the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 07 Jun 2019
| 07 Jun 2019
Effect of temperature on the formation of highly oxygenated organic molecules (HOMs) from alpha-pinene ozonolysis
Lauriane L. J. Quéléver
Kasper Kristensen
Louise Normann Jensen
Bernadette Rosati
Ricky Teiwes
Kaspar R. Daellenbach
Otso Peräkylä
Pontus Roldin
Rossana Bossi
Henrik B. Pedersen
Marianne Glasius
Merete Bilde
Highly oxygenated organic molecules (HOMs) are important contributors to secondary organic aerosol (SOA) and new-particle formation (NPF) in the boreal atmosphere. This newly discovered class of molecules is efficiently formed from atmospheric oxidation of biogenic volatile organic compounds (VOCs), such as monoterpenes, through a process called autoxidation. This process, in which peroxy-radical intermediates isomerize to allow addition of molecular oxygen, is expected to be highly temperature-dependent. Here, we studied the dynamics of HOM formation during α-pinene ozonolysis experiments performed at three different temperatures, 20, 0 and −15 ∘C, in the Aarhus University Research on Aerosol (AURA) chamber. We found that the HOM formation, under our experimental conditions (50 ppb α-pinene and 100 ppb ozone), decreased considerably at lower temperature, with molar yields dropping by around a factor of 50 when experiments were performed at 0 ∘C, compared to 20 ∘C. At −15 ∘C, the HOM signals were already close to the detection limit of the nitrate-based chemical ionization atmospheric pressure interface time-of-flight (CI-APi-TOF) mass spectrometer used for measuring gas-phase HOMs. Surprisingly, comparing spectra measured at 0 and 20 ∘C, ratios between HOMs of different oxidation levels, e.g., the typical HOM products C10H14O7, C10H14O9, and C10H14O11, changed considerably less than the total HOM yields. More oxidized species have undergone more isomerization steps; yet, at lower temperature, they did not decrease more than the less oxidized species. One possible explanation is that the primary rate-limiting steps forming these HOMs occur before the products become oxygenated enough to be detected by our CI-APi-TOF (i.e., typically seven or more oxygen atoms). The strong temperature dependence of HOM formation was observed under temperatures highly relevant to the boreal forest, but the exact magnitude of this effect in the atmosphere will be much more complex: the fate of peroxy radicals is a competition between autoxidation (influenced by temperature and VOC type) and bimolecular termination pathways (influenced mainly by concentration of reaction partners). While the temperature influence is likely smaller in the boreal atmosphere than in our chamber, both the magnitude and complexity of this effect clearly deserve more consideration in future studies in order to estimate the ultimate role of HOMs on SOA and NPF under different atmospheric conditions.
- Article
(2161 KB) - Full-text XML
- BibTeX
- EndNote





Aerosol particles impact Earth's climate by scattering and absorbing solar radiation and by influencing cloud properties when they act as cloud condensation nuclei (CCN; IPCC, 2013). Organic compounds contribute significantly to the chemical composition of aerosol, accounting from 20 % to 90 % of the total aerosol mass of submicrometer particles depending on their location on the globe (Jimenez et al., 2009). Submicron organic aerosol is dominantly secondary. Called secondary organic aerosol (SOA), it originates from gas-to-particle conversion from condensable vapors (Hallquist et al., 2009; Zhang et al., 2007). These vapors are mainly oxidation products of volatile organic compounds (VOCs), having sufficiently low vapor pressure (i.e., volatility) to condense onto aerosol particles (Hallquist et al., 2009).
In order to interact efficiently with solar radiation or to activate cloud droplets, aerosol particles need to be around 100 nm in diameter or larger (Dusek et al., 2006). If particles have formed through nucleation processes in the atmosphere (e.g., Kulmala et al., 2013), their ability to grow to climate-relevant sizes before being scavenged through coagulation is critically impacted by the rate at which low-volatile vapors will condense onto them (Donahue et al., 2013). Extremely low-volatile organic compounds (ELVOCs), introduced by Donahue et al. (2012), have the ability to condense irreversibly onto even the smallest aerosol particles and clusters and thus contribute to particle growth. Low-volatile organic compounds (LVOCs), typically more abundant in the atmosphere, are important for the growth of particles larger than a few nanometers (Tröstl et al., 2016).
Highly oxygenated organic molecules (HOMs; Ehn et al., 2014, 2017; Bianchi et al., 2019) were recently identified as a large contributor to ELVOCs and LVOCs and the growth of newly formed particles (Ehn et al., 2014; Tröstl et al., 2016). First observed in measurements of naturally charged ions in the boreal forest (Ehn et al., 2010, 2012) using the atmospheric pressure interface time-of-flight (APi-TOF) mass spectrometer (Junninen et al., 2010), HOM quantification only became possible through the application of nitrate ion chemical ionization (CI) mass spectrometry (Zhao et al., 2013; Ehn et al., 2014). Most studies have utilized the APi-TOF coupled to such a chemical ionization source (chemical ionization atmospheric pressure interface time-of-flight: CI-APi-TOF; Jokinen et al., 2012), and detailed laboratory studies have been able to elucidate the primary formation pathways of HOMs (Rissanen et al., 2014; Jokinen et al., 2014; Mentel et al., 2015). We also note that the HOM-related terminology has evolved over the last years, and here we define HOMs as organic molecules formed through gas-phase autoxidation, containing six or more oxygen atoms.
The main process in HOM formation is peroxy-radical (RO2) autoxidation (Crounse et al., 2013), which involves an intramolecular H abstraction by the peroxy-radical group to form a hydroperoxide and a carbon-centered radical to which molecular oxygen (O2) can rapidly add to form a new RO2 with a higher level of oxygenation. The efficiency of this process is mainly determined by the availability of easily “abstractable” H atoms, which are often formed in the ozonolysis of endocyclic alkenes (Rissanen et al., 2014, 2015; Berndt et al., 2015). This structural component can be found in many biogenic VOCs, such as monoterpenes, enhancing their role as SOA precursors through efficient autoxidation and HOM formation (Ehn et al., 2014; Jokinen et al., 2014; Berndt et al., 2016). Peroxy radicals are important intermediates in nearly all atmospheric oxidation processes. The RO2 that has undergone autoxidation will terminate to closed-shell species in similar ways as less oxidized RO2, taking place either by unimolecular processes leading to loss of OH or HO2 or bimolecular reactions with NO, HO2 or other RO2. The termination pathway strongly influences the type of HOMs that can be formed, with, for example, RO2+RO2 reactions being able to form ROOR dimers and RO2+NO often forming organic nitrates (Ehn et al., 2014; Berndt et al., 2018). All these bimolecular reactions of peroxy radicals, as well as the initial oxidant-VOC reaction, are temperature-dependent. For example, the reaction rate of ozone with α-pinene, a broadly studied SOA-forming system, is 6.2×1017 () cm3 molecules−1 s−1 at 3 ∘C and 8.3×1017 () cm3 molecules−1 s−1 at 22 ∘C (Atkinson et al., 1982). However, the intramolecular isomerization through H shifts is likely to have a much stronger temperature dependence, due to the higher energy barrier for the H shift (Seinfeld and Pandis, 2006; Otkjær et al., 2018). For example, Praske et al. (2018) reported theoretical estimates of different H shifts in hexane-derived RO2, which increased roughly by a factor of 5 to 10 when the temperature increased by 22 ∘C (from 23 to 45 ∘C). Possible changes in HOM formation as a function of temperature are thus expected to derive mainly from changes in the autoxidation process. However, a detailed mechanistic understanding the various autoxidation steps, let alone their temperature dependencies, is still lacking for most atmospheric VOC-oxidant systems, owing partly to the plethora and the complexity of the possible reaction pathways.
Despite recent work in determining the impact of temperature on aerosol formation (Kristensen et al., 2017; Stolzenburg et al., 2018), literature on corresponding HOM effects is extremely limited. At room temperature (i.e., 20 ∘C ± 5 ∘C), HOM molar yields have been estimated to be some percent for most monoterpenes in reactions with ozone or OH (Ehn et al., 2014; Jokinen et al., 2015). Only very recently, studies were presented with HOM formation experiments conducted at varying temperatures. Stolzenburg et al. (2018) showed that at lower temperatures, the CI-APi-TOF detects much lower HOM concentrations, though no quantitative values on the HOM yields were given. The impact of decreased HOMs on new-particle growth rates was compensated by less oxidized species being able to condense at the lower temperatures. In another study, Frege et al. (2018) also concluded that HOM formation decreased at lower temperatures, but the study was based on observations of naturally charged ions using APi-TOF, complicating the interpretation of HOM formation rates.
In this study, we directly evaluate the impact of temperature on HOM yields in a laboratory chamber during α-pinene ozonolysis experiments at 20, 0, and −15 ∘C. Relative changes in HOM formation are compared between temperatures both for total HOM yields as well as on a molecule-by-molecule basis. The more detailed impact of temperature on the molecular distribution of HOMs is expected to provide new insights into critical steps in the formation pathways.
2.1 The AURA chamber
A detailed description of the Aarhus University Research on Aerosol (AURA) chamber can be found in Kristensen et al. (2017). Essentially, it consists of a ∼5 m3 Teflon® bag contained in a temperature-controlled enclosure. Configured in batch sampling mode, the chamber was initially cleaned by flushing at 20 ∘C with purified ambient air (i.e., filtered air exempt of particles, water vapor, or VOCs, and with reduced NOx concentration), subsequently set to the desired temperature and finally filled with the necessary reagents. Over the course of the experiment, it was progressively emptied due to sampling by the measuring instrumentation. In our experiments, we first added ozone to a concentration of ∼100 ppb, provided by an ozone generator (Model 610, Jelight Company, Inc.); then, the oxidation reaction started after the VOC was introduced by vaporization of a calculated volume of liquid reagent (α-pinene or β-pinene) into a hot stream of nitrogen, reaching the desired VOC concentration (10 or 50 ppb).
2.2 The ACCHA experiment
The Aarhus chamber campaign on HOMs and aerosols (ACCHA) experiment aimed to explore oxidation processes and aerosol formation during dark monoterpene ozonolysis at different temperatures, from −15 to 20 ∘C. The experiments focused on α-pinene oxidation at two different concentrations (10 and 50 ppb) for three different temperatures: −15, 0 and 20 ∘C. Two additional experiments were conducted with temperatures ramped from the coldest to the warmest or reversely during experiments at 10 ppb of α-pinene. For comparison, fixed temperature runs were also performed using β-pinene, at a concentration of 50 ppb. Ozone (∼100 ppb) was used as the main oxidant, but hydroxyl radicals also took part in the oxidation reactions, as OH scavengers were not employed in the experiments discussed here. According to model simulations using the master chemical mechanism v3.3.1 (Jenkin et al., 1997, 2015; Saunders et al., 2003), ozonolysis accounted for approximately two-thirds and OH oxidation for one-third of the α-pinene oxidation. A table summarizing the experiments of the campaign can be found in the Appendix (Table A1).
2.3 Instrumentation
The ACCHA experiment involved a diverse set of instruments measuring both the gas phase and the particle phase. The gas-phase instrumentation included a proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF-MS; Model 8000-783, IONICON Inc.; Jordan et al., 2009) for measuring the concentrations of the injected VOCs (more data from the PTR-TOF-MS can be found in Rosati et al., 2019) and other volatile products as well as a nitrate-based CI-APi-TOF (TOFWERK AG and Aerodyne Research, Inc.; Jokinen et al., 2012) mass spectrometer, analyzing the highly oxidized organic products of lower volatility (e.g., HOMs). The CI-APi-TOF is described in more detail in the following section. The aerosol phase measurement was done using (1) a nano-condensation nuclei counter (nCNC), being a combination of a particle size magnifier (PSM; Model A10, Airmodus Ltd.) and a condensation particle counter (CPC; Model A20, Airmodus Ltd.), (2) a scanning mobility particle sizer (SMPS; Kr-85 neutralizer – Model 3077A TSI, electrostatic classifier – Model 3082, TSI, nano-water-based CPC – Model 3788, TSI), counting the size-resolved particles from 10 to 400 nm, and (3) a high-resolution time-of-flight aerosol mass spectrometer (HR-TOF-AMS; Aerodyne Research, Inc., Jayne et al., 2000) determining the chemical composition of non-refractory aerosol particles larger than ∼35 nm. The temperature and relative humidity inside the chamber were monitored using HC02-04 sensors (HygroFlex HF320, Rotronic AG), and the ozone concentration was measured with an ozone monitor (O3-42 Module, Environment S.A.).
2.4 Measuring highly oxygenated organic molecules in the gas phase
HOMs present in the gas phase were measured using a CI-APi-TOF mass spectrometer. The instrument sampled air about 80 cm from the wall of the chamber via a 3∕4 inch tube directly connected to the CI-APi-TOF, which was located outside the chamber enclosure (∼20 ∘C at all times). The sheath air (taken from a compressed air line) was 30 L min−1, and the total flow (generated by the house vacuum line) was 40 L min−1. The ∼1 m long inlet had a flow of 10 L min−1 generated by the difference between the sheath and total flows. With such a tube length and flow, roughly half of the HOMs are expected to be lost to the walls of the inlet lines. The CI-APi-TOF is described by Jokinen et al. (2012) but also briefly presented here. Strong acids and highly oxygenated organic molecules have been shown to cluster efficiently with nitrate ions (Ehn et al., 2014; Hyttinen et al., 2015). Nitrate ions (i.e., , , and (), produced by exposure of nitric acid vapors to soft X-ray radiation, were electrostatically introduced into the sample flow of 10 L min−1 with a reaction time of roughly 200 ms at atmospheric pressure.
The ions, clustered with , were sampled through a 300 µm critical orifice into the atmospheric pressure interface (APi), where they were guided and focused by two segmented quadrupole chambers with gradually decreasing pressures (∼2 and mbar). Finally, an ion lens assembly, at mbar, guided the ions into the time-of-flight (TOF) chamber ( mbar) where they were orthogonally extracted and their mass-to-charge ratios determined. The detected signal of each ion is then expressed as counts per second (cps) or counts per second normalized by the sum of reagent (nitrate) ions (norm. cps). More details about the APi-TOF itself can be found in Junninen et al. (2010). Quantification of HOMs remains challenging, and, in this work, we aim at explaining the relative changes of HOMs measured at different temperature rather than focusing on their absolute concentration. However, in some instances we also estimate absolute quantities by applying a calibration factor molecules cm−3, (see Jokinen et al., 2012, for details on C). This translates to ∼70 ppt of HOMs per normalized count. As no calibrations were performed during the ACCHA experiments, the value was taken from a sulfuric acid calibration (methodology according to Kürten et al., 2012) performed during an earlier measurement campaign. While associated with a large uncertainty (estimated to be at least −50 %/+100 %) using this value, we obtained HOM molar yields (as described in later sections) of a similar range as earlier studies (Jokinen et al., 2012; Ehn et al., 2014). We estimated a detection limit from our experimental data at the lowest temperature to be roughly 10−5 normalized counts, which corresponds to ∼104 molecules cm−3.
2.5 HOM dynamics in a batch mode chamber
Being configured in batch mode, without active mixing, the AURA chamber is a dynamic reactor where concentrations of products are a function of cumulative sources and cumulative sinks from the start of the experiment. In the case of HOMs, their lifetime in the gas phase must be short due to their low vapor pressure and, thus, their fast condensation. This means that the measured HOM concentrations are mainly the result of production and loss having occurred within the previous minutes, as described in more detail in the following section.
The temporal change in HOM concentrations (i.e., ) can be expressed as the sum of the production terms and loss terms. The HOM formation is governed by the VOC reaction rate, while the loss is dominated by condensation onto particles or walls. For the yield estimation analysis, we focus mainly on the high concentration experiments (i.e., [α-pinene] = 50 ppb), where the high condensation sink (CS; on the order of 0.1 s−1) will dominate over the wall loss rate. In a smaller chamber with active mixing, the wall loss rate for low-volatile species has been estimated to be around 10−2 s−1 (Ehn et al., 2014), and in the AURA chamber we expect it to be much slower, likely on the order of 10−3 s−1. Since experiments performed at lower temperatures would reduce the vapor pressure of the resulting oxidized products and form more SOA than in warmer conditions, the variation of the condensation sink was considered in our analysis, as we expect higher CS values at lower temperatures.
Therefore, we can formulate a simplified expression as in the following equations:
Herein, γHOM corresponds to the HOM yield. The temperature-dependent rate constant of α-pinene ozonolysis, k, was taken to be cm3 molecules−1 s−1, where T is the temperature in degrees Celsius (Atkinson, 2000; Calvert et al., 2002). Since the majority of HOMs are irreversibly lost upon contact with a surface (Ehn et al., 2014), the CS represents the total sink at a time t. The CS was estimated using the measured particle number size distributions from the SMPS (Dal Maso et al., 2005). The molecular properties that govern the CS are the mass accommodation coefficient, the molecular diffusion coefficient, and the mean molecular speed. Based on the work by Julin et al. (2014), the mass accommodation coefficient was set to unity. The molecular diffusion coefficient was calculated using Fuller's method (Tang et al., 2015), and the mean molecular speed was calculated using kinetic theory. Both the molecular diffusion and speed depend on molecular composition and on the absolute temperature during the experiments. C10H16O7 was taken as a reference for the CS estimation, being one of the most abundant HOMs. In comparison, the CSs calculated for the largest molecules (i.e., HOM dimers) were approximately 30 % lower. With the aforementioned assumptions, a distinct yield for each identified HOM of interest can be derived based on Eq. (2), as the slope of a linear fit to the data during an experiment, with on the x axis and on the y axis.
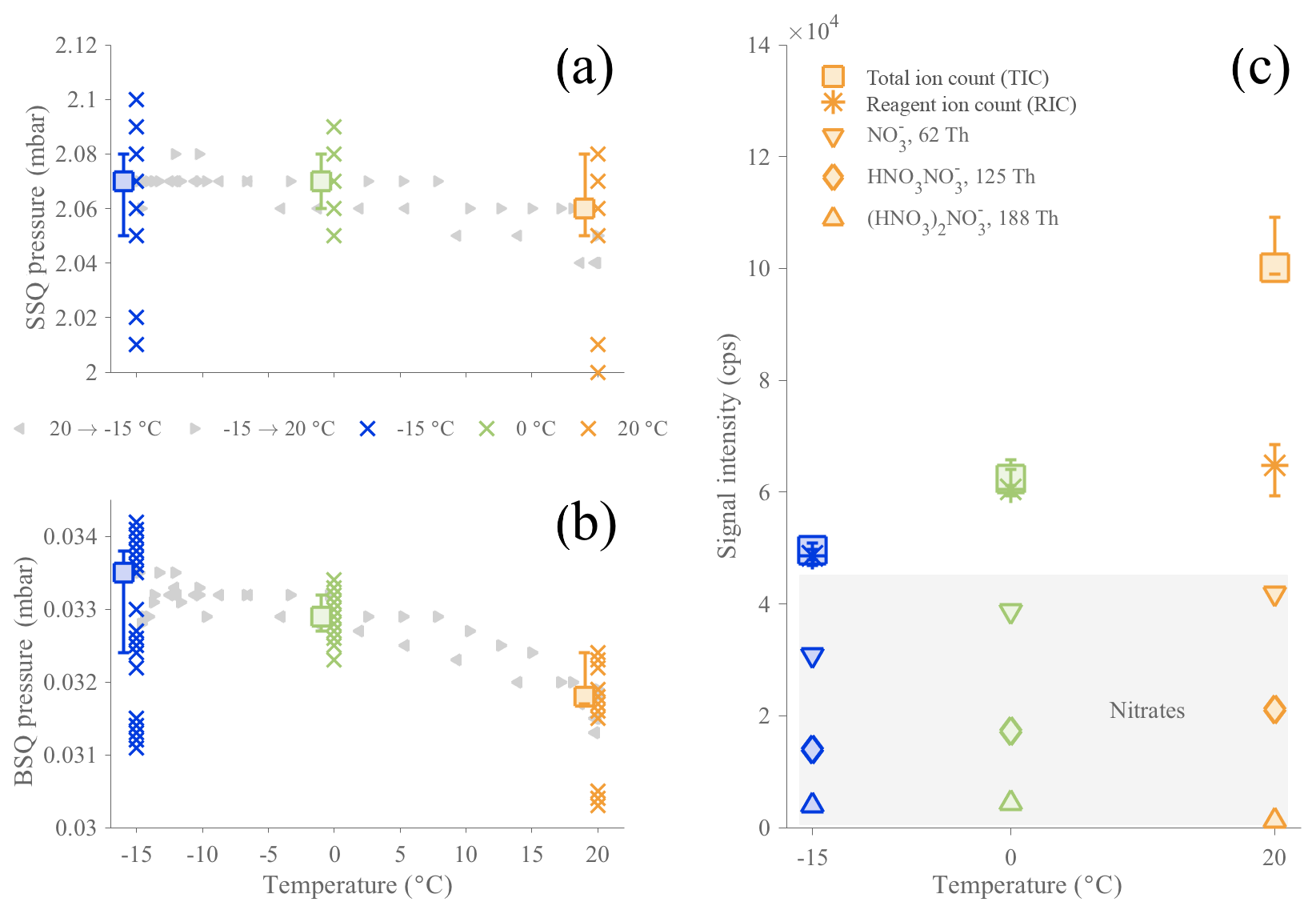
Figure 1Evolution of the CI-APi-TOF pressures in the first (a) and second (b) quadrupole chambers (SSQ and BSQ, respectively) and signal counts (c) as a function of temperature in the AURA chamber. The APi pressures (a, b) are represented by crosses, depicting 10 min averaged data points for all α-pinene ozonolysis experiments, colored by temperature (blue for −15 ∘C, green for 0 ∘C, and orange for 20 ∘C). The squares are the median values for each temperature with their 75th and 25th percentiles. Additionally, the gray triangles relate the data (10 min averages) of two temperature ramp experiments, from −15 to 20 ∘C (right-pointing triangles) or from 20 to −15 ∘C (left-pointing triangles). Panel (c) shows averages of the sum of all ion signals (TIC; square markers) and the sum of all reagent ion signals (RIC; asterisk markers). RIC markers also include 25th and 75th percentiles. Nitrate signal contributions are also included separately (markers in gray-shaded area: downward-pointing triangle for , diamond marker for , and triangle pointing upward for ().
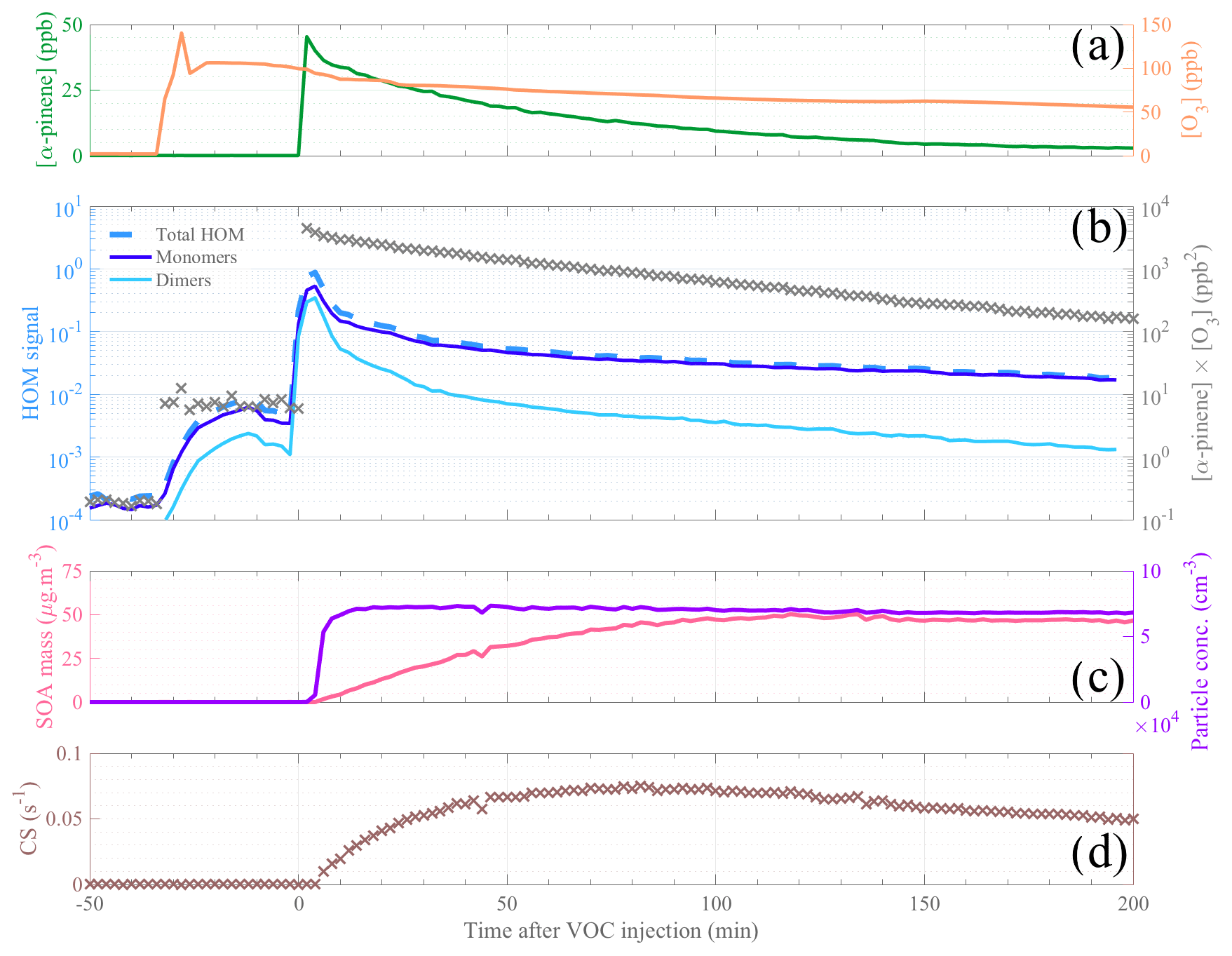
Figure 2Temporal evolution of the main parameters during a typical α-pinene ozonolysis experiment (initial conditions: [α-pinene] = 50 ppb, [O3] = 100 ppb, and T=20 ∘C). Reactant concentrations are shown in (a), with α-pinene concentration in dark green and ozone concentration in orange. HOM signals are plotted in (b), with a distinction between total HOMs (dashed medium-blue line), HOM monomers (, dark blue line), and HOM dimers (, light blue line) as well as the product [α-pinene] ⋅ [O3] represented by gray cross markers. Panel (c) depicts the SOA mass (pink line) and the particle concentration (purple line). Panel (d) shows the evolution of the condensation sink. The time span (in x axis) is expressed as minutes after α-pinene injection; thus the time zero represents the start of the experiment.
3.1 Effect of the temperature on the CI-APi-TOF
Since this work targets the variation of HOMs in relation to temperature, it is necessary to assess the reliability of the CI-APi-TOF measurement towards temperature variations. The sensitivity towards a certain molecule depends, by approximation, on the charging efficiency in the CI inlet and the transmission efficiency of the sampled ion in the APi-TOF. The charging efficiency of an HOM is primarily determined by the stability of the cluster relative to the cluster (Hyttinen et al., 2015), and we do not expect temperature to cause a large difference in this behavior. However, the transmission can be sensitive to small changes, and especially pressures inside the instrument are important to monitor, as the optimal voltages guiding the sampled ions through the instrument have been tuned for specific pressures. The pressures of the two quadrupole chambers (named SSQ and BSQ, where the pressure dependence is the largest) as well the total ion count (TIC; i.e., sum of all signals), the reagent ion count (RIC; i.e., sum of nitrate ion signals), and the contributions of each nitrate ion signal are presented in Fig. 1. The SSQ pressures (Fig. 1a) were found to be relatively stable (average: ∼2.07 mbar), and the BSQ averaged pressure (Fig. 1b) was mbar; these are typical values for this instrument. Unfortunately, the other instrumental pressures (i.e., ion lens assembly chamber or TOF chamber) were not recorded due to sensor failures. However, as these chambers are at low enough pressures that ion–gas collisions are very rare, any possible small variations in the pressures are unlikely to affect our results. When going from the coldest temperature (−15 ∘C) to the highest (20 ∘C), in a continuous temperature ramp, the SSQ pressure decreased by ∼0.01 mbar, corresponding to a relative change of 0.5 % (Fig. 1a). Over the same temperature range, the pressure within the second chamber (BSQ) decreased by mbar (∼4.5 %) when the temperature varied by 35 ∘C (Fig. 1a). The same characteristics were observed when comparing across experiments performed at constant temperatures and for the continuous temperature ramping experiments. The SSQ pressure values below 2.02 mbar at −15 and 20 ∘C, corresponding also to the lowest BSQ pressures measured, were related to particularly low ambient pressures (∼981.8 mbar). Thus, the effect of temperature within the AURA chamber caused only small variability in the internal pressures than ambient pressure changes.
The RIC signal (Fig. 1c) stayed within the range 5–7 × 104 cps, with its lowest values observed at −15 ∘C. The comparatively larger increase in TIC at the highest temperature is mainly explained by the fact that much higher HOM concentrations were formed at 20 ∘C compared to lower temperature experiments, and the transmission at the HOM mass range is generally higher than in the region of the reagent ions (Junninen et al., 2010; Ehn et al., 2011; Heinritzi et al., 2016). We conclude from the above investigations that changes on the order of tens of percent, based on the variation in RIC, occurred in our instrument as the AURA chamber temperature was varied and that only signal changes larger than this should be attributed to actual perturbations of the chemistry taking place in the chamber.
3.2 Ozonolysis reaction in the AURA chamber: a typical α-pinene experiment at 20 ∘C
Selected gas-phase precursors and products, including aerosols, for a high-load (i.e., 50 ppb) α-pinene oxidation experiment at 20 ∘C (during 12 January 2017) are shown in Fig. 2. The steep increase in α-pinene concentration, measured by PTR-TOF-MS, indicates the start (defined as time 0) of the oxidation reaction experiment (Fig. 2a). The formed aerosol products, i.e., the particle number and aerosol mass, are presented in Fig. 2c. Herein, we observe an increase in the aerosol mass over the first 2 h of the experiment, whereas the particle number concentration plateaued in the first 10 min after VOC injection. On the other hand, the HOM signals (Fig. 2b) show a large increase immediately as the VOC was injected. A smaller increase was also observed when the ozone was introduced, most likely due to residual volatiles reacting with ozone inside the chamber. After the first 10 min, HOM signals start to decrease as the CS (Fig. 2d) rapidly increases under these high aerosol loads. After the first half hour, the CS only changes by some tens of percents, while the VOC oxidation rate (gray crosses in Fig. 2b) decreases around 1 order of magnitude over the following hours of the experiment. Therefore, concentrations of low-volatile HOMs should largely track the decay rate of the VOC oxidation rate, which is also observed. We observe a slower decay of HOM monomers than dimers, suggesting that some of the monomers may be semi-volatile enough to not condense irreversibly upon every collision with a surface and/or that the VOC oxidation rate also influences the formation chemistry, as discussed in more detail in later sections.
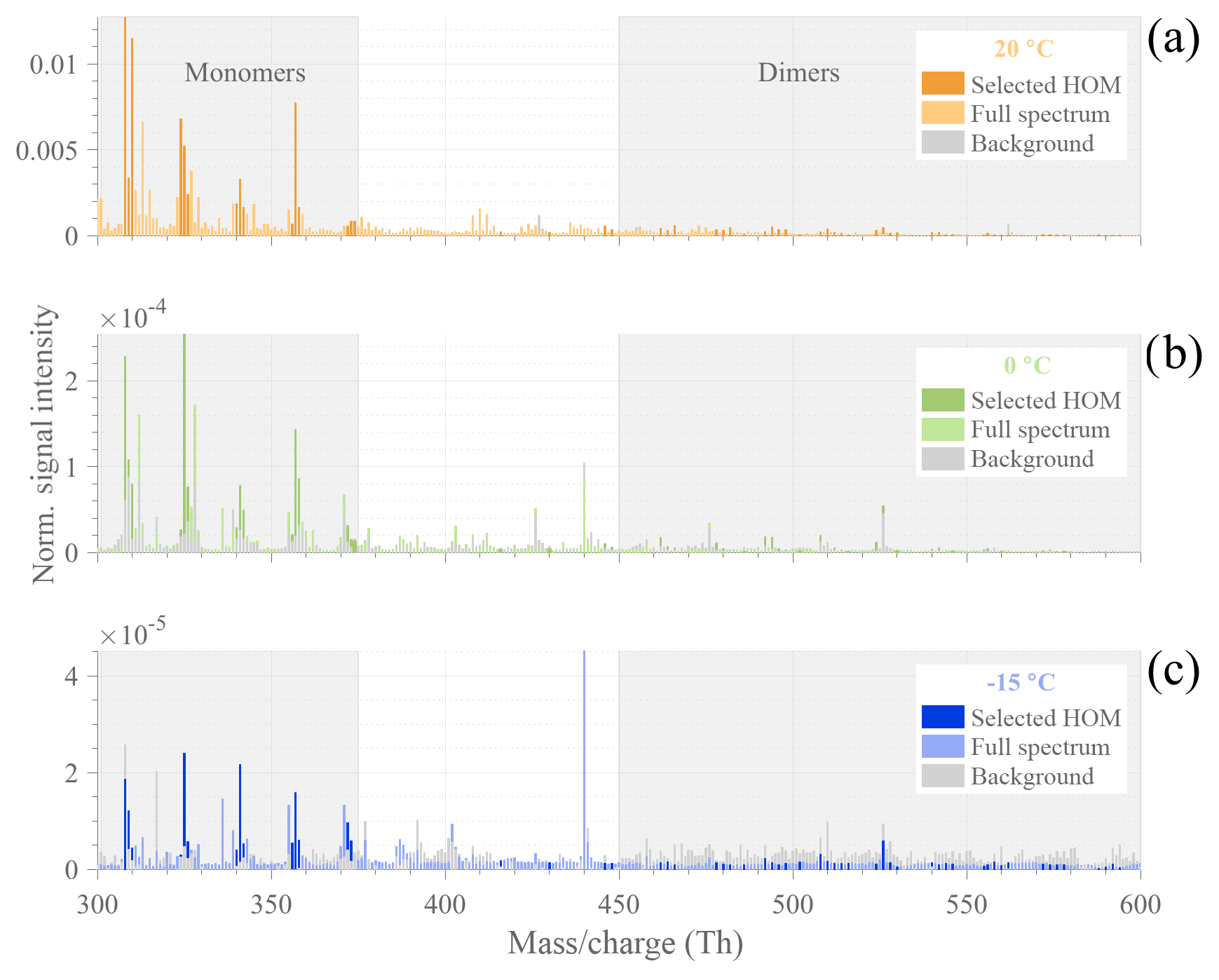
Figure 3Typical HOM mass spectra observed during α-pinene ozonolysis experiments (initial conditions: [α-pinene] = 50 ppb, [O3] = 100 ppb), with T=20 ∘C (a) in orange, T=0 ∘C (b) in green, and ∘C (c) in blue. The normalized signals were averaged over 5 min during background measurements before VOC injection (gray bars) and from 40 to 120 min after α-pinene injection (colored bars). Specific masses, selected for representing high-intensity HOMs, are highlighted in darker colors. Gray-shaded areas show HOM sub-ranges of monomers and dimers.
For a more detailed investigation at the HOM formation upon the reaction between ozone and α-pinene, we compare compounds observed in the range between 300–600 Th (Thomson) by the CI-APi-TOF during a background measurement before and from 40 to 120 min after α-pinene injection for each temperature (Fig. 3). The largest HOM signals, highlighted in darker colors, are primarily observed at the highest temperature in the monomer area (300–375 Th). The dimer signals (between 450–600 Th) are smaller but still contribute significantly to the total HOM concentration. With the exception of the −15 ∘C experiment where HOM dimers already reach the background level after 10 min, all molecules selected as representative HOMs are present in all spectra. The detailed peak list of HOM compounds, selected for their high signal intensity, including exact masses and elemental compositions, is provided in the Appendix (Table A2).

Figure 4Time series of HOMs measured during the ACCHA campaign. HOM monomer (a) and dimer (b) traces include compounds with chemical compositions of and , respectively. The series are colored based on temperature (orange for 20 ∘C experiments, green for 0 ∘C, and blue for −15 ∘C). Statistics over α-pinene (α in the legend) high-load (50 ppb, H) experiments are shown, with averaged values (av., in continuous line) and the maximum and minimum values of the measured HOM signal (ext., bounded shaded area). α-pinene low-load (10 ppb, L) experiments are symbolized with colored dotted lines and the β-pinene (“β”) experiments by dashed lines. The gray dotted line depicts the estimated background level of the CI-APi-TOF.
3.3 Effect of the temperature on measured HOMs
We performed a total of 12 α-pinene ozonolysis experiments, with seven at high loading (i.e., [α-pinene] = 50 ppb); out of these, two were conducted at 20 ∘C, two at 0 ∘C, and three at −15 ∘C. Three experiments were performed with [α-pinene] = 10 ppb – one for each aforementioned temperature. Experiments with 50 ppb of β-pinene were also performed at the same three temperatures (see Table A2). An overview of HOM measurements for the different experiments is shown in Fig. 4, with distinction between HOM monomers (Fig. 4a) and dimers (Fig. 4b) as defined earlier.
For a similar experiment type (i.e., same initial VOC concentrations), it can be seen that the resulting HOM concentrations were considerably impacted by the temperature at which the oxidation reaction occurred. The signal intensity for HOM monomers from α-pinene measured 30 min after the VOC injection was roughly 2 orders of magnitudes higher at 20 ∘C compared to 0 ∘C and about 3 orders of magnitude higher compared to the −15 ∘C experiment. Very similar behavior is observed with respect to temperature for the dimer species as well, but with the differences that (1) fewer dimers are found in comparison to the HOM monomers and (2) HOM dimer concentrations are found to decrease at a faster rate during the experiment. The faster decrease in dimers compared to monomers results either from a lower production or a higher loss of dimers towards the end of the experiments. We expect that the reduced [α-pinene] and [O3], leading to slower oxidation rates and consequently lower [RO2], will have a greater impact on the dimers than the monomers, as the formation rate of dimers is proportional to [RO2]2, while monomers can still be formed efficiently via other RO2 termination pathways, as discussed earlier.
When comparing the high (50 ppb) and low (10 ppb) loading α-pinene experiments, HOM signals were within the same range of concentration and even higher at 0 ∘C; the HOM were even more abundant in the low initial VOC concentration. Although this result may seem surprising at first, it only verifies our assumptions in Eq. (1) that the HOM concentration is a relatively simple function of formation and loss rates. Despite the fact that the low-concentration experiments had a [VOC] that was 5 times lower (and consequently an HOM formation rate that was 5 times lower), the condensation sink, being the primary loss for HOMs, was ∼8 times lower due to reduced aerosol formation. In other words, the loss rates decreased more than the formation rate when the precursor concentration was lowered, resulting in an increase in [HOM].
Finally, the use of β-pinene as the HOM precursor produced significantly fewer HOMs, with concentrations being more than a factor of 10 lower compared to experiments performed with α-pinene at the same conditions. This agrees with earlier studies (Jokinen et al., 2014; Ehn et al., 2014) which showed clearly lower HOM yields for β-pinene compared to α-pinene ozonolysis. The difference is primarily attributed to the exocyclic double bond in β-pinene. Note that the β-pinene HOM concentrations at the lowest temperature, −15 ∘C, were below the instrumental limit of detection.
3.4 Yield estimation and temperature influence for molecule-specific HOMs
We determined yield estimates, individually for each HOM of interest, from the results of a robust linear fit as described in the Methods section and Eq. (2), taking into account the difference in CS between the different temperatures. In fact, we considered the higher CS for lower temperature experiments. Examples of calculated CSs, from the measured particle size distribution data, are shown for few experiments in the Appendix (Fig. A1). The yield estimation was performed with a fit with data points averaged by 2 min from 40 to 120 min after the VOC injection. These results are shown in Fig. 5, with fit examples shown for C10H14O9 and C19H28O12 in the insets. As expected, based on Fig. 4, the retrieved yield (γHOM) values decrease considerably with colder reaction conditions, with a total HOM yield (i.e., sum of the individual yields for each temperature) found to be 5.2 % at 20 ∘C, 0.10 % at 0 ∘C, and % at −15 ∘C.
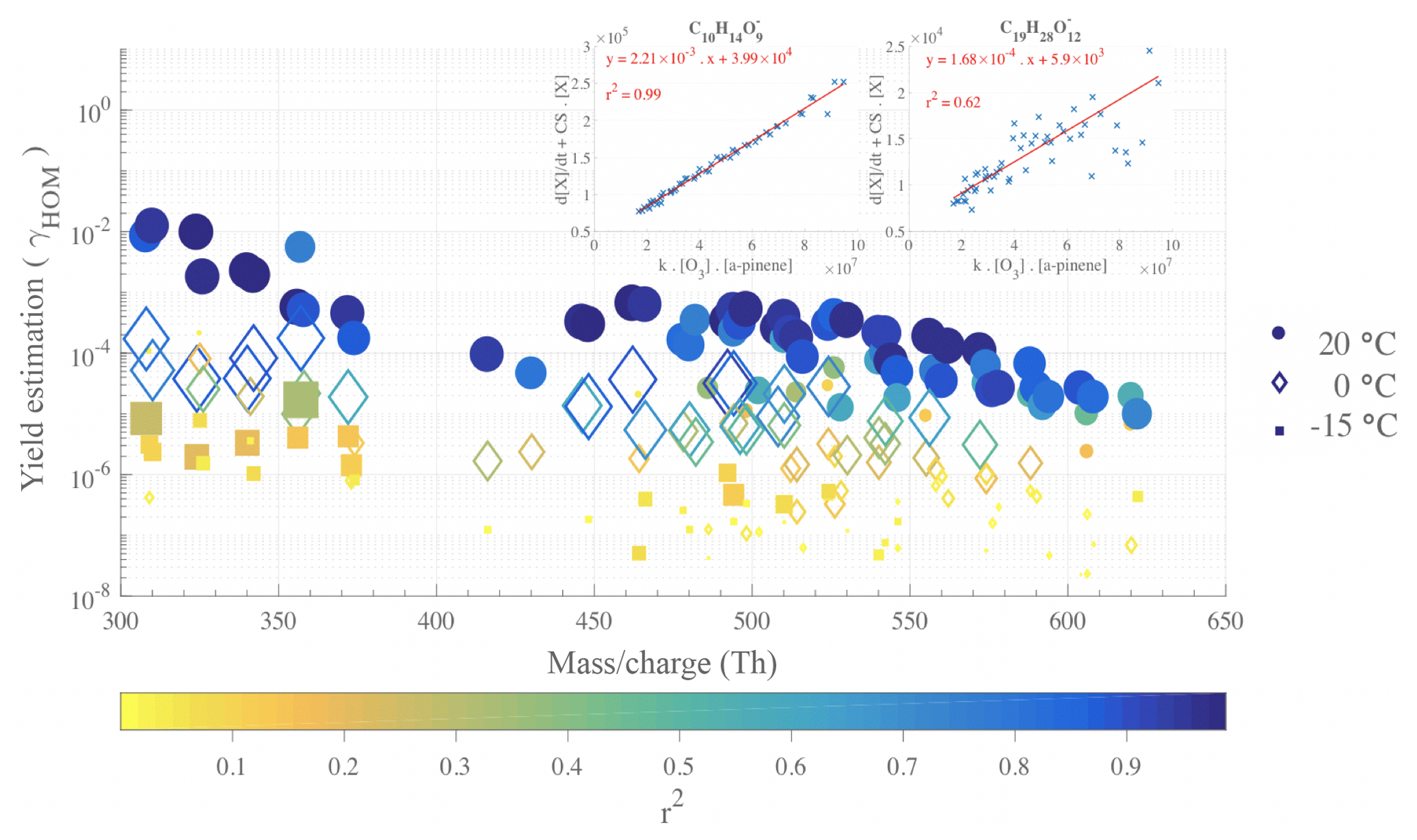
Figure 5Yield estimations for individual α-pinene HOMs from linear fits at 20, 0 and −15 ∘C, from 40 to 120 min after α-pinene injection. Filled circles symbolize data from a 20 ∘C experiment (12 January 2017), diamond symbols illustrate 0 ∘C data (16 January 2017), and the filled squares represent −15 ∘C data (13 January 2017). The markers are colored and sized by the r2 values, coefficient of determination, evaluating the goodness of the linear fit used to derive the yields. The top-right insets show two examples (for C10H14O9 and C19H28O12 at 20 ∘C) of the yield determination by robust linear fits to the variables described in the Methods section.
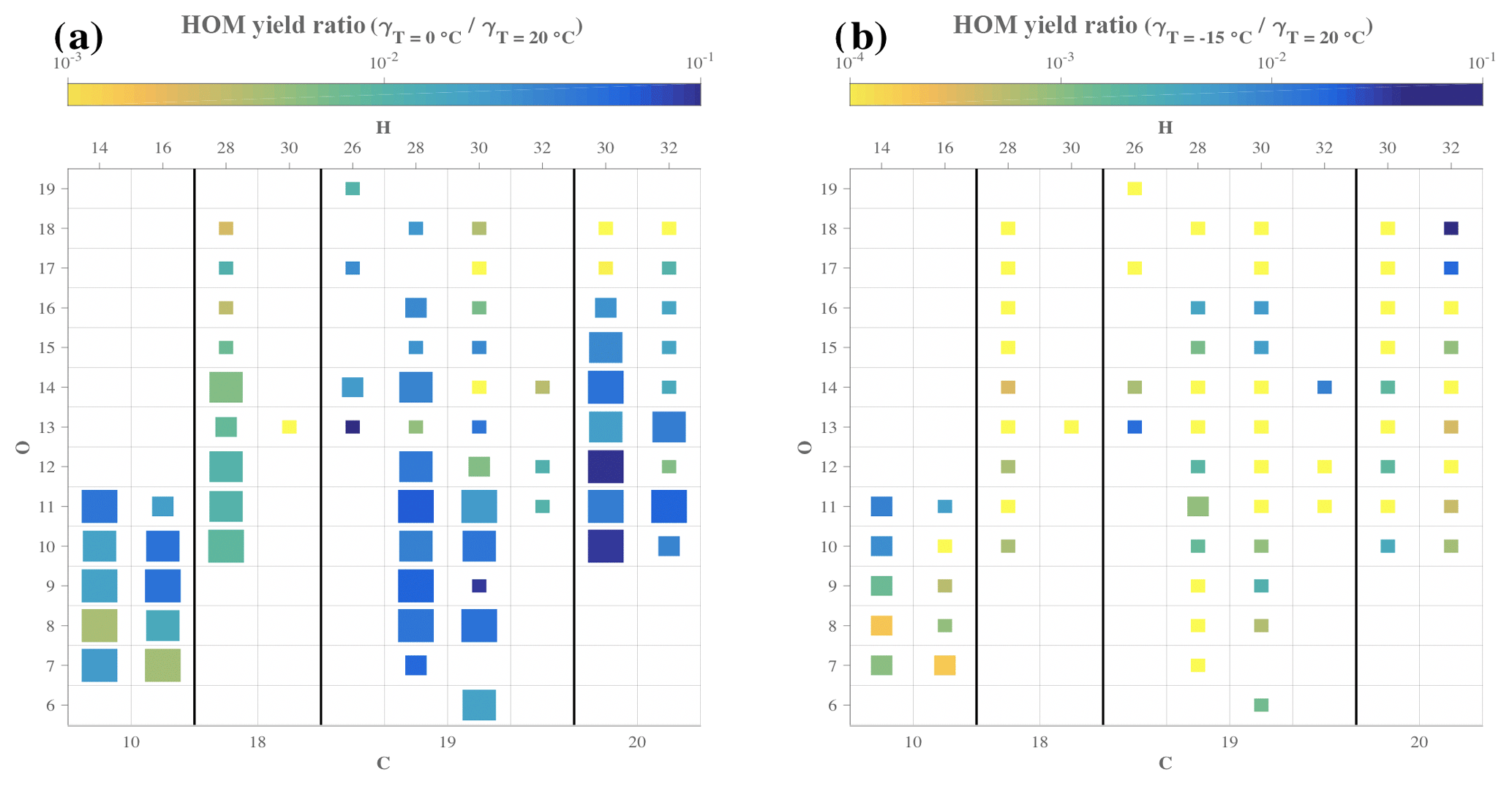
Figure 6Comparison of yields for specific HOM compositions at different temperatures. Each square symbolizes a specific HOM measured by the CI-APi-TOF. The elemental composition can be read by taking the number of C atoms from the bottom axis, the number of H atoms from the top axis, and the number of O atoms from the left axis. The size of the square depicts the goodness of fit (r2) used to derive the yields, and color shows the ratio of the yield at 0 ∘C (a) or −15 ∘C (b) compared to the yield estimate for 20 ∘C.

Figure 7Scatter plot of the HOM normalized signal intensity at 0 and at 20 ∘C. The data points are colored by the mass-to-charge ratio (a) or by oxygen-to-carbon ratio (b) with distinction between monomers (circle markers) and dimer compounds (diamond markers). Guiding lines were added as indicators: 1:1 line (in black), 1:50 line (in red), and 1:25 and 1:100 lines (in dotted gray).
We again emphasize the large uncertainties in these molar yield estimations, but the HOM yield values for T=20 ∘C agree with earlier reported values (e.g., Ehn et al., 2014; Jokinen et al., 2014; Sarnela et al., 2018). As the largest contribution to the HOM yield comes from the least oxidized monomers (e.g., high signal intensity at 308 and 310 Th for C10H14O7 and C10H16O7, respectively), the molar yield may be slightly overestimated, especially at 20 ∘C, due to the loss rates possibly being lower than assumed if these HOMs are not condensing irreversibly onto the aerosol. γHOM values are on average higher for HOM monomers than for dimers, with the overall shape of the distribution closely resembling the mass spectrum in Fig. 3. We performed the same calculation for the experiment where [α-pinene] = 10 ppb and found total HOM yields in the same range as the numbers found at 50 ppb, considering our estimated uncertainty: 8.8 % at 20 ∘C, 0.25 % at 0 ∘C, and % at −15 ∘C. The slightly higher values may indicate that at the higher loadings, bimolecular RO2 termination reactions are already occurring so quickly that autoxidation is hampered. The total HOM yield when going from 20 to 0 ∘C decreased by a factor 50 at the higher loading, while the corresponding value at lower loading was 35.
While Fig. 5 showed the estimated yields for every HOM at every temperature probed, specific chemical compositions cannot be read from the plot. In order to assess the impact of temperature on the yield of HOMs based on each elemental composition, Fig. 6 depicts, for each compound, the ratio of the yield at 0 ∘C (Fig. 6a) or −15 ∘C (Fig. 6b) compared to the yield at 20 ∘C for high-load experiment of α-pinene ozonolysis. In Fig. 6a, many larger squares are observable, indicating a good reliability of our comparison analysis, but in Fig. 6b, it is clear that the HOM concentrations at the lowest temperature were too low to provide much reliable compound-specific information. From Fig. 6a we see no clear trend in the yield change for any column (i.e., change in oxygen content HOMs with a given amount of C and H). The HOM yield ratios between the two temperatures are primarily within 10−2–10−1, meaning that the molecule-specific yields dropped to between 1 %–10 % when temperature decreased from 20 to 0 ∘C. If autoxidation of RO2 decreased this considerably, one could have expected the more oxygenated HOM to decrease more than the less oxygenated ones. However, this did not seem to be the case, as, for example, some of the most abundant HOMs, C10H14O7, C10H14O9, and C10H14O11, seemingly decreased by the same amounts.
In Fig. 7, we show the HOM signal intensities, molecule by molecule, based on m∕z (Fig. 7a) and on the O:C ratio (Fig. 7b) from the 20 ∘C experiment compared to the one at 0 ∘C. While there is scatter observable between individual HOMs, the vast majority of compounds fall close to the 1:50 line, when compared to the distance between the red and the black line. Additionally, the points with the largest scatter (e.g., >50 % from the 1:50 line) show no trends as a function of oxygen content, which also agrees with our observations from Fig. 6. One possible interpretation of this is that the rate-limiting step in the autoxidation chain takes place in RO2 radicals with six or fewer O atoms, which are not detected with our CI-APi-TOF, while the later H-shift reactions are fast enough that other reactions still do not become competitive. These “non-HOM” RO2 radicals may then also be key molecules for determining the final branching leading to the different observed HOMs with seven or more O atoms. This may shed light on one of the main open challenges (Ehn et al., 2017) in understanding HOM formation, namely how RO2 radicals with, for example, 6, 8, and 10 O atoms can form within a second, yet the relative distribution of these three does not change if the reaction time is allowed to increase (Berndt et al., 2015). Since the O10-RO2 (or its closed-shell products) are not seen accumulating over time, our results here provide support for a pathway where the O6-RO2 and O8-RO2 are to some extent “terminal” products incapable of further fast H-shift reactions, while the O10-RO2 has been formed via another branch of the reaction where the autoxidation is able to proceed further. In this branch, the O6-RO2 and O8-RO2 are likely only short-lived intermediates. While in no way conclusive, this highlights the need for fast measurements of HOM formation as well as improved techniques for observing less oxidized RO2 radicals.
The only compound group where a slight signal decrease can be seen as a function of O atom content is the C20H30 dimers. Interestingly, these also show some of the smallest yield ratios of all compounds. At the same time, the level of C18-dimers appears to drop for most of all compound groups, potentially suggesting that the mechanism through which carbon atoms were lost on the way to the C18 dimers was sensitive to temperature, and at 0 ∘C the fragmentation was less prominent. It is conceivable that the different branching at 0 ∘C caused some of the C18-dimer precursors to form C20-dimers instead. However, this issue would need more detailed experiments in order to be verified.
The decrease in HOM yield due to slower RO2 H-shift rates at lower temperatures was found to be very dramatic under our conditions. However, the exact magnitude of this decrease in HOM yield is determined by the processes competing with the H shifts. Under our conditions, the RO2 lifetime is kept quite short, both due to bimolecular (RO2+RO2 or RO2+HO2) reactions and collisions with particles, and therefore any reduction in H-shift rates can strongly reduce the HOM yield. Inversely, under very low loadings, the RO2 lifetime may be long enough that the temperature decreases from 20 to 0 ∘C may cause much smaller changes in the HOM yields. If the lifetime of RO2 radicals is clearly longer than the time needed for multiple consecutive H shifts to take place, HOM yields would decrease only marginally with temperature. In the atmosphere, the RO2 lifetime will often be governed by NO, which means that an intricate dependence of HOM yields as a function of temperature, VOC type, VOC oxidation rate, and NOx can exist.
We presented laboratory studies of HOM formation from monoterpene ozonolysis at different temperatures (20, 0, and −15 ∘C). Our main insight is that temperature in the studied range considerably impacted the HOM formation, decreasing the observed HOM yield by around 50-fold upon a decrease by 20 ∘C. The exact temperature dependence of HOM formation is likely both VOC- and loading-dependent, due to the competition between autoxidation and termination reactions, and will likely be smaller at lower loadings. While autoxidation is expected to decrease with temperature, our result is still striking, as it takes place over a temperature range which is atmospherically relevant to areas where monoterpene emissions are abundant, e.g., the boreal forest. One important observation when decreasing the temperature was that we found no clear trends of more oxygenated HOMs decreasing more than the less oxygenated ones. This, in turn, suggested that the autoxidation for the species with ∼6 oxygen atoms to species with ∼10 oxygen atoms was not strongly impacted by the colder temperature in our experiment. This meant that the total HOM yield, as well as the final HOM distribution, was mainly determined by the first H-shift steps, i.e., in the region where the CI-APi-TOF is unable to measure. This highlights the need for more comprehensive observations of autoxidation, allowing direct observations of the critical steps determining the HOM yields and, subsequently, the production rate of low-volatile organic compounds able to form secondary organic aerosol.
The data used in this study are available from the first author upon request: please contact Lauriane L. J. Quéléver (lauriane.quelever@helsinki.fi).
Table A1ACCHA experiment overview.

* Estimation based on model simulations using the Master Chemical Mechanism v3.3.2 (Jenkin et al., 1997, 2015; Saunders et al., 2003).
Table A2Main monoterpene ozonolysis HOM products: peak list.
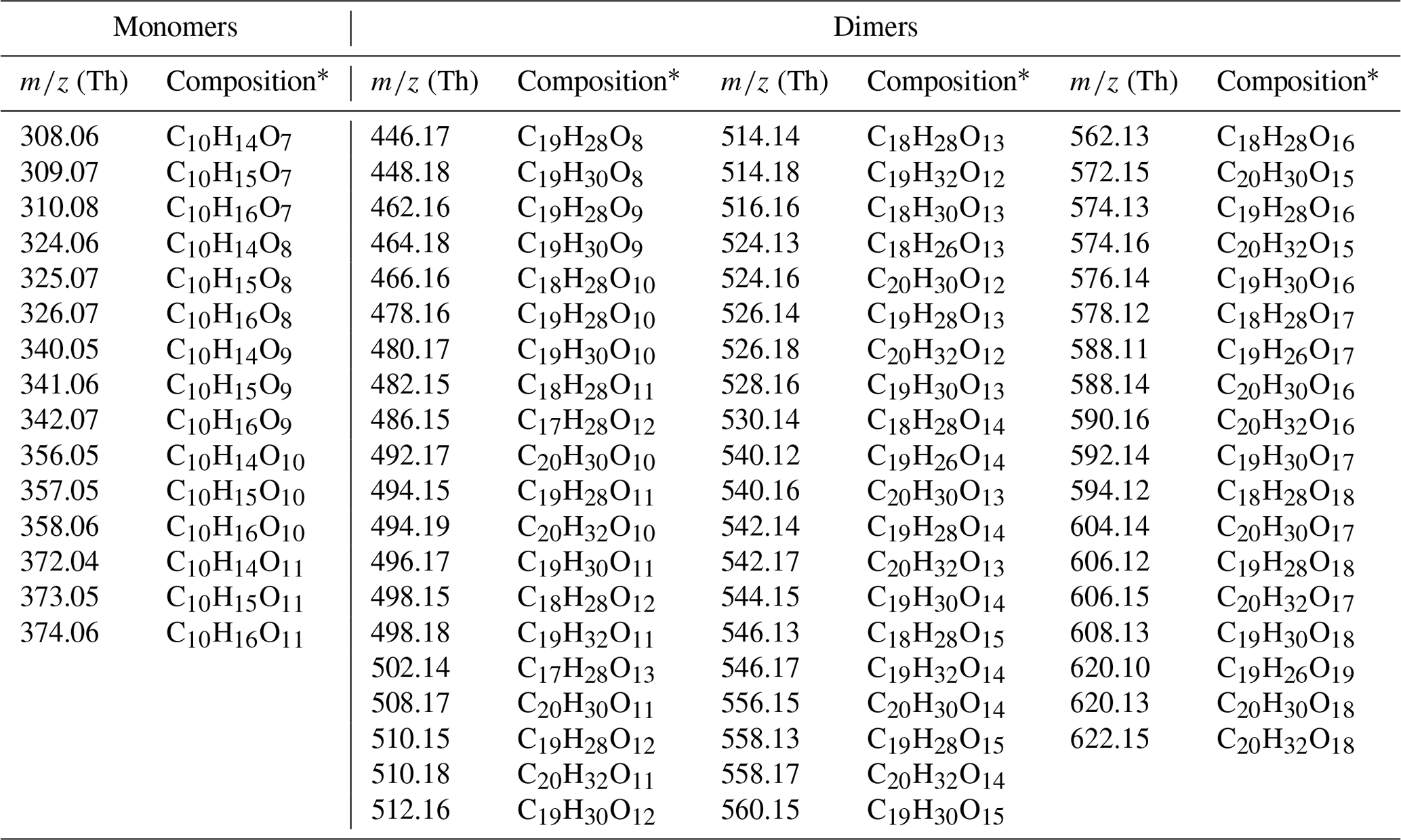
* Note that all compounds are detected as cluster with nitrate ion ().
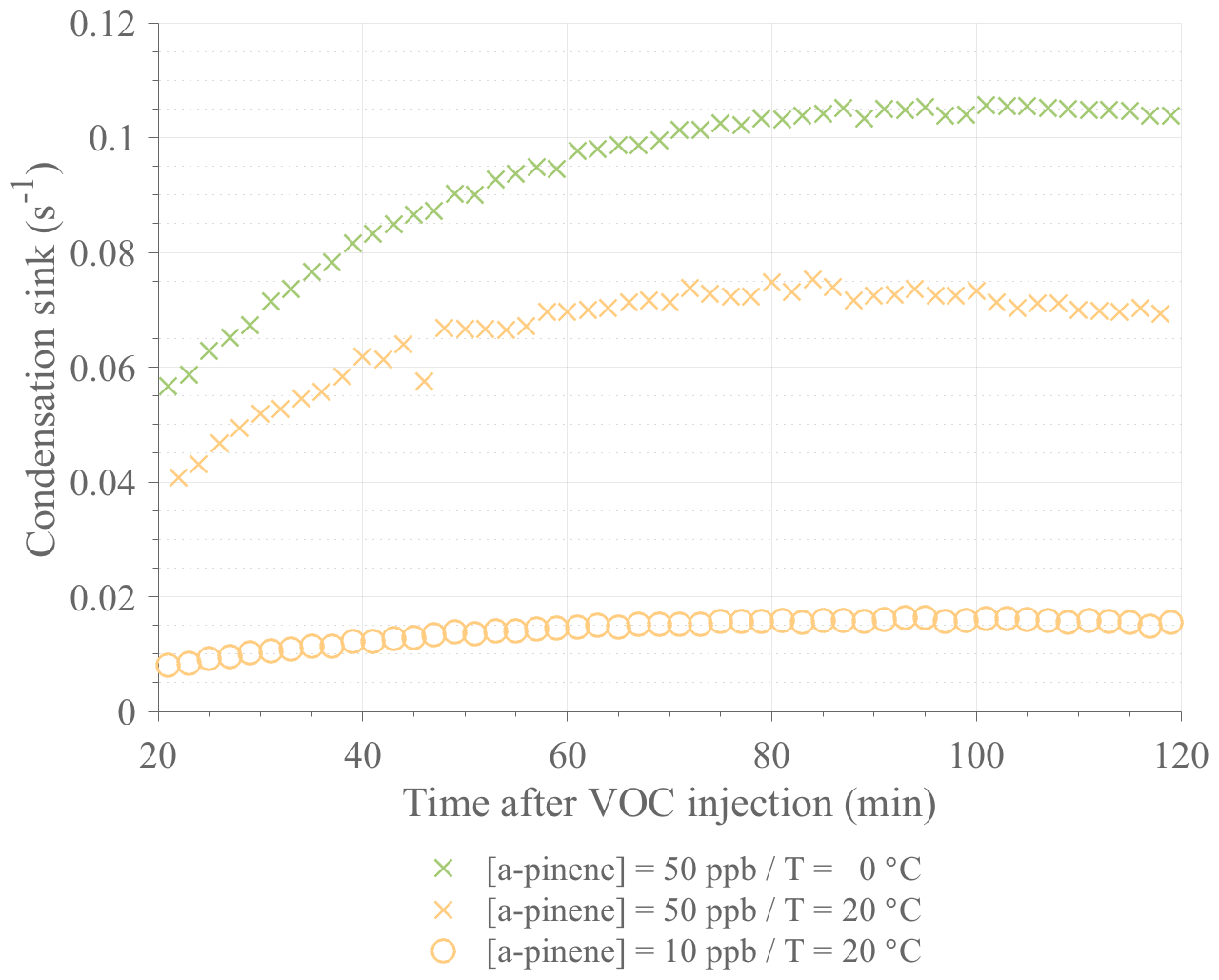
Figure A1Comparison of the calculated condensation sinks during selected ACCHA runs. Data are shown from 20 to 120 min after α-pinene injection for experiments performed at 50 ppb at 0 ∘C (16 January 2017; green crosses) and 20 ∘C (12 January 2017; orange crosses) and at 10 ppb at 20 ∘C (12 December 2016; orange circles).
MB, ME, and MG and HBP supervised the ACCHA campaign. LLJQ, ME, KK, and MB designed the experiments. KK and LNJ initialized the chamber for experiments. LLJQ performed the measurement and analyzed the gas-phase HOMs. KK and LNJ measured and analyzed the aerosol phase. KK, BR, and RT measured and analyzed the VOCs and their semi-volatile oxidation production, also supervised by RB. ME, KRD, OP, and PR guided and helped with the analysis of the HOM yields performed by LLJQ. LLJQ prepared the manuscript with the contributions from all co-authors.
The authors declare that they have no conflict of interest.
This work was funded by the European Research Council (grant no. 638703-COALA), the Academy of Finland Centre of Excellence program (grant no. 307331), Aarhus University, and the Aarhus University Research Foundation. We also thank Henrik Skov (Aarhus University) for the use of the PTR-TOF-MS. We thank Anders Feilberg (Aarhus University) for assistance in relation to the PTR-TOF-MS. We express our gratitude for the free use of the following mass spectrometry analysis tools: TofTools freeware provided by Heikki Junninen (University of Tartu). Otso Peräkylä thanks the Vilho, Yrjö & Kalle Väisälä Foundation. We finally thank Matti Rissanen (Tampere University and University of Helsinki) and Theo Kurtén (University of Helsinki) for their spontaneous input on this work.
Open access funding provided by Helsinki University Library.
This paper was edited by Nga Lee Ng and reviewed by three anonymous referees.
Atkinson, R.: Atmospheric chemistry of VOCs and NOx, Atmos. Environ., 34, 2063–2101, 2000.
Atkinson, R., Winer, A., and Pitts Jr., J.: Rate constants for the gas phase reactions of O3 with the natural hydrocarbons isoprene and α-and β-pinene, Atmo. Environ., 16, 1017–1020, 1982.
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 2019.
Berndt, T., Richters, S., Kaethner, R., Voigtländer, J., Stratmann, F., Sipilä, M., Kulmala, M., and Herrmann, H.: Gas-phase ozonolysis of cycloalkenes: formation of highly oxidized RO2 radicals and their reactions with NO, NO2, SO2, and other RO2 radicals, J. Phys. Chem. A, 119, 10336–10348, 2015.
Berndt, T., Richters, S., Jokinen, T., Hyttinen, N., Kurtén, T., Otkjær, R. V., Kjaergaard, H. G., Stratmann, F., Herrmann, H., Sipilä, M., Kulmala, M., and Ehn, M: Hydroxyl radical-induced formation of highly oxidized organic compounds, Nat. Commun., 7, 13677, https://doi.org/10.1038/ncomms13677, 2016.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Self-and Cross-Reactions of RO2 Radicals in the Atmosphere, Angew. Chem. Int. Edit., 57, 3820–3824, https://doi.org/10.1002/anie.201710989, 2018.
Calvert, J. G., Atkinson, R., Becker, K. H., Kamens, R. M., Seinfeld, J. H., Wallington, T. H., and Yarwood, G.: The mechanisms of atmospheric oxidation of the aromatic hydrocarbons, Oxford University Press, New York, 2002.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of organic compounds in the atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013.
Dal Maso, M., Kulmala, M., Riipinen, I., Wagner, R., Hussein, T., Aalto, P. P., and Lehtinen, K. E.: Formation and growth of fresh atmospheric aerosols: eight years of aerosol size distribution data from SMEAR II, Hyytiala, Finland, Boreal Environ. Res., 10, 323–336, 2005.
Donahue, N. M., Kroll, J. H., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set – Part 2: Diagnostics of organic-aerosol evolution, Atmos. Chem. Phys., 12, 615–634, https://doi.org/10.5194/acp-12-615-2012, 2012.
Donahue, N. M., Ortega, I. K., Chuang, W., Riipinen, I., Riccobono, F., Schobesberger, S., Dommen, J., Baltensperger, U., Kulmala, M., and Worsnop, D. R.: How do organic vapors contribute to new-particle formation?, Faraday Discuss., 165, 91–104, https://doi.org/10.1039/C3FD00046J, 2013.
Dusek, U., Frank, G., Hildebrandt, L., Curtius, J., Schneider, J., Walter, S., Chand, D., Drewnick, F., Hings, S., Jung, D., Borrmann, S., and Andreae, M. O.: Size matters more than chemistry for cloud-nucleating ability of aerosol particles, Science, 312, 1375–1378, https://doi.org/10.1126/science.1125261, 2006.
Ehn, M., Junninen, H., Petäjä, T., Kurtén, T., Kerminen, V.-M., Schobesberger, S., Manninen, H. E., Ortega, I. K., Vehkamäki, H., Kulmala, M., and Worsnop, D. R.: Composition and temporal behavior of ambient ions in the boreal forest, Atmos. Chem. Phys., 10, 8513–8530, https://doi.org/10.5194/acp-10-8513-2010, 2010.
Ehn, M., Junninen, H., Schobesberger, S., Manninen, H. E., Franchin, A., Sipilä, M., Petäjä, T., Kerminen, V.-M., Tammet, H., Mirme, A., Hõrrak, U., Kulmala, M., and Worsnop, D. R.: An instrumental comparison of mobility and mass measurements of atmospheric small ions, Aerosol Sci. Tech., 45, 522–532, https://doi.org/10.1080/02786826.2010.547890, 2011.
Ehn, M., Kleist, E., Junninen, H., Petäjä, T., Lönn, G., Schobesberger, S., Dal Maso, M., Trimborn, A., Kulmala, M., Worsnop, D. R., Wahner, A., Wildt, J., and Mentel, Th. F.: Gas phase formation of extremely oxidized pinene reaction products in chamber and ambient air, Atmos. Chem. Phys., 12, 5113–5127, https://doi.org/10.5194/acp-12-5113-2012, 2012.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I. H., Rissanen, M. P., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Dal Maso, M., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V. M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Ehn, M., Berndt, T., Wildt, J., and Mentel, T.: Highly Oxygenated Molecules from Atmospheric Autoxidation of Hydrocarbons: A Prominent Challenge for Chemical Kinetics Studies, Int. J. Chem. Kinet., 49, 821–831, 2017.
Frege, C., Ortega, I. K., Rissanen, M. P., Praplan, A. P., Steiner, G., Heinritzi, M., Ahonen, L., Amorim, A., Bernhammer, A.-K., Bianchi, F., Brilke, S., Breitenlechner, M., Dada, L., Dias, A., Duplissy, J., Ehrhart, S., El-Haddad, I., Fischer, L., Fuchs, C., Garmash, O., Gonin, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kirkby, J., Kürten, A., Lehtipalo, K., Leiminger, M., Mauldin, R. L., Molteni, U., Nichman, L., Petäjä, T., Sarnela, N., Schobesberger, S., Simon, M., Sipilä, M., Stolzenburg, D., Tomé, A., Vogel, A. L., Wagner, A. C., Wagner, R., Xiao, M., Yan, C., Ye, P., Curtius, J., Donahue, N. M., Flagan, R. C., Kulmala, M., Worsnop, D. R., Winkler, P. M., Dommen, J., and Baltensperger, U.: Influence of temperature on the molecular composition of ions and charged clusters during pure biogenic nucleation, Atmos. Chem. Phys., 18, 65–79, https://doi.org/10.5194/acp-18-65-2018, 2018.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Heinritzi, M., Simon, M., Steiner, G., Wagner, A. C., Kürten, A., Hansel, A., and Curtius, J.: Characterization of the mass-dependent transmission efficiency of a CIMS, Atmos. Meas. Tech., 9, 1449–1460, https://doi.org/10.5194/amt-9-1449-2016, 2016.
Hyttinen, N., Kupiainen-Määttä, O., Rissanen, M. P., Muuronen, M., Ehn, M., and Kurtén, T.: Modeling the charging of highly oxidized cyclohexene ozonolysis products using nitrate-based chemical ionization, J. Phys. Chem. A, 119, 6339–6345, 2015.
IPCC: Climate change 2013: the physical science basis. Contribution of the Working Group 1 to the Fifth Assessment Report (AR5) of the Intergovernmental Panel on Climate Change, edited by: Stocker, T. F., Qin, D., Plattner, G., Tignor, M., Allen, S., Boschung, J., Nauels, A., Xia, Y., Bex, V., and Midgley, P. M., Cambridge University Press, Cambridge, UK, New York, USA, 2013.
Jayne, J. T., Leard, D. C., Zhang, X., Davidovits, P., Smith, K. A., Kolb, C. E., and Worsnop, D. R.: Development of an aerosol mass spectrometer for size and composition analysis of submicron particles, Aerosol Sci. Tech., 33, 49–70, 2000.
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 31, 81–104, 1997.
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of organic aerosols in the atmosphere, Science, 326, https://doi.org/10.1126/science.1180353, 2009.
Jokinen, T., Sipilä, M., Junninen, H., Ehn, M., Lönn, G., Hakala, J., Petäjä, T., Mauldin III, R. L., Kulmala, M., and Worsnop, D. R.: Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF, Atmos. Chem. Phys., 12, 4117–4125, https://doi.org/10.5194/acp-12-4117-2012, 2012.
Jokinen, T., Sipilä, M., Richters, S., Kerminen, V. M., Paasonen, P., Stratmann, F., Worsnop, D., Kulmala, M., Ehn, M., Herrmann, H., and Berndt, T.: Rapid autoxidation forms highly oxidized RO2 radicals in the atmosphere, Angew. Chem. Int. Edit., 53, 14596–14600, 2014.
Jokinen, T., Berndt, T., Makkonen, R., Kerminen, V.-M., Junninen, H., Paasonen, P., Stratmann, F., Herrmann, H., Guenther, A. B., Worsnop, D. R., Kulmala, M., Ehn, M., and Sipilä, M.: Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications, P. Natl. Acad. Sci. USA, 112, 7123–7128, https://doi.org/10.1073/pnas.1423977112, 2015.
Jordan, A., Haidacher, S., Hanel, G., Hartungen, E., Märk, L., Seehauser, H., Schottkowsky, R., Sulzer, P., and Märk, T. D.: A high resolution and high sensitivity proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF-MS), Int. J. Mass Spectrom., 286, 122–128, 2009.
Julin, J., Winkler, P. M., Donahue, N. M., Wagner P. E., and Riipinen, I.: Near-unity mass accommodation coefficient of organic molecules of varying structure, Environ. Sci. Technol., 48, 12083–12089, 2014.
Junninen, H., Ehn, M., Petäjä, T., Luosujärvi, L., Kotiaho, T., Kostiainen, R., Rohner, U., Gonin, M., Fuhrer, K., Kulmala, M., and Worsnop, D. R.: A high-resolution mass spectrometer to measure atmospheric ion composition, Atmos. Meas. Tech., 3, 1039–1053, https://doi.org/10.5194/amt-3-1039-2010, 2010.
Kristensen, K., Jensen, L., Glasius, M., and Bilde, M.: The effect of sub-zero temperature on the formation and composition of secondary organic aerosol from ozonolysis of alpha-pinene, Environ. Sci.-Proc. Imp., 19, 1220–1234, 2017.
Kulmala, M., Kontkanen, J., Junninen, H., Lehtipalo, K., Manninen, H. E., Nieminen, T., Petäjä, T., Sipilä, M., Schobesberger, S., Rantala, P., Franchin, A., Jokinen, T., Järvinen, E., Äijälä, M., Kangasluoma J., Hakala, J., Aalto, P. P., Paasonen, P., Mikkilä, J., Vanhanen, J., Aalto, J., Hakola, H., Makkonen, H., Ruuskanen T., Mauldin, R. L., Duplissy, J., Vehkamäki, H., Bäck, J., Kortelainen, A., Riipinen, I., Kurtén, T., Johnston, M. V., Smith, J., N., Ehn, M., Mentel, T. F., Lehtinen, K. E. J., Laaksonen, A., Kerminen, V.-M, and Worsnop, D. R.: Direct observations of atmospheric aerosol nucleation, Science, 339, 943–946, 2013.
Kürten, A., Rondo, L., Ehrhart, S., and Curtius, J.: Calibration of a chemical ionization mass spectrometer for the measurement of gaseous sulfuric acid, J. Phys. Chem. A, 116, 6375–6386, 2012.
Mentel, T. F., Springer, M., Ehn, M., Kleist, E., Pullinen, I., Kurtén, T., Rissanen, M., Wahner, A., and Wildt, J.: Formation of highly oxidized multifunctional compounds: autoxidation of peroxy radicals formed in the ozonolysis of alkenes – deduced from structure–product relationships, Atmos. Chem. Phys., 15, 6745–6765, https://doi.org/10.5194/acp-15-6745-2015, 2015.
Otkjær, R. V., Jakobsen, H. H., Tram, C. M., and Kjaergaard, H. G.: Calculated Hydrogen Shift Rate Constants in Substituted Alkyl Peroxy Radicals, J. Phys. Chem. A, 122, 8665–8673, 2018.
Praske, E., Otkjær, R. V., Crounse, J. D., Hethcox, J. C., Stoltz, B. M., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric autoxidation is increasingly important in urban and suburban North America, P. Natl. Acad. Sci. USA, 115, 64–69, 2018.
Rissanen, M. P., Kurtén, T., Sipilä, M., Thornton, J. A., Kangasluoma, J., Sarnela, N., Junninen, H., Jørgensen, S., Schallhart, S., Kajos, M. K., Taipale, R., Springer, M., Mentel, T. M., Ruuskanen, T., Petäjä, T., Worsnop, D. R., Kjaergaard, H. G., and Ehn, M.: The formation of highly oxidized multifunctional products in the ozonolysis of cyclohexene, J. Am. Chem. Soc., 136, 15596–15606, 2014.
Rissanen, M. P., Kurtén, T., Sipilä, M., Thornton, J. A., Kausiala, O., Garmash, O., Kjaergaard, H. G., Petäjä, T., Worsnop, D. R., Ehn, M., and Kulmala, M.: Effects of chemical complexity on the autoxidation mechanisms of endocyclic alkene ozonolysis products: From methylcyclohexenes toward understanding α-pinene, J. Phys. Chem. A, 119, 4633–4650, 2015.
Rosati, B., Teiwes, R., Kristensen, K., Bossi, R., Skov, H., Glasius, M., Pedersen, H., and Bilde, M.: Factor analysis of chemical ionization experiments: Numerical simulation and an experimental case strudy of the ozonolysis of a-pinene using a PTR-TOF-MS, Atmos. Environ., 199, 15–13, https://doi.org/10.1016/j.atmosenv.2018.11.012, 2019.
Sarnela, N., Jokinen, T., Duplissy, J., Yan, C., Nieminen, T., Ehn, M., Schobesberger, S., Heinritzi, M., Ehrhart, S., Lehtipalo, K., Tröstl, J., Simon, M., Kürten, A., Leiminger, M., Lawler, M. J., Rissanen, M. P., Bianchi, F., Praplan, A. P., Hakala, J., Amorim, A., Gonin, M., Hansel, A., Kirkby, J., Dommen, J., Curtius, J., Smith, J. N., Petäjä, T., Worsnop, D. R., Kulmala, M., Donahue, N. M., and Sipilä, M.: Measurement–model comparison of stabilized Criegee intermediate and highly oxygenated molecule production in the CLOUD chamber, Atmos. Chem. Phys., 18, 2363–2380, https://doi.org/10.5194/acp-18-2363-2018, 2018.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics: From air pollution to climate change, 2nd Edn., John Willey & Sons, New York, 2006.
Stolzenburg, D., Fischer, L., Vogel, A. L., Heinritzi, M., Schervish, M., Simon, M., Wagner, A. C., Dada, L., Ahonen, L. R., Amorim, A., Baccarini, A., Bauer, P. S., Baumgartner, B., Bergen, A., Bianchi, F., Breitenlechner, M., Brilke, S., Buenrostro Mazon, S., Chen, D., Dias, A., Draper, D. C., Duplissy, J., El Haddad, I., , Finkenzeller, H., Frege, C., Fuchs, C., Garmash, O., Gordon, H., He, X., Helm, J., Hofbauer, V., Hoyle, C. H., Kim, C., Kirkby, J., Kontkanen, J., Kürten, A., Lampilahti, J., Lawler, M., Lehtipalo, K., Leiminger, M., Mai, H., Mathot, S., Mentler, B., Molteni, U., Nie, W., Nieminen, T., Nowak, J. B., Ojdanic, A., Onnela, A., Passananti, M., Petäjä, T., Quéléver, L. L. J., Rissanen, M. P., Sarnela, N., Schallhart, S., Tauber, S., Tomé, A., Wagner, R., Wang, M., Weitz, L., Wimmer, D., Xiao, M., Yan, C., Ye, P., Zha, Q., Baltensperger, U., Curtius, J., Dommen, J., Flagan, R. C., Kulmala, M., Smith, J. N., Worsnop, D. R., Hansel, A., Donahue, N. M., and Winkler, P. M.: Rapid growth of organic aerosol nanoparticles over a wide tropospheric temperature range, P. Natl. Acad. Sci. USA, 115, 9122–9127, 2018.
Tang, M. J., Shiraiwa, M., Pöschl, U., Cox, R. A., and Kalberer, M.: Compilation and evaluation of gas phase diffusion coefficients of reactive trace gases in the atmosphere: Volume 2. Diffusivities of organic compounds, pressure-normalised mean free paths, and average Knudsen numbers for gas uptake calculations, Atmos. Chem. Phys., 15, 5585–5598, https://doi.org/10.5194/acp-15-5585-2015, 2015.
Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., Wagner, R., Simon, M., Lehtipalo, K., Williamson, C., Craven, J. S., Duplissy, J., Adamov, A., Almeida, J., Bernhammer, A.-K., Breitenlechner, M., Brilke, S., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Claudia, F., Guida, R., Gysel, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Keskinen, H., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lawler, M., Leiminger, M., Mathot, S., Möhler, O., Nieminen, T., Onnela, A., Petäjä, T, Piel, F. M., Miettinen, P., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Sipilä, M., Smith, J. N., Steiner, G., Tomè, A., Virtanen, A., Wagner, A. C., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Carslaw, K. S., Curtius, J., Dommen, J., Kirkby, J., Kulmala, M., Riipinen, I., Worsnop, D. R., Donahue, N. M., and Baltensperger, U.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 533, 527–531, https://doi.org/10.1038/nature18271, 2016.
Zhang, Q., Jimenez, J. L., Canagaratna, M., Allan, J., Coe, H., Ulbrich, I., Alfarra, M., Takami, A., Middlebrook, A., Sun, Y., Dzepina, K., Dunlea, E., Docherty, K., DeCarlo, P. F., Salcedo, D., Onasch, T., Jayne, J. T., Miyoshi, T., Shimono, A., Hatakeyama, S., Takegawa, N., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Williams, P., Bower, K., Bahreini, R., Cottrell, L., Griffin, R. J., Rautiainen, J., Sun, J. Y., Zhang, Y. M., and Worsnop, D. R.: Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes, Geophys. Res. Lett., 34, L13801, https://doi.org/10.1029/2007GL029979, 2007.
Zhao, J., Ortega, J., Chen, M., McMurry, P. H., and Smith, J. N.: Dependence of particle nucleation and growth on high-molecular-weight gas-phase products during ozonolysis of α-pinene, Atmos. Chem. Phys., 13, 7631–7644, https://doi.org/10.5194/acp-13-7631-2013, 2013.