the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 27 Nov 2019
| 27 Nov 2019
Large-scale particulate air pollution and chemical fingerprint of volcanic sulfate aerosols from the 2014–2015 Holuhraun flood lava eruption of Bárðarbunga volcano (Iceland)
Olivier Favez
Véronique Riffault
Jean-Eudes Petit
Yunjiang Zhang
Colette Brogniez
Jean Sciare
Isabelle Chiapello
Lieven Clarisse
Shouwen Zhang
Nathalie Pujol-Söhne
Emmanuel Tison
Hervé Delbarre
Philippe Goloub
Volcanic sulfate aerosols play a key role in air quality and climate. However, the rate of oxidation of sulfur dioxide (SO2) precursor gas to sulfate aerosols () in volcanic plumes is poorly known, especially in the troposphere. Here we determine the chemical speciation as well as the intensity and temporal persistence of the impact on air quality of sulfate aerosols from the 2014–2015 Holuhraun flood lava eruption of Icelandic volcano Bárðarbunga. To do so, we jointly analyse a set of SO2 observations from satellite (OMPS and IASI) and ground-level measurements from air quality monitoring stations together with high temporal resolution mass spectrometry measurements of an Aerosol Chemical Speciation Monitor (ACSM) performed far from the volcanic source. We explore month/year long ACSM data in France from stations in contrasting environments, close and far from industrial sulfur-rich activities. We demonstrate that volcanic sulfate aerosols exhibit a distinct chemical signature in urban/rural conditions, with NO3:SO4 mass concentration ratios lower than for non-volcanic background aerosols. These results are supported by thermodynamic simulations of aerosol composition, using the ISORROPIA II model, which show that ammonium sulfate aerosols are preferentially formed at a high concentration of sulfate, leading to a decrease in the production of particulate ammonium nitrate. Such a chemical signature is however more difficult to identify at heavily polluted industrial sites due to a high level of background noise in sulfur. Nevertheless, aged volcanic sulfates can be distinguished from freshly emitted industrial sulfates according to their contrasting degree of anion neutralization. Combining AERONET (AErosol RObotic NETwork) sunphotometric data with ACSM observations, we also show a long persistence over weeks of pollution in volcanic sulfate aerosols, while SO2 pollution disappears in a few days at most. Finally, gathering 6-month long datasets from 27 sulfur monitoring stations of the EMEP (European Monitoring and Evaluation Programme) network allows us to demonstrate a much broader large-scale European pollution, in both SO2 and SO4, associated with the Holuhraun eruption, from Scandinavia to France. While widespread SO2 anomalies, with ground-level mass concentrations far exceeding background values, almost entirely result from the volcanic source, the origin of sulfate aerosols is more complex. Using a multi-site concentration-weighted trajectory analysis, emissions from the Holuhraun eruption are shown to be one of the main sources of SO4 at all EMEP sites across Europe and can be distinguished from anthropogenic emissions from eastern Europe but also from Great Britain. A wide variability in SO2:SO4 mass concentration ratios, ranging from 0.8 to 8.0, is shown at several stations geographically dispersed at thousands of kilometres from the eruption site. Despite this apparent spatial complexity, we demonstrate that these mass oxidation ratios can be explained by a simple linear dependency on the age of the plume, with a SO2-to-SO4 oxidation rate of 0.23 h−1. Most current studies generally focus on SO2, an unambiguous and more readily measured marker of the volcanic plume. However, the long persistence of the chemical fingerprint of volcanic sulfate aerosols at continental scale, as shown for the Holuhraun eruption here, casts light on the impact of tropospheric eruptions and passive degassing activities on air quality, health, atmospheric chemistry and climate.
- Article
(26268 KB) - Full-text XML
-
Supplement
(8387 KB) - BibTeX
- EndNote





Volcanic sulfate aerosols play a key role in climate. While the direct radiative forcing caused by scattering of incoming solar radiation by stratospheric sulfate aerosols from major eruptions is well known (Robock, 2000), the climate effect of sulfate aerosols from smaller eruptions, reaching the lower stratosphere or restricted to the troposphere, has been overlooked and underestimated. Indeed, moderate eruptions, which have a much greater frequency, may be capable of sporadically feeding the stratospheric aerosol load (Vernier et al., 2011; Neely et al., 2013; Ridley et al., 2014). The identification by CMIP5 (Coupled Model Intercomparison Project) of a systematic bias toward underestimation of the cooling of the lower stratosphere and overestimation of the troposphere warming (also called “warming hiatus”) over 1998–2012 in current global circulation models might be partly due to an inappropriate account of these smaller volcanic eruptions (Solomon et al., 2011; Santer et al., 2014; Schmidt et al., 2018). Hence, the impact of tropospheric eruptions on radiative forcing, generally neglected, deserves greater attention. Sulfate aerosols can be rapidly washed out by precipitation in the troposphere, which results in a shorter lifetime relative to stratospheric aerosols. However, sulfate aerosols reduce the nucleation rate of ice crystals, affecting the properties of the ubiquitous upper troposphere cirrus clouds that play a critical role in climate (Kuebbeler et al., 2012). The properties of low-altitude meteorological clouds, their formation, lifetime and precipitation can be also substantially affected by the presence of volcanic sulfate aerosols in the lower troposphere, that are issued from persistent passive degassing activity (Gassó, 2008; Schmidt et al., 2012; Ebmeier et al., 2014) or from effusive eruptions (Yuan et al., 2011; McCoy and Hartmann, 2015; Malavelle et al., 2017).
Volcanic sulfate aerosols in the troposphere, the topic of this paper, also have a detrimental impact on air quality and human health, as they represent a dominant component of fine particulate matter characterized by a diameter less than 2.5 µm. Owing to their small size, these aerosols have slow settling velocities and thus can accumulate in the boundary layer and penetrate deeply into the lung, exacerbating symptoms of asthma and cardiorespiratory diseases (Delmelle, 2003; Thordarson and Self, 2003; Longo et al., 2008; van Manen, 2014). They also adversely affect the environment, with deleterious effects on vegetation, agriculture, soils and groundwater (Delmelle, 2003; van Manen, 2014; Thordarson and Self, 2003; Oppenheimer et al., 2011). Last but not least, sulfate aerosols can damage aircraft engines (Carn et al., 2009), a poorly known impact especially under repeated aircraft encounters with diluted volcanic clouds as recently tolerated by legislation (ICAO, 2016).
Volcanic sulfate aerosols can be divided in two categories, either of primary or secondary nature. Primary sulfate aerosols are directly emitted at the vent, as observed at a few volcanoes worldwide (e.g. Allen et al., 2002; Mather et al., 2003b, 2004; Zelenski et al., 2015). On the other hand, secondary sulfate aerosols result from in-plume oxidation of sulfur dioxide (SO2), one of the most abundant gas species emitted by volcanoes, during transport downwind (Oppenheimer et al., 2011; Pattantyus et al., 2018). Dominant pathways have been identified for this SO2-to-sulfate conversion in the troposphere via both gas- and aqueous-phase processes. In the gas phase, SO2 oxidation predominantly occurs by reaction with hydroxyl radicals (OH) to form sulfuric acid (H2SO4) according to the reactions (Seinfeld and Pandis, 2012)
where M is another molecule (usually N2) that is required to absorb excess kinetic energy from the reactants. In presence of water vapour, SO3 is then rapidly converted to H2SO4(g):
Due to its highly hygroscopic nature, H2SO4(g) is efficiently taken up to the aqueous phase to form sulfate aerosols (Seinfeld and Pandis, 2012) following the reactions
As shown in volcanic clouds, H2SO4(g) can also nucleate to form new particles (Boulon et al., 2011).
Gas-phase SO2 oxidation takes place on a timescale of weeks in the troposphere. Much faster oxidation occurs, over hours or days, through heterogeneous reactions in the aqueous phase if SO2 is taken up to particles, either aerosols or cloud droplets. SO2 easily dissolves in water and can form three different chemical species depending on pH values: (1) bisulfite ion (), the preferential sulfur species for pH values in [2–7]; (2) hydrated SO2 (SO2•H2O), for low pH values (pH < 2); and (3) sulfite ion () for basic pH values (pH > 7), according to equilibrium reactions (Seinfeld and Pandis, 2012)
These three species have a sulfur oxidation state equal to 4, referred to as S(IV). Oxidation of these S(IV) species to sulfate aerosols (), whose sulfur oxidation state is equal to 6 (S(VI)), is mainly known to occur by reaction with dissolved ozone (O3) for pH > 5.5 and with hydrogen peroxide (H2O2) as follows (Seinfeld and Pandis, 2012; Stevenson et al., 2003):
In volcanic plumes as in other environments, S(IV) can also be oxidized in the aqueous phase by dissolved oxygen (O2) catalyzed by iron and manganese (Seinfeld and Pandis, 2012) and halogen-rich species (HOBr or HOCl) as shown by von Glasow and Crutzen (2003). More recently, the importance, if not dominance, of O2-catalyzed oxidation in volcanic environments has been highlighted (Galeazzo et al., 2018).
Therefore, SO2 oxidation to sulfate within volcanic clouds involves complex processes in the gas and aqueous phases, depending on many variables, including solar insolation, relative humidity, temperature, pH of aerosol/cloud droplets and concentrations of the co-existent ash particles and various gas species. As such, the rate of production of volcanic sulfate aerosols is still poorly known, with a large range of rates observed near-source in different volcanic environments in the world, as summarized in Pattantyus et al. (2018).
The chemical speciation of volcanic sulfate aerosols has been poorly studied until now and is also barely known. Some observations have been occasionally collected, using filter packs or cascade impactors, near the vent of a few volcanoes worldwide (e.g. Mather et al., 2003a; Martin et al., 2011; Ilyinskaya et al., 2017). However, such methods only provide an average representation of the chemical composition of aerosols over the duration of instrument exposure to volcanic emissions, which is usually limited to short campaigns. In addition to the low temporal resolution of these sparse and limited-time observations, a tedious and careful post-collection laboratory analysis is required to avoid biases. To our knowledge, one single study of Kroll et al. (2015) explored through near real-time quasi-continuous measurements the partitioning between SO2 and sulfate aerosols taking place near-source at the strongly degassing Kilauea volcano in 2013, showing the wide variability of sulfur partitioning linked to the complex atmospheric dynamics of the plume.
Volcanic aerosols may also affect the troposphere at a long distance. Various volcanic eruptions or persistent passive degassing activities (e.g. Laki/Iceland in 1783–1784, Miyake-jima/Japan in 2001, Erebus/Antarctica, Holuhraun eruption of Bárðarbunga volcano/Iceland in 2014–2015) have been shown to trigger, at a large scale, modifications of the atmospheric chemistry and air pollution episodes in SO2 (Tu et al., 2004; Schmidt et al., 2015; Ialongo et al., 2015; Steensen et al., 2016; Boichu et al., 2016) and sulfate aerosols (Radke, 1982; Thordarson and Self, 2003; Aas et al., 2015, 2016; Twigg et al., 2016). These studies demonstrate that volcanic SO2 and SO4 coexist in the troposphere at long distances indicating that the oxidation of SO2 to secondary sulfates operates on long timescales of several days. However, the kinetics of SO2-to-SO4 oxidation remains poorly constrained, especially within volcanic plumes transported over large distances in contrasting environments. Understanding the life cycle of sulfur in volcanic plumes is fundamental to better (1) understanding the rate of SO2 depletion (review in Pattantyus et al., 2018) and robustly describing it in volcanic plume dispersal models in order to rigorously evaluate volcanic SO2 emissions from satellite observations (e.g. Theys et al., 2013; Boichu et al., 2013; Flemming and Inness, 2013; Moxnes et al., 2014), and (2) assessing the rate of production of sulfate to rigorously estimate the intensity, geographical influence and temporal persistence of long-range volcanogenic particulate pollution and the impact of tropospheric eruptions on climate.
Understanding the factors controlling the oxidation of SO2 within volcanic plumes requires sampling of the composition of volcanic gases and particles over a broad range of plume residence time, which is only accessible by collecting observations over a vast spatial region. Furthermore, as chemical interactions of sulfate with co-existent aerosols of different type also affect the speciation and chemical partitioning of sulfur, these observations should include monitoring of inorganic and organic aerosol concentrations. A multi-parameter chemical analysis is also indispensable for distinguishing a specific signature of volcanogenic pollution, in particular in contexts where anthropogenic pollution may interfere.
In this paper, we propose to fill this gap by exploring the chemical signature of volcanic sulfate aerosols after long-range transport and by assessing the intensity of air pollution that these particles may generate at a large scale. We benefit here from a recently developed technology based on near real-time mass spectrometry, named Aerosol Chemical Speciation Monitor (ACSM), which provides mass and chemical composition of the non-refractory fraction of submicron particles at high temporal resolution.
By gathering a large set of ground-level in situ gas and aerosol data jointly analysed with satellite remote sensing observations from OMPS/Suomi NPP and IASI/MetOp-A sensors, this study aims first to quantify the intensity of air pollution in sulfur-rich particles caused by the Holuhraun eruption of the Bárðarbunga volcano (Iceland) in France (Sect. 4.1 and 4.2).
Secondly, we propose to explore whether the chemical signature of volcanic sulfate aerosols is distinct from those of background aerosols in industrial or urban environments, comparing observed patterns with ISORROPIA II thermodynamic model simulations of aerosol composition (Sect. 4.3). To achieve these goals, along with satellite SO2 observations, we exploit ground-level in situ observations of SO2 from regional air quality monitoring stations and ACSM measurements performed at two French research sites in contrasting environments, near to or far from industrial sulfur-rich emitting activities. Both sites were indeed impacted by sulfur dioxide and sulfate aerosols in relation with the Holuhraun eruption of Bárðarbunga volcano (Iceland) on repeated occasions in September 2014.
In a third stage, the joint analysis of in situ ACSM measurements with sunphotometry column-integrated observations from co-located stations of the AERONET AErosol RObotic NETwork allows us to evaluate the temporal persistence of particulate pollution in sulfur (Sect. 4.4).
Fourthly, to provide a broader picture, we explore 6-month long sulfur monitoring datasets (September 2014–February 2015) from 27 stations of the EMEP (European Monitoring and Evaluation Programme) network. Using a multi-site concentration-weighted trajectory analysis for selected EMEP stations, we evaluate the intensity of the large-scale chemical fingerprint of the Holuhraun eruption on gaseous SO2 and particulate sulfate in Europe, compared to other anthropogenic industrial sources (Sect. 4.5).
Finally, we assess the range of variability of SO2-to-SO4 mass concentration ratios according to the volcanic cloud history and derive for the first time an estimation of the oxidation rate from the eruption site to stations located a few thousands of kilometres away (Sect. 4.6).
2.1 Ground-level in situ observations
2.1.1 Aerosol chemical speciation monitor
The chemical composition of non-refractory submicron aerosols (NR-PM1), including sulfate (), nitrate (), ammonium () and organic (Org) species, are monitored with a time resolution of about 30 min and detection limits of 0.2 µg m−3, using quadrupole Aerosol Chemical Monitors (ACSM) at two French sites with contrasting background conditions (Dunkirk and SIRTA). Note that charges of inorganic species, determined as ions by ACSM, are not systematically indicated in text and figures hereafter, to ease readability.
For a detailed description of the ACSM, developed by Aerodyne Research Inc., the reader is referred to Ng et al. (2011). Briefly, aerosols are sampled into the instrument through a critical orifice mounted at the inlet of a PM1 aerodynamic lens and focused under vacuum to an oven at the temperature of 600 ∘C. Flash vaporized molecules are then ionized at 70 eV electron impact before being detected and quantified by the mass spectrometer. Raw data are corrected for aerosol collection efficiency following the protocol defined by Middlebrook et al. (2012). A specific ionization efficiency (relative to nitrate, RIE) should also be defined for each species. For the Dunkirk ACSM, a constant 0.55 SO4 RIE has been used, based on results obtained from calibrations performed regularly (typically, every 2 months) during the campaign. By the time of the measurement, a default SO4 RIE value was preferably taken as equal to 1.20 for the SIRTA ACSM (Ng et al., 2011; Crenn et al., 2015). Therefore, figures hereafter display ACSM data processed using these values of 0.55 and 1.20 for the Dunkirk and SIRTA datasets, respectively. However, it may be noted that recent optimizations of the ACSM calibration procedure currently allow us to reassess SO4 RIE values (Xu et al., 2018; Freney et al., 2019). In particular, a value of 0.86 was obtained in spring 2016 when applying the new calibration procedure for the first time to the ACSM at SIRTA (Freney et al., 2019). Note that the more recent calibrated RIE value (0.86) may not be relevant to correct older measurements, and standard practice is to keep the original value (1.2) for older measurements, which include 2014 (our period of study). For the sake of completeness, impacts of the choice of the RIE value on SO4 mass concentrations used in the present study are evaluated in Sect. 4.3.2 and 4.3.3. Such differences are still in the range of uncertainties (15 %–36 %) estimated for the measurements of major submicron chemical species using ACSM (Budisulistiorini et al., 2014; Crenn et al., 2015).
Standard diagnostics were used to clean up the ACSM data, such as spikes in the air beam and/or water signals, drop of inlet pressures indicative of clogging. No averaging was needed to compare the species obtained with the same instrument and therefore the original time resolution was kept.
Dunkirk located in northern France (latitude 51.041∘ N, longitude 2.312∘ E, map in the inset of Fig. 1c) hosts a large harbour, ranking seventh in Europe, with a developed manufacturing industry (map in Fig. A1) accounting for more than 80 % of total particulate matter (PM) emitted locally over 2009–2011 (Clerc et al., 2012). About 97 % of primary PM1 is emitted by metallurgy, steel and smelter activities (Figs. 1–7 of Zhang, 2016). A remarkable 14-month long 30 min resolved ACSM dataset was collected at the Port-East site (map in Fig. A1), with collocated ground-level SO2 measurements, from 15 July 2013 to 11 September 2014 (Zhang, 2016), allowing us to compare the chemical signature of industrial and volcanic sulfate aerosols.
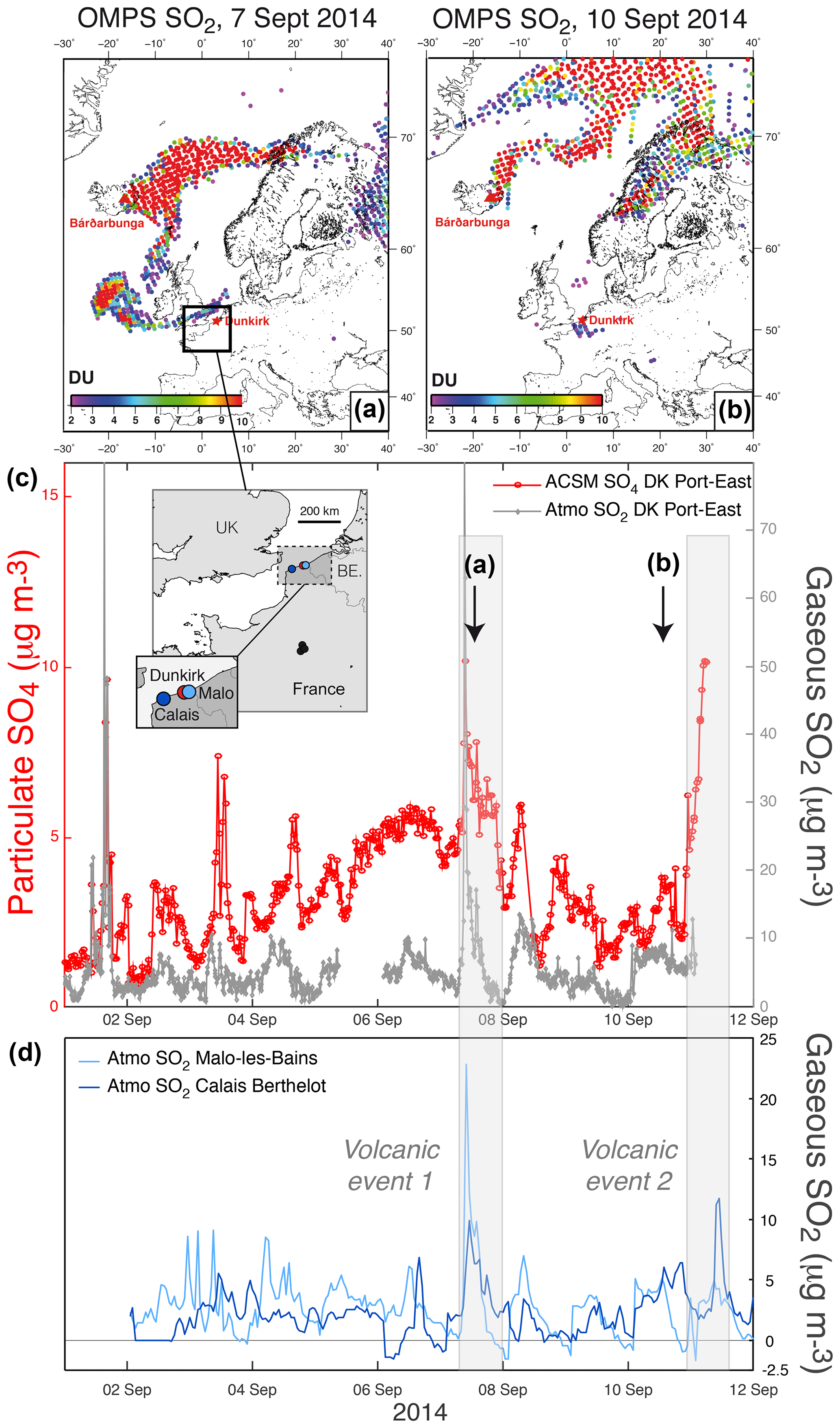
Figure 1(a, b) OMPS L2 PBL observations (13:30 local time at the Equator) showing volcanic SO2 from the Holuhraun eruption transported over northern France in early September 2014. (c) Time series of ground-level mass concentrations of (red) particulate SO4 from 30 min resolved ACSM and (grey) gaseous SO2 from 15 min resolved air quality measurements at Dunkirk (Port-East). Map of all stations in inset. (d) Hourly time series of SO2 mass concentration from the regional neighbouring stations of Malo-les-Bains (light blue) and Calais-Berthelot (dark blue) belonging to the Atmo Hauts-de-France air quality network. Note the end of ACSM SO4 data collection on 11 September 2014 at 05:50 UTC and the absence of valid SO2 data after 02:00 on the same day.
The SIRTA facility (Site Instrumental de Recherche par Télédétection Atmosphérique, http://sirta.ipsl.fr (last access: 14 November 2019), Haeffelin et al., 2005, latitude 48.713∘ N and longitude 2.214∘ E) is located about 20 km south-west of the Paris city centre (map in the inset of Fig. 2d). This atmospheric observatory is notably part of the European Aerosol, Clouds and Traces gases Research InfraStructure (ACTRIS, https://www.actris.eu, last access: 14 November 2019) as a peri-urban station for remote sensing and in situ measurements representative of background particulate matter (PM) levels of the Paris region. ACSM data have been routinely collected there since the end of 2011 (Petit et al., 2015). A 2-month hourly resolved dataset (September–October 2014) has been used for the purpose of the present study to investigate the speciation of volcanic sulfate aerosols, especially during the largest event of volcanogenic air pollution affecting France in late September 2014 (Boichu et al., 2016).
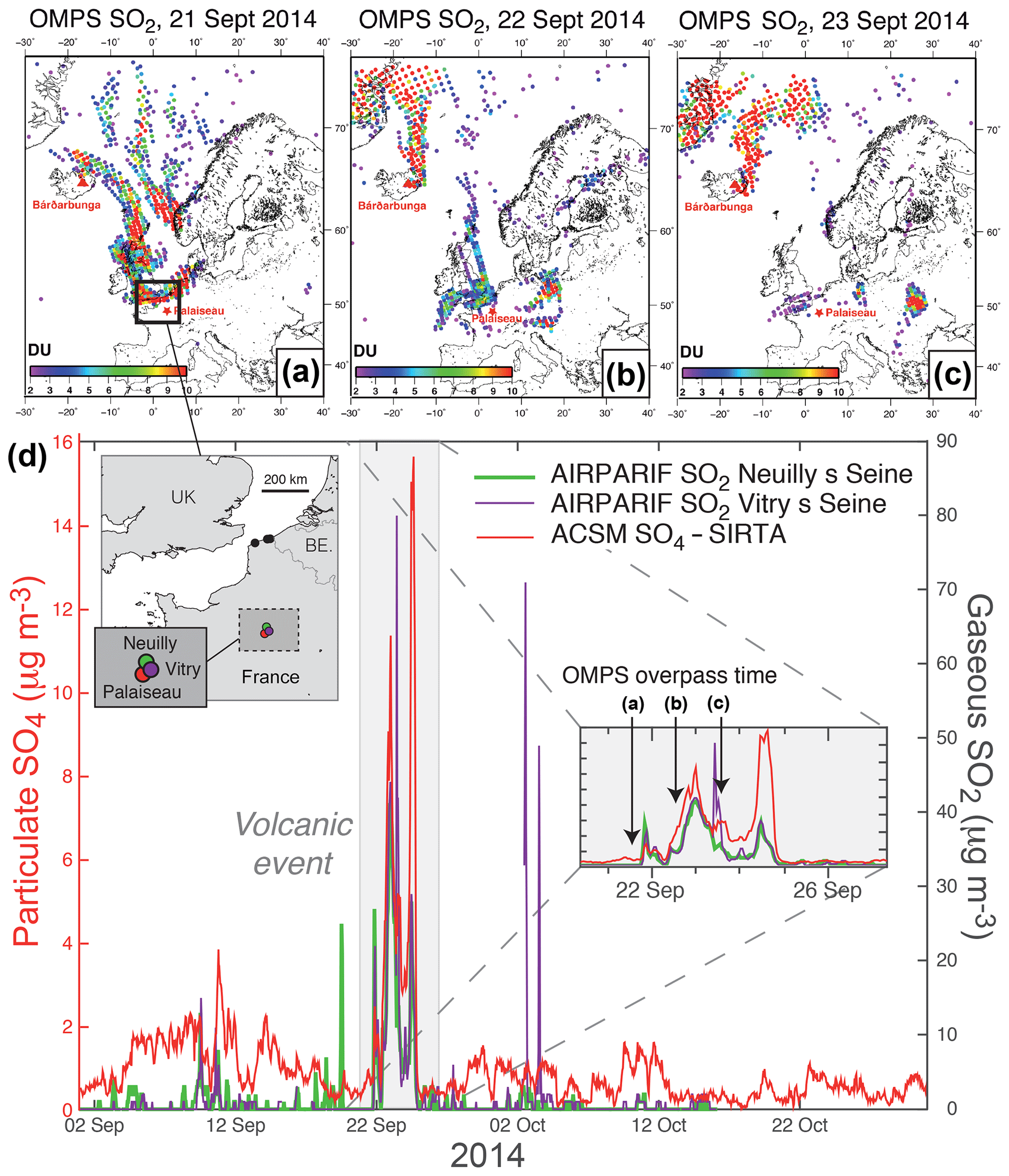
Figure 2(a, b, c) OMPS L2 PBL observations (13:30 local time at the Equator) showing volcanic SO2 from the Holuhraun eruption transported over northern France in late September 2014. (d) Hourly time series covering September–October 2014 of ground-level mass concentrations of (red) particulate SO4 from ACSM at SIRTA and (green and purple) gaseous SO2 from the regional neighbouring stations of Vitry-sur-Seine and Neuilly-sur-Seine belonging to the Airparif air quality monitoring network (station location indicated on the map). In the inset is a zoom on the period 19–26 September 2014, when the largest episode of volcanogenic air pollution in France took place.
2.1.2 SO2 mass concentrations from French air quality monitoring network
Ground-level mass concentrations of SO2 are routinely monitored using ultraviolet fluorescence analysers by regional air quality monitoring networks, with a detection limit of 5.3 µg m−3 and an uncertainty never exceeding 15 %. For the present study, data from Atmo Hauts-de-France and Airparif could be explored, corresponding to the following stations: Dunkirk Port-East site (latitude 51.041∘ N, longitude 2.312∘ E), Calais-Berthelot (latitude 50.947∘ N, longitude 1.843∘ E) and Malo-les-Bains (latitude 51.049∘ N, longitude 2.420∘ E) on the one hand, and Neuilly-sur-Seine (latitude 48.881∘ N, longitude 2.278∘ E) and Vitry-sur-Seine (latitude 48.775∘ N, longitude 2.376∘ E) on the other hand (maps in the inset of Figs. 1c and 2d). Hourly mean data have been used here for all stations but the Port-East one in Dunkirk with 15 min time resolution.
2.1.3 Filter pack and online ion chromatography measurements from the EMEP network
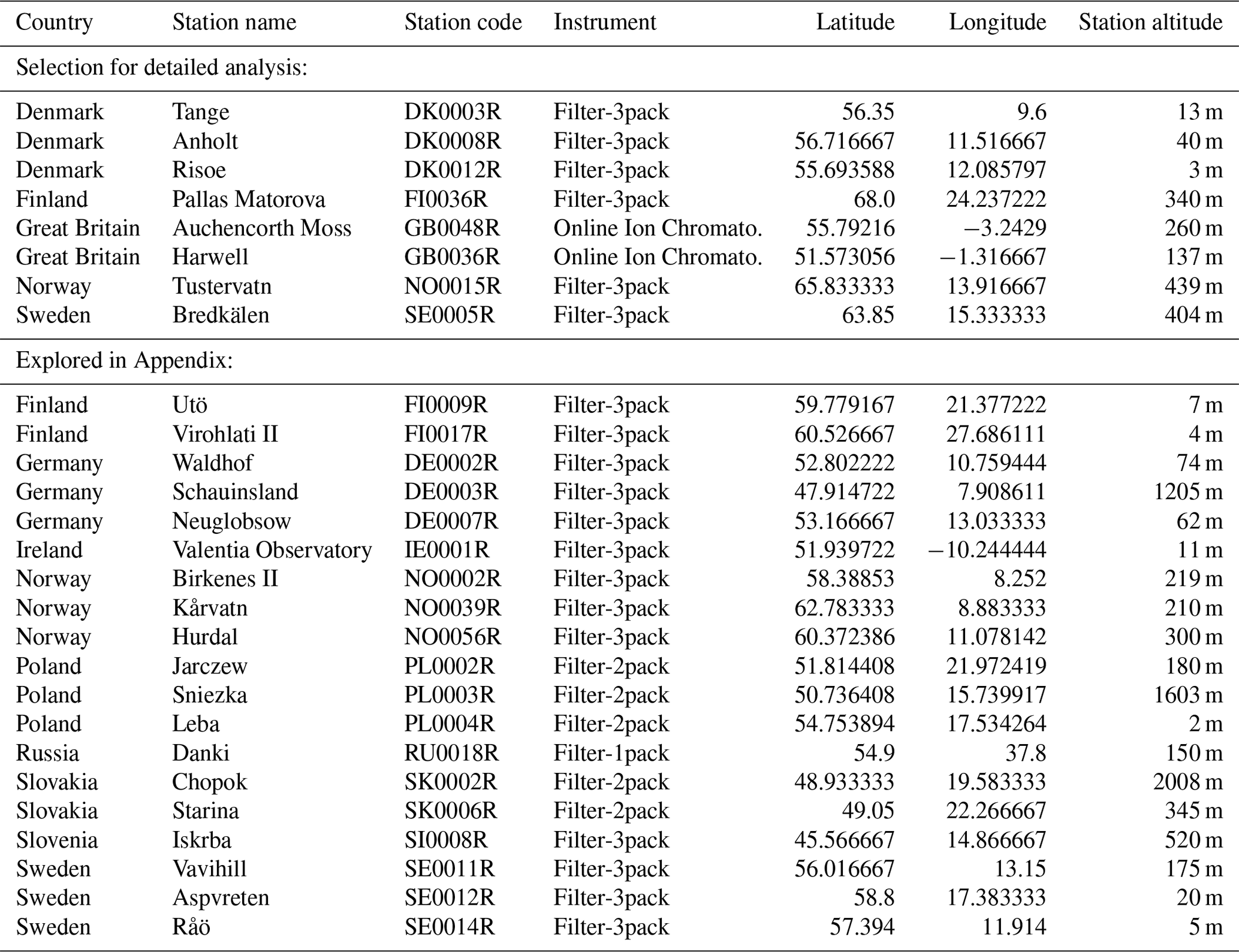
The EMEP (European Monitoring and Evaluation Programme, http://ebas.nilu.no, last access: 14 November 2019) network, in charge of monitoring air pollution and surface deposition with harmonized measurements across Europe, gathers ground stations that are weakly affected by local sources of pollution (Tørseth et al., 2012). We focus here on stations where measurements provide at the same temporal resolution ground-level mass concentrations of both gaseous SO2 and particulate SO4. More precisely, we exploit here data of the corrected sulfate mass concentration, i.e. the total sulfate minus sulfate originating from sea-salt particles, of the PM10 fraction of samples. Such observations are collected on a daily or hourly basis, using, respectively, either filter-pack measurements, the most common method, or online ion chromatography with a MARGA instrument. These latter observations, presenting the best time resolution, are only available at two stations in Great Britain at the time of the Bárðarbunga Holuhraun eruption in 2014–2015 (Twigg et al., 2016). Unfortunately, measurements providing mass concentrations of both SO2 and SO4 species simultaneously were not performed anymore at that time in many north-western European countries, including France, Belgium, and the Netherlands. The network still adequately covers Scandinavia (Finland, Sweden, Norway and Denmark) and only a few stations are left in Germany, Ireland, Poland, Slovakia and Slovenia. We consequently explore in this study data from 27 stations based in 11 countries (Great Britain, Finland, Norway, Sweden, Denmark, Germany, Ireland, Poland, Slovakia, Slovenia and Russia) as listed in Table 1. Details of sampling and chemical analyses are provided within the EMEP Standard Operating Protocol (NILU, 2014).
Table 1Details of the 27 EMEP stations explored in this study and shown on the map of Fig. 13.
2.2 Satellite observations of the volcanic SO2 cloud
Ultraviolet (UV) observations from the OMPS (Ozone Mapping and Profiler Suite)/SNPP (Suomi National Polar-orbiting Partnership) sensor, with pixel size at nadir of 50 km × 50 km and an Equator-crossing time of 13:30 local time (Carn et al., 2015), allow tracking of the large-scale dispersal of the Holuhraun SO2-rich cloud and identification of the dates it is transported over specific ground stations. According to the IASI (Infrared Atmospheric Sounding Interferometer) satellite observations described below (and also shown by Schmidt et al., 2015; Boichu et al., 2016; Carboni et al., 2019), the altitude of Holuhraun SO2 is most often lower than 6 km over France (see the animation in the Supplement). Consequently, the Level-2 planetary boundary layer (PBL) products for the SO2 total column are chosen to study the dispersal of the Holuhraun cloud over France.
IASI observations from polar-orbiting MetOp-A satellite, with a pixel footprint at nadir of 12 km diameter, full swath width of 2200 km and Equator crossing time at 09:30 and 21:30 local time are also presented. IASI observations are generally less sensitive than OMPS to SO2 below 5 km of altitude as shown in the study of the Holuhraun cloud dispersal (Boichu et al., 2016). However, IASI benefits from a shorter revisit interval (i.e. 12 h) and provides both column amount and altitude of SO2. After the retrieval of the SO2 altitude using the algorithm described in detail in Clarisse et al. (2014), an optimal estimation scheme with generalized noise covariance is used for SO2 column retrieval (Bauduin et al., 2014).
2.3 Column-integrated aerosol properties from the AERONET ground-based remote sensing network
Time series of daily averaged aerosol optical depth (AOD) at 500 nm, derived from Direct Sun photometer measurements (Version 3, Level 2.0, in cloud-free conditions) from the AErosol RObotic NETwork (AERONET) (Holben et al., 2001), are exploited at the two French sites of Dunkirk (map in Fig. A1 of the precise location of the station on Dunkirk Port) and SIRTA.
3.1 Thermodynamic modelling of aerosol composition and pH
Simulations with the ISORROPIA II thermodynamic model (Fountoukis and Nenes, 2007) are performed to evaluate inorganic aerosol composition and pH under our study conditions at SIRTA. The model is run in forward mode (Fountoukis and Nenes, 2007) along with an aerosol system of –––H2O and corresponding gas-phase species, including ammonia (NH3) and nitric acid (HNO3). The total concentrations of those inorganic species (i.e. , , and ) are set up as the model inputs for the calculation of gas-particle equilibrium concentrations. The particle , , and mass concentrations were measured by the PM1 ACSM in 2014; however, gaseous NH3 and HNO3 were not collected during the same period of time. To evaluate possible mass concentration range of NH3 and HNO3, we use the data observed in 2010 autumn in Paris using, respectively, an AiRRmonia monitor and a wet annular denuder coupled with ion chromatography (Petetin et al., 2016). The 10th and 90th percentiles of measured NH3 mass concentrations (0.74 and 7.40 µg m−3) were assumed as the comparable high and low concentration levels for the present study. Hence, we design two different model runs corresponding to poor or rich NH3 scenarios, with NH3 mass concentration held constant and equal to 0.74 or 7.40 µg m−3, respectively. The average HNO3 mass concentration (0.15 µg m−3) is used in both model runs. Ambient air relative humidity (RH) and temperature (T), also model input variables, were collected at the SIRTA ground-based meteorological platform.
To address the response of changes in sulfate concentration to particulate nitrate production under our study conditions in 2014, we perform a sensitivity test using again the ISORROPIA II model. The average values of T (15.8 ∘C), RH (79.3 %), (2.00 µg m−3) and (1.23 µg m−3) mass concentrations, measured over September–October 2014, as well as average NH3 (3.09 µg m−3) and HNO3 (0.15 µg m−3) mass concentrations taken from the 2010 autumn observations, are used as model inputs, while the mass concentration of is left as a free variable ranging from 0.5 to 30.0 µg m−3. This range encompasses the observed mass concentrations at SIRTA during the entire study period.
3.2 Multi-site concentration-weighted trajectory analysis
In order to evaluate the influence of the Holuhraun eruption on the ground-level concentrations of SO2 and over northern Europe, a trajectory analysis work has been undertaken for a selection of EMEP stations, whose coordinates are detailed in Table 1. First, a concentration-weighted trajectory analysis (CWT, Cheng et al., 2013) has been applied separately at each site for both pollutants, as follows:
where nij is the residence time of back-trajectories in the (i,j) cell and mij is the sum of concentrations going through each trajectory. Five-day back-trajectories, starting at an altitude of 500 m above ground level, were calculated every 3 h for each site using the Hybrid Single Particle Lagrangian Integrated Trajectory model (HYSPLIT, Stein et al., 2015), with the 1∘ × 1∘ Global Data Assimilation System (GDAS). Because of the statistically low representativeness of one back-trajectory to a daily concentration value, the data coverage has been increased by taking more back-trajectories into account for a particular day (Waked et al., 2014). Wet deposition has been estimated by cutting the trajectory where significant precipitation (≥1 mm h−1) occurred. For graphical purpose, a Gaussian smoothing has been applied.
Secondly, a multi-site (MS) approach was applied in order to take the spatial and temporal variabilities of all sites at once, which has been proven to take spatio-temporal variabilities of all sites into account (Biegalski and Hopke, 2004):
where ml and nl are the m and n matrices of site l. In order to retrieve quantitative information from the multi-site analysis, an edge-detection algorithm allows us to integrate CWT values over a particular hotspot. Compared to the total integration, this provides an estimation of the contribution of the selected zone for particulate SO4 and gaseous SO2.
This whole work has been performed with ZeFir (Petit et al., 2017), a user-friendly tool for wind and trajectory analysis.
First, we evaluate the intensity of air pollution in sulfur-rich particles induced by the Holuhraun eruption in France. We also propose to explore whether the chemical signature of sulfate aerosols is specific or not within volcanic plumes, by comparison with sulfate aerosols of industrial origin. We then define a methodology to discriminate volcanic vs. local industrial sulfur-rich compounds. To do so, we study several events of air pollution observed in France in September 2014 at two locations nearby (Dunkirk) and distant (SIRTA) from industrial activities. We show the volcanogenic origin of these episodes of atmospheric pollution that are characterized by elevated ground-level mass concentrations of both SO2 and SO4. Then, we investigate whether similar events of air pollution are also detected more broadly, at the European scale, by exploiting in situ data from the EMEP ground network. Finally, we identify, using a multi-site concentration-weighted trajectory analysis, the sources of gas and particulate pollution in sulfur and examine whether the sulfur partitioning in volcanic samples collected in France is similar at various other EMEP stations in Europe.
4.1 Volcanogenic short-term events of air pollution
SO2 is commonly used as a marker of volcanic plumes. Hence, OMPS satellite SO2 observations allow us to detect when the volcanic cloud passes over the two French sites equipped with ACSM (i.e. Dunkirk and SIRTA), bearing in mind that satellite ultraviolet observations of SO2, aside from their detection limit, have a lower sensitivity especially in the lower troposphere and the planetary boundary layer (Krotkov et al., 2008). Figure 1a and b indicate that a branch of the Holuhraun SO2 cloud passes close to Dunkirk in northern France on 7 September 2014 and air masses containing volcanic SO2 are still detected over Dunkirk on 10 September 2014. Concomitantly, elevated values in ground-level SO4 mass concentration up to ≈10 µg m−3 (Fig. 1c) are recorded by 30 min resolved ACSM measurements at Dunkirk, and large anomalies in SO2 mass concentration (up to 70 µg m−3) are regionally measured by various air quality stations of Nord-Pas de Calais (now Hauts-de-France), as exemplified here at Dunkirk Port-East with 15 min resolved measurements (Fig. 1c) and hourly observations at Malo-les-Bains and Calais Berthelot (Fig. 1d).
It should be pointed out that high peaks in both ground-level SO2 (up to ≈80 µg m−3) and SO4 (up to ≈9 µg m−3) mass concentrations, are also recorded at Dunkirk Port-East on 1 September 2014 before the arrival of the Holuhraun cloud over France. In contrast to other days in early September 2014 of intense air pollution in SO2, the meteorological station at Port-East also indicates that on 1 September 2014 local winds originate from the nearby industrial site before passing over Port-East station with a wind direction of about 270∘ (Fig. A2a). Hence, the ground-level concentration in volcanic sulfate aerosols on 7 September 2014, despite a transport and dispersion of emissions over a few thousands of kilometres from Iceland to France, is of a similar magnitude to the concentration in sulfate aerosols emitted on 1 September by a nearby industrial site hosting metallurgy activities.
To conclude, this joint analysis of complementary observations, from space and from the ground at a regional scale, allows us to demonstrate the volcanogenic origin of the two events of air pollution associated with elevated ground-level mass concentration in both SO2 and SO4, recorded in Dunkirk on 7 September between 07:36 and 23:19 UTC (hereafter named “DK volcanic event 1”) and the second between 10 September 20:00 and 11 September 2014 05:50 UTC (hereafter named “DK volcanic event 2”) (grey shaded areas in Fig. 1).
Similarly, exploiting OMPS satellite maps (Fig. 2a, b, c) and Airparif SO2 measurements (Fig. 2d) at various air quality monitoring stations of the Paris region (only Vitry-sur-Seine and Neuilly-sur-Seine are shown here) demonstrates the volcanic origin of the largest event of air pollution in sulfate aerosols (with a ground-level mass concentration up to 16 µg m−3), in terms of magnitude and duration, which is recorded with ACSM at SIRTA between 21 and 25 September 2014 (hereafter named “SI volcanic event”, grey shaded area in Fig. 2d). This particulate pollution is concomitant with a pronounced air pollution in SO2, with a ground-level mass concentration up to 80 µg m−3 in the Paris region (Fig. 2d) but also more broadly at various places in northern France as observed by Boichu et al. (2016). Nevertheless, despite these high SO2 ground-level mass concentrations measured regionally on 22–24 September (Fig. 2d), it is interesting to point out that, on 24 September, neither OMPS nor OMI satellite observations are sensitive enough to detect any SO2 over the northern part of France encompassing the Paris region (see the animation of OMPS observations in the Supplement, OMI satellite data not shown here). This demonstrates the necessity to combine both space and ground observations, especially when SO2 is confined in the boundary layer. Note that the two simultaneous anomalies observed on 9 and 10 September 2014 in both SO4 at SIRTA and SO2 mass concentrations at Airparif stations may also be volcanogenic. Nevertheless, this 2 d long episode of air pollution is not selected for further analysis as it is of lower intensity compared to the three other volcanogenic events already selected.
4.2 Background air pollution in sulfur-rich gas and aerosol species
At SIRTA, a 2-month average SO4 mass concentration of 1.0 µg m−3 is recorded with hourly resolved ACSM data during the September–October 2014 period, while the concentration rises up to 16.0 µg m−3 between 21 and 25 September 2014 during the largest event of volcanogenic pollution in SO2 recorded in France (Fig. 2d). Over the same period of time, air quality monitoring stations of the region record a mean mass concentration in SO2 of 1.4 and 1.9 µg m−3 at Neuilly-sur-Seine and Vitry-sur-Seine, respectively, which peaks at 80 and 42 µg m−3 during the volcanogenic pollution episode in late September 2014. Note that two other high peaks in SO2 mass concentration (up to about 70 and 50 µg m−3) are also observed in early October 2014, coincident with low SO4 mass concentration values. These anomalies are not of volcanic origin according to OMPS and IASI SO2 observations (see the animations of OMPS and IASI observations of the Holuhraun cloud dispersal in the Supplement). They are clearly associated with local emissions, since they are not recorded simultaneously at the three air quality stations of the Paris region and may be linked to heating systems turned on again before winter.
By comparison, Dunkirk Port-East is a much more polluted site in sulfur compounds, as revealed by mean mass concentrations in SO4 of 2.35 µg m−3 and in SO2 of 10.4 µg m−3 over a 14-month period (15 July 2013–11 September 2014) (Fig. 3a), which represent mass concentrations in SO4 and SO2, respectively, more than 2 and 5 times larger than at SIRTA.
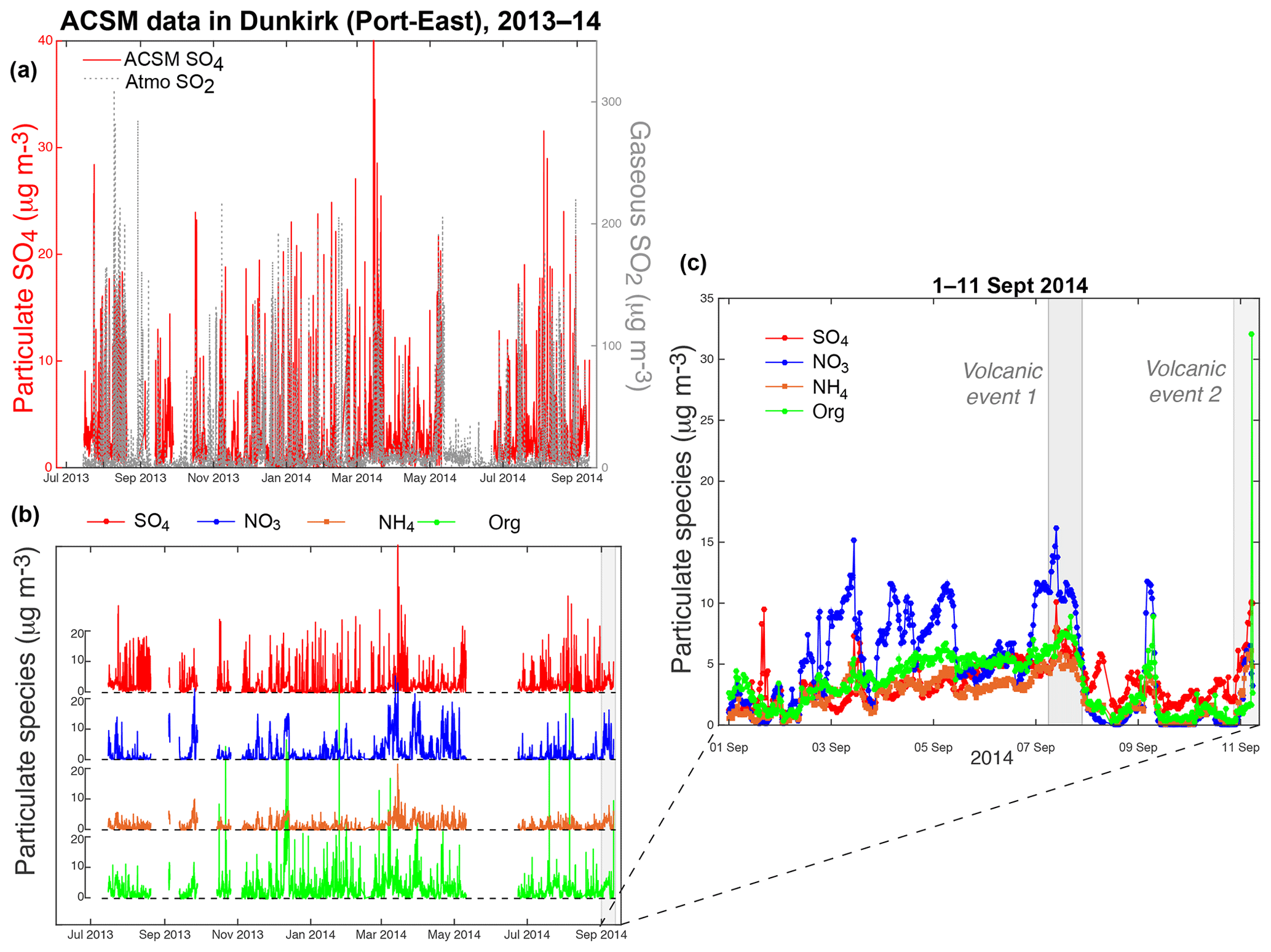
Figure 3(a, b) 14-month long time series of (a) (left red) particulate SO4 (ACSM), (right grey) gaseous SO2 (Atmo Hauts-de-France air quality station) and (b) ACSM species (sulfate (SO4), nitrate (NO3), ammonium (NH4) and organic (Org) aerosols) mass concentrations from 15 July 2013 until 11 September 2014, at Dunkirk Port-East station. (c) Focus on the period 1–11 September 2014 when events of air pollution induced by the Holuhraun eruption were recorded.
4.3 Chemical signature of volcanic sulfate aerosols
4.3.1 Chemical signature of volcanic and background aerosols at two contrasting sites
The 14-month long ACSM dataset with a resolution of 30 min collected between 15 July 2013 and 11 September 2014 in Dunkirk indicates large fluctuations, up to 40 µg m−3, in the mass concentration of sulfate aerosols at ground level (Fig. 3a). Large variations in ground-level SO2 mass concentrations, up to 340 µg m−3, are also recorded by Atmo Hauts-de-France air quality stations. However, no constant correlation is observed between SO2 and SO4 mass concentrations over the July 2013–September 2014 period of interest (Fig. 3a). Significant fluctuations in mass concentrations are also shown for NO3 (variations up to 30 µg m−3), NH4 (up to 20 µg m−3) and organic aerosols, the latter presenting the most important variations (up to 70 µg m−3) (Fig. 3b).
Although investigated here on a shorter period of 2 months (September–October 2014), variations in submicron particle mass concentrations at the SIRTA platform are much more limited, with peak values of 16, 13, 11 and 4 µg m−3 for SO4, organic, NO3 and NH4 aerosols, respectively (Fig. 4). At SIRTA, unlike nitrate and organics, the highest mass concentrations in ammonium aerosols are recorded between 21 and 25 September 2014, a period corresponding to the largest volcanogenic event of air pollution in sulfur-rich gas and particulate species in France (Sect. 4.1).
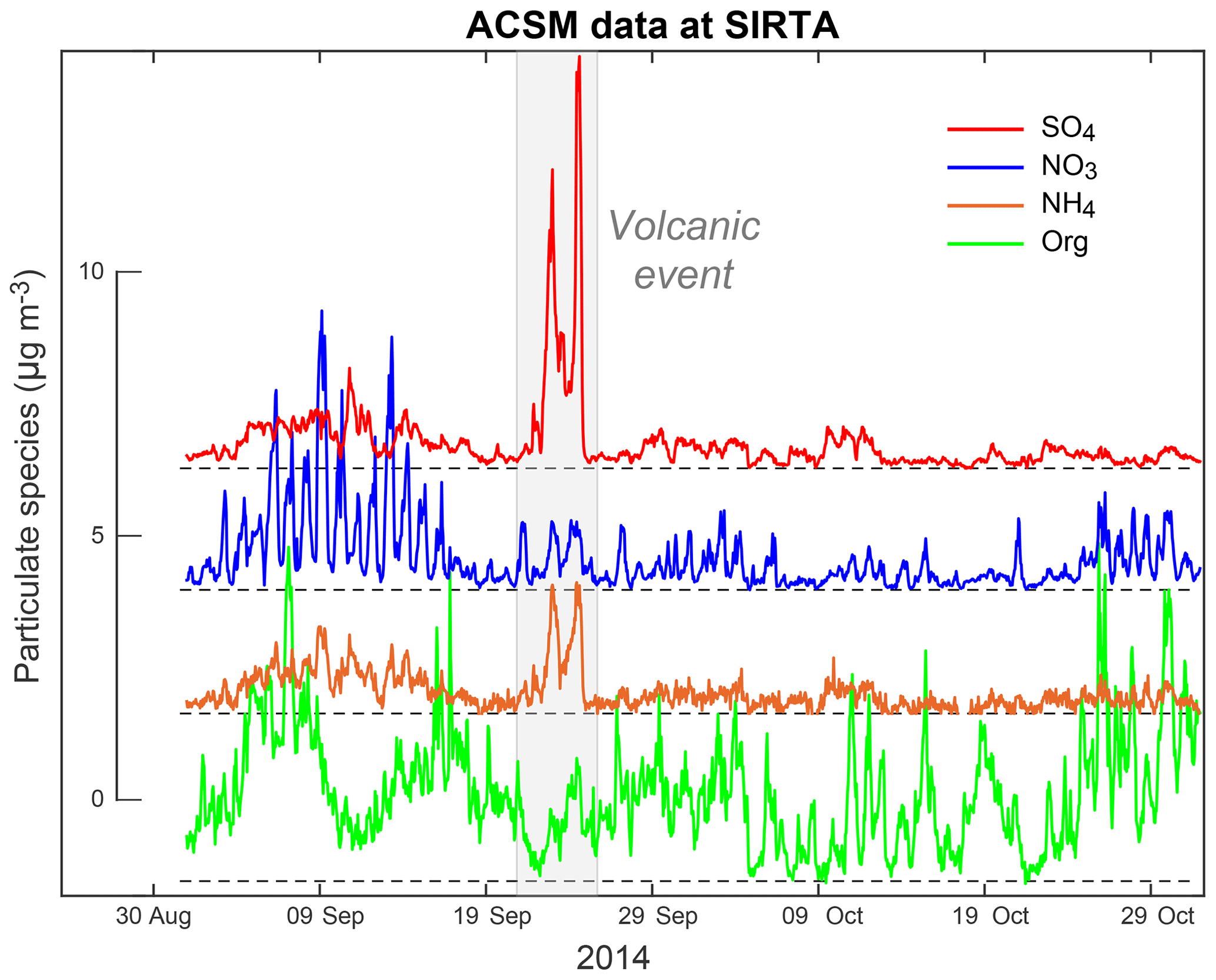
Figure 4Two-month long time series of the mass concentration of species measured with ACSM (SO4 in red, NO3 in blue, NH4 in orange, Org in green) at SIRTA covering the period September–October 2014 that is punctuated by a major volcanogenic event of air pollution in late September.
Scatter plots of the mass concentrations of gaseous SO2, measured by air quality stations, and of the various aerosol species (NH4, NO3, Org) measured with ACSM, vs. the mass concentration of sulfate aerosols, at the two sites of SIRTA and Dunkirk, display a wide dispersion of data (top of Figs. 5 and 6). As described previously in Sect. 4.1, three episodes of volcanogenic air pollution in SO2 have been highlighted at Dunkirk and SIRTA in September 2014. The ACSM data collected during the time period of occurrence of these volcanic events are marked specifically in the bottom of Figs. 5 and 6 (red squares for the largest event of air pollution in volcanic SO2 and SO4 that is recorded at SIRTA, green triangles and circles for DK volcanic events 1 and 2, respectively).
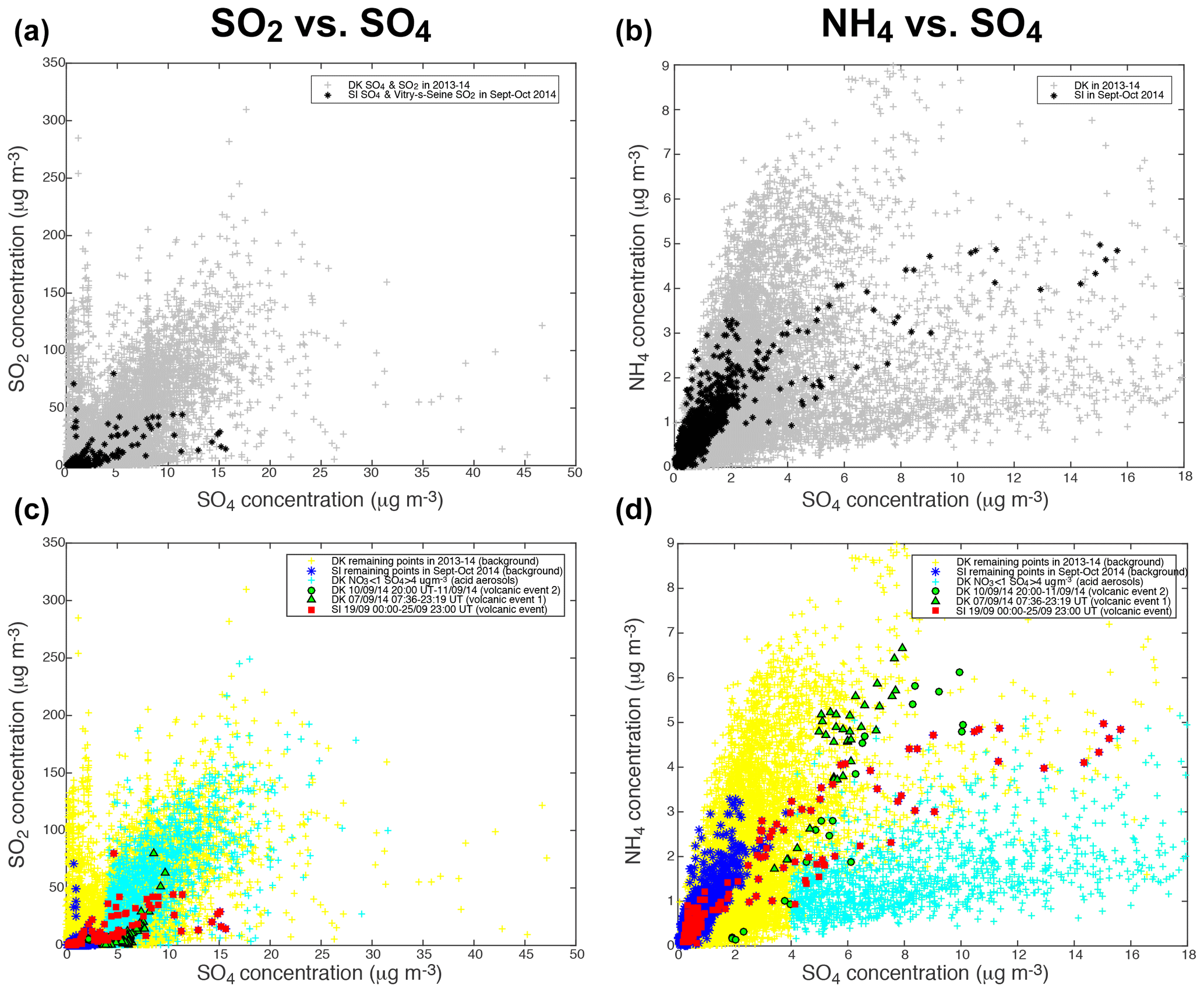
Figure 5Scatter plots of (a, c) SO2 (from Atmo Hauts-de-France station in Dunkirk or Airparif Vitry-sur-Seine station nearby SIRTA) and (b, d) ACSM NH4 vs. ACSM SO4 mass concentrations. (a, b) All available data at Dunkirk/Port-East (DK) over 15 July 2013–11 September 2014 (grey), and at SIRTA (SI) and nearby Vitry-sur-Seine Airparif station for SO2 over 1 September–31 October 2014 (black). (c, d) Red squares: SI data over 19 September 2014 00:00–25 September 2014 23:00 UT (volcanic event), green triangles: DK data over 7 September 2014 07:36–23:19 UT (volcanic event 1); green circles: DK data over 10 September 2014 20:00 UT–11 September 2014 (end of data) (volcanic event 2); cyan crosses: DK data with mass concentrations of NO3 < 1 and SO4 > 4 µg m−3 (acidic aerosols); blue stars: SI remaining data (background); yellow crosses: DK remaining data (background).
As Dunkirk is a much more polluted site than SIRTA, with various types and sources of aerosols, we start by comparing the signature of volcanic aerosols to SIRTA background. We observe that volcanic aerosols at both sites can be clearly distinguished from SIRTA (SI) background aerosols (in blue), especially in the scatter plots of SO2 (Fig. 5c), NO3 (Fig. 6c) and Org (Fig. 6d) vs. SO4 mass concentrations.
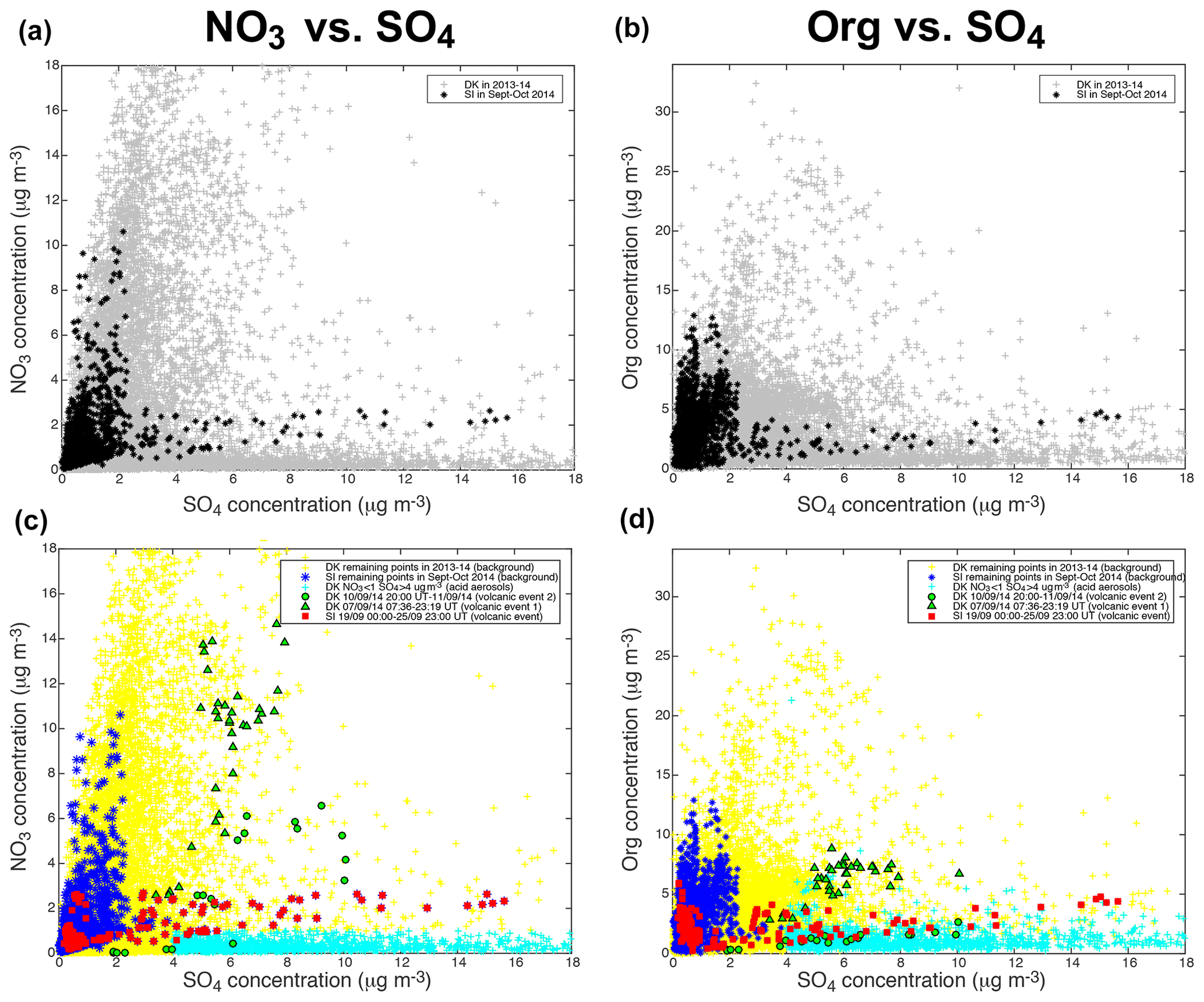
Figure 6Same as Fig. 5 but for (a, c) ACSM NO3 and (b, d) ACSM Org vs. ACSM SO4 mass concentrations.
Focusing on the NO3 vs. SO4 scatter plot (Fig. 6c), we observe that the mass concentrations of SO4 in SI background values are much lower (≤4 µg m−3) than during volcanic events at both sites (rising up to 16 µg m−3). A wider range of NO3 mass concentrations is also recorded during volcanic events, with a maximum of ≈15 µg m−3 during DK volcanic event 1 and lower values (<3 µg m−3) during the largest volcanic event, while background mass concentrations at SIRTA never exceed ≈11 µg m−3. Globally, we observe that volcanic aerosols at both sites display a lower NO3:SO4 mass concentration ratio than background aerosols at SIRTA, thus exhibiting a clearly distinct pattern. Similarly, it could be noted that a forecasted ammonium nitrate pollution event did not eventually occurred when Eyjafjallajökull volcanic emissions significantly impacted air quality over France in Spring 2010 (Colette et al., 2011).
In contrast to NO3, a narrower range of mass concentration in organics is observed during volcanic events (<9 µg m−3) than during background conditions at SIRTA with Org mass concentrations up to 13 µg m−3 (Fig. 6d). Again, volcanic aerosols present a distinct behaviour with a much lower Org:SO4 mass concentration ratio compared to SI background aerosols. Similarly, volcanic aerosols display a much lower SO2:SO4 mass concentration ratio than background aerosols (Fig. 5c).
However, isolating volcanic aerosols from SI background is less obvious in the scatter plot of NH4 vs. SO4 mass concentrations (Fig. 5d). This will be further analysed next in the text with thermodynamical simulations of aerosol composition. Whereas higher NH4 mass concentrations up to 7 µg m−3 are recorded during volcanic events, concentrations are about twice as low in SI background conditions. Nevertheless, volcanic aerosols do not present a NH4:SO4 mass concentration ratio significantly different from SI background characteristics (Fig. 5d).
4.3.2 Specific signature of freshly emitted industrial sulfate-rich aerosols
Particle mass concentrations at Dunkirk display a more complex behaviour with widely scattered values compared to SIRTA. We are especially intrigued by a group of ACSM data at Dunkirk that are associated with very low mass concentrations of NO3, hence presenting a signature close to the one of the largest volcanic event recorded at SIRTA (red squares) but showing a larger spread of SO4 mass concentration values up to 30 µg m−3 (Fig. 6c). For this reason, we colour in cyan these specific data associated with mass concentrations of NO3<1 and SO4>4 µg m−3 in the various scatter plots of Figs. 5c, d and 6c, d.
Polar plots in Dunkirk (Fig. A3) cover four sectors defined as follows: marine (271–70∘), urban (71–140∘), industrial-urban (141–225∘), and industrial (226–270∘). Figure A3d shows that most aerosols associated with NO3<1 and SO4>4 µg m−3 originate from the direction 225–270∘ corresponding to the industrial sector.
We demonstrate in the following that cyan data points, shown to be industrial aerosols, are not neutralized but acidic. To do so, we compare the predicted concentration of NH4 with the measured concentration of NH4 (Fig. 7). According to Seinfeld and Pandis (2012), the preferred form of sulfate is the neutral (NH4)2SO4 form in an ammonia–nitric acid–sulfuric acid–water system rich in ammonia and presenting a relatively elevated relative humidity. Under these assumptions, NH4,pred, the predicted concentration of NH4, is calculated assuming that has completely neutralized available sulfate, nitrate and chloride ions to form (NH4)2SO4, NH4NO3 and NH4Cl aerosols, which is written as
with molar masses of each species, , , and MCl, respectively equal to 18, 96, 62 and 35.5 g mol−1. In ACSM observations, the measured concentration of Cl is negligible compared to other species at both sites of SIRTA and Dunkirk that sits on the coast. Indeed, aerosol mass spectrometers flash vaporize particulate species impacted onto a heated surface. Instruments are classically operated with heaters set at 600 ∘C, which minimize the vaporization of sea salt. Ovadnevaite et al. (2012) recorded sea salt with a high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS) while operating the instrument at 650 ∘C. Nevertheless, some groups have reported issues of low vaporization in the instruments even at the temperature of 600 ∘C, leading in the case of ACSM observations to strongly negative chloride signals (since the chloride signal is then recorded while sampling filtered air and not ambient air and therefore subtracted from the “sample” signal). Our ACSM instrument at Dunkirk never displayed such a behaviour, thus confirming refractory chloride was not observed with our instrument in its normal operating conditions, the contribution of chloride species being only 0.3 % for an average NR-PM1 mass concentration of 8 µg m−3 in summer 2014. Given this negligible concentration of Cl, the last term in Eq. (3) is neglected.
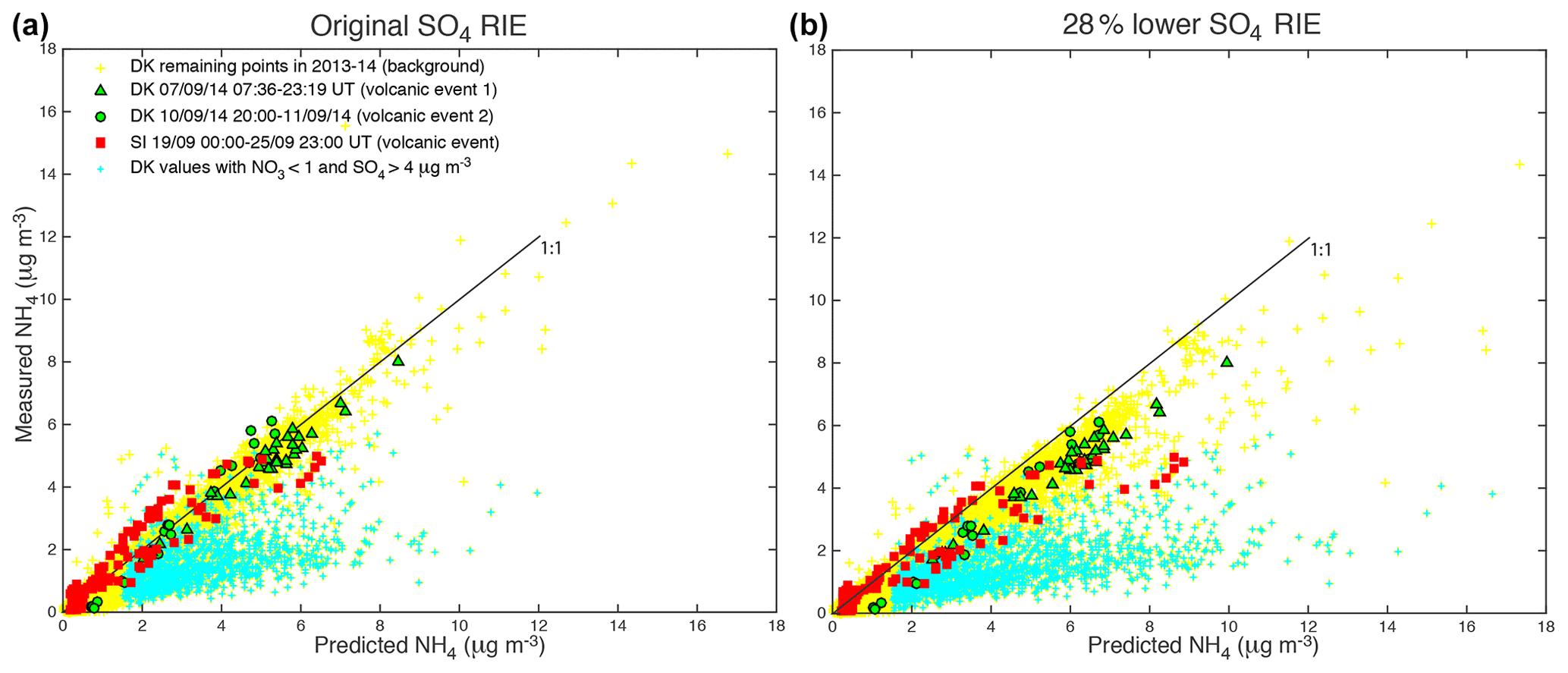
Figure 7Scatter plot of measured ACSM NH4 vs. predicted NH4 mass concentration for the three volcanic events of air pollution recorded at SIRTA (in red) and Dunkirk/Port-East (in green, triangles and circles for volcanic events 1 and 2, respectively) in September 2014. Data in cyan indicate values associated with aerosols with mass concentrations of NO3 < 1 and SO4 > 4 µg m−3. Yellow data correspond to the remaining ACSM values recorded in Dunkirk over 2013–2014, referring to background conditions. (a) Original and (b) 28 % lower sulfate RIE coefficients.
ACSM data associated with volcanic events and with background conditions in Dunkirk are roughly aligned in the scatter plot of measured vs. predicted concentrations of NH4 along the first bisector indicating their neutralization (Fig. 7). However, industrial aerosols coloured in cyan are widely scattered below the first bisector. This result demonstrates that, regarding these industrial aerosols, ions have not neutralized surrounding sulfate and nitrate ions. We assess in the following whether this absence of neutralization results from a lack of background NH3 or a lack of time available for neutralization.
The industrial sector in Dunkirk – where two main sulfur emitters (a refinery and a coke power plant) are located – expands between 500 m and 3 km from the sampling site. Winds blowing from this industrial sector often exhibit speeds above 5 m s−1 (Fig. A3a), thus residence times of industrial plumes in the atmosphere are generally well below one hour, and often only a few minutes, before reaching the sampling site.
On the other hand, wind sector analysis of the predicted vs. measured NH4 levels, or anion neutralization ratio (ANR), demonstrates that under urban or marine emissions, there is enough NH3 to neutralize both sulfate and nitrate aerosols on the same site, but that industrial emissions disturb the equilibrium (bottom of Fig. A3). Figure 3b shows the extent of ammonium mass concentrations over the 14 months of ACSM field observations, with levels often reaching up to 9 µg m−3. Most of the time in Dunkirk, sulfate mass concentration does not exceed 25 µg m−3 (Fig. 3a). Fully neutralizing such a substantial amount of sulfate requires about 9.5 µg m−3 of NH4 according to Eq. (3). To the best of our knowledge, there has not been any direct measurement of NH3 in Dunkirk. However, a rough estimation of the urban background level can be inferred from NH3 measurements in the middle-sized city of Douai, northern France (100 km away), over a year in 2015–2016 using a MARGA (Roig Rodelas et al., 2019). Mass concentrations were higher in the spring and summer seasons with averages of 4.3±2.9 and 4.0±2.8 µg m−3, reaching maxima of 11–12 µg m−3, respectively. In the Dunkirk area, we expect that local emissions – 50 % originating from the “'manufacturing industries, waste treatment and construction” according to the latest available inventory of AtmoHDF (2012), compared to 96 % from the agricultural sector when considering the entire Hauts-de-France region – will even increase this background level by a few µg m−3. Dunkirk atmosphere can consequently be considered to be sufficiently rich in NH3 to produce the concentration of ammonium required to neutralize the concentrations of industrial sulfate the most commonly measured. Local NH3 may generally not be lacking, but rather short residence times between the plume emission points and the sampling site are responsible for the acidity of these observed aerosols.
To summarize, we show that the group of ACSM data very poor in particulate nitrate, while rich in sulfate, originates from the industrial sector, is acidic and displays short residence time. We conclude that they represent freshly emitted aerosols of industrial origin, likely emitted by metallurgy and steel activities. We note that these aerosols are also relatively poor in ammonium and very poor in organic compared to background aerosols (Figs. 5d and 6d).
4.3.3 Best strategy to distinguish volcanic sulfate from other types of aerosols
We have shown in Sect. 4.3.1 and 4.3.2 that exploring the detailed chemical speciation of aerosols provided by ACSM measurements allows us to distinguish the signature of aged volcanic sulfate aerosols (e.g. aerosols already transported over a long distance from the eruption site) from those of freshly emitted industrial sulfate or background aerosols in various urban, marine or agricultural-influenced environments. As summarized in Fig. 8, angular sectors, which highlight the broad range of values associated with each type of aerosol, are more distinctively separated in the scatter plots of NO3 or Org vs. SO4 mass concentrations (Fig. 8c and d), which are thus more informative for identifying the aerosol source.
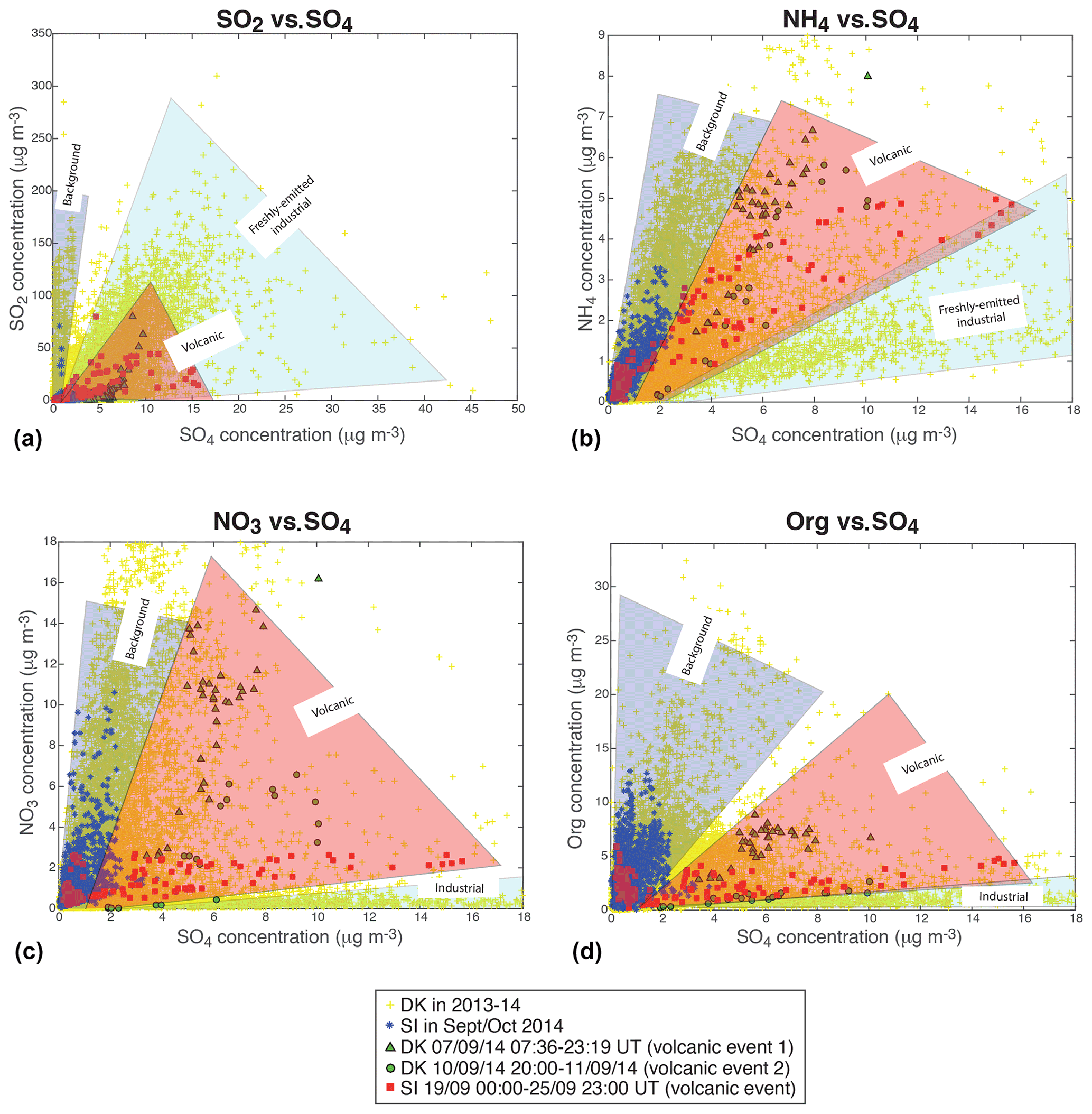
Figure 8Distinction of aerosol sources, either representative of background conditions at SIRTA (blue) or of volcanic (red) or industrial (cyan) origins, in the scatter plots of (a) gaseous SO2 from air quality stations, and various ACSM particulate species: (b) NH4, (c) NO3 and (d) Org, vs. sulfate mass concentrations. Sectors in colour, added to facilitate interpretation, represent an envelope roughly spanning the range of observed gas and particulate mass concentration values according to the type of aerosol.
To combine in a single plot the information on both the chemical signature of aerosols from these scatter plots as well as their degree of neutralization or acidity, we represent the variations of the NO3:SO4 (top of Fig. 9) or Org:SO4 (bottom of Fig. 9) mass concentration ratios vs. the ratio of measured to predicted NH4 mass concentrations. To avoid a noisy representation, we select ACSM values meeting the criteria µg m−3 for the top of Fig. 9 and µg m−3 for the bottom of Fig. 9.
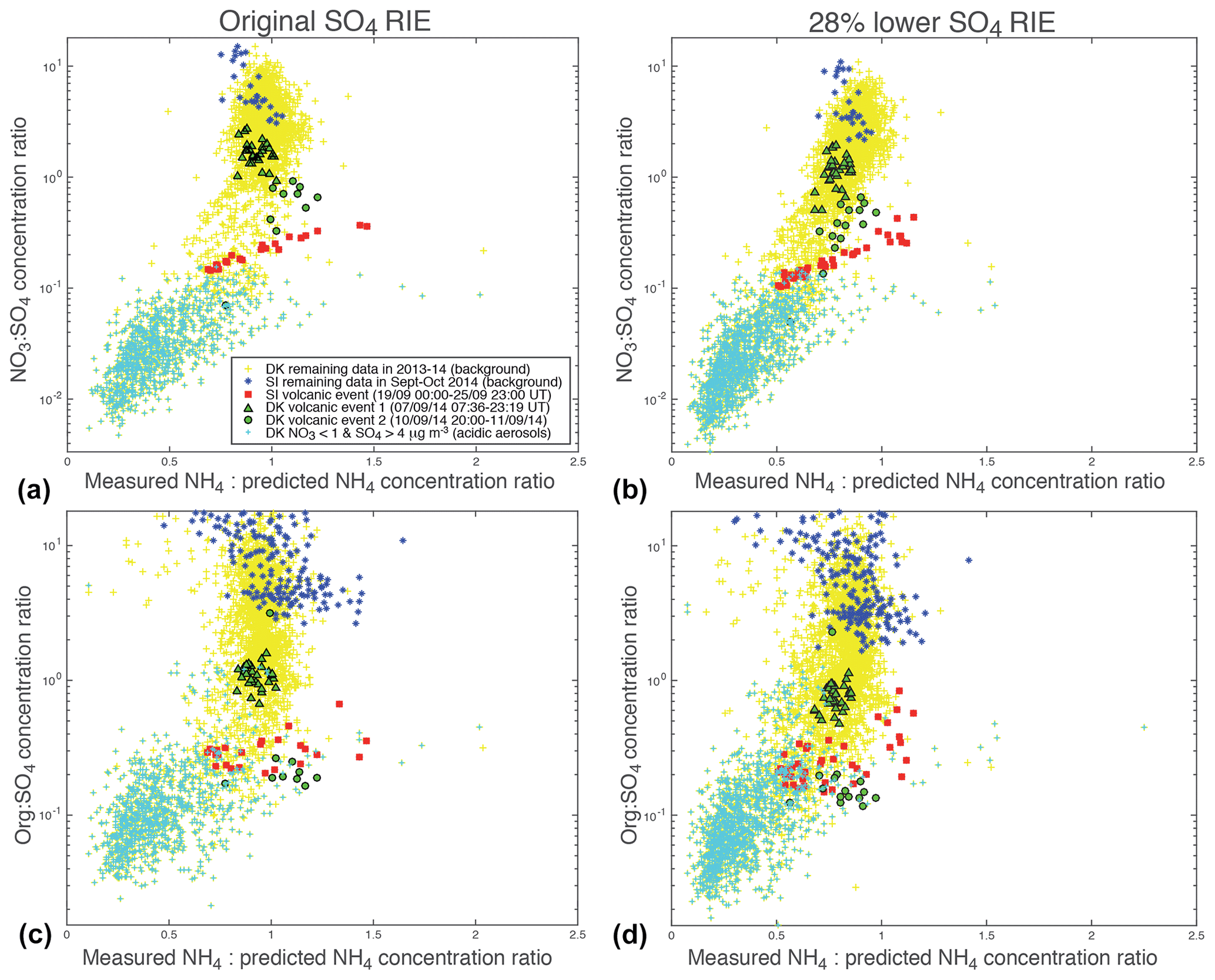
Figure 9Scatter plots of (a, b) NO3:SO4 or (c, d) Org:SO4 mass concentration ratios (in logarithmic scale) vs. the ratio of measured to predicted NH4 mass concentrations for (a, c) original and (b, d) 28 % lower sulfate RIE coefficients. Selected ACSM data meeting the criteria (a, b) µg m−3 and (c, d) µg m−3 are displayed.
All aerosols present values of the mass concentration ratio, or anion neutralization ratio (ANR) close to 1 indicating their neutralization, except freshly emitted industrial aerosols in Dunkirk (in cyan) with most values < 0.75 indicative of their strong acidity (left of Fig. 9). Nevertheless, we note a few values of the neutralization ratio exceeding 1 (up to 1.5) for both the largest volcanic event at SIRTA (in red) and some background aerosols in Dunkirk (in blue) (left of Fig. 9). This phenomenon could be linked with NH3 uptake onto particulate organic acids, as previously observed in north-western Europe (Schlag et al., 2017). It may also partly result from possible bias in the evaluation of the SO4 relative ionization efficiency (RIE), as explained in Sect. 2.1.1. Indeed, the chosen RIE value could lead to an underestimation of SO4 mass concentrations and subsequently NH4,pred values if indeed the true SO4 RIE was lower. Considering that a SO4 RIE value of 0.86 was obtained from the new calibration procedure applied for the first time to SIRTA ACSM in spring 2016 (Freney et al., 2019), we recalculated SO4 mass concentrations using RIE values lower than the chosen one by 28 % (i.e. 0.39 and 0.86 for Dunkirk and SIRTA ACSMs, respectively) to investigate the influence of this possible bias. While NO3:SO4 and Org:SO4 mass concentration ratios are weakly influenced by such a change (Fig. 9b, d), it weakly impacts aerosol acidity as ANR values are lower with a RIE equal to 0.86, independently of the type of aerosols (Figs. 7b and 9b, d). ANR values no longer greatly exceed the value of 1, reducing the bias above mentioned.
Concerning the NO3:SO4 mass concentration ratio, whichever the sulfate RIE coefficient, volcanic aerosols (in red and green) present values between 0.1 and 3, while background aerosols at SIRTA (in blue) are associated with the highest values (>3) and freshly emitted industrial aerosols in Dunkirk (in cyan) with the lowest values (<0.15) (top of Fig. 9).
Concerning the Org:SO4 mass concentration ratio, background aerosols at SIRTA are characterized by ratios greater than ∼2.5. In contrast, low values (mostly <1.6) are observed during the volcanic event (bottom of Fig. 9). Accordingly, these low ratios are primarily explained by a high concentration of SO4 (denominator). Nevertheless, we note that the volcanic event coincides with a period of relatively low concentration of organics (numerator). Although similarly low concentrations are observed in the months prior to or following the volcanic event (Fig. 4), one cannot exclude the possibility that this coincidence may also reflect a causal relationship between the low organic concentration and the high SO4 concentration. Indeed, Fig. 6d shows that the Org:SO4 mass concentration ratio at Dunkirk is remarkably impacted by the occurrence of industrial pollution events carrying acidic freshly emitted aerosols (detected by means of their anion neutralization ratio and trajectory analysis; see Sect. 4.3.2). Hence, such sulfur-rich industrial pollution events are generally characterized by a very low concentration of organics at Dunkirk, if not a quasi-complete depletion.
A depletion of organic aerosols in response to an increased acidity seems at odds with the findings of Zhang et al. (2007) and Pathak et al. (2011) who show an enhancement of secondary organic aerosols with acidity. Alternatively, this apparent decrease in organic aerosol mass concentrations may reflect the transformation of organic aerosols measured by ACSM into other species that are not resolved by our measurements. An hypothesis could be the formation of organosulfate aerosols, especially in presence of highly acidic sulfate aerosols, according to laboratory experiments (Surratt et al., 2008; Perri et al., 2010) and modelling studies (McNeill et al., 2012). Formation of organonitrates has also been observed under SO2 and NH3-rich conditions in both smog chamber (Chu et al., 2016) and ambient air (Zaveri et al., 2010) experiments. These transformation mechanisms, likely at play during industrial sulfur-rich pollution events as shown by Zaveri et al. (2010) in a coal-fired power plant plume, may also be active during the 2014 volcanic event. A thorough analysis of additional ACSM observations at other sites in Europe may allow for disentangling of the respective roles of sulfur-rich volcanogenic pollution vs. ambient air natural variability in leading to fluctuations of organic aerosol concentration.
To summarize, both NO3:SO4 and Org:SO4 mass concentration ratios allow us to distinguish volcanic aerosols from background aerosols at SIRTA. However, the NO3:SO4 mass concentration ratio seems the most powerful to also distinguish the chemical pattern of volcanic aerosols from those of freshly emitted industrial aerosols as shown in Dunkirk.
Nonetheless, Fig. 9 (as well as Figs. 5, 6 and 8) illustrates much more data scatter for background aerosols in Dunkirk (in yellow) compared to SIRTA (in blue), independently of the ratio of interest (NO3:SO4 or Org:SO4). It has to be recalled that the Dunkirk dataset covers a much longer time period (more than a year) than the SIRTA one (2 months), which may partly explain this observation. In addition to its coastal location implying the presence of sulfur-rich aerosols from marine or ship emissions (Zhang, 2016), that are naturally absent at SIRTA, Dunkirk hosts both intense harbor and industrial activities as previously mentioned (Sect. 4.2). Therefore, Dunkirk is a much more polluted site in sulfur-rich particles than SIRTA. This certainly explains the significantly broader range of both NO3:SO4 and Org:SO4 mass concentration ratios observed for Dunkirk background aerosols, with values much lower than for SIRTA background aerosols that even intersect those associated with volcanic aerosols (in red and green). Hence, such a result demonstrates the most challenging issue to discriminate the signature of volcanic aerosols among other types of aerosols at a heavily polluted site.
4.3.4 Thermodynamic modelling of aerosol composition
While the NH4:SO4 mass concentration ratio varies only slightly (Fig. 10a2 and b2), thermodynamic simulations of aerosol composition for the atmospheric conditions met at SIRTA reproduce a large decrease in the NO3:SO4 mass concentration ratio with an increasing concentration of total sulfate, whichever the background level of NH3 (Fig. 10a1 and b1). However, only the NH3-rich scenario allows us to best fit the NO3 observations during the volcanic event in late September 2014 which is characterized by large SO4 mass concentrations exceeding 4 µg m−3 (Fig. 10a1), with a determination coefficient between modelled and observed NO3 mass concentrations of 0.96. The NH3-poor scenario overestimates the decrease in particulate nitrate, with its almost complete depletion for a mass concentration of total sulfate exceeding 12 µg m−3 (Fig. 10b1) concomitant with a total depletion of NH3 (Fig. 10b3) and an increase in the mass concentration of nitric acid (Fig. 10b4). Interestingly, these thermodynamic simulations allow us to indirectly estimate the rich background mass concentration of ammonia at SIRTA in September–October 2014, showing no evidence of any lack of NH3 to neutralize the substantial load of sulfate aerosols (up to 16 µg m−3) during the large volcanic event in late September 2014.

Figure 10ISORROPIA II thermodynamic model simulations (red) of atmospheric composition (aerosol NO3 a1, b1 and NH4 a2, b2, gas-phase NH3 a3, b3 and HNO3 a4, b4) as well as pH (a5, b5) vs. SO4 mass concentration at SIRTA in September–October 2014 considering an environment either (a) rich (7.40 µg m−3) or (b) poor (0.74 µg m−3) in NH3. Comparison with ACSM observations of aerosols (blue). Inset in (a1) and (b1) shows ISORROPIA NO3 vs. ACSM NO3 coloured with the concentration of sulfate.
Therefore, thermodynamic model simulations suggest that the distinct chemical signature observed for Holuhraun volcanic aerosols, compared to background aerosols, results from the large abundance of sulfate within the volcanic plume. This is confirmed by model sensitivity tests addressing the impact on the production of particulate nitrate of an increasing concentration of sulfate, while all other parameters are kept constant (Fig. 11). At high concentrations of sulfate aerosols, simulations show that ammonia preferentially neutralizes sulfate rather than nitrate, favouring the formation of ammonium sulfate ((NH4)2SO4) rather than ammonium nitrate (NH4NO3). In these conditions, the decrease in particulate NO3 mass concentration with increasing sulfate concentration (Fig. 11a) coincides with an increase in gas-phase HNO3 (Fig. 11d), since pH has an impact on gas-particle partitioning of NO3–HNO3. In an atmosphere very rich in sulfate (e.g. a total sulfate exceeding 12 µg m−3 here), a complete depletion of gas-phase NH3 (Fig. 11e) and particulate NO3 (Fig. 11a) can occur, concomitantly with NH4 mass concentration reaching a plateau value (Fig. 11b). The preferred form of sulfate aerosols is no longer , but bisulfate () (Fig. 11c) and pH drastically decrease (Fig. 11f).
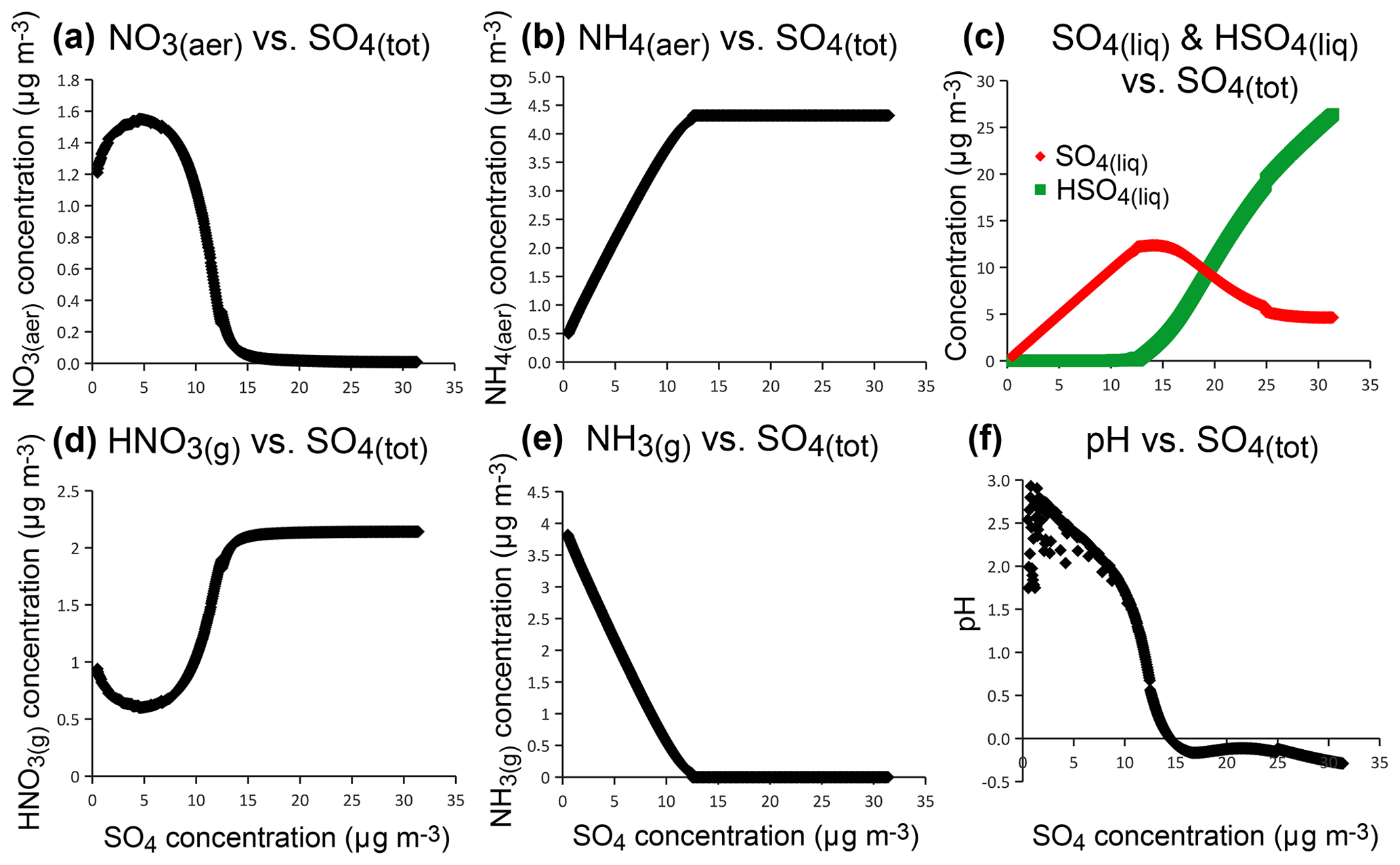
Figure 11Sensitivity tests of aerosol composition (particulate NO3 a, particulate NH4 b, liquid SO4 and HSO4 c), gas-phase composition (HNO3 d, NH3 e) and pH (f) with increasing concentration with increasing concentration of total sulfate aerosols, using ISORROPIA II thermodynamic model for conditions met at SIRTA in September–October 2014.
Thermodynamic simulations have been compared to ACSM observations with the original SO4 RIE of 1.20 (Fig. 10). Nevertheless, investigating the influence of SO4 RIE values, we find that while volcanic SO4 aerosols could be overall considered neutralized with a RIE of 1.20 (Fig. 7a), some volcanic aerosols are non-neutralized with a RIE = 0.86 (Fig. 7b), industrial aerosols remaining nevertheless still always more acidic than volcanic sulfates. We find that the three periods which are affected by the presence of acidic volcanic aerosols characterized by values of the neutralization ratio < 0.7 (22 September 2014 from 12:00 to 21:00, 23 September from 11:00 to 16:00 and 24 September from 10:00 to 17:00 UTC) are associated with periods of elevated mass concentrations of SO4 exceeding 5 µg m−3 (Fig. 2d). Note that the most acidic volcanic aerosols, characterized by a weak neutralization ratio of about 0.5, are recorded on 24 September and are associated with SO4 mass concentrations >15 µg m−3, the most substantial amount of volcanic SO4 recorded at ground level at SIRTA, which is also associated with a large SO2-to-SO4 mass concentration ratio (Fig. 2d). OMPS SO2 maps (in the Supplement) indicate that the queue of the Holuhraun cloud arrives over northern France on 22 September and does not seem to greatly move on the following days, when it gets diluted according to the observed decrease in SO2 column amounts with time. Simultaneously, an increase in mass concentrations of sulfur-rich species is recorded at ground level over northern France (Fig. 2d). This joint analysis of satellite and ground-level in situ observations suggests that the volcanic plume is captured within the boundary layer, hence being more unlikely to be detected by any satellite sensor. This stagnation of the Holuhraun plume within the boundary layer, preventing any more displacement, may explain an exceptional lack of local NH3 to fully neutralize volcanic sulfur-rich aerosols, which justifies the presence of remaining acidic H2SO4 aerosols within the volcanic cloud according to thermodynamic simulations in Fig. 11. We can wonder whether these specific transport and meteorological conditions explain the largest SO2-to-SO4 mass concentration ratio which is observed. Therefore, as suspected by model simulations of various Icelandic eruption scenarios in the UK atmosphere (Witham et al., 2015), our observations show here that, despite a very long transport and dispersion over thousands of kilometres from Iceland, the Holuhraun plume may exceptionally remain so rich in sulfur that the available amount of ammonia along its way is not sufficient to neutralize all volcanic sulfate aerosols.
4.4 Persistent weeks long air pollution by volcanic sulfate aerosols
We find from ACSM observations some strikingly elevated ground-level mass concentrations of sulfate aerosols, well in excess of mean values, in September 2014 at both French sites, at SIRTA, over a period of about 2 weeks from 4 to 18 September with [SO4] > 0.5 µg m−3 (Fig. 2d), and at Dunkirk, over at least 8 d from 3 to 11 September with [SO4] > 2 µg m−3 (Fig. 1c). As shown in Sect. 4.1, these periods of time are punctuated by a few episodes of volcanogenic pollution in SO2 from the Holuhraun eruption: two events at Dunkirk on 7 and 10–11 September and two events at SIRTA, a major one on 21–25 September but also a more minor episode on 9–10 September 2014. Interestingly, these episodes of volcanogenic air pollution in SO2 are short-lived, lasting less than a day or a few days at the most. We consequently wonder whether this persistent particulate pollution in SO4, which is broadly observed in France at locations a few hundreds of kilometres apart, could also be of volcanic origin.
To make progress on this issue, we jointly explore ACSM ground-level in situ measurements with sunphotometer observations from the AERONET (AErosol RObotic NETwork) ground-based remote sensing network (Holben et al., 2001) at the two stations of Dunkirk and SIRTA that provide column-integrated information on aerosols (Fig. 12). In the period of the persisting exceedance anomaly in ground-level SO4 mass concentration, we also observe elevated values of the aerosol optical depth at 500 nm, >0.2 at SIRTA (given a mean AOD value of 0.131±0.035 for September months between the start of AERONET measurements at SIRTA in 2008 and 2016, excluding 2014) and >0.3 in Dunkirk (given a mean AOD value of 0.175±0.047 for September months between the start of AERONET measurements in Dunkirk in 2006 and 2017, excluding 2014). Most importantly, we find a remarkable correlation between time series of SO4 ground-level mass concentration and of aerosol optical depth at SIRTA (Fig. 12a) and also at Dunkirk though to a lesser extent due to a shorter ACSM dataset (Fig. 12b). This result demonstrates that the aerosol optical depth, a column-integrated property, is mainly impacted by ground-level sulfate aerosols in these occasions. As observed on 1 September at Dunkirk (Sect. 4.1), industrial activities can only trigger short-term peaks, lasting a few hours, in both SO2 and SO4 ground-level mass concentrations (Fig. 1c). Therefore, we suggest that the persisting excess anomaly in both SO4 ground-level mass concentration and aerosol optical depth observed in September 2014 at a regional scale in France may result from the broad dispersion of sulfur-rich emissions, likely originating here from the Holuhraun eruption.
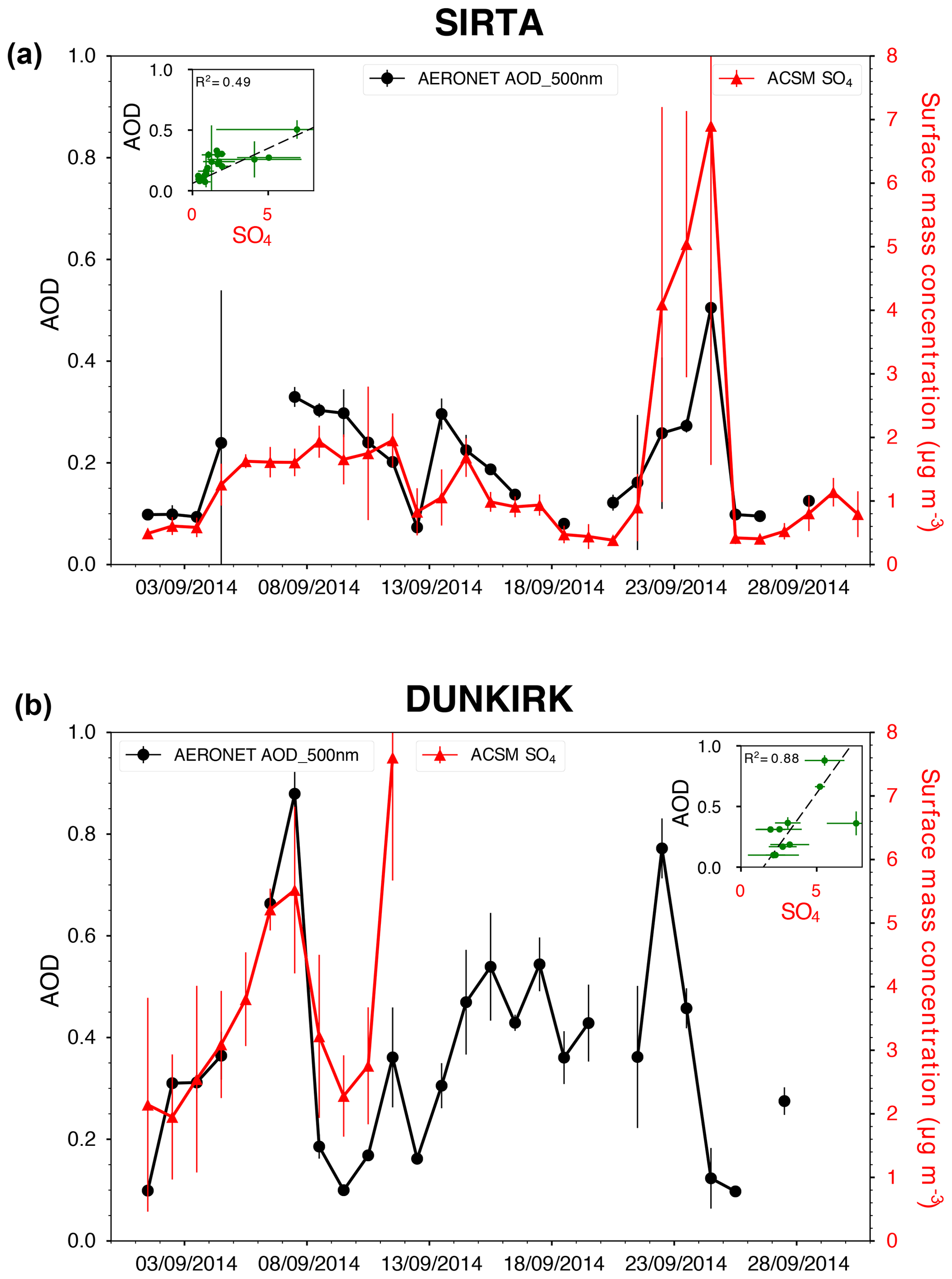
Figure 12Time series of daily averaged values of both AERONET AOD at 500 nm and ACSM SO4 mass concentration, with vertical bars indicating the dispersion of data over 24 h, at (a) SIRTA and (b) Dunkirk. In inset are included scatter plots and associated determination coefficients.
As suspected by the modelling study of Witham et al. (2015), this result illustrates the much longer atmospheric persistence (a few weeks) of volcanic sulfate aerosols compared to SO2 (a few days), even in the boundary layer, in a real case study. Meteorological conditions, without abundant long-lasting precipitations in September 2014, have likely favoured this persistence of aerosols in the atmosphere. Hence, the impact of the Holuhraun eruption on European air quality, mainly studied through observations and atmospheric modelling of SO2 (Schmidt et al., 2015; Ialongo et al., 2015; Boichu et al., 2016; Steensen et al., 2016) since SO2 represents a clear marker of volcanic emissions, could have been largely underestimated. This shows that a synergistic analysis of both SO2 and SO4 gas/particulate species, combining multi-instrumental and multi-parametric approaches, as developed in this paper, is fundamental to rigorously assess the large-scale impact of volcanic sulfur-rich emissions on atmospheric composition, air quality and health. Holuhraun sulfate aerosols have been shown to strongly affect the microphysical properties of low-altitude meteorological clouds above oceans (McCoy and Hartmann, 2015; Malavelle et al., 2017). This study demonstrates the need to extend such studies above continents to robustly estimate the global volcanic forcing on climate of tropospheric eruptions and persistent passive degassing activities.
4.5 Large-scale volcanogenic pollution in gas and particulate sulfur recorded by the EMEP network
To put into perspective our results showing a persistent atmospheric pollution in sulfate particles in France and assess more broadly the geographical impact of Holuhraun emissions on air quality, we explore daily and hourly datasets of sulfur monitoring by filter pack and online ion chromatography measurements from ground stations of the European EMEP network (map in Fig. 13) over the complete 6-month long eruption (September 2014–February 2015). We also examine the partitioning of volcanic sulfur species (e.g. the SO2:SO4 mass concentration ratio here) to see whether the one observed with ACSM and SO2 measurements in France is similarly found elsewhere.
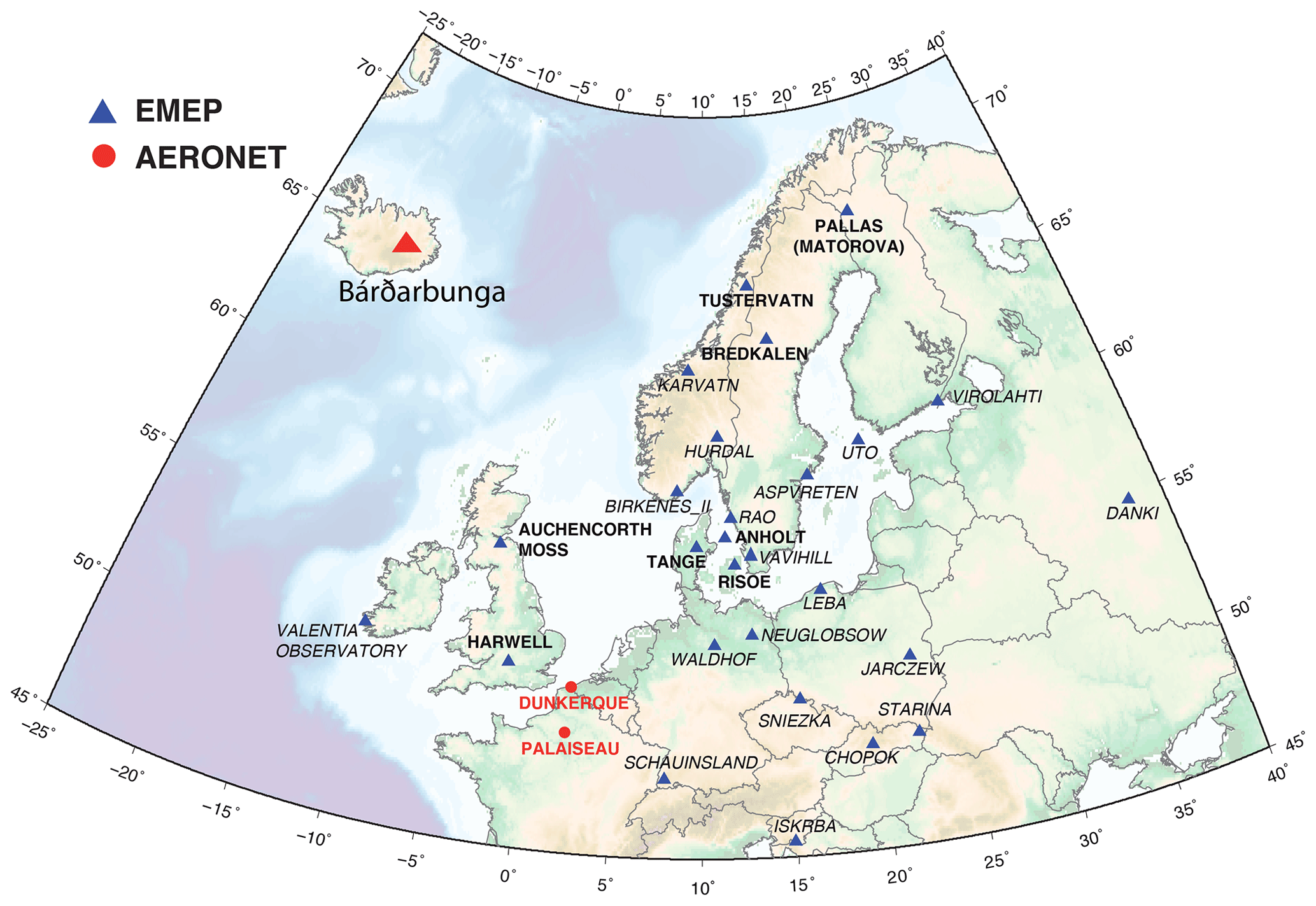
Figure 13Map of the 27 EMEP stations (blue triangles) explored in this study. Stations with name in bold, with a few daily SO2 mass concentrations higher than 3 µg m−3 over the period September 2014–February 2015 suggesting a clear impact of the Holuhraun eruption, are selected for a detailed multi-site concentration-weighted trajectory analysis, while stations in italic are not. Red circles indicate the AERONET network stations of Dunkirk and SIRTA (Palaiseau).
Unfortunately, the number of EMEP stations in Europe performing the monitoring, at the same temporal resolution, of ground-level mass concentrations of both SO2 and SO4 has significantly decreased in the last decade, and only 27 stations, listed in Table 1, are of interest for our study (time series covering the September 2014–February 2015 period of the eruption at each of these stations are displayed in the Supplement). Among these 27 stations, we investigate in details those presenting a few daily SO2 mass concentrations >3 µg m−3 over the September 2014–February 2015 period, a threshold well above noise level, which suggests a clear volcanic impact. The eight selected stations of interest, whose name appears in bold below blue triangles in Fig. 13 and details are listed at the top of Table 1, are located in Scandinavia (Pallas-Matorova in Finland, Tustervatn in Norway, Bredkälen in Sweden, Risoe, Anholt and Tange in Denmark) and in Great Britain (Auchencorth Moss and Harwell). The station of Starina in Slovakia is not selected as it presents some elevated daily mass concentrations of SO2 that are not correlated with SO4 nor recorded at neighbouring stations, indicating a local source of sulfur without long-range influence. Time series of SO2 and SO4 ground-level mass concentrations for selected stations are displayed in Fig. 14. Note that if a station does not meet this criterion and is consequently not selected for detailed analysis, it may nevertheless also be impacted by the eruption as a daily SO2 threshold of 3 µg m−3 is high.

Figure 14Time series (top) and scatter plot (bottom) of ground-level mass concentrations (in µg S m−3) of SO2 and corrected PM10 (i.e. non-marine SO4) covering the Holuhraun eruption from September 2014 to February 2015, at selected EMEP stations in Scandinavia and Great Britain clearly impacted by the eruption. Grey circles in scatter plot indicate data points selected for plume age estimation in Fig. 16.
Persistent week long elevated values in ground-level daily SO2 mass concentrations (up to >20 µg m−3), much in excess of background values, are recorded especially in September 2014 in Great Britain (Harwell and Auchencorth Moss), Finland (Pallas-Matorova), Sweden (Bredkälen) and Norway (Tustervatn) to a lesser extent as anomalies are shorter (Fig. 14). During these periods of elevated values in surface SO2 mass concentrations, increased levels in sulfate mass concentrations are always simultaneously recorded (up to 7 µg m−3). Note that Pallas-Matorova, Bredkälen and Tustervatn represent rural background stations with no significant local or regional air pollution sources, Pallas and Bredkälen being surrounded by coniferous forest or grasslands (Hatakka et al., 2003; Targino et al., 2013), while Tustervatn lies in an agricultural environment poor in sulfur (Aas et al., 2014). By comparison, stations in Denmark lie in a much more polluted environment, as shown by higher and noisier background values in ground-level sulfur mass concentrations (Fig. 14). Nevertheless, some elevated values in SO2 and SO4 mass concentrations (up to 5 and 5.5 µg m−3, respectively), well exceeding the SO2 noise level, are recorded simultaneously at all three Denmark stations (Risoe, Anholt and Tange), but also much more broadly at Bredkälen (Sweden), Pallas (Finland) and Auchencorth Moss (Great Britain) at the end of October 2014 over a few days.
These widespread anomalies in both gas and particulate sulfur concentration at ground level suggest the impact of long-range transported pollutants.
The volcanic origin of this large-scale atmospheric pollution in SO2 is attested by SO2 observations of OMPS and IASI satellite sensors (see the animations of OMPS and IASI SO2 observations of the Holuhraun SO2 cloud dispersal in the Supplement) showing the Holuhraun volcanic cloud, rich in SO2, transported repeatedly over Scandinavia and Great Britain in September and October 2014.
This is also confirmed by concentration-weighted trajectory analysis of EMEP ground-level data over September–October 2014 applied using a multi-site approach (Fig. 15a) or separately at seven out of eight stations studied individually (left of Figs. A4 and A5). The strong impact of icelandic emissions of volcanic SO2 is all the more remarkable given the relatively low number of back-trajectories leading to Iceland from each of the eight stations, as illustrated by trajectory density maps (right of Figs. A4b, c, d and A5a, b, c, d, e). The only exception is the result obtained at Tustervatn (Norway) (left of Fig. A4c), indicating a pollution by SO2 emissions from the polar Arctic region and Svalbard. Boreal biomass burning fires or industrial emissions from northern Russia may be hypothesized as distant sources of this northerly pollution (Law and Stohl, 2007), but are unlikely in our case since trajectory analysis from neighbouring stations (Bredkälen and Pallas) does not point to any source in the Arctic region (left of Fig. A4b and d). This suggests an inconsistency with the Tustervatn trajectory analysis. A tuning of altitude initialization in the trajectory analysis (here assumed identical for all stations) may be required to resolve this incoherence. For Denmark stations, we identify a supplementary weak influence of SO2 emissions from eastern Europe industry (left of Fig. A5a, b, c). These sources correspond to SO2 anthropogenic sources that have already been identified in the catalogue of large SO2 emissions in 2013 derived from OMI satellite sensor observations from Fioletov et al. (2016), represented in Fig. 15a, b. Hotspot integration shows the major contribution of emissions from the Iceland area that accounts for 25 % of ground-level SO2 measured over Europe, which contrasts with the 0.2 % contribution of eastern European sources (Fig. 15a).

Figure 15Multi-site concentration-weighted trajectory analysis for SO2 and SO4 mass concentrations measured in September–October 2014 at a set of eight selected EMEP stations in northern Europe (shown in Fig. 14): retrieved source mass concentrations (µg S m−3) of (a) SO2 and (b) corrected SO4 (i.e. non-marine SO4), (c) trajectory density (log of residence time, no unit) with the location of stations (light green circles). Contribution to the widespread atmospheric pollution of Icelandic volcanism (A and C green areas) and anthropogenic (B and D pink areas) sources is calculated in the white dashed rectangles, using an edge detection at 1 and 1.5 µg S m−3 for SO2 and SO4, respectively. SO2 emission sources for 2013 derived from OMI satellite sensor observations (from Fioletov et al., 2016) are indicated by dark green circles.
Contrary to SO2, the origin of sulfate aerosols measured by EMEP stations is more complex. Using a multi-site concentration-weighted trajectory analysis, emissions from the Holuhraun eruption are also identified as a major source of SO4 at all stations (except Tustervatn again) (Fig. 15b). In addition to this volcanic source, we also show the significant impact on SO4 of anthropogenic emissions from eastern Europe (especially from Ukraine) but also from Great Britain albeit to a lesser extent. As shown in Fig. 15b, these retrieved industrial sources of sulfate are in good agreement with the sources of anthropogenic SO2 emissions in 2013 from Fioletov et al. (2016). Interestingly, both volcanic and eastern Europe emissions contribute almost equally to SO4 over Europe (Fig. 15b), which contrasts with the volcanic specificity observed for SO2. Retrieved sources of SO4 are also found to be more geographically dispersed than SO2 sources (Fig. 15a, b), which is likely due to their longer atmospheric persistence as discussed in Sect. 4.4. These results attest to the interest in developing a multi-site approach as well as the importance of jointly analysing SO2 and SO4 species, as performed in this study, to better distinguish, among other anthropogenic sources of pollution, the volcanic impact on the concentration of aerosols.
Therefore, we demonstrate here the large-scale fingerprint of the Horuhraun eruption on both gas and particulate air pollution in SO2 and sulfate aerosols, affecting broadly Europe, not only France as shown in Sect. 4.1 and 4.4 but also vastly Great Britain and Scandinavia.
4.6 Evolution of SO2 to sulfate oxidation rates during plume aging
To understand the process of SO2 oxidation to sulfate in volcanic clouds, we investigate the SO2:SO4 mass concentration ratio observed during major volcanic events in the PM1 fraction collected by ACSM in France at SIRTA (Sect. 4.3, Fig. 2d) and in the PM10 fraction sampled at the eight EMEP stations studied in detail (Sect. 4.5, Fig. 14). For this purpose, we select maximum values of the SO2 mass concentration (and the corresponding SO4 mass concentration values) associated with back-trajectories leading to Iceland over September–October 2014 (these values are indicated by the grey circles at the bottom of Fig. 14). In addition, we also evaluate the age of the volcanic plume for these selected volcanic events.
The scatter plot of the SO2:SO4 mass concentration ratio with plume age (Fig. 16) indicates a wide array of SO2-to-SO4 mass ratios in the Holuhraun plume ranging from 0.8 to 8.0 at stations in five different countries of northern Europe (France, Great Britain, Denmark, Sweden and Finland). Elevated SO2:SO4 mass ratios observed at northern Scandinavia stations may suggest the impact on air quality of relatively young volcanic clouds (despite the traveled distance). Indeed, IASI satellite observations of the altitude of SO2 mostly indicate a high-altitude (up to 8 km) transport of the Holuhraun cloud at high latitudes, in broad agreement with Carboni et al. (2019) (see the animation of the IASI SO2 column amount and altitude observations of the Holuhraun cloud dispersal in September and October 2014 in the Supplement). Such high-altitude transport is expected to be faster and to cross an atmosphere poorer in solar radiation and OH- radicals favouring a lower SO2-to-SO4 oxidation. On the other hand, lower SO2:SO4 mass ratios may be associated with more aged and diluted volcanic clouds, hence providing more time for SO2 oxidation. These aged volcanic clouds have also probably resided a longer time at lower altitude thus meeting drastically different atmospheric conditions and more likely mixing with other types of aerosols.
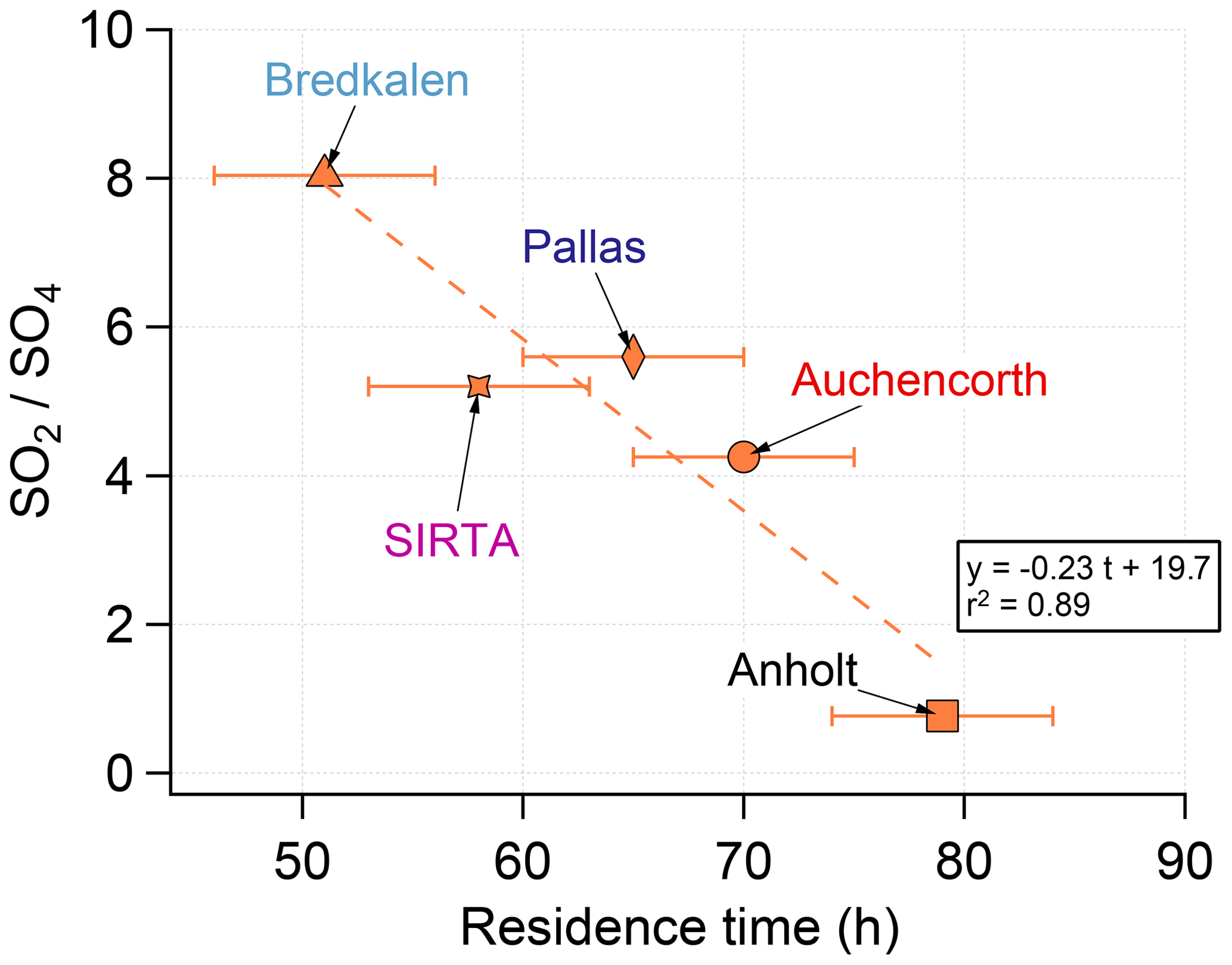
Figure 16Scatter plot of the SO2:SO4 mass concentration ratio (in PM1 fraction for ACSM data at SIRTA, PM10 for EMEP stations) with the residence time or plume age (in h) of the volcanic cloud at a selection of stations in five different countries of northern Europe displayed in Fig. 14.
To our knowledge, this dataset of SO2-to-SO4 mass ratios at a very long distance (a few thousand kilometres) from the volcanic source is unique. The significant variability in mass ratios that we observe attests of the complex atmospheric history and processes that control the oxidation of SO2 within a volcanic cloud. Nevertheless, despite this apparent complexity and the vast geographical area over which the volcanic plume is sampled, we show in Fig. 16 that the SO2-to-SO4 mass ratio evolves linearly (determination coefficient of 0.89) with t, the plume age (in hours), for stations located between 1200 and 2200 km from the eruption site, associated with plume age ranging between 50 and 80 h, as follows:
Hence, we estimate a nearly constant SO2-to-SO4 mass oxidation rate equal to 0.23 h−1. If we hypothesize that this linear relationship is also valid close to the volcanic source, we would expect a near-source SO2-to-SO4 mass ratio of ≈20. This result is in agreement with measurements performed at a few hundred kilometres from the eruption site by Ilyinskaya et al. (2017), indicating a molar ratio of S-bearing particulate matter to SO2 of 0.006–0.62 in Reykjahlíið (at 100 km distance) in January 2015 and 0.016–0.38 in Reykjavíik (at 250 km distance), corresponding to SO2-to-SO4 mass ratios of 2–250 and 4–94, respectively.
By jointly analysing OMPS and IASI satellite observations with time series of mass concentrations of SO2 and SO4 from ground-level air quality monitoring and ACSM stations, we identify the arrival of the Holuhraun SO2-rich cloud in France, triggering three noteworthy episodes of volcanogenic air pollution in September 2014. We explore the chemical signature of these volcanic events, associated with elevated values in both SO2 and SO4 surface mass concentrations, through ACSM observations at two distant French stations situated in contrasting environmental conditions. Indeed, Dunkirk hosts vigorous harbour and industrial sulfur-rich emitting activities whereas the SIRTA site, located in the Paris suburb, is influenced by urban and agricultural activities. We show that the chemical signature of Holuhraun sulfate particles is clearly distinct from background aerosols in an urban/agricultural environment. This volcanic signature is mainly characterized by a decreased concentration of particulate nitrate and organic relative to the sulfate concentration. Thermodynamic simulations with ISORROPIA II model demonstrate that the elevated concentration of sulfates recorded within volcanic clouds explains this distinct depletion in particulate nitrate as ammonia preferentially neutralizes sulfate rather than nitrate in a sulfur-rich environment. Volcanic sulfate aerosols in France are shown to be mostly neutralized by ammonium, except when recorded at a very high concentration. As a consequence, aged (neutralized) volcanic sulfates can be clearly distinguished from freshly emitted (acidic) industrial sulfates. Hence, representing scatter plots of NO3:SO4 and Org:SO4 mass concentration ratios vs. the degree of aerosol neutralization by ammonia allows for discrimination of volcanic sulfate aerosols from other types of surrounding particles, except in environments where heavy sulfur-rich pollution prevails.
Moreover, the joint analysis of ACSM sulfate ground-level mass concentration and aerosol optical depth from the AERONET sunphotometer network allows us to demonstrate in France a consecutive exceedance duration of sulfate particulate pollution of a few weeks, much longer than SO2 gas pollution (a few days at most).
In addition, the analysis of SO2 and SO4 ground-level mass concentrations from 27 stations of the EMEP network shows that the Holuhraun atmospheric pollution is not restricted to France but is spread more broadly in Europe up to the north of Scandinavia. Based on a multi-site concentration-weighted trajectory analysis, we identify the Holuhraun eruption as the major source of widespread persisting exceedance anomalies in SO2 and SO4 mass concentration at ground level. This volcanogenic pollution in SO4 is distinguished from the additional contribution of distant anthropogenic SO4 emissions from eastern Europe and Great Britain.
We describe a wide range of volcanic SO2 to sulfate mass concentration ratios at EMEP stations located at a distance of a few thousands of kilometres from the eruption site, reflecting the complex atmospheric history of volcanic clouds. In spite of an apparent spatial complexity, we highlight that the SO2-to-SO4 mass concentration ratio evolves following a simple linear dependency with the age of the plume, allowing us to estimate a SO2-to-SO4 mass oxidation rate of 0.23 h−1.
Low-tropospheric aerosols of volcanic origin can modify the microphysical properties of clouds (e.g. Schmidt et al., 2010; Yuan et al., 2011; Schmidt et al., 2012; Malavelle et al., 2017). As we show here that volcanic sulfate aerosol pollution can broadly persist over weeks in the lower troposphere, even in the planetary boundary layer, this volcanogenic indirect effect should be all the more important. While the Holuhraun eruption is of particular interest for studying such atmospheric effects given its 6-month long duration, many other tropospheric eruptions, albeit of lesser magnitude, and passive degassing activities of numerous volcanoes worldwide, are expected to collectively impact the background load of aerosols in the troposphere. More studies should address the cumulative effect of volcanoes emitting into the troposphere in order to better understand their influence on atmospheric chemistry, large-scale atmospheric pollution and climate.
OMPS satellite observations, data from air quality stations in France and in Europe (EMEP network) as well as AERONET measurements are publicly available from the NASA Goddard Earth Sciences Data and Information Services Center, Atmo Hauts-de-France, Airparif, EBAS and AERONET websites. IASI SO2 satellite observations can be provided on demand. While ACSM data for SIRTA are available on the EBAS website (http://ebas.nilu.no/, last access: 14 November 2019), data for Dunkirk can be provided on request.
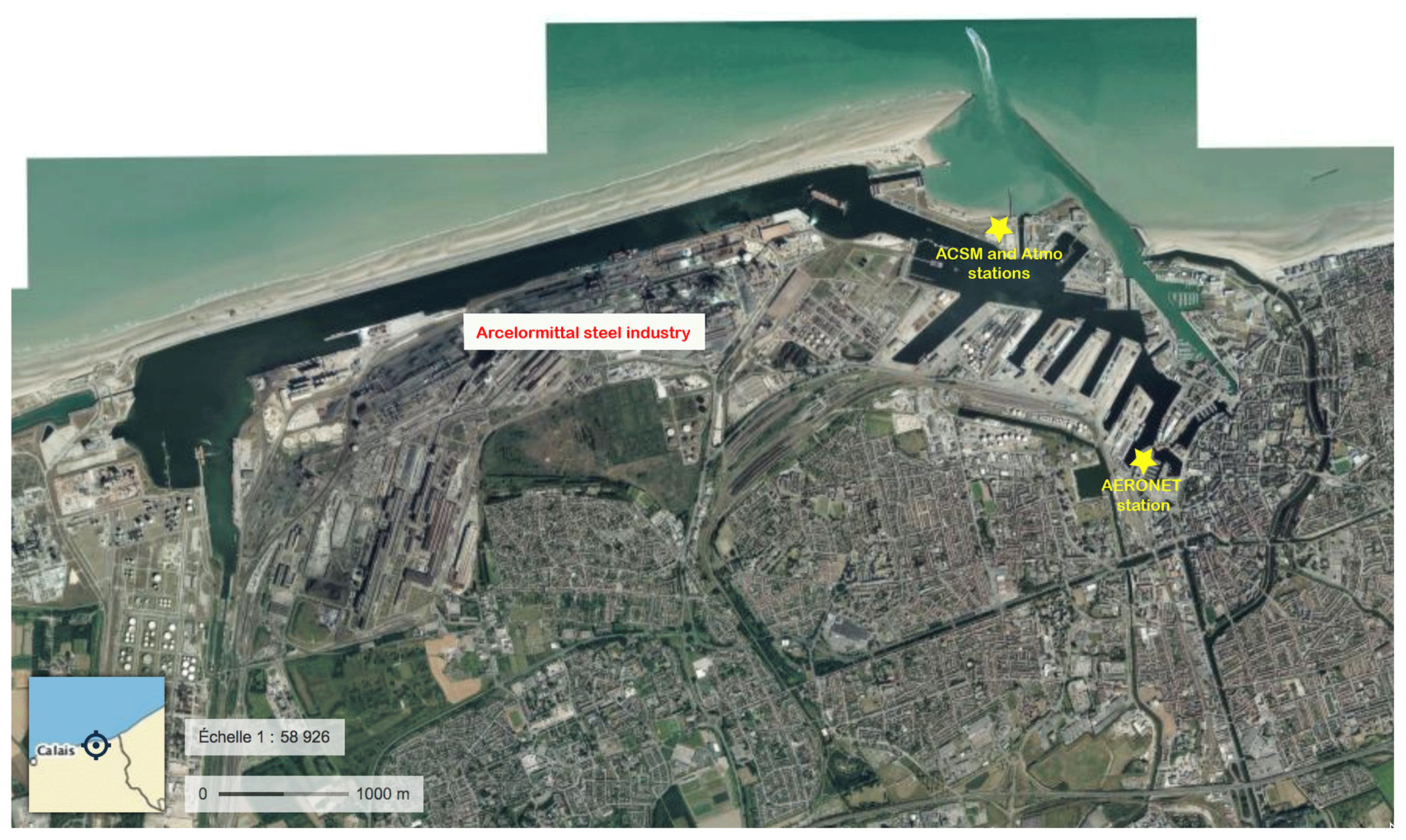
Figure A1Location in Dunkirk of the ACSM and Atmo stations at Port-East as well as the AERONET station. The aerial image used as a base map is from the Geoportail of the French government (https://www.geoportail.gouv.fr, last access: 14 November 2019). Note that the Arcelormittal site is the only one mentioned on the map as it represents the largest source of particles from the steel industry in Dunkirk, well ahead of the other industrial activities, according to Clerc et al. (2012).

Figure A2(a) Local wind speed and direction, mass concentrations of (b) black carbon and (c) ACSM sulfate, nitrate and organic aerosols in Dunkirk from 1 to 11 September 2014.
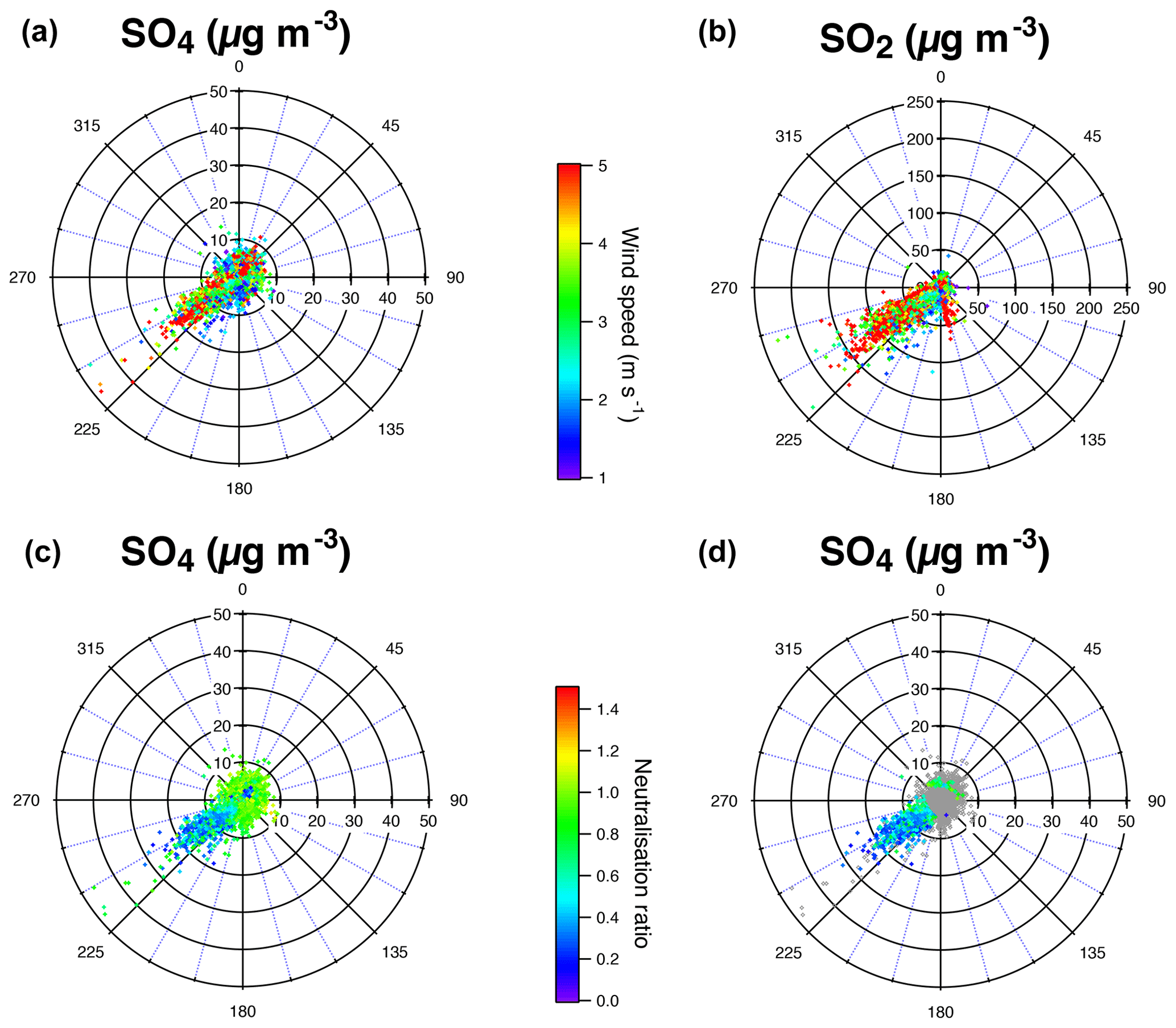
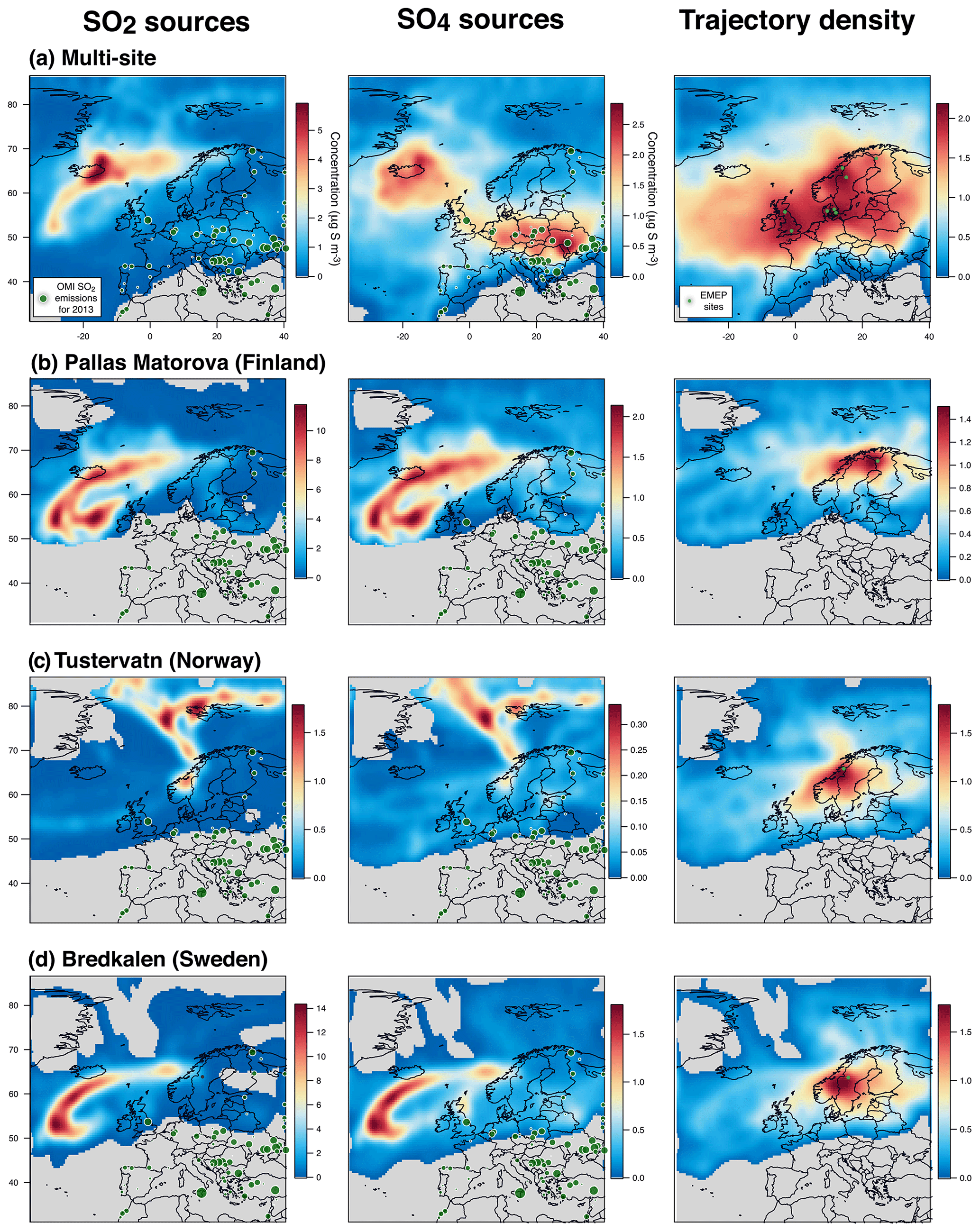
Figure A4Concentration-weighted trajectory analysis with either (a) a multi-site approach considering all eight selected EMEP stations in five countries of northern Europe listed in Table 1 or (b, c, d) each of the selected EMEP stations individually (here b – Pallas Matorova (Finland), c – Tustervatn (Norway), d – Bredkälen (Sweden), other stations in Fig. A5): retrieved source mass concentrations (µg S m−3) of (left) SO2 and (middle) SO4, (right) trajectory density (log of residence time, no unit) including station location (light green circles). SO2 emission sources for 2013 derived from OMI satellite sensor observations (from Fioletov et al., 2016) are indicated by dark green circles on the maps of SO2 and SO4 sources.
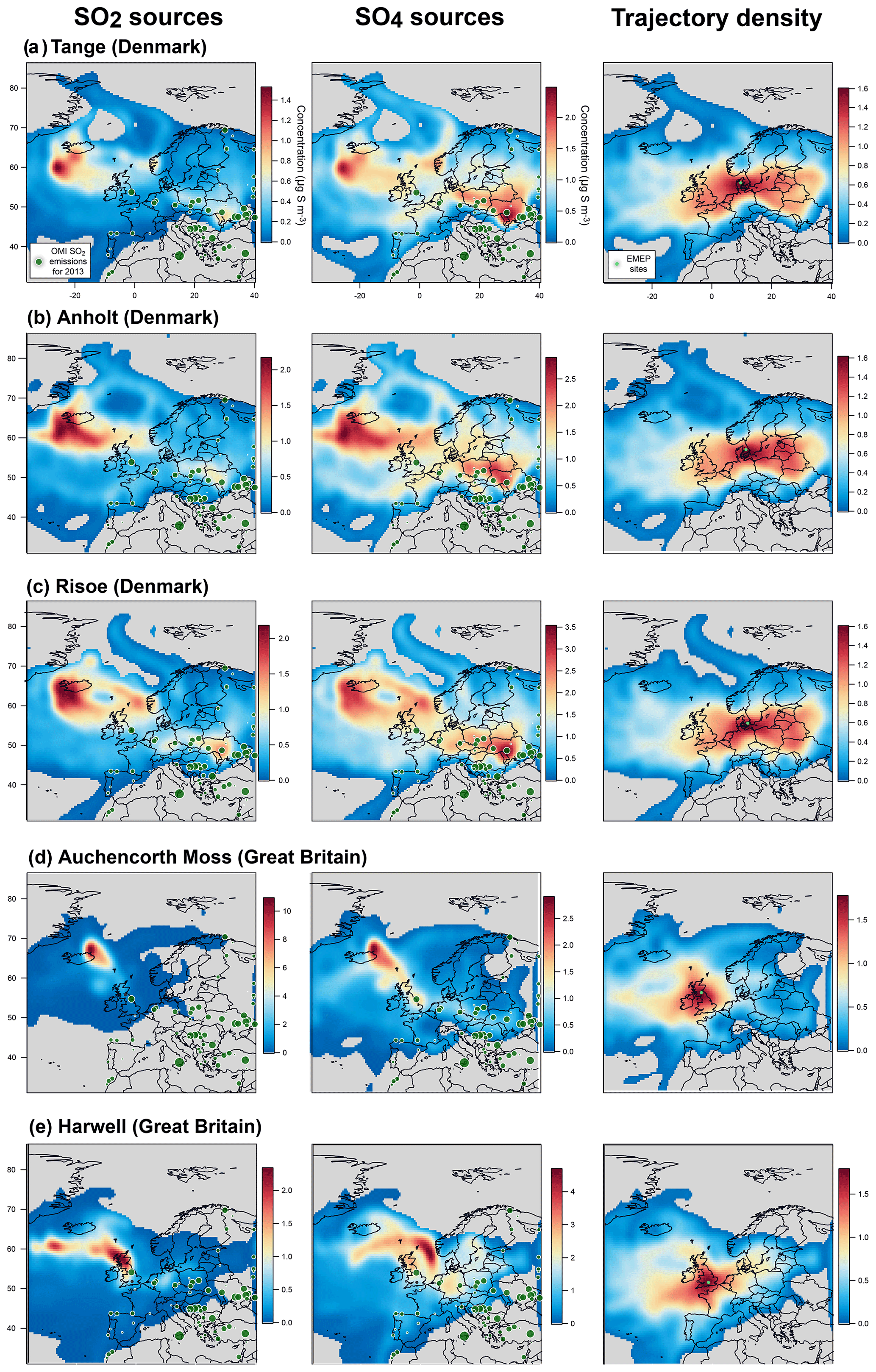
Figure A5Same as Fig. A4 for EMEP stations in Denmark (Tange – a, Anholt – b, Risoe – c) and Great Britain (Auchencorth Moss – d and Harwell – e).
Two movies illustrating the tropospheric dispersal of the Holuhraun volcanic cloud in September and October 2014 are available in the Supplement, the first including IASI SO2 column amount and altitude observations (https://doi.org/10.5446/40473, Boichu, 2019a) and the second OMPS SO2 satellite observations (https://doi.org/10.5446/40472, Boichu, 2019b).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-14253-2019-supplement.
MB (text, analysis and interpretation of satellite, ACSM, EMEP, air quality monitoring, AERONET data and thermodynamical model simulations), OF (ACSM data acquisition at SIRTA, interpretation and contribution to text), VR (ACSM data acquisition at Dunkirk and interpretation), CB (analysis of AERONET AOD time series and discussions on the overall manuscript), JEP (multi-site concentration-weighted trajectory analysis), YZ (ISORROPIA thermodynamical model simulations), JS (ACSM data acquisition at SIRTA), IC (participation to AERONET data analysis), LC (processing of IASI SO2 observations), SZ (initial processing of ACSM data in Dunkirk, air quality monitoring data acquisition at Dunkirk Port-East station), NPS (air quality monitoring data acquisition in Hauts-de-France region), ET (ACSM data acquisition at Dunkirk), HD and PG (PIs of the AERONET data acquisition at Dunkirk and SIRTA, respectively).
The authors declare that they have no conflict of interest.
Marie Boichu, Isabelle Chiapello and Colette Brogniez gratefully acknowledge support from the French National Research Agency (ANR) for funding the VOLCPLUME project (ANR-15-CE04-0003-01) and the Chantier Arctique for funding the PARCS project (Pollution in the ARCtic System). This work is a contribution to the CaPPA project (Chemical and Physical Properties of the Atmosphere), funded by the ANR through the PIA (Programme d'Investissement d'Avenir) under contract ANR-11-LABX-0005-01 and by the Regional Council of Hauts-de-France and the European Funds for Regional Economic Development (FEDER) and to the CPER Climibio program.
Aerosol measurements at SIRTA have been performed in the framework of the EU-FP7 and H2020 ACTRIS projects (grant agreement nos. 262254 and 654109). They are also supported by CNRS and the Ministry of Environment through the French reference laboratory for air quality monitoring (LCSQA). Lieven Clarisse is a research associate supported by the Belgian F.R.S.-FNRS.
Researchers and agencies in charge of air quality monitoring networks (Atmo NPDC (now Atmo Hauts-de-France), Airparif and EMEP) provided invaluable observations and are gratefully thanked. In particular, we thank the following EMEP data providers: Ulla Makkonen and Mika Vestenius (Finnish Meteorological Institute, FMI, Atmospheric Composition Unit, Helsinki, Finland) for Pallas-Matorova station in Finland; Wenche Aas and Anne Hjellbrekke (Norwegian Institute for Air Research, NILU, Atmosphere and Climate Department, Kjeller, Norway) for Tustervatn station in Norway; Karin Sjoberg (Swedish Environmental Research Institute, IVL, Göteborg, Sweden) for Bredkälen station in Sweden; Thomas Ellermann, Christian Monies and Rune Keller (Aarhus Universitet, ATAIR, ENVIS, Roskilde, Denmark) for Risoe, Anholt and Tange stations in Denmark; Christine Braban and Keith Vincent (Center for Ecology and Hydrology, Edinburgh) as well as Christopher Conolly (AEA Technology, National Environmental Techn. Centre) for Auchencorth Moss and Harwell stations in Great Britain.
We thank Hervé Delbarre and Philippe Goloub for their efforts in establishing and maintaining Dunkirk and Palaiseau AERONET sites. NASA Goddard Earth Sciences Data and Information Services Center (GES DISC) are acknowledged for providing OMPS satellite SO2 total column data. The French Government Geoportail is acknowledged for providing an aerial image of Dunkirk through its public website (https://www.geoportail.gouv.fr, last access: 14 November 2019). We thank the three anonymous reviewers for their detailed and useful reviews as well as co-editor Anja Schmidt for her comments on the manuscript.
This research has been supported by the Agence Nationale de la Recherche (VOLCPLUME project (grant no. ANR-15-CE04-0003)).
This paper was edited by Anja Schmidt and reviewed by three anonymous referees.
Aas, W., Solberg, S., and Yttri, K.: Monitoring of long-range transported air pollutants in Norway, Annual Report 2013, M-203, NILU, Kjeller, Norway, 2014. a
Aas, W., Platt, S., Solberg, S., and Yttri, K.: Monitoring of long-range transported air pollutants in Norway, Annual Report 2014, M-367, NILU, Kjeller, Norway, 2015. a
Aas, W., Fiebig, M., Platt, S., Solberg, S., and Yttri, K.: Monitoring of long-range transported air pollutants in Norway, Annual Report 2015, M-562, NILU, Kjeller, Norway, 2016. a
Allen, A. G., Oppenheimer, C., Ferm, M., Baxter, P. J., Horrocks, L. A., Galle, B., McGonigle, A. J. S., and Duffell, H. J.: Primary sulfate aerosol and associated emissions from Masaya Volcano, Nicaragua, J. Geophys. Res.-Atmos., 107, 4682, https://doi.org/10.1029/2002JD002120, 2002 a
AtmoHDF: Emission inventory of air pollutants/Inventaire des émissions de polluants de l'air, available at: https://www.atmo-hdf.fr/acceder-aux-donnees/emissions-de-polluants.html (last access: 14 November 2019), 2012. a
Bauduin, S., Clarisse, L., Clerbaux, C., Hurtmans, D., and Coheur, P.-F.: IASI observations of sulfur dioxide (SO2) in the boundary layer of Norilsk, J. Geophys. Res., 119, 4253–4263, https://doi.org/10.1002/2013JD021405, 2014. a
Biegalski, S. R. and Hopke, P. K.: Total Potential Source Contribution Function Analysis of Trace Elements Determined in Aerosol Samples Collected near Lake Huron, Environ. Sci. Technol., 38, 4276–4284, 2004. a
Boichu, M.: Animation of IASI SO2 column amount and altitude observations of the Holuhraun volcanic cloud dispersal in September and October 2014, TIB, https://doi.org/10.5446/40473, 2019a. a
Boichu, M.: Animation of OMPS SO2 satellite observations of the Holuhraun volcanic cloud dispersal in September and October 2014, TIB, https://doi.org/10.5446/40472, 2019b. a
Boichu, M., Menut, L., Khvorostyanov, D., Clarisse, L., Clerbaux, C., Turquety, S., and Coheur, P.-F.: Inverting for volcanic SO2 flux at high temporal resolution using spaceborne plume imagery and chemistry-transport modelling: the 2010 Eyjafjallajökull eruption case study, Atmos. Chem. Phys., 13, 8569–8584, https://doi.org/10.5194/acp-13-8569-2013, 2013. a
Boichu, M., Chiapello, I., Brogniez, C., Péré, J.-C., Thieuleux, F., Torres, B., Blarel, L., Mortier, A., Podvin, T., Goloub, P., Söhne, N., Clarisse, L., Bauduin, S., Hendrick, F., Theys, N., Van Roozendael, M., and Tanré, D.: Current challenges in modelling far-range air pollution induced by the 2014–2015 Bárðarbunga fissure eruption (Iceland), Atmos. Chem. Phys., 16, 10831–10845, https://doi.org/10.5194/acp-16-10831-2016, 2016. a, b, c, d, e, f
Boulon, J., Sellegri, K., Hervo, M., and Laj, P.: Observations of nucleation of new particles in a volcanic plume, P. Natl. Acad. Sci. USA, 108, 12223–12226, 2011. a
Budisulistiorini, S. H., Canagaratna, M. R., Croteau, P. L., Baumann, K., Edgerton, E. S., Kollman, M. S., Ng, N. L., Verma, V., Shaw, S. L., Knipping, E. M., Worsnop, D. R., Jayne, J. T., Weber, R. J., and Surratt, J. D.: Intercomparison of an Aerosol Chemical Speciation Monitor (ACSM) with ambient fine aerosol measurements in downtown Atlanta, Georgia, Atmos. Meas. Tech., 7, 1929–1941, https://doi.org/10.5194/amt-7-1929-2014, 2014. a
Carboni, E., Mather, T. A., Schmidt, A., Grainger, R. G., Pfeffer, M. A., Ialongo, I., and Theys, N.: Satellite-derived sulfur dioxide (SO2) emissions from the 2014–2015 Holuhraun eruption (Iceland), Atmos. Chem. Phys., 19, 4851–4862, https://doi.org/10.5194/acp-19-4851-2019, 2019. a, b
Carn, S., Krueger, A., Krotkov, N., Yang, K., and Evans, K.: Tracking volcanic sulfur dioxide clouds for aviation hazard mitigation, Nat. Hazards, 51, 325–343, https://doi.org/10.1007/s11069-008-9228-4, 2009. a
Carn, S. A., Yang, K., Prata, A. J., and Krotkov, N. A.: Extending the long-term record of volcanic SO2 emissions with the Ozone Mapping and Profiler Suite nadir mapper, Geophys. Res. Lett., 42, 925–932, https://doi.org/10.1002/2014GL062437, 2015. a
Cheng, I., Zhang, L., Blanchard, P., Dalziel, J., and Tordon, R.: Concentration-weighted trajectory approach to identifying potential sources of speciated atmospheric mercury at an urban coastal site in Nova Scotia, Canada, Atmos. Chem. Phys., 13, 6031–6048, https://doi.org/10.5194/acp-13-6031-2013, 2013. a
Chu, B., Zhang, X., Liu, Y., He, H., Sun, Y., Jiang, J., Li, J., and Hao, J.: Synergetic formation of secondary inorganic and organic aerosol: effect of SO2 and NH3 on particle formation and growth, Atmos. Chem. Phys., 16, 14219–14230, https://doi.org/10.5194/acp-16-14219-2016, 2016. a
Clarisse, L., Coheur, P.-F., Theys, N., Hurtmans, D., and Clerbaux, C.: The 2011 Nabro eruption, a SO2 plume height analysis using IASI measurements, Atmos. Chem. Phys., 14, 3095–3111, https://doi.org/10.5194/acp-14-3095-2014, 2014. a
Clerc, F., Douchez, C., Gibaux, J.-P., Dedourge, J.-M., Chanu, G., and Mathon, M.-P.: L'industrie au regard de l'environnement en Nord-Pas-de-Calais, Tech. rep., DREAL (Direction régionale de l'environnement, de l'aménagement et du logement), Nord-Pas-de-Calais, 2012. a, b
Colette, A., Favez, O., Meleux, F., Chiappini, L., Haeffelin, M., Morille, Y., Malherbe, L., Papin, A., Bessagnet, B., Menut, L., Leoz, M., and Rouil, L.: Assessing in near real time the impact of the April 2010 Eyjafjallajökull ash plume on air quality, Atmos. Environ., 45, 1217–1221, https://doi.org/10.1016/j.atmosenv.2010.09.064, 2011. a
Crenn, V., Sciare, J., Croteau, P. L., Verlhac, S., Fröhlich, R., Belis, C. A., Aas, W., Äijälä, M., Alastuey, A., Artiñano, B., Baisnée, D., Bonnaire, N., Bressi, M., Canagaratna, M., Canonaco, F., Carbone, C., Cavalli, F., Coz, E., Cubison, M. J., Esser-Gietl, J. K., Green, D. C., Gros, V., Heikkinen, L., Herrmann, H., Lunder, C., Minguillón, M. C., Močnik, G., O'Dowd, C. D., Ovadnevaite, J., Petit, J.-E., Petralia, E., Poulain, L., Priestman, M., Riffault, V., Ripoll, A., Sarda-Estève, R., Slowik, J. G., Setyan, A., Wiedensohler, A., Baltensperger, U., Prévôt, A. S. H., Jayne, J. T., and Favez, O.: ACTRIS ACSM intercomparison – Part 1: Reproducibility of concentration and fragment results from 13 individual Quadrupole Aerosol Chemical Speciation Monitors (Q-ACSM) and consistency with co-located instruments, Atmos. Meas. Tech., 8, 5063–5087, https://doi.org/10.5194/amt-8-5063-2015, 2015. a, b
Delmelle, P.: Environmental impacts of tropospheric volcanic gas plumes, Geol. Soc. Spec. Publ., 213, 381–400, 2003. a, b
Ebmeier, S. K., Sayer, A. M., Grainger, R. G., Mather, T. A., and Carboni, E.: Systematic satellite observations of the impact of aerosols from passive volcanic degassing on local cloud properties, Atmos. Chem. Phys., 14, 10601–10618, https://doi.org/10.5194/acp-14-10601-2014, 2014. a
Fioletov, V. E., McLinden, C. A., Krotkov, N., Li, C., Joiner, J., Theys, N., Carn, S., and Moran, M. D.: A global catalogue of large SO2 sources and emissions derived from the Ozone Monitoring Instrument, Atmos. Chem. Phys., 16, 11497–11519, https://doi.org/10.5194/acp-16-11497-2016, 2016. a, b, c, d
Flemming, J. and Inness, A.: Volcanic sulfur dioxide plume forecasts based on UV satellite retrievals for the 2011 Grimsvötn and the 2010 Eyjafjallajökull eruptions, J. Geophys. Res., 118, 10172–10189, https://doi.org/10.1002/jgrd.50753, 2013. a
Fountoukis, C. and Nenes, A.: ISORROPIA II: a computationally efficient thermodynamic equilibrium model for K+–Ca2+–Mg2+––Na+–––Cl−–H2O aerosols, Atmos. Chem. Phys., 7, 4639–4659, https://doi.org/10.5194/acp-7-4639-2007, 2007. a, b
Freney, E., Zhang, Y., Croteau, P., Amodeo, T., Williams, L., Truong, F., Petit, J.-E., Sciare, J., Sarda-Estève, R., Bonnaire, N., Crenn, V., Arumae, T., Aurela, M., Bougiatioti, K., Coz, E., Elste, T., Heikkinen, L., Minguillon, M.-C., Poulain, L., Priestman, M., Stavroulas, I., Tobler, A., Vasilescu, J., Zanca, N., Alastuey, A., Artinano, B., Carbone, C., Flentje, H., Green, D., Herrmann, H., Maasikmets, M., Marmureanu, L., Prévôt, A. S. H., Wiedensohler, A., Canagaratna, M., Gros, V., Jayne, J. T., and Favez, O.: The second ACTRIS inter-comparison (2016) for Aerosol Chemical Speciation Monitors (ACSM): Calibration protocols and instrument performance evaluations, Aerosol Sci. Tech., 53, 830–842, 2019. a, b, c
Galeazzo, T., Bekki, S., Martin, E., Savarino, J., and Arnold, S. R.: Photochemical box modelling of volcanic SO2 oxidation: isotopic constraints, Atmos. Chem. Phys., 18, 17909–17931, https://doi.org/10.5194/acp-18-17909-2018, 2018. a
Gassó, S.: Satellite observations of the impact of weak volcanic activity on marine clouds, J. Geophys. Res.-Atmos., 113, D14S19, https://doi.org/10.1029/2007JD009106, 2008. a
Haeffelin, M., Barthès, L., Bock, O., Boitel, C., Bony, S., Bouniol, D., Chepfer, H., Chiriaco, M., Cuesta, J., Delanoë, J., Drobinski, P., Dufresne, J.-L., Flamant, C., Grall, M., Hodzic, A., Hourdin, F., Lapouge, F., Lemaître, Y., Mathieu, A., Morille, Y., Naud, C., Noël, V., O'Hirok, W., Pelon, J., Pietras, C., Protat, A., Romand, B., Scialom, G., and Vautard, R.: SIRTA, a ground-based atmospheric observatory for cloud and aerosol research, Ann. Geophys., 23, 253–275, https://doi.org/10.5194/angeo-23-253-2005, 2005. a
Hatakka, J., Aalto, T., Aaltonen, V., Aurela, M., Hakola, H., Komppula, M., and Viisanen, Y.: Overview of the atmospheric research activities and results at Pallas GAW station, Boreal Environ. Res., 8, 365–384, 2003. a
Holben, B., Smirnov, A., Eck, T., Slutsker, I., Abuhassan, N., Newcomb, W., Schafer, J., Tanre, D., Chatenet, B., and Lavenu, F.: An emerging ground-based aerosol climatology- Aerosol optical depth from AERONET, J. Geophys. Res., 106, 12067–12097, 2001. a, b
Ialongo, I., Hakkarainen, J., Kivi, R., Anttila, P., Krotkov, N. A., Yang, K., Li, C., Tukiainen, S., Hassinen, S., and Tamminen, J.: Comparison of operational satellite SO2 products with ground-based observations in northern Finland during the Icelandic Holuhraun fissure eruption, Atmos. Meas. Tech., 8, 2279–2289, https://doi.org/10.5194/amt-8-2279-2015, 2015. a, b
ICAO: Volcanic Ash Contingency Plan – European and North Atlantic Regions, European and North Atlantic Office of the International Civil Organization, EUR Doc 019, NAT Doc 006, Part II, European and North Atlantic (EUR/NAT) Office of the International Civil Aviation Organization (ICAO), Neuilly-sur-Seine, France, 2016. a
Ilyinskaya, E., Schmidt, A., Mather, T. A., Pope, F. D., Witham, C., Baxter, P., Jóhannsson, T., Pfeffer, M., Barsotti, S., Singh, A., Sanderson, P., Bergsson, B., McCormick Kilbride, B., Donovan, A., Peters, N., Oppenheimer, C., and Edmonds, M.: Understanding the environmental impacts of large fissure eruptions: Aerosol and gas emissions from the 2014–2015 Holuhraun eruption (Iceland), Earth Planet. Sc. Lett., 472, 309–322, 2017. a, b
Kroll, J. H., Cross, E. S., Hunter, J. F., Pai, S., XII, T., TREX XI Wallace, M., Croteau, P., Jayne, J., Worsnop, D. R., Heald, C. L., Murphy, J., and Frankel, S.: Atmospheric evolution of volcanic smog (“vog”) from Kilauea: real-time measurements of oxidation, dilution and neutralization within a volcanic plume, Environ. Sci. Technol., 49, 4129–4137, 2015. a
Krotkov, N., McClure, B., Dickerson, R., Carn, S. A., Li, C., Bhartia, P. K., Yang, K., Krueger, A., and Li, Z.: Validation of SO2 retrievals from the Ozone Monitoring Instrument over NE China, J. Geophys. Res., 113, D16S40, https://doi.org/10.1029/2007JD008818, 2008. a
Kuebbeler, M., Lohmann, U., and Feichter, J.: Effects of stratospheric sulfate aerosol geo-engineering on cirrus clouds, Geophys. Res. Lett., 39, L23803, https://doi.org/10.1029/2012GL053797, 2012. a
Law, K. S. and Stohl, A.: Arctic air pollution: Origins and impacts, Science, 315, 1537–1540, 2007. a
Longo, B., Rossignol, A., and Green, J.: Cardiorespiratory health effects associated with sulphurous volcanic air pollution, Public Health, 122, 809–820, 2008. a
Malavelle, F. F., Haywood, J. M., Jones, A., Gettelman, A., Clarisse, L., Bauduin, S., Allan, R., Karset, I., Kristjansson, J., Oreopoulous, L., Cho, N., Lee, D., Bellouin, N., Boucher, O., Grosvenor, D., Carslaw, K., Dhomse, S., Mann, G., Schmidt, A., Coe, H., Hartley, M., Dalvi, M., Hill, A., Johnson, B. T., Johnson, C., Knight, J., O'Connor, F., Partridge, D., Stier, P., Myhre, G., Platnick, S., Stephens, G., Takahashi, H., and Thordarson, T.: Strong constraints on aerosol–cloud interactions from volcanic eruptions, Nature, 546, 485–491, https://doi.org/10.1038/nature22974, 2017. a, b, c
Martin, R. S., Ilyinskaya, E., Sawyer, G. M., Tsanev, V. I., and Oppenheimer, C.: A re-assessment of aerosol size distributions from Masaya volcano (Nicaragua), Atmos. Environ., 45, 547–560, 2011. a
Mather, T. A., Allen, A. G., Oppenheimer, C., Pyle, D. M., and McGonigle, A. J. S.: Size-resolved characterisation of soluble ions in the particles in the tropospheric plume of Masaya volcano, Nicaragua: Origins and plume processing, J. Atmos. Chem., 46, 207–237, 2003a. a
Mather, T. A., Pyle, D. M., and Oppenheimer, C.: Tropospheric volcanic aerosol, Geophys. Monogr. Ser., 139, 189–212, 2003b. a
Mather, T. A., Tsanev, V. I., Pyle, D. M., McGonigle, A. J. S., Oppenheimer, C., and Allen, A. G.: Characterization and evolution of tropospheric plumes from Lascar and Villarrica volcanoes, Chile, J. Geophys. Res.-Atmos., 109, D21303, https://doi.org/10.1029/2004JD004934, 2004. a
McCoy, D. T. and Hartmann, D. L.: Observations of a substantial cloud-aerosol indirect effect during the 2014–2015 Bárðarbunga-Veiðivötn fissure eruption in Iceland, Geophys. Res. Lett., 42, 10409–10414, 2015. a, b
McNeill, V. F., Woo, J. L., Kim, D. D., Schwier, A. N., Wannell, N. J., Sumner, A. J., and Barakat, J. M.: Aqueous-phase secondary organic aerosol and organosulfate formation in atmospheric aerosols: a modeling study, Environ. Sci. Technol., 46, 8075–8081, 2012. a
Middlebrook, A. M., Bahreini, R., Jimenez, J. L., and Canagaratna, M. R.: Evaluation of Composition-Dependent Collection Efficiencies for the Aerodyne Aerosol Mass Spectrometer using Field Data, Aerosol Sci. Tech., 46, 258–271, 2012. a
Moxnes, E., Kristiansen, N., Stohl, A., Clarisse, L., Durant, A., Weber, K., and Vogel, A.: Separation of ash and sulfur dioxide during the 2011 Grímsvötn eruption, J. Geophys. Res., 119, 7477–7501, https://doi.org/10.1002/2013JD021129, 2014. a
Neely, R., Toon, O., Solomon, S., Vernier, J.-P., Alvarez, C., English, J., Rosenlof, K., Mills, M., Bardeen, C., Daniel, J. S., and Thayer, J. P.: Recent anthropogenic increases in SO2 from Asia have minimal impact on stratospheric aerosol, Geophys. Res. Lett., 40, 999–1004, https://doi.org/10.1002/grl.50263, 2013. a
Ng, N. L., Herndon, S. C., Trimborn, A., Canagaratna, M. R., Croteau, P. L., Onasch, T. B., Sueper, D., Worsnop, D. R., Zhang, Q., Sun, Y. L., and Jayne, J. T.: An Aerosol Chemical Speciation Monitor (ACSM) for Routine Monitoring of the Composition and Mass Concentrations of Ambient Aerosol, Aerosol Sci. Tech., 45, 780–794, 2011. a, b
NILU: EMEP manual for sampling and chemical analysis, Tech. rep., Norwegian Institute for Air Research, available at: https://projects.nilu.no//ccc/manual/index.html (last access: 14 November 2019), 2014. a
Oppenheimer, C., Scaillet, B., and Martin, R.: Sulfur Degassing From Volcanoes: Source Conditions, Surveillance, Plume Chemistry and Earth System Impacts, Rev. Mineral. Geochem., 73, 363–421, 2011. a, b
Ovadnevaite, J., Ceburnis, D., Canagaratna, M., Berresheim, H., Bialek, J., Martucci, G., Worsnop, D. R., and O'Dowd, C.: On the effect of wind speed on submicron sea salt mass concentrations and source fluxes, J. Geophys. Res.-Atmos., 117, D16201, https://doi.org/10.1029/2011JD017379, 2012. a
Pathak, R. K., Wang, T., Ho, K., and Lee, S.: Characteristics of summertime PM2.5 organic and elemental carbon in four major Chinese cities: Implications of high acidity for water-soluble organic carbon (WSOC), Atmos. Environ., 45, 318–325, 2011. a
Pattantyus, A., Businger, S., and Howell, S.: Review of sulfur dioxide to sulfate aerosol chemistry at Kilauea Volcano, Hawai'i, Atmos. Environ., 185, 262–271, 2018. a, b, c
Perri, M. J., Lim, Y. B., Seitzinger, S. P., and Turpin, B. J.: Organosulfates from glycolaldehyde in aqueous aerosols and clouds: Laboratory studies, Atmos. Environ., 44, 2658–2664, 2010. a
Petetin, H., Sciare, J., Bressi, M., Gros, V., Rosso, A., Sanchez, O., Sarda-Estève, R., Petit, J.-E., and Beekmann, M.: Assessing the ammonium nitrate formation regime in the Paris megacity and its representation in the CHIMERE model, Atmos. Chem. Phys., 16, 10419–10440, https://doi.org/10.5194/acp-16-10419-2016, 2016. a
Petit, J.-E., Favez, O., Sciare, J., Crenn, V., Sarda-Estève, R., Bonnaire, N., Močnik, G., Dupont, J.-C., Haeffelin, M., and Leoz-Garziandia, E.: Two years of near real-time chemical composition of submicron aerosols in the region of Paris using an Aerosol Chemical Speciation Monitor (ACSM) and a multi-wavelength Aethalometer, Atmos. Chem. Phys., 15, 2985–3005, https://doi.org/10.5194/acp-15-2985-2015, 2015. a
Petit, J.-E., Favez, O., Albinet, A., and Canonaco, F.: A user-friendly tool for comprehensive evaluation of the geographical origins of atmospheric pollution: Wind and trajectory analyses, Environ. Modell. Softw., 88, 183–187, 2017. a
Radke, L.: Chlorine, fluorine, and sulfur emissions from Mount Erebus, Antarctica and estimated contributions to the Antarctic atmosphere, Nature, 299, 710–712, 1982. a
Ridley, D., Solomon, S., Barnes, J., Burlakov, V., Deshler, T., Dolgii, S., Herber, A. B., Nagai, T., Neely, R., Nevzorov, A. V., Ritter, C., Sakai, T., Santer, B. D., Sato, M., Schmidt, A., Uchino, O., and Vernier, J. P.: Total volcanic stratospheric aerosol optical depths and implications for global climate change, Geophys. Res. Lett., 41, 7763–7769, https://doi.org/10.1002/2014GL061541, 2014. a
Robock, A.: Volcanic eruptions and climate, Rev. Geophys., 38, 191–220, https://doi.org/10.1029/1998RG000054, 2000. a
Roig Rodelas, R., Perdrix, E., Herbin, B., and Riffault, V.: Characterization and variability of inorganic aerosols and their gaseous precursors at a suburban site in northern France over one year (2015–2016), Atmos. Environ., 200, 142–157, 2019. a
Santer, B. D., Bonfils, C., Painter, J. F., Zelinka, M. D., Mears, C., Solomon, S., Schmidt, G. A., Fyfe, J. C., Cole, J. N., Nazarenko, L., Taylor, K. E., and Wentz, F. J.: Volcanic contribution to decadal changes in tropospheric temperature, Nat. Geosci., 7, 185–189, 2014. a
Schlag, P., Rubach, F., Mentel, T. F., Reimer, D., Canonaco, F., Henzing, J. S., Moerman, M., Otjes, R., Prevot, A., Rohrer, F., Rosati, B., Tillmann, R., Weingartner, E., and Kiendler-Scharr, A.: Ambient and laboratory observations of organic ammonium salts in PM1, Faraday Discuss., 200, 331–351, 2017. a
Schmidt, A., Carslaw, K. S., Mann, G. W., Wilson, M., Breider, T. J., Pickering, S. J., and Thordarson, T.: The impact of the 1783–1784 AD Laki eruption on global aerosol formation processes and cloud condensation nuclei, Atmos. Chem. Phys., 10, 6025–6041, https://doi.org/10.5194/acp-10-6025-2010, 2010. a
Schmidt, A., Carslaw, K. S., Mann, G. W., Rap, A., Pringle, K. J., Spracklen, D. V., Wilson, M., and Forster, P. M.: Importance of tropospheric volcanic aerosol for indirect radiative forcing of climate, Atmos. Chem. Phys., 12, 7321–7339, https://doi.org/10.5194/acp-12-7321-2012, 2012. a, b
Schmidt, A., Leadbetter, S., Theys, N., Carboni, E., Witham, C. S., Stevenson, J. A., Birch, C. E., Thordarson, T., Turnock, S., Barsotti, S., Delaney, L., Feng, W., Grainger, R. G., Hort, M. C., Höskuldsson, Á., Ialongo, I., Ilyinskaya, E., Jóhannsson, T., Kenny, P., Mather, T. A., Richards, N. A. D., and Shepherd, J.: Satellite detection, long-range transport, and air quality impacts of volcanic sulfur dioxide from the 2014–2015 flood lava eruption at Bárðarbunga (Iceland), J. Geophys. Res.-Atmos., 120, 9739–9757, 2015. a, b, c
Schmidt, A., Mills, M. J., Ghan, S., Gregory, J. M., Allan, R. P., Andrews, T., Bardeen, C. G., Conley, A., Forster, P. M., Gettelman, A., Portmann, R. W., Solomon, S., and Toon, O. B.: Volcanic radiative forcing from 1979 to 2015, J. Geophys. Res.-Atmos., 123, 12–491, 2018. a
Seinfeld, J. and Pandis, S. N.: Atmospheric chemistry and physics: from air pollution to climate change, John Wiley and Sons, New Jersey, USA, 2012. a, b, c, d, e, f
Solomon, S., Daniel, J., Neely, R., Vernier, J.-P., Dutton, E., and Thomason, L.: The persistently variable background stratospheric aerosol layer and global climate change, Science, 333, 866–870, 2011. a
Steensen, B. M., Schulz, M., Theys, N., and Fagerli, H.: A model study of the pollution effects of the first 3 months of the Holuhraun volcanic fissure: comparison with observations and air pollution effects, Atmos. Chem. Phys., 16, 9745–9760, https://doi.org/10.5194/acp-16-9745-2016, 2016. a, b
Stein, A., Draxler, R. R., Rolph, G. D., Stunder, B. J., Cohen, M., and Ngan, F.: NOAA's HYSPLIT atmospheric transport and dispersion modeling system, B. Am. Meteorol. Soc., 96, 2059–2077, https://doi.org/10.1175/BAMS-D-14-00110.1, 2015. a
Stevenson, D., Johnson, C., Collins, W., and Derwent, R.: The tropospheric sulphur cycle and the role of volcanic SO2, Geol. Soc. Spec. Publ., 213, 295–305, 2003. a
Surratt, J. D., Gómez-González, Y., Chan, A. W., Vermeylen, R., Shahgholi, M., Kleindienst, T. E., Edney, E. O., Offenberg, J. H., Lewandowski, M., Jaoui, M., Maenhaut, W., Claeys, M., Flagan, R. C., and Seinfeld, J. H.: Organosulfate formation in biogenic secondary organic aerosol, J. Phys. Chem., 112, 8345–8378, 2008. a
Targino, A. C., Krecl, P., Johansson, C., Swietlicki, E., Massling, A., Coraiola, G. C., and Lihavainen, H.: Deterioration of air quality across Sweden due to transboundary agricultural burning emissions, Boreal Environ. Res., 18, 19–36, 2013. a
Theys, N., Campion, R., Clarisse, L., Brenot, H., van Gent, J., Dils, B., Corradini, S., Merucci, L., Coheur, P.-F., Van Roozendael, M., Hurtmans, D., Clerbaux, C., Tait, S., and Ferrucci, F.: Volcanic SO2 fluxes derived from satellite data: a survey using OMI, GOME-2, IASI and MODIS, Atmos. Chem. Phys., 13, 5945–5968, https://doi.org/10.5194/acp-13-5945-2013, 2013. a
Thordarson, T. and Self, S.: Atmospheric and environmental effects of the 1783–1784 Laki eruption: A review and reassessment, J. Geophys. Res., 108, 4011, https://doi.org/10.1029/2001JD002042, 2003. a, b, c
Tørseth, K., Aas, W., Breivik, K., Fjæraa, A. M., Fiebig, M., Hjellbrekke, A. G., Lund Myhre, C., Solberg, S., and Yttri, K. E.: Introduction to the European Monitoring and Evaluation Programme (EMEP) and observed atmospheric composition change during 1972–2009, Atmos. Chem. Phys., 12, 5447–5481, https://doi.org/10.5194/acp-12-5447-2012, 2012. a
Tu, F., Thornton, D., Bandy, A., Carmichael, G., Tang, Y., Thornhill, K., Sachse, G., and Blake, D.: Long-range transport of sulfur dioxide in the central Pacific, J. Geophys. Res.-Atmos., 109, D15S08, https://doi.org/10.1029/2003JD004309, 2004. a
Twigg, M. M., Ilyinskaya, E., Beccaceci, S., Green, D. C., Jones, M. R., Langford, B., Leeson, S. R., Lingard, J. J. N., Pereira, G. M., Carter, H., Poskitt, J., Richter, A., Ritchie, S., Simmons, I., Smith, R. I., Tang, Y. S., Van Dijk, N., Vincent, K., Nemitz, E., Vieno, M., and Braban, C. F.: Impacts of the 2014–2015 Holuhraun eruption on the UK atmosphere, Atmos. Chem. Phys., 16, 11415–11431, https://doi.org/10.5194/acp-16-11415-2016, 2016. a, b
van Manen, S. M.: Perception of a chronic volcanic hazard: persistent degassing at Masaya volcano, Nicaragua, J. Appl. Volcanol., 3, 1–16, 2014. a, b
Vernier, J.-P., Thomason, L. W., Pommereau, J.-P., Bourassa, A., Pelon, J., Garnier, A., Hauchecorne, A., Blanot, L., Trepte, C., Degenstein, D., and Vargas, F.: Major influence of tropical volcanic eruptions on the stratospheric aerosol layer during the last decade, Geophys. Res. Lett., 38, 12807, https://doi.org/10.1029/2011GL047563, 2011. a
von Glasow, R. and Crutzen, P.: Tropospheric Halogen Chemistry, in: Treatise on Geochemistry – The Atmosphere, edited by: Keeling, R., Holland, H., and Turekion, K., Vol. 4, p. 347, Elsevier/Pergamon, Oxford, 2003. a
Waked, A., Favez, O., Alleman, L. Y., Piot, C., Petit, J.-E., Delaunay, T., Verlinden, E., Golly, B., Besombes, J.-L., Jaffrezo, J.-L., and Leoz-Garziandia, E.: Source apportionment of PM10 in a north-western Europe regional urban background site (Lens, France) using positive matrix factorization and including primary biogenic emissions, Atmos. Chem. Phys., 14, 3325–3346, https://doi.org/10.5194/acp-14-3325-2014, 2014. a
Witham, C., Aspinall, W., Braban, C., Hall, J., Loughlin, S., Schmidt, A., Vieno, M., Bealey, B., Hort, M., Ilyinskaya, E., Kentisbeer, J., Roberts, E., and Rowe, E.: UK hazards from a large Icelandic effusive eruption, effusive eruption modelling project final report, Effusive eruption modelling project final report, Met Office, Exeter, UK, 2015. a, b
Xu, W., Lambe, A., Silva, P., Hu, W., Onasch, T., Williams, L., Croteau, P., Zhang, X., Renbaum-Wolff, L., Fortner, E., Jimenez, J. L., Jayne, J. T., Worsnop, D., and Canagaratna, M.: Laboratory evaluation of species-dependent relative ionization efficiencies in the Aerodyne Aerosol Mass Spectrometer, Aerosol Sci. Tech., 52, 626–641, 2018. a
Yuan, T., Remer, L. A., and Yu, H.: Microphysical, macrophysical and radiative signatures of volcanic aerosols in trade wind cumulus observed by the A-Train, Atmos. Chem. Phys., 11, 7119–7132, https://doi.org/10.5194/acp-11-7119-2011, 2011. a, b
Zaveri, R. A., Berkowitz, C. M., Brechtel, F. J., Gilles, M. K., Hubbe, J. M., Jayne, J. T., Kleinman, L. I., Laskin, A., Madronich, S., Onasch, T. B., Pekour, M. S., Springston, S. R., Thornton, J. A., Tivanski, A. V., and Worsnop, D. R.: Nighttime chemical evolution of aerosol and trace gases in a power plant plume: Implications for secondary organic nitrate and organosulfate aerosol formation, NO3 radical chemistry, and N2O5 heterogeneous hydrolysis, J. Geophys. Res.-Atmos., 115, D12304, https://doi.org/10.1029/2009JD013250, 2010. a, b
Zelenski, M., Taran, Y., and Galle, B.: High emission rate of sulfuric acid from Bezymianny volcano, Kamchatka, Geophys. Res. Lett., 42, 7005–7013, 2015. a
Zhang, Q., Jimenez, J. L., Worsnop, D. R., and Canagaratna, M.: A case study of urban particle acidity and its influence on secondary organic aerosol, Environ. Sci. Technol., 41, 3213–3219, 2007. a
Zhang, S.: Analyse dynamique, en champ proche et à résolution temporelle fine, de l'aérosol submicronique en situation urbaine sous influence industrielle, PhD thesis, Université du Littoral Côte d'Opale, 2016. a, b, c, d