the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 11 Oct 2019
| 11 Oct 2019
Chemical composition and hydrolysis of organic nitrate aerosol formed from hydroxyl and nitrate radical oxidation of α-pinene and β-pinene
Masayuki Takeuchi
Atmospheric organic nitrate (ON) is thought to play a crucial role in the formation potential of ozone and aerosol, which are the leading air pollutants of concern across the world. Limited fundamental knowledge and understanding of the life cycles of ON currently hinder the ability to quantitatively assess its impacts on the formation of these pollutants. Although hydrolysis is currently considered an important loss mechanism of ON based on prior field measurement studies, this process for atmospherically relevant ON has not been well constrained by fundamental laboratory studies. In this comprehensive study, we investigated the chemical composition and hydrolysis process of particulate ON (pON) formed from the oxidation of α-pinene and β-pinene by hydroxyl (OH•) and nitrate radicals (NO3•). For pON that undergoes hydrolysis, the hydrolysis lifetime is determined to be no more than 30 min for all systems explored. This is significantly shorter than those reported in previous chamber studies (i.e., 3–6 h) but is consistent with the reported lifetime from bulk solution measurement studies (i.e., 0.02–8.8 h). The discrepancy appears to stem from the choice of proxy used to estimate the hydrolysis lifetime. The measured hydrolyzable fractions of pON (FH) in the α-pinene + OH•, β-pinene + OH•, α-pinene + NO3•, and β-pinene + NO3• systems are 23 %–32 %, 27 %–34 %, 9 %–17 %, and 9 %–15 %, respectively. While a very low FH for the NO3• oxidation system is expected based on prior studies, FH for the OH• oxidation system is surprisingly lower than predicted in past studies. Overall, the hydrolysis lifetime as well as FH obtained in this study serve as experimentally constrained parameters that are required in regional and global chemical transport models to accurately evaluate the impacts of ON on nitrogen budget and formation of ozone and aerosol.
- Article
(4474 KB) - Full-text XML
-
Supplement
(1077 KB) - BibTeX
- EndNote





The oxidation of biogenic volatile organic compounds (BVOCs) by ozone (O3), hydroxyl radicals (OH•), and nitrate radicals (NO3•) is a major source of secondary organic aerosol (SOA) globally (Kanakidou et al., 2005; Goldstein and Galbally, 2007; Spracklen et al., 2011). Many studies have pointed to the synergistic effects of anthropogenic emissions on biogenic SOA formation in the atmosphere (Weber et al., 2007; Carlton et al., 2010; Hoyle et al., 2011; Shilling et al., 2013; Xu et al., 2015a; Shrivastava et al., 2019). The oxidation of BVOC in environments with anthropogenic NOx emissions is an important mechanism for coupled biogenic–anthropogenic interactions. In the presence of NOx, the oxidation of BVOC can lead to the formation of organic nitrate (ON), a large component of reactive oxidized nitrogen. Results from ambient field measurements have revealed the ubiquitous presence of particulate ON (pON), where it contributes to a large fraction of submicron organic aerosol at different sites worldwide (Fry et al., 2013; Xu et al., 2015b; Liu et al., 2012; Rollins et al., 2012, 2013; Lee et al., 2016; Kiendler-Scharr et al., 2016; Ng et al., 2017). These findings highlight the importance to understand the formation and fates of ON to accurately evaluate its roles in NOx recycling, O3 formation, and SOA formation.
Monoterpenes (C10H16) are a major class of BVOC with annual emissions of 157–177 Tg C yr−1 (Guenther et al., 2012). Laboratory studies have demonstrated that the NO3• oxidation of monoterpenes leads to a substantial formation of ON and SOA with ON yields up to ∼70 % (Wangberg et al., 1997; Berndt and Boge, 1997; Griffin et al., 1999; Hallquist et al., 1999; Spittler et al., 2006; Fry et al., 2009, 2014; Boyd et al., 2015, 2017; Nah et al., 2016; Slade et al., 2017; Claflin and Ziemann, 2018). For photooxidation of monoterpenes in the presence of NOx, ON yields as high as 26 % have been reported for α-pinene (Noziere et al., 1999; Aschmann et al., 2002; Rindelaub et al., 2015). Monoterpene emissions do not depend strongly on light and typically continue at night, making them important ON and SOA precursors at any time of day (daytime and nighttime) and throughout the year (different seasons). It has been shown that monoterpene-derived ON is prevalent in areas where there are substantial biogenic–anthropogenic interactions and oxidation of monoterpenes contributes to a large fraction of SOA observed in the Southeastern US (L. Xu et al., 2015a, b, 2018; Lee et al., 2016; Zhang et al., 2018).
One of the largest uncertainties in our understanding of monoterpene ON chemistry is the extent to which ON acts as a permanent sink versus temporary reservoir of NOx (Takeuchi and Ng, 2018). This would depend on the fates of ON as they can either retain or release NOx upon further reactions. Once formed, gas-phase ON can undergo photolysis or OH• oxidation to release NOx or partition into the particle phase. pON in turn can undergo further chemistry to release NOx or hydrolyze in the particle phase to form nitric acid (HNO3). Further, ON and HNO3 can be removed via dry and wet deposition. One important reaction of ON in the particle phase is hydrolysis in the presence of aerosol water, which is a mechanism for NOx loss (Day et al., 2010; Russell et al., 2011). Studies with bulk solutions showed that particle-phase hydrolysis of tertiary nitrate is fast with a lifetime of minutes, while primary and secondary nitrate is stable (Darer et al., 2011; Hu et al., 2011). However, the hydrolysis of pON in aerosol water is largely unconstrained. Results from field and modeling studies suggested a pON lifetime of a few hours (Pye et al., 2015; Lee et al., 2016; Fisher et al., 2016; Zare et al., 2018). A few recent laboratory chamber studies elicited a complex picture where pON formed from photochemical oxidation and NO3• oxidation of monoterpenes appears to experience different magnitudes of hydrolysis (Boyd et al., 2015, 2017; Rindelaub et al., 2015; Bean and Hildebrandt Ruiz, 2016), likely due to the difference in the relative amount of primary, secondary, and tertiary nitrate in these oxidation systems. Overall, there are very limited studies on the further evolutions of ON produced from the oxidation of monoterpenes.
Here, we present results from a laboratory chamber study on the chemical composition and hydrolysis process of pON formed from oxidation of α-pinene and β-pinene by OH• and NO3•. Specifically, we report the hydrolysis lifetimes and the fraction of hydrolyzable pON formed in the systems examined in this study. This comprehensive chamber study on the hydrolysis of pON produced from various oxidation pathways of monoterpenes and peroxy radical (RO2•) fates provides the fundamental data to better constrain the role of hydrolysis in modulating pON concentrations and lifetimes in the atmosphere; their potential as a NOx loss pathway; and their impacts on overall nitrogen budget, O3 formation, and SOA formation.
2.1 Chamber experiment design and procedure
A series of chamber experiments were performed in the Georgia Tech Environmental Chamber facility (Boyd et al., 2015) housing two 12 m3 Teflon reactors. Precursor volatile organic compounds (VOCs) were α-pinene (99 %, Sigma-Aldrich) and β-pinene (99 %, Sigma-Aldrich) and the oxidation conditions of interest were OH• and NO3• oxidation, which were represented as “daytime” and “nighttime” experiments, respectively. Specifically, four different systems of VOC and oxidation conditions were studied: α-pinene + OH•, β-pinene + OH•, α-pinene + NO3•, and β-pinene + NO3•. In order to infer the hydrolysis process, experiments were performed under low-RH (relative humidity; i.e., ∼5 %) or high-RH (i.e., ∼50 %–70 %) conditions and with effloresced or deliquesced seed particles for the same initial concentrations of precursor VOC and oxidant precursors. Temperature in the reactors was kept at room temperature (22–25 ∘C). Experimental conditions are summarized in Table 1.
Table 1Summary of experimental conditions considered in this study.

a The target for the initial reactor RH is ∼70 %.
However, the irradiation of UV light increases the reactor temperature by
several degrees Celsius and hence decreases RH.
b Ammonium sulfate
c Sulfuric acid and magnesium sulfate
Prior to every experiment, the reactor was flushed with zero air (Aadco, 747-14) for at least a day. A typical experiment began with the injection of seed aerosol into the reactor by atomizing dilute ammonium sulfate (AS; 0.015 M) or sulfuric acid + magnesium sulfate (SA + MS; 0.01+0.005 M) aqueous solution. The seed aerosol was either directly atomized into the reactor or passed through a dryer before entering the reactor. The difference between efflorescence RH (∼35 %) and deliquescence RH (∼80 %) for AS aerosol is fairly large (Seinfeld and Pandis, 2016). Taking advantage of this property, it is possible to vary the amount of water in aerosol under the same RH in the reactor. Initial seed number and volume concentrations upon atomization for 20 min were approximately 2×104 cm−3 and 2×1010 nm3 cm−3, respectively. A known amount of precursor VOC in the liquid form was transferred into a glass bulb, which was then evaporated and carried into the reactor by flowing zero air at 5 L min−1 through the bulb. Although the measurement of the precursor VOC concentration was not available for all experiments, the target and measured concentrations in the experiments when the measurements were available were consistent.
For daytime experiments, an oxidant precursor (i.e., H2O2) was introduced to the reactor in the same manner as VOC except that the glass bulb was gently heated by a heat gun to help evaporate faster. During the injection of H2O2, a desired amount of NO was introduced into the reactor from a cylinder containing 500 ppm of NO (Matheson). For Exps. 3–5, 5 ppm of NO at 5 L min−1 was continuously injected to the reactor until the SOA growth ceased. For Exps. 1, 2, 6, and 7, 15 ppm of NO at 5 L min−1 was injected for 5–20 min several times until the SOA growth ceased. The NO concentration was usually on the order of tens of ppb and always remained above a few ppb, making the bimolecular reaction with NO a favorable RO2• reaction pathway. The experiment was initiated by turning on the irradiation of UV light approximately 20 min after the end of the last injection to ensure that particles and vapors were mixed well inside the reactor.
The procedure for nighttime experiments was the same until the end of the precursor VOC injection. An oxidant precursor (i.e., N2O5) was pre-made in a flow tube by simultaneously injecting 500 ppm of NO2 (Matheson) at 0.4 L min−1 and ∼250 ppm of O3 (Jelight 610) at 0.5 L min−1. A simple kinetic box model was used to adjust the concentration of O3 and flow rates of both NO2 and O3 to maximize the production of N2O5 and minimize the concentration of O3, such that the VOC was dominantly oxidized by NO3•. Once N2O5 entered the reactor, it thermally decomposed to generate NO2 and NO3•. VOC was usually depleted within the first 15 min of the experiment. For Exps. 8 and 14, the injection order of precursor VOC and oxidant precursor was switched such that the injection of VOC marked the beginning of the experiment. For Exps. 12 and 13, 25 ppm of formaldehyde was added to the reactor to enhance the branching ratio of RO2•+HO2• (Schwantes et al., 2015; Boyd et al., 2015) by injecting an appropriate amount of formalin solution (37 % HCHO, Sigma-Aldrich) in the same manner as the injection of H2O2. We do not discuss the details of the effect of the injection order nor the effects of the RO2• fate here as they had negligible impact on the results concerning hydrolysis.
2.2 Instrumentation and data analysis
A high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS; Aerodyne Research Inc.) measured the concentrations of non-refractory organics (Org), sulfate (SO4), nitrate (NO3), ammonium (NH4), and chloride (Chl) (DeCarlo et al., 2006). The data were analyzed using PIKA v1.16I and the unity collection efficiency was applied to all datasets. For the majority of nitrate-containing aerosol regardless of the class (i.e., inorganic or organic), the nitrate moiety (i.e., -NO2, -ONO2, and -O2NO2) was known to be thermally fragmented into NO+ and and was measured as NO3 (Farmer et al., 2010). As many past studies have demonstrated the feasibility to separate the contribution of inorganic (NO3,Inorg) and organic nitrate (NO3,Org) to the measured NO3 based on the ratio of NO+ and (Fry et al., 2009, 2018; Farmer et al., 2010; Xu et al., 2015b; Kiendler-Scharr et al., 2016), we used Eq. (1) presented in Farmer et al. (2010) to obtain NO3,Org. RAN (i.e., from ammonium nitrate) was obtained from the routine ionization efficiency calibration of HR-ToF-AMS using 300 nm ammonium nitrate particles. The drawback of this method is that RON (i.e., from organic nitrate aerosol) could vary depending on the chemical composition (Xu et al., 2015b). In addition, a non-negligible contribution of ammonium nitrate could be expected in experiments with deliquesced seed aerosol owing to high solubility of HNO3. Thus, we obtained the RON measured in low-RH experiments for each system of VOC and oxidation condition. In order to account for changes in RAN over time, RON was scaled accordingly assuming that the ratio of RON to RAN in the same system was constant (Fry et al., 2013). RAN, RON, and RON∕RAN values obtained in this study were consistent with previously reported values (Fry et al., 2009; Bruns et al., 2010; Boyd et al., 2015; Nah et al., 2016) and are summarized in Table S1 in the Supplement.
A Filter Inlet for Gases and AEROsols (FIGAERO) (Lopez-Hilfiker et al., 2014) coupled to a high-resolution time-of-flight iodide chemical ionization mass spectrometer (HR-ToF-I-CIMS; Aerodyne Research Inc.) detected a suite of gaseous and particulate oxidized organic species as well as selected inorganic species (Bertram et al., 2011; Lee et al., 2014). The operation of FIGAERO-HR-ToF-I-CIMS was detailed in the previous studies (Nah et al., 2016; Sanchez et al., 2016). Reagent ions were generated by flowing a mixture of CH3I and dry N2 (Airgas) through a polonium-210 source (NRD; model P-2021). The instrument measured gaseous compounds by sampling air from the reactor at ∼1.7 L min−1 while collecting particles onto a Teflon filter. Upon completion of the collection period, collected particles were desorbed by temperature-programmed dry N2 flow and subsequently analyzed by HR-ToF-I-CIMS. Sensitivity could decrease if the amount of reagent ions was significantly depleted and/or if the secondary chemistry in the ion–molecule reaction (IMR) chamber occurred at a significant degree (Lee et al., 2014). To avoid changes in sensitivity among experiments, gas-phase sampling flow was diluted with zero air immediately before the inlet such that the evaporation of aerosol was minimal. The amount of aerosol collected on the filter was also adjusted by varying the sampling rate from 1 to 6 L min−1 depending on the aerosol mass concentrations inside the reactor. Overall, the fraction of reagent ions to the total ions was kept above 80 %. In addition, iodide ion chemistry has been known to be affected by the water vapor pressure inside the IMR owing to the difference in thermodynamics between I− and IH2O− to analyte compounds (Lee et al., 2014). In order to minimize changes in the water vapor pressure inside the IMR, a small continuous flow of humidified N2 (30–50 cm3) through a bubbler at a reduced pressure was continuously added to the IMR directly. Therefore, while the instrument was not calibrated to report the concentration of detected species, it was possible to quantitatively compare the measured signal of each ion among experiments because the instrument was operated under configurations that prevented undesired changes in sensitivity. The data were analyzed using Tofware v2.5.11 and all the masses presented in this study were I− adducts.
A scanning mobility particle sizer (SMPS) that consisted of a differential mobility analyzer (TSI 3040) and a condensation particle counter (TSI 3775) was operated under the low-flow mode with the sheath flow of 2 L min−1 to detect particles up to 1 µm in electrical mobility size. A cavity attenuated phase shift monitor (CAPS; Aerodyne Research Inc.) (Kebabian et al., 2005), an ultra-sensitive NOx analyzer (Teledyne M200EU), and an UV absorption O3 analyzer (Teledyne T400) measured NO2, NOx, and O3, respectively. In selected experiments, a gas chromatograph coupled to a flame ionization detector (GC-FID; Agilent) was used to make sure that a desired amount of a precursor VOC was injected. Except the CAPS, NOx analyzer, and O3 analyzer, all instruments had their own dedicated sampling line.
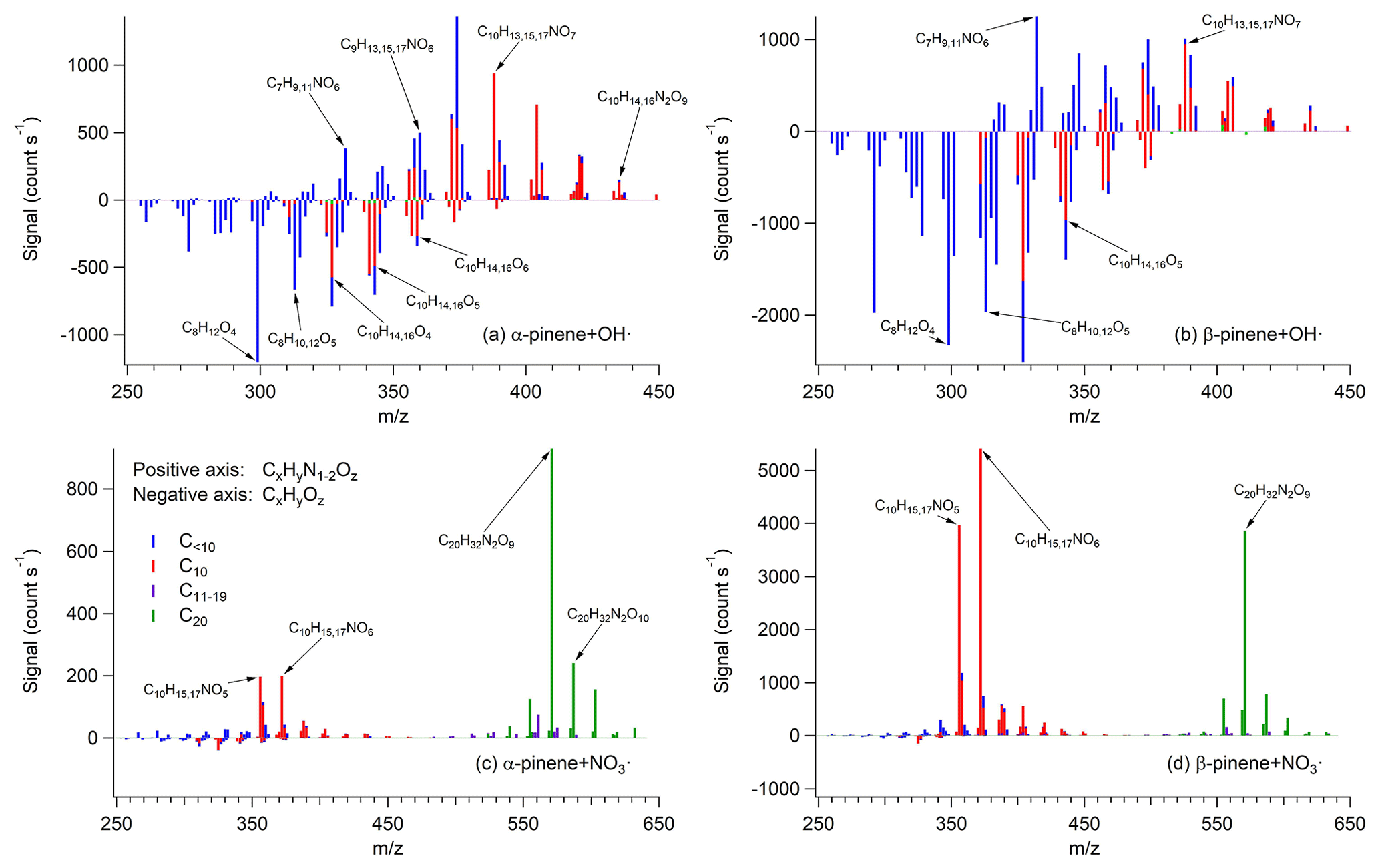
Figure 1FIGAERO-HR-ToF-I-CIMS mass spectra of SOA in (a) α-pinene + OH• from Exp. 3, (b) β-pinene + OH• from Exp. 6, (c) α-pinene + NO3• from Exp. 10, and (d) β-pinene + NO3• from Exp. 14. Top portion of each panel represents CxHyN1−2Oz, whereas bottom represents CxHyOz. Bars are colored by the number of carbon atoms as noted in the legend. Prominent masses are labeled with the corresponding chemical formulae without an iodide ion.
3.1 Chemical composition of secondary organic aerosol
Shown in Fig. 1 are the mass spectra of particle-phase species obtained from FIGAERO-HR-ToF-I-CIMS at peak SOA growth. Many of the major species detected in this study are previously reported using the same or different technique (Eddingsaas et al., 2012; Claflin and Ziemann, 2018; Boyd et al., 2015; Nah et al., 2016; Lee et al., 2016; Romonosky et al., 2017). Concerning the chemical composition of SOA from each system, a more distinct difference is observed between different oxidation conditions (i.e., OH• vs. NO3• oxidation) than between different precursor VOC (i.e., α-pinene vs. β-pinene). This is expected as α-pinene and β-pinene have the same chemical formula and very similar structures, while the oxidation conditions are distinctively different (Kroll and Seinfeld, 2008; Ziemann and Atkinson, 2012).
For daytime experiments where OH• are the dominant oxidants, the contribution of ON (i.e., CxHyN1,2Oz) and non-nitrated organics (i.e., CxHyOz) are comparable and their contributions are well spread out over a wide range of masses. A large contribution of CxHyOz is expected because the formation of ON is a minor pathway in RO2•+NO (Perring et al., 2013). In Eddingsaas et al. (2012), the major compounds reported in the α-pinene + OH• system include C8H12O4−6 and C10H16O4,6, which are also detected in our study. A suite of C10 ON from the chamber experiment of the α-pinene + OH• system are reported in Lee et al. (2016) with the chemical formula of . All of these masses are detected in this study, though we observe a considerable contribution of C<10 ON (i.e., C7H9,11NO6 and ) as well as a small fraction of C10 dinitrate (i.e., ) that has been rarely reported (Fig. 1a). This significant contribution from species containing C<10 indicates the large contribution of fragmentation process that is a preferred pathway in high NO conditions (Kroll and Seinfeld, 2008; Ziemann and Atkinson, 2012; Perring et al., 2013). It is possible that these species with C<10 are thermally decomposed products during the thermal desorption process (Stark et al., 2017). However, it is unlikely that thermal decomposition plays a significant role for the SOA generated via OH• oxidation of monoterpenes because the desorption temperature for these compounds (i.e., peaking at ∼50–70 ∘C) is much lower than the temperature at which decarboxylization or dehydration reactions (>120 ∘C) are expected to occur (Stark et al., 2017). SOA chemical composition of the β-pinene + OH• system is similar to that of the α-pinene + OH• system but with a larger contribution of CxHyOz.
Another interesting observation in the α-/β-pinene + OH• systems is that a selected class of compounds (i.e., ) with the same H∕C ratio exhibits the same time evolutions regardless of the number of oxygen (Fig. S1 in the Supplement). This observation is consistent with the autoxidation mechanism, in which highly oxidized molecules are formed in a short timescale (Ehn et al., 2014; Crounse et al., 2013; Jokinen et al., 2015). Based on FIGAERO-HR-ToF-I-CIMS data (Fig. S1), C10H17NO≥6 peak at 75 min. This is comparable to the lifetime of α-pinene at 53 min in the same experiment, which is related to the characteristic time of OH oxidation. This suggests that the aforementioned ON species are likely formed via one OH oxidation reaction, which is consistent with the autoxidation scheme to generate C10H17NO≥6 proposed in prior studies (Berndt et al., 2016; Xu et al., 2019; Pye et al., 2019). It is important to note that the concentration of NO in our experiments has been mostly kept on the order of tens of ppb over the course of the experiments by a continuous injection of dilute NO and, therefore, this result suggests that autoxidation is not a negligible pathway of RO2• fate even at a moderately high NO level in laboratory experiments and in polluted ambient environments. Indeed, recent studies (Berndt et al., 2016; Xu et al., 2019; Pye et al., 2019) suggest that the autoxidation rate constant for the α-pinene + OH• system could be up to a few per second, which is comparable to the NO level of ∼10 ppb, assuming a typical RO2•+NO reaction rate constant of (Orlando and Tyndall, 2012). The autoxidation rate constant as well as the role of NO in autoxidation based on this observation will be discussed in detail in a forthcoming publication.
In contrast, the signals of CxHyN1−2Oz are dominant in the NO3• oxidation condition, indicating that the production of ON is greatly favored over non-nitrated organics. This observation is consistent with a direct addition of a nitrate functional group to a double bond (Wayne et al., 1991; Ng et al., 2017), whereas the formation of ON in OH• oxidation condition is a minor channel of RO2•+NO reaction (Perring et al., 2013). Although this is generally true for many monoterpenes, the organic nitrate yield in α-pinene + NO3• has been known to be low (Fry et al., 2014) due to loss of a nitrate functional group followed by alkoxy radical bond scission (Kurten et al., 2017). Fry et al. (2014) observe no SOA formation in the same system, though another study has reported the formation of non-negligible SOA mass even at a relatively low initial concentration of α-pinene (Nah et al., 2016). Since the particle-phase compounds represent <10 % of overall α-pinene + NO3• products by mass, it is not necessarily inconsistent to observe a higher abundance of ON than non-nitrated organics in the particle phase if low-volatility compounds mainly consist of ON.
Moreover, the contribution from species containing C<10 is minimal in the NO3• oxidation condition. Once NO3• attacks a double bond in the initial oxidation reaction, the majority of the reaction products no longer contain any double bond. Unlike OH•, a hydrogen abstraction reaction by NO3• is slower by orders of magnitude (Atkinson and Arey, 2003). Therefore, multigeneration oxidation is unlikely to occur within the timescale of experiments (Wayne et al., 1991; Ng et al., 2017). This means that once the precursor VOC undergoes functionalization upon the initial NO3• oxidation, it is not likely to experience fragmentation during the experiment. Unlike daytime SOA, the distribution of masses is dominated by a few signature ions, such as C10H15NO5,6 and C20H32N2O8−10 (Fig. 1c and 1d). In Boyd et al. (2015), Nah et al. (2016), and Lee et al. (2016), the major species reported in the α-/β-pinene + NO3• systems are monomeric nitrate aerosol (i.e., ), while in this study a substantial contribution of dimeric species (i.e., C20H32N2O8−11) is observed. The abundant presence of dimeric compounds has been previously observed in some studies on NO3• oxidation of α-/β-pinene (Romonosky et al., 2017; Claflin and Ziemann, 2018) and particle-phase reactions to produce such dimers have been proposed by Claflin and Ziemann (2018). Many of the reported species in Claflin and Ziemann (2018) except the trimeric species (mass scan range not extended to trimeric species in our study) are observed in our study. One major difference between Claflin and Ziemann (2018) and this study is the substantial presence of monomeric nitrate aerosol (i.e., 30 %–60 % by signal) in this study. This difference may be attributed to the difference in the amount of available monomeric blocks to form dimer species. Assuming a reversible dimerization process, the concentration of dimer species shall be proportional to the square of monomer concentration, such that the monomer-to-dimer ratio increases in a quadratic manner as the available monomer concentration decreases. Since the amount of SOA formed in Claflin and Ziemann (2018) is approximately 2 orders of magnitude higher than that in this study, the concentration of monomers in the particle phase is higher, favoring a more efficient formation of dimeric species. Together, these results suggest that the contribution of dimeric nitrate aerosol could vary greatly depending on the concentrations of monomeric blocks at the specific time and location.
Previous field studies have reported the mass spectra of ambient C10 pON obtained by FIGAERO-HR-ToF-I-CIMS in a rural Alabama site during the Southern Oxidant and Aerosol Study (SOAS) (Lee et al., 2016) and in a rural forest in Germany (Zhang et al., 2018). A comparison of the ambient mass spectra with those obtained in this study reveals that average ambient pON resembles daytime pON more than nighttime pON (Fig. S2). pON from daytime experiments has a distribution of masses centered around , which is consistent with the ambient measurement data. On the other hand, NO3• oxidation does not seem to oxidize organic species enough such that the distribution of masses would be skewed towards a less-oxidized region (i.e., ). However, it is difficult to draw a quantitative conclusion simply based on this comparison because O3, another important oxidant at night, is not studied. Moreover, an average lifetime of aerosol could extend up to a week and thus ambient aerosol is continuously exposed to further oxidation while the experiments here are more applicable to freshly formed aerosol. In addition, the use of C10 pON alone may not be a good representative of monoterpene-derived pON as 42 %–74 % of pON in this study contains fewer or more than 10 carbon atoms (Table S2). Nonetheless, the chemical composition of aerosol generated in this study is comparable to those in the atmosphere and thus the results shall be directly applicable to relevant ambient conditions.
3.2 Hydrolysis of particulate organic nitrate
3.2.1 Proxy used to evaluate hydrolysis process
Various proxies using HR-ToF-AMS data have been used to infer ON hydrolysis in previous studies. In Bean and Hildebrandt Ruiz (2016), NO3 measured by an aerosol chemical speciation monitor (ACSM, practically similar to how AMS operates and measures aerosol species) (Ng et al., 2011) is normalized to SO4 as a means to account for particle wall loss and is fitted by an exponential function to estimate the ON hydrolysis rate. On the other hand, Boyd et al. (2015) normalize NO3 measured by HR-ToF-AMS to Org and attribute the relative decay of NO3∕Org between humid (RH ∼50 %) to dry (RH <5 %) conditions to hydrolysis. Other approaches include the SMPS-derived particle wall-loss correction of NO3 measured by HR-ToF-AMS followed by fitting its decay trend (Liu et al., 2012) and the determination of the fraction of total (i.e., gas and particle) ON to the precursor VOC consumed as a function of RH using Fourier-transform infrared spectroscopy (FTIR). Below, we systematically examine the use of different proxies using HR-ToF-AMS data to infer hydrolysis and discuss how the corresponding results are interpreted.

Figure 2(a, b) NO3 concentration and the molar ratio of NH4 to SO4 and (c, d) concentrations of Org, SO4, and NH4 measured by HR-ToF-AMS. Data presented in panels (a) and (c) are from Exp. 10 (low RH, dry AS), while those in panels (b) and (d) are from Exp. 11 (high RH, wet AS). These two experiments are essentially the same except for RH and phase state of seed aerosol.
Figure 2 shows the time series of Org, NO3, SO4, and NH4 measured by HR-ToF-AMS for the α-pinene + NO3• system. There is a substantial difference in NO3 for the same VOC system but under different reactor RH and phase state of seed particles (Exps. 10 and 11), while Org and SO4 concentrations are similar. The spike in NO3 in the high-RH wet-seed experiment (Fig. 2b) is attributed to the uptake of N2O5 and/or dissolution of HNO3 into aqueous aerosol followed by neutralization with ammonia to produce ammonium nitrate. This is consistent with the sharp increase in molar ratio of NH4/SO4 to higher than 2, which is the theoretical value for AS particles. It is also possible that inorganic nitrate is generated via hydrolysis of gaseous ON that is too volatile to condense but is soluble enough to dissolve in aqueous aerosol and thus only appears in high-RH experiments. Since we do not have a way to quantitatively differentiate the contribution of the aforementioned sources, the focus of this study is on hydrolysis of pON that partitions to the aerosol due to condensation rather than dissolution. However, further study is required to investigate the hydrolysis of volatile yet soluble gaseous ON, and the approach must be different from the comparison between low- and high-RH experiments to obtain meaningful results.
To evaluate the extent of pON hydrolysis, the contributions of inorganic nitrate (NO3,Inorg) and organic nitrate (NO3,Org) to the measured NO3 need to be calculated. Firstly, NO3,Org is estimated by subtracting NO3 associated with excess NH4. Secondly, NO3,Org is derived from approach (Sect. 2.2). Figure S3 shows the comparison of NO3,Org estimated by these two independent methods for the α-pinene + NO3• system. It is clear that NO3,Org from both methods are consistent and that there is a considerable contribution of NO3,Inorg to NO3 in the experiment. We note that the contribution of NO3,Inorg to NO3 can vary depending on experimental conditions with a range from to 28 % to 90 % for all experiments in this study. Nevertheless, these results demonstrate that for laboratory experiments with high RH and wet seeds, when using HR-ToF-AMS data to infer hydrolysis, it is important to separate the measured NO3 into NO3,Inorg and NO3,Org.
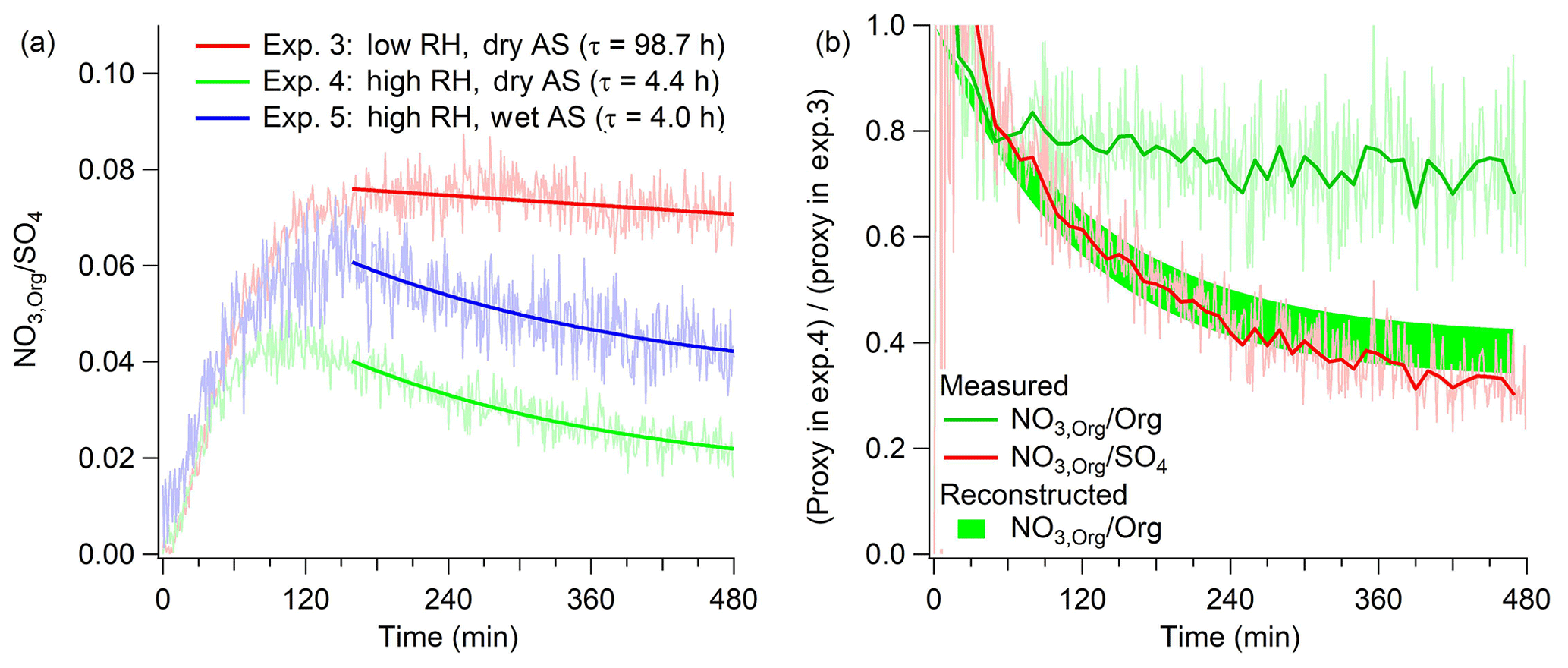
Figure 3(a) Time series data of from Exps. 3–5 (α-pinene + OH•) and the exponential fits with corresponding characteristic times. (b) , , and reconstructed based on the decay rate of . Each proxy in Exp. 4 is divided by that in Exp. 3 to determine the relative decay between high- and low-RH experiments.
Once NO3 is separated into NO3,Inorg and NO3,Org, we evaluate whether the normalization of NO3,Org to SO4 and Org provides a consistent decay trend. Photooxidation of α-pinene (Exps. 3–5) is used as a case system. As hydrolysis is a reaction in which liquid water is a reactant, it is expected that the rate of hydrolysis will change proportionally as a function of aerosol water content. Based on the hygroscopicity parameter for AS (κ=0.53) (Petters and Kreidenweis, 2007) and for ambient LO-OOA (κ=0.08) (Cerully et al., 2015) that has a substantial contribution from pON (Xu et al., 2015a), estimated aerosol water contents at peak SOA growth in Exps. 3–5 are approximately 0, 1, and 26 µg m−3, respectively. Figure S4 illustrates that AS seed particles are indeed effloresced in Exp. 4 (high RH, dry AS) but not in Exp. 5 (high RH, wet AS). These mass concentrations of aerosol water translate to 0, 6, and 36 mol L−1, respectively, under the assumption that SOA is miscible with liquid water. It is speculated that SOA formed in Exps. 3–5 are miscible with water because (1) the measured O∕C ratio in HR-ToF-AMS (Canagaratna et al., 2015) is close to 0.7, which is near the lower end but above the liquid–liquid phase separation condition (Song et al., 2012) and (2) there is evidence of aqueous-phase reactions which highly depend on the availability of aerosol water, as discussed in Sect. 3.3. Thus, the decay rate of NO3,Org normalized to SO4 and/or Org between Exp. 4 and Exp. 5 shall differ by a factor of 6 based on the molar concentrations of aerosol water.
Figure 3a shows the mass ratio of NO3,Org/SO4 and the decay rate as a characteristic time in Exps. 3–5. The characteristic times of Exps. 4 and 5 (4.4 vs. 4.0 h) do not differ regardless of the molar concentrations of aerosol water, suggesting that the decreasing trend in may not be due to changes in aerosol water content and pON hydrolysis, but arise from the difference in the reactor humidity alone. A comparison of and also reveals that these two proxies capture a different range of decay mechanisms. Figure 3b shows the relative decay trend of and between Exp. 4 (high RH) and Exp. 3 (low RH). If hydrolysis is a dominant decay mechanism of pON, the trend of would be identical to that of . This is because the organic moiety of hydrolysis product is generally considered to have a substituted alcohol group (Boschan et al., 1955) and to have a relatively similar vapor pressure and shall remain in the particle phase (Pankow and Asher, 2008). Similar to this is the formation of organic sulfate from hydrolysis of pON (Liggio and Li, 2006, 2008; Surratt et al., 2008), which has sufficiently low volatility to remain in the particle phase as with alcohol-substituted products. However, the measured decay trend of the two proxies is greatly different. It is possible that some organic moiety of hydrolysis product could be significantly more volatile and repartition back to the gas phase (Rindelaub et al., 2016; Bean and Hildebrandt Ruiz, 2016) and thus both organics and HNO3 formed from hydrolysis evaporate. In this case, not only NO3,Org but also some fraction of Org would decrease because Org measured by HR-ToF-AMS includes the contribution from the organic part of pON. This will lead to the relatively smaller decrease in NO3,Org/Org compared to . We can reconstruct the decay rate of assuming (1) the decay rate of is solely due to hydrolysis of pON and (2) the maximum contribution of pON to organic aerosol (OA) is 35 % (see Fig. 4 and discussions below). The reconstructed decay rate of is shown in Fig. 3b. As observed in the figure, the decay rate of the reconstructed is much larger than the measured . This suggests that hydrolysis is not the only loss process reflected in the decreasing trend of , while is likely a better proxy that isolates hydrolysis from other loss processes. The likely important loss process manifested in is the loss of organic vapors to the walls of the reactor (Matsunaga and Ziemann, 2010; Krechmer et al., 2016; Huang et al., 2018; Loza et al., 2010; Mcvay et al., 2014; Zhang et al., 2015, 2014; La et al., 2016). For example, Huang et al. (2018) observe that the decay of isoprene hydroxy nitrate depends on the reactor humidity. While SO4 is practically nonvolatile in the experimental condition of this study, both ON and non-nitrated organics could have some fractions of semi-volatile species whose vapors are subject to wall loss. Assuming a uniform loss rate of ON and non-nitrated organic vapors to the reactor walls, the effect of vapor wall loss could be effectively canceled out in , but not in . This assumption is likely reasonable because the vapor wall-loss rate is a function of saturation mass concentration (Zhang et al., 2015) and the average saturation mass concentrations of bulk ON and non-nitrated organic aerosol are similar based on the thermal desorption profiles in FIGAERO-HR-ToF-I-CIMS (Fig. S5). Therefore, is a better proxy to infer hydrolysis of pON than others.
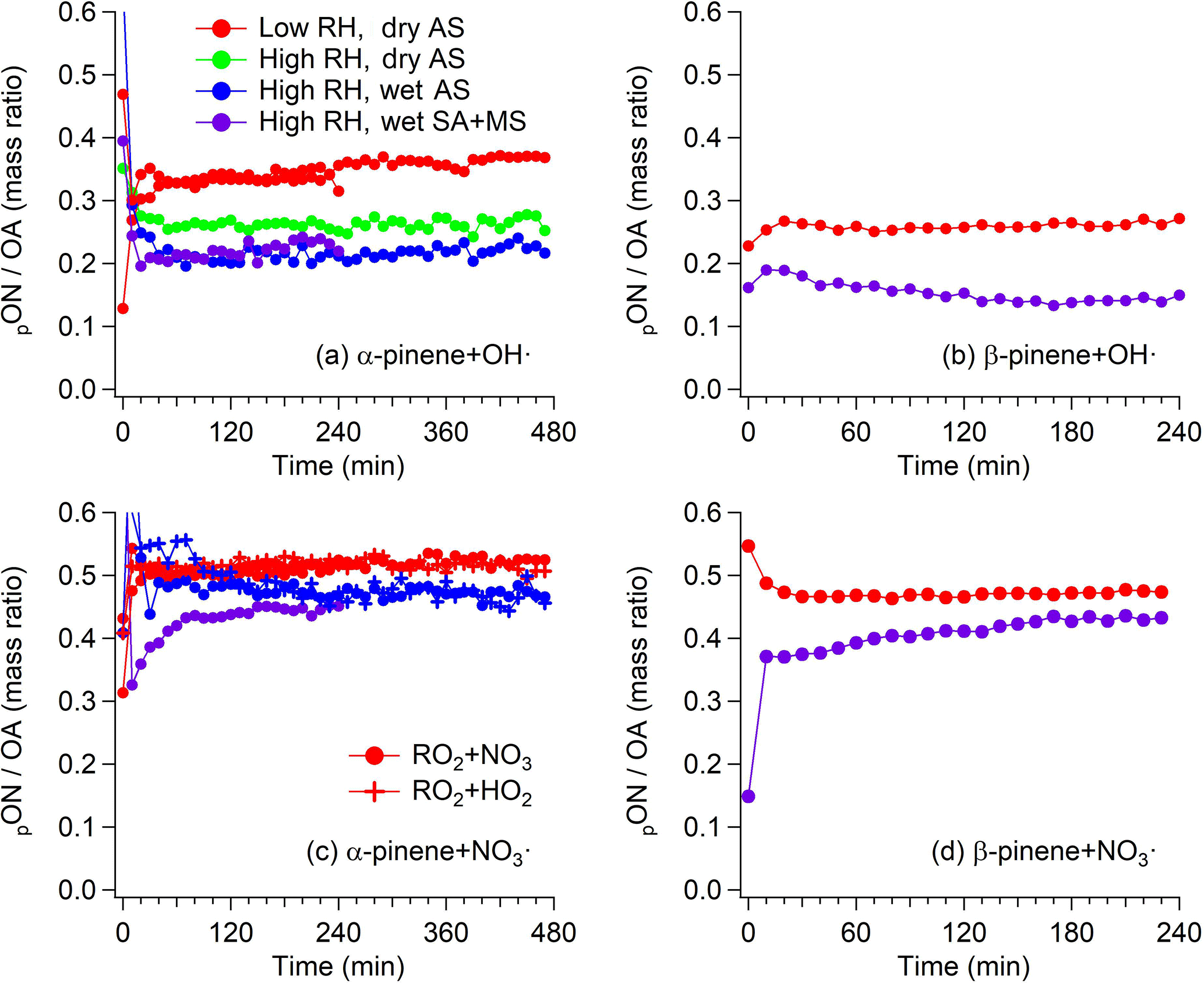
Figure 4Time series data of pON/OA in (a) α-pinene + OH• from Exps. 1–5, (b) β-pinene + OH• from Exps. 6–7, (c) α-pinene + NO3• from Exps. 8–13, and (d) β-pinene + NO3• from Exps. 14–15. Data points are colored by conditions concerning reactor RH and phase state of seed aerosol. For α-pinene + NO3•, data points are also shaped differently depending on the expected dominant RO2• fate.
3.2.2 Hydrolysis lifetime of particulate organic nitrate
In order for the data to be easily comparable with those reported in models or using other techniques, the use of general terms instead of the AMS-specific terms (i.e., NO3,Org and Org) can be convenient. We define pON as the total mass concentration of particulate organic nitrate (includes the organics part and nitrate part of the ON compounds) and OA as the total mass concentration of organic aerosol (includes nitrate and non-nitrated organics). The inclusion of nitrate mass concentration in OA is important as the contribution of nitrate functional groups to the total organic aerosol mass concentration is large. The conversion method from into pON∕OA is illustrated in Eq. (1).
MW represents the average molecular weight of pON per nitrate functional group estimated from FIGAERO-HR-ToF-I-CIMS data assuming a uniform sensitivity among detected species, and MW indicates the molecular weight of nitrate (i.e., 62 g mol−1). Since MW does not significantly vary during the course of experiments (i.e., relative standard deviation of <1.2 %), the average value is applied to each experiment. The variability in MW among different systems is also found to be small, ranging from 229 to 238 g mol−1 and thus an average MW is used for experiments where FIGAERO-HR-ToF-I-CIMS data are not available. Figure 4 shows the time series data of pON∕OA for all the systems investigated in this study.
For nighttime experiments, the relative ratio of CxHyN1−2Oz and CxHyOz obtained from FIGAERO-HR-ToF-I-CIMS data in Fig. 1 does not appear to match well with pON∕OA from HR-ToF-AMS data. For example, the signals are dominated by CxHyN1−2Oz in the β-pinene + NO3• system, as shown in Fig. 1d, while pON∕OA is at most 0.5, as shown in Fig. 4d. The discrepancy would be attributed to the overestimation of Org (in particular, CxHy family) in HR-ToF-AMS and/or underestimation of CxHyOz in FIGAERO-HR-ToF-I-CIMS. The relative ionization efficiency (RIE) of less-oxidized organic species in HR-ToF-AMS is experimentally measured to be higher at least by a factor of 2 (W. Xu et al., 2018). As previously reported (Boyd et al., 2015), the HR-ToF-AMS mass spectrum of SOA formed from β-pinene + NO3• contains significant amounts of CxHy fragments, indicating the less-oxidized nature of SOA from β-pinene + NO3•. For example, if the true RIE of Org by β-pinene + NO3• SOA were to be a factor of 2 higher than the default RIE of Org (i.e., 1.4), the reported concentration of Org would have been overestimated by a factor of 2, such that the actual pON∕OA would have been higher than reported in Fig. 4d. On the other hand, an iodide reagent ion is not quite selective enough to detect less-oxidized species, which could overestimate the contribution of pON to OA (Aljawhary et al., 2013). Nonetheless, this discrepancy between HR-ToF-AMS and FIGAERO-HR-ToF-I-CIMS data, however, should not affect the results regarding the hydrolysis lifetime and hydrolyzable fraction of pON presented later.
As illustrated in Fig. 4, the time series of pON∕OA stabilizes fairly quickly in most of the experiments, regardless of RH and/or the phase state of seed aerosol, supporting the appropriateness of the reconstructive approach shown in Fig. 3b. This suggests that the timescale of pON hydrolysis in the systems studied is significantly shorter or longer than the timescale of our experiments. It is also evident from Fig. 4 that pON∕OA in high-RH experiments are always lower than that in low-RH experiments. These two observations imply that the rate of pON hydrolysis may be fast enough such that the decay trend of pON compared to OA is not visibly manifested, though a clear difference of pON∕OA between low- and high-RH experiments is exhibited as a result of fast hydrolysis. Since no sudden, drastic change in pON∕OA is observed except for a few initial data points, we conclude that the hydrolysis lifetime of hydrolyzable pON for α-/β-pinene-derived ON shall be no more than 30 min (i.e., third data point in Fig. 4). Particle acidity is found to enhance hydrolysis rate of α-pinene hydroxy nitrate (Rindelaub et al., 2016), though no clear difference is observed here between experiments with AS and SA + MS seed particles (i.e., Exps. 5 and 2). It is worth noting that aqueous AS particles are not neutral but slightly acidic due to partitioning of ammonium to the gas phase once the particles enter the chamber (Gao et al., 2004). In Gao et al. (2004), the reported pH of aqueous AS particles is 4.6 and we expect a similar pH in our study. In Rindelaub et al. (2016), the reported hydrolysis lifetime is short at 1.3 h at a pH of 4.0. On the other hand, previous studies have shown that isoprene-derived hydroxy nitrates do not require low pH to undergo fast hydrolysis (Darer et al., 2011; Hu et al., 2011). Thus, pON formed from OH• and NO3• oxidation of α-pinene and β-pinene may not require a low pH to undergo hydrolysis at a rate comparable to the timescale of chamber experiments.
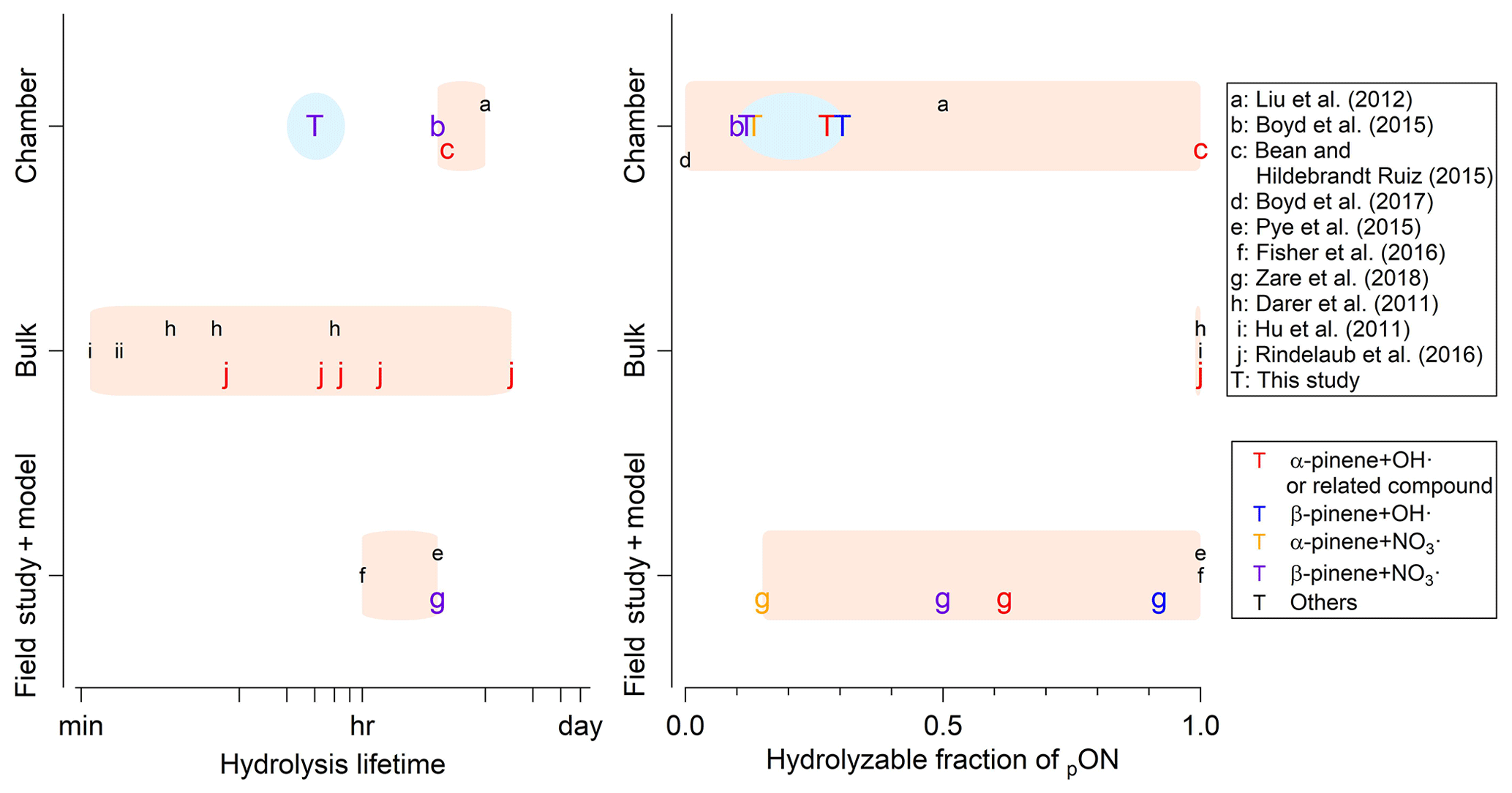
Figure 5Comparison of hydrolysis lifetime of organic nitrate and hydrolyzable fraction of pON in literature. “Chamber” refers to laboratory studies of organic nitrate aerosol via chamber experiments, “Bulk” refers to laboratory studies of organic nitrate compounds using bulk solution measurements, and “model” refers to modeling studies using ambient measurement data as constraints. Data points are colored by the system of VOC and oxidant condition and are alphabetized based on the reference. The relevant systems explored in this study are emphasized by enlarging the corresponding font size. Pink shaded regions are ranges reported in literature, while blue shaded regions are ranges reported in this study.
Comparing with the results from past chamber studies reporting a pON hydrolysis lifetime of 3 to 6 h (Liu et al., 2012; Bean and Hildebrandt Ruiz, 2016; Boyd et al., 2015), our estimated hydrolysis lifetime is substantially shorter, but is consistent with the range (i.e., 1 min to 8.8 h) reported in studies using the bulk solution method (Darer et al., 2011; Hu et al., 2011; Jacobs et al., 2014; Rindelaub et al., 2016) (Fig. 5). In Liu et al. (2012) and Bean and Hildebrandt Ruiz (2016), the hydrolysis lifetime is derived from the decay rate of NO3 corrected for the particle wall loss or normalized to SO4. As demonstrated in Sect. 3.2.1, this NO3∕SO4 decay rate is likely affected by other loss processes, such as vapor wall loss, and thus is not a good proxy to estimate the hydrolysis lifetime. The apparent discrepancy does not stem from the contradiction in the obtained data itself but rather in the data interpretation. Indeed, the lifetime estimated based on the decay of in our study is 4 h (Fig. 3a), which is consistent with 6 and 3 h reported in Liu et al. (2012) and Bean and Hildebrandt Ruiz (2016). On the other hand, in Boyd et al. (2015) the hydrolysis lifetime is estimated based on the decay rate of NO3∕Org. The discrepancy in the reported hydrolysis lifetime here could be attributed to the fact that NO3 is not separated into NO3,Org and NO3,Inorg. Figure S6 shows our data analyzed in the same manner as Boyd et al. (2015). The lifetime calculated based on the decay is 2.2 h, which is close to the reported 3–4.5 h (Boyd et al., 2015). The reduction of NO3∕Org in Fig. S6 (∼30 %) is greater than in Boyd et al. (2015) (10 %), which could be because (1) the amount of N2O5, a source of inorganic nitrate, used in our study is slightly larger and (2) RH in our study is higher by 10 %–20 %, which may have allowed for greater uptake of N2O5 to produce inorganic nitrate due to changes in aerosol viscosity (Gržinić et al., 2015).
In previous bulk solution studies where the concentration of interested organic nitrate (in particular hydroxy nitrate) in aqueous solution rather than in aerosol water is monitored over time, it is unlikely that the data interpretation is affected by other loss processes present in chamber experiments, such as vapor wall loss. It is also common to monitor the organic moiety of hydrolysis product (Darer et al., 2011), while it is extremely difficult in chamber experiments where hundreds of organic species are present at the same time, leading to the difficulty in accurately measuring the hydrolysis lifetime in chamber experiments. Based on the comprehensive analysis we demonstrate above on evaluating pON hydrolysis in chamber experiments, we recommend the use of the hydrolysis lifetime reported in this study, which is no more than 30 min, for pON formed from α-pinene and β-pinene.
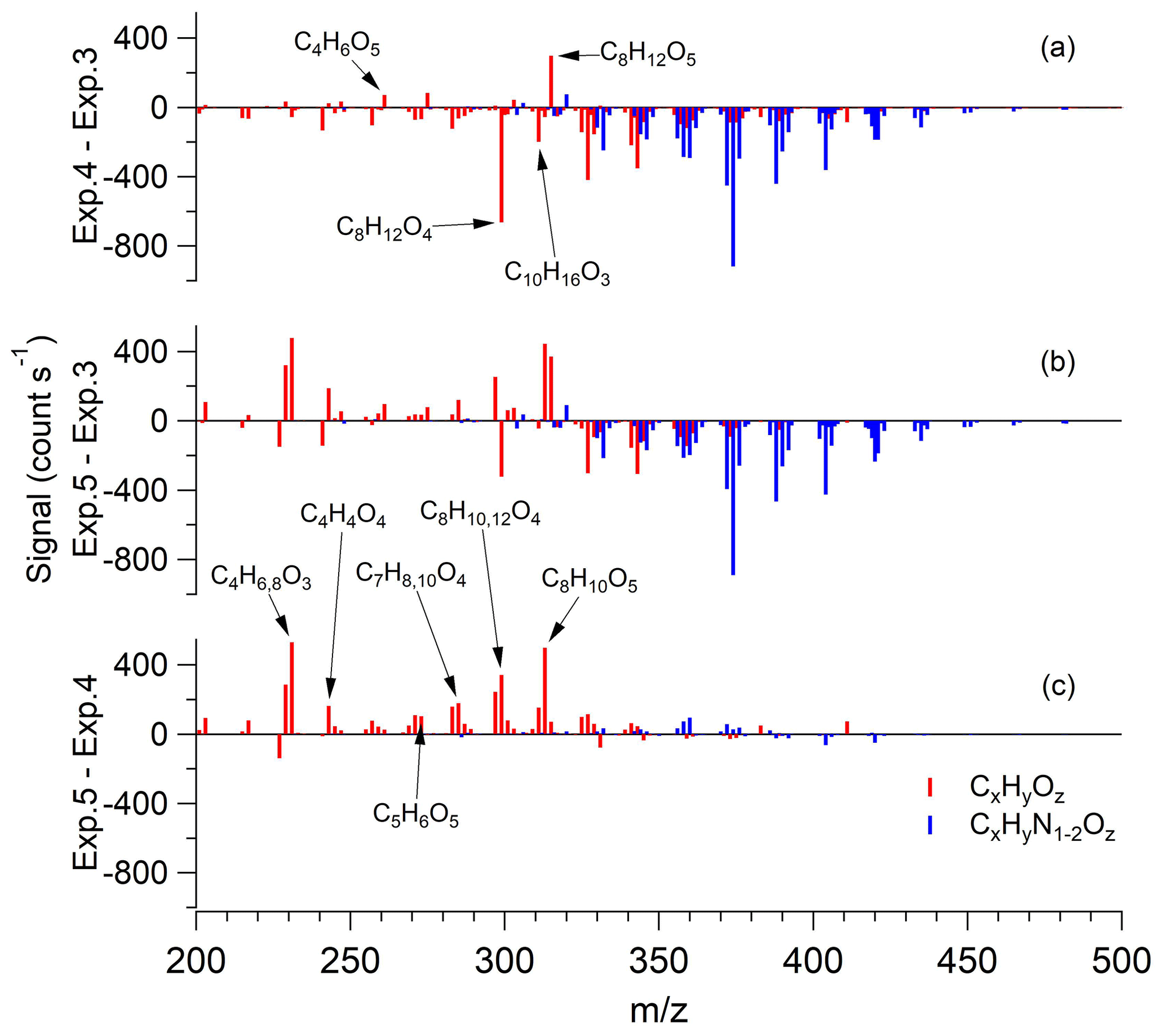
Figure 6FIGAERO-HR-ToF-I-CIMS difference mass spectra of SOA in α-pinene + OH•. (a) Exp. 4 (high RH, dry AS) minus Exp. 3 (low RH, dry AS). (b) Exp. 5 (high RH, wet AS) minus Exp. 3 (low RH, dry AS). (c) Exp. 5 (high RH, wet AS) minus Exp. 4 (high RH, dry AS). Bars are colored by the difference in chemical composition (i.e., red for CxHyOz and blue for CxHyN1−2Oz). Prominent masses are labeled with the corresponding chemical formulae without an iodide ion.
3.2.3 Hydrolyzable fraction of particulate organic nitrate
The fraction of hydrolyzable pON (FH) can be directly estimated from the difference in pON∕OA between low- and high-RH experiments shown in Fig. 4. Although we show that hydrolysis is substantially faster than the timescale of chamber experiments in our study, there still appears to be a clear difference in pON∕OA between high-RH experiments but with a different phase state of seed aerosol (i.e., Exps. 4 and 5). The difference in mass spectra among Exps. 3–5 obtained from FIGAERO-HR-ToF-I-CIMS reveal that the difference in pON∕OA between Exps. 4 and 5 does not arise from the reduction in pON but from the increase in non-nitrated organics (Fig. 6). The reason for this OA increase with the abundant presence of aerosol water is speculated to be uptake and other aqueous-phase reactions than hydrolysis and is briefly discussed in Sect. 3.3. Thus, the absolute difference in pON∕OA between low RH with dry seed and high RH with dry seed experiments best indicates FH. Depending on the fate of the organic moiety of the hydrolysis product (i.e., stay in the particle phase or repartition back to the gas phase), FH varies. Since we are unable to determine the fate of hydrolysis product, an upper and lower limit of FH are reported as a range of FH. For the α-pinene+OH• system, 23 %–32 % of pON formed undergoes hydrolysis within the timescale of the experiments. For the other systems with no experiment under high RH with dry seed aerosol, the same level of additional contribution from non-nitrated organics encountered in α-pinene + OH• system is assumed and FH is scaled accordingly. For the β-pinene + OH•, α-pinene + NO3•, and β-pinene + NO3• systems, 27 %–34 %, 9 %–17 %, and 9 %–15 %, respectively, of pON are found hydrolyzable within the timescale of the experiments. Table 2 summarizes the hydrolysis lifetime and FH in the systems explored in this study.
Table 2Hydrolysis lifetime and corresponding fraction of hydrolyzable pON.

a Lower or higher limit of hydrolyzable pON fraction is based on the assumptions that organic moiety of hydrolysis products remains in aerosol or partitions back to the gas phase.
FH has been only reported in a few studies (Liu et al., 2012; Boyd et al., 2015; Zare et al., 2018). The determination of FH is essential because the assumption that all pON hydrolyzes biases the relative importance of hydrolysis among the loss mechanisms of pON and NOx. Boyd et al. (2015) report that FH of pON formed via β-pinene + NO3• is ∼10 %, which is in a good agreement with our range of 9 %–15 %. Based on the proposed molecular structures of pON from Claflin and Ziemann (2018), <5 % of pON from β-pinene + NO3• is tertiary and thus hydrolyzable. In our study, a considerable contribution of monomeric (C10) pON is observed (Fig. 1d), while dimeric (C20) pON is dominant in Claflin and Ziemann (2018). This may indicate that monomeric pON is more susceptible to hydrolysis such that FH in this study is slightly higher than expected based on the proposed molecular structures of pON in Claflin and Ziemann (2018).
FH for α-/β-pinene + OH• systems are higher than those from α-/β-pinene + NO3• systems. This trend is consistent with the understanding that pON via OH• oxidation have a larger fraction of tertiary nitrate groups, which are significantly more susceptible to hydrolysis (Darer et al., 2011; Hu et al., 2011) than those formed via NO3• oxidation (Ng et al., 2017). Although the relative trend of FH between α-/β-pinene + OH• and α-/β-pinene + NO3• systems is consistent, the magnitude of FH in the α-/β-pinene + OH• system appears to be smaller than expected based on the tertiary nitrate fraction estimated via the explicit gas-phase chemical mechanism (Browne et al., 2013; Zare et al., 2018). In previous studies, FH is estimated to be 62 % and 92 % for α-/β-pinene + OH• systems, respectively. However, our results indicate FH is 23 %–32 % and 27 %–34 % for the same systems (Fig. 5). The chemical mechanism used in Browne et al. (2013) and Zare et al. (2018) is based on the Master Chemical Mechanism (MCM) that is well known in the degradation chemistry of VOC in the gas phase (Jenkin et al., 1997; Saunders et al., 2003). However, the same mechanism performs poorly in regards to the chemical composition of SOA (Faxon et al., 2018) as well as the prediction of SOA formation (Ruggeri et al., 2016; Boyd et al., 2017) when equipped with gas-particle partitioning modules based on the absorptive gas-particle partitioning theory (Pankow, 1994). It is, therefore, reasonable to argue that the chemical composition of pON could greatly differ from that of total ON predicted by the MCM. Thus, FH reported in this study provides the fundamental experimental constraints on pON hydrolysis that can be used in regional and global models for elucidating potential impacts of ON on nitrogen budget and formation of ozone and aerosol.
3.3 Signature of other aqueous-phase reactions besides hydrolysis
As briefly discussed in the above section, the presence of elevated levels of aerosol water seems to have enhanced the contribution of small (i.e., C<9) but more-oxidized organic species to SOA. As shown in Fig. 5c, the enhancement is observed for a variety of non-nitrated organic aerosol including C4H6,8O3, C4H4O4, C5H6O5, C7H8,10O4, and , while pON overall neither increase nor decrease. C5H6O5 has been reported as a product of the aqueous-phase reaction of α-pinene-derived compounds (Aljawhary et al., 2016). Other compounds, such as C4H4O4, C7H8,10O4, and C8H10O5, also appear to result from the aqueous processing because compounds with similar chemical formulae but with slightly higher degrees of oxidation (i.e., C4H4O5, C7H10O5, and C8H12O6) are reported in Aljawhary et al. (2016). The reason for this less-oxidized nature of SOA in this study may be attributed to our experiments being performed in moderately high NO conditions that promote a higher contribution of a carbonyl group than a hydroperoxyl group, which is preferred in low-NO conditions. In Aljawhary et al. (2016), the starting compound is a product in low-NO conditions (pinonic acid, C10H16O3). Thus, it is reasonable that products of the aqueous processing in this study are slightly less oxidized than observed in Aljawhary et al. (2016).
The enhancement of non-nitrated organic aerosol in FIGAERO-HR-ToF-I-CIMS may be due to aqueous processing of species in the particles in the presence of aerosol water. This can come from further reactions of semi-volatile and/or low-volatility species in the particle phase, or reactive uptake of volatile (but highly water soluble) species into the aerosol followed by subsequent aqueous-phase reactions to form low-volatility products (Ervens et al., 2011). A comparison of the average thermogram at peak SOA growth among Exps. 3–5 indicates a higher contribution of low-volatility compounds in Exp. 5 than in Exps. 3 and 4, as illustrated by the bimodal peaks (Fig. S7). Given the same degree of gas-phase oxidation expected among Exps. 3–5, these results show that aqueous chemistry in wet aerosol contributes to the further formation of low-volatility compounds. Overall, the highest average degree of oxidation () is observed in the high-RH wet-aerosol experiment (Exp. 5). The effect of particle water on monoterpene SOA formation warrants further studies.
3.4 Atmospheric implications
There is emerging evidence that monoterpene SOA greatly contributes to atmospheric aerosol in the Southeastern US (Zhang et al., 2018; L. Xu et al., 2018). ON is no exception; a substantial fraction of pON is considered to be from monoterpenes (Lee et al., 2016; Huang et al., 2019; Xu et al., 2015a). While C10 pON measured in FIGAERO-HR-ToF-I-CIMS is a good tracer of monoterpene-derived pON, we show that a fair amount of α-/β-pinene pON is found as C<10 or C20 depending on the oxidation condition. This implies that the contribution of monoterpene-derived pON could be substantially underestimated when only considering C10 pON. The fraction of pON with different number of carbon atoms reported in this study (Table S2) is a useful parameter to quantitatively determine the contribution of monoterpene-derived pON to total pON.
Many previous modeling studies using ambient measurement data as constraints report that the lifetime of pON is likely several hours (Pye et al., 2015; Fisher et al., 2016; Lee et al., 2016; Zare et al., 2018). Hydrolysis of pON is used as a dominant loss process with the lifetime of several hours to improve the concentration of modeled OA (Pye et al., 2015) and to improve the concentrations of gaseous and particulate ON (Fisher et al., 2016). If the ambient pON concentration is indeed governed by a loss process with a few hours of lifetime, our results imply that the particle-phase hydrolysis may not be the only dominant loss process because the hydrolysis lifetime reported in this study is significantly shorter. Other potential but less-studied loss mechanisms of ON and pON include deposition (Nguyen et al., 2015), photolysis (Müller et al., 2014), and aqueous photooxidation (Romonosky et al., 2017; Nah et al., 2016). For instance, an enhanced photolysis rate is observed for carbonyl nitrate derived from isoprene (Müller et al., 2014), while no similar study is available for monoterpene-derived ON in literature. Also, it has been demonstrated that different monoterpene pON can have drastically different photochemical fates (Nah et al., 2016). Taken together, results from this study highlight the importance to investigate other potential loss processes of monoterpene-derived pON.
Aside from the hydrolysis lifetime, many modeling studies assume FH as unity (Pye et al., 2015; Fisher et al., 2016; Lee et al., 2016). Even when FH is considered, the value of FH used in other studies is still substantially higher than estimated in this study (Browne et al., 2013; Zare et al., 2018). The use of higher FH would result in overestimating the contribution of hydrolysis as a loss process of pON and NOx. While hydrolysis is considered a permanent sink of NOx, many other loss processes, such as further OH• oxidation and photolysis, are considered a temporary reservoir of NOx. If the relative importance of pON fates in models was not accurate, the role of ON in NOx cycling and the formation potential of ozone could have been inaccurately interpreted. Therefore, results from this study regarding the hydrolysis lifetime and FH serve as experimentally constrained parameters for chemical transport models to accurately evaluate the role of ON in regards to nitrogen budget and the formation of ozone and fine aerosol.
Data are available upon request from the corresponding author (ng@chbe.gatech.edu).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-12749-2019-supplement.
MT designed and performed the research and analyzed the data with substantial inputs from NLN. MT and NLN wrote the article.
The authors declare that they have no conflict of interest.
The authors would like to acknowledge financial support by the National Science Foundation (NSF) CAREER AGS-1555034 and by the National Oceanic and Atmospheric Administration (NOAA) NA18OAR4310112. The FIGAERO-HR-ToF-CIMS has been purchased through NSF Major Research Instrumentation (MRI) grant 1428738.
This research has been supported by the National Science Foundation (CAREER (grant no. AGS-1555034)), the National Oceanic and Atmospheric Administration (grant no. NA18OAR4310112), and the National Science Foundation (grant no. 1428738).
This paper was edited by Alexander Laskin and reviewed by two anonymous referees.
Aljawhary, D., Lee, A. K. Y., and Abbatt, J. P. D.: High-resolution chemical ionization mass spectrometry (ToF-CIMS): application to study SOA composition and processing, Atmos. Meas. Tech., 6, 3211–3224, https://doi.org/10.5194/amt-6-3211-2013, 2013.
Aljawhary, D., Zhao, R., Lee, A. K. Y., Wang, C., and Abbatt, J. P. D.: Kinetics, Mechanism, and Secondary Organic Aerosol Yield of Aqueous Phase Photo-oxidation of alpha-Pinene Oxidation Products, J. Phys. Chem. A, 120, 1395–1407, https://doi.org/10.1021/acs.jpca.5b06237, 2016.
Aschmann, S. M., Atkinson, R., and Arey, J.: Products of reaction of OH radicals with alpha-pinene, J. Geophys. Res.-Atmos., 107, 4191, https://doi.org/10.1029/2001jd001098, 2002.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003.
Bean, J. K. and Hildebrandt Ruiz, L.: Gas–particle partitioning and hydrolysis of organic nitrates formed from the oxidation of α-pinene in environmental chamber experiments, Atmos. Chem. Phys., 16, 2175–2184, https://doi.org/10.5194/acp-16-2175-2016, 2016.
Berndt, T. and Boge, O.: Products and mechanism of the gas-phase reaction of NO3 radicals with alpha-pinene, J. Chem. Soc. Faraday T., 93, 3021–3027, https://doi.org/10.1039/a702364b, 1997.
Berndt, T., Richters, S., Jokinen, T., Hyttinen, N., Kurten, T., Otkjaer, R. V., Kjaergaard, H. G., Stratmann, F., Herrmann, H., Sipila, M., Kulmala, M., and Ehn, M.: Hydroxyl radical-induced formation of highly oxidized organic compounds, Nat. Commun., 7, 13677, https://doi.org/10.1038/ncomms13677, 2016.
Bertram, T. H., Kimmel, J. R., Crisp, T. A., Ryder, O. S., Yatavelli, R. L. N., Thornton, J. A., Cubison, M. J., Gonin, M., and Worsnop, D. R.: A field-deployable, chemical ionization time-of-flight mass spectrometer, Atmos. Meas. Tech., 4, 1471–1479, https://doi.org/10.5194/amt-4-1471-2011, 2011.
Boschan, R., Merrow, R. T., and Vandolah, R. W.: The Chemistry of Nitrate Esters, Chem. Rev., 55, 485–510, https://doi.org/10.1021/cr50003a001, 1955.
Boyd, C. M., Sanchez, J., Xu, L., Eugene, A. J., Nah, T., Tuet, W. Y., Guzman, M. I., and Ng, N. L.: Secondary organic aerosol formation from the β-pinene + NO3 system: effect of humidity and peroxy radical fate, Atmos. Chem. Phys., 15, 7497–7522, https://doi.org/10.5194/acp-15-7497-2015, 2015.
Boyd, C. M., Nah, T., Xu, L., Berkemeier, T., and Ng, N. L.: Secondary Organic Aerosol (SOA) from Nitrate Radical Oxidation of Monoterpenes: Effects of Temperature, Dilution, and Humidity on Aerosol Formation, Mixing, and Evaporation, Environ. Sci. Technol., 51, 7831–7841, https://doi.org/10.1021/acs.est.7b01460, 2017.
Browne, E. C., Min, K.-E., Wooldridge, P. J., Apel, E., Blake, D. R., Brune, W. H., Cantrell, C. A., Cubison, M. J., Diskin, G. S., Jimenez, J. L., Weinheimer, A. J., Wennberg, P. O., Wisthaler, A., and Cohen, R. C.: Observations of total RONO2 over the boreal forest: NOx sinks and HNO3 sources, Atmos. Chem. Phys., 13, 4543–4562, https://doi.org/10.5194/acp-13-4543-2013, 2013.
Bruns, E. A., Perraud, V., Zelenyuk, A., Ezell, M. J., Johnson, S. N., Yu, Y., Imre, D., Finlayson-Pitts, B. J., and Alexander, M. L.: Comparison of FTIR and Particle Mass Spectrometry for the Measurement of Particulate Organic Nitrates, Environ. Sci. Technol., 44, 1056–1061, https://doi.org/10.1021/es9029864, 2010.
Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R.: Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 15, 253–272, https://doi.org/10.5194/acp-15-253-2015, 2015.
Carlton, A. G., Pinder, R. W., Bhave, P. V., and Pouliot, G. A.: To What Extent Can Biogenic SOA be Controlled?, Environ. Sci. Technol., 44, 3376–3380, https://doi.org/10.1021/es903506b, 2010.
Cerully, K. M., Bougiatioti, A., Hite Jr., J. R., Guo, H., Xu, L., Ng, N. L., Weber, R., and Nenes, A.: On the link between hygroscopicity, volatility, and oxidation state of ambient and water-soluble aerosols in the southeastern United States, Atmos. Chem. Phys., 15, 8679–8694, https://doi.org/10.5194/acp-15-8679-2015, 2015.
Claflin, M. S. and Ziemann, P. J.: Identification and Quantitation of Aerosol Products of the Reaction of beta-Pinene with NO3 Radicals and Implications for Gas- and Particle-Phase Reaction Mechanisms, J. Phys. Chem. A, 122, 3640–3652, https://doi.org/10.1021/acs.jpca.8b00692, 2018.
Crounse, J. D., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett, 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013.
Darer, A. I., Cole-Filipiak, N. C., O'Connor, A. E., and Elrod, M. J.: Formation and Stability of Atmospherically Relevant Isoprene-Derived Organosulfates and Organonitrates, Environ. Sci. Technol., 45, 1895–1902, https://doi.org/10.1021/es103797z, 2011.
Day, D. A., Liu, S., Russell, L. M., and Ziemann, P. J.: Organonitrate group concentrations in submicron particles with high nitrate and organic fractions in coastal southern California, Atmos. Environ., 44, 1970–1979, https://doi.org/10.1016/j.atmosenv.2010.02.045, 2010.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006.
Eddingsaas, N. C., Loza, C. L., Yee, L. D., Chan, M., Schilling, K. A., Chhabra, P. S., Seinfeld, J. H., and Wennberg, P. O.: α-pinene photooxidation under controlled chemical conditions – Part 2: SOA yield and composition in low- and high-NOx environments, Atmos. Chem. Phys., 12, 7413–7427, https://doi.org/10.5194/acp-12-7413-2012, 2012.
Ehn, M., Thornton, J. A., Kleist, E., Sipila, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I. H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurten, T., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., Canagaratna, M., Dal Maso, M., Berndt, T., Petaja, T., Wahner, A., Kerminen, V. M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476, https://doi.org/10.1038/nature13032, 2014.
Ervens, B., Turpin, B. J., and Weber, R. J.: Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies, Atmos. Chem. Phys., 11, 11069–11102, https://doi.org/10.5194/acp-11-11069-2011, 2011.
Farmer, D. K., Matsunaga, A., Docherty, K. S., Surratt, J. D., Seinfeld, J. H., Ziemann, P. J., and Jimenez, J. L.: Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry, P. Natl. Acad. Sci. USA, 107, 6670–6675, https://doi.org/10.1073/pnas.0912340107, 2010.
Faxon, C., Hammes, J., Le Breton, M., Pathak, R. K., and Hallquist, M.: Characterization of organic nitrate constituents of secondary organic aerosol (SOA) from nitrate-radical-initiated oxidation of limonene using high-resolution chemical ionization mass spectrometry, Atmos. Chem. Phys., 18, 5467–5481, https://doi.org/10.5194/acp-18-5467-2018, 2018.
Fisher, J. A., Jacob, D. J., Travis, K. R., Kim, P. S., Marais, E. A., Chan Miller, C., Yu, K., Zhu, L., Yantosca, R. M., Sulprizio, M. P., Mao, J., Wennberg, P. O., Crounse, J. D., Teng, A. P., Nguyen, T. B., St. Clair, J. M., Cohen, R. C., Romer, P., Nault, B. A., Wooldridge, P. J., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Shepson, P. B., Xiong, F., Blake, D. R., Goldstein, A. H., Misztal, P. K., Hanisco, T. F., Wolfe, G. M., Ryerson, T. B., Wisthaler, A., and Mikoviny, T.: Organic nitrate chemistry and its implications for nitrogen budgets in an isoprene- and monoterpene-rich atmosphere: constraints from aircraft (SEAC4RS) and ground-based (SOAS) observations in the Southeast US, Atmos. Chem. Phys., 16, 5969–5991, https://doi.org/10.5194/acp-16-5969-2016, 2016.
Fry, J. L., Kiendler-Scharr, A., Rollins, A. W., Wooldridge, P. J., Brown, S. S., Fuchs, H., Dubé, W., Mensah, A., dal Maso, M., Tillmann, R., Dorn, H.-P., Brauers, T., and Cohen, R. C.: Organic nitrate and secondary organic aerosol yield from NO3 oxidation of β-pinene evaluated using a gas-phase kinetics/aerosol partitioning model, Atmos. Chem. Phys., 9, 1431–1449, https://doi.org/10.5194/acp-9-1431-2009, 2009.
Fry, J. L., Draper, D. C., Zarzana, K. J., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Brown, S. S., Cohen, R. C., Kaser, L., Hansel, A., Cappellin, L., Karl, T., Hodzic Roux, A., Turnipseed, A., Cantrell, C., Lefer, B. L., and Grossberg, N.: Observations of gas- and aerosol-phase organic nitrates at BEACHON-RoMBAS 2011, Atmos. Chem. Phys., 13, 8585–8605, https://doi.org/10.5194/acp-13-8585-2013, 2013.
Fry, J. L., Draper, D. C., Barsanti, K. C., Smith, J. N., Ortega, J., Winkle, P. M., Lawler, M. J., Brown, S. S., Edwards, P. M., Cohen, R. C., and Lee, L.: Secondary Organic Aerosol Formation and Organic Nitrate Yield from NO3 Oxidation of Biogenic Hydrocarbons, Environ. Sci. Technol., 48, 11944–11953, https://doi.org/10.1021/es502204x, 2014.
Fry, J. L., Brown, S. S., Middlebrook, A. M., Edwards, P. M., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Allen, H. M., Ryerson, T. B., Pollack, I., Graus, M., Warneke, C., de Gouw, J. A., Brock, C. A., Gilman, J., Lerner, B. M., Dubé, W. P., Liao, J., and Welti, A.: Secondary organic aerosol (SOA) yields from NO3 radical + isoprene based on nighttime aircraft power plant plume transects, Atmos. Chem. Phys., 18, 11663–11682, https://doi.org/10.5194/acp-18-11663-2018, 2018.
Gao, S., Ng, N. L., Keywood, M., Varutbangkul, V., Bahreini, R., Nenes, A., He, J. W., Yoo, K. Y., Beauchamp, J. L., Hodyss, R. P., Flagan, R. C., and Seinfeld, J. H.: Particle phase acidity and oligomer formation in secondary organic aerosol, Environ. Sci. Technol., 38, 6582–6589, https://doi.org/10.1021/es049125k, 2004.
Goldstein, A. H. and Galbally, I. E.: Known and unexplored organic constituents in the earth's atmosphere, Environ. Sci. Technol., 41, 1514–1521, https://doi.org/10.1021/es072476p, 2007.
Griffin, R. J., Cocker, D. R., Flagan, R. C., and Seinfeld, J. H.: Organic aerosol formation from the oxidation of biogenic hydrocarbons, J. Geophys. Res.-Atmos., 104, 3555–3567, https://doi.org/10.1029/1998jd100049, 1999.
Gržinić, G., Bartels-Rausch, T., Berkemeier, T., Türler, A., and Ammann, M.: Viscosity controls humidity dependence of N2O5 uptake to citric acid aerosol, Atmos. Chem. Phys., 15, 13615–13625, https://doi.org/10.5194/acp-15-13615-2015, 2015.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Hallquist, M., Wangberg, I., Ljungstrom, E., Barnes, I., and Becker, K. H.: Aerosol and product yields from NO3 radical-initiated oxidation of selected monoterpenes, Environ. Sci. Technol., 33, 553–559, https://doi.org/10.1021/es980292s, 1999.
Hoyle, C. R., Boy, M., Donahue, N. M., Fry, J. L., Glasius, M., Guenther, A., Hallar, A. G., Huff Hartz, K., Petters, M. D., Petäjä, T., Rosenoern, T., and Sullivan, A. P.: A review of the anthropogenic influence on biogenic secondary organic aerosol, Atmos. Chem. Phys., 11, 321–343, https://doi.org/10.5194/acp-11-321-2011, 2011.
Hu, K. S., Darer, A. I., and Elrod, M. J.: Thermodynamics and kinetics of the hydrolysis of atmospherically relevant organonitrates and organosulfates, Atmos. Chem. Phys., 11, 8307–8320, https://doi.org/10.5194/acp-11-8307-2011, 2011.
Huang, W., Saathoff, H., Shen, X. L., Ramisetty, R., Leisner, T., and Mohr, C.: Chemical Characterization of Highly Functionalized Organonitrates Contributing to Night-Time Organic Aerosol Mass Loadings and Particle Growth, Environ. Sci. Technol., 53, 1165–1174, https://doi.org/10.1021/acs.est.8b05826, 2019.
Huang, Y. L., Zhao, R., Charan, S. M., Kenseth, C. M., Zhang, X., and Seinfeld, J. H.: Unified Theory of Vapor-Wall Mass Transport in Teflon-Walled Environmental Chambers, Environ. Sci. Technol., 52, 2134–2142, https://doi.org/10.1021/acs.est.7b05575, 2018.
Jacobs, M. I., Burke, W. J., and Elrod, M. J.: Kinetics of the reactions of isoprene-derived hydroxynitrates: gas phase epoxide formation and solution phase hydrolysis, Atmos. Chem. Phys., 14, 8933–8946, https://doi.org/10.5194/acp-14-8933-2014, 2014.
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: A protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997.
Jokinen, T., Berndt, T., Makkonen, R., Kerminen, V. M., Junninen, H., Paasonen, P., Stratmann, F., Herrmann, H., Guenther, A. B., Worsnop, D. R., Kulmala, M., Ehn, M., and Sipila, M.: Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications, P. Natl. Acad. Sci. USA, 112, 7123–7128, https://doi.org/10.1073/pnas.1423977112, 2015.
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005.
Kebabian, P. L., Herndon, S. C., and Freedman, A.: Detection of nitrogen dioxide by cavity attenuated phase shift spectroscopy, Anal. Chem., 77, 724–728, https://doi.org/10.1021/ac048715y, 2005.
Kiendler-Scharr, A., Mensah, A. A., Friese, E., Topping, D., Nemitz, E., Prevot, A. S. H., Aijala, M., Allan, J., Canonaco, F., Canagaratna, M., Carbone, S., Crippa, M., Dall Osto, M., Day, D. A., De Carlo, P., Di Marco, C. F., Elbern, H., Eriksson, A., Freney, E., Hao, L., Herrmann, H., Hildebrandt, L., Hillamo, R., Jimenez, J. L., Laaksonen, A., McFiggans, G., Mohr, C., O'Dowd, C., Otjes, R., Ovadnevaite, J., Pandis, S. N., Poulain, L., Schlag, P., Sellegri, K., Swietlicki, E., Tiitta, P., Vermeulen, A., Wahner, A., Worsnop, D., and Wu, H. C.: Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol, Geophys. Res. Lett., 43, 7735–7744, https://doi.org/10.1002/2016gl069239, 2016.
Krechmer, J. E., Pagonis, D., Ziemann, P. J., and Jimenez, J. L.: Quantification of Gas-Wall Partitioning in Teflon Environmental Chambers Using Rapid Bursts of Low-Volatility Oxidized Species Generated in Situ, Environ. Sci. Technol., 50, 5757–5765, https://doi.org/10.1021/acs.est.6b00606, 2016.
Kroll, J. H. and Seinfeld, J. H.: Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 42, 3593–3624, https://doi.org/10.1016/j.atmosenv.2008.01.003, 2008.
Kurten, T., Moller, K. H., Nguyen, T. B., Schwantes, R. H., Misztal, P. K., Su, L. P., Wennberg, P. O., Fry, J. L., and Kjaergaard, H. G.: Alkoxy Radical Bond Scissions Explain the Anomalously Low Secondary Organic Aerosol and Organonitrate Yields From alpha-Pinene + NO3, J. Phys. Chem. Lett, 8, 2826–2834, https://doi.org/10.1021/acs.jpclett.7b01038, 2017.
La, Y. S., Camredon, M., Ziemann, P. J., Valorso, R., Matsunaga, A., Lannuque, V., Lee-Taylor, J., Hodzic, A., Madronich, S., and Aumont, B.: Impact of chamber wall loss of gaseous organic compounds on secondary organic aerosol formation: explicit modeling of SOA formation from alkane and alkene oxidation, Atmos. Chem. Phys., 16, 1417–1431, https://doi.org/10.5194/acp-16-1417-2016, 2016.
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurten, T., Worsnop, D. R., and Thornton, J. A.: An Iodide-Adduct High-Resolution Time-of-Flight Chemical-Ionization Mass Spectrometer: Application to Atmospheric Inorganic and Organic Compounds, Environ. Sci. Technol., 48, 6309–6317, https://doi.org/10.1021/es500362a, 2014.
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurten, T., Hu, W. W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H. Y., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J. M., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., D'Ambro, E. L., and Thornton, J. A.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci. USA, 113, 1516–1521, https://doi.org/10.1073/pnas.1508108113, 2016.
Liggio, J. and Li, S. M.: Reactive uptake of pinonaldehyde on acidic aerosols, J. Geophys. Res.-Atmos., 111, D24303, https://doi.org/10.1029/2005jd006978, 2006.
Liggio, J. and Li, S.-M.: Reversible and irreversible processing of biogenic olefins on acidic aerosols, Atmos. Chem. Phys., 8, 2039–2055, https://doi.org/10.5194/acp-8-2039-2008, 2008.
Liu, S., Shilling, J. E., Song, C., Hiranuma, N., Zaveri, R. A., and Russell, L. M.: Hydrolysis of Organonitrate Functional Groups in Aerosol Particles, Aerosol. Sci. Tech., 46, 1359–1369, https://doi.org/10.1080/02786826.2012.716175, 2012b.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Lutz, A., Hallquist, M., Worsnop, D., and Thornton, J. A.: A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 7, 983–1001, https://doi.org/10.5194/amt-7-983-2014, 2014.
Loza, C. L., Chan, A. W. H., Galloway, M. M., Keutsch, F. N., Flagan, R. C., and Seinfeld, J. H.: Characterization of Vapor Wall Loss in Laboratory Chambers, Environ. Sci. Technol., 44, 5074–5078, https://doi.org/10.1021/es100727v, 2010.
Matsunaga, A. and Ziemann, P. J.: Gas-Wall Partitioning of Organic Compounds in a Teflon Film Chamber and Potential Effects on Reaction Product and Aerosol Yield Measurements, Aerosol Sci. Tech., 44, 881–892, https://doi.org/10.1080/02786826.2010.501044, 2010.
Mcvay, R. C., Cappa, C. D., and Seinfeld, J. H.: Vapor-Wall Deposition in Chambers: Theoretical Considerations, Environ. Sci. Technol., 48, 10251–10258, https://doi.org/10.1021/es502170j, 2014.
Müller, J.-F., Peeters, J., and Stavrakou, T.: Fast photolysis of carbonyl nitrates from isoprene, Atmos. Chem. Phys., 14, 2497–2508, https://doi.org/10.5194/acp-14-2497-2014, 2014.
Nah, T., Sanchez, J., Boyd, C. M., and Ng, N. L.: Photochemical Aging of alpha-pinene and beta-pinene Secondary Organic Aerosol formed from Nitrate Radical Oxidation, Environ. Sci. Technol., 50, 222–231, https://doi.org/10.1021/acs.est.5b04594, 2016.
Ng, N. L., Herndon, S. C., Trimborn, A., Canagaratna, M. R., Croteau, P. L., Onasch, T. B., Sueper, D., Worsnop, D. R., Zhang, Q., Sun, Y. L., and Jayne, J. T.: An Aerosol Chemical Speciation Monitor (ACSM) for Routine Monitoring of the Composition and Mass Concentrations of Ambient Aerosol, Aerosol Sci. Technol., 45, 780–794, https://doi.org/10.1080/02786826.2011.560211, 2011.
Ng, N. L., Brown, S. S., Archibald, A. T., Atlas, E., Cohen, R. C., Crowley, J. N., Day, D. A., Donahue, N. M., Fry, J. L., Fuchs, H., Griffin, R. J., Guzman, M. I., Herrmann, H., Hodzic, A., Iinuma, Y., Jimenez, J. L., Kiendler-Scharr, A., Lee, B. H., Luecken, D. J., Mao, J., McLaren, R., Mutzel, A., Osthoff, H. D., Ouyang, B., Picquet-Varrault, B., Platt, U., Pye, H. O. T., Rudich, Y., Schwantes, R. H., Shiraiwa, M., Stutz, J., Thornton, J. A., Tilgner, A., Williams, B. J., and Zaveri, R. A.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 17, 2103–2162, https://doi.org/10.5194/acp-17-2103-2017, 2017.
Nguyen, T. B., Crounse, J. D., Teng, A. P., Clair, J. M. S., Paulot, F., Wolfe, G. M., and Wennberg, P. O.: Rapid deposition of oxidized biogenic compounds to a temperate forest, P. Natl. Acad. Sci. USA, 112, E392–E401, https://doi.org/10.1073/pnas.1418702112, 2015.
Noziere, B., Barnes, I., and Becker, K. H.: Product study and mechanisms of the reactions of alpha-pinene and of pinonaldehyde with OH radicals, J. Geophys. Res.-Atmos., 104, 23645–23656, https://doi.org/10.1029/1999jd900778, 1999.
Orlando, J. J. and Tyndall, G. S.: Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 41, 6294–6317, https://doi.org/10.1039/c2cs35166h, 2012.
Pankow, J. F.: An Absorption-Model of Gas-Particle Partitioning of Organic-Compounds in the Atmosphere, Atmos. Environ., 28, 185–188, https://doi.org/10.1016/1352-2310(94)90093-0, 1994.
Pankow, J. F. and Asher, W. E.: SIMPOL.1: a simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds, Atmos. Chem. Phys., 8, 2773–2796, https://doi.org/10.5194/acp-8-2773-2008, 2008.
Perring, A. E., Pusede, S. E., and Cohen, R. C.: An Observational Perspective on the Atmospheric Impacts of Alkyl and Multifunctional Nitrates on Ozone and Secondary Organic Aerosol, Chem. Rev., 113, 5848–5870, https://doi.org/10.1021/cr300520x, 2013.
Petters, M. D. and Kreidenweis, S. M.: A single parameter representation of hygroscopic growth and cloud condensation nucleus activity, Atmos. Chem. Phys., 7, 1961–1971, https://doi.org/10.5194/acp-7-1961-2007, 2007.
Pye, H. O. T., Luecken, D. J., Xu, L., Boyd, C. M., Ng, N. L., Baker, K. R., Ayres, B. R., Bash, J. O., Baumann, K., Carter, W. P. L., Edgerton, E., Fry, J. L., Hutzell, W. T., Schwede, D. B., and Shepson, P. B.: Modeling the Current and Future Roles of Particulate Organic Nitrates in the Southeastern United States, Environ. Sci. Technol., 49, 14195–14203, https://doi.org/10.1021/acs.est.5b03738, 2015.
Pye, H. O. T., D'Ambro, E. L., Lee, B., Schobesberger, S., Takeuchi, M., Zhao, Y., Lopez-Hilfiker, F., Liu, J. M., Shilling, J. E., Xing, J., Mathur, R., Middlebrook, A. M., Liao, J., Welti, A., Graus, M., Warneke, C., de Gouw, J. A., Holloway, J. S., Ryerson, T. B., Pollack, I. B., and Thornton, J. A.: Anthropogenic enhancements to production of highly oxygenated molecules from autoxidation, P. Natl. Acad. Sci. USA, 116, 6641–6646, https://doi.org/10.1073/pnas.1810774116, 2019.
Rindelaub, J. D., McAvey, K. M., and Shepson, P. B.: The photochemical production of organic nitrates from alpha-pinene and loss via acid-dependent particle phase hydrolysis, Atmos. Environ., 100, 193–201, 2015.
Rindelaub, J. D., Borca, C. H., Hostetler, M. A., Slade, J. H., Lipton, M. A., Slipchenko, L. V., and Shepson, P. B.: The acid-catalyzed hydrolysis of an α-pinene-derived organic nitrate: kinetics, products, reaction mechanisms, and atmospheric impact, Atmos. Chem. Phys., 16, 15425–15432, https://doi.org/10.5194/acp-16-15425-2016, 2016.
Rollins, A. W., Browne, E. C., Min, K. E., Pusede, S. E., Wooldridge, P. J., Gentner, D. R., Goldstein, A. H., Liu, S., Day, D. A., Russell, L. M., and Cohen, R. C.: Evidence for NOx Control over Nighttime SOA Formation, Science, 337, 1210–1212, https://doi.org/10.1126/science.1221520, 2012.
Rollins, A. W., Pusede, S., Wooldridge, P., Min, K. E., Gentner, D. R., Goldstein, A. H., Liu, S., Day, D. A., Russell, L. M., Rubitschun, C. L., Surratt, J. D., and Cohen, R. C.: Gas/particle partitioning of total alkyl nitrates observed with TD-LIF in Bakersfield, J. Geophys. Res.-Atmos., 118, 6651–6662, https://doi.org/10.1002/jgrd.50522, 2013.
Romonosky, D. E., Li, Y., Shiraiwa, M., Laskin, A., Laskin, J., and Nizkorodov, S. A.: Aqueous Photochemistry of Secondary Organic Aerosol of alpha-Pinene and alpha-Humulene Oxidized with Ozone, Hydroxyl Radical, and Nitrate Radical, J. Phys. Chem. A, 121, 1298–1309, https://doi.org/10.1021/acs.jpca.6b10900, 2017.
Ruggeri, G., Bernhard, F. A., Henderson, B. H., and Takahama, S.: Model–measurement comparison of functional group abundance in α-pinene and 1,3,5-trimethylbenzene secondary organic aerosol formation, Atmos. Chem. Phys., 16, 8729–8747, https://doi.org/10.5194/acp-16-8729-2016, 2016.
Russell, L. M., Bahadur, R., and Ziemann, P. J.: Identifying organic aerosol sources by comparing functional group composition in chamber and atmospheric particles, P. Natl. Acad. Sci. USA, 108, 3516–3521, https://doi.org/10.1073/pnas.1006461108, 2011.
Sanchez, J., Tanner, D. J., Chen, D., Huey, L. G., and Ng, N. L.: A new technique for the direct detection of HO2 radicals using bromide chemical ionization mass spectrometry (Br-CIMS): initial characterization, Atmos. Meas. Tech., 9, 3851–3861, https://doi.org/10.5194/amt-9-3851-2016, 2016.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Schwantes, R. H., Teng, A. P., Nguyen, T. B., Coggon, M. M., Crounse, J. D., St Clair, J. M., Zhang, X., Schilling, K. A., Seinfeld, J. H., and Wennberg, P. O.: Isoprene NO3 Oxidation Products from the RO2 + HO2 Pathway, J. Phys. Chem. A, 119, 10158–10171, https://doi.org/10.1021/acs.jpca.5b06355, 2015.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics : from air pollution to climate change, John Wiley & Sons, Inc., Hoboken, New Jersey, 2016.
Shilling, J. E., Zaveri, R. A., Fast, J. D., Kleinman, L., Alexander, M. L., Canagaratna, M. R., Fortner, E., Hubbe, J. M., Jayne, J. T., Sedlacek, A., Setyan, A., Springston, S., Worsnop, D. R., and Zhang, Q.: Enhanced SOA formation from mixed anthropogenic and biogenic emissions during the CARES campaign, Atmos. Chem. Phys., 13, 2091–2113, https://doi.org/10.5194/acp-13-2091-2013, 2013.
Shrivastava, M., Andreae, M. O., Artaxo, P., Barbosa, H. M. J., Berg, L. K., Brito, J., Ching, J., Easter, R. C., Fan, J. W., Fast, J. D., Feng, Z., Fuentes, J. D., Glasius, M., Goldstein, A. H., Alves, E. G., Gomes, H., Gu, D., Guenther, A., Jathar, S. H., Kim, S., Liu, Y., Lou, S. J., Martin, S. T., McNeill, V. F., Medeiros, A., de Sa, S. S., Shilling, J. E., Springston, S. R., Souza, R. A. F., Thornton, J. A., Isaacman-VanWertz, G., Yee, L. D., Ynoue, R., Zaveri, R. A., Zelenyuk, A., and Zhao, C.: Urban pollution greatly enhances formation of natural aerosols over the Amazon rainforest, Nat. Commun., 10, 1046, https://doi.org/10.1038/s41467-019-08909-4, 2019.
Slade, J. H., de Perre, C., Lee, L., and Shepson, P. B.: Nitrate radical oxidation of γ-terpinene: hydroxy nitrate, total organic nitrate, and secondary organic aerosol yields, Atmos. Chem. Phys., 17, 8635–8650, https://doi.org/10.5194/acp-17-8635-2017, 2017.
Song, M., Marcolli, C., Krieger, U. K., Zuend, A., and Peter, T.: Liquid-liquid phase separation in aerosol particles: Dependence on O:C, organic functionalities, and compositional complexity, Geophys. Res. Lett., 39, L19801, https://doi.org/10.1029/2012gl052807, 2012.
Spittler, M., Barnes, I., Bejan, I., Brockmann, K. J., Benter, T., and Wirtz, K.: Reactions of NO3 radicals with limonene and alpha-pinene: Product and SOA formation, Atmos. Environ., 40, S116–S127, https://doi.org/10.1016/j.atmosenv.2005.09.093, 2006.
Spracklen, D. V., Jimenez, J. L., Carslaw, K. S., Worsnop, D. R., Evans, M. J., Mann, G. W., Zhang, Q., Canagaratna, M. R., Allan, J., Coe, H., McFiggans, G., Rap, A., and Forster, P.: Aerosol mass spectrometer constraint on the global secondary organic aerosol budget, Atmos. Chem. Phys., 11, 12109–12136, https://doi.org/10.5194/acp-11-12109-2011, 2011.
Stark, H., Yatavelli, R. L. N., Thompson, S. L., Kang, H., Krechmer, J. E., Kimmel, J. R., Palm, B. B., Hu, W. W., Hayes, P. L., Day, D. A., Campuzano-Jost, P., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., and Jimenez, J. L.: Impact of Thermal Decomposition on Thermal Desorption Instruments: Advantage of Thermogram Analysis for Quantifying Volatility Distributions of Organic Species, Environ. Sci. Technol., 51, 8491–8500, https://doi.org/10.1021/acs.est.7b00160, 2017.
Surratt, J. D., Gomez-Gonzalez, Y., Chan, A. W. H., Vermeylen, R., Shahgholi, M., Kleindienst, T. E., Edney, E. O., Offenberg, J. H., Lewandowski, M., Jaoui, M., Maenhaut, W., Claeys, M., Flagan, R. C., and Seinfeld, J. H.: Organosulfate formation in biogenic secondary organic aerosol, J. Phys. Chem. A, 112, 8345–8378, https://doi.org/10.1021/jp802310p, 2008.
Takeuchi, M. and Ng, N. L.: Organic Nitrates and Secondary Organic Aerosol (SOA) Formation from Oxidation of Biogenic Volatile Organic Compounds, Acs. Sym. Ser., 1299, 105–125, 2018.
Wangberg, I., Barnes, I., and Becker, K. H.: Product and mechanistic study of the reaction of NO3 radicals with alpha-pinene, Environ. Sci. Technol., 31, 2130–2135, https://doi.org/10.1021/es960958n, 1997.
Wayne, R. P., Barnes, I., Biggs, P., Burrows, J. P., Canosamas, C. E., Hjorth, J., Lebras, G., Moortgat, G. K., Perner, D., Poulet, G., Restelli, G., and Sidebottom, H.: The Nitrate Radical – Physics, Chemistry, and the Atmosphere, Atmos. Environ. A-Gen., 25, 1–203, https://doi.org/10.1016/0960-1686(91)90192-A, 1991.
Weber, R. J., Sullivan, A. P., Peltier, R. E., Russell, A., Yan, B., Zheng, M., de Gouw, J., Warneke, C., Brock, C., Holloway, J. S., Atlas, E. L., and Edgerton, E.: A study of secondary organic aerosol formation in the anthropogenic-influenced southeastern United States, J. Geophys. Res.-Atmos., 112, D13302, https://doi.org/10.1029/2007jd008408, 2007.
Xu, L., Guo, H. Y., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., Knote, C., Olson, K., Koss, A., Goldstein, A. H., Hering, S. V., de Gouw, J., Baumann, K., Lee, S. H., Nenes, A., Weber, R. J., and Ng, N. L.: Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, P. Natl. Acad. Sci. USA, 112, E4509–E4509, https://doi.org/10.1073/pnas.1512279112, 2015a.
Xu, L., Suresh, S., Guo, H., Weber, R. J., and Ng, N. L.: Aerosol characterization over the southeastern United States using high-resolution aerosol mass spectrometry: spatial and seasonal variation of aerosol composition and sources with a focus on organic nitrates, Atmos. Chem. Phys., 15, 7307–7336, https://doi.org/10.5194/acp-15-7307-2015, 2015b.
Xu, L., Pye, H. O. T., He, J., Chen, Y., Murphy, B. N., and Ng, N. L.: Experimental and model estimates of the contributions from biogenic monoterpenes and sesquiterpenes to secondary organic aerosol in the southeastern United States, Atmos. Chem. Phys., 18, 12613–12637, https://doi.org/10.5194/acp-18-12613-2018, 2018.
Xu, L., Moller, K. H., Crounse, J. D., Otkjwr, R. V., Kjaergaard, H. G., and Wennberg, P. O.: Unimolecular Reactions of Peroxy Radicals Formed in the Oxidation of alpha-Pinene and beta-Pinene by Hydroxyl Radicals, J. Phys. Chem. A, 123, 1661–1674, https://doi.org/10.1021/acs.jpca.8b11726, 2019.
Xu, W., Lambe, A., Silva, P., Hu, W. W., Onasch, T., Williams, L., Croteau, P., Zhang, X., Renbaum-Wolff, L., Fortner, E., Jimenez, J. L., Jayne, J., Worsnop, D., and Canagaratna, M.: Laboratory evaluation of species-dependent relative ionization efficiencies in the Aerodyne Aerosol Mass Spectrometer, Aerosol Sci. Technol., 52, 626–641, https://doi.org/10.1080/02786826.2018.1439570, 2018.
Zare, A., Romer, P. S., Nguyen, T., Keutsch, F. N., Skog, K., and Cohen, R. C.: A comprehensive organic nitrate chemistry: insights into the lifetime of atmospheric organic nitrates, Atmos. Chem. Phys., 18, 15419–15436, https://doi.org/10.5194/acp-18-15419-2018, 2018.
Zhang, H. F., Yee, L. D., Lee, B. H., Curtis, M. P., Worton, D. R., Isaacman-VanWertz, G., Offenberg, J. H., Lewandowski, M., Kleindienst, T. E., Beaver, M. R., Holder, A. L., Lonneman, W. A., Docherty, K. S., Jaoui, M., Pye, H. O. T., Hu, W. W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Guo, H. Y., Weber, R. J., de Gouw, J., Koss, A. R., Edgerton, E. S., Brune, W., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Kreisberg, N. M., Spielman, S. R., Hering, S. V., Wilson, K. R., Thornton, J. A., and Goldstein, A. H.: Monoterpenes are the largest source of summertime organic aerosol in the southeastern United States, P. Natl. Acad. Sci. USA, 115, 2038–2043, https://doi.org/10.1073/pnas.1717513115, 2018.
Zhang, X., Cappa, C. D., Jathar, S. H., Mcvay, R. C., Ensberg, J. J., Kleeman, M. J., and Seinfeld, J. H.: Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol, P. Natl. Acad. Sci. USA, 111, 5802–5807, https://doi.org/10.1073/pnas.1404727111, 2014.
Zhang, X., Schwantes, R. H., McVay, R. C., Lignell, H., Coggon, M. M., Flagan, R. C., and Seinfeld, J. H.: Vapor wall deposition in Teflon chambers, Atmos. Chem. Phys., 15, 4197–4214, https://doi.org/10.5194/acp-15-4197-2015, 2015.
Ziemann, P. J. and Atkinson, R.: Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 41, 6582–6605, https://doi.org/10.1039/c2cs35122f, 2012.