the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 21 Feb 2018
| 21 Feb 2018
Understanding in situ ozone production in the summertime through radical observations and modelling studies during the Clean air for London project (ClearfLo)
Lisa K. Whalley
Daniel Stone
Rachel Dunmore
Jacqueline Hamilton
James R. Hopkins
James D. Lee
Alastair C. Lewis
Paul Williams
Jörg Kleffmann
Sebastian Laufs
Robert Woodward-Massey
Measurements of OH, HO2, RO2i (alkene and aromatic-related RO2) and total RO2 radicals taken during the ClearfLo campaign in central London in the summer of 2012 are presented. A photostationary steady-state calculation of OH which considered measured OH reactivity as the OH sink term and the measured OH sources (of which HO2+ NO reaction and HONO photolysis dominated) compared well with the observed levels of OH. Comparison with calculations from a detailed box model utilising the Master Chemical Mechanism v3.2, however, highlighted a substantial discrepancy between radical observations under lower NOx conditions ([NO] < 1 ppbv), typically experienced during the afternoon hours, and indicated that the model was missing a significant peroxy radical sink; the model overpredicted HO2 by up to a factor of 10 at these times. Known radical termination steps, such as HO2 uptake on aerosols, were not sufficient to reconcile the model–measurement discrepancies alone, suggesting other missing termination processes. This missing sink was most evident when the air reaching the site had previously passed over central London to the east and when elevated temperatures were experienced and, hence, contained higher concentrations of VOCs. Uncertainties in the degradation mechanism at low NOx of complex biogenic and diesel related VOC species, which were particularly elevated and dominated OH reactivity under these easterly flows, may account for some of the model–measurement disagreement. Under higher [NO] (> 3 ppbv) the box model increasingly underpredicted total [RO2]. The modelled and observed HO2 were in agreement, however, under elevated NO concentrations ranging from 7 to 15 ppbv.
The model uncertainty under low NO conditions leads to more ozone production predicted using modelled peroxy radical concentrations (∼ 3 ppbv h−1) versus ozone production from peroxy radicals measured (∼ 1 ppbv h−1). Conversely, ozone production derived from the predicted peroxy radicals is up to an order of magnitude lower than from the observed peroxy radicals as [NO] increases beyond 7 ppbv due to the model underprediction of RO2 under these conditions.
- Article
(8217 KB) - Full-text XML
-
Supplement
(989 KB) - BibTeX
- EndNote





With more than 50 % of the global population residing in urban conurbations, poor urban air quality has a demonstrable effect on human health. OH and HO2 radicals (collectively termed HOx), together with RO2 radicals, mediate virtually all of the oxidative chemistry in the atmosphere. The hydroxyl radical initiates the removal of primary emissions, including toxic gases such as CO and benzene, leading to the formation of peroxy radicals which, in the presence of NO, form secondary pollutants such as NO2, O3 and particulates. Public Health England (2014) reports that pollutants contribute to 29 000 deaths a year in the UK, with a reduction in life expectancy (by an average of 6 months) caused by long-term exposure to pollutants, and the cost to society is estimated at up to GBP 20 billion per year. In areas of London up to 1 in 12 deaths are at least partly attributable to air pollution, yet big uncertainties still remain relating to the chemistry, transformation and removal rate of primary emissions in large urban conurbations, meaning our ability to predict pollution episodes is compromised.
The EU air quality guidelines recommend that ozone concentrations do not exceed 60 ppbv for longer than an 8 h period (http://www.eea.europa.eu/themes/air/ozone), with a 10 ppbv increment in long-term exposure to ozone increasing the risk of death from respiratory causes by ∼ 3–4 % (Jerrett et al., 2009). Short-term exposure to elevated levels of tropospheric ozone have been associated with several adverse health effects including, for example, exacerbation of asthma in children (Thurston et al., 1997).
Despite successful reductions in many ozone precursors across Europe, ozone levels have increased at certain urban sites due to the long-term decrease in NOx emissions. For example, Bigi and Harrison (2010) report a steady increase in ozone between 1996 and 2008 in North Kensington, an urban background site in London.
To implement efficient reduction strategies for ozone, a detailed understanding of the factors controlling free radicals is critical since the reaction of HO2 and RO2 radicals with NO, forming NO2, followed by the subsequent photolysis of NO2 represents the only net formation pathway to tropospheric ozone:
Measurements of radicals have been made at various urban and suburban locations worldwide, both during the summer and winter (Stone et al. (2012) and references therein). Observations of OH and HO2 in the urban atmosphere have primarily been made using fluorescence assay by gas expansion (FAGE), and comparisons with predicted radical concentrations using chemistry box models constrained with co-located radical precursor measurements have revealed varying levels of success in replicating observations. Radical concentrations have been reported to be underpredicted by models (Ren et al., 2003; Martinez et al., 2003; Emmerson et al., 2005a; Chen et al., 2010; Lu et al., 2012, 2013), overpredicted (George et al., 1999; Konrad et al., 2003; Dusanter et al., 2009) and, at times, models and measurements have been reported to be in reasonable agreement, to within 40 % (Shirley et al., 2006; Emmerson et al., 2007; Kanaya et al., 2007; Sheehy et al., 2010; Elshorbany et al., 2012; Ren et al., 2013; Griffith et al., 2016). Often the level of agreement observed was found to be dependent on the time of day (Brune et al., 2016), with poorest agreement between modelled and measured OH concentrations generally observed at night. Griffith et al. (2016) found that the level of agreement between modelled and measured HO2 was dependent on whether it was a weekday or weekend: the model underpredicted HO by a factor of 3.4 during the week, when NO mixing ratio were greater than 4 ppbv but agreed well on weekends (observed to modelled HO) when NO concentrations were below 4 ppbv (where HO = [HO2] [RO2i], and αi is the mean fractional contribution of the RO2 species that interfere (RO2i)). In a number of studies, the model–measurement discrepancy was noted to increase as NOx levels increased beyond ∼ 1 ppbv (Martinez et al., 2003; Ren et al., 2013; Brune et al., 2016). This increasing underprediction of the free radicals (particularly for HO2) with increasing observed NOx concentrations may reflect inaccuracies in the radical propagation steps in the model, which cycle HO2 to OH. In light of the recently reported RO2 interference suffered by FAGE (Fuchs et al., 2011; Whalley et al., 2013) when detecting HO2, however, it is possible that the measured HO2 under these conditions may have been increasingly influenced by the presence of RO2 species. The extent of this interference will be dependent upon the level of interference suffered by the specific FAGE instrument utilised and the concentration of those RO2 species that interfere (principally aromatic, alkene and > C3 alkane-derived RO2 species) and were present in a particular environment. Similarly, two FAGE groups have reported interferences in their OH measurements made using wavelength modulation in the presence of ambient levels of ozone and alkenes (Mao et al., 2012; Novelli et al., 2014), whilst, in contrast, good agreement between OH measurements made using FAGE and differential optical absorption spectroscopy (DOAS) during chamber measurements suggests minimal interferences in the presence of ozone and alkenes for a third FAGE instrument (Fuchs et al., 2013). This lack of interference was further corroborated in recent laboratory tests (Fuchs et al., 2016), although an artefact signal under dark conditions (deriving from NO3 in the presence of H2O) was identified. These potential artefacts make it difficult to identify trends in earlier model–measurement comparisons and to assess how well the models are performing under a range of chemical conditions. Some of the more recently published radical measurements at urban sites include corrections for OH interferences (e.g. Ren et al., 2013; Brune et al., 2016; Griffith et al., 2016) and radical measurements from the MEGAPOLI project, which took place at a suburban site close to Paris employed the chemical ionisation mass spectrometry (CIMS) technique to make observations of OH and the sum of HO2 and RO2 species rather than FAGE (Michoud et al., 2012). HO model–measurement comparisons are now often reported (rather than HO2) to take into account contributions from RO2 species (Lu et al., 2013; Griffith et al., 2016) and very recently Tan et al. (2017) presented interference-free HO2 observations alongside RO2 observations which were made using the FAGE technique coupled to a flow reactor (Fuchs et al., 2008) at a rural site in Wangdu, China. In contrast to some of the earlier HO2 model–measurement comparisons which diverged at NO concentrations > 1ppbv with models increasingly underpredicting the levels of observed HO2, the predicted levels of HO2 were in good agreement with HO2 observations made in Wangdu over the whole range (0.1–4 ppbv) of NO encountered (Tan et al., 2017). However, the authors did report an increasing model underprediction of the observed RO2 with increasing NO (Tan et al., 2017).
Despite uncertainties in some of the earlier radical observations and discrepancies between observed and predicted radical concentrations, detailed modelling studies have demonstrated a number of common themes relevant to urban photochemistry:
-
The primary source of OH from the photolysis of ozone and subsequent reaction of the excited state oxygen atom with H2O, which is often considered the dominant radical source in many other environments (e.g in the remote marine atmosphere, Whalley et al., 2010), tends to only play a minor role in urban centres, with this source accounting for < 6 % of the total radical sources during MCMA-2006 campaign (Dusanter et al., 2009), which took place in Mexico City.
-
Owing to the prevalence of carbonyl and dicarbonyl species in the urban atmosphere, a number of studies have highlighted the role that the photolysis of these species play as key radical precursors (and, hence, ozone precursors) in the summertime: during the SHARP-2009 project that took place in Houston, Texas, the photolysis of formaldehyde accounted for 14 % of radical production, with the photolysis of other OVOCs contributing a further 15 % (Ren et al., 2013). During the CAREBeijing-2006, HCHO was estimated to contribute ∼ 30 % to the overall radical production (Lu et al., 2013).
-
Ozonolysis reactions have been reported as important primary radical sources in a number of studies, for example accounting for 67 % of the OH initiation in Birmingham during the PUMA campaign in winter (Emmerson et al., 2005b), whilst in Tokyo during the IMPACT campaign ozonolysis reactions were the dominant radical source at night-time in winter (Kanaya et al., 2007).
-
The photolysis of HONO, which takes place at longer wavelengths than ozone photolysis, has been demonstrated to act as an important OH source in the morning (Kleffmann, 2007). At urban sites (including London), significant concentrations of HONO (often several hundred pptv) have been reported to persist throughout the day (Lee et al., 2016), and so HONO should be considered an important OH source throughout sunlit hours, and not just at sunrise, in these environments. Dusanter et al. (2009) found that HONO photolysis contributed 35 % of daytime HOx production in Mexico City during MCMA-2006, whilst Tan et al. (2017) found that HONO photolysis was the most important primary radical source in Wangdu in the North China Plain.
There have now been several observations of total OH reactivity (kOH) in urban environments with some of the highest reactivities of > 120 s−1 recorded in megacities such as Mexico City, London and Paris. In many of the large cities (Houston, New York City, Mexico City), OH reactivity has been found to be dominated by anthropogenic hydrocarbons, CO and NOx. OVOCs have been highlighted as significant OH sinks in a number of urban studies, contributing between 11 and 24 % during summertime at these urban centres (Mao et al., 2010b), whilst we recently reported that the oxidation products of biogenic emissions contributed a significant fraction to the total OH reactivity observed in London (Whalley et al., 2016). A measurement of OH reactivity can provide an additional model target, with model–measurement comparisons helping to identify unmeasured primary emissions or unmeasured oxidised intermediates which may promote radical propagation. Furthermore, when coupled with OH (and HO2) observations, the closure of OH production (POH) and OH loss (DOH =kOH[OH]) terms can be critically assessed independent of a model. In an urban atmosphere the dominant OH sources include recycling from HO2+ NO, HONO photolysis, O(1D) (from ozone photolysis) +H2O and ozonolysis reactions. In the recent study in the Wangdu region of China, POH was found to equal DOH within uncertainties throughout the day (Tan et al., 2017), demonstrating consistency between the observed radical concentrations and observed OH reactivity (Fuchs et al., 2017). Several previous studies in urban regions, however, have found that POH is balanced by DOH during the afternoon but not in the mornings, with measured POH approximately twice DOH from sunrise to noon (Brune et al., 2016). This imbalance of POH and DOH suggests either a negative bias of OH reactivity measurements, an error in the HO2 measurement or uncertainties in the chemistry of other key OH sources, e.g. HONO (Brune et al., 2016).
Urban radical measurements can be used to estimate local ozone production (Kanaya et al., 2007; Ren et al., 2013; Brune et al., 2016) by approximating the rate of ozone production to the production rate of NO2 from the reaction of NO with HO2 and RO2 radicals and assuming instantaneous O3 production following photolysis of NO2 at wavelengths < 400 nm. Any loss of NO2 which does not yield O3, for example the reaction of OH or RO2 radicals with NO2, and also deposition, should also be considered:
Given the short lifetime of the radicals, this estimate provides a method of gauging the extent to which the fast local chemistry influences the net ozone levels observed relative to O3 generated during transport. A shortcoming of this approach in earlier studies is that often the RO2 concentration used in Eq. (1) is estimated or modelled, as traditionally the FAGE technique measures OH and HO2 only. In the Wangdu study, however, Tan et al. (2017) using observed RO2 demonstrated that models may underpredict ozone production at high NO due to an underestimation of the RO2 radical concentration.
In the present paper we utilise observations of OH and HO2 radicals made using the FAGE technique and RO2 radicals using the ROxLIF method (Whalley et al., 2013). The radical observations were made during the Clean air for London project (ClearfLo) during the summer of 2012 and are used to directly determine local ozone production. To assess the factors controlling the radical budget and in turn ozone production, we have employed a detailed box model based on the MCM v3.2 (Master Chemical Mechanism). By comparing model predictions to radical observations the key reactions taking place in London that are ultimately controlling the air quality are identified and uncertainties in our current understanding of urban oxidation chemistry are highlighted.
2.1 Site description
The ClearfLo intensive operation period (IOP) ran from 22 July to 18 August and overlapped with the London 2012 summer Olympics. An extensive suite of instrumentation was deployed and operated from the grounds of Sion Manning School in North Kensington (51∘31′16 N, 0∘12′48 W), which is located adjacent to a long-term air quality monitoring site in North Kensington (Bigi and Harrison, 2010). Further details on the campaign and location may be found in Bohnenstengel et al. (2015).
2.2 FAGE instrument description
The University of Leeds ground-based FAGE instrument was deployed to the North Kensington site and made measurements of OH, HO2 and RO2 radicals. Further details on the instrument for OH and HO2 detection can be found in Whalley et al. (2010) with only an outline of the specific set-up and running conditions during ClearfLo described here. The radical measurements were made from a 20 ft air-conditioned shipping container which had been converted into a mobile laboratory. The instrument consists of two FAGE detection cells which were located on the roof of the shipping container, in a weatherproof housing, at a height of 3.5 m. A Nd : YAG pumped Ti : Sapphire laser (Photonics Industries) generated pulsed (repetition rate of 5 kHz), tunable near-IR radiation, which was doubled in frequency and tripled to provide UV light at 308 nm and was used to excite OH via the Q1(1) transition of the A2Σ+, , = 0 band. On-resonance fluorescence was detected using a gated channel photomultiplier and photon counting for a period of 300 s. The laser was then scanned beyond the OH transition (by 0.004 nm) and a background signal collected for a further 75 s to determine the contribution of laser, solar scatter and detector noise to the total signal for subtraction (OHWAVE).
In previous configurations, the two detection cells were used to simultaneously detect OH by laser-induced fluorescence (LIF) (cell 1) and HO2 by NO titration to OH followed by LIF (cell 2). The UV laser light was split upon exiting the laser and focussed into fibre optics (5 m length) for delivery to each cell individually. During ClearfLo, the two cells were coupled together via a connecting side arm, which enabled light exiting cell 1 to pass into cell 2 and meant that light previously needed for the detection of HO2 in cell 2 could be used for other applications (for example OH reactivity measurements, as was the case during this deployment Stone et al., 2016). As in previous configurations, the light exiting the fibre optic passed through a collimator coupled to a baffled entrance arm. This arrangement produced a beam profile of ∼ 1 cm diameter, which remained well collimated as it passed through both cells. A UV anti-reflective coated window was placed in the centre of the connecting arm to effectively seal the cells from each other. A further modification to the previously deployed configuration involved the coupling of a flow reactor to detection cell 2 to enable an RO2 radical measurement. Further details on this approach are outlined below. Consequently, cell 1 was used for sequential measurements of OH and HO2, with NO (BOC, 99.5 %) injected into this cell during the second half of the online detection period.
2.3 ROxLIF description
An 83 cm long, 6.4 cm internal diameter flow reactor was coupled vertically to the second FAGE detection cell to facilitate detection of RO2 radicals by LIF using the approach described by Fuchs et al. (2008). This flow reactor was held at approximately 30 Torr, with ∼ 7.5 SLM ambient air drawn into the reactor via a 1 mm diameter pinhole. The flow reactor was operated in two modes. In the first, referred to as the HOx mode, 250 sccm CO (BOC, 5 % in N2) was mixed with the ambient air close to the inlet to promote conversion of ambient OH to HO2. In the second, referred to as the ROx mode, 25 sccm of NO in N2 (BOC, 500 ppmv) was also added to the CO flow, which led to the conversion of RO2 to OH. The CO present rapidly reconverted any OH formed (or any OH sampled) to HO2. Air (5 SLM) sampled by the flow reactor was transferred into the FAGE fluorescence detection cell (which was held at ∼ 1.5 Torr) via a 4 mm diameter pinhole and 100 sccm NO (BOC, 99.5 %) was injected into the fluorescence cell, converting HO2 to OH for subsequent detection by LIF. In ROx mode a measure of OH + HO2+ ΣRO2 was obtained.
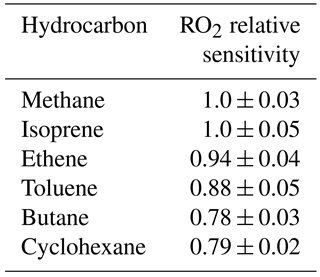
In laboratory tests, the relative sensitivity of the instrument to a range of different RO2 species was investigated (see Table 1). Similar sensitivities were determined for the RO2 species tested; therefore, we use the assumption that under ambient conditions individual RO2 species are converted and, hence, detected with the same efficiency as methane-derived RO2 radicals. The same assumption was drawn in the recent ROx study in Wangdu, China (Tan et al., 2017). This assumption means that the concentration of RO2 observed may be a lower estimate as certain RO2 species will not convert as efficiently as the methane-derived RO2 radical. For example, the MCM predicts that only ∼ 20 % of NO3-adduct RO2 radicals which derive from the reaction of simple alkenes (e.g. ethene and propene) with NO3 will convert to HO2 in the presence of NO at the reduced pressures of the flow reactor and so we expect ROxLIF to have low sensitivity to these RO2 types.
Table 1Relative efficiency of RO2 to HOx conversion for different RO2 species.
2.4 Calibration
The instrument was calibrated twice weekly on average using photolysis of a known concentration of water vapour at 185 nm within a turbulent flow tube to generate OH and HO2, with the product of the photon flux at 185 nm and the water vapour photolysis time measured using a chemical actinometer (Commane et al., 2010). For RO2, methane (BOC, CP grade, 99.5 %) was added to the humidified airflow in sufficient quantity to rapidly convert OH to CH3O2. The limit of detection (LOD) at a signal-to-noise ratio of one for one data acquisition cycle lasting 7 min was ∼ 4.5 × 105 molecule cm−3 for OH, ∼ 2.1 × 106 molecule cm−3 for HO2 and ∼ 6.9 × 106 molecule cm−3 for CH3O2 at a typical laser power of 13 mW in each cell. The measurements were recorded with 1 s time resolution, and the accuracy of the measurements was ∼ 26 % (2σ).
2.5 Potential radical artefacts and corrections
2.5.1 OH
A small OH artefact signal (OHINT) which is derived from photolysis of O3 by the 308 nm laser light, followed by the abstraction of an H atom from H2O vapour within the FAGE cell, has been observed in laboratory tests. This artefact has been observed in other FAGE systems (Griffith et al., 2016; Fuchs et al., 2016; Tan et al., 2017), and although the reported magnitude of the interference is variable for different systems, the signal scales linearly with both O3 and H2O and displays a quadratic dependence with laser power. The following correction has been applied to the OH data presented here which corresponds to 5.2 × 105 molecule cm−3 of OH at 50 ppbv O3, 2 % H2O and 10 mW laser power (determined after the campaign but under the same experimental conditions):
where
It should be noted that in later laboratory tests on the Leeds FAGE system with a modified nozzle design, the determined OHINT was slightly lower than reported here. Fuchs et al. (2016) also report a variable artefact signal for the Jülich FAGE system. This variability introduces a high level of uncertainty into this correction. The OHINT presented here should likely be considered an upper limit as any increase in the magnitude of this correction would lead to negative OH concentrations calculated during night-time periods.
Along with a full characterisation of the O3-H2O OH artefact signal, the Leeds FAGE system has subsequently been characterised with respect to other potential artefact signals, for example, an artefact deriving from reaction products of ozone and alkenes. Furthermore, in the most recent field campaigns, an inlet pre-injector (IPI) has been used to chemically scavenge ambient OH, and provides an alternative method for determining background signals (to generate OHCHEM) alongside the wavelength tuning approach discussed above (OHWAVE). The laboratory interference tests and field comparison of OHCHEM and OHWAVE in different environments will be the subject of a future publication (Woodward-Massey et al., 2018). In general, however, good agreement between OHCHEM and OHWAVE has been observed for the Leeds FAGE instrument (including during ambient measurements conducted in another urban environment in central Beijing with an OHWAVE to OHCHEM ratio of 1.04 and 1.07 in winter and summer respectively, Woodward-Massey et al., 2017) and no significant artefact signal was observed in the interference tests conducted to date, providing confidence in the OH measurements presented here.
2.5.2 HO2 and RO2i
Fuchs et al. (2011) and later Whalley et al. (2013) identified that specific RO2 radical classes (primarily those derived from alkene and aromatic hydrocarbons, defined here as RO2i) have the potential to decompose into OH in the presence of NO under typical FAGE cell conditions and, as a result, may be classed as an HO2 interference. Depending on the type of FAGE cell, pressures employed, and NO concentration used, the level of interference can be deliberately varied (Whalley et al., 2013). During ClearfLo, two different NO concentrations (1.0 and 9.0 × 1013 molecule cm−3) were introduced into cell 1 to promote detection of (a) mainly HO2 under low concentrations of added NO, and (b) HO2+ RO2i under high concentrations of added NO. With knowledge of the sensitivity to HO2 and RO2i at the two added NO concentrations, determined by adding a known concentration of HO2 and ethene-derived RO2 during calibration, and using the methodology outlined in Whalley et al. (2013), the concentration of RO2i and interference-free HO2 can be determined. In the following Results and Discussions we compare RO2i derived from measurements using αi= 0.72 ± 0.09 and αi= 0.19 ± 0.09 at the high and low NO flows to modelled RO2i.
2.5.3 RO2
Fuchs et al. (2008) described the potential of peroxy nitric acid and methyl peroxy nitric acid, HO2NO2 and CH3O2NO2 (the concentration of which will be most elevated at high NOx) to thermally decompose in the ROx-LIF flow reactor. In this urban setting, the RO2 signal that we attribute solely to non-interfering RO2 species (RO2ni) (determined by subtracting HO2+ RO2i measured in cell 1 from the total RO2 signal measured by ROxLIF in ROx mode) may also include a contribution from CH3O2NO2. Here, we refer to the measurement of non-interfering RO2 species (RO2ni), which includes a contribution from the thermal decomposition of CH3O2NO2 as RO2ni∗. If the concentration of RO2ni is dominated by CH3O2, it is possible to estimate the ambient concentration of CH3O2NO2 from the radical measurements themselves and, thus, make a correction for this artefact without relying on model predictions of CH3O2NO2. The methodology for this correction is outlined in the Supplement along with the RO2ni data corrected for this potential artefact. Owing to the unknown fraction of the total RO2ni that is CH3O2, we have left the data uncorrected in Sects. 3 and 4 below. It is worth noting, however, that this correction is most significant when NO concentrations peak during the morning, a time (as discussed in Sect. 3.3.2) at which the RO2ni observations are underestimated by model predictions.
Table 2Listing of the concurrent measurements made during ClearfLo.
2.6 Model description
A zero-dimensional box model based on the MCM v3.2 (Jenkin et al., 2012) was used to predict radical concentrations for comparison with those observed. Complete details of the kinetic and photochemical data used in the mechanism are available at the MCM website (http://mcm.leeds.ac.uk/MCMv3.2/). The model was run with a subset of the MCM and treated the degradation of simultaneously measured trace VOCs, CH4 and CO following oxidation by OH, O3 and NO3, and included ∼ 15 000 reactions and ∼ 3800 species. The model was constrained by measurements of NO, NO2, O3, CO and CH4; 62 individual VOC species measured by GC-FID; and also 2D-GC PAN, HCHO, HNO3, HONO, water vapour, temperature and pressure. The model was constrained with measured j(O1D), j(NO2), j(HONO), j(HCHO), j(CH3COCH3) and j(CH3CHO) made using a spectral radiometer. For further instrumental details relating to all the model constraints please refer to Table 2 (and the references therein). For all other photolabile species in the model, photolysis rates were scaled to the ratio of clear-sky j(O1D), calculated using a two-steam isotropic scattering model (Hayman, 1997), to observed j(O1D) to account for clouds. A constant H2 concentration of 500 ppbv was assumed (Forster et al., 2012). The model inputs were updated every 15 min. For species measured more frequently, data were averaged to 15 min intervals, whilst those measured at a lower time resolution were interpolated. The loss of all non-constrained, model-generated species by deposition or mixing was represented as a first-order loss rate equivalent to 1 cm s−1 in a boundary layer depth which varied from ∼ 300 m at night to 1800 m in the afternoon (estimated from vertical velocity variance, Barlow et al., 2015) leading to lifetimes of ∼ 8 h at night and ∼ 50 h during the afternoons.
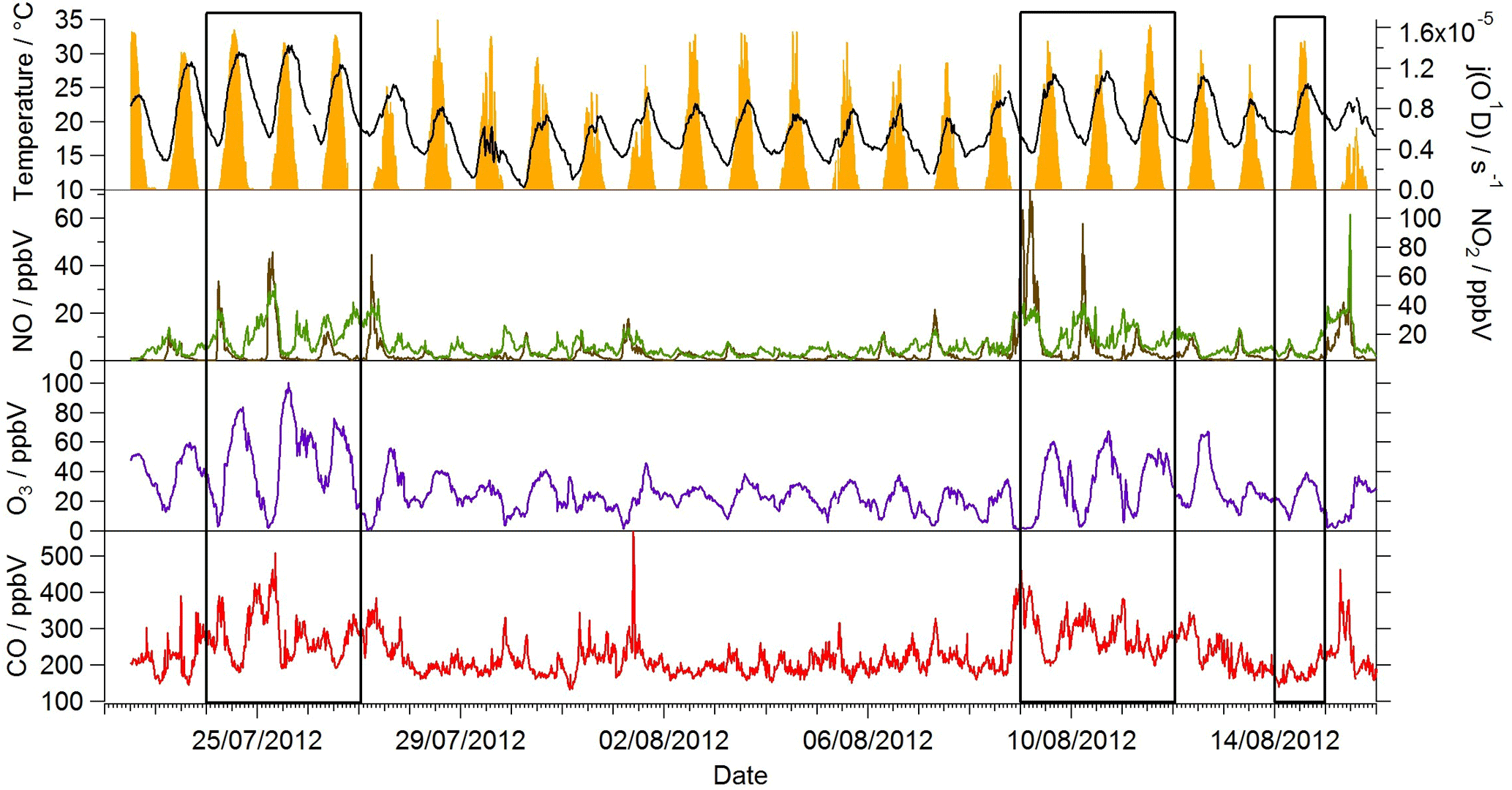
Figure 1Observed temperature (black line), j(O1D) (yellow area), NO (brown line), NO2 (green line), O3 (purple line) and CO (red line) mixing ratios during the summer ClearfLo IOP. Data time resolution is 15 min. Periods of easterly flow are highlighted with the black boxes.
The model was run for the entirety of the campaign in overlapping 7-day segments. To allow all the unmeasured, model-generated intermediate species time to reach steady-state concentrations, the model was initialised with inputs from the first measurement day (23 July) and spun-up for 5 days before comparison to measurements were made. Comparison of these 5 spin-up days demonstrated that the concentration of model-generated species rapidly converged and there was less than a 1 % difference in (for example) modelled OH concentration by the second spin-up day. As a result of this, the model segments were run so as to overlap for 2 days only to reduce the computing time.
In all model scenarios a first of loss ( for HO2 was included to represent heterogeneous removal (Ravishankara, 1997):
where ω is the mean molecular speed of HO2 (equal to 43 725 cm s−1 at 298 K), γ is the aerosol uptake coefficient and A is the aerosol surface area density in cm2 cm−3. A is calculated using data from an aerodynamic particle sizer instrument (TSI Inc, model 3321) which counts particles in 53 size bins ranging from 0.53 to 21.29 µm. For most of the scenarios considered γ was held constant at 0.1.
A series of distinct model scenarios were simulated to assess the sensitivity of the modelled radical concentrations to a number of model parameters. In the following results and discussions the radical measurements are compared (for the most part) to the base model scenario (MCM-BASE), which was run with the constraints outlined above.
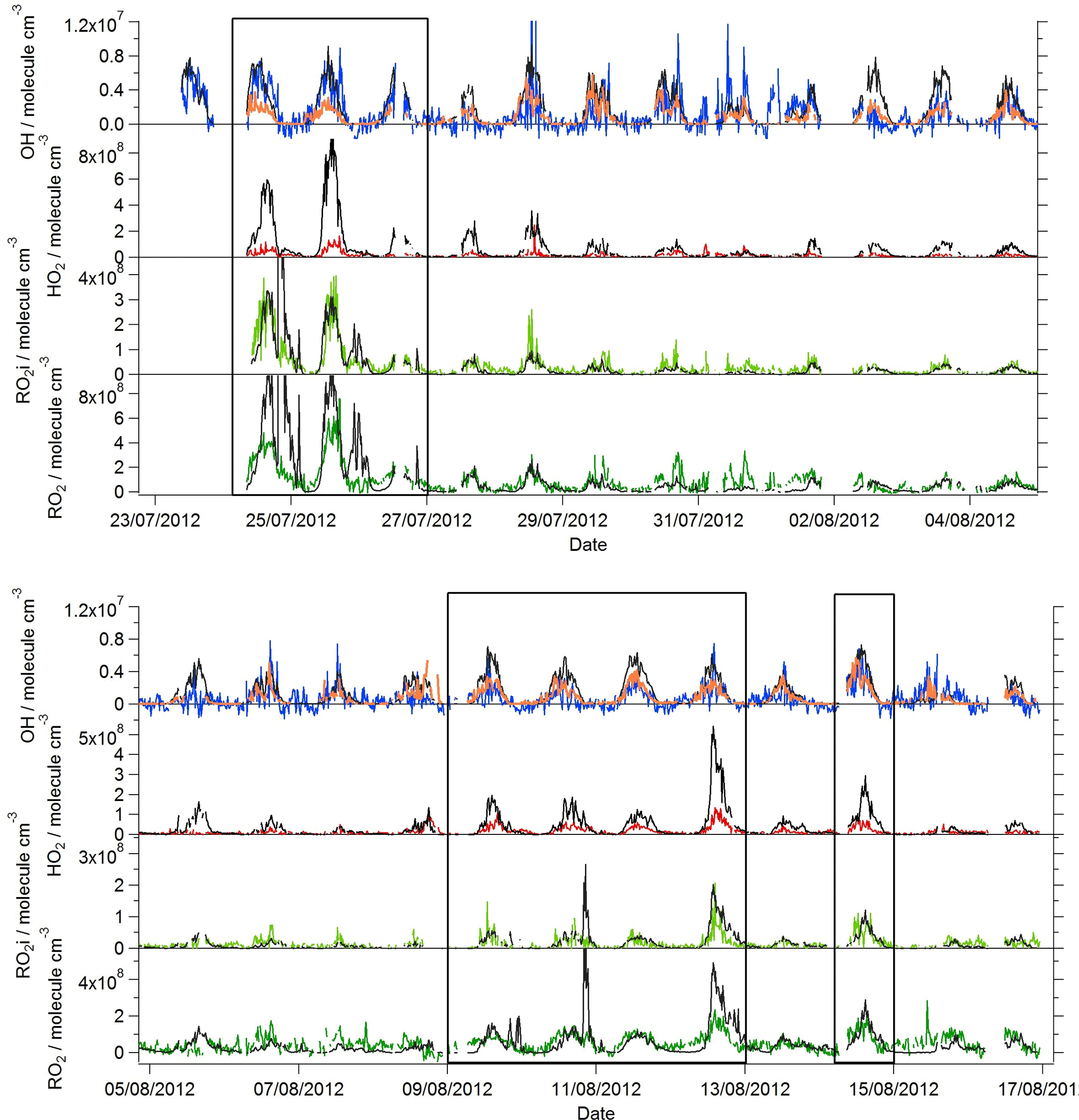
Figure 2Observed (coloured lines) and MCM-BASE modelled (black lines) OH, HO2, RO2i and RO2 during the summer ClearfLo IOP; steady-state [OH] ([OH]PSS) is displayed by the orange line. Periods of easterly flow are highlighted inside the black boxes.
3.1 Radical observations and model predictions during the summer ClearfLo IOP
Near-continuous radical measurements were made in London from 23 July to 17 August 2012. Typically, winds from the south-west, ranging from less than 1 ms−1 at night to between 4 and 6 ms−1 in the afternoon, were encountered. Close to the start of the campaign (24–27 July) and also later in the campaign (9–12, 14 August), however, the wind direction switched to an easterly flow, bringing air that had passed over central London to the site, and wind speeds dropped. Fine weather prevailed during these easterly flows, with enhancements in air temperature and solar radiation (Fig. 1, top panel). During these periods, radical concentrations (particularly the peroxy radicals) were elevated; the time series of OH, HO2, RO2i, and the sum of RO2 species (not including a HO2 contribution) is presented in Fig. 2. The concentration of a number of other species such as NOx, CO and O3 were also elevated (Fig. 1) during the easterly flows. Indeed, the concentration of the O3 was observed to increase rapidly on the warmer days from sunrise, peaking during the afternoon at concentrations between 60 and 100 pbbv, and was found, on the 25 July, to exceed EU air quality recommendations of 60 ppbv for greater than a 6 h period. The average diurnal profiles of the different radicals during south-westerly and easterly flows are presented in Fig. 3.
The short lifetime of OH (10–100 ms) measured directly in London from OH reactivity measurements (Whalley et al., 2016) dictates that OH exists in a photostationary steady state (PSS), where the rate of OH production is balanced by the rate of OH destruction (f is the fraction of O(1D) that reacts with H2O to form OH):
[OH]PSS may be estimated from the rate of production from the sum of co-measured OH sources; here the rate of OH production from the rate of reaction of HO2 with NO, HONO photolysis, O3 photolysis and the subsequent reaction of O1D with H2O vapour yielding two OH radicals, and ozonolysis is considered. The measured total OH reactivity (Whalley et al., 2016), which is representative of the sum of the concentration of all the individual OH sinks present multiplied by their bimolecular rate coefficients for reaction with OH, is used as the denominator Eq. (6).
Table 3Model constraints and their average and maximum noontime concentrations during south-westerly and easterly flows.
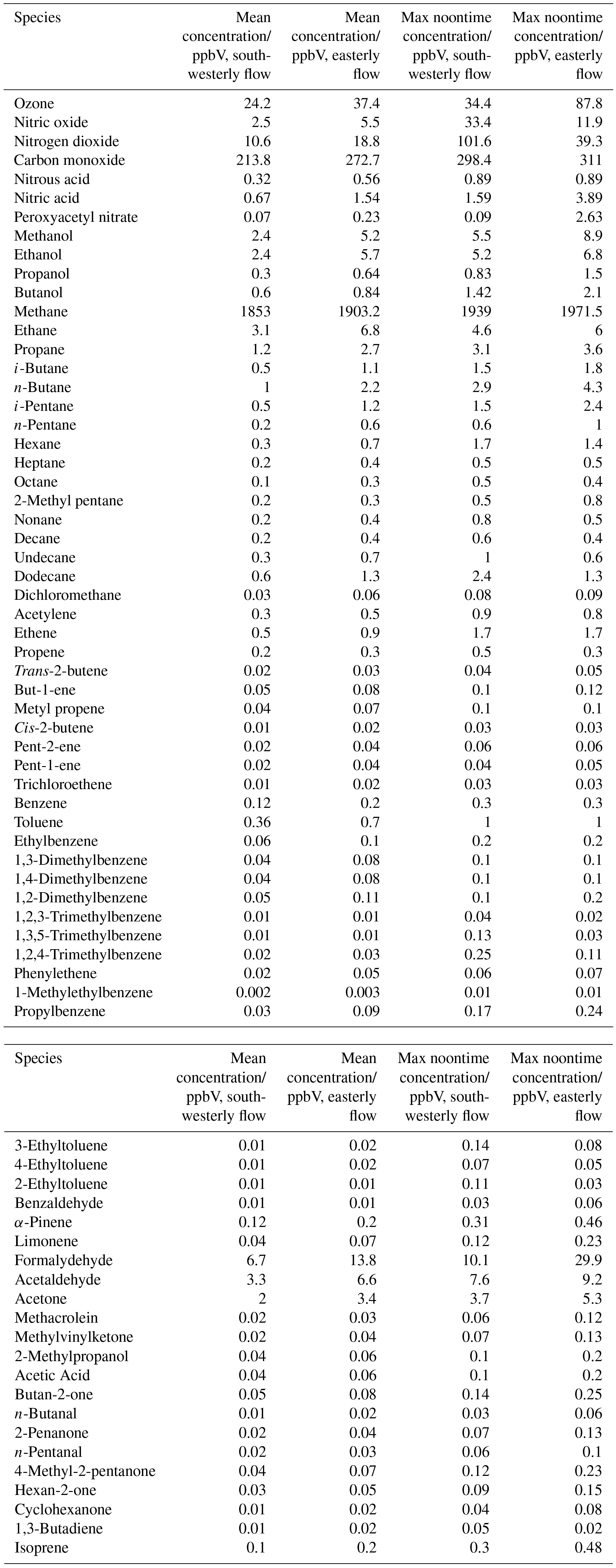
The [OH]PSS time series is overlaid with the OH observations in Figs. 2 and 3 and, on the whole, is able to predict the observed [OH] reasonably well, particularly during the south-westerly flows. The campaign median ratio of the rate of OH production to the turnover rate of OH (DOH), equal to the product of the total OH reactivity and the observed [OH] concentration, is close to 1 throughout the day (Fig. 4), highlighting consistency between the OH, HO2 and OH reactivity observations as well as the ancillary, co-located HONO (Lee et al., 2016) and NO observations (Lee et al., 2009). From late morning and throughout the afternoon, when NO concentrations dropped, the production rate of OH from HONO photolysis becomes competitive with the rate of production of OH from the secondary reaction of HO2 with NO. During the easterly conditions experienced at the beginning of the IOP, [OH]PSS does underpredict the observed [OH] between 10:00 and 18:00 (Fig. 2), however, suggesting that, if all the observed OH sources used in the PSS calculation are correct, there may be a missing OH source under these conditions (discussed further below). The small OH interference deriving from the photolysis of O3 within the FAGE cell (determined through laboratory tests) is corrected for in all OH data presented. Interestingly, when the wind direction switched for a second time to an easterly flow, [OH]PSS reproduces the observed [OH] well (Fig. 2, lower panel).
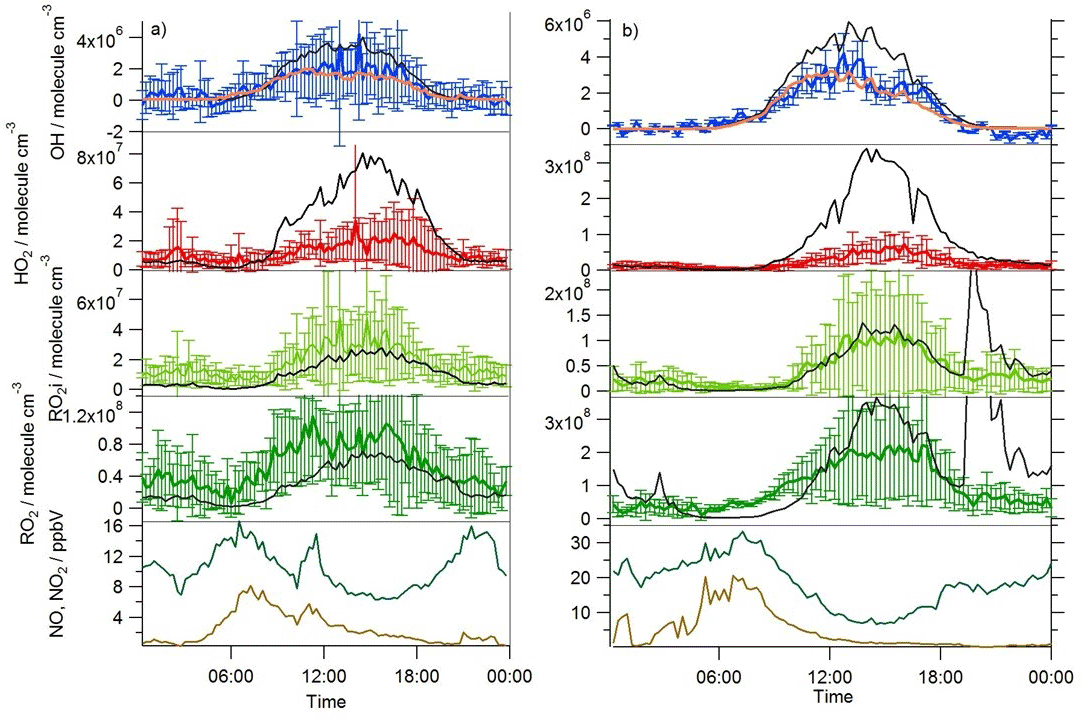
Figure 3Average diel observed (coloured lines with error bars) and MCM-BASE (black line) OH, HO2, RO2i and RO2 profiles during (a) south-westerly and (b) easterly flows; [OH]PSS is displayed by the orange line. The error bars represent the 1σ variability in the observations. The average diel observed NO (brown line) and NO2 (green line) are displayed in the bottom panels.

Figure 4Median diurnal profiles for the whole campaign of the observed DOH=kOH × [OH]. The summed rate of production of OH (POH) from the photolysis of HONO, the reaction of O(1D) with H2O, ozonolysis reactions and the reaction of HO2 with NO is overlaid. The dashed black line represents the median daytime (06:30–18:30) POH:DOH ratio; error bars highlight the 1σ standard deviation of this ratio. The red line represents a ratio of 1.
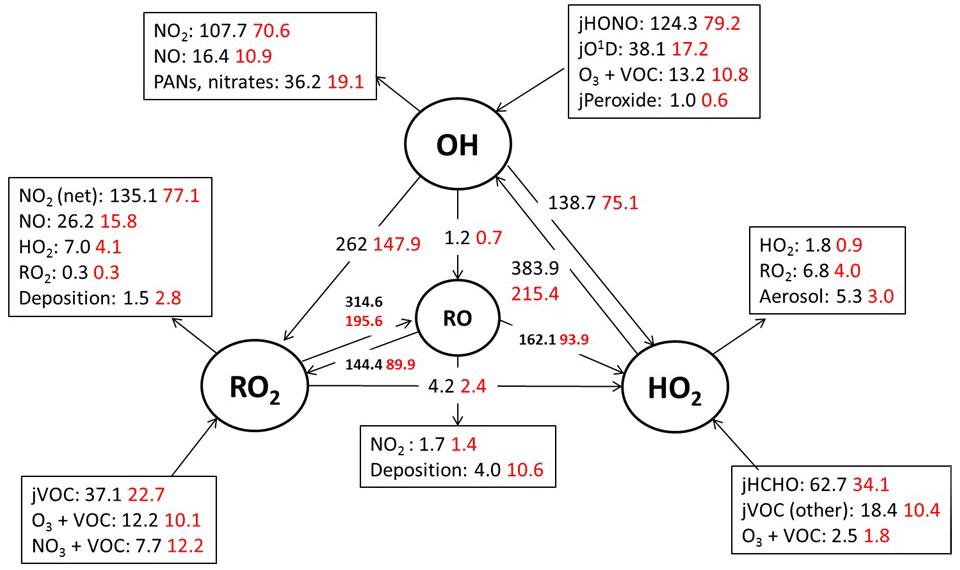
Figure 5Mean daytime (11:00–15:00 black number and 06:00–21:00, red number) rates of reaction for formation, propagation and termination of radicals in units of 105 molecule cm−3 s−1 for the whole campaign period.
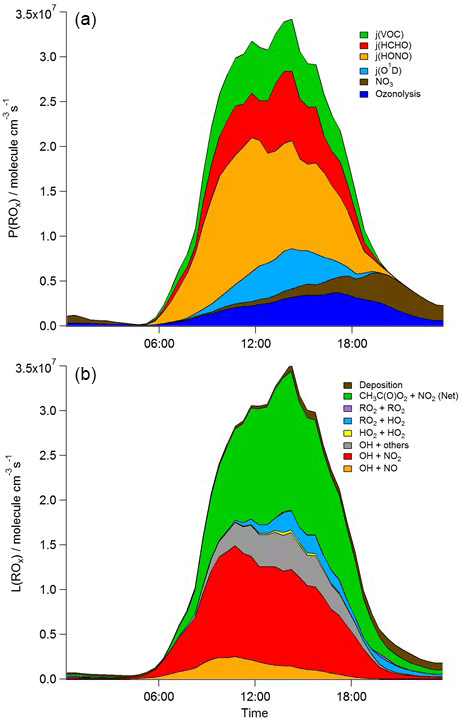
Figure 6Mean diurnal profiles of MCM-BASE modelled rates of ROx initiation (a) and termination (b) reactions for the whole campaign period. CH3C(O)O2+ NO2 (Net) represents the net (forward minus backward) CH3C(O)O2+ NO2 ↔ PAN species.
A zero-dimensional box model (MCM-BASE), which is run unconstrained to the radicals but constrained to all other measured OH sources and constrained to the very detailed VOC observations (Table 3 provides the mean and maximum noontime concentration for all model constraints both under south-westerly and easterly flows), performs much better than [OH]PSS during the first period of easterlies from late morning to late evening. However, under south-westerly conditions and also under the easterly conditions encountered during the second half of the campaign MCM-BASE overpredicts [OH] during the daytime by ∼ 25 % during the south-westerlies and by over a factor of 2 during the easterlies encountered at the end of the campaign. The box model also has a tendency to underpredict the observed [OH] during the morning rush hour (from dawn to 10:00) throughout the IOP. Since the model is able to reproduce the observed OH reactivity well during the easterly flows (Whalley et al., 2016), an underestimation of the total sink term for OH is not the cause of the daytime (10:00–18:00) discrepancy. Rather, as can be seen in Figs. 2 and 3, this model significantly overpredicts the observed [HO2] by close to a factor of 10 during the day under easterly conditions. The model overestimates the total RO2 concentration observed by close to a factor of 2 during the easterly flows but predicts RO2i well in this air mass. It should be noted that the model RO2 is simply the sum of all individual RO2 species that the model predicts from the VOCs it is constrained to, and no attempt is made to subtract the contribution of RO2 species that ROxLIF may have a low sensitivity to. This model–measurement RO2 discrepancy could, therefore, indicate the presence of RO2 species which do not readily convert to HO2 in the ROxLIF reactor in these easterly flows. Alternatively the modelled RO2 overestimate may either be due to the model overestimating the sources of RO2 or underestimating RO2 sinks. Previous work (Whalley et al., 2016) highlighted that in general the model was able to capture the observed OH reactivity (kOH) well during the easterly conditions encountered once the contribution to reactivity from monoterpenes and the heavier-weight alkanes which derive from diesel emissions (Dunmore et al., 2015) was considered. This agreement between modelled and observed kOH suggests that the production of RO2 from the oxidation of VOCs by OH should be reasonably well captured by the model and suggests that the model–measurement disagreement during the easterly flow may derive from an underestimation of the RO2 sinks. During the south-westerly flows, the model is able to capture the observed [RO2] and [RO2i] well during the afternoon on most days. However, the model underpredicts the observed [RO2] throughout the morning hours and into the early afternoon (Fig. 3a). Our previous work highlighted that this model slightly underpredicted the observed OH reactivity (by ∼ 25 %) during south-westerly flows (Whalley et al., 2016) and indicates, therefore, that a RO2 source (from the oxidation of VOCs by OH) may be missing from the model under these conditions.
Despite the factor of 5 increase in modelled [HO2] as the air mass arriving at the site switched from south-westerly to easterly (the [HO2] increase is driven to a large extent by the increase in [HCHO] a major source of HO2 under easterly flows), [OH] observed (and modelled) increased by only ∼ 35 % on average. This demonstrates that the increase in OH sources was almost entirely compensated for by an equivalent increase in OH sinks during these different flow regimes. The differences between [OH]PSS and [OH]MCM-BASE observed throughout the IOP reflects the impact of changing HO2, the dominant OH source (see Sect. 3.2) by an order of magnitude without changing the total OH sink term.
During 4 nights of the IOP, [RO2] is predicted to be elevated, reaching concentrations of > 1 × 1010 molecule cm−3 at 20:00 on 24 July. These high night-time [RO2] were not observed to the magnitude predicted by the model, and other radical types (OH and HO2) were not observed nor predicted to increase at the same time. These high modelled RO2 excursions correspond to evenings on which VOC concentrations were elevated and NO concentrations were low and which reflect periods of active nitrate chemistry in the model (see brown area, Fig. 6). The ROxLIF technique is likely insensitive to some NO3-adduct alkene peroxy radicals. Only around 20 % of the short-chain alkene-derived NO3-adduct peroxy radicals (e.g. those deriving from ethene and propene) are expected to convert to HO2 in the reactor with the dominant reaction pathway (around 80 %), instead leading to the formation of two aldehydes and NO2 (according to the MCM). For the NO3-adduct peroxy radical deriving from isoprene, however, the MCM assumes 100 % yield of HO2. The insensitivity of ROxLIF to certain NO3-adduct alkene peroxy radicals may explain the RO2 model–measurement discrepancy at night-time.
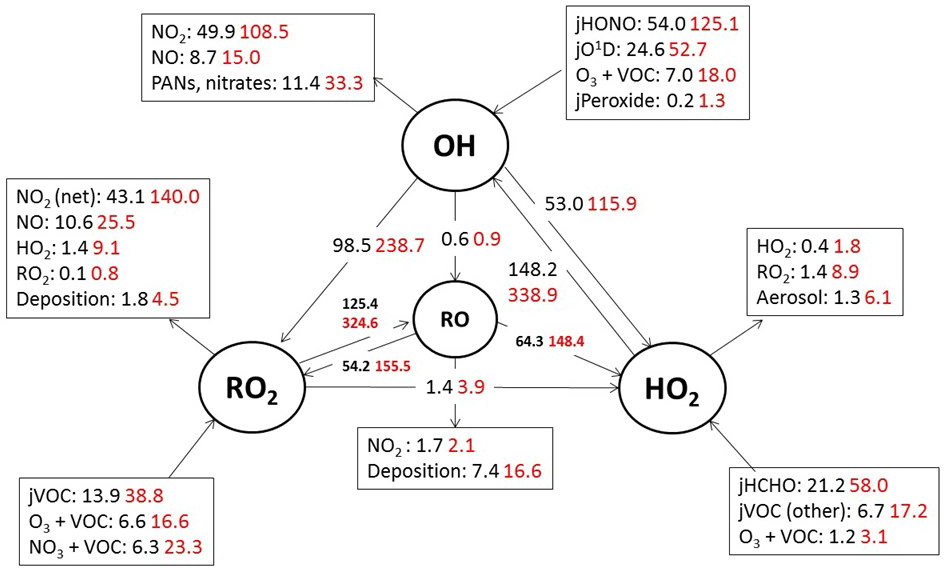
Figure 7Mean daytime (06:00–21:00) rates of reaction for formation, propagation and termination of radicals in units of 105 molecule cm−3 s−1 for south-westerly (black) and easterly (red) air masses.

Figure 8Observed and modelled HOx behaviour as a function of NO for the whole campaign period. (a) Median OH measured (blue squares), OH modelled (base MCM model is black squares, steady-state calculation is orange squares). (b) Median HO2 measured (red squares), HO measured (pink squares), HO2 modelled (black squares). Patterned areas represents the 25/75th percentiles. Data are filtered for daytime hours between 06:00 and 19:00 and binned by [NO] with a bin width of 1 ppbv for [NO] between 0 and 20 ppbv and bin width of 5 ppbv for [NO] between 20 and 45 ppbv. The number of points in each bin is displayed in the lower panel.
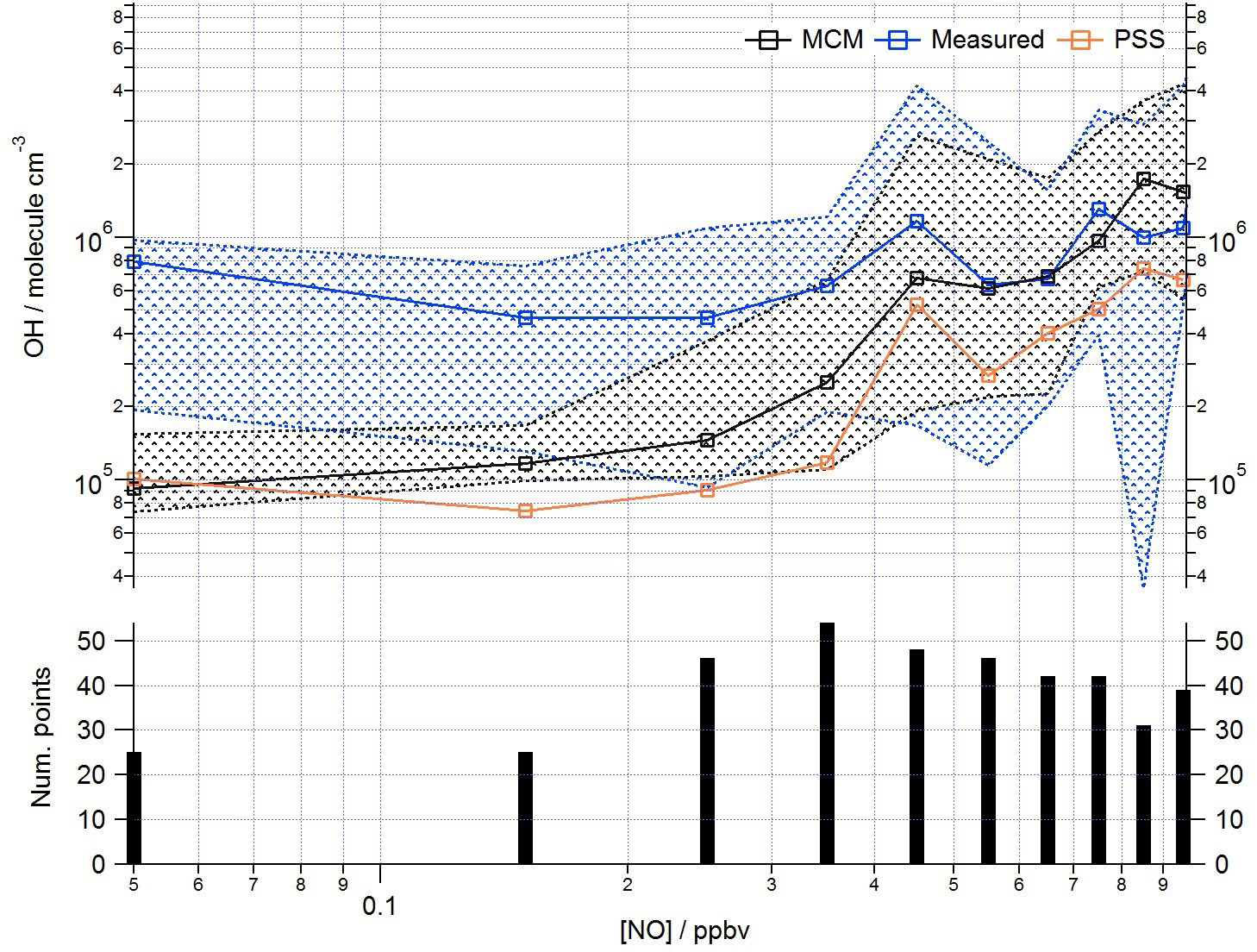
Figure 9Observed and modelled OH behaviour as a function of NO (< 1 ppbv) for the whole campaign period. Median OH measured (blue squares), OH modelled (base MCM model is black squares, steady-state calculation is orange squares). Patterned areas represents the 25/75th percentiles. Data are filtered for daytime hours between 06:00 and 19:00 and binned by [NO] with a bin width of 0.1 ppbv. The number of points in each bin is displayed in the lower panel.
3.2 Model radical budget analysis
Figure 5 shows the main initiation, propagation and termination pathways in the model, and the hourly mean diel profiles of the modelled rates of primary radical initiation and termination are shown in Fig. 6. Similar budget analyses have been conducted at other urban locations and may be compared and contrasted with the radical cycling here. During ClearfLo, the chain termination reaction of OH with NO2, which leads to a net loss of radicals, accounts for 24 % of the modelled loss of OH between 06:00 and 21:00 (21 % for 11:00 and 15:00 if morning and evening rush hours are excluded) (Whalley et al., 2016). For comparison, this reaction contributed 20 % to the total modelled OH loss in Los Angeles during CALNEX (Griffith et al., 2016), 19 % in Mexico City during MCMA-2006 (Dusanter et al., 2009), and just 11 % in Birmingham during PUMA in summer (11:00–15:00) (Emmerson et al., 2005b).
In terms of total radical destruction reactions, OH+NO2 accounts for 32 % (red area, Fig. 6, lower panel), with net (forward – backward) RO2+ NO2 to PAN species accounting for 35 % (green area, Fig. 6, lower panel). As shown in Fig. 6, the termination of ROx is dominated by loss of the OH radical (by reaction with NO2) in the morning, whilst during the afternoon, radical termination is dominated by the loss of RO2 species via PAN formation. In Birmingham, during the PUMA campaign (Emmerson et al., 2005b) net PAN formation reactions contributed close to 50 % of the total radical destruction pathways, reflecting the high OVOC fraction of total VOCs present, particularly aldehydes, in Birmingham. The photolysis of HONO is the dominant primary radical source in London, accounting for 40 % of the total radical initiation steps between 11:00 and 15:00 (Lee et al., 2016); the photolysis of formaldehyde that yields two HO2 radicals contributes 20 %, whilst O(1D) + H2O contributes 12 % and ozonolysis reactions only 9 %. HONO photolysis also contributed a significant fraction to radical initiation in the MCMA-2006 and CALNEX studies. In Birmingham the contribution of HONO photolysis as a primary radical source was likely underestimated as HONO was not measured directly. In London, the model significantly underestimated [HONO] if only gas phase reactions were considered (Lee et al., 2016). Ozonolysis reactions were identified as the most important primary source of radicals in Birmingham, with these reactions accounting for 25 % of the radical initiation, which is much more significant than for London and highlights the very different VOC profile that exists in these two major UK cities. The PUMA campaign took place in Birmingham in the 2000 and so the difference in the VOC speciation may reflect, in part, the change in VOC emissions in the UK over the past decade. As shown by Fig. 6, blue area, ozonolysis reactions form an increasingly significant fraction of the radical initiation reactions during the afternoon hours and, along with VOC + NO3 reactions, accounts for all the night-time radical initiation reactions. In London formaldehyde acts as a significant source of HO2 radicals via photolysis and its reaction with OH. The latter (OH to HO2) propagation step (including OH + HCHO but also OH + CO, OH + aromatics and OH + O3) accounts for 27 % of all the OH reactions in London. This OH to HO2 propagation step, which is lower at other urban sites (19 and 20 % during CALNEX and MCMA-2006 and 11 % in Birmingham) contributes, in part, to the high modelled HO2 concentration predicted for ClearfLo.
The relative importance of the individual formation, propagation and termination reactions under south-westerly and easterly flows remains similar. However, as highlighted by Fig. 7, the rate of many of the reactions are at least twice as fast under the easterly flows with HO2+ RO2 and RO2+ RO2 reactions approximately 6 and 8 times faster and NO3+VOC reactions close to 4 times faster.
3.3 Observed and modelled HOx radical behaviour as a function of NO
As highlighted in Figs. 2 and 3, the degree of model-to-measured agreement varies depending on the chemical conditions encountered, which changed as a function of the wind direction and time of day. To gain further insight into chemical regimes under which model performance becomes compromised, the observed and modelled radical trends as a function of NO are considered and are shown in Figs. 8 and 9.
3.3.1 Low NO
This analysis highlights that under low [NO] conditions (< 1 ppbv) the median [OH]MCM-BASE and [OH]obs agree reasonably well (black and blue squares, Fig. 8, upper panel), whilst the [OH]PSS (orange squares) underestimate the observations by ∼ 35 % (and up to a factor of 3 during the first easterlies). By expanding the number of bins representing the OH data at [NO] < 1 ppbv (Fig. 9) it is evident that both the MCM-BASE and PSS calculation underestimate the observed OH at [NO] < 0.5 ppbv, with the MCM-BASE agreeing with the observations between 0.5 and 1 ppbv [NO]. Beyond 1 ppbv [NO], [OH]obs and [OH]PSS are in good agreement.
In several other urban studies, during which a range of NOx conditions were encountered, a tendency to underpredict the observed [OH] at [NO] below 1 ppbv has been reported (Kanaya et al., 2007; Lu et al., 2012, 2013). As noted above in Sect. 3.1, the differences in the MCM-BASE and PSS model predictions observed for ClearfLo at low [NO] derives from the large overestimation of HO2 by the box model. If the box model is missing a large peroxy radical sink (discussed further in Sect. 4 below) and therefore a model constrained to the observed HO2 provides a better representation of the OH sources. Then, in agreement with the findings from Tokyo and China, an important OH source under low NOx conditions must be missing from the model mechanism for London. In both central Tokyo (Kanaya et al., 2007) and Beijing (Lu et al., 2013) an observed-to-modelled OH ratio of ∼ 2–3 was found at 200 pptv NO. At [NO] 0.2–1 ppbv, the median [OH]obs to [OH]PSS ratio was ∼ 1.5 for ClearfLo; the mean ratio was ∼ 3, reflecting the larger difference between the observed OH and [OH]PSS during the first easterlies. Lu et al. (2013) considered an additional recycling mechanism of HO2 to OH by an unknown species and found that the rate of recycling required to reconcile the modelled and measured OH in Beijing was roughly half that required in an earlier study conducted in the Pearl River Delta (Lu et al., 2012).
In contrast to the findings from these field observations made in China where modelled and observed [HO2] were generally in reasonable agreement, the peroxy radical concentrations measured in London, particularly HO2 concentrations, were greatly overestimated by MCM-BASE under lower NOx conditions. It is interesting to note that HO, which comprises HO2 and a fraction of RO2 radicals that rapidly decompose into HO2 within low pressure FAGE cells (RO2i) (Fuchs et al., 2011; Whalley et al., 2013), more closely follows the decrease in HO2 predicted by the box model under the low NO conditions, than the interference-free HO2 concentration that was observed (Fig. 8b). As shown in Fig. 8b, the HO2 and HO observations display the greatest deviation from each other at the lowest NO concentrations encountered, and as NO concentrations increased the two measurements merged (i.e. HO2 represented an increasing fraction of the HO signal). The lowest [NO] tended to occur during the daytime after the morning rush hour and so considering the diurnal profile of RO2 radicals (i.e. peak concentrations during the day, low concentrations at night and in the early morning) this trend is perhaps expected. There was variability in the day-to-day HO2 : HO ratio with a smaller ratio observed under easterly conditions compared to south-westerly conditions. The similarity in the HO2 and HO concentrations at high NO (Fig. 8b) demonstrates that the HO2 artefact signal from RO2 radicals is unlikely to contribute to any model measured discrepancies under high NOx conditions as discussed below. It is possible that previous HO2 measurements made at urban sites which did not correct for the RO2 artefact (Fuchs et al., 2011; Whalley et al., 2013) may have masked a problem with model predictions of HO2 under lower NO conditions. Recent radical observations made in Wangdu, a rural site in the North China Plain, however, which corrected HO2 for possible RO2 interferences, did not highlight any model deviation from measured HO2 under low NO conditions (Tan et al., 2017). The model overestimation of HO2 (and higher peroxy radicals) during ClearfLo, therefore, may be a reflection of the model's skill in predicting radical propagation in the presence of the complex VOC mix which was observed in central London. The VOCs observed in London included a range of long-chain hydrocarbons deriving from diesel emissions as well as a range of monoterpene emissions from biogenic sources. This breakdown in model performance will be discussed further in Sect. 4.
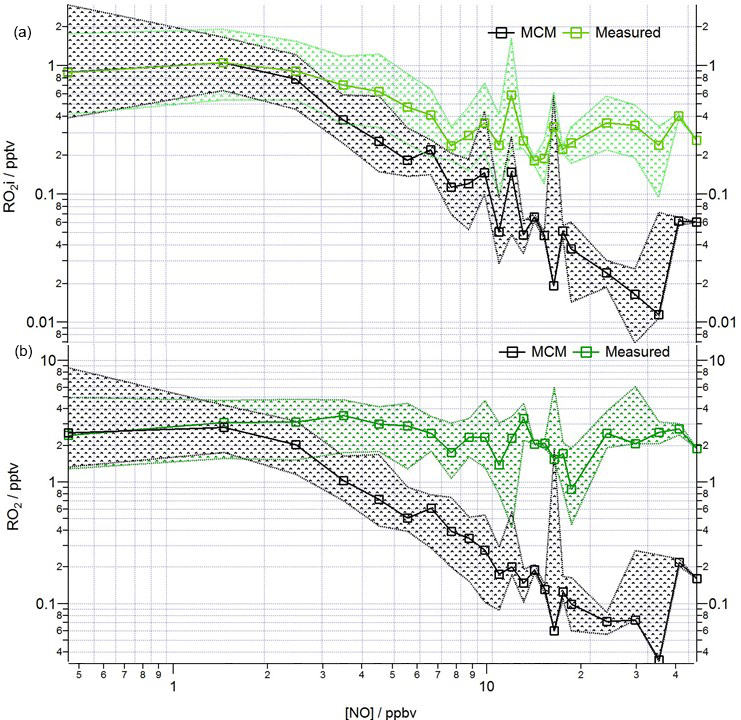
Figure 10Observed and modelled ROx behaviour as a function of NO for the whole campaign period. (a) Median RO2i measured (green squares), RO2i modelled (black squares). (b) Median RO2 measured (green squares), RO2 modelled (black squares). Patterned areas represents the 25/75 percentiles. Data are filtered for daytime hours between 06:00 and 19:00 and binned by [NO] with a bin width of 1 ppbv for [NO] between 0 and 20 ppbv and bin width of 5 ppbv for [NO] between 20 and 45 ppbv.

Figure 11Average diel observed and modelled HOx profiles during (a) south-westerly and (b) easterly flows. The base model predictions are represented by the black line. The model scenario run with standard VOC species only and missing reactivity represented by converting OH to C6H5O2 is represented by the pale blue line; the purple line represents the model scenario run with standard VOC species only and missing reactivity represented by converting OH to C6H5O2 and an enhanced heterogeneous loss of HO2, .
3.3.2 High NO
During ClearfLo, at [NO] > 15 ppbv, encountered primarily in the mornings, the modelled [OH]MCM-BASE underpredicted the observed [OH]. However, [OH]PSS was able to reproduce the OH measurements well (out to [NO] = 25 ppbv), as seen in Fig. 8. The underprediction in OH by the MCM-BASE corresponds to an underprediction in HO2 between 15 and 30 ppbv NO. An underprediction in HO2 at elevated [NO] has been highlighted during a number of earlier urban studies (Martinez et al., 2003; Ren et al., 2013; Brune et al., 2016). Brune et al. (2016) measured HO2 concentrations that were a factor of 10 greater than those predicted when NO concentrations reached 10 ppbv during the CalNex study which took place in Bakerfield, USA. During ClearfLo, the modelled and observed HO2 were in good agreement under NO concentrations ranging from 7 to 15 ppbv but beyond 15 ppbv the model began to underestimate the observations by approximately a factor of 3. It should be noted that the number of radical observations made under these elevated [NOx] were relatively few and in fact at [NO] concentrations greater than 30 ppbv, the model and observed HO2 converge once more (Fig. 8). At high NOx, Brune and co-workers report a measured OH production rate (POH) (determined by summing the rates of production from all measured OH sources), which was about twice the measured OH turnover rate (determined from the product of the total OH reactivity and observed [OH]), highlighting an inconsistency between the OH, HO2 and OH reactivity observations. In contrast to this, as demonstrated by the good agreement between the median observed OH production and OH loss rates during ClearfLo (Fig. 4), the [HO2] observed at the highest NO is supported by the observed [OH] and OH reactivity.
The median modelled and measured total RO2 and RO2i trend as a function of NO are shown in Fig. 10. The model predicts RO2 well at [NO] < 1 ppbv; the overprediction of total RO2 during the first easterlies does not bias the overall median model trend, which instead largely reflects the good agreement between modelled and measured RO2 under the dominating south-westerly conditions at low [NO]. In contrast to the reasonable agreement between modelled and observed HO2 at high [NO], the model increasingly underpredicts the total RO2 concentration (particularly RO2ni) at [NO] beyond ∼ 3 ppbv. As highlighted in Fig. S3 in the Supplement, applying a correction to the RO2 data to account for the possible decomposition of CH3O2NO2 with the ROxLIF flow reactor leads to an improved agreement between the model and observations for RO2 under high NO conditions, although the extent to which CH3O2NO2 decomposes within the flow reactor is highly uncertain. The photolysis of ClNO2 to Cl atoms may provide an additional source of RO2 radicals early in the morning as reported by Riedel et al. (2014). ClNO2 was measured during the ClearfLo project (Bannan et al., 2015) and, although Cl atom chemistry can increase the modelled RO2 concentrations in the morning when NOx levels are high, the predicted increase is modest at ∼ 20 %, and so cannot fully reconcile the model underprediction in RO2. For the more complex VOCs present (e.g. biogenics and the long-chain alkanes) the rate of RO2 propagation vs. RO2 termination may be faster than assumed in the model which would help to bring the model into better agreement with the observations.
4.1 Possible explanations for the differences between observed and modelled peroxy radical concentrations at low NO
A number of possible explanations for the differences between the observed and modelled peroxy radical concentrations under the low NO conditions have been explored through a series of model scenarios (detailed below and also in the Supplement). The impact of [NOx] deviations from a photostationary steady state in the real atmosphere as well as underestimation of the heterogeneous loss of HO2 to aerosol surfaces are discussed in the Supplement, and model runs highlighting the sensitivity of the modelled radical concentrations to these parameters are presented in Fig. S4. Enhancing the rate of HO2 termination in the model, e.g. by enhancing the uptake probability of HO2 to aerosols, only improves the HO2 modelled to measured agreement by a modest amount. So, given the dominant reactions involving HO2 are radical propagating, with the reaction of RO2 + NO acting as the largest source of HO2 and the reaction of HO2 with NO (recycling OH) acting as the dominant HO2 sink (Fig. 5), this raises the question of whether the model discrepancy relates to uncertainties in the RO2 oxidation chemistry and the cycling of RO2 to HO2. Of particular relevance are the reactions involving the complex RO2 species deriving from VOCs emitted from diesel and biogenic sources.
Hydrocarbon autoxidation processes which are known to readily occur in the liquid phase (Bolland, 1949) were, until recently, thought to be unimportant in the gas phase owing to the low probability of intermolecular H-atom abstraction. The low probability is due to the low concentration of hydrocarbons in the atmosphere and the competition between intramolecular H-shift reactions (from a C–H to an R–O–O bond forming a peroxide) and bimolecular reactions of the RO2 radical (e.g. with NO, HO2 or RO2). There is increasing evidence, however, that autoxidation processes are occurring in the atmosphere, which can quickly lower the volatility of VOCs and promote SOA formation. Laboratory studies have shown that monoterpenes, α-Pinene and limonene, following an initial attack by ozone or OH, form highly oxidised RO2 radicals within a few seconds via repeated H-shift from C–H to an R–O–O bond and subsequent O2 additions (Crounse et al., 2011; Jokinen et al., 2014; Ehn et al., 2014; Berndt et al., 2016). Mass spectrometric signals relating to these highly oxidised RO2 species have also been observed during field measurements (Jokinen et al., 2014). Autoxidation processes could be relevant during ClearfLo and omission of these processes in the model mechanism could account for some of the radical overprediction observed under the lower NO conditions, particularly under easterly flow conditions when elevated concentrations of monoterpenes were observed. At high NO, bimolecular RO2+ NO reactions likely outcompete intramolecular processes. Importantly here, autoxidation steps which involve intramolecular H-atom abstraction from a C–H to an O–O bond and subsequent addition of O2 to reform a more oxidised RO2 radical do not generate HO2. Jokinen et al. (2014) observed a high formation rate of organic nitrates (of the order of 30 %) when NO was added to experiments, which would serve to further decrease RO2 to HO2 propagation. For ClearfLo conditions, a model run unconstrained to the monoterpenes and the heavier-weight alkanes (MCM-VOC-STANDARD) underestimated OH reactivity (Whalley et al., 2016) with the missing OH reactivity fraction largely reconciled by the model-generated intermediates which derive from α-Pinene and limonene. If the current oxidation mechanism for these species is inaccurate, the reactivity attributed to these oxidation products could be wrong and instead may derive from other oxidised species. The missing reactivity in the MCM-VOC-STANDARD run can be included by adding a single OH to RO2 conversion to the model equivalent to the missing reactivity (in s−1) at each time stamp. To represent an autoxidation pathway, we convert OH to MCM species C6H5O2. This RO2 species is formed via a minor phenol +OH channel. C6H5O2 does not readily convert to HO2 by reaction with NO (due to the lack of available H on the alpha C) and, instead, following oxygen atom abstraction, C6H5O reacts with NO2 to form a nitrophenol, or reacts with ozone to reform C6H5O2, which reacts with further NO and so on. We do not consider the reactions of C6H5O2 to be representative of what is actually occurring but rather chose this species to represent a mechanism by which the propagation of RO2 to HO2 is inhibited. Inclusion of OH → C6H5O2 leads to a ∼ 30 % decrease in modelled HO2 (see pale blue vs. black diel profiles in Fig. 11) and close to a 50 % decrease in modelled HO2 if the heterogeneous loss to aerosol is enhanced also by increasing the HO2 uptake probability to aerosols from 0.1 to 1 (purple vs. black diel profiles in Fig. 11). Including autoxidation in the model improves the agreement with the observations during the daytime for all radical species apart from the total RO2 species observed during south-westerly flows. As discussed in Sect. 3.1, however, the model is, likely, missing VOCs under this air mass regime (implied from the underprediction of the observed kOH) and this may contribute to the model underprediction of RO2. The underprediction of RO2i when an autoxidation step is included is due to the choice of OH→RO2 conversion species (i.e. C6H5O2 is an RO2 species that does not decompose into HO2 in the presence of NO within a FAGE cell). The model underprediction of RO2i suggests that at least some of the RO2 species that undergo autoxidation are species that would decompose into HO2 within a FAGE cell.
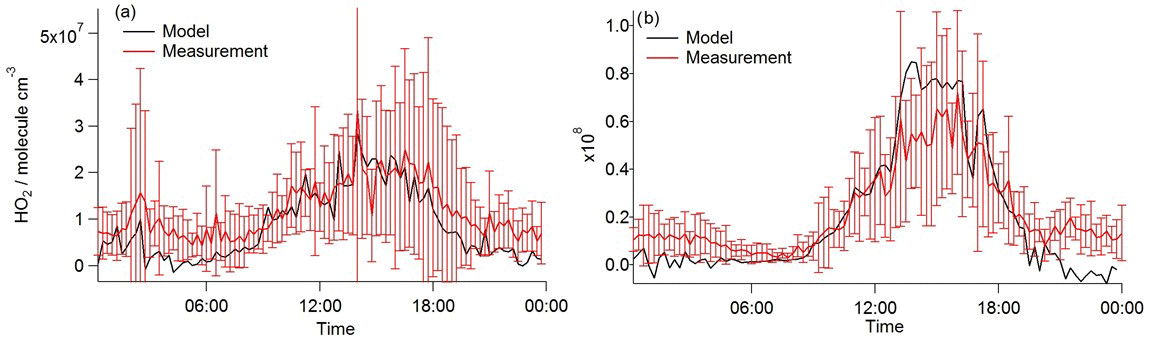
Figure 12Average diel profiles of HO2 concentration observed (red line with error bars) under (a) south-westerly and (b) easterly conditions. Overlaid is HO2 calculated using the solution to the HO2 quadratic expression Eq. (8) (represented by the black line) with α equal to 0.15.
The partitioning of larger, lower volatility OVOCs to the aerosol phase may be another important step which is not included in the model. The partitioning of gases to the aerosol phase will reduce RO2 to HO2 propagation and act as a net radical sink, and omitting this process may further contribute to some of the discrepancy between observed radical concentrations and MCM-BASE predictions.
As depicted by the model radical flux (Fig. 5) roughly half (162 × 105 molecule cm−3s−1 of 314 × 105 molecule cm−3 s−1 between 11:00 15:00) of the modelled RO2 radicals that react with NO eventually form HO2 (via an alkoxy radical, RO). If, however, RO2 to HO2 propagation is overestimated by the model due to the uncertainties outline above, modelled HO2 may become artificially high. A steady-state [HO2] can be estimated (without the use of a model) by balancing the dominant HO2 production and destruction reactions (first- and second-order loss processes) that occur:
α is equal to the fraction of RO2 radicals which propagate to HO2 (which is roughly half in MCM-BASE).
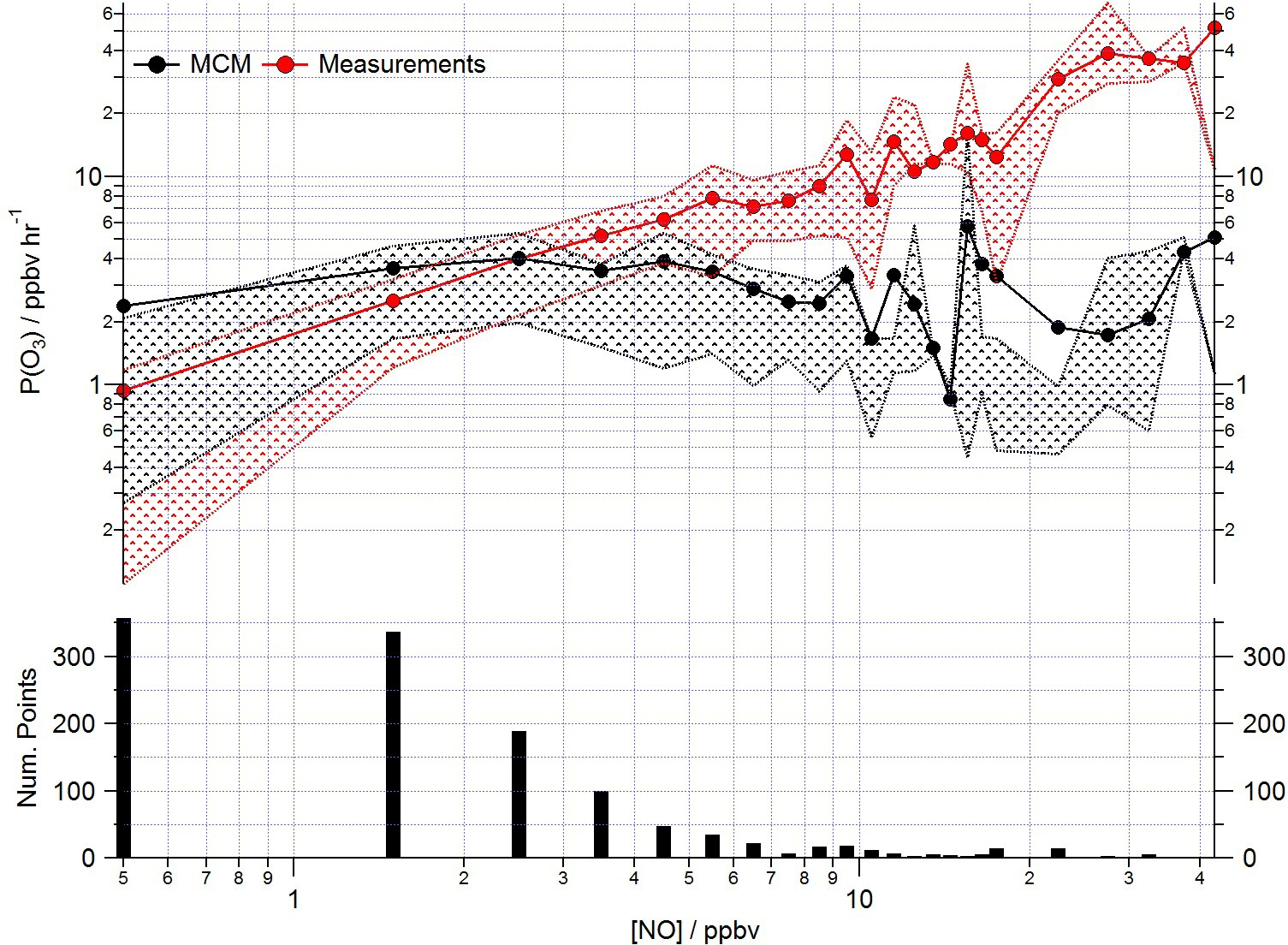
Figure 13Mean ozone production (ppbv h−1) calculated from observed (red squares) and modelled (black squares) ROx concentrations using Eq. (1) as a function of NO. Data are filtered for daytime hours between 06:00 and 19:00 and binned by [NO] with a bin width of 1 ppbv for [NO] between 0 and 20 ppbv and bin width of 5 ppbv for [NO] between 20 and 45 ppbv.
Equation (7) can be rewritten as a quadratic equation for HO2 and then solved for HO2 to yield the following solution:
where
Using the observed RO2 and OH concentrations in Eqs. (8)–(11) above to calculate [HO2], generally good agreement between HO2 observed and HO2 calculated can be achieved if α equal to 0.15 is assumed as shown in Fig. 12. Using α= 0.15 leads to a model underprediction of HO2 for the higher NOx conditions experienced in the early morning, however. This may indicate that α is dependent on NO concentrations and likely on the VOC speciation too. Furthermore, the value for α is sensitive to the rate coefficient, used with the [HO2] in Fig. 11 calculated using (= 7.7 × 10−12 cm3 molecule−1 s−1 at 298 K). If some of the RO2 species contributing to the total RO2 measured react faster with NO (as is the case for CH3CO•O2 radicals, 2 × 10−11 cm3 molecule−1 s−1 at 298 K), α would become < 0.15. This low fraction of RO2 to HO2 conversion (if this is the cause for the observed and modelled discrepancy) compared to α ∼ 0.5 in MCM-BASE highlights a significant misunderstanding in the oxidation chemistry mechanism of the larger more complex VOCs. This misunderstanding likely becomes increasingly important in low-NO and high-VOC environments such as forests.
4.2 Impact of the model uncertainties on predictions of in situ ozone production
Poor representation of the observed peroxy radical concentration leads to significantly more ozone production predicted by the model than is calculated from the observed concentrations under low NO conditions (Fig. 13) using Eq. (1) (which is repeated below for clarity). Conversely, significantly less ozone production is predicted by the modelled peroxy radicals than by the observed peroxy radicals as [NO] increases.
As highlighted in Sect. 3.3, the model's failure to predict the observed HO2 radical concentrations is most evident under low NOx conditions, typically experienced during the afternoon hours and particularly during easterly flows. At this time the observed ozone concentrations peaked (Fig. 13) due to reduced destruction by titration with NO. At NO concentrations < 3 ppbv, the ozone production rate determined from the modelled peroxy radical concentrations remains relatively constant at ∼ 3 ppbv h−1. P(O3) calculated with the observed peroxy radical, however, decreases to ∼ 1 ppbv h−1. Under higher NOx conditions, [NO] > 3 ppbv, the ozone production rate determined from the modelled peroxy radical concentrations is up to an order of magnitude lower than the ozone production rate calculated from the observations (which at the highest [NO] reaches ∼ 30 ppbv h−1). The calculation of ozone production from many earlier urban studies often relied on an inferred RO2 concentration estimated from the measured [HO2] and assumed value of RO2 : HO2, as measurements of total RO2 were not available (e.g. Ren et al., 2013; Brune et al., 2016). In these studies, under high NO conditions, the P(O3) calculated from the observed HO2 (and inferred RO2) was significantly greater than P(O3) calculated using the modelled HO2 and RO2, reflecting the model underestimation of HO2 at high NO reported from these studies. In the recent Wangdu study conducted in China, Tan et al. (2017), using observed RO2, demonstrated that models may underpredict ozone production at high NO due to an underestimation of the RO2 radical concentration rather than underestimation of HO2. In the Wangdu study modelled and measured HO2 were in good agreement at high NO. From the rate of ozone production calculated from the modelled and measured peroxy radicals for ClearfLo, we would draw similar conclusions as drawn by Tan and co-workers; i.e. there would be missing RO2 at high NO if the correction for decomposition of CH3O2NO2 was not applicable. Although there are some uncertainties surrounding the magnitude of CH3O2NO2 decomposition in the ROxLIF cell (which is experimentally difficult to determine), in agreement with Tan et al. we find no evidence from the ClearfLo HO2 data set that there is a significant model bias for HO2 which influences the model-predicted P(O3) under the elevated NO conditions encountered.
The discrepancy between model and observations at low NO may arise from the model uncertainties in the treatment of the oxidation and removal of the complex VOC species observed as discussed above. The oxidation of these complex species tend not to be included in air quality models used to predict ozone and other secondary pollutants. The MCM, however, is used as the benchmark mechanism against which simpler mechanisms used within air quality models are tested (Malkin et al., 2016) and so the chemistry of these complex VOCs present in the urban atmosphere and the impact they have on peroxy radical concentrations needs to be adequately resolved.
Measurement and model comparisons of OH, HO2, RO2i and total RO2 have displayed varying levels of agreement as a function of NOx. Under higher NOx conditions the box model increasingly underpredicted total [RO2] and, as a consequence, ozone production derived from the predicted peroxy radicals is up to an order of magnitude lower than from the observed peroxy radicals.
A large uncertainty in peroxy radical cycling, RO2→ HO2, has been identified under lower NO conditions experienced during the daytime. We hypothesise that uncertainties in the degradation mechanism of RO2 deriving from complex biogenic and diesel-related VOC, species which were particularly elevated and dominated the OH reactivity under easterly flows when the model–measurement discrepancy was largest, may account for the model–measurement disagreement. Autoxidation processes now known to play a role in the chemical oxidation of monoterpenes in the gas phase, and which can enhance SOA formation, may serve to reduce the rate of RO2 to HO2 propagation under lower NO conditions. Omission of this oxidation process from the model mechanism leads to more ozone production predicted using modelled peroxy radical concentrations versus those measured at a time when ozone destruction (by NO titration) is slow. Although air quality models do not typically consider these VOC types and tend to run with simplified chemistry schemes, the MCM is viewed as a benchmark mechanism against which these simpler chemistry schemes may be tested. Hence, these uncertainties in the mechanism identified here need to be critically assessed through further laboratory and field measurements.
Data deposited at the Centre for Environmental Data Analysis, CEDA, http://archive.ceda.ac.uk/.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-2547-2018-supplement.
The authors declare that they have no conflict of interest.
The work was supported by the National Environment Research Council (NERC)
through grant NE/H003193/1. We are also grateful to the National Centre for
Atmospheric Science, which is funded by NERC, for ongoing support. We would
like to thank all participants of ClearfLo for their help in supporting
these measurements.
Edited by: Ronald Cohen
Reviewed by: two anonymous referees
Barlow, J. F., Halios, C. H., Lane, S. E., and Wood, C. R.: Observations of urban boundary layer structure during a strong urban heat island event, Environ. Fluid Mech., 15, 373–398, 2015.
Berndt, T., Richters, S., Jokinen, T., Hyttinen, N., Kurten, T., Otkjaer, R. V., Kjaergaard, H. G., Stratmann, F., Herrmann, H., Sipila, M., Kulmala, M., and Ehn, M.: Hydroxyl radical-induced formation of highly oxidized organic compounds, Nat. Commun., 7, 13677, https://doi.org/10.1038/Ncomms13677, 2016.
Bigi, A. and Harrison, R. M.: Analysis of the air pollution climate at a central urban background site, Atmos. Environ., 44, 2004–2012, https://doi.org/10.1016/j.atmosenv.2010.02.028, 2010.
Bohn, B., Heard, D. E., Mihalopoulos, N., Plass-Dulmer, C., Schmitt, R., and Whalley, L. K.: Characterisation and improvement of j((O filter radiometers, Atmos. Meas. Tech., 9, 3455–3466, https://doi.org/10.5194/amt-9-3455-2016, 2016.
Bohnenstengel, S. I., Belcher, S. E., Aiken, A., Allan, J. D., Allen, G., Bacak, A., Bannan, T. J., Barlow, J. F., Beddows, D. C. S., Bloss, W. J., Booth, A. M., Chemel, C., Coceal, O., Di Marco, C. F., Dubey, M. K., Faloon, K. H., Fleming, Z. L., Furger, M., Gietl, J. K., Graves, R. R., Green, D. C., Grimmond, C. S. B., Halios, C. H., Hamilton, J. F., Harrison, R. M., Heal, M. R., Heard, D. E., Helfter, C., Herndon, S. C., Holmes, R. E., Hopkins, J. R., Jones, A. M., Kelly, F. J., Kotthaus, S., Langford, B., Lee, J. D., Leigh, R. J., Lewis, A. C., Lidster, R. T., Lopez-Hilfiker, F. D., McQuaid, J. B., Mohr, C., Monks, P. S., Nemitz, E., Ng, N. L., Percival, C. J., Prevot, A. S. H., Ricketts, H. M. A., Sokhi, R., Stone, D., Thornton, J. A., Tremper, A. H., Valach, A. C., Visser, S., Whalley, L. K., Williams, L. R., Xu, L., Young, D. E., and Zotter, P.: Meteorology, Air Quality, and Health in London the Clearflo Project, B. Am. Meteorol. Soc., 96, 779–804, https://doi.org/10.1175/Bams-D-12-00245.1, 2015.
Bolland, J. L.: Kinetics of olefin oxidation, Quarterly Reviews, Chemical Society, 3, 1–21, 1949.
Brune, W. H., Baier, B. C., Thomas, J., Ren, X., Cohen, R. C., Pusede, S. E., Browne, E. C., Goldstein, A. H., Gentner, D. R., Keutsch, F. N., Thornton, J. A., Harrold, S., Lopez-Hilfiker, F. D., and Wennberg, P. O.: Ozone production chemistry in the presence of urban plumes, Faraday Discuss., 189, 169–189, https://doi.org/10.1039/c5fd00204d, 2016.
Centre for Environmental Data Analysis, http://archive.ceda.ac.uk/, last access: 9 February 2018.
Chen, S. A., Ren, X. R., Mao, J. Q., Chen, Z., Brune, W. H., Lefer, B., Rappengluck, B., Flynn, J., Olson, J., and Crawford, J. H.: A comparison of chemical mechanisms based on TRAMP-2006 field data, Atmos. Environ., 44, 4116–4125, https://doi.org/10.1016/j.atmosenv.2009.05.027, 2010.
Commane, R., Floquet, C. F. A., Ingham, T., Stone, D., Evans, M. J., and Heard, D. E.: Observations of OH and HO2 radicals over West Africa, Atmos. Chem. Phys., 10, 8783–8801, https://doi.org/10.5194/acp-10-8783-2010, 2010.
Crounse, J. D., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Peroxy radical isomerization in the oxidation of isoprene, Phys. Chem. Chem. Phys., 13, 13607–13613, https://doi.org/10.1039/c1cp21330j, 2011.
Dunmore, R. E., Hopkins, J. R., Lidster, R. T., Lee, J. D., Evans, M. J., Rickard, A. R., Lewis, A. C., and Hamilton, J. F.: Diesel-related hydrocarbons can dominate gas phase reactive carbon in megacities, Atmos. Chem. Phys., 15, 9983–9996, https://doi.org/10.5194/acp-15-9983-2015, 2015.
Dusanter, S., Vimal, D., Stevens, P. S., Volkamer, R., Molina, L. T., Baker, A., Meinardi, S., Blake, D., Sheehy, P., Merten, A., Zhang, R., Zheng, J., Fortner, E. C., Junkermann, W., Dubey, M., Rahn, T., Eichinger, B., Lewandowski, P., Prueger, J., and Holder, H.: Measurements of OH and HO2 concentrations during the MCMA-2006 field campaign – Part 2: Model comparison and radical budget, Atmos. Chem. Phys., 9, 6655–6675, https://doi.org/10.5194/acp-9-6655-2009, 2009.
Ehn, M., Thornton, J. A., Kleist, E., Sipila, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I. H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurten, T., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., Canagaratna, M., Dal Maso, M., Berndt, T., Petaja, T., Wahner, A., Kerminen, V. M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Elshorbany, Y. F., Kleffmann, J., Hofzumahaus, A., Kurtenbach, R., Wiesen, P., Brauers, T., Bohn, B., Dorn, H. P., Fuchs, H., Holland, F., Rohrer, F., Tillmann, R., Wegener, R., Wahner, A., Kanaya, Y., Yoshino, A., Nishida, S., Kajii, Y., Martinez, M., Kubistin, D., Harder, H., Lelieveld, J., Elste, T., Plass-Dulmer, C., Stange, G., Berresheim, H., and Schurath, U.: HOx budgets during HOxComp: A case study of HOx chemistry under NOx-limited conditions, J. Geophys. Res.-Atmos., 117, D03307, https://doi.org/10.1029/2011jd017008, 2012.
Emmerson, K. M., Carslaw, N., Carpenter, L. J., Heard, D. E., Lee, J. D., and Pilling, M. J.: Urban atmospheric chemistry during the PUMA campaign 1: Comparison of modelled OH and HO2 concentrations with measurements, J. Atmos. Chem., 52, 143–164, https://doi.org/10.1007/s10874-005-1322-3, 2005a.
Emmerson, K. M., Carslaw, N., and Pilling, M. J.: Urban atmospheric chemistry during the PUMA campaign 2: Radical budgets for OH, HO2 and RO2, J. Atmos. Chem., 52, 165–183, https://doi.org/10.1007/s10874-005-1323-2, 2005b.
Emmerson, K. M., Carslaw, N., Carslaw, D. C., Lee, J. D., McFiggans, G., Bloss, W. J., Gravestock, T., Heard, D. E., Hopkins, J., Ingham, T., Pilling, M. J., Smith, S. C., Jacob, M., and Monks, P. S.: Free radical modelling studies during the UK TORCH Campaign in Summer 2003, Atmos. Chem. Phys., 7, 167–181, https://doi.org/10.5194/acp-7-167-2007, 2007.
Gowers, A. M., Miller, B. G., and Stedman, J. R.: Estimating local mortality burdens associated with particulate air pollution, Public Health England, PHE-CRCe-010, https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/332854/PHE_CRCE_010.pdf, 2014.
Fuchs, H., Holland, F., and Hofzumahaus, A.: Measurement of tropospheric RO2 and HO2 radicals by a laser-induced fluorescence instrument, Rev. Sci. Instrum., 79, 084104, https://doi.org/10.1063/1.2968712, 2008.
Fuchs, H., Bohn, B., Hofzumahaus, A., Holland, F., Lu, K. D., Nehr, S., Rohrer, F., and Wahner, A.: Detection of HO2 by laser-induced fluorescence: calibration and interferences from RO2 radicals, Atmos. Meas. Tech., 4, 1209–1225, https://doi.org/10.5194/amt-4-1209-2011, 2011.
Fuchs, H., Hofzumahaus, A., Rohrer, F., Bohn, B., Brauers, T., Dorn, H. P., Haseler, R., Holland, F., Kaminski, M., Li, X., Lu, K., Nehr, S., Tillmann, R., Wegener, R., and Wahner, A.: Experimental evidence for efficient hydroxyl radical regeneration in isoprene oxidation, Nat. Geosci., 6, 1023–1026, https://doi.org/10.1038/NGEO1964, 2013.
Fuchs, H., Tan, Z. F., Hofzumahaus, A., Broch, S., Dorn, H. P., Holland, F., Kunstler, C., Gomm, S., Rohrer, F., Schrade, S., Tillmann, R., and Wahner, A.: Investigation of potential interferences in the detection of atmospheric ROx radicals by laser-induced fluorescence under dark conditions, Atmos. Meas. Tech., 9, 1431–1447, https://doi.org/10.5194/amt-9-1431-2016, 2016.
Fuchs, H., Tan, Z. F., Lu, K. D., Bohn, B., Broch, S., Brown, S. S., Dong, H. B., Gomm, S., Haseler, R., He, L. Y., Hofzumahaus, A., Holland, F., Li, X., Liu, Y., Lu, S. H., Min, K. E., Rohrer, F., Shao, M., Wang, B. L., Wang, M., Wu, Y. S., Zeng, L. M., Zhang, Y. S., Wahner, A., and Zhang, Y. H.: OH reactivity at a rural site (Wangdu) in the North China Plain: contributions from OH reactants and experimental OH budget, Atmos. Chem. Phys., 17, 645–661, https://doi.org/10.5194/acp-17-645-2017, 2017.
George, L. A., Hard, T. M., and O'Brien, R. J.: Measurement of free radicals OH and HO2 in Los Angeles smog, J. Geophys. Res.-Atmos., 104, 11643–11655, https://doi.org/10.1029/1998jd100113, 1999.
Gerbig, C., Schmitgen, S., Kley, D., Volz-Thomas, A., Dewey, K., and Haaks, D.: An improved fast-response vacuum-UV resonance fluorescence CO instrument, J. Geophys. Res.-Atmos., 104, 1699–1704, https://doi.org/10.1029/1998jd100031, 1999.
Griffith, S. M., Hansen, R. F., Dusanter, S., Michoud, V., Gilman, J. B., Kuster, W. C., Veres, P. R., Graus, M., de Gouw, J. A., Roberts, J., Young, C., Washenfelder, R., Brown, S. S., Thalman, R., Waxman, E., Volkamer, R., Tsai, C., Stutz, J., Flynn, J. H., Grossberg, N., Lefer, B., Alvarez, S. L., Rappenglueck, B., Mielke, L. H., Osthoff, H. D., and Stevens, P. S.: Measurements of hydroxyl and hydroperoxy radicals during CalNex-LA: Model comparisons and radical budgets, J. Geophys. Res.-Atmos., 121, 4211–4232, https://doi.org/10.1002/2015JD024358, 2016.
Hayman, G. D.: Effect of pollution control on UV exposure, Oxfordshire, UK, 1997.
Heland, J., Kleffmann, J., Kurtenbach, R., and Wiesen, P.: A new instrument to measure gaseous nitrous acid (HONO) in the atmosphere, Environ. Sci. Technol., 35, 3207–3212, https://doi.org/10.1021/Es000303t, 2001.
Hopkins, J. R., Lewis, A. C., and Read, K. A.: A two-column method for long-term monitoring of non-methane hydrocarbons (NMHCs) and oxygenated volatile organic compounds (o-VOCs), J. Environ. Monitor., 5, 8–13, https://doi.org/10.1039/b202798d, 2003.
Jenkin, M. E., Wyche, K. P., Evans, C. J., Carr, T., Monks, P. S., Alfarra, M. R., Barley, M. H., McFiggans, G. B., Young, J. C., and Rickard, A. R.: Development and chamber evaluation of the MCM v3.2 degradation scheme for beta-caryophyllene, Atmos. Chem. Phys., 12, 5275–5308, https://doi.org/10.5194/acp-12-5275-2012, 2012.
Jerrett, M., Burnett, R. T., Pope, C. A., Ito, K., Thurston, G., Krewski, D., Shi, Y. L., Calle, E., and Thun, M.: Long-Term Ozone Exposure and Mortality, New Engl. J. Med., 360, 1085–1095, https://doi.org/10.1056/Nejmoa0803894, 2009.
Jokinen, T., Sipila, M., Richters, S., Kerminen, V. M., Paasonen, P., Stratmann, F., Worsnop, D., Kulmala, M., Ehn, M., Herrmann, H., and Berndt, T.: Rapid Autoxidation Forms Highly Oxidized RO2 Radicals in the Atmosphere, Angew. Chem. Int. Edit., 53, 14596–14600, https://doi.org/10.1002/anie.201408566, 2014.
Kanaya, Y., Cao, R. Q., Akimoto, H., Fukuda, M., Komazaki, Y., Yokouchi, Y., Koike, M., Tanimoto, H., Takegawa, N., and Kondo, Y.: Urban photochemistry in central Tokyo: 1. Observed and modeled OH and HO2 radical concentrations during the winter and summer of 2004, J. Geophys. Res.-Atmos., 112, D21312, https://doi.org/10.1029/2007jd008670, 2007.
Kleffmann, J.: Daytime sources of nitrous acid (HONO) in the atmospheric boundary layer, Chemphyschem, 8, 1137–1144, https://doi.org/10.1002/cphc.200700016, 2007.
Konrad, S., Schmitz, T., Buers, H. J., Houben, N., Mannschreck, K., Mihelcic, D., Musgen, P., Patz, H. W., Holland, F., Hofzumahaus, A., Schafer, H. J., Schroder, S., Volz-Thomas, A., Bachmann, K., Schlomski, S., Moortgat, G., and Grossmann, D.: Hydrocarbon measurements at Pabstthum during the BERLIOZ campaign and modeling of free radicals, J. Geophys. Res.-Atmos., 108, 8251, https://doi.org/10.1029/2001jd000866, 2003.
Lee, J. D., Moller, S. J., Read, K. A., Lewis, A. C., Mendes, L., and Carpenter, L. J.: Year-round measurements of nitrogen oxides and ozone in the tropical North Atlantic marine boundary layer, J. Geophys. Res.-Atmos., 114, D21302, https://doi.org/10.1029/2009jd011878, 2009.
Lee, J. D., Whalley, L. K., Heard, D. E., Stone, D., Dunmore, R. E., Hamilton, J. F., Young, D. E., Allan, J. D., Laufs, S., and Kleffmann, J.: Detailed budget analysis of HONO in central London reveals a missing daytime source, Atmos. Chem. Phys., 16, 2747–2764, https://doi.org/10.5194/acp-16-2747-2016, 2016.
Lidster, R. T., Hamilton, J. F., Lee, J. D., Lewis, A. C., Hopkins, J. R., Punjabi, S., Rickard, A. R., and Young, J. C.: The impact of monoaromatic hydrocarbons on OH reactivity in the coastal UK boundary layer and free troposphere, Atmos. Chem. Phys., 14, 6677–6693, https://doi.org/10.5194/acp-14-6677-2014, 2014.
Lu, K. D., Rohrer, F., Holland, F., Fuchs, H., Bohn, B., Brauers, T., Chang, C. C., Haseler, R., Hu, M., Kita, K., Kondo, Y., Li, X., Lou, S. R., Nehr, S., Shao, M., Zeng, L. M., Wahner, A., Zhang, Y. H., and Hofzumahaus, A.: Observation and modelling of OH and HO2 concentrations in the Pearl River Delta 2006: a missing OH source in a VOC rich atmosphere, Atmos. Chem. Phys., 12, 1541–1569, https://doi.org/10.5194/acp-12-1541-2012, 2012.
Lu, K. D., Hofzumahaus, A., Holland, F., Bohn, B., Brauers, T., Fuchs, H., Hu, M., Haseler, R., Kita, K., Kondo, Y., Li, X., Lou, S. R., Oebel, A., Shao, M., Zeng, L. M., Wahner, A., Zhu, T., Zhang, Y. H., and Rohrer, F.: Missing OH source in a suburban environment near Beijing: observed and modelled OH and HO2 concentrations in summer 2006, Atmos. Chem. Phys., 13, 1057–1080, https://doi.org/10.5194/acp-13-1057-2013, 2013.
Mao, J., Ren, X., Zhang, L., Van Duin, D. M., Cohen, R. C., Park, J. H., Goldstein, A. H., Paulot, F., Beaver, M. R., Crounse, J. D., Wennberg, P. O., DiGangi, J. P., Henry, S. B., Keutsch, F. N., Park, C., Schade, G. W., Wolfe, G. M., Thornton, J. A., and Brune, W. H.: Insights into hydroxyl measurements and atmospheric oxidation in a California forest, Atmos. Chem. Phys., 12, 8009–8020, https://doi.org/10.5194/acp-12-8009-2012, 2012.
Martinez, M., Harder, H., Kovacs, T. A., Simpas, J. B., Bassis, J., Lesher, R., Brune, W. H., Frost, G. J., Williams, E. J., Stroud, C. A., Jobson, B. T., Roberts, J. M., Hall, S. R., Shetter, R. E., Wert, B., Fried, A., Alicke, B., Stutz, J., Young, V. L., White, A. B., and Zamora, R. J.: OH and HO2 concentrations, sources, and loss rates during the Southern Oxidants Study in Nashville, Tennessee, summer 1999, J. Geophys. Res.-Atmos., 108, 4617, https://doi.org/10.1029/2003jd003551, 2003.
Michoud, V., Kukui, A., Camredon, M., Colomb, A., Borbon, A., Miet, K., Aumont, B., Beekmann, M., Durand-Jolibois, R., Perrier, S., Zapf, P., Siour, G., Ait-Helal, W., Locoge, N., Sauvage, S., Afif, C., Gros, V., Furger, M., Ancellet, G., and Doussin, J. F.: Radical budget analysis in a suburban European site during the MEGAPOLI summer field campaign, Atmos. Chem. Phys., 12, 11951–11974, https://doi.org/10.5194/acp-12-11951-2012, 2012.
Novelli, A., Hens, K., Ernest, C. T., Kubistin, D., Regelin, E., Elste, T., Plass-Dulmer, C., Martinez, M., Lelieveld, J., and Harder, H.: Characterisation of an inlet pre-injector laser-induced fluorescence instrument for the measurement of atmospheric hydroxyl radicals, Atmos. Meas. Tech., 7, 3413–3430, https://doi.org/10.5194/amt-7-3413-2014, 2014.
Peters, T. M. and Leith, D.: Concentration measurement and counting efficiency of the aerodynamic particle sizer 3321, J. Aerosol. Sci., 34, 627–634, https://doi.org/10.1016/S0021-8502(03)00030-2, 2003.
Ravishankara, A. R.: Heterogeneous and multiphase chemistry in the troposphere, Science, 276, 1058–1065, 1997.
Ren, X. R., Harder, H., Martinez, M., Lesher, R. L., Oliger, A., Simpas, J. B., Brune, W. H., Schwab, J. J., Demerjian, K. L., He, Y., Zhou, X. L., and Gao, H. G.: OH and HO2 chemistry in the urban atmosphere of New York City, Atmos. Environ., 37, 3639–3651, https://doi.org/10.1016/S1352-2310(03)00459-X, 2003.
Ren, X. R., van Duin, D., Cazorla, M., Chen, S., Mao, J. Q., Zhang, L., Brune, W. H., Flynn, J. H., Grossberg, N., Lefer, B. L., Rappengluck, B., Wong, K. W., Tsai, C., Stutz, J., Dibb, J. E., Jobson, B. T., Luke, W. T., and Kelley, P.: Atmospheric oxidation chemistry and ozone production: Results from SHARP 2009 in Houston, Texas, J. Geophys. Res.-Atmos., 118, 5770–5780, https://doi.org/10.1002/jgrd.50342, 2013.
Salmon, R. A., Bauguitte, S. J. B., Bloss, W., Hutterli, M. A., Jones, A. E., Read, K., and Wolff, E. W.: Measurement and interpretation of gas phase formaldehyde concentrations obtained during the CHABLIS campaign in coastal Antarctica, Atmos. Chem. Phys., 8, 4085–4093, https://doi.org/10.5194/acp-8-4085-2008, 2008.
Sheehy, P. M., Volkamer, R., Molina, L. T., and Molina, M. J.: Oxidative capacity of the Mexico City atmosphere – Part 2: A ROx radical cycling perspective, Atmos. Chem. Phys., 10, 6993–7008, https://doi.org/10.5194/acp-10-6993-2010, 2010.
Shirley, T. R., Brune, W. H., Ren, X., Mao, J., Lesher, R., Cardenas, B., Volkamer, R., Molina, L. T., Molina, M. J., Lamb, B., Velasco, E., Jobson, T., and Alexander, M.: Atmospheric oxidation in the Mexico City Metropolitan Area (MCMA) during April 2003, Atmos. Chem. Phys., 6, 2753–2765, https://doi.org/10.5194/acp-6-2753-2006, 2006.
Stone, D., Whalley, L. K., and Heard, D. E.: Tropospheric OH and HO2 radicals: field measurements and model comparisons, Chem. Soc. Rev., 41, 6348–6404, https://doi.org/10.1039/c2cs35140d, 2012.
Stone, D., Whalley, L. K., Ingham, T., Edwards, P. M., Cryer, D. R., Brumby, C. A., Seakins, P. W., and Heard, D. E.: Measurement of OH reactivity by laser flash photolysis coupled with laser-induced fluorescence spectroscopy, Atmos. Meas. Tech., 9, 2827–2844, https://doi.org/10.5194/amt-9-2827-2016, 2016.
Tan, Z., Fuchs, H., Lu, K., Hofzumahaus, A., Bohn, B., Broch, S., Dong, H., Gomm, S., Häseler, R., He, L., Holland, F., Li, X., Liu, Y., Lu, S., Rohrer, F., Shao, M., Wang, B., Wang, M., Wu, Y., Zeng, L., Zhang, Y., Wahner, A., and Zhang, Y.: Radical chemistry at a rural site (Wangdu) in the North China Plain: observation and model calculations of OH, HO2 and RO2 radicals, Atmos. Chem. Phys., 17, 663–690, https://doi.org/10.5194/acp-17-663-2017, 2017.
Thurston, G. D., Lippmann, M., Scott, M. B., and Fine, J. M.: Summertime haze air pollution and children with asthma, Am. J. Resp. Crit. Care, 155, 654–660, 1997.
Whalley, L. K., Lewis, A. C., McQuaid, J. B., Purvis, R. M., Lee, J. D., Stemmler, K., Zellweger, C., and Ridgeon, P.: Two high-speed, portable GC systems designed for the measurement of non-methane hydrocarbons and PAN: Results from the Jungfraujoch High Altitude Observatory, J. Environ. Monitor., 6, 234–241, https://doi.org/10.1039/b310022g, 2004.
Whalley, L. K., Furneaux, K. L., Goddard, A., Lee, J. D., Mahajan, A., Oetjen, H., Read, K. A., Kaaden, N., Carpenter, L. J., Lewis, A. C., Plane, J. M. C., Saltzman, E. S., Wiedensohler, A., and Heard, D. E.: The chemistry of OH and HO2 radicals in the boundary layer over the tropical Atlantic Ocean, Atmos. Chem. Phys., 10, 1555–1576, https://doi.org/10.5194/acp-10-1555-2010, 2010.
Whalley, L. K., Blitz, M. A., Desservettaz, M., Seakins, P. W., and Heard, D. E.: Reporting the sensitivity of laser-induced fluorescence instruments used for HO2 detection to an interference from RO2 radicals and introducing a novel approach that enables HO2 and certain RO2 types to be selectively measured, Atmos. Meas. Tech., 6, 3425–3440, https://doi.org/10.5194/amt-6-3425-2013, 2013.
Whalley, L. K., Stone, D., Bandy, B., Dunmore, R., Hamilton, J. F., Hopkins, J., Lee, J. D., Lewis, A. C., and Heard, D. E.: Atmospheric OH reactivity in central London: observations, model predictions and estimates of in situ ozone production, Atmos. Chem. Phys., 16, 2109–2122, https://doi.org/10.5194/acp-16-2109-2016, 2016.
Woodward-Massey, R., Slater, E., Ye, C., Seakins, P. W., Whalley, L. K., and Heard, D. E.: Assessment of interferences during field measurements of OH using a laser-induced fluorescence instrument equipped with a scavenger injector, EGU, Vienna, Austria, 2017.