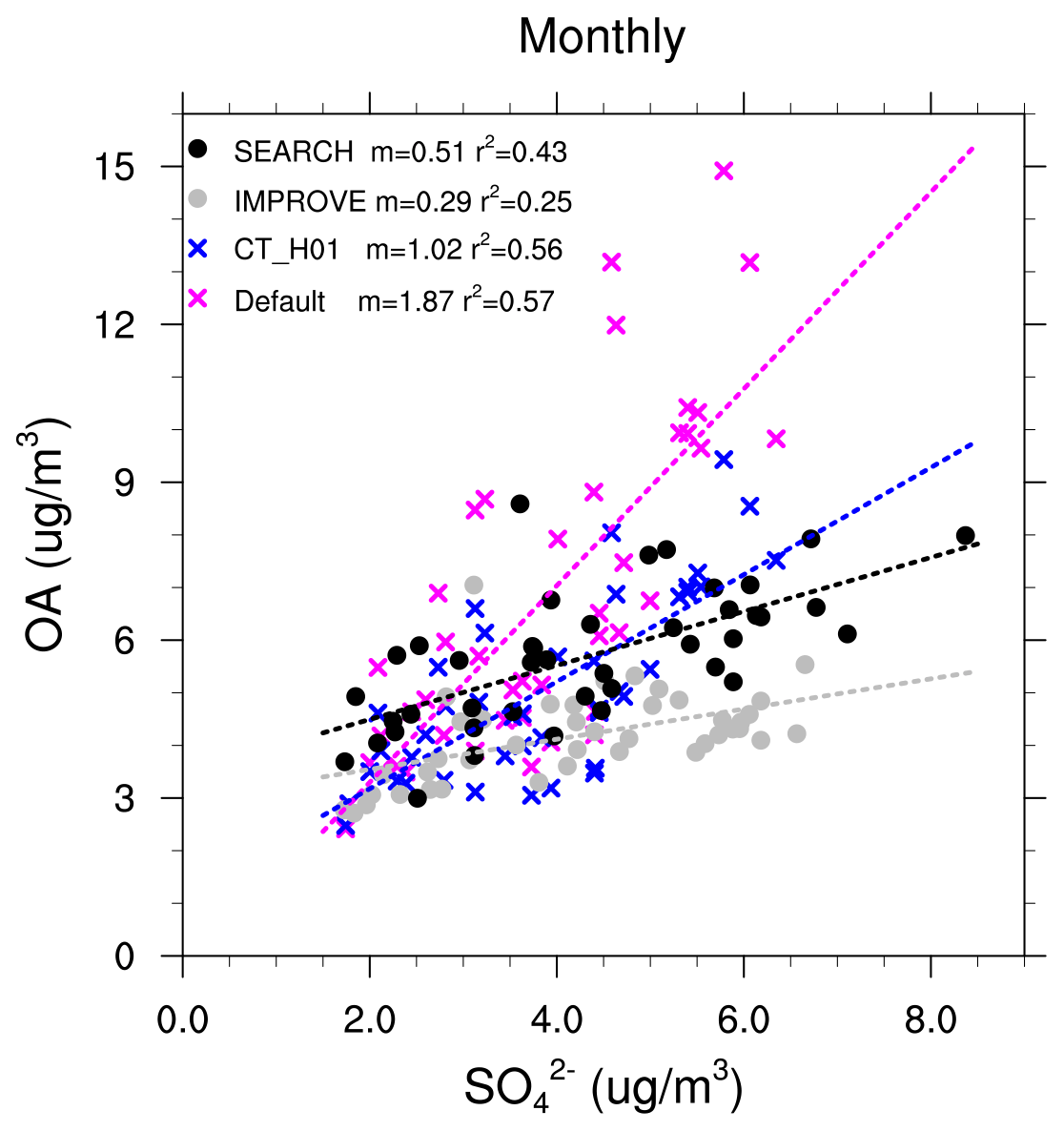
Figure 5Relationships between monthly OA and sulfate concentrations (µg m−3). Each dot represents monthly data averaged from all sites from each network within the southeast US.
The formation of aqueous SOA explicitly depends on sulfate aerosol and aerosol acidity which is also impacted by sulfate. The default model, in which a large fraction of simulated total OA mass is from aqueous SOA (mostly IEPOX SOA), shows a stronger dependence of total OA on sulfate than the observations (Fig. 5). The OA-to-sulfate regression slope calculated using monthly OA and sulfate (averaged from all sites beforehand for each network) is m=1.87 for the default simulation, which is much higher than m=0.29 from IMPROVE and m=0.51 from SEARCH. Such a strong dependence is clearly demonstrated by the MMV of IEPOX SOA (Fig. 2). Adding the coating effect and fixing = 0.1 mol L−1 substantially reduces the MMV of IEPOX SOA and the simulated monthly OA-to-sulfate slope (m=1.02).
Despite the model improvement against the observations in terms of OA and IEPOX-SOA magnitude and long-term relationship with sulfate, the CT_H01 scheme needs to be further improved. The rate of OA decreases per year in CT_H01 is about 0.8 times higher than the long-term observations, with modeled MMV still larger than observations in the early 2000s (Fig. 4d). Recent studies (Riva et al., 2019) suggested that the IEPOX-SOA production per unit mass of sulfate likely increases with decreasing sulfate due to changes in aerosol properties, such as acidity, morphology, phase state and viscosity, as well as formation of organosulfates, suggesting nonlinearity between IEPOX SOA and sulfate (Riva et al., 2019; Zhang et al., 2019a). Further modeling studies with separated IEPOX-SOA species and detailed aerosol properties are needed to achieve a better mechanistic understanding of the dependence of OA on inorganic aerosol.
Significant reductions of SO2 emissions, combined with monthly variations of sulfate and NH3 emissions, provide a unique dataset to test the sensitivity of biogenic SOA formation to inorganic species. Observations from two networks (IMPROVE and SEARCH) show a slowly decreasing trend in total OA mass from 2000 to 2013 in the southeast US (−1.7 % yr−1 from IMPROVE and −1.9 % yr−1 from SEARCH), in contrast to a much faster rate of sulfate reduction (−6.9 % yr−1 from IMPROVE and −6.7 % yr−1 from SEARCH). The standard version of the GEOS-Chem model was able to reproduce the long-term trend of sulfate (−6.7 % yr−1), but with a faster decrease in OA (−4.9 % yr−1) and larger interannual variability.
The MMV of total OA mass during summers provides a novel observational constraint on the SOA formation mechanism. Remarkably, we find little MMV of OA from all three surface networks (IMPROVE, SEARCH and CSN) during summer months in 2000–2013, despite larger MMV in sulfate and NH3 emissions. This is in contrast to the standard version of the GEOS-Chem model, which shows a much larger MMV of OA during 2000–2008. Large MMV of OA in the standard model is mainly due to the high sensitivity of modeled IEPOX SOA to sulfate and aerosol acidity (and NH3 emissions) when sulfate aerosol is abundant. The resulting strong correlation between OA and sulfate also appears to be at odds with long-term observations (Fig. 5). Incorporating a coating effect for IEPOX uptake and fixing aerosol acidity have together improved the model performance in terms of OA trend, variability, and the relationship between OA and sulfate, though further improvement is needed.
There are many uncertainties associated with the calculation of IEPOX-SOA formation. In the default scheme, the Henry law constant for IEPOX uptake was tuned using measurements from the SOAS 2013 campaign and was found to be 1.7 × 107 M atm−1, which is 10 times smaller than suggested by Gaston et al. (2014) based on laboratory experiments and about half of the suggested value (3 × 107 M atm−1) in some other studies (Budisulistiorini et al., 2017; Nguyen et al., 2014; Pye et al., 2017; Woo and McNeill, 2015; Y. Zhang et al., 2018). The default simulation agrees well with surface IEPOX-SOA data from SOAS 2013 and SEAC4RS 2013 aircraft campaigns (Marais et al., 2016) but overestimates OA magnitude and MMV against long-term observations from IMPROVE and SEARCH. The CT_newNH3 simulation reproduces the long-term OA trend but underestimates IEPOX SOA by a factor of 2 against SOAS 2013. The coating effect may be stronger than used here, as Gaston et al. (2014) used a low-viscosity organic material in the experiments. The NH3 emissions (which are critical for the calculation of aerosol acidity) are highly uncertain (Dammers et al., 2019), and the acidity calculation is further complicated by nonvolatile cations (Guo et al., 2018) and meteorological conditions (Guo et al., 2015; Tao and Murphy, 2019). Uncertainties are also associated with the volatility of IEPOX SOA. Some studies suggested a large fraction of IEPOX-SOA compounds (e.g., 2-methyltetrol) are semivolatile and can reevaporate back into the gas phase (Ambro et al., 2019; Isaacman-VanWertz et al., 2016), while other studies suggest IEPOX-SOA products are mostly nonvolatile or low volatility (Hu et al., 2016; Lopez-Hilfiker et al., 2016). As multiple parameters may be tuned in the model to fit observations, further laboratory, field and modeling studies are needed to integrate Henry's law constant, IEPOX-SOA yields, volatility, coating effect and acidity dependence for a better mechanistic understanding. The CT_H01 scheme lacks mechanical representation of detailed aerosol properties like phase state, acidity, viscosity and morphology but reasonably captures both the OA and IEPOX-SOA magnitude (compared to both the three filter measurement networks and the SOAS 2013 campaign), long-term variability and relationship with sulfate (Figs. 4, 5 and S6); therefore, it may serve as a simplified representation for climate models. Simulations in this study are conducted at a horizontal resolution of 2∘ × 2.5∘, which is comparable to most global climate models. However, as shown by Yu et al. (2016), from coarse to fine horizontal resolution, there will be a shift from a low-NOx to a high-NOx pathway for isoprene oxidation. Therefore, using a fine resolution may reduce the production of IEPOX and IEPOX SOA, which needs further investigation. For all kinds of models, long-term filter-based measurements, especially intraseasonal MMV, are important observational constraints that should be considered in model development.
Even with our improved model, the rate of OA decrease per year is still 0.8 times higher the long-term observations, and still shows a higher MMV than observations particularly in the early 2000s (Fig. 4d). Such discrepancies may suggest a more important role of SOA pathways that are less dependent on inorganic aerosol, such as terpene SOA formed by reversible gas-aerosol partitioning. Terpene SOA is included in GEOS-Chem (yellow color in Fig. 1) and contributes to 8 %–24 % of total OA, which might be underestimated compared to recent field studies. Xu et al. (2015b) finds a large MMV in IEPOX SOA, but the less-oxidized oxygenated OA (LO-OOA, an indicator for freshly formed monoterpene SOA) and the more-oxidized oxygenated OA (MO-OOA, also likely from biogenic sources) have little MMV in summer months, and they contribute to more than 50 % of total OA mass in the southeast US (Xu et al., 2018). The important role of monoterpene SOA is also confirmed by molecular-level characterization of organic aerosols (H. Zhang et al., 2018). Other pathways may contribute to SOA to some extent and may add to the predicted SOA formed by partitioning, including biogenic SOA from auto-oxidation (Bianchi et al., 2019; Pye et al., 2019), in-cloud SOA formation that may be less dependent on acidity than aqueous SOA (Tsui et al., 2019), a small but underestimated contribution of anthropogenic SOA (Schroder et al., 2018; Shah et al., 2019) and other possible mechanisms (Schwantes et al., 2019). Further quantifying the relative importance of the different pathways will allow a more accurate quantification of the anthropogenic influence on biogenic SOA and the associated radiative forcing.
The observational datasets from long-term filter measurement networks IMPROVE and CSN are available at http://views.cira.colostate.edu/fed/QueryWizard/Default.aspx (last access: 26 February 2019). The SEARCH observational datasets are available by contacting Eric Edgerton. The model code and modeling results are available by contacting Yiqi Zheng and Jingqiu Mao.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-13091-2020-supplement.
YZ and JM designed the research, performed the simulations and conducted the analysis. JAT and EAM provided guidance on aerosol coating parameterization. HC and DKH provided the CrIS-derived NH3 emission. NLN, WH and JLJ provided data from the SOAS 2013 field campaign. EEM provided data from the SEARCH network. YZ wrote the paper with all coauthors providing input.
The authors declare that they have no conflict of interest.
Yiqi Zheng and Jingqiu Mao acknowledge funding from NOAA NA18OAR4310114. Nga Lee Ng acknowledges funding from NOAA NA18OAR4310112. Hansen Cao and Daven K. Henze recognize support from NASA 80NSSC18K0689. Weiwei Hu and Jose L. Jimenez acknowledge funding from NSF AGS-1822664. Eloise A. Marais acknowledges funding from NERC/EPSRC (award number EP/R513465/1). The authors acknowledge the Electric Power Research Institute (EPRI) and Southern Company for support of the SEARCH network and Atmospheric Research & Analysis, Inc. IMPROVE and CSN data are accessed from the Federal Land Manger Environmental Database. Yiqi Zheng thanks Arlene M. Fiore, Róisín Commane and V. Faye McNeill for their helpful discussions.
This research has been supported by the US NOAA (grant no. NA18OAR4310114).
This paper was edited by Manabu Shiraiwa and reviewed by two anonymous referees.
Ambro, E. L., Schobesberger, S., Gaston, C. J., Lopez-Hilfiker, F. D., Lee, B. H., Liu, J., Zelenyuk, A., Bell, D., Cappa, C. D., Helgestad, T., Li, Z., Guenther, A., Wang, J., Wise, M., Caylor, R., Surratt, J. D., Riedel, T., Hyttinen, N., Salo, V.-T., Hasan, G., Kurtén, T., Shilling, J. E., and Thornton, J. A.: Chamber-based insights into the factors controlling IEPOX SOA yield, composition, and volatility, Atmos. Chem. Phys., 19, 11253–11265, https://doi.org/10.5194/acp-2019-271, 2019.
Anttila, T., Kiendler-Scharr, A., Tillmann, R., and Mentel, T. F.: On the reactive uptake of gaseous compounds by organic-coated aqueous aerosols: Theoretical analysis and application to the heterogeneous hydrolysis of N2O5, J. Phys. Chem. A, 110, 10435–10443, https://doi.org/10.1021/jp062403c, 2006.
Attwood, A. R., Washenfelder, R. A., Brock, C. A., Hu, W., Baumann, K., Campuzano-Jost, P., Day, D. A., Edgerton, E. S., Murphy, D. M., Palm, B. B., McComiskey, A., Wagner, N. L., De Sá, S. S., Ortega, A., Martin, S. T., Jimenez, J. L., and Brown, S. S.: Trends in sulfate and organic aerosol mass in the Southeast U.S.: Impact on aerosol optical depth and radiative forcing, Geophys. Res. Lett., 41, 7701–7709, https://doi.org/10.1002/2014GL061669, 2014.
Bey, I., Jacob, D. J., Yantosca, R. M., Logan, J. A., Field, B. D., Fiore, A. M., Li, Q.-B., Liu, H.-Y., Mickley, L. J., and Schultz, M. G.: Global Modeling of Tropospheric Chemistry with Assimilated Meteorology: Model Description and Evaluation, J. Geophys. Res., 106, 73–95, https://doi.org/10.1029/2001JD000807, 2001.
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Blanchard, C. L., Hidy, G. M., Shaw, S., Baumann, K., and Edgerton, E. S.: Effects of emission reductions on organic aerosol in the southeastern United States, Atmos. Chem. Phys., 16, 215–238, https://doi.org/10.5194/acp-16-215-2016, 2016.
Budisulistiorini, S. H., Li, X., Bairai, S. T., Renfro, J., Liu, Y., Liu, Y. J., McKinney, K. A., Martin, S. T., McNeill, V. F., Pye, H. O. T., Nenes, A., Neff, M. E., Stone, E. A., Mueller, S., Knote, C., Shaw, S. L., Zhang, Z., Gold, A., and Surratt, J. D.: Examining the effects of anthropogenic emissions on isoprene-derived secondary organic aerosol formation during the 2013 Southern Oxidant and Aerosol Study (SOAS) at the Look Rock, Tennessee ground site, Atmos. Chem. Phys., 15, 8871–8888, https://doi.org/10.5194/acp-15-8871-2015, 2015.
Budisulistiorini, S. H., Baumann, K., Edgerton, E. S., Bairai, S. T., Mueller, S., Shaw, S. L., Knipping, E. M., Gold, A., and Surratt, J. D.: Seasonal characterization of submicron aerosol chemical composition and organic aerosol sources in the southeastern United States: Atlanta, Georgia,and Look Rock, Tennessee, Atmos. Chem. Phys., 16, 5171–5189, https://doi.org/10.5194/acp-16-5171-2016, 2016.
Budisulistiorini, S. H., Nenes, A., Carlton, A. G., Surratt, J. D., McNeill, V. F., and Pye, H. O. T.: Simulating Aqueous-Phase Isoprene-Epoxydiol (IEPOX) Secondary Organic Aerosol Production during the 2013 Southern Oxidant and Aerosol Study (SOAS), Environ. Sci. Technol., 51, 5026–5034, https://doi.org/10.1021/acs.est.6b05750, 2017.
Butler, T., Vermeylen, F., Lehmann, C. M., Likens, G. E., and Puchalski, M.: Increasing ammonia concentration trends in large regions of the USA derived from the NADP/AMoN network, Atmos. Environ., 146, 132–140, https://doi.org/10.1016/j.atmosenv.2016.06.033, 2016.
Cao, H., Henze, D. K., Shephard, M. W., Dammers, E., Cady-Pereira, K., Alvarado, M. J., Lonsdale, C. R., Luo, G., Yu, F., Zhu, L., Danielson, C. G., and Edgerton, E. S.: Inverse modeling of NH3 sources using CrIS remote sensing measurements, Environ. Res. Lett., 15, 10, https://doi.org/10.1088/1748-9326/abb5cc, 2020.
Carlton, A. G., Pinder, R. W., Bhave, P. V., and Pouliot, G. A.: To what extent can biogenic SOA be controlled?, Environ. Sci. Technol., 44, 3376–3380, https://doi.org/10.1021/es903506b, 2010.
Dammers, E., McLinden, C. A., Griffin, D., Shephard, M. W., Van Der Graaf, S., Lutsch, E., Schaap, M., Gainairu-Matz, Y., Fioletov, V., Van Damme, M., Whitburn, S., Clarisse, L., Cady-Pereira, K., Clerbaux, C., Coheur, P. F., and Erisman, J. W.: NH3 emissions from large point sources derived from CrIS and IASI satellite observations, Atmos. Chem. Phys., 19, 12261–12293, https://doi.org/10.5194/acp-19-12261-2019, 2019.
Donahue, N. M., Robinson, A. L., Stanier, C. O., and Pandis, S. N.: Coupled partitioning, dilution, and chemical aging of semivolatile organics, Environ. Sci. Technol., 40, 2635–2643, https://doi.org/10.1021/es052297c, 2006.
Eddingsaas, N. C., Vandervelde, D. G., and Wennberg, P. O.: Kinetics and products of the acid-catalyzed ring-opening of atmospherically relevant butyl epoxy alcohols, J. Phys. Chem. A, 114, 8106–8113, https://doi.org/10.1021/jp103907c, 2010.
Edgerton, E. S., Hartsell, B. E., Saylor, R. D., Jansen, J. J., Hansen, D. A., and Hidy, G. M.: The southeastern aerosol research and characterization study: Part II. Filter-based measurements of fine and coarse particulate matter mass and composition, J. Air Waste Manage., 55, 1527–1542, https://doi.org/10.1080/10473289.2005.10464744, 2005.
EPA: The benefits and costs of the clean air act from 1990–2020, in: Better Air: Benefits and Costs of the Clean Air Act, available at: https://www.epa.gov/sites/production/files/2015-07/documents/summaryreport.pdf (last access: 12 February 2020), US Environmental Protection Agency, Washington D.C. 20460, 2011.
Ervens, B., Turpin, B. J., and Weber, R. J.: Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies, Atmos. Chem. Phys., 11, 11069–11102, https://doi.org/10.5194/acp-11-11069-2011, 2011.
Fisher, J. A., Jacob, D. J., Travis, K. R., Kim, P. S., Marais, E. A., Chan Miller, C., Yu, K., Zhu, L., Yantosca, R. M., Sulprizio, M. P., Mao, J., Wennberg, P. O., Crounse, J. D., Teng, A. P., Nguyen, T. B., St. Clair, J. M., Cohen, R. C., Romer, P., Nault, B. A., Wooldridge, P. J., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Shepson, P. B., Xiong, F., Blake, D. R., Goldstein, A. H., Misztal, P. K., Hanisco, T. F., Wolfe, G. M., Ryerson, T. B., Wisthaler, A., and Mikoviny, T.: Organic nitrate chemistry and its implications for nitrogen budgets in an isoprene- and monoterpene-rich atmosphere: constraints from aircraft (SEAC4RS) and ground-based (SOAS) observations in the Southeast US, Atmos. Chem. Phys., 16, 5969–5991, https://doi.org/10.5194/acp-16-5969-2016, 2016.
Fountoukis, C. and Nenes, A.: ISORROPIA II: a computationally efficient thermodynamic equilibrium model for K+-Ca2+-Mg2+--Na+---Cl−-H2O aerosols, Atmos. Chem. Phys., 7, 4639–4659, https://doi.org/10.5194/acp-7-4639-2007, 2007.
Fu, T. M., Jacob, D. J., Wittrock, F., Burrows, J. P., Vrekoussis, M., and Henze, D. K.: Global budgets of atmospheric glyoxal and methylglyoxal, and implications for formation of secondary organic aerosols, J. Geophys. Res.-Atmos., 113, D15303, https://doi.org/10.1029/2007JD009505, 2008.
Gaston, C. J., Riedel, T. P., Zhang, Z., Gold, A., Surratt, J. D., and Thornton, J. A.: Reactive uptake of an isoprene-derived epoxydiol to submicron aerosol particles, Environ. Sci. Technol., 48, 11178–11186, https://doi.org/10.1021/es5034266, 2014.
Guenther, A., Karl, T., Harley, P., Wiedinmyer, C., Palmer, P. I., and Geron, C.: Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature), Atmos. Chem. Phys., 6, 3181–3210, https://doi.org/10.5194/acp-6-3181-2006, 2006.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Guo, H., Xu, L., Bougiatioti, A., Cerully, K. M., Capps, S. L., Hite Jr., J. R., Carlton, A. G., Lee, S.-H., Bergin, M. H., Ng, N. L., Nenes, A., and Weber, R. J.: Fine-particle water and pH in the southeastern United States, Atmos. Chem. Phys., 15, 5211–5228, https://doi.org/10.5194/acp-15-5211-2015, 2015.
Guo, H., Nenes, A., and Weber, R. J.: The underappreciated role of nonvolatile cations in aerosol ammonium-sulfate molar ratios, Atmos. Chem. Phys., 18, 17307–17323, https://doi.org/10.5194/acp-18-17307-2018, 2018.
Hayes, P. L., Carlton, A. G., Baker, K. R., Ahmadov, R., Washenfelder, R. A., Alvarez, S., Rappenglück, B., Gilman, J. B., Kuster, W. C., de Gouw, J. A., Zotter, P., Prévôt, A. S. H., Szidat, S., Kleindienst, T. E., Offenberg, J. H., Ma, P. K., and Jimenez, J. L.: Modeling the formation and aging of secondary organic aerosols in Los Angeles during CalNex 2010, Atmos. Chem. Phys., 15, 5773–5801, https://doi.org/10.5194/acp-15-5773-2015, 2015.
Henze, D. K., Hakami, A., and Seinfeld, J. H.: Development of the adjoint of GEOS-Chem, Atmos. Chem. Phys., 7, 2413–2433, https://doi.org/10.5194/acp-7-2413-2007, 2007.
Hidy, G. M., Blanchard, C. L., Baumann, K., Edgerton, E., Tanenbaum, S., Shaw, S., Knipping, E., Tombach, I., Jansen, J., and Walters, J.: Chemical climatology of the southeastern United States, 1999–2013, Atmos. Chem. Phys., 14, 11893–11914, https://doi.org/10.5194/acp-14-11893-2014, 2014.
Hodshire, A. L., Akherati, A., Alvarado, M. J., Brown-Steiner, B., Jathar, S. H., Jimenez, J. L., Kreidenweis, S. M., Lonsdale, C. R., Onasch, T. B., Ortega, A. M., and Pierce, J. R.: Aging Effects on Biomass Burning Aerosol Mass and Composition: A Critical Review of Field and Laboratory Studies, Environ. Sci. Technol., 53, 10007–10022, https://doi.org/10.1021/acs.est.9b02588, 2019.
Hu, W., Palm, B. B., Day, D. A., Campuzano-Jost, P., Krechmer, J. E., Peng, Z., de Sá, S. S., Martin, S. T., Alexander, M. L., Baumann, K., Hacker, L., Kiendler-Scharr, A., Koss, A. R., de Gouw, J. A., Goldstein, A. H., Seco, R., Sjostedt, S. J., Park, J.-H., Guenther, A. B., Kim, S., Canonaco, F., Prévôt, A. S. H., Brune, W. H., and Jimenez, J. L.: Volatility and lifetime against OH heterogeneous reaction of ambient isoprene-epoxydiols-derived secondary organic aerosol (IEPOX-SOA), Atmos. Chem. Phys., 16, 11563–11580, https://doi.org/10.5194/acp-16-11563-2016, 2016.
Hu, W. W., Campuzano-Jost, P., Palm, B. B., Day, D. A., Ortega, A. M., Hayes, P. L., Krechmer, J. E., Chen, Q., Kuwata, M., Liu, Y. J., de Sá, S. S., McKinney, K., Martin, S. T., Hu, M., Budisulistiorini, S. H., Riva, M., Surratt, J. D., St. Clair, J. M., Isaacman-Van Wertz, G., Yee, L. D., Goldstein, A. H., Carbone, S., Brito, J., Artaxo, P., de Gouw, J. A., Koss, A., Wisthaler, A., Mikoviny, T., Karl, T., Kaser, L., Jud, W., Hansel, A., Docherty, K. S., Alexander, M. L., Robinson, N. H., Coe, H., Allan, J. D., Canagaratna, M. R., Paulot, F., and Jimenez, J. L.: Characterization of a real-time tracer for isoprene epoxydiols-derived secondary organic aerosol (IEPOX-SOA) from aerosol mass spectrometer measurements, Atmos. Chem. Phys., 15, 11807–11833, https://doi.org/10.5194/acp-15-11807-2015, 2015.
Isaacman-VanWertz, G., Yee, L. D., Kreisberg, N. M., Wernis, R., Moss, J. A., Hering, S. V., De Sá, S. S., Martin, S. T., Alexander, M. L., Palm, B. B., Hu, W., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Riva, M., Surratt, J. D., Viegas, J., Manzi, A., Edgerton, E., Baumann, K., Souza, R., Artaxo, P., and Goldstein, A. H.: Ambient Gas-Particle Partitioning of Tracers for Biogenic Oxidation, Environ. Sci. Technol., 50, 9952–9962, https://doi.org/10.1021/acs.est.6b01674, 2016.
Janssens-Maenhout, G., Crippa, M., Guizzardi, D., Dentener, F., Muntean, M., Pouliot, G., Keating, T., Zhang, Q., Kurokawa, J., Wankmüller, R., Denier van der Gon, H., Kuenen, J. J. P., Klimont, Z., Frost, G., Darras, S., Koffi, B., and Li, M.: HTAP_v2.2: a mosaic of regional and global emission grid maps for 2008 and 2010 to study hemispheric transport of air pollution, Atmos. Chem. Phys., 15, 11411–11432, https://doi.org/10.5194/acp-15-11411-2015, 2015.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of organic aerosols in the atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Jo, D. S., Hodzic, A., Emmons, L. K., Marais, E. A., Peng, Z., Nault, B. A., Hu, W., Campuzano-Jost, P., and Jimenez, J. L.: A simplified parameterization of isoprene-epoxydiol-derived secondary organic aerosol (IEPOX-SOA) for global chemistry and climate models: a case study with GEOS-Chem v11-02-rc, Geosci. Model Dev., 12, 2983–3000, https://doi.org/10.5194/gmd-12-2983-2019, 2019.
Kiendler-Scharr, A., Mensah, A. A., Friese, E., Topping, D., Nemitz, E., Prevot, A. S. H., Äijälä, M., Allan, J., Canonaco, F., Canagaratna, M., Carbone, S., Crippa, M., Dall Osto, M., Day, D. A., De Carlo, P., Di Marco, C. F., Elbern, H., Eriksson, A., Freney, E., Hao, L., Herrmann, H., Hildebrandt, L., Hillamo, R., Jimenez, J. L., Laaksonen, A., McFiggans, G., Mohr, C., O'Dowd, C., Otjes, R., Ovadnevaite, J., Pandis, S. N., Poulain, L., Schlag, P., Sellegri, K., Swietlicki, E., Tiitta, P., Vermeulen, A., Wahner, A., Worsnop, D., and Wu, H. C.: Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol, Geophys. Res. Lett., 43, 7735–7744, https://doi.org/10.1002/2016GL069239, 2016.
Kim, P. S., Jacob, D. J., Fisher, J. A., Travis, K., Yu, K., Zhu, L., Yantosca, R. M., Sulprizio, M. P., Jimenez, J. L., Campuzano-Jost, P., Froyd, K. D., Liao, J., Hair, J. W., Fenn, M. A., Butler, C. F., Wagner, N. L., Gordon, T. D., Welti, A., Wennberg, P. O., Crounse, J. D., St. Clair, J. M., Teng, A. P., Millet, D. B., Schwarz, J. P., Markovic, M. Z., and Perring, A. E.: Sources, seasonality, and trends of southeast US aerosol: an integrated analysis of surface, aircraft, and satellite observations with the GEOS-Chem chemical transport model, Atmos. Chem. Phys., 15, 10411–10433, https://doi.org/10.5194/acp-15-10411-2015, 2015.
Kroll, J. H., Ng, N. L., Murphy, S. M., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from isoprene photooxidation under high-NOx conditions, Geophys. Res. Lett., 32, 1–4, https://doi.org/10.1029/2005GL023637, 2005.
Kroll, J. H., Ng, N. L., Murphy, S. M., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from isoprene photooxidation, Environ. Sci. Technol., 40, 1869–1877, https://doi.org/10.1021/es0524301, 2006.
Li, J., Mao, J., Min, K. E., Washenfelder, R. A., Brown, S. S., Kaiser, J., Keutsch, F. N., Volkamer, R., Wolfe, G. M., Hanisco, T. F., Pollack, I. B., Ryerson, T. B., Graus, M., Gilman, J. B., Lerner, B. M., Warneke, C., de Gouw, J. A., Middlebrook, A. M., Liao, J., Welti, A., Henderson, B. H., Faye McNeill, V., Hall, S. R., Ullmann, K., Donner, L. J., Paulot, F., and Horowitz, L. W.: Observational constraints on glyoxal production from isoprene oxidation and its contribution to organic aerosol over the Southeast United States, J. Geophys. Res., 121, 9849–9861, https://doi.org/10.1002/2016JD025331, 2016.
Li, J., Mao, J., Fiore, A. M., Cohen, R. C., Crounse, J. D., Teng, A. P., Wennberg, P. O., Lee, B. H., Lopez-Hilfiker, F. D., Thornton, J. A., Peischl, J., Pollack, I. B., Ryerson, T. B., Veres, P., Roberts, J. M., Neuman, J. A., Nowak, J. B., Wolfe, G. M., Hanisco, T. F., Fried, A., Singh, H. B., Dibb, J., Paulot, F., and Horowitz, L. W.: Decadal changes in summertime reactive oxidized nitrogen and surface ozone over the Southeast United States, Atmos. Chem. Phys., 18, 2341–2361, https://doi.org/10.5194/acp-18-2341-2018, 2018.
Li, Y., Day, D. A., Stark, H., Jimenez, J. L., and Shiraiwa, M.: Predictions of the glass transition temperature and viscosity of organic aerosols from volatility distributions, Atmos. Chem. Phys., 20, 8103–8122, https://doi.org/10.5194/acp-20-8103-2020, 2020.
Liggio, J., Li, S. M., and McLaren, R.: Reactive uptake of glyoxal by particulate matter, J. Geophys. Res.-Atmos., 110, 1–13, https://doi.org/10.1029/2004JD005113, 2005.
Lin, Y.-H., Knipping, E. M., Edgerton, E. S., Shaw, S. L., and Surratt, J. D.: Investigating the influences of SO2 and NH3 levels on isoprene-derived secondary organic aerosol formation using conditional sampling approaches, Atmos. Chem. Phys., 13, 8457–8470, https://doi.org/10.5194/acp-13-8457-2013, 2013.
Lopez-Hilfiker, F. D., Mohr, C., D'Ambro, E. L., Lutz, A., Riedel, T. P., Gaston, C. J., Iyer, S., Zhang, Z., Gold, A., Surratt, J. D., Lee, B. H., Kurten, T., Hu, W. W., Jimenez, J., Hallquist, M., and Thornton, J. A.: Molecular Composition and Volatility of Organic Aerosol in the Southeastern U.S.: Implications for IEPOX Derived SOA, Environ. Sci. Technol., 50, 2200–2209, https://doi.org/10.1021/acs.est.5b04769, 2016.
Malm, W. C., Schichtel, B. A., Hand, J. L., and Collett, J. L.: Concurrent Temporal and Spatial Trends in Sulfate and Organic Mass Concentrations Measured in the IMPROVE Monitoring Program, J. Geophys. Res.-Atmos., 122, 10462–10476, https://doi.org/10.1002/2017JD026865, 2017.
Mao, J., Paulot, F., Jacob, D. J., Cohen, R. C., Crounse, J. D., Wennberg, P. O., Keller, C. A., Hudman, R. C., Barkley, M. P., and Horowitz, L. W.: Ozone and organic nitrates over the eastern United States: Sensitivity to isoprene chemistry, J. Geophys. Res.-Atmos., 118, 11256–11268, https://doi.org/10.1002/jgrd.50817, 2013.
Mao, J., Carlton, A., Cohen, R. C., Brune, W. H., Brown, S. S., Wolfe, G. M., Jimenez, J. L., Pye, H. O. T., Lee Ng, N., Xu, L., McNeill, V. F., Tsigaridis, K., McDonald, B. C., Warneke, C., Guenther, A., Alvarado, M. J., de Gouw, J., Mickley, L. J., Leibensperger, E. M., Mathur, R., Nolte, C. G., Portmann, R. W., Unger, N., Tosca, M., and Horowitz, L. W.: Southeast Atmosphere Studies: learning from model-observation syntheses, Atmos. Chem. Phys., 18, 2615–2651, https://doi.org/10.5194/acp-18-2615-2018, 2018.
Marais, E. A., Jacob, D. J., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Krechmer, J., Zhu, L., Kim, P. S., Miller, C. C., Fisher, J. A., Travis, K., Yu, K., Hanisco, T. F., Wolfe, G. M., Arkinson, H. L., Pye, H. O. T., Froyd, K. D., Liao, J., and McNeill, V. F.: Aqueous-phase mechanism for secondary organic aerosol formation from isoprene: application to the southeast United States and co-benefit of SO2 emission controls, Atmos. Chem. Phys., 16, 1603–1618, https://doi.org/10.5194/acp-16-1603-2016, 2016.
Marais, E. A., Jacob, D. J., Turner, J. R., and Mickley, L. J.: Evidence of 1991–2013 decrease of biogenic secondary organic aerosol in response to SO2 emission controls, Environ. Sci. Technol., 12, 12 054018 , https://doi.org/10.1088/1748-9326/aa69c8, 2017.
McNeill, V. F., Woo, J. L., Kim, D. D., Schwier, A. N., Wannell, N. J., Sumner, A. J., and Barakat, J. M.: Aqueous-phase secondary organic aerosol and organosulfate formation in atmospheric aerosols: A modeling study, Environ. Sci. Technol., 46, 8075–8081, https://doi.org/10.1021/es3002986, 2012.
Murphy, D. M., Cziczo, D. J., Froyd, K. D., Hudson, P. K., Matthew, B. M., Middlebrook, A. M., Peltier, R. E., Sullivan, A., Thomson, D. S., and Weber, R. J.: Single-peptide mass spectrometry of tropospheric aerosol particles, J. Geophys. Res.-Atmos., 111, 1–15, https://doi.org/10.1029/2006JD007340, 2006.
Ng, N. L., Brown, S. S., Archibald, A. T., Atlas, E., Cohen, R. C., Crowley, J. N., Day, D. A., Donahue, N. M., Fry, J. L., Fuchs, H., Griffin, R. J., Guzman, M. I., Herrmann, H., Hodzic, A., Iinuma, Y., Jimenez, J. L., Kiendler-Scharr, A., Lee, B. H., Luecken, D. J., Mao, J., McLaren, R., Mutzel, A., Osthoff, H. D., Ouyang, B., Picquet-Varrault, B., Platt, U., Pye, H. O. T., Rudich, Y., Schwantes, R. H., Shiraiwa, M., Stutz, J., Thornton, J. A., Tilgner, A., Williams, B. J., and Zaveri, R. A.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 17, 2103–2162, https://doi.org/10.5194/acp-17-2103-2017, 2017.
Nguyen, T. B., Coggon, M. M., Bates, K. H., Zhang, X., Schwantes, R. H., Schilling, K. A., Loza, C. L., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Organic aerosol formation from the reactive uptake of isoprene epoxydiols (IEPOX) onto non-acidified inorganic seeds, Atmos. Chem. Phys., 14, 3497–3510, https://doi.org/10.5194/acp-14-3497-2014, 2014.
Pai, S. J., Heald, C. L., Pierce, J. R., Farina, S. C., Marais, E. A., Jimenez, J. L., Campuzano-Jost, P., Nault, B. A., Middlebrook, A. M., Coe, H., Shilling, J. E., Bahreini, R., Dingle, J. H., and Vu, K.: An evaluation of global organic aerosol schemes using airborne observations, Atmos. Chem. Phys., 20, 2637–2665, https://doi.org/10.5194/acp-20-2637-2020, 2020.
Pankow, J. F.: An Absorption-Model of the Gas Aerosol Partitioning Involved in the Formation of Secondary Organic Aerosol, Atmos. Environ., 28, 189–193, https://doi.org/10.1016/j.atmosenv.2007.10.060, 1994.
Paulot, F., Crounse, J. D., Kjaergaard, H. G., Kürten, A., Clair, J. M. S., Seinfeld, J. H., and Wennberg, P. O.: Unexpected Epoxide Formation in the Gas-Phase Photooxidation of Isoprene, Science, 325, 730–734, https://doi.org/10.1126/science.1174251, 2009.
Paulot, F., Jacob, D. J., Pinder, R. W., Bash, J. O., Travis, K. R., and Henze, D. K.: Ammonia emissions in the United States, European Union, and China derived by high-resolution inversion of ammonium wet deposition data: Interpretation with a new agricultural emissions inventory (MASAGE_NH3), J. Geophys. Res.-Atmos., 119, 4343–4364, https://doi.org/10.1002/2013JD021130, 2014.
Presto, A. A., Huff Hartz, K. E., and Donahue, N. M.: Secondary organic aerosol production from terpene ozonolysis. 2. Effect of NOx concentration, Environ. Sci. Technol., 39, 7046–7054, https://doi.org/10.1021/es050400s, 2005.
Pye, H. O. T., Liao, H., Wu, S., Mickley, L. J., Jacob, D. J., Henze, D. J., and Seinfeld, J. H.: Effect of changes in climate and emissions on future sulfate-nitrate-ammonium aerosol levels in the United States, J. Geophys. Res.-Atmos., 114, 1–18, https://doi.org/10.1029/2008JD010701, 2009.
Pye, H. O. T., Chan, A. W. H., Barkley, M. P., and Seinfeld, J. H.: Global modeling of organic aerosol: the importance of reactive nitrogen (NOx and NO3), Atmos. Chem. Phys., 10, 11261–11276, https://doi.org/10.5194/acp-10-11261-2010, 2010.
Pye, H. O. T., Pinder, R. W., Piletic, I. R., Xie, Y., Capps, S. L., Lin, Y. H., Surratt, J. D., Zhang, Z., Gold, A., Luecken, D. J., Hutzell, W. T., Jaoui, M., Offenberg, J. H., Kleindienst, T. E., Lewandowski, M., and Edney, E. O.: Epoxide pathways improve model predictions of isoprene markers and reveal key role of acidity in aerosol formation, Environ. Sci. Technol., 47, 11056–11064, https://doi.org/10.1021/es402106h, 2013.
Pye, H. O. T., Murphy, B. N., Xu, L., Ng, N. L., Carlton, A. G., Guo, H., Weber, R., Vasilakos, P., Appel, K. W., Budisulistiorini, S. H., Surratt, J. D., Nenes, A., Hu, W., Jimenez, J. L., Isaacman-VanWertz, G., Misztal, P. K., and Goldstein, A. H.: On the implications of aerosol liquid water and phase separation for organic aerosol mass, Atmos. Chem. Phys., 17, 343–369, https://doi.org/10.5194/acp-17-343-2017, 2017.
Pye, H. O. T., D'Ambro, E. L., Lee, B. H., Schobesberger, S., Takeuchi, M., Zhao, Y., Lopez-Hilfiker, F., Liu, J., Shilling, J. E., Xing, J., Mathur, R., Middlebrook, A. M., Liao, J., Welti, A., Graus, M., Warneke, C., de Gouw, J. A., Holloway, J. S., Ryerson, T. B., Pollack, I. B., and Thornton, J. A.: Anthropogenic enhancements to production of highly oxygenated molecules from autoxidation, P. Natl. Acad. Sci. USA, 116, 6641–6646, https://doi.org/10.1073/pnas.1810774116, 2019.
Pye, H. O. T., Nenes, A., Alexander, B., Ault, A. P., Barth, M. C., Clegg, S. L., Collett Jr., J. L., Fahey, K. M., Hennigan, C. J., Herrmann, H., Kanakidou, M., Kelly, J. T., Ku, I.-T., McNeill, V. F., Riemer, N., Schaefer, T., Shi, G., Tilgner, A., Walker, J. T., Wang, T., Weber, R., Xing, J., Zaveri, R. A., and Zuend, A.: The acidity of atmospheric particles and clouds, Atmos. Chem. Phys., 20, 4809–4888, https://doi.org/10.5194/acp-20-4809-2020, 2020.
Randerson, J. T., van der Werf, G. R., Giglio, L., Collatz, G. J., and Kasibhatla, P. S.: Global Fire Emissions Database, Version 4, (GFEDv4), ORNL DAAC, https://doi.org/10.3334/ORNLDAAC/1293, 2015.
Ridley, D. A., Heald, C. L., Ridley, K. J., and Kroll, J. H.: Causes and consequences of decreasing atmospheric organic aerosol in the United States, P. Natl. Acad. Sci. USA, 115, 290–295, https://doi.org/10.1073/pnas.1700387115, 2018.
Riedel, T. P., Lin, Y. H., Budisulistiorini, S. H., Gaston, C. J., Thornton, J. A., Zhang, Z., Vizuete, W., Gold, A., and Surratt, J. D.: Heterogeneous reactions of isoprene-derived epoxides: Reaction probabilities and molar secondary organic aerosol yield estimates, Environ. Sci. Tech. Let., 2, 38–42, https://doi.org/10.1021/ez500406f, 2015.
Riedel, T. P., Lin, Y.-H., Zhang, Z., Chu, K., Thornton, J. A., Vizuete, W., Gold, A., and Surratt, J. D.: Constraining condensed-phase formation kinetics of secondary organic aerosol components from isoprene epoxydiols, Atmos. Chem. Phys., 16, 1245–1254, https://doi.org/10.5194/acp-16-1245-2016, 2016.
Riva, M., Bell, D. M., Hansen, A. M. K., Drozd, G. T., Zhang, Z., Gold, A., Imre, D., Surratt, J. D., Glasius, M., and Zelenyuk, A.: Effect of Organic Coatings, Humidity and Aerosol Acidity on Multiphase Chemistry of Isoprene Epoxydiols, Environ. Sci. Technol., 50, 5580–5588, https://doi.org/10.1021/acs.est.5b06050, 2016.
Riva, M., Chen, Y., Zhang, Y., Lei, Z., Olson, N. E., Boyer, H. C., Narayan, S., Yee, L. D., Green, H. S., Cui, T., Zhang, Z., Baumann, K., Fort, M., Edgerton, E., Budisulistiorini, S. H., Rose, C. A., Ribeiro, I. O., e Oliveira, R. L., dos Santos, E. O., Machado, C. M. D., Szopa, S., Zhao, Y., Alves, E. G., de Sá, S. S., Hu, W., Knipping, E. M., Shaw, S. L., Duvoisin Junior, S., de Souza, R. A. F., Palm, B. B., Jimenez, J.-L., Glasius, M., Goldstein, A. H., Pye, H. O. T., Gold, A., Turpin, B. J., Vizuete, W., Martin, S. T., Thornton, J. A., Dutcher, C. S., Ault, A. P., and Surratt, J. D.: Increasing Isoprene Epoxydiol-to-Inorganic Sulfate Aerosol Ratio Results in Extensive Conversion of Inorganic Sulfate to Organosulfur Forms: Implications for Aerosol Physicochemical Properties, Environ. Sci. Technol., 53, 8682–8694, https://doi.org/10.1021/acs.est.9b01019, 2019.
Schmedding, R., Ma, M., Zhang, Y., Farrell, S., Pye, H. O. T., Chen, Y., Wang, C. tsan, Rasool, Q. Z., Budisulistiorini, S. H., Ault, A. P., Surratt, J. D., and Vizuete, W.: α-Pinene-Derived organic coatings on acidic sulfate aerosol impacts secondary organic aerosol formation from isoprene in a box model, Atmos. Environ., 213, 456–462, https://doi.org/10.1016/j.atmosenv.2019.06.005, 2019.
Schroder, J. C., Campuzano-Jost, P., Day, D. A., Shah, V., Larson, K., Sommers, J. M., Sullivan, A. P., Campos, T., Reeves, J. M., Hills, A., Hornbrook, R. S., Blake, N. J., Scheuer, E., Guo, H., Fibiger, D. L., McDuffie, E. E., Hayes, P. L., Weber, R. J., Dibb, J. E., Apel, E. C., Jaeglé, L., Brown, S. S., Thornton, J. A., and Jimenez, J. L.: Sources and Secondary Production of Organic Aerosols in the Northeastern United States during WINTER, J. Geophys. Res.-Atmos., 123, 7771–7796, https://doi.org/10.1029/2018JD028475, 2018.
Schwantes, R. H., Charan, S. M., Bates, K. H., Huang, Y., Nguyen, T. B., Mai, H., Kong, W., Flagan, R. C., and Seinfeld, J. H.: Low-volatility compounds contribute significantly to isoprene secondary organic aerosol (SOA) under high-NOx conditions, Atmos. Chem. Phys., 19, 7255–7278, https://doi.org/10.5194/acp-19-7255-2019, 2019.
Shah, V., Jaeglé, L., Jimenez, J. L., Schroder, J. C., Campuzano-Jost, P., Campos, T. L., Reeves, J. M., Stell, M., Brown, S. S., Lee, B. H., Lopez-Hilfiker, F. D., and Thornton, J. A.: Widespread Pollution From Secondary Sources of Organic Aerosols During Winter in the Northeastern United States, Geophys. Res. Lett., 46, 2974–2983, https://doi.org/10.1029/2018GL081530, 2019.
Shephard, M. W. and Cady-Pereira, K. E.: Cross-track Infrared Sounder (CrIS) satellite observations of tropospheric ammonia, Atmos. Meas. Tech., 8, 1323–1336, https://doi.org/10.5194/amt-8-1323-2015, 2015.
Shrivastava, M., Cappa, C. D., Fan, J., Goldstein, A. H., Guenther, A. B., Jimenez, J. L., Kuang, C., Laskin, A., Martin, S. T., Ng, N. L., Petaja, T., Pierce, J. R., Rasch, P. J., Roldin, P., Seinfeld, J. H., Shilling, J., Smith, J. N., Thornton, J. A., Volkamer, R., Wang, J., Worsnop, D. R., Zaveri, R. A., Zelenyuk, A., and Zhang, Q.: Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 55, 509–559, https://doi.org/10.1002/2016RG000540, 2017.
Shrivastava, M., Andreae, M. O., Artaxo, P., Barbosa, H. M. J., Berg, L. K., Brito, J., Ching, J., Easter, R. C., Fan, J., Fast, J. D., Feng, Z., Fuentes, J. D., Glasius, M., Goldstein, A. H., Alves, E. G., Gomes, H., Gu, D., Guenther, A., Jathar, S. H., Kim, S., Liu, Y., Lou, S., Martin, S. T., McNeill, V. F., Medeiros, A., de Sá, S. S., Shilling, J. E., Springston, S. R., Souza, R. A. F., Thornton, J. A., Isaacman-VanWertz, G., Yee, L. D., Ynoue, R., Zaveri, R. A., Zelenyuk, A., and Zhao, C.: Urban pollution greatly enhances formation of natural aerosols over the Amazon rainforest, Nat. Commun., 10, 1046, https://doi.org/10.1038/s41467-019-08909-4, 2019.
Silvern, R. F., Jacob, D. J., Kim, P. S., Marais, E. A., Turner, J. R., Campuzano-Jost, P., and Jimenez, J. L.: Inconsistency of ammonium–sulfate aerosol ratios with thermodynamic models in the eastern US: a possible role of organic aerosol, Atmos. Chem. Phys., 17, 5107–5118, https://doi.org/10.5194/acp-17-5107-2017, 2017.
Solomon, P. A., Crumpler, D., Flanagan, J. B., Jayanty, R. K. M., Rickman, E. E., and McDade, C. E.: U.S. National PM2.5 chemical speciation monitoring networks – CSN and IMPROVE: Description of networks, J. Air Waste Manage., 64, 1410–1438, https://doi.org/10.1080/10962247.2014.956904, 2014.
Song, S., Gao, M., Xu, W., Shao, J., Shi, G., Wang, S., Wang, Y., Sun, Y., and McElroy, M. B.: Fine-particle pH for Beijing winter haze as inferred from different thermodynamic equilibrium models, Atmos. Chem. Phys., 18, 7423–7438, https://doi.org/10.5194/acp-18-7423-2018, 2018.
Surratt, J. D., Kroll, J. H., Kleindienst, T. E., Edney, E. O., Claeys, M., Sorooshian, A., Ng, N. L., Offenberg, J. H., Lewandowski, M., Jaoui, M., Flagan, R. C., and Seinfeld, J. H.: Evidence for organosulfates in secondary organic aerosol, Environ. Sci. Technol., 41, 517–527, https://doi.org/10.1021/es062081q, 2007a.
Surratt, J. D., Lewandowski, M., Offenberg, J. H., Jaoui, M., Kleindienst, T. E., Edney, E. O., and Seinfeld, J. H.: Effect of acidity on secondary organic aerosol formation from isoprene, Environ. Sci. Technol., 41, 5363–5369, https://doi.org/10.1021/es0704176, 2007b.
Surratt, J. D., Chan, A. W. H., Eddingsaas, N. C., Chan, M., Loza, C. L., Kwan, A. J., Hersey, S. P., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Reactive intermediates revealed in secondary organic aerosol formation from isoprene, P. Natl. Acad. Sci. USA, 107, 6640–6645, https://doi.org/10.1073/pnas.0911114107, 2010.
Tan, Y., Lim, Y. B., Altieri, K. E., Seitzinger, S. P., and Turpin, B. J.: Mechanisms leading to oligomers and SOA through aqueous photooxidation: insights from OH radical oxidation of acetic acid and methylglyoxal, Atmos. Chem. Phys., 12, 801–813, https://doi.org/10.5194/acp-12-801-2012, 2012.
Tao, Y. and Murphy, J. G.: The sensitivity of PM2.5 acidity to meteorological parameters and chemical composition changes: 10-year records from six Canadian monitoring sites, Atmos. Chem. Phys., 19, 9309–9320, https://doi.org/10.5194/acp-19-9309-2019, 2019.
Travis, K. R., Jacob, D. J., Fisher, J. A., Kim, P. S., Marais, E. A., Zhu, L., Yu, K., Miller, C. C., Yantosca, R. M., Sulprizio, M. P., Thompson, A. M., Wennberg, P. O., Crounse, J. D., St. Clair, J. M., Cohen, R. C., Laughner, J. L., Dibb, J. E., Hall, S. R., Ullmann, K., Wolfe, G. M., Pollack, I. B., Peischl, J., Neuman, J. A., and Zhou, X.: Why do models overestimate surface ozone in the Southeast United States?, Atmos. Chem. Phys., 16, 13561–13577, https://doi.org/10.5194/acp-16-13561-2016, 2016.
Tsui, W. G., Woo, J. L., and Mcneill, V. F.: Impact of Aerosol-Cloud Cycling on Aqueous Secondary Organic Aerosol Formation, Atmosphere-Basel, 10, 666, https://doi.org/10.3390/atmos10110666, 2019.
Wagner, N. L., Brock, C. A., Angevine, W. M., Beyersdorf, A., Campuzano-Jost, P., Day, D., de Gouw, J. A., Diskin, G. S., Gordon, T. D., Graus, M. G., Holloway, J. S., Huey, G., Jimenez, J. L., Lack, D. A., Liao, J., Liu, X., Markovic, M. Z., Middlebrook, A. M., Mikoviny, T., Peischl, J., Perring, A. E., Richardson, M. S., Ryerson, T. B., Schwarz, J. P., Warneke, C., Welti, A., Wisthaler, A., Ziemba, L. D., and Murphy, D. M.: In situ vertical profiles of aerosol extinction, mass, and composition over the southeast United States during SENEX and SEAC4RS: observations of a modest aerosol enhancement aloft, Atmos. Chem. Phys., 15, 7085–7102, https://doi.org/10.5194/acp-15-7085-2015, 2015.
Weber, R. J., Guo, H., Russell, A. G., and Nenes, A.: High aerosol acidity despite declining atmospheric sulfate concentrations over the past 15 years, Nat. Geosci., 9, 282–285, https://doi.org/10.1038/NGEO2665, 2016.
Woo, J. L. and McNeill, V. F.: simpleGAMMA v1.0 – a reduced model of secondary organic aerosol formation in the aqueous aerosol phase (aaSOA), Geosci. Model Dev., 8, 1821–1829, https://doi.org/10.5194/gmd-8-1821-2015, 2015.
Worton, D. R., Surratt, J. D., Lafranchi, B. W., Chan, A. W. H., Zhao, Y., Weber, R. J., Park, J. H., Gilman, J. B., De Gouw, J., Park, C., Schade, G., Beaver, M., Clair, J. M. S., Crounse, J., Wennberg, P., Wolfe, G. M., Harrold, S., Thornton, J. A., Farmer, D. K., Docherty, K. S., Cubison, M. J., Jimenez, J. L., Frossard, A. A., Russell, L. M., Kristensen, K., Glasius, M., Mao, J., Ren, X., Brune, W., Browne, E. C., Pusede, S. E., Cohen, R. C., Seinfeld, J. H., and Goldstein, A. H.: Observational insights into aerosol formation from isoprene, Environ. Sci. Technol., 47, 11403–11413, https://doi.org/10.1021/es4011064, 2013.
Xu, L., Guo, H., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., Knote, C., Olson, K., Koss, A., Goldstein, A. H., Hering, S. V., de Gouw, J., Baumann, K., Lee, S.-H., Nenes, A., Weber, R. J., and Ng, N. L.: Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, P. Natl. Acad. Sci. USA, 112, 37–42, https://doi.org/10.1073/pnas.1417609112, 2015a.
Xu, L., Suresh, S., Guo, H., Weber, R. J., and Ng, N. L.: Aerosol characterization over the southeastern United States using high-resolution aerosol mass spectrometry: spatial and seasonal variation of aerosol composition and sources with a focus on organic nitrates, Atmos. Chem. Phys., 15, 7307–7336, https://doi.org/10.5194/acp-15-7307-2015, 2015b.
Xu, L., Middlebrook, A. M., Liao, J., de Gouw, J. A., Guo, H., Weber, R. J., Nenes, A., Lopez-Hilfiker, F. D., Lee, B. H., Thornton, J. A., Brock, C. A., Neuman, J. A., Nowak, J. B., Pollack, I. B., Welti, A., Graus, M., Warneke, C., and Ng, N. L.: Enhanced formation of isoprene-derived organic aerosol in sulfur-rich power plant plumes during Southeast Nexus, J. Geophys. Res., 121, 11137–11153, https://doi.org/10.1002/2016JD025156, 2016.
Xu, L., Pye, H. O. T., He, J., Chen, Y., Murphy, B. N., and Ng, N. L.: Experimental and model estimates of the contributions from biogenic monoterpenes and sesquiterpenes to secondary organic aerosol in the southeastern United States, Atmos. Chem. Phys., 18, 12613–12637, https://doi.org/10.5194/acp-18-12613-2018, 2018.
Yu, K., Jacob, D. J., Fisher, J. A., Kim, P. S., Marais, E. A., Miller, C. C., Travis, K. R., Zhu, L., Yantosca, R. M., Sulprizio, M. P., Cohen, R. C., Dibb, J. E., Fried, A., Mikoviny, T., Ryerson, T. B., Wennberg, P. O., and Wisthaler, A.: Sensitivity to grid resolution in the ability of a chemical transport model to simulate observed oxidant chemistry under high-isoprene conditions, Atmos. Chem. Phys., 16, 4369–4378, https://doi.org/10.5194/acp-16-4369-2016, 2016.
Zhang, H., Yee, L. D., Lee, B. H., Curtis, M. P., Worton, D. R., Isaacman-VanWertz, G., Offenberg, J. H., Lewandowski, M., Kleindienst, T. E., Beaver, M. R., Holder, A. L., Lonneman, W. A., Docherty, K. S., Jaoui, M., Pye, H. O. T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Guo, H., Weber, R. J., de Gouw, J., Koss, A. R., Edgerton, E. S., Brune, W., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Kreisberg, N. M., Spielman, S. R., Hering, S. V., Wilson, K. R., Thornton, J. A., and Goldstein, A. H.: Monoterpenes are the largest source of summertime organic aerosol in the southeastern United States, P. Natl. Acad. Sci. USA, 115, 2038–2043, https://doi.org/10.1073/pnas.1717513115, 2018.
Zhang, Y., Chen, Y., Lambe, A. T., Olson, N. E., Lei, Z., Craig, R. L., Zhang, Z., Gold, A., Onasch, T. B., Jayne, J. T., Worsnop, D. R., Gaston, C. J., Thornton, J. A., Vizuete, W., Ault, A. P., and Surratt, J. D.: Effect of the Aerosol-Phase State on Secondary Organic Aerosol Formation from the Reactive Uptake of Isoprene-Derived Epoxydiols (IEPOX), ACS Earth Space Chem., 5, 167–174, https://doi.org/10.1021/acs.estlett.8b00044, 2018.
Zhang, Y., Chen, Y., Lei, Z., Olson, N. E., Riva, M., Koss, A. R., Zhang, Z., Gold, A., Jayne, J. T., Worsnop, D. R., Onasch, T. B., Kroll, J. H., Turpin, B. J., Ault, A. P., and Surratt, J. D.: Joint Impacts of Acidity and Viscosity on the Formation of Secondary Organic Aerosol from Isoprene Epoxydiols (IEPOX) in Phase Separated Particles, ACS Earth Space Chem., 3, 2646–2658, https://doi.org/10.1021/acsearthspacechem.9b00209, 2019a.
Zhang, Y., Nichman, L., Spencer, P., Jung, J. I., Lee, A., Heffernan, B. K., Gold, A., Zhang, Z., Chen, Y., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., Onasch, T. B., Surratt, J. D., Chandler, D., Davidovits, P., and Kolb, C. E.: The Cooling Rate- And Volatility-Dependent Glass-Forming Properties of Organic Aerosols Measured by Broadband Dielectric Spectroscopy, Environ. Sci. Technol., 53, 12366–12378, https://doi.org/10.1021/acs.est.9b03317, 2019b.
Zheng, Y., Unger, N., Hodzic, A., Emmons, L., Knote, C., Tilmes, S., Lamarque, J.-F., and Yu, P.: Limited effect of anthropogenic nitrogen oxides on secondary organic aerosol formation, Atmos. Chem. Phys., 15, 13487–13506, https://doi.org/10.5194/acp-15-13487-2015, 2015.
Ziemann, P. J. and Atkinson, R.: Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 41, 6582, https://doi.org/10.1039/c2cs35122f, 2012.