the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 06 Feb 2019
| 06 Feb 2019
Relative humidity effect on the formation of highly oxidized molecules and new particles during monoterpene oxidation
Xiaoxiao Li
Sabrina Chee
Jiming Hao
Jonathan P. D. Abbatt
Jingkun Jiang
It has been widely observed around the world that the frequency and intensity of new particle formation (NPF) events are reduced during periods of high relative humidity (RH). The current study focuses on how RH affects the formation of highly oxidized molecules (HOMs), which are key components of NPF and initial growth caused by oxidized organics. The ozonolysis of α-pinene, limonene, and Δ3-carene, with and without OH scavengers, were carried out under low NOx conditions under a range of RH (from ∼3 % to ∼92 %) in a temperature-controlled flow tube to generate secondary organic aerosol (SOA). A Scanning Mobility Particle Sizer (SMPS) was used to measure the size distribution of generated particles, and a novel transverse ionization chemical ionization inlet with a high-resolution time-of-fight mass spectrometer detected HOMs. A major finding from this work is that neither the detected HOMs nor their abundance changed significantly with RH, which indicates that the detected HOMs must be formed from water-independent pathways. In fact, the distinguished OH- and O3-derived peroxy radicals (RO2), HOM monomers, and HOM dimers could mostly be explained by the autoxidation of RO2 followed by bimolecular reactions with other RO2 or hydroperoxy radicals (HO2), rather than from a water-influenced pathway like through the formation of a stabilized Criegee intermediate (sCI). However, as RH increased from ∼3 % to ∼92 %, the total SOA number concentrations decreased by a factor of 2–3 while SOA mass concentrations remained relatively constant. These observations show that, while high RH appears to inhibit NPF as evident by the decreasing number concentration, this reduction is not caused by a decrease in RO2-derived HOM formation. Possible explanations for these phenomena were discussed.
- Article
(4420 KB) - Full-text XML
-
Supplement
(1123 KB) - BibTeX
- EndNote





New particle formation (NPF) is ubiquitous around the world (Kulmala et al., 2004). Newly formed particles contribute greatly to global particle populations and can grow further to act as cloud condensation nuclei (CCN), thereby influencing clouds and climate (Makkonen et al., 2012; Merikanto et al., 2009; Dunne et al., 2016). NPF characteristics vary from site to site because of varying precursors and atmospheric conditions. It has been widely observed that the intensity (Sihto et al., 2006; Dada et al., 2017) and frequency (Dada et al., 2017; Boy and Kulmala, 2002; Hyvönen et al., 2005) of continental NPF are reduced during periods of high relative humidity (RH), resulting in reduced ultrafine particle number concentrations during these periods (Weber et al., 1997). For example, 20 years of observations in the boreal forest at Hyytiälä, Finland, showed that NPF is more likely to happen during periods of low ambient RH (Dada et al., 2017). In urban areas, NPF also favors low RH (Cai et al., 2017; Shen et al., 2011). Despite the low continental NPF event frequency at high RH, NPF has still been observed in the free troposphere in the vicinity of clouds, where RH is extremely high (Weber et al., 1999) and in coastal and marine areas where RH is typically greater than 90 % (O'Dowd et al., 1998).
The widely observed anti-correlation between NPF and RH in the field experiments can be attributed to the indirect influence of water. For example, high RH often corresponds to greater cloud cover, which can lead to lower ground-level concentrations of photo-oxidized precursors such as H2SO4 and highly oxidized molecules (HOMs) as well as an increased condensation sink that leads to scavenging of precursors and clusters (Hamed et al., 2011). On the other hand, water vapor may also directly influence NPF by regulating the formation of gas-phase precursors or by participating in cluster formation. For example, chamber and model experiments on the binary sulfuric acid–water system have demonstrated positive relationships between particle formation rate and RH (Duplissy et al., 2016; Merikanto et al., 2016), while in the ternary (H2SO4∕MSA-H2O-amine∕NH3) system, H2O was reported to have either positive (Chen et al., 2015) or negative (Napari et al., 2002) effects on NPF. Some studies have hypothesized that high water content might suppress the formation of NPF-related organics from the oxidation of biogenic precursors (Hyvönen et al., 2005; Boy and Kulmala, 2002). However, no direct evidence of this has been provided.
Although sulfuric acid has been recognized as the most important precursor of new particle formation, it alone cannot explain the rapid formation and growth rates observed in the field (Kuang et al., 2008). Organic compounds, ammonia, amines, and water are also likely involved (Zhang et al., 2012; Chen et al., 2012; Yu et al., 2012). Organics have been shown to be very important for cluster formation and stabilization in theoretical studies (Ortega et al., 2016; Donahue et al., 2013), laboratory experiments (Tröstl et al., 2016; Schobesberger et al., 2013), and field measurements (Bianchi et al., 2016; Hoffmann et al., 2001; Metzger et al., 2010). Organics can either form clusters with sulfuric acid or purely with themselves (Zhao et al., 2013, 2009). They can also contribute significantly to the condensational growth of newly formed particles, determining particle growth rates, particle lifetime, and global particle and CCN concentrations (Donahue et al., 2011; Vehkamäki and Riipinen, 2012). The ability of organics to take part in particle formation and condensational growth depends on their volatility as well as reactivity. HOMs, such as extremely low-volatility organic compounds (ELVOCs; saturation mass concentration (C*) µg m−3) or low-volatility organic compounds (LVOCs; µg m−3), are likely contributors to NPF (Donahue et al., 2012; Ehn et al., 2014).
Despite their large contribution to NPF, the direct measurement of HOMs has long been a challenge because of their low atmospheric concentrations, low volatilities, and short lifetimes. Recently, the development of the high-resolution time-of-flight chemical ionization mass spectrometer (HRToF-CIMS) overcame this barrier and made the measurement and identification of HOMs feasible (Junninen et al., 2010; Jokinen et al., 2012). HOMs from both monoterpene and aromatic oxidation showed high O∕C ratios of >0.7–0.8, and were present as monomers, dimers, and even higher order clusters (Molteni et al., 2018; Ehn et al., 2012). These high O∕C ratios could not be explained by any of the formerly known oxidization pathways unless the autoxidation of RO2 was taken into consideration (Crounse et al., 2013; Barsanti et al., 2017). The autoxidation of RO2 includes intramolecular hydrogen shifts and O2 additions. Several repetitions of the autoxidation cycle lead to a rapid increase in oxygen content as well as a decrease in saturation vapor pressure. Autoxidation was widely observed in condensed phase reactions; however, it was not considered in the gas phase previously due to the perception of a high barrier for the intramolecular hydrogen shift. This was confirmed by the fact that at higher temperatures, more HOMs are formed than at low temperatures (Frege et al., 2018). Modeling studies now show that intramolecular hydrogen shifts are fast enough to compete with bimolecular sink reactions (Kurten et al., 2015).
Since most laboratory experiments related to the formation of HOMs have been conducted under conditions of constant RH, usually low or medium RH of less than 60 % (Ehn et al., 2012; Zhang et al., 2015), it was still unknown whether and how water vapor might impact HOM formation. High RH conditions are difficult to achieve in chamber experiments without significantly changing temperature and pressure. In addition, HOM detection by the current commercially available CIMS inlet based on the design of Eisele and Tanner is subject to water cluster influence at high RH (Kürten et al., 2016).
In this research, three different endocyclic monoterpenes, α-pinene, limonene, and Δ3-carene were reacted with ozone, with and without hydroxyl radical (OH) scavengers, in a reaction flow tube to generate secondary organic aerosol (SOA). RH influences on HOM formation and organic-driven NPF were studied under a range of RH from ∼3 % to ∼92 %. Generated closed-shell HOMs and RO2 were measured using a home-built CIMS inlet coupled to a HRToF mass spectrometer (LTOF mass analyzer, Tofwerk AG). The CIMS inlet effectively reduced water clustering onto ions sampled into vacuum, thus removing sample artifacts caused by high water vapor levels. Water vapor influence on the formation of RO2, HOM monomers, and HOM dimers was studied. The volatility of O3- and OH-derived closed-shell HOMs was estimated using a group-contribution-based model (SIMPOL) and a recently developed statistical model to study the potential contribution of O3- and OH-initiated chemistry on NPF.
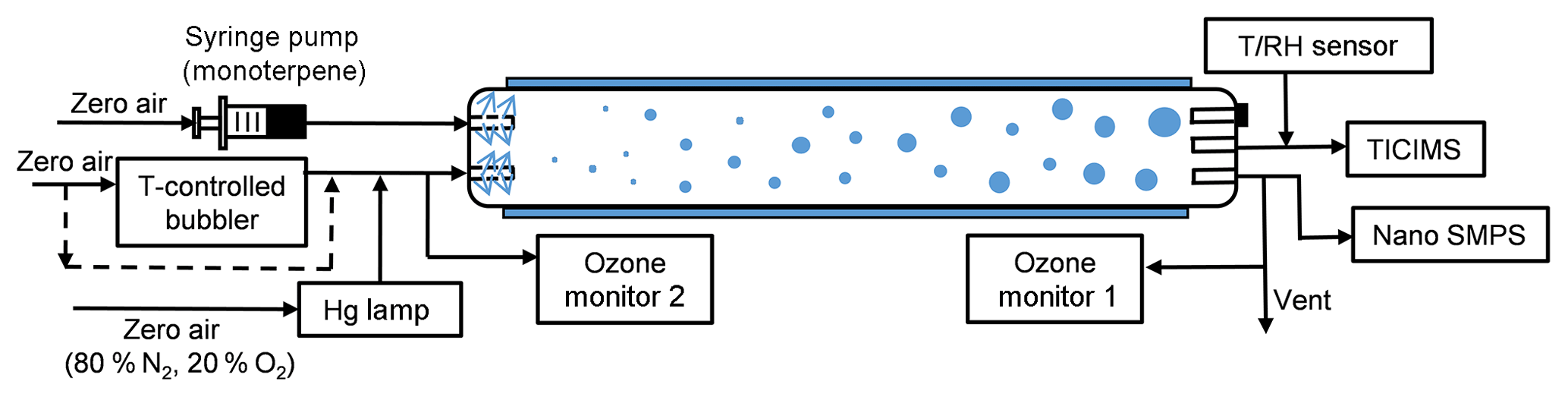
Figure 1Experiment setup for the flow tube experiments. The 8.5 L flow tube was placed at a temperature-controlled room (21±1 ∘C) and covered. The total flow rate was 8.5 L min−1. The RH was adjusted by mixing temperature controlled bubbler flow with dry zero air.
2.1 Flow tube reactor
The experiments were performed in a laminar flow tube reactor consisting of a 150 cm long Pyrex glass cylinder with a volume of 8.5 dm3 (Fig. 1). The flow tube was located in a temperature-controlled room ( K) and was covered so that all experiments were performed under dark conditions. For these experiments, dry “zero air” was generated with a zero air generator (model 747-30, Aadco Instruments), with NOx and SO2 concentrations each specified to be less than 0.5 ppbv. The monoterpenes were injected into the flow tube using a syringe pump (model NE-300, New Era Pump Systems, Inc.) evaporated into a 2.5 L min−1 flow of dry zero air. O3 was generated by passing 0.5 L min−1 dry zero air (79 % N2, 21 % O2) over a Hg UV lamp (model 90-0004-04, UVP, LLC) and then diluted with 6.5 L min−1 of humidity-controlled zero air. A temperature-controlled bubbler filled with deionized water was used to generate humid air, and the prescribed RH was achieved by controlling the temperature of the bubbler. An ozone analyzer, described below, sampled at 1 L min−1, resulting in a total flow rate of 8.5 L min−1 and a corresponding reaction time of ∼60 s for each experiment. Gas inlets to the flow tube were made from Teflon tubes with an outside diameter of 0.64 cm that were capped and drilled with ∼1 mm holes to distribute sample air uniformly into the flow tube, as described and modeled in Ball et al. (1999). The uniform distributions of O3 and H2O in the flow tube were confirmed by measuring [O3] and RH at the different locations prior to the experiments. In every experiment, RH was adjusted to be constant for at least 30 min for each of the four RH steps (3 %–5 %, 30 %–38 %, 58 %–65 %, 85 %–92 %). In order to achieve the highest relative humidity stage, the temperature of the humid flow was saturated at 299 K before being mixed with room temperature air from the monoterpene source. This resulted in a slightly higher temperature at the inlet of the flow tube, which could contribute to lower nucleation rates (Burkholder et al., 2007). Nevertheless, over the range of the first three humidity stages, up to 65 % RH, the gas temperature was constant before, during, and after the reaction. At the beginning of the experiments, the inner wall of the reactor was washed with ultra-pure water. All of the flow rates were calibrated before and during the experiments.
2.2 Instrumentation
2.2.1 Transverse ionization chemical ionization mass spectrometer
A self-designed and home-built chemical ionization inlet, called the transverse ionization (TI) inlet (Figs. 2 and S1 in the Supplement), was used in front of the LTOF mass analyzer. The TI design is similar to those of the Ambient-pressure Proton transfer Mass Spectrometer (AmPMS) (Hanson et al., 2011) and the cluster CIMS (Zhao et al., 2010). In the TI inlet, a 4–10 L min−1 flow of sample air is passed across the inlet orifice of the mass spectrometer, where it encounters an orthogonal, 1 L min−1 reagent ion gas flow consisting of N2-containing ionized nitrate ions () as well as potential cluster ions (HNO3)n with n=1–3. For the current study, the sample flow to the inlet was set to 4.5 L min−1. Chemical ionization occurs at atmospheric pressure and temperature. The reagent gas is generated by passing 3 ccm of N2 over a small vial containing nitric acid, which is then ionized by a 370 MBq Po210 radioactive source (model P-2021, NRD, LLC). An additional flow of N2 can be added to the reagent gas to change the reagent ion concentration, and the assembly can be adjusted to vary ion–molecule reaction time. The latter can be controlled by adjusting the sample and reagent gas flow rates or by applying different voltages to the ionization source and the main inlet block. To minimize the diffusion loss in sample lines, the inlet of the TI source was connected to the flow tube outlet by a short (∼10 cm) piece of electro-polished stainless steel tubing. Compared to the widely used commercial nitrate inlet patterned according to the design by Eisele and Tanner (1993) and marketed by Aerodyne, Inc., no additional sheath flow is required; thus any impurities potentially introduced by the sheath flow are eliminated. Some flow disturbance may occur when the sample flow encounters the transverse reagent flow, which may lead to nonideal behavior. However, even at the maximum total flow of 11 L min−1, the Reynolds number in this region is ∼500; thus turbulence is not expected to be significant.
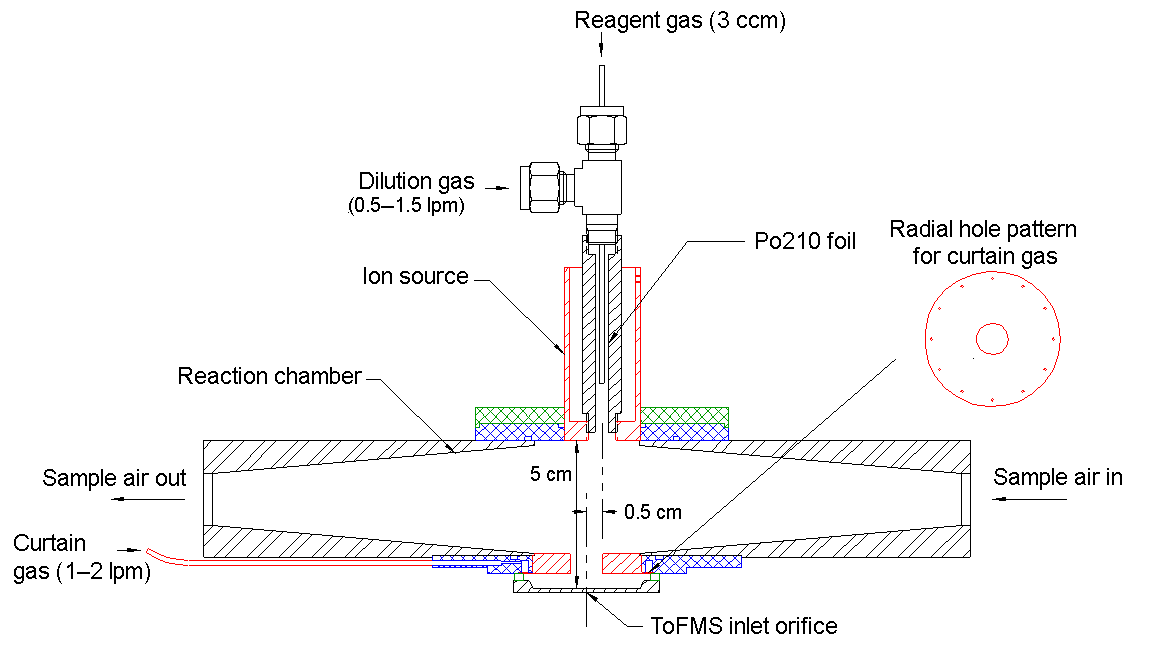
Figure 2Schematic of the transverse ionization (TI) inlet, showing the N2 curtain gas configuration. The relative position of the ion source to the inlet orifice is adjustable. The configuration shown here is the most sensitive in calibrations with H2SO4 (see Sect. 3.1).
Another unique aspect of the TI design is the use of an N2 curtain gas in front of the inlet orifice to the mass spectrometer to reduce water clustering on reagent and sample ions. Water clusters are expected to form at high RH, mainly during the free-jet expansion of the sampled gas on the vacuum side of the orifice plate (Thomson and Iribarne, 1979). The presence of these clusters makes the identification and quantification of both sample and reagent ions challenging (Kulmala et al., 2014; Lee et al., 2014; Kürten et al., 2016; Ehn et al., 2014). Figure 2 shows the details of the TI source that address this issue. Small holes drilled in a radial channel blow N2 uniformly in front of the orifice plate so that only sampled ions and this clean N2 gas pass into the vacuum chamber. Since the sampling flow rate of the mass spectrometer is ∼0.7 L min−1 when using the 0.3 mm orifice, the N2 curtain flow is set to be 1 L min−1 to overflow the region surrounding the orifice. By applying voltages to the ion source and the block, the ions can be efficiently guided into the mass spectrometer while neutral molecules such as water vapor are prevented from entering by the N2 curtain gas.
This TI inlet is suitable to all types of reagent ion chemistry, e.g., , I−, and H3O+. Nitrate ion chemistry was used as the reagent ion in these experiments, which is selective to highly oxidized molecules that have at least two hydroperoxy (-OOH) groups or some other H-bond-donating groups (Hyttinen et al., 2015). HOM monomers, HOM dimers, and highly oxidized RO2 radicals can also be measured using nitrate ion chemistry.
2.2.2 Other measurements
Ozone concentrations were measured with two ozone analyzers (model 106L, 2B Technology) at the inlet and outlet of the flow tube. The sampling flow of each analyzer is 1 L min−1. The two ozone analyzers were intercompared prior to the experiments and the difference was within 5 ppbv when [O3] <1000 ppbv. A Scanning Mobility Particle Sizer (SMPS), consisting of a Po210 bipolar neutralizer, a nano-differential mobility analyzer (nano-DMA; model 3081, TSI, Inc.), and a condensation particle counter (MCPC; model 1720, Brechtel Manufacturing) were used to measure the number size distribution of particles, which is later used to deduce the total particle number and mass concentrations (the latter assumes a uniform density for organic particles of 1.2 g cm−3). The sampling flow rate of the MCPC was 0.3 L min−1 and the sheath and excess flows of the nano DMA were set to 3 L min−1 . The flow tube particle-number size distribution was measured without further drying to get a more accurate measure of the actual particle surface area and volume, which are important for HOM partitioning, and also to prevent particle evaporation during the measurements.
2.3 Experimental conditions
Three monoterpenes were used in our experiments (see Table 1), α-pinene, limonene, and Δ3-carene. Oxidation by ozone is believed to dominate over other oxidation radicals (i.e., OH or NO3) in forming SOA under atmospheric conditions (Atkinson and Arey, 2003). Ozonolysis of alkenes generates a substantial amount of OH, leading to products that are produced by a combination of O3 and OH oxidation. For some experiments, in order to isolate oxidation by O3, cyclohexane (see Table 1 for mixing ratios) was premixed with the monoterpene and added to the flow tube as an OH scavenger. For other experiments, the combination of OH and O3 chemistry was investigated to study atmospheric oxidation chemistry more representative of ambient air. The “high concentration” experiments were conducted with similar mixing ratios of monoterpene (∼1100 ppb) and O3 (∼900 ppb). The “low concentration” experiments were conducted to study the particle-free chemical processes with initial concentrations of monoterpenes and O3 shown in Table 1. Since wall losses should be comparable for different precursors as a function of RH, it was not taken into consideration in our analysis of HOM production.
Table 1Experiment conditions and products.
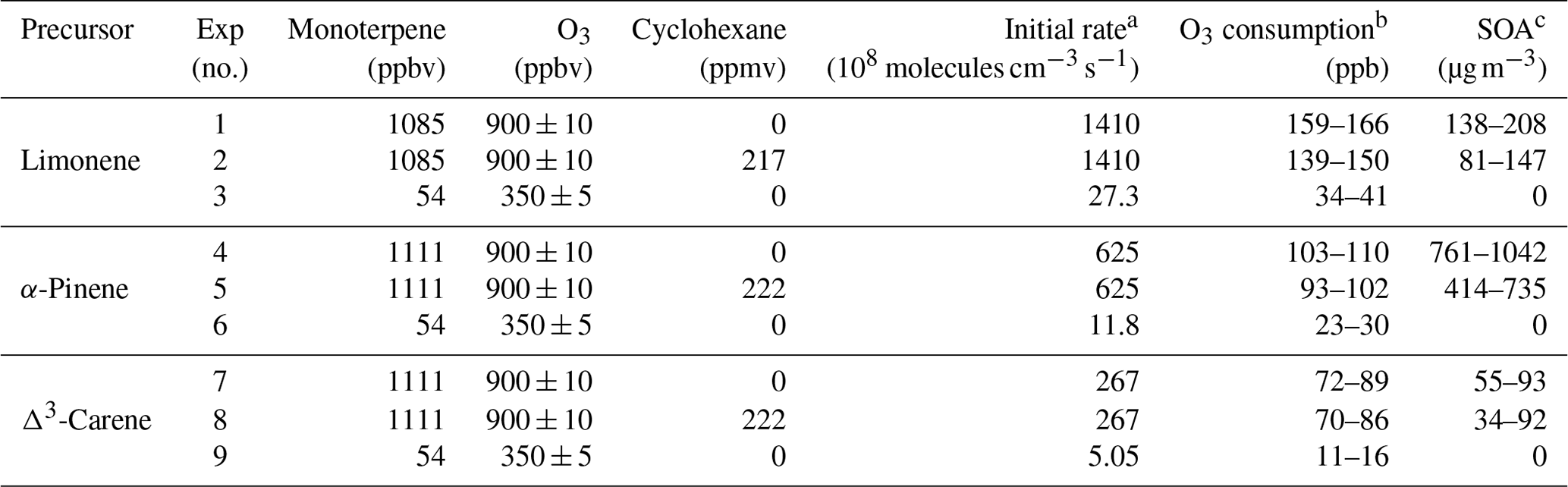
a At room temperature (298 K), the rate coefficients for limonene, α-pinene, and Δ3-carene to react with O3 were , , and cm3 molecule−1 s−1, respectively. b O3 consumption values were calculated from the difference between inlet and outlet O3 concentrations. c SOA mass concentrations were calculated from SMPS-measured volume concentrations and an assumed organic effective density (1.2 g cm−3).
2.4 HOM volatility predictions
The SIMPOL.1 method (Pankow and Asher, 2008) and the molecular corridor method (Li et al., 2016) were used to predict the saturation mass concentrations (C*) of some of the detected OH- and O3-related HOMs. SIMPOL.1 is a group contribution method and requires information on molecular structure, while the molecular corridor method only requires the molecular formulae. Both methods are semi-empirical and based on volatility data from hundreds or thousands of compounds. The calculated volatilities were then applied to the two-dimensional volatility basis set (2D-VBS) (Donahue et al., 2012) to explore the likelihood that the products participate in the initial stages of nanoparticle growth.
3.1 TI-CIMS performance
When comparing the TI inlet with the commercial nitrate inlet in measuring α-pinene ozonolysis products, both inlets produced identical mass spectra. The sensitivities of both inlets to H2SO4 were determined using a home-built H2SO4 calibration system (Fig. S2) based on the design of Kürten et al. (2012). Figure 3 summarizes the results of these calibrations. The position of the ion source assembly relative to the inlet orifice is critical for determining the sensitivity of the TI inlet. When the ion source is placed 0.5 cm upstream along the sample flow axis and 5 cm away from the inlet orifice along the reagent ion flow axis (configuration shown in Fig. 2), the instrument is at its most sensitive. The calibration factors, defined as (Eisele and Tanner, 1993), for the TI in this position and the commercial inlet were 3.25×1010 and 1.41×1010 molecules cm−3, respectively. The lower calibration factor for the TI inlet is attributed to the shorter reaction time (∼80 ms) compared to the commercial inlet (∼200 ms). We note that the reaction time of the TI inlet can be further increased by positioning the ion source assembly further upstream relative to the inlet orifice, which would require a slight modification of the current design. The total ion counts (TICs) of the TI inlet are more than 5 times higher than the commercial inlet, which we attribute to the more direct path of ions through the ion source as well as the use of a Po210 radioactive source as compared to the soft X-ray in the commercial nitrate inlet. The limit of detection for sulfuric acid, which is defined as 3 times the standard deviation of the background (Jokinen et al., 2012), is 9.3×104 and 1.26×105 molecules cm−3 for the TI and commercial inlets, respectively.
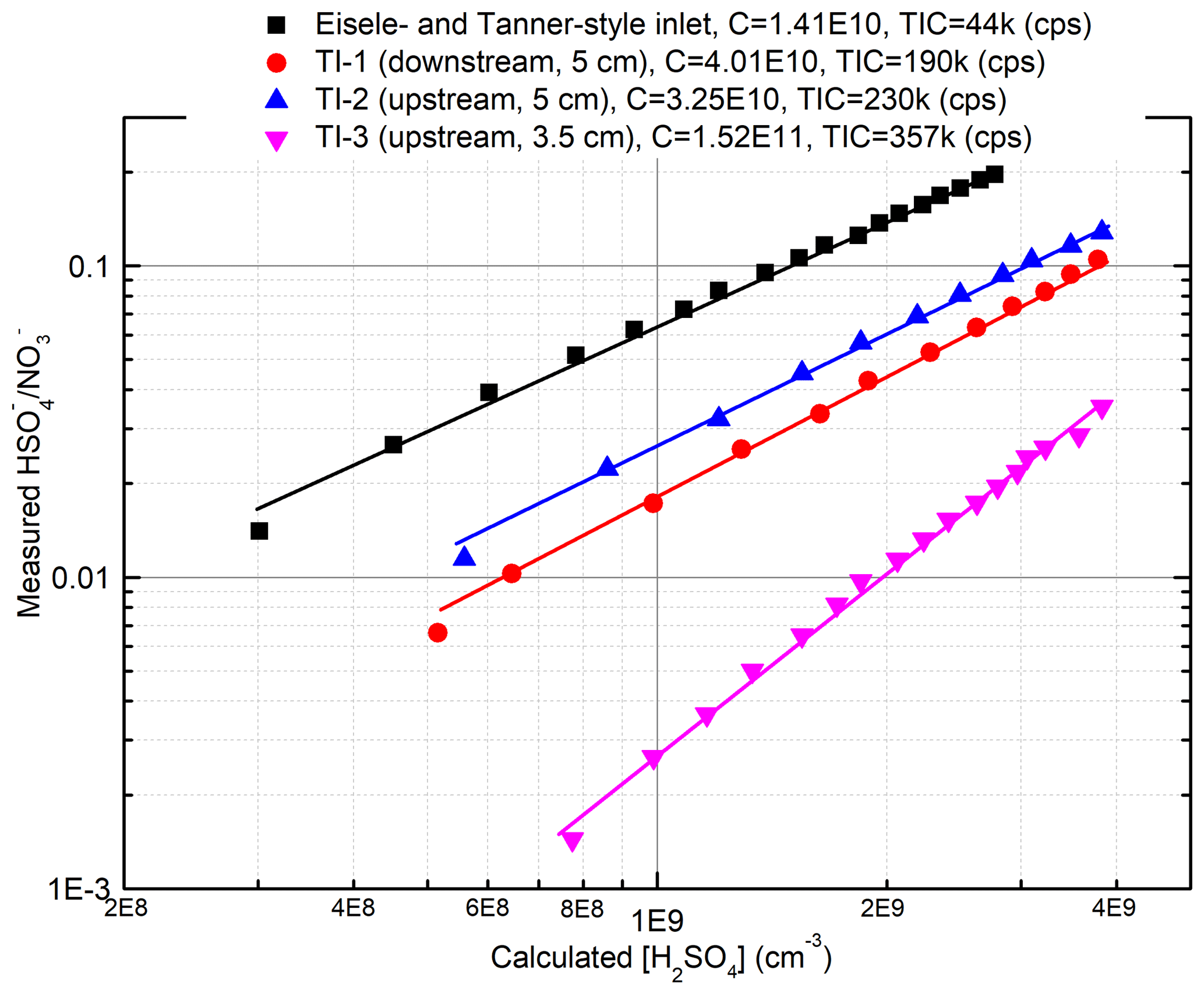
Figure 3Comparison of the sensitivities for the two inlets to H2SO4. The calibration process followed that reported by Kürten et al. (2012) and is discussed in detail in the Supplement. TI-1, 2, and 3 represent different locations of the ion source relative to the inlet orifice of the mass spectrometer. “Upstream” and “downstream” indicate 0.5 cm upstream or downstream along the sample flow axis, and “3.5 cm” and “5 cm” indicate 3.5 or 5 cm away from the inlet orifice along the reagent ion flow axis.
After applying the N2 curtain gas flow, the TICs recorded by the TI-CIMS decreased significantly. This was compensated for by increasing the ion source and reaction chamber voltages that direct ions to the orifice (Fig. S3). When RH ≈90 %, the reagent ion mass spectrum was dominated by water clusters (H2O)m (HNO3)n (m=0–30, n=0–2) if no N2 curtain flow was applied (Fig. 4a). The reagent ions , , and (HNO3)2 decreased as RH increased, with [(HNO3)2] and [] decreasing much faster than []. In contrast, after 1 L min−1 N2 curtain flow was applied to the inlet, most of the water clusters were removed (Fig. 4b). The reagent ions, sample ions, and TICs remained stable as RH increased, which resulted in a reliable measurement of HOM concentrations as a function of RH. The result that the N2 curtain flow eliminated water clustering to a large extent confirms that most of the water clusters in the spectrum were produced during the free-jet expansion into vacuum instead of being formed in the ion–molecular reagent region.

Figure 4Mass defect plots of α-pinene ozonolysis HOMs with 0 L min−1 (a) and 1 L min−1 (b) N2 curtain gas flow when RH ≈85 %, with monomer and dimer HOMs circled in green. The most intense ions comprising 60 % of the total ion count are plotted for clarity. H2O clusters (H2O)m(HNO3)n (m=1–35, n=0–2) are circled in red in (a) and are notably absent with the application of the N2 curtain gas. (HNO3)2 and are much more likely to cluster with H2O than .
3.2 Identification of HOM spectrum
Figure 5 shows the average mass spectra of the HOM dimers, and Fig. S4 shows the average mass spectra of the HOM monomers and RO2 radicals for each of the six particle generation experiments. More than 400 peaks were identified in each spectrum, the majority of which were clusters with or . [H2SO4], which arises from the oxidation of trace amounts of SO2 in the zero air, was ∼105 molecules cm−3 and was always less than 3 % of the most abundant C10 products, suggesting that sulfuric acid plays a negligible role in nucleation and cluster growth in our experiments. After subtracting the reagent ions ( or ), molecular formulae for organics with an odd number of H atoms were assigned to radicals, which are generally difficult to detect experimentally (Rissanen et al., 2015), and formulae with an even number of H atoms were assigned to closed-shell molecules. Most of the HOM products from the three endocyclic monoterpenes were very similar, while the relative abundance of different HOMs was quite different, indicating similar reaction pathways but different branching ratios in the reaction mechanisms. The main products were for closed-shell monomers and RO2 and for closed-shell dimers. Among these, C10 and C20 compounds were the most abundant. C5−9 products could be formed from O3 attack on the less reactive exocyclic carbon double bond or the decomposition of intermediate radicals. Some fragments were found to be unique for specific monoterpene precursors. For instance, C5H6O7 (m∕z 240) was much more abundant in α-pinene oxidation than for the other two precursors, which might be a tricarboxylic acid (Ehn et al., 2012).
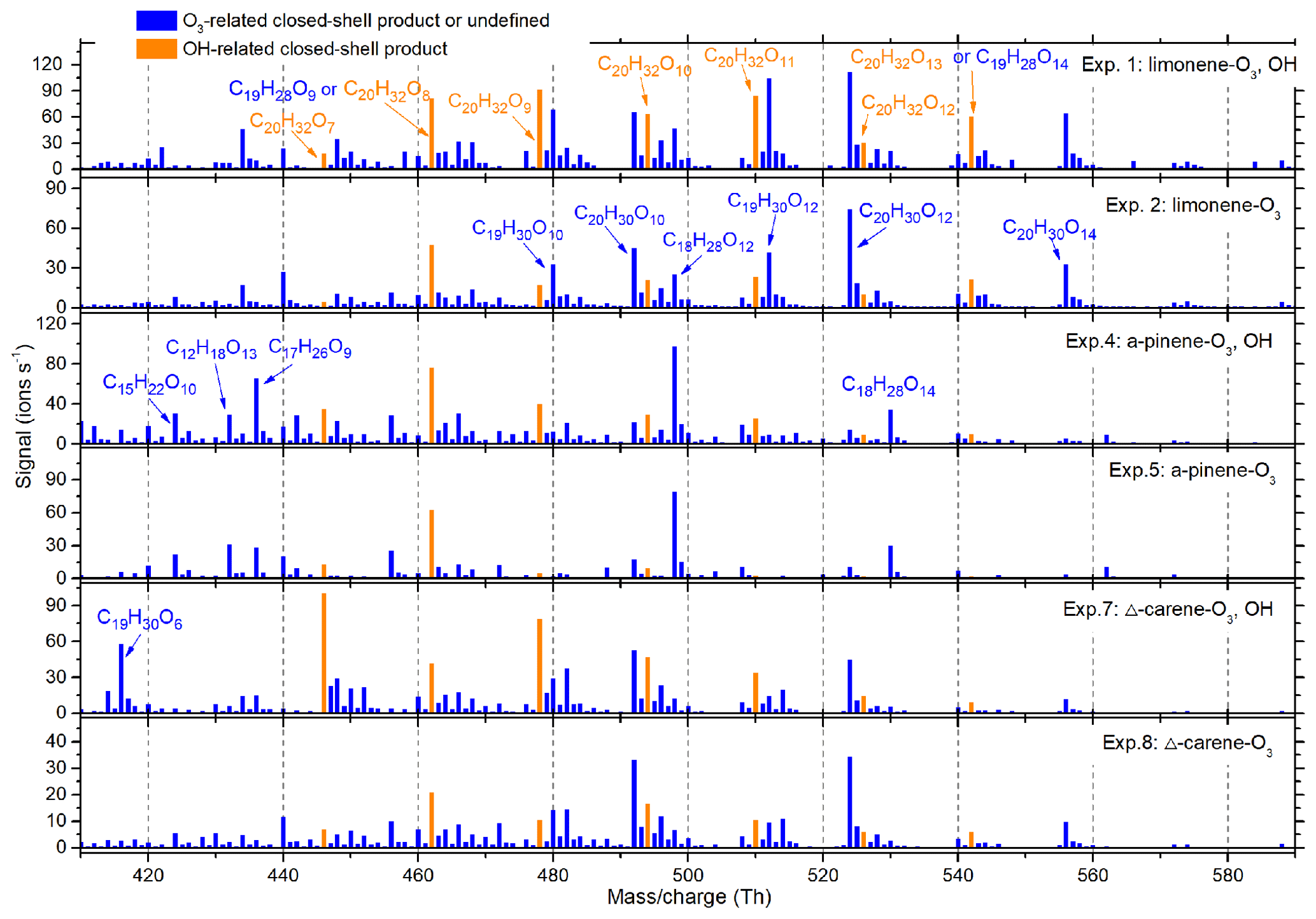
Figure 5Average dimer mass spectrum in each of the particle generation experiments. The OH- and O3-derived species were distinguished by comparing relative abundance of experiments with and without OH scavengers. All the peaks shown were in the form of adducts with or reagent ions.
Comparing total HOM abundance for the three monoterpene oxidation reactions, limonene created the most, followed by α-pinene and then Δ3-carene. This is in qualitative agreement with prior studies (Jokinen et al., 2014; Ehn et al., 2014). The total dimer signal intensity was 15–30 % of monomers for all three monoterpenes. Experiments with an OH scavenger generated fewer HOMs than those without OH scavengers.
As observed in previous studies, and comprised the O3- and OH-related RO2, respectively (Jokinen et al., 2014). comprised the O3-related closed-shell monomers, while and C10H18O6,7 comprised the OH-related closed-shell monomers (Ehn et al., 2014; Berndt et al., 2016). When comparing the average spectra with and without OH scavengers, no obvious differences were seen for OH-related RO2 or monomers (Fig. S4). In contrast, for dimers we found that C20H32O6−13 were more abundant in experiments without OH scavengers (Fig. 5). The formation of these dimers can be explained by the reaction of one OH-related RO2 with one O3-derived RO2 (see Sect. 3.5), and can therefore be considered as markers for combined OH and O3 chemistry. As HOM dimers are generally less volatile than monomers with an identical O∕C ratio, rapid production of dimers is believed to play a more important role in initial particle formation and growth (Zhang et al., 2015).
3.3 RH influence on HOM generation
Figure 6 shows a time series of experimental parameters, particle size distribution, and key ions from the limonene ozonolysis experiment with OH scavengers (Exp. 2 in Table 1). The O3 inlet and outlet concentrations were approximately constant with increasing RH (Fig. 6a), indicating that RH did not significantly change O3 levels in the flow tube. This also shows that the reactivity of the limonene with ozone does not change with RH. The number concentration of the generated particles decreased from 4.9×106 to 2.7×106 cm−3 with increasing RH, while the peak of the number size distribution increased slightly, due in part to water absorption. When RH was above 80 %, both the integrated number and mass concentrations, which were calculated from the number size distributions, decreased (Fig. 6b).
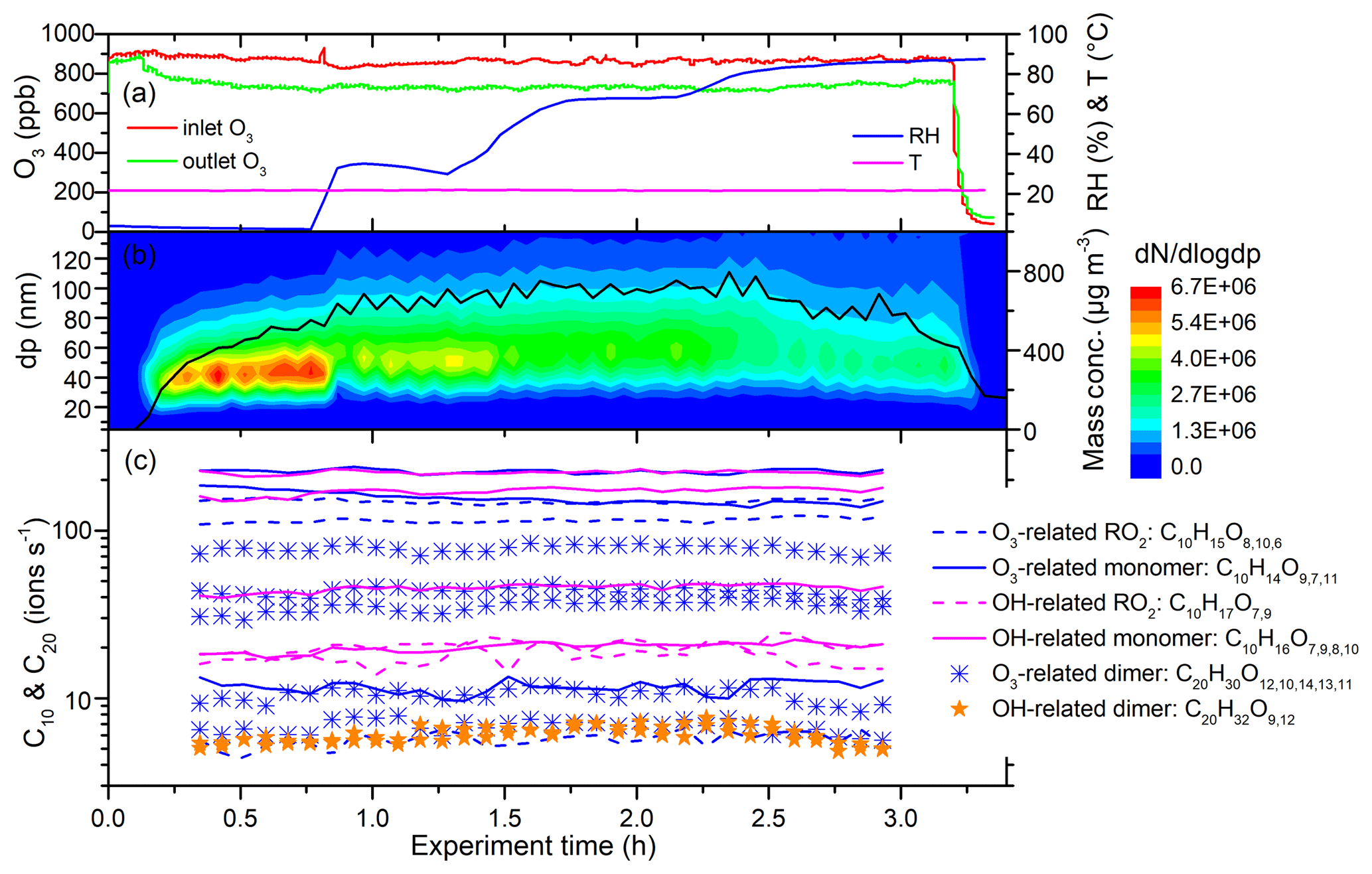
Figure 6Time series of experimental parameters, particle size distribution, and key ions during Exp. 2 (limonene oxidized by O3 without OH scavenger). (a) Inlet and outlet O3 concentrations, temperature, and RH. (b) Particle size distribution and integrated mass concentrations (assuming effective density is 1.2 g cm−3). (c) Some of the main HOMs detected by TI-CIMS with reagent ion. The subscript oxygen numbers in the formulae were ranked (left to right) according to signal abundance of the corresponding molecule.
Despite the change in particle number and mass concentrations with RH, the concentration of all the main HOMs, including RO2, monomers, and dimers, did not change for both OH- and O3-derived products (Fig. 6c). In fact, the only signals in the mass spectra that changed with RH corresponded to increases associated with water clusters. The variations in HOM concentrations can be explained by the competition between production and condensational losses. As almost all of the detected HOMs are ELVOCs or LVOCs (see Sect. 3.6), they are not likely to partition back to the gas phase after they encounter a surface. The condensation sink (CS) and wall loss rate for a compound with a diffusion coefficient of m2 s−1 (e.g., sulfuric acid) were estimated using established methods (Kürten et al., 2014; Crump and Seinfeld, 1981; Kulmala et al., 2001). The calculated CS varied between 0.1 and 3.5 s−1 in different SOA generation experiments, much larger than the wall loss rate (<0.01 s−1). There was about 5 %–30 % variation in CS in each SOA generation experiment from RH of 3 % to 92 %. This amount of variation in CS does not seem to have a noticeable influence on the final concentration of HOMs. To further test the hypothesis that variations in condensation sink do not impact final HOM concentrations, particle-free experiments were performed, and, again, detected HOM concentrations did not change with RH (Fig. 7).
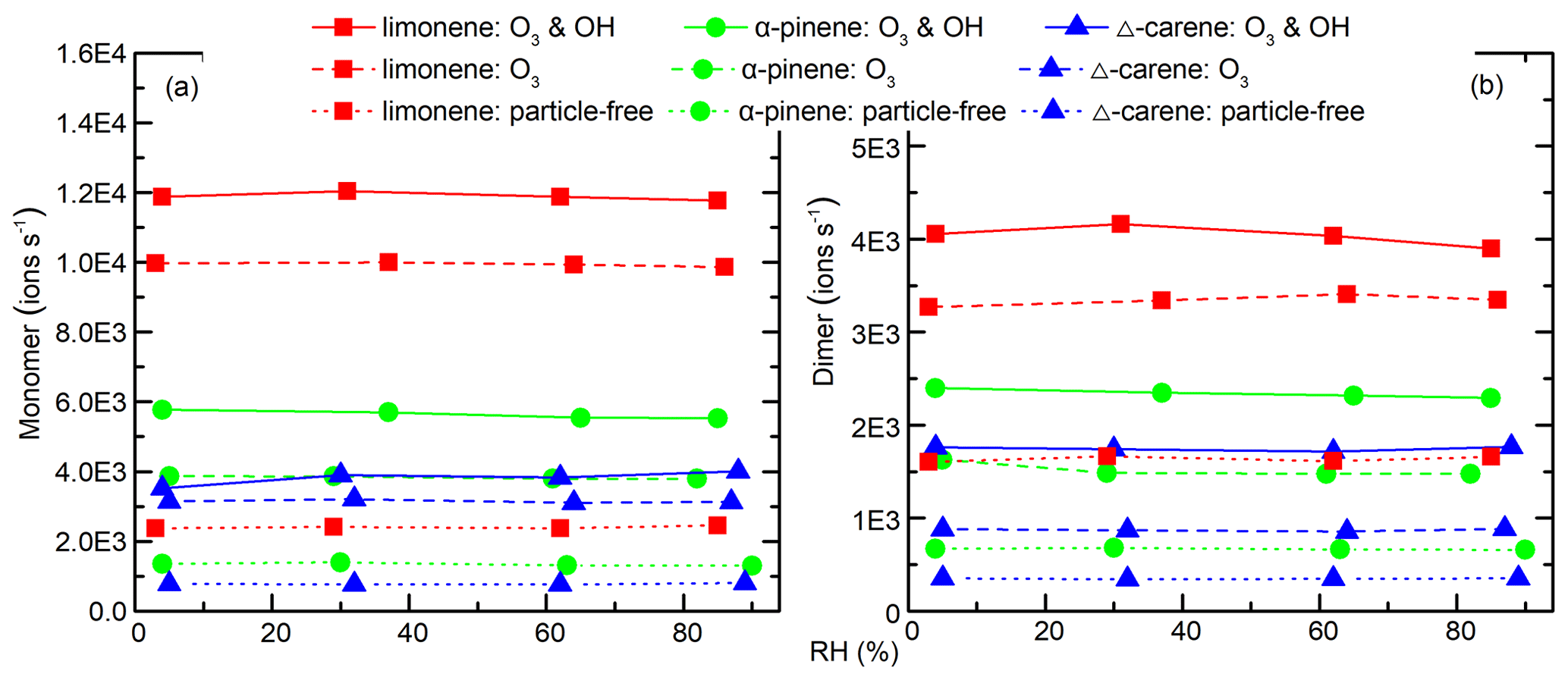
Figure 7Average (a) monomer and (b) dimer HOMs signal intensity (ions s−1) as a function of RH in each experiment. Monomer signals were the sum of C5−10 molecules, and dimer signals were the sum of C15−20 molecules. No obvious signal change was seen for increasing RH in any of the experiments.
3.4 RH influence on SOA generation
Figure 8 shows the integrated SOA particle number and mass concentrations over the observed diameter range of 10–100 nm. The generated SOA particle number and mass concentrations for limonene (2.2–6.0×106 cm−3 for number concentrations and 470–1025 µg m−3 for mass concentrations) were ∼3–12 times greater than for Δ3-carene (0.3–2.0×106 cm−3 for number concentrations and 56–86 µg m−3 for mass concentrations) and α-pinene (0.4–2.2×106 cm−3 for number concentrations and 61–130 µg m−3 for mass concentrations). This is because the theoretical ozone reactivity of limonene is 3–5 times higher than the last two and molar yield from limonene ozonolysis is also the highest. Peaks in the particle-number size distributions were between 40 and 70 nm (Fig. S5). In most of the experiments, generated SOA mass concentrations increased or decreased slightly when RH increased from ∼0 % to ∼60 % and decreased as RH further increased to ∼90 %. The variability in particle mass concentration as a function of RH for different experiments can be attributed to the combined effects of gas-phase reactions, condensed phase reactions, and the physical uptake of water, as well as the re-evaporation of semi-volatile compounds from the wall. We cannot accurately quantify these effects. As a result, although the measured SOA mass concentration remained relatively constant, we cannot draw conclusions from this observation. In contrast, while particle number concentrations may also be affected by the factors mentioned above, they decreased by a factor of 2–3 with increasing RH.
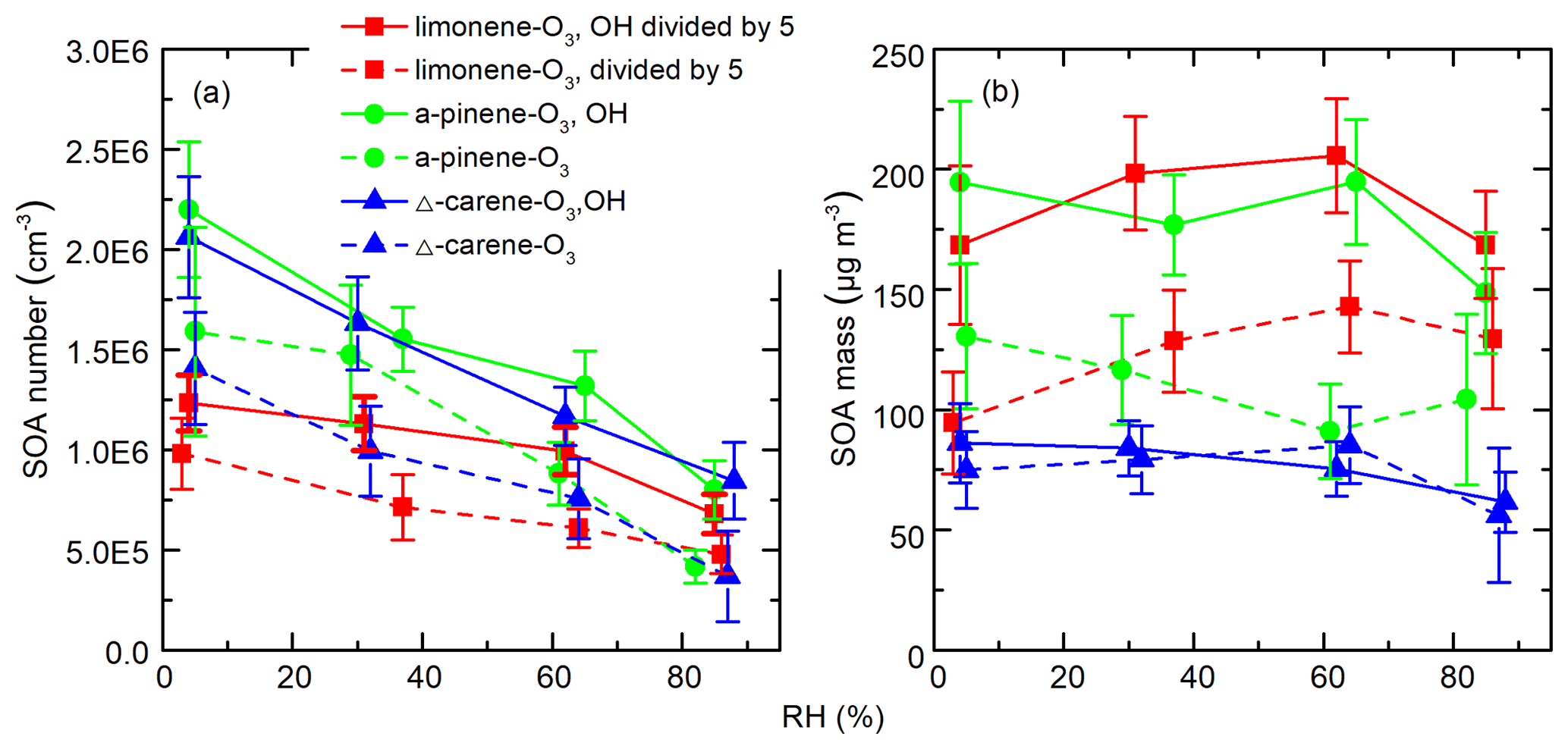
Figure 8SOA (a) number and (b) mass concentrations as a function of RH during different experiments. The error bars were calculated using both the statistical errors of all individual size distributions during each RH stage and assuming a systematic CPC counting error of 10 %.
A number of studies have demonstrated different water and OH influences on the ozonolysis products of exocyclic and endocyclic organic compounds. They have reported either suppressing (Bonn et al., 2002; Bonn and Moorgat, 2002) or promoting (Jonsson et al., 2006, 2008) effects of water vapor on the particle formation processes during ozonolysis of monoterpenes by measuring the number size distributions of generated SOA particles with the SMPS. The discrepancies between different results could be attributed to the different experiment setups, e.g., monoterpene and O3 concentration, temperature, RH range, OH scavengers, reaction time, and so on. Specifically, our results are in good agreement with those of Bonn et al. (2002), who studied SOA generation from the ozonolysis of endocyclic monoterpenes. In that study, SOA number concentrations decreased by a factor of 1.1–2.5 as RH increased, while the variation in volume concentrations was negligible (within ±10 %). They concluded that water's influence on non-volatile products, which are responsible for the initial steps of nucleation, was much larger than its influence on semi-volatile compounds which mainly determined the final volume concentrations of particles. Thus, it was highly suspected that water influenced new particle formation through influencing the generation of NPF precursors. However, our measurements indicate that at least the formation of the detected HOMs is independent of water vapor concentrations. There may be other species that are crucial to the initial steps of NPF and are affected by water vapor but are not detected by nitrate CIMS (see Sect. 3.5). Another possible explanation is that a fraction of HOMs cluster with water at high RH in such a way that they may no longer be able to participate in further cluster formation, thereby suppressing NPF. If the CIMS measurement only detected the declustered molecule, then such a mechanism may still be consistent with our observations.
3.5 Possible formation pathways of water-relevant C10 and C20 HOMs
Although the oxidation of BVOCs has been widely studied, it has mostly been constrained to the early stages (first- and second-generation intermediates), and many uncertainties still exist (Johnson and Marston, 2008; Isaacman-VanWertz et al., 2018; Atkinson and Arey, 2003). The first step of ozonolysis for the three BVOCs (α-pinene, Δ3-carene, and limonene) is ozone attack on the endocyclic carbon double bond to form a primary ozonide. Figure 9 shows the O3-initiated oxidation pathways of α-pinene that may be related to the detected C10 and C20 HOMs for representative isomers. The primary ozonide rapidly transforms to two excited Criegee intermediates (eCIs), one of which (branching ratio =0.4) (Kamens et al., 1999) is shown in Fig. 9. The reaction pathways of the eCI are complex; the most important two under ambient and most chamber conditions are the sCI channel (Reaction R1) and the hydroperoxide channel (Reaction R2) (Bonn et al., 2002). The sCI either reacts with aldehydes to form a secondary ozonide (SOZ; when the aldehyde is C10, then the formed SOZ is C20 and is marked as sCI-C10) or with water or other acidic compounds such as alcohols and carboxylic acids to form hydroxy-hydroperoxide, which then decomposes to carboxylic acids or aldehydes. For α-pinene, the main decomposition product is pinonic acid. In the hydroperoxide channel (Reaction R2), the formed hydroperoxide quickly decomposes to a first-generation alkyl radical (R) and OH (Johnson and Marston, 2008). R reacts with O2 immediately to form the first-generation RO2, which can undergo numerous reactions, including reaction with HO2, R′O2, and autoxidation. The reaction with HO2 mainly forms hydroperoxides, with a small fraction forming hydroxyl- or carbonyl-containing compounds. When reacted with another R′O2, either ROOR′ or an alkoxy radical (RO) or a carbonyl and a hydroxyl are formed. The RO can undergo isomerization, or form a carbonyl and HO2, for which the branching ratios are extremely difficult to evaluate. RO can also undergo decomposition, which is one of the pathways to form C5 ∼ C9. The autoxidation process is key to HOM formation. Each autoxidation step adds two O atoms to the molecule and thus increases the oxidation state very rapidly. The competition between autoxidation processes and bimolecular reactions (RO2 reactions with R′O2 or HO2) determines the ultimate oxidation state of the products (Barsanti et al., 2017; Crounse et al., 2013; Rissanen et al., 2015).
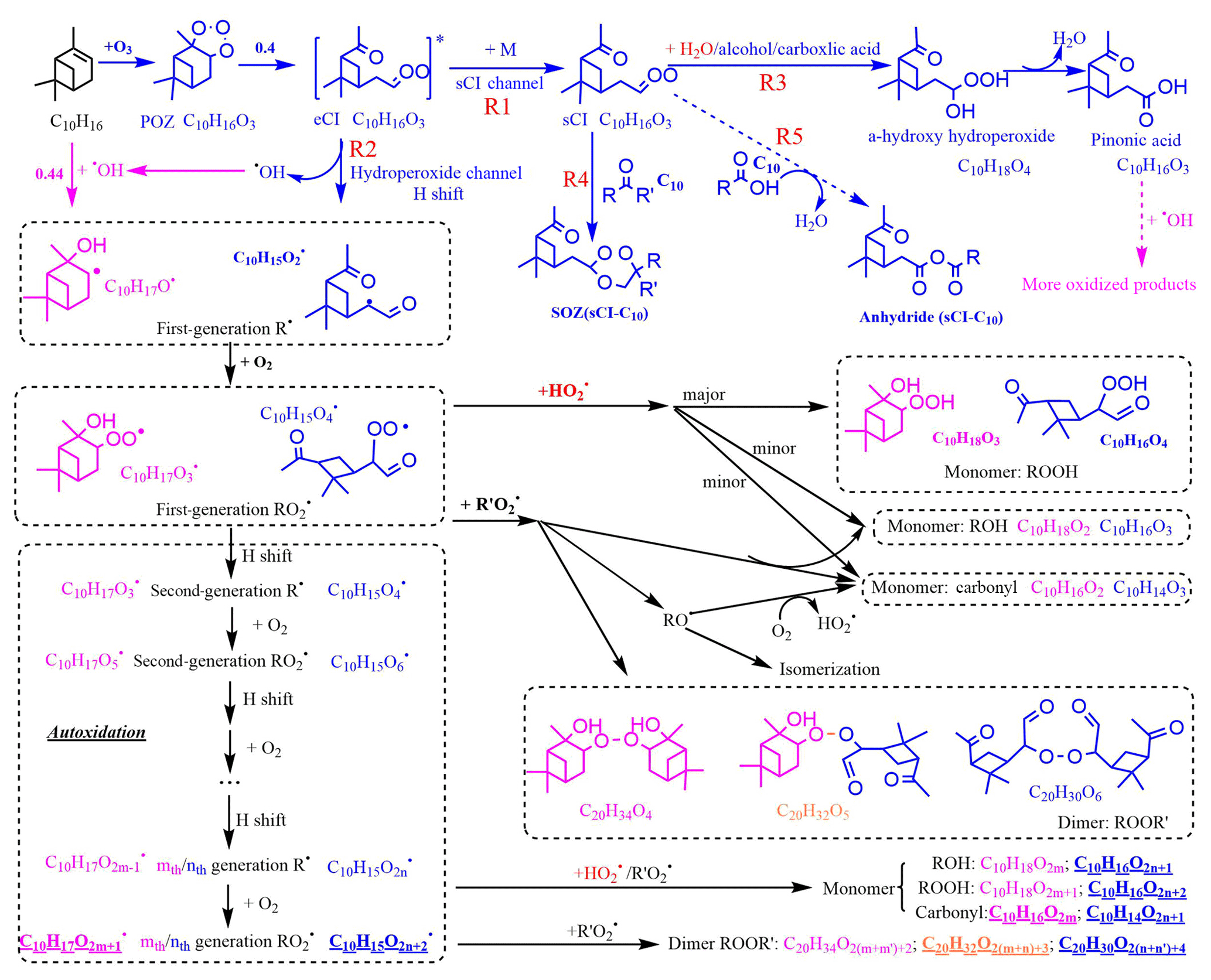
Figure 9Proposed key steps in the formation of the representative C10 and C20 closed-shell products from α-pinene oxidation and possible water vapor influence. Dashed lines represent pathways that may or may not happen, depending on the situation. Pink and blue colors represent the pathways or products from O3 and OH oxidation, respectively. Common pathways or products are indicated in black type. Orange colors represent the combined products of O3 and OH chemistry. Red colors highlight the direct or indirect influence of water. Underlined formulae are the main products observed from the mass spectrum. C10H18O2m and C20H34O were not observed in our spectrum, but they dominated the spectrum in other reported experiments in which extra OH was generated (Berndt et al., 2016).
OH can be generated in the ozonolysis of alkenes and the yield is near unity (Atkinson, 1997). The reaction of OH with α-pinene directly forms first-generation R and then RO2; one possible structure for this RO2 (branching ratio =0.44), formed from OH addition to the double bond (Berndt et al., 2016), is shown in Fig. 9. However, the RO2 formed (C10H17O2m+1) are not the same RO2 as those formed through ozonolysis (C10H15O2n+2) (McVay et al., 2016). Accordingly, the structure and composition of C10 and C20 HOMs formed from OH or O3 chemistry are different, and so too are their potential impacts on NPF. The combined OH- and O3-derived dimers (C10H32O), formed by collision of an OH-derived RO2 with an O3-derived RO2, were only observed in ozonolysis experiments without OH scavengers.
The RO2 autoxidation pathway explains most of the observed C10 and C20 compounds in the mass spectra. One exception to this is that C10H18O2m+1, C10H18O2m, and C20H34O were not observed in the spectrum, whereas in experiments performed by Berndt et al. (2016) in which OH oxidation for α-pinene was studied, C10H18O2m and C20H34O dominated the mass spectrum. This could be explained by a lower OH∕O3 ratio in our experiments, since unlike Berndt et al. (2016) we did not provide an extra source of OH to the flow tube. Also, Berndt et al. (2016) reported lower sensitivity of nitrate reagent ions to OH-related RO2 compared to other reagent ions such as acetate.
Despite this close agreement achieved by the RO2 autoxidation mechanism and the observed mass spectra in our study, prior studies suggest that other potential pathways cannot be excluded. An accretion product involving sCI is one possibility (Barsanti et al., 2017). It is possible that sCI reacts with long-chain carboxylic acids or carbonyls, such as those with 10 carbon atoms, forming in this instance anhydrides (sCI-C10; Reaction R5) or secondary ozonides (sCI-C10; Reaction R4) with vapor pressures lower than 10−15 torr (Kamens et al., 1999; Tobias and Ziemann, 2001; Bonn et al., 2002). The formation of anhydride is more likely in the condensed phase, whereas there is also a possibility it can also happen in the gas phase (Kamens et al., 1999). However, it is unknown whether these sCI-C10 can be detected using a nitrate CIMS as they may lack hydrogen bond donor moieties. The semi-volatile pinonic acid can also form HOMs after further oxidation by OH (Ehn et al., 2014), provided that excess α-pinene is not present to compete with pinonic acid for the generated OH.
Water vapor's influence on HOM formation can be direct or indirect. For monoterpene oxidation, the direct participation of water vapor is to react with sCI, favoring the formation of the hydroperoxide and its decomposition products (Reaction R3) over the secondary ozonides (sCI-C10; Reaction R4) or possible anhydrides (sCI-C10; Reaction R5). Since the formation of sCI-C10 is more likely to contribute to NPF than the products from sCI and water vapor (Kamens et al., 1999; Tobias et al., 2000), a decrease in low-volatility sCI-C10 with high RH could explain the decreasing SOA number concentrations in our experiment. It has been shown previously that OH yields from the reactions of O3 with a series of monoterpenes were not affected by the presence of water vapor (Atkinson et al., 1992; Aschmann et al., 2002), which implies that the hydroperoxide channel (Reaction R2) is similarly unaffected by water. Since the detected HOMs in our experiments were RH-independent, we conclude that all the detected HOMs were formed from the hydroperoxide channel (Reaction R2) and not via the sCI channel (Reaction R1). Similarly, the detected HOMs were not likely to form through the hydration reaction (Reaction R6; Ehn et al., 2012), which is supposed to increase with increasing RH.
The indirect water effect on HOM formation includes the water influence on HO2 fate. As water promotes HO2 self-reaction (Reaction R7), reaction of HO2 with RO2 should decrease and the related HOM monomers should likewise decrease with increasing RH. However, as the formation of both HOM monomers and dimers was not affected by H2O, it was likely that water does not significantly increase HO2 self-reaction or that HO2 chemistry was not important in our experiments.
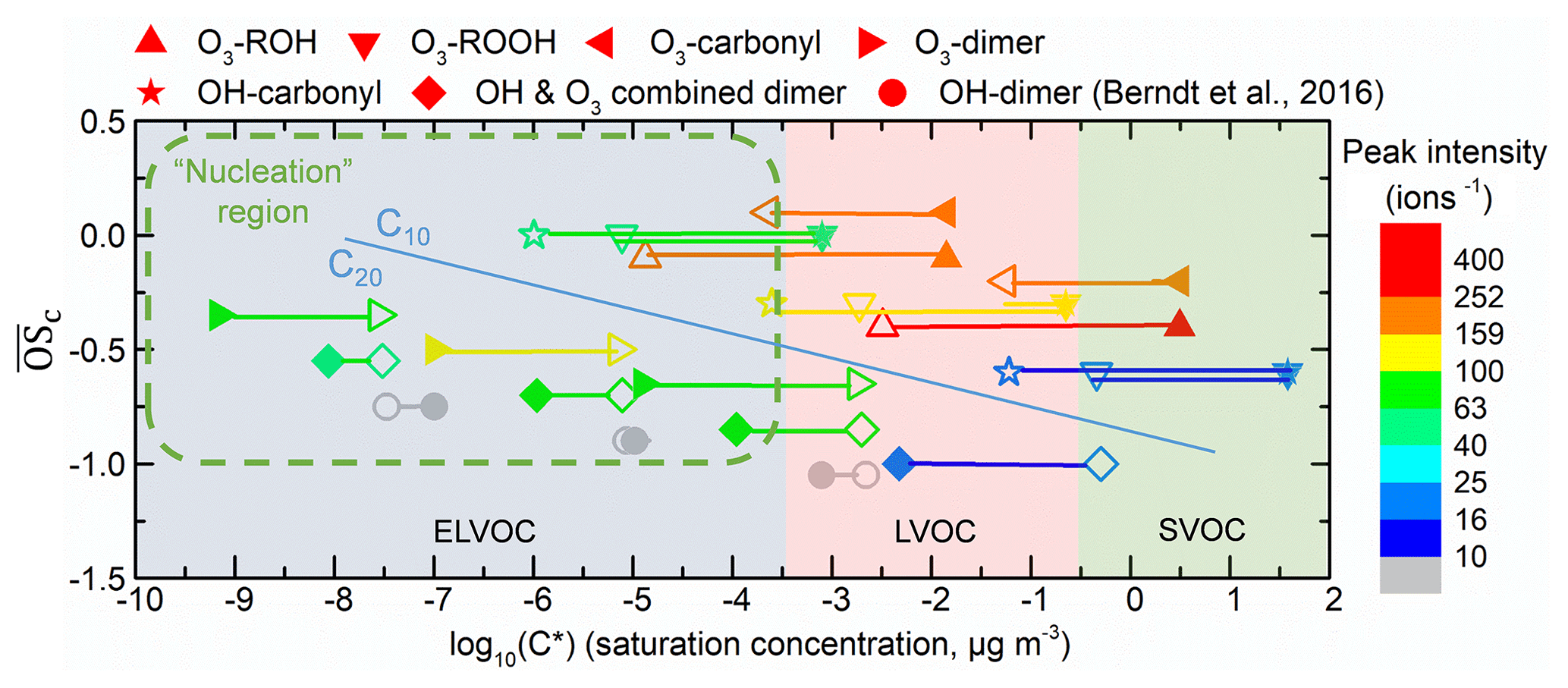
Figure 10Vapor saturation mass concentration C* (T=298 K) of the major C10 and C20 closed-shell products were predicted with SIMPOL.1 (open points) (Pankow and Asher, 2008) and molecular corridor method (filled points) (Li et al., 2016). Lines connecting the open points and filled points represent the difference between the two methods. O3- or OH-derived monomers and dimers are shown by different shapes. The peak intensity, represented by color, is from Exp.1 (limonene oxidation without an OH scavenger). The gray points, which represent OH-derived dimers, are dominating products in OH-initiated oxidation experiments (Berndt et al., 2016) while not observed in this study. Data used in this figure are given in Table S1. The nucleation region (green dashed rectangle) is from Donahue et al. (2013).
3.6 Volatility predictions
The volatility of the gas-phase products is one of the most important properties that determines whether a compound contributes to the formation, initial growth, or further growth of SOA particles (Donahue et al., 2012; Kroll et al., 2011). As the products with identical elemental composition can be formed from different bimolecular reactions of the intermediate RO2, it is difficult to predict their exact structures. For the current study, the number of different structural and functional groups (e.g., aromatic rings, aldehydes, ketones, hydroxyls, peroxides, hydro-peroxides) was estimated and used to derive saturation vapor pressure using SIMPOL.1 (Table S1 in the Supplement). To simplify the calculation, the functional groups used here were directly predicted from the proposed formation pathways in Fig. 10 and did not include intramolecular isomerization, although that may be important in some situations. For example, one of the ROOH can be replaced with an endoperoxide via ring closure of unsaturated RO2 (Berndt et al., 2016). It has to be noted that the group contribution methods very likely underestimate the volatility of HOM products from α-pinene autoxidation products due to ignoring intramolecular H bonding (Kurtén et al., 2016). There may be large uncertainties in the SIMPOL method as well as in our functional group estimation process. As a comparison, the molecular corridor method (Li et al., 2016), which does not require information on functional groups, was used to estimate the saturation vapor pressure as well. The average carbon oxidation state () was calculated with Eq. (3), in replacement of the commonly used formula (), as the second oxygen atom in the (hydro)peroxide group does not increase the carbon oxidation state. In Eq. (3), nO, nC, and nH are the oxygen, carbon, and hydrogen numbers in the molecule; n(hydro)peroxide is the number of (hydro)peroxide groups in the molecule.
Figure 10 shows the predicted saturation mass concentrations (C*) of the main C10 and C20 closed-shell products. The difference of C* predicted from the two methods was ∼1–4 orders of magnitudes. Despite these differences, most of the C20 HOMs can be classified as ELVOCs, while C10 products were mostly LVOCs. Typically, for those compounds with identical , such as C20H30O10 (O3-derived dimer, ), C20H32O11 (OH and O3 combined dimer, ), C20H34O12 (OH-derived dimer, ), and OH-derived HOMs, have lower volatilities than O3-derived HOMs due to a greater number of (hydro)peroxide groups.
The RH influence on HOM formation and NPF during monoterpene oxidation was explored in this study. HOMs were detected with a TI-CIMS, using nitrate as reagent ions; C10 and C20 dominated the spectra. There are mainly three potential paths for water vapor influence on the formation of C10 and C20 HOMs. One is water reacting with sCI, thereby influencing the branching ratio between the formation of more volatile compounds decomposed from hydroxyl hydroperoxide, such as pinonic acid, and accretion products with sCI such as secondary ozonide (sCI-C10) and anhydride (sCI-C10). The second hypothesized water influence is on the HOMs formed from hydration reactions. The third is that water increases the rate of self-reaction of HO2, thus indirectly impacting the loss pathways of RO2. Our experimental results, both with high particle loading and particle-free conditions, demonstrated that neither the detected HOM species nor their signal abundance changed significantly with RH. This indicates that the detected HOMs, which can mostly be explained by RO2 autoxidation, must be formed from water-independent pathways rather than by those reactions mentioned above. One implication of this result is that HO2 self-reaction was not significantly promoted by water or that the RO2 reaction with HO2 was not be significant in our system, but instead that RO2 reacts with another peroxy radical, R′O2, to generate both closed-shell monomers and dimers. Another implication is that the sCI pathway is not responsible for the generation of the detected HOMs, while the role of sCI-related HOMs' (SOZ or anhydride) formation by accretion with long-chain products, which may not be detected with nitrate CIMS, may be important in causing the decrease in SOA number concentrations with increased RH. Another possible explanation for the decreasing SOA number concentration is that water may cluster with HOMs and suppress NPF.
The detected HOMs, which could mostly be explained by autoxidation of RO2 followed by reactions with R′O2 or HO2, were distinguished as OH-related, O3-related RO2, closed-shell HOM monomers, and HOM dimers. The volatility of the identified products was estimated with the SIMPOL.1 group contribution method and with the molecular corridor technique. That analysis confirmed that C20 closed-shell products have significantly lower volatility compared to C10 products and are thus more likely to contribute to NPF. For the HOM products with identical , OH-derived HOMs have lower volatilities than O3-derived HOMs due to a greater number of (hydro)peroxide groups. As a result, OH chemistry is suspected to be more likely to lead to NPF than O3 chemistry, given the same level of oxidants and VOC precursors.
Data are publicly available and archived at https://doi.org/10.7280/D1B674 (Smith and Jiang, 2019).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-1555-2019-supplement.
XL, JPDA, JJ, JNS designed the experiments and XL and SC carried them out. JH and SC assisted XL in data analysis. XL prepared the manuscript with contributions from all co-authors.
The authors declare that they have no conflict of interest.
This research was supported by the US Department of Energy's Atmospheric
System Research program under grant no DE-SC0019000, the US National Science
Foundation under grant no. AGS-1762098, and the National Key R&D
Program of China under grant no. 2017YFC0209503. Xiaoxiao Li acknowledges the
financial support from the State Scholarship Fund managed by Chinese
Scholarship Council (CSC). We thank Hayley Glicker, Deanna Caroline Myers,
Michael Lawler, and Danielle Draper for their kind help.
Edited by: Alexander Laskin
Reviewed by: three anonymous referees
Aschmann, S. M., Arey, J., and Atkinson, R.: OH radical formation from the gas-phase reactions of O3 with a series of terpenes, Atmos Environ, 36, 4347–4355, https://doi.org/10.1016/s1352-2310(02)00355-2, 2002.
Atkinson, R.: Gas-phase tropospheric chemistry of volatile organic compounds: 1. Alkanes and alkenes, J. Phys. Chem. Ref. Data, 26, 215–290, 1997.
Atkinson, R. and Arey, J.: Gas-phase tropospheric chemistry of biogenic volatile organic compounds: a review, Atmos. Environ., 37, 197–219, 2003.
Atkinson, R., Aschmann, S. M., Arey, J., and Shorees, B.: Formation of OH radicals in the gas phase reactions of O3 with a series of terpenes, J. Geophys. Res., 97, 6065–6073, 1992.
Ball, S. M., Hanson, D. R., Eisele, F. L., and McMurry, P. H.: Laboratory studies of particle nucleation: Initial results for H2SO4, H2O, and NH3 vapors, J Geophys Res-Atmos, 104, 23709–23718, https://doi.org/10.1029/1999jd900411, 1999.
Barsanti, K. C., Kroll, J. H., and Thornton, J. A.: Formation of Low-Volatility Organic Compounds in the Atmosphere: Recent Advancements and Insights, J. Phys. Chem. Lett., 8, 1503–1511, https://doi.org/10.1021/acs.jpclett.7b02969, 2017.
Berndt, T., Richters, S., Jokinen, T., Hyttinen, N., Kurtén, T., Otkjær, R. V., Kjaergaard, H. G., Stratmann, F., Herrmann, H., and Sipilä, M.: Hydroxyl radical-induced formation of highly oxidized organic compounds, Nat. Commun., 7, 13677, https://doi.org/10.1038/ncomms13677, 2016.
Bianchi, F., Tröstl, J., Junninen, H., Frege, C., Henne, S., Hoyle, C. R., Molteni, U., Herrmann, E., Adamov, A., and Bukowiecki, N.: New particle formation in the free troposphere: A question of chemistry and timing, Science, 352, 1109–1112, https://doi.org/10.1126/science.aad5456, 2016.
Bonn, B. and Moorgat, G. K.: New particle formation during a- and b-pinene oxidation by O3, OH and NO3, and the influence of water vapour: particle size distribution studies, Atmos. Chem. Phys., 2, 183–196, https://doi.org/10.5194/acp-2-183-2002, 2002.
Bonn, B., Schuster, G., and Moortgat, G. K.: Influence of water vapor on the process of new particle formation during monoterpene ozonolysis, J. Phys. Chem. A, 106, 2869–2881, 2002.
Boy, M. and Kulmala, M.: Nucleation events in the continental boundary layer: Influence of physical and meteorological parameters, Atmos. Chem. Phys., 2, 1–16, https://doi.org/10.5194/acp-2-1-2002, 2002.
Burkholder, J. B., Baynard, T., Ravishankara, A. R., and Lovejoy, E. R.: Particle nucleation following the O3 and OH initiated oxidation of α-pinene and β-pinene between 278 and 320 K, J. Geophys. Res., 112, D10216, https://doi.org/10.1029/2006JD007783, 2007.
Cai, R., Yang, D., Fu, Y., Wang, X., Li, X., Ma, Y., Hao, J., Zheng, J., and Jiang, J.: Aerosol surface area concentration: a governing factor in new particle formation in Beijing, Atmos. Chem. Phys., 17, 12327–12340, https://doi.org/10.5194/acp-17-12327-2017, 2017.
Chen, H., Ezell, M. J., Arquero, K. D., Varner, M. E., Dawson, M. L., Gerber, R. B., and Finlayson-Pitts, B. J.: New particle formation and growth from methanesulfonic acid, trimethylamine and water, Phys. Chem. Chem. Phys., 17, 13699–13709, 2015.
Chen, M., Titcombe, M., Jiang, J., Jen, C., Kuang, C., Fischer, M. L., Eisele, F. L., Siepmann, J. I., Hanson, D. R., and Zhao, J.: Acid–base chemical reaction model for nucleation rates in the polluted atmospheric boundary layer, P. Natl. Acad. Sci. USA, 109, 18713–18718, 2012.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of organic compounds in the atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, 2013.
Crump, J. G. and Seinfeld, J. H.: Turbulent deposition and gravitational sedimentation of an aerosol in a vessel of arbitrary shape, J. Aerosol Sci., 12, 405–415, https://doi.org/10.1016/0021-8502(81)90036-7, 1981.
Dada, L., Paasonen, P., Nieminen, T., Buenrostro Mazon, S., Kontkanen, J., Peräkylä, O., Lehtipalo, K., Hussein, T., Petäjä, T., Kerminen, V.-M., Bäck, J., and Kulmala, M.: Long-term analysis of clear-sky new particle formation events and nonevents in Hyytiälä, Atmos. Chem. Phys., 17, 6227–6241, https://doi.org/10.5194/acp-17-6227-2017, 2017.
Donahue, N. M., Trump, E. R., Pierce, J. R., and Riipinen, I.: Theoretical constraints on pure vapor-pressure driven condensation of organics to ultrafine particles, Geophys. Res. Lett., 38, L16801, https://doi.org/10.1029/2011gl048115, 2011.
Donahue, N. M., Kroll, J. H., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set – Part 2: Diagnostics of organic-aerosol evolution, Atmos. Chem. Phys., 12, 615–634, https://doi.org/10.5194/acp-12-615-2012, 2012.
Donahue, N. M., Ortega, I. K., Chuang, W., Riipinen, I., Riccobono, F., Schobesberger, S., Dommen, J., Baltensperger, U., Kulmala, M., Worsnop, D. R., and Vehkamaki, H.: How do organic vapors contribute to new-particle formation?, Faraday Discuss., 165, 91–104, https://doi.org/10.1039/c3fd00046j, 2013.
Dunne, E. M., Gordon, H., Kürten, A., Almeida, J., Duplissy, J., Williamson, C., Ortega, I. K., Pringle, K. J., Adamov, A., Baltensperger, U., Barmet, P., Benduhn, F., Bianchi, F., Breitenlechner, M., Clarke, A., Curtius, J., Dommen, J., Donahue, N. M., Ehrhart, S., Flagan, R. C., Franchin, A., Guida, R., Hakala, J., Hansel, A., Heinritzi, M., Jokinen, T., Kangasluoma, J., Kirkby, J., Kulmala, M., Kupc, A., Lawler, M. J., Lehtipalo, K., Makhmutov, V., Mann, G., Mathot, S., Merikanto, J., Miettinen, P., Nenes, A., Onnela, A., Rap, A., Reddington, C. L. S., Riccobono, F., Richards, N. A. D., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Simon, M., Sipila, M., Smith, J. N., Stozkhov, Y., Tome, A., Trostl, J., Wagner, P. E., Wimmer, D., Winkler, P. M., Worsnop, D. R., and Carslaw, K. S.: Global atmospheric particle formation from CERN CLOUD measurements, Science, 354, 1119–1124, https://doi.org/10.1126/science.aaf2649, 2016.
Duplissy, J., Merikanto, J., Franchin, A., Tsagkogeorgas, G., Kangasluoma, J., Wimmer, D., Vuollekoski, H., Schobesberger, S., Lehtipalo, K., Flagan, R., Brus, D., Donahue, N. M., Vehkamäki, H., Almeida, J., Amorim, A., Barmet, P., Bianchi, F., Breitenlechner, M., Dunne, E. M., Guida, R., Henschel, H., Junninen, H., Kirkby, J., Kürten, A., Kupc, A., Määttänen, A., Makhmutov, V., Mathot, S., Nieminen, T., Onnela, A., Praplan, A. P., Riccobono, F., Rondo, L., Steiner, G., Tome, A., Walther, H., Baltensperger, U., Carslaw, K. S., Dommen, J., Hansel, A., Petäjä, T., Sipilä, M., Stratmann, F., Vrtala, A., Wagner, P. E., Worsnop, D. R., Curtius, J., and Kulmala, M.: Effect of ions on sulfuric acid-water binary particle formation: 2. Experimental data and comparison with QC-normalized classical nucleation theory, J. Geophys. Res.-Atmos., 121, 1752–1775, 2016.
Ehn, M., Kleist, E., Junninen, H., Petäjä, T., Lönn, G., Schobesberger, S., Dal Maso, M., Trimborn, A., Kulmala, M., Worsnop, D. R., Wahner, A., Wildt, J., and Mentel, Th. F.: Gas phase formation of extremely oxidized pinene reaction products in chamber and ambient air, Atmos. Chem. Phys., 12, 5113–5127, https://doi.org/10.5194/acp-12-5113-2012, 2012.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Dal Maso, M., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Eisele, F. L. and Tanner, D. J.: Measurement of the gas phase concentration of H2SO4 and methane sulfonic-acid and estimates of H2SO4 production and loss in the atmosphere, J. Geophys. Res., 98, 9001–9010, https://doi.org/10.1029/93jd00031, 1993.
Frege, C., Ortega, I. K., Rissanen, M. P., Praplan, A. P., Steiner, G., Heinritzi, M., Ahonen, L., Amorim, A., Bernhammer, A.-K., Bianchi, F., Brilke, S., Breitenlechner, M., Dada, L., Dias, A., Duplissy, J., Ehrhart, S., El-Haddad, I., Fischer, L., Fuchs, C., Garmash, O., Gonin, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kirkby, J., Kürten, A., Lehtipalo, K., Leiminger, M., Mauldin, R. L., Molteni, U., Nichman, L., Petäjä, T., Sarnela, N., Schobesberger, S., Simon, M., Sipilä, M., Stolzenburg, D., Tomé, A., Vogel, A. L., Wagner, A. C., Wagner, R., Xiao, M., Yan, C., Ye, P., Curtius, J., Donahue, N. M., Flagan, R. C., Kulmala, M., Worsnop, D. R., Winkler, P. M., Dommen, J., and Baltensperger, U.: Influence of temperature on the molecular composition of ions and charged clusters during pure biogenic nucleation, Atmos. Chem. Phys., 18, 65–79, https://doi.org/10.5194/acp-18-65-2018, 2018.
Hamed, A., Korhonen, H., Sihto, S. L., Joutsensaari, J., Järvinen, H., Petäjä, T., Arnold, F., Nieminen, T., Kulmala, M., Smith, J. N., Kari Lehtinen, E. J., and Laaksonen, A.: The role of relative humidity in continental new particle formation, J. Geophys. Res., 116, D03202, https://doi.org/10.1029/2010JD014186, 2011.
Hanson, D., McMurry, P., Jiang, J., Tanner, D., and Huey, L.: Ambient pressure proton transfer mass spectrometry: detection of amines and ammonia, Environ. Sci. Technol., 45, 8881–8888, 2011.
Hoffmann, T., O'Dowd, C. D., and Seinfeld, J. H.: Iodine oxide homogeneous nucleation: An explanation for coastal new particle production, Geophys. Res. Lett., 28, 1949–1952, 2001.
Hyttinen, N., Kupiainen-Maäättä, O., Rissanen, M. P., Muuronen, M., Ehn, M., and Kurtén, T.: Modeling the charging of highly oxidized cyclohexene ozonolysis products using nitrate-based chemical ionization, J. Phys. Chem. A, 119, 6339–6345, 2015.
Hyvönen, S., Junninen, H., Laakso, L., Dal Maso, M., Grönholm, T., Bonn, B., Keronen, P., Aalto, P., Hiltunen, V., Pohja, T., Launiainen, S., Hari, P., Mannila, H., and Kulmala, M.: A look at aerosol formation using data mining techniques, Atmos. Chem. Phys., 5, 3345–3356, https://doi.org/10.5194/acp-5-3345-2005, 2005.
Isaacman-VanWertz, G., Massoli, P., O'Brien, R., Lim, C., Franklin, J. P., Moss, J. A., Hunter, J. F., Nowak, J. B., Canagaratna, M. R., and Misztal, P. K.: Chemical evolution of atmospheric organic carbon over multiple generations of oxidation, Nat. Chem., 10, 462–468, 2018.
Johnson, D. and Marston, G.: The gas-phase ozonolysis of unsaturated volatile organic compounds in the troposphere, Chem. Soc. Rev., 37, 699–716, 2008.
Jokinen, T., Sipilä, M., Junninen, H., Ehn, M., Lönn, G., Hakala, J., Petäjä, T., Mauldin III, R. L., Kulmala, M., and Worsnop, D. R.: Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF, Atmos. Chem. Phys., 12, 4117–4125, https://doi.org/10.5194/acp-12-4117-2012, 2012.
Jokinen, T., Sipilä, M., Richters, S., Kerminen, V. M., Paasonen, P., Stratmann, F., Worsnop, D., Kulmala, M., Ehn, M., Herrmann, H., and Berndt, T.: Rapid autoxidation forms highly oxidized RO2 radicals in the atmosphere, Angew. Chem. Int. Edit., 53, 14596–14600, 2014.
Jonsson, Å., M., Hallquist, M., and Ljungström, E.: Impact of humidity on the ozone initiated oxidation of limonene, Δ3-carene, and α-pinene, Environ. Sci. Technol., 40, 188–194, 2006.
Jonsson, Å., M., Hallquist, M., and Ljungström, E.: Influence of OH scavenger on the water effect on secondary organic aerosol formation from ozonolysis of limonene, Δ3-carene, and α-pinene, Environ. Sci. Technol., 42, 5938–5944, 2008.
Junninen, H., Ehn, M., Petäjä, T., Luosujärvi, L., Kotiaho, T., Kostiainen, R., Rohner, U., Gonin, M., Fuhrer, K., Kulmala, M., and Worsnop, D. R.: A high-resolution mass spectrometer to measure atmospheric ion composition, Atmos. Meas. Tech., 3, 1039–1053, https://doi.org/10.5194/amt-3-1039-2010, 2010.
Kamens, R., Jang, M., Chien, C.-J., and Leach, K.: Aerosol Formation from the Reaction of α-Pinene and Ozone Using a Gas-Phase Kinetics-Aerosol Partitioning Model, Environ. Sci. Technol., 33, 1430–1438, https://doi.org/10.1021/es980725r, 1999.
Kroll, J. H., Donahue, N. M., Jimenez, J. L., Kessler, S. H., Canagaratna, M. R., Wilson, K. R., Altieri, K. E., Mazzoleni, L. R., Wozniak, A. S., Bluhm, H., Mysak, E. R., Smith, J. D., Kolb, C. E., and Worsnop, D. R.: Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol, Nat. Chem., 3, 133–139, https://doi.org/10.1038/nchem.948, 2011.
Kuang, C., McMurry, P. H., McCormick, A. V., and Eisele, F. L.: Dependence of nucleation rates on sulfuric acid vapor concentration in diverse atmospheric locations, J. Geophys. Res., 113, D10209, https://doi.org/10.1029/2007jd009253, 2008.
Kulmala, M., Dal Maso, M., Makela, J. M., Pirjola, L., Vakeva, M., Aalto, P., Miikkulainen, P., Hameri, K., and O'Dowd, C. D.: On the formation, growth and composition of nucleation mode particles, Tellus B, 53, 479–490, https://doi.org/10.1034/j.1600-0889.2001.d01-33.x, 2001.
Kulmala, M., Vehkamäki, H., Petäjä, T., Dal Maso, M., Lauri, A., Kerminen, V. M., Birmili, W., and McMurry, P. H.: Formation and growth rates of ultrafine atmospheric particles: a review of observations, J. Aerosol. Sci., 35, 143–176, https://doi.org/10.1016/j.jaerosci.2003.10.003, 2004.
Kulmala, M., Petäjä, T., Ehn, M., Thornton, J., Sipilä, M., Worsnop, D., and Kerminen, V.-M.: Chemistry of atmospheric nucleation: on the recent advances on precursor characterization and atmospheric cluster composition in connection with atmospheric new particle formation, Annu. Rev. Phys. Chem., 65, 21–37, 2014.
Kürten, A., Rondo, L., Ehrhart, S., and Curtius, J.: Calibration of a Chemical Ionization Mass Spectrometer for the Measurement of Gaseous Sulfuric Acid, J. Phys. Chem. A, 116, 6375–6386, 2012.
Kürten, A., Jokinen, T., Simon, M., Sipilä, M., Sarnela, N., Junninen, H., Adamov, A., Almeida, J., Amorim, A., Bianchi, F., Breitenlechner, M., Dommen, J., Donahue, N. M., Duplissy, J., Ehrhart, S., Flagan, R. C., Franchin, A., Hakala, J., Hansel, A., Heinritzi, M., Hutterli, M., Kangasluoma, J., Kirkby, J., Laaksonen, A., Lehtipalo, K., Leiminger, M., Makhmutov, V., Mathot, S., Onnela, A., Petäjä, T., Praplan, A. P., Riccobono, F., Rissanen, M. P., Rondo, L., Schobesberger, S., Seinfeld, J. H., Steiner, G., Tome, A., Troestl, J., Winkler, P. M., Williamson, C., Wimmer, D., Ye, P., Baltensperger, U., Carslaw, K. S., Kulmala, M., Worsnop, D. R., and Curtius, J.: Neutral molecular cluster formation of sulfuric acid-dimethylamine observed in real time under atmospheric conditions, P. Natl. Acad. Sci. USA, 111, 15019–15024, https://doi.org/10.1073/pnas.1404853111, 2014.
Kürten, A., Bergen, A., Heinritzi, M., Leiminger, M., Lorenz, V., Piel, F., Simon, M., Sitals, R., Wagner, A. C., and Curtius, J.: Observation of new particle formation and measurement of sulfuric acid, ammonia, amines and highly oxidized organic molecules at a rural site in central Germany, Atmos. Chem. Phys., 16, 12793–12813, https://doi.org/10.5194/acp-16-12793-2016, 2016.
Kurten, T., Rissanen, M. P., Mackeprang, K., Thornton, J. A., Hyttinen, N., Jorgensen, S., Ehn, M., and Kjaergaard, H. G.: Computational Study of Hydrogen Shifts and Ring-Opening Mechanisms in alpha-Pinene Ozonolysis Products, J. Phys. Chem. A, 119, 11366–11375, https://doi.org/10.1021/acs.jpca.5b08948, 2015.
Kurtén, T., Tiusanen, K., Roldin, P., Rissanen, M., Luy, J.-N., Boy, M., Ehn, M., and Donahue, N.: α-Pinene Autoxidation Products May Not Have Extremely Low Saturation Vapor Pressures Despite High O : C Ratios, J. Phys. Chem. A, 120, 2569–2582, https://doi.org/10.1021/acs.jpca.6b02196, 2016.
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurtén, T., Worsnop, D. R., and Thornton, J. A.: An iodide-adduct high-resolution time-of-flight chemical-ionization mass spectrometer: Application to atmospheric inorganic and organic compounds, Environ. Sci. Technol., 48, 6309–6317, 2014.
Li, Y., Pöschl, U., and Shiraiwa, M.: Molecular corridors and parameterizations of volatility in the chemical evolution of organic aerosols, Atmos. Chem. Phys., 16, 3327–3344, https://doi.org/10.5194/acp-16-3327-2016, 2016.
Makkonen, R., Asmi, A., Kerminen, V.-M., Boy, M., Arneth, A., Hari, P., and Kulmala, M.: Air pollution control and decreasing new particle formation lead to strong climate warming, Atmos. Chem. Phys., 12, 1515–1524, https://doi.org/10.5194/acp-12-1515-2012, 2012.
McVay, R. C., Zhang, X., Aumont, B., Valorso, R., Camredon, M., La, Y. S., Wennberg, P. O., and Seinfeld, J. H.: SOA formation from the photooxidation of α-pinene: systematic exploration of the simulation of chamber data, Atmos. Chem. Phys., 16, 2785–2802, https://doi.org/10.5194/acp-16-2785-2016, 2016.
Merikanto, J., Spracklen, D. V., Mann, G. W., Pickering, S. J., and Carslaw, K. S.: Impact of nucleation on global CCN, Atmos. Chem. Phys., 9, 8601–8616, https://doi.org/10.5194/acp-9-8601-2009, 2009.
Merikanto, J., Duplissy, J., Määttänen, A., Henschel, H., Donahue, N. M., Brus, D., Schobesberger, S., Kulmala, M., and Vehkamäki, H.: Effect of ions on sulfuric acid-water binary particle formation: 1. Theory for kinetic- and nucleation-type particle formation and atmospheric implications, J. Geophys. Res.-Atmos., 121, 1736–1751, 2016.
Metzger, A., Verheggen, B., Dommen, J., Duplissy, J., Prevot, A. S., Weingartner, E., Riipinen, I., Kulmala, M., Spracklen, D. V., Carslaw, K. S., and Baltensperger, U.: Evidence for the role of organics in aerosol particle formation under atmospheric conditions, P. Natl. Acad. Sci. USA, 107, 6646–6651, https://doi.org/10.1073/pnas.0911330107, 2010.
Molteni, U., Bianchi, F., Klein, F., El Haddad, I., Frege, C., Rossi, M. J., Dommen, J., and Baltensperger, U.: Formation of highly oxygenated organic molecules from aromatic compounds, Atmos. Chem. Phys., 18, 1909–1921, https://doi.org/10.5194/acp-18-1909-2018, 2018.
Napari, I., Noppel, M., Vehkamäki, H., and Kulmala, M.: Parametrization of ternary nucleation rates for vapors, J. Geophys. Res., 107, 4381, https://doi.org/10.1029/2002JD002132, 2002.
O'Dowd, C. D., Geever, M., Hill, M. K., Smith, M. H., and Jennings, S. G.: New particle formation: Nucleation rates and spatial scales in the clean marine coastal environment, Geophys. Res. Lett., 25, 1661–1664, 1998.
Ortega, I. K., Donahue, N. M., Kurtén, T., Kulmala, M., Focsa, C., and Vehkamaki, H.: Can Highly Oxidized Organics Contribute to Atmospheric New Particle Formation?, J. Phys. Chem. A, 120, 1452–1458, https://doi.org/10.1021/acs.jpca.5b07427, 2016.
Pankow, J. F. and Asher, W. E.: SIMPOL.1: a simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds, Atmos. Chem. Phys., 8, 2773–2796, https://doi.org/10.5194/acp-8-2773-2008, 2008.
Rissanen, M. P., Kurtén, T., Sipilä, M., Thornton, J. A., Kausiala, O., Garmash, O., Kjaergaard, H. G., Petäjä, T., Worsnop, D. R., and Ehn, M.: Effects of chemical complexity on the autoxidation mechanisms of endocyclic alkene ozonolysis products: From methylcyclohexenes toward understanding α-pinene, J. Phys. Chem. A, 119, 4633–4650, 2015.
Schobesberger, S., Junninen, H., Bianchi, F., Lonn, G., Ehn, M., Lehtipalo, K., Dommen, J., Ehrhart, S., Ortega, I. K., Franchin, A., Nieminen, T., Riccobono, F., Hutterli, M., Duplissy, J., Almeida, J., Amorim, A., Breitenlechner, M., Downard, A. J., Dunne, E. M., Flagan, R. C., Kajos, M., Keskinen, H., Kirkby, J., Kupc, A., Kürten, A., Kurtén, T., Laaksonen, A., Mathot, S., Onnela, A., Praplan, A. P., Rondo, L., Santos, F. D., Schallhart, S., Schnitzhofer, R., Sipilä, M., Tomé, A., Tsagkogeorgas, G., Vehkamäki, H., Wimmer, D., Baltensperger, U., Carslaw, K. S., Curtius, J., Hansel, A., Petäjä, T., Kulmala, M., Donahue, N. M., and Worsnop, D. R.: Molecular understanding of atmospheric particle formation from sulfuric acid and large oxidized organic molecules, P. Natl. Acad. Sci. USA, 110, 17223–17228, https://doi.org/10.1073/pnas.1306973110, 2013.
Shen, X. J., Sun, J. Y., Zhang, Y. M., Wehner, B., Nowak, A., Tuch, T., Zhang, X. C., Wang, T. T., Zhou, H. G., Zhang, X. L., Dong, F., Birmili, W., and Wiedensohler, A.: First long-term study of particle number size distributions and new particle formation events of regional aerosol in the North China Plain, Atmos. Chem. Phys., 11, 1565–1580, https://doi.org/10.5194/acp-11-1565-2011, 2011.
Sihto, S.-L., Kulmala, M., Kerminen, V.-M., Dal Maso, M., Petäjä, T., Riipinen, I., Korhonen, H., Arnold, F., Janson, R., Boy, M., Laaksonen, A., and Lehtinen, K. E. J.: Atmospheric sulphuric acid and aerosol formation: implications from atmospheric measurements for nucleation and early growth mechanisms, Atmos. Chem. Phys., 6, 4079–4091, https://doi.org/10.5194/acp-6-4079-2006, 2006.
Smith, J. and Jiang, J.: Supporting Data for Relative Humidity Effect on the Formation of Highly Oxidized Molecules and New Particles during Monoterpene Oxidation, UC Irvine Dash, Dataset, https://doi.org/10.7280/D1B674, 2019.
Thomson, B. and Iribarne, J.: Field induced ion evaporation from liquid surfaces at atmospheric pressure, J. Chem. Phys., 71, 4451–4463, 1979.
Tobias, H. J., Docherty, K. S., Beving, D. E., and Ziemann, P. J.: Effect of relative humidity on the chemical composition of secondary organic aerosol formed from reactions of 1-tetradecene and O3, Environ. Sci. Technol., 34, 2116–2125, 2000.
Tobias, H. J. and Ziemann, P. J.: Kinetics of the gas-phase reactions of alcohols, aldehydes, carboxylic acids, and water with the C13 stabilized Criegee intermediate formed from ozonolysis of 1-tetradecene, J. Phys. Chem. A, 105, 6129–6135, 2001.
Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., and Wagner, R.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 533, 527–531, 2016.
Vehkamäki, H. and Riipinen, I.: Thermodynamics and kinetics of atmospheric aerosol particle formation and growth, Chem. Soc. Rev., 41, 5160–5173, 2012.
Weber, R., Marti, J., McMurry, P., Eisele, F., Tanner, D., and Jefferson, A.: Measurements of new particle formation and ultrafine particle growth rates at a clean continental site, J. Geophys. Res., 102, 4375–4385, 1997.
Weber, R., McMurry, P. H., Mauldin, R., Tanner, D., Eisele, F., Clarke, A., and Kapustin, V.: New particle formation in the remote troposphere: A comparison of observations at various sites, Geophys. Res. Lett., 26, 307–310, 1999.
Yu, H., McGraw, R., and Lee, S.-H.: Effects of amines on formation of sub-3 nm particles and their subsequent growth, Geophys. Res. Lett., 39, L02807, https://doi.org/10.1029/2011gl050099, 2012.
Zhang, R., Khalizov, A., Wang, L., Hu, M., and Xu, W.: Nucleation and growth of nanoparticles in the atmosphere, Chem. Rev., 112, 1957–2011, https://doi.org/10.1021/cr2001756, 2012.
Zhang, X., McVay, R. C., Huang, D. D., Dalleska, N. F., Aumont, B., Flagan, R. C., and Seinfeld, J. H.: Formation and evolution of molecular products in α-pinene secondary organic aerosol, P. Natl. Acad. Sci. USA, 112, 14168–14173, 2015.
Zhao, J., Khalizov, A., Zhang, R., and McGraw, R.: Hydrogen-bonding interaction in molecular complexes and clusters of aerosol nucleation precursors, J. Phys. Chem. A, 113, 680–689, 2009.
Zhao, J., Eisele, F. L., Titcombe, M., Kuang, C., and McMurry, P. H.: Chemical ionization mass spectrometric measurements of atmospheric neutral clusters using the cluster-CIMS, J. Geophys. Res., 115, D08205, https://doi.org/10.1029/2009JD012606, 2010.
Zhao, J., Ortega, J., Chen, M., McMurry, P. H., and Smith, J. N.: Dependence of particle nucleation and growth on high-molecular-weight gas-phase products during ozonolysis of α-pinene, Atmos. Chem. Phys., 13, 7631–7644, https://doi.org/10.5194/acp-13-7631-2013, 2013.