the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 02 Apr 2019
| 02 Apr 2019
The distribution of sea-salt aerosol in the global troposphere
Daniel M. Murphy
Karl D. Froyd
Huisheng Bian
Charles A. Brock
Jack E. Dibb
Joshua P. DiGangi
Glenn Diskin
Maximillian Dollner
Agnieszka Kupc
Eric M. Scheuer
Gregory P. Schill
Bernadett Weinzierl
Christina J. Williamson
Pengfei Yu
We present the first data on the concentration of sea-salt aerosol throughout most of the depth of the troposphere and over a wide range of latitudes, which were obtained during the Atmospheric Tomography (ATom) mission. Sea-salt concentrations in the upper troposphere are very small, usually less than 10 ng per standard m3 (about 10 parts per trillion by mass) and often less than 1 ng m−3. This puts stringent limits on the contribution of sea-salt aerosol to halogen and nitric acid chemistry in the upper troposphere. Within broad regions the concentration of sea-salt aerosol is roughly proportional to water vapor, supporting a dominant role for wet scavenging in removing sea-salt aerosol from the atmosphere. Concentrations of sea-salt aerosol in the winter upper troposphere are not as low as in the summer and the tropics. This is mostly a consequence of less wet scavenging in the drier, colder winter atmosphere. There is also a source of sea-salt aerosol over pack ice that is distinct from that over open water. With a well-studied and widely distributed source, sea-salt aerosol provides an excellent test of wet scavenging and vertical transport of aerosols in chemical transport models.
- Article
(2724 KB) - Full-text XML
- BibTeX
- EndNote





Sea-salt particles are the largest aerosol component in the atmosphere by mass (Liao et al., 2006). They represent about 30 % of global column optical depth due to aerosols (Bellouin et al., 2013), a somewhat smaller percentage than for mass because of their relatively large size compared to other aerosols. Global climate models disagree on future changes in sea-salt aerosol due to changes in wind speed and sea ice in a warming climate (Liao et al., 2006; Jones et al., 2007; Boucher et al., 2013). Given the large contribution of sea salt to the global aerosol optical depth, this could represent a significant climate feedback, with uncertainty even between different scenarios in the same model (Hoose et al., 2008). Sulfuric acid, nitric acid, and some other acids can displace halogens in salt particles. This provides both a sink for sulfate and nitrate and a source of reactive chlorine, bromine, and iodine to the atmosphere (Chameides and Stelson, 1992; Finlayson-Pitts and Hemminger, 2000).
There is a large literature on the source of sea-salt aerosol as a function of wind speed (e.g., Gong, 2003; Lewis and Schwartz, 2004; Grythe et al., 2014). There has been increased recognition of the importance of submicron salt particles to aerosol number (Kreidenweis et al., 1998; Clarke et al., 2003, 2006). These submicron sea-salt particles are enriched in organics compared to sea water, although the amount of enrichment is not consistent and may vary under differing conditions (Middlebrook et al., 1998; Modini et al., 2010; Vignati et al., 2010; Ovadnevaite et al., 2011; Gantt and Meskhidze, 2013).
Almost all of the sea-salt aerosol literature considers measurements within the marine boundary layer and even there consists mostly of surface measurements. There have been few measurements of how sea salt varies with altitude. Shinozuka et al. (2004) presented profiles of nonvolatile aerosol, presumed to be sea salt, up to about 2 km altitude for one region in the Southern Ocean and one region in the tropical Pacific.
We present here the first measurements of the concentration of sea-salt aerosol over a wide range of altitudes and latitudes. We consider the sea-salt vertical transport, wet removal, and compositional variability. These data provide strong constraints on aerosol transport efficiency out of the marine boundary layer and are a useful tool in evaluating aerosol removal in large-scale models.
We quantify sea-salt aerosol by merging measured size distributions with the fraction of particles in each size range identified as sea salt by single particle mass spectrometry. Sea-salt particles were identified from mass spectra of single aerosol particles using the Particle Analysis by Laser Mass Spectrometry (PALMS) instrument (Thomson et al., 2000). Particles enter a vacuum and cross a split continuous laser beam. The transit time between the beams provides the particle velocity, used to determine its aerodynamic diameter. The aerosol inlet to PALMS is controlled to about 35 mbar, with a small dependence on outside pressure because the pressure transducer used for control was not positioned to capture the full effect of a jet downstream of the first critical orifice. Transit times were calibrated to known particle sizes at laboratory pressure (about 820 mbar) before and after every field deployment. An excimer laser is triggered when a particle arrives at the second laser beam and ions are produced when the 193 nm pulse hits the particle. Either positive or negative ions are analyzed with a time-of-flight mass spectrometer, with the polarity switched every few minutes. For these data, about 60 % of the time was spent acquiring positive ion spectra.
The PALMS instrument was flown on the NASA DC-8 during the Atmospheric Tomography Mission (ATom) as well as earlier missions. The ATom mission consisted of several series of flights between about 85∘ N and 65–80∘ S latitude over both the Pacific and Atlantic oceans. Flights consisted of successive en route ascents and descents from about 160 m to 12 km with 5 to 15 min at the bottom and top of each profile. An extensive payload characterized both gas-phase and aerosol species (ATom, 2017; https://daac.ornl.gov/ATOM/guides/ATom_merge.html, last access: 25 September 2018). Two deployments are considered here: ATom1 from 29 July to 22 August 2016, in northern summer, and ATom2 26 January to 21 February 2017, in northern winter. Some preliminary data from one ATom3 flight (14 October 2017) are included to extend the latitude range further south. During ATom1 and ATom2, PALMS acquired about 510 000 positive ion spectra and 350 000 negative ion spectra of individual particles.
Particle size distributions were measured by an ultra-high-sensitivity aerosol spectrometer (UHSAS; Kupc et al., 2018) and a laser aerosol spectrometer (LAS, TSI Incorporated). In the merged size distribution, UHSAS data were used for particles less than 0.51 µm diameter during ATom1 and 0.97 µm during ATom2; the LAS was used for larger particles. The two instruments agreed extremely well in the overlap region (about 0.4 to 0.97 µm) for all of ATom1 and at low altitudes during ATom2. A leak in the sheath flow of the LAS, traced to the threads of the set screws that center the inlet inside the sheath flow, led to the LAS under-sampling particles during ATom2 at high altitudes, when the aircraft cabin pressure was much higher than the sample pressure. The extra sheath flow reduced the sampling rate but did not introduce false counts, as checked occasionally in-flight with filtered air. A correction was derived by comparing the LAS to the UHSAS, a printed optical particle spectrometer (POPS), and cloud aerosol spectrometer (CAS) data.
The PALMS instrument has substantial biases in sampling efficiency for different size particles, with higher efficiency for particles about 1 to 3 µm diameter compared to about 300 nm by a factor of about 10 and steeply falling efficiency for sizes smaller than 300 nm. In addition, a custom virtual impactor in the PALMS sampling line deliberately enriched the concentration of super-micron particles in order to get better statistics for dust and sea salt. To some extent the bias toward sampling larger particles is beneficial because it enhances the rate of sampling particles with more mass, but it complicates interpretation. Overall, PALMS is much better at measuring fractional rather than absolute abundances of species such as sea salt. Combining such fractions with independently measured size distributions allows quantitative concentration measurements. Rather than directly calibrating the PALMS and virtual impactor sampling efficiencies, data here are normalized to the UHSAS and LAS particle size distributions. Conceptually, if a given percentage of, for example, 1 to 2 µm diameter particles are sea salt, then the absolute concentration can be determined by multiplying the concentration of particles of that size from the LAS by the percentage of sea salt determined by PALMS. Although simple in concept, this normalization is complicated in detail because the UHSAS and LAS measure optical diameter, which depends on refractive index, whereas PALMS measures aerodynamic diameter, which depends on density. The PALMS vacuum inlet is pressure-controlled so its transmission is mostly independent of aircraft altitude. A separate publication (Froyd et al., 2019) is in preparation with more details about the normalization.
PALMS, the LAS, and the UHSAS all sampled from a University of Hawaii inlet owned by NASA Langley on the DC-8 (McNaughton et al., 2007). About 1.5 m of 0.25 in. outside diameter stainless steel tubing with a usual volumetric flow rate of 3.5 to 7 L min−1 led to a custom virtual impactor with a 1 µm cut point mounted on the PALMS instrument. The virtual impactor design closely followed Loo and Cork (1988) except it was slightly scaled for our flow and desired cut point. After the virtual impactor about 20 cm of 0.125 in. stainless steel followed by 20 cm of 0.125 in. conductive Teflon tubing led to the PALMS focusing inlet. The volumetric flow after the virtual impactor was 0.7 L min−1. The tubing was at aircraft cabin temperature except that about the last 5 cm was taped to a heat pipe connected to the PALMS ion source region, which was temperature controlled at 35 ∘C. This slight warming to >25 ∘C was done to avoid condensation in the aerodynamic focusing vacuum inlet on PALMS at low altitudes in the tropics. With the inlet tubing warmer than outside the aircraft, the relative humidity in the inlet was less than 40 % for the majority of boundary layer sampling so the water content of the sea-salt particles was reduced. Correlations between aerodynamic diameter and light scattering suggest that the salt particles did not effloresce in the inlet. There was probably insufficient residence time for efflorescence even when the relative humidity in the inlet was very low. For sea-salt aerosol mass concentrations we assume that the sea-salt particles were deliquesced, except if the outside relative humidity was less than 35 %, when we assume they were dry. At low temperatures sea salt partially effloresces at about 40 % relative humidity (Koop et al., 2000).
The University of Hawaii inlet on the DC-8 has been shown to quantitatively transmit particles as large as 3.1 µm, at least at low altitude (McNaughton et al., 2007). Both the focusing inlet on PALMS and the LAS inlet tubing do not transmit particles larger than about 4 or 5 µm, so results here represent particles smaller than about 3 µm (dry geometric diameter). Note that the measured size range of ∼0.18 to 3 µm often represents a minority of sea-salt aerosol by mass, as indicated by the size distribution of large sea-salt particles detected by the cloud probes in the cloud-free marine boundary. When comparing to other data or model results, the size range must be considered.
In-cloud data are excluded from the results shown here. Huebert et al. (1990) showed that up to 90 % of the largest sea-salt particles in the marine boundary layer can deposit to the walls of an inlet. Cloud droplets or ice crystals impacting a forward-facing aircraft inlet can act like a high-pressure washer to dislodge some of that salt, potentially leading to large sea-salt artifacts in clouds. During both ATom and previous missions (Murphy et al., 2004), PALMS observed anomalous particles in clouds, both sea salt and other compositions, reinforcing the decision to exclude in-cloud data.

Figure 1Mass spectra of (a) a typical sea-salt particle and (b) a highly processed sea-salt particle where the chloride has been replaced by nitrate and sulfate. The spectrum in panel (a) is chosen to be similar to the average of all low-altitude spectra of sea salt.
Sea-salt particles were identified in the positive ion mass spectra using a combination of peaks, starting with a large Na+ peak (greater than at least 17 % or 30 % of the ion current from a particle, depending on other peaks in the spectrum). Identification also required a potassium peak of appropriate size. Particles were excluded as sea salt if they contained aluminum (from clay minerals), barium (from fly ash and other minerals), or a variety of other metals. A hierarchical cluster analysis (Murphy et al., 2003) was also used. Although the cluster analysis generated clusters of mass spectra that matched sea salt, criteria based on peak height were more reliable than the cluster analysis for identifying salt. Instead, the cluster analysis was most useful for excluding a few uncommon clusters that passed the peak height criteria but were not sea salt. For the great majority of sea-salt particles the identification was very clear. Figure 1 shows the mass spectrum of a single particle, chosen to be close to the average of all sea-salt mass spectra.
The main concern in identification is that at extremely low concentrations of sea-salt aerosol, as found over land or at high altitude, there may be a contribution from other particles, particularly dust from salt flats that is chemically similar to oceanic sea salt. A manual review was made of ∼100 particles automatically classified as sea salt and a similar number automatically classified as not sea salt. This was done for high-altitude coarse particles, the hardest region for the automatic classification. No definite sea-salt particles were missed and there was one definite false positive. More than 20 particles were borderline, mostly classified as sea salt by the automated algorithm. These were mostly spectra with sodium and potassium in a reasonable ratio for sea salt but magnesium at the wrong ratio. Note that if particles effloresce into inhomogeneous crystals, then the PALMS laser can sometimes preferentially ionize just one region of a particle with varying amounts of magnesium. There was a much smaller percentage of borderline identification in the marine boundary layer where higher humidity led to mostly liquid particles that ionize more uniformly. The Na+ peaks from sea-salt particles were often sufficiently large that they either saturated the detector or were broadened due to some sort of repulsion in the ion source. This tended not to affect the identification of a mass spectrum as sea salt but only the quantification of the Na peak.
Sea-salt particles can also be identified from negative ion spectra using chloride ions and cluster ions containing Na. Data in this paper are from positive ion spectra because in aged sea-salt particles the chloride can be almost entirely displaced by sulfate and nitrate, making identification more difficult with negative ions. In regions with fresh sea salt, results from negative ion spectra were nearly identical to those from positive ion spectra.
The number and mass of sea-salt particles were calculated for every 5 min of flight time in order to acquire enough mass spectra for a statistically significant normalization to the UHSAS and LAS size distributions. During climbs and descents, 5 min represents about 2.5 km in altitude. Mass spectra of sea-salt particles were typically acquired at a rate of more than one per second in the marine boundary layer and less than one per minute at high altitude.
Figure 2 shows concentrations of sea-salt aerosol at low altitudes measured by the PALMS and LAS combination compared to filter measurements of sodium (Dibb et al., 1999). The filter samples indicate more sea-salt mass, which is expected because the inlet to the filter sampler transmitted larger particles than the inlet to PALMS. The good correlation adds confidence to the PALMS measurements in the upper troposphere, which are much more sensitive than the filter sampler. Because PALMS samples each particle at a particular time, its composition can be associated with a particular altitude, aerosol concentration, water vapor concentration, and so forth. This allows for very long averaging times in similar air. For example, the average concentration of sea-salt aerosol in air with 10 to 20 ppmv of water can be calculated from collecting many such stretches of flight data even though they are not contiguous in time and might even be on different days. Furthermore, the PALMS single particle data are easier to screen for short periods of cloud and other artifacts than the extended filter samples are. One can eliminate short cloud encounters without losing the data before and after the cloud. The internal consistency of the data suggests that the detection limit for sea salt is better than 10 ng m−3 over a few minutes and better than 1 ng m−3 when hours of data are available. Exact detection limits depend on the size distribution and the amount of internal mixing. In favorable circumstances the detection limits can be ≪1 ng m−3.
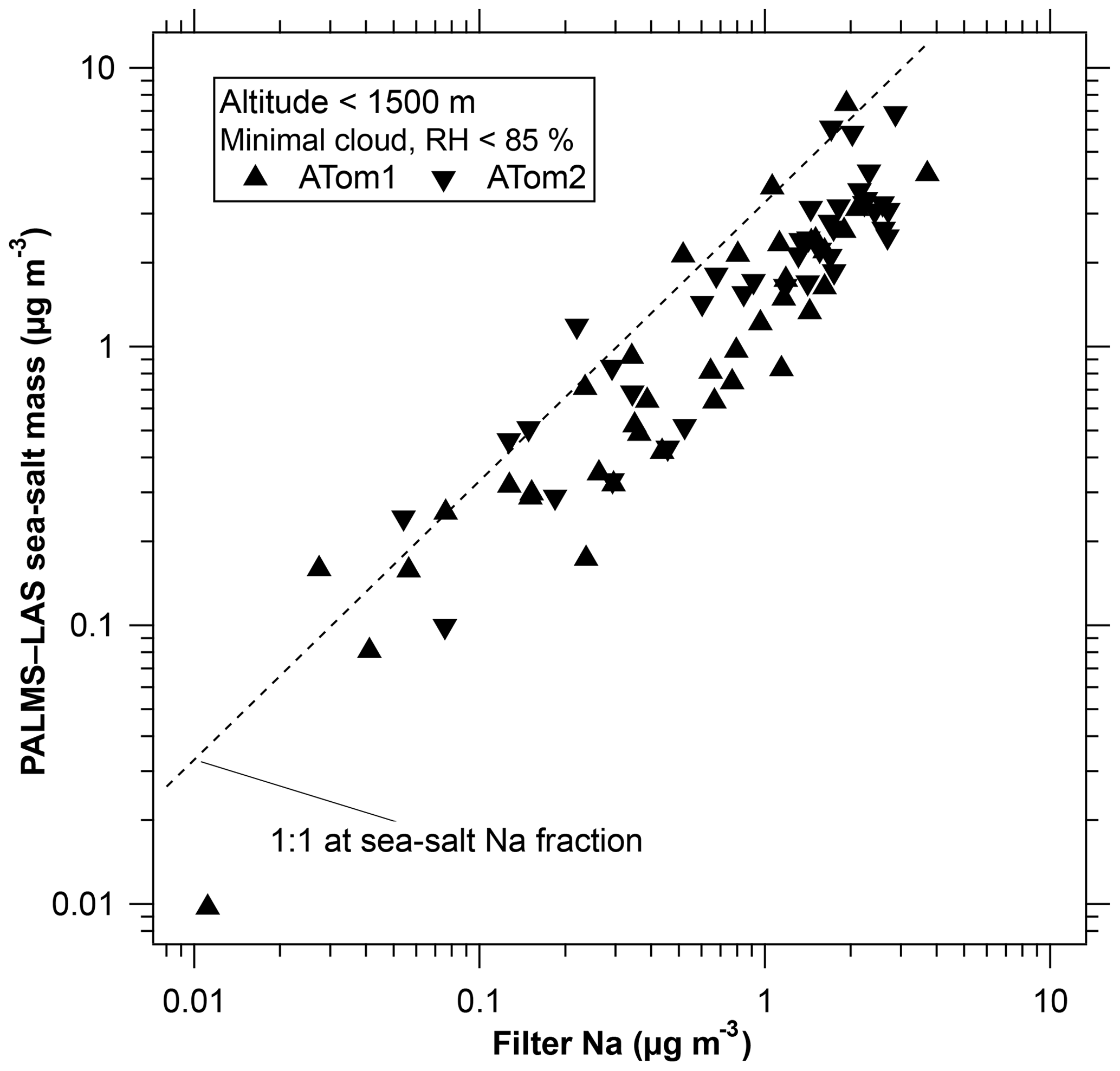
Figure 2Sea-salt aerosol mass at low altitudes derived from PALMS compared to filter samples of aerosol sodium. The inlet for the filter sampler transmits larger particles than the PALMS inlet; hence, PALMS is expected to sample somewhat less mass. The 1 : 1 line is displaced because not all sea-salt mass is sodium. The cutoff at 85 % relative humidity is imposed because comparing the different inlet cut points becomes especially problematic when the particles are enlarged due to water uptake at high humidity.
The concentration of sea-salt aerosol in the marine boundary layer measured by PALMS was usually between 0.3 and 3 µg m−3. Such concentrations are reasonable considering the upper cut point of about 3 µm diameter. The concentrations in the marine boundary layer were highly variable and modestly positively correlated with the local wind speed at flight altitude. This is consistent with Shinozuka et al. (2004). Although the sea-salt aerosol production rate can be closely related to wind speed, the correlation with local wind speed is modest because the concentration also depends on the wind fetch and whether or not there has been recent precipitation. Sea-salt aerosol concentrations near the surface were also correlated with relative humidity. This is expected since low relative humidity would indicate that the air was not well mixed from the ocean surface up to the altitude of the DC-8. For example, during one boundary layer sampling leg over the ocean there was almost no sea salt at 160 m altitude, but the relative humidity was much less than 50 %, indicating limited surface influence for that particular leg.
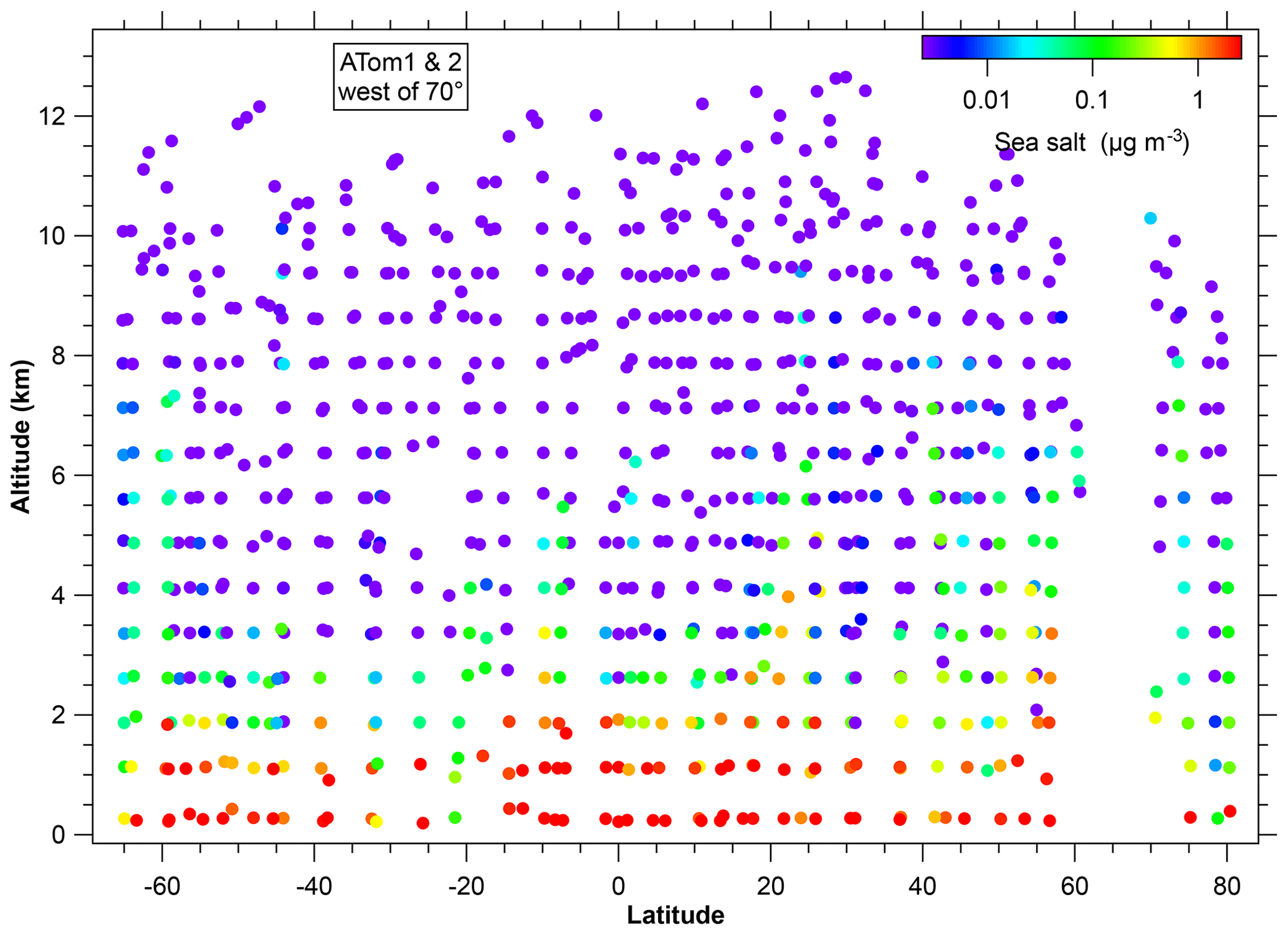
Figure 3Concentration of sea-salt aerosol during the ATom1 and ATom2 flights over the Arctic, Pacific, and Southern oceans. The blank region between about 60 to 70∘ N is because of flying over land (Alaska). The color scale is from 2.5 ng m−3 to 2.5 µg m−3 (at standard conditions). These concentrations include sea-salt particles between about 180 nm and 3 µm diameter. Data are averaged over bins of 750 m vertically and about 4∘ latitude; points are plotted at the midpoint location of the data within each bin.
There are already extensive measurements of sea-salt aerosol in the marine boundary layer (Lewis and Schwartz, 2004). The novel data here are the concentrations at higher altitudes. Figure 3 shows a latitude–altitude cross section of sea-salt aerosol concentrations over the Pacific Ocean. Even though they were in different seasons, systematic differences between ATom1 and ATom2 are not visible on a log color scale so they are combined in Fig. 3. More subtle differences depending on season are discussed below. The Pacific Ocean is shown because there was less mineral dust to complicate the analysis of very low concentrations; concentrations over the Atlantic Ocean were similar. Shinozuka et al. (2004) inferred sea-salt aerosol concentrations from nonvolatile particles. The results shown here agree that this was valid for their measurements at <2 km over the Pacific and Southern oceans. However, it is not true in general that most nonvolatile particles are sea salt. For example, at higher altitudes (e.g., 10 km) than Shinozuka et al. (2004) measured, nonvolatile particles would be many times more likely to be dust than sea salt, even over the middle of the ocean. Dust concentrations measured during ATom will be discussed in future publications.
A salient property of the distribution of sea-salt aerosol in Fig. 3 is that the concentration falls off rapidly with altitude, by about a factor of 10 for every 2 km. Above 6 km, the concentrations were almost always less than 10 ng m−3. The decrease at the top of the marine boundary layer was often very sharp in individual profiles. This was more apparent on past missions because the ATom mission ascents were relatively rapid. During the 2002 Intercontinental Transport and Chemical Transformation Experiment (ITCT) off the coast of California, PALMS was on the NOAA P-3 that often flew just above and below the top of the boundary layer. There, the sea-salt aerosol concentration could change by more than a factor of 10 in less than 100 m altitude at the top of the marine boundary layer. The few sea-salt particles in the upper troposphere were similar in size and more chemically processed than those in the marine boundary layer.
Figure 4 shows size distributions of sea-salt particles from PALMS during ATom1. Most of the sea-salt mass is in particles larger than 1 µm, most of the number is smaller than 1 µm. In the lower panel the size distribution from the upper troposphere is multiplied by 500 to get it on the same scale as the size distribution in the marine boundary layer, emphasizing the strong removal of sea salt. The number distribution is shown from the Southern Hemisphere (SH) because for submicron particles in the Northern Hemisphere (NH) both the number of particles and the fraction of sea salt are very steep functions of diameter, leading to large uncertainties when they are multiplied. The mass distributions and the Southern Hemisphere number distribution are less steep. The high and low altitude size distributions (Fig. 4b) show some differences of a factor of ∼3 as a function of diameter. Such detailed differences in the size distribution with altitude vary by region. We would emphasize instead that an overall large removal factor is present across the entire 0.3 to 3 µm diameter range. This may indicate that much of the removal of sea-salt aerosol was by nucleation scavenging in cloud rather than impaction by precipitation. Particles large enough to be measured by PALMS are all large enough to be condensation nuclei, whereas impaction scavenging is much more efficient for coarse particles than submicron particles (Pruppacher and Klett, 1997).
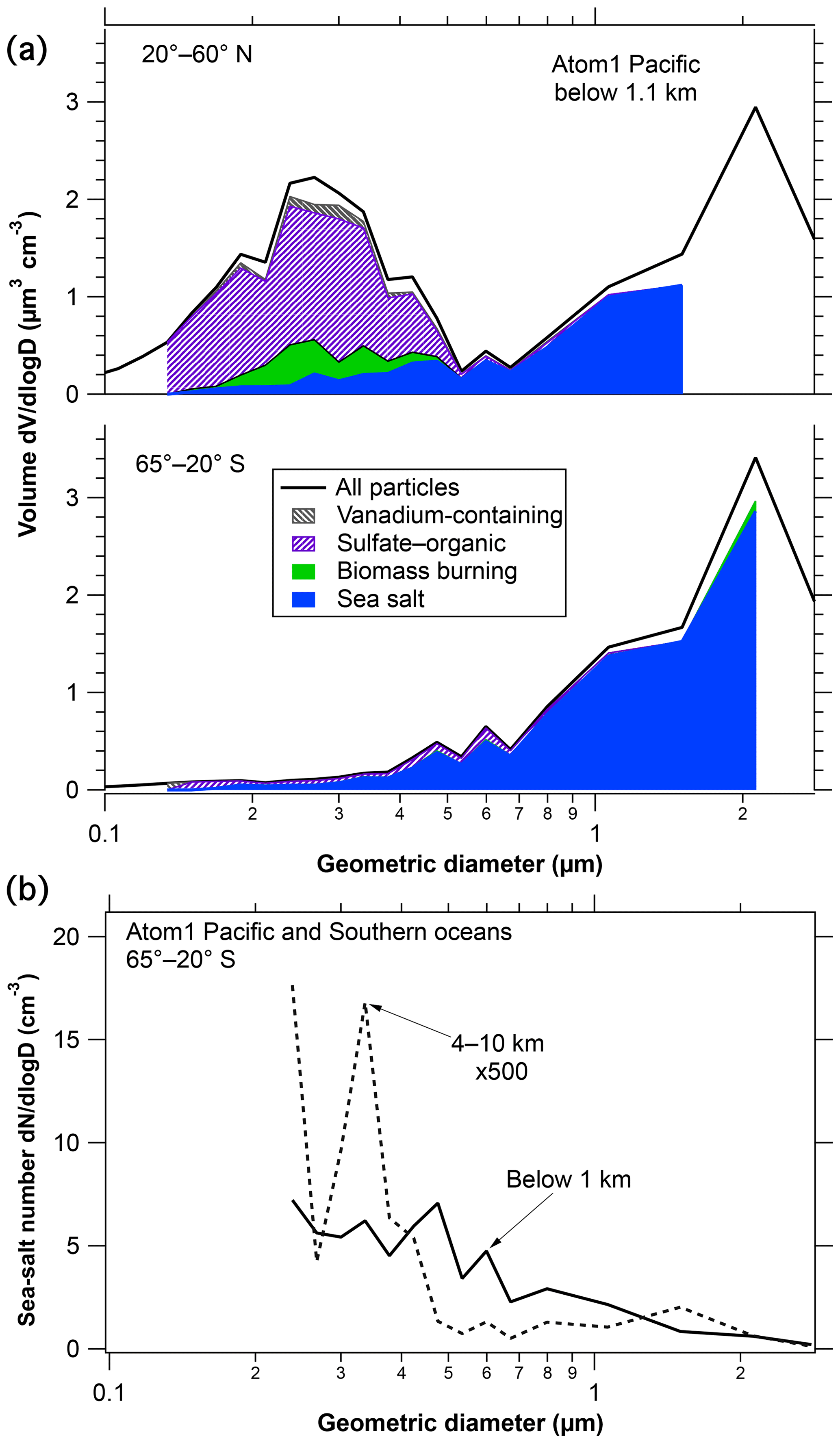
Figure 4Size distributions of sea-salt particles in the marine boundary layer. Panel (a) shows how the total size distribution (black line) is multiplied by PALMS composition at each size to obtain the volume of sea-salt particles. White areas below the size distribution curves represent unknown compositions or, at the largest and smallest diameters, insufficient data to assign composition. The reduced concentration above about 3 µm diameter is because the aircraft inlet does not efficiently transmit larger particles. Panel (b) compares the number size distributions at low and high altitudes.
3.1 Sea-salt aerosol over pack ice
Sea-salt aerosols were not confined to areas with open water. Significant concentrations of sea-salt aerosol were also observed over ice-covered regions of the Arctic Ocean during ATom. For a portion of a flight north of Alaska on 19 February 2017, the nearest large areas of open water were about 1000 km away. Yet significant concentrations of sea-salt aerosol (∼1 µg m−3 below 3 µm diameter) were observed at altitudes below 400 m. The sea-salt aerosol was only at low altitude, supporting a local source. Similar concentrations were also observed by PALMS on flights north of Alaska during the Aerosol, Radiation, and Cloud Processes affecting Arctic Climate (ARCPAC) mission in March–April 2008. Significant sea-salt aerosol concentrations have also been measured during winter at ground-based Arctic stations (Quinn et al., 2002). Data from Utqiaġvik (formerly Barrow), Alaska, indicate some sea-salt aerosol production from leads but also show high concentrations of sea salt at times with no nearby open water in leads (May et al., 2016).
Mass spectra of sea-salt particles over the ice-covered Arctic Ocean were depleted in Na relative to Mg, K, and Ca compared to particles at lower latitudes (Fig. 5). The Na depletion over the Arctic was due to an increased population of particles with low Na rather than every particle having less Na. It is robust in the following sense: we used the NH Pacific as a reference case for ATom2 because that is the region with the most data in the marine boundary layer. We then compared data from other ocean regions with the NH Pacific. The magnitude of the difference in the average Na signal between the Arctic Ocean and the NH Pacific was much larger than the differences between the NH Pacific and any of the other open ocean regions, showing that the Arctic sea-salt aerosol is distinct from open ocean sea-salt aerosol.
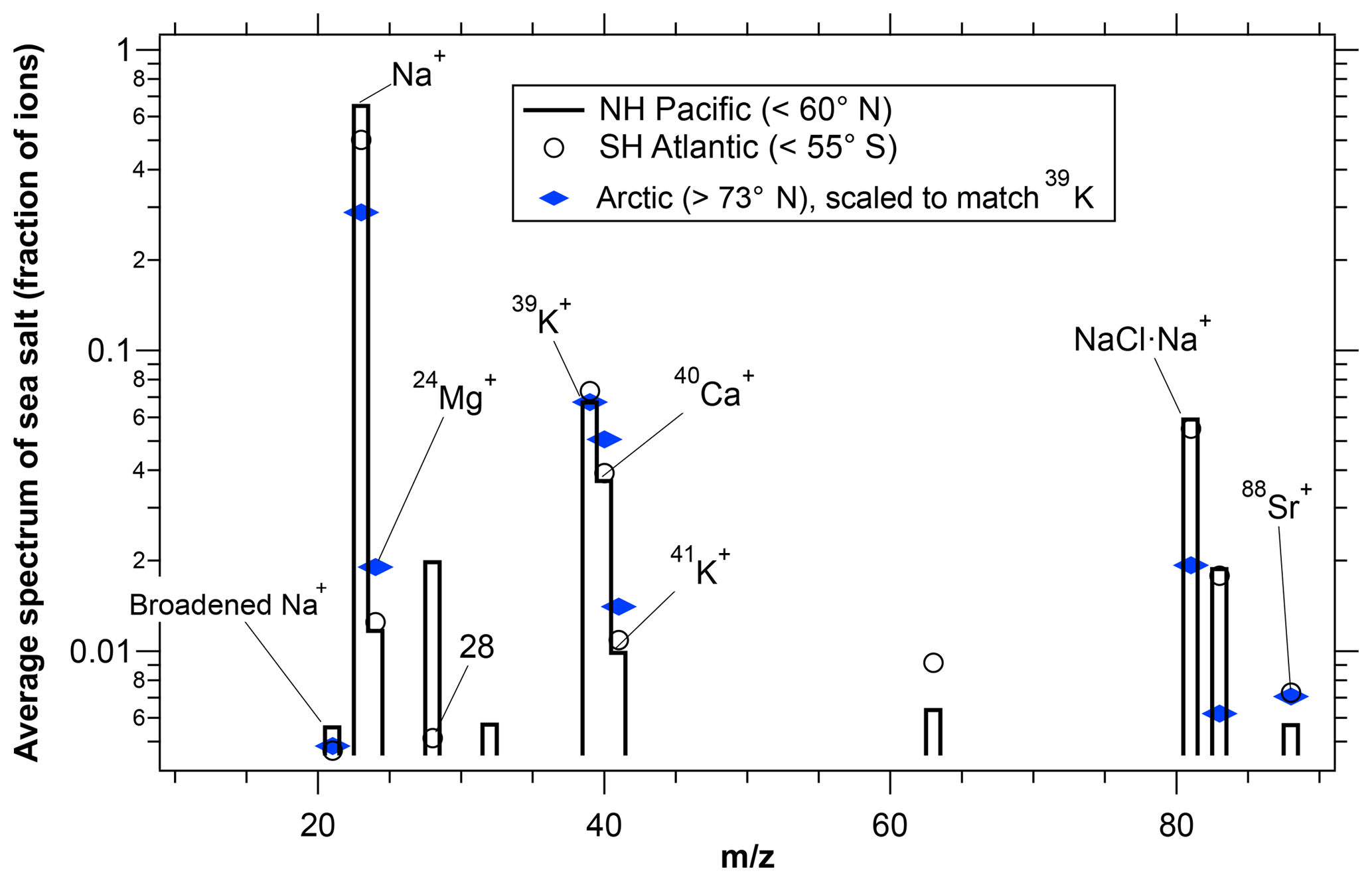
Figure 5Average spectra of sea-salt particles larger than 1 µm diameter for the Arctic and lower latitude regions during ATom2. Na peaks were smaller over the Arctic Ocean than over the Pacific Ocean in the Northern Hemisphere . The South Atlantic is included to provide an estimate of consistency between regions that should be similar. Spectra were selected that have ion intensities large enough to measure minor peaks but that will not significantly saturate the Na peak. n≈5500 for the NH Pacific, 700 for the SH Atlantic, and 300 for the Arctic.
Similar Na depletion was also observed during ARCPAC when comparing mass spectra of particles over Arctic Ocean compared to test flights over the Gulf of Mexico. In contrast, significant Na depletion over the Arctic Ocean was not observed during ATom1, which was flown during August when much of the Arctic Ocean had some open water. Unfortunately, occasional detector saturation by the Na+ peak, as well as changing ionization patterns (such as the presence or absence of cluster ions), makes it impossible to be more precise about the amount of Na depletion other than to say it was between about 20 % and 50 %. During both ARCPAC and ATom, sea-salt particles were also somewhat smaller over the ice-covered Arctic Ocean than other regions.
Na depletion in polar sea-salt aerosol is consistent with Wagenbach et al. (1998) and Hara et al. (2012), who found ≥10 % depletion of Na relative to Mg and K in sea-salt aerosol presumed to be formed from sea ice around Antarctica. The PALMS data support the idea that some form of ice brine, whether it be frost flowers and/or blowing briny snow, is an important source of sea-salt aerosol in the Arctic and Antarctic (Domine et al., 2004; Alvarez-Aviles et al., 2008; Huang and Jaeglé, 2017).
3.2 Sea-salt aerosol as a diagnostic for aerosol removal
Simple altitude profiles of PALMS sea-salt aerosol in various latitude bands will be presented in model–measurement comparison papers (Yu et al., 2019; Bian et al., 2019; Zhang et al., 2019). An alternative way of presenting the concentration of sea-salt aerosol is as a correlation with water vapor (Fig. 6). Sea salt is water soluble, so one might expect that its removal would approximately scale with removal of water via precipitation. Figure 6 shows that this is the case, at least when considered as an average over many vertical profiles. Because the concentration of water vapor falls off rapidly with altitude, the correlations in Fig. 6 are in a sense vertical profiles. However, the concentration of sea-salt aerosol is often better correlated with water vapor than with altitude itself.
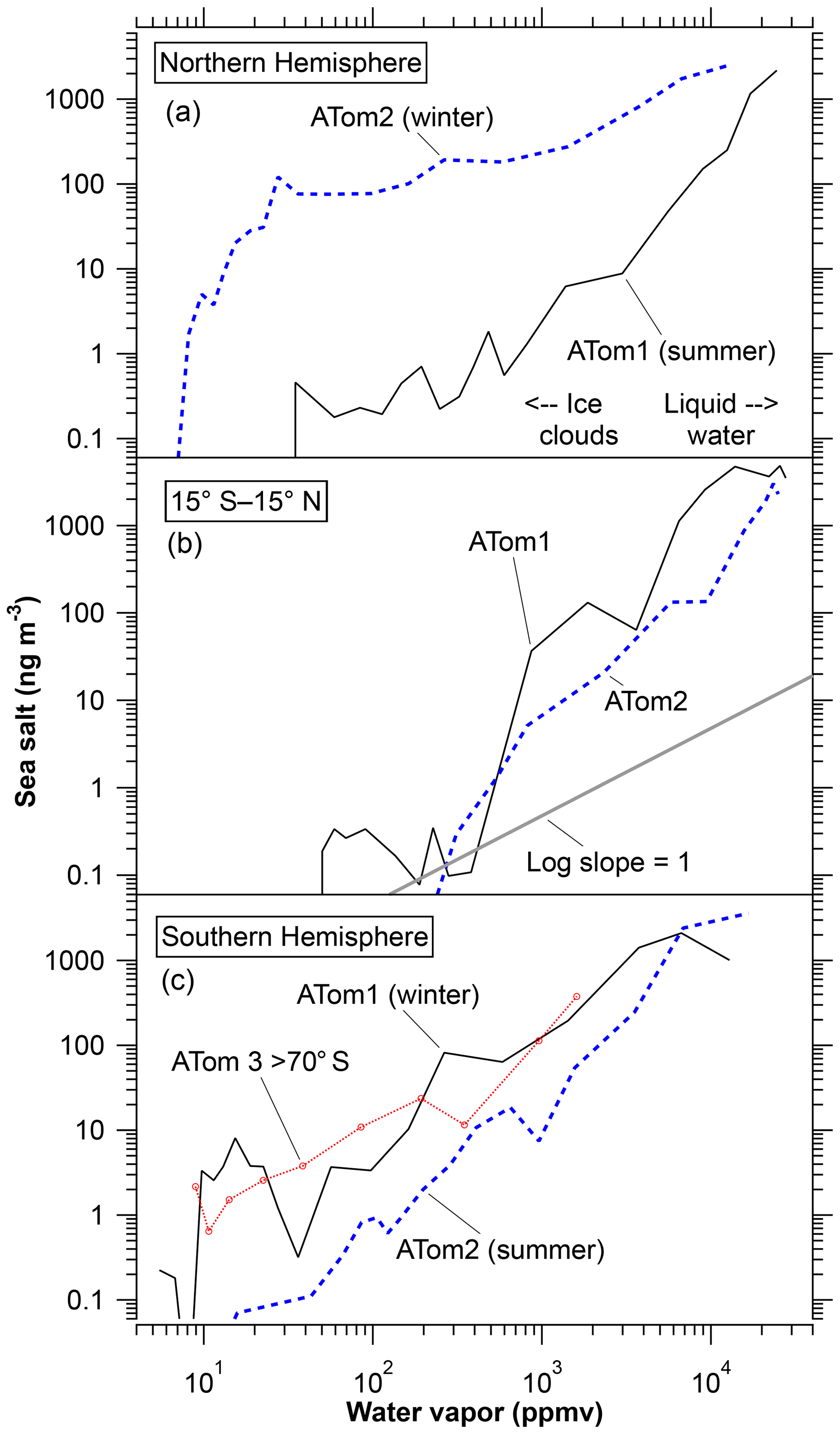
Figure 6Measured average sea-salt mass concentrations as a function of water vapor. Latitude bands for the Northern and Southern hemispheres are 20 to 65∘ except for the labeled data over Antarctica. All data except those beyond 70∘ S are from the Pacific Ocean side of North and South America. Arrows in panel (a) show approximate water mixing ratios above which most clouds are liquid water and below which most clouds are ice. One way to view the graph is to consider water vapor as a vertical scale with wet air at low altitudes and dry air at high altitudes. In both hemispheres the winter data show more sea-salt aerosol in the upper troposphere at a given amount of water vapor than either the summer hemisphere or the tropics.
The log–log slopes are not far from one, indicating that sea-salt aerosol is removed with water: when 90 % of the water is removed about 90 % of the sea-salt aerosol is removed. Labels indicate water mixing ratios beyond which most clouds are ice or most are liquid water, based on saturation vapor pressures of −15 to −5 ∘C at 850 to 500 mbar. There is some indication, especially in the Northern Hemisphere data, that sea-salt aerosol removal is more efficient in liquid water clouds than in ice clouds. The log slopes are greater than 1 at high water mixing ratios and close to or less than 1 at low water mixing ratios. Some proportionality between sea-salt aerosol and water vapor continues to very low water mixing ratios where the clouds must be ice rather than liquid water. This is somewhat surprising, since the Bergeron process whereby ice crystals grow at the expense of water droplets in a mixed-phase cloud could leave many salt particles behind even if those particles had originally acted as condensation nuclei. There are several possible explanations for the continued removal of sea-salt aerosol at very low temperatures. Sodium chloride dihydrate can be an effective ice nucleus below 227 K (Wagner and Möhler, 2013). Nonspherical ice crystals can scavenge aerosols by impaction more efficiently than spherical droplets (Pruppacher and Klett, 1997, chap. 17) A less microphysical explanation could be that the driest air in the upper troposphere is the result of very deep convection. If such intense convective clouds scavenge nearly all sea-salt particles, then the observed correlation of sea-salt aerosol with water vapor at low concentrations could be due to mixing very dry air with nearly all of sea-salt aerosol removed with mid-tropospheric air containing more of both water and salt.
In many ways Fig. 6 shows a remarkably simple picture of sea-salt aerosol concentrations by season and hemisphere. Since the ATom1 and ATom2 deployments were roughly 6 months apart and covered both hemispheres, it is possible to distinguish seasonal and hemispheric differences. Concentrations in the tropical atmosphere show little seasonal dependence. The two summer hemispheres are fairly similar to the tropics. The two winter curves are shifted up and to the left. Especially in the Northern Hemisphere, the sea-salt concentration near the ocean surface (at the top right of each curve) is not all that different in winter and summer. Instead, a similar amount of sea-salt aerosol is emitted into a lower absolute humidity in the colder winter air. This suggests two reasons more sea-salt aerosol can reach the upper troposphere in winter than in summer. The main reason is that more sea-salt particles can survive into the upper troposphere in winter simply because in winter there is less water available to wash out the aerosol. Second, removal of sea-salt aerosol in ice clouds may be less efficient than in liquid water clouds.
Model results for the correlation between sea-salt aerosol and water vapor are shown in Fig. 7. For simplicity only a subset of the regions in Fig. 6 are shown. The Community Earth System Model with the Continuous-time Autoregressive Moving Average (CESM-CARMA) couples a sectional aerosol model (Yu et al., 2015; Toon et al., 1988) with the National Science Foundation and Department of Energy CESM. CARMA uses 20 size bins for sea spray aerosols which are composed of salt, marine sulfate, and marine organics. The Goddard Earth Observing System Model version 5 (GEOS-5) simulates meteorological fields to drive the online Goddard Chemistry Aerosol Radiation and Transport (GOCART) aerosol model (Colarco et al., 2010; Bian et al., 2013). GOCART sea-salt aerosol is emitted using an upgraded emission algorithm (Gong, 2003; Bian et al., 2019) and removed by warm cloud from convective updraft and large-scale rainout and washout, as well as by dry deposition and sedimentation (Chin et al., 2002). A humidified sea-salt particle size (Gerber, 1985) is used for computations of particle fall velocity, deposition velocity, and optical parameters. The detailed description of the GEOS-5–GOCART sea-salt aerosol simulation for this work is given in Bian et al. (2019).
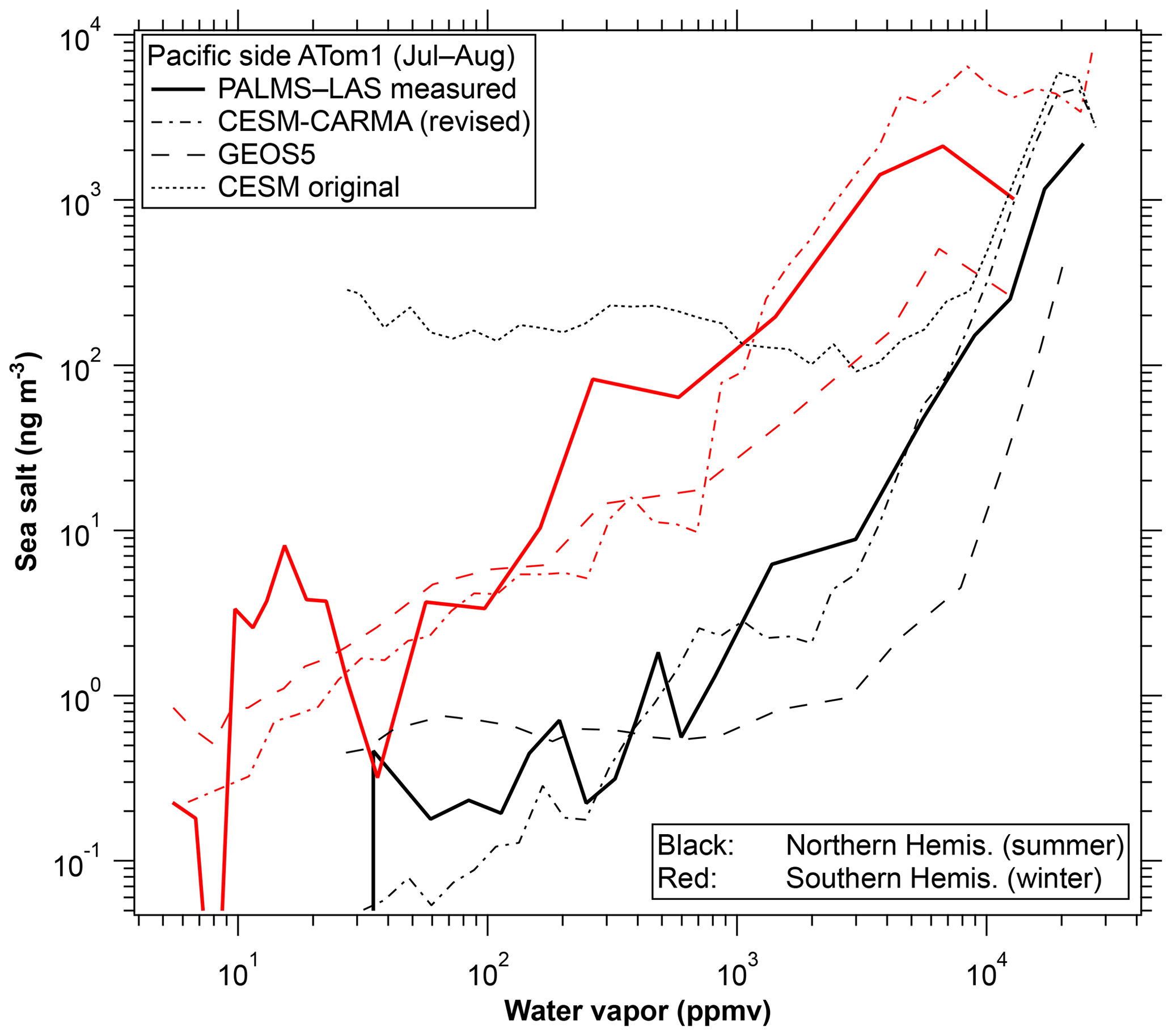
Figure 7Model correlations for the CESM-CARMA and GEOS-5 models. ATom2 and tropical curves are omitted for simplicity. GEOS-5 model output was sampled along the flight tracks and CESM-CARMA at all altitudes in a curtain along the flight tracks. Also shown is one curve from the CESM-CARMA model before a revised convective aerosol removal scheme was implemented (Yu et al., 2019).
Both models reproduce the strong correlation between sea-salt aerosol and water vapor. Both models also capture the difference between the winter and summer hemispheres in the correlation. The GEOS-5 model may be removing aerosol too slowly in ice clouds. The comparison to these data uncovered an error in aerosol removal in the CESM model in which sea-salt aerosol was originally overestimated by a factor of over 100 in the upper troposphere. One example is shown in Fig. 7. The overestimate was traced to aerosols not being properly removed from air transported in the sub-grid convective parameterization. A detailed analysis and new removal parameterization are described by Yu et al. (2019). An interaction between removal and entrainment parameterizations was also identified as an issue in the Community Atmosphere Model version 5 (CAM5) by Wang et al. (2013), based on black carbon data in the upper troposphere. The CARMA bin microphysics also reproduces the similar shape of the size distribution of sea-salt aerosol at different altitudes (Fig. 4b). In the model, only particles larger than about 3 µm decrease strongly with altitude due to gravitational sedimentation.
3.3 Implications for reactive gases
One consequence of the small concentrations of sea-salt aerosol in the upper troposphere is that it contributes little to chemical reactivity. In particular, Wang et al. (2015) postulated an important role for upward transport of sea-salt aerosol followed by release of bromine into the upper troposphere. These data show that de-bromination of sea salt cannot be a significant source in the upper troposphere. There was almost always less than 10 ppt of sea-salt aerosol by mass and often less than 1 ppt. Given that sea salt is very roughly 0.1 % by mole bromine, there would be parts per quadrillion of bromine available from transported sea-salt aerosol. If bromine from sea-salt aerosol is to significantly affect the upper troposphere, it would have to be released at low altitude and transported in the gas phase, although it is not clear if there are any suitable gas-phase bromine compounds that would survive wet scavenging.
The small concentrations of sea-salt aerosol in the upper troposphere also provide a strong constraint on the influence of salt on the gas-phase reactive-nitrogen budget. Even complete replacement at altitude of sea-salt chlorine by nitrate would be a very small sink for nitrate compared to the hundreds of pptv of NOy in the upper troposphere (Weinheimer et al., 1994; Emmons et al., 1997). NOy measurements are available as part of the ATom data set (ATom, 2017). A similar argument applies to sulfate: in the upper troposphere the maximum amount of sulfate in sea-salt aerosol is far less than the amount of sulfate in mixed sulfate and organic particles. Note that the minimal chemical importance of sea-salt aerosol is for the upper troposphere only: in the marine boundary layer reactions with sea-salt particles can significantly modify the gas-phase bromine, nitrate, and sulfate budgets.
These are the first measurements of sea-salt aerosol over a wide range of altitudes and latitudes. Data are available from near the surface to about 12 km altitude from about 65∘ S to 80∘ N. The final ATom data set will add two more seasons and extend the data to beyond 80∘ S. Detailed comparisons with chemical transport models are underway (Bian et al., 2019; Zhang et al., 2019). One of these comparisons identified a problem in aerosol vertical transport in the CESM model (Yu et al., 2019).
Sea-salt aerosol's only source is at the surface and its only sink is by scavenging (i.e., sea-salt particles do not evaporate). That makes sea-salt aerosol a powerful tool to study wet removal of aerosol. The data here indicate that removal of sea-salt aerosol is very approximately proportional to the removal of water over a wide range of absolute humidity, with possibly more efficient removal in liquid water clouds than in ice clouds.
Data are publicly available at https://doi.org/10.5067/Aircraft/ATom/TraceGas_Aerosol_Global_Distribution (ATom, 2017) and http://esrl.noaa.gov/csd/projects/atom/data.php (last access: 28 March 2019).
KDF, GPS, and DMM provided PALMS data. CAB, AK, MD, BW, and CJW provided size distribution data. JED and EMS provided filter data. JPD and GD provided water vapor data. HB and PY provided model results. DMM wrote the paper with assistance from all authors.
The authors declare that they have no conflict of interest.
The participation of PALMS in the Atmospheric Tomography Mission flights (ATom1 and ATom2) was supported by NOAA climate funding. The mission as a whole was supported by NASA's Earth System Science Pathfinder Program EVS-2 funding. Charles A. Brock and Christina J. Williamson were supported by award NNH15AB12I and by NOAA's Health of the Atmosphere and Atmospheric Chemistry, Carbon Cycle, and Climate Programs. Agnieszka Kupc was supported by the Austrian Science Fund's Erwin Schrodinger Fellowship J-3613. The CESM project is supported by the National Science Foundation and the Office of Science (BER) of the US Department of Energy. Bernadett Weinzierl and Maximillian Dollner have received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program under grant agreement no. 640458 (A-LIFE) and from the University of Vienna. We thank the ATom team and crews for making the flights possible, and Bruce Anderson of NASA Langley for the use of the University of Hawaii inlet.
This paper was edited by Yafang Cheng and reviewed by two anonymous referees.
Alvarez-Aviles, L., Simpson, W. R., Douglas, T. A., Sturm, M., Perovich, D., and Domine, F.: Frost flower chemical composition during growth and its implications for aerosol production and bromine activation, J. Geophys. Res., 113, D21304, https://doi.org/10.1029/2008JD010277, 2008.
ATom: Measurements and modeling results from the NASA Atmospheric Tomography Mission, https://doi.org/10.5067/Aircraft/ATom/TraceGas_Aerosol_Global_Distribution, 2017.
Bellouin, N., Quaas, J., Morcrette, J.-J., and Boucher, O.: Estimates of aerosol radiative forcing from the MACC re-analysis, Atmos. Chem. Phys., 13, 2045–2062, https://doi.org/10.5194/acp-13-2045-2013, 2013.
Bian, H., Colarco, P. R., Chin, M., Chen, G., Rodriguez, J. M., Liang, Q., Blake, D., Chu, D. A., da Silva, A., Darmenov, A. S., Diskin, G., Fuelberg, H. E., Huey, G., Kondo, Y., Nielsen, J. E., Pan, X., and Wisthaler, A.: Source attributions of pollution to the Western Arctic during the NASA ARCTAS field campaign, Atmos. Chem. Phys., 13, 4707–4721, https://doi.org/10.5194/acp-13-4707-2013, 2013.
Bian, H., Froyd, K., Murphy, D. M., Dibb, J., Chin, M., Colarco, P. R., Darmenov, A., da Silva, A., Kucsera, T. L., Schill, G., Yu, H., Bui, P., Dollner, M., Weinzierl, B., and Smirnov, A.: Observationally constrained analysis of sea salt aerosol in the marine atmosphere, Atmos. Chem. Phys. Discuss., https://doi.org/10.5194/acp-2019-18, in review, 2019.
Boucher, O., Randall, D., Artaxo, P., Bretherton, C., Feingold, G., Forster, P., Kerminen, V.-M., Kondo, Y., Liao, H., Lohmann, U., Rasch, P., Satheesh, S. K., Sherwood, S., Stevens B., and Zhang, X. Y.: Clouds and Aerosols, in: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, edited by: Stocker, T. F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S. K., Boschung, J., Nauels, A., Xia, Y., Bex, V., and Midgley, P. M., Cambridge University Press, Cambridge, UK and New York, NY, USA, 2013.
Chameides, W. L. and Stelson, A. W.: Aqueous-phase chemical processes in deliquescent sea-salt aerosols: A mechanism that couples the atmospheric cycles of S and sea salt, J. Geophys. Res., 97, 20565–20580, 1992.
Chin, M., Ginoux, P., Kinne, S., Torres, O., Holben, B. N., Duncan, B. N., Martin, R. V., Logan, J. A., Higurashi, A., and Nakajima, T.: Tropospheric aerosol optical thickness from the GOCART model and comparisons with satellite and sun photometer measurements, J. Atmos. Sci., 59, 461–483, 2002.
Clarke, A., Kapustin, V. Howell, S., Moore, K., Lienert, B., Masonis, S., Anderson, T., and Covert, D.: Sea-salt size distributions from breaking waves: Implications for marine aerosol production and optical extinction measurements during SEAS, J. Atmos. Ocean. Tech., 20, 1362–1374, 2003.
Clarke, A. D., Owens, S. R., and Zhou, Z.: An ultrafine sea-salt flux from breaking waves: Implications for cloud condensation nuclei in the remote marine atmosphere, J. Geophys. Res., 111, D06202, https://doi.org/10.1029/2005JD006565, 2006.
Colarco, P., da Silva, A., Chin, M., and Diehl, T.: On- line simulations of global aerosol distributions in the NASA GEOS-4 model and comparisons to satellite and ground based aerosol optical depth, J. Geophys. Res., 115, D14207, https://doi.org/10.1029/2009JD012820, 2010.
Dibb, J. E, Talbot, R. W., Scheuer, E. M., Blake, D. R., Blake, N. J., Gregory, G. L., Sachse, G. W., and Thornton, D. C.: Aerosol chemical composition and distribution during the Pacific Exploratory Mission (PEM) Tropics, J. Geophys. Res., 104, 5785–5800, 1999.
Domine, F., Sparapani, R., Ianniello, A., and Beine, H. J.: The origin of sea salt in snow on Arctic sea ice and in coastal regions, Atmos. Chem. Phys., 4, 2259–2271, https://doi.org/10.5194/acp-4-2259-2004, 2004.
Emmons, L. K., Carroll, M. A., Hauglustaine, D. A., Brasseur, G. P., Atherton, C., Penner, J., Sillman, S., Levy II., H., Rohrer, F., Wauben, W. M. F., Van Velthoven, P. F. J., Wang, Y., Jacob, D., Bakwin, P., Dickerson, R., Doddridge, B., Gerbig, C., Honrath, R., Hübler, G., Jaffe, D., Kondo, Y., Munger, J. W., Torres, A., and Volz-Thomas, A.: Climatologies of NOx and NOy: A comparison of sata and models, Atmos. Environ., 12, 1851–1904, 1997.
Finlayson-Pitts, B. J. and Hemminger, J. C.: Physical chemistry of airborne sea salt particles and their components, J. Phys. Chem. A, 104, 11463–11477, 2000.
Froyd, K. D., Murphy, D. M., Ziemba. L. D., Brock, C. A., Kupc, A., Williamson, C., Schill, G. P., Middlebrook, A. M., Campuzano-Jost, P., Dibb, J. E., and Wilson, J. C.: A new method to quantify mineral dust and other aerosol species from aircraft platforms using single particle mass spectrometry, in preparation, 2019.
Gantt, B. and Meskhidze, N.: The physical and chemical characteristics of marine primary organic aerosol: a review, Atmos. Chem. Phys., 13, 3979–3996, https://doi.org/10.5194/acp-13-3979-2013, 2013.
Gerber, H. E.: Relative-humidity parameterization of the Navy aerosol model (NAM), NRL Rep. 8956, Naval Res. Lab., Washington, D.C., USA, 1985.
Gong, S. L.: A parameterization of sea-salt aerosol source function for sub- and super-micron particles, Global Biogeochem. Cy., 17, 1097, https://doi.org/10.1029/2003GB002079, 2003.
Grythe, H., Ström, J., Krejci, R., Quinn, P., and Stohl, A.: A review of sea-spray aerosol source functions using a large global set of sea salt aerosol concentration measurements, Atmos. Chem. Phys., 14, 1277–1297, https://doi.org/10.5194/acp-14-1277-2014, 2014.
Hara, K., Osada, K., Yabuki, M., and Yamanouchi, T.: Seasonal variation of fractionated sea-salt particles on the Antarctic coast, Geophys. Res. Lett., 39, L18801, https://doi.org/10.1029/2012GL052761, 2012.
Hoose, C., Lohmann, U., Erdin, R., and Tegen, I.: The global influence of dust mineralogical composition on heterogeneous ice nucleation in mixed-phase clouds, Environ. Res. Lett., 3, 025003, https://doi.org/10.1088/1748-9326/3/2/025003, 2008.
Huang, J. and Jaeglé, L.: Wintertime enhancements of sea salt aerosol in polar regions consistent with a sea ice source from blowing snow, Atmos. Chem. Phys., 17, 3699–3712, https://doi.org/10.5194/acp-17-3699-2017, 2017.
Huebert, B. J., Lee, G., and Warren, W. L.: Airborne aerosol inlet passing efficiency measurement, J. Geophys. Res., 95, 16369–16381, 1990.
Jones, A., Haywood, J. M., and Boucher, O.: Aerosol forcing, climate response and climate sensitivity in the Hadley Centre climate model, J. Geophys. Res., 112, D20211, https://doi.org/10.1029/2007JD008688, 2007.
Koop, T., Kapilashrami, A., Molina, L. T., and Molina, M. J.: Phase transitions of sea-salt/water mixtures at low temperatures: Implications for ozone chemistry in the polar marine boundary layer, J. Geophys. Res., 105, 26393–26402, 2000.
Kreidenweis, S. M., McInnes, L. M., and Brechtel, F. J.: Observations of aerosol volatility and composition at Macquarie Island during the First Aerosol Characterization Experiment (ACE 1), J. Geophys. Res., 103, 16511–16524, 1998.
Kupc, A., Williamson, C., Wagner, N. L., Richardson, M., and Brock, C. A.: Modification, calibration, and performance of the Ultra-High Sensitivity Aerosol Spectrometer for particle size distribution and volatility measurements during the Atmospheric Tomography Mission (ATom) airborne campaign, Atmos. Meas. Tech., 11, 369–383, https://doi.org/10.5194/amt-11-369-2018, 2018.
Lewis, E. R. and Schwartz, S. E.: Sea salt aerosol production: mechanisms, methods, measurements, and models – A critical review, Geophysical Monograph Series, vol. 152, American Geophysical Union, Washington, D.C., USA, 2004.
Liao, H., Chen, W. T., and Seinfeld, J. H.: Role of climate change in global predictions of future tropospheric ozone and aerosols, J. Geophys. Res., 111, D12304, https://doi.org/10.1029/2005JD006852, 2006.
Loo, B. W. and Cork, C. P.: Development of high efficiency virtual impactors, Aerosol Sci. Tech., 9, 167–176, 1988.
May, N. W., Quinn, P. K., McNamara, S. M., and Pratt, K. A.: Multiyear study of the dependence of sea salt aerosol on wind speed and sea ice conditions in the coastal Arctic, J. Geophys. Res.-Atmos., 121, 9208–9219, https://doi.org/10.1002/2016JD025273, 2016.
McNaughton, C. S., Clarke, A. D., Howell, S. G., Pinkerton, M., Anderson, B., Thornhill, L., Hudgins, C., Winstead, E., Dibb, J. E., Scheuer, E., and Maring, H.: Results from the DC-8 Inlet Characterization Experiment (DICE): Airborne versus surface sampling of mineral dust and sea salt aerosols, Aerosol Sci. Tech., 41, 136–159, 2007.
Middlebrook, A. M., Murphy, D. M., and Thomson, D. S.: Observations of organic material in individual marine particles at Cape Grim during the First Aerosol Characterization Experiment (ACE 1), J. Geophys. Res., 103, 16475–16483, 1998.
Modini, R. L., Harris, B., and Ristovski, Z. D.: The organic fraction of bubble-generated, accumulation mode Sea Spray Aerosol (SSA), Atmos. Chem. Phys., 10, 2867–2877, https://doi.org/10.5194/acp-10-2867-2010, 2010.
Murphy, D. M., Middlebrook, A. M., and Warshawsky, M.: Cluster analysis of data from the Particle Analysis by Laser Mass Spectrometry (PALMS) instrument, Aerosol Sci. Tech., 37, 382–391, 2003.
Murphy, D. M., Cziczo, D. J., Hudson, P. K., Thomson, D. S., Wilson, J. C., Kojima, T., and Buseck, P. R.: Particle generation and resuspension in aircraft inlets when flying in clouds, Aerosol Sci. Tech., 38, 400–408, 2004.
Ovadnevaite, J., O'Dowd, C., Dall'Osto, M., Ceburnis, D., Worsnop, D. R., and Berresheim, H.: Detecting high contributions of primary organic matter to marine aerosol: A case study, Geophys. Res. Lett., 38, L02807, https://doi.org/10.1029/2010GL046083, 2011.
Pruppacher, H. R. and Klett, J. D.: Microphysics of Clouds and Precipitation, 2nd ed., Kluwer Acad., Dordrecht, the Netherlands, 1997.
Quinn, P. K., Miller, T. L., Bates, T. S., Ogren, J. A., Andrews, E., and Shaw, G. E.: A 3-year record of simultaneously measured aerosol chemical and optical properties at Barrow, Alaska, J. Geophys. Res., 107, 4130, https://doi.org/10.1029/2001JD001248, 2002.
Shinozuka, Y., Clarke, A. D., Howell, S. G., Kapustin, V. N., and Huebert, B. J.: Sea-salt vertical profiles over the Southern and tropical Pacific oceans: Microphysics, optical properties, spatial variability, and variations with wind speed, J. Geophys. Res., 108, D24201, https://doi.org/10.1029/2004JD004975, 2004.
Thomson, D. S., Schein, M. E., and Murphy, D. M.: Particle analysis by laser mass spectrometry WB-57F instrument overview, Aerosol Sci. Tech., 33, 153–169, 2000.
Toon, O. B., Turco, R. P., Westphal, D., Malone, R., and Liu, M. S.: A multidimensional model for aerosols – Description of computational analogs, J. Atmos. Sci., 45, 2123–2144, https://doi.org/10.1175/1520-0469(1988)045<2123:AMMFAD>2.0.CO;2, 1988.
Vignati, E., Facchini, M. C., Rinaldi, M., Scannell, C., Ceburnis, D., Sciare, J., Kanakidou, M., Myriokefalitakis, S., Dentener, F., and O'Dowd, C. D.: Global scale emission and distribution of sea-spray aerosol: Sea-salt and organic enrichment, Atmos. Environ., 44, 670–677, 2010.
Wagenbach, D., Ducroz, F., Mulvaney, R., Keck, L., Legrand, M., Hall, J. S., and Wolff, E. W.: Sea-salt aerosol in coastal Antarctic regions, Geophys. Res. Lett., 39, L18801, https://doi.org/10.1029/2012GL052761, 1998.
Wagner, R. and Möhler, O.: Heterogeneous ice nucleation ability of crystalline sodium chloride dihydrate particles, J. Geophys. Res.-Atmos., 118, 4610–4622, https://doi.org/10.1002/jgrd.50325, 2013.
Wang, H., Easter, R. C., Rasch, P. J., Wang, M., Liu, X., Ghan, S. J., Qian, Y., Yoon, J.-H., Ma, P.-L., and Vinoj, V.: Sensitivity of remote aerosol distributions to representation of cloud–aerosol interactions in a global climate model, Geosci. Model Dev., 6, 765–782, https://doi.org/10.5194/gmd-6-765-2013, 2013.
Wang, S., Schmidt, J. A., Baidar, S., Coburn, S., Dix, B., Koenig, T. K., Apel, D., Bowdalo, D., Campos, T. L., Eloranta, E., Evans, M. J., DiGangi, J. P., Zondlo, M. A., Gao, R.-S., Haggerty, J. A., Hall, S. R., Hornbrook, R. S., Jacob, D., Morley, B., Pierce, B., Reeves, M., Romashkin, P., ter Schure, A., and Volkamer, R.: Active and widespread halogen chemistry in the tropical and subtropical free troposphere, P. Natl. Acad. Sci. USA, 112, 9281–9286, 2015.
Weinheimer, A. J., Walega, J. G., Ridley, B. A., Gary, B. L., Black, D. R., Blake, N. J., Rowland, R. S., Sachse, G. W., Anderson, B. E., and Collins, J. E.: Meridional distributions of NOx, NOy, and other species in the lower stratosphere and upper troposphere during AASE II, Geophys. Res. Lett., 21, 2583–2586, 1994.
Yu, P., Toon, O. B., Bardeen, C. G., Mills, M. J., Fan, T., English, J. M., and Neely, R. R.: Evaluations of tropospheric aerosol properties simulated by the Community Earth System Model with a sectional aerosol microphysics scheme, J. Adv. Model. Earth Syst., 7, 865–914, https://doi.org/10.1002/2014MS000421, 2015.
Yu, P., Froyd, K. D., Portmann, R. W., Toon, O. B., Freitas, S. R., Bardeen, C. G., Brock, C., Fan, T., Gao, R.-S., Katich, J. M., Kupc, A., Liu, S., Maloney, C., Murphy, D. M., Rosenlof, K. H., Schill, G., Schwarz, J. P., and Williamson, C.: Efficient In-cloud removal of aerosols by deep convection, Geophys. Res. Lett., 46, 1061–1069, https://doi.org/10.1029/2018GL080544, 2019.
Zhang et al.: The development of different complexity in aerosol and chemistry modules within an online global weather forecast modeling system FIM-Chem and the evaluation using ATom-1 observations, in preparation, 2019.