the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 29 Jun 2018
| 29 Jun 2018
Tropospheric HONO distribution and chemistry in the southeastern US
Xianliang Zhou
Dennis Pu
Jochen Stutz
James Festa
Max Spolaor
Catalina Tsai
Christopher Cantrell
Roy L. Mauldin III
Andrew Weinheimer
Rebecca S. Hornbrook
Eric C. Apel
Alex Guenther
Lisa Kaser
Bin Yuan
Thomas Karl
Julie Haggerty
Samuel Hall
Kirk Ullmann
James Smith
John Ortega
Here we report the measurement results of nitrous acid (HONO) and a suite of relevant parameters on the NCAR C-130 research aircraft in the southeastern US during the NOMADSS 2013 summer field study. The daytime HONO concentration ranged from low parts per trillion by volume (pptv) in the free troposphere (FT) to mostly within 5–15 pptv in the background planetary boundary layer (PBL). There was no discernible vertical HONO gradient above the lower flight altitude of 300 m in the PBL, and the transport of ground surface HONO was not found to be a significant contributor to the tropospheric HONO budget. The total in situ HONO source mean (±1 SD) was calculated as 53 (±21) pptv h−1 during the day. The upper-limit contribution from NOx-related reactions was 10 (±5) pptv h−1, and the contribution from photolysis of particulate nitrate (pNO3) was 38 (±23) pptv h−1, based on the measured pNO3 concentrations and the median pNO3 photolysis rate constant of 2.0 × 10−4 s−1 determined in the laboratory using ambient aerosol samples. The photolysis of HONO contributed to less than 10 % of the primary OH source. However, a recycling NOx source via pNO3 photolysis was equivalent to ∼ 2.3 × 10−6 mol m−2 h−1 in the air column within the PBL, a considerable supplementary NOx source in the low-NOx background area. Up to several tens of parts per trillion by volume of HONO were observed in power plant and urban plumes during the day, mostly produced in situ from precursors including NOx and pNO3. Finally, there was no observable accumulation of HONO in the nocturnal residual layer and the nocturnal FT in the background southeastern US, with an increase in the HONO ∕ NOx ratio of ≤ 3 × 10−4 h−1 after sunset.
- Article
(3116 KB) - Full-text XML
-
Supplement
(387 KB) - BibTeX
- EndNote




Extensive field studies at ground sites have shown that gas-phase nitrous acid (HONO) exists at much higher levels than expected during the day, with a mixing ratio of HONO of up to several parts per billion by volume (ppbv) in the urban atmosphere (Acker et al., 2006; Villena et al., 2011) and up to several hundred parts per trillion by volume (pptv) in rural environments (Acker et al., 2006; Kleffmann et al., 2003; Zhang et al., 2009; Zhou et al., 2002, 2011). At the observed concentrations, HONO photolysis (Reaction R1) becomes an important or even a major OH primary source in both urban (Elshorbany et al., 2010; Villena et al., 2011) and rural environments near the ground surface (Acker et al., 2006; He et al., 2006; Kleffmann et al., 2003; Zhou et al., 2002, 2011).
The OH radical is responsible for the removal of primary pollutants and plays a crucial role in the formation of secondary pollutants, such as O3 and aerosol (Finlayson-Pitts and Pitts Jr., 2000), and thus HONO plays an important role in atmospheric chemistry.
The removal processes of HONO from the troposphere are relatively well understood, including mainly photolysis, reaction with OH radicals and surface deposition. Photolysis is the dominant sink for HONO during the day (Kleffmann et al., 2003; Oswald et al., 2015; Zhang et al., 2009, 2012), and dry deposition is the major HONO loss pathway at night, especially over wet surfaces (He et al., 2006; VandenBoer et al., 2015). However, HONO sources in the planetary boundary layer (PBL) are numerous. HONO is directly emitted from combustion processes, such as automobile emissions (Y. Q. Li et al., 2008) and biomass burning (Burling et al., 2010; Trentmann et al., 2003). Soil emission via nitrification or denitrification is another source of HONO, which might be important in agricultural regions (Maljanen et al., 2013; Oswald et al., 2013; Su et al., 2011). Due to the relatively short photolytic lifetime of HONO, on the order of 10 min around summer noontime, the impacts of the direct emission on HONO distribution and chemistry is highly localized and limited to the source region.
HONO is a unique species that is produced through heterogeneous reactions of different precursors, such as NO2 and HNO3, on surfaces (Reactions R2–R3):
Heterogeneous reactions of NO2 with organics (Reaction R2) on the surfaces have been found to be greatly accelerated by sunlight through photosensitization (George et al., 2005; Kleffmann, 2007; Stemmler et al., 2006, 2007) and these reactions on ground surfaces are likely the major daytime HONO source in urban environments (Acker et al., 2006; Villena et al., 2011; Wong et al., 2011). Laboratory studies have confirmed that HNO3 undergoes photolysis in sunlight at rates 2–3 orders of magnitude greater on the surface than in the gas phase (Baergen and Donaldson, 2013; Du and Zhu, 2011; Ye et al., 2016a, b; Zhou et al., 2003; Zhu et al., 2008), producing NOx and HONO. In low-NOx environments, photolysis of nitric acid/nitrate deposited on the surface has been proposed to be the major daytime HONO source near the ground surface (Ye et al., 2016b; Zhou et al., 2003, 2011).
Several processes within an air mass may lead to volume, or in situ, production of HONO. The OH+NO reaction ('R1) in the gas phase may be a significant HONO source in high NOx and photochemically reactive atmospheres (Kleffmann, 2007; Neuman et al., 2016; Villena et al., 2011) but becomes negligible in low-NOx environments (Li et al., 2014; Ye et al., 2016b). Two additional gas-phase reactions have been proposed to produce HONO within the air column: excited NO2 (NO2∗) with water vapor (Reaction R4) (S. P. Li et al., 2008), and NO2 with the hydroperoxyl–water complex (HO2 ⋅ H2O) (Reaction R5) (Li et al., 2014):
However, further laboratory evidence suggests that Reaction (R4) is too slow to be important (Carr et al., 2009; Wong et al., 2011). And recent airborne observations have demonstrated that the HONO yield (α) from Reaction (R5) is less than 0.03 (Ye et al., 2015). Heterogeneous reactions of NO2 (Reaction R2) on aerosol surfaces and photolysis of particulate nitrate (pNO3) similar to Reaction (R3) also contribute to in situ HONO production in the air column.
Table 1Measurements from the NOMADSS 2013 summer study used in this analysis.
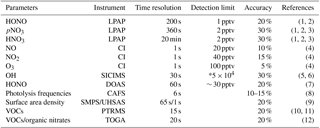
* In molecules per cubic centimeter. LPAP: long-path absorption photometric systems; CI: four-channel chemiluminescence instrument; SICIMS: selected-ion chemical-ionization mass spectrometer; DOAS: differential optical absorption spectroscopy; CAFS: charged-coupled device actinic flux spectroradiometer; SMPS: scanning mobility particle sizer; UHSAS: ultra-high-sensitivity aerosol spectrometer; PTRMS: proton-transfer-reaction mass spectrometry; TOGA: Trace Organic Gas Analyzer. (1) Zhang et al., 2012. (2) Ye et al., 2016b. (3) Huang et al., 2002. (4) Ridley et al., 2004. (5) Hornbrook et al., 2011b. (6) Mauldin et al., 2010. (7) Platt and Stutz, 2008. (8) Shetter et al., 2002. (9) Flagan, 2002. (10) Karl et al., 2003. (11) de Gouw and Warneke, 2007. (12) Hornbrook et al., 2011a.
Most HONO measurements have been made at ground stations. The observed HONO concentrations reported in the literature represent the HONO levels in the lower PBL under the significant but varying influence of ground surface processes. Thus, it is difficult to distinguish the ground surface HONO sources from the in situ HONO sources. Measurements of vertical HONO concentration profiles and HONO fluxes have suggested that ground surfaces can be major HONO sources for the overlying atmosphere in many cases (He et al., 2006; Kleffmann et al., 2003; Stutz et al., 2002; Zhou et al., 2011; VandenBoer et al., 2013; Young et al., 2012), but not in some other cases (Villena et al., 2011). A recent HONO flux measurement has suggested that the HONO source from the forest canopy contributed ∼ 60 % of the measured HONO budget at the measurement height of 11 m above the forest canopy, and the in situ HONO production contributed the remaining ∼ 40 % (Zhou et al., 2011). Similarly, observational and modeling studies implied the presence of a volume HONO source at 130 m of altitude above Houston, TX (Wong et al., 2012, 2013). The relative importance of in situ HONO production would be expected to increase with altitude due to decreasing influence of the ground surface, at least during the day. Airborne measurements in the air mass above the altitude influenced directly by ground HONO sources should provide more direct and quantitative evidence for in situ HONO production in the troposphere. Indeed, the limited number of airborne measurements available have shown that HONO exists not only in substantial amounts in combustion and urban plumes (Neuman et al., 2016) but also throughout the troposphere (Li et al., 2014; Ye et al., 2015; Zhang et al., 2009).
Here we report airborne HONO measurement results and findings from five research flights in the southeastern US during the NOMADSS (Nitrogen, Oxidants, Mercury and Aerosol Distributions, Sources and Sinks) 2013 summer field campaign aboard the NSF/NCAR C-130 research aircraft.
NOMADSS was an airborne field study under the umbrella of SAS (Southeast Atmosphere Study). It consisted of 19 research flights onboard the NSF/NCAR C-130 aircraft from 1 June 2013 to 15 July 2013. Parameters observed included HONO, HNO3, particulate nitrate, NOx, O3, BrO, OH radicals, HO2 radicals, RO2 radicals, aerosol surface area densities (size < 1 µm), VOCs, photolysis frequencies and other meteorology parameters. Table 1 summarizes the instrumentation, time resolution, detection limit, accuracy, and references for the measurements. The results from 5 out of 19 flights are presented here to discuss vertical HONO distribution and HONO chemistry in the southeastern US. The flight tracks are shown in Fig. 1.
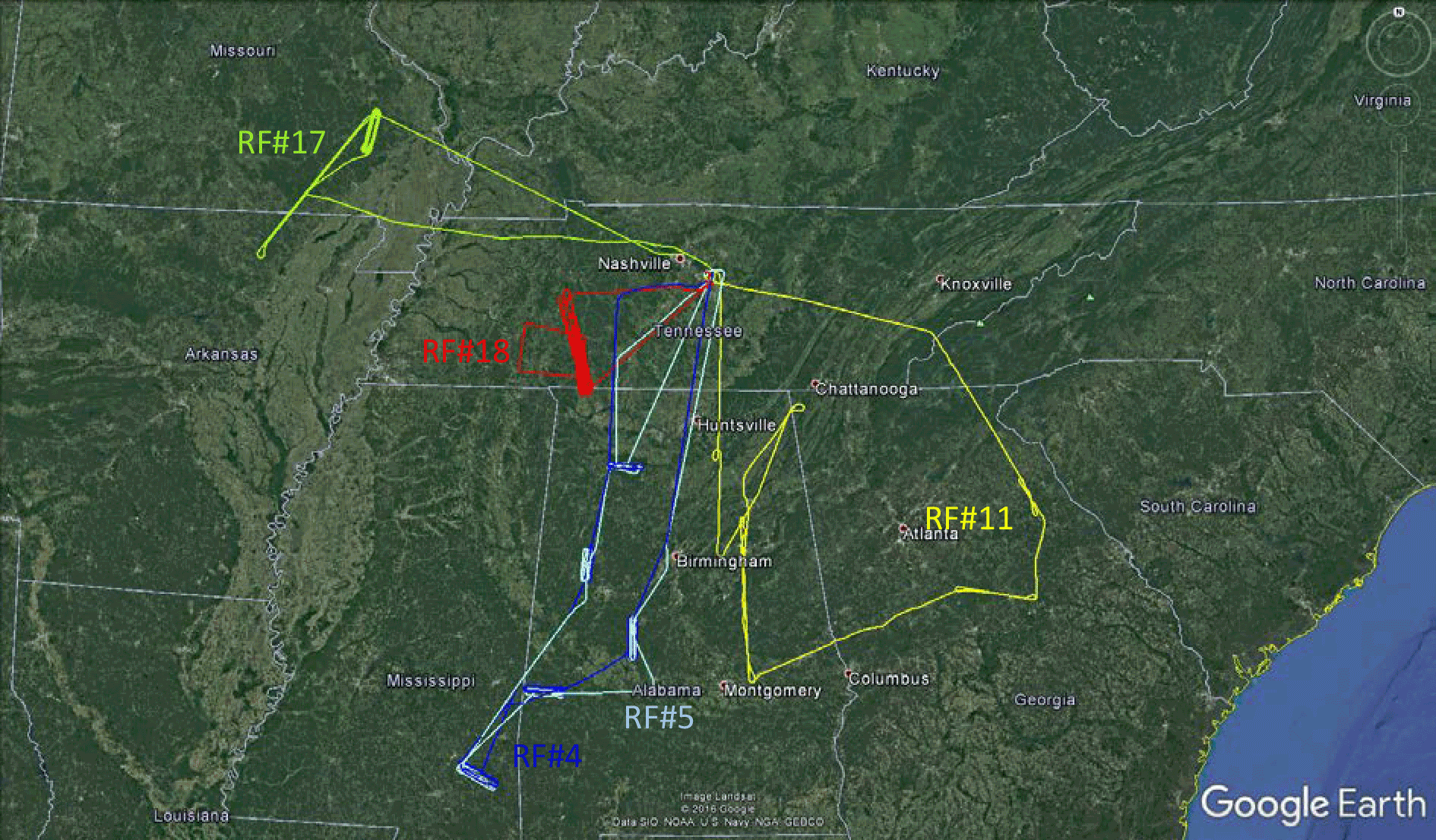
Figure 1Flight tracks in the southeastern US during the NOMADSS 2013 summer study. The flight start time and end time in UTC (= EDT +4) are for RF 4 (blue): 15:12 and 22:30, 12 June 2013; RF 5 (light blue): 15:04 and 21:52, 14 June 2013; RF 11 (yellow): 15:20 and 21:02, 29 June 2013; RF 17 (green): 15:07 and 21:57, 11 July 2013; and RF 18 (red): 20:32, 12 July 2013 and 03:37, 13 July 2013.
2.1 LPAP measurements of HONO and pNO3
HONO was measured by two long-path absorption photometric (LPAP) systems based on the Griess–Saltzman reaction (Zhang et al., 2012; Ye et al., 2016b). Briefly, ambient air was first brought into the aircraft through a heated PFA inlet, with a residence time of 0.14 s. HONO was scrubbed using deionized (DI) water in two 10-turn glass coil samplers and the collected nitrite was then derivatized with 5 mM of sulfanilamide (SA) and 0.5 mM of N-(1-Naphthyl)ethylenediamine (NED) in 40 mM of HCl to form an azo dye. The azo dye was then detected by light absorption at 540 nm using a four-channel optic fiber spectrometer (LEDspec, WPI) with two 1 m liquid waveguide capillary flow cells (WPI). Each LPAP system ran a 30 min measurement and zero cycle, with 20 min sampling ambient air and 10 min sampling “zero-HONO” air for baseline correction, and with a 15 min time offset between the two sampling cycles. The combination of the data from the two systems provides continuous HONO concentration measurement. The zero-HONO air was generated by directing the sample stream through a Na2CO3-coated denuder to remove HONO while allowing most interfering species, such as NOx, peroxyacetyl nitrate (PAN) and particulate nitrite, to pass through. The absorbance signals were sampled at a rate of 1 Hz, and were averaged into 1 or 3 min data for analysis.
Interference from NOx, PAN and particulate nitrite was corrected by subtracting the baseline from the ambient air signal. Due to the low collecting efficiency of these interfering species in the sampling coil and their low concentrations, the combined interference was estimated to be less than 10 % of the total signal. Potential interference from peroxynitric acid (HO2NO2) was suppressed by heating the PFA sampling line to 50 ∘C. The HO2NO2 steady state concentration ([HO2NO2]SS) was estimated to be less than 1 pptv at temperatures of 20–30 ∘C in the background PBL (Gierczak et al., 2005), and thus interference from HO2NO2 was negligible. By contrast in power plant and urban plumes in the PBL or biomass burning plumes in the upper free troposphere (FT), HO2NO2 interference was not negligible and thus a correction for the HONO measurement was made. An upper-limit HO2NO2 response efficiency was estimated to be 0.2 for our HONO measurement systems. The estimation was made from the lowest ratio of the measured HONO to the corresponding [HO2NO2]SS in cold air masses at high altitude, assuming no HONO existed. HONO concentrations were then corrected by subtracting a term of 0.2 × [HO2NO2]SS. The correction was below 10 % of the measured HONO concentrations in the PBL plumes. However, there may be overcorrections in the cold FT.
The lower detection limit of LPAP HONO measurement was estimated to be ≤ 1 pptv, based on 3 times the standard deviation of the zero air signal (N > 10). An overall uncertainty of ±(1 + 0.2 [HONO]) pptv was estimated, combining the uncertainties in signal acquisition and processing, air and liquid flow rates, standard preparation, and baseline correction. The accuracy of HONO measurements was confirmed by comparison with a limb-scanning differential optical absorption spectroscopy (DOAS) instrument (Platt and Stutz, 2008) during the NOMADSS 2013 summer field study onboard the C-130 aircraft (Ye et al., 2016b). When measuring in wide power plant plumes in which HONO mixing ratios exceeded the lower detection limits of both instruments, the agreement between these two instruments was very good, within the assessed uncertainties (Ye et al., 2016b).
Particulate nitrate was quantitatively collected with a frit disc sampler after a NaCl-coated denuder to remove HNO3 (Huang et al., 2002). The collected nitrate was reduced to nitrite by a Cd column and determined using a LPAP system (Zhang et al., 2012). “Zero-pNO3” air was generated to establish measurement baselines for pNO3 by passing the ambient air through a Teflon filter to remove aerosol particles and then a NaCl-coated denuder to remove HNO3 before reaching the sampling unit of LPAP. Potential interferences from HONO, NOx and PAN were corrected by subtracting the baselines from the ambient air signals. The lower detection limit of pNO3 was estimated to be 2 pptv, based on 3 times the standard deviation of the zero-air signal (N > 10). An overall uncertainty of ±(2 + 0.3 [pNO3]) pptv was estimated.
Noisy baselines were observed when the C-130 was flying in the clouds, due to the sampling of cloud droplets by our sampling systems. Because of the lack of a valid way to correct for this interference, all in-cloud measurement data of HONO and pNO3 have been excluded from the data analysis. In addition, large baseline shifts were observed sometimes when the flow state of the scrubbing solution was disturbed by rapid pressure changes during the aircraft's rapid ascent to or descent from high altitudes. The data were excluded from analysis if the baseline shifts caused by rapid pressure changes could not be reasonably corrected, regardless of the sign or magnitude of the data.
2.2 Supporting measurements
The mixing ratios of a large number of non-methane organic compounds (NMOCs) were measured by Trace Organic Gas Analyzer (TOGA) (Hornbrook et al., 2011a) and proton-transfer-reaction mass spectrometry (PTR-MS) (Karl et al., 2003; de Gouw and Warneke, 2007). The surface area density of fine particles was obtained by the measurement of a scanning mobility particle sizer (SMPS), under the assumption of preface sphere of aerosol particle. The photolysis frequencies were determined by a charged-coupled device actinic flux spectroradiometer (CAFS) instrument (Shetter et al., 2002). The mixing ratios of HOx and RO2 radicals were measured using a method based on selected-ion chemical-ionization mass spectrometry (SICIMS) (Hornbrook et al., 2011b; Mauldin et al., 2010). The mixing ratios of ozone and NOx were measured using chemiluminescence instruments (Ridley et al., 2004). Meteorology parameters were provided by state parameter measurements onboard the C-130.
3.1 General data description
Figure 2 shows the time series of HONO, NOx and pNO3 concentrations and the measurement altitude for five selected research flights in the southeastern US during the NOMADSS 2013 summer field study. Research flight (RF) 4, RF 5 and RF 17 are racetrack flights in the background terrestrial areas designed to establish HONO distribution and explore HONO chemistry in background air masses. RF 11 is a racetrack flight designed to intercept plumes from local power plants and urban areas and explore HONO chemistry therein. All four flights were conducted in the daytime, roughly from 14:00 to 22:00 UTC (10:00 to 18:00 EDT). RF 18 is a racetrack flight conducted from 20:30 on 12 July to 03:30 on 13 July UTC (16:30 to 23:30 on 12 July 2013 EDT), aiming to study the potential nighttime HONO accumulation in both the PBL and the FT.
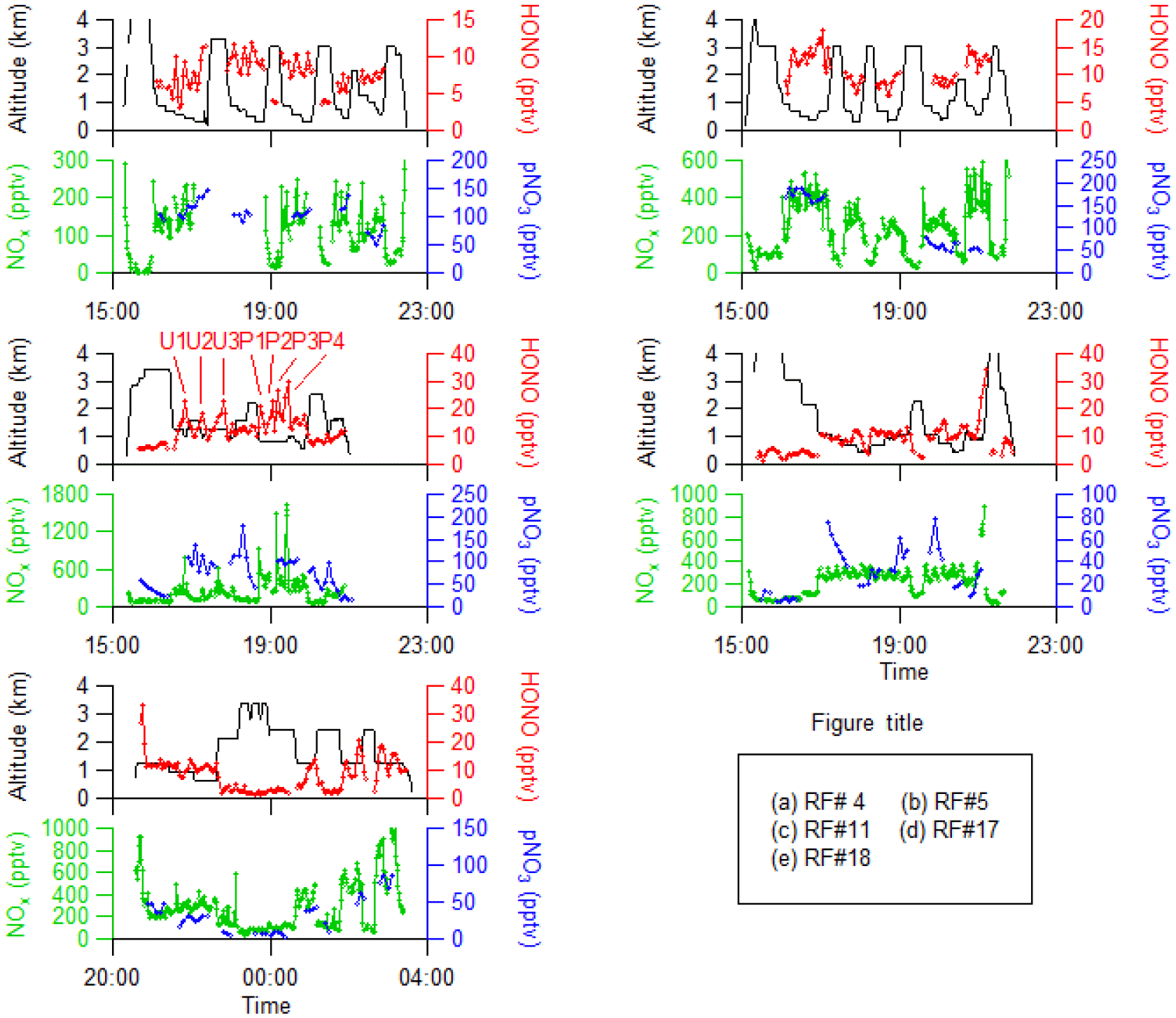
Figure 2Time series of altitude, HONO, NOx and pNO3 in five flights (RF 4, RF 5, RF 11, RF 17 and RF 18) in the southeastern US during the NOMADSS 2013 summer study. In RF 11, plumes U1 and U3 were from Birmingham, AL; plume U2 was from Montgomery, AL; plumes P1–P3 were from a power plant in Monroe County, GA; and plume P4 was from a power plant in Putnam County, GA.
Table 2Data statistics for HONO, NOx and pNO3 measurements in both the PBL and the FT from the five southeastern US research flights during the NOMADSS 2013 summer field study. The statistics analysis is based on 1 min NOx data, 3 min HONO data and 6 min pNO3 data.
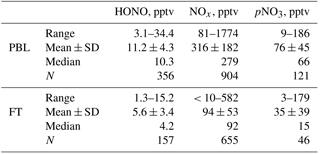
Table 2 summarizes the data statistics for HONO, NOx and pNO3 measurements in the PBL and the FT, and Fig. 3 shows composite vertical distributions of HONO, NOx and pNO3 concentrations from the five flights in the southeastern US during the NOMADSS 2013 summer field study. HONO, NOx and pNO3 concentrations show horizontal gradients in every racetrack flight and vary in different racetrack flights, reflecting the inhomogeneity of air masses in the region. However, there was no significant vertical gradient in HONO, NOx and pNO3 concentrations, which will be further discussed below. Except in a few power plant plumes and urban plumes mostly encountered in RF 11, most of the data are representative of background terrestrial air masses. The range of the mixing ratio of HONO is 3.1–34.4 pptv. The mean (±1 SD) and median values of HONO concentration are 5.6 (±3.4) pptv and 4.2 pptv in the FT and 11.2 (±4.3) and 10.3 pptv in the PBL. HONO levels at ∼ 4 pptv are typically found in the background FT, but high HONO concentrations of up to 15.2 pptv are also observed in the elevated biomass burning plumes. HONO levels at ∼ 11 pptv are representative of background conditions in the PBL. High HONO levels of up to 34.4 pptv are observed in the power plant plumes and urban plumes in RF 11 (see Sect. 3.4) and RF 17. These measured HONO values are consistent with the range of 4–74 pptv in the troposphere over northern Michigan (Zhang et al., 2009) but are significantly lower than 100–150 pptv in the morning residual layer over an industrial region of northern Italy (Li et al., 2014), where the levels of HONO precursors, such as NOx and pNO3, were much higher. The HONO concentrations were also consistent with the levels reported for the same region during the Southeast Nexus Experiment on the NOAA WP-3D aircraft (Neuman et al., 2016), that is, “indistinguishable from zero within the 15 pptv measurement uncertainty” in the background air and up to 150 pptv in the power plant plumes during the day. The lower HONO concentrations measured in the power plant plumes in this study than the daytime values reported by Neuman et al. (2016) probably reflect greater dilution of smaller plumes encountered by the C-130 than by the WP-3D, as indicated by much lower NOx levels observed, up to 1.8 ppbv vs. up to 60 ppbv.
The range of the mixing ratio of NOx is from several pptv to around 1.8 ppbv. The mean (±1 SD) and median values of NOx concentration are 94 (±53) and 92 pptv in the FT and 316 (±182) and 279 pptv in the PBL. The mixing ratios of NOx are mostly between 50 and 150 pptv in the background conditions in the FT and between 200 and 500 pptv in the background conditions in the PBL. Similar to HONO, high values of NOx also occur in the urban and power plant plumes in the PBL (up to 1.8 ppbv) and in the biomass burning plumes in the FT (up to 0.6 ppbv).
Fewer measurement data points are available for pNO3, compared to those for NOx and HONO, due to air bubble formation in the flow cell of the pNO3 system, especially at high altitudes. The range of the mixing ratio of pNO3 is from 3 to 186 pptv, with mean (±1 SD) and median values of 35 (±39) and 15 pptv in the FT and 76 (±45) and 66 pptv in the PBL. The pNO3 levels were highly variable in both the FT and the PBL. In the FT, the pNO3 levels were often under 10 pptv, but high concentrations of up to 179 pptv were also observed in elevated biomass burning plumes. In the PBL, high pNO3 levels were sometimes observed in relatively clean conditions, whereas low pNO3 levels were observed in high HONO and NOx power plant plumes. Both the N(V) level (= [HNO3] + [pNO3]) and the partitioning between HNO3 and pNO3 seem to play roles in determining the pNO3 level.
3.2 HONO contribution from ground-level sources
There are several ground-level HONO sources that may contribute to the HONO budget in the overlying atmosphere. They include anthropogenic sources, such as power plant and automobile emissions (Y. Q. Li et al., 2008; Neuman et al., 2016), and natural processes, such as soil emission (Maljanen et al., 2013; Oswald et al., 2013; Su et al., 2011), heterogeneous reactions of NO2 (Acker et al., 2006; George et al., 2005; Ndour et al., 2008, 2009; Ramazan et al., 2006) and surface HNO3 photolysis (Ye et al., 2016b; Zhou et al., 2003, 2011). Since HONO photolytic lifetime is relatively short, e.g., 8–16 min in RF 4, RF 5, RF 11 and RF 17, a steep negative vertical gradient of HONO concentration would be expected if a significant contribution originated from the ground. The lack of a significant vertical gradient in the measured HONO concentrations (Fig. 3a) thus suggests that the ground contribution is either limited to the shallow layer of the boundary layer near the ground, below the lowest C-130 flight altitude of 300 m, or small relative to the in situ production of HONO in the air column (Ye et al., 2017).
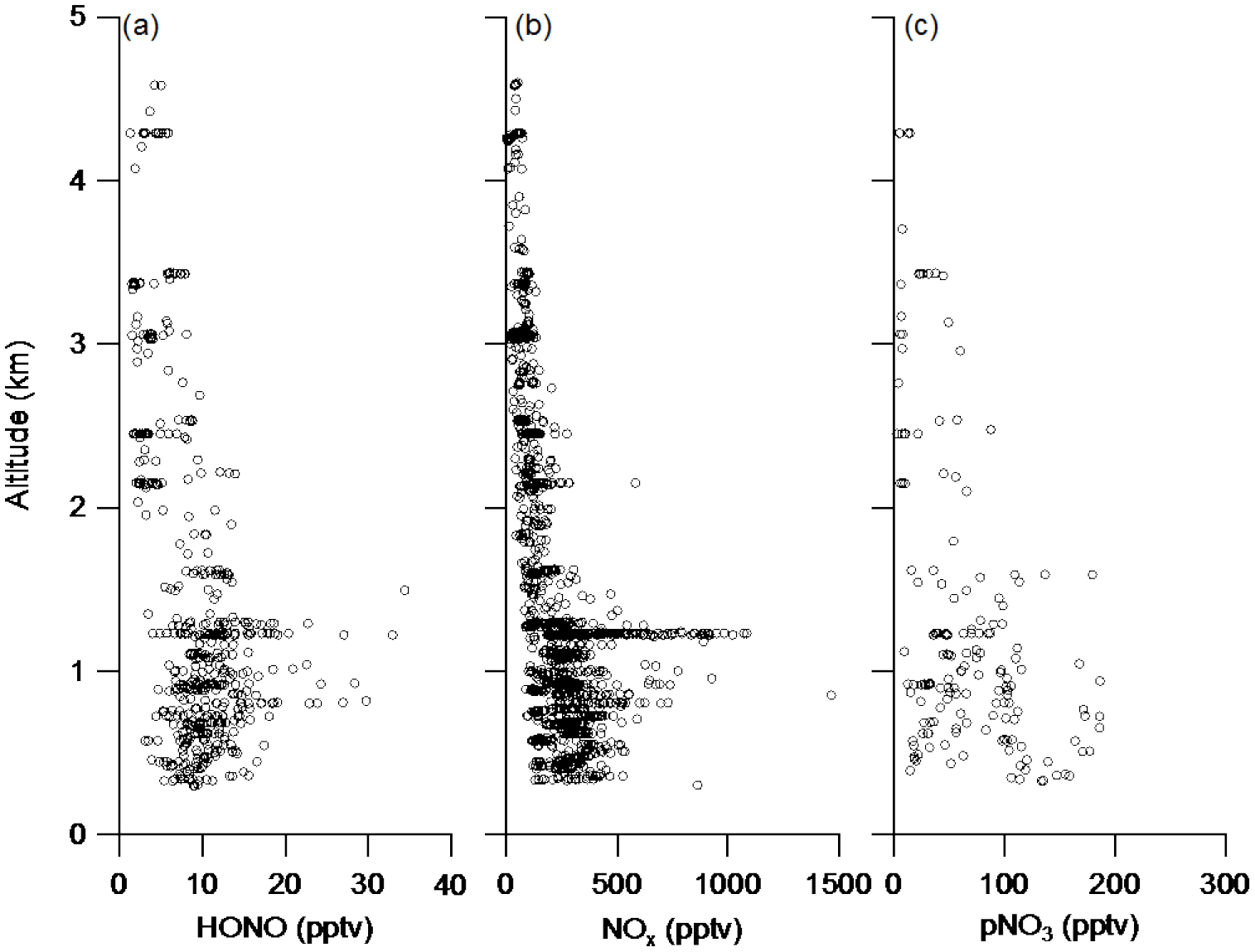
Figure 3Vertical distributions of concentrations of HONO (a), NOx (b) and pNO3 (c) in the five selected flights in the southeastern US during the NOMADSS 2013 summer study.

Figure 4Vertical distributions of concentrations of HONO (a), NOx (b), pNO3 (c), isoprene (d) and potential temperature (e) in the PBL during the first racetrack of RF 4 from 11:00 to 12:15 LT (16:00–17:15 UTC), 12 June 2013. The small open circles represent the time-averaged data of 1 min for NOx, isoprene and potential temperature, 3 min for HONO, and 6 min for pNO3. The large solid circles the mean values for each racetrack measurement altitude. The line in (d) is the best fit of Eq. (1) to the isoprene data: C=4700 e, r2=0.93.
To further examine the potential HONO contribution from the ground sources, vertical profiles of HONO, NOx and pNO3 are compared with those of potential temperature (K) and isoprene measured, for example, in the first racetrack of RF 4 from 11:00 to 12:15 LT (Fig. 4). The PBL height (H) was ∼ 1200 m as indicated by the constant potential temperature (Fig. 4e). The vertical distribution of isoprene originating from the ground can be expressed with the following Eq. (1):
where Co and C are its concentrations near the ground and at the altitude h, k is the pseudo-first-order degradation rate constant, τ is the average mixing time in the PBL, and h∗ (=H/(kτ)) is its characteristic transport height within one degradation lifetime of isoprene. According to the best fit of (Eq. 1) to the observed isoprene data (Fig. 4d), its characteristic transport height h∗ is estimated to be 692 m for isoprene. Assuming isoprene is mainly oxidized by the OH radical whose average concentration was found to be ∼ 3 × 106 mole cm−3 in the PBL in the southeastern US during the NOMADSS study (Kaser et al., 2015), the pseudo-first-order degradation rate constant of ∼ 3.0 × 10−4 s−1 (or the degradation rate of ∼ 0.93 h−1) is determined for isoprene. Based on a boundary layer height of ∼ 1.2 km (Fig. 4e), an average PBL mixing time τ is estimated to be ∼ 1.6 h between 11:00 and 12:15 LT of RF 4. With a photolytic lifetime of ∼ 11 min for HONO, about 11 % of the HONO originated from the ground level is expected to reach the altitude of 300 m, the lowest flight altitude of the C-130 aircraft between 11:00 and 12:15 LT in RF 4. The vertical mixing of the PBL is enhanced from the morning to the afternoon, as the ground surface is heated by solar radiation gradually during the day. The average mixing time in the PBL is reduced from ∼ 3 h in the morning to ∼ 1.5 h around noontime and to ∼ 30 min in the afternoon, determined from isoprene gradients from RF 4, RF 5 and RF 17. About 50 % of the ground-emitted HONO could survive and be transported to lower measurement altitudes. Again, if this ground source contribution were significant, the HONO concentration profile should exhibit a significant gradient, probably more pronounced than that of isoprene due to its shorter lifetime. The lack of such a vertical HONO gradient in the measured HONO concentration profiles (Fig. 3a) suggests that the contribution from ground HONO sources to the observed HONO concentrations in the PBL above 300 m is insignificant.
The NOx level was ≤ 0.5 ppbv in the PBL over the southeastern US (Fig. 2), excluding the power plant plumes. Based on an upper-limit HONO ∕ NOx ratio of 0.05 for the urban atmosphere at ground level (Villena et al., 2011), the initial HONO concentration would be ≤ 25 pptv in the source air mass on the ground level. With a transport time of ≥ 0.5 h, i.e., ≥ 3 times the HONO photolysis lifetime, the contribution from the ground HONO source would be ≤ 1 pptv. This analysis supports the conclusion that contribution of surface HONO source to the PBL HONO budget is insignificant.

Figure 5Correlation analysis of the main HONO sink (“HONO × JHONO”) with the contribution from NOx-related reactions (a) and with the contribution from particulate nitrate photolysis, pNO3× (b) in the southeastern US during the NOMADSS 2013 summer study. The line represents the Deming least-squares regression (Wu and Yu, 2018) (r2=0.40, intercept = −0.51 and slope = 0.19 for (a); r2=0.34, intercept = −5.0 and slope = 0.69 for (b)).
3.3 Daytime HONO chemistry in low-NOx areas
After removing the data measured in the urban and power plant plumes, the daytime HONO concentrations are within the range of 5–15 pptv throughout the PBL in the background terrestrial areas in the five racetrack research flights. Photolysis is the dominant sink for HONO, with a photolysis lifetime of 8–16 min during these four daytime flights (RF 4, RF 5, RF 11 and RF 17). Therefore, there must be a significant volume HONO source, up to 173 pptv h−1, within the air mass to sustain the observed HONO concentrations.
Both NOx and pNO3 are potential HONO precursors in the air column. While HONO correlates moderately with NOx (r2=0.45; Fig. S1 in the Supplement), it only weakly correlates with pNO3 (R2=0.17). It appears at first that NOx is a more important HONO precursor than pNO3. However, the detailed analysis below suggests that NOx is only a minor precursor to the observed HONO, and photolysis of pNO3 is the major in situ HONO source.
The upper limit of the photostationary-state HONO concentration ([HONO]pss) was calculated using Eq. (2), which takes into account all the known HONO source contributions from NOx-related reactions, including gaseous reactions of OH and NO (Reaction 'R1), excited NO2 (NO2*) and water vapor (Reaction R4) (Carr et al., 2009; S. P. Li et al., 2008), NO2, and the hydroperoxyl–water complex (HO2 ⋅ H2O) with an upper-limit HONO yield of 3 % (Reaction R5a) (Li et al., 2014; Ye et al., 2015). It also takes into account the heterogeneous reaction of NO2 on aerosol surfaces (Reaction R2) using an upper-limit uptake coefficient of 10−4 reported in the literature (George et al., 2005; Monge et al., 2010; Ndour et al., 2008, 2009; Stemmler et al., 2006, 2007):
where Saerosol is the aerosol surface area density. It should be noted that the upper-limit values of rate constants were used in the calculation to avoid the underestimation of the [HONO]pss value. Under typical daytime conditions in the PBL with the median measured values of reactants, the upper-limit [HONO]pss value is less than 2 pptv, much lower than the median measured HONO concentration of ∼ 11 pptv. Figure 5a shows the relationship (r2=0.40) between the photolytic HONO loss rate and the sum of HONO production rates from all the NOx-related reactions calculated with upper-limit reaction rate constants. A slope of about 0.19 indicates that the contribution from these NOx-related reactions to the volume HONO source is minor in the background troposphere, despite the good correlation between HONO and NOx.
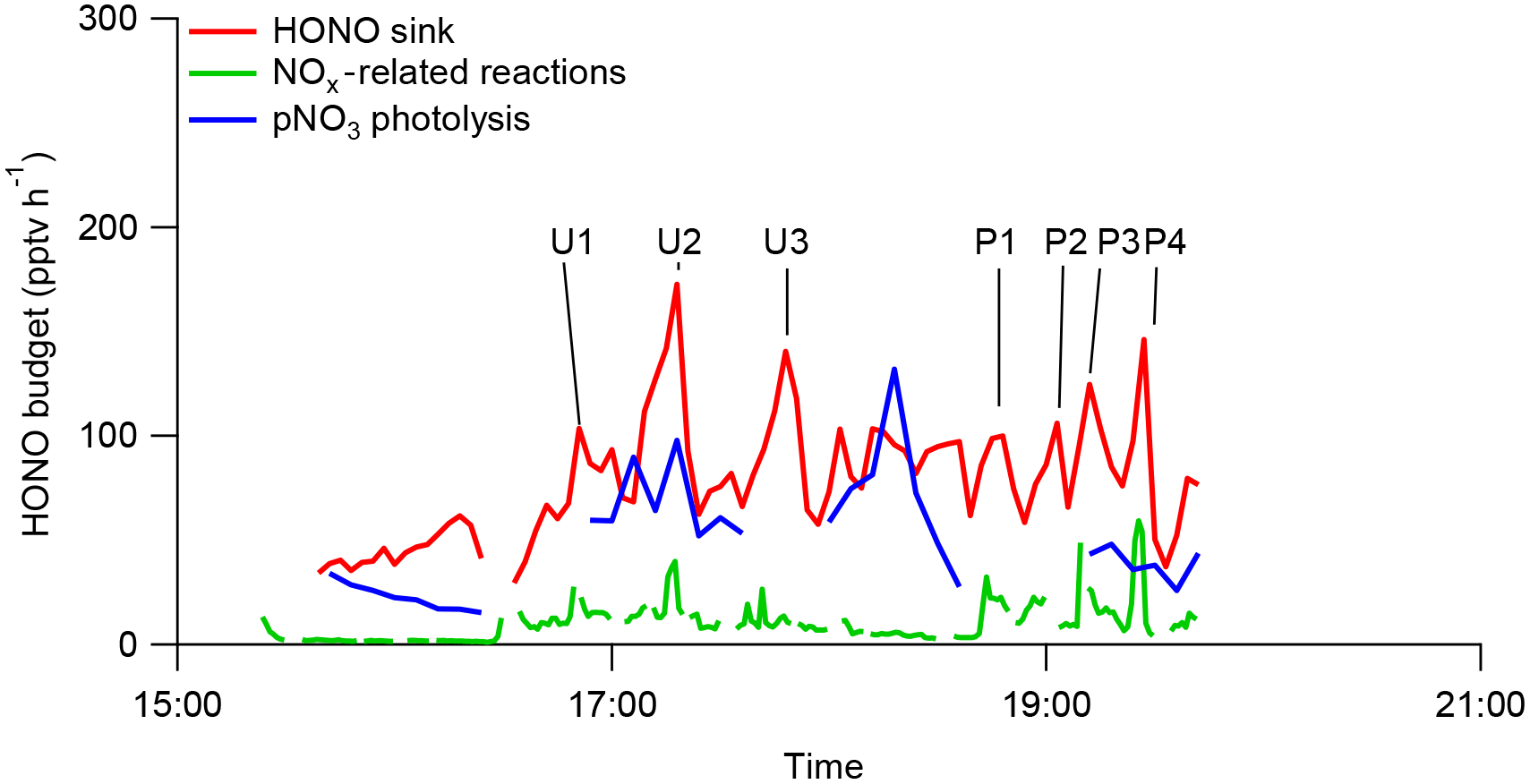
Figure 6HONO budget analysis in RF 11 in the southeastern US during the NOMADSS 2013 summer study. “HONO sink” is the HONO loss rate contributed by photolysis and the reaction of HONO with OH radicals, “NOx related reactions” is the sum of HONO production rates from all known NOx reactions and “pNO3 photolysis” is the HONO production rate from photolysis of pNO3. The calculations are based on 1 min NOx data, 3 min HONO data and 6 min pNO3 data.
Photolysis of HNO3 on surfaces has been found to proceed at a much higher rate than in the gas phase (Baergen and Donaldson, 2013; Du and Zhu, 2011; Ramazan et al., 2004; Ye et al., 2016b; Zhou et al., 2003; Zhu et al., 2008), with HONO as the major product on environmental surfaces (Ye et al., 2016a, 2017). Furthermore, photolysis of particulate nitrate has been found to be the major daytime HONO source in the marine boundary layer (Ye et al., 2016b). To examine the role of particulate nitrate as a potential HONO source in the troposphere, aerosol samples were collected and used in the light-exposure experiments to determine the photolysis rate constants for particulate nitrate in the laboratory. The determined pNO3 photolysis rate constant () varies over a wide range, from 8.3 × 10−5 s−1 to 3.1 × 10−4 s−1, with a median of 2.0 × 10−4 s−1 and a mean (±1 SD) of 1.9 (±1.2) × 10−4 s−1, when normalized to tropical noontime conditions at ground level (solar zenith angle = 0∘), and the average HONO to NO2 relative yield is 2.0 (Ye et al., 2017). Figure 5b shows the relationship between the photolytic HONO loss rate (JHONO × [HONO]) and the volume HONO production rates from pNO3 photolysis (2/3 × × [pNO3]). The median of ∼ 2.0 × 10−4 s−1 was used to calculate the ambient by scaling to :
where is the photolysis rate constant of gas-phase HNO3 calculated from light intensity measurement on the C-130 aircraft, and 7.0 × 10−7 s−1 is the photolysis rate constant of gas-phase HNO3 under the tropical noontime condition at ground level (solar zenith angle = 0∘). A slope of 0.69 can be derived from Fig. 5b, suggesting that pNO3 photolysis is the major volume HONO source. However, the r2 of 0.34 is not as strong as expected from pNO3 photolysis being the major volume HONO source. It may be in part due to the use of a single median value of ∼ 2.0 × 10−4 s−1 in the calculations of the ambient and the production rates of HONO in Fig. 5b; the actual values are highly variable, ranging from 8.3 × 10−5 s−1 to 3.1 × 10−4 s−1 (Ye et al., 2017). HONO source contribution from particulate nitrate photolysis in Fig. 5b is thus estimates of the in situ HONO production rates from pNO3 photolysis in different air masses.
HONO photolysis has been found to be an important or even a major OH primary source in the atmosphere near the ground surface (Elshorbany et al., 2010; He et al., 2006; Kleffmann et al., 2003; Villena et al., 2011; Zhou et al., 2011). However, HONO is not a significant daytime OH precursor in the background troposphere away from the ground surface. Based on the measurement results in this study, the mean (±SD) contribution of HONO photolysis to the OH source budget is 53 (±21) pptv h−1 in the PBL and 44 (±26) pptv h−1 in the FT (Table S1), respectively, less than 10 % of the OH production contributed by O3 photolysis. Since HONO is mainly produced from photolysis of particulate nitrate, it becomes an important intermediate product of a photochemical renoxification process recycling nitric acid and nitrate back to NOx. The regenerating rate of NOx of about 38 (±23) pptv h−1 via pNO3 photolysis (Table S1) is equivalent to an air column NOx source of ∼ 2.3 × 10−6 mol m−2 h−1 in the 1.5 km PBL, a considerable supplementary NOx source in the low-NOx background area.
It should be pointed out that particulate nitrate is in a dynamic equilibrium with gas-phase HNO3; the latter accounts for a larger (or even dominant) fraction of total nitrate (pNO3 + HNO3) and is photochemically inert. The overall photolysis of pNO3 + HNO3 would be much slower than indicated by In addition, oxidation of NOx via several reactions will replenish the pNO3 + HNO3 reservoir. The results reported here and in earlier papers (Reed et al., 2017; Ye et al., 2016a) suggest that there is a rapid cycling in reactive nitrogen species in the low-NOx atmosphere, sustaining the observed levels of HONO, NOx and pNO3.
3.4 HONO chemistry in plumes
One of the objectives of RF 11 was to study the chemistry of HONO in urban and coal-fired power plant plumes. The arrows and corresponding labels in Fig. 2 indicate the urban plumes (U1–U3) and power plant plumes (P1–P4). CO and benzene were used to identify influence from urban plumes, SO2 to identify influence from power plant plumes and acetonitrile to identify the influence of biomass burning plumes (Fig. S2). Plumes U1–U3 were identified as urban plumes from cities of Birmingham (U1, U3) and Montgomery (U2) in Alabama and plumes P1–P4 were identified as power plant plumes from power plants in Monroe County (P1–P3) and Putnam County (P4) in Georgia, respectively. The influence of biomass burning plumes was negligible as acetonitrile concentration was low and stable. The power plant plumes were generated from high-intensity point sources, and thus had features of narrow but high peaks of both HONO and NOx concentrations in the time series plots (Figs. 2, 6 and S2). In contrast, the urban plumes were generated from area sources and thus were shown as broad peaks of HONO and NOx in the time series plots with low levels of NOx (mostly below 500 pptv) (Figs. 2, 6 and S2). There were a few sharp but small NOx peaks within the broad urban plumes, reflecting the contributions of some point sources in the urban areas. The observed HONO ∕ NOx ratio was 0.019 (±0.004) in the power plant plumes (e.g., P4) and 0.057 (±0.0019) in urban plumes, significantly higher than the typical HONO ∕ NOx emission ratio of ∼ 0.002 in the fresh power plant plumes (Neuman et al., 2016) and ≤ 0.01 in automobile exhaust (Kurtenbach et al., 2001; Y. Q. Li et al., 2008). The elevated HONO ∕ NOx ratios observed in the plumes suggest that the observed HONO was mostly produced from precursors within the air mass during the transport. Based on the distances between measurement locations from the power plants or the centers of urban areas and wind speed, the transport times of these plumes were estimated to be ≥ 1 h, ∼ 5 times longer than HONO photolysis lifetime of 8–16 min, again suggesting that most of the observed HONO in the plumes was produced in situ within the air masses.
Figure 6 shows the time series plot of HONO budgets within the air masses sampled by the C-130 aircraft during flight RF 11, comparing its photolysis loss rate with its production rates from pNO3 photolysis and from all the NOx-related reactions combined. Photolysis of particulate nitrate appears to be the major volume HONO source in all urban plumes and in most of the power plant plumes except for plume P4 observed here. NOx was generally more important as a HONO precursor in the power plant plumes than in the urban plumes and in low-NOx background air masses, due to higher levels of NOx (up to 1.8 ppb in Figs. 2c and S2), OH radical and aerosol surface density. For example, all the NOx-related reactions combined contributed up to 52 % of the total volume HONO source required to sustain the observed HONO concentration in plume P4 (Fig. 6). In fresh and larger power plant plumes encountered during RF 7 to the Ohio River valley (X. Zhou, unpublished data), over 20 ppb NOx was detected, and the NOx-related reactions, mainly the NO+OH reaction, were found to account for almost all the required HONO source strength to sustain the observed HONO, in agreement with Neuman et al. (2016). The power plant plumes undergo rapid physical and photochemical evolution during the day, such as dilution and NOx-to-HNO3 conversion. Thus, the relative contributions from NOx-related reactions and particulate nitrate photolysis as HONO sources change rapidly as the plumes age.
3.5 Nighttime HONO chemistry
Nighttime HONO accumulation has been widely observed at the ground level (Kleffmann et al., 2003; Oswald et al., 2015; Stutz et al., 2002, 2010; VandenBoer et al., 2013, 2014, 2015), contributed by various anthropogenic and natural HONO sources on the ground. The main objective of RF 18 was to study the nighttime HONO evolution in both the nocturnal residual layer and the nocturnal FT. After sunset, the surface cooling promotes the formations of an inversion layer near the ground surface and a nocturnal residual layer above; the contribution from ground HONO sources then becomes negligible to the air masses beyond the surface inversion layer. Meanwhile, no effective HONO sinks, such as photolysis, oxidation by OH and dry deposition, exist in the nocturnal residual layer. Thus the HONO accumulation, if any, is a net contribution from dark heterogeneous NO2 reaction on aerosol surfaces (Reaction R2).
The C-130 flew in an elongated racetrack pattern along a north–south direction, about 140 km from Nashville, TN (Fig. 1), alternating between the PBL (1200 m) and the FT (2500 m), from late afternoon to midnight local time (Fig. 2). In the FT, HONO and NOx concentrations were relatively stable throughout the afternoon and the night, staying around 4 ppt and 90 pptv, respectively. The lack of nighttime HONO accumulation is expected from the low levels of HONO precursors, mostly NO2, and surface area of aerosol particles in the FT (Fig. 2).
The conditions in the PBL were far more variable and complicated. There were strong horizontal gradients of NOx, pNO3 and HONO in the PBL, with higher concentrations at the southern end and lower concentrations at the northern end of the flight track. Back-trajectory analysis using NOAA's HYSPLIT model (Stein et al., 2015) indicates that the encountered air masses in the PBL at the southern end passed over Nashville, about 140 km northeast of the sample area, with a transport time of about 6 h (Fig. S3a), while the air masses at the northern end stayed to the north of Nashville (Fig. S3b). Therefore, the anthropogenic emissions from the metropolitan area of Nashville contributed to the higher concentrations of pollutants observed at the southern end of the flight track. There were also trends in increasing concentrations of NOx, pNO3 and HONO with time after the sunset (Fig. 2). This was probably a result of less dispersion and dilution of anthropogenic pollutants, including NOx, as the PBL became more stable after sunset. Furthermore, as time progressed from late afternoon into evening and night, the air masses were less photochemically aged during the transport from the source areas due to the decreasing solar light intensity and shorter solar light exposure time.
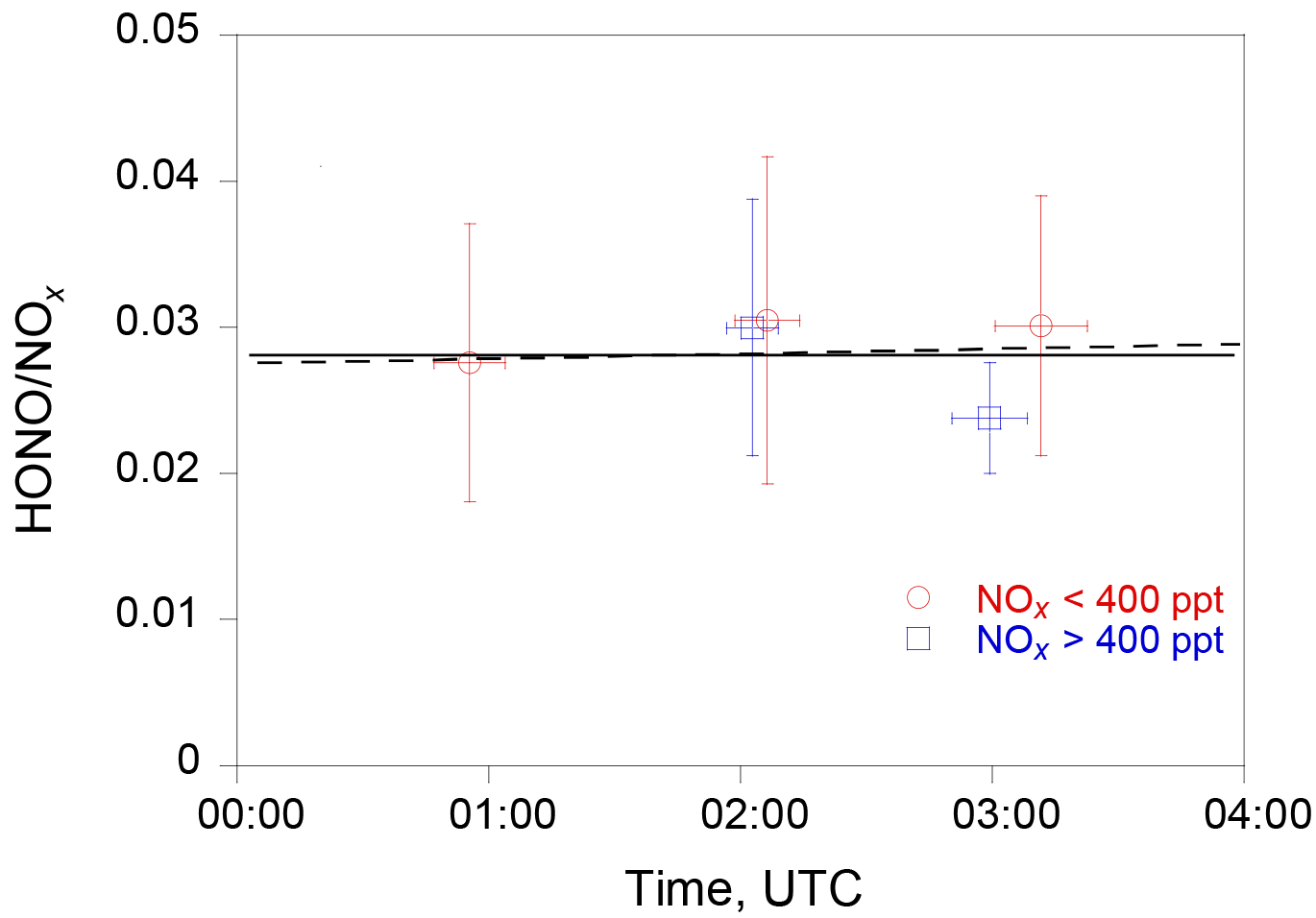
Figure 7The evolution of the HONO ∕ NOx ratio in the nocturnal boundary layer during RF 18. The red circles and blue squares are the median HONO ∕ NOx values under the conditions of NOx ≤ 400 pptv and NOx > 400 pptv, respectively. The horizontal bars indicate the averaging time periods and the vertical bars the 1 standard deviation of HONO ∕ NOx ratios. The solid line is the least-squared fit to the data, and the dashed line indicates a slope of 3 × 10−4 h−1. The sunset time at the sampling location was 00:40 UTC.
Because of the large spatial and temporal variations in the concentrations of HONO and its precursors in the PBL (Fig. 2), it is difficult to directly evaluate the nighttime HONO accumulation from HONO measurements alone. The concentration ratio of HONO and its dominant nighttime precursor, NO2, can be used as an indicator of nighttime HONO accumulation. As the air masses at a measurement altitude of 1200 m decoupled from the ground-level processes after sunset, the HONO production (P(HONO)) from heterogeneous NO2 (Reaction R2) on aerosol surface becomes the only HONO source, and can be expressed using the following Eqs. (4, 5):
where is the specific aerosol surface area density, R is the gas constant, K the absolute temperature, M the molecular weight of NO2, and γ the dark uptake coefficient of NO2 leading to HONO production. The NO2-normalized HONO accumulation over time, , can then be calculated using Eq. (6):
Assuming a dark uptake coefficient γ of 1 × 10−5 of NO2 on aerosol (George et al., 2005; Monge et al., 2010; Ndour et al., 2008; Stemmler et al., 2006, 2007) with a value of ∼ 10−4 m−1, a relative HONO accumulation rate, , of ∼ 0.0003 h−1 is estimated using the equation (Eq. 6), equivalent to a HONO accumulation of 0.13 pptv h−1 at a constant NO2 concentration of 400 pptv. Such a low HONO accumulation rate is below our measurement detection limit. Indeed, the calculated HONO-to-NOx ratio using the measurement data stayed almost unchanged with time (Fig. 7), well within the observational variability after the sunset, suggesting no significant volume production of HONO in the nocturnal boundary layer.
Substantial levels of HONO existed during the day in both the PBL (median ∼ 11 pptv) and the FT (median ∼ 4 pptv) over the southeastern US during the NOMADSS 2013 summer field study. The ground HONO sources did not significantly contribute to the HONO budget in the PBL above the minimum measurement heights of 300 m. HONO budget analysis suggests that photolysis of particulate nitrate was the major volume HONO source (∼ 69 %), while the sum of known NOx-related reactions was a minor HONO source (∼ 19 %) in the low-NOx background air masses. HONO was not a significant daytime OH precursor in the PBL away from the ground surface; however, HONO was an important intermediate product of photolysis of particulate nitrate in the renoxification process. Up to several tens of parts per trillion by volume of HONO was observed in power plant plumes and urban plumes during the day, mostly produced in situ from precursors including NOx and pNO3. No significant nighttime HONO accumulation was observed in the nocturnal residual layer and the free troposphere due to low levels of NOx and specific aerosol surface area.
The data are available in our project data archive (http://data.eol.ucar.edu/master_list/?project=SAS).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-9107-2018-supplement.
The authors declare that they have no conflict of interest.
This research is funded by National Science Foundation (NSF) grants
(AGS-1216166, AGS-1215712 and AGS-1216743). We would like to acknowledge
operational, technical and scientific support provided by NCAR, sponsored
by the National Science Foundation.
Any opinions, findings and conclusions or
recommendations expressed in this paper are those of the authors and do not
necessarily reflect the views of NSF.
Edited by: Anne Perring
Reviewed by: two anonymous referees
Acker, K., Moller, D., Wieprecht, W., Meixner, F. X., Bohn, B., Gilge, S., Plass-Dulmer, C., and Berresheim, H.: Strong daytime production of OH from HNO2 at a rural mountain site, Geophys. Res. Lett., 33, L02809, https://doi.org/10.1029/2005gl024643, 2006.
Baergen, A. M. and Donaldson, D. J.: Photochemical renoxification of nitric scid on real urban grime, Environ. Sci. Technol., 47, 815–820, https://doi.org/10.1021/es3037862, 2013.
Burling, I. R., Yokelson, R. J., Griffith, D. W. T., Johnson, T. J., Veres, P., Roberts, J. M., Warneke, C., Urbanski, S. P., Reardon, J., Weise, D. R., Hao, W. M., and de Gouw, J.: Laboratory measurements of trace gas emissions from biomass burning of fuel types from the southeastern and southwestern United States, Atmos. Chem. Phys., 10, 11115–11130, https://doi.org/10.5194/acp-10-11115-2010, 2010.
Carr, S., Heard, D. E., and Blitz, M. A.: Comment on “Atmospheric hydroxyl radical production from electronically excited NO2 and H2O”, Science, 324, 336b, 2009.
de Gouw, J. and Warneke, C.: Measurements of volatile organic compounds in the earths atmosphere using proton-transfer-reaction mass spectrometry, Mass Spectrom. Rev., 26, 223–257, 2007.
Du, J. and Zhu, L.: Quantification of the absorption cross sections of surface-adsorbed nitric acid in the 335–365 nm region by Brewster angle cavity ring-down spectroscopy, Chem. Phys. Lett., 511, 213–218, https://doi.org/10.1016/j.cplett.2011.06.062, 2011.
Elshorbany, Y. F., Kleffmann, J., Kurtenbach, R., Lissi, E., Rubio, M., Villena, G., Gramsch, E., Rickard, A. R., Pilling, M. J., and Wiesen, P.: Seasonal dependence of the oxidation capacity of the city of Santiago de Chile, Atmos. Environ., 44, 5383–5394, https://doi.org/10.1016/j.atmosenv.2009.08.036, 2010.
Finlayson-Pitts, B. J. and Pitts Jr., J. N.: Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications, Academic Press, San Diego, California, 2000.
Flagan, R. C.: Electrical mobility methods for sub-micrometer particle characterization, in: Aerosol Measurement: Principles, Techniques, and Applications, Third Edition, edited by: Kulkarni, P., Baron, P. A., and Willeke, K., 339–364, John Wiley & Sons, New York, 2002.
George, C., Strekowski, R. S., Kleffmann, J., Stemmler, K., and Ammann, M.: Photoenhanced uptake of gaseous NO2 on solid-organic compounds: a photochemical source of HONO?, Faraday Discuss., 130, 195–210, 2005.
Gierczak, T., Jimenez, E., Riffault, V., Burkholder, J. B., and Ravishankara, A. R.: Thermal decomposition of HO2NO2 (peroxynitric acid, PNA): Rate coefficient and determination of the enthalpy of formation, J. Phys. Chem. A, 109, 586–596, 2005.
He, Y., Zhou, X. L., Hou, J., Gao, H. L., and Bertman, S. B.: Importance of dew in controlling the air-surface exchange of HONO in rural forested environments, Geophys. Res. Lett., 33, L02813, https://doi.org/10.1029/2005GL024348, 2006.
Hornbrook, R. S., Blake, D. R., Diskin, G. S., Fried, A., Fuelberg, H. E., Meinardi, S., Mikoviny, T., Richter, D., Sachse, G. W., Vay, S. A., Walega, J., Weibring, P., Weinheimer, A. J., Wiedinmyer, C., Wisthaler, A., Hills, A., Riemer, D. D., and Apel, E. C.: Observations of nonmethane organic compounds during ARCTAS – Part 1: Biomass burning emissions and plume enhancements, Atmos. Chem. Phys., 11, 11103–11130, https://doi.org/10.5194/acp-11-11103-2011, 2011a.
Hornbrook, R. S., Crawford, J. H., Edwards, G. D., Goyea, O., Mauldin III, R. L., Olson, J. S., and Cantrell, C. A.: Measurements of tropospheric HO2 and RO2 by oxygen dilution modulation and chemical ionization mass spectrometry, Atmos. Meas. Tech., 4, 735–756, https://doi.org/10.5194/amt-4-735-2011, 2011b.
Huang, G., Zhou, X., Deng, G., Qiao, H., and Civerolo, K.: Measurements of atmospheric nitrous acid and nitric acid, Atmos. Environ., 36, 2225–2235, 2002.
Karl, T., Jobson, T., Kuster, W. C., Williams, E., Stutz, J., Shetter, R., Hall, S. R., Goldan, P., Fehsenfeld, F., and Lindinger, W.: The use of Proton-Transfer-Reaction Mass Spectrometry to Characterize VOC Sources at the La Porte Super Site during the Texas Air Quality Study 2000, J. Geophys. Res., 108, 4508, https://doi.org/10.1029/2002JD003333, 2003.
Kaser, L., Karl, T., Yuan, B., Mauldin, R. L. III, Cantrell, C. A., Guenther, A. B., Patton, E. G., Weinheimer, A. J., Knote, C., Orlando, J., Emmons, L., Apel, E., Hornbrook, Shertz, R., S., Ullmann, K., Hall, S., Graus, M., de Gouw, J., Zhou, X., and Ye, C.: chemistry-turbulence interactions and mesoscale variability influence the cleansing efficiency of the atmosphere, Geophys. Res. Lett., 42, 10894–10903, https://doi.org/10.1002/2015GL066641, 2015.
Kleffmann, J.: Daytime sources of nitrous acid (HONO) in the atmospheric boundary layer, Chem. Phys. Chem., 8, 1137–1144, https://doi.org/10.1002/cphc.200700016, 2007.
Kleffmann, J., Kurtenbach, R., Lorzer, J., Wiesen, P., Kalthoff, N., Vogel, B., and Vogel, H.: Measured and simulated vertical profiles of nitrous acid – Part I: Field measurements, Atmos. Environ., 37, 2949–2955, 2003.
Kurtenbach, R., Becker, K. H., Gomes, J. A. G., Kleffmann, J., Lorzer, J. C., Spittler, M., Wiesen, P., Ackermann, R., Geyer, A., and Platt, U.: Investigations of emissions and heterogeneous formation of HONO in a road traffic tunnel, Atmos. Environ., 35, 3385–3394, https://doi.org/10.1016/S1352-2310(01)00138-8, 2001.
Li, S. P., Matthews, J., and Sinha, A.: Atmospheric hydroxyl radical production from electronically excited NO2 and H2O, Science, 319, 1657–1660, 2008.
Li, Y. Q., Schwab, J. J., and Demerjian, K. L.: Fast time response measurements of gaseous nitrous acid using a tunable diode laser absorption spectrometer: HONO emission source from vehicle exhausts, Geophys. Res. Lett., 35, L04803, https://doi.org/10.1029/2007GL031218, 2008.
Li, X., Rohrer, F., Hofzumahaus, A., Brauers, T., Haseler, R., Bohn, B., Broch, S., Fuchs, H., Gomm, S., Holland, F., Jager, J., Kaiser, J., Keutsch, F. N., Lohse, I., Lu, K. D., Tillmann, R., Wegener, R., Wolfe, G. M., Mentel, T. F., Kiendler-Scharr, A., and Wahner, A.: Missing gas-phase source of HONO inferred from Zeppelin measurements in the troposphere, Science, 344, 292–296, 2014.
Maljanen, M., Yli-Pirila, P., Hytonen, J., Joutsensaari, J., and Martikainen, P. J.: Acidic northern soils as sources of atmospheric nitrous acid (HONO), Soil. Biol. Biochem., 67, 94–97, https://doi.org/10.1016/j.soilbio.2013.08.013, 2013.
Mauldin, R., Kosciuch, E., Eisele, F., Huey, G., Tanner, D., Sjostedt, S., Blake, D., Chen, G., Crawford, J., and Davis, D.: South Pole Antarctica observations and modeling results: New insights on HOx radical and sulfur chemistry, Atmos. Environ., 44, 572–581, 2010.
Monge, M. E., D'Anna, B., Mazri, L., Giroir-Fendler, A., Ammann, M., Donaldson, D. J., and George, C.: Light changes the atmospheric reactivity of soot, P. Natl. Acad. Sci. USA, 107, 6605–6609, 2010.
Ndour, M., D'Anna, B., George, C., Ka, O., Balkanski, Y., Kleffmann, J., Stemmler, K., and Ammann, M.: Photoenhanced uptake of NO2 on mineral dust: Laboratory experiments and model simulations, Geophys. Res. Lett., 35, L05812, https://doi.org/10.1029/2007GL032006, 2008.
Ndour, M., Nicolas, M., D'Anna, B., Ka, O., and George, C.: Photoreactivity of NO2 on mineral dusts originating from different locations of the Sahara desert, Phys. Chem. Chem. Phys., 11, 1312–1319, 2009.
Neuman, J. A., Trainer, M., Brown, S. S., Min, K.-E., Nowak, J. B., Parrish, D. D., Peischl, J., Pollack, I. B., Roberts, J. M., Ryerson, T. B., and Veres, P. R.: HONO emission and production determined from airborne measurements over the Southeast U.S., J. Geophys. Res.-Atmos., 121, 9237–9250, 2016.
Oswald, R., Behrendt, T., Ermel, M., Wu, D., Su, H., Cheng, Y., Breuninger, C., Moravek, A., Mougin, E., Delon, C., Loubet, B., Pommerening-Roser, A., Sorgel, M., Poschl, U., Hoffmann, T., Andreae, M. O., Meixner, F. X., and Trebs, I.: HONO emissions from soil bacteria as a major source of atmospheric reactive nitrogen, Science, 341, 1233–1235, https://doi.org/10.1126/science.1242266, 2013.
Oswald, R., Ermel, M., Hens, K., Novelli, A., Ouwersloot, H. G., Paasonen, P., Petäjä, T., Sipilä, M., Keronen, P., Bäck, J., Königstedt, R., Hosaynali Beygi, Z., Fischer, H., Bohn, B., Kubistin, D., Harder, H., Martinez, M., Williams, J., Hoffmann, T., Trebs, I., and Sörgel, M.: A comparison of HONO budgets for two measurement heights at a field station within the boreal forest in Finland, Atmos. Chem. Phys., 15, 799–813, https://doi.org/10.5194/acp-15-799-2015, 2015.
Platt, U. and Stutz, J: Differential Optical Absorption Spectroscopy: Principles and Applications, Springer, Berlin, 2008.
Ramazan, K. A., Syomin, D., and Finlayson-Pitts, B. J.: The photochemical production of HONO during the heterogeneous hydrolysis of NO2, Phys. Chem. Chem. Phys., 6, 3836–3843, https://doi.org/10.1039/b402195a, 2004.
Ramazan, K. A., Wingen, L. M., Miller, Y., Chaban, G. M., Gerber, R. B., Xantheas, S. S., and Finlayson-Pitts, B. J.: New experimental and theoretical approach to the heterogeneous hydrolysis of NO2: Key role of molecular nitric acid and its complexes, J. Phys. Chem. A, 110, 6886–6897, https://doi.org/10.1021/jp056426n, 2006.
Reed, C., Evans, M. J., Crilley, L. R., Bloss, W. J., Sherwen, T., Read, K. A., Lee, J. D., and Carpenter, L. J.: Evidence for renoxification in the tropical marine boundary layer, Atmos. Chem. Phys., 17, 4081–4092, https://doi.org/10.5194/acp-17-4081-2017, 2017.
Ridley, B., Ott, L., Pickering, K., Emmons, L., Montzka, D., Weinheimer, A., Knapp, D., Grahek, F., Li, L., Heymsfield, G., McGill, M., Kucera, P., Mahoney, M. J., Baumgardner, D., Schultz, M., and Brasseur, G.: Florida thunderstorms: A faucet of reactive nitrogen to the upper troposphere, J. Geophys. Res.-Atmos., 109, D17305, https://doi.org/10.1029/2004JD004769, 2004.
SAS Southeast Atmosphere Study: SAS Data Sets, available at: http://data.eol.ucar.edu/master_list/?project=SAS, 2018.
Shetter, R. E., Cinquini, L., Lefer, B. L., Hall, S. R., and Madronich, S.: Comparison of airborne measured and calculated spectral actinic flux and derived photolysis frequencies during the PEM Tropics B mission, J. Geophys. Res.-Atmos., 108, 8234, 2002.
Stein, A. F., Draxler, R. R., Rolph, G. D., Stunder, B. J. B., Cohen, M. D., and Ngan, F.: NOAA's HYSPLIT atmospheric transport and dispersion modeling system, B. Am. Meteorol. Soc., 96, 2059–2077, https://doi.org/10.1175/BAMS-D-14-00110.1, 2015.
Stemmler, K., Ammann, M., Donders, C., Kleffmann, J., and George, C.: Photosensitized reduction of nitrogen dioxide on humic acid as a source of nitrous acid, Nature, 440, 195–198, https://doi.org/10.1038/nature04603, 2006.
Stemmler, K., Ndour, M., Elshorbany, Y., Kleffmann, J., D'Anna, B., George, C., Bohn, B., and Ammann, M.: Light induced conversion of nitrogen dioxide into nitrous acid on submicron humic acid aerosol, Atmos. Chem. Phys., 7, 4237–4248, https://doi.org/10.5194/acp-7-4237-2007, 2007.
Stutz, J., Alicke, B., and Neftel, A.: Nitrous acid formation in the urban atmosphere: Gradient measurements of NO2 and HONO over grass in Milan, Italy, J. Geophys. Res.-Atmos., 107, 8192, https://doi.org/10.1029/2001jd000390, 2002.
Stutz, J., Oh, H. J., Whitlow, S. I., Anderson, C., Dibbb, J. E., Flynn, J. H., Rappengluck, B., and Lefer, B.: Simultaneous DOAS and mist-chamber IC measurements of HONO in Houston, TX, Atmos. Environ., 44, 4090–4098, 2010.
Su, H., Cheng, Y. F., Oswald, R., Behrendt, T., Trebs, I., Meixner, F. X., Andreae, M. O., Cheng, P., Zhang, Y., and Poschl, U.: Soil Nitrite as a Source of Atmospheric HONO and OH Radicals, Science, 333, 1616–1618, https://doi.org/10.1126/science.1207687, 2011.
Trentmann, J., Andreae, M. O., and Graf, H. F.: Chemical processes in a young biomass-burning plume, J. Geophys. Res.-Atmos., 108, 4705, 2003.
VandenBoer, T. C., Brown, S. S., Murphy, J. G., Keene, W. C., Young, C. J., Pszenny, A. A. P., Kim, S., Warneke, C., de Gouw, J. A., Maben, J. R., Wagner, N. L., Riedel, T. P., Thornton, J. A., Wolfe, D. E., Dube, W. P., Ozturk, F., Brock, C. A., Grossberg, N., Lefer, B., Lerner, B., Middlebrook, A. M., and Roberts, J. M.: Understanding the role of the ground surface in HONO vertical structure: High resolution vertical profiles during NACHTT-11, J. Geophys. Res.-Atmos., 118, 10155–10171, https://doi.org/10.1002/jgrd.50721, 2013.
VandenBoer, T. C., Markovic, M. Z., Sanders, J. E., Ren, X., Pusede, S. E., Browne, E. C., Cohen, R. C., Zhang, L., Thomas, J., Brune, W. H., and Murphy, J. G.: Evidence for a nitrous acid (HONO) reservoir at the ground surface in Bakersfield, CA, during CalNex 2010, J. Geophys. Res.-Atmos., 119, 9093–9106, https://doi.org/10.1002/2013JD020971, 2014.
VandenBoer, T. C., Young, C. J., Talukdar, R. K., Markovic, M. Z., Brown, S. S., Roberts, J. M., and Murphy, J. G.: Nocturnal loss and daytime source of nitrous acid through reactive uptake and displacement, Nat. Geosci., 8, 55–60, 2015.
Villena, G., Kleffmann, J., Kurtenbach, R., Wiesen, P., Lissi, E., Rubio, M. A., Croxatto, G., and Rappengluck, B.: Vertical gradients of HONO, NOx and O3 in Santiago de Chile, Atmos. Environ., 45, 3867–3873, 2011.
Wong, K. W., Oh, H.-J., Lefer, B. L., Rappenglück, B., and Stutz, J.: Vertical profiles of nitrous acid in the nocturnal urban atmosphere of Houston, TX, Atmos. Chem. Phys., 11, 3595–3609, https://doi.org/10.5194/acp-11-3595-2011, 2011.
Wong, K. W., Tsai, C., Lefer, B., Haman, C., Grossberg, N., Brune, W. H., Ren, X., Luke, W., and Stutz, J.: Daytime HONO vertical gradients during SHARP 2009 in Houston, TX, Atmos. Chem. Phys., 12, 635–652, https://doi.org/10.5194/acp-12-635-2012, 2012.
Wong, K. W., Tsai, C., Lefer, B., Grossberg, N., and Stutz, J.: Modeling of daytime HONO vertical gradients during SHARP 2009, Atmos. Chem. Phys., 13, 3587–3601, https://doi.org/10.5194/acp-13-3587-2013, 2013.
Wu, C. and Yu, J. Z.: Evaluation of linear regression techniques for atmospheric applications: the importance of appropriate weighting, Atmos. Meas. Tech., 11, 1233–1250, https://doi.org/10.5194/amt-11-1233-2018, 2018.
Ye, C. X., Zhou, X. L., Pu, D., Stutz, J., Festa, J., Spolaor, M., Cantrell, C., Mauldin, R. L., Weinheimer, A., and Haggerty, J.: Comment on “Missing gas-phase source of HONO inferred from Zeppelin measurements in the troposphere”, Science, 348, 1326, https://doi.org/10.1126/science.aaa1992, 2015.
Ye, C. X., Gao, H. L., Zhang, N., and Zhou, X.: Photolysis of nitric Acid and nitrate on natural and artificial surfaces, Environ. Sci. Technol., 50, 3530–3536, 2016a.
Ye, C. X., Zhou, X. L., Pu, D., Stutz, J., Festa, J., Spolaor, M., Tsai, C., Cantrell, C., Mauldin, R. L., Campos, T., Weinheimer, A., Hornbrook, R. S., Apel, E. C., Guenther, A., Kaser, L., Yuan, B., Karl, T., Haggerty, J., Hall, S., Ullmann, K., Smith, J. N., Ortega, J., and Knote, C.: Rapid cycling of reactive nitrogen in the marine boundary layer, Nature, 532, 489–491, 2016b.
Ye, C., Zhang, N., Gao, H., and Zhou, X.: Photolysis of particulate nitrate as a source of HONO and NOx, Environ. Sci. Technol., 51, 6849–6856, https://doi.org/10.1021/acs.est.7b00387, 2017.
Young, C. J., Washenfelder, R. A., Roberts, J. M., Mielke, L. H., Osthoff, H. D., Tsai, C., Pikelnaya, O., Stutz, J., Veres, P. R., Cochran, A. K., VandenBoer, T. C., Flynn, J., Grossberg, N., Haman, C. L., Lefer, B., Stark, B., Martin, G., Gouw, Joost de., Gilman, J. B., Kuster, W. C., and Brown, S. S.: Vertically Resolved Measurements of Nighttime Radical Reservoirs in Los Angeles and Their Contribution to the Urban Radical Budget, Environ. Sci. Technol., 46, 10965–10973, 2012.
Zhang, N., Zhou, X., Shepson, P. B., Gao, H., Alaghmand, M., and Stirm, B.: Aircraft measurement of HONO vertical profiles over a forested region, Geophys. Res. Lett., 36, L15820, https://doi.org/10.1029/2009gl038999, 2009.
Zhang, N., Zhou, X., Bertman, S., Tang, D., Alaghmand, M., Shepson, P. B., and Carroll, M. A.: Measurements of ambient HONO concentrations and vertical HONO flux above a northern Michigan forest canopy, Atmos. Chem. Phys., 12, 8285–8296, https://doi.org/10.5194/acp-12-8285-2012, 2012.
Zhou, X., Civerolo, K., Dai, H., Huang, G., Schwab, J., and Demerjian, K.: Summertime nitrous acid chemistry in the atmospheric boundary layer at a rural site in New York State, J. Geophys. Res., 107, 4590, https://doi.org/10.1029/2001JD001539, 2002.
Zhou, X., Gao, H., He, Y., Huang, G., Bertman, S. B., Civerolo, K., and Schwab, J.: Nitric acid photolysis on surfaces in low-NOx environments: Significant atmospheric implications, Geophys. Res. Lett., 30, 2217, https://doi.org/10.1029/2003gl018620, 2003.
Zhou, X., Zhang, N., TerAvest, M., Tang, D., Hou, J., Bertman, S., Alaghmand, M., Shepson, P. B., Carroll, M. A., Griffith, S., Dusanter, S., and Stevens, P. S.: Nitric acid photolysis on forest canopy surface as a source for tropospheric nitrous acid, Nat. Geosci., 4, 440–443, https://doi.org/10.1038/NGEO1164, 2011.
Zhu, C. Z., Xiang, B., Zhu, L., and Cole, R.: Determination of absorption cross sections of surface-adsorbed HNO(3) in the 290–330 nm region by Brewster angle cavity ring-down spectroscopy, Chem. Phys. Lett., 458, 373–377, 2008.