the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 16 Jul 2019
| 16 Jul 2019
Trends in N2O and SF6 mole fractions in archived air samples from Cape Meares, Oregon (USA), 1978–1996
Terry C. Rolfe
Andrew L. Rice
Quantifying historical trends in atmospheric greenhouse gases (GHGs) is important to understanding changes in their budgets and for climate modeling, which simulates historic and projects future climate. Archived samples analyzed using updated measurement techniques and calibration scales can reduce uncertainties in historic records of GHG mole fractions and their trends in time. Here, we present historical measurements of two important GHGs, nitrous oxide (N2O) and sulfur hexafluoride (SF6), collected at the midlatitude Northern Hemisphere station Cape Meares, Oregon (USA, 45.5∘ N, 124∘ W), between 1978 and 1996 in archived air samples from the Oregon Health and Science University – Portland State University (OHSU–PSU) air archive. N2O is the third most important anthropogenically forced GHG behind carbon dioxide (CO2) and methane (CH4). SF6 has a low abundance in the atmosphere, but is one of the most powerful GHGs known. Measurements of atmospheric N2O made during this period are available for select locations, but before mid-1990 they have larger uncertainties than more recent periods due to advancements made in gas chromatography (GC) methods. Few atmospheric SF6 measurements exist pre-1990, particularly in the Northern Hemisphere. The GC system used to measure N2O and SF6 mixing ratios in this work is designed to be fully automated, and is capable of running up to 15 samples per batch. Measurement precision (1σ) of N2O and SF6 is 0.16 % and 1.1 %, respectively (evaluated at 328.7 ppb and 8.8 ppt). Samples were corrected for detector response nonlinearity when measured against our reference standard, with the corrections determined to be 0.14 ppb ppb−1 in N2O and 0.03 ppt ppt−1 in SF6. The mixing ratio of N2O in archived samples is found to be 301.5±0.3 ppb in 1980 and rises to 313.5±0.3 ppb in 1996. The average growth rate over this period is 0.78±0.03 ppb yr−1 (95 % CI). The seasonal amplitude is statistically robust, with a maximum anomaly of 0.3 ppb near April and a minimum near November of −0.4 ppb. Measurements of N2O match well with previously reported values for Cape Meares and other comparable locations. The mixing ratio of SF6 in analyzed samples is found to be 0.85±0.03 ppt in 1980 and rises to 3.83±0.03 ppt in 1996. The average growth rate over this period is 0.17±0.01 ppt yr−1 (95 % CI). The seasonality is statistically robust and has an annual peak amplitude of 0.04 ppt near January and a minimum amplitude of −0.03 ppt near July. These are unique SF6 results from this site and represent a significant increase in the SF6 data available during the 1980s and early 1990s. The mixing ratio and growth rate of SF6 measured compares well to other Northern Hemisphere measurements over this period. From these N2O and SF6 measurements, we conclude that sample integrity is generally robust in the OHSU-PSU air archive for N2O and SF6.
- Article
(2405 KB) - Full-text XML
-
Supplement
(356 KB) - BibTeX
- EndNote





Anthropogenic sources of greenhouse gases (GHGs) have altered the atmospheric composition, resulting in a significant climate forcing of approximately 3 W m−2 since 1750 (Myhre et al., 2013). Measurements of GHG mixing ratios since the industrial revolution constrain global budget uncertainties and interpret recent changes to source and sink processes (Prinn et al., 2000; Khalil et al., 2002; Saikawa et al., 2014). When projecting future GHG mixing ratios, many additional factors must be included in models such as climate feedback effects and possible changes in transport processes. Uncertainties in model predictions can be minimized if GHG measurements are precise and span many different latitudes (Meinshausen et al., 2017).
When historical time series records are not available, past atmospheric GHG abundance can be evaluated using either archived air samples or by analyzing ice core and firn air. One significant advantage of using ice core and firn air for measuring past atmospheric mixing ratios of GHGs is that samples may be collected today that represent past conditions. However, ice core and firn samples are difficult to obtain due to the remoteness of the locations where the samples are collected (Greenland and Antarctica) and provide limited spatial information. Temporal uncertainties must also be evaluated when measuring ice core and firn samples due to diffusion and gravitational separation (Ishijima et al., 2007); samples are best represented by a mean age, limiting temporal resolution. By contrast, archived air samples are discrete in time and space, making them very valuable for evaluating past atmospheric abundance at specific periods in time. However, few air archives are available today. The most well-known air archive is that of Cape Grim, Tasmania (41∘ S, 145∘ E), in the Southern Hemisphere, which contains samples dating back to 1978 (Vollmer et al., 2018). However, archive samples may contain storage artefacts that can contaminate historical records and must be stored carefully to prevent damage or loss.
Nitrous oxide (N2O) is the third most important GHG with anthropogenic sources after carbon dioxide (CO2) and methane (CH4). The global mean mixing ratio of N2O in 2017 was 329.8 ppb with a mean annual trend of 0.85 ppb yr−1 over the last 20 years (Dlugokencky et al., 2018). N2O has a large global warming potential (GWP), 298 times that of CO2 over a 100-year period and a global radiative forcing estimated at 0.19 W m−2 since 1750 (Myhre et al., 2013). The long lifetime of this species (∼120 years) results in most emitted N2O reaching the stratosphere, where photooxidation is the primary source of stratospheric NOx (“active nitrogen”). NOx is the main natural catalyst of ozone (O3) destruction (Crutzen, 1970).
Anthropogenic sources of N2O account for roughly 40 % of all N2O emissions, with natural sources accounting for the other 60 % (Ciais et al., 2013). Bottom-up calculations estimate anthropogenic production of 6.9 (2.7–11.1) TgN yr−1 and natural production of 11 (5.4–19.6) TgN yr−1. The uncertainty in these estimations is large, with 1σ error nearly ±50 %. Together with atmospheric measurements, top-down modeling better constrains the N2O budget and reduces uncertainty in the global source. Sources of N2O calculated this way estimate anthropogenic and natural source production of 6.5 (5.2–7.8) TgN yr−1 and 9.1 (8.1–10.1) TgN yr−1, respectively (Prather et al., 2012).
There are three major natural sources and six major anthropogenic sources of N2O. Natural sources of N2O are natural soils (3.3–9.0 TgN yr−1), oceans (1.8–9.4 TgN yr−1), and atmospheric chemistry (0.3–1.2 TgN yr−1) (note: sources include the minimum and maximum estimates provided from bottom-up calculations in Ciais et al., 2013). By far, the largest anthropogenic source is agriculture, which produces 1.7–4.8 TgN yr−1, followed by industrial and fossil fuel sources (0.2–1.8 TgN yr−1), biomass burning (0.2–1 TgN yr−1), rivers and estuaries (0.1–2.9 TgN yr−1), atmospheric deposition (0.4–1.3 TgN yr−1), and human excreta (0.1–0.3 TgN yr−1) (Ciais et al., 2013). More constraints on source production provided via atmospheric measurements are needed to improve estimates of individual source magnitudes.
The main loss mechanism for N2O is destruction in the stratosphere through photolysis and the reaction with O(1D) (Prather et al., 2015). Soils and the oceans can act as sinks for N2O through microbial processes; however, because the production of N2O is greater than what is consumed, the global net flux is positive. Estimates of the stratospheric sink account for 11.9 (11.0–12.8) TgN yr−1 (Ciais et al., 2013).
Rising global mixing ratios of N2O are due to the imbalance between the sources and the sinks. Based on a top-down constraint, the imbalance between sources and sinks is 3.6 (3.5–3.8) TgN yr−1 (Ciais et al., 2013).
Models have shown that future climate conditions will likely amplify N2O production via positive climate feedback effects, meaning a linear increase in time may underpredict future mixing ratios based on the current rate of change (Khalil and Rasmussen 1983; Stocker et al., 2013). To minimize uncertainty in the N2O budget and in model projections, precise measurements of current and past atmospheric conditions from multiple global locations are needed. Measurements of atmospheric N2O made prior to mid-1990 have larger uncertainties than more recent periods due to advancements made in gas chromatography (GC) methods (Prinn et al., 2000; Jiang et al., 2007; Hall et al., 2011). To reduce uncertainty during this period, archived samples may be analyzed using updated measurement techniques. Additionally, measurements of the isotopic composition of N2O in archived samples can constrain the N2O budget and changes in time due to characteristic isotopic effects in sources and sinks (Park et al., 2012; Snider et al., 2015).
Sulfur hexafluoride (SF6) is an extremely potent GHG. Recent estimates calculated the GWP to be 22 800 (over 100 years compared to CO2) and an atmospheric lifetime of 580–1475 years (Kovács et al., 2017; Ray et al., 2017). While SF6 is one of the strongest GHGs controlled under emission regulations, it has a low global mixing ratio (9.3 ppt in the Northern Hemisphere in 2017), so it does not add significantly to climate forcing by itself (Prinn et al., 2018).
Sources of SF6 are anthropogenic, with main uses being high-voltage insulation, magnesium production, and semiconductor manufacture (Maiss and Brenninkmeijer 1998; Olivier et al., 2005). Global production in 2008 was estimated to be 7.16 Gg yr−1 (Levin et al., 2010). With a very low solubility and no reactivity in the lower atmosphere, the only known sink for SF6 is loss in the mesosphere.
With almost all of the SF6 that has been emitted since the industrial revolution to the atmosphere still present, global emissions can be accurately determined from observations of the atmospheric mixing ratio. Due to its long lifetime and anthropogenic origins, SF6 is used as a validity check for atmospheric transport models (Levin and Hesshaimer, 1996; Patra et al., 2009). It has been estimated that 94 % of all SF6 emissions originate in the Northern Hemisphere (Maiss et al., 1996), explaining a north–south hemispheric gradient in the SF6 mixing ratio of about 0.4 ppt (Levin et al., 2010).
Observations of the growth rate have been reported by several studies (Levin et al., 2010; Rigby et al., 2010; Hall et al., 2011). The trend in SF6 has varied over the last 30+ years and while the magnitude of the growth rate differs slightly between sample locations, several features are prominent. From the early 1970s to the mid-1990s, the trend steadily increased from 0.1 ppt yr−1 and peaked near 0.26 ppt yr−1. The trend then slowly declined to ∼0.20 ppt yr−1 until the early 2000s, when the trend increased again. The inferred global emission of SF6 from the trend increases nearly linearly from 2 Gg yr−1 in the late 1970s to over 6 Gg yr−1 in 1994–1995 (Levin et al., 2010; Rigby et al., 2010).
Reported atmospheric measurements of SF6 before the year 1987 are few. In the Southern Hemisphere, Cape Grim, Tasmania (41∘ S, 145∘ E), archive measurements date back to 1978 (Levin et al., 2010). Northern Hemisphere measurements are reported dating from 1973 from Trinidad Head, CA (41∘ N, 121∘ W), but few are prior to 1990 (Rigby et al., 2010). A more complete record of past SF6 atmospheric mixing ratios is desirable.
The Oregon Health and Science University–Portland State University (OHSU-PSU) air archive includes archived air samples collected from Cape Meares, Oregon (45.5∘ N, 124.0∘ W), in the late 1970s, 1980s, and 1990s by the Department of Environmental and Bimolecular Systems, Oregon Graduate Institute of Science and Engineering (currently OHSU). The samples were collected by air liquefaction, where ∼1000 L (STP) of dried air (using a condenser) was compressed to 3000 kPa into 33 L electropolished stainless steel canisters. Today, archive samples are stored at Portland State University and contain pressures ranging from 60 to 2000 kPa (Rice et al., 2016). Here, we present details of the analytical technique employed and results from the analysis of 159 Cape Meares air samples from the OHSU-PSU air archive.
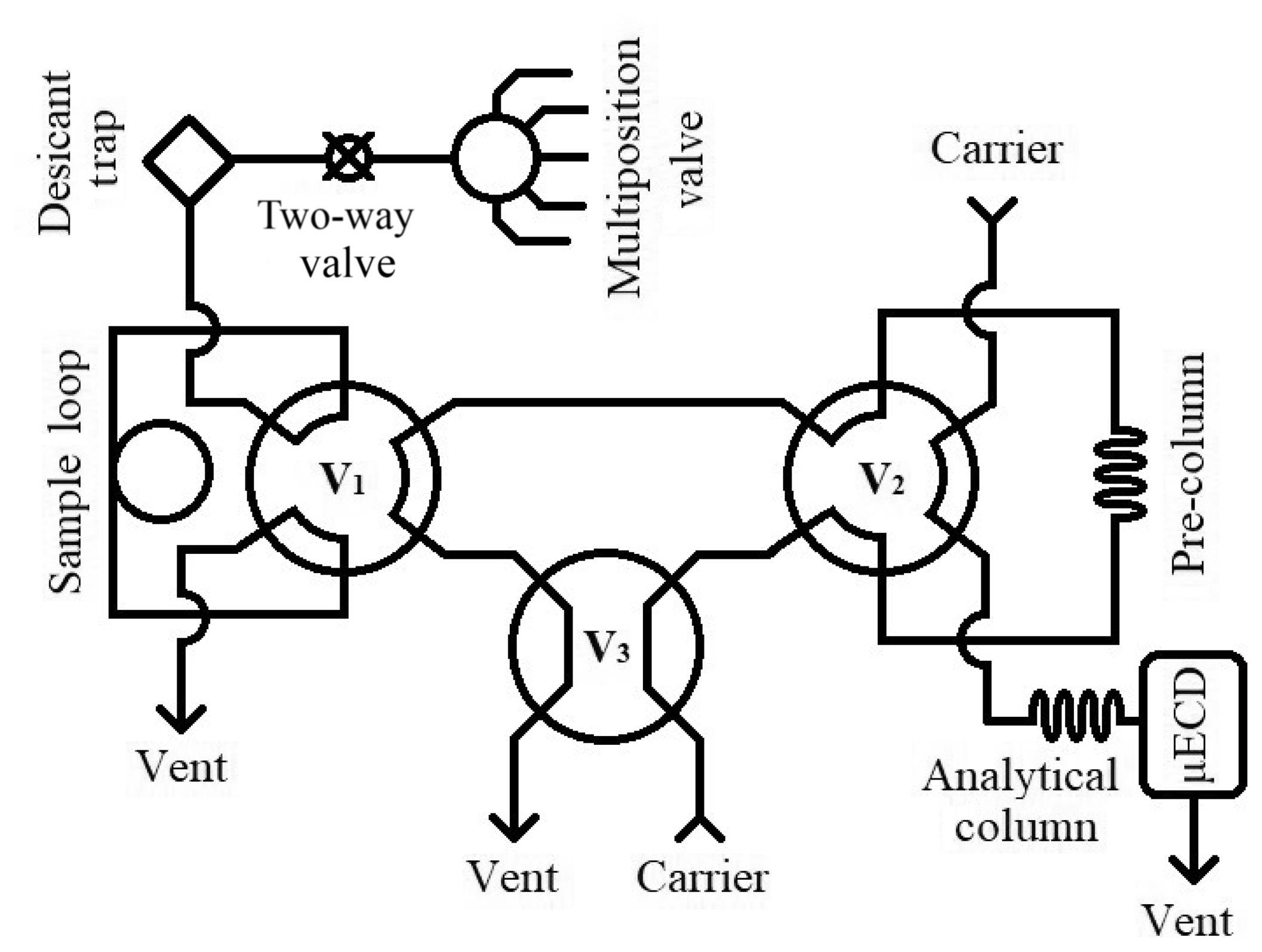
Figure 1Schematic view of the analytical system for sample evaluation. The system is shown in “back-flush” mode. V1 represents Valve 1, V2 represents Valve 2, and V3 represents Valve 3.
2.1 Gas chromatography analytical system
The gas chromatography (GC) analytical system (Fig. 1) employed at Portland State University for measuring N2O and SF6 in archived air samples is based on the configuration used by Hall et al. (2007) and references therein. We use an Agilent 6890N gas chromatograph fitted with a micro-electron capture detector (µECD, Agilent Technologies, Santa Clara, CA). Two Poropak Q 80/100 mesh columns (1.8 m × 2 mm i.d. pre-column, 3.7 × 2 mm i.d. analytical column) achieve peak separation. The carrier gas is P5 (99.999 %, Airgas, Portland, OR) equipped with O2 and hydrocarbon traps (Restek, Bellefonte, PA) to further reduce impurities and to significantly improve baseline signal stability. Two 6-port switching valves (V1 and V2), a 4-port switching valve (V3), and a 16-port multi-position valve (Valvo Instrument Company Inc., Houston, TX) are controlled by Chemstation (V1.A, Agilent Technologies Inc., Santa Clara, CA).
A sample run begins in “back-flush” mode, with the carrier gas flushing the pre-column in the reverse analytical direction to remove the buildup of water on the analytical column that would otherwise eventually elute to the µECD and affect signal baseline. A 16-port multi-position valve is used to introduce pressurized samples into the system; a two-way electric valve (Clippard, Cincinnati, OH) is used to stop sample flow to the sample loop and prevent sample loss. Samples initially pass through a desiccant trap (Perma Pure, Toms River, NJ), before flushing a 10 mL sample loop at 60 mL min−1 for 1.5 min. At this time, V3 rotates, which places the system in “front-cut” mode and allows the sample loop to equilibrate. V1 rotates at 1.75 min and allows the carrier gas to carry the sample N2O and SF6 to the pre-column where separation from O2 and H2O occurs. After O2 elutes through the pre-column to vent, at 3 min V2 rotates and places the pre-column in line with the analytical column, transferring N2O and SF6 to the analytical column. At 4.25 min, the sample has reached the analytical column and V1, V2, and V3 rotate. This begins the back-flush of the pre-column while the analytes are carried to the µECD on the analytical column.

Figure 2Sample chromatogram showing N2O peak at a retention time of 6.1 min and the SF6 peak at a retention time of 7.0 min. The inlay in the upper-right corner shows an enlarged plot of the SF6 peak.
Oven and detector temperatures are maintained at 56 and 310 ∘C, respectively. Carrier gas flow rates are 40 mL min−1 and are maintained by the electronic pressure control of the 6890N. The N2O peak retention time is 6.1 min and the SF6 peak retention time is 7.0 min (Fig. 2). Peak integration is accomplished via Chemstation based on peak height.
All measurements of N2O and SF6 are made relative to a calibrated whole air sample on the NOAA-06A N2O scale and NOAA-14 SF6 scale (NOAA Tank CB11406-A, 328.71±0.5 ppb N2O, 8.76±0.06 ppt SF6), hereafter referred to as the NOAA reference gas. Each sample is analyzed six times and bracketed by six reference gas runs used to measure instrument response and track signal drift. The GC-µECD analytical system was evaluated for precision, reproducibility, and linearity prior to its application to measure mole fractions in historic archive samples.
2.2 Precision and reproducibility of analytical system
Precision of measurement was determined by repeated analysis of the reference standard. Histogram distributions in Fig. 3 show 180 residuals (expressed as a percent relative standard deviation) collected from 30 sets of 6 measurements of N2O (Fig. 3a) and SF6 (Fig. 3b) of the NOAA reference gas. Both N2O and SF6 compare well to a normal distribution (black dashed lines), with chi-square goodness of fit p values of 0.16 and 0.35, respectively. For N2O, 1σ=0.16 % whereas for SF6, 1σ=1.1 %. This corresponds to an uncertainty of ±0.52 ppb for N2O and ±0.10 ppt for SF6. The mean measurement uncertainty (1σ) of OHSU-PSU air archive samples for N2O is 0.23 %. The mean measurement uncertainty (1σ) of SF6 in the OHSU-PSU air archive samples ranges between 6.5 % for samples below 1 ppt and 2.5 % for samples at 4 ppt.
Measurement reproducibility was evaluated by repeatedly measuring a dry air sample (Breathing Air, Airgas, Portland, OR) against the NOAA reference gas and evaluating consistency from the standard deviation of the results. The sample was measured 18 times over 2 weeks with mean measured mixing ratios of N2O and SF6 of 390.9 ppb and 13.2 ppt, respectively. The standard deviations in N2O and SF6 measurements are 0.46 ppb and 0.11 ppt, respectively, which are indistinguishable from 1σ precision for a set of six NOAA reference gas measurements.
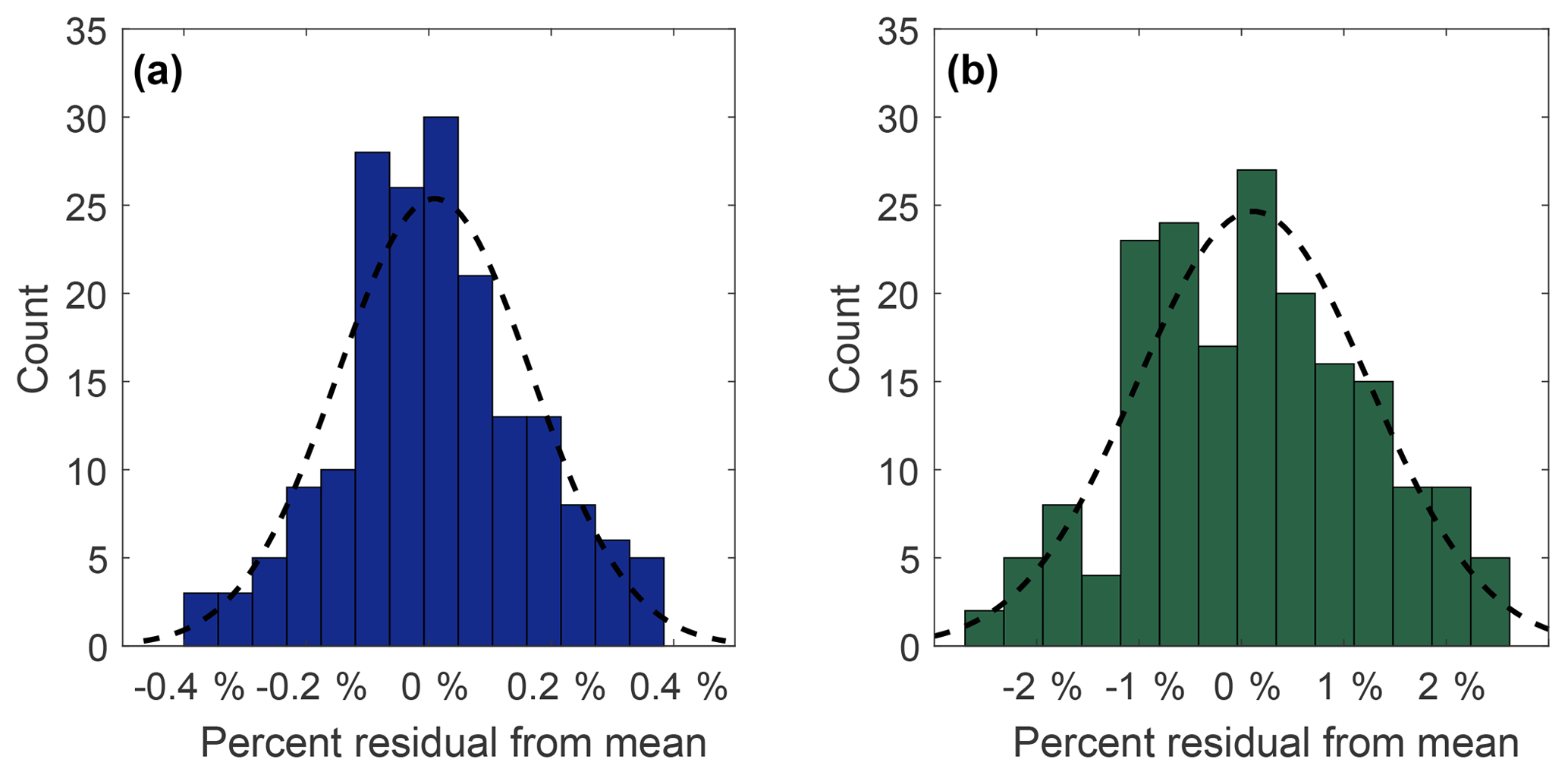
Figure 3Measurement precision for N2O (a) and SF6 (b) expressed as percent relative standard deviation from 30 sets of 6 measurements of the NOAA reference gas. The black dotted line represents a normal distribution curve with the same mean and standard deviation. The standard deviation for N2O and SF6 is 0.16 % and 1.1 %, respectively.
2.3 Linearity of the GC-µECD system
To ensure accurate results for this work, the detector response was evaluated over the mole fraction range expected for N2O and SF6 in the OHSU-PSU air archive. The range in the Northern Hemisphere N2O mole fraction between 1978 and 1996 is between 295 and 314 ppb (Prinn et al., 2000; Ciais et al., 2013). Archived air sample measurements of the Northern Hemisphere SF6 mole fraction from Trinidad Head, CA, measure below 1 ppt in the 1970s and rise to nearly 4 ppt in 1997; Southern Hemisphere measurements from Cape Grim, Tasmania, and the South Pole show a similar range (Levin et al., 2010; Rigby et al., 2010).
A series of manometric dilutions were prepared from the NOAA reference gas at Portland State University to evaluate the µECD response over the historical N2O and SF6 mole fraction sample range. To characterize the N2O response, the N2O reference gas was diluted with ultrapure air (zero grade, Airgas, Portland, OR; N2O and SF6 at mixing ratios below detection limits) using capacitance manometers (MKS Instruments, Andover, MA; range 0–10 and 0–1000 torr) into 3 L electropolished stainless steel canisters (precision ±0.01 %). The range of N2O mixing ratios produced in the 3 L canisters was 32.2–321.4 ppb. The error introduced from the manometric process is small when compared with measurement uncertainty (maximum 1σ error of ±0.07 ppb for N2O).
Table 1Characteristics of 12 manometric N2O dilution samples prepared at Portland State University.

a PRef is the NOAA reference gas pressure (in kPa) introduced to the canister. b PTotal is the final pressure (in kPa) of the canister after balancing with ultrapure air. c Expected response is calculated from the PRef∕PFinal fraction. d Measured N2O response of the µECD. e Measured N2O in parts per billion ( ppb). f N2O 1σ (ppb) is from the combined uncertainty of the sample and the surrounding NOAA reference.
Table 2Characteristics of nine manometric SF6 dilution samples prepared at Portland State University.
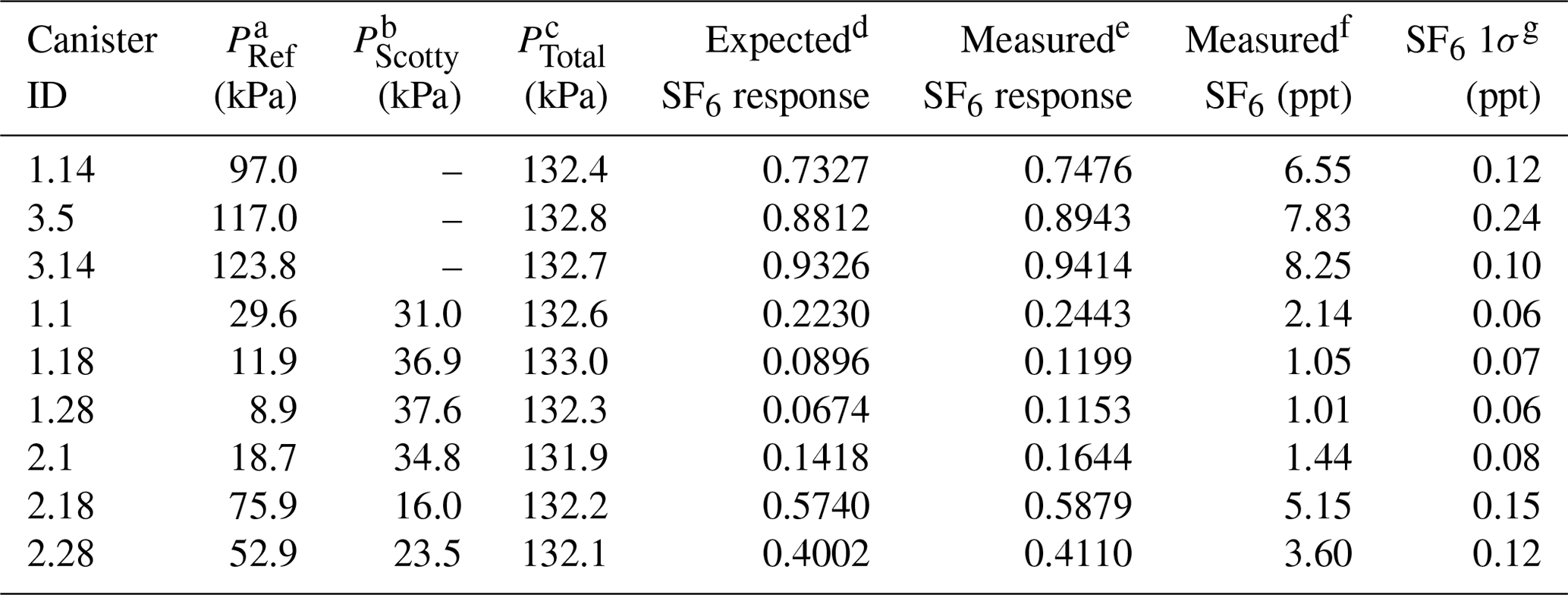
a PRef is the NOAA reference gas pressure (in kPa) introduced to the canister. b PScott is the 1 ppm N2O balanced with He (in kPa) introduced to the canister. c PTotal is the final pressure (in kPa) of the canister after balancing with ultrapure air. d Expected SF6 response is calculated from the PRef∕PFinal fraction. e Measured SF6 response of the µECD. f Measured SF6 in parts per trillion (ppt). g SF6 1σ (ppt) is from the combined uncertainty of the sample and the surrounding NOAA reference.
To characterize the SF6 response at low part per trillion ( ppt) mixing ratios requires consideration of the effect of the falling N2O tail on the chromatogram baseline. To properly account for this interference, SF6 dilutions at low mixing ratios (0.6–6.0 ppt) must have N2O mole fractions that reflect expected mixing ratios in archived samples (300–315 ppb). Prepared dilutions of SF6 included the addition of an aliquot of 1 ppm N2O (±5 %, Scott Specialty Gases, St. Louis, MO) into the canister prior to dilution with ultrapure air. The maximum error (1σ) in SF6 introduced from the manometric process is small (0.001 ppt) compared with the measurement uncertainty. However, SF6 present in either ultrapure air dilution gas or the N2O aliquot at trace levels below the detection limit of our measurement (<0.1 ppt) contribute to the uncertainty in prepared samples. All dilution samples were measured at PSU on the GC-µECD system over several weeks to account for instrument drift. Tables 1 and 2 provide dilution sample pressures, calculated and observed µECD response, and measured N2O and SF6 mole fractions with the error in measurement used to characterize the GC-µECD linearity.
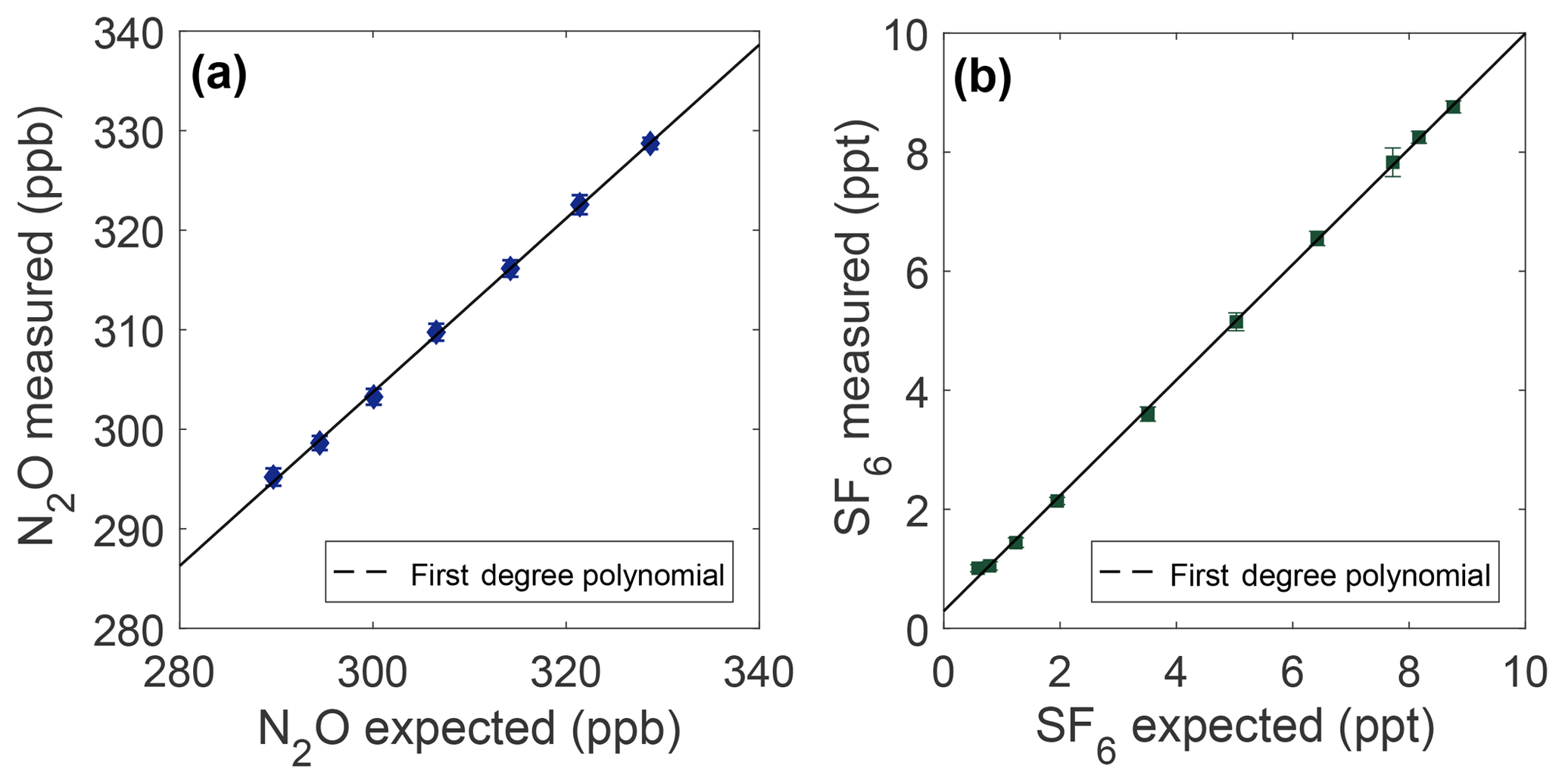
Figure 4Measurement linearity from plots of the measured mole fraction vs. the expected mole fraction of N2O (a) and SF6 (b). The expected mole fraction is calculated from the NOAA reference mole fraction (328.71 ppb N2O and 8.76 ppt SF6) after dilution with ultrapure air. Error bars represent 1σ total uncertainty.
Results of linearity experiments are shown in Fig. 4. For N2O, a slope of 0.8747±0.028 (95 % CI) is found over the data range from 289.7 to 328.7 ppb, which is most relevant for this work. A linear fit is a good model for the deviation from expected over this range (R2=0.964); additional polynomial terms are not statistically robust. This results in sample measurements deviating from expected by ∼0.14 ppb ppb−1 N2O difference from the NOAA reference. For the range of the N2O in the OHSU-PSU air archive, all N2O samples are adjusted for a linear correction of the following form:
where [N2O]Y is the response evaluated N2O mole fraction and [N2O]X is the corrected value. The slope and y intercept, as well as their 95 % confidence intervals, are represented by a1 and a2, respectively. This correction is applied to all sample N2O measurements (corrected values ranging between 298.9 and 314.8 ppb).
The entire NOAA reference gas dilution range for N2O (32–321 ppb) results in a deviation that can be adequately modeled using a third degree polynomial. The linear fit discussed above is indistinguishable from the full third degree polynomial over the N2O mixing ratio range of the OHSU-PSU air archive. However, if measuring N2O samples with a difference of more than 80 ppb compared with the NOAA reference gas, the full third degree polynomial is necessary to correct for the nonlinear response in the µECD.
For SF6, the prepared sample range over which the linear correction is applied is 0.59–8.76 ppt, which is most relevant for this work. The slope of the SF6 linear fit is 0.9728±0.017 (95 % CI) and is a good model for the deviation from expected over this range (R2=0.9995). This results in a deviation from expected of ∼0.03 ppt ppt−1 SF6 difference from the NOAA reference when measuring samples. All SF6 measurements are adjusted for a linear correction of the following form:
where [SF6]Y is the response evaluated SF6 mole fraction and [SF6]Y is the corrected value. The slope and y intercept, as well as their 95 % confidence intervals, are represented by b1 and b2, respectively. This correction is applied to all sample SF6 measurements (corrected values ranging between 0.6 and 4.3 ppt).
Detector response nonlinearity has been evaluated in previous work by other groups on GC-ECD systems. For N2O, deviations from expected of ∼0.2 ppb ppb−1 difference from the reference gas are are typical when in the linear range (Schmidt et al., 2001; Hall et al., 2007). These are similar to the value reported here for the µECD. Over larger ranges, a similar nonlinear response curve is also reported. SF6 nonlinearity reported in Levin et al. (2010) has a similar curvature to the full N2O nonlinear response previously discussed. However, this curvature is not observed to be significant over the range of SF6 dilutions conducted here.
3.1 Air archive mole fractions of N2O and SF6
Measurements of N2O and SF6 mole fractions from 159 samples of the OHSU-PSU air archive were initially filtered for analysis using a 7 median absolute deviation (7MAD) noise filter to remove far outliers. Polynomial fits (first degree for N2O and second degree for SF6) were then applied to the data. Residual values outside of 2σ for N2O and 3σ for SF6 were removed for further data analysis. The entire process removed 12 data points for N2O and 4 data points for SF6 used in the analysis.

Figure 5Deseasonalized measurements of mole fraction vs. date of collection for N2O (a) and SF6 (b), and annual trends in time from Cape Meares, Oregon, for N2O (c) SF6 (d). Error bars are 1σ uncertainty. The solid black lines are LOWESS fit to the data using a smoothing window of 3 years, and shaded areas are 95 % confidence intervals in the LOWESS fit calculated from bootstrapping residual variability 1000 times.
Deseasonalized measurements of N2O and SF6 from Cape Meares are shown in Fig. 5a and b, respectively. A locally weighted linear regression (LOWESS) is utilized to smooth the data using a 3 year smoothing window (Cleaveland and Devlin 1988). The confidence intervals around regressions are calculated by bootstrapping residual variability 1000 times. The regression results in a N2O mole fraction of 301.5±0.3 ppb (1σ) in 1980 that increases roughly linearly to the mid-1990s, where the mixing ratio is 313.5±0.3 ppb (1σ) in 1996.
Observations of the N2O mole fraction match well with previously published measurements of N2O from Cape Meares between 1978 and 1998 of 301.2 ppb in 1980 and 313–314.5 ppb in 1996 on the SIO-1998 N2O scale (Prinn et al., 1990, 2000; Khalil et al., 2002). The N2O scale difference between SIO-1998 and NOAA-06 is minimal (Hall et al., 2007). Additional measurements by the Advanced Global Atmospheric Gases Experiment (AGAGE) and NOAA/ESRL (on the SIO-1998 N2O and NOAA-06 N2O scales, respectively) are reported from comparable sample locations. Trinidad Head, CA (41∘ N, 121∘ W), Mace Head, Ireland (53∘ N, 10∘ W), and Niwot Ridge, CO (40∘ N, 106∘ W), all measure ∼313 ppb in 1996 (Prinn et al., 2000; Hall et al., 2007). Together, these comparisons indicate that the N2O in the archived samples has stored well.
The measured SF6 mixing ratio in archived Cape Meares samples is determined to be 0.85±0.03 ppt (1σ) in 1980 and increases to a mixing ratio of 3.83±0.03 ppt (1σ) in 1996. Cape Meares does not have previously reported measurements of SF6 to compare with directly. Measurements of SF6 from Trinidad Head, CA, are reported to be ∼0.85 ppt in 1980 and ∼3.73 ppt in 1996 on the SIO-2005 SF6 scale (Rigby et al., 2010). To convert to the NOAA-06 SF6 scale, values measured on the SIO-2005 SF6 scale are divided by a conversion factor of 0.9991 (Hall et al., 2014). In 1996, values of 3.87, 3.87, and 3.78 ppt are reported for Alert, Canada (82∘ N, 62∘ W), Barrow, AK (71∘ N, 157∘ W), and Niwot Ridge, CO, respectively, on the NOAA-06 SF6 scale (Hall et al., 2011). At these SF6 mixing ratios, the difference between the NOAA-06 scale and the NOAA-14 scale is minimal. Cape Meares SF6 measured values compare well with these Northern Hemisphere locations.
In the Northern Hemisphere, maximum background mixing ratio measurements of SF6 are reported from mid-to-high latitudes (Geller et al., 1997). For the year 1994, measurements from Fraserdale, Canada (50∘ N, 82∘ W), are reported to be 0.14 ppt higher than samples measured from Izaña, Tenerife (28∘ N, 16∘ W) (Maiss et al., 1996). This difference is explained by the vast majority of SF6 emissions coming from the midlatitudes in the Northern Hemisphere (Maiss and Brenninkmeijer 1998; Levin et al., 2010; Rigby et al., 2010). The measured SF6 mixing ratios from Cape Meares, also a midlatitude Northern Hemisphere site, appear to fit in well with the expected meridional gradient when comparing to previously mentioned reported values.
Southern Hemisphere measurements of SF6 from archived atmospheric samples from Cape Grim, Tasmania (41∘ S, 145∘ E), and Neumayer, Antarctica (70∘ S, 8∘ W), are ∼0.6–0.7 ppt in 1980 and ∼3.4–3.5 ppt in 1996 on the SIO-2005 and University of Heidelberg SF6 scales (Levin et al., 2010, Rigby et al., 2010). As with the SIO-2005 SF6 scale, the NOAA-06 and University of Heidelberg scale differences are small. To convert to the NOAA-06 SF6 scale, values measured on the University of Heidelberg SF6 scale are divided by a conversion factor of 0.9954 (Hall et al., 2014). Including a scale correction, Cape Meares SF6 measurements are higher than Cape Grim and Neumayer during this period by 0.2–0.4 ppt. Much or all of this difference can be explained by an interhemispheric north–south difference of 0.3–0.4 ppt (Levin et al., 2010).
3.2 Growth rate in N2O and SF6
The mean secular trend between 1978 and 1996 for N2O and SF6 is 0.78±0.03 ppb yr−1 (95 % CI) and 0.17±0.01 ppt yr−1 (95 % CI), respectively, and is determined by applying a linear fit to deseasonalized data over this time period. These trends translate to annual increases of ∼0.25 % and ∼0.1 % for N2O and SF6, respectively. Annual trends for N2O and SF6 at Cape Meares, Oregon, are determined from the derivative of the deseasonalized localized regression (Fig. 5c, d). Uncertainty bands are generated from regressions of bootstrapped variability. Data points represent the mean annual trend with error bars equal to ±1σ of the trend over the year.
The mean annual trend in N2O (Fig. 5c) ranges between 0.6 and 1.2 ppb yr−1. All years between 1980 and 1996 show a positive rate of change significant at the 95 % confidence level. The uncertainty in the annual trend is smallest in the early 1980s, at ±0.15 ppb yr−1 (95 % CI), where the largest amount of data is available (∼50 % of samples are between 1980 and 1985). After 1985, uncertainty in the annual trend becomes ±0.5 ppb yr−1 (95 % CI). This relatively large uncertainty results in an annual growth rate that is statistically indistinguishable between years.
A previously reported secular trend of N2O found between 1978 and 1998 for Cape Meares is 0.74±0.02 ppb yr−1, which is indistinguishable from our result (Prinn et al., 2000). The global secular trend of N2O for the period from 1985 to 1996 reported by Khalil et al. (2002) is 0.69±0.03 ppb yr−1, which is also compatible with our trend at Cape Meares.
The SF6 annual trend (Fig. 5d) from the Cape Meares analysis increases from 0.07±0.03 ppt yr−1 (95 % CI) in 1980 to 0.26±0.05 ppt yr−1 (95 % CI) in 1994. The average rate of change in the growth rate (second derivative of mole fraction vs. time) over this period is 0.014 ppt yr−2. The increase in the growth rate over this period is statistically significant at high levels of confidence (95 %). After 1994, we measure a decrease in the growth rate, although this decline is not statistically significant at high levels of confidence over this short time interval.
Comparable trends in SF6 measured at other locations are available for the mid-1990s. The average global growth rate of SF6 in 1994 was reported at 0.23 ppt yr−1 in the Northern Hemisphere (Maiss et al., 1996). Alert, Canada, and Izaña, Tenerife, are observed to have maximum trends of 0.26 ppt yr−1 in mid-1994 and at the beginning of 1995 (Levin et al., 2010), respectively, which are compatible with results presented here. This localized maximum in the growth rate is also present in some Southern Hemisphere observations of SF6 at a similar time; Neumayer, Antarctica shows a maximum trend of 0.25 ppt yr−1 in 1995–1996 (Levin et al., 2010). This finding is consistent with a peak in SF6 emissions as reported by the European Database for Global Atmospheric Research (EDGAR, v4.2).
Another feature observed in the SF6 trend from Cape Meares is a local maximum in the growth rate near 1987 (Fig. 5d). Notably, however, not all datasets agree. The growth rate reported from Neumayer, Antarctica, has this feature during a similar period (Levin et al., 2010), but the trend reported at Cape Grim, Tasmania, does not show this local maximum (Rigby et al., 2010). Due to the large uncertainty from the few archived samples available during that time period, this local maximum is not statistically distinguishable from surrounding years at high levels of confidence in the Cape Meares analysis; thus, this result is merely suggestive. Additional evidence is needed to corroborate this finding.
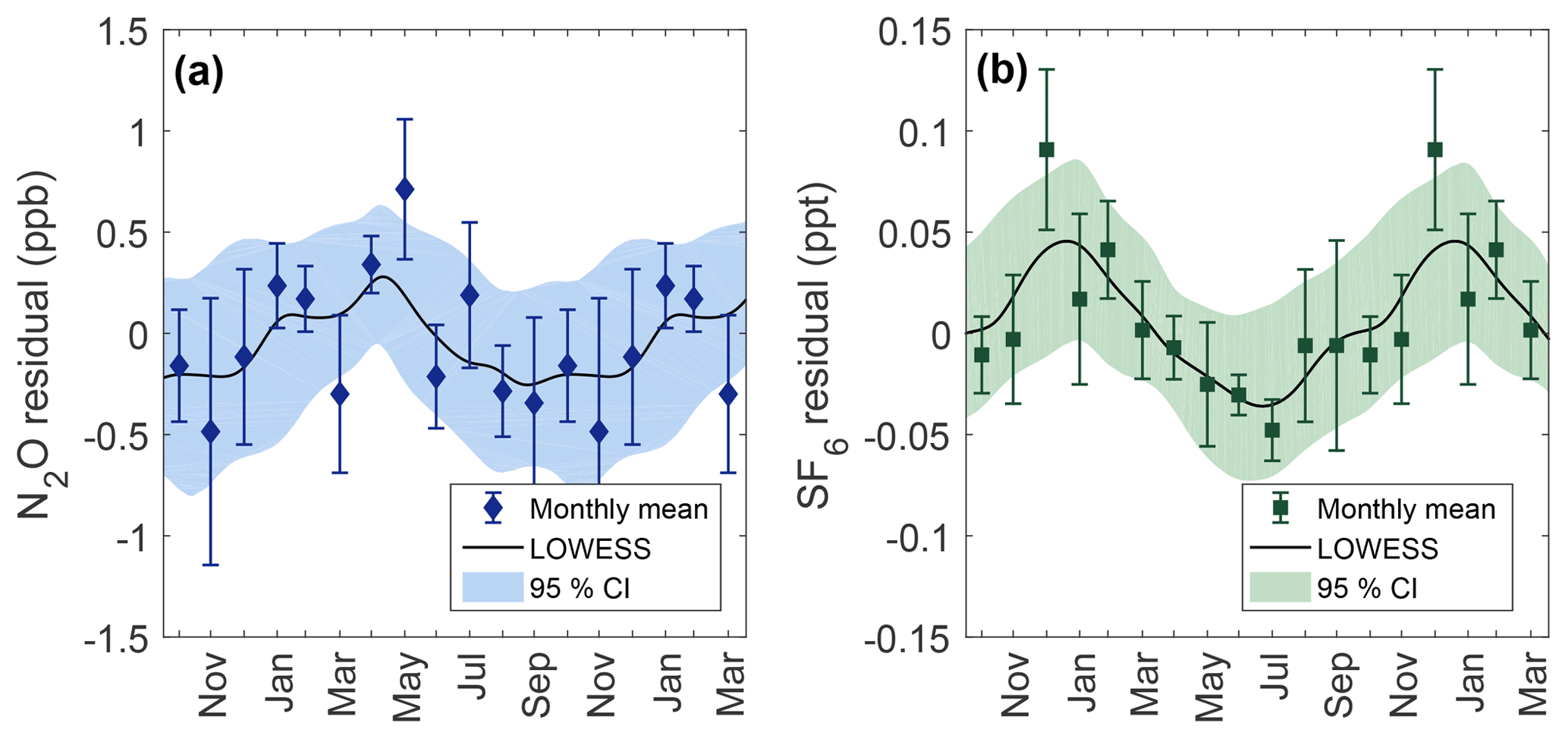
Figure 6Seasonality for N2O (a) and SF6 (b) calculated from the residuals of observed data points to the secular trend. The black line is a LOWESS fit to residuals with a smoothing window of 1 month. Data points show the observed monthly mean residual after binning by month with error bars representing the standard error within the month. Shaded areas are 95 % CI calculated from 1000 bootstrapped LOWESS fits while including the measurement uncertainty to each data point.
3.3 Seasonality in the N2O and SF6 mole fraction
Seasonal behavior for N2O and SF6 are shown in Fig. 6 and are determined from residuals to the secular trend. The N2O seasonal cycle at Cape Meares shows a maximum near April and May of 0.3 ppb and an extended minimum from September through December of −0.4 ppb. Although there is considerable uncertainty surrounding monthly means, the difference between the spring maximum and fall minimum is statistically robust at high levels of confidence (two-sample KS test, p value = 0.003).
The seasonal amplitude matches well with previously reported Northern Hemisphere magnitudes of ±0.4 ppb (Liao et al., 2004). Other midlatitude Northern Hemisphere sites also show a seasonal phase similar to that observed at Cape Meares. N2O seasonality reported at Mace Head, Ireland, has a maximum near April and a minimum near August and September (Nevison et al., 2004; Jiang et al., 2007), and Trinidad Head, CA, seasonality has a maximum near late May and a broad minimum from September to January (Nevison et al., 2007).
In general, the N2O seasonal amplitude is known to vary strongly with latitude, e.g., 0.29 ppb at the South Pole (90∘ S, 102∘ W) and 1.15 ppb at Alert, Canada (Jiang et al., 2007). This is attributed in part to the stronger branch of the Brewer–Dobson circulation in the Northern Hemisphere which also explains the high-latitude minimums in late-summer months related to the influx of N2O-depleted air from the stratosphere during the spring (Liao et al., 2004; Nevison et al., 2004). Aside from atmospheric circulation, N2O seasonality may also be influenced by regional sources. Lueker et al. (2003) suggested that local maximums at Trinidad Head may reflect the influence of strong coastal upwelling. Similarly located in the eastern Pacific, Cape Meares may also be subject to coastal upwelling influences. Isotopic analysis or modeling of transport effects and source influence would be useful to help interpret seasonal behavior of N2O at Cape Meares.
Seasonality for SF6 shows a maximum between December and February of 0.04 ppt and a minimum near July of −0.03 ppt. The difference between the winter maximum and summer minimum is statistically significant (two-sample KS test, p value = 0.004). SF6 seasonality has not previously been reported for Cape Meares.
Some seasonality in Northern Hemisphere observations of SF6 is reported in the literature at select locations. Barrow, AK, has a minimum in September and October with a broad maximum from December to June (Patra et al., 2009). Alert, Canada, shows a strong minimum in October, although a maximum is not clearly defined (Wilson et al., 2014). Continental sites such as Niwot Ridge show large interannual variability (IAV) but have little distinguishable seasonality (Patra et al., 2009).
SF6 seasonality at Cape Grim has been reported to have an amplitude of ±0.01 ppt with a maximum in September and October and a minimum near February (Nevison et al., 2007; Wilson et al., 2014). The seasonality phase of Cape Grim is nearly antiphase of the Cape Meares results reported here, although the amplitude is a factor of 4 smaller at Cape Grim. Similar to N2O, the seasonal amplitude is expected to be larger in the Northern Hemisphere than in the Southern Hemisphere (Nevison et al., 2007). Because sources of SF6 are aseasonal and sinks are essentially zero in the troposphere; the driving force behind the observed seasonality in SF6 is considered to be atmospheric transport (Patra et al., 2009). Processes such as convection, boundary layer mixing, stratosphere–troposphere exchange (STE), and shifts in the intertropical convergence zone can potentially influence the observed seasonality at a location. Seasonal transport from STE adds relatively depleted SF6 air into the troposphere from the stratosphere. The seasonal phase of SF6 observed at Cape Meares closely reflects seasonality phasing observed in CFCs in the Northern Hemisphere driven by STE (Liang et al., 2008). Modeling atmospheric transport effects on SF6 at Cape Meares could help confirm amplitude and phase reported here.
We measured 159 samples from the OHSU-PSU air archive from Cape Meares, Oregon (45.5∘ N, 124.0∘ W), for N2O and SF6 mole fraction using GC-µECD spanning the period from April 1978 to December 1996. The GC-µECD system is designed to be fully automated, and is capable of running multiple pressurized samples per run. Measurement precision of N2O and SF6 is 0.16 % and 1.1 %, respectively. Sample mixing ratios were also corrected for detector response nonlinearity when measured against our reference gas. The linearity correction was found to be 0.14 ppb ppb−1 and 0.03 ppt ppt−1 for N2O and SF6, respectively.
The analysis of archived air samples gives a mole fraction of N2O in 1980 of 301.5±0.3 ppb (1σ), which rises to 313.5±0.3 ppb (1σ) in 1996. The average growth rate over this period is 0.78±0.03 ppb yr−1 (95 % CI). Seasonality shows a peak amplitude of 0.3 ppb near April and a minimum amplitude of −0.4 ppb near November and is statistically robust. Our measurements of N2O were found to match well with previously reported values for Cape Meares and other comparable Northern Hemisphere midlatitude locations.
For SF6, the mixing ratio in 1980 is found to be 0.85±0.03 ppt (1σ), increasing to 3.83±0.03 ppt (1σ) in 1996. The average growth rate over this period is 0.17±0.01 ppt yr−1 (95 % CI). Seasonality shows a peak amplitude of 0.04 ppb near January and a a minimum amplitude of −0.03 ppt near July. There are no previous reported measurements of SF6 from Cape Meares to compare against directly. SF6 measurements compare well to other Northern Hemisphere measurements from Levin et al. (2010), Rigby et al. (2010), and Hall et al. (2011) over similar time periods when including spatial variability. From these N2O and SF6 measurements, we can conclude the sample integrity is robust within the OHSU-PSU air archive from Cape Meares, Oregon. The resulting dataset of SF6, in particular, contributes to a better characterization of historic SF6 growth rate and its atmospheric variability over this period of dramatic growth.
A supplementary dataset of N2O and SF6 mole fractions at Cape Meares, Oregon, measured for this work from the OSHU-PSU air archive are available to the scientific community (upon publication) and may be obtained by contacting the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-8967-2019-supplement.
Both authors (TR and AR) worked closely together in the development and implementation of the GC technique used to make the mole fraction measurements of N2O and SF6. Data curation and analysis were also completed by both authors. The original draft of this paper was prepared by TR; AR was responsible for the review and editing of the paper.
The authors declare that they have no conflict of interest.
This study was supported by the US National Science Foundation (Atmospheric and Geospace Sciences grant no. 0952307). The authors would like to recognize Johnathan Radda for his assistance developing the GC-µECD technique used to measure the samples, Reinhold A. Rasmussen for help with establishing the OHSU-PSU air archive, and Christopher Butenhoff and M. Aslam Khan Khalil for their assistance with data analysis and interpretation.
This research has been supported by the US National Science Foundation (grant no. 0952307).
This paper was edited by Gabriele Stiller and reviewed by Andreas Engel, Brad Hall, Eric Ray, and one anonymous referee.
Ciais, P., Sabine, C., Bala, G., Bopp, L., Brovkin, V., Canadell, J., Chhabra, A., DeFries, R., Galloway, J., Heimann, M., Jones, C., Le Quéré, C., Myneni, R. B., Piao, S., and Thornton, P.: Carbon and Other Biogeochemical Cycles, in: Climate Change 2013: The Physical Science Basis, Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, edited by: Stocker, T. F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S. K., Boschung, J., Nauels, A., Xia, Y., Bex, V., and Midgley, P. M., Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 465–570, 2013.
Crutzen, P.: The influence of nitrogen oxides on the atmospheric ozone content, Q. J. Roy. Meteor. Soc., 96, 320–325, 1970.
Cleveland, W. S. and Devlin, S. J.: Locally weighted regression: An approach to regression analysis by local fitting, J. Am. Stat. Assoc., 83, 596–610, 1988.
Dlugokencky, E. J., Hall, B. D., Montzka, S. A., Dutton, G., Mühle, J., and Elkins, J. W.: Atmospheric composition: Long-lived Greenhouse Gases, in: State of the Climate in 2017, B. Am. Meteorol. Soc., 99, 46–49, https://doi.org/10.1175/2018BAMSStateoftheClimate.1, 2018.
EDGAR, Emission Database for Global Atmospheric Research (EDGAR): European Commission, Joint Research Centre (JRC)/Netherlands Environmental Assessment Agency (PBL), release version 4.2., available at: http://edgar.jrc.ec.europa.eu (last access: 28 December 2018), 2013.
Geller, L., Elkins, J., Lobert, J., Clarke, A., Hurst, D., Butler, J., and Myers, R.: Tropospheric SF6: Observed latitudinal distribution and trends, derived emissions and interhemispheric exchange time, Geophys. Res. Lett., 24, 675–678, 1997.
Hall, B. D., Dutton, G. S., and Elkins, J. W.: The NOAA nitrous oxide standard scale for atmospheric observations, J. Geophys. Res.-Atmos., 112, D09305, https://doi.org/10.1029/2006JD007954, 2007.
Hall, B. D., Dutton, G. S., Mondeel, D. J., Nance, J. D., Rigby, M., Butler, J. H., Moore, F. L., Hurst, D. F., and Elkins, J. W.: Improving measurements of SF6 for the study of atmospheric transport and emissions, Atmos. Meas. Tech., 4, 2441–2451, https://doi.org/10.5194/amt-4-2441-2011, 2011.
Hall, B. D., Engel, A., Mühle, J., Elkins, J. W., Artuso, F., Atlas, E., Aydin, M., Blake, D., Brunke, E.-G., Chiavarini, S., Fraser, P. J., Happell, J., Krummel, P. B., Levin, I., Loewenstein, M., Maione, M., Montzka, S. A., O'Doherty, S., Reimann, S., Rhoderick, G., Saltzman, E. S., Scheel, H. E., Steele, L. P., Vollmer, M. K., Weiss, R. F., Worthy, D., and Yokouchi, Y.: Results from the International Halocarbons in Air Comparison Experiment (IHALACE), Atmos. Meas. Tech., 7, 469–490, https://doi.org/10.5194/amt-7-469-2014, 2014.
Ishijima, K., Sugawara, S., Kawamura, K., Hashida, G., Morimoto, S., Murayama, S., Aoki, S., and Nakazawa, T.: Temporal variations of the atmospheric nitrous oxide concentration and its δ15N and δ18O for the latter half of the 20th century reconstructed from firn air analyses, J. Geophys. Res.-Atmos., 112, D03305, https://doi.org/10.1029/2006JD007208, 2007.
Jiang, X., Ku, W. L., Shia, R.-L., Li, Q., Elkins, J. W., Prinn, R. G., and Yung, Y. L.: Seasonal cycle of N2O: Analysis of data, Global Biogeochem. Cy., 21, GB1006, https://doi.org/10.1029/2006GB002691, 2007.
Khalil, M. A. K. and Rasmussen, R. A.: Increase and seasonal cycles of nitrous oxide in the earth's atmosphere, Tellus, 35, 161–169, 1983.
Khalil, M. A. K., Rasmussen, R. A., and Shearer, M. J.: Atmospheric nitrous oxide: patterns of global change during recent decades and centuries, Chemosphere, 47, 807–821, 2002.
Kovács, T., Feng, W., Totterdill, A., Plane, J. M. C., Dhomse, S., Gómez-Martín, J. C., Stiller, G. P., Haenel, F. J., Smith, C., Forster, P. M., García, R. R., Marsh, D. R., and Chipperfield, M. P.: Determination of the atmospheric lifetime and global warming potential of sulfur hexafluoride using a three-dimensional model, Atmos. Chem. Phys., 17, 883–898, https://doi.org/10.5194/acp-17-883-2017, 2017.
Levin, I. and Hesshaimer, V.: Refining of atmospheric transport model entries by the globally observed passive tracer distributions of 85 krypton and sulfur hexafluoride (SF6), J. Geophys. Res.-Atmos., 101, 16745–16755, https://doi.org/10.1029/96JD01058, 1996.
Levin, I., Naegler, T., Heinz, R., Osusko, D., Cuevas, E., Engel, A., Ilmberger, J., Langenfelds, R. L., Neininger, B., Rohden, C. v., Steele, L. P., Weller, R., Worthy, D. E., and Zimov, S. A.: The global SF6 source inferred from long-term high precision atmospheric measurements and its comparison with emission inventories, Atmos. Chem. Phys., 10, 2655–2662, https://doi.org/10.5194/acp-10-2655-2010, 2010.
Liao, T., Camp, C., and Yung, Y.: The seasonal cycle of N2O, Geophys. Res. Lett., 31, L17108, https://doi.org/10.1029/2004GL020345, 2004.
Liang, Q., Stolarski, R., Douglass, A., Newman, P., and Nielsen, J.: Evaluation of emissions and transport of CFCs using surface observations and their seasonal cycles and the GEOS CCM simulation with emissions-based forcing, J. Geophys. Res.-Atmos., 113, D14302, https://doi.org/10.1029/2007JD009617, 2008.
Lueker, T., Walker, S., Vollmer, M., Keeling, R., Nevison, C., Weiss, R., and Garcia, H: Coastal upwelling air-sea fluxes revealed in atmospheric observations of O2∕N2, CO2, and N2O, Geophys. Res. Lett., 30, 1292, https://doi.org/10.1029/2002GL016615, 2003.
Maiss, M. and Brenninkmeijer, C.: Atmospheric SF6: Trends, sources, and prospects, Environ. Sci. Technol., 3, 3077–3086, 1998.
Maiss, M., Steele, L. P., Francey, R., Fraser, P., Langenfelds, R., Trivett, N., and Levin, I.: Sulfur hexafluoride – a powerful new atmospheric tracer, Atmos. Environ., 30, 1621–1629, https://doi.org/10.1016/1352-2310(95)00425-4, 1996.
Meinshausen, M., Vogel, E., Nauels, A., Lorbacher, K., Meinshausen, N., Etheridge, D. M., Fraser, P. J., Montzka, S. A., Rayner, P. J., Trudinger, C. M., Krummel, P. B., Beyerle, U., Canadell, J. G., Daniel, J. S., Enting, I. G., Law, R. M., Lunder, C. R., O'Doherty, S., Prinn, R. G., Reimann, S., Rubino, M., Velders, G. J. M., Vollmer, M. K., Wang, R. H. J., and Weiss, R.: Historical greenhouse gas concentrations for climate modelling (CMIP6), Geosci. Model Dev., 10, 2057–2116, https://doi.org/10.5194/gmd-10-2057-2017, 2017.
Myhre, G., Shindell, D., Bréon, F.-M., Collins, W., Fuglestvedt, J., Huang, J., Koch, D., Lamarque, J.-F., Lee, D., Mendoza, B., Nakajima, T., Robock, A., Stephens, G., Takemura, T., and Zhang, H.: Anthropogenic and Natural Radiative Forcing, in: Climate Change 2013: The Physical Science Basis, Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, in: Stocker, T. F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S. K., Boschung, J., Nauels, A., Xia, Y., Bex, V., and Midgley, P. M., Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2013.
Nevison, C., Kinninson, D., and Weiss, R.: Stratospheric influences on the tropospheric seasonal cycles of nitrous oxide and chlorofluorocarbons, Geophys. Res. Lett., 31, L20103, https://doi.org/10.1029/2004GL020398, 2004.
Nevison, C., Mahowald, N., Weiss, R., and Prinn, R.: Interannual and seasonal variability in atmospheric N2O, Global Biogeochem. Cy., 21, GB3017, https://doi.org/10.1029/2006GB002755, 2007.
Olivier, J., Van Aardenne, J., Dentener, F., Pagliari, V., Ganzeveld, L., and Peters, J.: Recent trends in global greenhouse gas emissions:regional trends 1970–2000 and spatial distribution of key sources in 2000, Environ. Sci., 2, 81–99, https://doi.org/10.1080/15693430500400345, 2005.
Park, S., Croteau, P. Boering, K. A., Etheridge, D. M., Ferretti, D., Fraser, P. J., Kim, K-R., Krummel, P. B., Langenfelds, R. L., van Ommen, T. D., Steele, L. P., and Trudinger, C. M.: Trends and seasonal cycles in the isotopic composition of nitrous oxide since 1940, Nat. Geosci., 5, 261–265, https://doi.org/10.1038/NGEO1421, 2012.
Patra, P. K., Takigawa, M., Dutton, G. S., Uhse, K., Ishijima, K., Lintner, B. R., Miyazaki, K., and Elkins, J. W.: Transport mechanisms for synoptic, seasonal and interannual SF6 variations and “age” of air in troposphere, Atmos. Chem. Phys., 9, 1209–1225, https://doi.org/10.5194/acp-9-1209-2009, 2009.
Prather, M. J., Holmes, C. D., and Hsu, J.: Reactive green house gas scenarios: Systematic exploration of uncertainties and the role of atmospheric chemistry, Geophys. Res. Lett., 39, L09803, https://doi.org/10.1029/2012GL051440, 2012.
Prather, M. J., Hsu, J., DeLuca, N. M., Jackman, C. H., Oman, L. D., Douglass, A. R., Fleming, E. L., Strahan, S. E., Steenrod, S. D., Søvde, O. A., Isaksen, I. S. A., Froidevaux, L., and Funke, B.: Measuring and modeling the lifetime of nitrous oxide including its variability, J. Geophys. Res.-Atmos., 120, 5693–5705, https://doi.org/10.1002/ 2015JD023267, 2015.
Prinn, R., Cunnold, D., Rasmussen, R., Simmonds, P., Alyea, F., Crawford, A., Fraser, P., and Rosen, R.: Atmospheric emissions and trends of nitrous oxide deduced from 10 years of ALE-GAGE data, J. Geophys. Res., 95, 18369–18385, 1990.
Prinn, R., Weiss, R., Fraser, P., Simmonds, P., Cunnold, D., Alyea, F., O'Doherty, S., Salameh, P., Miller, B. R., Huang, J., Wang, R. H. J., Hartley, D. E., Harth, C., Steele, L. P., Sturrock, G., Midgley, P. M., and McCulloch, A.: A history of chemically and radiatively important gases in air deducted from ALE/GAGE/AGAGE, J. Geophys. Res., 105, 17751–17792, https://doi.org/10.1029/2000JD900141, 2000.
Prinn, R. G., Weiss, R. F., Arduini, J., Arnold, T., DeWitt, H. L., Fraser, P. J., Ganesan, A. L., Gasore, J., Harth, C. M., Hermansen, O., Kim, J., Krummel, P. B., Li, S., Loh, Z. M., Lunder, C. R., Maione, M., Manning, A. J., Miller, B. R., Mitrevski, B., Mühle, J., O'Doherty, S., Park, S., Reimann, S., Rigby, M., Saito, T., Salameh, P. K., Schmidt, R., Simmonds, P. G., Steele, L. P., Vollmer, M. K., Wang, R. H., Yao, B., Yokouchi, Y., Young, D., and Zhou, L.: History of chemically and radiatively important atmospheric gases from the Advanced Global Atmospheric Gases Experiment (AGAGE), Earth Syst. Sci. Data, 10, 985–1018, https://doi.org/10.5194/essd-10-985-2018, 2018.
Ray, E., Moore, F. L., Elkins, J. W., Rosenlof, K. H., Laube, J. C., Röckmann, T., Marsh, D. R., and Andrews, A. E.: Quantification of the SF6 Lifetime Based on Mesospheric Loss Measured in the Stratospheric Polar Vortex, J. Geophys. Res.-Atmos., 122, 4626–4638, https://doi.org/10.1002/2016JD026198, 2017.
Rice, A. L., Butenhoff, C. L., Teama, D. G., Röger, F. H., Khalil, M. A. K., and Rasmussen, R. A.: Atmospheric methane isotopic record favors fossil sources flat in 1980s and 1990s with recent increase, P. Natl. Acad. Sci. USA, 113, 10791–10796, 2016.
Rigby, M., Mühle, J., Miller, B. R., Prinn, R. G., Krummel, P. B., Steele, L. P., Fraser, P. J., Salameh, P. K., Harth, C. M., Weiss, R. F., Greally, B. R., O'Doherty, S., Simmonds, P. G., Vollmer, M. K., Reimann, S., Kim, J., Kim, K.-R., Wang, H. J., Olivier, J. G. J., Dlugokencky, E. J., Dutton, G. S., Hall, B. D., and Elkins, J. W.: History of atmospheric SF6 from 1973 to 2008, Atmos. Chem. Phys., 10, 10305–10320, https://doi.org/10.5194/acp-10-10305-2010, 2010.
Saikawa, E., Prinn, R. G., Dlugokencky, E., Ishijima, K., Dutton, G. S., Hall, B. D., Langenfelds, R., Tohjima, Y., Machida, T., Manizza, M., Rigby, M., O'Doherty, S., Patra, P. K., Harth, C. M., Weiss, R. F., Krummel, P. B., van der Schoot, M., Fraser, P. J., Steele, L. P., Aoki, S., Nakazawa, T., and Elkins, J. W.: Global and regional emissions estimates for N2O, Atmos. Chem. Phys., 14, 4617–4641, https://doi.org/10.5194/acp-14-4617-2014, 2014.
Schmidt, M., Glatzel-Mattheier, H., Sartorius, H., Worthy, D. E., and Levin, I.: Western European N2O emissions: A top-down approach based on atmospheric observations, J. Geophys. Res., 106, 5507–5515, 2001.
Snider, D. M., Venkiteswaran, J. J., Schiff, S. L., and Spoelstra, J.: From the ground up: Global nitrous oxide sources are constrained by stable isotope values, PLOS ONE, 10, E0118954, https://doi.org/10.1371/journal.pone.011895, 2015.
Stocker, B. D., Roth, R., Joos, F., Spahni, R., Steinacher, M., Zeahle, S., Bouwman, L., and Prentice, I. C.: Multiple greenhouse-gas feedbacks from the land biosphere under future climate change scenarios, Nat. Clim. Change, 3, 666–672, https://doi.org/10.1038/nclimate1864, 2013.
Sulfur Hexafluoride (SF6) WMO Scale, available at: https://www.esrl.noaa.gov/gmd/ccl/sf6_scale.html, last access: 17 June 2018.
Vollmer, M. K., Young, D., Trudinger, C. M., Mühle, J., Henne, S., Rigby, M., Park, S., Li, S., Guillevic, M., Mitrevski, B., Harth, C. M., Miller, B. R., Reimann, S., Yao, B., Steele, L. P., Wyss, S. A., Lunder, C. R., Arduini, J., McCulloch, A., Wu, S., Rhee, T. S., Wang, R. H. J., Salameh, P. K., Hermansen, O., Hill, M., Langenfelds, R. L., Ivy, D., O'Doherty, S., Krummel, P. B., Maione, M., Etheridge, D. M., Zhou, L., Fraser, P. J., Prinn, R. G., Weiss, R. F., and Simmonds, P. G.: Atmospheric histories and emissions of chlorofluorocarbons CFC-13 (CClF3), ΣCFC-114 (C2Cl2F4), and CFC-115 (C2ClF5), Atmos. Chem. Phys., 18, 979–1002, https://doi.org/10.5194/acp-18-979-2018, 2018.
Wilson, C., Chipperfield, M. P., Gloor, M., and Chevallier, F.: Development of a variational flux inversion system (INVICAT v1.0) using the TOMCAT chemical transport model, Geosci. Model Dev., 7, 2485–2500, https://doi.org/10.5194/gmd-7-2485-2014, 2014.