the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 22 Nov 2019
| 22 Nov 2019
Novel approaches to improve estimates of short-lived halocarbon emissions during summer from the Southern Ocean using airborne observations
Elizabeth Asher
Rebecca S. Hornbrook
Britton B. Stephens
Doug Kinnison
Eric J. Morgan
Ralph F. Keeling
Elliot L. Atlas
Sue M. Schauffler
Simone Tilmes
Eric A. Kort
Martin S. Hoecker-Martínez
Matt C. Long
Jean-François Lamarque
Alfonso Saiz-Lopez
Kathryn McKain
Colm Sweeney
Alan J. Hills
Eric C. Apel
Fluxes of halogenated volatile organic compounds (VOCs) over the Southern Ocean remain poorly understood, and few atmospheric measurements exist to constrain modeled emissions of these compounds. We present observations of CHBr3, CH2Br2, CH3I, CHClBr2, CHBrCl2, and CH3Br during the O2∕N2 Ratio and CO2 Airborne Southern Ocean (ORCAS) study and the second Atmospheric Tomography mission (ATom-2) in January and February of 2016 and 2017. Good model–measurement correlations were obtained between these observations and simulations from the Community Earth System Model (CESM) atmospheric component with chemistry (CAM-Chem) for CHBr3, CH2Br2, CH3I, and CHClBr2 but all showed significant differences in model : measurement ratios. The model : measurement comparison for CH3Br was satisfactory and for CHBrCl2 the low levels present precluded us from making a complete assessment. Thereafter, we demonstrate two novel approaches to estimate halogenated VOC fluxes; the first approach takes advantage of the robust relationships that were found between airborne observations of O2 and CHBr3, CH2Br2, and CHClBr2. We use these linear regressions with O2 and modeled O2 distributions to infer a biological flux of halogenated VOCs. The second approach uses the Stochastic Time-Inverted Lagrangian Transport (STILT) particle dispersion model to explore the relationships between observed mixing ratios and the product of the upstream surface influence of sea ice, chl a, absorption due to detritus, and downward shortwave radiation at the surface, which in turn relate to various regional hypothesized sources of halogenated VOCs such as marine phytoplankton, phytoplankton in sea-ice brines, and decomposing organic matter in surface seawater. These relationships can help evaluate the likelihood of particular halogenated VOC sources and in the case of statistically significant correlations, such as was found for CH3I, may be used to derive an estimated flux field. Our results are consistent with a biogenic regional source of CHBr3 and both nonbiological and biological sources of CH3I over these regions.
- Article
(9454 KB) - Full-text XML
-
Supplement
(3152 KB) - BibTeX
- EndNote




Emissions of halogenated volatile organic compounds (VOCs) influence regional atmospheric chemistry and global climate. Through the production of reactive halogen radicals at high latitudes, halogenated VOCs contribute to tropospheric and stratospheric ozone destruction and alter the sulfur, mercury, nitrogen oxide, and hydrogen oxide cycles (e.g., Schroeder et al., 1998; Boucher et al., 2003; Bloss et al., 2005; von Glasow and Crutzen, 2004; Saiz-Lopez et al., 2007; Obrist et al., 2010; Engel et al., 2018). In the marine boundary layer and lower troposphere, sea salt is the main source of reactive bromine (Finlayson-Pitts, 2003; Simpson et al., 2015). Yet halogenated VOCs may also be a more important source of inorganic bromine to the whole atmosphere than previously thought according to a recent study, which indicates that sea salt is scarce and insufficient to control the bromine budget in the middle and upper troposphere (Murphy et al., 2019).
Phytoplankton and macroalgae in the ocean are the main sources to the atmosphere of several very short-lived bromocarbons, including bromoform (CHBr3), dibromomethane (CH2Br2), dibromochloromethane (CHClBr2), and bromodichloromethane (CHBrCl2) (Moore et al., 1996; Carpenter et al., 2003; Butler et al., 2007; Raimund et al., 2011). Other halogenated VOCs, such as methyl iodide (CH3I) and methyl bromide (CH3Br), have many natural sources, such as coastal macroalgae, phytoplankton, temperate forest soil and litter, and biomass burning (e.g., Bell et al., 2002; Sive et al., 2007; Colomb et al., 2008; Drewer et al., 2008). CH3I is also formed through nonbiological reactions in surface seawater, and CH3Br is emitted as a result of quarantine and pre-shipment activities, which are not regulated by the Montreal Protocol (e.g., Moore and Zafiriou, 1994; Engel et al., 2018). Over the Southern Ocean specifically, hypothesized sources of halogenated VOCs include coastal macroalgae, phytoplankton, sea-ice algae, and photochemical or dust-stimulated nonbiological production at the sea surface (e.g., Abrahamsson et al., 2018; Manley and Dastoor, 1998; Moore and Zafiriou, 1994; Moore et al., 1996; Richter and Wallace, 2004; Williams et al., 2007; Tokarczyk and Moore, 1994; Sturges et al., 1992).
We largely owe our current understanding of marine halogenated VOC emissions over the Southern Ocean to ship-based field campaigns and laboratory process studies (e.g., Abrahamsson et al., 2004a, b; Atkinson et al., 2012; Carpenter et al., 2007; Moore et al., 1996; Chuck, 2005; Butler et al., 2007; Raimund et al., 2011; Hughes et al., 2009, 2013; Mattsson et al., 2013). These studies have reported surface water and sea-ice halogenated VOC supersaturation and corresponding elevated levels of halogenated VOCs in the marine boundary layer (MBL) in summer and have identified numerous biological and nonbiological ocean sources for these compounds. Mattsson et al. (2013) noted that the ocean also acts as a sink for halogenated VOCs when undersaturated surface waters equilibrate with air masses transported from halogenated VOC source regions. The spatially heterogeneous ocean sources of CHBr3 and CH2Br2 at high latitudes in the Southern Hemisphere are often underestimated in global atmospheric models (Hossaini et al., 2013; Ordoñez et al., 2012; Ziska et al., 2013). Ship-based and Lagrangian float observations provide invaluable information on the sources and temporal variability of compounds in the surface ocean. These methods offer the advantage of simultaneous measurements of both air and seawater to evaluate the gases' saturation state in the surface ocean and calculate fluxes. Yet ship-based measurements onboard these slow-moving platforms also have drawbacks: they under-sample the spatial variability of halogenated VOCs (e.g., Butler et al., 2007) and require assumptions about gas exchange rates to estimate fluxes.
Disentangling the roles of the atmospheric transport and spatial variability of emissions in halogenated VOC distributions requires large-scale atmospheric observations. At low latitudes, large-scale convection at the intertropical convergence zone carries bromocarbons and other halogenated VOCs into the free troposphere and lower stratosphere (e.g., Liang et al., 2014; Navarro et al., 2015). Polar regions are characterized by stable boundary layers in summer. Wind shear, frontal systems, and internal gravity waves create turbulence and control vertical mixing within and across a stable polar boundary layer (e.g., Anderson et al., 2008), and small, convective plumes may form over the marginal sea-ice zone, related to sea-ice leads as well as winds from ice-covered to open-ocean waters (e.g., Schnell et al., 1989). As a result of limited vertical transport in these regions, however, air–sea fluxes lead to strong vertical gradients. Zonal transport from lower latitudes has a large impact on the vertical gradients of trace gas mixing ratios over polar regions (Salawitch et al., 2010). Given their extended photochemical lifetimes at high latitudes (see Sect. 2.3 for a brief discussion), many halogenated VOC distributions are particularly sensitive to zonal transport at altitude.
Aircraft observations can rapidly map basin-wide vertical distributions, support quantitative flux estimates, and provide spatial constraints to atmospheric models (e.g., Xiang et al., 2013; Stephens et al., 2018; Wofsy, 2011). Few airborne observations of halogenated VOCs exist at high latitudes in the Southern Hemisphere. Two earlier aircraft campaigns that measured summertime halogenated VOCs in this region are the first Aerosol Characterization Experiment (ACE-1; Bates, 1999) and the first High-performance Instrumented Airborne Platform for Environmental Research (HIAPER) Pole-to-Pole Observations (HIPPO; Wofsy, 2011) campaign. For these two aircraft campaigns, whole-air samples were collected onboard the NSF/NCAR C-130 and the NSF/NCAR Gulfstream V (GV) during latitudinal transects over the Pacific Ocean as far south as 60 and 67∘ S, respectively. However, the ACE-1 and HIPPO campaigns obtained relatively few whole-air samples in this region, with ≤100 samples poleward of 60∘ S combined (e.g., Blake et al., 1999; Hossaini et al., 2013). ACE-1 measurements of CH3I in the MBL indicate a strong ocean source between 40 and 50∘ S in austral summer, with mixing ratios above 1.2 pmol below ∼1 km (Blake et al., 1999).
Halogenated VOC emissions are frequently incorporated into Earth system models, using either climatologies or parameterizations based on satellite observations of chlorophyll and geographical region, and evaluated using mixing ratio comparisons with airborne observations. In Sect. 3.1 and 3.2, we report new airborne observations of CHBr3, CH2Br2, CH3I, CHClBr2, CHBrCl2, and CH3Br from high latitudes in the Southern Hemisphere, where data are scarce, and large-scale regional mixing ratio comparisons for halogenated VOCs with the Community Earth System Model (CESM) atmospheric component with chemistry (CAM-Chem). In Sect. 3.4, we present two novel approaches to estimate regional fluxes of halogenated VOCs for comparison with global climate model parameterizations or climatologies. One approach uses correlations of halogenated VOCs with oxygen (O2) of marine origin, as measured by deviations in the ratio of O2 to nitrogen (N2) (δ(O2∕N2); see Sects. 2.1.2 and 3.1.2). We exploit robust ratios of halogenated VOCs to oxygen (O2) determined from linear regressions (i.e., the enrichment ratio), and the ocean flux of O2 from CESM's ocean component, to estimate the marine biogenic flux of several halogenated VOCs. The second approach relies on observed halogenated VOC mixing ratios, the Stochastic Time-Inverted Lagrangian Transport (STILT) particle dispersion model, and geophysical datasets (see Sects. 2.3 and 3.3). We assess contributions from previously hypothesized regional sources for the Southern Ocean and estimate halogenated VOC fluxes based on regressions between upstream influences, observed mixing ratios, and distributions of remotely sensed data.
2.1 Measurements
Atmospheric measurements for this study were collected at high latitudes in the Southern Hemisphere as part of the O2∕N2 Ratio and CO2 Airborne Southern Ocean (ORCAS) study (Stephens et al., 2018) and the second NASA Atmospheric Tomography Mission (ATom-2) near Punta Arenas, Chile (Fig. 1). The ORCAS field campaign took place from 15 January to 29 February 2016 onboard the NSF/NCAR GV. On 10 and 13 February 2017 the sixth and seventh ATom-2 research flights passed over the eastern Pacific sector poleward of 60∘ S (defined here as Region 1) and over the Patagonian Shelf between 40 and 55∘ S and between 70 and 50∘ W (defined here as Region 2), respectively. The two regions for this study are defined based loosely on dynamic biogeochemical provinces identified using bathymetry, algal biomass, sea surface temperature, and salinity (Reygondeau et al., 2013).
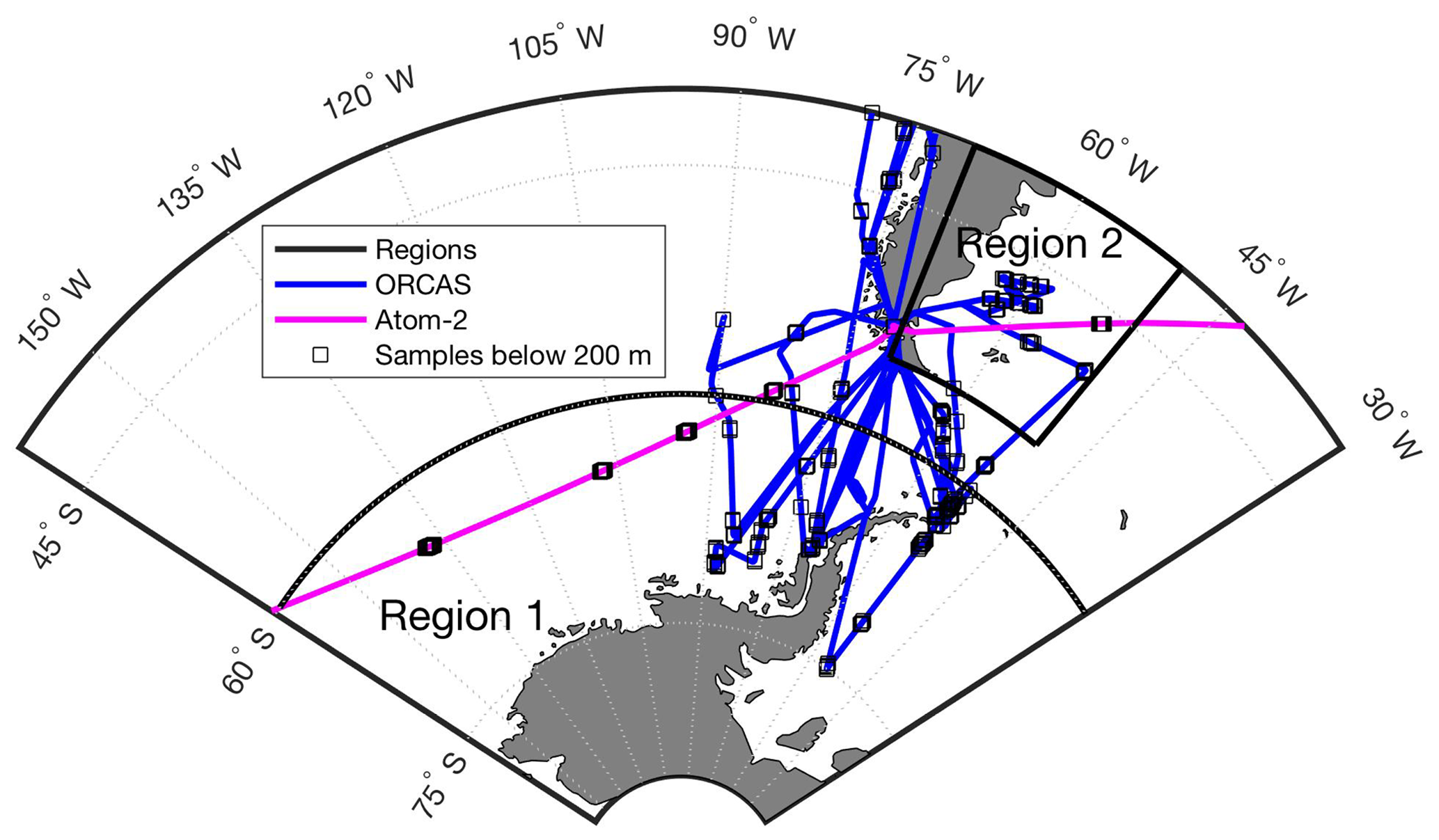
Figure 1Overview map of ORCAS and ATom-2 flight tracks in the study regions: (1) high latitudes in the Southern Hemisphere poleward of 60∘ S and (2) the Patagonian Shelf. The ORCAS and ATom-2 aircraft flights and dips below 200 m that took place within these regions are also shown.
Both projects featured en route vertical profiling from near the ocean surface (∼150 m) to the upper troposphere, with 74 ORCAS and seven ATom-2 (during the sixth and seventh flights) low-altitude level legs in the MBL. These campaigns shared a number of instruments, including the NCAR Trace Gas Organic Analyzer (TOGA), the NCAR Atmospheric Oxygen (AO2) instrument, and a Picarro cavity ring-down spectrometer operated by NOAA, discussed below. More information about individual instruments may be found in Stephens et al. (2018) and at https://www.eol.ucar.edu/field_projects/orcas (last access: 12 January 2019) and https://espo.nasa.gov/atom/content/ATom (last access: 25 October 2019).
2.1.1 Halogenated VOCs
During ORCAS and ATom-2 TOGA provided mixing ratios of over 60 organic compounds, including halogenated VOCs. The instrument, described in Apel et al. (2015), continuously collects and analyzes samples for CHBr3, CH2Br2, CHClBr2, CHBrCl2, and CH3I among other compounds, with a 35 s sampling period and repeats the cycle every 2 min using online fast gas chromatography and mass spectrometry. This study also leverages measurements of CH3Br from whole-air samples from the U. Miami/NCAR Advanced Whole Air Sampler (AWAS; Schauffler et al., 1999) onboard the GV during the ORCAS campaign and the UC Irvine Whole Air Sampler (WAS; Blake et al., 2001) onboard the DC-8 during the ATom-2 campaign. Halogenated VOCs reported here have an overall ±15 % accuracy and ±3 % relative precision, with detection limits of 0.03 ppt for CH3I, 0.2 ppt for CHBr3, 0.03 ppt for CH2Br2, 0.03 ppt for CHClBr2, 0.05 ppt for CHBrCl2, and 0.2 ppt for CH3Br – 0.2 ppt. In addition, comparisons between onboard collected whole-air samples and in-flight TOGA measurements, when sharing over half of their sampling period with TOGA measurements, showed good correlations for CHBr3, CH2Br2, CH3I, and CHClBr2, although there were some calibration differences (Figs. S1 and S2 in the Supplement). In addition to the comparison between colocated atmospheric measurements, we also conducted a lab intercomparison following the campaign between NOAA's programmable flask package (PFP) and TOGA (Table S1; see the Supplement for details).
2.1.2 δ(O2∕N2) and CO2
The AO2 instrument measures variations in atmospheric O2, which are reported as relative deviations in the oxygen-to-nitrogen ratio (δ(O2∕N2)), following a dilution correction for CO2 (Keeling et al., 1998; Stephens et al., 2018). The instrument's precision is ±2 per meg unit (one in 1 million relative) for a 5 s measurement (Stephens et al., 2003). Anthropogenic, biogenic, and oceanic processes introduce O2 perturbations that are superimposed on the background concentrations of O2 in air (XO2; in dry air 0.2095). Air–sea O2 fluxes are driven by both the biological production and consumption of O2 and by the heating and cooling of surface waters. O2 is consumed when fossil fuels are burned and produced and consumed during terrestrial photosynthesis and respiration. Seasonal changes in the ocean heat content lead to small changes in atmospheric N2. As others have done, we isolated the air–sea O2 signal by subtracting model estimates of the terrestrial O2, fossil fuel O2, and air–sea N2 flux influences from the δ(O2∕N2) measurements (Eq. 1; Keeling et al., 1998; Garcia and Keeling, 2001; Stephens et al., 2018). The difference of the δ(O2∕N2) measurement and these modeled components is multiplied by XO2 to convert to parts per million equivalent as needed (ppm eq; Keeling et al., 1998; Eq. 1).
We obtained the modeled δ(O2∕N2) signal terrestrial influences from the land model component of the CESM, the fossil fuel combustion influences from the Carbon Dioxide Information Analysis Center (CDIAC; Boden et al., 2017), and the air–sea N2 influences from the oceanic component of CESM. These fluxes were all advected through the specified dynamics version of CESM's atmosphere component, as described below in Sect. 2.2 and in Stephens et al. (2018).
CO2 measurements were provided by NOAA's Picarro G2401-m cavity ring-down spectrometer modified to have a ∼1.2 s measurement interval and a lower cell pressure of 80 Torr, which enabled the instrument to function at the full range of GV altitudes (Karion et al., 2013). Dry-air mole fractions were calculated using empirical corrections to account for dilution and pressure-broadening effects as determined in the laboratory before and after the campaign deployments, and in-flight calibrations were used to determine an offset correction for each flight. Corrected CO2 data have a total average uncertainty of 0.07 ppm (Karion et al., 2013). To merge them with the TOGA data, these faster O2 and CO2 measurements were arithmetically averaged over TOGA's 35 s sampling periods (Stephens, 2017, and https://espo.nasa.gov/atom/content/ATom, last access: 20 December 2018).
2.2 CAM-Chem model configuration
The CESM version 1 atmospheric component with chemistry (CAM-Chem) is a global three-dimensional chemistry climate model that extends from the Earth's surface to the stratopause. CAM-Chem version 1.2 includes all the physical parameterizations of Neale et al. (2013) and a finite-volume dynamical core (Lin, 2003) for tracer advection. The model has a horizontal resolution of 0.9∘ latitude × 1.25∘ longitude, with 56 vertical hybrid levels and a time step of 30 min. Meteorology is specified using the NASA Global Modeling and Assimilation Office (GMAO) Goddard Earth Observing System Model version 5 (GEOS-5; Rienecker et al., 2008) (GEOS-5), following the specified dynamic procedure described by Lamarque et al. (2012). Winds, temperatures, surface pressure, surface stress, and latent and sensible heat fluxes are nudged using a 5 h relaxation timescale to GEOS-5 1∘ × 1∘ meteorology. The sea surface temperature boundary condition is derived from the Merged Hadley–NOAA Optimal Interpolation Sea Surface Temperature and Sea-Ice Concentration product (Hurrell et al., 2008). The model uses chemistry described by Tilmes et al. (2016), biomass burning and biogenic emissions from the Fire INventory of NCAR (FINN; Wiedinmyer et al., 2011) and MEGAN (Model of Emissions of Gases and Aerosols from Nature) 2.1 products (Guenther et al., 2012), and additional tropospheric halogen chemistry described in Fernandez et al. (2014) and Saiz-Lopez et al. (2014). These include ocean emissions of CHBr3, CH2Br2, CHBr2Cl, and CHBrCl2, with parameterized emissions based on chlorophyll a (chl a) concentrations and scaled by a factor of 2.5 over coastal regions, as opposed to open-ocean regions (Ordóñez et al., 2012). The model used an existing CH3I flux climatology (Bell et al., 2002), and CH3Br was constrained to a surface lower boundary condition, also described by Ordóñez et al. (2012). This version of the model was run for the period of the ORCAS field campaign (January and February 2016), following a 24-month spin-up. To facilitate comparisons to ORCAS observations, output included vertical profiles of modeled constituents from the two nearest latitude and two nearest longitude model grid points (four profiles in total) to the airborne observations at every 30 min model time step. Following the run, simulated constituent distributions were linearly interpolated to the altitude, latitude and longitude along the flight track, yielding colocated modeled constituents and airborne observations. This version of the model has not yet been run for the ATom-2 period.
2.3 STILT model configuration
The Stochastic Time-Inverted Lagrangian Transport (STILT; Lin, 2003) particle dispersion model uses a receptor-oriented framework to infer surface sources or sinks of trace gases from atmospheric observations collected downstream, thus simulating the upstream influences that are ultimately measured at the receptor site. The model tracks ensembles of particle trajectories backward in time and the resulting distributions of these particles can be used to define surface influence maps for each observation. STILT was run using 0.5∘ Global Data Assimilation System (GDAS) reanalysis winds to investigate the transport history of air sampled along the flight track (Stephens et al., 2018). For each TOGA observation, an ensemble of 4096 particles was released from the sampling location and followed over a backwards simulation period of 7 d. Particles in the lower half of the simulated MBL are assigned a surface influence value, which quantitatively links observed mixing ratios to surface sources (Lin, 2003). The average surface influence of all 4096 particles per sampling location yields an hourly and spatially gridded surface influence value (ppt m2 s pmol−1) at a spatial resolution of 0.25∘ × 0.25∘ for each sample point.
Uncertainty in the surface influence value is strongly influenced by the accuracy of the underlying meteorological transport, as discussed in Xiang et al. (2013). We evaluated the GDAS reanalysis winds by comparing model winds interpolated in space and averaged between corresponding time points and pressure levels to match aircraft observations. By evaluating observed winds compared with modeled winds along the flight tracks we can estimate uncertainty in the surface influence values. We consider the observation–model differences in both wind speed and direction to approximate errors in surface influence strength and location. For wind speed, a small bias may be present; we find a median difference between observations and reanalysis of 0.68 m s−1, a 5 % relative bias. The 1σ of the wind speed difference is 2.3 m s−1, corresponding to a 19 % 1σ uncertainty in wind speed. In its simplest approximation, the surface influence strength error is perfectly correlated with the wind speed error, and thus we take 19 % as an approximation of the surface influence strength uncertainty. The uncertainty in surface influence location depends on the error in the wind direction. We find a 1σ error of 14∘ in wind speed, which corresponds to a possible error of 260 km d−1.
Finally, we note that photochemical loss during transport is not accounted for in this analysis. Low OH mixing ratios, cold temperatures, and lower photolysis rates due to angled sunlight at high latitudes lead to longer than average halogenated VOC lifetimes. For instance, assuming an average diurnal OH concentration of 0.03 ppt and average photochemical loss according to the Tropospheric Ultraviolet and Visible (TUV) radiation model and the Mainz Spectral data site (http://satellite.mpic.de/spectral_atlas, last access: 9 January 2019) for 29 January under clear-sky conditions at 60∘ S, CHBr3 has a lifetime of 30 d, CH2Br2 has a lifetime of 270 d, CH3I has a lifetime of 7 d, and CHClBr2 has a lifetime of 63 d. As such, the photochemical lifetimes of these gases are greater than or equal to the time of our back-trajectory analysis. Moreover, OH concentrations in this region have large uncertainties, the inclusion of which would lead to more, not less, uncertainty in surface-influence-based regression coefficients and estimated fluxes (see Sects. 2.3 and 3.3 for details).
2.3.1 STILT surface influence functions
For this study, we used STILT surface influence distributions with remotely sensed ocean surface and reanalysis data (i.e., surface source fields) in linear and multi-linear regressions to generate empirical STILT influence functions. Surface influence functions can help explain observed mixing ratios of CHBr3, CH2Br2, CH3Br, and CH3I, evaluate the likelihood of particular halogenated VOC sources, and in the case of statistically significant correlations may be used to derive an estimated flux field (see Sect. 3.3 and 3.4.2 for details).
We tested whether observed mixing ratios (Z) could be explained by a linear relationship in which the predictor variable is a surface influence function equal to the product of the surface influence (H) and a potential geophysical surface source field(s), such as chl a, as well as an intercept (b), a slope (a), and error term ξ (Eq. 2; Fig. S5). This relationship can be generalized as a multiple linear regression with multiple surface influence functions (Hs1, Hs2…) and slope coefficients (a1, a2; Eq. 3) when multiple sources contribute to observed halogenated VOC mixing ratios. The multiple linear regression may also include an interaction term (Hs2) between predictor variables (e.g., Hs1 and Hs2) with a slope coefficient (a3) to improve the fit. Statistical correlations between mixing ratios and surface influence functions may be used to support or reject hypothesized sources. A flux (µmol m−2 s−1) may then be estimated for each grid cell based on the product of the slopes (a1, a2…) and the potential source fields (s1, s2…). Grid cell fluxes are averaged over a geographical region to yield the average regional flux. We used the standard deviation of the regression coefficients and the relative uncertainty in the surface source, added in quadrature, to estimate the uncertainty in the flux (see Sect. 3.4.2 for fractional uncertainties). We note that the uncertainty in STILT transport (see Sect. 2.3 for details) is inherently reflected in the relative uncertainty of the regression coefficients (a1, a2…).
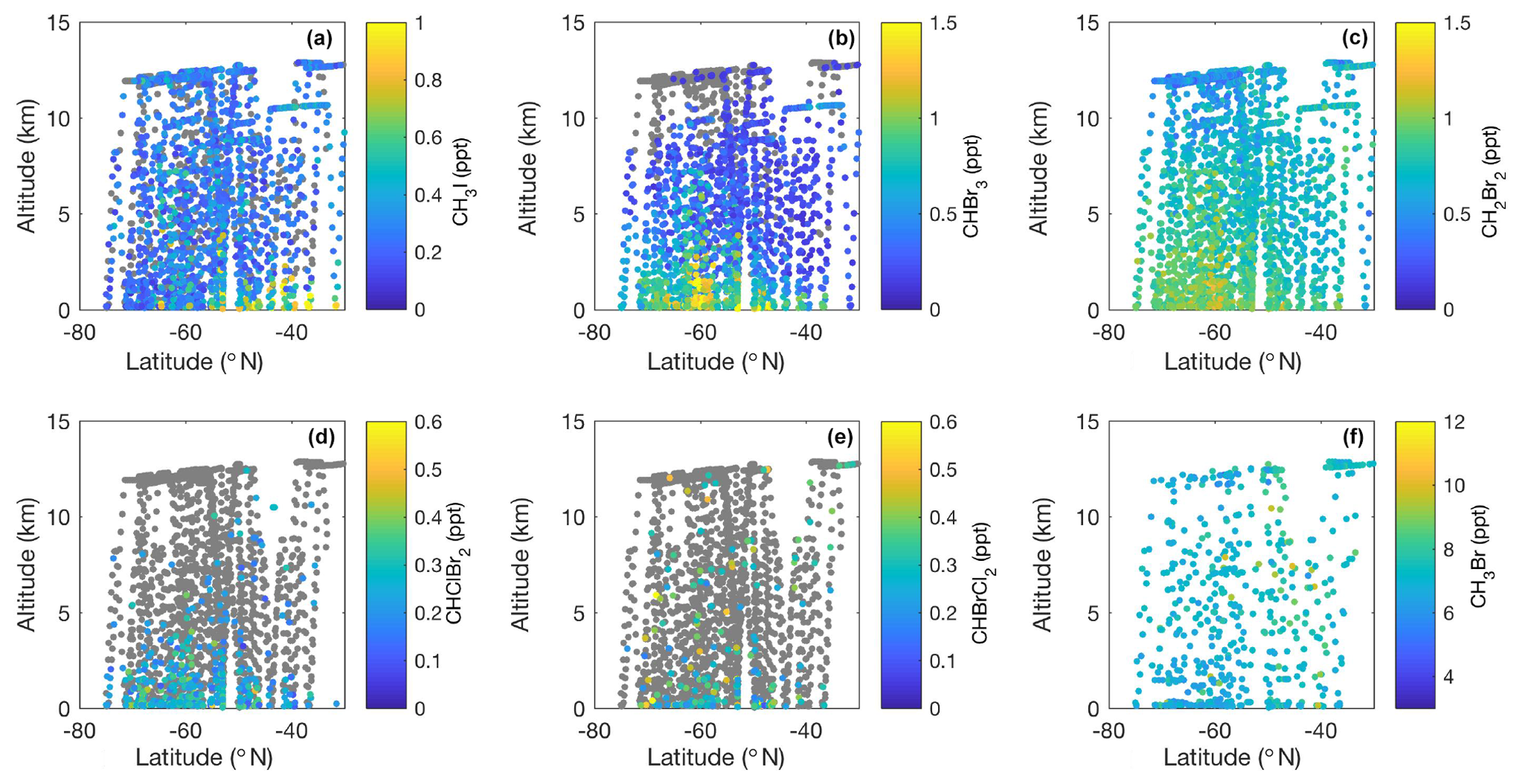
Figure 2Meridional–altitudinal cross sections of mixing ratios of (a) CH3I, (b) CHBr3, (c) CH2Br2, (d) CHClBr2, and (e) CHBrCl2 from the TOGA and mixing ratios of (f) CH3Br from AWAS and WAS in 2016 and 2017, respectively, during the ORCAS and ATom-2 campaigns over the Southern Ocean in austral summer. Note the different color bar scales. Gray points denote measurements below the detection limit of each species (CH3I – 0.03 ppt, CHBr3 – 0.2 ppt, CH2Br2 – 0.03 ppt, CHClBr2 – 0.03 ppt, CHBrCl2 – 0.05 ppt, CH3Br – 0.2 ppt).
2.3.2 Surface source fields
Geophysical surface source fields of remotely sensed and reanalysis data included a combination of sea-ice concentration, chl a, absorption due to ocean detrital material, and downward shortwave radiation at the ocean surface.
We used daily sea-ice concentration data (https://nsidc.org/data/nsidc-0081, last access: 7 January 2019) at a 25 km × 25 km spatial resolution between 39.23– 90∘ S and 180∘ W–180∘ E from the NASA National Snow and Ice Data Center Distributed Active Archive Center (NSIDC; Maslanik, 1999). These data report the fraction of sea-ice cover, land-ice cover, and open water. Unfortunately, these data do not provide any information on sea-ice thickness or the presence of brine channels or melt ponds, which may modulate emissions from sea-ice-covered regions. Sea-ice concentration data were calculated using measurements of near-real-time passive microwave brightness temperature from the Special Sensor Microwave Image/Sounder (SSMIS) on the Defense Meteorological Satellite Program (DMSP) satellites. NSIDC sea-ice concentration data were arithmetically averaged to yield a 0.25∘ × 0.25∘ binned sea-ice fraction for use with gridded surface influences.
Due to persistent cloud cover over the Southern Ocean, which often precludes the retrieval of remotely sensed ocean color data, we used 8 d mean composite Aqua MODIS L3 distributions of chl a from the Ocean Color Index (OCI) algorithm and absorption due to gelbstoff and detrital material at 443 nm from the Generalized Inherent Optical Properties (GIOP) model (NASA Goddard Space Flight Center, 2014). Absorption due to gelbstoff and detrital material at 443 nm is used as a proxy for colored dissolved organic matter (CDOM; https://oceancolor.gsfc.nasa.gov/atbd/giop/, last access: 3 October 2018). CDOM is hypothesized to be an important source of carbon for the photochemical production of CH3I (Moore and Zafiriou, 1994). The GIOP model also publishes an uncertainty in the absorption due to gelbstoff and detrital material at 443 nm. Raw 4 km × 4 km data were geometrically averaged, based on lognormal probability density functions, to a spatial resolution of 0.25∘ × 0.25∘ for use with gridded surface influences. We used the ratio of the 0.25∘ × 0.25∘ gridded uncertainty in the detrital material absorption to the absorption as the relative uncertainty for flux calculations (see Sect. 3.4.2).
The National Center for Environmental Prediction (NCEP) provides Final Global Data Assimilation System (GDAS/FNL) global data of downward shortwave radiation at the surface at 0.25∘ and 6 h resolution (NCEP, 2015). We chose downward shortwave radiation for use with gridded surface influences because the photo-production of CH3I has been observed at all visible wavelengths (Moore and Zafiriou, 1994). These reanalysis data are available at a higher temporal resolution and better spatial coverage than satellite retrievals of photosynthetically active radiation (PAR) or temperature.
3.1 Observed halogenated VOC patterns and relationships
Zonal cross sections of halogenated VOC data collected on ORCAS and ATom-2 illustrate unprecedented spatial sampling across our study area between the surface and 12 km (Fig. 2). Above average mixing ratios of CH3I, CHBr3, and CHClBr2 typically remain confined to the lower ∼ 2–4 km of the atmosphere (Fig. 2a, b, d). These compounds have lifetimes of approximately 2 months or less. Conversely, weak sources and longer lifetimes (≥3 months) may have contributed to similar concentrations of CH2Br2 and CHBrCl2 throughout the troposphere and above average mixing ratios as high as 8 km (Fig. 2c, e). Unfortunately, the availability of data above the detection limit and absence of BL enhancements for CHBrCl2 preclude the identification of strong regional sources at this time. Meridional distributions also indicate lower-latitude sources of CH3I and CH3Br (<50∘ S), potentially resulting from terrestrial and anthropogenic contributions, and higher-latitude sources (>60∘ S) of CHBr3, CH2Br2, and CHClBr2 (Fig. 2a–d, f).
3.1.1 Observed halogenated VOC interrelationships
Across our study area in both 2016 and 2017, we found that CHBr3 and CH2Br2 exhibit a consistent enhancement ratio with each other in the bottom 2 km of the atmosphere both in Region 1 and Region 2, which suggests that these bromocarbon fluxes are closely coupled. Previous studies have documented colocated source regions of CHBr3 and CH2Br2 in the Southern Ocean (e.g., Hughes et al., 2009; Nightingale et al., 1995; Laturnus 1996), and laboratory studies have demonstrated that phytoplankton and their associated bacteria cultures, including a cold-water diatom isolated from coastal waters along the Antarctic Peninsula and common to the Southern Ocean, produce both CHBr3 and CH2Br2 (Hughes et al., 2013; Tokarczyk and Moore 1994; Sturges et al., 1992). The nonlinearity observed in ratios of these two gases at low CHBr3 may reflect the different rates of their production or loss in seawater or, possibly, the influence of air masses from distant, more productive low-latitude source regions. Several studies have documented the bacterially mediated loss of CH2Br2, but not CHBr3, and report distinct ratios of CH2Br2 to CHBr3 in seawater during the growth and senescent phases of a phytoplankton bloom (e.g., Carpenter et al., 2009; Hughes et al., 2013). Although this analysis is restricted to the bottom 2 km of the atmosphere, zonal transport of air masses with lower ratios of CH2Br2 to CHBr3, as have been observed in the MBL over productive, low-latitude regions, may also have influenced our observations (Yokouchi et al., 2005). Mixing ratios of CHBr3 and CHClBr2 were also correlated (Fig. 3d) in Region 2, and a similar, weaker relationship was observed in Region 1 (Fig. 3b). CHClBr2 is a less well-studied compound than CH2Br2. Yet these consistent relationships suggest that CHBr3 and CHClBr2 may either share some of the same sources or have sources that covary.
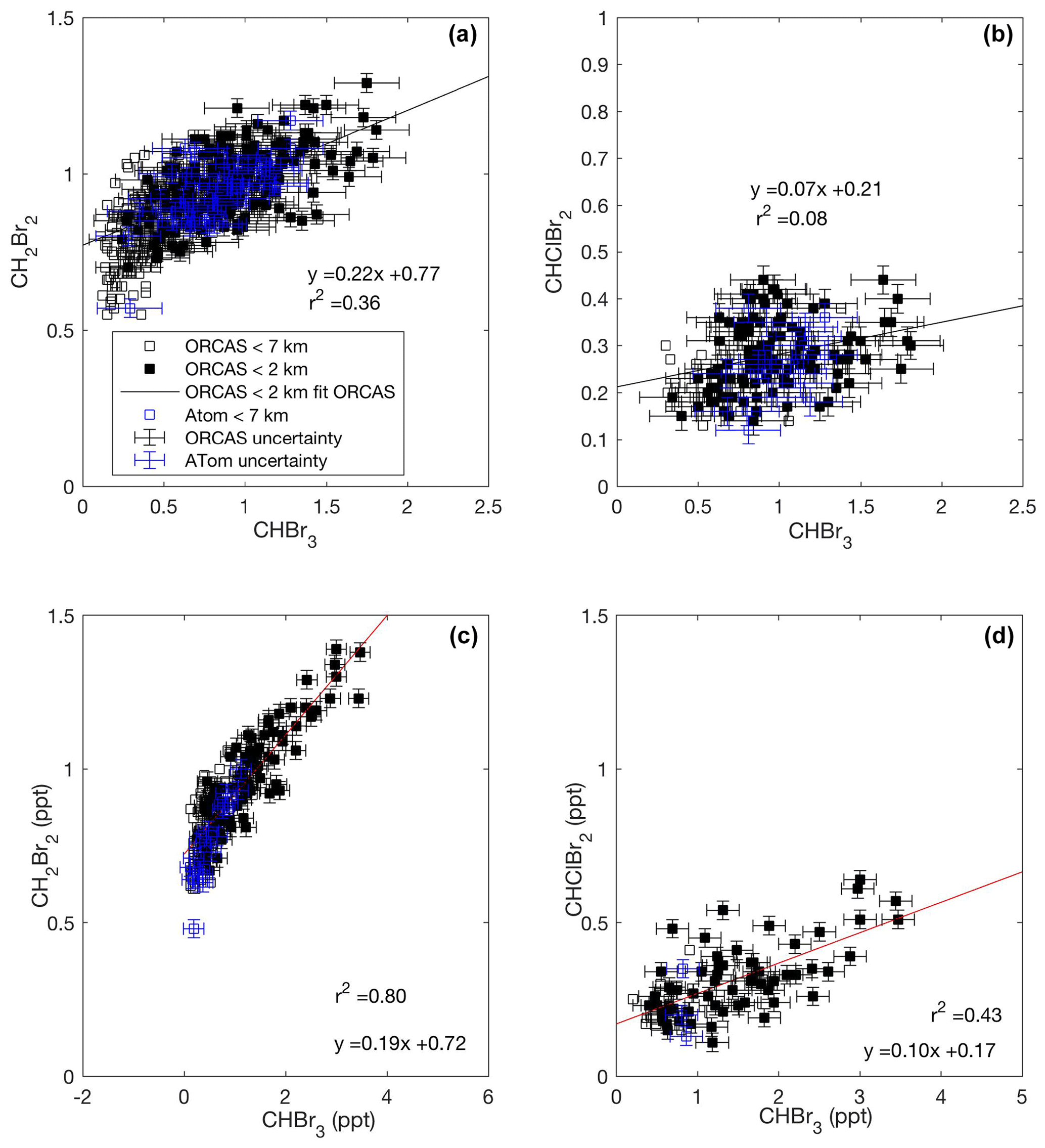
Figure 3Mixing ratios of CHBr3 vs. CH2Br2 and CHClBr2 across the ORCAS and ATom-2 campaigns in Region 1 (a, b) and in Region 2 (c, d), respectively. Type II major axis regression models (bivariate least squares regressions) are based on ORCAS data below 2 km and illustrate regional enhancement ratios. Error bars represent the uncertainty in halogenated VOC measurements.
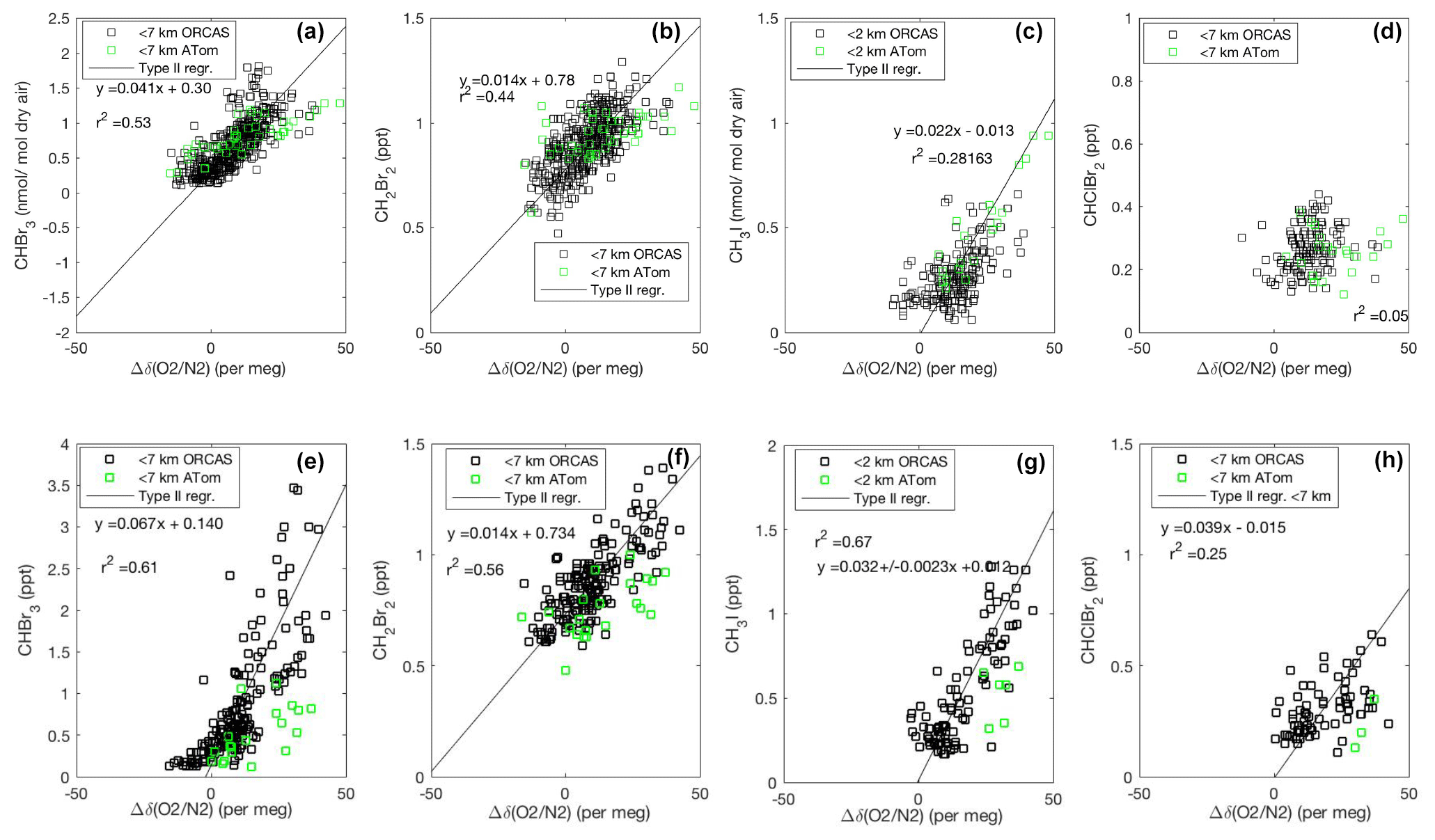
Figure 4Mixing ratios of CHBr3, CH2Br2, and CH3I vs. O2 on ORCAS and ATom-2 in Region 1 poleward of 60∘ S (a–d) and Region 2 over the Patagonian Shelf (e–h). Slopes ± standard errors from type II major axis regression model (bivariate least squares regression) fits of ORCAS data for regressions with r2>0.2 (fits were calculated on variables scaled to their full range). The slopes reported in the figure are converted to picomole-to-mole ratios prior to estimating biogenic halogenated VOC fluxes based on modeled CESM O2 fluxes. Data from above 7 km were excluded due to the influence of air masses transported from further north.
3.1.2 Observed halogenated VOC relationships to δ(O2∕N2) and CO2
We sought to test if the biologically mediated production of bromocarbons and oxygen results in similar atmospheric distributions. Conversely, we expected halogenated VOC atmospheric distributions and CO2 distributions to anticorrelate because CO2 fixation in surface waters is proportional to the production of oxygen.
For these comparisons, both O2 and CO2 mixing ratios from the upper troposphere (5–7 km) were subtracted from the data to detrend for seasonal and interannual variability (Figs. 4; S3). To isolate the contribution of ocean O2 fluxes, the ORCAS δ(O2∕N2) values reported here represent the Δδ(O2∕N2) to observed values between 5 and 7 km adjusted for CESM O2 land and fossil fuel contributions and the influence of air–sea N2 fluxes. In Fig. 4 we present type II major axis regression fits to data (fits were calculated using data scaled to their full range) between the ocean surface and the lowest 7 km for bromocarbons with photochemical lifetimes of ≥1 month and from the lowest 2 km for CH3I with a photochemical lifetime of ∼1 week. We used a type II major axis regression model to balance the influences of uncorrelated processes and measurement uncertainty in halogenated VOCs (on the y axis) and uncorrelated processes and measurement uncertainty in O2 and CO2 (on the x axis) on the regression slope (Ayers, 2001; Glover et al., 2011). As noted by previous studies, simple least squares linear regressions fail to account for uncertainties in predictor variables (e.g., Cantrell, 2008).
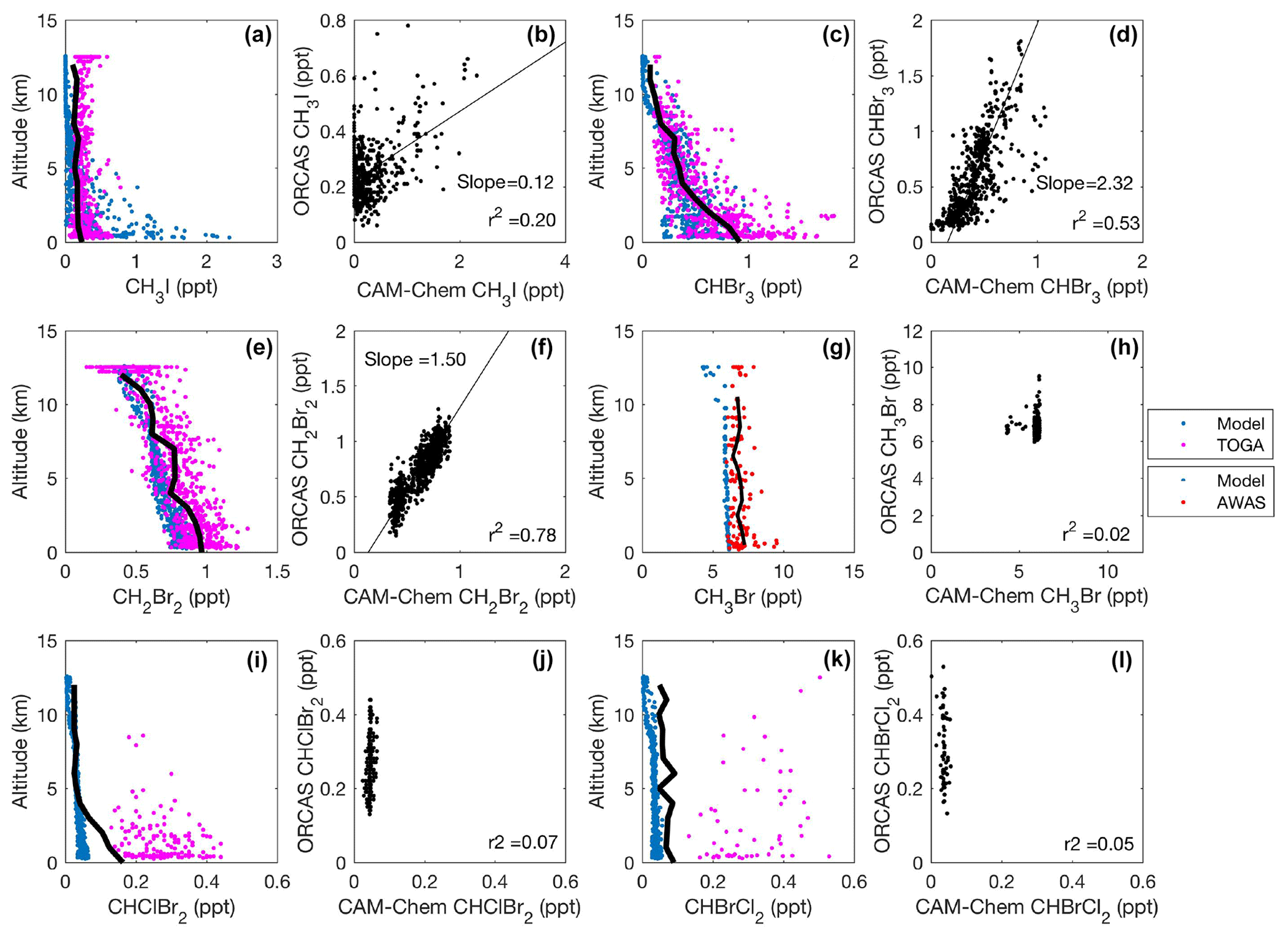
Figure 5CAM-Chem1.2 model–aircraft measurement comparison during the ORCAS campaign between 1 and 12 km in Region 1 high latitudes in the Southern Hemisphere poleward of 60∘ S. All regressions are type II major axis regression models with bivariate least squares regressions (slopes are shown when r2≥0.2). The bold black line in each vertical profile represents the binned (mean) mixing ratio of halogenated VOC measurements at that altitude. The binned mean includes measurements below the detection limit (DL), which for this calculation are assigned a value equal to the DL multiplied by the percentage of data below detection. Modeled values include locations where observations were below the DL.
The robust correlations of CHBr3 and CH2Br2 with δ(O2∕N2), in both 2016 and 2017 and in Region 1 and Region 2, provide support for a regional biogenic source of these two halogenated VOCs (Fig. 4a, b, e, and f). The air–sea exchange of O2 during summer in the Southern Ocean is driven by net community production (the excess of photosynthesis over respiration) in the surface mixed layer, surface warming, and to a lesser extent ocean advection and mixing (e.g., Stephens et al., 1998; Tortell and Long 2009; Tortell et al., 2014). Note that we adjust for influences on the δ(O2∕N2) from thermal N2 fluxes (see Eq. 1, Sect. 2.1.2 for details). Biological O2 supersaturation in the surface mixed layer develops quickly in the first few days of a phytoplankton bloom and diminishes as community respiration increases and air–sea gas exchange equilibrates the surface layer with the atmosphere on a timescale of ∼1 week. CHBr3 and CH2Br2 are emitted from phytoplankton during the exponential growth phase (Hughes et al., 2013), which often coincides with high net community production and the accumulation of O2 in surface waters. The bulk air–sea equilibration time for an excess of CHBr3 and other halogenated VOCs is less than 2 weeks, although the photochemical loss of halogenated VOCs will alter their ratio over time (see the Supplement for details on calculations of bulk sea–air equilibration times).
Our observations suggest a biological source for CHBr3 and CH2Br2 in both Region 1 and Region 2 (Fig. 4). Interestingly, the slope of the regression between CHBr3 and O2 appears distinct in Region 1 and Region 2, but for CH2Br2 it is the same. Molar enrichment ratios are 0.20±0.01 and 0.07±0.004 pmol : mol for CHBr3 and CH2Br2 to O2 in Region 1, and they are 0.32±0.02 and 0.07±0.004 pmol : mol in Region 2. We observe a weaker relationship between CH3I and CHClBr2 and O2 in Region 1 (Fig. 4c, d), consistent with the existence of other nonbiological sources of CH3I in this region. Figure 4g and h illustrate a strong relationship between CH3I and O2, as well as CHClBr2 and O2, in Region 2, however, which implies that the dominant sources of CH3I and CHClBr2 emissions over the Patagonian Shelf are biological. The corresponding molar enrichment ratios of CH3I to O2 and CHClBr2 to O2 in Region 2 are 0.38±0.03 pmol : mol and 0.19±0.04 pmol : mol, respectively.
In contrast to O2, air–sea fluxes of CO2 over the Southern Ocean during summer reflect the balance of opposing thermal and biological drivers (e.g., Stephens et al., 1998, 2018). Ocean buffering chemistry results in CO2 equilibration across the air–sea interface on a timescale of several months. ORCAS observations showed a depletion of CO2 in the MBL, indicating that uptake driven by net photosynthesis dominated over thermally driven outgassing during the few months preceding the campaign (Stephens et al., 2018). CHBr3 and CH2Br2 in the lowest 7 km were negatively correlated with CO2 in both years in Region 1 and Region 2 (Fig. S3a, b, e, f). Interestingly, CH3I was not correlated with CO2 in Region 1, likely due to the long air–sea equilibration timescale of CO2 compared with a 9 d air–sea equilibration time and a ∼7 d photochemical lifetime for CH3I. For longer-lived species, correlations for halogenated VOCs to CO2 have similar r2 values as those for halogenated VOCs to δ(O2∕N2), but model and climatological estimates of Southern Ocean CO2 fluxes are much less certain than for O2 (Anav et al., 2015; Nevison et al., 2016). As a result, we use modeled O2 fluxes as the basis for our halogenated VOC flux estimates (see Sect. 3.4.1 for details).
3.2 Model–observation comparisons
The ORCAS dataset provides an exceptional opportunity to evaluate the CAM-Chem halogenated VOC emission scheme (Ordóñez et al., 2012) at high latitudes in the Southern Hemisphere. We compared modeled halogenated VOC constituents to corresponding observations along the ORCAS flight track (Figs. 5; 6). In these figures, we used type II major axis regression models to balance the measurement uncertainty (on the y axis) and the inherent, yet difficult to quantify representativeness and errors in a global atmospheric chemistry model (on the x axis). We note that this comparison may favor constituents with longer photochemical lifetimes when transport and mixing dominate over source heterogeneity.

Figure 6CAM-Chem 1.2 model–aircraft measurement (TOGA and AWAS) comparison during the ORCAS campaign between 1 and 12 km in Region 2 for the Patagonian Shelf. All regressions are type II major axis regression models with bivariate least squares regressions (slopes are shown when the r2≥0.2). The bold black line in each vertical profile represents the binned (mean) mixing ratio of halogenated VOC measurements at that altitude. Again, the binned mean includes measurements below the detection limit (DL), which for this calculation are assigned a value equal to the DL multiplied by the percentage of data below detection. Modeled values include locations where observations were below the DL.
In Region 1 and Region 2, both the model and observations indicate that elevated mixing ratios of CH3I remain confined to the MBL (Figs. 5a and 6a), presumably due to its relatively short photochemical lifetime. Modeled and observed CH3I are poorly correlated in Region 1 (r2=0.20; Fig. 5b) and better correlated in Region 2 (r2=0.70; Fig. 6b). In both regions, the model most likely underpredicts CH3I in the upper troposphere and lower stratosphere (UTLS), likely stemming from the poleward transport of lower-latitude air masses, for which CAM-Chem also exhibits a negative bias. Mixing ratio comparisons with CAM-Chem over the tropics (see Fig. 10 in Ordóñez et al., 2012) depict similar or larger discrepancies and have been attributed to stronger than anticipated convective cells in the tropics. We found strong correlations and agreement to within a factor of ∼2 between modeled and observed CHBr3 and CH2Br2 (Figs. 5c–f and 6c–f). Relatively long lifetimes (≥1 month) in Region 1 likely enable the vertical and zonal transport of CHBr3 and CH2Br2 to the middle and upper troposphere (Fig. 5c and e). The model was biased low with respect to measurements of CH3Br by ∼25 % in Region 1 and Region 2 (Figs. 5g–h and 6g–h), potentially as a result of an incorrect surface lower boundary condition. The model underpredicted the mean vertical gradient in CHClBr2, although it did a reasonable job of representing the mean vertical gradient in CHBrCl2 in both Region 1 and Region 2. In both cases, however, the model failed to capture the spatial variability in both CHClBr2 and CHBrCl2 observations (Figs. 5i–l and 6i–l). Region 2 contains stronger sources of halogenated VOCs than Region 1, which has been documented in numerous ship-based campaigns and archived in the Halocarbons in the Ocean and Atmosphere database (HalOcAt; https://halocat.geomar.de/, last access: 8 January 2019). Region 2 also has much higher chl a (Fig. S4), supporting biogenic sources for these gases.
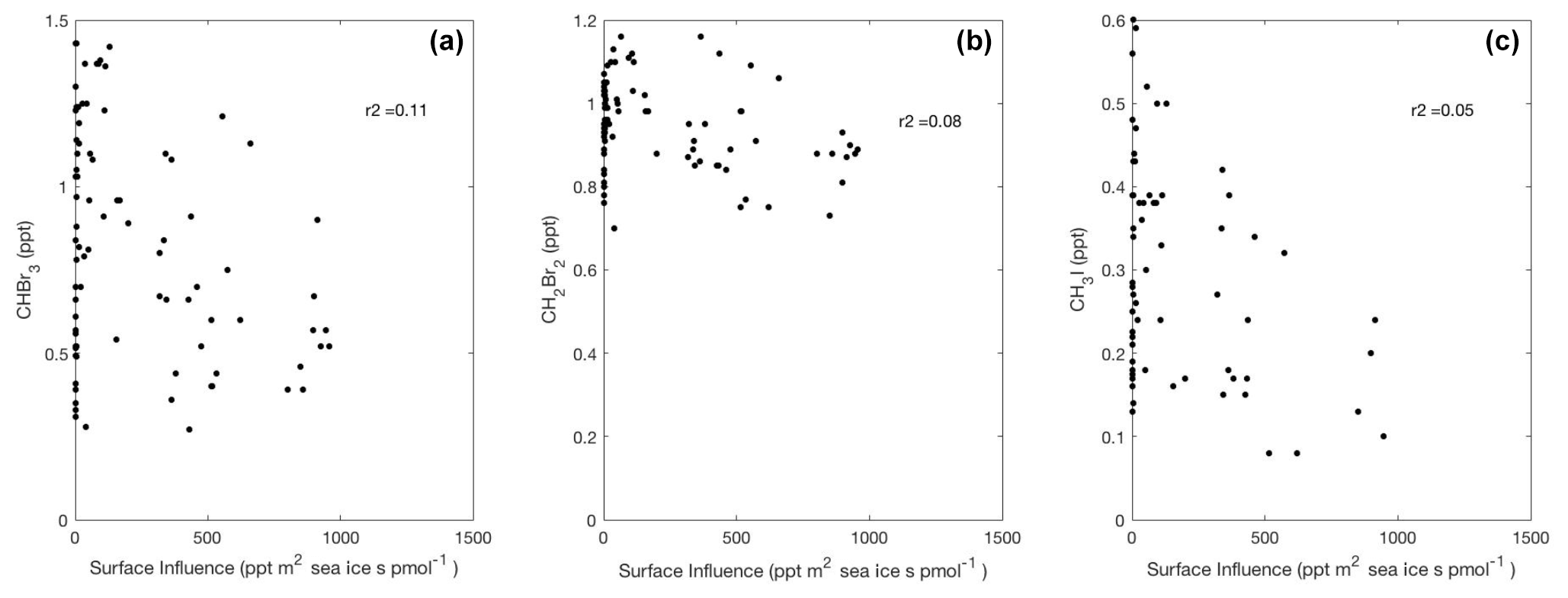
Figure 7Linear type II regressions between influence functions convolved with sea-ice distributions, which exclude land ice, and mixing ratios for CHBr3, CH2Br2, and CH3I in Region 1 poleward of 60∘ S. Surface influence (ppt m2 s pmol−1) in each grid cell was multiplied by fractional sea-ice concentration surface field, which is unitless, yielding sea-ice surface influence function units (ppt m2 s pmol−1), as shown on the x axis. Linear regression lines are not shown, as p≥0.001.
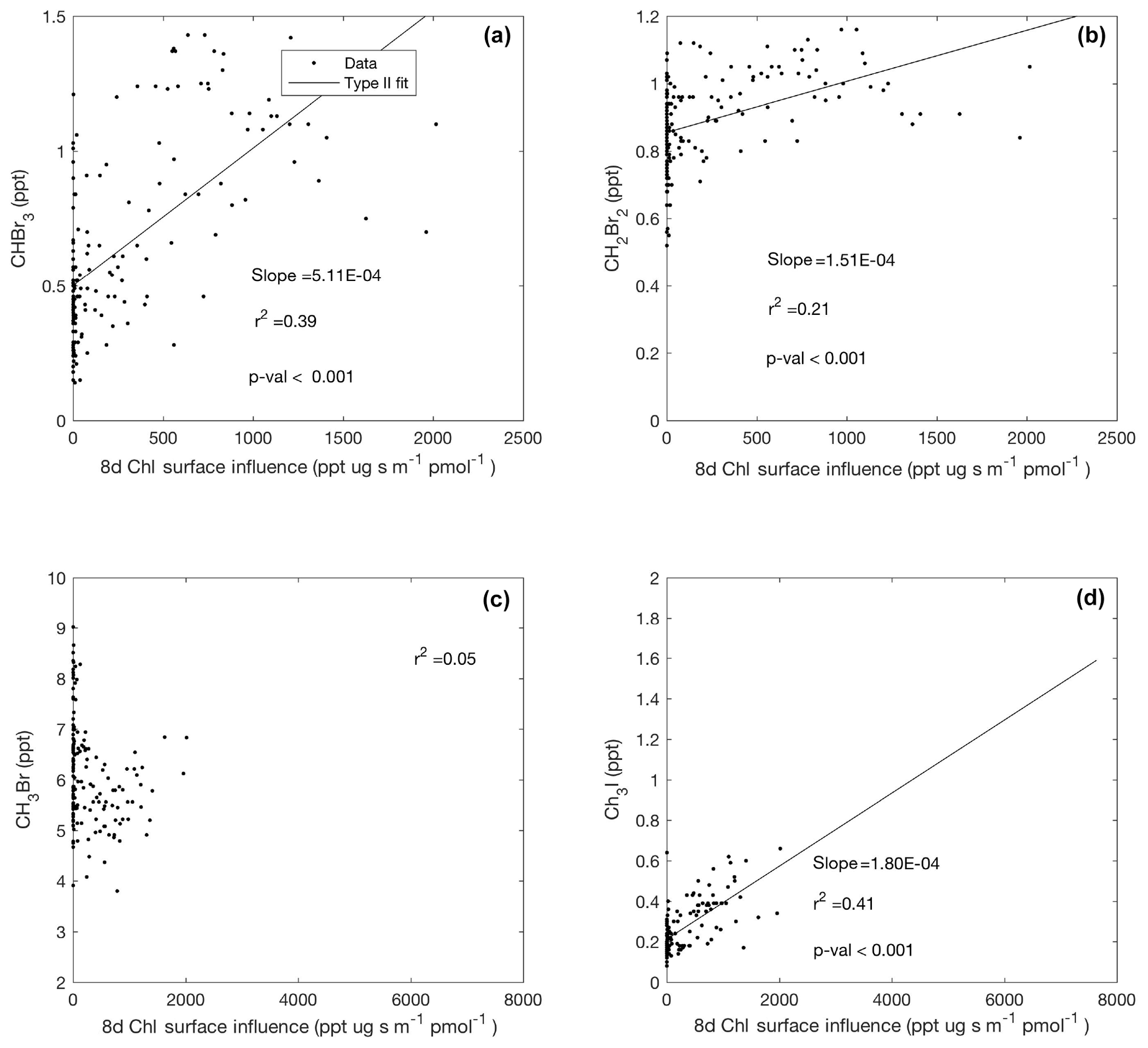
Figure 8Linear type II regressions between influence functions of 8 d composites of chl a and mixing ratios of halogenated VOCs (a–d) poleward of 60∘ S (Region 1). Surface influence (ppt m2 s pmol−1) in each grid cell was multiplied by the chl a (µg m−3) surface field, resulting in surface influence function units (µg ppt s pmol−1 m−1) shown on the x axis. Linear regression lines are shown where when p<0.001.
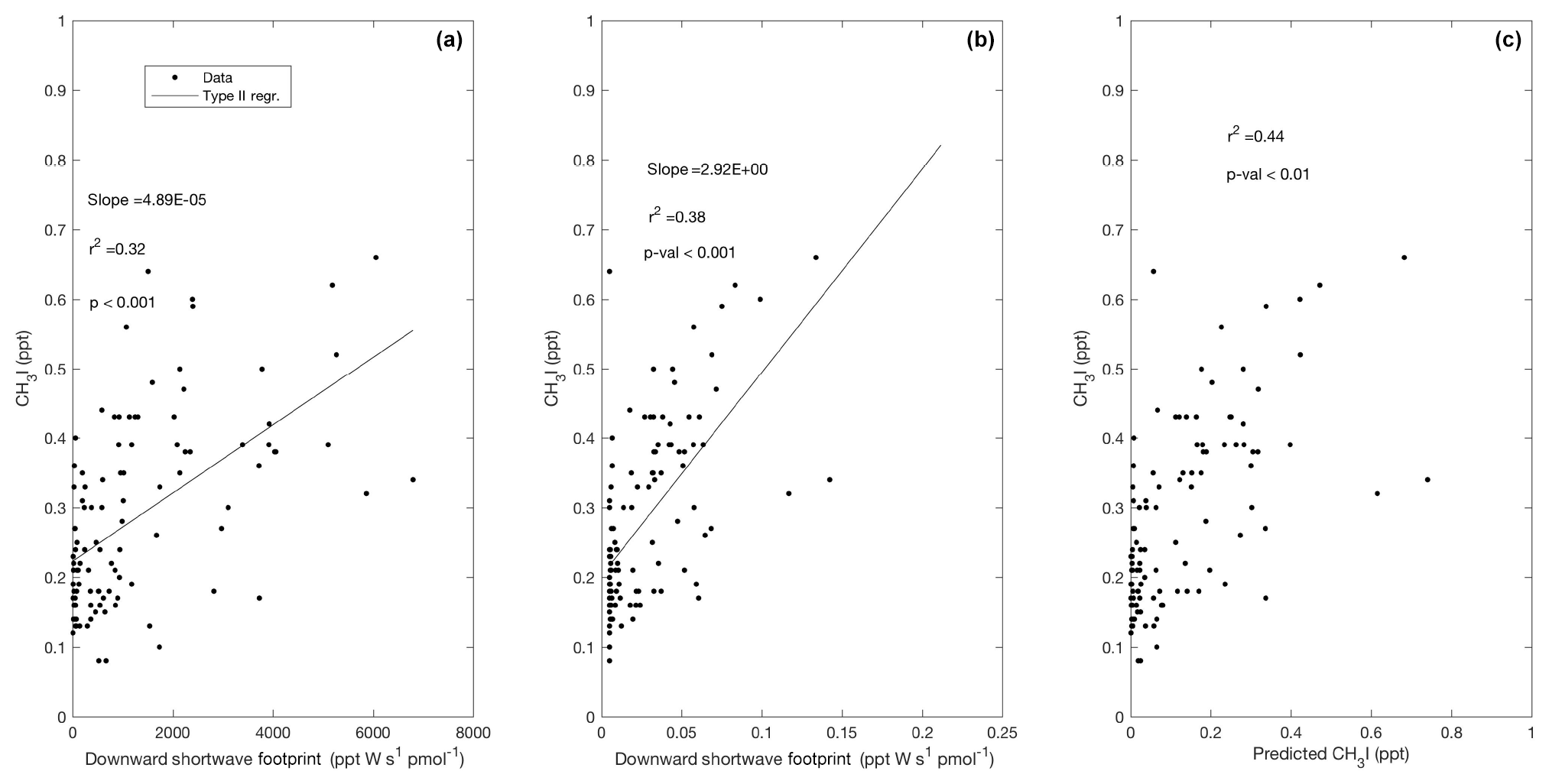
Figure 9Observed CH3I plotted against the surface influence functions of downward shortwave radiation (a) and absorption due to detritus (b). Predicted mixing ratios of CH3I based on a multiple linear regressions (MLRs) using these two predictors in Region 1 are shown in (c) according to Eq. (3). Surface influence (ppt m2 s pmol−1) in each grid cell was multiplied by the surface source field, such as shortwave radiation at the surface (W m−2; ppt W s pmol−1), and the surface ocean's detrital absorption (m−1; ppt m s pmol−1), shown on the x axes.
3.3 Relationships between STILT surface influence functions and observations
We used the STILT model to explore the relationships between observed mixing ratios and the upstream surface influence functions (Eqs. 2–3) of sea ice, chl a, absorption due to detritus, and downward shortwave radiation at the surface, which relate to various regional hypothesized sources of halogenated VOCs such as marine phytoplankton, phytoplankton in sea-ice brines, and decomposing organic matter in surface seawater (e.g., Moore and Zafiriou 1994; Moore et al., 1996; Tokarczyk and Moore, 1994; Sturges et al., 1992).
We found no positive relationships between upstream sea-ice influence and any measured halogenated VOC in Region 1 (Fig. 7). We interpret this result to mean that increased summertime sea ice acts either to reduce the production of halogenated VOCs by blocking sunlight or as a physical barrier to oceanic emissions of halogenated VOCs from under-ice algae. Both of these mechanisms are also consistent with a link between enhanced CHBr3 and CH2Br2 emissions due to sea-ice retreat and surface sea-ice meltwater (Carpenter et al., 2007).

Figure 10Resulting mean January–February 2016 O2-based (parameterized) CHBr3, CH2Br2, and CHClBr2 fluxes (pmol m−2 s−1) in Region 1 (a–c) poleward of 60∘ S and Region 2 (d–f) over the Patagonian Shelf. CESM modeled O2 fluxes are scaled by the slope between the oceanic contribution to δ(O2∕N2) and CHBr3, CH2Br2, and CHClBr2 reported in Fig. 4. Note that these fluxes represent mean estimated biogenic fluxes in January–February 2016 (see Sect. 3.4.1 for details).
In other studies, it has also been proposed that sea ice could be an important source for CHBr3 and other halogenated VOCs, since high mixing ratios of CHBr3 have been observed at the sea-ice and ice–snow interface in the austral winter (Abrahamsson et al., 2018) and in under-ice algae in the austral spring (Sturges et al., 1992). At present, CAM-Chem v1.2 with very short-lived halogen chemistry does not include a regional flux of halogenated VOCs over sea-ice-covered waters in summer, and our results do not indicate a need to include one. Our data, which were collected in January and February, however, cannot assess the importance of sea ice as a source of halogenated VOCs in other seasons, such as winter or spring (Abrahamsson et al., 2018; Sturges et al., 1992). More field campaigns are needed to further study the seasonality and regional strength of sea-ice-related halogenated VOC emissions.
We observed a statistically significant positive correlation between the surface influence function of 8 d satellite composites of chl a concentration, which is widely used as a proxy for near-surface phytoplankton biomass, and mixing ratios of CHBr3 and CH2Br2 in Region 1 (Fig. 8a, b). This corroborates previous findings from shipborne field campaigns and laboratory studies that have suggested a biogenic source for these two bromocarbons (e.g., Moore et al., 1996; Hughes et al., 2013) and further substantiates the current CAM-Chem parameterization of regional bromocarbon emissions using satellite retrievals of chl a in polar regions. CH3Br mixing ratios were not significantly correlated with chl a surface influence functions (Fig. 8c). Although potentially suggesting that marine phytoplankton and microalgae were not a strong regional source of CH3Br during ORCAS, it is also possible that the relatively long lifetime of CH3Br precludes a definitive analysis of its origin based on chl a using 7 d back trajectories. Neither CHClBr2 nor CHBrCl2 was significantly correlated with chl a composite surface influence functions (data not shown); however, more observations of these short-lived species in the remote MBL are needed to substantiate this result.
Similar to Lai et al. (2011), we observed a significant correlation between mixing ratios of CH3I and total weekly upstream influence functions of 8 d chl a composites (Fig. 8d). Weaker correlations were observed with upstream influence functions on timescales shorter than 7 d. We found that CH3I, particularly in Region 1, was better explained by a multi-linear regression with two predictors: (1) the influence function of downward shortwave radiation at the surface (Fig. 9a) and (2) the absorption of light due to detrital material (Fig. 9b), yielding improved agreement between predicted and observed CH3I (Fig. 9c). Several previous studies have correlated mixing ratios of CH3I with satellite retrievals of PAR and surface ocean temperature, revealing a link to solar radiation (e.g., Happell et al., 1996; Yokouchi et al., 2001).
Although certain species of phytoplankton are capable of producing CH3I (e.g., Manley and de la Cuesta, 1997; Hughes et al., 2011), several studies also indicate a nonbiological source for CH3I in the surface ocean. This nonbiological source, though not fully understood, requires light, a humic-like substance at the surface ocean supplying a carbon source and methyl group, and reactive iodine (Moore and Zarifou, 1994; Richter and Wallace, 2004). Thus far, two chemical mechanisms have been proposed for the nonbiological production of methyl iodide; the first is a radical recombination of a methyl group and iodine involving UV photolysis (e.g., Moore and Zarifou, 1994), and the second is a substitution reaction involving the reduction of an oxidant, such as iron III (e.g., Williams et al., 2007).
3.4 Flux estimation
3.4.1 O2-based emission estimates
We present a novel approach that facilitates a basin-wide halogenated VOC flux estimate using the robust relationship between airborne observations of O2 and halogenated VOCs combined with modeled O2 fluxes. Unlike the existing CAM-Chem halogenated VOC biogenic flux parameterization, this method does not rely on weekly retrievals of chl a at high latitudes, which are often patchy. In addition, our study indicates that CHBr3, CH2Br2, and CHClBr2 and CH3I are better correlated with marine-derived O2 than the upstream influence of chl a.
For CHBr3, CH2Br2, and CHClBr2 we construct ocean emission inventories for January and February using a scaled version of gridded modeled air–sea O2 fluxes and the slopes (i.e., molar ratios) of linear correlations between δ(O2∕N2) and halogenated VOC mixing ratios (Fig. 10). O2 fluxes were obtained from simulations using a configuration of CESM nudged to reanalysis temperatures and winds as described in Stephens et al. (2018). An earlier free-running version of CESM was one of the best evaluated for reproducing the seasonal cycle of δ(O2∕N2) over the Southern Ocean (Nevinson et al., 2015, 2016). To date, the north–south gradient in atmospheric O2 has not been well reproduced by any models (Resplandy et al., 2016). Vertical gradients in O2 on ORCAS indicate that CESM overestimated gradients by 47 % on average; accordingly, O2 fluxes were adjusted downward by 47 % to better match the observations. This is obviously a very simple adjustment to the modeled fluxes, and the actual air–sea O2 flux biases in CESM likely have a great deal of spatial and temporal heterogeneity. We calculated an uncertainty for the CESM flux using a second independent estimate of O2 fluxes based on dissolved O2 measurements in surface seawater. The Garcia and Keeling (2001) climatology has much smoother temporal and spatial patterns than CESM flux estimates but also results in overestimated atmospheric O2 spatial gradients. We calculate the relative uncertainty in O2 flux as the ratio of the mean absolute difference between gridded Garcia and Keeling values (2001; also adjusted down by 51 % everywhere to better match ORCAS observations) to the CESM flux estimates in Regions 1 and 2 (adjusted down by 47 % everywhere). These disagreements were 7.3 % and 3.4 % for Regions 1 and 2, respectively. Based on the ratios of halogenated VOC to O2 mixing ratios in bivariate least squares regressions and these adjusted O2 fluxes, we estimate mean emissions of CHBr3 and CH2Br2 in Region 1 and Region 2. Relative uncertainty in the slopes (i.e., the standard deviation of the slopes) from these regressions and the mean relative uncertainties in regional O2 fluxes were added in quadrature to yield uncertainties in calculated halogenated VOC emission rates.
Figure 10 shows the mean emissions for January and February of CHBr3, CH2Br2, and CHClBr2 in Region 1 and Region 2. Mean regional emissions of CHBr3, CH2Br2, and CHClBr2 are 91±8, 31±17, and 11±4 pmol m−2 h−1 in Region 1 and 329±23, 69±5, and 24±5 pmol m−2 h−1 in Region 2 (Table 1). The mean flux of CH3I in Region 2 is 392±32 (Table 1). Table 1 also lists the mean January and February CAM-Chem emissions from Region 1 and Region 2, as well as emissions from several other observational and modeling Antarctic polar studies. Our estimates fall within the range of these other studies, which span every month of the year and whose estimated fluxes range from negative (i.e., from the atmosphere into the ocean) to 3500 pmol m−2 h−1 CHBr3 in a coastal bay during its peak in primary production. CAM-Chem emissions for all species are significantly lower than our observationally derived values in Region 1, with the exception of CH3I. Conversely, CAM-Chem emissions are significantly higher than our estimated emissions in Region 2, with the exception of CHClBr2 in Region 1, which remains underpredicted by the model (Table 1). We note that in Region 2, CAM-Chem fluxes of CHBr3 and CH2Br2, although still significantly different, are more similar to our estimated fluxes.
Table 1Mean ± uncertainty (see Sect. 3.4.1 and 3.4.2 for details) of halogenated VOC emission estimates (pmol m−2 h−1) in Region 1 and Region 2 calculated in this study (with the method indicated below each value), from CAM-Chem (Ordóñez et al., 2012), and from several other modeling and ship-based observational studies.

3.4.2 STILT-based emission estimates
Similar to our O2-based emission estimates, we used the relationship between surface influence functions and CH3I mixing ratios (Fig. 9) to predict a flux field in Region 1 (Fig. 11). We used a multiple linear regression (±1 standard deviations; Eq. 2), wherein Hs1 and Hs2 are the downward shortwave radiation and detrital absorption surface influence functions, respectively, with an intercept , influence coefficients and , and an interaction term with the coefficient (Fig. 9c). These regression coefficients and interaction term were used to estimate an average nonbiological flux of CH3I (Fig. 11; Table 1). This method could be used in place of the current Bell et al. (2002) climatology to update near weekly (∼8 d) emissions of CH3I in future versions of CAM-Chem. Our estimated mean CH3I flux in Region 1 (35±29 pmol m−2 h−1) is significantly lower than the current CAM-Chem estimated emissions (Table 1). As noted in Sect. 3.2, our observations of CH3I are also much lower than the modeled mixing ratios. As discussed above, the strong correlations between CH3I and O2 in Region 2 also suggest a dominant biological source for this compound in this region. As a result, we have not used this relationship to parameterize a flux for CH3I in Region 2 (see Sect. 3.1.2 and 3.4.1 for details). We note that although it would be possible to provide STILT-based emission estimates for other halogenated VOCs (e.g., CHBr3 and CH2Br2), the correlations of these compounds were less strong with surface influence functions than those with O2∕N2.

Figure 11Mean estimated CH3I fluxes for January–February. The multi-linear regression in Fig. 9 between CH3I mixing ratios and geophysical influence functions related to shortwave radiation and detrital material at the sea surface was used to derive a mean flux field in January–February 2016 for Region 1.
Our work combined TOGA and AWAS halogenated VOC airborne observations from the ORCAS and ATom-2 campaigns, with coincident measurements of O2 and CO2, geophysical datasets, and numerical models, including the global atmospheric chemistry model CAM-Chem and the Lagrangian transport model STILT. We evaluated model predictions, calculated molar enrichment ratios, inferred regional sources, and provided novel means of parameterizing ocean fluxes. We found that the Southern Ocean MBL is enriched in halogenated VOCs but that these MBL enhancements are less pronounced at higher latitudes, i.e., poleward of 60∘ S (Region 1), than over the productive Patagonian Shelf (Region 2). Overall, our results indicated that the Southern Ocean is a moderate regional source of CHBr3, CH2Br2, and CH3I and a weak source of CHClBr2 and CHBrCl2 in January and February. Good model–measurement correlations were obtained between our observations and simulations from the Community Earth System Model (CESM) atmospheric component with chemistry (CAM-Chem) for CHBr3, CH2Br2, CH3I, and CHClBr2 but all showed significant differences in model : measurement ratios. The model : measurement comparison for CH3Br was satisfactory and for CHBrCl2 the low levels present precluded us from making a complete assessment.
CHBr3 and CH2Br2 exhibited strong and robust correlations with each other and with O2 and weaker but statistically significant correlations with the influence of chl a, which is a proxy for phytoplankton biomass. CHClBr2 and CHBr3 were well correlated with one another, particularly in Region 2. Together, these correlations suggested a biological source for these gases over the Southern Ocean. We found that CH3I mixing ratios in Region 1 were best correlated with a nonbiological surface influence function, although biogenic CH3I emissions appear important in Region 2.
Our flux estimates based on the relationship of halogenated VOC mixing ratios to O2 and remotely sensed parameters (for CH3I) were compared with those derived from global models and ship-based studies (Table 1). Our emission estimates of CHBr3, CH2Br2, and CHClBr2 are significantly higher than CAM-Chem's globally prescribed emissions in Region 1, where halogenated VOC mixing ratios are underpredicted (Table 1; Fig. 5). Similarly, our estimate of CHClBr2 emissions is also significantly higher than CAM-Chem's in Region 2, where CHClBr2 mixing ratios remained underpredicted. Yet, to the best of our knowledge, CAM-Chem's global parameterization of halogenated VOC fluxes has not been compared with data at high latitudes. Indeed, our emission estimates of CHBr3, CH2Br2, and CH3I fall within a range of CAM-Chem's estimates (on the low end) and most prior estimates based on either other models or localized studies using seawater-side measurements from the Antarctic polar region in summer (on the high end). In the case of CH3I, our estimated emissions suggest that the prescribed emissions in CAM-Chem may be too high in Region 1 and Region 2. Our parameterizations of the CH3I flux could be used to explore interannual variability in emissions, which is not captured by the Bell et al. (2002) CH3I climatology currently employed in CAM-Chem.
To extend these relationships to year-round and global parameterizations for use in global climate models, they must be studied using airborne observations in other seasons and regions. These approaches may help parameterize emissions of new species that can be correlated with surface influence functions or the biological production of oxygen, or they may improve existing emissions where persistent biases exist. Finally, future airborne observations of halogenated VOCs have the potential to further improve our understanding of air–sea flux rates and their drivers for these chemically and climatically important gases over the Southern Ocean.
Data used in this publication are publicly available at https://doi.org/10.5065/D6639N5B (Apel, 2017) and https://doi.org/10.3334/ORNLDAAC/1581 (Wofsy et al., 2018).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-14071-2019-supplement.
EA is responsible for the bulk of the conceptualization, formal analysis, writing, review, and editing with contributions from all authors. BBS and ECA were instrumental in the investigation and supervision related to this paper. RSH contributed to the conceptualization, as well as the investigation and halogenated VOC data curation for this project. BBS, EJM, and RFK were responsible for the curation of δ(O2∕N2) data and contributed to formal analysis involving these data. MSHM and EAK were responsible for STILT data curation and formal analysis, as well as the conceptualization and formal analysis of SITLT-based geostatistical influence functions; flux estimates were also informed by these two. DK, ST, JFL, and ASL were responsible for constructing CAM halogenated VOC emissions and conducting CAM runs. MCL was responsible for CESM simulations yielding O2 fluxes and comparing this product alongside the Garcia and Keeling O2 climatology in CAM. KMC and CM were responsible for the data curation of CO2 observations. AJH contributed to the investigation for halogenated VOC data.
The authors declare that they have no conflict of interest.
We would like to thank the ORCAS and ATom-2 science teams as well as the NCAR Research Aviation Facility and NASA DC-8 pilots, technicians, and mechanics for their support during the field campaigns. In addition, we appreciate the NCAR EOL staff, who facilitated computing and data archival. In particular, we thank Tim Newberger for his help in supporting the NOAA Picarro CO2 observations and Andrew Watt for his help in supporting the AO2 O2 observations. This work was made possible by grants from NSF Polar Programs (1501993, 1501997, 1501292, 1502301, 1543457), NSF Atmospheric Chemistry grants 1535364, 1623745, and 1623748, and NASA funding of the EVS2 Atmospheric Tomography (ATom) project, as well as the support of the NCAR Advanced Study Program (ASP) Postdoctoral Fellowship Program and computing support from Yellowstone, provided by NCAR's Computational and Information Systems Laboratory. The National Center for Atmospheric Research is sponsored by the National Science Foundation.
This research has been supported by the NSF (grant nos. 1501993, 1501997, 1501292, 1502301, and 1543457) and NSF Atmospheric Chemistry (grant nos. 1535364, 1623745, and 1623748).
This paper was edited by Andreas Engel and reviewed by three anonymous referees.
Abrahamsson, K., Lorén, A., Wulff, A., and Wängberg, S.-Å.: Air–sea exchange of halocarbons: the influence of diurnal and regional variations and distribution of pigments, Deep-Sea Res. Pt. II, 51, 2789–2805, https://doi.org/10.1016/j.dsr2.2004.09.005, 2004a.
Abrahamsson, K., Bertilsson, S., Chierici, M., Fransson, A., Froneman, P. W., Lorén, A. and Pakhomov, E. A.: Variations of biochemical parameters along a transect in the Southern Ocean, with special emphasis on volatile halogenated organic compounds, Deep-Sea Res. Pt. II, 51, 2745–2756, https://doi.org/10.1016/j.dsr2.2004.09.004, 2004b.
Abrahamsson, K., Granfors, A., Ahnoff, M., Cuevas, C. A., and Saiz-Lopez, A.: Organic bromine compounds produced in sea ice in Antarctic winter, Nat. Commun., 9, 5291, https://doi.org/10.1038/s41467-018-07062-8, 2018.
Anav, A., Friedlingstein, P., Beer, C., Ciais, P., Harper, A., Jones, C., Murray-Tortarolo, G., Papale, D., Parazoo, N. C., Peylin, P., Piao, S., Sitch, S., Viovy, N., Wiltshire, A., and Zhao, M.: Spatiotemporal patterns of terrestrial gross primary production: A review: GPP Spatiotemporal Patterns, Rev. Geophys., 53, 785–818, https://doi.org/10.1002/2015RG000483, 2015.
Anderson, P. S. and Neff, W. D.: Boundary layer physics over snow and ice, Atmos. Chem. Phys., 8, 3563–3582, https://doi.org/10.5194/acp-8-3563-2008, 2008.
Apel, E.: ORCAS Trace Organic Gas Analyzer (TOGA) VOC Data. Version 1.0, available at: https://data.eol.ucar.edu/dataset/490.018 (last access: 29 January 2019), 2017.
Apel, E. C., Hornbrook, R. S., Hills, A. J., Blake, N. J., Barth, M. C., Weinheimer, A., Cantrell, C., Rutledge, S. A., Basarab, B., Crawford, J., Diskin, G., Homeyer, C. R., Campos, T., Flocke, F., Fried, A., Blake, D. R., Brune, W., Pollack, I., Peischl, J., Ryerson, T., Wennberg, P. O., Crounse, J. D., Wisthaler, A., Mikoviny, T., Huey, G., Heikes, B., O'Sullivan, D., and Riemer, D. D.: Upper tropospheric ozone production from lightning NOx-impacted convection: Smoke ingestion case study from the DC3 campaign, J. Geophys. Res.-Atmos., 120, 2505–2523, https://doi.org/10.1002/2014JD022121, 2015.
Atkinson, H. M., Huang, R.-J., Chance, R., Roscoe, H. K., Hughes, C., Davison, B., Schönhardt, A., Mahajan, A. S., Saiz-Lopez, A., Hoffmann, T., and Liss, P. S.: Iodine emissions from the sea ice of the Weddell Sea, Atmos. Chem. Phys., 12, 11229–11244, https://doi.org/10.5194/acp-12-11229-2012, 2012.
Ayers, G. P.: Comment on regression analysis of air quality data, Atmos. Environ., 35, 2423–2425, https://doi.org/10.1016/S1352-2310(00)00527-6, 2001.
Bates, T. S.: Preface [to special section on First Aerosol Characterization Experiment (AGE 1)], J. Geophys. Res.-Atmos., 104, 21645–21647, https://doi.org/10.1029/1999JD900365, 1999.
Bell, N., Hsu, L., Jacob, D. J., Schultz, M. G., Blake, D. R., Butler, J. H., King, D. B., Lobert, J. M., and Maier-Reimer, E.: Methyl iodide: Atmospheric budget and use as a tracer of marine convection in global models: GLOBAL ATMOSPHERIC METHYL IODIDE, J. Geophys. Res.-Atmos., 107, ACH 8-1–ACH 8-12, https://doi.org/10.1029/2001JD001151, 2002.
Blake, N. J., Blake, D. R., Wingenter, O. W., Sive, B. C., Kang, C. H., Thornton, D. C., Bandy, A. R., Atlas, E., Flocke, F., Harris, J. M., and Rowland, F. S.: Aircraft measurements of the latitudinal, vertical, and seasonal variations of NMHCs, methyl nitrate, methyl halides, and DMS during the First Aerosol Characterization Experiment (ACE 1), J. Geophys. Res.-Atmos., 104, 21803–21817, https://doi.org/10.1029/1999JD900238, 1999.
Bloss, W. J., Lee, J. D., Johnson, G. P., Sommariva, R., Heard, D. E., Saiz-Lopez, A., Plane, J. M. C., McFiggans, G., Flynn, M., Williams, P., Rickard, A. R., and Fleming, Z. L.: Impact of halogen monoxide chemistry upon boundary layer OH and HO2 concentrations at a coastal site, Geophys. Res. Lett., 32, L06814, https://doi.org/10.1029/2004GL022084, 2005.
Boden, T., Andres, R., and Marland, G.: Global, Regional, and National Fossil-Fuel CO2 Emissions (1751–2014) (V. 2017), available at: https://www.osti.gov/servlets/purl/1389331/ (last access: 25 November 2018), 2017.
Boucher, O., Moulin, C., Belviso, S., Aumont, O., Bopp, L., Cosme, E., von Kuhlmann, R., Lawrence, M. G., Pham, M., Reddy, M. S., Sciare, J., and Venkataraman, C.: DMS atmospheric concentrations and sulphate aerosol indirect radiative forcing: a sensitivity study to the DMS source representation and oxidation, Atmos. Chem. Phys., 3, 49–65, https://doi.org/10.5194/acp-3-49-2003, 2003.
Butler, J. H., King, D. B., Lobert, J. M., Montzka, S. A., Yvon-Lewis, S. A., Hall, B. D., Warwick, N. J., Mondeel, D. J., Aydin, M., and Elkins, J. W.: Oceanic distributions and emissions of short-lived halocarbons: OCEANIC EMISSIONS OF SHORT-LIVED HALOCARBONS, Global Biogeochem. Cy., 21, GB1023, https://doi.org/10.1029/2006GB002732, 2007.
Cantrell, C. A.: Technical Note: Review of methods for linear least-squares fitting of data and application to atmospheric chemistry problems, Atmos. Chem. Phys., 8, 5477–5487, https://doi.org/10.5194/acp-8-5477-2008, 2008.
Carpenter, L. J., Liss, P. S., and Penkett, S. A.: Marine organohalogens in the atmosphere over the Atlantic and Southern Oceans: MARINE ORGANOHALOGENS IN THE ATMOSPHERE, J. Geophys. Res.-Atmos., 108, 4256, https://doi.org/10.1029/2002JD002769, 2003.
Carpenter, L. J., Wevill, D. J., Palmer, C. J., and Michels, J.: Depth profiles of volatile iodine and bromine-containing halocarbons in coastal Antarctic waters, Mar. Chem., 103, 227–236, https://doi.org/10.1016/j.marchem.2006.08.003, 2007.
Carpenter, L. J., Jones, C. E., Dunk, R. M., Hornsby, K. E., and Woeltjen, J.: Air-sea fluxes of biogenic bromine from the tropical and North Atlantic Ocean, Atmos. Chem. Phys., 9, 1805–1816, https://doi.org/10.5194/acp-9-1805-2009, 2009.
Chuck, A. L.: Oceanic distributions and air-sea fluxes of biogenic halocarbons in the open ocean, J. Geophys. Res., 110, C10022, https://doi.org/10.1029/2004JC002741, 2005.
Colomb, A., Yassaa, N., Williams, J., Peeken, I., and Lochte, K.: Screening volatile organic compounds (VOCs) emissions from five marine phytoplankton species by head space gas chromatography/mass spectrometry (HS-GC/MS), J. Environ. Monitor., 10, 325, https://doi.org/10.1039/b715312k, 2008.
Drewer, J., Heal, K. V., Smith, K. A., and Heal, M. R.: Methyl bromide emissions to the atmosphere from temperate woodland ecosystems, Glob. Change Biol., 14, 2539–2547, https://doi.org/10.1111/j.1365-2486.2008.01676.x, 2008.
Engel, A., Rigby, M., Burkholder, J. B., Fernandez, R. P., Froidevaux, L., Hall, B. D., Hossaini, R., Saito, T., Vollmer, M. K., and Yao, B.: Update on Ozone-Depleteing Substances (ODSs) and Other Gases of Interest to the Montreal Protocol, chap. 1, in: Scientific Assessment of Ozone Depletion: 2018, Global Ozone Research and Monitoring Project-Report No. 58, World Meteorological Organization, Geneva, Switzerland, 2018.
Fernandez, R. P., Salawitch, R. J., Kinnison, D. E., Lamarque, J.-F., and Saiz-Lopez, A.: Bromine partitioning in the tropical tropopause layer: implications for stratospheric injection, Atmos. Chem. Phys., 14, 13391–13410, https://doi.org/10.5194/acp-14-13391-2014, 2014.
Finlayson-Pitts, B. J.: The Tropospheric Chemistry of Sea Salt: A Molecular-Level View of the Chemistry of NaCl and NaBr, Chem. Rev., 103, 4801–4822, https://doi.org/10.1021/cr020653t, 2003.
Garcia, H. E. and Keeling, R. F.: On the global oxygen anomaly and air-sea flux, J. Geophys. Res.-Oceans, 106, 31155–31166, https://doi.org/10.1029/1999JC000200, 2001.
Glover, D. M., Jenkins, W. J. and Doney, S. C.: Modeling Methods for Marine Science, Cambridge University Press, Cambridge, 2011.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Happell, J. D., Wallace, D. W. R., Wills, K. D., Wilke, R. J., and Neill, C. C.: A purge-and-trap capillary column gas chromatographic method for the measurement of halocarbons in water and air, available at: http://www.osti.gov/servlets/purl/366493-84sOfy/webviewable/ (last access: 26 July 2018), 1996.
Hossaini, R., Mantle, H., Chipperfield, M. P., Montzka, S. A., Hamer, P., Ziska, F., Quack, B., Krüger, K., Tegtmeier, S., Atlas, E., Sala, S., Engel, A., Bönisch, H., Keber, T., Oram, D., Mills, G., Ordóñez, C., Saiz-Lopez, A., Warwick, N., Liang, Q., Feng, W., Moore, F., Miller, B. R., Marécal, V., Richards, N. A. D., Dorf, M., and Pfeilsticker, K.: Evaluating global emission inventories of biogenic bromocarbons, Atmos. Chem. Phys., 13, 11819–11838, https://doi.org/10.5194/acp-13-11819-2013, 2013.
Hughes, C., Chuck, A. L., Rossetti, H., Mann, P. J., Turner, S. M., Clarke, A., Chance, R., and Liss, P. S.: Seasonal cycle of seawater bromoform and dibromomethane concentrations in a coastal bay on the western Antarctic Peninsula: BROMOCARBON SEASONALITY ANTARCTICA, Global Biogeochem. Cy., 23, GB2024, https://doi.org/10.1029/2008GB003268, 2009.
Hughes, C., Franklin, D. J., and Malin, G.: Iodomethane production by two important marine cyanobacteria: Prochlorococcus marinus (CCMP 2389) and Synechococcus sp. (CCMP 2370), Mar. Chem., 125, 19–25, doi:10.1016/j.marchem.2011.01.007, 2011.
Hughes, C., Johnson, M., Utting, R., Turner, S., Malin, G., Clarke, A., and Liss, P. S.: Microbial control of bromocarbon concentrations in coastal waters of the western Antarctic Peninsula, Mar. Chem., 151, 35–46, https://doi.org/10.1016/j.marchem.2013.01.007, 2013.
Hurrell, J. W., Hack, J. J., Shea, D., Caron, J. M., and Rosinski, J.: A New Sea Surface Temperature and Sea Ice Boundary Dataset for the Community Atmosphere Model, J. Climate, 21, 5145–5153, https://doi.org/10.1175/2008JCLI2292.1, 2008.
Karion, A., Sweeney, C., Wolter, S., Newberger, T., Chen, H., Andrews, A., Kofler, J., Neff, D., and Tans, P.: Long-term greenhouse gas measurements from aircraft, Atmos. Meas. Tech., 6, 511–526, https://doi.org/10.5194/amt-6-511-2013, 2013.
Keeling, R. F., Manning, A. C., McEvoy, E. M., and Shertz, S. R.: Methods for measuring changes in atmospheric O2 concentration and their application in southern hemisphere air, J. Geophys. Res.-Atmos., 103, 3381–3397, https://doi.org/10.1029/97JD02537, 1998.
Lai, S. C., Williams, J., Arnold, S. R., Atlas, E. L., Gebhardt, S., and Hoffmann, T.: Iodine containing species in the remote marine boundary layer: A link to oceanic phytoplankton: IODINE SPECIES AND PHYTOPLANKTON, Geophys. Res. Lett., 38, L20801, https://doi.org/10.1029/2011GL049035, 2011.
Lamarque, J.-F., Emmons, L. K., Hess, P. G., Kinnison, D. E., Tilmes, S., Vitt, F., Heald, C. L., Holland, E. A., Lauritzen, P. H., Neu, J., Orlando, J. J., Rasch, P. J., and Tyndall, G. K.: CAM-chem: description and evaluation of interactive atmospheric chemistry in the Community Earth System Model, Geosci. Model Dev., 5, 369–411, https://doi.org/10.5194/gmd-5-369-2012, 2012.
Laturnus, F.: Volatile halocarbons released from Arctic macroalgae, Mar. Chem., 55, 359–366, https://doi.org/10.1016/S0304-4203(97)89401-7, 1996.
Liang, Q., Atlas, E., Blake, D., Dorf, M., Pfeilsticker, K., and Schauffler, S.: Convective transport of very short lived bromocarbons to the stratosphere, Atmos. Chem. Phys., 14, 5781–5792, https://doi.org/10.5194/acp-14-5781-2014, 2014.
Lin, J. C.: A near-field tool for simulating the upstream influence of atmospheric observations: The Stochastic Time-Inverted Lagrangian Transport (STILT) model, J. Geophys. Res., 108, ACH 2-1–ACH 2-17, https://doi.org/10.1029/2002JD003161, 2003.
Manley, S. L. and Dastoor, M. N.: Methyl iodide (CH3I) production by kelp and associated microbes, Mar. Biol., 98, 477–482, https://doi.org/10.1007/BF00391538, 1988.
Manley, S. L. and de la Cuesta, J. L.: Methyl iodide production from marine phytoplankton cultures, Limnol. Oceanogr., 42, 142–147, https://doi.org/10.4319/lo.1997.42.1.0142, 1997.
Maslanik, J.: Near-Real-Time DMSP SSM/I-SSMIS Daily Polar Gridded Sea Ice Concentrations, Version 1, NASA National Snow and Ice Data Center Distributed Archive Center, Boulder, Colorado USA, https://doi.org/10.5067/U8C09DWVX9LM, 1999.
Mattsson, E., Karlsson, A., and Abrahamsson, K.: Regional sinks of bromoform in the Southern Ocean: REGIONAL SINKS OF CHBR3 IN THE ANTARCTIC, Geophys. Res. Lett., 40, 3991–3996, https://doi.org/10.1002/grl.50783, 2013.
Moore, R. M. and Zafiriou, O. C.: Photochemical production of methyl iodide in seawater, J. Geophys. Res., 99, 16415, https://doi.org/10.1029/94JD00786, 1994.
Moore, R. M., Webb, M., Tokarczyk, R., and Wever, R.: Bromoperoxidase and iodoperoxidase enzymes and production of halogenated methanes in marine diatom cultures, J. Geophys. Res.-Oceans, 101, 20899–20908, https://doi.org/10.1029/96JC01248, 1996.
Murphy, D. M., Froyd, K. D., Bian, H., Brock, C. A., Dibb, J. E., DiGangi, J. P., Diskin, G., Dollner, M., Kupc, A., Scheuer, E. M., Schill, G. P., Weinzierl, B., Williamson, C. J., and Yu, P.: The distribution of sea-salt aerosol in the global troposphere, Atmos. Chem. Phys., 19, 4093–4104, https://doi.org/10.5194/acp-19-4093-2019, 2019.
NASA Goddard Space Flight Center, O. E. L.: Sea-viewing Wide Field-of-view Sensor (SeaWiFS) Ocean Color Data, NASA OB.DAAC, https://doi.org/10.5067/ORBVIEW-2/SEAWIFS_OC.2014.0, 2014.
NCEP (National Centers For Environmental Prediction/National Weather Service/NOAA/U.S. Department Of Commerce): Research Data Archive at the National Center for Atmospheric Research, Computational and Information Systems Laboratory, https://doi.org/10.5065/D65Q4T4Z, 2015.
Navarro, M. A., Atlas, E. L., Saiz-Lopez, A., Rodriguez-Lloveras, X., Kinnison, D. E., Lamarque, J.-F., Tilmes, S., Filus, M., Harris, N. R. P., Meneguz, E., Ashfold, M. J., Manning, A. J., Cuevas, C. A., Schauffler, S. M., and Donets, V.: Airborne measurements of organic bromine compounds in the Pacific tropical tropopause layer, P. Natl. Acad. Sci. USA, 112, 13789–13793, https://doi.org/10.1073/pnas.1511463112, 2015.
Neale, R. B., Richter, J., Park, S., Lauritzen, P. H., Vavrus, S. J., Rasch, P. J., and Zhang, M.: The Mean Climate of the Community Atmosphere Model (CAM4) in Forced SST and Fully Coupled Experiments, J. Climate, 26, 5150–5168, https://doi.org/10.1175/JCLI-D-12-00236.1, 2013.
Nevison, C. D., Manizza, M., Keeling, R. F., Kahru, M., Bopp, L., Dunne, J., Tiputra, J., Ilyina, T., and Mitchell, B. G.: Evaluating the ocean biogeochemical components of Earth system models using atmospheric potential oxygen and ocean color data, Biogeosciences, 12, 193–208, https://doi.org/10.5194/bg-12-193-2015, 2015.
Nevison, C. D., Manizza, M., Keeling, R. F., Stephens, B. B., Bent, J. D., Dunne, J., Ilyina, T., Long, M., Resplandy, L., Tjiputra, J., and Yukimoto, S.: Evaluating CMIP5 ocean biogeochemistry and Southern Ocean carbon uptake using atmospheric potential oxygen: Present-day performance and future projection: CMIP5 APO AND SOUTHERN OCEAN CARBON FLUX, Geophys. Res. Lett., 43, 2077–2085, https://doi.org/10.1002/2015GL067584, 2016.
Nightingale, P. D., Malin, G., and Liss, P. S.: Production of chloroform and other low molecular-weight halocarbons by some species of macroalgae, Limnol. Oceanogr., 40, 680–689, https://doi.org/10.4319/lo.1995.40.4.0680, 1995.
Obrist, D., Tas, E., Peleg, M., Matveev, V., Faïn, X., Asaf, D., and Luria, M.: Bromine-induced oxidation of mercury in the mid-latitude atmosphere, Nat. Geosci., 4, 22, https://doi.org/10.1038/ngeo1018, 2010.
Ordóñez, C., Lamarque, J.-F., Tilmes, S., Kinnison, D. E., Atlas, E. L., Blake, D. R., Sousa Santos, G., Brasseur, G., and Saiz-Lopez, A.: Bromine and iodine chemistry in a global chemistry-climate model: description and evaluation of very short-lived oceanic sources, Atmos. Chem. Phys., 12, 1423–1447, https://doi.org/10.5194/acp-12-1423-2012, 2012.
Raimund, S., Quack, B., Bozec, Y., Vernet, M., Rossi, V., Garçon, V., Morel, Y., and Morin, P.: Sources of short-lived bromocarbons in the Iberian upwelling system, Biogeosciences, 8, 1551–1564, https://doi.org/10.5194/bg-8-1551-2011, 2011.
Resplandy, L., Keeling, R. F., Stephens, B. B., Bent, J. D., Jacobson, A., Rödenbeck, C., and Khatiwala, S.: Constraints on oceanic meridional heat transport from combined measurements of oxygen and carbon, Clim. Dynam., 47, 3335–3357, https://doi.org/10.1007/s00382-016-3029-3, 2016.
Reygondeau, G., Longhurst, A., Martinez, E., Beaugrand, G., Antoine, D., and Maury, O.: Dynamic biogeochemical provinces in the global ocean: DYNAMIC BIOGEOCHEMICAL PROVINCES, Global Biogeochem. Cy., 27, 1046–1058, https://doi.org/10.1002/gbc.20089, 2013.
Richter, U. and Wallace, D. W. R.: Production of methyl iodide in the tropical Atlantic Ocean: PRODUCTION OF METHYL IODIDE, Geophys. Res. Lett., 31, L23S03, https://doi.org/10.1029/2004GL020779, 2004.
Rienecker, M. M., Suarez, M. J., Todling, R., Bacmeister, J., Takacs, L., Liu, H. C., Gu, W., Sienkiewicz, M., Koster, R. D., Gelaro, R., Stajner, I., and Nielsen, J. E.: The GEOS-5 Data Assimilation System – Documentation of Versions 5.0.1, 5.1.0, and 5.2.0, NASA/TM-2008-104606, 2008.
Saiz-Lopez, A., Mahajan, A. S., Salmon, R. A., Bauguitte, S. J.-B., Jones, A. E., Roscoe, H. K., and Plane, J. M. C.: Boundary Layer Halogens in Coastal Antarctica, Science, 317, 348–351, https://doi.org/10.1126/science.1141408, 2007.
Saiz-Lopez, A., Fernandez, R. P., Ordóñez, C., Kinnison, D. E., Gómez Martín, J. C., Lamarque, J.-F., and Tilmes, S.: Iodine chemistry in the troposphere and its effect on ozone, Atmos. Chem. Phys., 14, 13119–13143, https://doi.org/10.5194/acp-14-13119-2014, 2014.
Salawitch, R. J., Canty, T., Kurosu, T., Chance, K., Liang, Q., da Silva, A., Pawson, S., Nielsen, J. E., Rodriguez, J. M., Bhartia, P. K., Liu, X., Huey, L. G., Liao, J., Stickel, R. E., Tanner, D. J., Dibb, J. E., Simpson, W. R., Donohoue, D., Weinheimer, A., Flocke, F., Knapp, D., Montzka, D., Neuman, J. A., Nowak, J. B., Ryerson, T. B., Oltmans, S., Blake, D. R., Atlas, E. L., Kinnison, D. E., Tilmes, S., Pan, L. L., Hendrick, F., Van Roozendael, M., Kreher, K., Johnston, P. V., Gao, R. S., Johnson, B., Bui, T. P., Chen, G., Pierce, R. B., Crawford, J. H., and Jacob, D. J.: A new interpretation of total column BrO during Arctic spring: FRONTIER, Geophys. Res. Lett., 37, L21805, https://doi.org/10.1029/2010GL043798, 2010.
Schauffler, S. M., Atlas, E. L., Blake, D. R., Flocke, F., Lueb, R. A., Lee-Taylor, J. M., Stroud, V. and Travnicek, W.: Distributions of brominated organic compounds in the troposphere and lower stratosphere, J. Geophys. Res.-Atmos., 104, 21513–21535, https://doi.org/10.1029/1999JD900197, 1999.
Schroeder, W. H., Anlauf, K. G., Barrie, L. A., Lu, J. Y., Steffen, A., Schneeberger, D. R., and Berg, T.: Arctic springtime depletion of mercury, Nature, 394, 331, https://doi.org/10.1038/28530, 1998.
Schnell, R. C., Barry, R. G., Miles, M. W., Andreas, E. L., Radke, L. F., Brock, C. A., McCormick, M. P., and Moore, J. L.: Lidar detection of leads in Arctic sea ice, Nature, 339, 530–532, https://doi.org/10.1038/339530a0, 1989.
Simpson, W. R., Brown, S. S., Saiz-Lopez, A., Thornton, J. A., and von Glasow, R.: Tropospheric Halogen Chemistry: Sources, Cycling, and Impacts, Chem. Rev., 115, 4035–4062, https://doi.org/10.1021/cr5006638, 2015.
Sive, B. C., Varner, R. K., Mao, H., Blake, D. R., Wingenter, O. W., and Talbot, R.: A large terrestrial source of methyl iodide, Geophys. Res. Lett., 34, L17808, https://doi.org/10.1029/2007GL030528, 2007.
Stephens, B.: ORCAS Merge Products, Version 1.0, available at: https://data.eol.ucar.edu/dataset/490.024 (last access: 31 December 2018), 2017.
Stephens, B. B., Keeling, R. F., Heimann, M., Six, K. D., Murnane, R., and Caldeira, K.: Testing global ocean carbon cycle models using measurements of atmospheric O2 and CO2 concentration, Global Biogeochem. Cy., 12, 213–230, https://doi.org/10.1029/97GB03500, 1998.
Stephens, B. B., Keeling, R. F., and Paplawsky, W. J.: Shipboard measurements of atmospheric oxygen using a vacuum-ultraviolet absorption technique, Tellus B, 55, 857–878, https://doi.org/10.1046/j.1435-6935.2003.00075.x, 2003.
Stephens, B. B., Long, M. C., Keeling, R. F., Kort, E. A., Sweeney, C., Apel, E. C., Atlas, E. L., Beaton, S., Bent, J. D., Blake, N. J., Bresch, J. F., Casey, J., Daube, B. C., Diao, M., Diaz, E., Dierssen, H., Donets, V., Gao, B.-C., Gierach, M., Green, R., Haag, J., Hayman, M., Hills, A. J., Hoecker-Martínez, M. S., Honomichl, S. B., Hornbrook, R. S., Jensen, J. B., Li, R.-R., McCubbin, I., McKain, K., Morgan, E. J., Nolte, S., Powers, J. G., Rainwater, B., Randolph, K., Reeves, M., Schauffler, S. M., Smith, K., Smith, M., Stith, J., Stossmeister, G., Toohey, D. W., and Watt, A. S.: The O2∕N2 Ratio and CO2 Airborne Southern Ocean Study, B. Am. Meteorol. Soc., 99, 381–402, https://doi.org/10.1175/BAMS-D-16-0206.1, 2018.
Sturges, W. T., Cota, G. F., and Buckley, P. T.: Bromoform emission from Arctic ice algae, Nature, 358, 660–662, 1992.
Tilmes, S., Lamarque, J.-F., Emmons, L. K., Kinnison, D. E., Marsh, D., Garcia, R. R., Smith, A. K., Neely, R. R., Conley, A., Vitt, F., Val Martin, M., Tanimoto, H., Simpson, I., Blake, D. R., and Blake, N.: Representation of the Community Earth System Model (CESM1) CAM4-chem within the Chemistry-Climate Model Initiative (CCMI), Geosci. Model Dev., 9, 1853–1890, https://doi.org/10.5194/gmd-9-1853-2016, 2016.
Tokarczyk, R. and Moore, R. M.: Production of volatile organohalogens by phytoplankton cultures, Geophys. Res. Lett., 21, 285–288, https://doi.org/10.1029/94GL00009, 1994.
Tortell, P. D. and Long, M. C.: Spatial and temporal variability of biogenic gases during the Southern Ocean spring bloom, Geophys. Res. Lett., 36, L01603, https://doi.org/10.1029/2008GL035819, 2009.
Tortell, P. D., Asher, E. C., Ducklow, H. W., Goldman, J. A. L., Dacey, J. W. H., Grzymski, J. J., Young, J. N., Kranz, S. A., Bernard, K. S., and Morel, F. M. M.: Metabolic balance of coastal Antarctic waters revealed by autonomous pCO2 and ΔO2∕Ar measurements: metabolic balence of Antarctic waters, Geophys. Res. Lett., 41, 6803–6810, https://doi.org/10.1002/2014GL061266, 2014.
von Glasow, R. and Crutzen, P. J.: Model study of multiphase DMS oxidation with a focus on halogens, Atmos. Chem. Phys., 4, 589–608, https://doi.org/10.5194/acp-4-589-2004, 2004.
Wiedinmyer, C., Akagi, S. K., Yokelson, R. J., Emmons, L. K., Al-Saadi, J. A., Orlando, J. J., and Soja, A. J.: The Fire INventory from NCAR (FINN): a high resolution global model to estimate the emissions from open burning, Geosci. Model Dev., 4, 625–641, https://doi.org/10.5194/gmd-4-625-2011, 2011.
Williams, J., Gros, V., Atlas, E., Maciejczyk, K., Batsaikhan, A., Schöler, H. F., Forster, C., Quack, B., Yassaa, N., Sander, R., and Van Dingenen, R.: Possible evidence for a connection between methyl iodide emissions and Saharan dust, J. Geophys. Res., 112, D07302, https://doi.org/10.1029/2005JD006702, 2007.
Wofsy, S. C.: HIAPER Pole-to-Pole Observations (HIPPO): fine-grained, global-scale measurements of climatically important atmospheric gases and aerosols, Philos. T. R. Soc. A, 369, 2073–2086, https://doi.org/10.1098/rsta.2010.0313, 2011.
Wofsy, S. C., Afshar, S., Allen, H. M., Apel, E., Asher, E. C., Barletta, B., Bent, J., Bian, H., Biggs, B. C., Blake, D. R., Blake, N., Bourgois, I., Brock, C. A., Brune, W. H., Budney, J. W., Bui, T. P., Butler, A., Campuzano-Jost, P., Chang, C. S., Chin, M., Commane, R., Correa, G., Crounse, J. D., Cullis, P. D., Daube, B. C., Day, D. A., Dean-Day, J. M., Dibb, J. E., Digangi, J. P., Diskin, G. S., Dollner, M., Elkins, J. W., Erdesz, F., Fiore, A. M., Flynn, C. M., Froyd, K., Gesler, D. W., Hall, S. R., Hanisco, T. F., Hannun, R. A., Hills, A. J., Hintsa, E. J., Hoffmann, A., Hornbrook, R. S., Huey, L. G., Hughes, S., Jimenez, J. L., Johnson, B. J., Katich, J. M., Keeling, R., Kim, M. J., Kupc, A., Lait, L. R., Lamarque, J.-F., Liu, H. B., McKain, K., Mclaughlin, R. J., Meinardi, S., Miller, D. O., Montzka, S. A., Moore, F. L., Morgan, E. J., Murphy, D. M., Murray, L. T., Nault, B. A., Neuman, J. A., Newman, P. A., Nicely, J. M., Pan, X., Paplawsky, W., Peischl, J., Prather, M. J., Price, D. J., Ray, E., Reeves, J. M., Richardson, M., Rollins, A. W., Rosenlof, K. H., Ryerson, T. B., Scheuer, E., Schill, G. P., Schroder, J. C., Schwarz, J. P., St.Clair, J. M., Steenrod, S. D., Stephens, B. B., Strode, S. A., Sweeney, C., Tanner, D., Teng, A. P., Thames, A. B., Thompson, C. R., Ullmann, K., Veres, P. R., Vizenor, N., Wagner, N. L., Watt, A., Weber, R., Weinzierl, B., Wennberg, P., Williamson, C. J., Wilson, J. C., Wolfe, G. M., Woods, C. T., and Zeng, L. H.: ATom: Merged Atmospheric Chemistry, Trace Gases, and Aerosols, ORNL DAAC, Oak Ridge, Tennessee, USA, https://doi.org/10.3334/ORNLDAAC/1581, 2018.
Xiang, B., Miller, S. M., Kort, E. A., Santoni, G. W., Daube, B. C., Commane, R., Angevine, W. M., Ryerson, T. B., Trainer, M. K., Andrews, A. E., Nehrkorn, T., Tian, H., and Wofsy, S. C.: Nitrous oxide (N2O) emissions from California based on 2010 CalNex airborne measurements: California N2O emissions, J. Geophys. Res.-Atmos., 118, 2809–2820, https://doi.org/10.1002/jgrd.50189, 2013.
Yokouchi, Y., Nojiri, Y., Barrie, L. A., Toom-Sauntry, D., and Fujinuma, Y.: Atmospheric methyl iodide: High correlation with surface seawater temperature and its implications on the sea-to-air flux, J. Geophys. Res.-Atmos., 106, 12661–12668, https://doi.org/10.1029/2001JD900083, 2001.
Yokouchi, Y., Hasebe, F., Fujiwara, M., Takashima, H., Shiotani, M., Nishi, N., Kanaya, Y., Hashimoto, S., Fraser, P., Toom-Sauntry, D., Mukai, H., and Nojiri, Y.: Correlations and emission ratios among bromoform, dibromochloromethane, and dibromomethane in the atmosphere, J. Geophys. Res., 110, D23309, https://doi.org/10.1029/2005JD006303, 2005.
Ziska, F., Quack, B., Abrahamsson, K., Archer, S. D., Atlas, E., Bell, T., Butler, J. H., Carpenter, L. J., Jones, C. E., Harris, N. R. P., Hepach, H., Heumann, K. G., Hughes, C., Kuss, J., Krüger, K., Liss, P., Moore, R. M., Orlikowska, A., Raimund, S., Reeves, C. E., Reifenhäuser, W., Robinson, A. D., Schall, C., Tanhua, T., Tegtmeier, S., Turner, S., Wang, L., Wallace, D., Williams, J., Yamamoto, H., Yvon-Lewis, S., and Yokouchi, Y.: Global sea-to-air flux climatology for bromoform, dibromomethane and methyl iodide, Atmos. Chem. Phys., 13, 8915–8934, https://doi.org/10.5194/acp-13-8915-2013, 2013.