the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 23 May 2019
| 23 May 2019
Mercury and trace metal wet deposition across five stations in Alaska: controlling factors, spatial patterns, and source regions
Christopher Pearson
Dean Howard
Christopher Moore
Daniel Obrist
A total of 1360 weeks of mercury (Hg) wet deposition data were collected by the state of Alaska Department of Environmental Conservation and the U.S. National Park Service across five stations spanning up to 8 years. Here, we analyze concentration patterns, source regions, and seasonal and annual Hg deposition loadings across these five sites in Alaska, along with auxiliary trace metals including Cr, Ni, As, and Pb.
We found that Hg concentrations in precipitation at the two northernmost stations, Nome (64.5∘ N) along the coast of the Bering Sea and the inland site of Gates of the Arctic (66.9∘ N), were statistically higher (average of 5.3 and 5.5 ng L−1, respectively) than those at the two lowest-latitude sites, Kodiak Island (57.7∘ N, 2.7 ng L−1) and Glacier Bay (58.5∘ N, 2.6 ng L−1). These differences were largely explained by different precipitation regimes, with higher precipitation at the lower-latitude stations leading to dilution effects. The highest annual Hg deposition loads were consistently observed at Kodiak Island (4.80±1.04 µg m−2), while the lowest annual deposition was at Gates of the Arctic (2.11±0.67 µg m−2). Across all stations and collection years, annual precipitation strongly controlled annual Hg deposition, explaining 73 % of the variability in observed annual Hg deposition. The data further showed that annual Hg deposition loads across all five Alaska sites were consistently among the lowest in the United States, ranking in the lowest 1 % to 5 % of over 99 monitoring stations.
Detailed back-trajectory analyses showed diffuse source regions for most Hg deposition sites suggesting largely global or regional Hg sources. One notable exception was Nome, where we found increased Hg contributions from the western Pacific Ocean downwind of East Asia. Analysis of other trace elements (As, Cr, Cu, Ni, Pb, Se, Zn) from Dutch Harbor, Nome, and Kodiak Island showed generally higher trace metal concentrations at the northern station Nome compared to Kodiak Island further to the south, with concentrations at Dutch Harbor falling in between. Across all sites, we find two distinct groups of correlating elements: Cr and Ni and As and Pb. We attribute these associations to possibly different source origins, whereby sources of Ni and Cr may be derived from crustal (e.g., dust) sources while As and Pb may include long-range transport of anthropogenic pollution. Hg was not strongly associated with either of these two groups.
- Article
(6902 KB) - Full-text XML
- BibTeX
- EndNote





The land surface area of the state of Alaska is approximately one-fifth of that of the contiguous United States, but little spatial information is available on pollutant deposition and impacts affecting local ecosystems, wildlife, and humans. Most contaminant studies have focused on the Arctic domain, including studies conducted by large international collaborative efforts such as the Arctic Monitoring and Assessment Program (AMAP), a working group of the Arctic Council (AMAP, 2009b, a, 2011). The interests in northern-latitude pollutant studies are driven by reports that show significant neurotoxicity, as well as immunological, cardiovascular, and reproductive effects, in Arctic populations and wildlife from exposure to contaminants (AMAP, 2011). Important pollutants found in northern arctic and boreal areas include mercury (Hg) – which is the focus of the current analysis – as well as persistent organic pollutants (POPs), polycyclic aromatic hydrocarbons (PAHs), and other trace metals such as lead (Pb) and cadmium (Cd) that are primarily supplied to the region by atmospheric transport and deposition (AMAP, 2005, 2009a, 2011). The major concerns of these pollutants are their toxicity and persistency in the environment. Delivery of contaminants to the high Arctic is expected to increase in the future due to changes in synoptic atmospheric transport patterns and an expected increase in contaminant source fluxes related to increased development, resource extraction, and transportation activities within northern regions (Jaeglé, 2010; Streets et al., 2011).
Mercury is a neurotoxic pollutant significantly affecting northern latitudes, with human exposure mainly derived from consumption of seafood and marine mammals that are part of traditional diets based on hunting and fishing (Stow et al., 2011). Risks associated with long-term exposure to Hg, particularly to the organic monomethylmercury (MeHg), include neurodevelopmental delays in children exposed in utero, impaired cardiovascular health in adults, and disruption of immunological and endocrine functions (Karagas et al., 2012; Tan et al., 2009). Hg biomagnifies in aquatic and terrestrial food webs and is present at elevated concentrations in northern wildlife such as seals, polar bears, beluga whales, Arctic foxes, birds, and fish (Lawson and Mason, 1998; Watras and Bloom, 1992; Baeyens et al., 2003; Loseto et al., 2008; Evans et al., 2005; Dietz et al., 2009; Walker et al., 2006; Outridge et al., 2008; Douglas et al., 2012; Macdonald and Bewers, 1996; Leitch et al., 2007; Braune et al., 2014; Bocharova et al., 2013; Laird et al., 2013; Ackerman et al., 2016; Eagles-Smith et al., 2016). Long-range transport via the atmosphere is considered the primary source of Hg deposition to the high latitudes (Dommergue et al., 2010; Steffen et al., 2008). In addition, springtime photochemical reactions, termed atmospheric mercury depletion events (AMDEs), lead to additional Hg deposition to snow and ice, particularly along the Arctic Ocean coast (Douglas and Sturm, 2004; Lindberg et al., 2002). Regional and local sources of atmospheric Hg exist in Alaska from both natural and anthropogenic emissions. The EPA Toxics Release Inventory (TRI) reported total air emissions of Hg and Hg compounds in Alaska of 24 kg (53 lbs) from both fugitive and point-source air sources in 2014 (Table 1; U.S. EPA, 2014). Other sources include natural emissions from wildfires (Mitchell et al., 2012; Wiedinmyer et al., 2006; Turetsky et al., 2006; Friedli et al., 2001; Brunke et al., 2001; Webster et al., 2016; Obrist et al., 2008), volcanic emissions (Mather and Pyle, 2004; Pyle and Mather, 2003; Nriagu and Becker, 2003; Ferrara et al., 2000), and degassing from Hg-enriched soils and possibly background soils (Agnan et al., 2015a; Gustin et al., 2008), although many of these sources are poorly constrained across Alaska. Jaeglé (2010) conducted a detailed study of atmospheric sources of Hg contamination over Alaska using GEOS-Chem model simulations and suggested that anthropogenic emissions contribute approximately 57 % of Hg deposition over Alaska, with other sources dominated by natural land (i.e., volcanos, wild fires) and ocean-based emissions. The estimated anthropogenic contributions seem quite high given that globally anthropogenic Hg emissions are estimated to account for approximately 30 % of total atmospheric sources (i.e., total anthropogenic and natural emissions plus re-emission) (UNEP, 2013).

Here we analyze Alaska Hg wet deposition data collected by the state of Alaska and the National Park Service between 2007 and 2015 across five Hg wet deposition stations in Alaska. Deposition sites are Gates of the Arctic, Nome, Glacier Bay National Park, Kodiak Island, and Dutch Harbor (Table 1; Fig. 4). The dataset contains 1360 weeks of total measurements across the five stations, with the longest record lasting almost 8 years, allowing for analysis of temporal, seasonal, and spatial patterns of Hg wet deposition across Alaska. All measurements were conducted according to trace metal sampling protocols following the National Atmospheric Deposition Program (NADP) Mercury Deposition Network (MDN) standards. We use statistical tests to compare deposition concentrations, loads, and seasonal and inter-annual patterns to assess variables controlling Hg deposition. In addition, we performed detailed back-trajectory analyses for full annual datasets at select stations to quantify source regions that contribute to annual Hg deposition loads. We further use deposition data of auxiliary trace metals, including Cr, Ni, As, and Pb, to identify associations with Hg and source patterns. Finally, we performed spatial scaling and mapping of annual wet deposition of Hg throughout all of Alaska based on observed concentration gradients and precipitation distributions.
2.1 Collection and analysis of Hg deposition data and 10 additional trace elements
Weekly Hg deposition data were collected from five wet deposition stations in Alaska operated by the state of Alaska Division of Environmental Conservation and the National Park Service. Deposition stations include Gates of the Arctic, Nome, Glacier Bay National Park, Kodiak Island, and Dutch Harbor (Table 1; Fig. 4). Gates of the Arctic is a protected wilderness area in northern Alaska consisting of multiple mountain ranges and sparse boreal forests. Nome is at sea level located on the western coast of Alaska off the Bering Sea. Nome's landscape is characterized as arctic tundra with ground brush vegetation and little tree cover. Glacier Bay National Park is a coastal site located in the northern section of the Alaska panhandle. Glacier Bay consists of wet tundra and dense coastal forest. Kodiak is a mountainous island located off the southern coast of Alaska. Low-elevation vegetation consist of shrubs and grasses, while alpine tundra exists at higher elevations. Finally, Dutch Harbor is located on Amaknak Island in the Aleutian Islands. Most of the islands consist of dense shrubs with very little conifer growth. Sample collections were performed on a weekly basis using trace metal wet deposition collectors (model MDN 00-125-4; N-Con Inc., Crawford, GA, USA), following MDN protocols for collection of Hg in precipitation (Mercury Deposition Network: Field Methods, 2017). In summary, the protocols include weekly collection using a specially modified NADP sampler. Analysis of samples for Hg were performed by the Mercury Analytical Laboratory (HAL, Eurofins Frontier Global Sciences, Inc., Seattle, Washington, USA) according to EPA Methods 1669 and 1631.
Data coverage varied by site and ranged from September 2007 to September 2015. The longest dataset is from Kodiak (2007 to 2015), followed by Gates of the Arctic (2008 to 2015), and Dutch Harbor (2009 to 2015). Both Glacier Bay (2010 to 2013) and Nome (2013 to 2015) had shorter datasets (less than 3 years). All sites had intermittent data gaps ranging from weeks to months. The most significant data gaps occurred at Kodiak during the summers of 2009 and 2010 and Dutch Harbor during 2010, 2013, and 2014.
Detailed quality assurance and control of data followed the protocols of the NADP MDN. Each sample was assigned a quality rating (A, B, or C) based on collector performance, sample quality, and analytical measurement excellence. A ratings were assigned to samples of the “highest quality” with no issues during collection or analysis, B ratings referred to data with minor problems, and C ratings referred to samples with significant defects. Samples with a rating of C were removed prior to our data analysis. During weeks with missing rain gage data, the measured volume of water collected in the sample bottle was used as the precipitation measurement. Trace samples (unmeasurable by the rain gage) were assigned a precipitation value of 3.23 mm (following NADP MDN protocol).
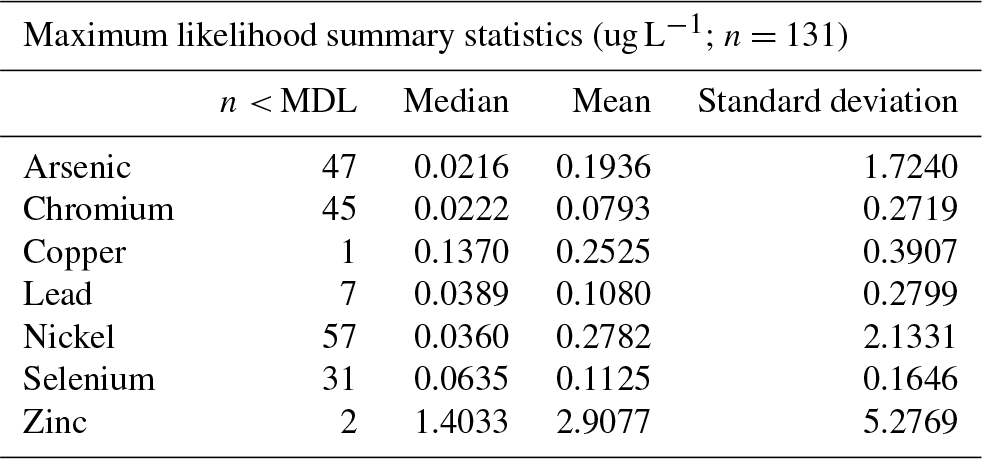
In addition to the Hg deposition data, we analyzed corresponding data of additional trace elements collected at three of the five Hg deposition stations: Dutch Harbor, Kodiak Island, and Nome. Available analysis of trace elements includes the following 10 additional elements: arsenic (As), beryllium (Be), cadmium (Cd), chromium (Cr), copper (Cu), lead (Pb), nickel (Ni), selenium (Se), and zinc (Zn). Trace metal analysis was performed with inductively coupled plasma mass spectrometry following EPA Method 200.8 (Brockhoff et al., 1999). Seven of these trace elements are listed on the EPA's list of hazardous air pollutants (U.S. EPA, 2016), including As, Be, Cd, Cr, Pb, Ni, and Se. In total, there were 132 trace element deposition samples. Dutch Harbor had a total of 24 samples collected from 24 September 2013 through 28 April 2015, Nome had a total of 42 samples between 30 October 2013 and 28 April 2015, and Kodiak Island had a total of 65 samples between 17 September 2013 and 28 April 2015. We assumed that large gaps in data coverage were mainly due to low sample volumes collected during sampling, which often did not allow measurement of these trace metals. A significant number of samples showed trace element concentrations below the analytical detection limit. The percentage of samples below detection limits were as follows, from highest to lowest percentage: Cd: (80 %), Be (56 %), Ni (44 %), As (36 %), and Cr (34 %). All other elements had observations below reported detection limits less than 6 %. Be and Cd were not included in this current analysis due to their large percentage of missing values (>50 %).
2.2 Statistical analyses, spatial interpolation, and mapping
All statistical analyses were performed using the statistical software program “R” (R-Core-Team, 2014). Specific R packages utilized throughout the analysis included ade4, ggplot, NADA, corrplot, and psych (Lee, 2013; Dray and Dufour, 2007; Revelle, 2014; Wickham, 2009). Outliers for both the Trace Metal Dataset and NADP Hg Deposition dataset were determined using the 1.5× interquartile range (IQR) rule. The trace metal dataset contained several elements where significant portions of the data fell below detection limits (BDLs). For these data, maximum likelihood estimation (MLE) summary statistics were calculated using the NADA package (Lee, 2013) in addition to 1/2 MDL substitution. In general, MLE and nonparametric techniques are preferred to be used for non-detected values since replacement of data (e.g., using 1/2 detection limits) can be problematic (Helsel, 2012). A lognormal distribution was assumed for MLE estimation. Analysis of variance (ANOVA) and analysis of covariance (ANCOVA) were performed on Log10-transformed data using a type-III ANOVA to account for unbalanced factor levels in the dataset. Trace metal ANOVAs were performed on the 1/2 MDL substituted datasets.
A PCA (principal component analysis) using all trace metals was performed on ranked data to account for BDL values and non-normal distributions. Season was defined as follows for both datasets: spring (March, April, May), summer (June, July, August), fall (September, October, November), and winter (December, January, February).
Mapping and spatial interpolation and extrapolation of Hg deposition loads were performed in ArcGIS®. Inverse distance weighting (n=5; p=0.5) was used to interpolate and extrapolate precipitation-weighted mean (PWM) concentrations across Alaska. Precipitation-weighted mean Hg concentrations were used from NADP MDN annual estimates and averaged for all available years. MDN annual estimates were only available for years with (i) Hg sampling covering ≥75 % of the sampling period, (ii) Hg measurements of >75 % of annual precipitation events, and (iii) >90 % coverage of total annual precipitation (either gage or sample bottle). The estimated PWM Hg concentrations were then combined with annual normal precipitation averaged for the period of 2007–2015 from the NCEP Climate Forecast System Version 2 (CFSv2) 6-hourly products (Saha, 2011) to estimate deposition totals across the state of Alaska. Precipitation data were accessed and compiled using Google Earth Engine (Google Earth Engine Team, 2015).
2.3 Back-trajectory analysis and modeling and determination of deposition source areas
Back-trajectory analyses utilized the National Oceanic and Atmospheric Administration Air Resources Laboratory's Hybrid Single Particle Lagrangian Integrated Trajectory (HYSPLIT) Version 4 model (http://ready.arl.noaa.gov/hyreg/HYSPLIT_pchysplit.php, last access: 24 August 2016). Global Data Assimilation System (GDAS) 0.5 degree meteorological data from NOAA were used within the HYSPLIT model (http://ready.arl.noaa.gov/archives.php, last access: 24 August 2016). We performed back-trajectory analyses for each individual precipitation event for the entire year of 2014 for each station with available Hg wet deposition for that year (Nome, Gates of the Arctic, and Kodiak Island). Individual precipitation events were considered when measurable precipitation data were present at the 1 h time resolution. If a storm lasted several hours without an interruption of storm activity, the event was classified as a single precipitation event; if there was an interruption in measurable precipitation >2 h in duration, the storm was separated into two events. Using this method, the number of precipitation events identified for 2014 was 247 (Kodiak Island), 182 (Nome), and 148 (Gates of the Arctic). A single air parcel trajectory was calculated for each precipitation event, with the end time initialized to coincide with the center of each precipitation period along with the end latitude and longitude set to each monitoring station. The altitude at which all back trajectories were initiated was set to 2000 m a.s.l.
In order to delineate source regions for seasonal and annual Hg wet deposition for all stations, each back trajectory was weighted according to its contribution to annual Hg deposition as a fraction of the total annual Hg deposition sum. In other words, each of the 240 3-D coordinates of each trajectory output was allocated a scalar value representing the measured Hg deposition value for that precipitation event. We then calculated the residence time of each weighed back trajectory in 2∘ × 2∘ grid cells, with the fractional residence time in a particular grid cell based on the time of the entire trajectory. The weighting of a back-trajectory residence time in a grid cell was then combined with the weighting of that trajectory as a fraction of total annual deposition, so that both the contribution to annual deposition as well as the residence time in grid cells were fully weighted. Finally, the sum of all back-trajectory weights for each grid cell were summed up and represented as a normalized frequency for each grid cell. We performed this for 2014 whereby the weighting occurred as the contribution of each trajectory to annual Hg wet deposition.
3.1 Spatial, seasonal, and temporal patterns of Hg wet deposition concentrations
Minimum Hg concentrations at all stations were equal to the detection limit of the analysis (0.3 ng L−1), and maximum concentrations strongly varied among stations. By far the highest Hg concentrations were reported for Gates of the Arctic with concentrations of up to 396 ng L−1. Statistical analysis determined such high values as outliers, and for further analysis, we eliminated outlier concentrations (>26.14 ng L−1) determined by an outlier analysis (values above and below 1.5×IQR were removed, 1.9 % of all data). For consistency among the deposition stations, we selected a common outlier concentration across all stations, rather than delineate an outlier concentration for each station separately. Median Hg wet deposition concentrations measured across the five deposition stations were in the following order, from highest to lowest (Table 2): Gates of the Arctic (3.6 ng L−1) > Nome (3.5 ng L−1) > Dutch Harbor (2.3 ng L Kodiak Island and Glacier Bay (both 1.8 ng L−1). The distribution of mean values followed the same general order (Table 2), although mean values were higher compared to median values due to skewed distribution in concentration data.
Table 2Summary statistics of Hg concentrations observed at the five deposition stations, and ANOVA and post hoc comparisons to test for statistical differences in Hg concentrations among different stations. ANOVAs were performed after removal of outliers (concentrations > 26.14 ng L−1).
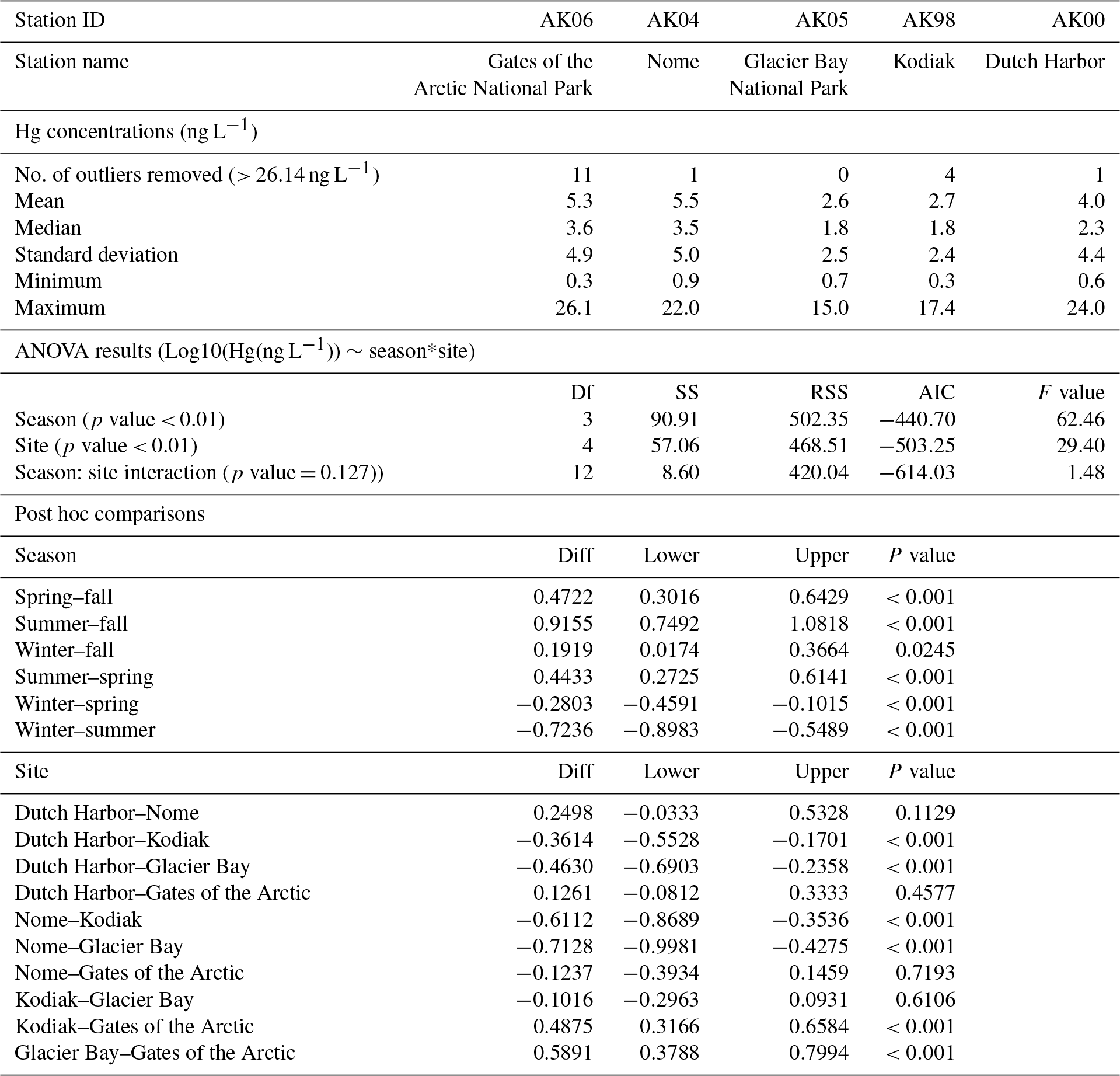
The highest wet Hg deposition concentrations (Table 2) were observed at the two northernmost sites Gates of the Arctic and Nome, with median values almost double and statistically higher compared to concentrations of the two lower-latitude stations (Kodiak Island and Glacier Bay). A third station located at lower latitudes, Dutch Harbor, the westernmost station located on the Aleutian Islands, was similar in Hg concentrations to the two northern stations and showed statistically higher concentrations (+28 %) compared to the other two lower-latitude stations.
We determined that the major reason for higher Hg concentrations at northern sites was a lower dilution (or “washout” effect) of Hg concentrations by smaller storm sizes (Fig. 1 and discussion below). It is known that large precipitation events (i.e., bigger storms or increasing duration of storms) lead to lower Hg wet deposition concentrations compared to small events due to initially higher scavenging of airborne Hg, in particular of particulate-bound Hg (HgP) or gaseous oxidized Hg (GOM) (washout effect: Poissant and Pilote, 1998; Ferrara et al., 1986; Lamborg et al., 1995; Mason et al., 1997; Landis et al., 2002; Lyman and Gustin, 2008; Faïn et al., 2011). Such washout effects also occur in individual storms during which Hg concentrations are highest at the beginning of an event and decrease over time (Glass and Sorensen, 1999; Ferrara et al., 1986). However, a washout effect cannot explain higher Hg concentrations at Dutch Harbor, which were similar to those at the more northern stations (see discussion below). Figure 1a shows the presence of this washout effect evident by inverse linear regressions between storm sizes (total weekly precipitation amounts) and respective measured weekly wet deposition Hg concentrations. All five stations showed statistically significant inverse correlations between the two variables. The slopes of the linear regressions, using log10-transformed Hg concentrations (ng L−1) and log10-transformed precipitation (mm), varied between −0.28 and −0.46, but were not statistically different between stations (based on ANCOVA). Overall, weekly precipitation totals explained 28 % of the variability in Hg concentrations (r2=0.28, p value < 0.01, all sites). The common relationship between wet deposition concentrations and precipitation among all stations was best described by the following inverse linear relationship:
Cumulative distribution of daily storm sizes (Fig. 1b) shows that higher precipitation amounts occurred at lower-latitude stations and were a driving factor leading to lower wet deposition concentrations. For example, precipitation totals at Gates of the Arctic and Nome were 3 to 5 times lower compared to the three lower-latitude sites. Similar to Hg concentrations, differences in precipitation totals were statistically significant between the northern and lower-latitude sites, but not between the two northern or among the three lower-latitude sites (based on post hoc comparison tests, not shown). The figure highlights a dominance of small precipitation events at Gates of the Arctic and Nome, where a high fraction of precipitation events were below 1 mm. In comparison, the three lower-latitude sites consistently experienced much higher daily storm totals, e.g., above 2 mm. We propose that the washout effect largely accounts for higher Hg deposition concentrations at the dryer, northern sites compared to the lower-latitude sites Glacier Bay and Kodiak Island. As mentioned, this analysis, however, fails to explain why Dutch Harbor showed similarly high levels to the northern, more mesic sites.
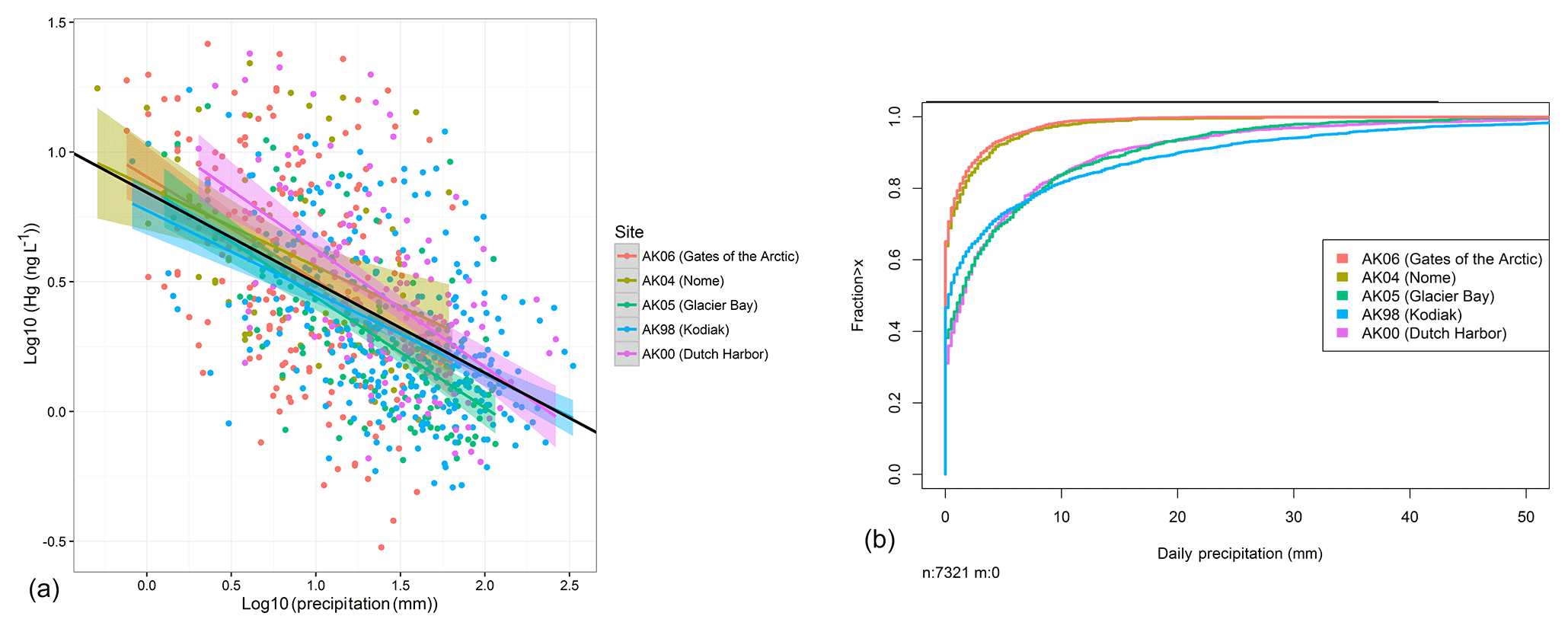
Figure 1(a) Scatterplots of observed Hg concentrations and precipitation amounts, separated by station. The black line shows the overall regression using all sites/all data. A clear dilution effect is observed at all sites. (b) Empirical cumulative distribution plot of daily precipitation at five monitoring stations in Alaska (plot has been cropped at 50 mm). Distinct differences in storm size are observed between the northern (red and gold lines) and southern stations (green, blue, and purple lines).
Figure 2 shows a pronounced seasonality of Hg wet deposition concentrations across all stations, with the highest Hg concentrations in summer, followed by spring, winter, and fall. ANOVA across all sites resulted in statistically significant seasonal effects (variable “season”: p value < 0.01), and post hoc comparisons showed that Hg concentrations differed among all seasons. Seasonal patterns were consistent among the five stations with no significant differences among stations (i.e., no statistically significant interaction of “season” × “station”). In general, median concentrations followed the order summer > spring > winter > fall, with one exception being Dutch Harbor where fall concentrations were slightly above those in winter (2.0 versus 1.9 ng L−1). Such seasonal patterns have been attributed to enhanced summertime GOM concentrations due to increased photochemical formation of oxidized mercury in summer that leads to higher Hg concentrations in precipitation (Pirrone and Mason, 2009; Selin and Jacob, 2008). We found, however, that observed seasonal patterns were also affected by storm sizes and dilution effects since precipitation amounts were generally lowest in summer and highest in fall and winter. We performed analysis on seasonal differences by “detrending” data for different storm sizes, i.e., adjusting Hg concentrations by deducting the linear trend of the washout effect (Eq. 1). ANOVA and post hoc Bonferroni comparisons of detrended Hg concentrations, however, showed that differences among seasons persisted after correcting for different precipitation sizes per season and that the order of Hgcorr concentrations followed the same order as the untrended concentrations (summer > spring > fall/winter). Hence, we propose a combination of Hg oxidation processes (Pirrone and Mason, 2009; Selin and Jacob, 2008) along with precipitation sizes contributing to seasonal differences.
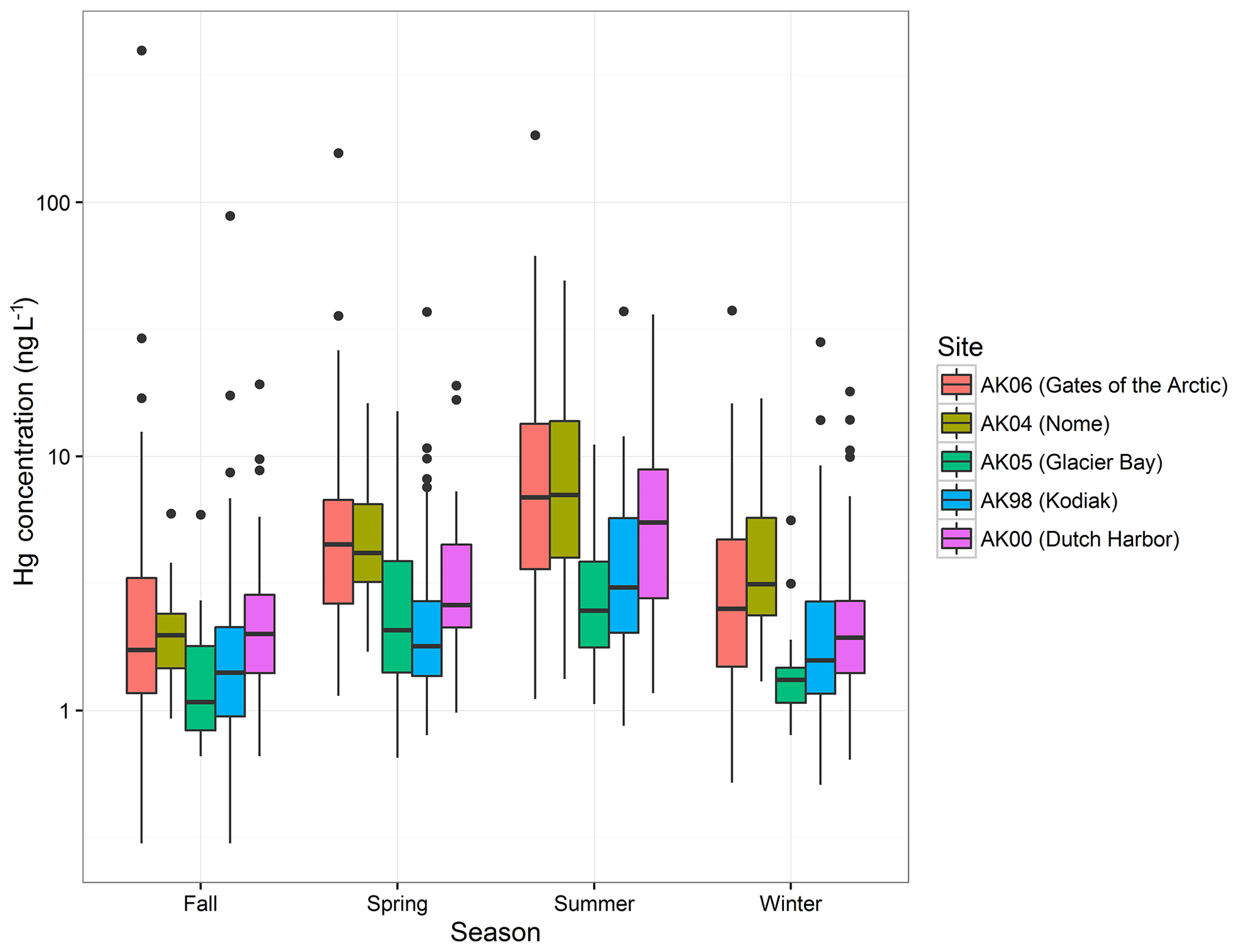
Figure 2Summary box plot of Hg concentrations separated by monitoring station and collection season. Similar seasonal trends are observed at each site with the highest concentrations occurring in summer and lowest concentrations in fall. Spring (March, April, May), summer (June, July, August), fall (September, October, November), and winter (December, January, February).
3.2 Spatial, seasonal, and annual patterns of Hg wet deposition loads
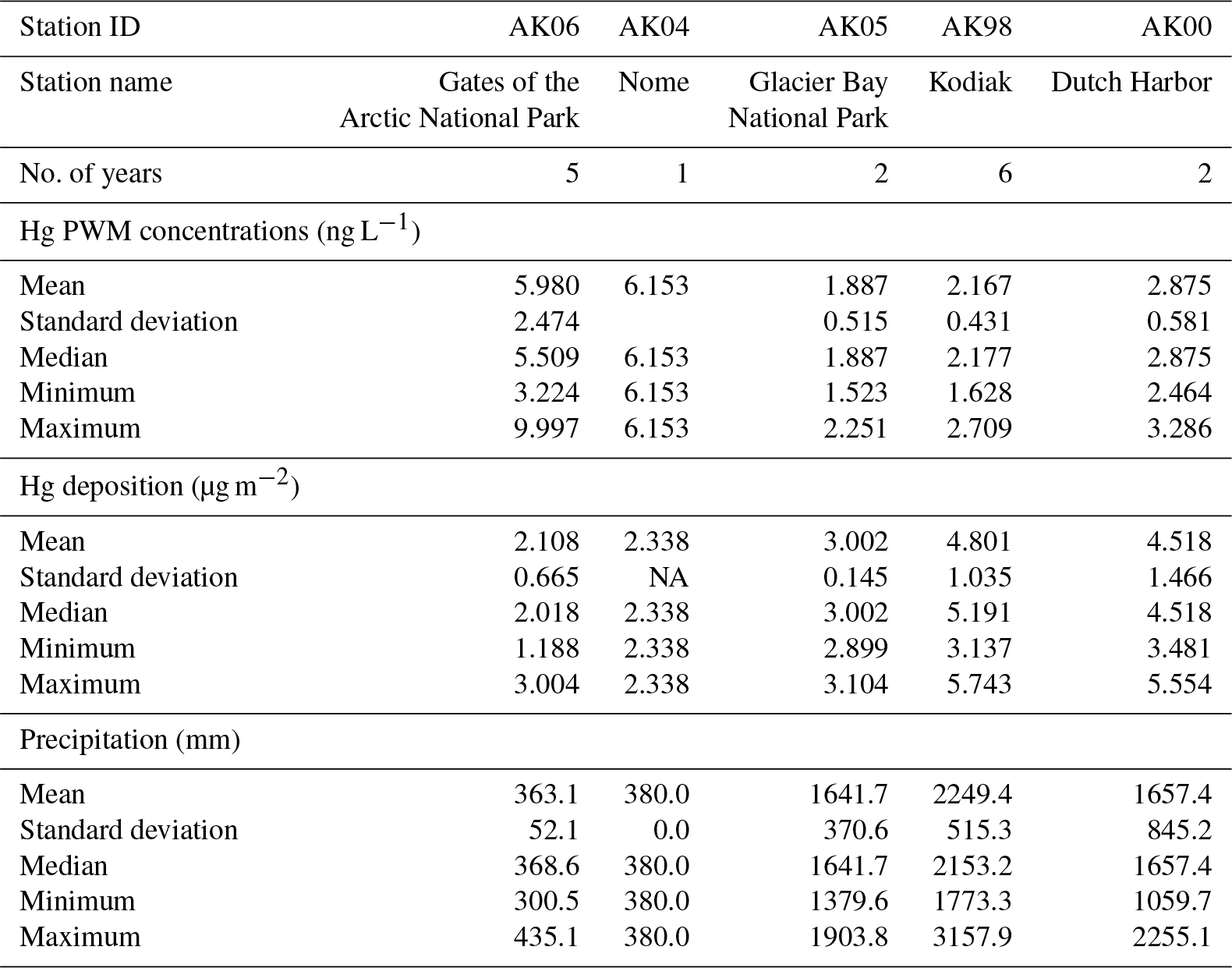
In order to calculate annual deposition loads, NADP MDN protocols use multiplication of precipitation-weighted annual Hg concentration by annual precipitation records, and require that the percentage of valid Hg samples exceeds 75 %, the percentage for which precipitation amounts were available exceeds 90 %, and the percentage of total measured precipitation associated with valid samples exceeds 75 %. Following these constraints, data coverage allowed for a total of 16 years of annual wet deposition estimates across the five stations (Table 3). Annual Hg deposition values across the five stations averaged 3.55±1.48 µg m−2, with a minimum of 1.94 µg m−2 at Gates of the Arctic in 2012 and a maximum of 5.74 µg m−2 at Kodiak Island in 2011. The highest Hg deposition loads consistently occurred at Kodiak Island and the lowest at Gates of the Arctic. Differences in wet Hg deposition loads between stations were large: for example, in the 4 years of corresponding data, Kodiak Island deposition exceeded that at Dutch Harbor by a factor of 2.6 (in 2009), 2.4 (in 2011), 2.0 (in 2012), and 2.6 (in 2014). The second highest deposition loads were consistently observed at Dutch Harbor, with loads that were slightly below those on Kodiak Island in the 2 years of corresponding measurements. Statistical tests showed that annual deposition was statistically different among stations, and a post hoc Bonferroni comparison showed that this difference was driven largely by a statistically significant difference between the highest (Kodiak Island) and lowest (Gates of the Arctic) stations (p value < 0.05). When using a statistical significance level of 10 % as opposed to 5 %, we also observed significant differences between Kodiak Island and Glacier Bay and Kodiak Island and Nome. Hence, annual deposition loads can be summarized as follows: the highest deposition occurred at Kodiak Island (4.80±1.04 µg m−2), but was not statistically different from Dutch Harbor (4.52±1.47 µg m−2), but was statistically different from all other stations. The lowest deposition was observed at Gates of the Arctic (2.11±0.67 µg m−2), and intermediate values were observed for Glacier Bay (3.00±0.14 µg m−2) and Nome (2.34 µg m−2). Although we did not observe significant effects of year among station deposition data (p value = 0.138), we observed substantial inter-annual variability in Hg deposition loads at individual stations. For example, using the 5 years of measurements at Kodiak Island, values ranged from 3.14 µg m−2 (in 2009) to 5.61 µg m−2 (in 2013), or a factor of 1.8 difference and a coefficient of variation of 22 % (CV: SD divided by mean). The inter-annual comparison of Gates of the Arctic showed values from 1.19 µg m−2 (2009) to 3.00 µg m−2 (2010), or a factor of 2.5 difference and a CV of 32 % (Table 3).
Table 3Summary statistics of NADP MDN annual estimates of precipitation-weighted mean concentration, deposition, and precipitation for five monitoring sites in Alaska.
3.3 Annual deposition loads and relationships to annual precipitation
In order to characterize what drives annual deposition loads, we analyzed PWM annual Hg concentrations and annual precipitation, the two factors which together constitute annual deposition. Figure 3a shows a scatterplot and linear regression between PWM Hg and precipitation amounts using data of all years and all stations (16 values), showing a strong linear relationship between PWM Hg and precipitation. The regression explains 59 % of the variability in PWM Hg, and a slope of −0.0189 suggests that with each 100 mm increase in annual precipitation, PWM Hg concentration decreased on average by 1.9 ng L−1. These patterns support a strong dependence of Hg concentrations on precipitation amounts, in agreement with the weekly Hg concentration data showing dilution effects as discussed above. Yet, an important difference is that the annual relationships between PWM Hg concentrations and precipitation are strongly linear, compared to nonlinear functions between weekly Hg wet deposition concentrations and weekly precipitation (i.e., log10–log10 relationships). The major control in determining Hg wet deposition loads across the five Alaska stations was annual precipitation, which alone explained 71 % of the variability in observed annual deposition loads.
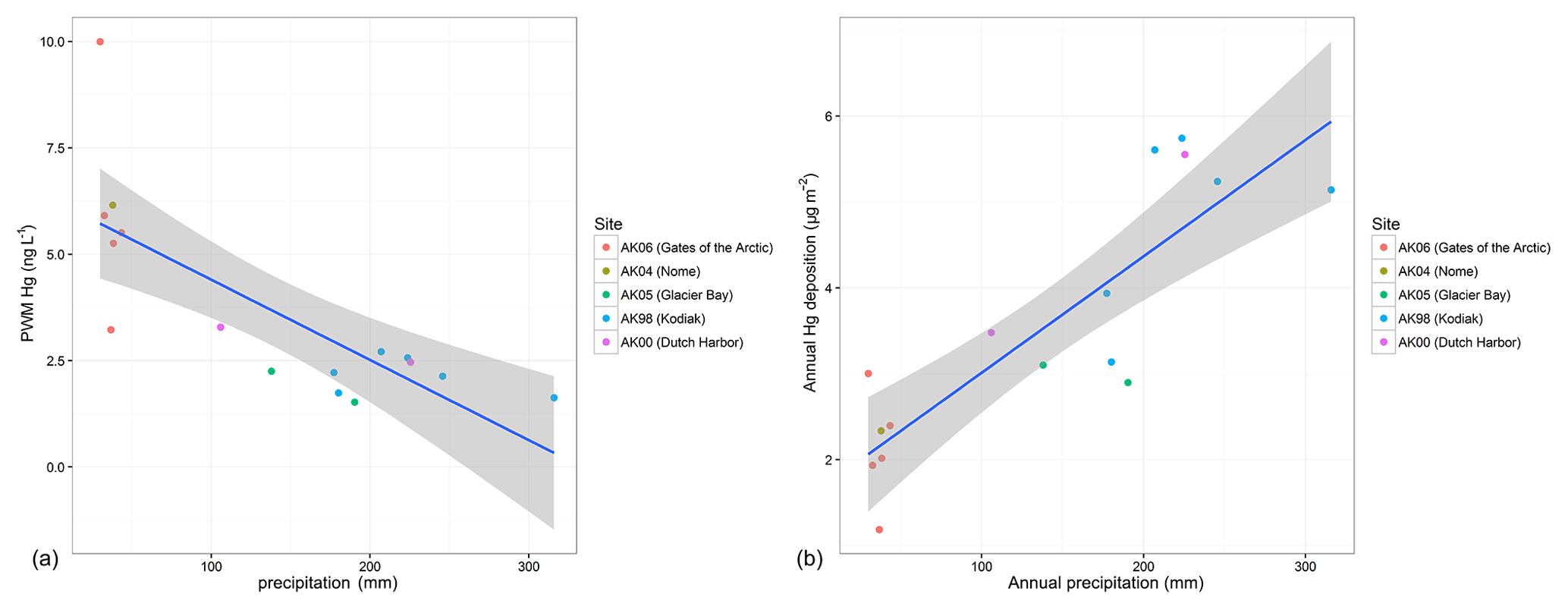
Figure 3(a) Plot of annual precipitation-weighted mean (PWM) Hg concentration versus annual precipitation (mm). Linear regression analysis shows a significant relationship with a correlation coefficient R2=0.557 and p value = < 0.001. (b) Plot of annual Hg deposition versus annual precipitation (mm). Linear regression analysis shows a significant relationship with a correlation coefficient of R2=0.7141 and p value = < 0.001.
Compared to annual deposition observed across the contiguous US (http://nadp.sws.uiuc.edu/mdn/, last access: 22 August 2016), Hg deposition in Alaska was extremely low. For example, Hg deposition across 99 deposition sites of the contiguous US in 2014 averaged 9.7±3.9 µg m−2, with a median value of 9.0 µg m−2. For example, Hg deposition across 99 sites in the contiguous US in 2014 averaged 9.7±3.9 µg m−2, with a median value of 9.0 µg m−2, while deposition at Gates of the Arctic, Kodiak Island, and Nome averaged 2.0, 5.1, and 2.4 µg m−2, respectively. When comparing the multi-year average annual Hg deposition of the Alaska stations to the 2014 deposition across the contiguous US, three Alaska stations (Nome: 2.3 µg m−2; Glacier Bay: 3.0 µg m−2; Gates of the Arctic: 2.1 µg m−2) showed annual deposition below all of the lower 48 contiguous US states in 2014 (lowest value in the contiguous US of 3.1 µg m−2 observed at CA94, Converse Flats, San Bernardino). Only Dutch Harbor (4.5 µg m−2) and Kodiak Island (4.8 µg m−2) exceeded the lowest deposition loads of the lower 48 states, yet even these two stations fell below the fifth percentile of annual deposition observed at the 99 sites in the lower 48 states in 2014 (5.3 µg m−2).
Similarly, PWM Hg concentrations were very low in Alaska compared to the rest of the US, which for the year 2014 averaged 10.6±9.1 ng L−1 with a median value of 8.9 ng L−1. Three Alaskan stations showed annual PWM Hg concentrations below the minimum concentrations (3.0 ng L−1) of any of the stations in the contiguous US, including Dutch Harbor (2.9 ng L−1), Glacier Bay (1.9 ng L−1), and Kodiak Island (2.2. ng L−1). Nome with an annual PWM concentration of 6.2 ng L−1 and Gates of the Arctic (6.0 ng L−1) were below the 15th percentile of concentrations of the lower 48 states. We conclude that low deposition values observed across coastal regions in Alaska were driven largely by very low wet deposition concentrations below concentrations at any of the contiguous US deposition stations. For the two northern stations, Gates of the Arctic and Nome, extremely low wet Hg deposition was driven by a combination of low deposition concentrations and very low annual precipitation.
In summary, very low concentrations and deposition totals were observed throughout Alaska. It is important, however, to note that wet deposition as reported in this study constitutes only part of the total Hg deposition. In terrestrial ecosystems, there is ample evidence that dry deposition of Hg constitutes a major Hg source, in large parts including gaseous elemental Hg, which accounts for 57 %–94 % of Hg found in soils (Obrist et al., 2018). This evidence is based on stable Hg isotope studies (e.g., Demers, 2013, and subsequent studies) along with earlier forest studies that showed that litterfall and throughfall deposition dominate as major dry deposition sources in forests (Iverfeldt, 1991; Munthe et al., 1995). Deposition modeling by Jaeglé (2010) showed that the relative contributions of wet and dry deposition throughout Alaska varied spatially, with wet deposition accounting for approximately 28 % on average. In agreement, a recent study in the northern Tundra (Obrist et al., 2017) showed that wet deposition accounted for a minor fraction of overall Hg deposition, with the largest source to the tundra deriving from atmospheric gaseous Hg deposition cycled through arctic vegetation.
3.4 Spatial scaling of Hg deposition to the entire state of Alaska
We used spatial interpolation and extrapolation techniques to create maps of deposition concentrations and deposition loads across the state of Alaska, following interpolation protocols described by the National Atmospheric Deposition Program (NADP, 2016). Limitations of deposition maps, as stated by the NADP network, include that “stations and maps represent regional trends (rather than local sources) and that uncertainty with maps varies geographically, have not been quantified, and high levels of uncertainty can occur due to topographic variability, near urban and industrial areas, and in regions isolated from deposition sites”. The NADP network specifically cautions making decisions based on projected maps when no direct measurements are available. Concentration maps shown in Fig. 4 are based on the inverse distance weighting interpolation method of average PWM Hg concentrations for each station (Fig. 4a), and as such represent different collection years and number of years for each station. The resulting Hg concentration maps (shown in Fig. 4) present a coarse spatial representation of the concentrations patterns (i.e., only five measurement stations) that relate to precipitation gradients, with the highest concentrations observed at the northern two stations that show low annual precipitation and small storm sizes. Inverse weighting procedures resulted in interpolated Hg concentrations that were not fully in accordance with the observed relationships to precipitation patterns. For example, precipitation maps show strong gradients in annual precipitation from the southern coast of Alaska to inland and northern locations and relatively consistently low precipitation values across much of central, northern, and eastern Alaska. In contrast, the interpolated Hg concentration map shows that interior and eastern Alaskan concentrations follow north-to-south gradients between the lower-latitude and higher-latitude stations, but do not account for east–west gradients. While we could have used precipitation-based estimates of Hg concentrations across the state (based on strong relationships of PWM Hg and annual precipitation), we decided not to deviate from common NADP mapping procedures.
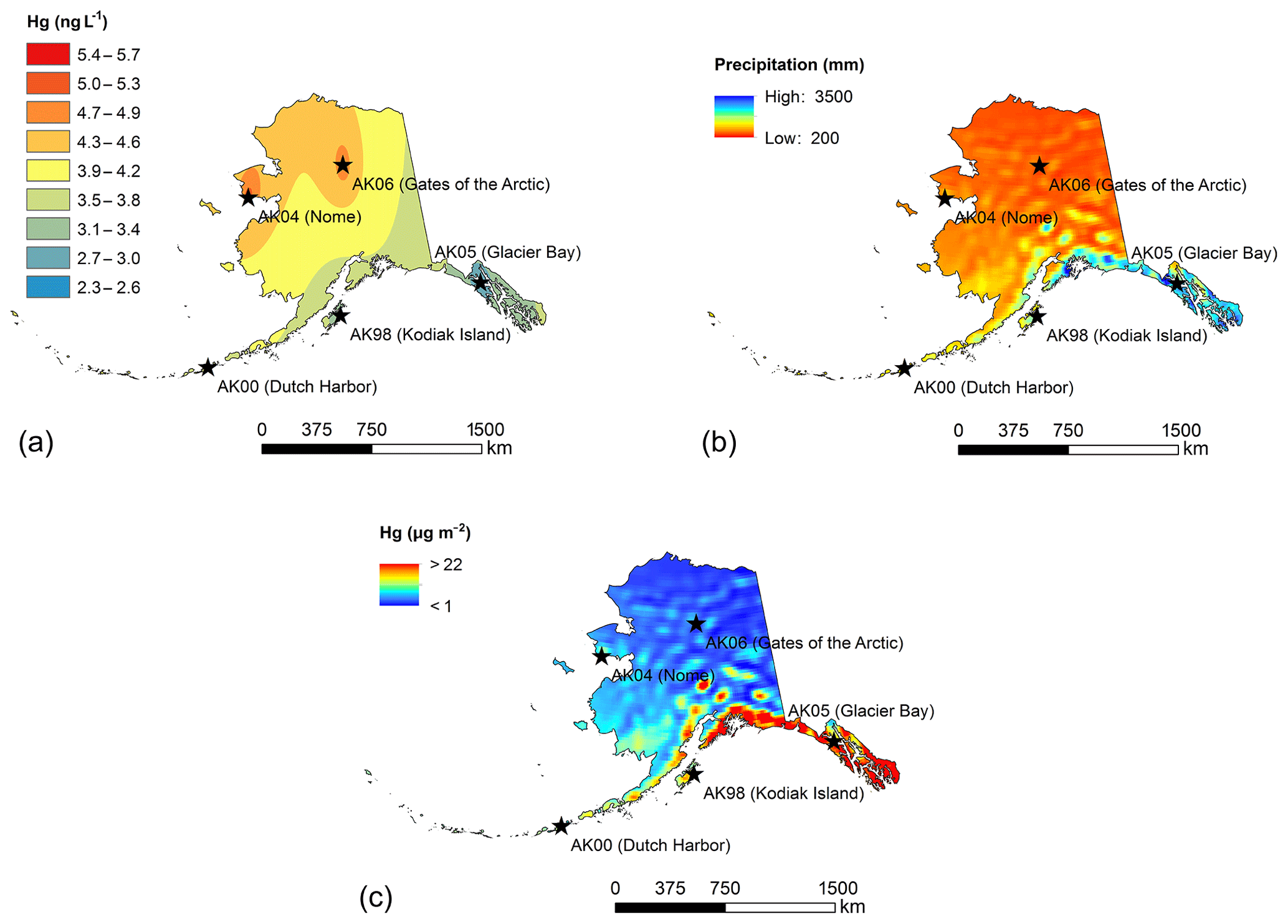
Figure 4Average annual Hg deposition maps for the state of Alaska: (a) inverse distance weighted concentration layer; (b) NCEP climate forecast system precipitation 2007–2015 annual average; (c) Hg deposition estimates. Maps show large-scale concentration gradients (north–south) largely due to precipitation differences with total loading being highly dependent on precipitation.
Figure 4b shows precipitation maps across Alaska based on precipitation data averaged for the years 2007 to 2015. The long-term NOAA precipitation maps show strong gradients from the southern coastal locations to interior and northern Alaska, with very strong precipitation changes within short distance (50–100 miles). The highest annual precipitation was observed along the southeastern and south-central coasts, with maximum precipitation of approximately 610 cm yr−1, and high precipitation amounts (200 to 300 cm yr−1) were also observed at Kodiak Island and Bristol Bay and the Aleutian and Probilof islands. In the interior and far north regions of Alaska, annual precipitation sums were low and generally below 100 cm yr−1. The resulting annual Hg deposition maps shown in Fig. 4c project distinct zones of the highest Hg deposition in Alaska along the southern and southeastern coasts, with annual Hg deposition exceeding 20 µg m−2 yr−1. The zones of the highest annual Hg deposition, based on the estimated map, were confined to narrow zones of approximately 75–150 km inland. Similarly, high Hg deposition may have occurred in isolated mountain areas near the southern coast such as in the Alaskan Range. For example, in the Denali National Park region, estimated Hg wet deposition of up to 15 µg m−2 yr−1 was at a similar magnitude of the highest deposition amounts along the southern and southeastern coast. Lower Hg deposition amounts were projected, and in fact observed, along the southwestern coastal region, including Kodiak Island and the western and eastern Aleutians. Here, estimated annual Hg deposition was in the range from 5 to 10 µg m−2 yr−1. Our estimated deposition maps indicated that in much of the state of Alaska, in particular in the interior and far northern regions, Hg deposition was very low, with annual Hg deposition generally below 4 µg m−2 yr−1 and in many areas (e.g., north of the Brooks Range) only in the range of 1–2 µg m−2 yr−1.
As stated above, maps of estimated annual deposition should be considered with caution and may include a variety of errors. Compared to measured deposition at the five stations, estimated deposition fell well within 10 % of observations at Gates of the Arctic and Kodiak Island. At other stations, we found larger discrepancies between observed and modeled deposition, and at Glacier Bay and Nome discrepancies were over 100 %. We attributed the larger biases to discrepancies in annual precipitation: for example, at Glacier Bay and Nome, the model strongly overestimated precipitation (by 90 % and 117 %, respectively), which accounted for the main part of the bias. Reasons for precipitation errors were mainly due to the large grid size of the modeled precipitation combined with strong coastal gradients. Another possible reason for differences between observed and predicted deposition may include issues of precipitation fetch during deposition measurements. Precipitation gages generally show a strong bias towards under-catch of precipitation caused by wind, even with precipitation gages that are designed with wind protection (Savina et al., 2012; Yang et al., 2000). Snowfall, which accounts for a very important fraction of annual precipitation in this area, can lead to under-catch ranging from 20 to 50 % during windy conditions (Rasmussen et al., 2012).
3.5 Back-trajectory-determined source regions of Hg deposition
We performed comprehensive back-trajectory analyses for the year 2014 and included data from the highest (Kodiak Island), lowest (Gates of the Arctic), and intermediate (Nome) deposition stations. As described in methods, back-trajectory modeling was performed for all individual precipitation events (total of 247 events for Kodiak Island, 182 events for Nome, and 148 events for Gates of the Arctic) and subsequently each deposition event was weighted by its contribution to annual deposition load. Figure 5 shows normalized back-trajectory frequency maps for Hg deposition for the year 2014. For Kodiak Island, trajectory frequency maps showed the highest trajectory frequencies in close vicinity of the deposition station and to the south of the station. These patterns were attributed to the fact that each trajectory passed through adjacent station grid cells prior to arriving at the deposition station. In addition, major source origins for Hg wet deposition stemmed from the Gulf of Alaska with additional contributions further south in the eastern Pacific Ocean up to a distance of 2500 km south of Kodiak Island. A similar pattern was observed for Gates of the Arctic where high contributions to annual Hg deposition were observed again in the vicinity of the station, with additional source regions from the center of the Bering Sea and the Gulf of Alaska. There were only a few occasions when storms or deposition events were tracked far into the western Pacific. We conclude that Gates of the Arctic experienced source regions that were predominantly located in the Bering Sea and the Gulf of Alaska. A different pattern was evident for Nome. Here, the frequency distributions of trajectories showed increased contributions in the western Pacific downwind of East Asia, indicating significant contributions from East Asia where known high-Hg-emission sources such as mining, industrial emissions, and coal burning have led to increased atmospheric Hg levels (Wong et al., 2006). A recent study by Pacyna et al. (2016) identified East Asia and India as the dominant source areas of global anthropogenic Hg emission from 2005 to 2010. Evidence that Hg pollution in East Asia contributes to elevated wet deposition Hg levels in downwind areas is also seen by the recently established Asia Pacific Mercury Monitoring Network (APMMN) where preliminary data show average wet deposition concentrations ranging from 7 to 23 ng L−1 in samples covering areas from Vietnam to Korea (Sheu, 2017).
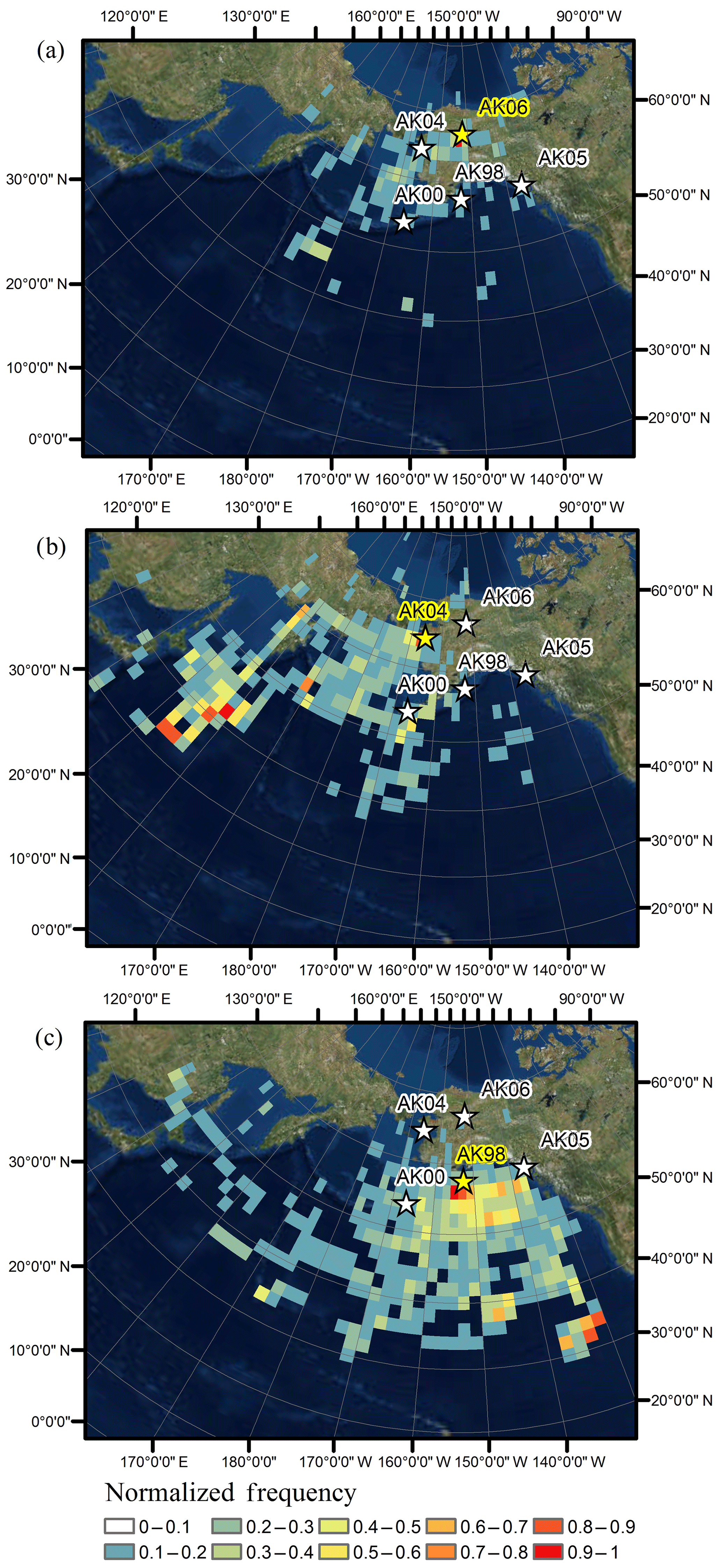
Figure 5Normalized-frequency maps weighted by Hg deposition for (a) Gates of the Arctic (AK06), (b) Nome (AK04), and (c) Kodiak Island (AK98).
Table 4Trace metal summary statistics using both maximum likelihood estimation and 1/2 MDL substitution techniques.
3.6 Auxiliary trace metal concentrations at Dutch Harbor (AK00), Kodiak Island (AK98), and Nome (AK04)
Across the three stations with data and deposition samples, we found the following order of median concentrations (MLE based) of trace elements (Table 4): Zn (1.40 µg L−1) > As (0.19 µg L−1) > Cu (0.14 µg L−1) > Se (0.06 µg L−1) > Ni (0.04 µg L−1) > Pb (0.04 µg L−1) > Cr (0.02 µg L−1) > Hg (0.002 µg L−1). The highest concentrations were always observed for Zn, which exceeded concentrations of all other elements by over an order of magnitude. Similarly, the lowest concentrations were always observed for Hg, which was below concentrations of all other trace metals by at least 1 order of magnitude. Similar patterns of trace element concentrations, although generally higher in concentrations, have been observed in snow samples at lower latitudes, such as in Utah snowpack where Carling et al. (2012) observed the highest bulk (unfiltered) concentrations of Zn in the range of 3–4 µg L−1, with concentrations that exceeded that of other trace metals severalfold, and similarly, Hg concentrations were about 1 order of magnitude below concentrations of other trace elements. In the Everest region in the Himalayas, Lee et al. (2008) observed high concentrations of Zn (0.48 mug L−1) as well compared to other trace elements (e.g., 0.11 for Cr, 0.08 for Pb and Ni, <0.01 for As). In fresh snow in the French Alps, Veysseyre et al. (2001) observed concentrations of Zn up to 0.75 µg L−1, again the highest compared to other trace metals (e.g., up to Cu: 0.2 µg L−1), although Pb showed some high values in that study as well (max. of 1.76 µg L−1).
3.7 Principal component analyses of the full trace element concentration dataset
In this section, we use PCAs to explore commonalities of trace metals using both the entire dataset available across the three stations as well as individual stations. Figure 6 shows PCAs using all elements for all sites (panel a) and for each individual site separately (panels b–d), with a graphical representation showing the two main components (first component: x axis; second component: y axis). Similar patterns appeared in both analyses. All elements showed a strong negative correlation with component one, suggesting that all element concentrations increased and decreased together. Component 1 likely represents precipitation with washout effects leading to lower chemical concentrations with larger storms. Interestingly, Hg showed the weakest correlation with component one, possibly related to Hg's highly volatile nature relative to other trace metals. Ni, Cr, and Hg consistently fell on the negative side of a second component, while Pb and As fell on the positive side of a second component. Weaker associations were observed for Se, Pb, and As. We propose that the second principal component may represent differences in the trace metals origins. Ni and Cr were likely associated due to a similar crustal and/or natural sources (Carling et al., 2012; Agnan et al., 2015b; Veysseyre et al., 2001). In the opposite loading of this second principal component we found Pb and As, possibly due to their different source origins. As and Pb are primarily driven by anthropogenic, industrial emission sources such as smelters and combustion processes (Tchounwou et al., 2012), and these may in some part be derived from long-range transport from Asia. Hg's association with Ni and Cr supports a more background/natural source rather than local or point-driven pollution source, or its distinctly different atmospheric behavior due to its presence in the gaseous phase.
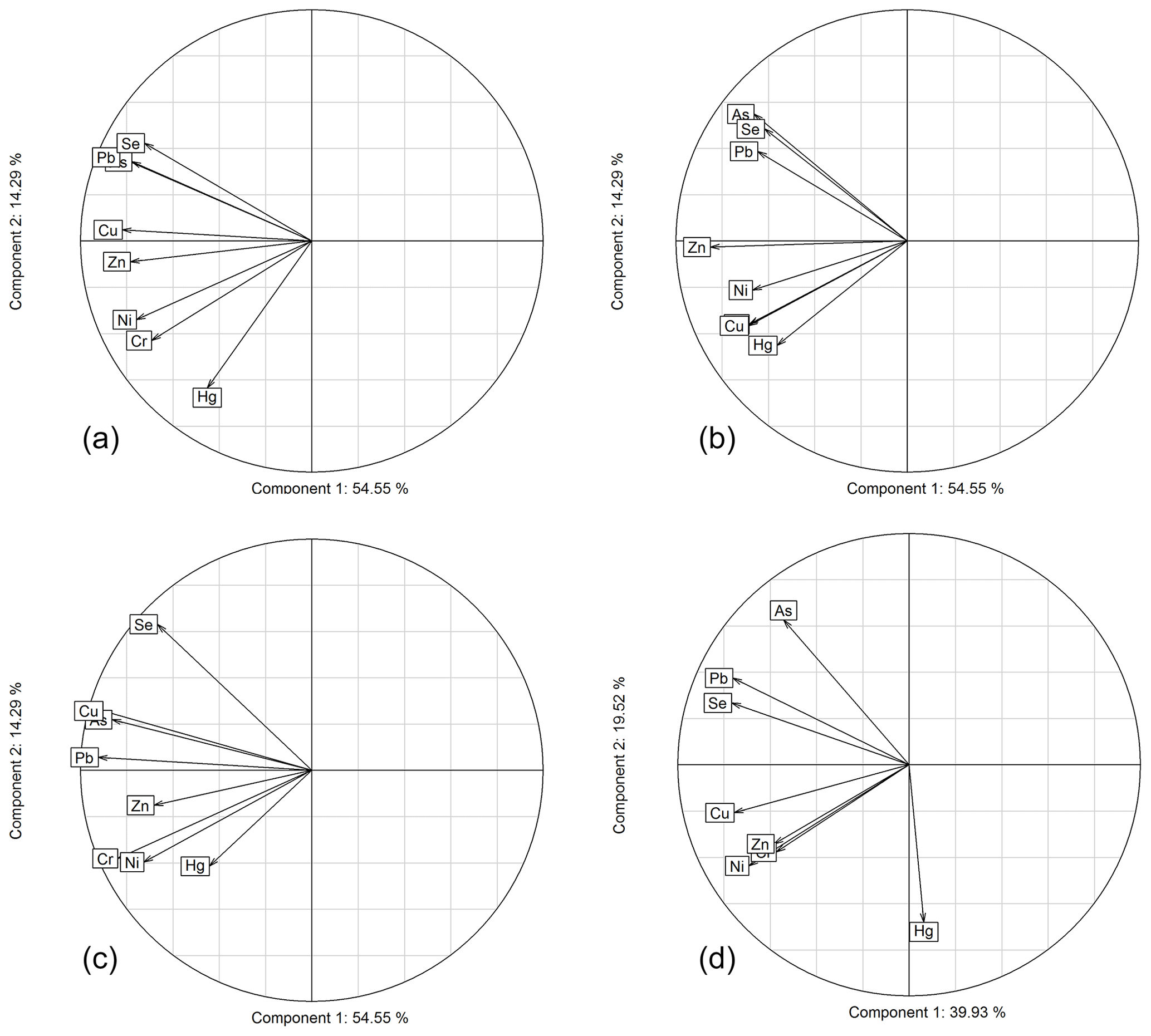
Figure 6Trace metal rank-based principal component analysis biplots of component 1 (x axis) versus component 2 (y axis) for (a) all sites, (b) Dutch Harbor, (c) Nome, and (d) Kodiak Island. Values represent the percentage of variance explained by each relative component.
Our analysis of wet deposition data from five stations in Alaska found that Hg concentrations in precipitation at the two northern stations (Nome and Gates of the Arctic) were consistently and significantly higher than the two lowest-latitude sites (Kodiak Island and Glacier Bay). These differences were largely explained by different precipitation regimes, with high amounts of precipitation at the lower-latitude stations leading to washout effects compared to dryer, northern deposition sites. Differences in Hg concentrations between sites still existed after the effects of precipitation differences were removed, with Gates of the Arctic (AK06) and Nome (AK04) showing the highest Hg concentrations and Kodiak Island statistically still had the lowest Hg concentrations, which suggests that other factors contributed to observed differences in Hg concentrations as well.
The highest annual Hg deposition loads were always observed at Kodiak Island (AK98), and the lowest deposition loads were always observed at Gates of the Arctic (AK06), and these differences were substantial. For example, Kodiak Island (AK98) exceeded deposition at Dutch Harbor (AK00) by a factor of 2.6 (in 2009), 2.4 (in 2011), 2.1 (in 2012), and 2.6 (in 2014). These patterns were also explained to a large degree by precipitation differences, whereby across all stations and collection years precipitation overwhelmingly controlled annual Hg deposition, explaining 73 % of the variability in observed deposition. Annual Hg deposition loads for all of Alaska were among the lowest anywhere in the United States, falling into the lowest fifth percentile of all observed annual deposition. Based on observations and spatial interpolations, we found distinct zones of higher Hg deposition in Alaska along the southern and southeastern coasts (confined to 75–150 km inland), and similarly high Hg depositions in isolated mountain areas near the southern coast, due to orographic precipitation enhancement. Lower Hg deposition amounts were observed along the southwestern coastal region, including Kodiak Island and the western and eastern Aleutians. For most of the state, particularly in the interior and far northern regions, Hg deposition was estimated to be very low.
Back-trajectory analysis of 2014 deposition data showed largely diffuse and regional Hg sources at Gates of the Arctic (AK06) and Kodiak Island (AK98). Origins of Hg wet deposition at Nome (AK04) were quite different with increased source contributions relative in the western Pacific Ocean near the East Asian continent, possibly due to long-range transport from East Asian emissions. PCA revealed two distinct associations of trace elements: Cr and Ni were clustered, and so were As and Pb, which was attributable to different source origins. Sources of Ni and Cr are often considered driven by crustal (e.g., dust) sources, while As and Pb are attributable to anthropogenic inputs, including by long-range transport from Asia. Mercury, nor any of the other trace elements analyzed, did not consistently associate with any of these four elements, suggesting more diffuse and possibly different source origins or a distinctly different behavior in the atmosphere.
Given the low wet deposition amounts of Hg across most of Alaska, we recommend additional focus on dry deposition monitoring, as recent research suggests that deposition of gaseous elemental Hg (Hg0) dominates Hg loading in arctic tundra ecosystems and across other vegetated ecosystems (Jiskra et al., 2018; Obrist et al., 2017, 2018). Finally, global warming is a particular concern in northern latitudes, with warming occurring at a rate almost twice the worldwide average in the Arctic (Polyakov et al., 2002). Global warming will change precipitation patterns and associated deposition dynamics, including shortening of winter and changes in the fraction of snow and rain. In addition, climate change will have profound consequences on other ecosystem processes, including loss of permafrost and increased active layer depths of soils (Schuur and Abbott, 2011) and profound changes in hydrology (Vonk et al., 2015), with complex, yet poorly understood, consequences for exposure to Hg (Obrist et al., 2018).
All Hg Deposition Data can be accessed via the National Atmospheric Deposition Program website at: http://nadp.slh.wisc.edu/mdn/ (last access: 22 August 2016, NADP, 2016). Auxiliary Trace Metal Data was provided by the Alaska Department of Environmental Conservation and is available upon request.
CP and DO designed this analysis. CP analyzed the data and generated figures. DH performed the HYSPLIT back-trajectory modeling. CP and DO prepared the paper with contributions from all co-authors.
The authors declare that they have no conflict of interest.
This work was funded by the Alaska Department of Environmental Conservation grant no. USFSW/15.668/F12AF00730. Funding was also provided by a collaborative research project by the US National Science Foundation (award nos. 1739567 and 1304305).
This research has been supported by the Alaska Department of Environmental Conservation (grant no. USFSW/15.668/F12AF00730) and the US National Science Foundation (grant nos. 1739567 and 1304305).
This paper was edited by Leiming Zhang and reviewed by two anonymous referees.
Ackerman, J. T., Eagles-Smith, C. A., Herzog, M. P., Hartman, C. A., Peterson, S. H., Evers, D. C., Jackson, A. K., Elliott, J. E., Vander Pol, S. S., and Bryan, C. E.: Avian mercury exposure and toxicological risk across western North America: a synthesis, Sci. Total Environ., 568, 749–769, https://doi.org/10.1016/j.scitotenv.2016.03.071, 2016.
Agnan, Y., LeDantec, T., Moore, C., Edwards, G., and Obrist, D.: New constraints on terrestrial surface-atmosphere fluxes of gaseous elemental mercury using a global database, Environ. Sci. Technol., 19, 507–524, 2015a.
Agnan, Y., Sejalon-Delmas, N., Claustres, A., and Probst, A.: Investigation of spatial and temporal metal atmospheric deposition in France through lichen and moss bioaccumulation over one century, Sci. Total Environ., 529, 285–296, 10.1016/j.scitotenv.2015.05.083, 2015b.
AMAP: AMAP Assessment 2002: Heavy Metals in the Arctic, Arctic Monitoring and Assessment Programme (AMAP), Oslo, Norway, xvi + 265 pp., 2005.
AMAP: Arctic Pollution 2009, Arctic Monitoring and Assessment Programme (AMAP), Oslo, Norway, 83, 2009a.
AMAP: Assessment 2009: Human Health in the Arctic, Oslo, Norway, 2009b.
AMAP: Arctic Pollution 2011, Arctic Monitoring and Assessment Programme (AMAP), Oslo, Norway, 38, 2011.
Baeyens, W., Leermakers, M., Papina, T., Saprykin, A., Brion, N., Noyen, J., De Gieter, M., Elskens, M., and Goeyens, L.: Bioconcentration and biomagnification of mercury and methylmercury in North Sea and Scheldt estuary fish, Arch. Environ. Con. Tox., 45, 498–508, https://doi.org/10.1007/s00244-003-2136-4, 2003.
Bocharova, N., Treu, G., Czirják, G. Á., Krone, O., Stefanski, V., Wibbelt, G., Unnsteinsdóttir, E. R., Hersteinsson, P., Schares, G., Doronina, L., Goltsman, M., and Greenwood, A. D.: Correlates between Feeding Ecology and Mercury Levels in Historical and Modern Arctic Foxes, PLoS ONE, 8, e60879, https://doi.org/10.1371/journal.pone.0060879, 2013.
Braune, B. M., Gaston, A. J., Gilchrist, H. G., Mallory, M. L., and Provencher, J. F.: A geographical comparison of mercury in seabirds in the eastern Canadian Arctic, Environ. Int., 66, 92–96, https://doi.org/10.1016/j.envint.2014.01.027, 2014.
Brockhoff, C., Creed, J., Martin, T., Martin, E., and Long, S.: EPA Method 200.8, Revision 5.5: Determination of trace metals in waters and wastes by inductively coupled plasma-mass spectrometry, EPA-821R-99-017, 1999.
Brunke, E. G., Labuschagne, C., and Slemr, F.: Gaseous mercury emissions from a fire in the Cape Peninsula, South Africa, during January 2000, Geophys. Res. Lett., 28, 1483–1486, 2001.
Carling, G. T., Fernandez, D. P., and Johnson, W. P.: Dust-mediated loading of trace and major elements to Wasatch Mountain snowpack, Sci. Total Environ., 432, 65–77, https://doi.org/10.1016/j.scitotenv.2012.05.077, 2012.
Demers, J. D., Blum, J. D., and Zak, D. R.: Mercury isotopes in a forested ecosystem: Implications for air-surface exchange dynamics and the global mercury cycle, Global Biogeochem. Cy., 27, 222–238, https://doi.org/10.1002/gbc.20021, 2013.
Dietz, R., Outridge, P. M., and Hobson, K. A.: Anthropogenic contributions to mercury levels in present-day Arctic animals-A review, Sci. Total Environ., 407, 6120–6131, https://doi.org/10.1016/j.scitotenv.2009.08.036, 2009.
Dommergue, A., Sprovieri, F., Pirrone, N., Ebinghaus, R., Brooks, S., Courteaud, J., and Ferrari, C. P.: Overview of mercury measurements in the Antarctic troposphere, Atmos. Chem. Phys., 10, 3309–3319, https://doi.org/10.5194/acp-10-3309-2010, 2010.
Douglas, T. A. and Sturm, M.: Arctic haze, mercury and the chemical composition of snow across northwestern Alaska, Atmos. Environ., 38, 805–820, 2004.
Douglas, T. A., Loseto, L. L., Macdonald, R. W., Outridge, P., Dommergue, A., Poulain, A., Amyot, M., Barkay, T., Berg, T., Chetelat, J., Constant, P., Evans, M., Ferrari, C., Gantner, N., Johnson, M. S., Kirk, J., Kroer, N., Larose, C., Lean, D., Nielsen, T. G., Poissant, L., Rognerud, S., Skov, H., Sorensen, S., Wang, F., Wilson, S., and Zdanowicz, C. M.: The fate of mercury in Arctic terrestrial and aquatic ecosystems, a review, Environ. Chem., 9, 321–355, https://doi.org/10.1071/en11140, 2012.
Eagles-Smith, C. A., Ackerman, J. T., Willacker, J. J., Tate, M. T., Lutz, M. A., Fleck, J. A., Stewart, A. R., Wiener, J. G., Evers, D. C., Lepak, J. M., Davis, J. A., and Pritz, C. F.: Spatial and temporal patterns of mercury concentrations in freshwater fish across the western United States and Canada. , Sci. Total Environ., 568, 1171–1184, https://doi.org/10.1016/j.scitotenv.2016.03.229, 2016.
Evans, M. S., Lockhart, W. L., Doetzel, L., Low, G., Muir, D., Kidd, K., Stephens, G., and Delaronde, J.: Elevated mercury concentrations in fish in lakes in the Mackenzie River Basin: The role of physical, chemical, and biological factors, Sci. Total Environ., 351, 479–500, https://doi.org/10.1016/j.scitotenv.2004.12.086, 2005.
Faïn, X., Obrist, D., Pierce, A., Barth, C., Gustin, M. S., and Boyle, D. P.: Whole-watershed mercury balance at Sagehen Creek, Sierra Nevada, CA, Geochim. Cosmochim. Acta, 75, 2379–2392, 2011.
Ferrara, R., Maserti, B., Petrosino, A., and Bargagli: Mercury levels in rain and air and the subsequent washout mechanism in a central Italian region, Atmos. Environ., 20, 125–128, 1986.
Ferrara, R., Mazzolai, B., Lanzillotta, E., Nucaro, E., and Pirrone, N.: Volcanoes as emission sources of atmospheric mercury in the Mediterranean basin, Sci. Total Environ., 259, 115–121, 2000.
Friedli, H., Radke, L., and Lu, J.: Mercury in smoke from biomass fires, Geophys. Res. Lett., 28, 3223–3226, 2001.
Glass, G. E. and Sorensen, J. A.: Six-year trend (1990–1995) of wet mercury deposition in the Upper Midwest, USA, Environ. Sci. Technol., 33, 3303–3312, 1999.
Google Earth Engine: A planetary-scale geospatial analysis platform, available at: https://earthengine.google.com (last access: 24 August 2016), 2015.
Gustin, M. S., Lindberg, S. E., and Weisberg, P. J.: An update on the natural sources and sinks of atmospheric mercury, Appl. Geochem., 23, 482–493, 2008.
Helsel, D.: Statistics for Censored Environmental Data Using Mintab and R, 2nd Edition Ed., Wiley Publishing, 2012.
Iverfeldt, Å.: Mercury in forest canopy throughfall water and its relation to atmospheric deposition, Water Air Soil Pollut., 56, 553–564, 1991.
Jaeglé, L.: Atmospheric long-range transport and deposition of mercury to Alaska, Department of Atmospheric Sciences, University of Washington, Seattle, A report to the Alaska Department of Environmental Conservation, 2010.
Jiskra, M., Sonke, J. E., Obrist, D., Bieser, J., Ebinghaus, R., Myhre, C. L., Pfaffhuber, K. A., Wängberg, I., Kyllönen, K., and Worthy, D.: A vegetation control on seasonal variations in global atmospheric mercury concentrations, Nat. Geosci., 11, 244–250, 2018.
Karagas, M. R., Choi, A. L., Oken, E., Horvat, M., Schoeny, R., Kamai, E., Cowell, W., Grandjean, P., and Korrick, S.: Evidence on the Human Health Effects of Low-Level Methylmercury Exposure, Environ. Health Persp., 120, 799–806, https://doi.org/10.1289/Ehp.1104494, 2012.
Laird, B. D., Goncharov, A. B., Egeland, G. M., and Chan, H. M.: Dietary Advice on Inuit Traditional Food Use Needs to Balance Benefits and Risks of Mercury, Selenium, and n3 Fatty Acids, J. Nutr., 143, 923–930, https://doi.org/10.3945/jn.112.173351, 2013.
Lamborg, C. H., Fitzgerald, W. F., Vandal, G. M., and Rolfhus, K. R.: Atmospheric mercury in Northern Wisconsin: sources and species, Water Air Soil Pollut., 80, 189–198, 1995.
Landis, M. S., Vette, A. F., and Keeler, G. J.: Atmospheric Mercury in the Lake Michigan Basin:? Influence of the Chicago/Gary Urban Area, Environ. Sci. Technol., 36, 4508–4517, https://doi.org/10.1021/es011216j, 2002.
Lawson, N. M. and Mason, R. P.: Accumulation of mercury in estuarine food chains, Biogeochemistry, 40, 235–247, https://doi.org/10.1023/A:1005959211768, 1998.
Lee, K., Hur, S. D., Hou, S., Hong, S., Qin, X., Ren, J., Liu, Y., Rosman, K. J. R., Barbante, C., and Boutron, C. F.: Atmospheric pollution for trace elements in the remote high-altitude atmosphere in central Asia as recorded in snow from Mt. Qomolangma (Everest) of the Himalayas, Sci. Total Environ., 404, 171–181, https://doi.org/10.1016/j.scitotenv.2008.06.022, 2008.
Leitch, D. R., Carrie, J., Lean, D., Macdonald, R. W., Stern, G. A., and Wang, F. Y.: The delivery of mercury to the Beaufort Sea of the Arctic Ocean by the Mackenzie River, Sci. Total Environ., 373, 178–195, https://doi.org/10.1016/j.scitotenv.2006.10.041, 2007.
Lindberg, S. E., Brooks, S., Lin, C.-J., Scott, K. J., Landis, M. S., Stevens, R. K., Goodsite, M., and Richter, A.: Dynamic Oxidation of Gaseous Mercury in the Arctic Troposphere at Polar Sunrise, Environ. Sci. Technol., 36, 1245–1256, 2002.
Lopaka, L.: NADA: Nondetects And Data Analysis for environmental data, R package version 1.5-6, available at: https://CRAN.R-project.org/package=NADA (last access: 19 September 2016), 2013.
Loseto, L. L., Stern, G. A., and Ferguson, S. H.: Size and biomagnification: How habitat selection explains beluga mercury levels, Environ. Sci. Technol., 42, 3982–3988, https://doi.org/10.1021/Es7024388, 2008.
Lyman, S. N. and Gustin, M. S.: Speciation of atmospheric mercury at two sites in northern Nevada, USA, Atmos. Environ., 42, 927–939, 2008.
Macdonald, R. W. and Bewers, J. M.: Contaminants in the arctic marine environment: Priorities for protection, ICES J. Mar. Sci., 53, 537–563, https://doi.org/10.1006/jmsc.1996.0077, 1996.
Mason, R. P., Lawson, N. M., and Sullivan, K. A.: The concentration, speciation and sources of mercury in Chesapeake Bay precipitation, Atmos. Environ., 31, 3541–3550, 1997.
Mather, T. A. and Pyle, D. M.: Volcanic emissions of mercury to the atmosphere: global and regional inventories. Comment, Sci. Total Environ., 327, 323–329, https://doi.org/10.1016/j.scitotenv.2003.08.031, 2004.
Mitchell, C. P. J., Kolka, R. K., and Fraver, S.: Singular and Combined Effects of Blowdown, Salvage Logging, and Wildfire on Forest Floor and Soil Mercury Pools, Environ. Sci. Technol., 46, 7963–7970, https://doi.org/10.1021/Es300133h, 2012.
Munthe, J., Hultberg, H., and Iverfeldt, A.: Mechanisms of Deposition of Methylmercury and Mercury to Coniferous Forests, Water Air Soil Pollut., 80, 363–371, https://doi.org/10.1007/Bf01189686, 1995.
NADP (National Atmospheric Deposition Program): about the NADP concentration and deposition maps, available at: http://nadp.sws.uiuc.edu/data/mapProcess.aspx, last access: 24 August 2016.
Nriagu, J. and Becker, C.: Volcanic emissions of mercury to the atmosphere: global and regional inventories, Sci. Total Environ., 304, 3–12, 2003.
Obrist, D., Moosmueller, H., Schuermann, R., Chen, L. W. A., and Kreidenweis, S. M.: Particulate-phase and gaseous elemental mercury emissions during biomass combustion: Controlling factors and correlation with particulate matter emissions, Environ. Sci. Technol., 42, 721–727, https://doi.org/10.1021/es071279n, 2008.
Obrist, D., Agnan, Y., Jiskra, M., Olsen, C. L., Colegrove, D. P., Hueber, J., Moore, C. W., Sonke, J. E., and Helmig, D.: Tundra uptake of atmospheric elemental mercury drives arctic mercury pollution, Nature, 574, 201–204, 2017.
Obrist, D., Kirk, J. L., Zhang, L., Sunderland, E. M., Jiskra, M. and Selin, N. E.: A review of global environmental mercury processes in response to human and natural perturbations: Changes of emissions, climate, and land use, Ambio, 47, 116–140, 2018.
Outridge, P. M., Macdonald, R. W., Wang, F., Stern, G. A., and Dastoor, A. P.: A mass balance inventory of mercury in the Arctic Ocean, Environ. Chem., 5, 89–111, 2008.
Pacyna, J. M., Travnikov, O., De Simone, F., Hedgecock, I. M., Sundseth, K., Pacyna, E. G., Steenhuisen, F., Pirrone, N., Munthe, J., and Kindbom, K.: Current and future levels of mercury atmospheric pollution on a global scale, Atmos. Chem. Phys., 16, 12495–12511, https://doi.org/10.5194/acp-16-12495-2016, 2016.
Pirrone, N. and Mason, R.: Mercury fate and transport in the global atmosphere, Dordrecht, The Netherlands: Springer, 10, 978–970, 2009.
Poissant, L. and Pilote, M.: Mercury concentrations in single event precipitation in southern Quebec, Sci. Total Environ., 213, 65–72, 1998.
Polyakov, I. V., Alekseev, G. V., Bekryaev, R. V., Bhatt, U., Colony, R. L., Johnson, M. A., Karklin, V. P., Makshtas, A. P., Walsh, D., and Yulin, A. V.: Observationally based assessment of polar amplification of global warming, Geophys. Res. Lett., 29, 25-21–25-24, 2002.
Pyle, D. M. and Mather, T. A.: The importance of volcanic emissions for the global atmospheric mercury cycle, Atmos. Environ., 37, 5115–5124, 2003.
Rasmussen, R., Baker, B., Kochendorfer, J., Meyers, T., Landolt, S., Fischer, A. P., Black, J., Theriault, J. M., Kucera, P., Gochis, D., Smith, C., Nitu, R., Hall, M., Ikeda, K., and Gutmann, E.: HOW WELL ARE WE MEASURING SNOW? The NOAA/FAA/NCAR Winter Precipitation Test Bed, B. Am. Meteorol. Soc., 93, 811–829, https://doi.org/10.1175/Bams-D-11-00052.1, 2012.
R Core Team: A language and environment for statistical computing, available at: http://www.R-project.org/ (last access: 7 April 2019), 2014.
Revelle, W.: psych: Procedures for Personality and Psychological Research, Northwestern University, Evanston, Illinois, USA, available at: http://CRAN.R-project.org/package=psych Version=1.6.6 (last access: 19 September 2016), 2016.
Saha, S., Moorthi, S., Wu, X., Wang, J., Nadiga, S., Tripp, P., and Ek, M.: NCEP climate forecast system version 2 (CFSv2) 6-hourly products. Research Data Archive at the National Center for Atmospheric Research, Computational and Information Systems Laboratory, 2011.
Savina, M., Schäppi, B., Molnar, P., Burlando, P., and Sevruk, B.: Comparison of a tipping-bucket and electronic weighing precipitation gage for snowfall, Atmos. Res., 103, 45–51, https://doi.org/10.1016/j.atmosres.2011.06.010, 2012.
Schuur, E. A. and Abbott, B.: Climate change: High risk of permafrost thaw, Nature, 480, 32–33, 2011.
Selin, N. E. and Jacob, D. J.: Seasonal and spatial patterns of mercury wet deposition in the United States: Constraints on the contribution from North American anthropogenic sources, Atmos. Environ., 42, 5193–5204, https://doi.org/10.1016/j.atmosenv.2008.02.069, 2008.
Sheu, G. R.: Update on the APMMN and Progress on Wet Deposition Sample Analysis, 2017.
Steffen, A., Douglas, T., Amyot, M., Ariya, P., Aspmo, K., Berg, T., Bottenheim, J., Brooks, S., Cobbett, F., Dastoor, A., Dommergue, A., Ebinghaus, R., Ferrari, C., Gardfeldt, K., Goodsite, M. E., Lean, D., Poulain, A. J., Scherz, C., Skov, H., Sommar, J., and Temme, C.: A synthesis of atmospheric mercury depletion event chemistry in the atmosphere and snow, Atmos. Chem. Phys., 8, 1445–1482, https://doi.org/10.5194/acp-8-1445-2008, 2008.
Stow, J., Krummel, E., Leech, T., and Donaldson, S.: What is the impact of mercury contamination on human health in the Arctic, AMAP assessment 2011: mercury in the Arctic, 159–170, 2011.
Streets, D. G., Devane, M. K., Lu, Z. F., Bond, T. C., Sunderland, E. M., and Jacob, D. J.: All-Time Releases of Mercury to the Atmosphere from Human Activities, Environ. Sci. Technol., 45, 10485–10491, https://doi.org/10.1021/Es202765m, 2011.
Tan, S. W., Meiller, J. C., and Mahaffey, K. R.: The endocrine effects of mercury in humans and wildlife, Crit. Rev. Toxicol., 39, 228–269, https://doi.org/10.1080/10408440802233259, 2009.
Tchounwou, P. B., Yedjou, C. G., Patlolla, A. K., and Sutton, D. J.: Heavy metal toxicity and the environment, in: Molecular, clinical and environmental toxicology, Springer, 133–164, 2012.
Turetsky, M. R., Harden, J. W., Friedli, H. R., Flannigan, M., Payne, N., Crock, J., and Radke, L.: Wildfires threaten mercury stocks in northern soils, Geophys. Res. Lett., 33, 6 pp, https://doi.org/10.1029/2005GL025595, 2006.
UNEP: Global Mercury Assessment 2013: Sources, Emissions, Releases and Environmental Transport, 2013.
U.S. EPA (Environmental Protection Agency): Initial List of Hazardous Air Pollutants with Modifications, available at: https://www.epa.gov/haps/initial-list-hazardous-air-pollutants-modifications (last access: 22 August 2016), 2016.
U.S. EPA: Toxics Release Inventory 2014, available at: https://www.epa.gov/toxics-release-inventory-tri-program (last access: 27 August 2016), 2014.
Veysseyre, A., Moutard, K., Ferrari, C., Van de Velde, K., Barbante, C., Cozzi, G., Capodaglio, G., and Boutron, C.: Heavy metals in fresh snow collected at different altitudes in the Chamonix and Maurienne valleys, French Alps: initial results, Atmos. Environ., 35, 415–425, https://doi.org/10.1016/s1352-2310(00)00125-4, 2001.
Vonk, J. E., Tank, S. E., Bowden, W. B., Laurion, I., Vincent, W. F., Alekseychik, P., Amyot, M., Billet, M. F., Canário, J., Cory, R. M., Deshpande, B. N., Helbig, M., Jammet, M., Karlsson, J., Larouche, J., MacMillan, G., Rautio, M., Walter Anthony, K. M., and Wickland, K. P.: Reviews and syntheses: Effects of permafrost thaw on Arctic aquatic ecosystems, Biogeosciences, 12, 7129–7167, https://doi.org/10.5194/bg-12-7129-2015, 2015.
Walker, J. B., Houseman, J., Seddon, L., McMullen, E., Tofflemire, K., Mills, C., Corriveau, A., Weber, J. P., LeBlanc, A., Walker, M., Donaldson, S. G., and Van Oostdam, J.: Maternal and umbilical cord blood levels of mercury, lead, cadmium, and essential trace elements in Arctic Canada, Environ. Res., 100, 295–318, https://doi.org/10.1016/j.envres.2005.05.006, 2006.
Watras, C. J. and Bloom, N. S.: Mercury and Methylmercury in Individual Zooplankton – Implications for Bioaccumulation, Limnol. Oceanogr., 37, 1313–1318, 1992.
Webster, J. P., Kane, T. J., Obrist, D., Ryan, J. N., and Aiken, G. R.: Estimating mercury emissions resutlign from wildfires in forests of the Western United States, Sci. Total Environ., 568, 578–586, https://doi.org/10.1016/j.scitotenv.2016.01.166, 2016.
Wickham, H.: ggplot2: Elegant Graphics for Data Analysis, Springer-Verlag, New York, 2009.
Wiedinmyer, C., Quayle, B., Geron, C., Belote, A., McKenzie, D., Zhang, X., O'Neill, S., and Wynne, K.: Estimating emissions from fires in North America for air quality modeling, Atmos. Environ., 40, 3419–3432, 2006.
Wong, C. S., Duzgoren-Aydin, N. S., Aydin, A., and Wong, M. H.: Sources and trends of environmental mercury emissions in Asia, Sci. Total Environ., 368, 649–662, 2006.
Yang, D., Kane, D. L., Hinzman, L. D., Goodison, B. E., Metcalfe, J. R., Louie, P. Y. T., Leavesley, G. H., Emerson, D. G., and Hanson, C. L.: An evaluation of the Wyoming Gauge System for snowfall measurement, Water Resour. Res., 36, 2665–2677, https://doi.org/10.1029/2000WR900158, 2000.