the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 12 Jul 2019
| 12 Jul 2019
Temperature effects on sulfuric acid aerosol nucleation and growth: initial results from the TANGENT study
Lee Tiszenkel
Chris Stangl
Justin Krasnomowitz
Qi Ouyang
Huan Yu
Michael J. Apsokardu
Murray V. Johnston
New particle formation (NPF) consists of two steps: nucleation and subsequent growth. At present, chemical and physical mechanisms that govern these two processes are not well understood. Here, we report initial results obtained from the TANGENT (Tandem Aerosol Nucleation and Growth Environment Tube) experiments. The TANGENT apparatus enables us to study these two processes independently. The present study focuses on the effects of temperature on sulfuric acid nucleation and further growth. Our results show that lower temperatures enhance both the nucleation and growth rate. However, under temperatures below 268 K the effects of temperature on the nucleation rate become less significant and the nucleation rate becomes less dependent on relative humidity, indicating that particle formation in the conditions of our flow tube takes place via barrierless nucleation at lower temperatures. We also examined the growth of newly formed particles under differing temperature conditions for nucleation and further growth. Our results show that newly nucleated clusters formed at low temperatures can indeed survive evaporation and grow in a warmer environment in the presence of SO2 and ozone and potentially other contaminant vapors. These results imply that some heterogeneous reactions involving nanoparticles affect nucleation and growth of newly formed particles.
- Article
(2356 KB) - Full-text XML
- BibTeX
- EndNote





Atmospheric nanoparticles affect human health and air quality. Newly formed particles can contribute to approximately 30 %–70 % of cloud condensation nuclei (CCN) in the atmosphere (Merikanto et al., 2009; Wang and Penner, 2009; Yu and Luo, 2009; Gordon et al., 2017). New particle formation (NPF) takes place via two steps: initial nucleation (formation of critical clusters) and subsequent growth of nucleated clusters (Kulmala et al., 2013). At present, chemical and physical mechanisms that govern these two processes, as well as the identity of chemical precursors involved in these processes, are still not well understood (Zhang et al., 2012; Yu et al., 2017b; Lee et al., 2019). Current global models fail to represent NPF in the atmosphere for a wide range of temperature and relative humidity (RH) conditions and for different emissions of biogenic and anthropogenic precursors, due to a lack of observations. For example, models predict frequent NPF during the summer in mixed deciduous forests in the United States (Yu et al., 2015), while field observations show an absence of NPF in this region (Kanawade et al., 2011; Lee et al., 2016). Also, current NPF theories are unable to explain the frequent NPF observed in extremely polluted megacities (Yu et al., 2017b; Kulmala et al., 2017).
Temperature and RH are the key thermodynamic properties of aerosol formation and growth (Seinfeld and Pandis, 2016). Nucleation rate (J) is a function of temperature and the Gibbs free energy barrier of cluster formation. At lower temperatures, Gibbs free energy barriers become lower and critical cluster diameters become smaller. Condensational species can affect aerosol growth differently at different temperatures because their saturation vapor pressures are dependent on temperature. For water, RH is the same as saturation ratio and chemical activity. Laboratory experiments of aerosol nucleation and growth as a function of temperature and RH remain limited, although these observations are critical in global models to correctly parameterize NPF under various altitude, latitude and seasonal conditions. Aerosol nucleation experiments are extremely challenging due to various experimental difficulties, including contamination of base compounds (Erupe et al., 2011; Yu et al., 2012). At present there is a lack of consistency between different experiments from different groups and even from the same groups using the same experimental setup (Lee et al., 2019). The lack of reproducibility and consistency of the nucleation experiments greatly hinders our understanding of nucleation mechanisms.
Duplissy et al. (2016) conducted studies of binary homogeneous nucleation of sulfuric acid and water, with and without ions in the CLOUD (Cosmics Leaving OUtdoor Droplets) chamber at different temperatures ranging from 207 to 299 K and RH between 11 % and 58 %. At lower temperatures both ion nucleation and neutral binary nucleation are at the kinetic regime, while at higher temperatures J is strongly dependent on [H2SO4], indicating there are high Gibbs free energy barriers at these temperatures. At the nucleation regime, nucleation rates are strongly dependent on RH. Kürten et al. (2016) reported the temperature dependence of ternary nucleation in the CLOUD chamber, at temperatures from 208 to 298 K, [H2SO4] between 105 and 109 cm−3 and [NH3] up to ∼1400 pptv. At 208 K, J reached the threshold of 1 cm−3 s−1 at [H2SO4] of cm−3 for the binary case and at [H2SO4] of cm−3 for the ternary case with [NH3] of 5 pptv. At 298 K, RH has strong effects on the measured J for both charged and neutral ternary nucleation because this increase in RH could lead to a displacement of NH3 from the stainless steel walls in the CLOUD chamber and lead to an elevated NH3 background level and consequently to higher J.
Laboratory experiments of growth rates (GRs) of newly nucleated particles are even more sparse. Skrabalova et al. (2014) studied GRs of newly formed particles in a flow tube at temperatures between 283 to 303 K and RH of 1 % and 30 %, designated as “dry” and “wet” conditions, respectively. [H2SO4] was varied between 2×108 and 1.4×1010 cm−3. They found different effects of RH on GR: at [H2SO4] below 109 cm−3, growth is promoted in drier conditions, whereas at [H2SO4] higher than 109 cm−3, growth favors wetter conditions. Yu et al. (2017a) performed flow tube experiments of sulfuric acid aerosol nucleation at temperatures from 248 to 313 K and RH from 1 % to 79 % under minimal base concentrations ([NH3] <23 pptv, methylamine <1.5 pptv and dimethylamine <0.52 pptv). Their study provides, for the first time, the temperature and RH dependence of both J and GR. J shows the following dependence within the experimental conditions:
where RA is relative acidity (or saturation ratio) of sulfuric acid and T is temperature. Their results show that GR is independent of temperature below 290 K but significantly decreases at temperatures above 290 K. RH has a moderate effect on GR.
At present, there is still no clear evidence from atmospheric observations that demonstrates “causal effects” of temperature and RH on NPF (Lee et al., 2019). Recent CLOUD studies have shown that oxygenated organics formed from biogenic volatile organic compounds (BVOCs) can grow newly nucleated particles in a wide range of tropospheric temperatures (Stolzenburg et al., 2018). This is because gas-phase autooxidation reactions involved in the formation of highly oxidized molecules (HOMs) usually have strong temperature dependencies, with higher reaction rates at higher temperatures (Frege et al., 2018), whereas nucleation is favored at lower temperatures.
Another important perspective of temperature effects on NPF is the effects of temperature on evaporation of newly nucleated clusters and nanoparticles that have undergone transport to differing temperature conditions for further growth. Particle concentrations in the free troposphere in marine regions have been shown to be due to sulfuric-acid-driven nucleation of nanoparticles as a result of oxidation of upwardly transported and oceanic dimethylsulfide. These particles then undergo downward transport to become reservoirs of nanoparticles in the boundary layer. This has been observed and modeled by several groups over the previous decades (Russell et al., 1994; Raes, 1995; Clarke, 1993).
The lack of local NPF events in Amazon forests has been a confounding observation for many years (Varanda Rizzo et al., 2018) and yet there are reservoirs of nuclei-mode particles at the surface which do not form typical “banana” plots of aerosol size distributions observed elsewhere (Martin et al., 2016). Understanding the origin of these nuclei-mode particles has been a subject of recent studies. Wang et al. (2016) reported that while NPF does not take place in Amazon forests at the surface level during the dry and wet season, NPF takes place in the colder free troposphere; these newly formed particles can be transported down to the boundary layer to become a reservoir of nanoparticles at the surface. This downward transport process is very similar to the above-mentioned marine boundary layer nanoparticle processes (Russell et al., 1994; Raes, 1995; Clarke, 1993). A subsequent question is thus whether newly nucleated clusters and nanoparticles in the free troposphere can survive evaporation during their transport to the warmer boundary region.
At present, the temperature effects on the growth of newly formed particles crossing different temperature regions have not been examined in a controlled laboratory environment. In order to address this shortcoming in laboratory experiments of this relatively common phenomenon, we have constructed the TANGENT apparatus, consisting of a temperature- and RH-controlled nucleation flow tube that permits study of gas-to-particle conversion over a wide parameter space. This nucleation region is connected to a room temperature growth tube where conditions for further growth of these nanoparticles can differ dramatically from those of their nucleation. With the suite of instruments monitoring precursor gases as well as particle size distributions from sub-3 nm up to CCN sizes at key areas of the flow apparatus, the critical processes that atmospheric nanoparticles undergo can be studied and parameterized with TANGENT.
Here, we present the initial results of the TANGENT experiments conducted during the Intensive Observation Period (IOP) study in June and July 2018. In the present study, we report the temperature effects on J and GR of sub-2 nm particles in the nucleation tube. We also discuss how temperature differences in the nucleation and growth tube affect the potential evaporation of newly nucleated clusters while these clusters are transported from the colder to the warmer temperature region.
2.1 The TANGENT experimental setup
Figure 1 shows the schematic diagram of the TANGENT apparatus. The TANGENT consists of the two flow tubes (FTs) to enable studies of nucleation (FT-1) and subsequent growth (FT-2). The nucleation tube was built by the University of Alabama in Huntsville, and the growth tube was built by University of Delaware. TANGENT allows for nucleation to be observed as a separate process from growth. With a short residence time (45 s), precise temperature control and the ability to produce H2SO4 at concentrations spanning 106 to 109 cm−3, FT-1 is able to nucleate and monitor particles from precursor gases at a variety of conditions. After particles are nucleated in FT-1 where size distributions are recorded, particles are then transported to the growth tube (FT-2), where the particles undergo further growth at room temperature for a longer residence time (4 min) with the ability to precisely control other trace gases in the growth environment. According to our knowledge, this is the first flow tube experimental setup that allows for aerosol nucleation and growth processes to be investigated independently and simultaneously.
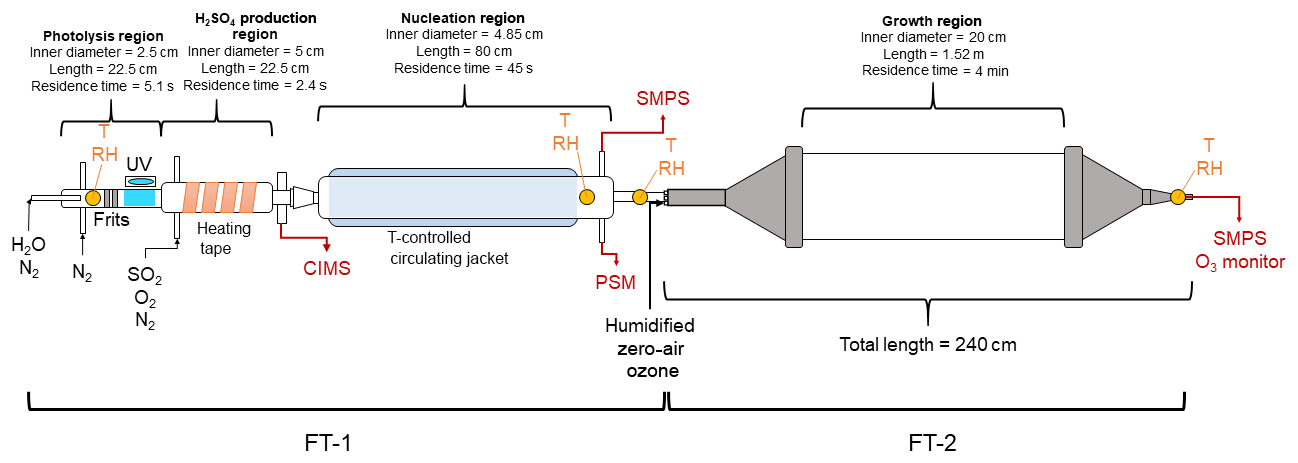
Figure 1Schematic diagram of the TANGENT experimental setup. This setup consists of two flow tubes (FTs). T indicates temperature. FT-1 is used as the nucleation region and FT-2 as the growth region. Table 1 shows the typical experimental conditions used during the 2018 IOP study.
The experimental setup of the nucleation region (FT-1) was based on several previous studies (Benson et al., 2008, 2009, 2011; Erupe et al., 2011; Young et al., 2008; Yu et al., 2012, 2017a). It consists a photolysis region where H2SO4 is generated photochemically and monitored and followed by a temperature-controlled nucleation tube. In the photolysis region, OH radicals were produced via photodissociation of water vapor in a quartz tube using a mercury lamp (Pen-Ray model 11SC-1). The mercury lamp was located in a temperature-controlled enclosure filled with a constant nitrogen flow. UV intensity was adjusted with an aperture over the slit in the enclosure, exposing the quartz tube to the UV lamp. UV intensity was monitored with a cesium iodide (CsI) phototube (Hamamatsu model R5764) and picoammeter (Keithley 6732). Measurements of UV intensity were taken to ensure consistency between experimental trials. SO2, O2 and N2 gases were introduced to the flow tube immediately after the photolysis region. H2SO4 forms from the SO2+OH reaction. Heating tape was applied to the H2SO4 production region to suppress nucleation prior to entering the temperature-controlled nucleation zone. The H2SO4 production region was monitored with a condensation particle counter (CPC, TSI 3776) and particle size magnifier (PSM, Airmodus A09) to ensure no particles were formed before the flow entered the nucleation region.
H2SO4 concentrations at the beginning of the nucleation tube were measured with a nitrate-based chemical ionization mass spectrometer (CIMS), based on Eisele and Tanner (1993), continuously during the experiments. Calibration of [H2SO4] with the nitrate-based CIMS was described previously by Young et al. (2008). The CIMS was operated with an inlet flow of 5.0 slpm and an ion–molecule reaction time of 0.05 s. The lower limit of detection was cm−3.
No base compounds were added, but base compounds were still present in the flow tube as impurities likely generated from deionized water used for H2SO4 production and RH control (Erupe et al., 2011; Yu et al., 2012). NH3 and amines were not measured during the 2018 IOP, but they were measured under very similar experimental conditions during the entire 2017 IOP with an ethanol-based CIMS at the beginning of the nucleation tube (You et al., 2014; Yu and Lee, 2012). Detection limits of NH3 and amines in our CIMS were parts-per-trillion by volume (pptv) or sub-pptv with a 1 min integration, as previously discussed elsewhere (Benson et al., 2010; Erupe et al., 2011; You et al., 2014; Yu and Lee, 2012).
The nucleation tube is an 80 cm long Pyrex glass tube with an inner diameter of 4.85 cm. The temperature of the nucleation tube was controlled with a circulating bath and a water-based potassium formate heat transfer fluid (Dynalene HC-50, Dynalene, Inc.) to adjust the temperature between 258 and 297 K. RH was adjusted by directing some of the dry nitrogen makeup flow through deionized water in a water bubbler. Thus, in our experimental setup, changes in RH in the nucleation tube did not affect the OH radical concentrations in the photolysis region. Temperature and RH probes (CS-215, Campbell Scientific) were used to monitor the conditions at the beginning of the photolysis region, as well as at the end of the nucleation region. An additional temperature and RH probe (Traceable, Fisher Scientific) was applied inside the nucleation tube to confirm the RH. Residence time in the nucleation region (FT-1) was 45 s.
Particle concentrations at the exit of FT-1 were measured with a PSM (Vanhanen et al., 2011). The PSM saturator flow was operated with a 240-step cycle between 0.1 and 0.9 slpm at a rate of 1 s per step, giving saturator-flow-dependent cutoff sizes between 1.26 and 2.85 nm. These cutoffs were resolved to six size bins in an inversion method based on Lehtipalo et al. (2014), producing size distributions with six size bins: 1.26–1.53, 1.53–1.79, 1.79–2.06, 2.06–2.32, 2.32–2.59 and 2.59–2.85 nm. Particle concentrations were also monitored with a scanning mobility particle sizer (SMPS) consisting of a differential mobility analyzer (DMA, TSI 3080) and a CPC (TSI 3776). However, under the typical experimental conditions, particles above 3 nm in diameter did not appear even with the most favorable conditions for nucleation and growth (e.g., high [H2SO4], high RH and low temperature).
During the experiments, [H2SO4] was varied by adjusting the aperture on the mercury lamp housing (hence varying [OH]) at a fixed [SO2], allowing for a range of [H2SO4] spanning roughly 1 order of magnitude for a given dilution of SO2. [H2SO4] was further varied by adjusting the SO2 dilution, allowing for measurements spanning [H2SO4] of 106 to 109 cm−3. The PSM measurements showed that each experimental condition was “stabilized” typically after ∼30 min for a specific set of [H2SO4], RH and temperature.
The photolysis and nucleation tubes were cleaned thoroughly with deionized water, citric acid solution and ethanol and allowed to dry for 24 h while heated to 60 ∘C with pure N2 flowing through the flow tube. Between experiments, the photolysis and nucleation tubes were continuously flushed with dry vaporized liquid nitrogen. A constant flow of N2 was passed through the experimental apparatus at all times during the IOP to ensure that the conditions inside the tube would remain constant and there would be no intrusion of room air.
The nucleated clusters (smaller than 2 nm) were transported to the growth tube (FT-2) for further growth with an extended residence time (4 min). FT-1 and FT-2 were coupled with an 0.2 m stainless steel tube with additional inlet ports for injection of ozone, zero air and SO2. The growth tube was described by Krasnomowitz et al. (2019) and Stangl et al. (2019). The growth tube consists of a 1.52 m long and 0.2 m inner diameter fused quartz tube fitted with stainless steel funnels on each end that reduce the inner diameter down to 0.051 m. The total volume of the tube and entrance and exit funnels is 52.4 L, giving a surface-to-volume ratio of 0.24 cm−1. The 0.2 m straight tube fitting allows carrier and reactant gases to enter the tube via an axial inlet port and continuous flow through the entire length of the reactor during the course of an experiment. The end of the tube was attached to an ozone monitor (Thermo Fisher Scientific 49i), a hygrometer (Traceable, Fisher Scientific) and an SMPS (TSI 3938, 3788).
FT-2 has several ports at the inlet to inject additional gases into the system to observe their effects on further growth of freshly nucleated particles. Ozone was added to FT-2 using the calibration ozone generator on a Thermo Fisher Scientific 49i. Ozone was varied in FT-2 by adjusting the UV intensity of the calibration lamp. The residence time in FT-2 could be changed by varying the flow of zero air into FT-2. During a typical experimental run using both FT-1 and FT-2, conditions would only be changed in FT-2, with temperature, RH and precursor species in FT-1 held constant throughout the entire experiment. The experiments undertaken in this study measured two effects in the system: the effect of changing temperature in the nucleation region as well as the effect of varying ozone (in the copresence of SO2) in the growth region.
2.2 Calculations of nucleation (J) and growth rate (GR)
Calculations of J were made based on Yu et al. (2017a). Briefly, J was calculated according to the following approximation:
where J0 represents the nucleation rate corresponding to the initial sulfuric acid concentration ([H2SO4]0) measured at the beginning of the nucleation tube, Ntot the total number concentration of particles detected at the end of the nucleation region, n the nucleation theorem power and kL the diffusion-limited, pseudo-first-order wall loss coefficient (Hanson and Eisele, 2000). The nucleation theorem power n was experimentally determined for each set of experiments by the linear fit of log Ntot vs. log [H2SO4]0, Our kL was typically 0.01 s−1. Equation (2) allows us to obtain J0 at different [H2SO4]0 using n and Ntot of the formed clusters (all smaller than 2 nm in the present study).
To calculate GR, the critical cluster size was determined experimentally, with the critical cluster size corresponding to the y intercept of the linear fit between the mean particle diameter, Dp, and the [H2SO4]0 (e.g., Fig. 3). Dp was obtained using the inversion of the PSM size distribution measured at the end of FT-1. GR was calculated by the difference between the critical size and Dp divided by the nucleation time.
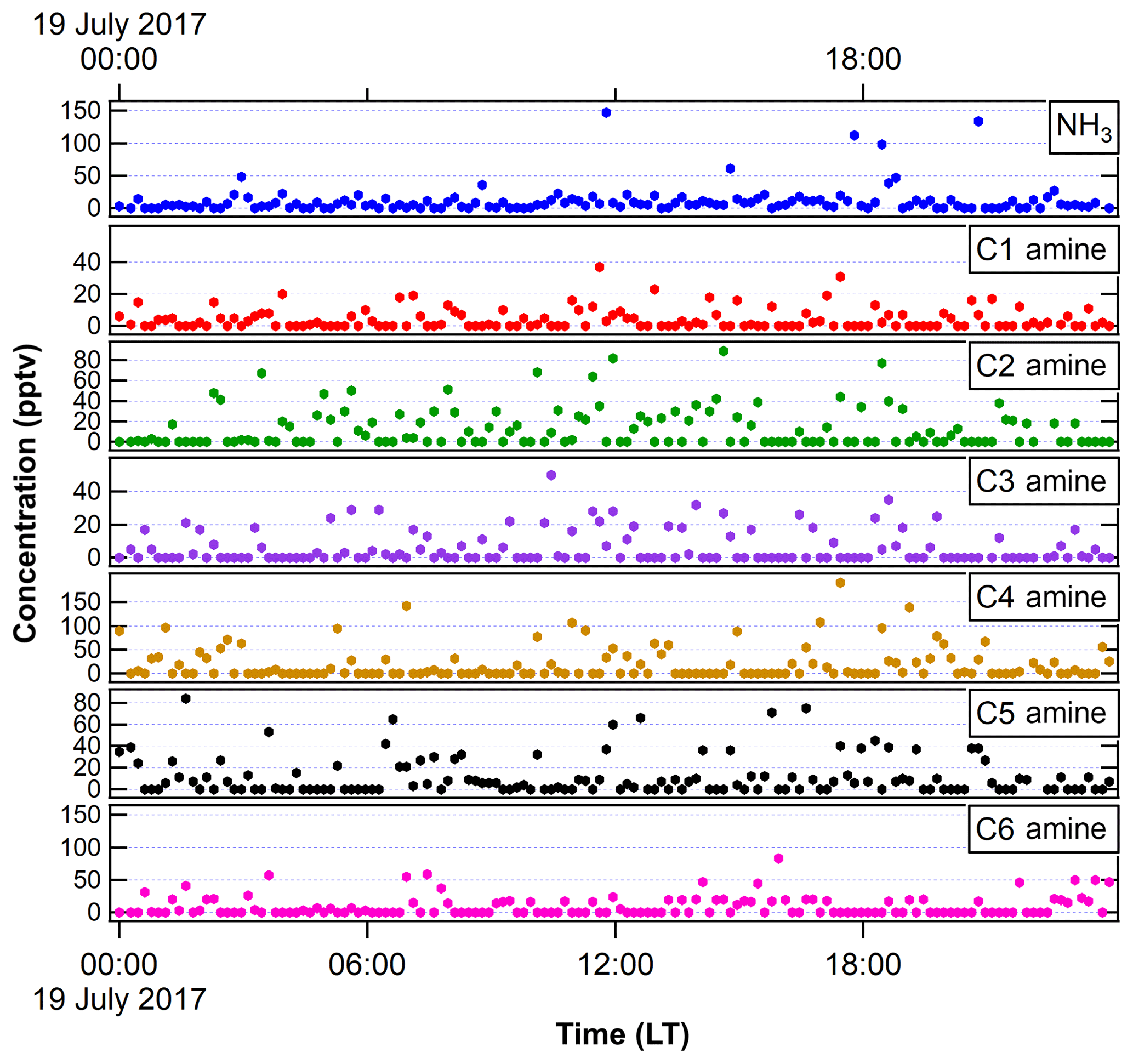
Figure 2NH3 and amines measured with the ethanol-based CIMS in FT-1 during the 2017 IOP in a very similar experimental condition as in 2018 IOP. We show 1 d of measurements (19 July 2017) here as an example.
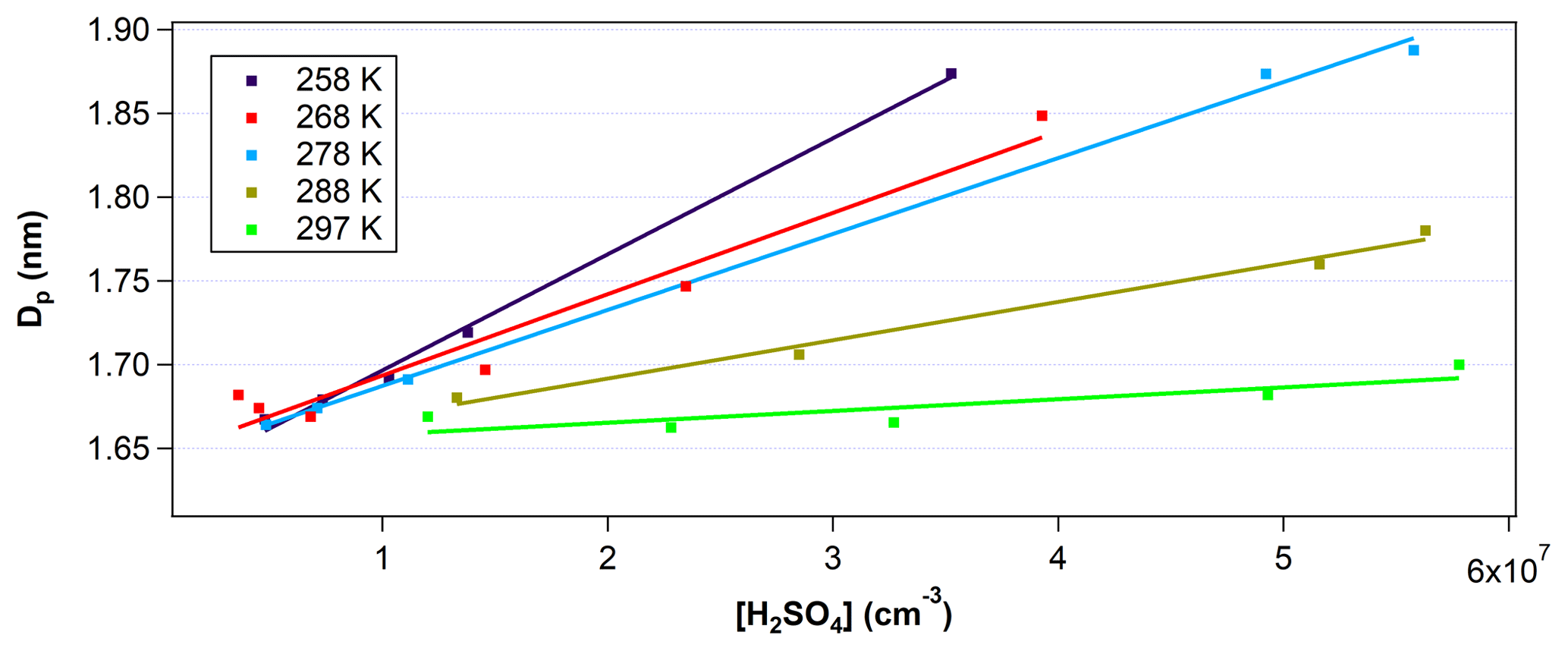
Figure 3Mean diameter of particles (Dp) inverted from the PSM measurements at the end of the nucleation tube as a function of [H2SO4] and temperature. Data points were taken at RH between 20 % and 30 %. Solid lines are linear fittings of the measurement data (colored squares) under different temperatures. Error in [H2SO4] is estimated to be ±60 %. Error in Dp at this size range is estimated to be ±0.2 nm or approximately ±12 %.
The growth rate factor kG is defined as the growth rate enhancement over 1 pptv H2SO4, causing 1 nm h−1 of growth. The collision limited condensation of H2SO4 of 1 pptv contributes roughly 1 nm h−1 of growth rate at a temperature of 298 K (Nieminen et al., 2011). Thus, the kG is an indicator of the deviation of the actual growth rate compared to this conventional collision-limited GR. The kG were determined by the expression derived by Yu et al. (2017a):
where ΔDp,tr represents the change in diameter over the residence time (tr) in the nucleation tube (FT-1). was experimentally determined from the slope of the Dp vs. [H2SO4]0 plot (e.g., Fig. 3). can be calculated from n and kL values. The kG is the product of this slope and . Extrapolation to the y axis of Fig. 3 gives a value for the critical cluster diameter. for the critical cluster, and thus Dp,critical will be equal to the y intercept of the linear fit of each data set.
2.3 Uncertainty analysis
The uncertainties in the J0 calculation arise from uncertainties in CIMS H2SO4 measurements, measurements of particle concentrations in the PSM and the size inversion from PSM-measured number concentrations. The uncertainty in the H2SO4 CIMS is approximately 60 % (Erupe et al., 2010; Eisele and Tanner, 1993; Benson et al., 2008, 2009; Kürten et al., 2012; Petäjä et al., 2009). The wall loss calculation has ±7 % uncertainty (Hanson and Eisele, 2000). The measurement error in the PSM size distribution, based on the standard error calculated between run-to-run experiments under identical conditions, is ±26 %. The uncertainties in inversion of the particle diameter from the PSM measurement are estimated to be around 12 % (or ±0.2 nm) for inorganic particles in the size range observed in this experiment (Lehtipalo et al., 2014, 2016; Kulmala et al., 2013; Yu et al., 2017a). Propagation of the errors in sulfuric acid measurement, wall loss and the PSM measurements results in an overall uncertainty of ±65.5 % in the calculation of J0.
3.1 Nucleation and growth in FT-1
Table 1 shows the typical experimental conditions used in FT-1 (nucleation tube) and FT-2 (growth tube) during the 2018 IOP study. In the nucleation tube, temperature was varied from 258 to 297 K and RH from 4 % to 85 %. [H2SO4] spanned from 106 to 108 cm−3, corresponding to RA of 10−5 to 10−2. RA was calculated using the sulfuric acid saturation vapor pressures provided by Vehkamaki et al. (2002). NH3 and amine measurements were not taken during this study, but they were undertaken during the 2017 IOP campaign using the same experimental apparatus, precursor gases and cleaning schedule as the 2018 campaign that is the concern of this study. The CIMS-measured NH3 (during the 2017 IOP) was 14.2±6.7 pptv (Fig. 2). Thus, the ratio of [NH3]∕[H2SO4] ranged from 0.6 to 268. According to Schobesberger et al. (2015) and Dunne et al. (2016), these ratios represent ternary nucleation and some nucleation in a transition regime between binary and ternary, when only considering the effects of NH3 (without amines). Amine concentrations were detected at levels ranging from 4.5±2.60 pptv for C1 amines to 44.8±41.8 pptv for C2 amines (Fig. 2). Overall, the measured nucleation rates ranged from 10 to 105 cm−3 at higher temperatures and lower [H2SO4] up to 105 cm−3 at lower temperatures and higher [H2SO4]. The observed GR ranged from 1 to 80 nm h−1. The growth tube (FT-2) was kept at room temperature (297 K) and dry conditions (RH of 10 %). SO2 was added in the range from 100 ppbv (parts-per-billion by volume) to 5 ppmv (part-per-million by volume) and ozone from 0 to 248 ppbv.
Table 1Typical experimental conditions used in FT-1 and FT-2. J in FT-1 is the nucleation rate for sub-2 nm particles. J in FT-2 is the formation rate of total particles in the size range from 3 to 60 nm. The particle mean diameter ranged from 2.7 to 3.4 nm depending on the ozone concentration. GR in FT-1 is the growth rate of sub-3 nm particles. GR in FT-2 is the growth rate of total particles from 3 to 60 nm.

Figure 3 shows the measured Dp with the PSM at the end of FT-1 as a function of the initial [H2SO4]0 at temperatures between 258 and 297 K. [H2SO4]0 was varied from 8×106 to 7×107 cm−3. The RH was kept in relatively narrow range between 20 % and 30 %. The y intercept in Fig. 3 indicates the critical cluster diameter was estimated to be between 1.6 and 1.7 nm depending on temperature, with lower temperatures resulting in smaller critical cluster diameters. However, since the PSM inversion that determines Dp has an uncertainty of ±0.2 nm, it is difficult to discuss the temperature trends of the critical cluster diameter. This critical size is consistent with the studies of Kulmala et al. (2013) and Almeida et al. (2013), which determined critical cluster diameters of 1.5±0.3 and 1.7 nm, respectively. For a given [H2SO4]0, the mean Dp at the end of the 45 s residence time in FT-1 was larger for lower temperatures. Previously, Glasoe et al. (2015) and Yu et al. (2017a) have also shown increasing GR with increasing [H2SO4] from flow tube experiments. The slope of Dp vs. [H2SO4]0 increased with each 10∘C decrease in temperature over the course of these experiments. Thus, the growth rate factor kG also increased with subsequent temperature decreases (e.g., from 1.27 at 297 K to 12.6 at 258 K). These results indicate that lower temperatures promote the faster growth of particles due to the reduction in saturation vapor pressures of H2SO4 at lower temperatures.
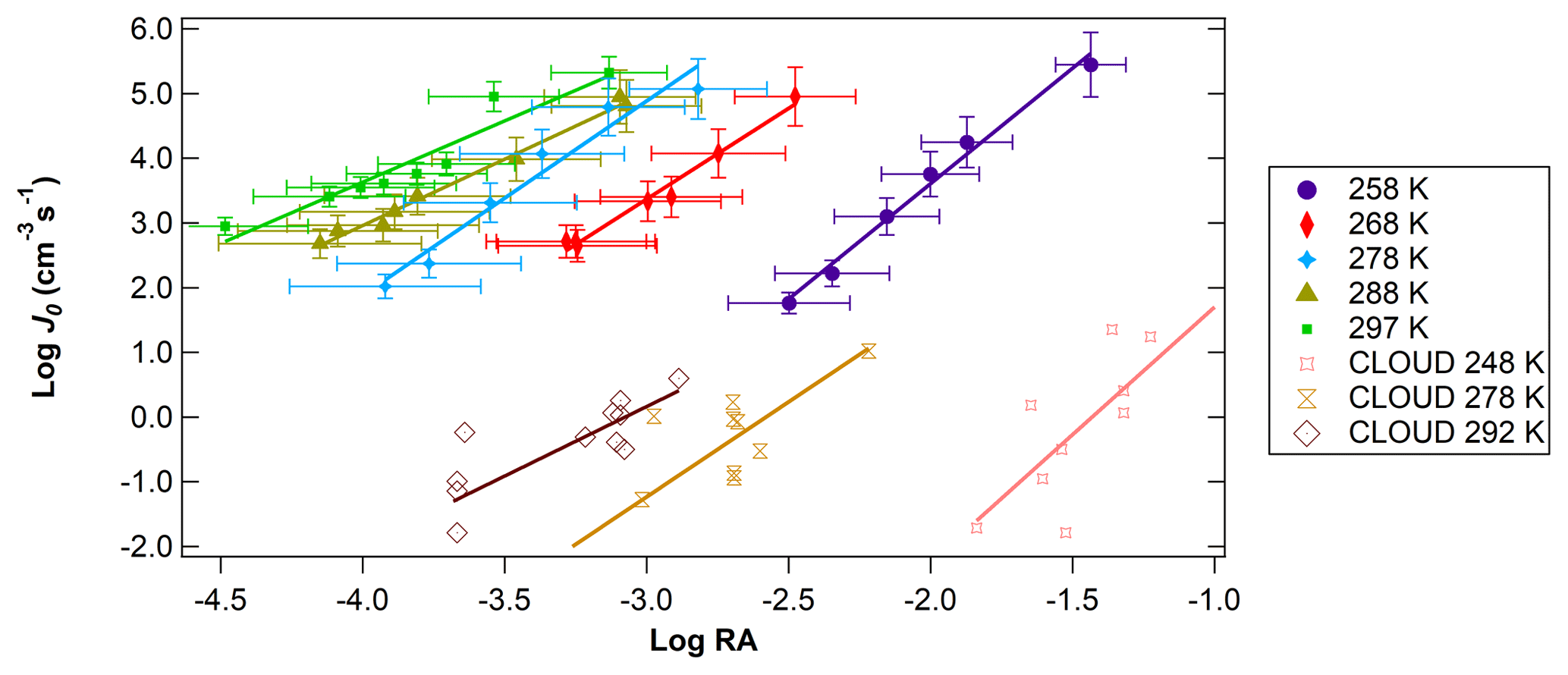
Figure 4Log J0 vs. log RA for different temperatures at a relatively constant RH (41 %–45 %). Temperatures ranged from 258 to 297 K. CLOUD data for similar RA and temperature conditions from Dunne et al. (2016) are also shown for comparison.
Figure 4 shows the relationship of log J vs. log RA for different temperatures. Experiments were conducted at 10 K intervals, starting from 297 K down to 258 K for RH between 41 % and 45 %. Across all temperature and RH experiments conducted, in general, J values were shifted 2 to 3 orders of magnitude above previous literature values of flow tube nucleation studies (Brus et al., 2010; Yu et al., 2017a) and 4 to 5 orders of magnitude above CLOUD measurements of H2SO4 nucleation as shown in the figure using CLOUD data from Dunne et al. (2016). This upward shift was consistent across trials. Based on our measured NH3 and amine concentrations (Fig. 2), this upward shift is consistent with the nucleation rate enhancement due to synergistic effects of NH3 concentrations on the order of 20 to 30 pptv and dimethylamine concentrations on the order of 5 pptv reported by other studies (Glasoe et al., 2015). There was a consistent relationship between log J and log RA. Except for 288 and 297 K, where the slope of log J vs. log RA was approximately 2, and the slope was 3 for all trials, with the best-fit lines shifting towards higher values of RA as temperature decreased. Hanson and colleagues (Glasoe et al., 2015; Zollner et al., 2012) provided comprehensive analysis of log J vs. log RA (or log [H2SO4]) obtained from flow tube studies. In general, flow tube studies from various groups have shown slopes between 3 and 6 for the ternary system (Glasoe et al., 2015; Zollner et al., 2012; Brus et al., 2010; Berndt et al., 2014; Erupe et al., 2011; Yu et al., 2012; Hanson et al., 2017). Using the CLOUD experiments (Dunne et al., 2016; Almeida et al., 2013; Kirkby et al., 2011) also showed the slope of 3 for the ternary system. This slope is consistent with the base-stabilization mechanism provided by Chen et al. (2012) and Jen et al. (2014, 2016), who concluded that the bottleneck clusters contain 3–4 H2SO4 molecules with at least one base molecule. It was previously believed that the slope of log J vs. log RA dictates the amount of H2SO4 molecules present in the critical cluster based on classical nucleation theory (CNT) (Kashchiev, 1982; McGraw and Zhang, 2008), which then would imply here that the critical cluster contains three H2SO4 molecules for the ternary system. However, more recent work by Malila et al. (2013) and Vehkamäki et al. (2012) has shown that this conclusion may be an oversimplification of the mechanism of particle formation resulting from an application of CNT with an incomplete understanding of the free energy maxima and minima.
Figure 5 shows the measured J as a function of temperature for [H2SO4] between 2×107 and 3×107 cm−3 and RH between 15 % and 45 %. J increased with decreasing temperatures in the temperature range above 268 K. The higher J at lower temperatures is consistent with predictions from CNT (Seinfeld and Pandis, 2016). However, a shift in slope is visible around 268 K, indicating that the dependence of J on temperature becomes less significant at low temperatures. The variation seen across RH at higher temperatures also becomes negligible below this temperature. Thus, these results indicate that at temperatures below 268 K, Gibbs free energy barriers are reduced significantly.
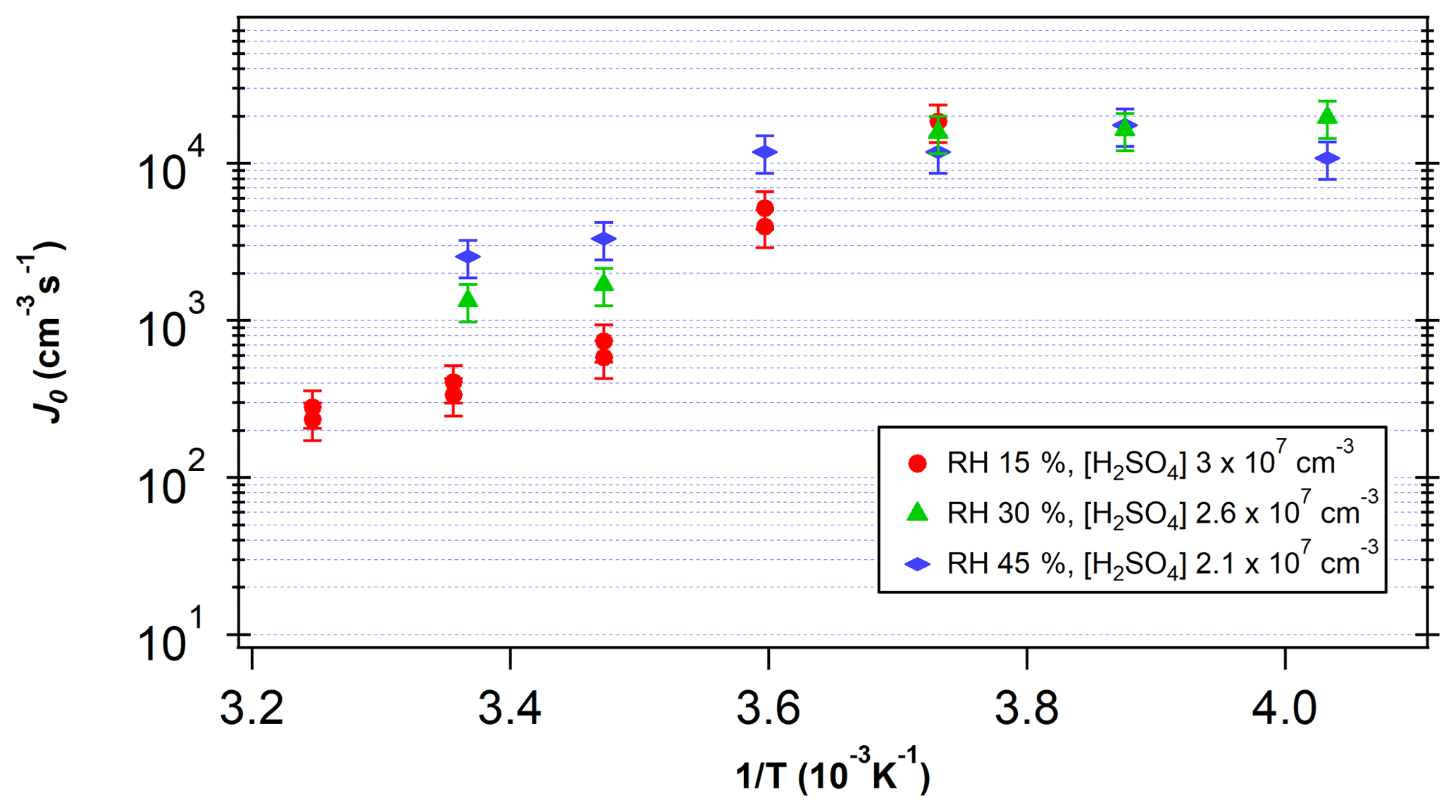
Figure 5Log J0 vs. 1∕T for [H2SO4] between 2×107 and 3×107 cm−3. T is the temperature in FT-1. RH ranged from 15 % to 45 %. Vertical bars indicate one standard deviation of the measured nucleation rates.
3.2 TANGENT experiments: further growth of clusters in FT-2
In order to determine whether newly formed particles nucleated at lower temperatures can survive downward transport to warmer temperature conditions, we conducted two tests (Figs. 6 and 7). The first test had a much lower temperature in FT-1 (268 K) than in FT-2 (297 K) and the second test had the same temperature (297 K) in both tubes. Figure 8 shows the GR and particle survival percentage, calculated by dividing the particle concentration at the end of FT-2 by the particle concentration at the end of FT-1. During the first test (Fig. 6), the average particle concentration coming out of FT-1 was 2×105 cm−3 with a median diameter of 1.9 nm at [H2SO4]0 of 6×107 cm−3 and RH of 10 %. These newly formed particles were further mixed with an additional zero air flow at a 1:6 dilution. The [SO2] was 83 ppbv and the ozone level was varied from 0 to 248 ppbv in FT-2. No particles were observed coming out of FT-2 when ozone was absent, indicating that SO2 alone does not cause nucleation and growth of clusters. However, in the presence of ozone and SO2 together, continuous nucleation and further growth of transported clusters took place in FT-2. The particle concentration measured at the end of FT-2 was closely correlated with the ozone concentration: the particle concentration ranged from 3×102 cm−3 (with Dp of 2.8 nm) at the lowest ozone of 28 ppbv up to 5×104 cm−3 (Dp of 3.4 nm) at the maximum ozone of 248 ppbv. Thus, the particle concentration in FT-2 at the highest ozone was even greater than that coming out of FT-1 after dilution, indicating that a high ozone load resulted in additional nucleation and the further growth of clusters in FT-2; this is indicated by the greater than 100 % particle survival shown in Fig. 8b. This could be the result of the remaining H2SO4 vapor passing through to FT-2, but we excluded this possibility. After considering wall loss in FT-1 and the 1:6 dilution FT-2, [H2SO4] in FT-2 was estimated to be 1.15×106 cm−3, which can result in J only on the order of 101 cm−3 s−1 at room temperature and dry conditions, as shown from the results obtained in FT-1 (e.g., Fig. 4). However, the measured formation rate (J) of particles in FT-2 was 190 cm−3 s−1, in contrast to this estimation. There may be another possibility that ozone reacted with possible organic impurities in FT-2 to produce OH, which oxidized SO2 to produce H2SO4 that nucleated in FT-2, but no organics were added in our experiments. At present it is not clear what is the cause of nucleation in FT-2 and this requires future study. However, it was clear that the copresence of ozone and SO2 was an important factor in preventing evaporation of newly formed particles and facilitated their further growth at a higher temperature. The overall GR of particles in FT-2 was from 14.9 to 23.1 nm h−1, depending on the ozone concentration. These results may imply some heterogeneous reactions occurring on acidic sulfuric acid clusters, in a similar way to how sulfate is formed from oxidation reactions of SO2 on acidic particles, as proposed by Hung and Hoffmann (2015).

Figure 6(a) The PSM-inverted size distribution and (b) [H2SO4] measured in FT-1 during the temperature gradient TANGENT experiment. FT-1 was at 268 K with a residence time of 45 s. Total concentration at the end of FT-1 was 1.79×105 cm−3 with a mean Dp of 1.9 nm. (c) SMPS-measured particle size distribution, (d) total number concentration, (e) the particle median diameter Dp and (f) ozone concentrations in FT-2. FT-2 was kept at 297 K and the residence time was 4 min. The red line in (e) indicates the average values of Dp. SO2 was 500 and 83 ppbv in FT-1 and FT-2, respectively. H2SO4 was not measured in FT-2; however, after considering wall loss in FT-1 and the 1:6 dilution FT-2, [H2SO4] in FT-2 was estimated to be 1.15×106 cm−3.
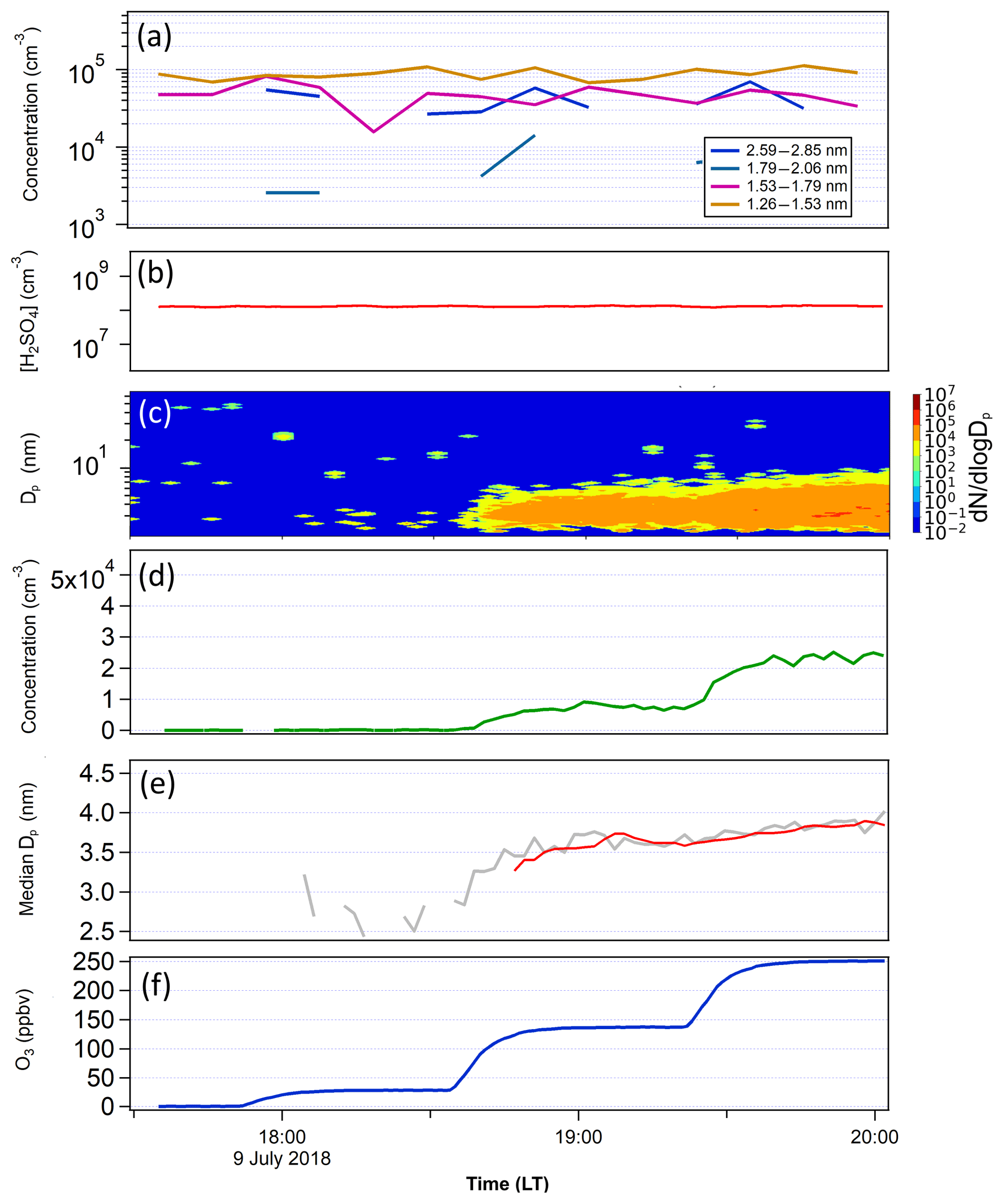
Figure 7The same as Fig. 6, except that FT-1 and FT-2 had the same temperature in this test. Total particle concentration at the end of FT-1 was 1.7×105 cm−3, with a mean Dp of 1.9 nm and [H2SO4] in FT-1 was 1.3×108 cm−3. In FT-2, the [H2SO4] was estimated to be 2.5×106 cm−3.
The second test was conducted with FT-1 and FT-2 both at a constant temperature of 297 K (Fig. 7). FT-1 was held at a constant [H2SO4]0 of 1.3×108 cm−3. The total particle concentration out of FT-1 for this experiment was 1.7×105 cm−3, with a mean diameter of 1.9 nm. Aside from the higher [H2SO4]0, which was necessary to produce the same particle concentration in the higher temperature FT-1, particle count and mean diameter were similar to the experiment with a cold FT-1 (Fig. 6). However, the size distributions in Figs. 6a and 7a indicate that there were more particles in the higher diameter size bins when the temperature in FT-1 was lower. At the end of FT-2, particle concentration and size were again closely correlated with ozone concentration, ranging from 8.6×101 cm−3 (Dp=2.7 nm) at the lowest ozone concentration up to 2.3×104 cm−3 (Dp=3.8 nm) at the highest ozone load. These concentrations and sizes result in growth rates ranging from 12.0 to 28.1 nm h−1, indicating that the growth at the highest ozone concentrations was faster during the constant temperature experiment. This was due to a greater concentration of smaller particles formed at higher [H2SO4]0 in the second test.
Examining the differences between the temperature gradient (Fig. 6) and constant temperature (Fig. 7) experiments, we see notable effects of temperature gradient on the survival of particles to the end of FT-2. While the average size at the end of the nucleation tube was similar in each experiment (Dp=1.9 nm with 268 K FT-1; Dp=1.9 nm with 298 K FT-1), we can see from the size distributions in each experiment that the low-temperature FT-1 resulted in a greater concentration of particles in the larger size bins. These particles (once they are stabilized, e.g., by base compounds) are more thermodynamically stable and therefore have a greater ability to survive evaporation when transported to the warmer environment of FT-2. Particles formed in warmer environments have a greater energy barrier to overcome before they become stable enough to grow spontaneously (Lovejoy et al., 2004). Figure 8 further shows that both the particle growth rate and the tendency for the particles to survive to the end of FT-2 were clearly correlated with ozone concentration, indicating that the concentration of ozone in FT-2 was a critical factor in the persistence, growth and nucleation that occurred in FT-2 during these experiments.

Figure 8(a) Particle growth rate as a function of ozone concentration in FT-2 during each experiment and (b) the particle survival percentage as a function of ozone concentration in FT-2. Particle survival percentage over 100 % indicates nucleation occurring in FT-2 due to an unknown heterogeneous process.
The additional nucleation occurring in FT-2 in the copresence of SO2 and ozone cannot be explained by current knowledge. These results suggest some possible heterogeneous reactions involving SO2 and ozone on acidic clusters. Understanding how SO2 and ozone interact each other on the acidic nanoparticles to facilitate nucleation and growth require future studies.
We examined the additional effects of organics and base compounds on GR in FT-2. First, to see the effects of possible impurities of organics, we used the growth rate parameterization from Tröstl et al. (2016):
Solving Eq. (4) for [HOM] using our observed growth rates of 12.0 to 28.1 nm h−1, Dp=1.9 nm, and p=1.424 (Tröstl et al., 2016) results in a [HOM] of 2.8 to 3.8 pptv necessary to account for these growth rates. The amount of monoterpene required to produce that amount of HOMs was then calculated using Equation 16 from Trostl et al. (2016):
Using the yield of HOMs from ozonolysis of monoterpenes Y1=2.9 % (Kirkby et al., 2016), temperature-dependent reaction rate of ozone and α-pinene (Khamaganov and Hites, 2001), and 10−3 for condensation sink (CS, calculated from the measured size distributions in FT-2), it would require between 100 and 1000 ppbv of monoterpenes to account for that concentration of HOMs. These high concentrations were very unlikely to be present in FT-2 considering the rigorous cleaning of the TANGENT apparatus and the lack of added monoterpenes to the system.
Second, to see the possible multicomponent growth due to the presence of ammonia in the system, we utilized the parameterization of multicomponent growth from Eq. (4) of Lehtipalo et al. (2018):
where k1 and k2 are and , respectively (Lehtipalo et al., 2018), and . Assuming [Org] =0 for simplicity and [H2SO4] and [NH3] values from the temperature gradient experiment (1.15×106 cm−3 H2SO4 and 14 pptv NH3 estimated from 2017 IOP measurements), we should expect a GR of 0.24 nm h−1. However, this calculation only considers the presence of NH3 in the system, when it is very likely that there were amines present at pptv levels (e.g., Fig. 2). Glasoe et al. (2015) and Yu et al. (2012) found synergistic effects on GR when both NH3 and methylamine and dimethylamine are present, which can enhance GR by 1 to 2 orders of magnitude and would put these calculated GR values in range of the observed 23.1 nm h−1 GR seen in FT-2.
Furthermore, cluster–cluster collision in the presence of stabilizing base compounds such as trace amines that were present in TANGENT may enhance growth by an order of magnitude (Lehtipalo et al., 2016). Thus, it seems that the observed high GRs in FT-2 were in some part due to multicomponent effects of base compounds and cluster–cluster collisional growth effects on GR, in addition to potential heterogeneous processes speculated above.
We have conducted experiments to study the temperature dependence of aerosol nucleation and growth using the TANGENT setup. This setup consists of two flow tubes that enables us to study nucleation and subsequent growth independently. In the nucleation tube, temperature was varied from 258 to 297 K and RH from 4 % to 85 %. [H2SO4] spanned 106 to 108 cm−3, which corresponds to RA of 10−5 to 10−2. Based on the measured [NH3] to [H2SO4] ratios, it was most likely that nucleation took place via the ternary process. The growth tube was kept at room temperature and the dry condition (RH of 10 %). SO2 was present at 100 ppbv to 5 ppmv and ozone was present at 0 to 248 ppbv.
Our results indicate that lower temperatures enhance both nucleation and growth rates as predicted by CNT. However, the temperature effects on nucleation rates become less important at lower temperatures (below 268 K), consistent with CLOUD studies (Duplissy et al., 2016), which found that sulfuric acid nucleation takes place at the kinetic limit without a Gibbs free energy barrier when temperature was low. These results emphasize the importance of NH3 and other ternary species at warmer temperatures, for example, especially in the conditions present in the boundary layer.
Our results demonstrate that clusters formed at lower temperatures, while being transported to warmer temperatures, can indeed survive evaporation and even grow further in the presence of SO2 and ozone. We are providing the first experimental evidence that supports the mechanism of downward transport of newly formed particles from the free troposphere to the marine boundary layer as proposed by several field and modeling studies (Russell et al., 1994; Raes, 1995; Clarke, 1993). Similarly, based on our results, it is reasonable to conclude that the new particles formed in the free troposphere over the Amazon forest are transferred downward to the warmer surface to act as a reservoir of nuclei-mode particles (Wang et al., 2016), though the mechanism of growth is different from the one studied here (SO2 + ozone).
Our results also show that the further growth is strongly dependent on the ozone level, implying that some unknown heterogeneous reaction processes involving SO2 and ozone on sulfuric clusters may play important roles in NPF. These results can open a new research avenue for future studies to create a better understanding of the roles of heterogeneous reactions involving nanoparticles and the effects of SO2 on the nanoparticle growth (Stangl et al., 2019). At present, it is not known why NPF takes place with high frequency and strong magnitude in extremely polluted megacities in China under the conditions with exceedingly high loadings of preexisting aerosol particles (Guo et al., 2014; Yao et al., 2018; Yu et al., 2017b; Lee et al., 2019). The heterogeneous reactions involving SO2 in nanoparticles proposed here, if they can be repeated and further understood in future studies, may provide some key insights into understanding the frequent nucleation and fast growth observed in these regions, where there are also very high concentrations of SO2 and ozone.
All data from this work can be obtained from Lee Tiszenkel (lt0021@uah.edu) and Shanhu Lee (shanhu.lee@uah.edu).
SL and MVJ designed the experiments and LT, CS, JK, QO and MJA carried them out. LT, QO and HY developed code used in the data analysis. LT, CS and QO performed the data analysis. LT and SHL prepared the manuscript with contributions from all co-authors.
The authors declare that they have no conflict of interest.
This research has been supported by the National Science Foundation (grant nos. AGS-1649719 and AGS-1649694).
This paper was edited by Kari Lehtinen and reviewed by two anonymous referees.
Almeida, J., Schobesberger, S., Kurten, A., Ortega, I. K., Kupiainen-Maatta, O., Praplan, A. P., Adamov, A., Amorim, A., Bianchi, F., Breitenlechner, M., David, A., Dommen, J., Donahue, N. M., Downard, A., Dunne, E., Duplissy, J., Ehrhart, S., Flagan, R. C., Franchin, A., Guida, R., Hakala, J., Hansel, A., Heinritzi, M., Henschel, H., Jokinen, T., Junninen, H., Kajos, M., Kangasluoma, J., Keskinen, H., Kupc, A., Kurten, T., Kvashin, A. N., Laaksonen, A., Lehtipalo, K., Leiminger, M., Leppa, J., Loukonen, V., Makhmutov, V., Mathot, S., McGrath, M. J., Nieminen, T., Olenius, T., Onnela, A., Petaja, T., Riccobono, F., Riipinen, I., Rissanen, M., Rondo, L., Ruuskanen, T., Santos, F. D., Sarnela, N., Schallhart, S., Schnitzhofer, R., Seinfeld, J. H., Simon, M., Sipila, M., Stozhkov, Y., Stratmann, F., Tome, A., Trostl, J., Tsagkogeorgas, G., Vaattovaara, P., Viisanen, Y., Virtanen, A., Vrtala, A., Wagner, P. E., Weingartner, E., Wex, H., Williamson, C., Wimmer, D., Ye, P., Yli-Juuti, T., Carslaw, K. S., Kulmala, M., Curtius, J., Baltensperger, U., Worsnop, D. R., Vehkamaki, H., and Kirkby, J.: Molecular understanding of sulphuric acid-amine particle nucleation in the atmosphere, Nature, 502, 359–363, https://doi.org/10.1038/nature12663, 2013.
Benson, D. R., Young, L. H., Kameel, R., and Lee, S. H.: Laboratory-measured sulfuric acid and water homogeneous nucleation rates from the SO2+OH reaction, Geophys. Res. Lett., 35, L11801, https://doi.org/10.1029/2008GL033387, 2008.
Benson, D. R., Erupe, M. E., and Lee, S. H.: Laboratory-measured ternary homogeneous nucleation rates: initial observations Geophys. Res. Lett., 36, L15818, https://doi.org/10.1029/2009GL038728, 2009.
Benson, D. R., Markovich, A., Al-Refai, M., and Lee, S.-H.: A Chemical Ionization Mass Spectrometer for ambient measurements of Ammonia, Atmos. Meas. Tech., 3, 1075–1087, https://doi.org/10.5194/amt-3-1075-2010, 2010.
Benson, D. R., Yu, J. H., Markovich, A., and Lee, S.-H.: Ternary homogeneous nucleation of H2SO4, NH3, and H2O under conditions relevant to the lower troposphere, Atmos. Chem. Phys., 11, 4755–4766, https://doi.org/10.5194/acp-11-4755-2011, 2011.
Berndt, T., Sipilä, M., Stratmann, F., Petäjä, T., Vanhanen, J., Mikkilä, J., Patokoski, J., Taipale, R., Mauldin III, R. L., and Kulmala, M.: Enhancement of atmospheric H2SO4∕H2O nucleation: organic oxidation products versus amines, Atmos. Chem. Phys., 14, 751–764, https://doi.org/10.5194/acp-14-751-2014, 2014.
Brus, D., Hyvärinen, A.-P., Viisanen, Y., Kulmala, M., and Lihavainen, H.: Homogeneous nucleation of sulfuric acid and water mixture: experimental setup and first results, Atmos. Chem. Phys., 10, 2631–2641, https://doi.org/10.5194/acp-10-2631-2010, 2010.
Chen, M., Titcombe, M., Jiang, J. K., Jen, C., Kuang, C. A., Fischer, M. L., Eisele, F. L., Siepmann, J. I., Hanson, D. R., Zhao, J., and McMurry, P. H.: Acid-base chemical reaction model for nucleation rates in the polluted atmospheric boundary layer, P. Natl. Acad. Sci. USA, 109, 18713–18718, https://doi.org/10.1073/pnas.1210285109, 2012.
Clarke, A. D.: Atmospheric nuclei in the Pacific mid-troposphere: their nature, concentration, and evolution, J. Geophys. Res., 98, 20633–20647, 1993.
Dunne, E. M., Gordon, H., Kürten, A., Almeida, J., Duplissy, J., Williamson, C., Ortega, I. K., Pringle, K. J., Adamov, A., Baltensperger, U., Barmet, P., Benduhn, F., Bianchi, F., Breitenlechner, M., Clarke, A., Curtius, J., Dommen, J., Donahue, N. M., Ehrhart, S., Flagan, R. C., Franchin, A., Guida, R., Hakala, J., Hansel, A., Heinritzi, M., Jokinen, T., Kangasluoma, J., Kirkby, J., Kulmala, M., Kupc, A., Lawler, M. J., Lehtipalo, K., Makhmutov, V., Mann, G., Mathot, S., Merikanto, J., Miettinen, P., Nenes, A., Onnela, A., Rap, A., Reddington, C. L. S., Riccobono, F., Richards, N. A. D., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Simon, M., Sipilä, M., Smith, J. N., Stozkhov, Y., Tomé, A., Tröstl, J., Wagner, P. E., Wimmer, D., Winkler, P. M., Worsnop, D. R., and Carslaw, K. S.: Global atmospheric particle formation from CERN CLOUD measurements, Science, 354, 1119–1123, 2016.
Duplissy, J., Merikanto, J., Franchin, A., Tsagkogeorgas, G., Kangasluoma, J., Wimmer, D., Vuollekoski, H., Schobesberger, S., Lehtipalo, K., Flagan, R. C., Brus, D., Donahue, N. M., Vehkamäki, H., Almeida, J., Amorim, A., Barmet, P., Bianchi, F., Breitenlechner, M., Dunne, E. M., Guida, R., Henschel, H., Junninen, H., Kirkby, J., Kürten, A., Kupc, A., Määttänen, A., Makhmutov, V., Mathot, S., Nieminen, T., Onnela, A., Praplan, A. P., Riccobono, F., Rondo, L., Steiner, G., Tome, A., Walther, H., Baltensperger, U., Carslaw, K. S., Dommen, J., Hansel, A., Petäjä, T., Sipilä, M., Stratmann, F., Vrtala, A., Wagner, P. E., Worsnop, D. R., Curtius, J., and Kulmala, M.: Effect of ions on sulfuric acid-water binary particle formation: 2. Experimental data and comparison with QC-normalized classical nucleation theory, J. Geophys. Res., 121, 1752–1775, https://doi.org/10.1002/2015JD023539, 2016.
Eisele, F. L. and Tanner, D. J.: Measurements of gas phase concentrations of H2SO4 and methane sulfonic acid and estimates of H2SO4 production and loss in the atmosphere, J. Geophys. Res., 98, 9001–9010, 1993.
Erupe, M. E., Benson, D. R., Li, J., Young, L. H., Verheggen, B., Al-Refai, M., Tahboub, O., Cunningham, V., Frimpong, F., Viggiano, A. A., and Lee, S. H.: Correlation of aerosol nucleation rate with sulfuric acid and ammonia in Kent, Ohio: An atmospheric observation, J. Geophys. Res., 115, D23216, https://doi.org/10.1029/2010JD013942, 2010.
Erupe, M. E., Viggiano, A. A., and Lee, S.-H.: The effect of trimethylamine on atmospheric nucleation involving H2SO4, Atmos. Chem. Phys., 11, 4767–4775, https://doi.org/10.5194/acp-11-4767-2011, 2011.
Frege, C., Ortega, I. K., Rissanen, M. P., Praplan, A. P., Steiner, G., Heinritzi, M., Ahonen, L., Amorim, A., Bernhammer, A.-K., Bianchi, F., Brilke, S., Breitenlechner, M., Dada, L., Dias, A., Duplissy, J., Ehrhart, S., El-Haddad, I., Fischer, L., Fuchs, C., Garmash, O., Gonin, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kirkby, J., Kürten, A., Lehtipalo, K., Leiminger, M., Mauldin, R. L., Molteni, U., Nichman, L., Petäjä, T., Sarnela, N., Schobesberger, S., Simon, M., Sipilä, M., Stolzenburg, D., Tomé, A., Vogel, A. L., Wagner, A. C., Wagner, R., Xiao, M., Yan, C., Ye, P., Curtius, J., Donahue, N. M., Flagan, R. C., Kulmala, M., Worsnop, D. R., Winkler, P. M., Dommen, J., and Baltensperger, U.: Influence of temperature on the molecular composition of ions and charged clusters during pure biogenic nucleation, Atmos. Chem. Phys., 18, 65–79, https://doi.org/10.5194/acp-18-65-2018, 2018.
Glasoe, W. A., Volz, K., Panta, B., Freshour, N., Bachman, R., Hanson, D. R., McMurry, P. H., and Jen, C.: Sulfuric acid nucleation: an experimental study of the effect of seven bases, J. Geophys. Res., 120, 1933–1950, https://doi.org/10.1002/2014JD022730, 2015.
Gordon, H., Kirkby, J., Baltensperger, U., Bianchi, F., Breitenlechner, M., Curtius, J., Dias, A., Dommen, J., Donahue, N. M., Dunne, E. M., Duplissy, J., Ehrhart, S., Flagan, R. C., Frege, C., Fuchs, C., Hansel, A., Hoyle, C. R., Kulmala, M., Kurten, A., Lehtipalo, K., Makhmutov, V., Molteni, U., Rissanen, M. P., Stozkhov, Y., Trostl, J., Tsagkogeorgas, G., Wagner, R., Williamson, C., Wimmer, D., Winkler, P. M., Yan, C., and Carslaw, K. S.: Causes and importance of new particle formation in the present-day and preindustrial atmospheres, J. Geophys. Res.-Atmos., 122, 8739–8760, https://doi.org/10.1002/2017jd026844, 2017.
Guo, S., Hua, M., Zamor, M. L., Peng, J., Shang, D., Zheng, J., Du, Z., Wu, Z., Shao, M., Zeng, L., Molina, M. J., and Zhang, R.: Elucidating severe urban haze formation in China, P. Natl. Acad. Sci. USA, 49, 17373–17378, 2014.
Hanson, D. R. and Eisele, F. L.: Diffusion of H2SO4 in humidified nitrogen: hydrated H2SO4, J. Phys. Chem., 104, 1715–1719, 2000.
Hanson, D. R., Bier, I., Panta, B., Jen, C. N., and McMurry, P. H.: Computational Fluid Dynamics Studies of a Flow Reactor: Free Energies of Clusters of Sulfuric Acid with NH3 or Dimethyl Amine, J. Phys. Chem. A, 121, 3976–3990, https://doi.org/10.1021/acs.jpca.7b00252, 2017.
Hung, H.-M. and Hoffmann, M. R.: Oxidation of gas-phase SO2 on the surfaces of acidic microdroplets: Implications for sulfate and sulfate radical anion formation in the atmospheric liquid phase, Environ. Sci. Technol., 49, 13768–13776, 2015.
Jen, C. N., McMurry, P. H., and Hanson, D. R.: Stabilization of sulfuric acid dimers by ammonia, methylamine, dimethylamine, and trimethylamine, J. Geophys. Res., 119, 7502–7514, https://doi.org/10.1002/2014jd021592, 2014.
Jen, C. N., Zhao, J., McMurry, P. H., and Hanson, D. R.: Chemical ionization of clusters formed from sulfuric acid and dimethylamine or diamines, Atmos. Chem. Phys., 16, 12513–12529, https://doi.org/10.5194/acp-16-12513-2016, 2016.
Kanawade, V. P., Jobson, B. T., Guenther, A. B., Erupe, M. E., Pressley, S. N., Tripathi, S. N., and Lee, S.-H.: Isoprene suppression of new particle formation in a mixed deciduous forest, Atmos. Chem. Phys., 11, 6013–6027, https://doi.org/10.5194/acp-11-6013-2011, 2011.
Kashchiev, D.: On the relation between nucleation work, nucleus size, and nucleation rate, J. Phys. Chem., 76, 5098–5102, https://doi.org/10.1063/1.442808, 1982.
Khamaganov, V. G. and Hites, R. A.: Rate Constants for the Gas-Phase Reactions of Ozone with Isoprene, α- and β-Pinene, and Limonene as a Function of Temperature, J. Phys. Chem. A, 105, 815–822, https://doi.org/10.1021/jp002730z, 2001.
Kirkby, J., Curtius, J., Almeida, J., Dunne, E., Duplissy, J., Ehrhart, S., Franchin, A., Gagne, S., Ickes, L., Kurten, A., Kupc, A., Metzger, A., Riccobono, F., Rondo, L., Schobesberger, S., Tsagkogeorgas, G., Wimmer, D., Amorim, A., Bianchi, F., Breitenlechner, M., David, A., Dommen, J., Downard, A., Ehn, M., Flagan, R. C., Haider, S., Hansel, A., Hauser, D., Jud, W., Junninen, H., Kreissl, F., Kvashin, A. N., Laaksonen, A., Lehtipalo, K., Lima, J., Lovejoy, E. R., Makhmutov, V., Mathot, S., Mikkila, J., Minginette, P., Mogo, S., Nieminen, T., Onnela, A., Pereira, P., Petaja, T., Schnitzhofer, R., Seinfeld, J. H., Sipila, M., Stozhkov, Y., Stratmann, F., Tome, A., Vanhanen, J., Viisanen, Y., Vrtala, A., Wagner, P. E., Walther, H., Weingartner, E., Wex, H., Winkler, P. M., Carslaw, K. S., Worsnop, D. R., Baltensperger, U., and Kulmala, M.: Role of sulphuric acid, ammonia and galactic cosmic rays in atmospheric aerosol nucleation, Nature, 476, 429–433, 2011.
Kirkby, J., Duplissy, J., Sengupta, K., Frege, C., Gordon, H., Williamson, C., Heinritzi, M., Simon, M., Yan, C., Almeida, J., Tröstl, J., Nieminen, T., Ortega, I. K., Wagner, R., Adamov, A., Amorim, A., Bernhammer, A.-K., Bianchi, F., Breitenlechner, M., Brilke, S., Chen, X., Craven, J., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Hakala, J., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Mathot, S., Molteni, U., Onnela, A., Peräkylä, O., Piel, F., Petäjä, T., Praplan, A. P., Pringle, K., Rap, A., Richards, N. A. D., Riipinen, I., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Scott, C. E., Seinfeld, J. H., Sipilä, M., Steiner, G., Stozhkov, Y., Stratmann, F., Tomé, A., Virtanen, A., Vogel, A. L., Wagner, A. C., Wagner, P. E., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Zhang, X., Hansel, A., Dommen, J., Donahue, N. M., Worsnop, D. R., Baltensperger, U., Kulmala, M., Carslaw, K. S., and Curtius, J.: Ion-induced nucleation of pure biogenic particles, Nature, 533, 521–526, https://doi.org/10.1038/nature17953, 2016.
Krasnomowitz, J. M., Apsokardu, M. J., Stangl, C. M., Tiszenkel, L., Ouyang, Q., Lee, S., and Johnston, M. V.: Growth of Aitken mode ammonium sulfate particles by α-pinene ozonolysis, Aerosol Sci. Tech., 53, 406–418, https://doi.org/10.1080/02786826.2019.1568381, 2019.
Kulmala, M., Kontkanen, J., Junninen, H., Lehtipalo, K., Manninen, H. E., Nieminen, T., Petäjä, T., Sipilä, M., Schobesberger, S., Rantala, P., Franchin, A., Jokinen, T., Järvinen, E., Äijälä, M., Kangasluoma, J., Hakala, J., Aalto, P. P., Paasonen, P., Mikkilä, J., Vanhanen, J., Aalto, J., Hakola, H., Makkonen, U., Ruuskanen, T., Mauldin, R. L., Duplissy, J., Vehkamäki, H., Bäck, J., Kortelainen, A., Riipinen, I., Kurtén, T., Johnston, M. V., Smith, J. N., Ehn, M., Mentel, T. F., Lehtinen, K. E. J., Laaksonen, A., Kerminen, V. M., and Worsnop, D. R.: Direct observations of atmospheric aerosol nucleation, Science, 339, 943–946, https://doi.org/10.1126/science.1227385, 2013.
Kulmala, M., Kerminen, V.-M., Petaja, T., Ding, A., and Wang, L.: Atmospheric gas-to-particle conversion: why NPF events are observed in megacities?, Faraday Discuss., 200, 271–288, https://doi.org/10.1039/c6fd00257a, 2017.
Kürten, A., Rondo, L., Ehrhart, S., and Curtius, J.: Calibration of a chemical ionization mass spectrometer for the measurement of gaseous sulfuric acid, J. Phys. Chem. A, 116, 6375–6386, https://doi.org/10.1021/jp212123n, 2012.
Kürten, A., Jokinen, T., Simon, M., Sipilä, M., Sarnela, N., Junninen, H., Adamov, A., Almeida, J., Amorim, A., Bianchi, F., Breitenlechner, M., Dommen, J., Donahue, N. M., Duplissy, J., Ehrhart, S., Flagan, R. C., Franchin, A., Hakala, J., Hansel, A., Heinritzi, M., Hutterli, M., Kangasluoma, J., Kirkby, J., Laaksonen, A., Lehtipalo, K., Leiminger, M., Makhmutov, V., Mathot, S., Onnela, A., Petäjä, T., Praplan, A. P., Riccobono, F., Rissanen, M. P., Rondo, L., Schobesberger, S., Seinfeld, J. H., Steiner, G., Tomé, A., Tröstl, J., Winkler, P. M., Williamson, C., Wimmer, D., Ye, P., Baltensperger, U., Carslaw, K. S., Kulmala, M., Worsnop, D. R., and Curtius, J.: Neutral molecular cluster formation of sulfuric acid–dimethylamine observed in real time under atmospheric conditions, P. Natl. Acad. Sci. USA, 111, 15019–15024, https://doi.org/10.1073/pnas.1404853111, 2014.
Kürten, A., Bianchi, F., Almeida, J., Kupiainen-Määttä, O., Dunne, E. M., Duplissy, J., Williamson, C., Barmet, P., Breitenlechner, M., Dommen, J., Donahue, N. M., Flagan, R. C., Franchin, A., Gordon, H., Hakala, J., Hansel, A., Heinritzi, M., Ickes, L., Jokinen, T., Kangasluoma, J., Kim, J., Kirkby, J., Kupc, A., Lehtipalo, K., Leiminger, M., Makhmutov, V., Onnela, A., Ortega, I. K., Petäjä, T., Praplan, A. P., Riccobono, F., Rissanen, M. P., Rondo, L., Schnitzhofer, R., Schobesberger, S., Smith, J. N., Steiner, G., Stozhkov, Y., Tomé, A., Tröstl, J., Tsagkogeorgas, G., Wagner, P. E., Wimmer, D., Ye, P., Baltensperger, U., Carslaw, K., Kulmala, M., and Curtius, J.: Experimental particle formation rates spanning tropospheric sulfuric acid and ammonia abundances, ion production rates, and temperatures, J. Geophys. Res., 121, 12377–12400, https://doi.org/10.1002/2015JD023908, 2016.
Lawler, M. J., Winkler, P. M., Kim, J., Ahlm, L., Tröstl, J., Praplan, A. P., Schobesberger, S., Kürten, A., Kirkby, J., Bianchi, F., Duplissy, J., Hansel, A., Jokinen, T., Keskinen, H., Lehtipalo, K., Leiminger, M., Petäjä, T., Rissanen, M., Rondo, L., Simon, M., Sipilä, M., Williamson, C., Wimmer, D., Riipinen, I., Virtanen, A., and Smith, J. N.: Unexpectedly acidic nanoparticles formed in dimethylamine–ammonia–sulfuric-acid nucleation experiments at CLOUD, Atmos. Chem. Phys., 16, 13601–13618, https://doi.org/10.5194/acp-16-13601-2016, 2016.
Lee, S.-H., Uin, J., Guenther, A. B., Gouw, J. A. d., Yu, F., Nadykto, A. B., Herb, J., Ng, N. L., Koss, A., Brune, W. H., Baumann, K., Kanawad, V. P., Keutsch, F. N., Nenes, A., Olsen, K., Goldstein, A., and Qi, O.: Isoprene suppression of new particle formation: Potential mechanism and implications, J. Geophys. Res., 121, 14621–14635, 10.1029/2016JD024844, 2016.
Lee, S.-H., Gordon, H., Yu, H., Lehtipalo, K., Haley, R., Li, Y., and Zhang, R.: New particle formation in the atmosphere: From molecular clusters to global climate, J. Geophys. Res., 124, https://doi.org/10.1029/2018JD029356, online first, 2019.
Lehtipalo, K., Leppä, J., Kontkanen, J., Kangasluoma, J., Franchin, A., Wimmer, D., Schobesberger, S., Junninen, H., Petäjä, T., Sipilä, M., Mikkilä, J., Vanhanen, J., Worsnop, D. R., and Kulmala, M.: Methods for determining particle size distribution and growth rates between 1 and 3 nm using the Particle Size Magnifier, Boreal Environ. Res., 19, 215–236, 2014.
Lehtipalo, K., Rondo, L., Kontkanen, J., Schobesberger, S., Jokinen, T., Sarnela, N., Kürten, A., Ehrhart, S., Franchin, A., Nieminen, T., Riccobono, F., Sipilä, M., Yli-Juuti, T., Duplissy, J., Adamov, A., Ahlm, L., Almeida, J., Amorim, A., Bianchi, F., Breitenlechner, M., Dommen, J., Downard, A. J., Dunne, E. M., Flagan, R. C., Guida, R., Hakala, J., Hansel, A., Jud, W., Kangasluoma, J., Kerminen, V.-M., Keskinen, H., Kim, J., Kirkby, J., Kupc, A., Kupiainen-Määttä, O., Laaksonen, A., Lawler, M. J., Leiminger, M., Mathot, S., Olenius, T., Ortega, I. K., Onnela, A., Petäjä, T., Praplan, A., Rissanen, M. P., Ruuskanen, T., Santos, F. D., Schallhart, S., Schnitzhofer, R., Simon, M., Smith, J. N., Tröstl, J., Tsagkogeorgas, G., Tomé, A., Vaattovaara, P., Vehkamäki, H., Vrtala, A. E., Wagner, P. E., Williamson, C., Wimmer, D., Winkler, P. M., Virtanen, A., Donahue, N. M., Carslaw, K. S., Baltensperger, U., Riipinen, I., Curtius, J., Worsnop, D. R., and Kulmala, M.: The effect of acid–base clustering and ions on the growth of atmospheric nano-particles, Nat. Commun., 7, 11594, https://doi.org/10.1038/ncomms11594, 2016.
Lehtipalo, K., Yan, C., Dada, L., Bianchi, F., Xiao, M., Wagner, R., Stolzenburg, D., Ahonen, L. R., Amorim, A., Baccarini, A., Bauer, P. S., Baumgartner, B., Bergen, A., Bernhammer, A.-K., Breitenlechner, M., Brilke, S., Buchholz, A., Mazon, S. B., Chen, D., Chen, X., Dias, A., Dommen, J., Draper, D. C., Duplissy, J., Ehn, M., Finkenzeller, H., Fischer, L., Frege, C., Fuchs, C., Garmash, O., Gordon, H., Hakala, J., He, X., Heikkinen, L., Heinritzi, M., Helm, J. C., Hofbauer, V., Hoyle, C. R., Jokinen, T., Kangasluoma, J., Kerminen, V.-M., Kim, C., Kirkby, J., Kontkanen, J., Kürten, A., Lawler, M. J., Mai, H., Mathot, S., Mauldin, R. L., Molteni, U., Nichman, L., Nie, W., Nieminen, T., Ojdanic, A., Onnela, A., Passananti, M., Petäjä, T., Piel, F., Pospisilova, V., Quéléver, L. L. J., Rissanen, M. P., Rose, C., Sarnela, N., Schallhart, S., Schuchmann, S., Sengupta, K., Simon, M., Sipilä, M., Tauber, C., Tomé, A., Tröstl, J., Väisänen, O., Vogel, A. L., Volkamer, R., Wagner, A. C., Wang, M., Weitz, L., Wimmer, D., Ye, P., Ylisirniö, A., Zha, Q., Carslaw, K. S., Curtius, J., Donahue, N. M., Flagan, R. C., Hansel, A., Riipinen, I., Virtanen, A., Winkler, P. M., Baltensperger, U., Kulmala, M., and Worsnop, D. R.: Multicomponent new particle formation from sulfuric acid, ammonia, and biogenic vapors, J. Sci. Adv., 4, eaau5363, https://doi.org/10.1126/sciadv.aau5363, 2018.
Lovejoy, E. R., Curtius, J., and Froyd, K. D.: Atmospheric ion-induced nucleation of sulfuric acid and water J. Geophys. Res., 109, D08204, https://doi.org/10.1029/2003JD004460, 2004.
Malila, J., McGraw, R., Laaksonen, A., and Lehtinen, K. E. J.: Repairing the first nucleation theorem: Precritical cluster losses, AIP Conf. Proc., 1527, 31–34, https://doi.org/10.1063/1.4803197, 2013.
Martin, S. T., Artaxo, P., Machado, L., Manzi, A. O., Souza, R. A. F., Schumacher, C., Wang, J., Biscaro, T., Brito, J., Calheiros, A., Jardine, K., Medeiros, A., Portela, B., de Sá, S. S., Adachi, K., Aiken, A. C., Albrecht, R., Alexander, L., Andreae, M. O., Barbosa, H. M. J., Buseck, P., Chand, D., Comstock, J. M., Day, D. A., Dubey, M., Fan, J., Fast, J., Fisch, G., Fortner, E., Giangrande, S., Gilles, M., Goldstein, A. H., Guenther, A., Hubbe, J., Jensen, M., Jimenez, J. L., Keutsch, F. N., Kim, S., Kuang, C., Laskin, A., McKinney, K., Mei, F., Miller, M., Nascimento, R., Pauliquevis, T., Pekour, M., Peres, J., Petäjä, T., Pöhlker, C., Pöschl, U., Rizzo, L., Schmid, B., Shilling, J. E., Dias, M. A. S., Smith, J. N., Tomlinson, J. M., Tóta, J., and Wendisch, M.: The Green Ocean Amazon Experiment (GoAmazon2014/5) Observes Pollution Affecting Gases, Aerosols, Clouds, and Rainfall over the Rain Forest, B. Am. Meteorol. Soc., 98, 981–997, https://doi.org/10.1175/BAMS-D-15-00221.1, 2016.
McGraw, R. and Zhang, R.: Multivariate analysis of homogeneous nucleation rate measurements, Nucleation in the p-toluic acid/sulfuric acid/water system, J. Chem. Phys., 128, 064508, https://doi.org/10.1063/1.2830030, 2008.
Merikanto, J., Spracklen, D. V., Mann, G. W., Pickering, S. J., and Carslaw, K. S.: Impact of nucleation on global CCN, Atmos. Chem. Phys., 9, 8601–8616, https://doi.org/10.5194/acp-9-8601-2009, 2009.
Nieminen, T., Paasonen, P., Manninen, H. E., Sellegri, K., Kerminen, V.-M., and Kulmala, M.: Parameterization of ion-induced nucleation rates based on ambient observations, Atmos. Chem. Phys., 11, 3393–3402, https://doi.org/10.5194/acp-11-3393-2011, 2011.
Petäjä, T., Mauldin III, R. L., Kosciuch, E., McGrath, J., Nieminen, T., Paasonen, P., Boy, M., Adamov, A., Kotiaho, T., and Kulmala, M.: Sulfuric acid and OH concentrations in a boreal forest site, Atmos. Chem. Phys., 9, 7435–7448, https://doi.org/10.5194/acp-9-7435-2009, 2009.
Raes, F.: Entrainment of free tropospheric aerosols as a regulating mechanism for cloud condensation nuclei in the remote marine boundary layer, J. Geophys. Res.-Atmos., 100, 2893–2903, https://doi.org/10.1029/94jd02832, 1995.
Russell, L. M., Pandis, S. N., and Seinfeld, J. H.: Aerosol production and growth in the marine boundary layer, J. Geophys. Res.-Atmos., 99, 20989–21003, https://doi.org/10.1029/94jd01932, 1994.
Schobesberger, S., Franchin, A., Bianchi, F., Rondo, L., Duplissy, J., Kürten, A., Ortega, I. K., Metzger, A., Schnitzhofer, R., Almeida, J., Amorim, A., Dommen, J., Dunne, E. M., Ehn, M., Gagné, S., Ickes, L., Junninen, H., Hansel, A., Kerminen, V.-M., Kirkby, J., Kupc, A., Laaksonen, A., Lehtipalo, K., Mathot, S., Onnela, A., Petäjä, T., Riccobono, F., Santos, F. D., Sipilä, M., Tomé, A., Tsagkogeorgas, G., Viisanen, Y., Wagner, P. E., Wimmer, D., Curtius, J., Donahue, N. M., Baltensperger, U., Kulmala, M., and Worsnop, D. R.: On the composition of ammonia–sulfuric-acid ion clusters during aerosol particle formation, Atmos. Chem. Phys., 15, 55–78, https://doi.org/10.5194/acp-15-55-2015, 2015.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics: From air pollution to climate change, John Wiley and Sons, Inc., New Jersey, 2016.
Skrabalova, L., Brus, D., Anttila, T., Zdimal, V., and Lihavainen, H.: Growth of sulphuric acid nanoparticles under wet and dry conditions, Atmos. Chem. Phys., 14, 6461–6475, https://doi.org/10.5194/acp-14-6461-2014, 2014.
Stangl, C. M., Krasnomowitz, J. M., Apsokardu, M. J., Tiszenkel, L., Ouyang, Q., Lee, S., and Johnston, M. V.: Sulfur Dioxide Modifies Aerosol Particle Formation and Growth by Ozonolysis of Monoterpenes and Isoprene, J. GEophys. Res.-Atmos., 124, 4800–4811, https://doi.org/10.1029/2018jd030064, 2019.
Stolzenburg, D., Fischer, L., Vogel, A. L., Heinritzi, M., Schervish, M., Simon, M., Wagner, A. C., Dada, L., Ahonen, L. R., Amorim, A., Baccarini, A., Bauer, P. S., Baumgartner, B., Bergen, A., Bianchi, F., Breitenlechner, M., Brilke, S., Mazon, S. B., Chen, D. X., Dias, A., Draper, D. C., Duplissy, J., Haddad, I., Finkenzeller, H., Frege, C., Fuchs, C., Garmash, O., Gordon, H., He, X., Helm, J., Hofbauer, V., Hoyle, C. R., Kim, C., Kirkby, J., Kontkanen, J., Kuerten, A., Lampilahti, J., Lawler, M., Lehtipalo, K., Leiminger, M., Mai, H., Mathot, S., Mentler, B., Molteni, U., Nie, W., Nieminen, T., Nowak, J. B., Ojdanic, A., Onnela, A., Passananti, M., Petaja, T., Quelever, L. L. J., Rissanen, M. P., Sarnela, N., Schallhart, S., Tauber, C., Tome, A., Wagner, R., Wang, M., Weitz, L., Wimmer, D., Xiao, M., Yan, C., Ye, P., Zha, Q., Baltensperger, U., Curtius, J., Dommen, J., Flagan, R. C., Kulmala, M., Smith, J. N., Worsnop, D. R., Hansel, A., Donahue, N. M., and Winkler, P. M.: Rapid growth of organic aerosol nanoparticles over a wide tropospheric temperature range, P. Natl. Acad. Sci. USA, 115, 9122–9127, https://doi.org/10.1073/pnas.1807604115, 2018.
Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., Wagner, R., Simon, M., Lehtipalo, K., Williamson, C., Craven, J. S., Duplissy, J., Adamov, A., Almeida, J., Bernhammer, A.-K., Breitenlechner, M., Brilke, S., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Gysel, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Keskinen, H., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lawler, M., Leiminger, M., Mathot, S., Möhler, O., Nieminen, T., Onnela, A., Petäjä, T., Piel, F. M., Miettinen, P., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Sipilä, M., Smith, J. N., Steiner, G., Tomè, A., Virtanen, A., Wagner, A. C., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Carslaw, K. S., Curtius, J., Dommen, J., Kirkby, J., Kulmala, M., Riipinen, I., Worsnop, D. R., Donahue, N. M., and Baltensperger, U.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 533, 527–531, https://doi.org/10.1038/nature18271, 2016.
Vanhanen, J., Mikkila, J., Lehtipalo, K., Sipila, M., Manninen, H. E., Siivola, E., Petaja, T., and Kulmala, M.: Particle size magnifier for nano-CN detection, Aerosol Sci. Tech., 45, 533–542, https://doi.org/10.1080/02786826.2010.547889, 2011.
Varanda Rizzo, L., Roldin, P., Brito, J., Backman, J., Swietlicki, E., Krejci, R., Tunved, P., Petäjä, T., Kulmala, M., and Artaxo, P.: Multi-year statistical and modeling analysis of submicrometer aerosol number size distributions at a rain forest site in Amazonia, Atmos. Chem. Phys., 18, 10255–10274, https://doi.org/10.5194/acp-18-10255-2018, 2018.
Vehkamäki, H., Kulmala, M., Napari, I., Lehtinen, E. J., Timmreck, C., Noppel, M., and Laaksonen, A.: An improved parameterization for sulfuric acid-water nucleation rates for tropospheric and stratospheric conditions, J. Geophys. Res., 107, D22, https://doi.org/10.1029/2002JD002184, 2002.
Vehkamäki, H., McGrath, M. J., Kurtén, T., Julin, J., Lehtinen, K. E. J., and Kulmala, M.: Rethinking the application of the first nucleation theorem to particle formation, J. Chem. Phys., 136, 094107, https://doi.org/10.1063/1.3689227, 2012.
Wang, J., Krejci, R., Giangrande, S., Kuang, C., Barbosa, H. M. J., Brito, J., Carbone, S., Chi, X., Comstock, J., Ditas, F., Lavric, J., Manninen, H. E., Mei, F., Moran-Zuloaga, D., Pöhlker, C., Pöhlker, M. L., Saturno, J., Schmid, B., Souza, R. A. F., Springston, S. R., Tomlinson, J. M., Toto, T., Walter, D., Wimmer, D., Smith, J. N., Kulmala, M., Machado, L. A. T., Artaxo, P., Andreae, M. O., Petäjä, T., and Martin, S. T.: Amazon boundary layer aerosol concentration sustained by vertical transport during rainfall, Nature, 539, 416–419, https://doi.org/10.1038/nature19819, 2016.
Wang, M. and Penner, J. E.: Aerosol indirect forcing in a global model with particle nucleation, Atmos. Chem. Phys., 9, 239–260, https://doi.org/10.5194/acp-9-239-2009, 2009.
Yao, L., Garmash, O., Bianchi, F., Zheng, J., Yan, C., Kontkanen, J., Junninen, H., Mazon, S. B., Ehn, M., Paasonen, P., Sipila, M., Wang, M. Y., Wang, X. K., Xiao, S., Chen, H. F., Lu, Y. Q., Zhang, B. W., Wang, D. F., Fu, Q. Y., Geng, F. H., Li, L., Wang, H. L., Qiao, L. P., Yang, X., Chen, J. M., Kerminen, V. M., Petaja, T., Worsnop, D. R., Kulmala, M., and Wang, L.: Atmospheric new particle formation from sulfuric acid and amines in a Chinese megacity, Science, 361, 278–281, https://doi.org/10.1126/science.aao4839, 2018.
You, Y., Kanawade, V. P., de Gouw, J. A., Guenther, A. B., Madronich, S., Sierra-Hernández, M. R., Lawler, M., Smith, J. N., Takahama, S., Ruggeri, G., Koss, A., Olson, K., Baumann, K., Weber, R. J., Nenes, A., Guo, H., Edgerton, E. S., Porcelli, L., Brune, W. H., Goldstein, A. H., and Lee, S.-H.: Atmospheric amines and ammonia measured with a chemical ionization mass spectrometer (CIMS), Atmos. Chem. Phys., 14, 12181–12194, https://doi.org/10.5194/acp-14-12181-2014, 2014.
Young, L. H., Benson, D. R., Kameel, F. R., Pierce, J. R., Junninen, H., Kulmala, M., and Lee, S.-H.: Laboratory studies of H2SO4∕H2O binary homogeneous nucleation from the SO2+OH reaction: evaluation of the experimental setup and preliminary results, Atmos. Chem. Phys., 8, 4997–5016, https://doi.org/10.5194/acp-8-4997-2008, 2008.
Yu, F. and Luo, G.: Simulation of particle size distribution with a global aerosol model: contribution of nucleation to aerosol and CCN number concentrations, Atmos. Chem. Phys., 9, 7691–7710, https://doi.org/10.5194/acp-9-7691-2009, 2009.
Yu, F., Luo, G., Pryor, S. C., Pillai, P. R., Lee, S. H., Ortega, J., Schwab, J. J., Hallar, A. G., Leaitch, W. R., Aneja, V. P., Smith, J. N., Walker, J. T., Hogrefe, O., and Demerjian, K. L.: Spring and summer contrast in new particle formation over nine forest areas in North America, Atmos. Chem. Phys., 15, 13993–14003, https://doi.org/10.5194/acp-15-13993-2015, 2015.
Yu, H. and Lee, S. H.: A chemical ionization mass spectrometer for the detection of atmospheric amines, Environ. Chem., 9, 190–201, 2012.
Yu, H., McGraw, R., and Lee, S. H.: Effects of amines on formation of sub-3 nm particles and their subsequent growth, Geophys. Res. Lett., 39, L02807, https://doi.org/10.1029/2011gl050099, 2012.
Yu, H., Dai, L., Zhao, Y., Kanawade, V. P., Tripathi, S. N., Ge, X., Chen, M., and Lee, S.-H.: Laboratory observations of temperature and humidity dependencies of nucleation and growth rates of sub-3 nm particles, J. Geophys. Res., 122, 1919–1929, https://doi.org/10.1002/2016JD025619, 2017a.
Yu, H., Ren, L., and Kanawade, V. P.: New Particle Formation and Growth Mechanisms in Highly Polluted Environments, Current Pollution Reports, 3, 245–253, https://doi.org/10.1007/s40726-017-0067-3, 2017b.
Zhang, R., Khalizov, A. F., Wang, L., Hu, M., and Xu, W.: Nucleation and growth of nanoparticles in the atmosphere, Chem. Rev., 112, 957–2011, 2012.
Zollner, J. H., Glasoe, W. A., Panta, B., Carlson, K. K., McMurry, P. H., and Hanson, D. R.: Sulfuric acid nucleation: power dependencies, variation with relative humidity, and effect of bases, Atmos. Chem. Phys., 12, 4399–4411, https://doi.org/10.5194/acp-12-4399-2012, 2012.