the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 20 Jun 2019
| 20 Jun 2019
Significant source of secondary aerosol: formation from gasoline evaporative emissions in the presence of SO2 and NH3
Tianzeng Chen
Yongchun Liu
Peng Zhang
Changgeng Liu
Jun Liu
Gasoline evaporative emissions have become an important anthropogenic source of urban atmospheric volatile organic compounds (VOCs) and secondary organic aerosol (SOA). These emissions have a significant impact on regional air quality, especially in China where car ownership is growing rapidly. However, the contribution of evaporative emissions to secondary aerosol (SA) is not clear in an air pollution complex in which a high concentration of SO2 and NH3 was present. In this study, the effects of SO2 and NH3 on SA formation from unburned gasoline vapor were investigated in a 30 m3 indoor smog chamber. It was found that an increase in SO2 and NH3 concentrations (0–151 and 0–200 ppb, respectively) could linearly promote the formation of SA, which could be enhanced by a factor of 1.6–2.6 and 2.0–2.5, respectively. Sulfate was most sensitive to the SO2 concentration, followed by organic aerosol, which was due not only to the acid catalytic effect, but was also related to the formation of organic sulfur-containing compounds. In the case of an increasing NH3 concentration, ammonium nitrate increased more significantly than organic aerosol, and nitrogen-containing organics were also enhanced, as revealed by the results of positive matrix factorization (PMF) analysis. New particle formation (NPF) and particle size growth were also significantly enhanced in the presence of SO2 and NH3. This work indicates that gasoline evaporative emissions will be a significant source of SA, especially in the presence of high concentrations of SO2 and NH3. Meanwhile, these emissions might also be a potential source of sulfur- and nitrogen-containing organics. Our work provides a scientific basis for the synergistic emission reduction of secondary aerosol precursors, including NOx, SO2, NH3, and particularly VOCs, to mitigate particulate matter (PM) pollution in China.
- Article
(1591 KB) - Full-text XML
-
Supplement
(1614 KB) - BibTeX
- EndNote





Many areas in China such as the Beijing–Tianjin–Hebei region (BTH), Yangtze River Delta (YRD), Sichuan Basin, and Pearl River Delta (PRD) are suffering from severe haze events (Li et al., 2017; Sun et al., 2016; Shen et al., 2015; He et al., 2014; Huang et al., 2014; Guo et al., 2014; Tan et al., 2009). Haze pollution has attracted widespread attention in recent years because of its adverse effects on human health, climate change, and visibility (Thalman et al., 2017; Davidson et al., 2005; Pöschl, 2005).
During haze events, high concentrations of SO2, NH3, and volatile organic compounds (VOCs) have always been observed (Zou et al., 2015; Liu et al., 2013; Meng et al., 2011; Yang et al., 2009), which are the precursors of secondary aerosol (SA). Although the emission of SO2 has decreased continuously since 2005 (Lu et al., 2010), China is still the largest contributor of SO2 emissions in the world, mainly owing to the great demand for coal combustion (Bauduin et al., 2016). Also, high concentrations of SO2 of more than 100 ppb (parts per billion) have been observed in northern China, especially during the heating period (Hou et al., 2016; Tong et al., 2016; Yang et al., 2009). As for atmospheric NH3, as an alkaline inorganic gas, its main emission source is agricultural practices in China (Zhang et al., 2018; Fu et al., 2015). Vehicles equipped with three-way catalytic converters also contribute to NH3 emissions in urban areas (Sun et al., 2017). Sometimes, high concentrations of NH3 of up to 100 ppb have been observed in Beijing, China (Ianniello et al., 2010), which are mainly derived from regional transportation for agricultural activity and fertilizer use, but the influence of local traffic emissions in Beijing cannot be excluded (Pan et al., 2016; Kang et al., 2016). With respect to VOCs, aromatics from anthropogenic sources (especially vehicle-related sources in urban areas) are critical secondary organic aerosol (SOA) precursors (T. Liu et al., 2015a; Gordon et al., 2014; Platt et al., 2013; Calvert et al., 2002). These aromatics could react with oxidants (e.g., O3, OH, and NO3 radicals) and undergo multi-step oxidative processes to form multifunctional products, which have sufficiently low volatility to contribute to SOA via gas–particle partitioning (Hallquist et al., 2009; Atkinson and Arey, 2003).
Research has shown that SA makes a significant contribution (30 %–77 %) to PM2.5 (particles with a diameter less than 2.5 µm) during severe haze events in China (Huang et al., 2014; Guo et al., 2014; Jimenez et al., 2009). However, there is still a significant gap between the predicted SA derived from current atmospheric quality models and that observed in field observations (Zhao et al., 2018a; Yang et al., 2018; Zheng et al., 2015). Therefore, considering the characteristics of complex pollution in China, it is crucial to study the synergistic effects of SO2 and NH3 on the formation of SA, which have been considered an important potential source of SA formation (Zhao et al., 2018b; Chu et al., 2016; Liu et al., 2016; Santiago et al., 2012; Na et al., 2007).
A few studies have focused on the influence of SO2 or NH3 on SA formation. Jang and Kamens (2001) first reported the acid catalytical effect of acidic H2SO4 on the oxidation of atmospheric carbonyls. The promotion effect of SO2 was further found in SA formation from typical biogenic (e.g., isoprene and α-pinene) (Lin et al., 2013; Jaoui et al., 2008; Kleindienst et al., 2006; Edney et al., 2005) and anthropogenic (e.g., toluene, o-xylene, 1,3,5-trimethylbenzene, and gasoline vehicle exhaust) precursors (Chu et al., 2016; Liu et al., 2016; Santiago et al., 2012) through acid-catalyzed heterogeneous reactions (Jang et al., 2002, 2003a, b; Czoschke et al., 2003), which promote the reactive uptake process of organic species or enhance the formation of high-molecular-weight compounds (Liggio and Li, 2008, 2006; Liggio et al., 2007). With regard to the role of NH3 in SA formation, knowledge is still limited. In previous studies, inconsistent impacts of NH3 on SA formation have been reported under different precursor systems. For example, NH3 could elevate SA formation in the α-pinene–ozone oxidation system through acid–base reactions (Na et al., 2007), while the effects of NH3 neutralization were masked by other multiple factors and did not show significant influence on isoprene-derived SOA formation (Lin et al., 2013). The addition of NH3 even significantly reduced SA formation in the styrene–ozone system, which was caused by nucleophilic attack from the NH3 molecule, leading to rapid decomposition of the major aerosol products (Na et al., 2006). For the photo-oxidation of aromatic VOCs (e.g., toluene, o-, m-, and p-xylene), the presence of NH3 could facilitate new particle formation (NPF) and particle growth, subsequently leading to increased SA formation (Li et al., 2018; T. Liu et al., 2015b).
At the present time, the effects of SO2 and NH3 on SA formation have rarely been studied under highly complex pollution conditions (Chu et al., 2016). Vehicular evaporative emissions have been reported to be non-negligible contributors (39.20 %) to ambient VOCs from anthropogenic sources compared with vehicular tailpipe emissions (H. Liu et al., 2017). In addition to short-chain alkanes, a certain proportion of aromatics and alkanes (C6 to C12) were also contained in the evaporative emissions (Liu et al., 2008; Zhang et al., 2013). Previous studies have reported that aromatics and long-chain (C6 to C19) alkanes, which are intermediate-volatility organic compounds (IVOCs) (Donahue et al., 2006), could contribute to SOA formation (Pye and Pouliot, 2012; Tkacik et al., 2012; Lim and Ziemann, 2005). Therefore, it is necessary to study the influence of SO2 and NH3 on SA formation from evaporative emissions.
In this study, unburned gasoline vapor was used as a substitute for evaporative emissions, and the roles of SO2 and NH3 in SA formation from the photo-oxidation of unburned gasoline vapor were investigated in a 30 m3 indoor smog chamber, in order to understand the formation potential of SA from the oxidation of gasoline vapor in the cocktail of pollutants in Beijing. The respective influences of SO2 and NH3 on both the microphysics and chemistry of SA formation were examined. Meanwhile, the chemical compositions of the formed SOA in the presence of SO2 and NH3 were further explored by applying positive matrix factorization (PMF) analysis. The formation potentials of SA and sulfur- and nitrogen-containing organics from vehicular evaporative emissions in the presence of SO2 and NH3 were evaluated and discussed.
2.1 Gasoline fuel
The utilized gasoline fuel with grade 92 was collected (refer to the standard method for manual sampling of petroleum liquids, GB/T 4756-2015) from a gas station located in Beijing. The gasoline complies with the China V gasoline fuel standard. It contains 65.1 % (v∕v) alkanes (C6 to C12), 22.8 % (v∕v) aromatics (mainly including benzene, toluene, xylene, and trimethylbenzene), and 12.1 % (v∕v) alkenes. The composition of the gasoline is similar to the gasoline collected in northern China reported by Tang et al. (2015) and could represent the gasoline used in most areas of China for studying SA formation potential. Details on the gasoline composition are given in Table S1 in the Supplement.
2.2 Smog chamber facility
A series of photochemical experiments with unburned gasoline vapor in the absence or presence of SO2 or NH3 were performed in a 30 m3 indoor smog chamber at the Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences (RCEES-CAS). The detailed schematic structure of the indoor smog chamber is given in Fig. S1 in the Supplement and described elsewhere (Chen et al., 2019a, b). Briefly, the cuboid chamber reactor ( m, m−1) was irradiated by 120 UV lamps (Philips) with peak intensity at 365 nm, providing an NO2 photolysis rate of 0.55 min−1. The interior was coated with 125 µm thick FEP100 film (DuPont™, US) and the chamber was located in a temperature-controlled room in which the temperature (T) and relative humidity (RH) could be controlled mechanically. A three-wing stainless-steel fan coated with Teflon was installed inside the reactor to guarantee that the gas- and particle-phase species mix sufficiently before photochemical reaction.
The chamber was also equipped with a series of gas- and particle-phase monitoring instruments. For gaseous NOx, O3, and SO2, a chemiluminescence analyzer (model 42i-TL, Thermo Fisher Scientific, USA), a UV photometric analyzer (model 49i, Thermo Fisher Scientific, USA), and a pulsed fluorescence analyzer (model 43i, Thermo Fisher Scientific, USA) were used to monitor the concentrations in real time, respectively. The VOC species in gasoline were measured with a gas chromatograph (7890B GC, Agilent, USA) equipped with a DB-624 column (60 m × 0.25 mm × 1.40 µm; Agilent, USA) and a mass spectrometry detector (5977A MS, Agilent, USA) (GC-MS). In addition, proton-transfer reaction time-of-flight mass spectrometry (PTR-ToF) (Ionicon Analytik GmbH, Austria) was also used for the measurement of gas-phase hydrocarbons and their intermediate products (Yuan et al., 2017). The size distribution and number concentration of the formed particulate matter (PM) were measured using a scanning mobility particle sizer (SMPS; TSI, USA), which was composed of a differential mobility analyzer (DMA; 3080 Classifier, TSI, USA) coupled with a condensation particle counter (CPC; 3776, TSI, USA). The mass concentration was estimated based on the volume concentration and the density of PM calculated from the equation , where dva is the mean vacuum aerodynamic diameter measured by an Aerodyne high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS) and dm is the mean electrical mobility diameter measured by SMPS (DeCarlo et al., 2004). The calculated density of PM ranged from 1.5 to 1.6 g cm−3 in the different reaction systems, which was in the range of density of SOA derived from aromatic hydrocarbons (1.24–1.48 g cm−3) (Sato et al., 2010) and ammonium nitrate (NH4NO3, 1.72 g cm−3) (Bahreini et al., 2005) and was comparable with previous studies (Li et al., 2018). The mass concentration and chemical composition of PM were simultaneously monitored using a high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS; Aerodyne Research Inc. USA). For all experiments, the HR-ToF-AMS operated in a cycle including two modes, 3 min V mode and 2 min W mode. Specifically, the V mode (higher signal) can obtain the mass concentrations of the aerosols and the W mode (higher resolution) can obtain high-resolution mass spectral data. The inlet flow rate, ionization efficiency (IE), and particle sizing were calibrated according to the standard protocols (Drewnick et al., 2005; Jimenez et al., 2003; Jayne et al., 2000) using the size-selected pure ammonium nitrate (AN) particles. All HR-ToF-AMS data were analyzed with the ToF-AMS analysis toolkit SQUIRREL 1.57I/PIKA 1.16I version in Igor Pro Version 6.37. HR-ToF-AMS results were also corrected using the mass concentration derived from SMPS according to the same method as Gordon et al. (2014); the details of this correction are shown in the Supplement. As for the RH control system, it is achieved by vaporizing Milli-Q ultrapure water contained in a 5.0 L high-pressure-resistant container, and the water vapor is flushed with purified dry zero air into the chamber. T and RH were monitored in real time using a hydro-thermometer (Vaisala HMP110) during the entirety of each experiment.
2.3 Wall loss corrections
The measured particle concentration was corrected in accordance with the relationship between the deposition rate (kdep) and particle diameter (Dp, nm) (i.e., ), which was described by Takekawa et al. (2003). The wall loss rates of NO2, NO, O3, SO2, and VOC species were determined to be , , , , and min−1, respectively. Therefore, the wall loss of gas-phase species was evaluated to be less than 5 % of their maximum concentration in this study.
Wall losses of semi-volatile organic compounds (SVOCs) and low-volatility organic compounds (LVOCs) would lead to a substantial underestimation of SA formation (Krechmer et al., 2016; Ye et al., 2016; Zhang et al., 2015, 2014), which is caused by the competition between vapor types condensing onto particles versus onto chamber walls. This competition could be evaluated by the corresponding timescales associated with reaching gas-to-particle partitioning equilibrium () and vapor wall loss (τg-w) (Zhang et al., 2014), and this underestimation of SA formation could be approximately quantified by the ratio of these two timescales (i.e., ). According to the methods described by Zhang et al. (2014), and τg-w could be estimated assuming an upper bound and a lower bound of the molecular mass of organic vapor (MW) (100–300 g mol−1) (as discussed in the Supplement). In order to accurately quantify the SA formation, the underestimation caused by the loss of SVOCs and LVOCs (including gaseous H2SO4) to the chamber walls was taken into account in this study. In this study, the SA yields were underestimated by a factor of 1.97–2.82 when considering the ratio of these two timescales (i.e., ), which showed a decreasing trend with the increase in SO2 and NH3 initial concentrations, suggesting that an increasing proportion of vapor is partitioned onto the suspended particle surface rather than the chamber wall.
2.4 Experimental conditions
Prior to each experiment, the chamber reactor was flushed by purified and dry zero air for about 24–36 h at a flow rate of 100 L min−1 until almost no gas-phase species (i.e., NOx, O3, and SO2) could be detected (<1 ppb) and the particle number concentration was <10 cm−3. Before the experiments, the chamber was humidified to ∼50 % RH by passing purified zero air through ultrapure water (18.2 MΩ; Millipore Milli-Q). After that, a known volume of liquid gasoline (100 µL) was injected into the chamber through a heated Teflon line system (∼100 ∘C) carried by purified dry zero air to ensure that all were evaporated into the chamber. Subsequently, NO, SO2, and/or NH3 were successively injected into the chamber from standard gas cylinders using mass flow controllers. The initial VOCs∕NOx ratio (ppb C ppb−1) was kept constant (Table 1). In order to reduce the adsorption of NH3 in the pipeline, the NH3 flow in a bypass line was balanced for about 30 min before it was injected into the chamber. The concentrations of NO and SO2 were continuously monitored until they were stable, ensuring that the gaseous species mixed well in the chamber. For the concentration of NH3, the value was estimated according to the amount of NH3 introduced and the volume of the reactor chamber. The experiment was then conducted for about 8 h after turning off the fan and turning on the UV lights. All the experiments were performed at a temperature of 26±1 ∘C and wet conditions (RH %). The detailed experimental conditions are listed in Table 1. The letters in the abbreviations represent the reactants introduced into the chamber reactor for each experiment. For example, SGN is an experiment with the presence of sulfur dioxide (S), gasoline vapor (G), and nitrogen oxides (N). Four experiments (Exps. SGN1, SGN2, SGN3, and SGN4) were carried out at different SO2 initial concentrations. AGN is an experiment with the presence of ammonia (A), gasoline vapor (G), and nitrogen oxides (N). Two experiments (Exps. AGN1 and AGN2) were carried out at different NH3 initial concentrations.
Table 1Summary of experimental conditions in this study.
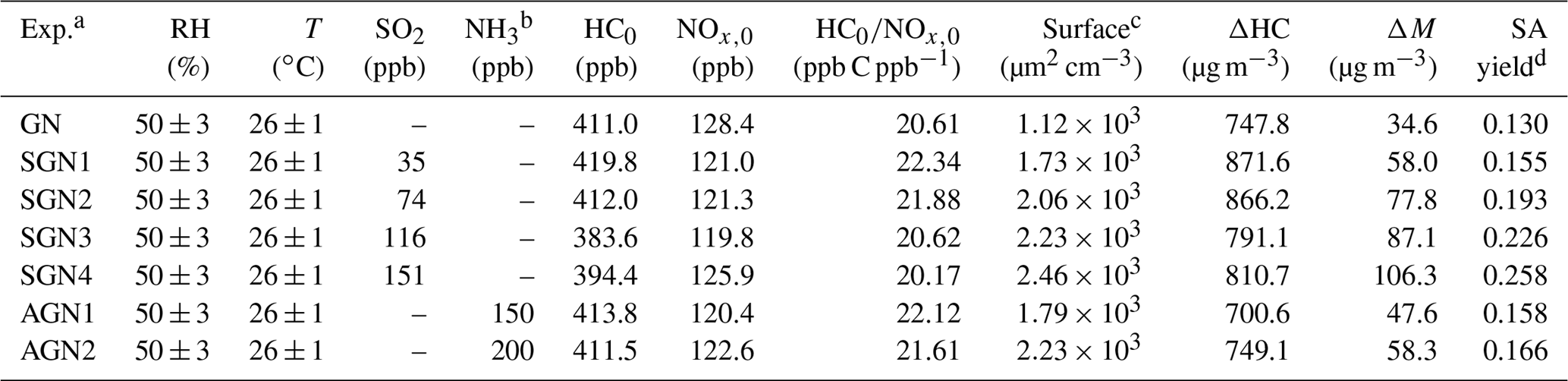
a Letters in the abbreviations represent the reactants introduced into the chamber reactor; i.e., “G” represents gasoline, “N” represents nitrogen oxides, “S” represents sulfur dioxide, and “A” represents ammonia. b The concentration of NH3 is estimated by the amount of NH3 added and the volume of the smog chamber. c The surface area of aerosol particles measured by SMPS after 480 min of each experiment. d SA yield was calculated after taking vapor and particle wall loss into account.
3.1 Effect of SO2 and NH3 on the gas-phase species
Time-resolved concentrations of inorganic and organic gas-phase species during the photo-oxidation of gasoline ∕ NOx in the absence or presence of SO2 and NH3 are shown in Figs. S2 and S3 in the Supplement, respectively. After turning on the UV lights, NO was rapidly converted to NO2. At the same time, O3 was gradually generated, with a maximum concentration of up to 350 ppb (Fig. S2). As shown in Fig. S2, there was no obvious difference in the variation of NOx and O3 in the presence of SO2 or NH3. Additionally, the decay of typical VOC precursors (e.g., benzene, toluene, methylcyclopentane, methylcyclohexane) measured by PTR-ToF and GC-MS are given in Fig. S3, which traced very closely with each other (Fig. S4 in the Supplement). There were also no observable differences in these precursor VOCs among these experiments. According to the decay curves of aromatic hydrocarbons, the OH radical concentrations were estimated to be (7.54–8.40) ×106 molec. cm−3, which were also similar among these experiments. This was consistent with the previous study conducted by Chu et al. (2016), who found that the presence of SO2 and NH3 did not significantly impact the OH concentration during the photo-oxidation of toluene in the presence of NOx.
However, the gas-phase intermediates formed during the photo-oxidation of gasoline ∕ NOx under different conditions, such as small-molecule oxygenated VOCs (OVOCs), could also be measured by PTR-ToF. The time series of OVOC concentration can vary with the concentration of SO2 and NH3. For example, we observed that acetic acid concentration decreased with the increased concentration of SO2 (Fig. S5 in the Supplement), suggesting that the uptake of acetic acid may be enhanced. This phenomenon was consistent with that reported by Liggio and Li (2006), who observed that the uptake of organic compounds under acidic conditions was enhanced significantly. Moreover, the presence of high concentrations of SO2 would generate gaseous H2SO4, which would contribute to the formation of the particle phase, as discussed in the next section. Similarly, the concentration of acetic acid also showed an obviously decreased trend in the presence of NH3 (Fig. S5 in the Supplement), which could be caused by the acid–base reaction or the uptake of acetic acid in the presence of NH3 (Y. Liu et al., 2015).
3.2 Role of SO2 in secondary aerosol formation
To investigate the effects of SO2 on SA formation from the photo-oxidation of gasoline ∕ NOx, smog chamber experiments with different SO2 initial concentrations were carried out (Table 1). As shown in Fig. 1, compared to the experiments without the addition of SO2, the SA concentration was enhanced to different degrees (1.6–2.6 times) in the presence of different SO2 concentrations (35–151 ppb; i.e., 100–431 µg m−3). As for each chemical species (i.e., organics, nitrate, sulfate, and ammonium), they all showed a trend of linear increase with the increase in SO2 concentration (Fig. 2), especially for the sulfate () and organic aerosol (). Previous studies have also revealed its promoting role in SA formation from different precursors (Zhao et al., 2018a; S. Liu et al., 2017; Liu et al., 2016; Díaz-de-Mera et al., 2017; Chu et al., 2016).
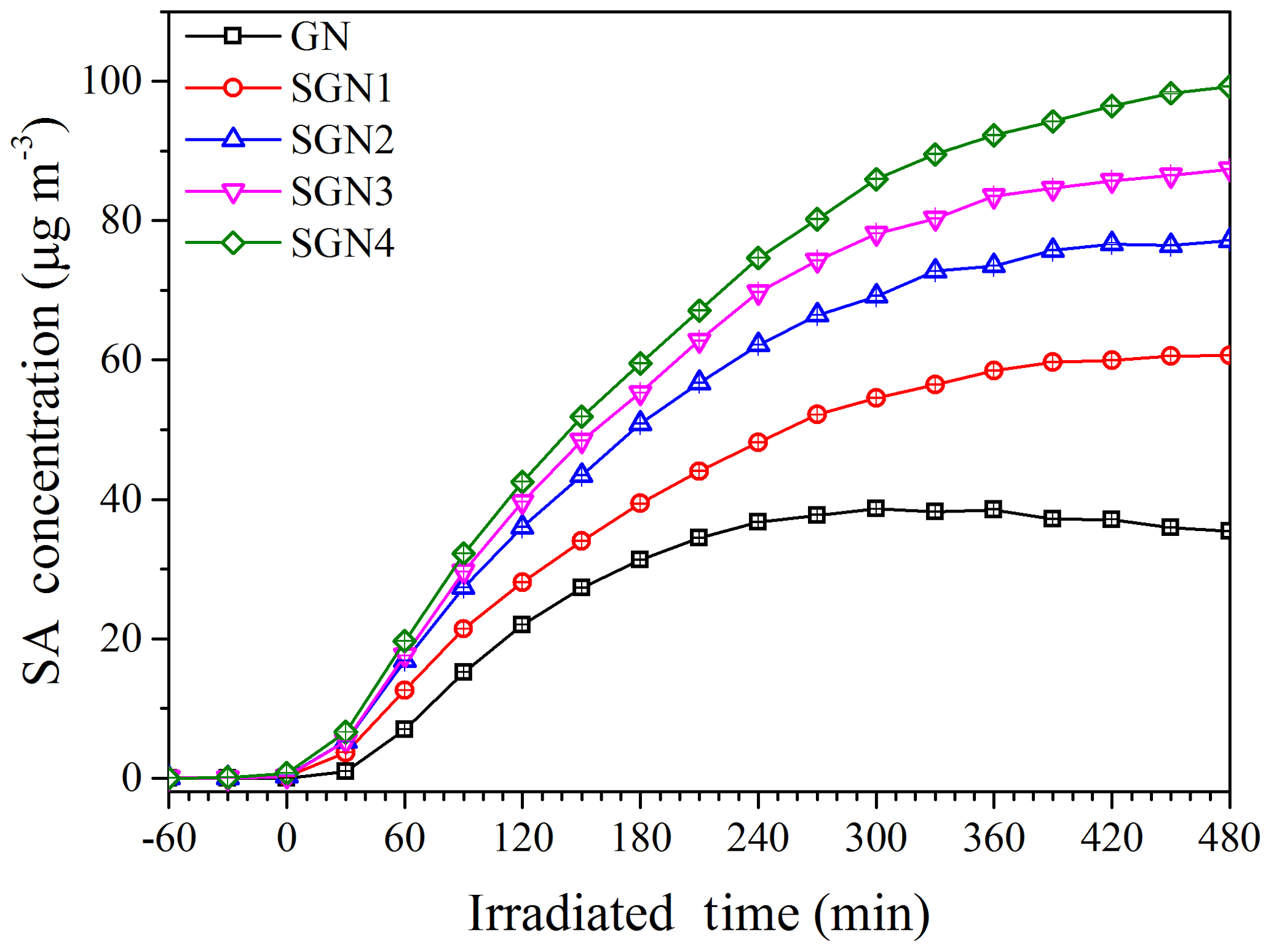
Figure 1Time series of secondary aerosol concentrations during the photo-oxidation experiments with different SO2 concentrations (Exps. GN, SGN1, SGN2, SGN3, and SGN4).
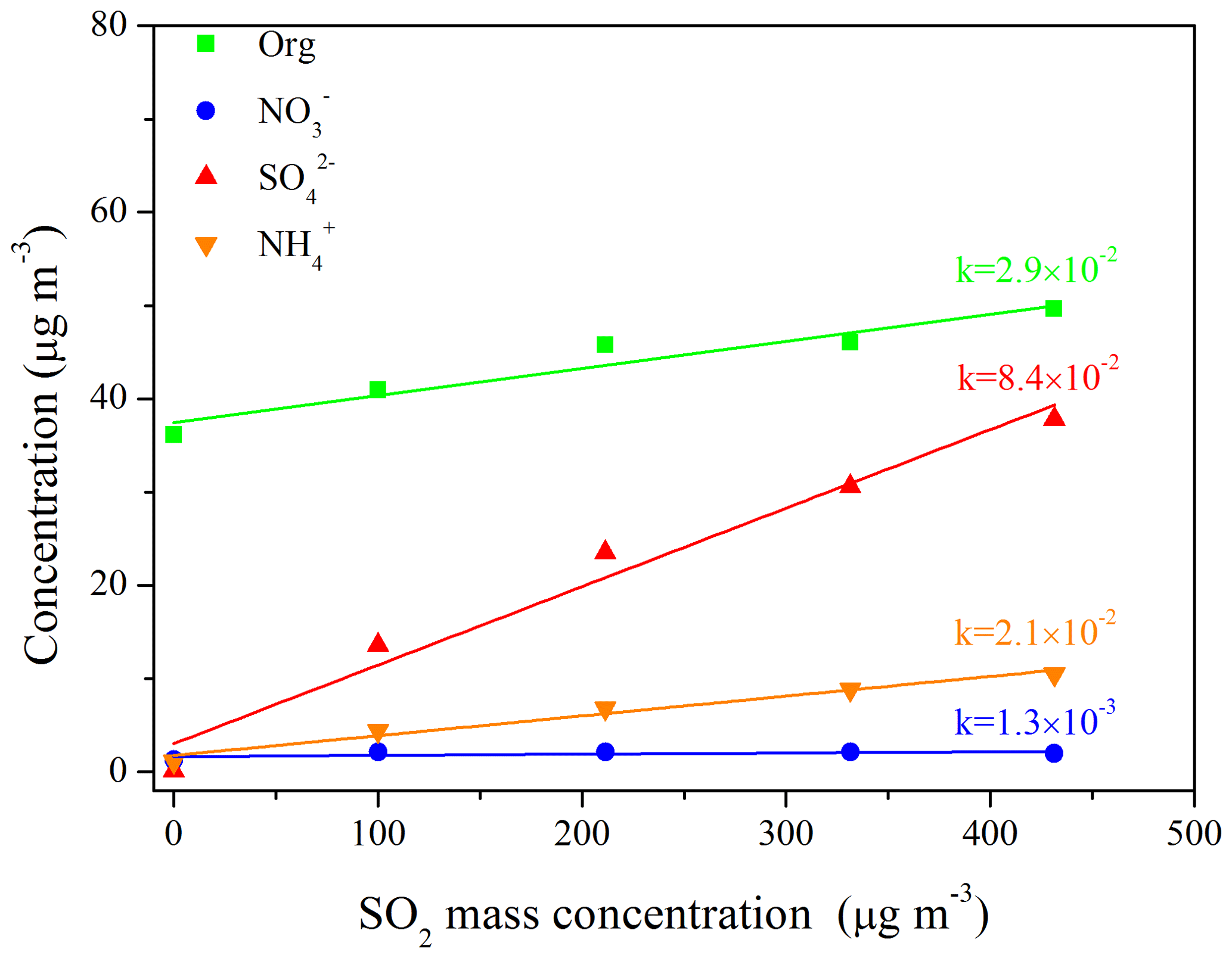
Figure 2Linear relationship between the concentration of chemical species (i.e., organic (green), nitrate (blue), sulfate (red), and ammonium (orange)) and SO2 under different SO2 initial concentration conditions (Exps. GN, SGN1, SGN2, SGN3, and SGN4). Each line represents a linear fitting, and the k values are the corresponding slopes for each chemical species.
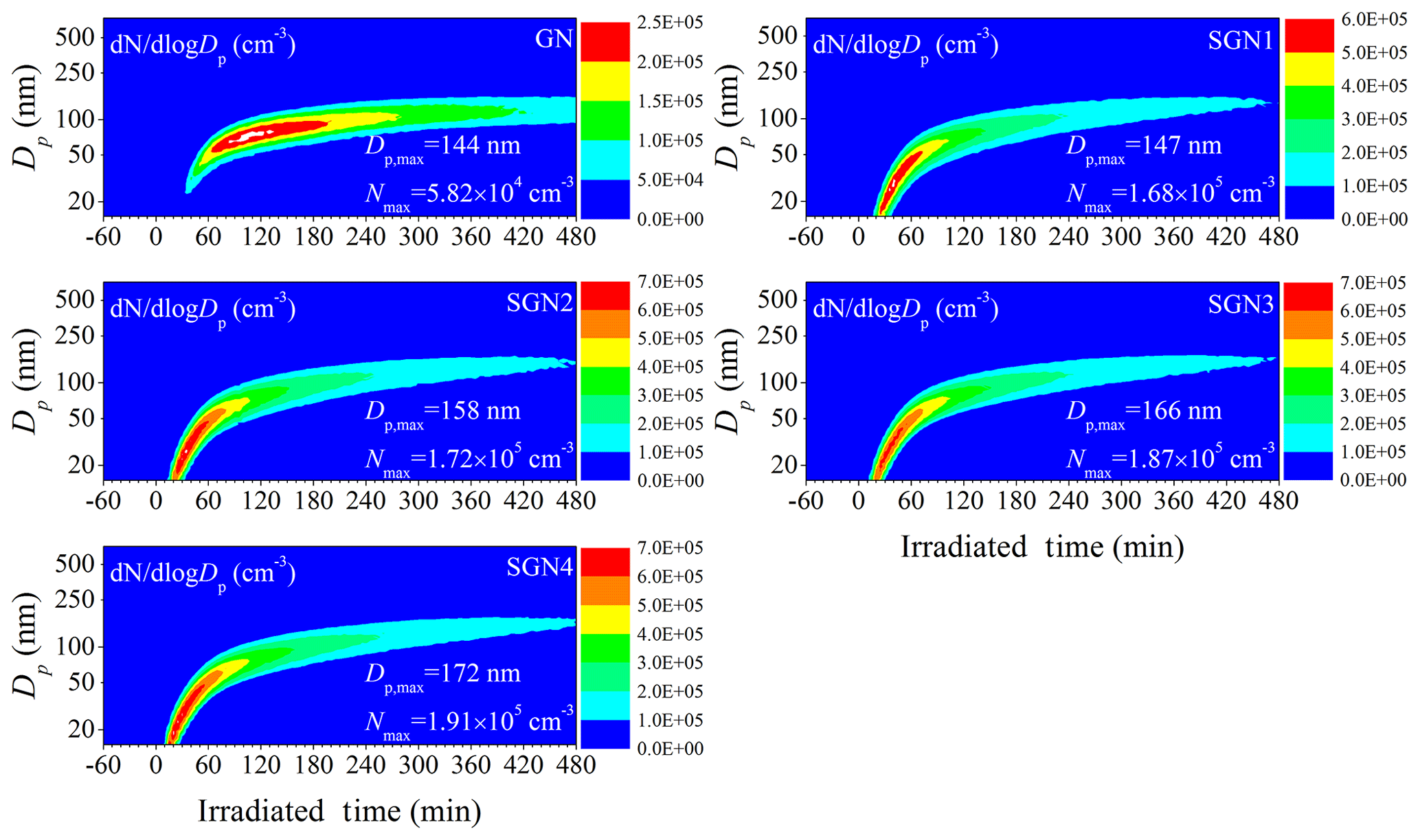
Figure 3Time series of the size distributions for the generated secondary aerosol during the photo-oxidation experiments with different SO2 initial concentrations (Exps. GN, SGN1, SGN2, SGN3, and SGN4). and Nmax represent the maximal diameter and number concentration of generated secondary aerosol, respectively, during each photo-oxidation experiment.
Additionally, the particle number concentrations and size growth were greatly enhanced by the presence of SO2. As evident from Fig. 3, the corresponding maximal particle number concentrations (5.82×104–1.91×105 cm−3) were significantly enhanced by a factor of 2.9–3.3 in the presence of SO2. This universal phenomenon has been reported by many studies (Díaz-de-Mera et al., 2017; S. Liu et al., 2017; Liu et al., 2016; Chu et al., 2016). For example, the maximal particle number concentrations were enhanced by the presence of SO2 (∼130 ppb) to 1 order of magnitude in the photo-oxidation of high concentrations of toluene ∕ NOx (Chu et al., 2016). For complex precursor systems (gasoline vehicle exhaust), Liu et al. (2016) have also found that under high SO2 concentration (∼150 ppb) conditions, the maximum particle number concentrations increased by 5.4–48 times compared to those without SO2 during the photo-oxidation of gasoline vehicle exhaust. This higher magnification of SO2 might be related to the different VOC composition between evaporative emissions and gasoline vehicle exhaust, especially the aromatic and IVOCs (H. Liu et al., 2017). Our recent study demonstrated that SOA formation could be significantly enhanced by the increase in aromatic content (Chen et al., 2019b). Those unspeciated organic emissions (e.g., IVOCs) from gasoline vehicle exhaust would also make a significant contribution to SOA formation (Jathar et al., 2014; Gordon et al., 2014). Moreover, a small amount of POA was present in the initial reaction systems in Liu et al. (2016). This enhanced SOA formation and the preexisting POA would provide larger surface areas for the condensation and heterogeneous uptake of low-volatility vapor (e.g., gaseous H2SO4), thus promoting a higher magnification in particle number concentrations in the presence of SO2. Higher initial mixing ratios of precursors (2.2–4.3 ppm) were also present in the reaction systems conducted by Liu et al. (2016), which would be further beneficial to SOA formation. In addition, size distributions of generated SA in smaller size ranges (4–160 nm) were also determined using another SMPS equipped with a nanometer differential mobility analyzer (nano-DMA), indicating that the new particle formation (NPF) phenomenon was enhanced significantly when the SO2 concentration increased (Fig. S6). The presence of high concentrations of SO2 would generate H2SO4, which would contribute to nucleation and increase the total particle number concentrations (Zhao et al., 2018a; Sipilä et al., 2010). As the SO2 concentration increased from 35 to 151 ppb, the maximal particle diameters (144–172 nm) became larger, which will have a direct impact on the scattering and absorption of light (Seinfeld and Pandis, 2016). An enhancement effect of SO2 on the surface area of particles was also observed. As shown in Table 1, the surface area of aerosol particles at the end of each experiment increased from 1.12×103 to 2.46×103 µm2 cm−3 when the SO2 concentration increased from 0 to 151 ppb. The larger surface area would be beneficial to the condensation and heterogeneous uptake of low-volatility vapor (Chapleski et al., 2016), consequently leading to higher SA yield in the presence of SO2 (Table 1) (Santiago et al., 2012). Additionally, it is worth noting that there was a discrepancy between the magnification of particle number concentrations, surface areas, and SO2 concentrations. On the one hand, there might be some particles, especially nanoclusters, lost to the chamber wall and not detected; on the other hand, the initial size of nanoclusters contributed from gaseous H2SO4 was small (sub-3 nm) (Chu et al., 2019; Sipilä et al., 2010) and could not be detected by our general SMPS. That is to say that the particle number concentrations and surface areas measured by our SMPS might be the particles after growing by collision. This could be supported by the enhancement in the particle diameters (144–172 nm) and sulfate concentrations (13–38 µg m−3) in the presence of SO2. After considering the underestimation of particle formation (factor of 1.97–2.82; Sect. 2.3), the sulfate concentrations will be enhanced by a factor of 5.8 when comparing experiments SGN 1 and SGN 4.
In order to further investigate the role of SO2 in the chemistry of SOA formation, the particle acidities were estimated using the E-AIM model (Model II: H+ – – – – H2O) (Clegg and Brimblecombe, 2005; Wexler and Clegg, 2002; Clegg et al., 1998). The concentrations of chemical components (i.e., , , and ) at the time when the SOA formation rate reached its peak were used as the inputs of the model. As shown in Fig. 4, the H+ concentration was increased from 8.5 to 32.5 nmol m−3 with the increase in SO2 concentration under moderate humidity conditions (RH = 50 %), and the higher SOA concentration and SOA yield could be well explained by the enhancement of the particle acidities (R2=0.960 and R2=0.986, respectively). The higher SOA concentration and SOA yield were related to the acid-catalyzed reactions of multifunctional aldehydes (e.g., glyoxal and methylglyoxal), which were the products of aromatic hydrocarbons in the gasoline vapor through gas-phase photo-oxidation. Hemiacetals, acetals, and alcohols could be generated through the acid-catalyzed heterogeneous reactions of glyoxal (Czoschke et al., 2003; Jang et al., 2002). These low-vapor-pressure products generated from heterogeneous reactions preferentially contribute to the SOA formation (Kroll and Seinfeld, 2008; Cao and Jang, 2007; Casale et al., 2007; Jang et al., 2002).
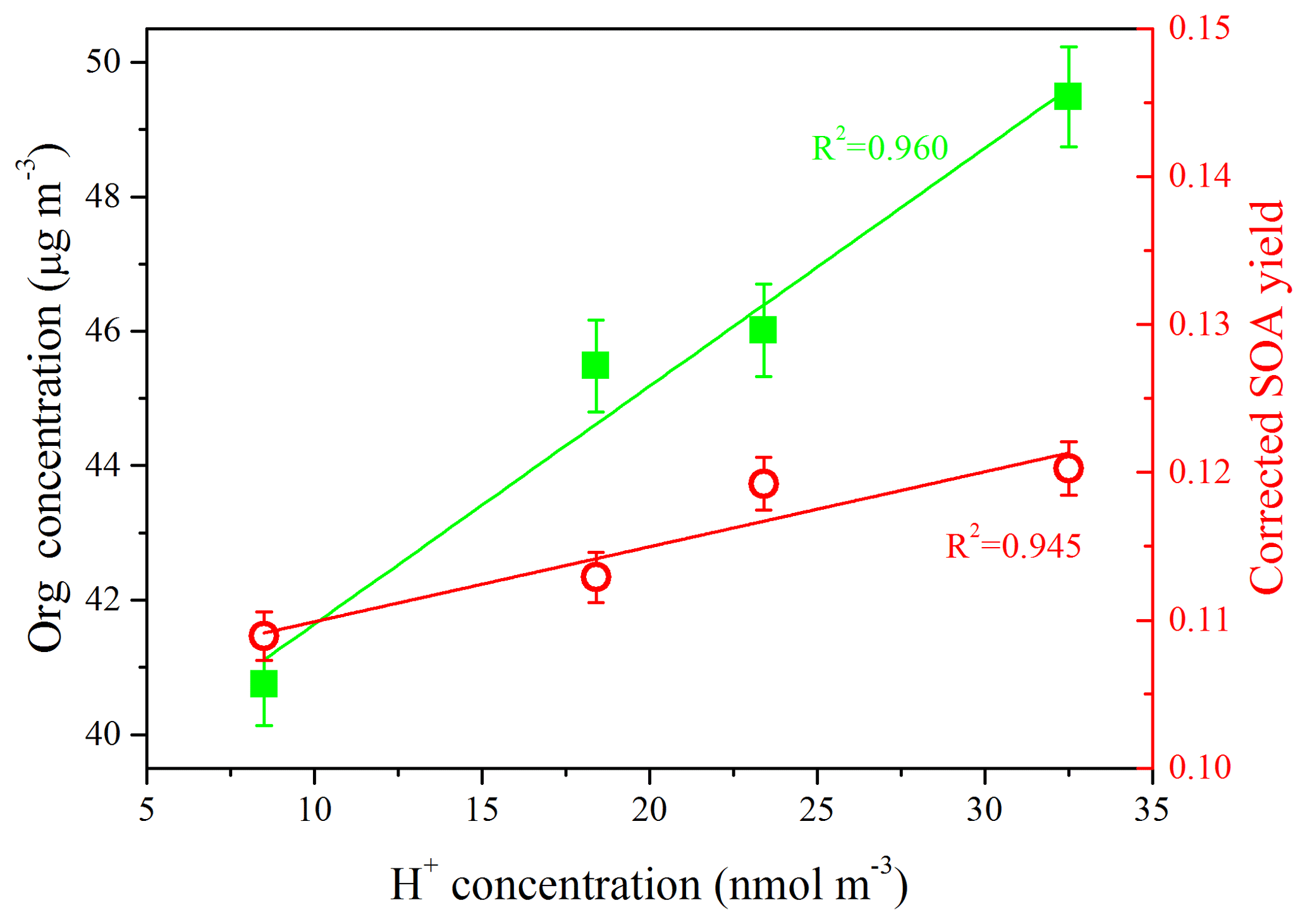
Figure 4Relationship between SOA concentration (left y axis), corrected SOA yield (right y axis), and H+ concentration, which was used to characterize the particle acidities. The H+ concentration presented in this plot was the value when the SOA formation rate reached the peak during each experiment (Exps. SGN1, SGN2, SGN3, and SGN4).
In addition, the sulfur-containing organics formed in the presence of SO2 might be another reason for the increase in SOA yield (Kundu et al., 2013; Liggio et al., 2005). Jaoui et al. (2008) have reported that the acidic aerosol generated in the presence of SO2 could lead to sulfur-incorporating reactions in the particle phase during the photo-oxidation of α-pinene ∕ toluene ∕ NOx mixtures. Sulfur-containing organics could be generated via reactions of organic species (e.g., polycyclic aromatic hydrocarbons (PAHs), C10–C12 alkanes, alcohols, epoxides) with sulfate, bisulfate, or sulfuric acid, especially under high relative humidity and acidity conditions (Riva et al., 2015, 2016; Huang et al., 2015; Hatch et al., 2011; Surratt et al., 2007; Liggio et al., 2005). Huang et al. (2015) have revealed that sulfur-containing organics with R–O– functional groups will yield S-bearing organic fragments (CxHyOzS) during ionization, which subsequently could be detected by HR-ToF-AMS and used as marker ions to quantify them. In our gasoline ∕ NOx experiments in the presence of SO2, the ions CSO+, , and could be separated (Fig. S7), although uncertainty might be induced in the peak fitting of the highly abundant ions , , and . These characteristic ions (i.e., CSO+, , and ) have also been observed from sulfur-containing organics in previous field measurements (Huang et al., 2015; Farmer et al., 2010). According to the estimation method for sulfur-containing organics mentioned in Huang et al. (2015), we found that the signal of these ions and the concentrations of sulfur-containing organics increased with the SO2 initial concentration (Fig. 5). The conservative lower-bound-estimated concentrations of sulfur-containing organics (13–26 ng m−3) were comparable to those (∼20 ng m−3) observed in the mid-Atlantic United States, which were derived from biogenic and anthropogenic hydrocarbons (Meade et al., 2016). Additionally, it should be noted that the sulfur-containing organics concentration in this study might be underestimated by the HR-ToF-AMS when considering that one cannot resolve all the sulfur-containing fragments that may exist, and some of the sulfur-containing organics might fragment into masses that do not contain sulfur and are thus quantified as organic. Furthermore, the relative ionization efficiency (RIE) for the sulfur-containing organics fragments was assumed to be equivalent to the remainder of the organics (1.3), since the RIE value for sulfur-containing organics is unknown. This may introduce an additional uncertainty to the quantitation of sulfur-containing organics. Therefore, photo-oxidation of gasoline vapor in the presence of SO2 might be a noteworthy source of sulfur-containing organics, although the concentration was low compared to that of generated (∼0.1 % of ).
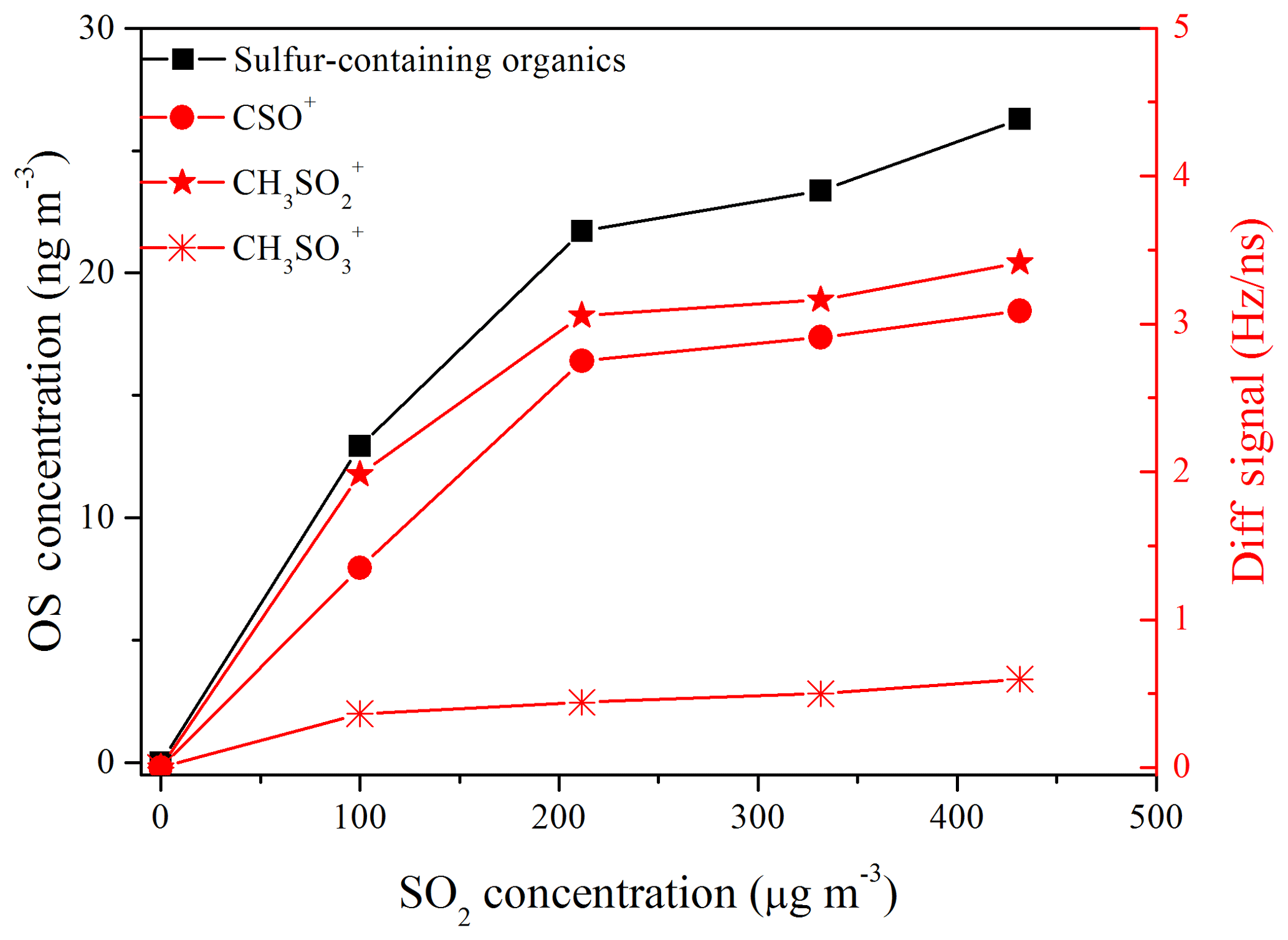
Figure 5Signal of fitted peaks, i.e., CSO+, , (right y axis), and sulfur-containing organics concentration (left y axis), as a function of SO2 initial concentration.
3.3 Role of NH3 in secondary aerosol formation
Similarly, the role of NH3 in SA formation was examined. It is worth noting that ammonium aerosols were formed without the addition of gaseous NH3 (Fig. S8 in the Supplement), which signified that some NH3 was present in the background air in the chamber or introduced during the humidification process of the chamber (Y. Liu et al., 2015). Unfortunately, appropriate instruments are unavailable to measure the exact concentration of background NH3 in the chamber. According to the concentration of generated ammonium aerosols, the concentration of background NH3 was estimated to be ∼15 ppb using the E-AIM model (Clegg and Brimblecombe, 2005; Wexler and Clegg, 2002; Clegg et al., 1998). Therefore, for the experiments with the presence of NH3, the concentration of injected NH3 (150–200 ppb) was much higher than this value to identify the effect of NH3 on SA formation. The SA concentration was enhanced by a factor of 2.0–2.5 in the presence of NH3, as shown in Fig. S9a. The formation of SOA, , and was enhanced to varying degrees. The increase in and could be attributed to the formation of inorganic NH4NO3 in the presence of NH3. The ratio, which could be derived from HR-ToF-AMS, has often been used as a proxy for the identification of inorganic nitrate and organic nitrogen compounds (Farmer et al., 2010; Sato et al., 2010; Rollins et al., 2009). Generally, the ratio of inorganic nitrate (1.08–2.81) is lower than that of organic nitrogen compounds (3.82–5.84) (Liu et al., 2016). In this study, the ratio became substantially lower (∼2.00) in the presence of NH3 compared with that in the absence of NH3 (∼5.46). Therefore, NH4NO3 was the dominant nitrate species in the presence of NH3. As for the reason for SOA enhancement, the NH3 present could react with some organic acids and subsequently contribute to SOA formation (Na et al., 2007, 2006), which could be supported by the increase in N∕C (from 0.016 to 0.033) with an increasing NH3 concentration at similar concentrations of NOx. In addition, we have found that the presence of NH3 readily increased the particle diameter and number concentration of SA generated in the photo-oxidation of gasoline (Fig. S9b and c), which revealed that NH3 played an important role in new particle formation (NPF). These are consistent with simulation results finding that NH3 promotes atmospheric NPF and also the conversion of SO2 and NO2 (Jiang and Xia, 2017). The increased surface area of particles was also observed (Table 1; 2.07×103 and 2.48×103 µm2 cm−3) as the NH3 concentration increased from 0 to 150 and 200 ppb. Similarly, the larger surface area would favor the partitioning of low-volatility vapor to the particle phase, leading to the higher SA yield (Table 1).
Previous studies have reported that the reaction of carbonyl compounds (e.g., glyoxal) could be catalyzed by ions through a Bronsted acid pathway or an iminium pathway, which could generate N-containing products and oligomers (Nozière et al., 2009), and then contribute a substantial fraction to SOA (Y. Liu et al., 2015; Farmer et al., 2010; Cheng et al., 2006). Researchers have identified the characteristic fragments of nitrogen-containing organics as CxHyNn and CxHyOzNn using HR-ToF-AMS (Lee et al., 2013; Farmer et al., 2010; Galloway et al., 2009). In this study, the typical normalized mass spectra of N-containing fragments in SOA after 480 min of photo-oxidation reaction at different concentrations of NH3 are given in Fig. 6. The prominent peaks in the CxHyNn family were at m∕z 27 (CHN+), 30 (CH4N+), 40 (C2H2N+), 41 (, C2H3N+), 42 (C2H4N+), 43 (C2H5N+), 54 (, C3H4N+), 55 (C3H5N+), and 68 (, C4H6N+); the CxHyOzNn fragments were dominated by 45 (CH3ON+), 46 (CH4ON+), 59 (C2H5ON+), 63 (CH5O2N+), 73 (, C3H7ON+), 86 (C3H4O2N+, ), 91 (C3H9O2N+), 97 (), and 104 (C3H6O3N+, C4H10O2N+). The N-containing fragments observed in the experiment without added NH3 could be attributed to the reactions between organic peroxy (RO2) radicals and NOx (Arey et al., 2001) or the uptake of background NH3 by SOA. Additionally, it was obvious that the signal intensities of most N-containing fragments became significantly stronger as the NH3 concentration increased (150–200 ppb). Therefore, a considerable amount of nitrogen-containing organics (the ratio of nitrogen-containing organics to SOA was about 6.7 %–7.7 %) was formed during the photo-oxidation of gasoline vapor in the presence of NH3. This was consistent with the previous study conducted by Y. Liu et al. (2015), who observed the formation of organic nitrogen compounds in the SOA generated from the OH oxidation of m-xylene. The promoting role of NH3 in the formation of N-containing species was also observed in the reaction system of ozonolysis and the photo-oxidation of α-pinene (Babar et al., 2017).
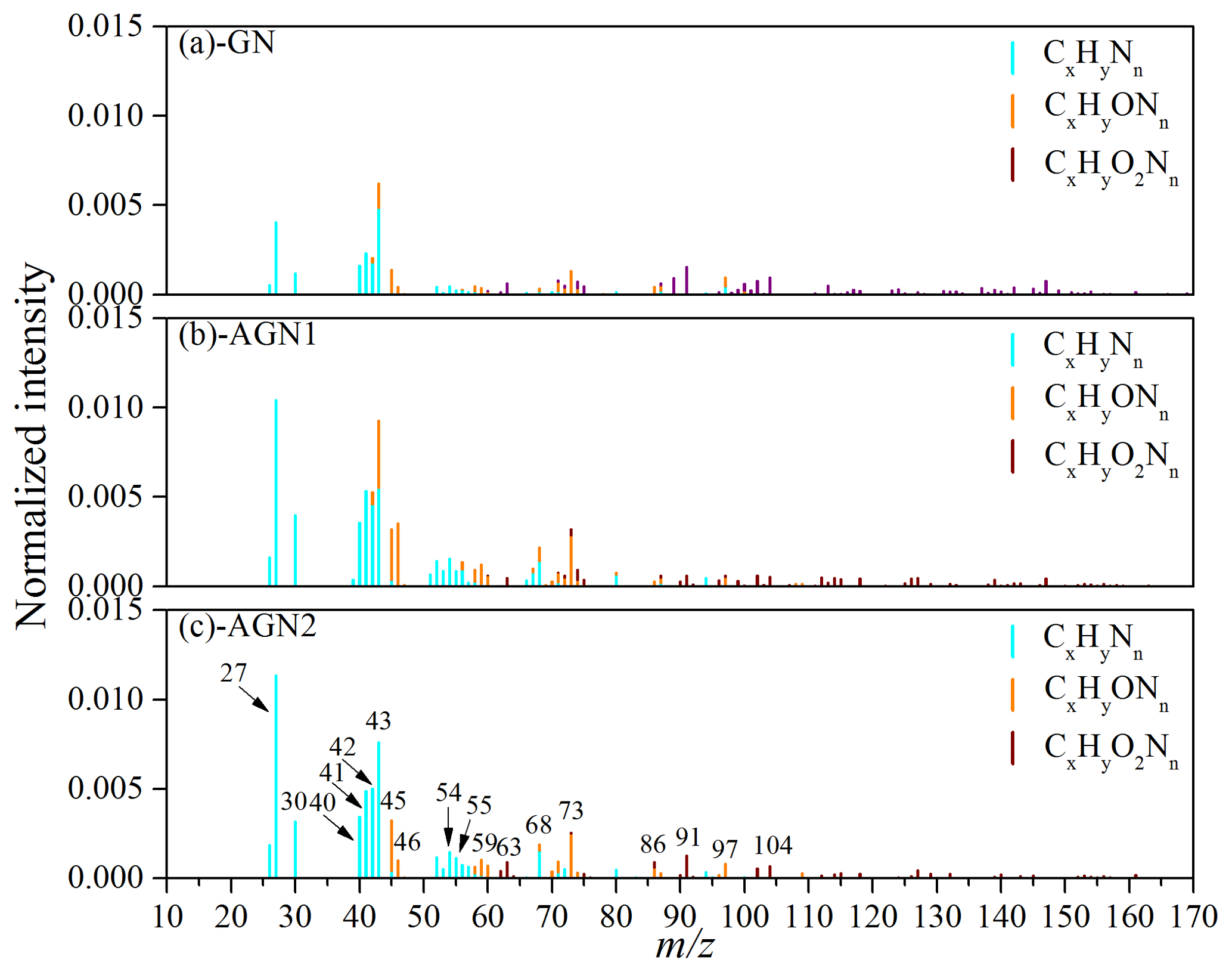
Figure 6Typical normalized mass spectra of N-containing fragments in SOA formed from the photo-oxidation of gasoline vapor at different concentrations of NH3 (Exps. GN, AGN1, and AGN2).
In addition, elemental analysis was also carried out to elucidate the SOA chemical composition and SOA formation mechanisms (Chhabra et al., 2011; Heald et al., 2010) at different concentrations of NH3. The time evolution of H∕C and O∕C in SOA formed from the photo-oxidation of gasoline vapor at different concentrations of NH3 is shown in Fig. 7. As evident from Fig. 7, all data points are located in the triangular area for a slope between −1 and 0, which suggests that SOA formation from the photo-oxidation of gasoline vapor is a combination of carboxylic acid and alcohol–peroxide (Heald et al., 2010). Moreover, in the presence of NH3, as shown in Fig. 8, N∕C increased as the reaction proceeded in the initial oxidation stage (0–120 min), accompanied by a rapid increase in O∕C (0.12–0.67), a decrease in H∕C (2.12–1.61), and rapid formation of SOA. During this stage, the photo-oxidation of VOC precursors leads to a rapid increase in O∕C and a rapid decrease in H∕C. The termination chemistry of NOx with free radicals and the NH3 uptake result in a rapid increase in N∕C. As the reaction proceeded further (120–300 min), an increase in H∕C, which should be caused by NH3 uptake, resulted in an almost constant oxidation state of SOA in continuous photo-oxidation, accompanied by an increase in the SOA concentration. Nozière et al. (2009) have reported that N-containing products are generated from carbonyl compound (e.g., glyoxal) self-reactions catalyzed by ions, which will have a dramatic impact on the volatility of oxidation products and the yield of SOA (Ortiz-Montalvo et al., 2014). In the last stage of the reaction (360–480 min), NH3 uptake might reach saturation; therefore, H∕C and N∕C are almost constant. Comparing experiments with different concentrations of NH3, the average H∕C shows an obvious increase (1.53–1.70), while the average O∕C (0.70–0.78) shows a slight increase with the increase in NH3 concentration (0–200 ppb), as seen in Fig. S10. The slope in the van Krevelen diagram shows a trend from slope to slope =0 (Fig. S10), indicating that the formed carboxylic acid could further react with NH3 via an acid–base reaction to generate an ammonium salt of a carboxylate anion in the presence of NH3 (Na et al., 2007). Xu et al. (2018) recently found that imidazole products containing multiple oxygen atoms could be generated through heterogeneous reactions between NH3 and carbonyl compounds (e.g., glyoxal), which might also contribute to the increase in the O∕C of the SOA.
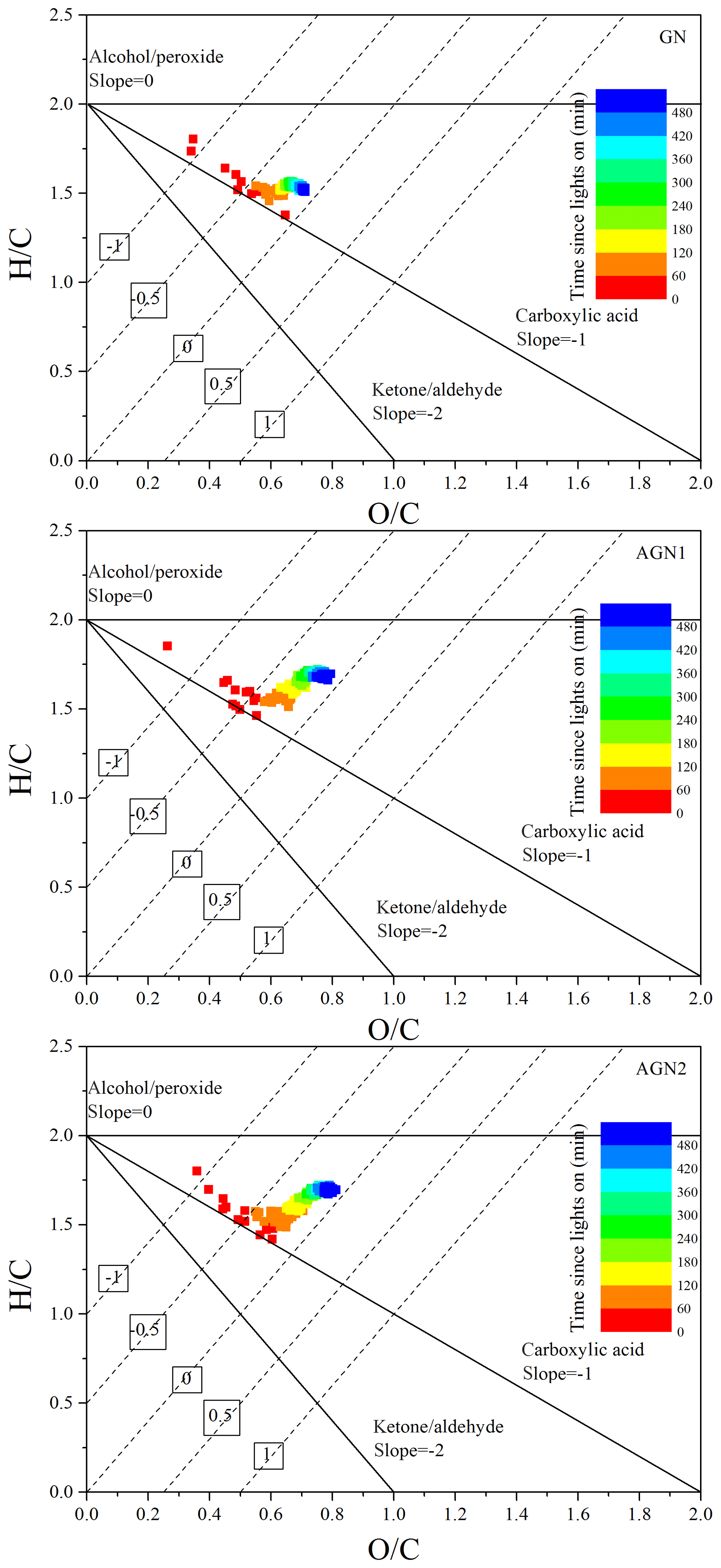
Figure 7Time evolution of H∕C and O∕C in SOA formed from the photo-oxidation of gasoline vapor at different concentrations of NH3 (Exp. GN, AGN1, and AGN2). The numbers (i.e., −1, −0.5, 0, 0.5, and 1) labeling the dashed lines show the average carbon oxidation state () (Kroll et al., 2011). The black lines represent the addition of functional groups to an aliphatic carbon (Heald et al., 2010).
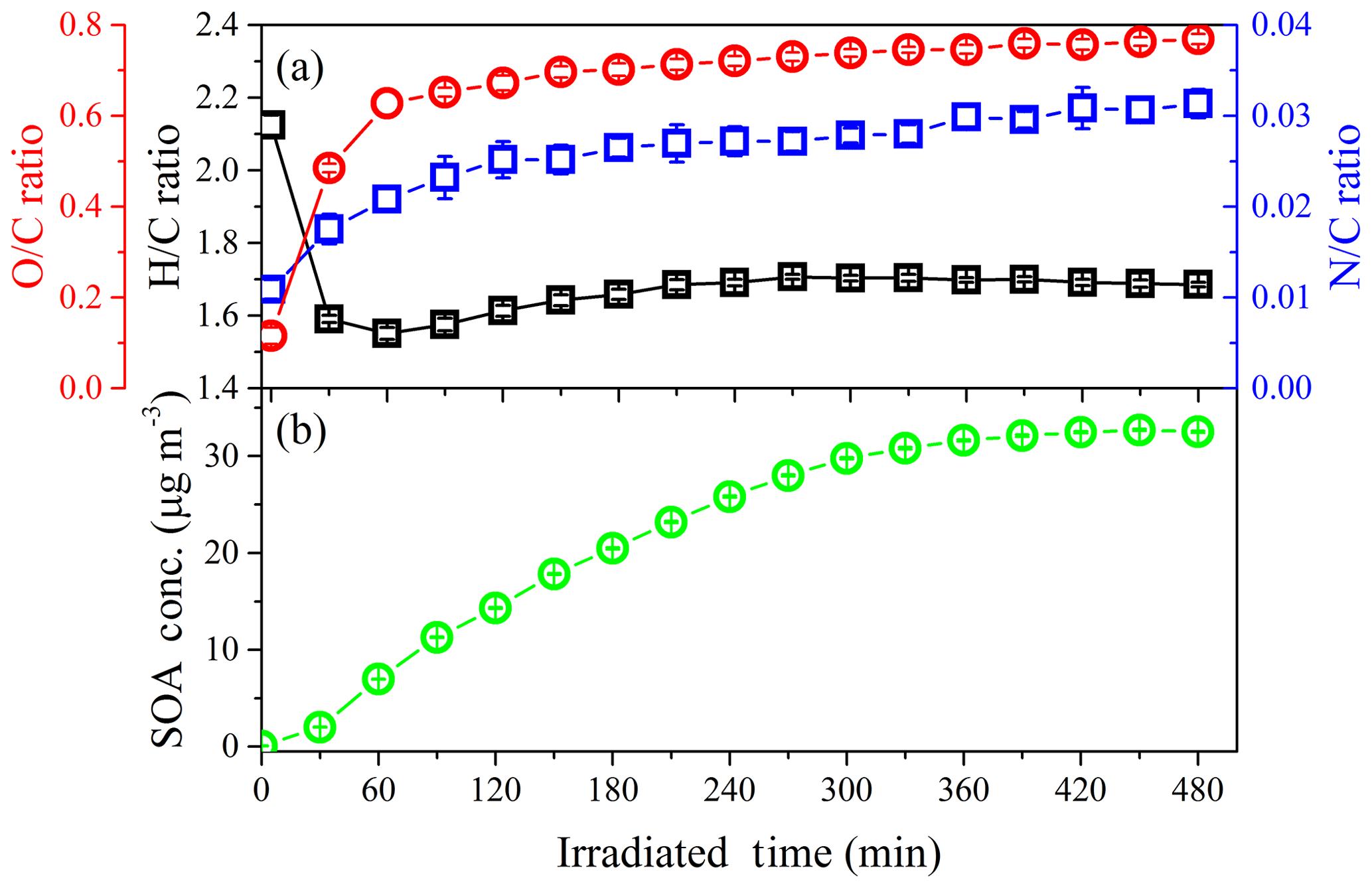
Figure 8Time evolution of (a) O∕C, H∕C, N∕C, and (b) SOA concentration in the photo-oxidation of gasoline vapor in the presence of 150 ppb NH3 (Exp. AGN1).
3.4 Different roles of SO2 and NH3 in SOA chemical properties
The chemical properties of the SOA generated under different concentrations of SO2 or NH3 were further compared by applying positive matrix factorization (PMF) analysis to the HR-ToF-AMS data (Chu et al., 2016; Liu et al., 2014). The details of the PMF analysis are given in the Supplement. For the experiments under different SO2 concentration conditions (i.e., Exps. GN, SGN1, SGN2, SGN3, and SGN4), two factors (Factor 1-S and Factor 2-S; Fig. S11a) were identified from the PMF analysis, and the different mass spectra (m∕z 12–170) between the two factors and the time series of the mass concentrations are shown in Fig. 9. The intensity of CxHy and S-bearing organic fragments (CxHyOzS) in Factor 1-S was obviously stronger than that in Factor 2-S. Meanwhile, fragments in the high m∕z range (>110 Da) were more abundant in Factor 1-S (Fig. 9a, marked by the red box). By contrast, the fragments containing oxygen in Factor 2-S were more abundant than in Factor 1-S, such as the typical fragment (m∕z 44). Therefore, Factor 1-S was tentatively assigned to the less-oxygenated organic aerosol and oligomers, while Factor 2-S was more-oxygenated organic aerosol (Ulbrich et al., 2009). Similarly, for the experiments at different NH3 concentrations (i.e., Exps. GN, AGN1, and AGN2), two factors (Factor 1-N and Factor 2-N; Fig. S11b) were also identified in the same way. According to Fig. 10, Factor 1-N was tentatively assigned to the less-oxygenated organic aerosol and oligomers, while Factor 2-N was more-oxygenated organic aerosol and nitrogen-containing organics.
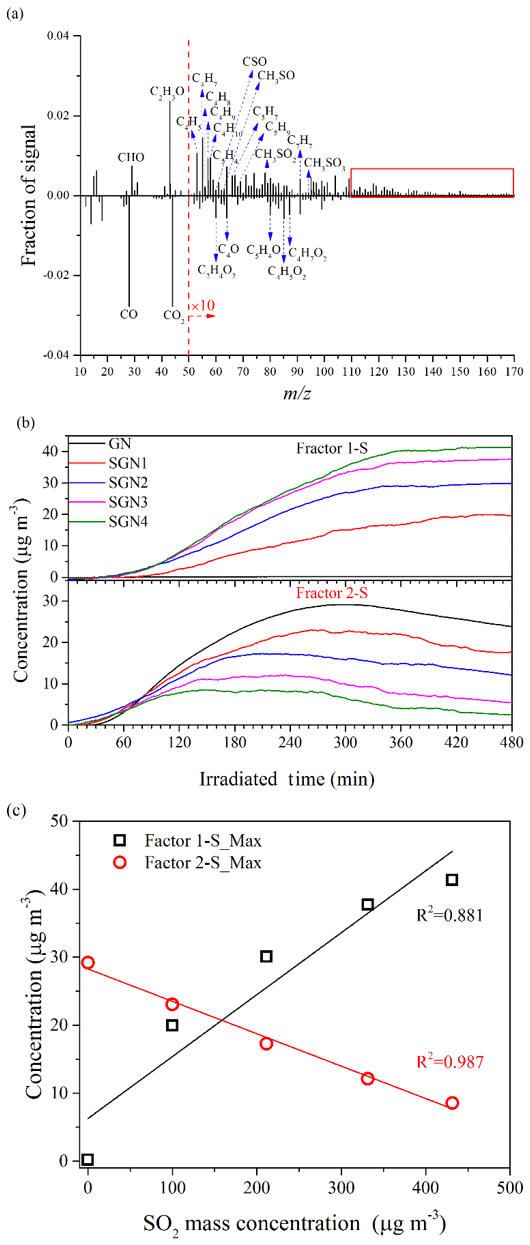
Figure 9(a) Different mass spectra (Factor 1-S–Factor 2-S) between the two factors, (b) time series of the mass concentration, and (c) relationship between the concentration of SO2 and the maximum concentration of the two factors identified by applying PMF analysis to the AMS data derived from the experiments at different concentrations of SO2 (Exps. GN, SGN1, SGN2, SGN3, and SGN4).
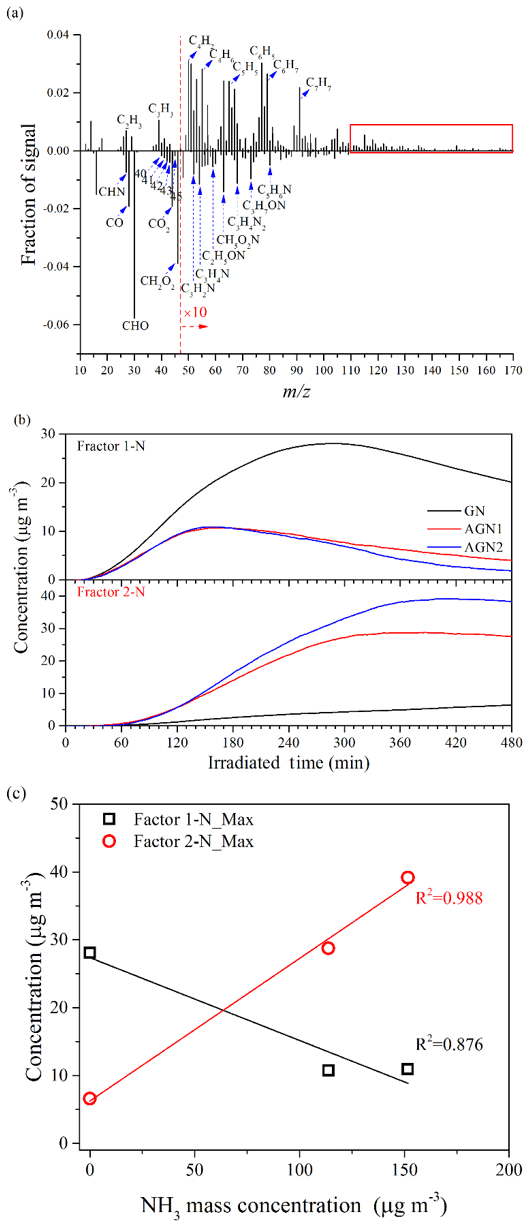
Figure 10(a) Different mass spectra (Factor 1-N–Factor 2-N) between the two factors, (b) time series of the mass concentration, and (c) relationship between the concentration of NH3 and the maximum concentration of the two factors identified by applying PMF analysis to the AMS data derived from the experiments at different concentrations of NH3 (Exps. GN, AGN1, and AGN2).
As shown in Figs. 9b and 10b, these two factors both had different time series during the entire reaction. With respect to Exps. GN, SGN1, SGN2, SGN3, and SGN4, Factor 1-S was formed later (∼30 min) than Factor 2-S and then continuously increased during the entire reaction. Comparing experiments with different SO2 concentrations, the maximum concentration of Factor 1-S, which was related to the less-oxygenated organic aerosol and oligomers, was enhanced with an increased SO2 concentration (R2=0.881; Fig. 9c). This suggested that the presence of SO2 was prone to decrease the oxidation state of organic aerosol via acid-catalyzed reactions and enhance the formation of oligomers (Liu et al., 2016), which was consistent with the evolution of O∕C vs. H∕C shown in Fig. S12. Moreover, the gradually increasing concentration of Factor 1-S was related to the formation of sulfur-containing organics in the presence of SO2 (Blair et al., 2017). By contrast, Factor 2-S was first gradually increased with the progress of the reaction and then decreased after reaching a peak (i.e., inflection point). The time to reach the inflection point was affected by the SO2 concentration (Fig. 9b). As the initial concentration of SO2 increased from 0 to 151 ppb, the time corresponding to the inflection point decreased, which indicated that the adverse influence of acid catalysis on Factor 2-S was gradually enhanced. In addition, the maximum concentration of Factor 2-S was negatively related to the SO2 concentration (R2=0.987; Fig. 9c); this suggested that the presence of SO2 and acid catalysis was adverse to the formation of more-oxygenated organic aerosol, leading to the decrease in the oxidation state of organic aerosol (Fig. S12).
By contrast, for Exps. GN, AGN1, and AGN2, Factor 1-N was first increased with the progress of the reaction and then gradually decreased after reaching a peak (Fig. 10b), while Factor 2-N was formed later (∼30 min) than Factor 1-N and then continuously increased during the entire reaction. This phenomenon was consistent with the expected behavior that less-oxidized organic aerosol would be further oxidized to form more-oxidized organic aerosol. When comparing experiments with different NH3 concentrations, it was observed that the concentration of Factor 2-N increased with an increasing NH3 concentration. Meanwhile, Factor 2-N, which was related to the more-oxidized organic aerosol and nitrogen-containing organics, was a dominant factor in the presence of NH3, and its maximum concentration was enhanced with the increase in NH3 concentration (R2=0.988; Fig. 10c). Thus, the formation of more-oxygenated organic aerosol and nitrogen-containing organics will be enhanced with an increase in NH3 concentration. In contrast, a negative correlation was observed between the maximum concentration of Factor 1-N and NH3 concentration (R2=0.876; Fig. 10c); this revealed that less-oxygenated organic aerosol was gradually transformed to more-oxidized species and nitrogen-containing organics in the presence of NH3.
In this study, SA formation from the photo-oxidation of gasoline ∕ NOx in the presence of SO2 or NH3 was investigated. Our experimental results demonstrated that SA was enhanced by a factor of 1.6–2.6 or 2.0–2.5, respectively, with the increase in SO2 or NH3 concentration (0–151 and 0–200 ppb, respectively). Meanwhile, both secondary organic aerosol (SOA) and secondary inorganic aerosol (SIA) were increased by varying degrees. In the presence of SO2, was the most sensitive linear increase with the increase in SO2 concentration, and SOA was also greatly enhanced due to the acid catalytic effect and the formation of sulfur-containing organics. In the presence of NH3, NH4NO3 was most enhanced, following by SOA. The formation of nitrogen-containing organics was also promoted by the presence of NH3. Meanwhile, conspicuous new particle formation (NPF) and particle size growth were enhanced in the presence of SO2 or NH3.
In this study, a linear relationship between the SA yield and SO2 or NH3 concentration was also obtained (Fig. S13). Considering the typical concentrations of SO2 and NH3 of 40 and 23 ppb in haze pollution in the North China Plain (Cheng et al., 2016) and the lower aromatics content (∼10 %) in vehicular evaporative emissions (Zhang et al., 2013), the SA yield is roughly estimated to be about 0.20. Recently, an updated emission inventory of vehicular evaporative emissions was reported to be 1.65 Tg yr−1 (H. Liu et al., 2017). Then, the SA formed from the photo-oxidation of VOCs emitted by vehicular evaporation in the presence of SO2 and NH3 is roughly estimated to be 0.33 Tg yr−1, which is about 1.5 times as much as the primary PM2.5 emissions from transportation (0.21 Tg yr−1) in China (Jing et al., 2015; Zhang et al., 2007), accounting for about 21 % of SOA production (1.6 Tg yr−1) from anthropogenic precursors estimated by global chemical transport models (Farina et al., 2010). In addition, the photo-oxidation of long-chain alkanes (>C6, IVOCs) contained in evaporative emissions also contributes to SOA formation (Pye and Pouliot, 2012; Tkacik et al., 2012; Presto et al., 2009; Lim and Ziemann, 2005; Zhao et al., 2016). This estimate suggests that vehicular evaporative emissions will be a significant source of SA in the presence of SO2 and NH3, although the estimate might have a high uncertainty due to the fact that SA yield might vary considerably under different atmospheric conditions. Meanwhile, in the presence of NOx, SO2, and NH3, vehicular evaporative emissions may be a potential source of sulfur- and nitrogen-containing organics according to the results obtained from our study. Sulfur- and nitrogen-containing organics will have an adverse influence on the climate by light absorption and/or by affecting aerosol hygroscopicity (Staudt et al., 2014; Nguyen et al., 2012), and they also make a significant contribution to SOA and nitrogen or sulfur budgets (Lee et al., 2016; Shang et al., 2016).
Therefore, under the compound pollution conditions of SO2 and NH3, the synergistic emission reduction of vehicular evaporative emissions, SO2 (e.g., coal-fired flue gas), and NH3 (e.g., emitted from agricultural nonpoint sources and traffic) should be taken into consideration by policy makers for future management, which will contribute to reducing the burden of PM2.5 and then cut the environmental, economic, and health costs caused by PM pollution. Our work will provide a scientific basis for taking corresponding control measures to relieve haze events in China. Additionally, there might be some differences between the VOC composition of gasoline vapor directly injected to the smog chamber and vehicular evaporative emissions. Thus, further work should be focused on SA formation directly from vehicular evaporative emissions under coexisting SO2 and NH3 conditions to shed light on the formation mechanism of SA under more atmospherically relevant conditions.
The experimental data are available upon request to the corresponding authors.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-8063-2019-supplement.
TC and YL contributed equally to this work and should be considered first coauthors. HH, QM, YL, and TC proposed the initial idea. YL and TC designed and led the study. YL, BC, QM, PZ, and TC conducted the data analyses. TC, YL, BC, PZ, CL, and JL interpreted the data. TC, YL, JL, and QM wrote the paper with input from all coauthors.
The authors declare that they have no conflict of interest.
This work was financially supported by the National Key R&D Program of China (2016YFC0202700, 2018YFC0506901), the National Natural Science Foundation of China (21876185, 41877306, 41877304, and 91744205), the special fund of the State Key Joint Laboratory of Environment Simulation and Pollution Control (17L01ESPC), the Youth Innovation Promotion Association (CAS; 2018055, 2018060, and 2017064), the Key Research Program of Frontier Sciences (CAS; QYZDB-SSW-DQC018), the national research program for key issues in air pollution control (DQGG0103), and the Young Talent Project of the Center for Excellence in Regional Atmospheric Environment (CAS; CERAE201801).
This research has been supported by the Ministry of Science and Technology of the People's Republic of China (grant nos. 2016YFC0202700 and 2018YFC0506901), the National Natural Science Foundation of China (grant nos. 41877306, 41877304, 21876185, and 91744205), the State Key Joint Laboratory of Environment Simulation and Pollution Control (grant no. 17L01ESPC), and the Chinese Academy of Sciences (grant nos. 2018060, 2018055, 2017064, CERAE201801, and QYZDB-SSW-DQC018).
This paper was edited by Jason Surratt and reviewed by two anonymous referees.
Arey, J., Aschmann, S. M., Kwok, E. S. C., and Atkinson, R.: Alkyl nitrate, hydroxyalkyl nitrate, and hydroxycarbonyl formation from the NOx-air photooxidations of C5−C8 n-alkanes, J. Phys. Chem. A, 105, 1020–1027, https://doi.org/10.1021/jp003292z, 2001.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003.
Babar, Z. B., Park, J.-H., and Lim, H.-J.: Influence of NH3 on secondary organic aerosols from the ozonolysis and photooxidation of α-pinene in a flow reactor, Atmos. Environ., 164, 71–84, https://doi.org/10.1016/j.atmosenv.2017.05.034, 2017.
Bahreini, R., Keywood, M. D., Ng, N. L., Varutbangkul, V., Gao, S., Flagan, R. C., Seinfeld, J. H., Worsnop, D. R., and Jimenez, J. L.: Measurements of secondary organic aerosol from oxidation of cycloalkenes, terpenes, and m-xylene using an Aerodyne aerosol mass spectrometer, Environ. Sci. Technol., 39, 5674–5688, https://doi.org/10.1021/es048061a, 2005.
Bauduin, S., Clarisse, L., Hadji-Lazaro, J., Theys, N., Clerbaux, C., and Coheur, P.-F.: Retrieval of near-surface sulfur dioxide (SO2) concentrations at a global scale using IASI satellite observations, Atmos. Meas. Tech., 9, 721–740, https://doi.org/10.5194/amt-9-721-2016, 2016.
Blair, S. L., MacMillan, A. C., Drozd, G. T., Goldstein, A. H., Chu, R. K., Paša-Tolić, L., Shaw, J. B., Tolić, N., Lin, P., Laskin, J., Laskin, A., and Nizkorodov, S. A.: Molecular characterization of organosulfur compounds in biodiesel and diesel fuel secondary organic aerosol, Environ. Sci. Technol., 51, 119–127, https://doi.org/10.1021/acs.est.6b03304, 2017.
Calvert, J. G., Atkinson, R., Becker, K. H., Kamens, R. M., Seinfeld, J. H., Wallington, T. H., and Yarwood, G.: The mechanisms of atmospheric oxidation of the aromatic hydrocarbons, Oxford University Press, New York, USA, 2002.
Cao, G. and Jang, M.: Effects of particle acidity and UV light on secondary organic aerosol formation from oxidation of aromatics in the absence of NOx, Atmos. Environ., 41, 7603–7613, https://doi.org/10.1016/j.atmosenv.2007.05.034, 2007.
Casale, M. T., Richman, A. R., Elrod, M. J., Garland, R. M., Beaver, M. R., and Tolbert, M. A.: Kinetics of acid-catalyzed aldol condensation reactions of aliphatic aldehydes, Atmos. Environ., 41, 6212–6224, https://doi.org/10.1016/j.atmosenv.2007.04.002, 2007.
Chapleski, R. C., Zhang, Y., Troya, D., and Morris, J. R.: Heterogeneous chemistry and reaction dynamics of the atmospheric oxidants, O3, NO3, and OH, on organic surfaces, Chem. Soc. Rev., 45, 3731–3746, https://doi.org/10.1039/C5CS00375J, 2016.
Chen, T., Liu, Y., Chu, B., Liu, C., Liu, J., Ge, Y., Ma, Q., Ma, J., and He, H.: Differences of the oxidation process and secondary organic aerosol formation at low and high precursor concentrations, J. Environ. Sci., 79, 256–263, https://doi.org/10.1016/j.jes.2018.11.011, 2019a.
Chen, T., Liu, Y., Liu, C., Liu, J., Chu, B., and He, H.: Important role of aromatic hydrocarbons in SOA formation from unburned gasoline vapor, Atmos. Environ., 201, 101–109, https://doi.org/10.1016/j.atmosenv.2019.01.001, 2019b.
Cheng, Y., Li, S.-M., and Leithead, A.: Chemical characteristics and origins of nitrogen-containing organic compounds in PM2.5 aerosols in the Lower Fraser Valley, Environ. Sci. Technol., 40, 5846–5852, https://doi.org/10.1021/es0603857, 2006.
Cheng, Y., Zheng, G., Wei, C., Mu, Q., Zheng, B., Wang, Z., Gao, M., Zhang, Q., He, K., Carmichael, G., Pöschl, U., and Su, H.: Reactive nitrogen chemistry in aerosol water as a source of sulfate during haze events in China, Sci. Adv., 2, e1601530, https://doi.org/10.1126/sciadv.1601530, 2016.
Chhabra, P. S., Ng, N. L., Canagaratna, M. R., Corrigan, A. L., Russell, L. M., Worsnop, D. R., Flagan, R. C., and Seinfeld, J. H.: Elemental composition and oxidation of chamber organic aerosol, Atmos. Chem. Phys., 11, 8827–8845, https://doi.org/10.5194/acp-11-8827-2011, 2011.
Chu, B., Zhang, X., Liu, Y., He, H., Sun, Y., Jiang, J., Li, J., and Hao, J.: Synergetic formation of secondary inorganic and organic aerosol: effect of SO2 and NH3 on particle formation and growth, Atmos. Chem. Phys., 16, 14219–14230, https://doi.org/10.5194/acp-16-14219-2016, 2016.
Chu, B., Kerminen, V.-M., Bianchi, F., Yan, C., Petäjä, T., and Kulmala, M.: Atmospheric new particle formation in China, Atmos. Chem. Phys., 19, 115–138, https://doi.org/10.5194/acp-19-115-2019, 2019.
Clegg, S. L. and Brimblecombe, P.: Comment on the “Thermodynamic Dissociation Constant of the Bisulfate Ion from Raman and Ion Interaction Modeling Studies of Aqueous Sulfuric Acid at Low Temperatures”, J. Phys. Chem. A, 109, 2703–2706, https://doi.org/10.1021/jp0401170, 2005.
Clegg, S. L., Brimblecombe, P., and Wexler, A. S.: Thermodynamic model of the system H+----H2O at tropospheric temperatures, J. Phys. Chem. A, 102, 2137–2154, https://doi.org/10.1021/jp973042r, 1998.
Czoschke, N. M., Jang, M., and Kamens, R. M.: Effect of acidic seed on biogenic secondary organic aerosol growth, Atmos. Environ., 37, 4287–4299, https://doi.org/10.1016/S1352-2310(03)00511-9, 2003.
Davidson, C. I., Phalen, R. F., and Solomon, P. A.: Airborne particulate matter and human health: a review, Aerosol Sci. Tech., 39, 737–749, https://doi.org/10.1080/02786820500191348, 2005.
DeCarlo, P. F., Slowik, J. G., Worsnop, D. R., Davidovits, P., and Jimenez, J. L.: Particle morphology and density characterization by combined mobility and aerodynamic diameter measurements. Part 1: Theory, Aerosol Sci. Tech., 38, 1185–1205, https://doi.org/10.1080/027868290903907, 2004.
Díaz-de-Mera, Y., Aranda, A., Martínez, E., Rodríguez, A. A., Rodríguez, D., and Rodríguez, A.: Formation of secondary aerosols from the ozonolysis of styrene: effect of SO2 and H2O, Atmos. Environ., 171, 25–31, https://doi.org/10.1016/j.atmosenv.2017.10.011, 2017.
Donahue, N. M., Robinson, A. L., Stanier, C. O., and Pandis, S. N.: Coupled partitioning, dilution, and chemical aging of semivolatile organics, Environ. Sci. Technol., 40, 2635–2643, https://doi.org/10.1021/es052297c, 2006.
Drewnick, F., Hings, S. S., DeCarlo, P., Jayne, J. T., Gonin, M., Fuhrer, K., Weimer, S., Jimenez, J. L., Demerjian, K. L., Borrmann, S., and Worsnop, D. R.: A new time-of-flight aerosol mass spectrometer (TOF-AMS) – instrument description and first field deployment, Aerosol Sci. Tech., 39, 637–658, https://doi.org/10.1080/02786820500182040, 2005.
Edney, E. O., Kleindienst, T. E., Jaoui, M., Lewandowski, M., Offenberg, J. H., Wang, W., and Claeys, M.: Formation of 2-methyl tetrols and 2-methylglyceric acid in secondary organic aerosol from laboratory irradiated isoprene/NOx/SO2/air mixtures and their detection in ambient PM2.5 samples collected in the eastern United States, Atmos. Environ., 39, 5281–5289, https://doi.org/10.1016/j.atmosenv.2005.05.031, 2005.
Farina, S. C., Adams, P. J., and Pandis, S. N.: Modeling global secondary organic aerosol formation and processing with the volatility basis set: Implications for anthropogenic secondary organic aerosol, J. Geophys. Res.-Atmos., 115, D09202, https://doi.org/10.1029/2009jd013046, 2010.
Farmer, D. K., Matsunaga, A., Docherty, K. S., Surratt, J. D., Seinfeld, J. H., Ziemann, P. J., and Jimenez, J. L.: Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry, P. Natl. Acad. Sci. USA, 107, 6670–6675, https://doi.org/10.1073/pnas.0912340107, 2010.
Fu, X., Wang, S. X., Ran, L. M., Pleim, J. E., Cooter, E., Bash, J. O., Benson, V., and Hao, J. M.: Estimating NH3 emissions from agricultural fertilizer application in China using the bi-directional CMAQ model coupled to an agro-ecosystem model, Atmos. Chem. Phys., 15, 6637–6649, https://doi.org/10.5194/acp-15-6637-2015, 2015.
Galloway, M. M., Chhabra, P. S., Chan, A. W. H., Surratt, J. D., Flagan, R. C., Seinfeld, J. H., and Keutsch, F. N.: Glyoxal uptake on ammonium sulphate seed aerosol: reaction products and reversibility of uptake under dark and irradiated conditions, Atmos. Chem. Phys., 9, 3331–3345, https://doi.org/10.5194/acp-9-3331-2009, 2009.
Gordon, T. D., Presto, A. A., May, A. A., Nguyen, N. T., Lipsky, E. M., Donahue, N. M., Gutierrez, A., Zhang, M., Maddox, C., Rieger, P., Chattopadhyay, S., Maldonado, H., Maricq, M. M., and Robinson, A. L.: Secondary organic aerosol formation exceeds primary particulate matter emissions for light-duty gasoline vehicles, Atmos. Chem. Phys., 14, 4661–4678, https://doi.org/10.5194/acp-14-4661-2014, 2014.
Guo, S., Hu, M., Zamora, M. L., Peng, J., Shang, D., Zheng, J., Du, Z., Wu, Z., Shao, M., Zeng, L., Molina, M. J., and Zhang, R.: Elucidating severe urban haze formation in China, P. Natl. Acad. Sci. USA, 111, 17373–17378, https://doi.org/10.1073/pnas.1419604111, 2014.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Hatch, L. E., Creamean, J. M., Ault, A. P., Surratt, J. D., Chan, M. N., Seinfeld, J. H., Edgerton, E. S., Su, Y., and Prather, K. A.: Measurements of isoprene-derived organosulfates in ambient aerosols by aerosol time-of-flight mass spectrometry – Part 2: temporal variability and formation mechanisms, Environ. Sci. Technol., 45, 8648–8655, https://doi.org/10.1021/es2011836, 2011.
He, H., Wang, Y., Ma, Q., Ma, J., Chu, B., Ji, D., Tang, G., Liu, C., Zhang, H., and Hao, J.: Mineral dust and NOx promote the conversion of SO2 to sulfate in heavy pollution days, Sci. Rep., 4, 4172, https://doi.org/10.1038/srep04172, 2014.
Heald, C. L., Kroll, J. H., Jimenez, J. L., Docherty, K. S., DeCarlo, P. F., Aiken, A. C., Chen, Q., Martin, S. T., Farmer, D. K., and Artaxo, P.: A simplified description of the evolution of organic aerosol composition in the atmosphere, Geophys. Res. Lett., 37, L08803, https://doi.org/10.1029/2010GL042737, 2010.
Hou, S., Tong, S., Ge, M., and An, J.: Comparison of atmospheric nitrous acid during severe haze and clean periods in Beijing, China, Atmos. Environ., 124, 199–206, https://doi.org/10.1016/j.atmosenv.2015.06.023, 2016.
Huang, D. D., Li, Y. J., Lee, B. P., and Chan, C. K.: Analysis of organic sulfur compounds in atmospheric aerosols at the HKUST supersite in Hong Kong using HR-ToF-AMS, Environ. Sci. Technol., 49, 3672–3679, https://doi.org/10.1021/es5056269, 2015.
Huang, R.-J., Zhang, Y., Bozzetti, C., Ho, K.-F., Cao, J.-J., Han, Y., Daellenbach, K. R., Slowik, J. G., Platt, S. M., Canonaco, F., Zotter, P., Wolf, R., Pieber, S. M., Bruns, E. A., Crippa, M., Ciarelli, G., Piazzalunga, A., Schwikowski, M., Abbaszade, G., Schnelle-Kreis, J., Zimmermann, R., An, Z., Szidat, S., Baltensperger, U., Haddad, I. E., and Prevot, A. S. H.: High secondary aerosol contribution to particulate pollution during haze events in China, Nature, 514, 218–222, https://doi.org/10.1038/nature13774, 2014.
Ianniello, A., Spataro, F., Esposito, G., Allegrini, I., Rantica, E., Ancora, M. P., Hu, M., and Zhu, T.: Occurrence of gas phase ammonia in the area of Beijing (China), Atmos. Chem. Phys., 10, 9487–9503, https://doi.org/10.5194/acp-10-9487-2010, 2010.
Jang, M. and Kamens, R. M.: Atmospheric secondary aerosol formation by heterogeneous reactions of aldehydes in the presence of a sulfuric acid aerosol catalyst, Environ. Sci. Technol., 35, 4758–4766, https://doi.org/10.1021/es010790s, 2001.
Jang, M., Czoschke, N. M., Lee, S., and Kamens, R. M.: Heterogeneous atmospheric aerosol production by acid-catalyzed particle-phase reactions, Science, 298, 814–817, https://doi.org/10.1126/science.1075798, 2002.
Jang, M., Carroll, B., Chandramouli, B., and Kamens, R. M.: Particle growth by acid-catalyzed heterogeneous reactions of organic carbonyls on preexisting aerosols, Environ. Sci. Technol., 37, 3828–3837, https://doi.org/10.1021/es021005u, 2003a.
Jang, M., Lee, S., and Kamens, R. M.: Organic aerosol growth by acid-catalyzed heterogeneous reactions of octanal in a flow reactor, Atmos. Environ., 37, 2125–2138, https://doi.org/10.1016/S1352-2310(03)00077-3, 2003b.
Jaoui, M., Edney, E. O., Kleindienst, T. E., Lewandowski, M., Offenberg, J. H., Surratt, J. D., and Seinfeld, J. H.: Formation of secondary organic aerosol from irradiated α-pinene/toluene/NOx mixtures and the effect of isoprene and sulfur dioxide, J. Geophys. Res.-Atmos., 113, D09303, https://doi.org/10.1029/2007jd009426, 2008.
Jathar, S. H., Gordon, T. D., Hennigan, C. J., Pye, H. O. T., Pouliot, G., Adams, P. J., Donahue, N. M., and Robinson, A. L.: Unspeciated organic emissions from combustion sources and their influence on the secondary organic aerosol budget in the United States, P. Natl. Acad. Sci. USA, 111, 10473–10478, https://doi.org/10.1073/pnas.1323740111, 2014.
Jayne, J. T., Leard, D. C., Zhang, X., Davidovits, P., Smith, K. A., Kolb, C. E., and Worsnop, D. R.: Development of an aerosol mass spectrometer for size and composition analysis of submicron particles, Aerosol Sci. Tech., 33, 49–70, https://doi.org/10.1080/027868200410840, 2000.
Jiang, B. and Xia, D.: Role identification of NH3 in atmospheric secondary new particle formation in haze occurrence of China, Atmos. Environ., 163, 107–117, https://doi.org/10.1016/j.atmosenv.2017.05.035, 2017.
Jimenez, J. L., Jayne, J. T., Shi, Q., Kolb, C. E., Worsnop, D. R., Yourshaw, I., Seinfeld, J. H., Flagan, R. C., Zhang, X., Smith, K. A., Morris, J. W., and Davidovits, P.: Ambient aerosol sampling using the aerodyne aerosol mass spectrometer, J. Geophys. Res.-Atmos., 108, 8425, https://doi.org/10.1029/2001JD001213, 2003.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of organic aerosols in the atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Jing, M., Junfeng, L., Yuan, X., and Shu, T.: Tracing primary PM2.5 emissions via Chinese supply chains, Environ. Res. Lett., 10, 054005, https://doi.org/10.1088/1748-9326/10/5/054005, 2015.
Kang, Y., Liu, M., Song, Y., Huang, X., Yao, H., Cai, X., Zhang, H., Kang, L., Liu, X., Yan, X., He, H., Zhang, Q., Shao, M., and Zhu, T.: High-resolution ammonia emissions inventories in China from 1980 to 2012, Atmos. Chem. Phys., 16, 2043–2058, https://doi.org/10.5194/acp-16-2043-2016, 2016.
Kleindienst, T. E., Edney, E. O., Lewandowski, M., Offenberg, J. H., and Jaoui, M.: Secondary organic carbon and aerosol yields from the irradiations of isoprene and α-pinene in the presence of NOx and SO2, Environ. Sci. Technol., 40, 3807–3812, https://doi.org/10.1021/es052446r, 2006.
Krechmer, J. E., Pagonis, D., Ziemann, P. J., and Jimenez, J. L.: Quantification of gas-wall partitioning in Teflon environmental chambers using rapid bursts of low-volatility oxidized species generated in situ, Environ. Sci. Technol., 50, 5757–5765, https://doi.org/10.1021/acs.est.6b00606, 2016.
Kroll, J. H. and Seinfeld, J. H.: Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 42, 3593–3624, https://doi.org/10.1016/j.atmosenv.2008.01.003, 2008.
Kroll, J. H., Donahue, N. M., Jimenez, J. L., Kessler, S. H., Canagaratna, M. R., Wilson, K. R., Altieri, K. E., Mazzoleni, L. R., Wozniak, A. S., Bluhm, H., Mysak, E. R., Smith, J. D., Kolb, C. E., and Worsnop, D. R.: Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol, Nat. Chem., 3, 133–139, https://doi.org/10.1038/nchem.948, 2011.
Kundu, S., Quraishi, T. A., Yu, G., Suarez, C., Keutsch, F. N., and Stone, E. A.: Evidence and quantitation of aromatic organosulfates in ambient aerosols in Lahore, Pakistan, Atmos. Chem. Phys., 13, 4865–4875, https://doi.org/10.5194/acp-13-4865-2013, 2013.
Lee, A. K. Y., Zhao, R., Li, R., Liggio, J., Li, S.-M., and Abbatt, J. P. D.: Formation of light absorbing organo-nitrogen species from evaporation of droplets containing glyoxal and ammonium sulfate, Environ. Sci. Technol., 47, 12819–12826, https://doi.org/10.1021/es402687w, 2013.
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurtén, T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., D'Ambro, E. L., and Thornton, J. A.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci. USA, 113, 1516–1521, https://doi.org/10.1073/pnas.1508108113, 2016.
Li, K., Chen, L., White, S. J., Yu, H., Wu, X., Gao, X., Azzi, M., and Cen, K.: Smog chamber study of the role of NH3 in new particle formation from photo-oxidation of aromatic hydrocarbons, Sci. Total Environ., 619–620, 927–937, https://doi.org/10.1016/j.scitotenv.2017.11.180, 2018.
Li, L., Tan, Q., Zhang, Y., Feng, M., Qu, Y., An, J., and Liu, X.: Characteristics and source apportionment of PM2.5 during persistent extreme haze events in Chengdu, southwest China, Environ. Pollut., 230, 718–729, https://doi.org/10.1016/j.envpol.2017.07.029, 2017.
Liggio, J. and Li, S. M.: Reactive uptake of pinonaldehyde on acidic aerosols, J. Geophys. Res.-Atmos., 111, D24303, https://doi.org/10.1029/2005JD006978, 2006.
Liggio, J. and Li, S.-M.: Reversible and irreversible processing of biogenic olefins on acidic aerosols, Atmos. Chem. Phys., 8, 2039–2055, https://doi.org/10.5194/acp-8-2039-2008, 2008.
Liggio, J., Li, S.-M., and McLaren, R.: Heterogeneous reactions of glyoxal on particulate matter: identification of acetals and sulfate esters, Environ. Sci. Technol., 39, 1532–1541, https://doi.org/10.1021/es048375y, 2005.
Liggio, J., Li, S. M., Brook, J. R., and Mihele, C.: Direct polymerization of isoprene and α-pinene on acidic aerosols, Geophys. Res. Lett., 34, L05814, https://doi.org/10.1029/2006GL028468, 2007.
Lim, Y. B. and Ziemann, P. J.: Products and mechanism of secondary organic aerosol formation from reactions of n-alkanes with OH radicals in the presence of NOx, Environ. Sci. Technol., 39, 9229–9236, https://doi.org/10.1021/es051447g, 2005.
Lin, Y.-H., Knipping, E. M., Edgerton, E. S., Shaw, S. L., and Surratt, J. D.: Investigating the influences of SO2 and NH3 levels on isoprene-derived secondary organic aerosol formation using conditional sampling approaches, Atmos. Chem. Phys., 13, 8457–8470, https://doi.org/10.5194/acp-13-8457-2013, 2013.
Liu, H., Man, H., Cui, H., Wang, Y., Deng, F., Wang, Y., Yang, X., Xiao, Q., Zhang, Q., Ding, Y., and He, K.: An updated emission inventory of vehicular VOCs and IVOCs in China, Atmos. Chem. Phys., 17, 12709–12724, https://doi.org/10.5194/acp-17-12709-2017, 2017.
Liu, S., Jia, L., Xu, Y., Tsona, N. T., Ge, S., and Du, L.: Photooxidation of cyclohexene in the presence of SO2: SOA yield and chemical composition, Atmos. Chem. Phys., 17, 13329–13343, https://doi.org/10.5194/acp-17-13329-2017, 2017.
Liu, T., Wang, X., Deng, W., Hu, Q., Ding, X., Zhang, Y., He, Q., Zhang, Z., Lü, S., Bi, X., Chen, J., and Yu, J.: Secondary organic aerosol formation from photochemical aging of light-duty gasoline vehicle exhausts in a smog chamber, Atmos. Chem. Phys., 15, 9049–9062, https://doi.org/10.5194/acp-15-9049-2015, 2015a.
Liu, T., Wang, X., Deng, W., Zhang, Y., Chu, B., Ding, X., Hu, Q., He, H., and Hao, J.: Role of ammonia in forming secondary aerosols from gasoline vehicle exhaust, Sci. China Chem., 58, 1377–1384, https://doi.org/10.1007/s11426-015-5414-x, 2015b.
Liu, T., Wang, X., Hu, Q., Deng, W., Zhang, Y., Ding, X., Fu, X., Bernard, F., Zhang, Z., Lü, S., He, Q., Bi, X., Chen, J., Sun, Y., Yu, J., Peng, P., Sheng, G., and Fu, J.: Formation of secondary aerosols from gasoline vehicle exhaust when mixing with SO2, Atmos. Chem. Phys., 16, 675–689, https://doi.org/10.5194/acp-16-675-2016, 2016.
Liu, X. G., Li, J., Qu, Y., Han, T., Hou, L., Gu, J., Chen, C., Yang, Y., Liu, X., Yang, T., Zhang, Y., Tian, H., and Hu, M.: Formation and evolution mechanism of regional haze: a case study in the megacity Beijing, China, Atmos. Chem. Phys., 13, 4501–4514, https://doi.org/10.5194/acp-13-4501-2013, 2013.
Liu, Y., Shao, M., Fu, L., Lu, S., Zeng, L., and Tang, D.: Source profiles of volatile organic compounds (VOCs) measured in China: Part I, Atmos. Environ., 42, 6247–6260, https://doi.org/10.1016/j.atmosenv.2008.01.070, 2008.
Liu, Y., Li, S.-M., and Liggio, J.: Technical Note: Application of positive matrix factor analysis in heterogeneous kinetics studies utilizing the mixed-phase relative rates technique, Atmos. Chem. Phys., 14, 9201–9211, https://doi.org/10.5194/acp-14-9201-2014, 2014.
Liu, Y., Liggio, J., Staebler, R., and Li, S.-M.: Reactive uptake of ammonia to secondary organic aerosols: kinetics of organonitrogen formation, Atmos. Chem. Phys., 15, 13569–13584, https://doi.org/10.5194/acp-15-13569-2015, 2015.
Lu, Z., Streets, D. G., Zhang, Q., Wang, S., Carmichael, G. R., Cheng, Y. F., Wei, C., Chin, M., Diehl, T., and Tan, Q.: Sulfur dioxide emissions in China and sulfur trends in East Asia since 2000, Atmos. Chem. Phys., 10, 6311–6331, https://doi.org/10.5194/acp-10-6311-2010, 2010.
Meade, L. E., Riva, M., Blomberg, M. Z., Brock, A. K., Qualters, E. M., Siejack, R. A., Ramakrishnan, K., Surratt, J. D., and Kautzman, K. E.: Seasonal variations of fine particulate organosulfates derived from biogenic and anthropogenic hydrocarbons in the mid-Atlantic United States, Atmos. Environ., 145, 405–414, https://doi.org/10.1016/j.atmosenv.2016.09.028, 2016.
Meng, Z. Y., Lin, W. L., Jiang, X. M., Yan, P., Wang, Y., Zhang, Y. M., Jia, X. F., and Yu, X. L.: Characteristics of atmospheric ammonia over Beijing, China, Atmos. Chem. Phys., 11, 6139–6151, https://doi.org/10.5194/acp-11-6139-2011, 2011.
Na, K., Song, C., and Cocker, D. R.: Formation of secondary organic aerosol from the reaction of styrene with ozone in the presence and absence of ammonia and water, Atmos. Environ., 40, 1889–1900, https://doi.org/10.1016/j.atmosenv.2005.10.063, 2006.
Na, K., Song, C., Switzer, C., and Cocker, D. R.: Effect of ammonia on secondary organic aerosol formation from α-pinene ozonolysis in dry and humid conditions, Environ. Sci. Technol., 41, 6096–6102, https://doi.org/10.1021/es061956y, 2007.
Nguyen, T. B., Lee, P. B., Updyke, K. M., Bones, D. L., Laskin, J., Laskin, A., and Nizkorodov, S. A.: Formation of nitrogen- and sulfur-containing light-absorbing compounds accelerated by evaporation of water from secondary organic aerosols, J. Geophys. Res.-Atmos., 117, D01207, https://doi.org/10.1029/2011jd016944, 2012.
Nozière, B., Dziedzic, P., and Córdova, A.: Products and kinetics of the liquid-phase reaction of glyoxal catalyzed by ammonium ions (), J. Phys. Chem. A, 113, 231–237, https://doi.org/10.1021/jp8078293, 2009.
Ortiz-Montalvo, D. L., Häkkinen, S. A. K., Schwier, A. N., Lim, Y. B., McNeill, V. F., and Turpin, B. J.: Ammonium addition (and aerosol pH) has a dramatic impact on the volatility and yield of glyoxal secondary organic aerosol, Environ. Sci. Technol., 48, 255–262, https://doi.org/10.1021/es4035667, 2014.
Pan, Y., Tian, S., Liu, D., Fang, Y., Zhu, X., Zhang, Q., Zheng, B., Michalski, G., and Wang, Y.: Fossil fuel combustion-related emissions dominate atmospheric ammonia sources during severe haze episodes: Evidence from 15N-stable isotope in size-resolved aerosol ammonium, Environ. Sci. Technol., 50, 8049–8056, https://doi.org/10.1021/acs.est.6b00634, 2016.
Platt, S. M., El Haddad, I., Zardini, A. A., Clairotte, M., Astorga, C., Wolf, R., Slowik, J. G., Temime-Roussel, B., Marchand, N., Ježek, I., Drinovec, L., Močnik, G., Möhler, O., Richter, R., Barmet, P., Bianchi, F., Baltensperger, U., and Prévôt, A. S. H.: Secondary organic aerosol formation from gasoline vehicle emissions in a new mobile environmental reaction chamber, Atmos. Chem. Phys., 13, 9141–9158, https://doi.org/10.5194/acp-13-9141-2013, 2013.
Pöschl, U.: Atmospheric aerosols: composition, transformation, climate and health effects, Angew. Chem. Int. Ed., 44, 7520–7540, https://doi.org/10.1002/anie.200501122, 2005.
Presto, A. A., Miracolo, M. A., Kroll, J. H., Worsnop, D. R., Robinson, A. L., and Donahue, N. M.: Intermediate-volatility organic compounds: A potential source of ambient oxidized organic aerosol, Environ. Sci. Technol., 43, 4744–4749, https://doi.org/10.1021/es803219q, 2009.
Pye, H. O. T. and Pouliot, G. A.: Modeling the role of alkanes, polycyclic aromatic hydrocarbons, and their oligomers in secondary organic aerosol formation, Environ. Sci. Technol., 46, 6041–6047, https://doi.org/10.1021/es300409w, 2012.
Riva, M., Tomaz, S., Cui, T., Lin, Y.-H., Perraudin, E., Gold, A., Stone, E. A., Villenave, E., and Surratt, J. D.: Evidence for an unrecognized secondary anthropogenic source of organosulfates and sulfonates: gas-phase oxidation of polycyclic aromatic hydrocarbons in the presence of sulfate aerosol, Environ. Sci. Technol., 49, 6654–6664, https://doi.org/10.1021/acs.est.5b00836, 2015.
Riva, M., Da Silva Barbosa, T., Lin, Y.-H., Stone, E. A., Gold, A., and Surratt, J. D.: Chemical characterization of organosulfates in secondary organic aerosol derived from the photooxidation of alkanes, Atmos. Chem. Phys., 16, 11001–11018, https://doi.org/10.5194/acp-16-11001-2016, 2016.
Rollins, A. W., Kiendler-Scharr, A., Fry, J. L., Brauers, T., Brown, S. S., Dorn, H.-P., Dubé, W. P., Fuchs, H., Mensah, A., Mentel, T. F., Rohrer, F., Tillmann, R., Wegener, R., Wooldridge, P. J., and Cohen, R. C.: Isoprene oxidation by nitrate radical: alkyl nitrate and secondary organic aerosol yields, Atmos. Chem. Phys., 9, 6685–6703, https://doi.org/10.5194/acp-9-6685-2009, 2009.
Santiago, M., Garcia Vivanco, M., and Stein, A. F.: SO2 effect on secondary organic aerosol from a mixture of anthropogenic VOCs: experimental and modelled results, Int. J. Environ. Pollut., 50, 224–233, https://doi.org/10.1504/ijep.2012.051195, 2012.
Sato, K., Takami, A., Isozaki, T., Hikida, T., Shimono, A., and Imamura, T.: Mass spectrometric study of secondary organic aerosol formed from the photo-oxidation of aromatic hydrocarbons, Atmos. Environ., 44, 1080–1087, https://doi.org/10.1016/j.atmosenv.2009.12.013, 2010.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics: from air pollution to climate change, John Wiley & Sons, Hoboken, NJ, USA, 2016.
Shang, J., Passananti, M., Dupart, Y., Ciuraru, R., Tinel, L., Rossignol, S., Perrier, S., Zhu, T., and George, C.: SO2 uptake on oleic acid: a new formation pathway of organosulfur compounds in the atmosphere, Environ. Sci. Technol. Lett., 3, 67–72, https://doi.org/10.1021/acs.estlett.6b00006, 2016.
Shen, X. J., Sun, J. Y., Zhang, X. Y., Zhang, Y. M., Zhang, L., Che, H. C., Ma, Q. L., Yu, X. M., Yue, Y., and Zhang, Y. W.: Characterization of submicron aerosols and effect on visibility during a severe haze-fog episode in Yangtze River Delta, China, Atmos. Environ., 120, 307–316, https://doi.org/10.1016/j.atmosenv.2015.09.011, 2015.
Sipilä, M., Berndt, T., Petäjä, T., Brus, D., Vanhanen, J., Stratmann, F., Patokoski, J., Mauldin, R. L., Hyvärinen, A.-P., Lihavainen, H., and Kulmala, M.: The role of sulfuric acid in atmospheric nucleation, Science, 327, 1243–1246, https://doi.org/10.1126/science.1180315, 2010.
Staudt, S., Kundu, S., Lehmler, H.-J., He, X., Cui, T., Lin, Y.-H., Kristensen, K., Glasius, M., Zhang, X., Weber, R. J., Surratt, J. D., and Stone, E. A.: Aromatic organosulfates in atmospheric aerosols: synthesis, characterization, and abundance, Atmos. Environ., 94, 366–373, https://doi.org/10.1016/j.atmosenv.2014.05.049, 2014.
Sun, K., Tao, L., Miller, D. J., Pan, D., Golston, L. M., Zondlo, M. A., Griffin, R. J., Wallace, H. W., Leong, Y. J., Yang, M. M., Zhang, Y., Mauzerall, D. L., and Zhu, T.: Vehicle emissions as an important urban ammonia source in the United States and China, Environ. Sci. Technol., 51, 2472–2481, https://doi.org/10.1021/acs.est.6b02805, 2017.
Sun, Y., Chen, C., Zhang, Y., Xu, W., Zhou, L., Cheng, X., Zheng, H., Ji, D., Li, J., Tang, X., Fu, P., and Wang, Z.: Rapid formation and evolution of an extreme haze episode in Northern China during winter 2015, Sci. Rep., 6, 27151, https://doi.org/10.1038/srep27151, 2016.
Surratt, J. D., Kroll, J. H., Kleindienst, T. E., Edney, E. O., Claeys, M., Sorooshian, A., Ng, N. L., Offenberg, J. H., Lewandowski, M., Jaoui, M., Flagan, R. C., and Seinfeld, J. H.: Evidence for organosulfates in secondary organic aerosol, Environ. Sci. Technol., 41, 517–527, https://doi.org/10.1021/es062081q, 2007.
Takekawa, H., Minoura, H., and Yamazaki, S.: Temperature dependence of secondary organic aerosol formation by photo-oxidation of hydrocarbons, Atmos. Environ., 37, 3413–3424, https://doi.org/10.1016/S1352-2310(03)00359-5, 2003.
Tan, J.-H., Duan, J.-C., Chen, D.-H., Wang, X.-H., Guo, S.-J., Bi, X.-H., Sheng, G.-Y., He, K.-B., and Fu, J.-M.: Chemical characteristics of haze during summer and winter in Guangzhou, Atmos. Res., 94, 238–245, https://doi.org/10.1016/j.atmosres.2009.05.016, 2009.
Tang, G., Sun, J., Wu, F., Sun, Y., Zhu, X., Geng, Y., and Wang, Y.: Organic composition of gasoline and its potential effects on air pollution in North China, Sci. China Chem., 58, 1416–1425, https://doi.org/10.1007/s11426-015-5464-0, 2015.
Thalman, R., de Sá, S. S., Palm, B. B., Barbosa, H. M. J., Pöhlker, M. L., Alexander, M. L., Brito, J., Carbone, S., Castillo, P., Day, D. A., Kuang, C., Manzi, A., Ng, N. L., Sedlacek III, A. J., Souza, R., Springston, S., Watson, T., Pöhlker, C., Pöschl, U., Andreae, M. O., Artaxo, P., Jimenez, J. L., Martin, S. T., and Wang, J.: CCN activity and organic hygroscopicity of aerosols downwind of an urban region in central Amazonia: seasonal and diel variations and impact of anthropogenic emissions, Atmos. Chem. Phys., 17, 11779–11801, https://doi.org/10.5194/acp-17-11779-2017, 2017.
Tkacik, D. S., Presto, A. A., Donahue, N. M., and Robinson, A. L.: Secondary organic aerosol formation from intermediate-volatility organic compounds: cyclic, linear, and branched alkanes, Environ. Sci. Technol., 46, 8773–8781, https://doi.org/10.1021/es301112c, 2012.
Tong, S., Hou, S., Zhang, Y., Chu, B., Liu, Y., He, H., Zhao, P., and Ge, M.: Exploring the nitrous acid (HONO) formation mechanism in winter Beijing: direct emissions and heterogeneous production in urban and suburban areas, Faraday Discuss., 189, 213–230, https://doi.org/10.1039/c5fd00163c, 2016.
Ulbrich, I. M., Canagaratna, M. R., Zhang, Q., Worsnop, D. R., and Jimenez, J. L.: Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data, Atmos. Chem. Phys., 9, 2891–2918, https://doi.org/10.5194/acp-9-2891-2009, 2009.
Wexler, A. S. and Clegg, S. L.: Atmospheric aerosol models for systems including the ions H+, , Na+, , , Cl−, Br−, and H2O, J. Geophys. Res.-Atmos., 107, 4207, https://doi.org/10.1029/2001jd000451, 2002.
Xu, J., Huang, M.-Q., Cai, S.-Y., Liao, Y.-M., Hu, C.-J., Zhao, W.-X., Gu, X.-J., and Zhang, W.-J.: Chemical composition and reaction mechanisms for aged p-xylene secondary organic aerosol in the presence of ammonia, J. Chin. Chem. Soc.-Taip, 65, 578–590, https://doi.org/10.1002/jccs.201700249, 2018.
Yang, S., Yuesi, W., and Changchun, Z.: Measurement of the vertical profile of atmospheric SO2 during the heating period in Beijing on days of high air pollution, Atmos. Environ., 43, 468–472, https://doi.org/10.1016/j.atmosenv.2008.09.057, 2009.
Yang, W., Li, J., Wang, M., Sun, Y., and Wang, Z.: A case study of investigating secondary organic aerosol formation pathways in Beijing using an observation-based SOA Box Model, Aerosol Air Qual. Res., 18, 1606–1616, https://doi.org/10.4209/aaqr.2017.10.0415, 2018.
Ye, P., Ding, X., Hakala, J., Hofbauer, V., Robinson, E. S., and Donahue, N. M.: Vapor wall loss of semi-volatile organic compounds in a Teflon chamber, Aerosol Sci. Tech., 50, 822–834, https://doi.org/10.1080/02786826.2016.1195905, 2016.
Yuan, B., Koss, A. R., Warneke, C., Coggon, M., Sekimoto, K., and de Gouw, J. A.: Proton-transfer-reaction mass spectrometry: Applications in atmospheric sciences, Chem. Rev., 117, 13187–13229, https://doi.org/10.1021/acs.chemrev.7b00325, 2017.
Zhang, L., Chen, Y., Zhao, Y., Henze, D. K., Zhu, L., Song, Y., Paulot, F., Liu, X., Pan, Y., Lin, Y., and Huang, B.: Agricultural ammonia emissions in China: reconciling bottom-up and top-down estimates, Atmos. Chem. Phys., 18, 339–355, https://doi.org/10.5194/acp-18-339-2018, 2018.
Zhang, Q., Streets, D. G., He, K., and Klimont, Z.: Major components of China's anthropogenic primary particulate emissions, Environ. Res. Lett., 2, 045027, https://doi.org/10.1088/1748-9326/2/4/045027, 2007.
Zhang, X., Cappa, C. D., Jathar, S. H., McVay, R. C., Ensberg, J. J., Kleeman, M. J., and Seinfeld, J. H.: Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol, P. Natl. Acad. Sci. USA, 111, 5802–5807, https://doi.org/10.1073/pnas.1404727111, 2014.
Zhang, X., Schwantes, R. H., McVay, R. C., Lignell, H., Coggon, M. M., Flagan, R. C., and Seinfeld, J. H.: Vapor wall deposition in Teflon chambers, Atmos. Chem. Phys., 15, 4197–4214, https://doi.org/10.5194/acp-15-4197-2015, 2015.
Zhang, Y., Wang, X., Zhang, Z., Lü, S., Shao, M., Lee, F. S. C., and Yu, J.: Species profiles and normalized reactivity of volatile organic compounds from gasoline evaporation in China, Atmos. Environ., 79, 110–118, https://doi.org/10.1016/j.atmosenv.2013.06.029, 2013.
Zhao, B., Wang, S., Donahue, N. M., Jathar, S. H., Huang, X., Wu, W., Hao, J., and Robinson, A. L.: Quantifying the effect of organic aerosol aging and intermediate-volatility emissions on regional-scale aerosol pollution in China, Sci. Rep., 6, 28815, https://doi.org/10.1038/srep28815, 2016.
Zhao, D., Schmitt, S. H., Wang, M., Acir, I.-H., Tillmann, R., Tan, Z., Novelli, A., Fuchs, H., Pullinen, I., Wegener, R., Rohrer, F., Wildt, J., Kiendler-Scharr, A., Wahner, A., and Mentel, T. F.: Effects of NOx and SO2 on the secondary organic aerosol formation from photooxidation of α-pinene and limonene, Atmos. Chem. Phys., 18, 1611–1628, https://doi.org/10.5194/acp-18-1611-2018, 2018a.
Zhao, D., Song, X., Zhu, T., Zhang, Z., Liu, Y., and Shang, J.: Multiphase oxidation of SO2 by NO2 on CaCO3 particles, Atmos. Chem. Phys., 18, 2481–2493, https://doi.org/10.5194/acp-18-2481-2018, 2018b.
Zheng, B., Zhang, Q., Zhang, Y., He, K. B., Wang, K., Zheng, G. J., Duan, F. K., Ma, Y. L., and Kimoto, T.: Heterogeneous chemistry: a mechanism missing in current models to explain secondary inorganic aerosol formation during the January 2013 haze episode in North China, Atmos. Chem. Phys., 15, 2031–2049, https://doi.org/10.5194/acp-15-2031-2015, 2015.
Zou, Y., Deng, X. J., Zhu, D., Gong, D. C., Wang, H., Li, F., Tan, H. B., Deng, T., Mai, B. R., Liu, X. T., and Wang, B. G.: Characteristics of 1 year of observational data of VOCs, NOx and O3 at a suburban site in Guangzhou, China, Atmos. Chem. Phys., 15, 6625–6636, https://doi.org/10.5194/acp-15-6625-2015, 2015.