the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 04 Mar 2019
| 04 Mar 2019
Arctic marine secondary organic aerosol contributes significantly to summertime particle size distributions in the Canadian Arctic Archipelago
Randall V. Martin
W. Richard Leaitch
Julia Burkart
Rachel Y.-W. Chang
Douglas B. Collins
Patrick L. Hayes
Anna L. Hodshire
Lin Huang
John K. Kodros
Alexander Moravek
Emma L. Mungall
Jennifer G. Murphy
Sangeeta Sharma
Samantha Tremblay
Gregory R. Wentworth
Megan D. Willis
Jonathan P. D. Abbatt
Jeffrey R. Pierce
Summertime Arctic aerosol size distributions are strongly controlled by natural regional emissions. Within this context, we use a chemical transport model with size-resolved aerosol microphysics (GEOS-Chem-TOMAS) to interpret measurements of aerosol size distributions from the Canadian Arctic Archipelago during the summer of 2016, as part of the “NETwork on Climate and Aerosols: Addressing key uncertainties in Remote Canadian Environments” (NETCARE) project. Our simulations suggest that condensation of secondary organic aerosol (SOA) from precursor vapors emitted in the Arctic and near Arctic marine (ice-free seawater) regions plays a key role in particle growth events that shape the aerosol size distributions observed at Alert (82.5∘ N, 62.3∘ W), Eureka (80.1∘ N, 86.4∘ W), and along a NETCARE ship track within the Archipelago. We refer to this SOA as Arctic marine SOA (AMSOA) to reflect the Arctic marine-based and likely biogenic sources for the precursors of the condensing organic vapors.
AMSOA from a simulated flux (500 , north of 50∘ N) of precursor vapors (with an assumed yield of unity) reduces the summertime particle size distribution model–observation mean fractional error 2- to 4-fold, relative to a simulation without this AMSOA. Particle growth due to the condensable organic vapor flux contributes strongly (30 %–50 %) to the simulated summertime-mean number of particles with diameters larger than 20 nm in the study region. This growth couples with ternary particle nucleation (sulfuric acid, ammonia, and water vapor) and biogenic sulfate condensation to account for more than 90 % of this simulated particle number, which represents a strong biogenic influence. The simulated fit to summertime size-distribution observations is further improved at Eureka and for the ship track by scaling up the nucleation rate by a factor of 100 to account for other particle precursors such as gas-phase iodine and/or amines and/or fragmenting primary particles that could be missing from our simulations. Additionally, the fits to the observed size distributions and total aerosol number concentrations for particles larger than 4 nm improve with the assumption that the AMSOA contains semi-volatile species: the model–observation mean fractional error is reduced 2- to 3-fold for the Alert and ship track size distributions. AMSOA accounts for about half of the simulated particle surface area and volume distributions in the summertime Canadian Arctic Archipelago, with climate-relevant simulated summertime pan-Arctic-mean top-of-the-atmosphere aerosol direct (−0.04 W m−2) and cloud-albedo indirect (−0.4 W m−2) radiative effects, which due to uncertainties are viewed as an order of magnitude estimate. Future work should focus on further understanding summertime Arctic sources of AMSOA.
- Article
(6051 KB) - Full-text XML
- BibTeX
- EndNote





Aerosols play important roles in the summertime Arctic climate system. Similar to their effects in other regions, aerosols interact directly with incoming solar radiation via scattering and absorption (Charlson et al., 1992; Hegg et al., 1996; Yu et al., 2006; Shindell and Faluvegi, 2009; Yang et al., 2014) and indirectly through the modification of cloud properties by acting as the seeds for cloud droplet formation (Lohmann and Feichter, 2005; McFarquhar et al., 2011). In the summertime Arctic, efficient wet removal by precipitation and the smaller extent of the polar dome limit the transport of pollution from lower latitudes and maintain an atmosphere that is more pristine than in the Arctic winter and springtime (Barrie, 1995; Polissar et al., 2001; Quinn et al., 2002; Stohl, 2006; Garrett et al., 2011; Brock et al., 2011; Fisher et al., 2011; Sharma et al., 2013; Xu et al., 2017; Willis et al., 2018). As a result, natural regional Arctic sources make strong contributions to summertime Arctic aerosol, to the related radiative effects, and to associated uncertainties (Korhonen et al., 2008; Leck and Bigg, 2010; Heintzenberg and Leck, 2012; Karl et al., 2013; Carslaw et al., 2013; Heintzenberg et al., 2015; Croft et al., 2016b; Willis et al., 2016, 2017; Burkart et al., 2017a; Mungall et al., 2017; Dall'Osto et al., 2017, 2018a; Breider et al., 2017; Leaitch et al., 2018).
Observations indicate that aerosol particle formation and growth events occur frequently in the summertime Canadian Arctic Archipelago region between 60–100∘ W and 66–85∘ N (Chang et al., 2011b; Leaitch et al., 2013; Willis et al., 2016, 2017; Croft et al., 2016b; Burkart et al., 2017a, b; Collins et al., 2017; Tremblay et al., 2019). These events contribute towards shaping a summertime aerosol number size distribution that is characterized by a dominant Aitken mode (particles with diameters between 10 and 100 nm) in this region (Croft et al., 2016a), similar to observations at other pan-Arctic sites (Tunved et al., 2013; Asmi et al., 2016; Nguyen et al., 2016; Freud et al., 2017; Gunsch et al., 2017; Heintzenberg et al., 2017; Kolesar et al., 2017). Summertime Arctic aerosol size distributions are also characterized by a suppressed accumulation mode (particles with diameters between 100 and 1000 nm) due to the efficient wet removal processes in frequently drizzling low clouds (Browse et al., 2014) and the limited transport from lower latitudes (Stohl, 2006; Law and Stohl, 2007; Korhonen et al., 2008).
Evidence points to a strong marine biogenic influence on summertime Arctic aerosols (Leck and Bigg, 2010; Chang et al., 2011a; Heintzenberg et al., 2015; Dall'Osto et al., 2018a). The oceans provide the atmosphere with many particle-relevant trace gases (Carpenter et al., 2012; Carpenter and Nightingale, 2015; Ghahremaninezhad et al., 2017; Mungall et al., 2017), as well as primary particles (Gantt and Meskhidze, 2013; Grythe et al., 2014; Wilson et al., 2015). Arctic melt ponds and melting ice are also sources of vapors such as dimethyl sulfide (DMS) (Hayashida et al., 2017; Gourdal et al., 2018), which yield condensable products following oxidation (Barnes et al., 2006) that can form and grow particles (Kirkby et al., 2011). Terrestrial volatile organic compounds (VOCs) from tundra and lakes are an additional biogenic influence (Potosnak et al., 2013; Lindwall et al., 2016; Steinke et al., 2018). Furthermore, observations suggest a key role for Arctic marine secondary organic aerosol (AMSOA) in the Canadian Arctic Archipelago (Willis et al., 2017; Burkart et al., 2017a; Köllner et al., 2017; Leaitch et al., 2018). The condensing vapors that contribute to particle growth via the formation of secondary organics in the Canadian Arctic Archipelago may be more volatile than at lower latitudes because smaller modes (particle diameters around 20 nm) grow more slowly than larger modes (particle diameters around 90 nm) (Burkart et al., 2017a). However, these vapors are still capable of growing newly formed particles, and the details regarding the origin and composition of AMSOA precursors are not well understood.
In this study, the terminology AMSOA indicates SOA formed from any organic precursor vapors emitted from ice-free seawater north of 50∘ N, excluding methane sulfonic acid (MSA), which we treat as a separate aerosol component, for consistency with most filter-based aerosol species mass measurements. In the Canadian Arctic Archipelago, AMSOA is likely strongly controlled by marine biogenic activity (Willis et al., 2017; Leaitch et al., 2018). Due to the spatial and temporal variability, and diversity of organic precursor vapor sources and chemistry, the chemical character of AMSOA is not necessarily the same as marine secondary organic aerosol arising from precursors originating in other marine regions. Other areas may have differing levels and cycles of marine biogenic activity (Facchini et al., 2008; Rinaldi et al., 2010) and/or different ship traffic emissions with VOCs that differ from natural sources (Endresen et al., 2003). In addition, under our definition of AMSOA, the presence of AMSOA is not limited to the atmospheric marine boundary layer or marine environment due to the transport of precursors and AMSOA to continental regions.
There are few measurements of size-resolved aerosol mass concentrations in the summertime Arctic (Zábori et al., 2015; Giamarelou et al., 2016; Tremblay et al., 2019). Such measurements can provide insight into the processes that control the size distributions. Limited observations indicate that growing Aitken-mode particles with diameters between 50 and 80 nm in the Canadian Arctic Archipelago are composed almost entirely of organics, suggesting a strong role for secondary organics (Tremblay et al., 2019). Conversely, observations from the Svalbard region (between 74–81∘ N and 10–35∘ E) indicate that the smaller sub-12 nm particles are composed primarily of ammonium sulfate, suggesting that ternary nucleation and early growth involving gas-phase water, ammonia (NH3), and sulfuric acid (H2SO4) play a key role in the development of nucleation-mode aerosols (particle diameters smaller than 10 nm) in the region (Giamarelou et al., 2016). In the Canadian Arctic Archipelago, summertime gas-phase NH3 concentrations have been observed to be in the range of a few hundred parts per trillion volume (Wentworth et al., 2016), levels that could contribute to initial particle formation (Napari et al., 2002; Kirkby et al., 2011; Almeida et al., 2013). Sources for NH3 in this region are not yet fully understood, but contributors include Arctic seabird colonies, biomass burning, and possibly other terrestrial sources such as tundra ecosystems that can contribute to bidirectional exchange (Skrzypek et al., 2015; Croft et al., 2016b; Lutsch et al., 2016; Wentworth et al., 2016). In addition to NH3, H2SO4, and gas-phase water, other components of nucleation-mode particles (diameters less than 10 nm) could include, but are not limited to, iodine (Allan et al., 2015; Dall'Osto et al., 2018b), amines (Almeida et al., 2013), and fragmentation of primary particles as clouds and fog evaporate (Leck and Bigg, 2010).
Given the complexity of the interacting processes and source-related uncertainties described above, a coupled model–measurement-based approach enables the exploration of the role of particles of biogenic origin in the development of summertime aerosol size distributions in the Canadian Arctic Archipelago. In this study, we use the GEOS-Chem-TOMAS model (http://geos-chem.org, last access: 26 February 2019) with size-resolved aerosol microphysics to interpret aerosol measurements taken during the summer of 2016 in the Canadian Arctic Archipelago, at both Alert and Eureka, in Nunavut, Canada, and also along the 2016 CCGS Amundsen ship track. These measurements include aerosol mass loading, total aerosol number, and aerosol size distributions, some of which were made as part of the NETwork on climate and aerosols: Addressing key uncertainties in Remote Canadian Environments (NETCARE) project (Abbatt et al., 2019). Section 2 describes our methodology, including further details regarding the observations, a model description, and a summary of simulations. Section 3 interprets simulations and observations to explore the contribution of both marine primary organic aerosol (arising from sea spray) and AMSOA in shaping the summertime aerosol size distributions in the Canadian Arctic Archipelago. We also consider the role of ternary nucleation in the simulated particle nucleation events and size distributions, and comparison with observations. Furthermore, Sect. 3 presents sensitivity studies to explore the role of the volatility of the AMSOA during growth events, and in shaping aerosol size distributions. Finally, this section also displays estimates of the contribution of AMSOA to summertime Arctic direct and indirect aerosol effects. Section 4 presents a summary and highlights key directions for future research.
2.1 Aerosol measurements in the Canadian Arctic Archipelago
Figure 1 shows the locations of aerosol measurements, taken at both Alert (82.5∘ N, 62.3∘ W) and Eureka (80.1∘ N, 86.4∘ W), in Nunavut, Canada and along the 2016 ship track of the CCGS Amundsen through the Canadian Arctic Archipelago; we interpret these measurements using the GEOS-Chem-TOMAS model. The measurements at Alert, Nunavut, Canada are made at the Global Atmospheric Watch (GAW) observatory. Since 2011, hourly mean size distributions for particles with diameters from 20 to 500 nm have been measured at Alert using a TSI 3034 scanning mobility particle system (SMPS), which is verified for sizing on site using monodisperse particles of polystyrene latex and of ammonium sulfate size selected with a Brechtel Manufacturing Incorporated scanning electrical mobility spectrometer. Total particle number concentration for particles larger than 10 nm in diameter is measured at Alert using a TSI 3772 condensation particle counter (CPC). The CPC and the SMPS agree to within 10 % when all particles are large enough to be counted by both instruments. Data that could be influenced by local camp activities are filtered from the data set by removing data under the following conditions: (1) when the wind direction is within a 45∘ angle centered on the Alert camp; (2) during line zeroes to check the connections to the instruments and any other repetitive occurrence that might influence the measurements; (3) when logs indicate potential sources nearby (e.g. trucks); and (4) when unknown factors must be accounted for – when data spikes remain that lasted 2 h hours or less.
At Eureka, Nunavut, Canada, aerosol measurements are taken at the Ridge Lab of the Polar Environment Atmospheric Research Laboratory (PEARL) (Fogal et al., 2013), which is located on Ellesmere Island at 610 m a.s.l. and about 480 km southwest of Alert. Since 2015, size distributions for particles with diameters between 10 and 500 nm have been measured at the Ridge Lab using a TSI 3034 SMPS. Data are recorded every 3 min and averaged to hourly means. Further details about the instrument and sampling are presented in Tremblay et al. (2019).

Figure 1CCGS Amundsen 2016 cruise track through Canadian Arctic Archipelago color coded by the number of days after 24 July 2016. Land is shaded in grey. The locations of Alert and Eureka are shown using red and cyan stars, respectively.
During the summer of 2016, the research icebreaker CCGS Amundsen traveled through the Canadian Arctic Archipelago as a part of the NETCARE project (Abbatt et al., 2019). Figure 1 shows the cruise track for the period from 24 July to 23 August 2016. During the cruise, the total number concentrations of aerosols with diameters larger than 4 nm were measured with a TSI 3776 ultrafine condensation particle counter (UCPC). Aerosol size distributions for particles with diameters between 10 and 430 nm were measured using a TSI 3080/3087 SMPS. Data collected while the wind direction was less than 60∘ to port and less than 90∘ to starboard of the ship's orientation were accepted for further analysis. We consider all measurements taken over the 23 July 2016–24 August 2016 period, when the ship was north of 66∘ N. Further details regarding the instrumental setup and sampling are available in Collins et al. (2017). Measurements of NH3 were also taken during the cruise with a quantum cascade tunable infrared laser differential absorption spectroscopy (QC-TILDAS) analyzer (Ellis et al., 2010). The instrument has a fast response time that enabled measurements at 1 Hz during the cruise, with measurements taken from 29 July 2016 to 23 August 2016. NH3 data were also filtered for wind direction, ship speed, and measured aerosol number concentrations to exclude periods that indicated influence from the ship exhaust.
Tundra samples were collected in triplicate from three sites near Alert on 14 and 15 July 2016 to estimate tundra NH3 emission potential. The sites ranged from approximately 1 to 9 km west of the GAW observatory. Sampling and subsequent analysis for ammonium concentration, [], and pH were carried out according to Wentworth et al. (2014). From mid-June to the end of July 2016, hourly measurements of tundra temperature were recorded adjacent to the GAW observatory using commercially available soil temperatures sensors (iButtons, Maxim Integrated). Tundra [], pH (both based on the 14 and 15 July 2016 soil samples), and hourly temperature were used to calculate the hourly NH3 tundra compensation, which reflects the predicted equilibrium NH3 concentration in the atmosphere above the tundra (Wentworth et al., 2014). A tundra–air exchange velocity was calculated using a resistance-in-series scheme with parameterizations from Wesely (1989) and Walker et al. (2014). The average NH3 emissions at the three sites were then calculated as the mean of the products of the exchange velocity and compensation point, resulting in estimated emission rates of 0.12, 1.4, and 2.2 . Here, we adopt the highest value to provide an upper estimate of the contribution of the tundra to atmospheric NH3. It should be noted that extrapolating calculated emissions from discrete tundra samples to the entire Canadian Arctic Archipelago carries a very large degree of uncertainty. However, the paucity of necessary tundra measurements and the lack of terrestrial Arctic NH3 flux data prevent a more rigorous approach.
2.2 Model description
The GEOS-Chem (GC) chemical transport model version 10-01 (http://geos-chem.org, last access: 26 February 2019) coupled to the TwO-Moment Aerosol Sectional (TOMAS) microphysics model (Adams and Seinfeld, 2002; Kodros et al., 2016; Kodros and Pierce, 2017) is employed in this study. Our model version has 47 vertical levels and a horizontal resolution. The GEOS-FP reanalysis (https://gmao.gsfc.nasa.gov/GMAO_products/NRT_products.php, last access: 26 February 2019) provides the meteorological fields. We use a TOMAS version with 15 size sections, including dry diameters ranging from 3 nm to 10 µm (Lee and Adams, 2012). Tracers within each size bin include particle number and mass of sulfate, black carbon (hydrophobic and hydrophilic), organic carbon (hydrophobic and hydrophilic), sea salt, dust, and water. All simulations are for the months of July and August 2016, with a 1-month spin-up during June that is not included in our analysis.
2.2.1 TOMAS aerosol microphysics
The TOMAS aerosol microphysics scheme employed in our simulations has 13 logarithmically spaced size sections for aerosol dry diameters from approximately 3 nm to 1 µm, and 2 additional size sections to represent aerosol dry diameters from 1 to 10 µm (Lee and Adams, 2012). Particle formation is treated according to the ternary H2SO4–NH3–H2O nucleation scheme described by Baranizadeh et al. (2016). The formation rate of particles at about 1.2 nm in mass diameter is determined from a full kinetics simulation by the Atmospheric Cluster Dynamics Code (Olenius et al., 2013) using particle evaporation rates based on quantum chemistry. This ternary nucleation scheme is implemented as a lookup table as a function of gas-phase H2SO4 and NH3 concentrations, relative humidity, temperature, and condensation sink for condensable vapors. Growth and loss of particles smaller than 3 nm are approximated with the Kerminen et al. (2004) scheme. Implementation of the ternary scheme is supported by the findings of Giamarelou et al. (2016) that 12 nm-diameter particles in the summertime Arctic are predominantly ammoniated sulfates. All simulations use the Brownian coagulation scheme of Fuchs (1964) and consider coagulation between all particle sizes, an important sink for particle number, particularly for those particles with diameters smaller than 100 nm. Coagulation between aerosols contained in cloud hydrometeors and interstitial aerosols is parameterized as described in Pierce et al. (2015). An overview of the condensational-growth assumptions follows the discussion of inventories and the secondary organic aerosol (SOA) scheme below.
2.2.2 Natural emissions
Several natural emission inventories and parameterizations are used in our study. Emissions of dimethyl sulfide (DMS) are based on the seawater DMS climatology of Lana et al. (2011) with modifications for the Canadian Arctic Archipelago region as described in Mungall et al. (2016), who found that the climatology seawater DMS was biased low relative to observations from summer 2014. The air–water transfer velocities for DMS are based on the scheme of Johnson (2010). Natural sources of NH3, along with biofuel and anthropogenic sources are from the Global Emissions InitiAtive (GEIA) (Bouwman et al., 1997).
For simulations with Arctic seabird colony NH3 emissions, these emissions are implemented following Riddick et al. (2012a, b) for the entire Arctic and near Arctic north of 50∘ N, with modifications and the spatial distribution of the colony-specific emissions, as described in Croft et al. (2016b) and Wentworth et al. (2016). The total summertime seabird colony NH3 emissions north of 50∘ N of 36 Gg are spread uniformly in period between 1 May and 30 September and the point source emissions from the individual colonies are treated as well-mixed within the respective grid box on emission.
Our simulations also implement an NH3 source from ice- and snow-free tundra for the entire Arctic, with a fixed emission rate of 2.2 . Due to knowledge gaps, these emissions are not temperature dependent. This source is an upper estimate based on inferred bidirectional exchange fluxes calculated using soil measurements made during the summer of 2016 near Alert, which found the tundra can act as a source of NH3 to the atmosphere (Murphy et al., 2019). Given the uncertainty in the tundra source, this source can be viewed as a surrogate for the missing emissions needed to bring the simulated NH3 mixing ratios to agreement with measurements as discussed in Sect. 3.1. For the regions between 60 and 100∘ W, with varying southward extent, the total implemented summertime tundra NH3 emissions range from about 1.5- to 7-fold greater than the total summertime seabird colony emissions, with respect to 72–90 and 50–90∘ N, respectively.
Additionally, natural sources of NH3 and organic carbon (OC) aerosol are included in the biomass burning emissions from the 3-hourly Global Fire Emissions Database, version 4 (GFED4) for 2016 (Giglio et al., 2013; Van Der Werf et al., 2017), which is employed in all simulations. Dust emissions employ the Dust Entrainment and Deposition (DEAD) scheme of Zender et al. (2003), developed in GEOS-Chem by Fairlie et al. (2007).
Emissions of sea spray in our simulations are based on the Mårtensson et al. (2003) parameterization. Comparisons with the Jaeglé et al. (2011) parameterization, employed in the standard GEOS-Chem-TOMAS model, indicate that the Mårtensson et al. (2003) parameterization yields greater sub-100 nm fluxes by up to a few orders of magnitude (Jaeglé et al., 2011). This choice of emission inventory enables evaluation of the contribution of sea spray to simulated ultrafine particle concentration with an inventory that is extremely favorable to ultrafine sea-spray primary particle production. Additionally, as opposed to assuming that all sea spray is sodium chloride, we emit sea-spray particles with diameters smaller than 100 nm as hydrophobic organic carbon aerosol and particles larger than 100 nm as sodium chloride. This modification was introduced based on measurements indicating that sub-100 nm sea-spray particles are composed mostly of hydrophobic organics, whereas larger particles have a progressively more dominant salt component (Facchini et al., 2008; Collins et al., 2013; Prather et al., 2013; Gantt and Meskhidze, 2013; Quinn et al., 2015). However, knowledge gaps remain in relation to the spatial distribution of sea-spray composition and hygroscopicity (Collins et al., 2016).
2.2.3 Anthropogenic emissions
Our simulations also include global anthropogenic emissions from the Emissions Database for Global Atmospheric Research (http://edgar.jrc.ec.europa.eu/archived_datasets.php, last access: 14 March 2016) (EDGAR) inventory. The EDGAR inventory is overwritten by regional inventories for Europe (European Monitoring and Evaluation Program – EMEP); Crippa et al., 2016), Canada (Criteria Air Contaminant Inventory), the United States (National Emission Inventory – NEI), and Asia (MIX inventory; Li et al., 2017). In addition, the Bond et al. (2007) inventory overwrites the EDGAR fossil fuel and biofuel emissions for black and organic carbon.
2.2.4 Chemical mechanism
The GEOS-Chem-TOMAS chemical mechanism represents the reactions of more than 100 gas-phase species, including particle-relevant reactions such as DMS oxidation by the hydroxyl radical (OH) to produce sulfur dioxide (SO2) by both the addition and abstraction channels, and also reaction with the nitrate radical (NO3) (Chatfield and Crutzen, 1990; Chin et al., 1996; Alexander et al., 2012). SO2 then undergoes either gas-phase reactions with OH to produce H2SO4 or aqueous oxidation with either hydrogen peroxide (H2O2) or ozone (O3) to produce particulate sulfate. For the aerosol microphysics scheme, gas-phase H2SO4 can join with water vapor and gas-phase NH3 for ternary nucleation of nascent particles, and it can also condense to grow preexisting particles. The sulfate produced by aqueous-phase reactions is added to particles that are large enough to have activated to form cloud droplets, only contributing to the growth of these larger particles. In general, particles with diameters of 50 nm or larger activate in our simulations, although observations from the Canadian Arctic Archipelago indicate that particles as small as 20 nm could activate in clean summertime atmospheric layers above 200 m altitude when low concentrations of larger particles (diameters greater than 100 nm) enable relatively high supersaturations (Leaitch et al., 2016). MSA that is produced by the DMS–OH-addition channel can contribute to condensational growth of existing particles (Chen et al., 2015; Hoffmann et al., 2016; Willis et al., 2016; Hodshire et al., 2019). In our simulations, MSA contributes to particle condensational growth, but not to particle nucleation. In this study, we did not include additional chemistry related to the production of dimethylsulfoxide (DMSO), which could increases the yield of MSA and reduce sulfate concentrations (Breider et al., 2014; Hoffmann et al., 2016). Future studies are needed to quantify the impact of multiphase DMS chemistry.
2.2.5 Secondary organic aerosol scheme
SOA is treated with the simplified SOA scheme developed by Kim et al. (2015), which includes SOA precursors from non-marine sources associated with terrestrial biogenic, fossil fuel, biofuel, and biomass burning emissions for all simulations. An AMSOA source is added for some simulations and is described further below. The SOA scheme introduces two additional tracers, a gas-phase SOA precursor, and a SOA tracer that immediately condenses on the preexisting particles. The gas-phase SOA precursor oxidizes to form the immediately condensed SOA tracer on a fixed timescale of 1 day. For biogenic sources, the emissions are distributed between these two tracers with a 50∕50 split to represent the fast oxidation timescale of biogenic precursors of typically shorter than 1 day. The model employed for this study does not include the explicit aqueous-phase production of SOA, which could further increase the SOA production and change the shape of the particle size distribution.
AMSOA-precursors are emitted in the entire Arctic and near Arctic north of 50∘ N over open seawater. Like other biogenic SOA sources, these vapors are emitted with a 50∕50 split between the gas-phase precursor and a vapor that is immediately condensed. Given knowledge gaps, these AMSOA precursor emissions are not dependent on other parameters such as temperature or marine biologic activity.
Justification for this AMSOA source draws upon measurements presented by Mungall et al. (2017) indicating that the marine microlayer is a source of oxygenated volatile organic compounds (OVOCs), key precursors to secondary organic aerosol. Furthermore, Willis et al. (2017) identified a positive relationship between the ratio of organic to sulfate aerosol mass concentrations and time spent over open water, suggesting a marine SOA source. Studies from other regions also identified biogenic VOCs of marine origin, but their marine sources are not fully understood (Carpenter and Nightingale, 2015; Tokarek et al., 2017; Chiu et al., 2017). Likewise for the Arctic, the emission rates for these vapors are not well understood (Burkart et al., 2017a). Given this uncertainty and the lack of a marine SOA source in our standard simulations, we introduced and tuned a simulated fixed AMSOA-precursor vapor source flux (AMSOA formed with a mass yield of unity) from the ice-free seawater in the Arctic and near Arctic (north of 50∘ N) for simulations with seabird and tundra NH3. We tuned to a satisfactory model–measurement agreement for the first four moments of the aerosol size distributions for Alert, Eureka, and the ship track.
We define the aerosol number distribution (zeroth moment) as
The aerosol integrated diameter (length) distribution (first moment) is
The aerosol surface area (second moment) is
The aerosol volume (third moment) is
We calculate the mean fractional error (MFE) (Boylan and Russell, 2006) between our simulations and observed aerosol size distributions using the following equation:
where Cm(i) is the integrated value for the ith moment of the simulated aerosol size distribution, and Co(i) is the integrated value for the ith moment of the observed size distribution, for the N values, in this case the zeroth to third moments. MFE ranges from 0 to +2. Following Boylan and Russell (2006), we treat a MFE value below 0.50 as indicating satisfactory model performance, with the MFE closest to zero indicating the best model performance among the simulation set. We include the four moments to yield a complete evaluation that gives equal weighting to aerosol number, integrated diameter, surface area, and volume. The absolute value in the MFE numerator prevents cancelations of overpredictions and underpredictions between the moments. Mean fractional bias (MFB) is similarly defined, but without the absolute value in the numerator, and ranges from −2 to +2. We consider a MFB between −0.3 and +0.3 as indicating satisfactory model performance. Fractional error (FE) and fractional bias (FB) are similarly defined with N=1.
The top-down estimate of the flux (500 ; north of 50∘ N) for our simulations is adopted by tuning the VOC flux in a simulation set (with the seabird colony and tundra NH3 emissions) until a MFE below 0.5 is achieved for the three measurement platforms. Further details on the related results are presented in Sect. 3. To put the implemented flux in context, this value exceeds either the estimated isoprene flux from a north temperate deep lake (Steinke et al., 2018) or tundra VOC emissions (Lindwall et al., 2016) by a factor of about 5–10. As this flux was tuned specifically to yield model–measurement agreement for our study, it should not be overinterpreted as being fully representative of Arctic marine VOC emissions. Future measurements of marine VOC concentration, fluxes, and volatility are needed for a bottom-up estimate of the marine SOA-precursor source flux.
Our simulations include growth of particles by condensation of the oxidized gas-phase SOA precursor, as well as by condensation of gas-phase H2SO4 and MSA, but do not allow initial formation of nascent particles by clusters of organic vapors arising from the oxidation of the gas-phase SOA precursor. In the standard model, all vapors condense proportional to the Fuchs-corrected aerosol surface area distribution, behaving like a nonvolatile condensing gas (Donahue et al., 2011; Pierce et al., 2011; Riipinen et al., 2011; Liu et al., 2016; Tröstl et al., 2016; Ye et al., 2016). The important role of organic condensation was found at lower latitude continental sites (Riipinen et al., 2011). Our simulations investigate this role for the Arctic. We also conduct additional sensitivity simulations (described in Sect. 2.3), which allow condensation of a fraction of the vapors according to the mass distribution, behavior like a semi-volatile as opposed to nonvolatile condensing organic. For all simulations regardless of the volatility treatment, the AMSOA-source emissions are split 50∕50 between the precursor vapors and the vapors that immediately condense.
2.2.6 Wet and dry deposition
Removal of simulated aerosol mass and number occurs by both wet and dry deposition. The wet deposition parameterization includes both in-cloud and below-cloud scavenging as developed by Liu et al. (2001) and Wang et al. (2011), with modifications as described in Croft et al. (2016a) to more closely link the wet removal to the meteorological fields for cloud liquid, ice water content, and cloud fraction. To represent the impact of drizzle from summertime Arctic low-level clouds, we implemented wet removal from all Arctic clouds below 500 m using a fixed efficiency of 0.01 s−1, similar to the approach of Browse et al. (2012). In-cloud wet removal in GEOS-Chem-TOMAS is limited to the size range that can activate to form cloud hydrometeors. Size-dependent dry deposition uses the resistance-in-series approach of Wesely (1989). Simulated gas-phase species are also removed by dry and wet deposition as described in Amos et al. (2012). Removal depends on solubility such that aerosol precursors including ammonia and sulfur dioxide are removed by precipitation, whereas SOA precursors and dimethyl sulfide are not.
2.2.7 Radiative calculations
The following radiative transfer calculations are conducted off-line using the simulated summertime-mean aerosol mass and number concentrations to examine the effects of organic condensation. For calculation of the direct radiative effect (DRE) attributed to AMSOA, the aerosol optical depth, single-scattering albedo, and asymmetry factor are calculated with Mie code (Bohren and Huffman, 1983) and use refractive indices from the Global Aerosol Dataset (GADS) (Koepke et al., 1997). These optical properties, along with cloud fraction and surface albedo from GEOS-FP assimilated meteorology are input to the Rapid Radiative Transfer Model for Global Climate Models (RRTMG) (Iacono et al., 2008), to determine the change in top-of-the-atmosphere solar flux between two simulations, treating all particles except black carbon as internally mixed and spherical. Kodros et al. (2018) found that the Arctic springtime DRE for all aerosol is less negative than the external mixing-state assumption by 0.05 W m−2 when constraining by coating thickness of the mixed particles and by 0.19 W m−2 when constraining by BC-containing particle number fraction. The radiative-effect sensitivity to the assumed black carbon mixing state is expected to be less for the Arctic summer than in springtime as changes in transport and wet removal, along with low regional sources limit the summertime black carbon concentrations (Xu et al., 2017).
We also calculate the cloud-albedo aerosol indirect effect (AIE) attributed to AMSOA using the method described in Croft et al. (2016b) and Kodros et al. (2016). The cloud droplet number concentration (CDNC) is calculated off-line using the parameterization of Abdul-Razzak and Ghan (2002), again utilizing the summertime mean aerosol mass and number concentrations from our simulations. We assume an updraft velocity of 0.5 m s−1 and treat all aerosols as internally mixed to determine the hygroscopicity parameter of Petters and Kreidenweis (2007). For each model grid box, we assume cloud droplet radii (r) of 10 µm and perturb this value with the ratio of summertime-mean CDNC from simulations (acronyms described in Table 1 and simulations described in Sect. 2.3), following Rap et al. (2013), Scott et al. (2014), and Kodros et al. (2016):
The RRTMG is then used to determine the change in top-of-the-atmosphere solar flux attributed to the change in effective cloud droplet radii, again using the summertime meteorological data from GEOS-FP. Our AIE calculation is limited to this single aerosol indirect effect; the impact of AMSOA on additional indirect effects (Lohmann and Feichter, 2005) requires further investigation in future studies.
2.3 Overview of simulations
Our simulations are conducted with a focus on interpreting the summertime 2016 aerosol measurements from the Canadian Arctic Archipelago. These simulations are used to explore the role of biogenic sources in shaping the aerosol size distributions by the processes of nucleation of particles from gas-phase molecules followed by growth, with a focus on AMSOA. We also consider the role of marine primary particle emissions.
Table 1 presents simulation acronyms used in the following discussion. The BASE simulation employs the standard GEOS-Chem-TOMAS model described in Sect. 2.2. We examine the potential contribution of regional terrestrial NH3 sources to aerosol size distributions with the BASE + BIRDS, BASE + TUNDRA and BASE + TUNDRA + BIRDS simulations. The BASE + BIRDS simulation implements the seabird colony NH3 emissions described in Sect. 2.2.2. The BASE+TUNDRA simulation adds NH3 emissions from all Arctic tundra as discussed in Sect. 2.2.2. The BASE + TUNDRA + BIRDS simulation uses both the seabird colony and tundra NH3 sources. The BASE + TUNDRA + BIRDS + AMSOAnv simulation adds a source of AMSOA as described in Sect. 2.2.5. At the point of condensation, we assume the vapors to be effectively nonvolatile, with condensation according to the Fuchs-corrected surface area.
Table 1Description of acronyms that are used in the simulation names. Simulations are described in more detail by full simulation name in Sect. 2.3.

The BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv simulation examines the impact of particle precursors in addition to H2SO4, NH3, and water that could nucleate nascent particle clusters in the Arctic. These precursors could include (but are not limited to) gas-phase iodine (Allan et al., 2015; Dall'Osto et al., 2018b), amines (Almeida et al., 2013), and organics (Riccobono et al., 2014). It is unclear if marine biological activity creates organic vapors that participate in particle nucleation. Disintegration of larger particles from evaporating clouds and fog could contribute to the number of nascent particles (Leck and Bigg, 2010). Unfortunately, a nucleation parameterization does not exist that is suitable for including interactions of all of these materials simultaneously (Riccobono et al., 2014; Dunne et al., 2016; Gordon et al., 2017). To explore these effects, we scale up the ternary nucleation 100-fold to represent the potential effects of particle precursors with a similar spatial origin to those involved in ternary nucleation. Almeida et al. (2013) and Riccobono et al. (2014) observed increases in nucleation rates of about 100-fold above the sulfate–ammonia–water vapor system when amines or monoterpene-oxidation products were added. We treat the 100-fold increase as an estimate of how additional materials could enhance nucleation. We also conduct the BASE + TUNDRA + BIRDS + 100xnuc simulation, which is otherwise identical to the simulation, but without the condensable marine organic vapors.
Finally, we conduct simulations to examine the impact of AMSOA volatility. Burkart et al. (2017a) found that condensing gas-phase materials in the Canadian Arctic Archipelago were surprisingly more volatile than at lower latitudes. The BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv/sv simulation is identical to the simulation, except that 30 % of the AMSOA behaves as nonvolatile compounds and condenses according to the Fuchs-corrected surface area, whereas 70 % of the AMSOA behaves as idealized semi-volatile compounds and condenses according to the particle mass distribution (quasi-equilibrium condensation). This is a larger fraction of semi-volatile vapors than the 50∕50 semi-∕nonvolatile split employed by Riipinen et al. (2011) for lower latitude continental sites, and consistent with the findings of Burkart et al. (2017a) for the Canadian Archipelago region which showed that the condensing vapors were more semi-volatile than at lower latitudes. We also conducted simulations with the assumption that 100 % of the AMSOA behaved as semi-volatile compounds and found excessively suppressed growth of the sub-40 nm particles relative to observed size distributions. Thus, for our simulations, we settled on 70 % of the AMSOA being semi-volatile as a reasonable intermediate between 50 % and 100 % (the range from Riipinen et al., 2011). The BASE + TUNDRA + BIRDS + 100xnuc + 2xAMSOAnv/sv simulation is identical to the simulation, except that for the former, we double the flux of marine organic vapors to examine the impact of a change in flux as the source rate is highly uncertain.
3.1 Total aerosol number concentrations along the 2016 ship track and at Alert
Figure 2 shows time series measurements during August 2016 of the total particle number concentration from condensation particle counter (CPC) measurements for particles with diameters larger than 4 nm conducted from the CCGS Amundsen (Collins et al., 2017) and for particles with diameters larger than 10 nm at Alert. Stand-alone CPC measurements were not available at Eureka. The measurement time series shows episodic bursts of particle number concentrations greater than 500 cm−3. These episodic bursts in number concentration are indicative of particle formation and growth events. Figure 2 also shows the time series of coincidently sampled simulated number concentrations for five of the simulations described in Table 1 and Sect. 2.3. The simulations have episodic bursts in total number concentration similar to the observations. However, the simulated grid-box mean total number concentration may not always represent the measurement site well, such that simulating the exact timing of the bursts is a greater challenge than simulating the time-averaged magnitude of the number concentration. The simulations may perform better for large-scale (few hundred km) growth events in the Canadian Arctic Archipelago, such as those shown by Tremblay et al. (2019). As an evaluation of the magnitude of the simulated total particle number, we calculated the model–measurement fractional bias (FB) using the period-averaged number concentrations for the first 22 days of August (Eq. 5, N=1 and removing the absolute value in numerator). The BASE simulation is associated with the greatest FB values for the ship track (−1.93) and Alert (−1.86).
The simulations better capture the total particle number when they include NH3 sources from seabird colonies and tundra, with FB values of +0.12 (ship track) and +0.34 (Alert), similar to the findings of Croft et al. (2016b). In addition, relative to measurements taken during the summer 2016 cruise track (not shown), the BASE simulation also underpredicts grid-box mean NH3 mixing ratios with a MFB of −1.98, which is reduced for the BASE + BIRDS (−1.23), BASE + TUNDRA (−0.22), and BASE + TUNDRA + BIRDS (+0.06) simulations.
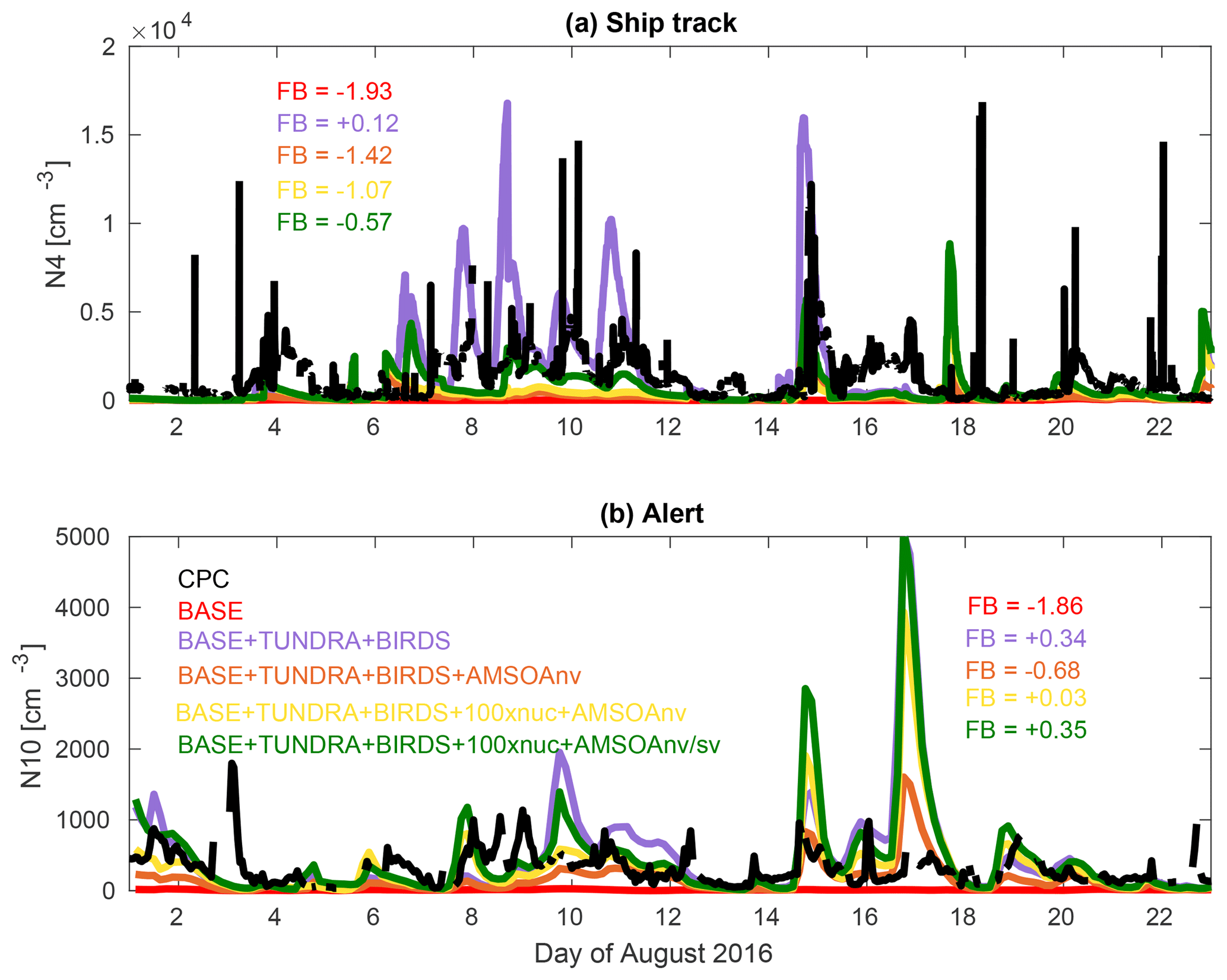
Figure 2Time series for the August 2016 observed number concentration from the condensation particle counter (CPC) for aerosols with (a) diameters larger than 4 nm (N4) along the Amundsen ship track (Fig. 1) and (b) diameters larger than 10 nm (N10) at Alert (described in Sect. 2.1) as well as for the simulations as described in Table 1 and Sect. 2.3 (color coded as shown in the legend). FB refers to the fractional bias (defined in Sect. 2.2.5) between observations and simulations, color coded to match simulation names.
Implementation of AMSOA in the BASE + TUNDRA + BIRDS + AMSOAnv simulation increases the FB magnitude relative to the BASE + TUNDRA + BIRDS simulation to −1.42 (ship track) and −0.68 (Alert). This magnitude increase occurs because more vapors are available to condense onto the particle surface area, building the condensation sink for H2SO4, which reduces the simulated formation of nascent particles by ternary nucleation with H2SO4. These effects reduce the number of ultrafine particles, similar to that described by D'Andrea et al. (2013) for a set of sites distributed around the world. Scaling the nucleation rate 100-fold reduces the FB magnitude to −1.07 for the ship track and +0.03 at Alert, for the BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv simulation. This increased nucleation rate enables ultrafine particles to become more numerous, despite the increased condensation sink associated with the implemented AMSOA source.
The total number concentration is strongly sensitive to the assumed volatility of the condensing vapors. For the BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv/sv simulation, the FB is −0.57 (ship) and +0.35 (Alert). Higher volatility condensing vapors in this simulation relative to the BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv simulation enable slower simulated growth of the nascent particles and faster growth of the larger particles. As the newly formed particles grow more slowly with semi-volatile relative to nonvolatile AMSOA, this lowers the condensation and coagulation sinks of ultrafine particles, and increases the total number of particles. There is relatively more condensation of the semi-volatile AMSOA to larger particles, which contribute proportionately less to surface area and more to aerosol mass. These larger particles are efficiently removed by the frequent low-cloud drizzle in the summertime Arctic in our simulations. As shown on Fig. 2, the net effect is an increase in the number of ultrafine particles that better matches the observed time series of total number concentration for the ship track among the simulations with AMSOA, and still yields a reasonable simulation for Alert.
3.2 Moments of the aerosol size distribution for Alert, Eureka, and the ship track
Figures 3, 4, and 5 show the 2016 summertime (July and August) median aerosol size distributions from SMPS measurements at Alert (Fig. 3), Eureka (Fig. 4), and for the 2016 ship track (Fig. 5). The figure panels show the zeroth through third moments of the aerosol size distribution, aerosol number, integrated diameter (length), surface area, and volume.
The observed distributions are similar between the three measurement sets. The number distributions peak in the Aitken mode at the particle diameter of 30–50 nm, which is similar to summertime observations at other pan-Arctic sites (Tunved et al., 2013; Asmi et al., 2016; Nguyen et al., 2016; Freud et al., 2017; Gunsch et al., 2017; Heintzenberg et al., 2017; Kolesar et al., 2017) and also in the central Arctic marine boundary layer (Heintzenberg and Leck, 2012; Karl et al., 2013; Heintzenberg et al., 2015). Interestingly, the value for the mode for the number distributions (dN∕dlog 10Dp) has its smallest magnitude of about 200 cm−3 at the most northerly site (Alert), and increases moving southward to about 300 cm−3 at Eureka and 400 cm−3 for the ship track, which includes the most southward extent. This pattern is consistent with the hypothesis of an important role for open water in building summertime Arctic size distributions (Heintzenberg et al., 2015; Willis et al., 2017; Dall'Osto et al., 2018a), along with the contribution of the more prominent continental influence at lower latitudes. A similar pattern is noted for the other three moments of the aerosol distribution. The integrated diameter distribution has a maximum between 50 and 150 nm for the three measurement platforms, whereas the surface area maximum is between 100 and 200 nm and the volume maximum is at about 200 nm or larger. For the ship track, the volume distribution peak extends towards 500 nm, reflecting the emission of larger sea-spray particles, which are susceptible to rapid sedimentation and are not as abundant at Alert and Eureka.
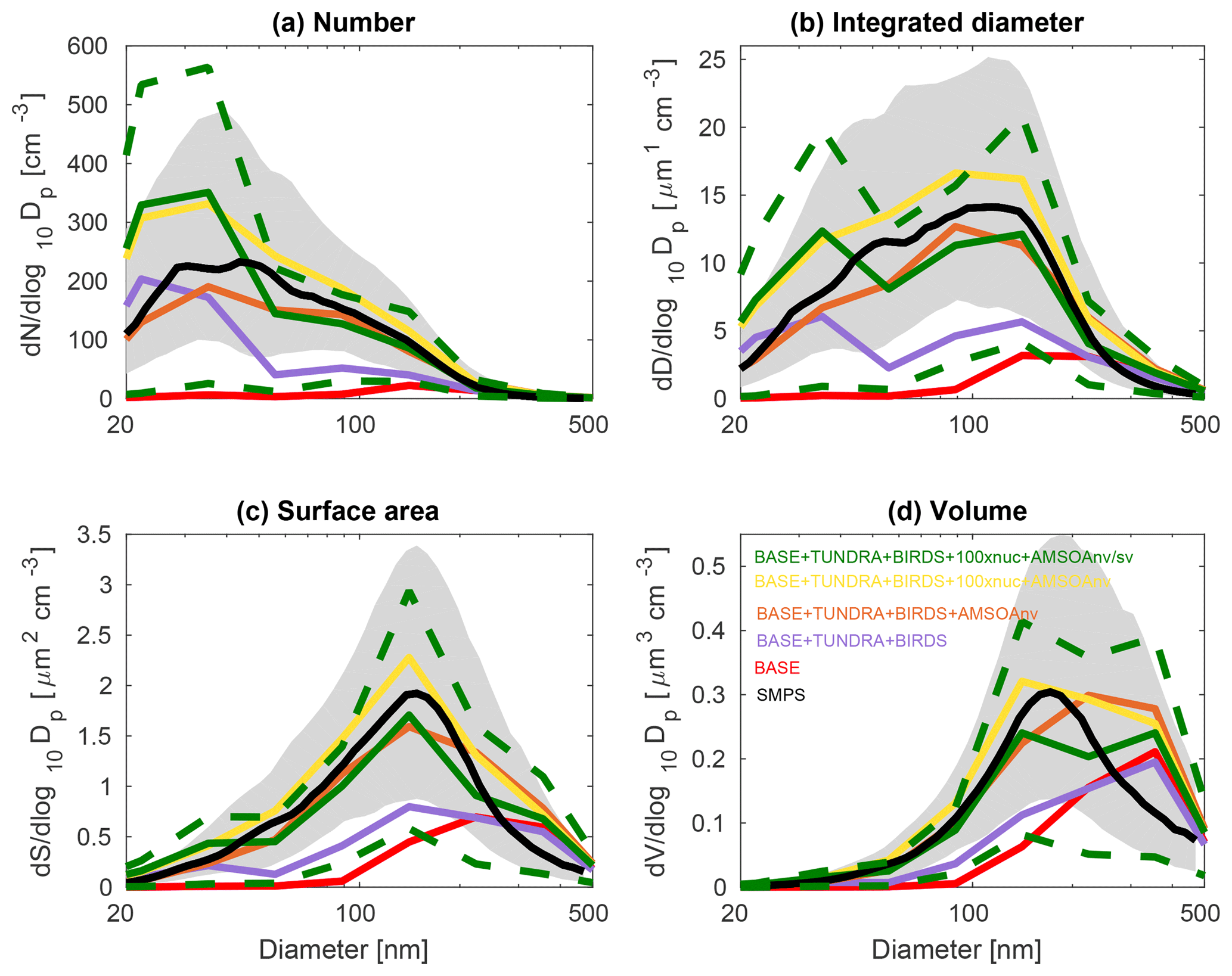
Figure 3July and August 2016 median aerosol size distributions from scanning mobility particle sizer (SMPS) measurements at Alert (82.5∘ N, 62.3∘ W; black; described in Sect. 2.1) and for five GEOS-Chem-TOMAS simulations (color coded as shown in the legend). Grey shading shows the SMPS 20th to 80th percentile and the green dashed lines show the 20th to 80th percentile for the BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv/sv simulation. Simulations are described in Table 1 and Sect. 2.3. The panels show the aerosol distribution moments with respect to (a) the aerosol number, (b) the integrated aerosol diameter, (c) the aerosol surface area, and (d) the aerosol volume distributions. Note the different vertical scale relative to Figs. 4 and 5.
Figures 3, 4, and 5 also show the simulated moments for the three sets of aerosol distributions. The BASE simulation strongly underpredicts all four moments of the distribution relative to all three of the measurement sets. Table 2 shows the MFE (Eq. 5) between the simulations and measurements, using integrated values from the four moments of the distributions, similar to the approach employed by Hodshire et al. (2018). The MFEs are 1.17, 1.36, and 1.34 for Alert, Eureka, and the ship track, respectively, for the BASE simulation. Implementation of sources of NH3 from seabird colonies (BASE + BIRDS simulation) reduces the MFE for all sites, and additional NH3 from a tundra source for the BASE + TUNDRA + BIRDS simulation further lowers the MFE at all sites (0.53 for Alert, 0.80 for Eureka, and 0.97 for the ship track).
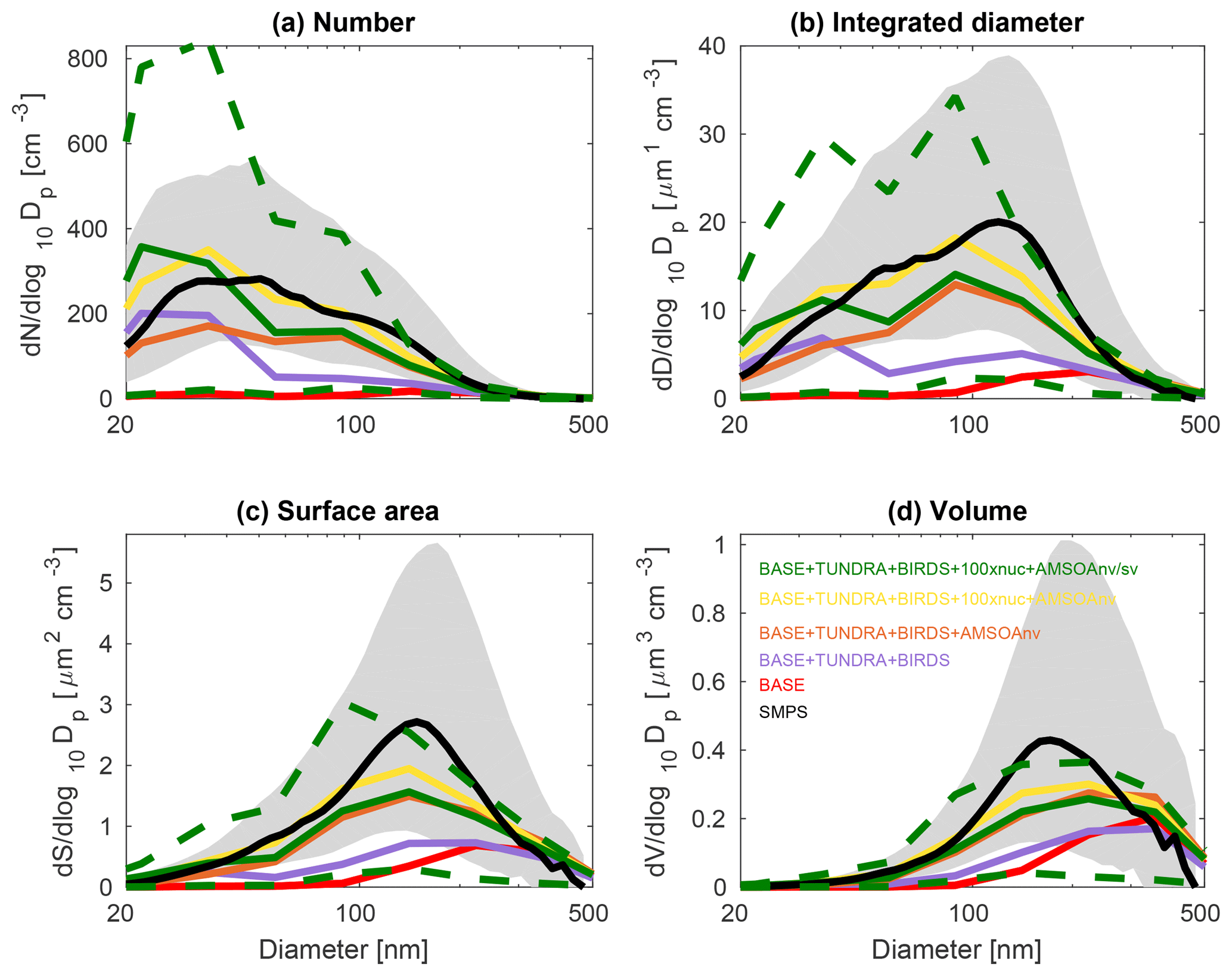
Figure 4July and August 2016 median aerosol size distributions from scanning mobility particle sizer (SMPS) measurements at Eureka (80.1∘ N, 86.4∘ W; black; described in Sect. 2.1) and for five GEOS-Chem-TOMAS simulations (color coded as shown in the legend). Grey shading shows the SMPS 20th to 80th percentile and the green dashed lines show the 20th to 80th percentile for the BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv/sv simulation. Simulations are described in Table 1 and Sect. 2.3. The panels show the aerosol distribution moments with respect to (a) the aerosol number, (b) the integrated aerosol diameter, (c) the aerosol surface area, and (d) the aerosol volume distributions. Note the different vertical scale relative to Figs. 3 and 5.
Figures 3–5 also show that with the NH3 from seabird colonies and tundra (BASE + TUNDRA + BIRDS simulation), an Aitken-mode peak develops around 20–30 nm, but there is an underprediction of the number of aerosols with diameters between 30 and 200 nm, and a strong underprediction of the aerosol diameter, surface area, and volume moments.
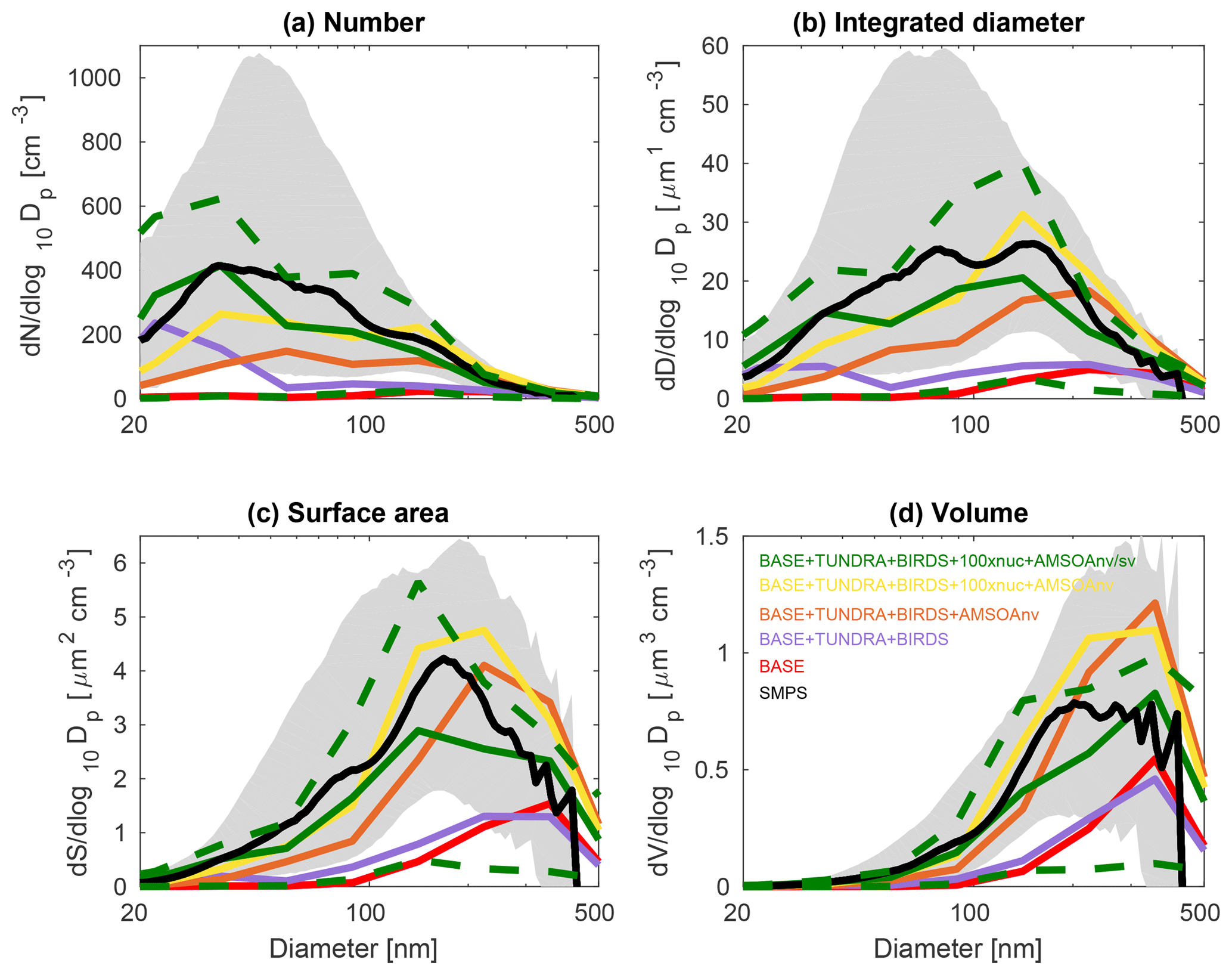
Figure 5July and August 2016 median aerosol size distributions from scanning mobility particle sizer (SMPS) measurements for the CCGS Amundsen 2016 ship track (black; described in Sect. 2.1) and for five GEOS-Chem-TOMAS simulations (color coded as shown in the legend). Grey shading shows the SMPS 20th to 80th percentile and the green dashed lines show the 20th to 80th percentile for the BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv/sv simulation. Simulations are described in Table 1 and Sect. 2.3. The panels show the aerosol distribution moments with respect to (a) the aerosol number, (b) the integrated aerosol diameter, (c) the aerosol surface area, and (d) the aerosol volume distributions. Note the different vertical scale relative to Figs. 3 and 4.
We also conducted comparisons of mass concentrations with filter measurements at Alert (not shown), and all simulations with seabird and tundra NH3 matched the sulfate + ammonium + MSA mass within 20 % (and contributions of other measured species, e.g., nitrate, were minor) so organic aerosol mass was likely the most uncertain species. This suggests that condensation of H2SO4 and MSA alone do not yield sufficient particle growth to match observations from the Canadian Arctic Archipelago, which show frequent particle growth events (Willis et al., 2016; Collins et al., 2017; Burkart et al., 2017b; Tremblay et al., 2019) and suggest a key role for growth by organic vapor condensation (Burkart et al., 2017a; Willis et al., 2017; Mungall et al., 2017). Marine primary organic aerosols could contribute to the Aitken mode as investigated further in the following (Sect. 3.4).
Table 2Mean fractional error (MFE; Eq. 5) between the nine GEOS-Chem-TOMAS simulations (described in Table 1 and Sect. 2.3) and the SMPS measurements (described in Sect. 2.1) for summertime- (July and August 2016) median aerosol size distributions at Alert, Eureka, and during the CCGS Amundsen cruise shown in Figs. 3, 4, and 5, respectively.
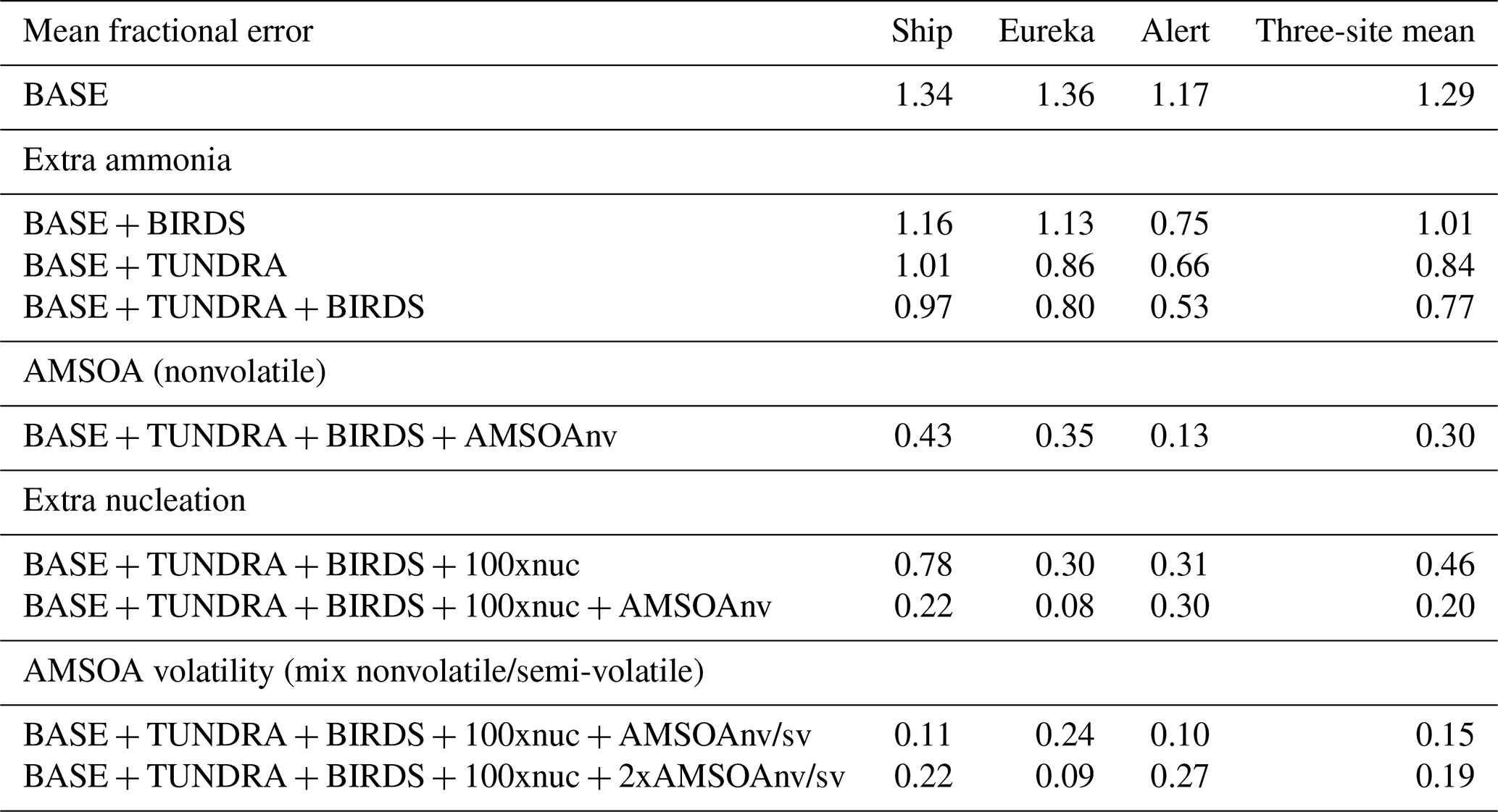
With the implementation of AMSOA (BASE + TUNDRA + BIRDS + AMSOAnv simulation), all four moments of the simulated aerosol distributions are more consistent with the measurements. The MFE is reduced for the ship track (0.43), Eureka (0.35), and Alert (0.13). These additional vapors condense on the simulated particles and build the aerosol diameter, surface area, and volume distributions to better represent the observations. For the ship track and at Eureka, scaling the nucleation rate up further reduces the MFE (BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv simulation) by maintaining the number of ultrafine particles despite the increase in the condensation sink that arises with the growth from the AMSOA. This scaling acts as a surrogate for nucleating vapors that could be missing in our simulations such as iodine (Allan et al., 2015; Dall'Osto et al., 2018b) and amines (Almeida et al., 2013), and also the possible contribution from primary particle fragmentation (Leck and Bigg, 2010). For Alert, the MFE deteriorates with nucleation scaling suggesting that the standard ternary scheme yields sufficient particle formation for that portion of the Canadian Arctic Archipelago under the assumption of growth by nonvolatile vapors.
The simulation with a 30∕70 mix of nonvolatile and semi-volatile AMSOA, respectively, (BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv/sv simulation) yielded the lowest MFE for the ship track (0.11) and for Alert (0.10). We find a similarly low MFE for Eureka (0.09) with a doubling of the AMSOA source under the assumption of a 30∕70 mixed volatility (BASE + TUNDRA + BIRDS + 100xnuc + 2xAMSOAnv/sv simulation). This inter-site difference in the AMSOA precursor source flux magnitude that yields a MFE of 0.1 suggests that the development of a parameterization for the precursors' volatility-dependent spatial distribution could be of benefit. Such a parameterization could also help to better capture the increase in the magnitude of the mode for the number, diameter, area, and volume distributions between Alert and Eureka. However, our current parameterizations do capture the larger magnitude of the mode value for all four moments for the ship track relative to those for Alert and Eureka (BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv/sv simulation). As shown on Figs. 3–5, this simulation also has a range of variability between the 20th and 80th percentiles that is similar to that of the measurements for all four moments.
Our finding that a mixture of nonvolatile and semi-volatile AMSOA gives a closer fit between the simulations and observations is in agreement with the measurement-based findings of Burkart et al. (2017a) which showed that the condensing vapors were surprisingly more volatile than at lower latitudes. As discussed by Burkart et al. (2017a), these semi-volatile (as opposed to nonvolatile) vapors enable slower growth of the smallest mode of particles with diameters around 20 nm and faster growth of the larger mode with diameters around 90 nm. This larger mode is more efficiently removed by precipitation, maintaining a relatively pristine environment with lower particle mass concentrations that favors particle formation and growth.
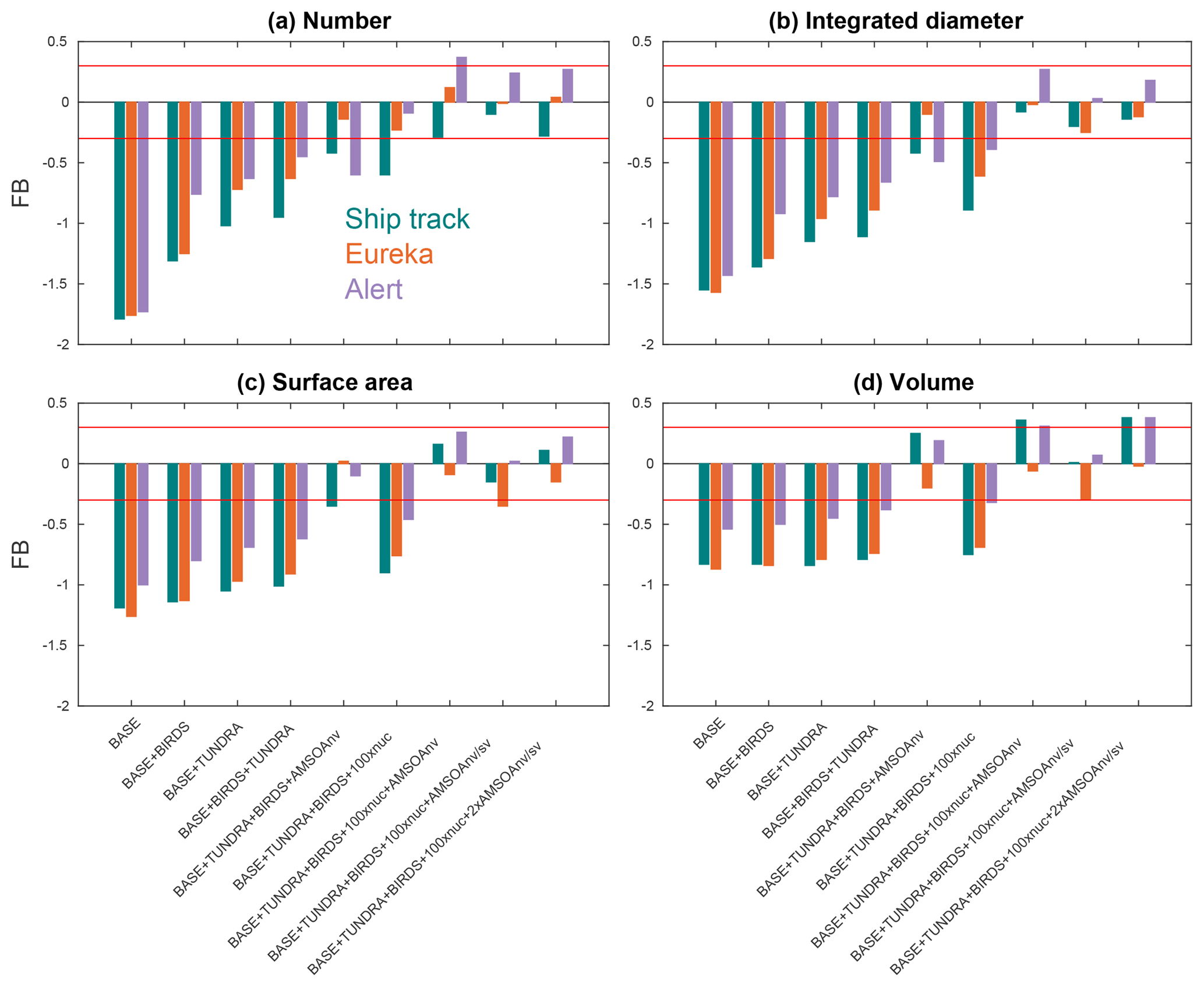
Figure 6Fractional bias (FB; as defined in Sect. 2.2.5) between the nine GEOS-Chem-TOMAS simulations (described in Table 1 and Sect. 2.3) and the SMPS measurements (described in Sect. 2.1) for the first four moments of the summertime- (July and August 2016) median aerosol size distributions at Alert, Eureka, and during the CCGS Amundsen cruise shown in Figs. 3, 4, and 5, respectively. Color coded by geographic site (see the legend in panel (a)). Red lines show the FB values of −0.3 and +0.3, which are the bounds for satisfactory model performance.
Considering each moment separately, Fig. 6 shows the model–measurement FB (defined in Sect. 2.2.5) for the first four moments of the size distributions, for the three measurement platforms, and for all simulations. Among the moments, the zeroth moment (number) is most sensitive to the addition of the seabird colony and tundra NH3 emissions, whereas the third moment (volume) shows the least sensitivity. The first and second moments show an intermediate sensitivity to the NH3 source. The volume distribution shows the highest sensitivity to the AMSOA source with relatively less sensitivity towards the lower moments. Figures 3–5 show that AMSOA contributes about half of the simulated total surface area and volume distributions. Figure 6 shows that the combination of NH3, nucleation scaling, and mixed volatility AMSOA is required to simultaneously bring all four moments within the range of satisfactory model performance at all three measurement platforms (BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv/sv simulation), except for a small exceedance for Eureka's second moment. The volume moment provides a year-matched constraint on the total aerosol mass concentrations in our simulations. The BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv/sv simulation has the lowest volume distribution FB for both Alert (+0.07) and the ship track (+0.01), whereas for Eureka two simulations had the lowest FB, BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv (−0.06) and (+0.06). For all three sites, the implementation of AMSOA reduced the volume fractional bias within the bounds of satisfactory model performance relative to an otherwise similar simulation without AMSOA. These general improvements of the simulations with the addition of AMSOA offers support for a key role of marine biogenic emissions in shaping the Arctic size distributions.
3.3 Role of AMSOA during a growth event in Canadian Arctic Archipelago
Figure 7 provides an example of a particle growth event from the summer 2016 CCGS Amundsen ship track through the Canadian Arctic Archipelago. The observations during the period from 14 to 15 August 2016 show the growth of particles from about 15 to about 40 nm over a duration of about 8 h. Collins et al. (2017) and Burkart et al. (2017a) also report growth rates of about 2–4 nm h−1 for similar size aerosols during other growth events observed from the CCGS Amundsen during the 2016 cruise. Figure 7b shows that without the source of AMSOA (BASE + TUNDRA + BIRDS + 100xnuc simulation), the nascent particles do not exhibit sufficient growth beyond about 15 nm by condensation of H2SO4 and MSA alone. Figure 7c shows that with the source of nonvolatile AMSOA for the BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv simulation, there is growth from about 10 to about 40 nm over 8 h, a growth rate that is slightly faster than observed for this event and faster than reported by Burkart et al. (2017a).
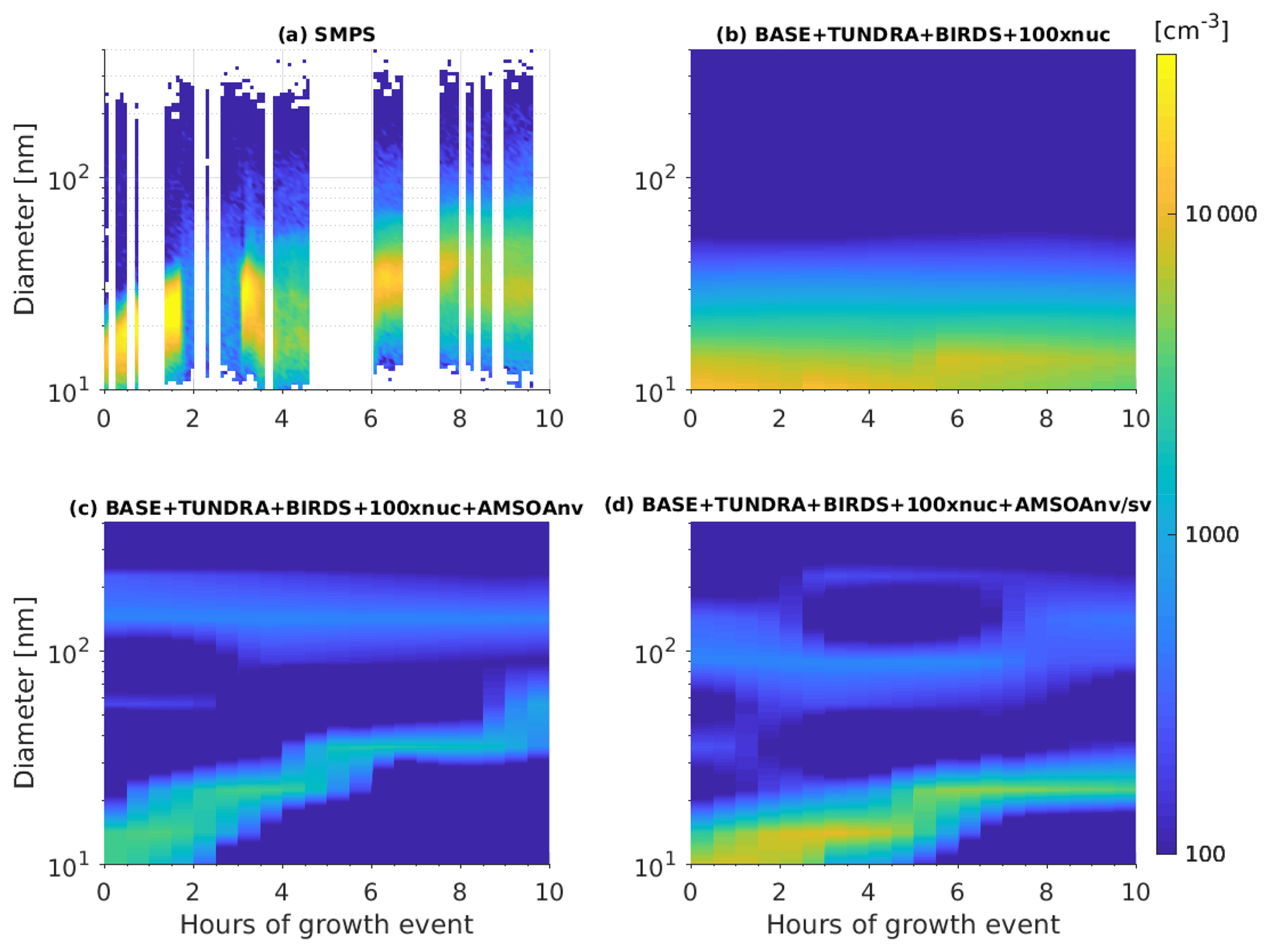
Figure 7Time series of size-resolved aerosol number distributions (color contours show dN∕dlog 10Dp) for the growth event from 14 to 15 August 2016 as (a) observed along the Amundsen ship track (described in Sect. 2.1) and for the GEOS-Chem-TOMAS simulations along the ship track: (b) BASE + TUNDRA + BIRDS + 100xnuc, (c) BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv, and (d) BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv/sv. Simulations are described in Table 1 and Sect. 2.3.
Figure 7d shows that particles grow from about 10 to 20 nm over about 8 h for the BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv/sv simulation, which is slightly slower than the observed rate and slower than the BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv simulation, which assumed nonvolatile AMSOA. Semi-volatile AMSOA also enables faster growth of the larger mode around 90 nm, in agreement with the observations of Burkart et al. (2017a) that the larger mode grew faster. This key role for semi-volatile AMSOA during the frequent summertime growth events in the Canadian Arctic Archipelago is consistent with measurement-based studies for this region (Willis et al., 2017; Leaitch et al., 2018; Tremblay et al., 2019). Based on this single-case model–measurement comparison, we can not infer the actual volatility of the condensing vapors in the region as the simulated grid-box mean might not be fully representative of the observations at the measurement site. However, Fig. 7 serves to demonstrate the impact of AMSOA and its volatility on particle growth.
3.4 Size-resolved aerosol composition
Few measurements are available regarding the composition of the summertime Arctic Aitken mode due to insufficient instrument detection limits to detect the extremely low mass concentrations in this size range (less than 100 ng m−3). However, the limited information available does provide insight into the processes that shape the size distribution. For example, Giamarelou et al. (2016) found using volatility analysis that 12 nm-diameter particles in the Svalbard region were primarily ammoniated sulfates, pointing to the importance of particle formation by ternary nucleation of gas-phase NH3, H2SO4 and water, and initial growth by low volatility sulfur-containing vapors.
Figure 8 shows the size-resolved mass fractions for the various aerosol components for the BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv/sv simulation. For the simulated sub-10 nm particles, the simulated summertime (July and August) mean mass fractions at Alert, Eureka, and for the ship track are primarily biogenic sulfate and MSA that arise from oxidation of DMS, which is released to the atmosphere by marine biological activity. Thus, the simulated composition exhibits similarities with the Svalbard measurements, with the additional identification of a biogenic source. Figure 8 is also consistent with the strong summertime biogenic sulfate component observed in the Canadian Arctic Archipelago by Ghahremaninezhad et al. (2016).
Limited measurements of the composition of particles with diameters between 50 and 80 nm during growth events at Eureka show that these particles are almost entirely composed of organic compounds, which could also include a minor contribution from MSA (Tremblay et al., 2019). Unfortunately, these measurements were limited to a few growth events and cannot be directly compared with the simulated summertime mean mass fractions shown in Fig. 8. Burkart et al. (2017a) calculated a cloud condensation nuclei (CCN) hygroscopicity parameter (Petters and Kreidenweis, 2007) for the particles during a growth event in the Canadian Arctic Archipelago and found a value also indicating a mostly organic composition for those particles large enough to act as CCN. Figure 8 shows that our simulation captures an increasing contribution of organics with particle diameters around 50–100 nm (sizes that can act as CCN), reflecting the key role of AMSOA in the growth of particles to sizes that can be climate-relevant by acting as seeds for cloud droplet formation, or directly scattering and absorbing radiation (diameters larger than about 100 nm). Semi-volatile organic vapors have also been shown to play a role in the growth of particles after they reach diameters of about 5 nm (Tröstl et al., 2016). However, as noted by Karl et al. (2013), lower volatility vapors are needed for initial growth over the first few nanometers. Thus, semi-volatile organic vapors are likely only important in later growth beyond 10–20 nm.
Figure 8 shows that the simulated contribution of organics is greatest for the ship track, reflecting the marine source of the condensable organics in our simulation. The ship track also has the strongest contribution of “other organics” in the sub-100 nm range, with a peak contribution for particle diameters of 10–30 nm. This sub-100 nm organic contribution (shaded in dark green in Fig. 8) represents the mass fraction contribution of marine primary organic aerosol (POA) in our simulation. The primary aerosol, particularly in the marine boundary layer, is climate-relevant as it grows due to condensation of AMSOA towards sizes of 50–100 nm. As described in Sect. 2.2, all sea-spray emissions with diameters smaller than 100 nm are treated as hydrophobic organic carbon. We use the Mårtensson et al. (2003) parameterization, which in comparison with other parameterizations yields among the largest sub-100 nm diameter sea-spray particle production fluxes for temperatures near 273 K (de Leeuw et al. 2011, Fig. 9).
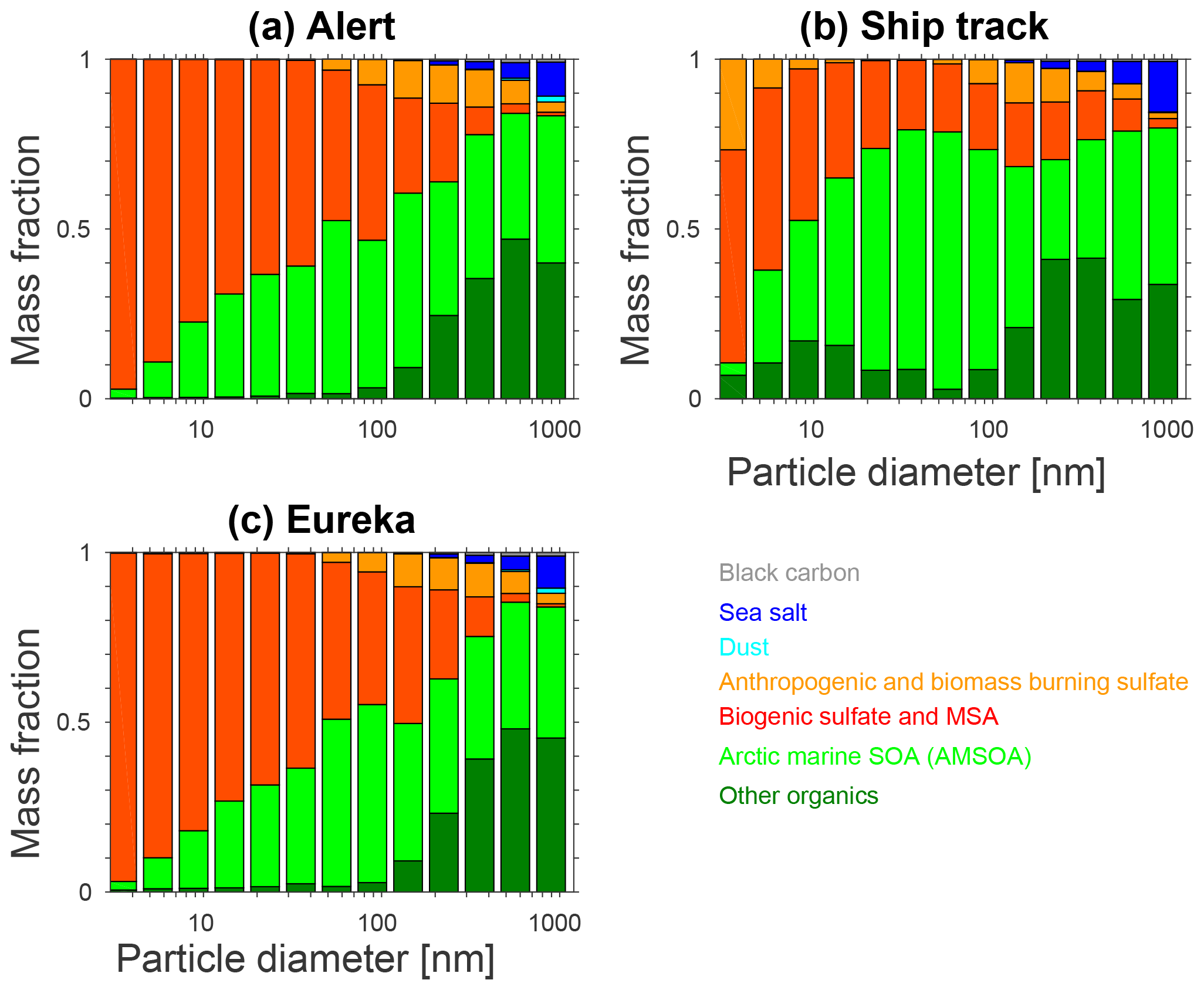
Figure 8Simulated summertime- (July and August 2016) mean size-resolved aerosol component mass fractions for (a) Alert, (b) the Amundsen ship track, and (c) Eureka, for the BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv/sv simulation as described in Table 1 and Sect. 2.3. Other organics includes all organic aerosol except the AMSOA. Biogenic sulfate includes all sulfate derived from the oxidation of DMS.
Furthermore, for particle diameters from 100 to 500 nm, the Mårtensson et al. (2003) parameterization exceeds the uncertainty ranges identified by Lewis and Schwartz (2004); therefore, the role of primary marine emissions is likely overestimated by this parameterization for this size range. There is evidence that primary organics could contribute 10 %–20 % of the mass of particles with diameters less than 500 nm (de Leeuw et al., 2011). Thus, a portion of the mass fraction labeled as sea salt on Fig. 8 for the 100– 500 nm size range could be organics that are misrepresented as sea salt. However as the sea-spray fraction in Fig. 8 indicates, this potential primary-organic contribution is considerably smaller than the AMSOA mass fraction. As a result, any missing POA for 100 to 500 nm diameter particles is likely not sufficient to yield a match for the volume distributions shown in Figs. 3–5. The dark green shading (“other organics”) in Fig. 8 for sizes larger than 100 nm represents contributions to the mass fractions by organics that have been transported from lower latitudes, including those primary and secondary aerosols from biomass burning and other non-marine lower-latitude sources. Sulfate transported from lower latitudes is included in the anthropogenic and biomass burning category (shown in orange shading in Fig. 8).
3.5 Impact of AMSOA on climate-relevant aerosol number concentrations as well as direct and indirect radiative effects
In this section, we consider the role of AMSOA on the simulated total number concentration of aerosols with diameter larger than 50 nm (N50) and 100 nm (N100) and the associated radiative effects using our simulation with the lowest overall model–measurement MFE (BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv/sv simulation) relative to the simulation without AMSOA (BASE + TUNDRA + BIRDS + 100xnuc). These simulations include particle precursor emissions for the entire Arctic as described in Sect. 2.2.2 and 2.2.5. Figure 9 shows the pan-Arctic distribution of the simulated summertime- (July and August) mean surface-layer N50 and N100 for the BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv/sv simulation. In the Canadian Arctic Archipelago region, the simulated summertime-mean N50 (50 to 100 cm−3) and N100 (10 to 30 cm−3) ranges are consistent with monthly mean values from observations at Alert presented in Croft et al. (2016a). Figure 9b and e show that the addition of AMSOA (the BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv/sv simulation relative to the BASE + TUNDRA + BIRDS + 100xnuc simulation) yields a N50 increase of about 50–75 cm−3 and a N100 increase of about 20 cm−3 in the Canadian Arctic Archipelago. These differences in the simulated N50 and N100 are attributed to the process of growth by the condensation of AMSOA, and will have climate-relevant impacts on aerosol radiative effects.
Figure 9 also shows the geographic distribution of the top-of-the-atmosphere DRE and cloud-albedo AIE (described in Sect. 2.2) for AMSOA (comparing between the BASE + TUNDRA + BIRDS + 100xnuc and BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv/sv simulations). The pan-Arctic mean DRE attributed to condensational growth by AMSOA is −0.04 W m−2. The simulated AMSOA effect is largest (about −0.1 to −0.2 W m−2) over the regions of open water such as Baffin Bay, east of Greenland, and the Bering Sea. These are also regions of the largest N100 change as particles with diameters larger than about 100 nm contribute strongly to the scattering of solar radiation.
The pan-Arctic mean cloud-albedo AIE attributed to AMSOA is about −0.4 W m−2. The AIE shows a similar geographic distribution to the changes in the N50, with the largest values of −1 to −2 W m−2 in the Canadian Arctic Archipelago and east of Greenland, again related to the open water regions associated with the AMSOA-precursor vapor flux implemented in our simulations.
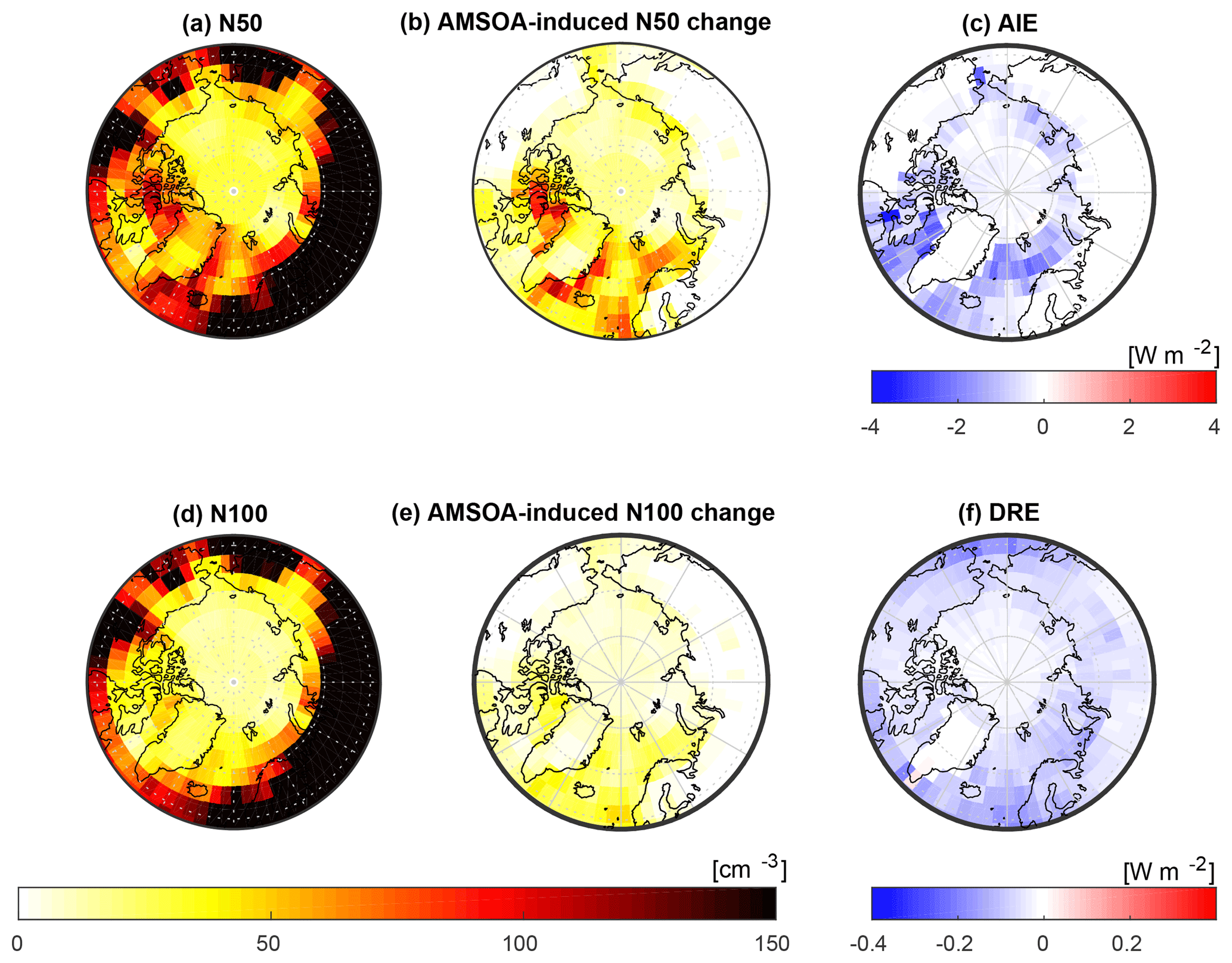
Figure 9Impact of AMSOA simulated summertime- (July and August 2016) mean geographic distribution of surface-layer aerosol number concentrations with respect to (a) particles with diameters larger than 50 nm (N50) for the BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv/sv simulation; (b) the surface-layer N50 difference for the BASE + TUNDRA + BIRDS + 100xnuc + AMSOAnv/sv simulation relative to the BASE + TUNDRA + BIRDS + 100xnuc simulation; (c) the aerosol indirect effect (AIE) at top of the atmosphere (methodology described in Sect. 2.2) between these two simulations, attributed to AMSOA; (d) similar to (a) but for N100; (e) similar to (b) but for N100 difference; and (f) direct aerosol effect (DRE) at top of the atmosphere (methodology described in Sect. 2.2) between these two simulations, attributed to AMSOA.
We caution that several uncertainties are associated with our quantification of the DRE and AIE. The sources for AMSOA precursor vapors, and also for the seabird colony and tundra ammonia are uncertain. In addition, there are uncertainties in the DRE and AIE due to the simulated cloud fields, surface albedo, and particle size distributions in the absence of AMSOA. Future work is needed to improve the emission parameterizations for Arctic particle precursors. Our simulations include AMSOA and tundra NH3 emissions that vary spatially with land type, but additional factors such as temperature and biological activity could also control these emissions and could be investigated in future studies. Further work is also needed to better understand the source and nature of AMSOA-precursor vapors. Additionally, work to examine the impact of a sub-grid plume processing parameterization for the seabird colony NH3 emissions could be beneficial. These effects could change the spatial distribution and magnitudes of the radiative effects attributed to AMSOA, and reduce associated uncertainty. As a result of these uncertainties and knowledge gaps, we consider the values presented for the DRE and AIE as an indication of the order of magnitude that AMSOA may contribute to the DRE and AIE. However, we view these calculations as identification that the impact of condensational growth by AMSOA is expected to be relevant for the Arctic climate.
We used the GEOS-Chem-TOMAS chemical transport model with size-resolved aerosol microphysics to interpret measurements conducted during the summertime of 2016 in the Canadian Arctic Archipelago, some as part of the NETwork on Climate and Aerosols: Addressing key uncertainties in Remote Canadian Environments (NETCARE) project (Abbatt et al., 2019). Three measurement platforms were considered. These platforms were located at Alert and Eureka, both in Nunavut, in Canada and also onboard the CCGS Amundsen. We focused on examining the key processes that build summertime aerosol size distributions in this region, particularly the role of Arctic marine secondary organic aerosol (AMSOA) condensation. The terminology AMSOA was used to indicate secondary organic aerosol formed from precursors from marine (ice-free seawater) sources north of 50∘ N, excluding MSA, which we treated as a separate aerosol component. In the Canadian Arctic Archipelago, AMSOA is likely strongly controlled by emissions from marine biogenic activity (Willis et al., 2017; Leaitch et al., 2018).
We find that AMSOA contributes strongly to the summertime particle size distributions in the Canadian Arctic Archipelago. Building on measurement-based studies from the NETCARE project, we implemented a flux of condensable AMSOA-precursor vapors into our GEOS-Chem-TOMAS simulations. This fixed flux of 500 of AMSOA-precursor vapors (with a yield of unity) emitted from open seawater in the Arctic and near Arctic (north of 50∘ N) was determined by tuning the simulated flux to achieve model–measurement agreement for the first four moments of the aerosol size distributions at the three measurements platforms in the Canadian Arctic Archipelago. This was a crude representation of the source function due to the lack of knowledge about the nature and source of AMSOA. However, implementation of condensable AMSOA in our simulation reduced the model–measurement MFE for the summertime median aerosol size distributions by a factor of 2–4 across the three measurement platforms, indicating a strong sensitivity of the simulated size distributions to growth by AMSOA. Without AMSOA, particle growth to diameters of 50–200 nm was strongly underpredicted in our simulations. Increasing the particle nucleation rate 100-fold further reduced the MFE for Eureka and the ship track, indicating that additional materials such as (but not limited to) gas-phase iodine, and/or amines, and/or possibly extremely low volatility organics may be participating in nucleation, and/or other mechanisms such as particle fragmentation, leading to faster rates than our ternary scheme.
Introduction of a 30∕70 nonvolatile/semi-volatile split for the simulated AMSOA reduced the model–measurement MFE 2- to 3-fold for the summertime aerosol size distributions for Alert (0.10) and the ship track (0.10), and also yielded the lowest MFE for Eureka (0.09) if the AMSOA-precursor vapor source flux was doubled. These findings offer support to the fact that the condensing AMSOA contributing to the growth of particles with diameters larger than about 20 nm in the Canadian Arctic Archipelago could contain a large fraction of semi-volatile species.
Size-resolved mass fractions indicated that the initial growth of simulated nascent sub-10 nm particles (arising from ternary nucleation of ammonia, sulfuric acid, and water vapors) occurred primarily by condensation involving biogenic sulfate and MSA, both derived from the oxidation of dimethyl sulfide of marine origin. AMSOA contributed about 20 %–80 % to size-resolved particle mass for diameters between 10 and 100 nm, with the largest contributions for the ship track simulation. The simulated contribution of primary organics of sea-spray origin to sub-100 nm particle mass fractions was largest for the ship track simulation in the marine boundary layer, with mass fractions approaching 20 % for particles with diameters around 10 to 20 nm, and was likely overestimated by the sea-spray parameterization.
By comparing our best (lowest MFE) simulations with and without the AMSOA formed from precursors with marine sources north of 50∘ N, we found that AMSOA had a strong summertime- and pan-Arctic-mean top-of-the-atmosphere aerosol DRE of −0.04 W m−2, and cloud-albedo AIE of −0.4 W m−2. The comparison of these simulations with and without AMSOA suggested a strong sensitivity of climate-relevant effects to AMSOA. However, we caution that a high level of uncertainty is associated with our quantification of these effects, due to uncertainty about the composition, and source fluxes for these condensing vapors. Future studies are needed to reduce these uncertainties.
Many knowledge gaps remain regarding the role of organics within the processes that shape particle size distributions in the Arctic climate system. For example, Willis et al. (2017) found that the organics in the aerosol in the summertime Canadian Arctic Archipelago were not like typical biogenic SOA, having instead a character with a long hydrocarbon chain, implying a fatty-acid-type precursor, which is a common component of the marine microlayer. Additionally, Mungall et al. (2017) found that the marine microlayer in the Canadian Arctic Archipelago was a source of OVOCs, which could also be related to AMSOA. Further measurements are needed to identify and quantify the fluxes of the organic vapors that yield AMSOA through condensational particle growth, along with their sources, chemistry, and spatial distribution within the Arctic. Additionally, given the climate relevance of NH3 through the formation of nascent particles, measurements are needed to better identify and quantify its sources across the summertime Arctic, and to further examine the spatial distribution of the subsequent Arctic particle growth events. Further, size-resolved particle concentrations and composition measurements (particularly for sulfate and organic aerosol), would constrain the controlling processes for all sub-micron particle diameters. Such work could also reduce the uncertainty related to aerosol effects within the Arctic climate system. This work will also lay a foundation for the prediction of future aerosol effects within the context of a rapidly changing and warming Arctic, as sea ice extent and biological and anthropogenic activity are altered.
The NETCARE atmospheric measurements are publicly available through the Government of Canada open data portal (https://open.canada.ca, last access: 27 February 2019), and the oceanic measurements are available via the Polar Data Catalogue (https://www.polardata.ca/, last access: 27 February 2019).
BC developed code and conducted the GEOS-Chem-TOMAS simulations. RVM and JRP provided advice on the simulations. ALH and JKK contributed to code developments. WRL, LH, SS, and GRW contributed measurements from Alert. RYWC, ST, and PLH contributed measurements from Eureka. DBC, AM, and JGM contributed measurements from the cruise track. JB, ELM, and MDW helped with the interpretation of NETCARE measurements. BC led the writing of the paper with contributions from all coauthors. JPDA was the lead PI of the NETCARE project.
The authors declare that they have no conflict of interest.
This article is part of the special issue “NETCARE (Network on Aerosols and Climate: Addressing Key Uncertainties in Remote Canadian Environments) (ACP/AMT/BG inter-journal SI)”. It is not associated with a conference.
We thank the observatory operators at Alert, Desiree Toom, Alina Chivulescu, Dan Veber, Wendy Zhang, Darrell Ernst, as well as Andrew Platt and Carrie Taylor, for their support of the Environment and Climate Change Canada (ECCC) aerosol programme at Alert and Eureka. We are grateful for the hard work and dedication of the CCGS Amundsen crew and for the help from our colleagues onboard. We thank Oona Kupiainen-Määttä, Tinja Olenius, Jan Julin, Hanna Vehkamäki, Benjamin Murphy, and Ilona Riipinen for their support of this project via the provision of the Atmospheric Cluster Dynamics Code. The authors also thank Bonne Ford and Katelyn O'Dell for preparing the 2016 GFED4 files in the format required for input to the GEOS-Chem-TOMAS model.
This work was supported by the Climate Change and Atmospheric Research
programme at NSERC, as part of the NETCARE project. This work was also
supported by Environment and Climate Change Canada (ECCC) and by the Ocean
Frontier Institute. Rachel Y.-W. Chang, Samantha Tremblay, and Patrick L. Hayes acknowledge support
from the NSERC CCAR project “Probing the Atmosphere of the High Arctic” (PAHA)
led by PI James R. Drummond, as well as support from the NSERC Discovery
Grants Program (RGPIN-05002-2014, RGPIN-05173-2014), and CANDAC (Canadian
Network for the Detection of Atmospheric Change). Colorado State University
researchers were supported by the US Department of Energy's Atmospheric
System Research, an Office of Science, Office of Biological and
Environmental Research program, under grant no. DE-SC0011780, the U.S. National Science Foundation, Atmospheric Chemistry program, under grant no. AGS-1559607, and by the U.S. National Oceanic and Atmospheric Administration,
an Office of Science, Office of Atmospheric Chemistry, Carbon Cycle, and
Climate Program, under the cooperative agreement award no. NA17OAR430001. Alexander Moravek's
work was supported by the NSERC CREATE program IACPES postdoctoral
fellowship.
Edited by: Lynn M. Russell
Reviewed by: three anonymous referees
Abbatt, J. P. D., Leaitch, W. R., Aliabadi, A. A., Bertram, A. K., Blanchet, J.-P., Boivin-Rioux, A., Bozem, H., Burkart, J., Chang, R. Y. W., Charette, J., Chaubey, J. P., Christensen, R. J., Cirisan, A., Collins, D. B., Croft, B., Dionne, J., Evans, G. J., Fletcher, C. G., Galí, M., Ghahremaninezhad, R., Girard, E., Gong, W., Gosselin, M., Gourdal, M., Hanna, S. J., Hayashida, H., Herber, A. B., Hesaraki, S., Hoor, P., Huang, L., Hussherr, R., Irish, V. E., Keita, S. A., Kodros, J. K., Köllner, F., Kolonjari, F., Kunkel, D., Ladino, L. A., Law, K., Levasseur, M., Libois, Q., Liggio, J., Lizotte, M., Macdonald, K. M., Mahmood, R., Martin, R. V., Mason, R. H., Miller, L. A., Moravek, A., Mortenson, E., Mungall, E. L., Murphy, J. G., Namazi, M., Norman, A.-L., O'Neill, N. T., Pierce, J. R., Russell, L. M., Schneider, J., Schulz, H., Sharma, S., Si, M., Staebler, R. M., Steiner, N. S., Thomas, J. L., von Salzen, K., Wentzell, J. J. B., Willis, M. D., Wentworth, G. R., Xu, J.-W., and Yakobi-Hancock, J. D.: Overview paper: New insights into aerosol and climate in the Arctic, Atmos. Chem. Phys., 19, 2527–2560, https://doi.org/10.5194/acp-19-2527-2019, 2019.
Abdul-Razzak, H. and Ghan, S. J.: A parameterization of aerosol activation 3. Sectional representation, J. Geophys. Res., 107, 4026, https://doi.org/10.1029/2001JD000483, 2002.
Adams, P. J. and Seinfeld, J. H.: Predicting global aerosol size distributions in general circulation models, J. Geophys. Res., 107, 4310–4370, 2002.
Alexander, B., Allman, D. J., Amos, H. M., Fairlie, T. D., Dachs, J., Hegg, D. A., and Sletten, R. S.: Isotopic constraints on the formation pathways of sulfate aerosol in the marine boundary layer of the subtropical northeast Atlantic Ocean, J. Geophys. Res.-Atmos., 117, 1–17, https://doi.org/10.1029/2011JD016773, 2012.
Allan, J. D., Williams, P. I., Najera, J., Whitehead, J. D., Flynn, M. J., Taylor, J. W., Liu, D., Darbyshire, E., Carpenter, L. J., Chance, R., Andrews, S. J., Hackenberg, S. C., and McFiggans, G.: Iodine observed in new particle formation events in the Arctic atmosphere during ACCACIA, Atmos. Chem. Phys., 15, 5599–5609, https://doi.org/10.5194/acp-15-5599-2015, 2015.
Almeida, J., Schobesberger, S., Kürten, A., Ortega, I. K., Kupiainen-Määttä, O., Praplan, A. P., Adamov, A., Amorim, A., Bianchi, F., Breitenlechner, M., David, A., Dommen, J., Donahue, N. M., Downard, A., Dunne, E., Duplissy, J., Ehrhart, S., Flagan, R. C., Franchin, A., Guida, R., Hakala, J., Hansel, A., Heinritzi, M., Henschel, H., Jokinen, T., Junninen, H., Kajos, M., Kangasluoma, J., Keskinen, H., Kupc, A., Kurtén, T., Kvashin, A. N., Laaksonen, A., Lehtipalo, K., Leiminger, M., Leppä, J., Loukonen, V., Makhmutov, V., Mathot, S., McGrath, M. J., Nieminen, T., Olenius, T., Onnela, A., Petäjä, T., Riccobono, F., Riipinen, I., Rissanen, M., Rondo, L., Ruuskanen, T., Santos, F. D., Sarnela, N., Schallhart, S., Schnitzhofer, R., Seinfeld, J. H., Simon, M., Sipilä, M., Stozhkov, Y., Stratmann, F., Tomé, A., Tröstl, J., Tsagkogeorgas, G., Vaattovaara, P., Viisanen, Y., Virtanen, A., Vrtala, A., Wagner, P. E., Weingartner, E., Wex, H., Williamson, C., Wimmer, D., Ye, P., Yli-Juuti, T., Carslaw, K. S., Kulmala, M., Curtius, J., Baltensperger, U., Worsnop, D. R., Vehkamäki, H., and Kirkby, J.: Molecular understanding of sulphuric acid-amine particle nucleation in the atmosphere, Nature, 502, 359–363, https://doi.org/10.1038/nature12663, 2013.
Amos, H. M., Jacob, D. J., Holmes, C. D., Fisher, J. A., Wang, Q., Yantosca, R. M., Corbitt, E. S., Galarneau, E., Rutter, A. P., Gustin, M. S., Steffen, A., Schauer, J. J., Graydon, J. A., Louis, V. L. St., Talbot, R. W., Edgerton, E. S., Zhang, Y., and Sunderland, E. M.: Gas-particle partitioning of atmospheric Hg(II) and its effect on global mercury deposition, Atmos. Chem. Phys., 12, 591–603, https://doi.org/10.5194/acp-12-591-2012, 2012.
Asmi, E., Kondratyev, V., Brus, D., Laurila, T., Lihavainen, H., Backman, J., Vakkari, V., Aurela, M., Hatakka, J., Viisanen, Y., Uttal, T., Ivakhov, V., and Makshtas, A.: Aerosol size distribution seasonal characteristics measured in Tiksi, Russian Arctic, Atmos. Chem. Phys., 16, 1271–1287, https://doi.org/10.5194/acp-16-1271-2016, 2016.
Baranizadeh, E., Murphy, B. N., Julin, J., Falahat, S., Reddington, C. L., Arola, A., Ahlm, L., Mikkonen, S., Fountoukis, C., Patoulias, D., Minikin, A., Hamburger, T., Laaksonen, A., Pandis, S. N., Vehkamäki, H., Lehtinen, K. E. J., and Riipinen, I.: Implementation of state-of-the-art ternary new-particle formation scheme to the regional chemical transport model PMCAMx-UF in Europe, Geosci. Model Dev., 9, 2741–2754, https://doi.org/10.5194/gmd-9-2741-2016, 2016.
Barnes, I., Hjorth, J., and Mihalopoulos, N.: Dimethyl Sulfide and Dimethyl Sulfoxide and Their Oxidation in theirAtmosphere, Chem. Rev., 106, 940–975, https://doi.org/10.1021/cr020529+, 2006.
Barrie, L. A.: Arctic Aerosols: Composition, Sources and Transport BT – Ice Core Studies of Global Biogeochemical Cycles, edited by: Delmas, R. J., 1–22, Springer, Berlin, Heidelberg, 1995.
Bohren, C. F. and Huffman, D. R.: Absorption and scattering of light by small particles, Wiley Interscience, New York, USA, 1983.
Bond, T. C., Bhardwaj, E., Dong, R., Jogani, R., Jung, S., Roden, C., Streets, D. G., and Trautmann, N. M.: Historical emissions of black and organic carbon aerosol from energy-related combustion, 1850–2000, Global Biogeochem. Cy., 21, 1–16, https://doi.org/10.1029/2006GB002840, 2007.
Bouwman, A. F., Lee, D. S., Asman, W. A. H., Dentener, F. J., Van Der Hoek, K. W., Olivier, J. G. J., and Tg, N.: A global high-resolution emission inventory for ammonia, Global Biogeochem. Cy., 11, 561–587, 1997.
Boylan, J. W. and Russell, A. G.: PM and light extinction model performance metrics, goals, and criteria for three-dimensional air quality models, Atmos. Environ., 40, 4946–4959, https://doi.org/10.1016/j.atmosenv.2005.09.087, 2006.
Breider, T. J., Mickley, L. J., Jacob, D. J., Wang, Q. Q., Fisher, J. A., Chang, R. Y., and Alexander, B.: Annual distributions and sources of Arctic aerosol components, aerosol optical depth, and aerosol absorption, J. Geophys. Res.-Atmos., 119, 4107–4124, https://doi.org/10.1002/2013JD020996, 2014.
Breider, T. J., Mickley, L. J., Jacob, D. J., Ge, C., Wang, J., Sulprizio, M. P., Croft, B., Ridley, D. A., Mcconnell, J. R., Sharma, S., Husain, L., Dutkiewicz, V. A., Eleftheriadis, K., Skov, H., and Hopke, P. K.: Multidecadal trends in aerosol radiative forcing over the Arctic: Contribution of changes in anthropogenic aerosol to Arctic warming since 1980, J. Geophys. Res.-Atmos., 122, 1–22, https://doi.org/10.1002/2016JD025321, 2017.
Brock, C. A., Cozic, J., Bahreini, R., Froyd, K. D., Middlebrook, A. M., McComiskey, A., Brioude, J., Cooper, O. R., Stohl, A., Aikin, K. C., de Gouw, J. A., Fahey, D. W., Ferrare, R. A., Gao, R.-S., Gore, W., Holloway, J. S., Hübler, G., Jefferson, A., Lack, D. A., Lance, S., Moore, R. H., Murphy, D. M., Nenes, A., Novelli, P. C., Nowak, J. B., Ogren, J. A., Peischl, J., Pierce, R. B., Pilewskie, P., Quinn, P. K., Ryerson, T. B., Schmidt, K. S., Schwarz, J. P., Sodemann, H., Spackman, J. R., Stark, H., Thomson, D. S., Thornberry, T., Veres, P., Watts, L. A., Warneke, C., and Wollny, A. G.: Characteristics, sources, and transport of aerosols measured in spring 2008 during the aerosol, radiation, and cloud processes affecting Arctic Climate (ARCPAC) Project, Atmos. Chem. Phys., 11, 2423–2453, https://doi.org/10.5194/acp-11-2423-2011, 2011.
Browse, J., Carslaw, K. S., Arnold, S. R., Pringle, K., and Boucher, O.: The scavenging processes controlling the seasonal cycle in Arctic sulphate and black carbon aerosol, Atmos. Chem. Phys., 12, 6775–6798, https://doi.org/10.5194/acp-12-6775-2012, 2012.
Browse, J., Carslaw, K. S., Mann, G. W., Birch, C. E., Arnold, S. R., and Leck, C.: The complex response of Arctic aerosol to sea-ice retreat, Atmos. Chem. Phys., 14, 7543–7557, https://doi.org/10.5194/acp-14-7543-2014, 2014.
Burkart, J., Hodshire, A. L., Mungall, E. L., Pierce, J. R., Collins, D. B., Ladino, L. A., Lee, A. K. Y., Irish, V., Wentzell, J. J. B., Liggio, J., Papakyriakou, T., Murphy, J., and Abbatt, J.: Organic Condensation and Particle Growth to CCN Sizes in the Summertime Marine Arctic Is Driven by Materials More Semivolatile Than at Continental Sites, Geophys. Res. Lett., 44, 10725–10734, https://doi.org/10.1002/2017GL075671, 2017a.
Burkart, J., Willis, M. D., Bozem, H., Thomas, J. L., Law, K., Hoor, P., Aliabadi, A. A., Köllner, F., Schneider, J., Herber, A., Abbatt, J. P. D., and Leaitch, W. R.: Summertime observations of elevated levels of ultrafine particles in the high Arctic marine boundary layer, Atmos. Chem. Phys., 17, 5515–5535, https://doi.org/10.5194/acp-17-5515-2017, 2017b.
Carpenter, L. J. and Nightingale, P. D.: Chemistry and Release of Gases from the Surface Ocean, Chem. Rev., 115, 4015–4034, https://doi.org/10.1021/cr5007123, 2015.
Carpenter, L. J., Archer, S. D., and Beale, R.: Ocean-atmosphere trace gas exchange, Chem. Soc. Rev., 41, 6473–6506, https://doi.org/10.1039/c2cs35121h, 2012.
Carslaw, K. S., Lee, L. A., Reddington, C. L., Pringle, K. J., Rap, A., Forster, P. M., Mann, G. W., Spracklen, D. V., Woodhouse, M. T., Regayre, L. A., and Pierce, J. R.: Large contribution of natural aerosols to uncertainty in indirect forcing, Nature, 503, 67, https://doi.org/10.1038/nature12674, 2013.
Chang, R. Y.-W., Leck, C., Graus, M., Müller, M., Paatero, J., Burkhart, J. F., Stohl, A., Orr, L. H., Hayden, K., Li, S.-M., Hansel, A., Tjernström, M., Leaitch, W. R., and Abbatt, J. P. D.: Aerosol composition and sources in the central Arctic Ocean during ASCOS, Atmos. Chem. Phys., 11, 10619–10636, https://doi.org/10.5194/acp-11-10619-2011, 2011a.
Chang, R. Y. W., Sjostedt, S. J., Pierce, J. R., Papakyriakou, T. N., Scarratt, M. G., Michaud, S., Levasseur, M., Leaitch, W. R., and Abbatt, J. P. D.: Relating atmospheric and oceanic DMS levels to particle nucleation events in the Canadian Arctic, J. Geophys. Res.-Atmos., 116, 1–10, https://doi.org/10.1029/2011JD015926, 2011b.
Charlson, R. J., Schwartz, S. E., Hales, J. M., Cess, R. D., Coakley, J. A., Hansen, J. E., and Hofmann, D. J.: Climate Forcing by Anthropogenic Aerosols, Science, 255, 423–430, 1992.
Chatfield, R. B. and Crutzen, P. J.: Are There Interactions of Iodine and Sulfur Species in Marine Air Photochemistry, J. Geophys. Res., 95, 22319–22341, https://doi.org/10.1029/JD095iD13p22319, 1990.
Chen, H., Ezell, M. J., Arquero, K. D., Varner, M. E., Dawson, M. L., Gerber, R. B., and Finlayson-Pitts, B. J.: New Particle Formation and Growth from Methanesulfonic Acid, Trimethylamine and Water, Phys. Chem. Chem. Phys., 17, 13699–13709, 2015.
Chin, M., Jacob, D. J., Gardner, G. M., Foreman-Fowler, M. S., Spiro, P. A., and Savoie, D. L.: A global three-dimensional model of tropospheric sulfate, J. Geophys. Res.-Atmos., 101, 18667–18690, https://doi.org/10.1029/96JD01221, 1996.
Chiu, R., Tinel, L., Gonzalez, L., Ciuraru, R., Bernard, F., George, C., and Volkamer, R.: UV photochemistry of carboxylic acids at the air-sea boundary: A relevant source of glyoxal and other oxygenated VOC in the marine atmosphere, Geophys. Res. Lett., 44, 1079–1087, https://doi.org/10.1002/2016GL071240, 2017.
Collins, D. B., Ault, A. P., Moffet, R. C., Ruppel, M. J., Cuadra-Rodriguez, L. A., Guasco, T. L., Corrigan, C. E., Pedler, B. E., Azam, F., Aluwihare, L. I., Bertram, T. H., Roberts, G. C., Grassian, V. H., and Prather, K. A.: Impact of marine biogeochemistry on the chemical mixing state and cloud forming ability of nascent sea spray aerosol, J. Geophys. Res.-Atmos., 118, 8553–8565, https://doi.org/10.1002/jgrd.50598, 2013.
Collins, D. B., Bertram, T. H., Sultana, C. M., Lee, C., Axson, J. L., and Prather, K. A.: Phytoplankton blooms weakly influence the cloud forming ability of sea spray aerosol, Geophys. Res. Lett., 43, 9975–9983, https://doi.org/10.1002/2016GL069922, 2016.
Collins, D. B., Burkart, J., Chang, R. Y.-W., Lizotte, M., Boivin-Rioux, A., Blais, M., Mungall, E. L., Boyer, M., Irish, V. E., Massé, G., Kunkel, D., Tremblay, J.-É., Papakyriakou, T., Bertram, A. K., Bozem, H., Gosselin, M., Levasseur, M., and Abbatt, J. P. D.: Frequent ultrafine particle formation and growth in Canadian Arctic marine and coastal environments, Atmos. Chem. Phys., 17, 13119–13138, https://doi.org/10.5194/acp-17-13119-2017, 2017.
Crippa, M., Janssens-Maenhout, G., Dentener, F., Guizzardi, D., Sindelarova, K., Muntean, M., Van Dingenen, R., and Granier, C.: Forty years of improvements in European air quality: regional policy–industry interactions with global impacts, Atmos. Chem. Phys., 16, 3825–3841, https://doi.org/10.5194/acp-16-3825-2016, 2016.
Croft, B., Martin, R. V., Leaitch, W. R., Tunved, P., Breider, T. J., D'Andrea, S. D., and Pierce, J. R.: Processes controlling the annual cycle of Arctic aerosol number and size distributions, Atmos. Chem. Phys., 16, 3665–3682, https://doi.org/10.5194/acp-16-3665-2016, 2016a.
Croft, B., Wentworth, G. R., Martin, R. V., Leaitch, W. R., Murphy, J. G., Murphy, B. N., Kodros, J. K., Abbatt, J. P. D., and Pierce, J. R.: Contribution of Arctic seabird-colony ammonia to atmospheric particles and cloud-albedo radiative effect, Nature Commun., 7, 1–10, https://doi.org/10.1038/ncomms13444, 2016b.
Dall'Osto, M., Beddows, D. C. S., Tunved, P., Krejci, R., Ström, J., Hansson, H. C., Yoon, Y. J., Park, K. T., Becagli, S., Udisti, R., Onasch, T., Ódowd, C. D., Simó, R., and Harrison, R. M.: Arctic sea ice melt leads to atmospheric new particle formation, Sci. Rep.-UK, 7, 1–10, https://doi.org/10.1038/s41598-017-03328-1, 2017.
Dall'Osto, M., Geels, C., Beddows, D. C. S., Boertmann, D., Lange, R., Nøjgaard, J. K., Harrison, R. M., Simo, R., Skov, H., and Massling, A.: Regions of open water and melting sea ice drive new particle formation in North East Greenland, Sci. Rep.-UK, 8, 6109, https://doi.org/10.1038/s41598-018-24426-8, 2018a.
Dall'Osto, M., Simo, R., Harrison, R. M., Beddows, D. C. S., Saiz-Lopez, A., Lange, R., Skov, H., Nøjgaard, J. K., Nielsen, I. E., and Massling, A.: Abiotic and biotic sources influencing spring new particle formation in North East Greenland, Atmos. Environ., 190, 126–134, https://doi.org/10.1016/J.ATMOSENV.2018.07.019, 2018b.
D'Andrea, S. D., Häkkinen, S. A. K., Westervelt, D. M., Kuang, C., Levin, E. J. T., Kanawade, V. P., Leaitch, W. R., Spracklen, D. V., Riipinen, I., and Pierce, J. R.: Understanding global secondary organic aerosol amount and size-resolved condensational behavior, Atmos. Chem. Phys., 13, 11519–11534, https://doi.org/10.5194/acp-13-11519-2013, 2013.
de Leeuw, G., Andreas, E. L., Anguelova, M. D., Fairall, C. W., Lewis, E. R., O'Dowd, C., Schulz, M., and Schwartz, S. E.: Production flux of sea spray aerosol, Rev. Geophys., 49, RG2001, https://doi.org/10.1029/2010RG000349, 2011.
Donahue, N. M., Epstein, S. A., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set: 1. organic-aerosol mixing thermodynamics, Atmos. Chem. Phys., 11, 3303–3318, https://doi.org/10.5194/acp-11-3303-2011, 2011.
Dunne, E. M., Gordon, H., Kurten, A., Almeida, J., Duplissy, J., Williamson, C., Ortega, I. K., Pringle, K. J., Adamov, A., Baltensperger, U., Barmet, P., Benduhn, F., Bianchi, F., Breitenlechner, M., Clarke, A., Curtius, J., Dommen, J., Donahue, N. M., Ehrhart, S., Flagan, R. C., Franchin, A., Guida, R., Hakala, J., Hansel, A., Heinritzi, M., Jokinen, T., Kangasluoma, J., Kirkby, J., Kulmala, M., Kupc, A., Lawler, M. J., Lehtipalo, K., Reddington, C. L. S., Riccobono, F., Richards, N. A. D., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Simon, M., Sipilä, M., Smith, J. N., Stozkhov, Y., Tomé, A., Tröstl, J., Wagner, P. E., Williamson, C., Wimmer, D., Winkler, P. M., Yan, C., and Carslaw, K. S.: Global atmospheric particle formation from CERN CLOUD measurements, Science, 354, 1119–1124, 2016.
Ellis, R. A., Murphy, J. G., Pattey, E., van Haarlem, R., O'Brien, J. M., and Herndon, S. C.: Characterizing a Quantum Cascade Tunable Infrared Laser Differential Absorption Spectrometer (QC-TILDAS) for measurements of atmospheric ammonia, Atmos. Meas. Tech., 3, 397–406, https://doi.org/10.5194/amt-3-397-2010, 2010.
Endresen, Ø., Sørgard, E., Sundet, J. K., Dalsøren, S. B., Isaksen, I. S. A., Berglen, T. F., and Gravir, G.: Emission from international sea transportation and environmental impact, J. Geophys. Res., 108, 4560, https://doi.org/10.1029/2002JD002898, 2003.
Facchini, M. C., Decesari, S., Rinaldi, M., Carbone, C., Finessi, E., Mircea, M., Fuzzi, S., Moretti, F., Tagliavini, E., Ceburnis, D., and O'Dowd, C. D.: Important Source of Marine Secondary Organic Aerosol from Biogenic Amines, Environ. Sci. Technol., 42, 9116–9121, https://doi.org/10.1021/es8018385, 2008.
Fairlie, T. D., Jacob, D. J., and Park, R. J.: The impact of transpacific transport of mineral dust in the United States, Atmos. Environ., 41, 1251–1266, https://doi.org/10.1016/j.atmosenv.2006.09.048, 2007.
Fisher, J. A., Jacob, D. J., Wang, Q., Bahreini, R., Carouge, C. C., Cubison, M. J., Dibb, J. E., Diehl, T., Jimenez, J. L., Leibensperger, E. M., Lu, Z., Meinders, M. B. J., Pye, H. O. T., Quinn, P. K., Sharma, S., Streets, D. G., van Donkelaar, A., and Yantosca, R. M.: Sources, distribution, and acidity of sulfate-ammonium aerosol in the Arctic in winter–spring, Atmos. Environ., 45, 7301–7318, https://doi.org/10.1016/j.atmosenv.2011.08.030, 2011.
Fogal, P. F., LeBlanc, L. M., and Drummond, J. R.: The Polar Environment Atmospheric Research Laboratory (PEARL): Sounding the Atmosphere at 80 North, Arctic, 66, 377–386, 2013.
Freud, E., Krejci, R., Tunved, P., Leaitch, R., Nguyen, Q. T., Massling, A., Skov, H., and Barrie, L.: Pan-Arctic aerosol number size distributions: seasonality and transport patterns, Atmos. Chem. Phys., 17, 8101–8128, https://doi.org/10.5194/acp-17-8101-2017, 2017.
Fuchs, N. A.: The mechanics of aerosols, translated by: Daisley, R. E. and Fuchs, M., edited by: Davies, C. N., Pergamon Press, London, Q. J. Roy. Meteorol. Soc., 91, 249, https://doi.org/10.1002/qj.49709138822, 1964.
Gantt, B. and Meskhidze, N.: The physical and chemical characteristics of marine primary organic aerosol: a review, Atmos. Chem. Phys., 13, 3979–3996, https://doi.org/10.5194/acp-13-3979-2013, 2013.
Garrett, T. J., Brattström, S., Sharma, S., Worthy, D. E. J., and Novelli, P.: The role of scavenging in the seasonal transport of black carbon and sulfate to the Arctic, Geophys. Res. Lett., 38, 1–6, https://doi.org/10.1029/2011GL048221, 2011.
Ghahremaninezhad, R., Norman, A.-L., Abbatt, J. P. D., Levasseur, M., and Thomas, J. L.: Biogenic, anthropogenic and sea salt sulfate size-segregated aerosols in the Arctic summer, Atmos. Chem. Phys., 16, 5191–5202, https://doi.org/10.5194/acp-16-5191-2016, 2016.
Ghahremaninezhad, R., Norman, A.-L., Croft, B., Martin, R. V., Pierce, J. R., Burkart, J., Rempillo, O., Bozem, H., Kunkel, D., Thomas, J. L., Aliabadi, A. A., Wentworth, G. R., Levasseur, M., Staebler, R. M., Sharma, S., and Leaitch, W. R.: Boundary layer and free-tropospheric dimethyl sulfide in the Arctic spring and summer, Atmos. Chem. Phys., 17, 8757–8770, https://doi.org/10.5194/acp-17-8757-2017, 2017.
Giamarelou, M., Eleftheriadis, K., Nyeki, S., Tunved, P., Torseth, K., and Biskos, G.: Indirect evidence of the composition of nucleation mode particles in the high Arctic, J. Geophys. Res.-Atmos., 121, 965–975, https://doi.org/10.1002/2015JD023646, 2016.
Giglio, L., Randerson, J. T., and Van Der Werf, G. R.: Analysis of daily, monthly, and annual burned area using the fourth-generation global fire emissions database (GFED4), J. Geophys. Res.-Biogeo., 118, 317–328, https://doi.org/10.1002/jgrg.20042, 2013.
Gordon, H., Kirkby, J., Baltensperger, U., Bianchi, F., Breitenlechner, M., Curtius, J., Dias, A., Dommen, J., Donahue, N. M., Dunne, E. M., Duplissy, J., Ehrhart, S., Flagan, R. C., Frege, C., Fuchs, C., Hansel, A., Hoyle, C. R., Kulmala, M., Kürten, A., Lehtipalo, K., Makhmutov, V., Molteni, U., Rissanen, M. P., Stozkhov, Y., Tröstl, J., Tsagkogeorgas, G., Wagner, R., Williamson, C., Wimmer, D., Winkler, P. M., Yan, C., and Carslaw, K. S.: Causes and importance of new particle formation in the present-day and preindustrial atmospheres, J. Geophys. Res.-Atmos., 122, 8739–8760, https://doi.org/10.1002/2017JD026844, 2017.
Gourdal, M., Lizotte, M., Massé, G., Gosselin, M., Poulin, M., Scarratt, M., Charette, J., and Levasseur, M.: Dimethyl sulfide dynamics in first-year sea ice melt ponds in the Canadian Arctic Archipelago, Biogeosciences, 15, 3169–3188, https://doi.org/10.5194/bg-15-3169-2018, 2018.
Grythe, H., Ström, J., Krejci, R., Quinn, P., and Stohl, A.: A review of sea-spray aerosol source functions using a large global set of sea salt aerosol concentration measurements, Atmos. Chem. Phys., 14, 1277–1297, https://doi.org/10.5194/acp-14-1277-2014, 2014.
Gunsch, M. J., Kirpes, R. M., Kolesar, K. R., Barrett, T. E., China, S., Sheesley, R. J., Laskin, A., Wiedensohler, A., Tuch, T., and Pratt, K. A.: Contributions of transported Prudhoe Bay oil field emissions to the aerosol population in Utqiagvik, Alaska, Atmos. Chem. Phys., 17, 10879–10892, https://doi.org/10.5194/acp-17-10879-2017, 2017.
Hayashida, H., Steiner, N., Monahan, A., Galindo, V., Lizotte, M., and Levasseur, M.: Implications of sea-ice biogeochemistry for oceanic production and emissions of dimethyl sulfide in the Arctic, Biogeosciences, 14, 3129–3155, https://doi.org/10.5194/bg-14-3129-2017, 2017.
Hegg, D. A., Hobbs, P. V, Gass, S., Nance, J. D., and Rangno, A. L.: Aerosol measurements in the Arctic relevant to direct and indirect radiative forcing, J. Geophys. Res., 101, 23349–23363, 1996.
Heintzenberg, J. and Leck, C.: The summer aerosol in the central Arctic 1991–2008: did it change or not?, Atmos. Chem. Phys., 12, 3969–3983, https://doi.org/10.5194/acp-12-3969-2012, 2012.
Heintzenberg, J., Leck, C., and Tunved, P.: Potential source regions and processes of aerosol in the summer Arctic, Atmos. Chem. Phys., 15, 6487–6502, https://doi.org/10.5194/acp-15-6487-2015, 2015.
Heintzenberg, J., Tunved, P., Galí, M., and Leck, C.: New particle formation in the Svalbard region 2006–2015, Atmos. Chem. Phys., 17, 6153–6175, https://doi.org/10.5194/acp-17-6153-2017, 2017.
Hodshire, A. L., Palm, B. B., Alexander, M. L., Bian, Q., Campuzano-Jost, P., Cross, E. S., Day, D. A., de Sá, S. S., Guenther, A. B., Hansel, A., Hunter, J. F., Jud, W., Karl, T., Kim, S., Kroll, J. H., Park, J.-H., Peng, Z., Seco, R., Smith, J. N., Jimenez, J. L., and Pierce, J. R.: Constraining nucleation, condensation, and chemistry in oxidation flow reactors using size-distribution measurements and aerosol microphysical modeling, Atmos. Chem. Phys., 18, 12433–12460, https://doi.org/10.5194/acp-18-12433-2018, 2018.
Hodshire, A. L., Campuzano-Jost, P., Kodros, J. K., Croft, B., Nault, B. A., Schroder, J. C., Jimenez, J. L., and Pierce, J. R.: The potential role of methanesulfonic acid (MSA) in aerosol formation and growth and the associated radiative forcings, Atmos. Chem. Phys., https://doi.org/10.5194/acp-2018-1022, accepted, 2019.
Hoffmann, E. H., Tilgner, A., Schrödner, R., Braüer, P., Wolke, R., and Herrmann, H.: An Advanced Modeling Study on the Impacts and Atmospheric Implications of Multiphase Dimethyl Sulfide Chemistry, P. Natl. Acad. Sci. USA, 113, 11776–11781, 2016.
Iacono, M. J., Delamere, J. S., Mlawer, E. J., Shephard, M. W., Clough, S. A., and Collins, W. D.: Radiative forcing by long-lived greenhouse gases: Calculations with the AER radiative transfer models, J. Geophys. Res.-Atmos., 113, 2–9, https://doi.org/10.1029/2008JD009944, 2008.
Jaeglé, L., Quinn, P. K., Bates, T. S., Alexander, B., and Lin, J.-T.: Global distribution of sea salt aerosols: new constraints from in situ and remote sensing observations, Atmos. Chem. Phys., 11, 3137–3157, https://doi.org/10.5194/acp-11-3137-2011, 2011.
Johnson, M. T.: A numerical scheme to calculate temperature and salinity dependent air–water transfer velocities for any gas, Ocean Sci., 6, 913–932, https://doi.org/10.5194/os-6-913-2010, 2010.
Karl, M., Leck, C., Coz, E., and Heintzenberg, J.: Marine nanogels as a source of atmospheric nanoparticles in the high Arctic, Geophys. Res. Lett., 40, 3738–3743, https://doi.org/10.1002/grl.50661, 2013.
Kerminen, V. M., Anttila, T., Lehtinen, K. E. J., and Kulmala, M.: Parameterization for atmospheric new-particle formation: Application to a system involving sulfuric acid and condensable water-soluble organic vapors, Aerosol Sci. Technol., 38, 1001–1008, https://doi.org/10.1080/027868290519085, 2004.
Kim, P. S., Jacob, D. J., Fisher, J. A., Travis, K., Yu, K., Zhu, L., Yantosca, R. M., Sulprizio, M. P., Jimenez, J. L., Campuzano-Jost, P., Froyd, K. D., Liao, J., Hair, J. W., Fenn, M. A., Butler, C. F., Wagner, N. L., Gordon, T. D., Welti, A., Wennberg, P. O., Crounse, J. D., St. Clair, J. M., Teng, A. P., Millet, D. B., Schwarz, J. P., Markovic, M. Z., and Perring, A. E.: Sources, seasonality, and trends of southeast US aerosol: an integrated analysis of surface, aircraft, and satellite observations with the GEOS-Chem chemical transport model, Atmos. Chem. Phys., 15, 10411–10433, https://doi.org/10.5194/acp-15-10411-2015, 2015.
Kirkby, J., Curtius, J., Almeida, J., Dunne, E., Duplissy, J., Ehrhart, S., Franchin, A., Gagné, S., Ickes, L., Kürten, A., Kupc, A., Metzger, A., Riccobono, F., Rondo, L., Schobesberger, S., Tsagkogeorgas, G., Wimmer, D., Amorim, A., Bianchi, F., Breitenlechner, M., David, A., Dommen, J., Downard, A., Ehn, M., Flagan, R. C., Haider, S., Hansel, A., Hauser, D., Jud, W., Junninen, H., Kreissl, F., Kvashin, A., Laaksonen, A., Lehtipalo, K., Lima, J., Lovejoy, E. R., Makhmutov, V., Mathot, S., Mikkilä, J., Minginette, P., Mogo, S., Nieminen, T., Onnela, A., Pereira, P., Petäjä, T., Schnitzhofer, R., Seinfeld, J. H., Sipilä, M., Stozhkov, Y., Stratmann, F., Tomé, A., Vanhanen, J., Viisanen, Y., Vrtala, A., Wagner, P. E., Walther, H., Weingartner, E., Wex, H., Winkler, P. M., Carslaw, K. S., Worsnop, D. R., Baltensperger, U., and Kulmala, M.: Role of sulphuric acid, ammonia and galactic cosmic rays in atmospheric aerosol nucleation, Nature, 476, 429–435, https://doi.org/10.1038/nature10343, 2011.
Kodros, J. K. and Pierce, J. R.: Important global and regional differences in aerosol cloud-albedo effect estimates between simulations with and without prognostic aerosol microphysics, J. Geophys. Res., 122, 4003–4018, https://doi.org/10.1002/2016JD025886, 2017.
Kodros, J. K., Cucinotta, R., Ridley, D. A., Wiedinmyer, C., and Pierce, J. R.: The aerosol radiative effects of uncontrolled combustion of domestic waste, Atmos. Chem. Phys., 16, 6771–6784, https://doi.org/10.5194/acp-16-6771-2016, 2016.
Kodros, J. K., Hanna, S. J., Bertram, A. K., Leaitch, W. R., Schulz, H., Herber, A. B., Zanatta, M., Burkart, J., Willis, M. D., Abbatt, J. P. D., and Pierce, J. R.: Size-resolved mixing state of black carbon in the Canadian high Arctic and implications for simulated direct radiative effect, Atmos. Chem. Phys., 18, 11345–11361, https://doi.org/10.5194/acp-18-11345-2018, 2018.
Koepke, P., Hess, M., Schult, I., and Shettle, E. P.: Global Aerosol Data Set, MPI Meteorologie Hamburg Report No. 243, 44 pp., 1997.
Kolesar, K. R., Cellini, J., Peterson, P. K., Jefferson, A., Tuch, T., Birmili, W., Wiedensohler, A., and Pratt, K. A.: Effect of Prudhoe Bay emissions on atmospheric aerosol growth events observed in Utqiagvik (Barrow), Alaska, Atmos. Environ., 152, 146–155, https://doi.org/10.1016/j.atmosenv.2016.12.019, 2017.
Köllner, F., Schneider, J., Willis, M. D., Klimach, T., Helleis, F., Bozem, H., Kunkel, D., Hoor, P., Burkart, J., Leaitch, W. R., Aliabadi, A. A., Abbatt, J. P. D., Herber, A. B., and Borrmann, S.: Particulate trimethylamine in the summertime Canadian high Arctic lower troposphere, Atmos. Chem. Phys., 17, 13747–13766, https://doi.org/10.5194/acp-17-13747-2017, 2017.
Korhonen, H., Carslaw, K. S., Spracklen, D. V., Ridley, D. A., and Ström, J.: A global model study of processes controlling aerosol size distributions in the Arctic spring and summer, J. Geophys. Res., 113, D08211, https://doi.org/10.1029/2007JD009114, 2008.
Lana, A., Bell, T. G., Simó, R., Vallina, S. M., Ballabrera-Poy, J., Kettle, A. J., Dachs, J., Bopp, L., Saltzman, E. S., Stefels, J., Johnson, J. E., and Liss, P. S.: An updated climatology of surface dimethlysulfide concentrations and emission fluxes in the global ocean, Global Biogeochem. Cy., 25, 1–17, https://doi.org/10.1029/2010GB003850, 2011.
Law, K. S. and Stohl, A.: Arctic Air Pollution: Orgins and Impacts, Science, 315, 1537–1540, 2007.
Leaitch, W. R., Sharma, S., Huang, L., Toom-Sauntry, D., Chivulescu, A., Macdonald, A. M., von Salzen, K., Pierce, J. R., Bertram, A. K., Schroder, J. C., Shantz, N. C., Chang, R. Y.-W., and Norman, A.-L.: Dimethyl sulfide control of the clean summertime Arctic aerosol and cloud, Elem. Sci. Anthr., 1, 000017, https://doi.org/10.12952/journal.elementa.000017, 2013.
Leaitch, W. R., Korolev, A., Aliabadi, A. A., Burkart, J., Willis, M. D., Abbatt, J. P. D., Bozem, H., Hoor, P., Köllner, F., Schneider, J., Herber, A., Konrad, C., and Brauner, R.: Effects of 20–100 nm particles on liquid clouds in the clean summertime Arctic, Atmos. Chem. Phys., 16, 11107–11124, https://doi.org/10.5194/acp-16-11107-2016, 2016.
Leaitch, W. R., Russell, L. M., Liu, J., Kolonjari, F., Toom, D., Huang, L., Sharma, S., Chivulescu, A., Veber, D., and Zhang, W.: Organic functional groups in the submicron aerosol at 82.5∘ N, 62.5∘ W from 2012 to 2014, Atmos. Chem. Phys., 18, 3269–3287, https://doi.org/10.5194/acp-18-3269-2018, 2018.
Leck, C. and Bigg, E. K.: New particle formation of marine biological origin, Aerosol Sci. Technol., 44, 570–577, https://doi.org/10.1080/02786826.2010.481222, 2010.
Lee, Y. H. and Adams, P. J.: A fast and efficient version of the TwO-Moment Aerosol Sectional (TOMAS) global aerosol microphysics model, Aerosol Sci. Technol., 46, 678–689, https://doi.org/10.1080/02786826.2011.643259, 2012.
Lewis, E. R. and Schwartz, S. E.: Sea Salt Aerosol Production: Mechanisms, Methods, Measurements and Models – A Critical Review, Geophys. Monogr. Ser., 152, 413 pp., AGU, Washington, D.C., 2004.
Li, M., Zhang, Q., Kurokawa, J.-I., Woo, J.-H., He, K., Lu, Z., Ohara, T., Song, Y., Streets, D. G., Carmichael, G. R., Cheng, Y., Hong, C., Huo, H., Jiang, X., Kang, S., Liu, F., Su, H., and Zheng, B.: MIX: a mosaic Asian anthropogenic emission inventory under the international collaboration framework of the MICS-Asia and HTAP, Atmos. Chem. Phys., 17, 935–963, https://doi.org/10.5194/acp-17-935-2017, 2017.
Lindwall, F., Schollert, M., Michelsen, A., Blok, D., and Rinnan, R.: Fourfold higher tundra volatile emissions due to arctic summer warming, J. Geophys. Res.-Biogeo., 121, 895–902, https://doi.org/10.1002/2015JG003295, 2016.
Liu, H., Jacob, D. J., Bey, I., and Yantosca, R. M.: Constraints from 210Pb and 7Be on wet deposition and transport in a global three-dimensional chemical tracer model driven by assimilated meteorological fields, J. Geophys. Res.-Atmos., 106, 12109–12128, https://doi.org/10.1029/2000JD900839, 2001.
Liu, P., Li, Y. J., Wang, Y., Gilles, M. K., Zaveri, R. A., Bertram, A. K., and Martin, S. T.: Lability of secondary organic particulate matter, P. Natl. Acad. Sci. USA, 113, 12643–12648, https://doi.org/10.1073/pnas.1603138113, 2016.
Lohmann, U. and Feichter, J.: Global indirect aerosol effects: a review, Atmos. Chem. Phys., 5, 715–737, https://doi.org/10.5194/acp-5-715-2005, 2005.
Lutsch, E., Dammers, E., Conway, S., and Strong, K.: Long-range Transport of NH3, CO, HCN and C2H6 from the 2014 Canadian Wildfires, Geophys. Res. Lett., 43, 1–12, https://doi.org/10.1002/2016GL070114, 2016.
Mårtensson, E. M., Nilsson, E. D., de Leeuw, G., Cohen, L. H., and Hansson, H.-C.: Laboratory simulations and parameterization of the primary marine aerosol production, J. Geophys. Res.-Atmos., 108, 4297, https://doi.org/10.1029/2002JD002263, 2003.
McFarquhar, G. M., Ghan, S., Verlinde, J., Korolev, A., Strapp, J. W., Schmid, B., Tomlinson, J. M., Wolde, M., Brooks, S. D., Cziczo, D., Dubey, M. K., Fan, J., Flynn, C., Gultepe, I., Hubbe, J., Gilles, M. K., Laskin, A., Lawson, P., Leaitch, W. R., Liu, P., Liu, X., Lubin, D., Mazzoleni, C., MacDonald, A. M., Moffet, R. C., Morrison, H., Ovchinnikov, M., Shupe, M. D., Turner, D. D., Xie, S., Zelenyuk, A., Bae, K., Freer, M., and Glen, A.: Indirect and semi-direct aerosol campaign: The impact of arctic aerosols on clouds, B. Am. Meteorol. Soc., 92, 183–201, https://doi.org/10.1175/2010BAMS2935.1, 2011.
Mungall, E. L., Croft, B., Lizotte, M., Thomas, J. L., Murphy, J. G., Levasseur, M., Martin, R. V., Wentzell, J. J. B., Liggio, J., and Abbatt, J. P. D.: Dimethyl sulfide in the summertime Arctic atmosphere: measurements and source sensitivity simulations, Atmos. Chem. Phys., 16, 6665–6680, https://doi.org/10.5194/acp-16-6665-2016, 2016.
Mungall, E. L., Abbatt, J. P. D., Wentzell, J. J. B., Lee, A. K. Y., Thomas, J. L., Blais, M., Gosselin, M., Miller, L. A., Papakyriakou, T., Willis, M. D., and Liggio, J.: Microlayer source of oxygenated volatile organic compounds in the summertime marine Arctic boundary layer, P. Natl. Acad. Sci. USA, 114, 6203–6208, https://doi.org/10.1073/pnas.1620571114, 2017.
Murphy, J. G., Moravek, A., Wentworth, G. R., Croft, B., Martin, R. V., Pierce, J., Mungall, E. L., and Abbatt, J. P. D.: Observational constraints on the atmospheric ammonia budget in the Canadian Arctic Archipelago, in preparation, 2019.
Napari, I., Noppel, M., Vehkamaki, H., and Kulmala, M.: Parametrization of ternary nucleation rates for H2SO4-NH3-H2O vapors, J. Geophys. Res.-Atmos., 107, 2–7, https://doi.org/10.1029/2002JD002132, 2002.
Nguyen, Q. T., Glasius, M., Sørensen, L. L., Jensen, B., Skov, H., Birmili, W., Wiedensohler, A., Kristensson, A., Nøjgaard, J. K., and Massling, A.: Seasonal variation of atmospheric particle number concentrations, new particle formation and atmospheric oxidation capacity at the high Arctic site Villum Research Station, Station Nord, Atmos. Chem. Phys., 16, 11319–11336, https://doi.org/10.5194/acp-16-11319-2016, 2016.
Olenius, T., Kupiainen-Määttä, O., Ortega, I. K., Kurtén, T., and Vehkamäki, H.: Free energy barrier in the growth of sulfuric acid-ammonia and sulfuric acid-dimethylamine clusters, J. Chem. Phys., 139, 084312, https://doi.org/10.1063/1.4819024, 2013.
Petters, M. D. and Kreidenweis, S. M.: A single parameter representation of hygroscopic growth and cloud condensation nucleus activity – Part 3: Including surfactant partitioning, Atmos. Chem. Phys., 13, 1081–1091, https://doi.org/10.5194/acp-13-1081-2013, 2013.
Pierce, J. R., Riipinen, I., Kulmala, M., Ehn, M., Petäjä, T., Junninen, H., Worsnop, D. R., and Donahue, N. M.: Quantification of the volatility of secondary organic compounds in ultrafine particles during nucleation events, Atmos. Chem. Phys., 11, 9019–9036, https://doi.org/10.5194/acp-11-9019-2011, 2011.
Pierce, J. R., Croft, B., Kodros, J. K., D'Andrea, S. D., and Martin, R. V.: The importance of interstitial particle scavenging by cloud droplets in shaping the remote aerosol size distribution and global aerosol-climate effects, Atmos. Chem. Phys., 15, 6147–6158, https://doi.org/10.5194/acp-15-6147-2015, 2015.
Polissar, A. V, Hopke, P. K., and Harris, J. M.: Source Regions for Atmospheric Aerosol Measured at Barrow, Alaska, Environ. Sci. Technol., 35, 4214–4226, https://doi.org/10.1021/es0107529, 2001.
Potosnak, M. J., Baker, B. M., LeStourgeon, L., Disher, S. M., Griffin, K. L., Bret-Harte, M. S., and Starr, G.: Isoprene emissions from a tundra ecosystem, Biogeosciences, 10, 871–889, https://doi.org/10.5194/bg-10-871-2013, 2013.
Prather, K. A., Bertram, T. H., Grassian, V. H., Deane, G. B., Stokes, M. D., DeMott, P. J., Aluwihare, L. I., Palenik, B. P., Azam, F., Seinfeld, J. H., Moffet, R. C., Molina, M. J., Cappa, C. D., Geiger, F. M., Roberts, G. C., Russell, L. M., Ault, A. P., Baltrusaitis, J., Collins, D. B., Corrigan, C. E., Cuadra-Rodriguez, L. A., Ebben, C. J., Forestieri, S. D., Guasco, T. L., Hersey, S. P., Kim, M. J., Lambert, W. F., Modini, R. L., Mui, W., Pedler, B. E., Ruppel, M. J., Ryder, O. S., Schoepp, N. G., Sullivan, R. C., and Zhao, D.: Bringing the ocean into the laboratory to probe the chemical complexity of sea spray aerosol, P. Natl. Acad. Sci. USA, 110, 7550–7555, https://doi.org/10.1073/pnas.1300262110, 2013.
Quinn, P. K., Miller, T. L., Bates, T. S., Ogren, J. A., Andrews, E., and Shaw, G. E.: A 3-year record of simultaneously measured aerosol chemical and optical properties at Barrow, Alaska, J. Geophys. Res.-Atmos., 107, 4130, https://doi.org/10.1029/2001JD001248, 2002.
Quinn, P. K., Collins, D. B., Grassian, V. H., Prather, K. A., and Bates, T. S.: Chemistry and Related Properties of Freshly Emitted Sea Spray Aerosol, Chem. Rev., 115, 4383–4399, https://doi.org/10.1021/cr500713g, 2015.
Rap, A., Scott, C. E., Spracklen, D. V., Bellouin, N., Forster, P. M., Carslaw, K. S., Schmidt, A., and Mann, G.: Natural aerosol direct and indirect radiative effects, Geophys. Res. Lett., 40, 3297–3301, https://doi.org/10.1002/grl.50441, 2013.
Riccobono, F., Schobesberger, S., Scott, C. E., Dommen, J., Ortega, I. K., Rondo, L., Almeida, J., Amorim, A., Bianchi, F., Breitenlechner, M., David, A., Downard, A., Dunne, E. M., Duplissy, J., Ehrhart, S., Flagan, R. C., Franchin, A., Hansel, A., Junninen, H., Kajos, M., Keskinen, H., Kupc, A., Kürten, A., Kvashin, A. N., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Mathot, S., Nieminen, T. Onnela, A., Petäjä, T., Praplan, A. P., Santos, F. D., Schallhart, S., Seinfeld, J. H., Sipilä, M., Spracklen, D. V., Stozhkov, Y., Stratmann, F., Tomé, A., Tsagkogeorgas, G., Vaattovaara, P., Viisanen, Y., Vrtala, A., Wagner, P. E., Weingartner, E., Wex, H., Wimmer, D., Carslaw, K. S., Curtius, J., Donahue, N. M., Kirkby, J., Kulmala, M., Worsnop, D. R., and Baltensperger, U.: Oxidation Products of Biogenic Emissions Contribute to Nucleation of Atmospheric Particles, Science, 344, 717–721, 2014.
Riddick, S. N., Dragosits, U., Blackall, T. D., Daunt, F., Wanless, S., and Sutton, M. A.: The global distribution of ammonia emissions from seabird colonies, Atmos. Environ., 55, 319–327, https://doi.org/10.1016/j.atmosenv.2012.02.052, 2012a.
Riddick, S. N., Dragosits, U., Blackall, T. D., Daunt, F., Wanless, S., and Sutton, M. A.: Global ammonia emissions from seabirds, NERC Environmental Information Data Centre, Centre for Ecology and Hydrology, UK, https://doi.org/10.5285/c9e802b3-43c8-4b36-a3a3-8861d9da8ea9, 2012b.
Riipinen, I., Pierce, J. R., Yli-Juuti, T., Nieminen, T., Häkkinen, S., Ehn, M., Junninen, H., Lehtipalo, K., Petäjä, T., Slowik, J., Chang, R., Shantz, N. C., Abbatt, J., Leaitch, W. R., Kerminen, V.-M., Worsnop, D. R., Pandis, S. N., Donahue, N. M., and Kulmala, M.: Organic condensation: a vital link connecting aerosol formation to cloud condensation nuclei (CCN) concentrations, Atmos. Chem. Phys., 11, 3865–3878, https://doi.org/10.5194/acp-11-3865-2011, 2011.
Rinaldi, M., Decesari, S., Finessi, E., Giulianelli, L., Carbone, C., Fuzzi, S., O'Dowd, C. D., Ceburnis, D., and Facchini, M. C.: Primary and Secondary Organic Marine Aerosol and Oceanic Biological Activity: Recent Results and New Perspectives for Future Studies, Adv. Meteorol., 2010, 310682, https://doi.org/10.1155/2010/310682, 2010.
Scott, C. E., Rap, A., Spracklen, D. V., Forster, P. M., Carslaw, K. S., Mann, G. W., Pringle, K. J., Kivekäs, N., Kulmala, M., Lihavainen, H., and Tunved, P.: The direct and indirect radiative effects of biogenic secondary organic aerosol, Atmos. Chem. Phys., 14, 447–470, https://doi.org/10.5194/acp-14-447-2014, 2014.
Sharma, S., Ishizawa, M., Chan, D., Lavoué, D., Andrews, E., Eleftheriadis, K., and Maksyutov, S.: 16-year simulation of arctic black carbon: Transport, source contribution, and sensitivity analysis on deposition, J. Geophys. Res.-Atmos., 118, 943–964, https://doi.org/10.1029/2012JD017774, 2013.
Shindell, D. and Faluvegi, G.: Climate response to regional radiative forcing during the twentieth century, Nat. Geosci., 2, 294–300, https://doi.org/10.1038/ngeo473, 2009.
Skrzypek, G., Wojtuñ, B., Richter, D., Jakubas, D., Wojczulanis-Jakubas, K., and Samecka-Cymerman, A.: Diversification of nitrogen sources in various tundra vegetation types in the high arctic, PLoS One, 10, 1–21, https://doi.org/10.1371/journal.pone.0136536, 2015.
Steinke, M., Hodapp, B., Subhan, R., Bell, T. G., and Martin-Creuzburg, D.: Flux of the biogenic volatiles isoprene and dimethyl sulfide from an oligotrophic lake, Sci. Rep.-UK, 8, 1–10, https://doi.org/10.1038/s41598-017-18923-5, 2018.
Stohl, A.: Characteristics of atmospheric transport into the Arctic troposphere, J. Geophys. Res.-Atmos., 111, 1–17, https://doi.org/10.1029/2005JD006888, 2006.
Tokarek, T. W., Brownsey, D. K., Jordan, N., Garner, N. M., Ye, C. Z., Assad, F. V., Peace, A., Schiller, C. L., Mason, R. H., Vingarzan, R., and Osthoff, H. D.: Biogenic Emissions and Nocturnal Ozone Depletion Events at the Amphitrite Point Observatory on Vancouver Island, Atmosphere-Ocean, 55, 121–132, https://doi.org/10.1080/07055900.2017.1306687, 2017.
Tremblay, S., Picard, J.-C., Bachelder, J. O., Lutsch, E., Strong, K., Fogal, P., Leaitch, W. R., Sharma, S., Kolonjari, F., Cox, C. J., Chang, R. Y.-W., and Hayes, P. L.: Characterization of aerosol growth events over Ellesmere Island during the summers of 2015 and 2016, Atmos. Chem. Phys., https://doi.org/10.5194/acp-2018-428, accepted, 2019.
Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., Wagner, R., Simon, M., Lehtipalo, K., Williamson, C., Craven, J. S., Duplissy, J., Adamov, A., Almeida, J., Bernhammer, A. K., Breitenlechner, M., Brilke, S., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Gysel, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Keskinen, H., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lawler, M., Leiminger, M., Mathot, S., Möhler, O., Nieminen, T., Onnela, A., Petäjä, T., Piel, F. M., Miettinen, P., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Sipilä, M., Smith, J. N., Steiner, G., Tomè, A., Virtanen, A., Wagner, A. C., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Carslaw, K. S., Curtius, J., Dommen, J., Kirkby, J., Kulmala, M., Riipinen, I., Worsnop, D. R., Donahue, N. M., and Baltensperger, U.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 533, 527–531, https://doi.org/10.1038/nature18271, 2016.
Tunved, P., Ström, J., and Krejci, R.: Arctic aerosol life cycle: linking aerosol size distributions observed between 2000 and 2010 with air mass transport and precipitation at Zeppelin station, Ny-Ålesund, Svalbard, Atmos. Chem. Phys., 13, 3643–3660, https://doi.org/10.5194/acp-13-3643-2013, 2013.
van der Werf, G. R., Randerson, J. T., Giglio, L., van Leeuwen, T. T., Chen, Y., Rogers, B. M., Mu, M., van Marle, M. J. E., Morton, D. C., Collatz, G. J., Yokelson, R. J., and Kasibhatla, P. S.: Global fire emissions estimates during 1997–2016, Earth Syst. Sci. Data, 9, 697–720, https://doi.org/10.5194/essd-9-697-2017, 2017.
Walker, J. T., Robarge, W. P., and Austin, R.: Modeling of ammonia dry deposition to a pocosin landscape downwind of a large poultry facility, Agric. Ecosyst. Environ., 185, 161–175, https://doi.org/10.1016/j.agee.2013.10.029, 2014.
Wang, Q., Jacob, D. J., Fisher, J. A., Mao, J., Leibensperger, E. M., Carouge, C. C., Le Sager, P., Kondo, Y., Jimenez, J. L., Cubison, M. J., and Doherty, S. J.: Sources of carbonaceous aerosols and deposited black carbon in the Arctic in winter–spring: implications for radiative forcing, Atmos. Chem. Phys., 11, 12453–12473, https://doi.org/10.5194/acp-11-12453-2011, 2011.
Wentworth, G. R., Murphy, J. G., Gregoire, P. K., Cheyne, C. A. L., Tevlin, A. G., and Hems, R.: Soil–atmosphere exchange of ammonia in a non-fertilized grassland: measured emission potentials and inferred fluxes, Biogeosciences, 11, 5675–5686, https://doi.org/10.5194/bg-11-5675-2014, 2014.
Wentworth, G. R., Murphy, J. G., Croft, B., Martin, R. V., Pierce, J. R., Côté, J.-S., Courchesne, I., Tremblay, J.-É., Gagnon, J., Thomas, J. L., Sharma, S., Toom-Sauntry, D., Chivulescu, A., Levasseur, M., and Abbatt, J. P. D.: Ammonia in the summertime Arctic marine boundary layer: sources, sinks, and implications, Atmos. Chem. Phys., 16, 1937–1953, https://doi.org/10.5194/acp-16-1937-2016, 2016.
Wesely, M. L.: Parameterization of surface resistances to gaseous dry deposition in regional-scale numerical models, Atmos. Environ., 23, 1293–1304, 1989.
Willis, M. D., Burkart, J., Thomas, J. L., Köllner, F., Schneider, J., Bozem, H., Hoor, P. M., Aliabadi, A. A., Schulz, H., Herber, A. B., Leaitch, W. R., and Abbatt, J. P. D.: Growth of nucleation mode particles in the summertime Arctic: a case study, Atmos. Chem. Phys., 16, 7663–7679, https://doi.org/10.5194/acp-16-7663-2016, 2016.
Willis, M. D., Köllner, F., Burkart, J., Bozem, H., Thomas, J. L., Schneider, J., Aliabadi, A. A., Hoor, P. M., Schulz, H., Herber, A. B., Leaitch, W. R., and Abbatt, J. P. D.: Evidence for marine biogenic influence on summertime Arctic aerosol, Geophys. Res. Lett., 44, 6460–6470, https://doi.org/10.1002/2017GL073359, 2017.
Willis, M. D., Leaitch, W. R., and Abbatt, J. P.: Processes controlling the composition and abundance of Arctic aerosol, Rev. Geophys., 56, 621–671, https://doi.org/10.1029/2018RG000602, 2018.
Wilson, T. W., Ladino, L. A., Alpert, P. A., Breckels, M. N., Brooks, I. M., Browse, J., Burrows, S. M., Carslaw, K. S., Huffman, J. A., Judd, C., Kilthau, W. P., Mason, R. H., McFiggans, G., Miller, L. A., Najera, J. J., Polishchuk, E., Rae, S., Schiller, C. L., Si, M., Temprado, J. V., Whale, T. F., Wong, J. P. S., Wurl, O., Yakobi-Hancock, J. D., Abbatt, J. P. D., Aller, J. Y., Bertram, A. K., Knopf, D. A., and Murray, B. J.: A marine biogenic source of atmospheric ice-nucleating particles, Nature, 525, 234–238, https://doi.org/10.1038/nature14986, 2015.
Xu, J.-W., Martin, R. V., Morrow, A., Sharma, S., Huang, L., Leaitch, W. R., Burkart, J., Schulz, H., Zanatta, M., Willis, M. D., Henze, D. K., Lee, C. J., Herber, A. B., and Abbatt, J. P. D.: Source attribution of Arctic black carbon constrained by aircraft and surface measurements, Atmos. Chem. Phys., 17, 11971–11989, https://doi.org/10.5194/acp-17-11971-2017, 2017.
Yang, Q., Bitz, C. M., and Doherty, S. J.: Offsetting effects of aerosols on Arctic and global climate in the late 20th century, Atmos. Chem. Phys., 14, 3969–3975, https://doi.org/10.5194/acp-14-3969-2014, 2014.
Ye, Q., Robinson, E. S., Ding, X., Ye, P., Sullivan, R. C., and Donahue, N. M.: Mixing of secondary organic aerosols versus relative humidity, P. Natl. Acad. Sci. USA, 113, 12649–12654, https://doi.org/10.1073/pnas.1604536113, 2016.
Yu, H., Kaufman, Y. J., Chin, M., Feingold, G., Remer, L. A., Anderson, T. L., Balkanski, Y., Bellouin, N., Boucher, O., Christopher, S., DeCola, P., Kahn, R., Koch, D., Loeb, N., Reddy, M. S., Schulz, M., Takemura, T., and Zhou, M.: A review of measurement-based assessments of the aerosol direct radiative effect and forcing, Atmos. Chem. Phys., 6, 613–666, https://doi.org/10.5194/acp-6-613-2006, 2006.
Zábori, J., Rastak, N., Yoon, Y. J., Riipinen, I., and Ström, J.: Size-resolved cloud condensation nuclei concentration measurements in the Arctic: two case studies from the summer of 2008, Atmos. Chem. Phys., 15, 13803–13817, https://doi.org/10.5194/acp-15-13803-2015, 2015.
Zender, C. S., Huisheng, B., and Newman, D.: Mineral Dust Entrainment and Deposition (DEAD) model: Description and 1990s dust climatology, J. Geophys. Res., 108, 4416, https://doi.org/10.1029/2002JD002775, 2003.