the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 03 May 2018
| 03 May 2018
Secondary organic aerosol production from pinanediol, a semi-volatile surrogate for first-generation oxidation products of monoterpenes
Penglin Ye
Yunliang Zhao
Wayne K. Chuang
Allen L. Robinson
We have investigated the production of secondary organic aerosol (SOA) from pinanediol (PD), a precursor chosen as a semi-volatile surrogate for first-generation oxidation products of monoterpenes. Observations at the CLOUD facility at CERN have shown that oxidation of organic compounds such as PD can be an important contributor to new-particle formation. Here we focus on SOA mass yields and chemical composition from PD photo-oxidation in the CMU smog chamber. To determine the SOA mass yields from this semi-volatile precursor, we had to address partitioning of both the PD and its oxidation products to the chamber walls. After correcting for these losses, we found OA loading dependent SOA mass yields from PD oxidation that ranged between 0.1 and 0.9 for SOA concentrations between 0.02 and 20 µg m−3, these mass yields are 2–3 times larger than typical of much more volatile monoterpenes. The average carbon oxidation state measured with an aerosol mass spectrometer was around −0.7. We modeled the chamber data using a dynamical two-dimensional volatility basis set and found that a significant fraction of the SOA comprises low-volatility organic compounds that could drive new-particle formation and growth, which is consistent with the CLOUD observations.
- Article
(4441 KB) - Full-text XML
-
Supplement
(478 KB) - BibTeX
- EndNote





Particulate matter (PM) in the atmosphere affects human health and life expectancy (Pope et al., 2009) and also influences Earth's climate by absorbing and scattering radiation (Solomon et al., 2007). Organic compounds constitute a large fraction of that PM, making up around 20–90 % of the aerosol mass in the lower troposphere (Kanakidou et al., 2005). Secondary organic aerosol (SOA), formed from oxidation of gas-phase organic compounds in the atmosphere, accounts for a significant fraction of the organic aerosol (OA) in PM (Zhang et al., 2007). In the atmosphere, OA is dynamic due to constant photo-oxidation and associated evolution in thermodynamic properties (Seinfeld and Pandis, 2006; Donahue et al., 2005). However, classical smog-chamber experiments encompass only the early stages of SOA formation, including one generation or at most a few generations of oxidation chemistry (Pandis et al., 1991; Odum et al., 1996a). While those experiments may include some later-generation chemistry, the commonly used two-product model (Odum et al., 1996a) treats the (quasi-)first-generation products as effectively non-reactive.
Further oxidation (aging) of SOA may add more functional groups to the carbon backbone, causing the second-generation oxidation products to be even less volatile and more water soluble than the first-generation products, which will also enhance the SOA mass (Donahue et al., 2005). However, ongoing oxidation must eventually fragment products and drive down the SOA mass because the end state of organic oxidation is CO2 formation (Kroll et al., 2009; Chacon-Madrid et al., 2012; Donahue et al., 2013). There is considerable evidence that the ongoing oxidation chemistry can increase SOA mass and oxidation state, both from smog-chamber experiments (Donahue et al., 2012a; Henry and Donahue, 2012; Qi et al., 2012) and also from flow tubes that simulate many days of oxidation using intense UV radiation to drive photochemistry (Lambe et al., 2011; Wong et al., 2011; Cubison et al., 2011). The flow-tube results also confirm that oxidation will eventually cause mass loss via fragmentation (Tkacik et al., 2014). The volatility basis set (VBS) was developed to treat this ongoing chemistry by condensing the enormous ensemble of organic compounds involved onto a basis grid described by volatility and the carbon oxidation state (Donahue et al., 2006, 2011a, 2012b; Chuang and Donahue, 2016; Tröstl et al., 2016), with coupling constants constrained by chemical behavior of representative or average compounds (Chacon-Madrid et al., 2012; Donahue et al., 2013).
Bulk SOA aging experiments show that later-generation chemistry will influence SOA properties, but those experiments provide limited mechanistic insight due to the extreme complexity of the chemistry involving multiple generations of multiple products. A complementary approach is to use selected first-generation products from SOA formation to probe second-generation chemistry systematically, and to proceed through representative later-generation products. For example, the known products of α-pinene oxidation include pinonaldehyde, which is one of the most volatile products, and acids such as cis-pinonic acid and pinic acid which are some of the least volatile monomer products (Jang and Kamens, 1999; Jaoui and Kamens, 2001). Smog-chamber experiments at Carnegie Mellon have shown that pinonaldehyde is a modest but significant source of SOA at both high NO (Chacon-Madrid and Donahue, 2011) and low NO (Chacon-Madrid et al., 2013) conditions. Aldehyde chemistry is dominated by OH radical attack on the terminal –CHO moiety, causing fragmentation (Chacon-Madrid et al., 2010), but OH attack along the carbon backbone leads to functionalized products that condense to enhance SOA formation from the first-generation parent α-pinene, with mass yields of roughly 10 % under atmospherically relevant conditions. If the most volatile α-pinene product can enhance SOA production, it stands to reason that less volatile organic compound products would have an even greater effect. Indeed, we have observed very low-volatility products from cis-pinonic acid oxidation, such as MBTCA (Müller et al., 2012), but we have not systematically explored the SOA mass yields from first-generation SVOC products. Here we use pinanediol (PD) as a surrogate for semi-volatile first-generation oxidation products of monoterpenes to study this aging chemistry. PD has a volatility similar to cis-pinonic acid (C* ∼ 300 µg m−3) but it is commercially available and easier to handle.
One reason that SOA mass yields from SVOCs are not commonly reported is that SVOCs are hard to handle and measure, and mass-yield determinations require accurate values for the amount of oxidized precursor because the mass yield by definition is the ratio of formed SOA to oxidized precursor mass. There are two reasons why this is challenging for SVOCs. First, they are sticky and hard to measure. Second, and more challenging, SVOCs may be lost to Teflon chamber walls (Matsunaga and Ziemann, 2010) and may even return from the chamber walls as oxidation perturbs a putative gas–Teflon equilibrium. This means any measured change in the SVOC concentration, even if an instrument is well characterized, may not reflect the actual amount of oxidized SVOC.
Sorption of SVOCs into Teflon chamber walls has recently become a matter of significant concern. Matsunaga and Ziemann (2010) showed that various organic compounds broadly in the intermediate volatility range (IVOCs; Donahue et al., 2011a) appear to sorb reversibly to Teflon chamber walls, and more recent work has confirmed this finding. The fraction of organic vapors left in gas phase appears to depend on the volatility and the molecular structure of the organics, but Matsunaga and Ziemann suggested that IVOCs partition into a disrupted surface layer of the Teflon as if the Teflon had an equivalent mass of between 2 and 10 mg m−3, depending on molecular structure (for a several-cubic-meter chamber). As an example, an 8 m3 chamber has a surface area of 24 m2, and if the disrupted Teflon surface layer postulated by Matsunaga and Ziemann were 1 µm thick, it would have a volume of m3 and thus a mass of roughly 10 g considering the density of the Teflon is 0.8 g cm−3; projected to the chamber volume this gives an equivalent mass concentration of roughly 1 g m−3. To have an effective “partitioning mass” of 1–10 mg m−3 this material would thus need to have a mass-based activity coefficient of 100–1000 (Trump et al., 2016). This is consistent with weak interactions involving non-polarizable Teflon and also a low degree of interactions among sorbed organics within the walls at the Henry's law, low-concentration limit. However, we must stress that the exact mechanism of organic sorption to Teflon chamber walls remains unclear.
More recently, Ye et al. (2016a) and Krechmer et al. (2016) showed that SVOCs are lost to the Teflon walls steadily, with a time constant of roughly 15 min (again for a several-cubic-meter chamber). The SVOCs in these studies had µg m−3 and so would be expected to leave only a small fraction (≪ 10 %) in the gas phase; this quasi-irreversible loss is thus broadly consistent with the reversible equilibration reported earlier for IVOCs.
We expect PD to partition substantially to the walls of a Teflon chamber. Even 2-decanol showed significant vapor loss (Matsunaga and Ziemann, 2010), and the additional OH group in PD decreases the vapor saturation concentration of PD by around 2.3 decades (Donahue et al., 2011a). This should cause larger mass loss to the chamber walls. In order to get an accurate SOA mass yield from oxidation of PD, we need to determine how much PD exists in the gas phase vs the chamber walls, and ultimately how much PD reacts during SOA formation experiments.
Another reason we are interested in SOA formation from PD is that it has already been used as a surrogate for the first-generation terpene oxidation products to explore the role of gas-phase aging in new-particle formation, and we wish to compare SOA formation with new-particle formation. The Cosmics Leaving OUtdoor Droplets (CLOUD) facility at CERN is designed to study the effects of cosmic rays on new-particle formation (nucleation and growth) (Kirkby et al., 2011; Duplissy et al., 2016). Early experiments focused on sulfuric acid vapor and different stabilizing species that include the ammonia, amines, and oxidation products of organic precursors (Kirkby et al., 2011; Schobesberger et al., 2013; Riccobono et al., 2014). PD was used to mimic first-generation oxidation products of monoterpene formed in the atmosphere (Schobesberger et al., 2013). Specifically the experiments addressed the hypothesis that oxidation of these first-generation products by OH radicals could produce later-generation products with sufficient supersaturation to participate in nucleation (Donahue et al., 2011c). The PD oxidation experiments were among the first to observe highly oxidized, extremely low-volatility organic compounds (ELVOCs) (Donahue et al., 2011a), with the original 10 carbon atoms decorated by up to 12 oxygen atoms (Schobesberger et al., 2013; Riccobono et al., 2014). The composition of these highly oxidized organic molecules (HOMs) and possible mechanisms for their formation remains an active research topic (Ehn et al., 2014).
In this study, we focus on SOA formation following oxidation of PD by OH radicals. Our first objective is to extend our understanding of SOA aging via experiments addressing carefully selected first-generation products from common SOA precursors. Our second objective is to compare the properties of bulk SOA produced at relatively high concentrations (0.3–30 µg m−3) with the PD oxidation products observed condensing onto particles during the CLOUD nucleation experiment. Our third objective is to use PD as a model compound to explore the complications of precursor losses to Teflon walls in smog-chamber SOA formation experiments. We explore the wall sorption of PD by comparing the total amount of PD injected into the chamber to the PD concentration observed in the gas phase. We also investigate the release of sorbed PD from the chamber walls by heating or diluting the chamber. We then calculate the SOA mass yields, accounting for the loss of PD and also the loss of oxidation products to the Teflon chamber walls. Finally, we describe the elemental composition of the formed SOA. We analyze the SOA volatility distribution and oxidation state within the two-dimensional volatility–oxidation set (2D-VBS) and compare the properties of bulk SOA to the ELVOCs observed in CLOUD.
We conducted experiments in the Carnegie Mellon University (CMU) smog chamber, a 10 m3 Teflon bag suspended in a temperature-controlled room. The chamber and our methodology have been described extensively in the literature (Hildebrandt et al., 2009). Before each experiment, we cleaned the bag by flushing it with clean, dry air and exposing it to UV irradiation at ∼ 35 ∘C. We subsequently maintained the chamber at a constant temperature unless otherwise noted.
For the experiments in this paper, we introduced organic compounds into the chamber via a flash vaporizer (Robinson et al., 2013). We used a small, resistive metal heater enclosed in a stainless-steel sheath to evaporate the organics inside the chamber, placing the organics into an indentation on the stainless-steel surface before inserting the heater into the chamber on the end of a long stainless-steel tube. With a flow of clean, dry dispersion air flowing through the tube for mixing, we power-cycled the heater until the organics completely evaporated. For various experiments, we used n-tridecane, 1-tridecene, 2-nonanone, 2-nonanol, oxy-pinocamphone, and pinanediol (Sigma-Aldrich, 99 %). For SOA formation experiments we used ammonium sulfate seed particles ((NH4)2SO4, Sigma Aldrich, 99.99 %), which we formed by atomizing a 1 g L−1 (NH4)2SO4 solution in ultrapure deionized water to produce droplets that passed through a diffusion dryer and a Po-210 neutralizer before they entered the chamber. These seed particles served as a condensation sink for condensable vapors in order to reduce vapor wall losses. To form OH radicals during oxidation experiments we added nitrous acid (HONO) to the chamber by bubbling filtered air through a HONO solution for 20 min.
We measured gas-phase organic species using both a proton-transfer-reaction mass spectrometer (PTRMS, Ionicon Analytik) and a gas chromatograph–mass spectrometer (GC/MS) (Agilent, 6890 GC/5975 MS) equipped with a thermal desorption and injection system (TDGC/MS, Gerstel, MA) and a capillary column (Agilent HP-5MS, 30 m × 0.25 mm) (Zhao et al., 2014). We maintained the temperature of the PTRMS inlet line at 60 ∘C to minimize line losses. For the thermal desorption GC measurements, we collected samples by drawing chamber air through Tenax® TA-filled glass tubes (Gerstel 6 mm OD, 4.5 mm ID glass tube filled with ∼ 290 mg of Tenax TA) at a flow rate of 0.5 L min−1 for 2 min. We tracked the recovery of organics during analysis using C12, C16, C20, C24, C30, C32, and C36 deuterated n-alkanes as standards that we spiked into each Tenax tube prior to the thermal desorption.
We measured particle number and volume concentrations inside the chamber using a scanning mobility particle sizer (SMPS, TSI classifier model 3080, CPC model 3772 or 3010). We measured size-resolved and bulk particle composition and mass concentrations with a high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS, Aerodyne Research, Inc.). We operated the HR-ToF-AMS following the common protocol with the vaporizer temperature at 600 ∘C and electron ionization at 70 eV. We collected mass spectra and particle time-of-flight (pToF) measurements in V mode, which provides high mass resolution (2000 m ∕ Δm) and excellent transmission efficiency. We analyzed the AMS data using the SQUIRREL V1.53G and PIKA 1.12G.
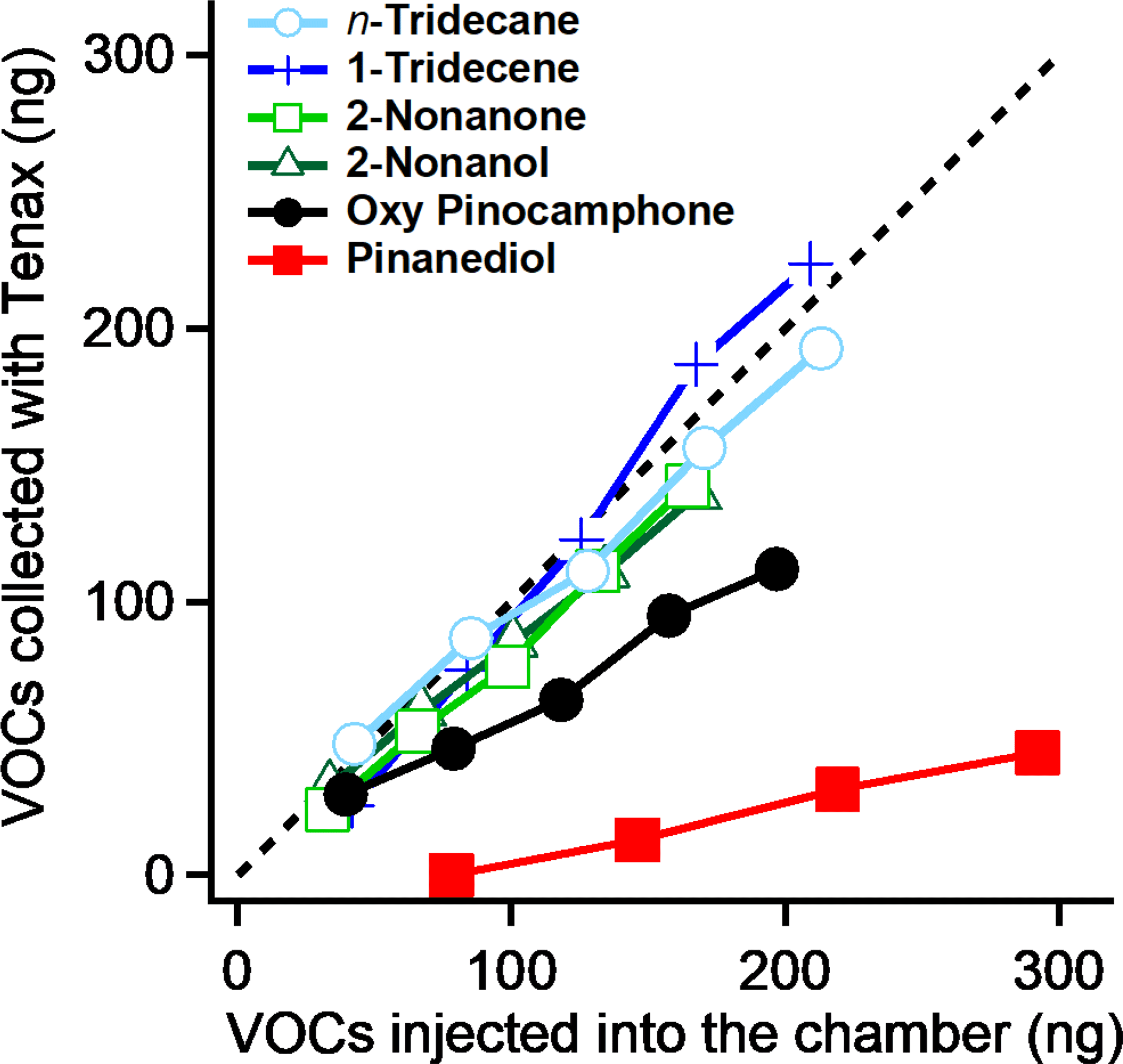
Figure 1The gas phase concentrations of n-tridecane, 1-tridecene, 2-nonanone, 2-Nonanol, oxy-pinocamphone, and pinanediol in the chamber measured by TDGC/MS. Compared to the amount of organics injected into the chamber, n-tridecane, 1-tridecene, 2-nonanone, and 2-nonanol show almost no vapor wall loss. Oxy-pinocamphone and pinanediol show 43 and 86 % loss, respectively.
3.1 Correction for the loss of the precursor, pinanediol, to the Teflon chamber walls
Because SVOCs should sorb to the Teflon walls, we expect a portion of PD to be lost after PD was injected into our chamber. To constrain this, we injected equal quantities of six compounds into our chamber simultaneously: PD, oxy-pinocamphone, n-tridecane, 1-tridecene, 2-nonanone, and 2-nonanol. The first two are an SVOC and an IVOC, while the last four are VOCs that should have very limited wall partitioning at equilibrium. We then measured the resulting gas-phase concentrations in the chamber using both TD-GC/MS and PTRMS and compared the observed signals to those we expected based on the injected amounts. We finished the injection in 15 min and collected Tenex tube samples at 15 min after the injections were completed.
In Fig. 1, we compare the TD-GC/MS measurements with the amounts of organics we injected. We averaged the concentrations from the time when the gas concentration became stable to right before the next injection. The VOCs, n-tridecane, 1-tridecene, 2-nonanone, and 2-nonanol, all fall along the 1:1 line, demonstrating that they have minimal wall losses and excellent recovery, consistent with our expectations. However, PD and oxy-pinocamphone show large discrepancies between the measured and injected amounts. The recovered gas-phase values show that 43 % of the injected oxy-pinocamphone and 86 % of the PD were lost; only 14 % of the PD remained in the gas phase.
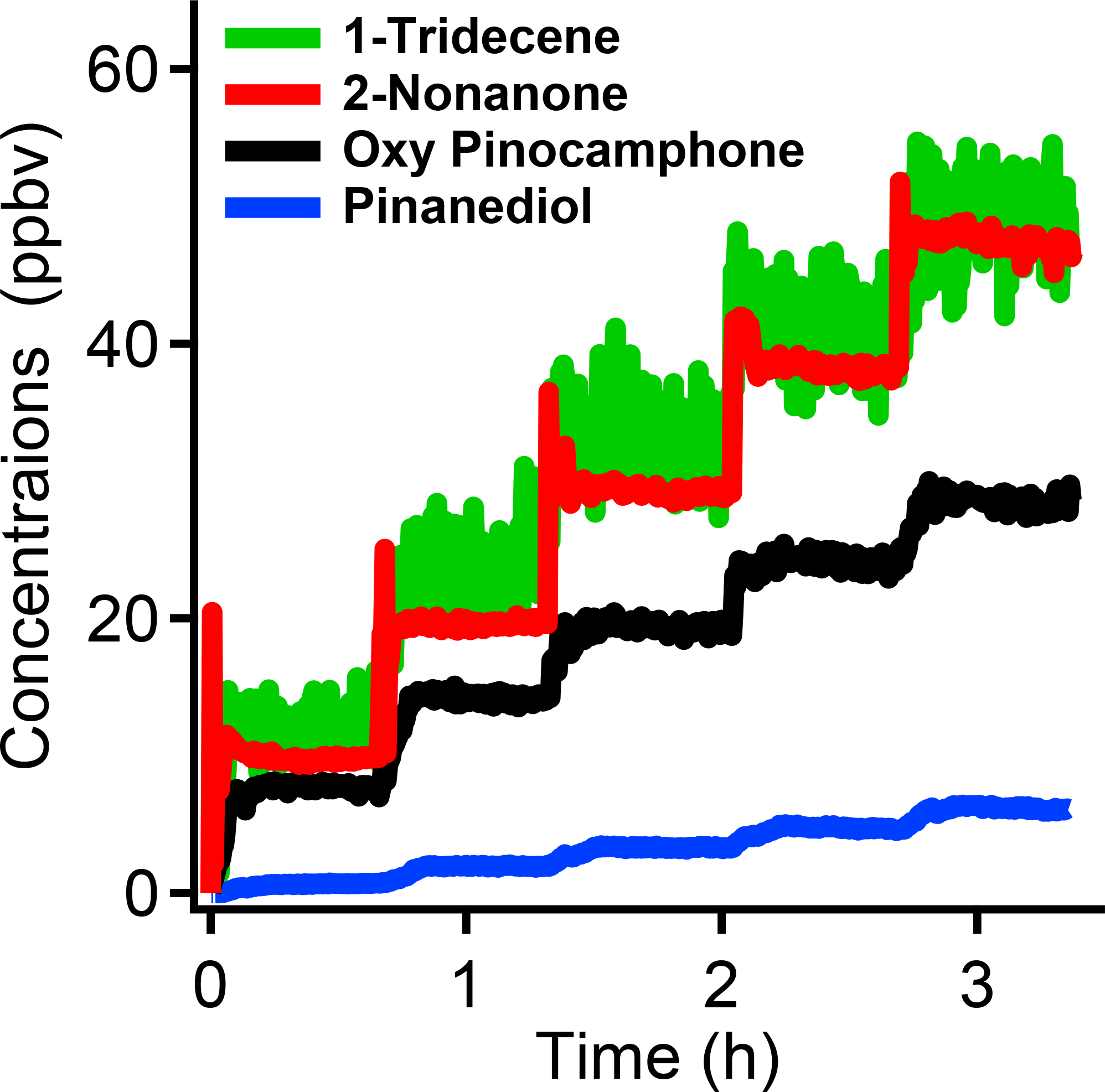
Figure 2The temporal concentrations of the organics after we injected a series of aliquots of 1-tridencene, 2-nonanone, oxy-pinocamphone, and pinanediol into the chamber in increments of 11 ppbv (at 100 % injection efficiency). Each injection resulted in a similar increase in all organics. The similar increase indicates that oxy-pinocamphone and pinanediol may have constant wall loss factors in the concentration range studied in this work.
In Fig. 2, we show the results of an experiment where we injected a succession of aliquots of 1-tridecene, 2-nonanone, oxy-pinocamphone and PD into the chamber, with expected stepwise incremental increases of 11 ppbv each, and measured the gas-phase concentrations with a PTRMS. We put the mixture the compounds in a flash vaporizer consisting of a stainless-steel tip with a machined trough for compounds containing a resistive heating element, all inserted well into the chamber at the end of a stainless-steel tube through which we passed purified, heated air. We used the purified air flow to transfer the vapors into the chamber while heating the mixture. We observed that the PTRMS signal stabilized after each injection, and each injection with the same amount of organics resulted in a similar step-wise vapor concentration increase. The two VOCs, 1-tridecene, and 2-nonanone, both showed concentration increases consistent with expectations. The PTRMS sensitivity to nonanone is higher than its sensitivity to 1-tridecene, and so the signal to noise is substantially higher. The 2-nonanone shows nearly square-wave response with a brief (∼ 5 to 10 min) overshoot related to the chamber mixing timescale, and the 1-tridecene signal displayed the same behavior. Oxy-pinocamphone and PD show lower than expected stepwise increases in concentration with a longer rise time. The step-wise increases for oxy-pinocamphone and PD are consistent with near-constant wall-loss factors in the concentration range in this study, but the signals are not consistent with instantaneous evaporation and subsequent wall partitioning. If that was the case we would expect a large initial spike similar and equal in magnitude to the spike in 2-nonanone (i.e., we would expect the full 11 ppb to appear initially in the gas phase); we would then expect the SVOC signal to drop to an equilibrium value on the equilibrium timescale for wall interactions – 10–15 min for our chamber (Ye et al., 2016a), as observed by Krechmer et al. using a core-flow inlet chemical ionization mass spectrometer and nitrate chemical ionization (Krechmer et al., 2016). The slow increase in signal we observe may be the convolution of two effects: less than instantaneous evaporation from the flash vaporizer for the SVOCs and slow equilibration of the PTRMS sampling line. Regardless, the signals in the PTRMS stabilize to values consistent with the TD-GC/MS results; these experiments are both consistent with relatively rapid, reversible equilibration of SVOCs (represented by the PD) and IVOCs (represented by the oxy-pinocamphone) between the gases and the Teflon chamber walls.
In order to calculate SOA mass yields, we must determine the amount of precursor oxidized based on the change in precursor signals (e.g., the gas-phase PTRMS measurements). This is straightforward for a VOC with minimal wall interactions, but for the SVOCs we must account for their significant interaction with the Teflon walls. It is not sufficient to simply measure the change in the gas-phase PD concentration, because of the apparently rapid equilibration suggested by the theory put forward by Matsunaga and Ziemann and supported by the rapid change in the SVOC concentration change in the gas phase due to the saturation concentration change caused by the temperature vibration in our previous paper (Ye et al., 2016a). If PD were in equilibrium with the walls there would be a substantial source of PD to the gas phase from the Teflon walls as PD was lost from the gas phase due to oxidization or any other sink. Simply put, the results suggest that, at equilibrium, for every 10 units of PD in the gas phase, roughly 100 units are sorbed in or on the Teflon walls. Therefore, removal of a small amount from the gas phase (say 1 unit) should result in replenishment of 90 % by the walls to maintain the equilibrium. Consequently, if we observe a decrease of 1 unit of PD vapor, that implies that 10 units are actually lost from the gas phase since the evaporation of PD from the Teflon walls re-establish the equilibrium. This, obviously, has large implications for the calculated SOA mass yields above and beyond any possible wall losses for products of the PD oxidation.
We use two methods, heating and isothermal dilution, to test whether the Teflon chamber walls in fact serve as an accessible reservoir of PD. Increasing the chamber temperature raises the saturation concentration of PD and thus decreases the activity of PD vapors. Heating by 30 ∘C should raise the saturation concentration of PD by a factor of 30 and lower the gas-phase activity (the concentration divided by the saturation concentration) by the same factor. Some PD sorbed to the Teflon should then evaporate to lower the condensed-phase activity. To test this, we injected 866 µg m−3 (118 ppbv, the middle value we measured to make sure it is not saturated in the gas phase) of PD vapor into the chamber at 13 ∘C and subsequently increased the chamber temperature to 44 ∘C. As shown in Fig. 3, the PD vapor concentration measured by the PTRMS increased rapidly after heating and reached a steady value after the temperature stabilized at 44 ∘C. The concentration rose by a factor of 2.5–3. To be certain that desorption from the walls was the only possible source, we also monitored the suspended aerosol mass using an HR-AMS. The total organic mass in particles was around 5 µg m−3, far less than the increase in the PD vapor concentration. Particle evaporation thus contributed negligibly to the increase in PD vapors; therefore, the PD adsorbed or absorbed by the Teflon chamber walls was the only possible source of the increased gas-phase burden.
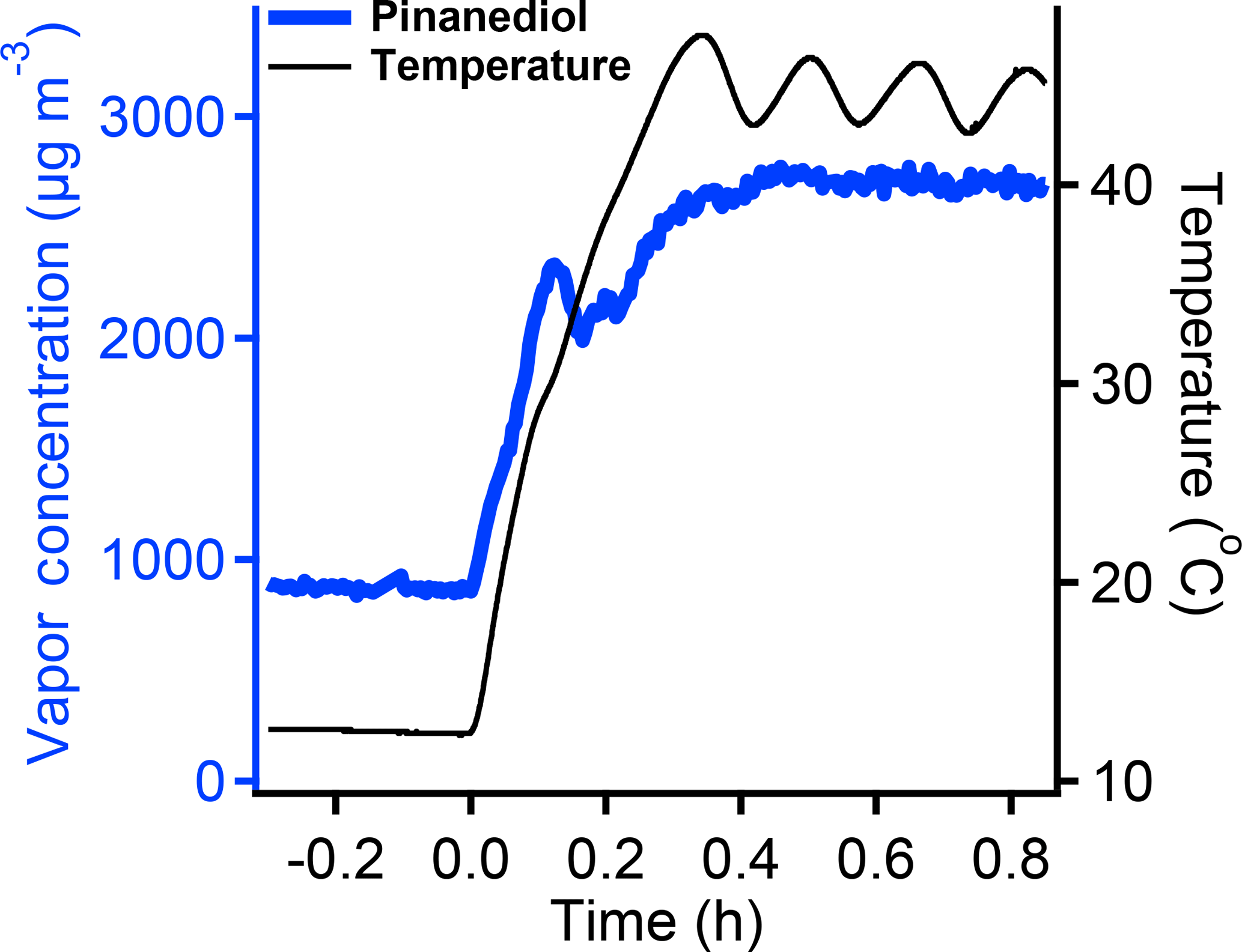
Figure 3The increase in the pinanediol vapor concentration after increasing the chamber temperature from 13 to 44 ∘C. The concentration of PD increased 2.5–3 times and reached a constant value after temperature stabilized at 44 ∘C. The increase in the PD concentration shows that PD can come out from the chamber walls at higher temperature.
Increasing temperature by 30 ∘C should increase the saturation concentration (C∗) of PD by roughly a factor of 30 (May et al., 2012). All else being equal, this should cause a 30-fold increase in the activity ratio of the sorbed PD to the gas-phase PD and thus drive a large return flux to the gas phase, with the equilibrium vapor fraction increasing from 13 % to around 80 %. This is consistent with our observations though we observe a factor of 2–3 less than this simple calculation would suggest. However, if PD is absorbed into the Teflon walls, it is likely that the activity coefficient of the PD in Teflon walls would drop substantially upon heating, so this would allow the activities to equilibrate with a smaller net change in absolute concentration. Acknowledging these large uncertainties, the heating experiment is broadly consistent with the postulated reversible equilibration of PD between the gas phase and the Teflon chamber walls.
Our SOA formation experiments are isothermal, but during the experiments the gas-phase PD concentration (and thus activity) drops due to oxidation. To reproduce these conditions, we used isothermal dilution to mimic the PD loss during SOA formation. We maintained the chamber temperature at 22 ∘C and injected PD along with acetonitrile into the chamber, and then measured their concentration ratio using the PTRMS. We used acetonitrile as a passive tracer because it is highly volatile, should not have wall losses, and it is readily measured with the PTRMS. In 1 h after injecting PD and acetonitrile into the chamber, we turned on a slow flow of dilution air, initially at a rate of 100 L min−1 (1 % min−1) and later at a rate of 300 L min−1 (3 % min−1). These rates roughly bracket the loss rate of PD via OH oxidation in our SOA formation experiments. We tracked the ratio of PD to acetonitrile. If the PD sorbed to the Teflon chamber wall were released continuously because it was in (a necessarily reversible) equilibrium, the PD concentration should fall more slowly than acetonitrile, and the ratio of PD to acetonitrile should rise steadily. The bottom plot in Fig. 4 shows a simulation of the expected signals. As shown in the top plot, the concentrations of both PD and acetonitrile steadily decreased after we started to flush the chamber. However, we did not observe any increase in the PD to acetonitrile ratio; instead, the ratio remained almost constant, and even showed a slight decrease. This suggests that PD does not return to the gas phase from the Teflon walls at 22 ∘C but instead still shows a modest loss to the chamber walls. This indicates slow diffusion into the bulk Teflon, and is inconsistent with the observations in Zhang et al. (2015).
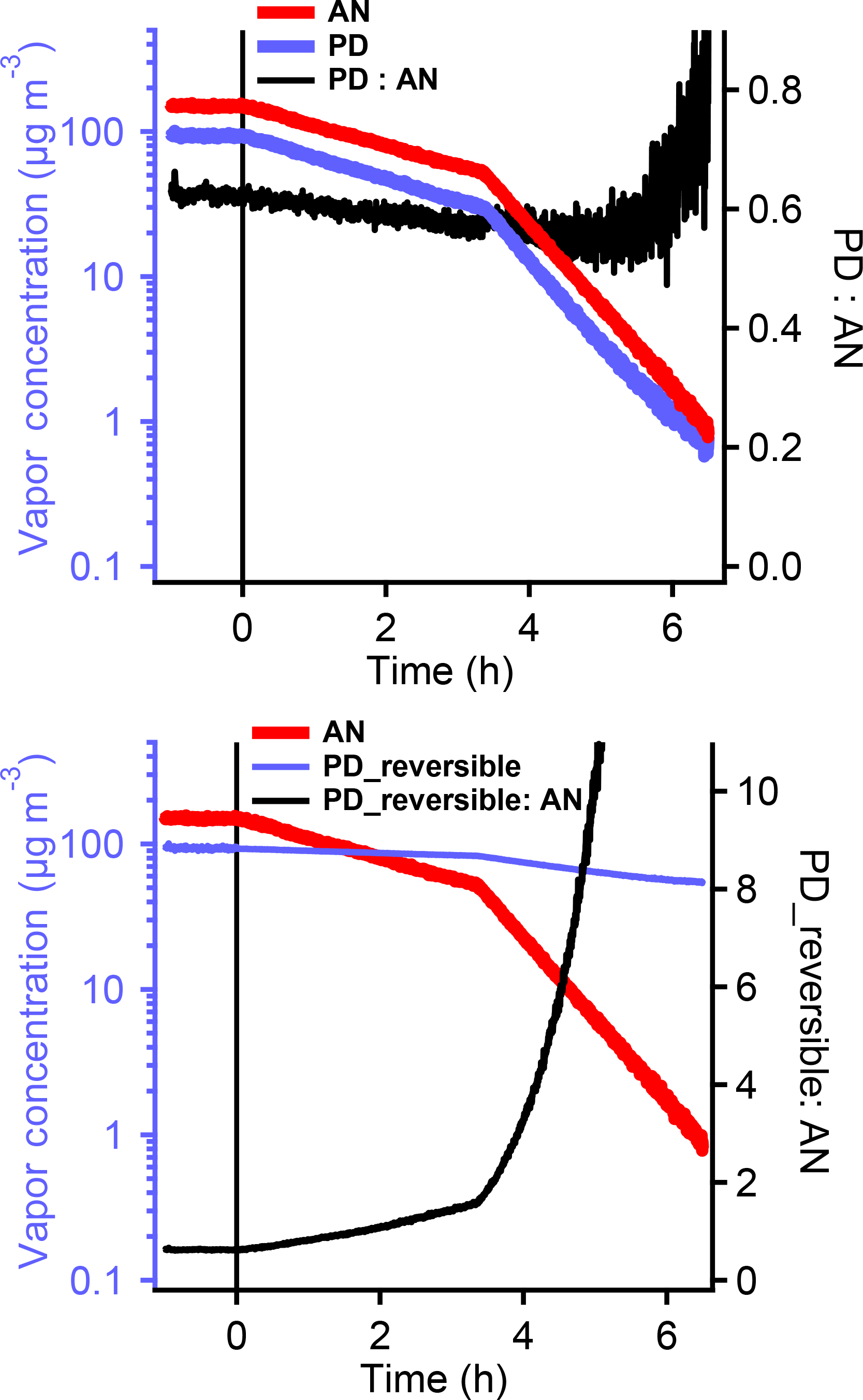
Figure 4The change in PD and AN concentrations during isothermal dilution of the chamber with fresh air, which mimics the depletion of PD during the SOA formation (top). The ratio of PD to AN shows very small change until the PD concentration dropped below 2 µg m−3. When considering the deposition of PD on the Teflon chamber walls as reversible partitioning, the predicted PD and AN concentration change during the dilution was shown at the bottom. The decrease in the predicted PD concentration should be slower than the decrease in AN. The ratio of the predicted PD to AN concentration should keep increasing. These indicate that PD does not return to the gas phase from the Teflon at 22 ∘C but instead still shows a modest loss to the chamber walls. So no further correction for the release or loss of PD is necessary when studying the SOA formation.
During the dilution experiments, only after the PD concentration reached 2 µg m−3 (2 % of the initial concentration), 5.5 h after we started dilution, did the ratio of PD to acetonitrile start to increase. This confirms that PD can return to the gas phase from the chamber walls even during isothermal dilution (or any other isothermal loss from the gas phase), but only after substantial depletion of gas-phase concentrations of PD. Thus, while reversible partitioning to the walls is the most straightforward explanation for the losses of PD we have presented, and even the results of chamber heating are broadly consistent with this explanation, we see no sign of reversibility under the conditions of our SOA formation experiments. This is a paradox, for which we have no explanation.
Therefore, based on the empirical evidence we conclude that the measured decrease in PD from PTRMS during SOA formation experiments is equal to the amount of PD oxidation, and that no further correction for wall equilibration is necessary. There is no reason for the PD to “know” whether its gas-phase concentration is decreasing because of reaction or isothermal dilution, and so we conclude that the dilution experiment accurately simulates the PD response to reactive loss. However, as a precaution against return flux after substantial PD depletion, we shall limit our analysis to the first 1.5 e-folding lifetimes in PD oxidation (we only use the data where the PD concentration is above 22 % of its initial value).
3.2 Correction for particle wall loss.
We conducted experiments to measure the SOA production from oxidation of PD by OH radicals generated via HONO photolysis at five different initial PD concentrations: 1, 2, 4, 5, and 6 ppbv. We used Eq. (1) to calculate SOA mass yields (Y).
where CSOA is the measured mass concentration of SOA, and ΔCPD is the mass concentration of the reacted PD. We measured the PD concentration using PTRMS with a unique mass fragment, , and then calculated the ΔCPD. As we have discussed, we do not correct the measured concentration change in PD for any interaction with the chamber walls. However, in order to calculate the CSOA, we must also account for wall losses of both particles and the condensable SOA products.
We employed three traditional methods to correct the particle wall loss, based on the assumption that particles deposited to the chamber walls function the same as the suspended particles for the SOA condensation. The corrected SOA production, CSOA, is determined by the ratio of suspended SOA ( to suspended ammonium sulfate seed ( and the initial concentration of ammonium sulfate seed particles at time 0 h (, as shown in Eq. (2) (Hildebrandt et al., 2009).
The essential term is the SOA-to-seed ratio, . We calculated this ratio directly from the organic and seed (sulfate + ammonium) concentrations measured by the HR-AMS (method 1). We also used the SMPS data. We determined the by applying an exponential function to fit the measured decay of the pure ammonium sulfate seeds before photo-oxidation and then extrapolating that decay for the duration of each experiment (method 2). We also calculated by scaling the total particle number concentration (method 3). Because both coagulation and nucleation were minimal during the experiments, we can correct for particle wall losses based on either mass or number loss. is proportional to the total suspended particle number concentration. We demonstrate method 2 and 3 in Fig. S1 in the Supplement. We calculated as the difference between the total particle mass and the after correcting with the SOA density, 1.4 g cm−3, which we calculated following the method of Nakao et al. (2013). As shown in Fig. S2, the SOA-to-seed ratios from these three methods agree to within roughly 20 %. Consequently, we focused on the HR-AMS data (method 1) to perform the particle wall-loss correction. We demonstrate one example of the temporal depletion of PD and SOA formation in Fig. S3. Around 80 % of PD reacted in the first hour. As mentioned previously, we excluded all data where the PD concentration was less than 22 % of its initial value from the analysis; those data are plotted in gray.
3.3 Correction for vapor wall loss
In addition to correcting for the loss of SOA as suspended particles, we also determine the amount of condensable SOA vapors that condense directly to the Teflon chamber walls after PD oxidation. This also reduces the observed SOA mass (Ye et al., 2016a; Krechmer et al., 2016). If the condensing species are functionally non-volatile (their saturation ratios are much larger than their particle-phase activity; Donahue et al., 2011b), then condensation to the suspended particles will be quasi-irreversible. Furthermore, for the relatively low saturation concentration values required, there should be efficient wall losses of the vapors. We thus assume that vapor wall losses are the same per unit condensation sink as condensation to the suspended particles.
The condensation sink (CS) represents the loss frequency of vapors to the suspended aerosol surface (Donahue et al., 2014); it can be thought of as the mean speed of the vapors multiplied by the aerosol surface area, but modified for the gas-phase diffusion near the particle surface and accounting for accommodation from the gas phase to the condensed phase when that is rate limiting. We calculated the CSP using Eq. (3) (Trump et al., 2014),
where k refers to a particle size bin, Nk is the number concentration of particles in this bin, v is the mean thermal speed of the gas phase molecules, dP,k is the particle diameter, and βk is the transition-regime correction factor (Seinfeld and Pandis, 2006), which is a function of the mass accommodation coefficient (α) and the mean free path of the organic vapor in air. We used two accommodation coefficient values, 0.1 and 1, as limiting cases as the available evidence suggests that 0. (Saleh et al., 2013). When α=1, the condensation sink will be the same as the collision frequency between the gas molecules and suspended particles.
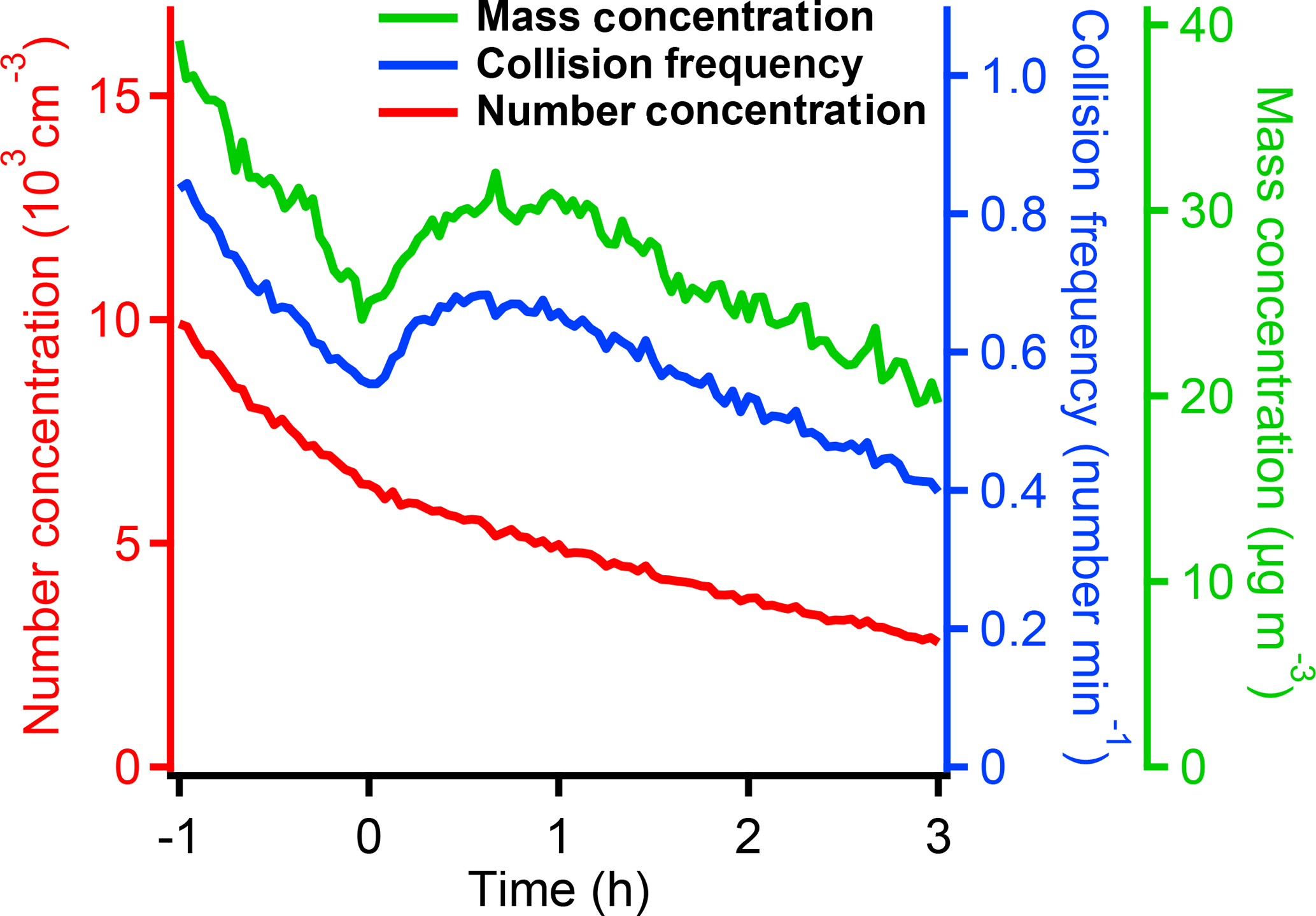
Figure 5The change in collision frequency and number, as well as mass concentration, of the suspended particles during the SOA formation. The collision frequency has the same value as condensation sink when α=1. After the SOA formation started at 0 h, the SOA mass condensed on the particles increased the particle surface areas and increased the collision frequency. We also observed the increase in the total mass concentrations. The particle number concentration always followed the exponential decay which indicated the nucleation may be minimal.
Figure 5 shows the suspended collision frequency versus time together with the number and mass concentration of the suspended particles during an SOA formation experiment. The collision frequency decreased initially due to particle wall losses. However, when the SOA formation started, the SOA condensation increased the particle surface area and thus increased the collision frequency. Later in the experiment, after the SOA formation was almost complete, the particle wall loss again dominated and the collision frequency decreased.
As shown in Scheme 1, the fraction of the oxidation products that initially condenses on the suspended particles versus the chamber walls is determined by the ratio of the suspended-particle condensation sink to the wall loss frequency (the wall condensation sink). We previously measured a wall condensation sink for SVOCs in the CMU chamber of 0.063 min−1 (Ye et al., 2016a). In Fig. 6 we compare the suspended-particle condensation sink to the wall condensation sink for the two limiting values of the mass accommodation coefficient: 0.1 and 1. When α=1, the suspended-particle condensation sink is much larger than the wall condensation sink. In this case, only a very small fraction of the condensable vapors are lost to the walls, at least initially. When α=0.1, the condensation sink of the suspended particles and the chamber wall are comparable, which makes vapor wall loss significant.
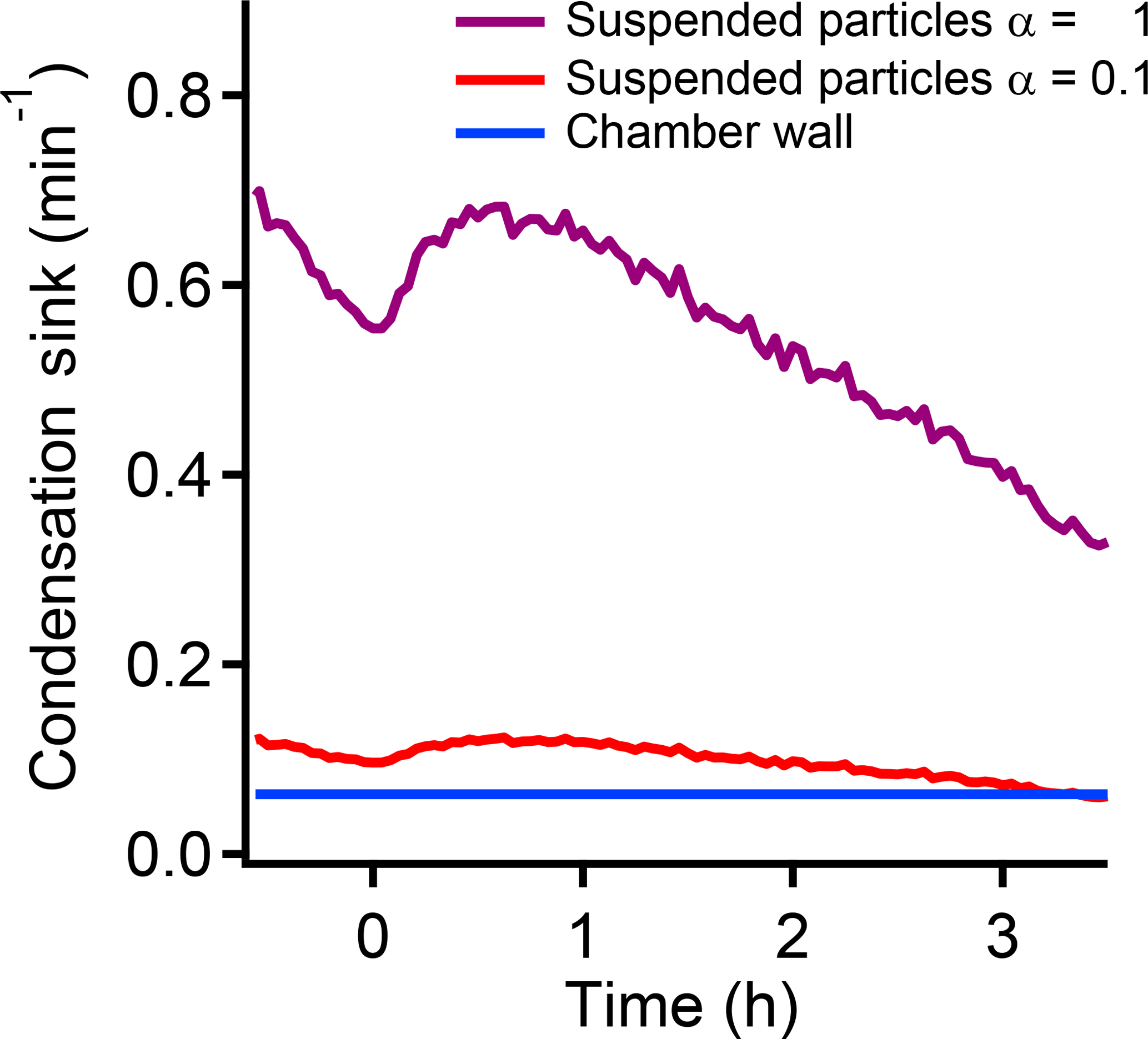
Figure 6The difference of the condensation sink between the chamber wall with the suspended particles when the mass accommodation coefficient is 0.1 or 1. When α=1, the condensation sink of the suspended particles is much larger than the wall condensation sink. When α=0.1, the two values are on a similar level, which indicates that the vapor wall loss may be very significant.
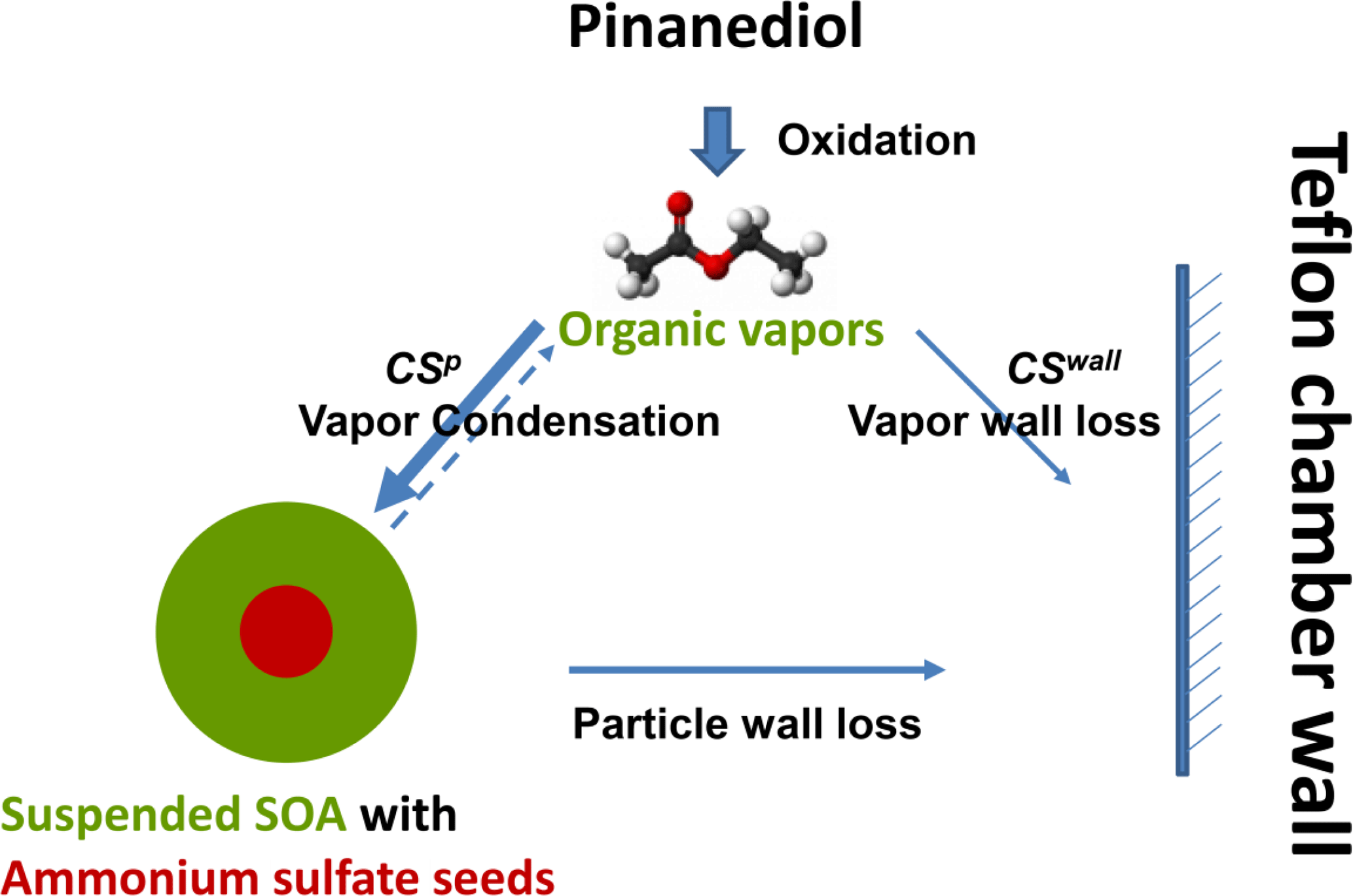
Scheme 1The competition of vapor deposition on the suspended particles and the Teflon chamber walls. The fraction of the oxidation products deposited on the suspended particles and the chamber wall are determined by the condensation sink to the particles and the chamber walls.
The interactions of semi-volatile oxidation products with the two different sinks (suspended particles and the walls) can be complex, but products that are effectively nonvolatile (with very high steady-state saturation ratios while the PD is being oxidized; Donahue et al., 2011b) should simply condense in proportion to the two condensation sinks. In this case the mass that condenses on the walls is given by the mass observed to condense on the suspended particles multiplied by the ratio of the wall condensation sink to the suspended condensation sink. In Fig. 7 we show the products lost to the chamber walls together with the SOA mass on the suspended particles and the particles lost to the chamber walls. The direct deposition of the product vapors to the chamber wall may have been as much as one-third of the total SOA mass at the lower limit of α=0.1 or as little as a few percent if α=1. This vapor wall loss correction is thus significant but not excessively large.
3.4 Correction for delayed condensation
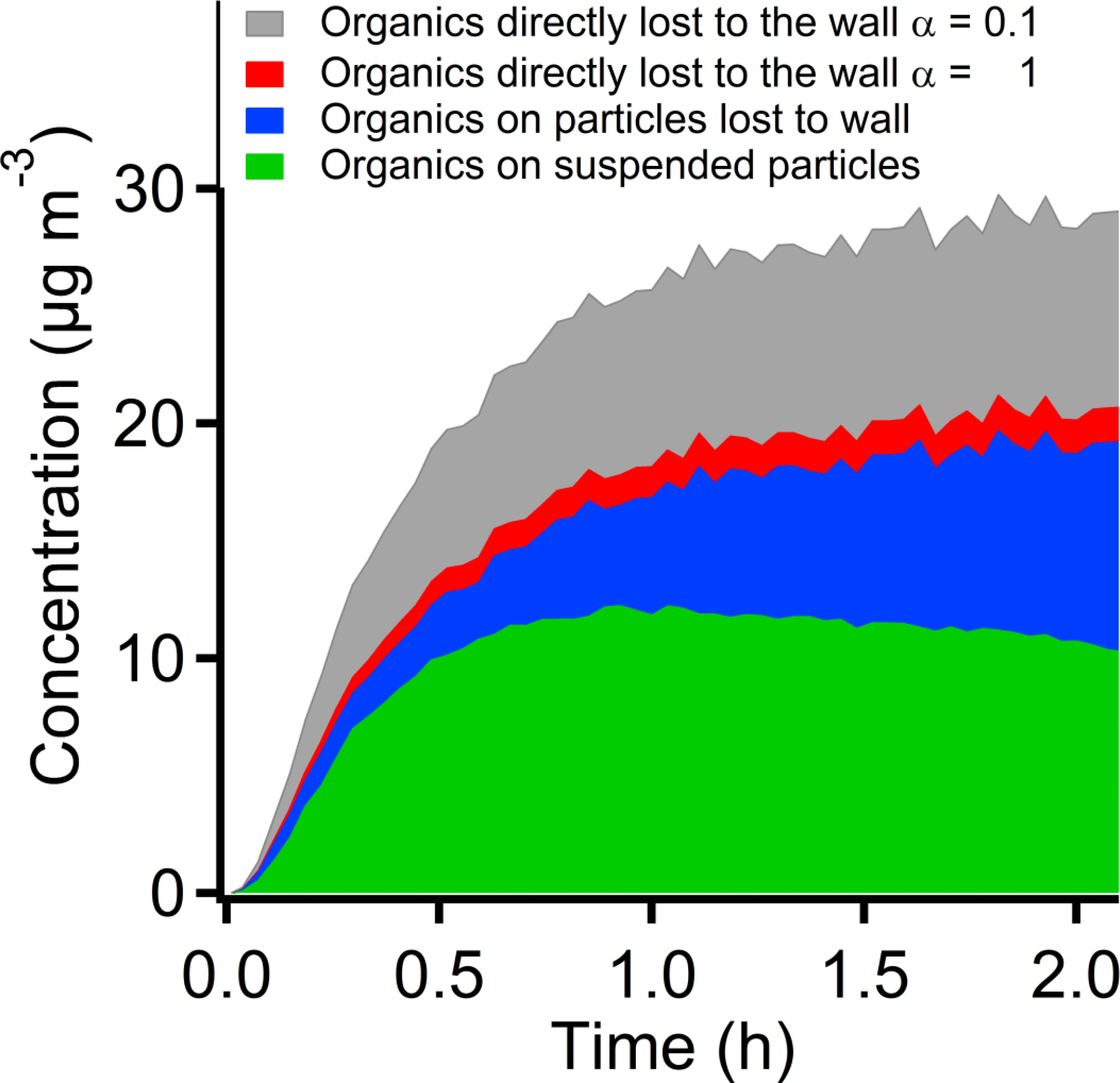
Figure 7The SOA mass on the suspended particles, lost to chamber wall due to particle wall loss and direct vapor deposition on the chamber wall. When α=0.1, the SOA mass lost to the chamber wall through the direct vapor deposition may have one third of the total SOA mass. When α=1, the vapor wall loss may not be significant.
Some condensable products will be accumulated in the gas phase in a steady state between production and loss even if they have a very low saturation concentration. This is especially significant early in an experiment when the oxidation rate (and thus production rate of condensable vapors) is high (Donahue et al., 2011b). We can estimate this simply by assuming that the condensable vapors are produced with a constant mass yield during PD oxidation (that the mechanism is invariant) and that their saturation concentrations are very low. We then apply a constant mass fraction to the amount of oxidized PD to estimate the total concentration of condensable products in any phase. In Fig. 8, we show an example calculation for α=0.1 and a constant mass yield of 0.88 as a dashed black curve; except for early in the reaction, this provides a good match to the total condensed organics, but for times less than 2 condensation lifetimes (21 min, indicated with the vertical dashed red line) the observed SOA concentration is substantially less than 0.88 times the oxidized PD (shown with the gray fill). The SOA mass yields during the first 10–20 min thus may be underestimated if delayed condensation is ignored (Donahue et al., 2011b). On the other hand, lower mass yields at lower OA concentrations can be interpreted in terms of semi-volatile partitioning (Odum et al., 1996b; Donahue et al., 2005).
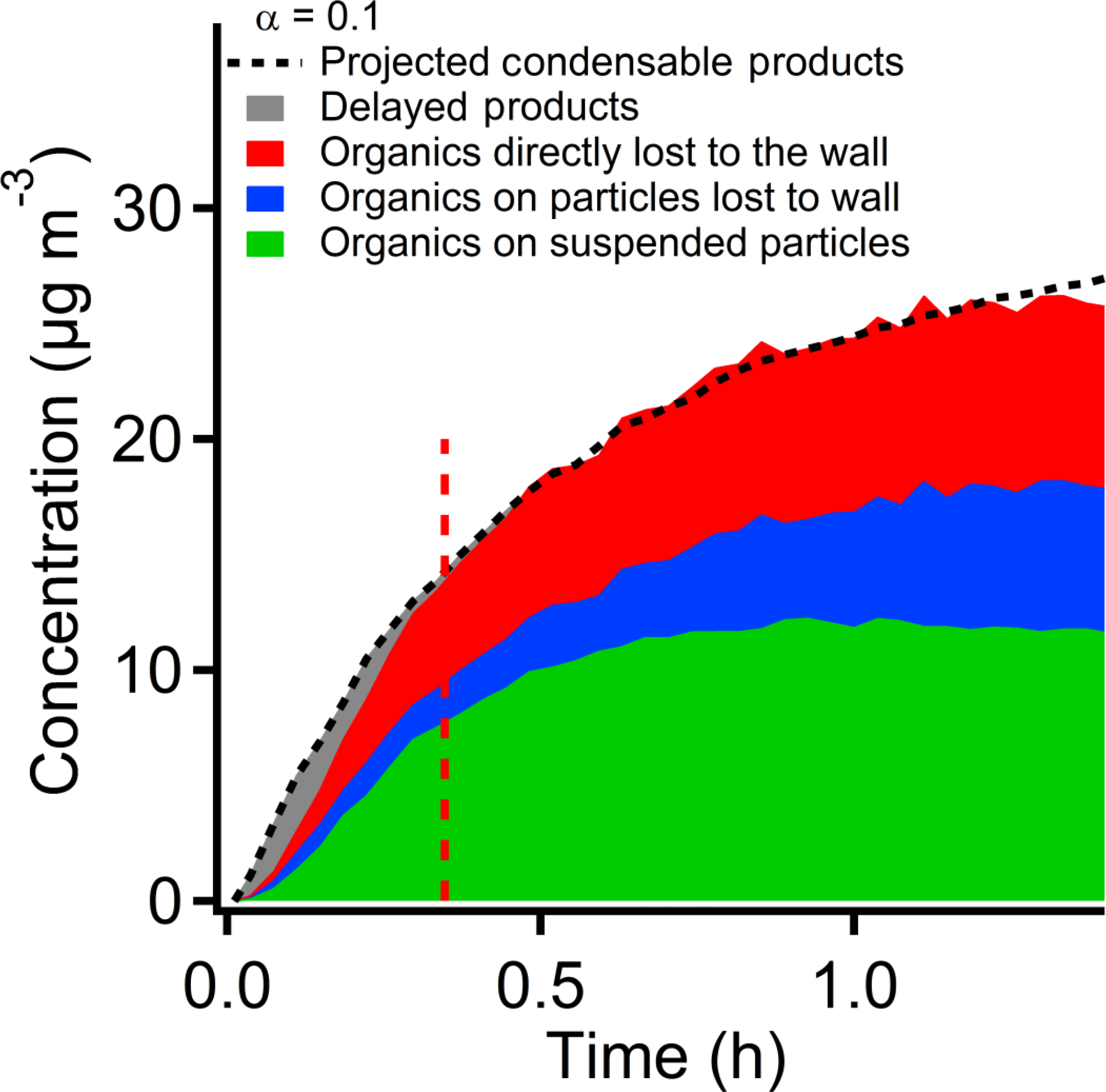
Figure 8The discrepancy of the observed SOA caused by the condensation delay. The black dash line shows the estimated concentration of condensable vapors from the reacted PD. The dashed area at 0–0.3 h shows the difference between formed vapors and the observed SOA. This gap may be caused by the diffusion time of vapor molecules to reach the surface of the particles or the chamber walls. This delay may result in a lower measured SOA mass yield at the early stage of the experiment.
3.5 SOA mass yields from PD oxidation by OH radicals
In Fig. 9 we show calculated SOA mass yields from the 6 ppb PD experiment for three cases, first considering only particle wall loss, and then treating both particle and vapor wall loss for α=1 and for α=0.1. When α=1, the difference with and without vapor wall losses (i.e., the first two cases) is very small. However, the mass yield increases by 30 % after correcting for vapor wall loss with α=0.1. We further estimate the delayed condensation of ELVOC and LVOC products by finding the mass yield after two condensation lifetimes, as illustrated in Fig. 8. The dashed horizontal lines indicate these values. The true equilibrium SOA mass yields may be closer to the dashed lines than the observed values due to delayed condensation.
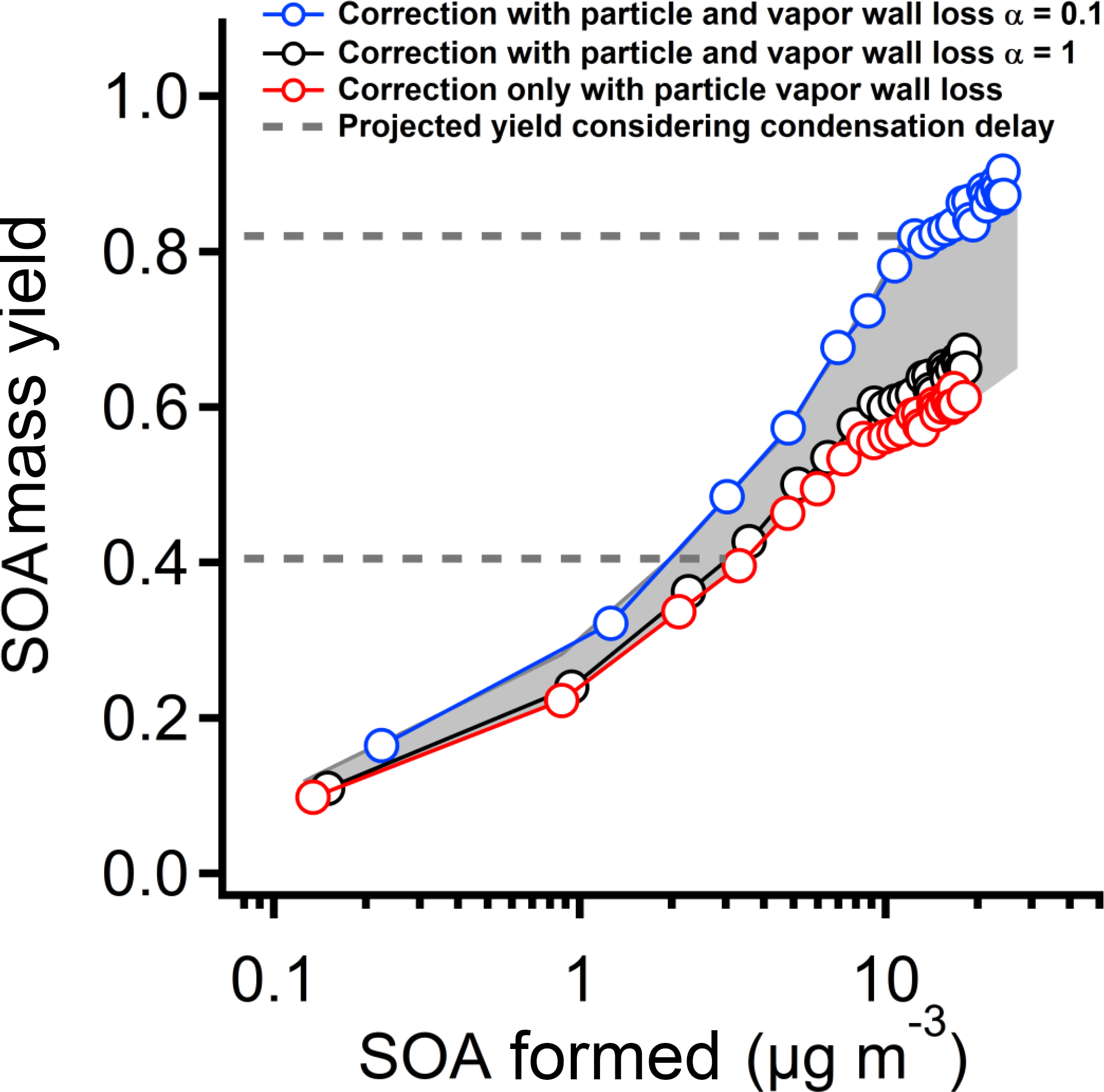
Figure 9The SOA yield from pinanediol photo-oxidation after correction for particle wall loss and vapor wall loss using three different methods: correction for particle wall-loss only; correction for vapor wall loss with α=1; and correction for vapor wall loss with α=0.1. For the first two methods the mass yields are similar. For the third, when α=0.1, the mass yield is 30 % higher than for the other two methods. The horizontal dashed lines indicate the mass yields at a time equal to twice the gas-phase lifetime of vapors due to condensation or wall loss. Before this time (below the lines) the measured SOA yields may be biased low due to the delay between production and condensation to the suspended particles.
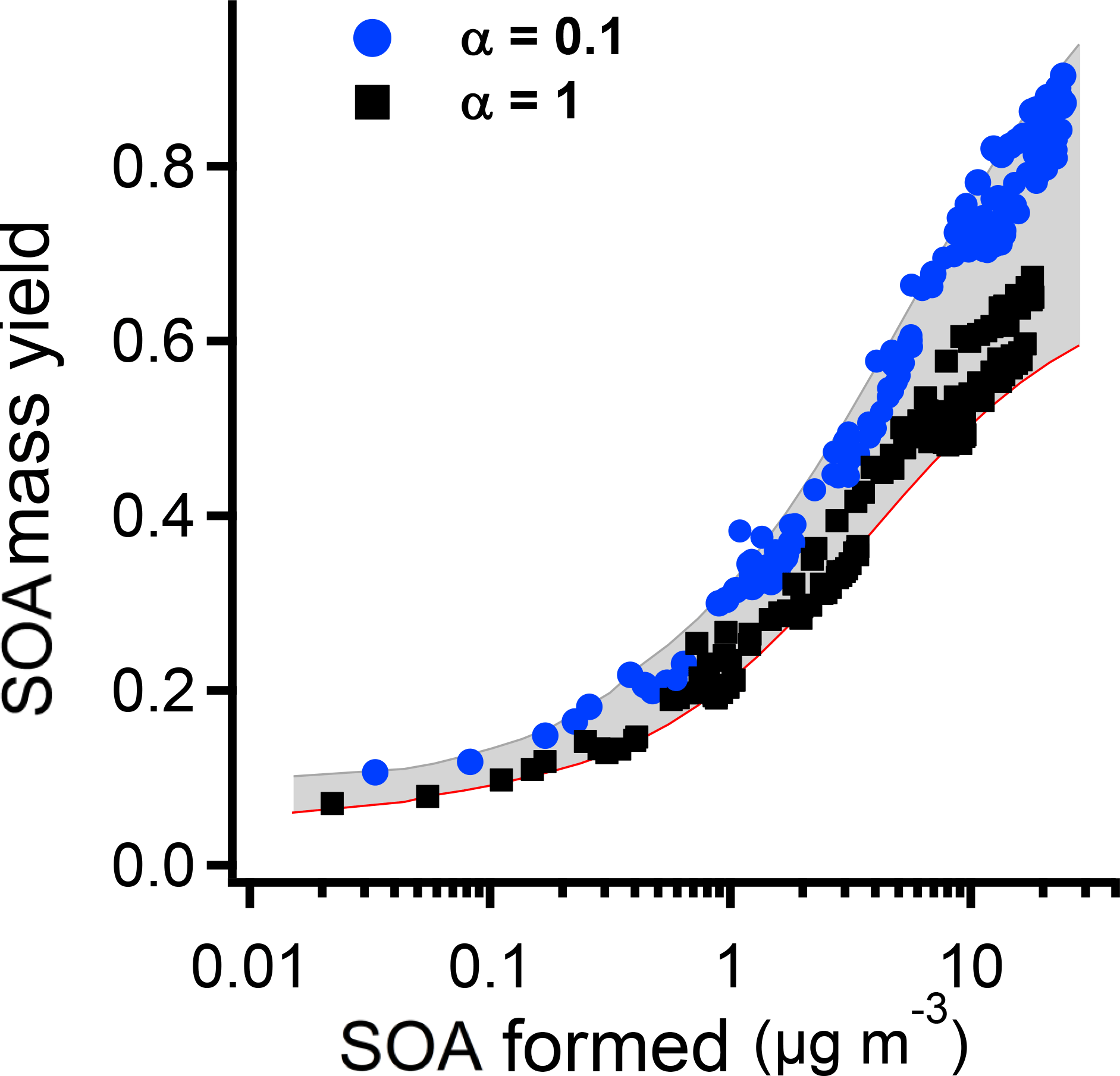
Figure 10The summary of all the SOA mass yield after correcting both particle and vapor wall loss. The initial PD concentrations are 1, 2, 4, 5, and 6 ppbv. The shaded area shows the yield range when α varies from 0.1 to 1.
In Fig. 10 we summarize data from five experiments with five different initial PD concentrations: 1, 2, 4, 5, and 6 ppbv. The shaded area shows the range of SOA yields when α values vary from 0.1 to 1. The instantaneous SOA mass yields are from 0.1 to 0.9 under the different SOA concentrations. As with the single case we present in Fig. 9, accounting for delayed condensation introduces a low-concentration asymptotic mass yield between 0.4 and 0.8. The bottom line is that, regardless of the mass accommodation coefficient, the SOA mass yields are high, with yields above 0.5 for COA>10 µg m−3. PD oxidation by OH is thus a very efficient source of second-generation SOA.
The yields for α=0.1 accounting for delayed condensation are implausibly high, implying that essentially all of the oxidation products have extremely low volatility and thus the only reason for the observed rising mass yields is the dynamical delay early in the experiment (which lasts for a relatively long time, ∼ 20 min, due to the low condensation sink associated with the low mass accommodation coefficient). On the other hand, the yields for α=1 are plausible, implying that approximately half of the condensable oxidation products consist of highly oxidized products formed via “auto-oxidation” (Ehn et al., 2014) while the other half are SVOCs that partition reversibly into the particles (Ye et al., 2016b, c).
PD oxidation has much higher SOA mass yields than α-pinene oxidation. When COA=20 µg m−3, the SOA mass yields from α-pinene oxidation (by ozone or OH) are in the range 0.1–0.2 (Hallquist et al., 2009), whereas the SOA mass yields from PD oxidation by OH are in the range 0.6–0.9, roughly 5 times larger. This finding holds regardless of wall effects or other complications to quantitative interpretation of the product volatility distribution, as those issues should be shared in common for each system. PD is a much more effective source of SOA than α-pinene. This can be well explained by the structure of PD. PD has two OH groups replacing the C=C double bond in α-pinene and yet it retains the bicyclic backbone of that monoterpene. PD can be considered as a first-generation of oxidation product of α-pinene; the likely atmospheric formation mechanism is hydrolysis of a β-hydroxy nitrate formed after OH addition to the double bond in high-NOx conditions. When PD is oxidized, C–C bond cleavage is unlikely because of the bicyclic backbone. Therefore, most PD oxidation products will be less volatile than PD and so more condensable compared to comparable products from α-pinene. One exception to this is that a major oxidation product of PD is oxy-pinocamphone, which is formed when OH abstracts a hydrogen atom from the hydroxymethylene moiety in PD and O2 immediately abstracts the second hydrogen from the OH group, analogous to acetone formation from 2-propanol. All of the other oxidation products of PD are plausibly condensable. It is thus sensible that the molar yields of condensable products from PD oxidation are in the range 0.5–0.8 and that the corresponding mass yields are significantly higher due to the added oxygen.
3.6 Elemental analysis of the SOA
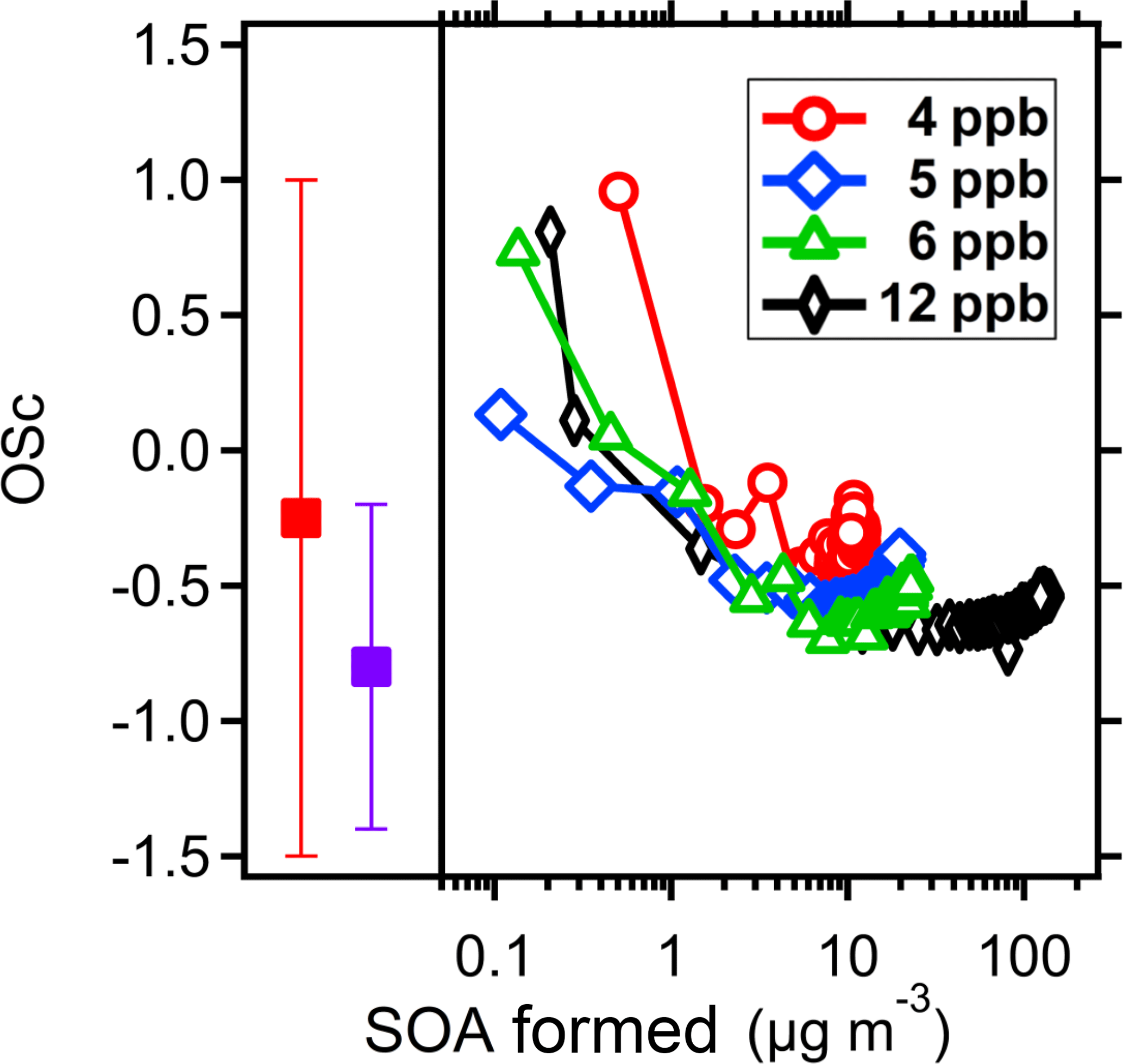
Figure 11The of the SOA from PD with initial concentrations at 4, 5, 6 and 12 ppb on the right panel. The left panel shows the of the oxidation products from PD in the clusters observed in the CLOUD experiments, which contained 1 (red solid square) and 4 (blue solid square) C10 organics. The SOA formed at the very early stage (low yields) shows highly oxidized. The in this study is comparable to the results from the CLOUD experiments.
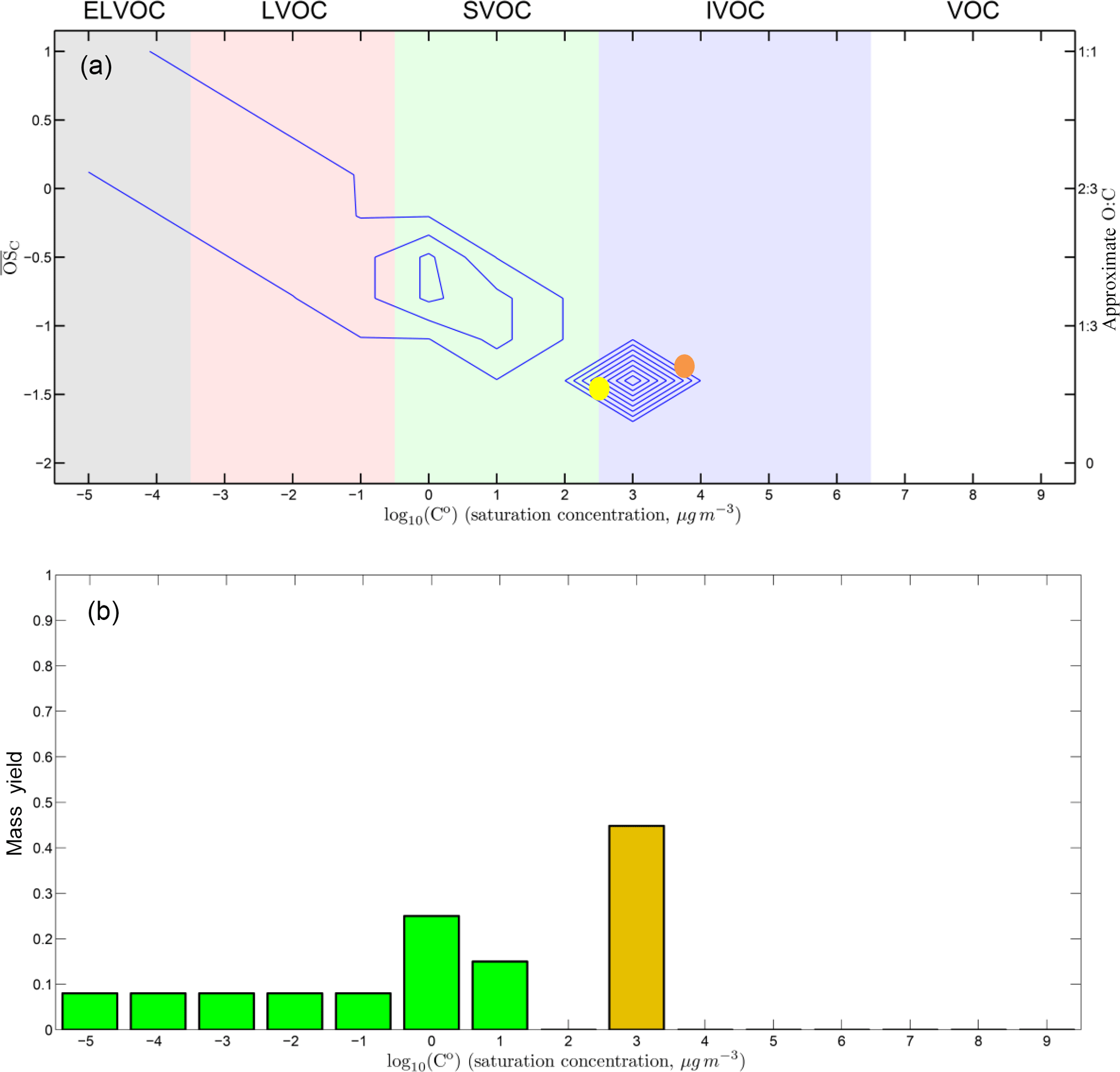
Figure 12Representation of the oxidation products from PD in the two-dimensional volatility–oxidation space for a mass accommodation coefficient α=1. We group organics in the broad classes of ELVOCs, LVOCs, SVOCs, or IVOCs. Panel (a) is a 2-D representation. PD is shown as a yellow dot. The blue contours show the oxidation products from PD, with higher values indicating higher yields. Panel (b) is a 1-D consolidation of the 2-D product contours, showing the total mass yields in each volatility bin. The major products spread toward the upper left from PD, with increased oxidation state and decreased volatility. The products near to the upper left corner, in the ELVOC region, may contribute to new-particle formation observed in the CLOUD experiments. They constitute around 15 % of the total SOA mass. Some products may undergo fragmentation or functional group change, such as converting an alcohol group to a carbonyl group, as with oxy-pinocamphone, which is shown in orange.
In Fig. 11, we plot the observed average carbon oxidation state, , of the SOA formed from PD as a function of the SOA mass concentration. decreases as the SOA mass increases, consistent with other studies of biogenic SOA (Donahue et al., 2006; Shilling et al., 2009). The SOA that condenses very early in the experiment (at low COA) is also highly oxidized. These promptly condensing organic products are ELVOCs or LVOCs, with sufficiently low volatility to build up a high saturation ratio early in the experiment. We also consistently observe a slight increase in at the end of each experiment. This may be due to the further oxidation (aging) of the products. The SOA formed from a lower initial PD concentration also shows a higher at the same SOA concentration than the SOA formed from a higher initial PD charge. When the initial PD concentration is low, the oxidation products may have more chance to react with OH radicals and become more oxidized. However, it is also possible that the higher absolute oxidation rate with higher PD concentrations drives up the gas-phase activity of SVOCs with relatively lower . Finally, it is possible that relatively more volatile (and less oxidized) products are lost from SOA particles near the end of each experiment due to sorption to the Teflon walls. As shown in Fig. S4, the ratio of organic to sulfate mass decreased slightly after 2 h, consistent with some SOA mass loss from the particles.
The composition findings are thus consistent with the mass-yield results for a relatively high mass accommodation coefficient; there is a substantial mass yield of ELVOC and LVOC products with very high but also a significant yield of SVOC products, probably with 1, that dilute the (E)LVOC condensate once conditions favor their condensation.
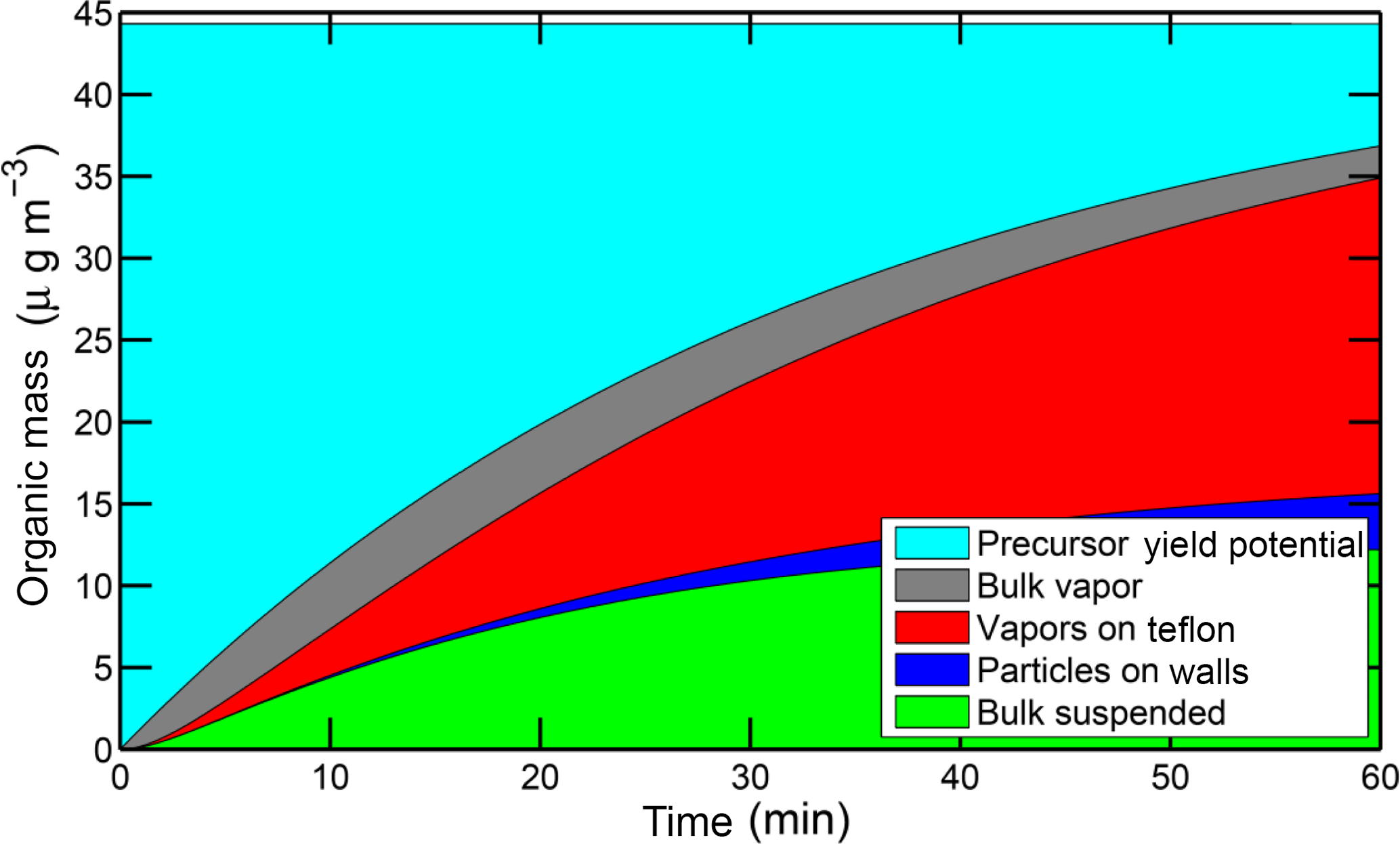
Figure 13Dynamical simulation of the SOA production from 6 ppb of PD with a mass accommodation coefficient α=1. The simulation treats five different reservoirs: unreacted precursor, vapors, suspended particles, deposited particles, and sorption to Teflon, as shown in the legend. The simulation reproduces the SOA observed on the suspended particles.
In Fig. 11 we also compare the of the SOA formed from PD in these experiments with the of PD oxidation products observed to participate in nucleation in the CLOUD experiment. We plot values for CLOUD for molecular clusters with a single C10 molecule and clusters with four C10 molecules; these values are based on molecular formulas in negatively charged clusters measured with an atmospheric pressure interface time-of-flight mass spectrometer (APITOF) where the negative charge resides on a bisulfate anion clustering with the (presumably neutral) C10 organic molecules formed from PD oxidation (Schobesberger et al., 2013). The CLOUD values are thus based on a much different technique than the highly fragmenting bulk particle electron ionization used in the AMS. Despite these differences, the values we observe are similar to those seen in the CLOUD experiments. The oxidized organics observed in the CLOUD experiments have molecular compositions C10HxOy, where x=12, 14, 16 and y = 2–12 (Schobesberger et al., 2013). They appear in four progressive bands from growing clusters, which contained 1–4 C10 organic molecules, respectively. The in the first band is relatively high, −0.2, but this decreases to −0.8 for the fourth band. The decrease in with increasing cluster size is consistent with what we observed in this study. We observed the of the bulk SOA at relatively high loading was around −0.7, which corresponds to the value measured in the CLOUD experiments for larger clusters.
A self-consistent interpretation of these observations is that the least-volatile, early condensing species forming SOA at low COA in our experiments are ELVOCs that also help form the smallest clusters in the CLOUD experiments, while the later condensing species are LVOCs and SVOCs that also contribute to cluster growth in the CLOUD experiment after initial nucleation.
3.7 Representation of PD SOA in the two-dimensional volatility–oxidation space
Following the procedures in the literature (Presto and Donahue, 2006; Donahue et al., 2011a), we mapped the distribution of volatility and in the two-dimensional volatility–oxidation space (2D-VBS). The constraints are relatively crude – just the observed mass concentrations and bulk composition – and so we present 2D-VBS yield distribution that is consistent with those constraints but still coarse grained. Specifically, we assume a long “tail” toward extremely low volatility with roughly constant mass yield, a cluster of products with slightly lower volatility than PD, and a large yield of oxy-pinocamphone, while is more volatile than PD. We present the full yield distribution, which conserves carbon, in the following section.
In Fig. 12 we show the product distribution, classifying organics in the broad classes of ELVOCs, LVOCs, SVOCs or IVOCs when α=1. The top panel is a 2-D representation. We show PD as a filled yellow circle. The blue contours show the oxidation products from PD, with higher values indicating higher yields. The lower panel is a consolidation of the two-dimensional product contours into a 1D-VBS, showing the total mass yields in each decadally spaced volatility bin. A majority of the condensed products fall to the upper left of PD, with a lower volatility and higher than PD. These compounds are produced mostly by the addition of oxygen containing moieties to the PD backbone. However, some products located on the right of PD show slightly higher , but also higher volatility. They may be formed by two possible reaction pathways. One is fragmentation, which breaks the carbon backbone and produces smaller molecules with higher volatility than the reactants. Another pathway is formation of oxy-pinocamphone, as discussed above.
The products at the end of the low-volatility tail extending toward the upper left in the top panel of Fig. 12 may contribute to the new-particle formation observed in the CLOUD experiments. These ELVOCs, with log Co<−3.5 are the most likely to form new particles because with constant mass yields the saturation ratio in each progressively less volatile bin will grow by an order of magnitude. The of these LVOC products ranges from 0 to 1, and they represent around 15 % of total SOA mass. This is consistent with CLOUD observations showing that ∼ 10 % of the PD oxidation products could drive new-particle formation (Schobesberger et al., 2013; Riccobono et al., 2014).
Employing the method of Chuang and Donahue (2017), we conducted a dynamical simulation of SOA production following oxidation of 6 ppb PD in the CMU chamber, assuming a mass accommodation coefficient α=1. As shown in Fig. 13, the simulation describes the formation of condensable vapors and subsequent production of SOA mass. The suspended SOA mass in the simulation matches the smog-chamber data very well. The particle mass and SOA vapors lost to the Teflon chamber wall are also comparable with the calculated values from the experimental data. Especially during the first 15 min, the simulation shows there is a large fraction of condensable SOA vapors in the gas phase. This agrees with the observed condensation delay due to the condensation sink timescale.
Our studies show that oxidation of pinanediol, a semi-volatile surrogate for first-generation oxidation products of monoterpenes, can produce SOA with very high mass yields. The SOA is also highly oxidized. This is thus a model system to describe chemical aging of first-generation SOA. Along with previously studied model systems for first-generation products, this shows that aging of semi-volatile SOA is a significant source of additional SOA mass, with higher mass yields typical of less volatile first-generation products. The second-generation oxidation products with sufficiently low volatility represent 15 % of the total SOA mass in a 2D-VBS model that reproduces the chamber data; these may contribute to new-particle formation. The oxidation state of the chamber SOA produced from oxidation of PD is also consistent with the observations during new-particle formation experiments at CERN. Thus, while first-generation oxidation is a substantial source of both SOA mass and new-particle formation, ongoing oxidation of first-generation vapors, which typically comprise the large majority of the first-generation oxidation products from common precursors, should also be considered as a significant source of both particle number and mass.
Data are available from the corresponding author upon request.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-6171-2018-supplement.
The authors declare that they have no conflict of interest.
This research was supported by grant AGS1136479 and AGS1447056, from the
National Science Foundation. The high-resolution aerosol mass spectrometer
was purchased with Major Research Instrumentation funds from NSF CBET0922643
and the Wallace Research Foundation.
Edited by:
Jason Surratt
Reviewed by: two anonymous referees
Chacon-Madrid, H. J. and Donahue, N. M.: Fragmentation vs. functionalization: chemical aging and organic aerosol formation, Atmos. Chem. Phys., 11, 10553–10563, https://doi.org/10.5194/acp-11-10553-2011, 2011.
Chacon-Madrid, H. J., Presto, A. A., and Donahue, N. M.: Functionalization vs. fragmentation: n-aldehyde oxidation mechanisms and secondary organic aerosol formation, Phys. Chem. Chem. Phys., 12, 13975–13982, https://doi.org/10.1039/C0CP00200C, 2010.
Chacon-Madrid, H. J., Murphy, B. N., Pandis, S. N., and Donahue, N. M.: Simulations of smog-chamber experiments using the two-dimensional volatility basis set: linear oxygenated precursors, Environ. Sci. Technol., 46, 11179–11186, https://doi.org/10.1021/es3017232, 2012.
Chacon-Madrid, H. J., Henry, K. M., and Donahue, N. M.: Photo-oxidation of pinonaldehyde at low NOx: from chemistry to organic aerosol formation, Atmos. Chem. Phys., 13, 3227–3236, https://doi.org/10.5194/acp-13-3227-2013, 2013.
Chuang, W. K. and Donahue, N. M.: A two-dimensional volatility basis set – Part 3: Prognostic modeling and NOx dependence, Atmos. Chem. Phys., 16, 123–134, https://doi.org/10.5194/acp-16-123-2016, 2016.
Chuang, W. K. and Donahue, N. M.: Dynamic consideration of smog chamber experiments, Atmos. Chem. Phys., 17, 10019–10036, https://doi.org/10.5194/acp-17-10019-2017, 2017.
Cubison, M. J., Ortega, A. M., Hayes, P. L., Farmer, D. K., Day, D., Lechner, M. J., Brune, W. H., Apel, E., Diskin, G. S., Fisher, J. A., Fuelberg, H. E., Hecobian, A., Knapp, D. J., Mikoviny, T., Riemer, D., Sachse, G. W., Sessions, W., Weber, R. J., Weinheimer, A. J., Wisthaler, A., and Jimenez, J. L.: Effects of aging on organic aerosol from open biomass burning smoke in aircraft and laboratory studies, Atmos. Chem. Phys., 11, 12049–12064, https://doi.org/10.5194/acp-11-12049-2011, 2011.
Donahue, N., Robinson, A., Trump, E., Riipinen, I., and Kroll, J.: Volatility and Aging of Atmospheric Organic Aerosol, in: Atmospheric and Aerosol Chemistry, edited by: McNeill, V. F. and Ariya, P. A., Topics in Current Chemistry, Springer Berlin Heidelberg, 97–143, 2014.
Donahue, N. M., Huff Hartz, K. E., Chuong, B., Presto, A. A., Stanier, C. O., Rosenhorn, T., Robinson, A. L., and Pandis, S. N.: Critical factors determining the variation in SOA yields from terpene ozonolysis: A combined experimental and computational study, Faraday Discuss., 130, 295–309, https://doi.org/10.1039/B417369D, 2005.
Donahue, N. M., Robinson, A. L., Stanier, C. O., and Pandis, S. N.: Coupled Partitioning, Dilution, and Chemical Aging of Semivolatile Organics, Environ. Sci. Technol., 40, 2635–2643, https://doi.org/10.1021/es052297c, 2006.
Donahue, N. M., Epstein, S. A., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set: 1. organic-aerosol mixing thermodynamics, Atmos. Chem. Phys., 11, 3303–3318, https://doi.org/10.5194/acp-11-3303-2011, 2011a.
Donahue, N. M., Trump, E. R., Pierce, J. R., and Riipinen, I.: Theoretical constraints on pure vapor-pressure driven condensation of organics to ultrafine particles, Geophys. Res. Lett., 38, L16801, https://doi.org/10.1029/2011GL048115, 2011b.
Donahue, N. M., Trump, E. R., Pierce, J. R., and Riipinen, I.: Theoretical constraints on pure vapor-pressure driven condensation of organics to ultrafine particles, Geophys. Res. Lett., 38, L16801, https://doi.org/10.1029/2011GL048115, 2011c.
Donahue, N. M., Henry, K. M., Mentel, T. F., Kiendler-Scharr, A., Spindler, C., Bohn, B., Brauers, T., Dorn, H. P., Fuchs, H., Tillmann, R., Wahner, A., Saathoff, H., Naumann, K.-H., Möhler, O., Leisner, T., Müller, L., Reinnig, M.-C., Hoffmann, T., Salo, K., Hallquist, M., Frosch, M., Bilde, M., Tritscher, T., Barmet, P., Praplan, A. P., DeCarlo, P. F., Dommen, J., Prévôt, A. S. H., and Baltensperger, U.: Aging of biogenic secondary organic aerosol via gas-phase OH radical reactions, P. Natl. Acad. Sci. USA, 109, 13503–13508, https://doi.org/10.1073/pnas.1115186109, 2012a.
Donahue, N. M., Kroll, J. H., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set – Part 2: Diagnostics of organic-aerosol evolution, Atmos. Chem. Phys., 12, 615–634, https://doi.org/10.5194/acp-12-615-2012, 2012b.
Donahue, N. M., Chuang, W., Epstein, S. A., Kroll, J. H., Worsnop, D. R., Robinson, A. L., Adams, P. J., and Pandis, S. N.: Why do organic aerosols exist? Understanding aerosol lifetimes using the two-dimensional volatility basis set, Environ. Chem., 10, 151–157, 2013.
Duplissy, J., Merikanto, J., Franchin, A., Tsagkogeorgas, G., Kangasluoma, J., Wimmer, D., Vuollekoski, H., Schobesberger, S., Lehtipalo, K., Flagan, R. C., Brus, D., Donahue, N. M., Vehkamäki, H., Almeida, J., Amorim, A., Barmet, P., Bianchi, F., Breitenlechner, M., Dunne, E. M., Guida, R., Henschel, H., Junninen, H., Kirkby, J., Kürten, A., Kupc, A., Määttänen, A., Makhmutov, V., Mathot, S., Nieminen, T., Onnela, A., Praplan, A. P., Riccobono, F., Rondo, L., Steiner, G., Tome, A., Walther, H., Baltensperger, U., Carslaw, K. S., Dommen, J., Hansel, A., Petäjä, T., Sipilä, M., Stratmann, F., Vrtala, A., Wagner, P. E., Worsnop, D. R., Curtius, J., and Kulmala, M.: Effect of ions on sulfuric acid-water binary particle formation: 2. Experimental data and comparison with QC-normalized classical nucleation theory, J. Geophys. Res.-Atmos., 121, 1752–1775, https://doi.org/10.1002/2015JD023539, 2016.
Ehn, M., Thornton, J. A., Kleist, E., Sipila, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurten, T., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petaja, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Henry, K. M. and Donahue, N. M.: Photochemical aging of α-pinene secondary organic aerosol: Effects of OH radical sources and photolysis, J. Phys. Chem. A, 116, 5932–5940, https://doi.org/10.1021/jp210288s, 2012.
Hildebrandt, L., Donahue, N. M., and Pandis, S. N.: High formation of secondary organic aerosol from the photo-oxidation of toluene, Atmos. Chem. Phys., 9, 2973–2986, https://doi.org/10.5194/acp-9-2973-2009, 2009.
Jang, M. and Kamens, R. M.: Newly characterized products and composition of secondary aerosols from the reaction of α-pinene with ozone, Atmos. Environ., 33, 459–474, 1999.
Jaoui, M. and Kamens, R. M.: Mass balance of gaseous and particulate products analysis from α-pinene/NOx/air in the presence of natural sunlight, J. Geophys. Res.-Atmos., 106, 12541–12558, https://doi.org/10.1029/2001JD900005, 2001.
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005.
Kirkby, J., Curtius, J., Almeida, J., Dunne, E., Duplissy, J., Ehrhart, S., Franchin, A., Gagne, S., Ickes, L., Kurten, A., Kupc, A., Metzger, A., Riccobono, F., Rondo, L., Schobesberger, S., Tsagkogeorgas, G., Wimmer, D., Amorim, A., Bianchi, F., Breitenlechner, M., David, A., Dommen, J., Downard, A., Ehn, M., Flagan, R. C., Haider, S., Hansel, A., Hauser, D., Jud, W., Junninen, H., Kreissl, F., Kvashin, A., Laaksonen, A., Lehtipalo, K., Lima, J., Lovejoy, E. R., Makhmutov, V., Mathot, S., Mikkila, J., Minginette, P., Mogo, S., Nieminen, T., Onnela, A., Pereira, P., Petaja, T., Schnitzhofer, R., Seinfeld, J. H., Sipila, M., Stozhkov, Y., Stratmann, F., Tome, A., Vanhanen, J., Viisanen, Y., Vrtala, A., Wagner, P. E., Walther, H., Weingartner, E., Wex, H., Winkler, P. M., Carslaw, K. S., Worsnop, D. R., Baltensperger, U., and Kulmala, M.: Role of sulphuric acid, ammonia and galactic cosmic rays in atmospheric aerosol nucleation, Nature, 476, 429–433, 2011.
Krechmer, J. E., Pagonis, D., Ziemann, P. J., and Jimenez, J. L.: Quantification of Gas-Wall Partitioning in Teflon Environmental Chambers Using Rapid Bursts of Low-Volatility Oxidized Species Generated in Situ, Environ. Sci. Technol., 50, 5757–5765, https://doi.org/10.1021/acs.est.6b00606, 2016.
Kroll, J. H., Smith, J. D., Che, D. L., Kessler, S. H., Worsnop, D. R., and Wilson, K. R.: Measurement of fragmentation and functionalization pathways in the heterogeneous oxidation of oxidized organic aerosol, Phys. Chem. Chem. Phys., 11, 8005–8014, https://doi.org/10.1039/B905289E, 2009.
Lambe, A. T., Ahern, A. T., Williams, L. R., Slowik, J. G., Wong, J. P. S., Abbatt, J. P. D., Brune, W. H., Ng, N. L., Wright, J. P., Croasdale, D. R., Worsnop, D. R., Davidovits, P., and Onasch, T. B.: Characterization of aerosol photooxidation flow reactors: heterogeneous oxidation, secondary organic aerosol formation and cloud condensation nuclei activity measurements, Atmos. Meas. Tech., 4, 445–461, https://doi.org/10.5194/amt-4-445-2011, 2011.
Matsunaga, A. and Ziemann, P. J.: Gas-Wall Partitioning of Organic Compounds in a Teflon Film Chamber and Potential Effects on Reaction Product and Aerosol Yield Measurements, Aerosol Sci. Tech., 44, 881–892, https://doi.org/10.1080/02786826.2010.501044, 2010.
May, A. A., Saleh, R., Hennigan, C. J., Donahue, N. M., and Robinson, A. L.: Volatility of Organic Molecular Markers Used for Source Apportionment Analysis: Measurements and Implications for Atmospheric Lifetime, Environ. Sci. Technol., 46, 12435–12444, https://doi.org/10.1021/es302276t, 2012.
Müller, L., Reinnig, M.-C., Naumann, K. H., Saathoff, H., Mentel, T. F., Donahue, N. M., and Hoffmann, T.: Formation of 3-methyl-1,2,3-butanetricarboxylic acid via gas phase oxidation of pinonic acid – a mass spectrometric study of SOA aging, Atmos. Chem. Phys., 12, 1483–1496, https://doi.org/10.5194/acp-12-1483-2012, 2012.
Nakao, S., Tang, P., Tang, X., Clark, C. H., Qi, L., Seo, E., Asa-Awuku, A., and Cocker Iii, D.: Density and elemental ratios of secondary organic aerosol: Application of a density prediction method, Atmos. Environ., 68, 273–277, 2013.
Odum, J. R., Hoffmann, T., Bowman, F., Collins, D., Flagan, R. C., and Seinfeld, J. H.: Gas/particle partitioning and secondary organic aerosol yields, Environ. Sci. Technol., 30, 2580–2585, 1996a.
Odum, J. R., Hoffmann, T., Bowman, F., Collins, D., Flagan, R. C., and Seinfeld, J. H.: Gas/Particle Partitioning and Secondary Organic Aerosol Yields, Environ. Sci. Technol., 30, 2580–2585, https://doi.org/10.1021/es950943+, 1996b.
Pandis, S. N., Paulson, S. E., Seinfeld, J. H., and Flagan, R. C.: Aerosol formation in the photooxidation of isoprene and β-pinene, Atmos. Environ., 25A, 997–1008, 1991.
Pope, C. A., Ezzati, M., and Dockery, D. W.: Fine-Particulate Air Pollution and Life Expectancy in the United States, New Engl. J. Med., 360, 376–386, https://doi.org/10.1056/NEJMsa0805646, 2009.
Presto, A. A. and Donahue, N. M.: Investigation of α-Pinene + Ozone Secondary Organic Aerosol Formation at Low Total Aerosol Mass, Environ. Sci. Technol., 40, 3536–3543, https://doi.org/10.1021/es052203z, 2006.
Qi, L., Nakao, S., and Cocker, D. R.: Aging of secondary organic aerosol from α-pinene ozonolysis: Roles of hydroxyl and nitrate radicals, JAPCA J. Air Waste Ma., 62, 1359–1369, https://doi.org/10.1080/10962247.2012.712082, 2012.
Riccobono, F., Schobesberger, S., Scott, C. E., Dommen, J., Ortega, I. K., Rondo, L., Almeida, J., Amorim, A., Bianchi, F., Breitenlechner, M., David, A., Downard, A., Dunne, E. M., Duplissy, J., Ehrhart, S., Flagan, R. C., Franchin, A., Hansel, A., Junninen, H., Kajos, M., Keskinen, H., Kupc, A., Kürten, A., Kvashin, A. N., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Mathot, S., Nieminen, T., Onnela, A., Petäjä, T., Praplan, A. P., Santos, F. D., Schallhart, S., Seinfeld, J. H., Sipilä, M., Spracklen, D. V., Stozhkov, Y., Stratmann, F., Tomé, A., Tsagkogeorgas, G., Vaattovaara, P., Viisanen, Y., Vrtala, A., Wagner, P. E., Weingartner, E., Wex, H., Wimmer, D., Carslaw, K. S., Curtius, J., Donahue, N. M., Kirkby, J., Kulmala, M., Worsnop, D. R., and Baltensperger, U.: Oxidation Products of Biogenic Emissions Contribute to Nucleation of Atmospheric Particles, Science, 344, 717–721, https://doi.org/10.1126/science.1243527, 2014.
Robinson, E. S., Saleh, R., and Donahue, N. M.: Organic aerosol mixing observed by single-particle mass spectrometry, J. Phys. Chem. A, 117, 13935–13945, https://doi.org/10.1021/jp405789t, 2013.
Saleh, R., Donahue, N. M., and Robinson, A. L.: Time Scales for Gas-Particle Partitioning Equilibration of Secondary Organic Aerosol Formed from Alpha-Pinene Ozonolysis, Environ. Sci. Technol., 47, 5588–5594, https://doi.org/10.1021/es400078d, 2013.
Schobesberger, S., Junninen, H., Bianchi, F., Lönn, G., Ehn, M., Lehtipalo, K., Dommen, J., Ehrhart, S., Ortega, I. K., Franchin, A., Nieminen, T., Riccobono, F., Hutterli, M., Duplissy, J., Almeida, J., Amorim, A., Breitenlechner, M., Downard, A. J., Dunne, E. M., Flagan, R. C., Kajos, M., Keskinen, H., Kirkby, J., Kupc, A., Kürten, A., Kurtén, T., Laaksonen, A., Mathot, S., Onnela, A., Praplan, A. P., Rondo, L., Santos, F. D., Schallhart, S., Schnitzhofer, R., Sipilä, M., Tomé, A., Tsagkogeorgas, G., Vehkamäki, H., Wimmer, D., Baltensperger, U., Carslaw, K. S., Curtius, J., Hansel, A., Petäjä, T., Kulmala, M., Donahue, N. M., and Worsnop, D. R.: Molecular understanding of atmospheric particle formation from sulfuric acid and large oxidized organic molecules, P. Natl. Acad. Sci. USA, 110, 17223–17228, https://doi.org/10.1073/pnas.1306973110, 2013.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics – from air pollution to climate change, 2nd Edn., John Wiley & Sons, New York, 2006.
Shilling, J. E., Chen, Q., King, S. M., Rosenoern, T., Kroll, J. H., Worsnop, D. R., DeCarlo, P. F., Aiken, A. C., Sueper, D., Jimenez, J. L., and Martin, S. T.: Loading-dependent elemental composition of a-pinene SOA particles, Atmos. Chem. Phys., 9, 771–782, https://doi.org/10.5194/acp-9-771-2009, 2009.
Solomon, S. Q. D., Manning, M., Alley, R. B., Berntsen, T., Bindoff, N. L.; Chen, Z., Chidthaisong, A., Gregory, J. M., and Hegerl, G. C.: Climate change 2007: The physical science basis, contribution of working group 1 to the fourth assessment report of the Intergovernmental Panel on Climate Change, 2007.
Tkacik, D. S., Lambe, A. T., Jathar, S., Li, X., Presto, A. A., Zhao, Y., Blake, D., Meinardi, S., Jayne, J. T., Croteau, P. L., and Robinson, A. L.: Secondary Organic Aerosol Formation from in-Use Motor Vehicle Emissions Using a Potential Aerosol Mass Reactor, Environ. Sci. Technol., 48, 11235–11242, https://doi.org/10.1021/es502239v, 2014.
Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., Wagner, R., Simon, M., Lehtipalo, K., Williamson, C., Craven, J. S., Duplissy, J., Adamov, A., Almeida, J., Bernhammer, A.-K., Breitenlechner, M., Brilke, S., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Gysel, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Keskinen, H., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lawler, M., Leiminger, M., Mathot, S., Möhler, O., Nieminen, T., Onnela, A., Petäjä, T., Piel, F. M., Miettinen, P., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Sipilä, M., Smith, J. N., Steiner, G., Tomè, A., Virtanen, A., Wagner, A. C., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Carslaw, K. S., Curtius, J., Dommen, J., Kirkby, J., Kulmala, M., Riipinen, I., Worsnop, D. R., Donahue, N. M., and Baltensperger, U.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 533, 527–531, https://doi.org/10.1038/nature18271, 2016.
Trump, E. R., Riipinen, I., and Donahue, N. M.: Interactions between atmospheric ultrafine particles and secondary organic aerosol mass: a model study, Boreal Environ. Res., 19, 352–362, 2014.
Trump, E. R., Epstein, S. A., Riipinen, I., and Donahue, N. M.: Wall effects in smog chamber experiments: A model study, Aerosol Sci. Technol., 50, 1180–1200, https://doi.org/10.1080/02786826.2016.1232858, 2016.
Wong, J. P. S., Lee, A. K. Y., Slowik, J. G., Cziczo, D. J., Leaitch, W. R., Macdonald, A., and Abbatt, J. P. D.: Oxidation of ambient biogenic secondary organic aerosol by hydroxyl radicals: Effects on cloud condensation nuclei activity, Geophys. Res. Lett., 38, L22805, https://doi.org/10.1029/2011GL049351, 2011.
Ye, P., Ding, X., Hakala, J., Hofbauer, V., Robinson, E. S., and Donahue, N. M.: Vapor wall loss of semi-volatile organic compounds in a Teflon chamber, Aerosol Sci. Tech., 50, 822–834, https://doi.org/10.1080/02786826.2016.1195905, 2016a.
Ye, P., Ding, X., Ye, Q., Robinson, E. S., and Donahue, N. M.: Uptake of Semivolatile Secondary Organic Aerosol Formed from α-Pinene into Nonvolatile Polyethylene Glycol Probe Particles, J. Phys. Chem. A, 120, 1459–1467, https://doi.org/10.1021/acs.jpca.5b07435, 2016b.
Ye, Q., Robinson, E. S., Ding, X., Ye, P., Sullivan, R. C., and Donahue, N. M.: Mixing of secondary organic aerosols versus relative humidity, P. Natl. Acad. Sci. USA, 113, 12649–12654, https://doi.org/10.1073/pnas.1604536113, 2016c.
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Allan, J. D., Coe, H., Ulbrich, I., Alfarra, M. R., Takami, A., Middlebrook, A. M., Sun, Y. L., Dzepina, K., Dunlea, E., Docherty, K., DeCarlo, P. F., Salcedo, D., Onasch, T., Jayne, J. T., Miyoshi, T., Shimono, A., Hatakeyama, S., Takegawa, N., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Williams, P., Bower, K., Bahreini, R., Cottrell, L., Griffin, R. J., Rautiainen, J., Sun, J. Y., Zhang, Y. M., and Worsnop, D. R.: Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes, Geophys. Res. Lett., 34, L13801, https://doi.org/10.1029/2007GL029979, 2007.
Zhang, X., Schwantes, R. H., McVay, R. C., Lignell, H., Coggon, M. M., Flagan, R. C., and Seinfeld, J. H.: Vapor wall deposition in Teflon chambers, Atmos. Chem. Phys., 15, 4197–4214, https://doi.org/10.5194/acp-15-4197-2015, 2015.
Zhao, Y., Hennigan, C. J., May, A. A., Tkacik, D. S., de Gouw, J. A., Gilman, J. B., Kuster, W. C., Borbon, A., and Robinson, A. L.: Intermediate-Volatility Organic Compounds: A Large Source of Secondary Organic Aerosol, Environ. Sci. Technol., 48, 13743–13750, https://doi.org/10.1021/es5035188, 2014.