the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 05 Nov 2018
| 05 Nov 2018
Physical state of 2-methylbutane-1,2,3,4-tetraol in pure and internally mixed aerosols
Jörn Lessmeier
Hans Peter Dette
Adelheid Godt
2-Methylbutane-1,2,3,4-tetraol (hereafter named tetraol) is an important oxidation product of isoprene and can be considered as a marker compound for isoprene-derived secondary organic aerosols (SOAs). Little is known about this compound's physical phase state, although some field observations indicate that isoprene-derived secondary organic aerosols in the tropics tend to be in a liquid rather than a solid state. To gain more knowledge about the possible phase states of tetraol and of tetraol-containing SOA particles, we synthesized tetraol as racemates as well as enantiomerically enriched materials. Subsequently the obtained highly viscous dry liquids were investigated calorimetrically by differential scanning calorimetry revealing subambient glass transition temperatures Tg. We also show that only the diastereomeric isomers differ in their Tg values, albeit only by a few kelvin. We derive the phase diagram of water–tetraol mixtures over the whole tropospheric temperature and humidity range from determining glass transition temperatures and ice melting temperatures of aqueous tetraol mixtures. We also investigated how water diffuses into a sample of dry tetraol. We show that upon water uptake two homogeneous liquid domains form that are separated by a sharp, locally constrained concentration gradient. Finally, we measured the glass transition temperatures of mixtures of tetraol and an important oxidation product of α-pinene-derived SOA: 3-methylbutane-1,2,3-tricarboxylic acid (3-MBTCA). Overall, our results imply a liquid-like state of isoprene-derived SOA particles in the lower troposphere at moderate to high relative humidity (RH), but presumably a semisolid or even glassy state at upper tropospheric conditions, particularly at low relative humidity, thus providing experimental support for recent modeling calculations.
- Article
(1652 KB) - Full-text XML
-
Supplement
(1352 KB) - BibTeX
- EndNote





Aerosol particles have been identified as a key factor for answering what drives our climate and, in particular, what is the anthropogenic contribution to climate change (Carslaw et al., 2013; Gordon et al., 2016; IPCC, 2013; Seinfeld et al., 2016). Aerosol particles influence our climate (i) directly through scattering and reflection as well as absorption of sunlight and of infrared radiation emitted from the earth's surface and (ii) indirectly by acting as nuclei for water and ice cloud formation (Andreae, 1997; Haywood and Boucher, 2000; Lohmann and Feichter, 2005; Pöschl, 2005). These direct and indirect effects are governed by the particles' chemical and physical properties such as their chemical composition and phase state, both of which affect important aerosol processes such as gas–particle partitioning, the rate of surface or bulk chemical reactions, water uptake kinetics, their light-scattering properties, and their ability to act as cloud condensation nuclei or ice nuclei (Baustian et al., 2013; Berkemeier et al., 2013, 2014; Kidd et al., 2014; Kuwata and Martin, 2012; Martin, 2000; Moise et al., 2015; Perraud et al., 2012; Saukko et al., 2012b; Schill et al., 2014; Shiraiwa et al., 2012; Zarzana et al., 2012). Despite considerable progress in our understanding of atmospheric aerosol processes, aerosols are still considered one of the largest uncertainties when predicting the current and future climate through model calculations (IPCC, 2013). This is especially true for secondary organic aerosol (SOA) particles that originate from the oxidation of biogenic and anthropogenic volatile organic compounds (VOCs) (Hallquist et al., 2009; Jimenez et al., 2009; Kanakidou et al., 2005; Kroll and Seinfeld, 2008; Shrivastava et al., 2017). The great variability in volatile precursors, atmospheric oxidants, and possible reaction pathways leads to a multitude of SOA compounds (Goldstein and Galbally, 2007), thus complicating the prediction of the chemical and physical properties of SOA particles (Donahue et al., 2011; Hallquist et al., 2009; Koop et al., 2011; Kroll et al., 2011; Li et al., 2016).
One of the major uncertainties is the aerosol particle phase state. Atmospheric in situ measurements of the phase state of aerosol particles are scarce and often consist of indirect methods such as deducing the phase state from the bouncing behavior of aerosol particles upon collision onto a solid surface (Bateman et al., 2014; Li et al., 2017; Saukko et al., 2012a, b; Virtanen et al., 2010, 2011). This lack of direct in situ measurements emphasizes the importance of laboratory studies that focus on the phase state and phase changes as well as viscosity measurements of representative atmospheric SOA compounds as a function of humidity, temperature, and mixing state (Bateman et al., 2014; Dette et al., 2014; Dette and Koop, 2015; Maclean et al., 2017; Saukko et al., 2012b; Song et al., 2015; Zobrist et al., 2008). The results of such measurements can be compared to results of atmospheric observations (Bateman et al., 2015; Virtanen et al., 2010) and they can be incorporated into models of atmospheric aerosol phase states (Shiraiwa et al., 2017; Shrivastava et al., 2017).
For a long time SOA particles had been assumed to be always in a liquid-phase state at atmospheric conditions (Hallquist et al., 2009; Jimenez et al., 2009; Pankow, 2007). More recently, however, several studies showed that they may indeed occur as liquids but can also be present as highly viscous semisolids or even in a solid glassy state, depending on the ambient relative humidity (RH), temperature, and precursor substances (Bateman et al., 2015; Koop et al., 2011; Kuwata and Martin, 2012; Mikhailov et al., 2009; Saukko et al., 2012a; Virtanen et al., 2010). For example, Virtanen et al. showed that SOA particles derived from α-pinene under the moderate temperature and dry humidity conditions over Scandinavian boreal forests are in a semisolid or even glassy state, whereas Bateman et al. showed that particles derived from isoprene in the warm and humid conditions of the Amazonian rain forests are often present in a liquid state. In the study presented here, we focus on the phase change behavior of 2-methylbutane-1,2,3,4-tetraol (hereafter named tetraol). Tetraol forms in the atmosphere from isoprene oxidation and has been identified as one prominent marker compound for isoprene oxidation products, which together with isoprene epoxydiols and other polyols make up a significant portion of isoprene-derived SOA (Claeys, 2004; Ebben et al., 2014; Isaacman-VanWertz et al., 2016; Nozière et al., 2015; Surratt et al., 2010; Zhang et al., 2012). Besides monoterpenes such as α-pinene, isoprene is one of the most abundant biogenic volatile organic compounds emitted into the atmosphere, especially over tropical forests, where it is oxidized to a variety of compounds (Claeys, 2004; Edney et al., 2005; Guenther et al., 1995, 2012; Martin et al., 2010; Pöschl et al., 2010). Here, we investigated through calorimetric measurements at which temperatures tetraol transforms from a liquid into a glass, both as a pure substance and in mixtures with water. Furthermore, we investigated whether the presence of stereoisomers affects the phase state of tetraol. This is of particular interest because recent studies have shown that tetraol can occur as an enantiomerically enriched mixture in the atmosphere due to its direct emission by plants that produce an excess of one of the four stereoisomer of tetraol in addition to the atmospheric non-stereoselective oxidation pathway from isoprene (Ebben et al., 2014; González et al., 2011; Nozière et al., 2011). Therefore, we synthesized enantiomerically enriched samples of all of the four stereoisomers and investigated these samples in addition to the two racemates. The synthesis is described in detail in this paper. Finally, we investigated changes in the glass transition temperature of tetraol when it is mixed with 3-methylbutane-1,2,3-tricarboxylic acid (3-MBTCA), which is a SOA marker compound for α-pinene oxidation products (Dette et al., 2014; Kostenidou et al., 2018; Szmigielski et al., 2007) and was chosen because isoprene and α-pinene are also found in the same air masses (Bateman et al., 2015; Kuhn, 2002) and their oxidation products in the corresponding SOA.
2.1 Measurement devices
2.1.1 MARBLES
Some samples were transformed into dry glassy samples by way of spray-drying using the MARBLES (metastable aerosol by low-temperature evaporation of solvent) technique that we developed recently (Dette et al., 2014). In short, an aqueous solution of the water-soluble compound is atomized into an aerosol, which is subsequently dried in several consecutive diffusion-drying tubes filled with silica gel beads (Silica Gel Orange, Roth). The dried aerosol particles are collected by impaction directly into a differential scanning calorimeter (DSC) sample pan, which is sealed afterwards within a glove box at a relative humidity of ≤0.1 %.
2.1.2 Differential scanning calorimetry
A differential scanning heat flow calorimeter (Q100, TA Instruments) with a temperature accuracy of ±0.4 K at heating or cooling rates of 10 K min−1 was used. Instrument calibration was performed using nine different calibration standards at variable heating and cooling rates between −10 and +10 K min−1 (Riechers et al., 2013). For these studies, an aluminum pan was filled with 3–10 mg of a material (see Sect. 2.2) and sealed hermetically. The sample was transferred into the DSC and cooled or heated at a rate of 10 K min−1 while measuring the heat flow difference between the sample and an air-filled reference pan. The glass transition of a sample is detected as a nearly stepwise change of the heat flow signal. The corresponding glass transition temperature Tg is determined as the intersection of the extrapolation of the baseline with the line of maximum slope in the heat flow signal, following a convention by Angell (Angell, 2002). This so-called onset is measured at a heating rate of 10 K min−1. Because of small ambiguities in the baseline extrapolation and the determination of the point of maximum slope as well as small differences between repeated measurements, we estimate an accuracy in Tg of about ±2 K, despite the calorimeter's principal accuracy of ±0.4 K. The melting of a sample is detected as a peak in the heat flow signal. The melting temperature is determined as the point of maximum slope in the low-temperature part of the melting peak.
2.1.3 Water activity measurements
The water activity of several aqueous samples was measured using a commercial water activity meter (Decagon model series 3 TE) at 25 ∘C. This device measures the dew point of the gas phase above an aqueous solution, from which the corresponding water activity is obtained with an absolute accuracy of ±0.03.
2.2 Sample preparation
2-Methylbutane-1,2,3,4-tetraol (tetraol 1) was obtained as described in Sect. 3.1 and, in more detail, in the Supplement. 3-Methylbutane-1,2,3-tricarboxylic acid (3-MBTCA) was prepared as published (Dette et al., 2014).
The water used for the preparation of aqueous mixtures was doubly distilled deionized water.
2.2.1 Pure tetraols
(2R,3S)65-Tetraol and (2S,3S)64-tetraol, which were available only in small amounts, were dried by heating to 80 ∘C for 120 min. The dried samples were transferred into a DSC sample pan at room atmosphere. The racemates rac-(2R,3R)-tetraol and rac-(2R,3S)-tetraol, and (2S,3R)86-tetraol and (2R,3R)97-tetraol, which were available in larger amounts, were dried using the spray-drying technique MARBLES as described above.
2.2.2 Two-component mixtures
Mixtures of stereoisomers were prepared by mixing their dilute aqueous solutions in the desired ratio. The mixture was dried with the MARBLES setup. Mixtures of tetraol and 3-MBTCA were prepared in the same manner.
2.2.3 Aqueous tetraol samples
Aqueous tetraol samples of particular mass fractions were prepared by weighing vacuum-dried tetraol and water directly into DSC sample pans at the desired ratio under room atmosphere. The pans were hermetically sealed immediately after sample preparation and were stored for at least 7 days at room temperature prior to DSC analysis to ensure a complete diffusive mixing of the components within the sample pan (see discussion below).
For the investigation of water uptake, a vacuum-dried tetraol was weighed into a DSC sample pan inside a glove box (≤0.1 % RH). The sample was then quickly transferred into another temperature- and humidity-controlled chamber (see Sect. 3.4, below). This chamber consists of a double-walled glass container, in which the walls and the bottom are cooled by a temperature-controlled circulating water flow, closed with a lid. In the center of the chamber a 15 mm diameter copper rod is placed vertically as a support for the DSC sample pan. The chamber is filled with a saturated aqueous NaCl solution up to about 1 cm below the upper end of the rod. Therefore, the air above that bath experienced by the sample in the pan is at a relative humidity of ∼75 %, when the cooling water temperature and, hence, the bath and rod temperature is set to 0.5 ∘C (Wexler and Hasegawa, 1954). In the experiment described, a sample of tetraol in a DSC sample pan was exposed to these conditions for 250 min. Thereafter, the sample pan was sealed immediately, weighed, and then directly transferred into the DSC for calorimetric measurement.

Scheme 1Synthesis of the racemic and enantiomerically enriched tetraols. NMO indicates N-methylmorpholine-N-oxide. The reaction was performed at room temperature, unless another temperature is given in the scheme.
3.1 Synthesis of 2-methylbutane-1,2,3,4-tetraol (tetraol)
For our study, we needed 2-methylbutane-1,2,3,4-tetraol (tetraol) in gram amounts as racemates and as enantiomerically enriched materials of all of its four stereoisomers. Because of their importance in biological and atmospheric research the syntheses of the stereoisomers of tetraol have been devised already (Cole-Filipiak et al., 2010; Duvold et al., 1997; Ebben et al., 2014; Enders et al., 2007; Fontana et al., 2000; Ghosh et al., 2012; Giner et al., 2002; Hoeffler et al., 2000; Koumbis et al., 2007; Moen et al., 2007; Robinson et al., 2009; Sharma et al., 2008; Urbansky et al., 2004). Among the reported syntheses, the reports of Ghosh et al. (2012), Moen et al. (2007), and Sharma et al. (2008) appeared most attractive to us as they show an access to both diastereomers through the same reaction sequence and give the four stereoisomers as stereochemically highly homogeneous materials with an enantiomeric excess (ee) of 80 %–99 %. We decided to use the strategy of Moen et al. (2007) because it requires the fewest number of steps. It takes up the procedures described by Anthonsen et al. (1976, 1980) for the preparation of the two diastereomeric tetraols as racemates.
In brief (Scheme 1), we started with the prochiral (E)-methylbut-2-enedioic acid (mesaconic acid, (E)-1) and the prochiral (Z)-methylbut-2-enedioic acid (citraconic acid, (Z)-1) which were converted into the diesters (E)- and (Z)-2. The syn-dihydroxylation of these diesters gave the two diastereomeric diols 3 as the racemates rac-(2R,3R)-3 and rac-(2R,3S)-4. Subsequent reduction provided the two diastereomeric tetraols as the racemates rac-(2R,3R)-tetraol and rac-(2R,3S)-tetraol. The enantiomerically enriched tetraols were obtained through a kinetic resolution at the stage of the diols 3 via enzymatic esterification. For purification of the tetraols, they were temporarily converted into the diacetonides 5. In the following, details of the individual synthetic steps and on product analysis are given.
One-step esterifications of the diacids (Z)- and (E)-1 on a scale of up to 100 g were achieved with methanol and sulfuric acid as the reagents (Anthonsen et al., 1980; Kar and Argade, 2002; Klimovica et al., 2011; Tripp et al., 2005) and through heating the reaction mixture to reflux in a Soxhlet extractor with a thimble filled with MgSO4 for trapping the evolving water. The diesters (Z)- and (E)-2 were isolated by washing with base and subsequent distillation.
Following the report of Moen et al. (2007) syn-dihydroxylations of the diesters (E)- and (Z)-2 were achieved through applying K2OsO2(OH)4 and N-methylmorpholine-N-oxide with the addition of citric acid in a mixture of tert-butanol and water. The reported work-up had to be modified for running this reaction on a multigram scale, such as starting with 38 g of diester (E)-2 and 107 g of diester (Z)-2. Moen et al. (2007) described the removal of tert-butanol by distillation followed by an extraction of the residual aqueous phase with diethyl ether. Applying this procedure to isolate diol rac-(2R,3S)-3 on a multigram scale, we found this procedure to be impractical because several liters of diethyl ether would be needed for extraction of this diol due to its low solubility. Therefore, when thin-layer chromatography proofed the reaction to be complete, the organic and aqueous phases were separated and the aqueous phase was extracted with a mixture of diethyl ether and tetrahydrofuran. Filtering of the combined organic phases through silica gel provided diol rac-(2R,3R)-3. We did not test the solubility of the diastereomeric diol rac-(2R,3S)-3 in diethyl ether. We simply applied the same work-up procedure to obtain diol rac-(2R,3S)-3, except for substituting the filtration through silica gel with recrystallization.
In order to obtain enantiomerically enriched materials, kinetic chiral resolutions of rac-(2R,3S)-diol 3 and rac-(2R,3R)-diol 3 were carried out making use of lipase A from Candida antarctica (CAL-A). In the following, if the text applies to enantiomerically enriched materials, the figure of ee is denoted as a subscript; e.g., (2S,3S)97-4 indicates a material consisting of (2S,3S)-4 and (2R,3R)-4 in a ratio of 98.5 to 1.5 and therefore having a 97 % excess of (2S,3S)-4. Likewise (2S,3S)x-4 indicates an enantiomerically enriched material with undetermined ee. CAL-A catalyzes the esterification of the secondary hydroxyl groups of diols 3 with vinyl butanoate and thereby favors the diols (2R,3S)-3 and (2S,3S)-3 as reaction partners over their enantiomers (Moen et al., 2007). The attempt of a chiral resolution of rac-(2R,3R)-diol 3 according to the conditions described by Moen et al. (2007), however, on a scale of 3 g instead of 1 g of diol and with CAL-A immobilized on Immobead 150 instead of Novozym 735, failed. No reaction occurred within 3 days. Increasing the temperature from 7 ∘C to room temperature and adding more CAL-A caused no change within 6 days. However, esterification occurred when less toluene and more vinyl butanoate were used. Under these conditions, kinetic resolution of diol rac-(2R,3R)-3 for 44 h at 14 ∘C resulted in a conversion of about 44 % and gave diol (2R,3R)64-3 and butanoate (2S,3S)97-4. Kinetic resolution of the diastereomeric rac-(2R,3S)-diol 3 for 19.5 h under otherwise identical conditions caused a conversion of about 31 % and gave diol (2S,3R)7-3 and butanoate (2R,3S)86-4. The conversion was deliberately kept lower in the latter case because of the reported lower enantioselectivity of this reaction (Moen et al., 2007). To obtain diol (2S,3R)-3 with a sufficiently high ee value, diol (2S,3R)7-3 was submitted to a kinetic resolution for about 10 days providing diol (2S,3R)65-3 and butanoate (2R,3S)75-4. The reduced solvent volume as compared to the procedure of Moen et al. (2007) resulted in partial separation of the diols as a second liquid phase. Because of this heterogeneity in phase, the monitoring of the conversion by taking samples for 1H NMR spectroscopic analysis was not very exact. Nevertheless, this monitoring was sufficiently indicative of when to stop the reaction. Please note that we did not optimize the reaction conditions, neither with respect to solvent volume and amount of vinyl butanoate nor with respect to temperature and reaction time. The butanoates 4 and leftover diols 3 were separated through column chromatography. The butanoates were coeluted with a compound that was, based on its 1H and 13C NMR spectroscopic data (see Supplement), identified as butanoic acid. The presence of this compound was considered irrelevant because the next step is the reduction of the diols 3 to the tetraols.
To determine the ee values of the diols 3 and butanoates 4, gas chromatography (GC) and 1H NMR spectroscopy using (S)-1,1′-bi-2-naphthol as the chiral shift agent were applied (Table S4). NMR spectroscopy was needed because the enantiomeric diols (2R,3S)-3 and (2S,3R)-3 were inseparable with the available GC equipment. The 1H NMR signals of the enantiomers were not well separated, therefore deconvolution was applied to allow the determination of the signal intensities. The analysis of (2R,3R)x-diol 3 revealed that both methods provide about the same ee values (Table S4). Because in the following synthetic steps no reaction takes place at the stereogenic centers, the enantiomeric excesses determined for the diols and butanoates are identical to those of the tetraols derived thereof.
The reduction of the diols 3 to the tetraols with lithium aluminium hydride proceeded smoothly; however, we did not succeed in removing the inorganic salts. Quantitative 1H NMR spectroscopy revealed a tetraol content in the isolated material of only 70 wt %–85 wt %. Moen et al. appear to have faced troubles here, too. Their work-up included three precipitations and the yields ranged from 20 %–70 % (Moen et al., 2007). Our solution to this problem was to convert the tetraols into the diacetonides 5 in order to mask the highly polar hydroxyl groups and therefore allow standard column chromatography on silica gel to be applied for the removal of the inorganic salts as well as organic side products. The synthesis of the diacetonide (2S,3R)100-5 has been described (Anthonsen et al., 1980; Proteau et al., 1999). Just as Proteau et al. (1999) we used 2,2-dimethoxypropane in the presence of toluene sulfonic acid. Our observations indicate that (2R,3S)-tetraol and (2S,3R)-tetraol react slower than their diastereomers, which may simply be the consequence of lower solubility in 2,2-dimethoxypropane but also may be caused by the difference in configuration. Because we used an acidic ion exchange resin as the catalyst for demasking, the catalyst could be removed from the tetraols simply by filtration. In this way materials with a tetraol content of 98–99 wt %–±3 wt % were obtained.
Table 1Glass transition temperatures Tg of vacuum-dried and spray-dried racemic and enantiomerically enriched tetraols and melting temperatures Tm for crystallized samples. The enantiomeric excess is given as subscript. The spray-dried samples were handled and sealed in a dry glove box before DSC measurement and, therefore, provide the most accurate Tg values, which is why they are given in boldface.

Please note that we synthesized the enantiomerically enriched tetraols before the racemic tetraols. Therefore, the procedures reported for the latter are the more refined, albeit not optimized ones, which we, if we needed more material, would apply to the enantiomerically enriched materials. The procedure of reduction and work-up to obtain pure tetraol can possibly be slimmed down to (1) hydrolysis of the reduction reaction mixture, neutralization or mild acidification with sulfuric acid, and removal of solvent and water; (2) reaction with 2,2-dimethoxypropane; (3) chromatography on silica gel; and (4) acetonide cleavage. We expect the masking as acetonide and subsequent demasking to be applicable also to the isolation of other polyols from mixtures with polar substances.
3.2 Glass transition temperatures of the tetraols
The samples available for the calorimetric study are summarized in Table 1. All materials – two racemates and four enantiomerically enriched materials – were obtained as highly viscous colorless liquids, in agreement with previous notes of a highly viscous or gel-like state (Ebben et al., 2014). When these liquids were cooled in the bulk state, none of them crystallized but all exhibited a liquid-to-glass transition. For each of the six materials, the glass transition temperature was determined by differential scanning calorimetry. Because of their hygroscopic nature, the samples required drying before the DSC measurements. All materials were dried through heating at reduced pressure. When sufficient material was available, it was spray-dried using the MARBLES technique.
For racemic and enantiomerically enriched tetraols, Tg values for vacuum-dried and spray-dried samples were obtained (see Table 1). Vacuum-dried samples always showed slightly lower Tg values than spray-dried samples. The most likely reason for this difference is that vacuum-dried samples were transferred into DSC sample pans and weighed at room atmosphere. Therefore, most probably, uptake of trace amounts of water occurred, which acted as a plasticizer, thus reducing the Tg values of these samples. The amount of water taken up by the sample can vary from experiment to experiment, for example caused by slightly varying times needed for the sample preparation. This explains as well why there is a difference in the Tg of (2R,3R)97-tetraol and (2S,3S)64-tetraol, which should otherwise be identical due to their enantiomeric nature. We conclude that the Tg values of spray-dried samples handled and sealed in a dry glove box are much less affected by traces of water and, thus, are more reliable (and therefore written boldfaced in Table 1). These measured Tg values of 238 and 245 K are considerably lower than the value of 283 K predicted by Berkemeier et al. (2014), which was based on a structure–property relationship modeling approach. This difference emphasizes the importance of laboratory measurements. While structure–property relationship predictions can often provide reasonable estimates for average phase transition temperatures of a large number of substances, they may show significant deviations on an individual compound basis.
The spray-dried samples showed no difference in Tg between rac-(2R,3S)-tetraol and (2S,3R)86-tetraol and between rac-(2R,3R)-tetraol and (2R,3R)97-tetraol. Due to a lack of sufficient amount of material, spray-drying was possible for only one enantiomer of each diastereomer. It is to be expected that the Tg values of enantiomers are equal because enantiomers share the same physical properties (Jabrane et al., 1995). Accordingly, our data strongly suggest that there is no difference between the Tg values of the enantiomers and their racemic mixtures of both diastereomers. There is, however, a difference of 7 K between the Tg values of the two diastereomers. This difference was further investigated by mixing the racemates in different mass ratios in an aqueous solution and subsequently spray-drying them with the MARBLES technique. The Tg values of each of these mixtures were determined with DSC measurements. The resulting Tg values show a nearly linear relationship with mass ratio, as shown in Fig. 1.

Figure 1Glass transition temperatures of mixtures of rac-(2R,3S)-tetraol and rac-(2R,3R)-tetraol. The Tg values show a nearly linear relationship with a Gordon–Taylor constant kGT of 0.92.
A Gordon–Taylor fit (Gordon and Taylor, 1952) of the Tg data (Eq. 1) results in a Gordon–Taylor constant kGT of 0.92±0.09, implying a nearly linear relationship (i.e., kGT=1), which is expected for chemically similar compounds. In general, the Gordon–Taylor equation is used to describe the glass transition temperature Tg of binary mixtures, with w1 and w2 as their respective mass fractions, Tg1 and Tg2 as the glass transition temperatures of the pure compounds, and an empirical fitting constant kGT.
While preparing DSC samples of rac-(2R,3S)-tetraol, the bulk sample crystallized, whereas none of the other samples crystallized, even during weeks of storage. The crystals of rac-(2R,3S)-tetraol were used as seeds for triggering the crystallization of the corresponding enantiomers. We note that all attempts of seeding the diastereomers with any of the crystallized compounds were unsuccessful. This may be due to different crystal structures of the two diastereomers. The melting points Tm of the crystallized compounds were also determined, see Table 1, and the corresponding ratios Tg∕Tm are very close to the value of 0.7 derived for a large number of molecular compounds (Koop et al., 2011). We note that the racemate rac-(2R,3S)-tetraol shows a Tm significantly higher than those of the individual enantiomers, which indicates the formation of a racemic crystal, i.e., a crystal that contains the two enantiomers in a 1:1 ratio in the elementary cell (Roozeboom, 1899).
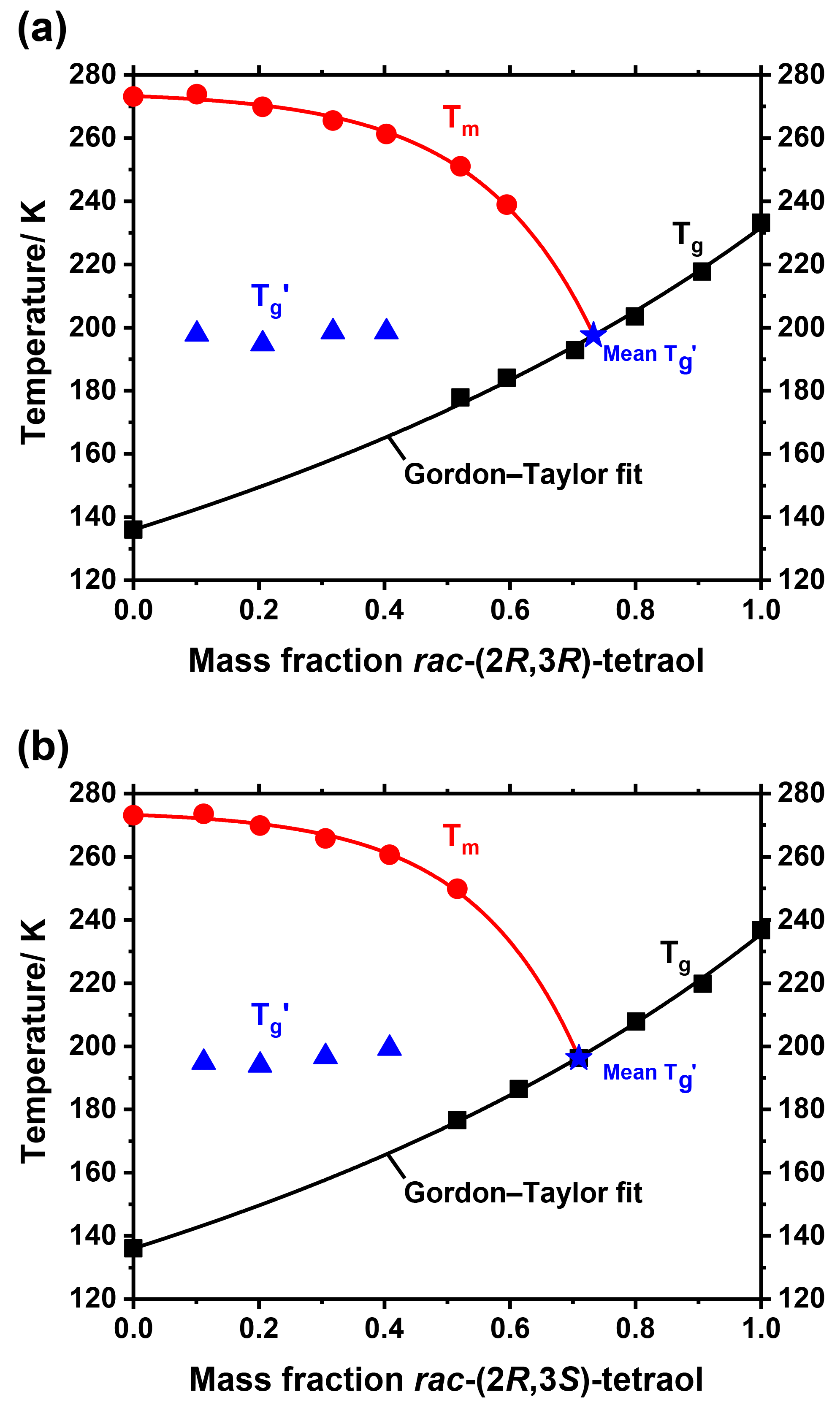
Figure 2Partial phase diagram of mixtures of rac-(2R,3R)-tetraol with water (a) and rac-(2R,3S)-tetraol with water (b). Shown are the ice melting temperatures Tm (red circles), glass transition temperatures Tg (black squares), and maximally freeze-concentrated glass transition temperatures (blue triangles). The melting temperatures are fitted with an exponential function and the glass transition temperatures are fitted with the Gordon–Taylor equation yielding a Gordon–Taylor constant of (a) and (b), respectively. The values are plotted at the overall mass fraction of the solution before cooling. The composition of the amorphous fraction of a sample after the water partly crystallized is depicted as the mean shifted towards the Gordon–Taylor fit (blue star) with a value of 197 K for the rac-(2R,3R)-tetraol and 196 K for the rac-(2R,3S)-tetraol.
3.3 Phase diagrams of mixtures of tetraol with water
Dry tetraol does not exist in the atmosphere because water is always present. Therefore, it is important to investigate possible phase states and transitions of tetraol–water mixtures as a function of temperature and water content. For example, how does the tetraol glass transition change with water content and how strongly is the ice melting point reduced as a function of tetraol mass fraction? Because tetraol and water are totally miscible over the entire concentration range, it is possible to investigate mixtures at any mass ratio between 0 and 1. We prepared nine different mixtures by weighing appropriate amounts of racemic tetraols and water directly into DSC sample pans, which were sealed immediately. Thereafter, the samples were equilibrated at room temperature for at least 7 days to ensure complete diffusive mixing of the two components. That a homogeneous mixture was reached within this time period was proven by DSC measurements which revealed only a single glass transition (see Sect. 3.4 below). Figure 2 shows for both racemates partial phase diagrams of tetraol–water mixtures, constructed from the measured phase transition temperatures. The diagrams include the glass transition temperatures Tg (black squares), the ice melting points Tm (red circles), and the glass transition temperature of the maximally freeze-concentrated solution (blue triangles). The of an aqueous mixture can be measured when, in samples with high to medium water mass fractions, ice crystallization is the first phase transition that occurs upon cooling. In these cases, some of the water nucleates and forms small ice crystals. Because of this ice formation the remaining solution is depleted in water and becomes enriched in tetraol until the system reaches the ice–solution equilibrium, i.e., where the current temperature matches the freezing point of the remaining solution. When the temperature is decreased further, as is done in the experiments described above, the ice crystals continue to grow and the solution becomes more and more concentrated in tetraol, thus increasing in viscosity. At some point the temperature is so low and the tetraol concentration becomes so large that the remaining solution forms a glass and no further ice crystal growth occurs due to the hindered macroscopic diffusion of water molecules in the now glassy phase. This temperature is termed and marks the intersection of the ice-melting-point curve with the glass transition curve. Note that it is common to plot the values at the overall mass fraction of the samples before ice formation. The actual mass fraction of the maximally freeze-concentrated solution after the water partly crystallized can be derived by determining the mean of the values, plotting this mean as a horizontal line and obtaining its intersection with the glass transition curve from the Gordon–Taylor fit (blue star).
The experimental glass transition temperatures were fitted with the Gordon–Taylor equation, resulting in a kGT of 1.53±0.08 for rac-(2R,3R)-tetraol (Fig. 2a). The preparation of the tetraol–water mixtures could be done under room atmosphere conditions only; thus, traces of water may have been taken up by the vacuum-dried tetraol during this preparation. For this reason, the tetraol mass fraction calculated for each tetraol–water mixture may be a little too high. Based on the data obtained for vacuum-dried and spray-dried samples (Table 1), the maximum error in the water mass fraction is estimated to be 0.03 for Fig. 2a and 0.05 for Fig. 2b, with smaller errors at smaller tetraol mass fractions. For that reason, we used the Tg values of the vacuum-dried tetraols for the Gordon–Taylor analysis shown in Fig. 2 to allow for a better comparability. Using the Tg derived from spray-dried samples of pure tetraol, changes the derived Gordon–Taylor curve only slightly yields a kGT of 1.68±0.13.
The partial phase diagram of rac-(2R,3S)-tetraol (Fig. 2b) is very similar to that of the diastereomeric racemate. The glass transition curve fitting results in a kGT of 1.58±0.05 (1.84±0.15 when using the Tg derived from a spray-dried sample of pure tetraol). The enantiomerically enriched materials (2S,3R)86-tetraol and (2R,3R)97-tetraol gave identical phase diagrams as their corresponding racemates within experimental uncertainty and are therefore not shown.
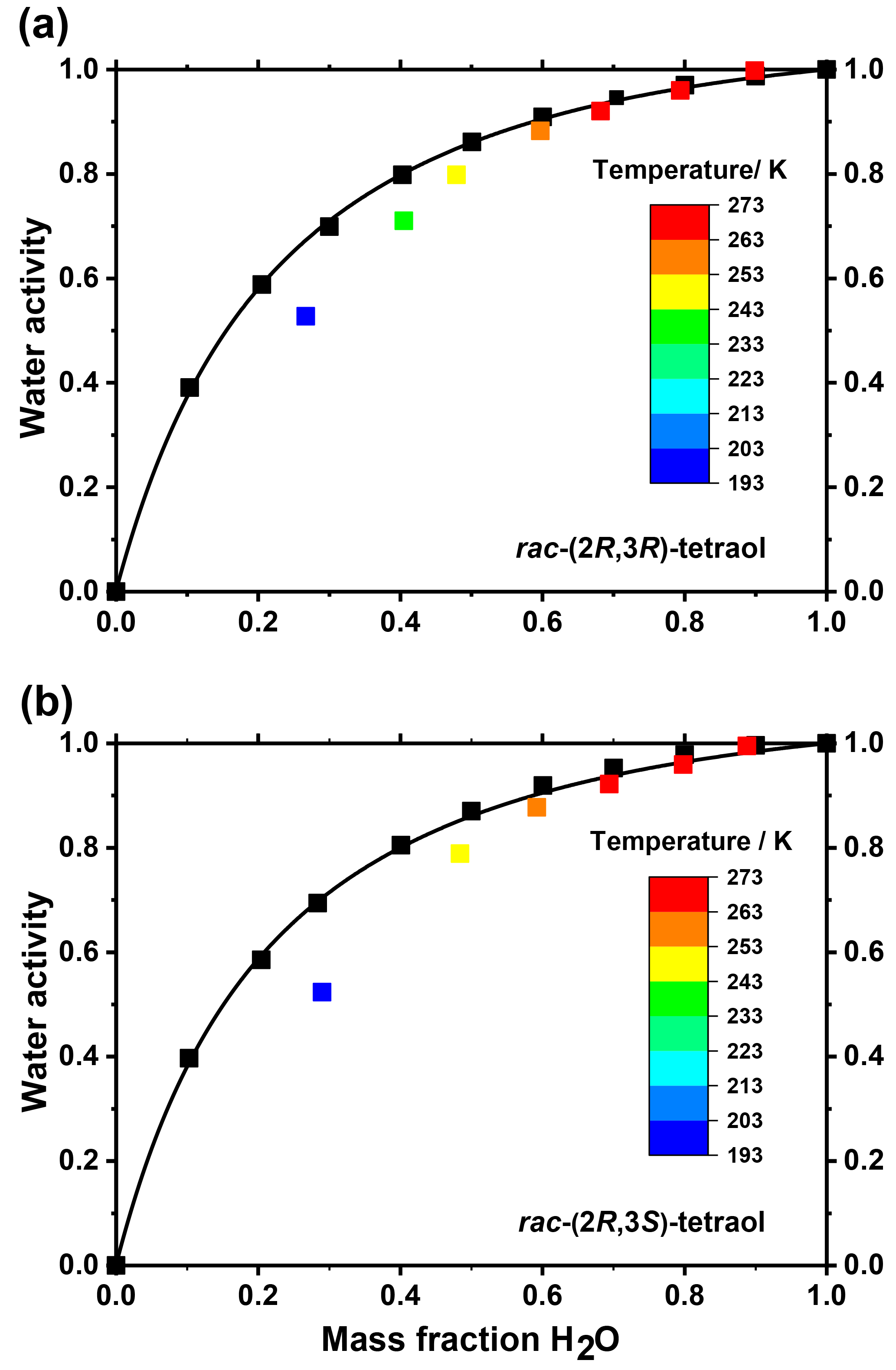
Figure 3Measured water activity of mixtures of (a) rac-(2R,3R)-tetraol with water and (b) rac-(2R,3S)-tetraol with water at 298.15 K (black squares) and at their ice melting points (colored squares). The curves are fits to the data: for details see main text.
From the two phase diagrams in Fig. 2 some conclusions can be drawn regarding the atmospheric phase state of tetraols. Aerosol particles that consist predominantly of racemic or enantiomerically enriched tetraol are likely to be amorphous rather than crystalline. At low water mass fractions, i.e., at low atmospheric relative humidity, the particles may be in a glassy or a highly viscous liquid state at the low temperatures of the upper troposphere. At medium to high water mass fractions, corresponding to moderate or high relative humidity, two scenarios are possible. First, the particles may be in a homogeneous, highly viscous liquid state. Second, when water crystallizes, they are likely to be in a heterogeneous state, consisting of a crystalline ice core that is surrounded by a tetraol-rich shell. This shell would be in a liquid state above the glass transition temperature of the maximally freeze-concentrated tetraol solution at about K for both diastereomers, i.e., over nearly the entire tropospheric temperature range.
In order to apply the above data better to atmospheric conditions, it is helpful to plot the glass transition temperatures as a function of water activity (aw) rather than mass fraction, because for particles in equilibrium with the surrounding air, aw directly corresponds to the ambient relative humidity. Therefore, we measured the water activity of tetraol–water mixtures with mass fractions ranging from 0 to 1 at a temperature of 298.15 K. The resulting data are shown in Fig. 3 together with a parameterization curve derived by fitting the data according to an approach developed previously (Zobrist et al., 2008). As expected, the water mass ratio dependence of aw is practically the same for both racemates within experimental uncertainty.
We further measured ice melting points for the tetraol–water mixtures and calculated the water activity of a corresponding liquid solution at this temperature according to Koop and Zobrist (2009). With these data we were able to describe the temperature dependence of aw as a function of tetraol concentration. By this procedure, the glass transition curve of Fig. 2 can be transferred from the mass fraction into the water activity regime. The result of this analysis is given in Fig. 4, where the grey shaded area indicates the temperature range at which an aqueous tetraol aerosol particle most likely transitions from a liquid to a glass upon cooling at a certain relative humidity. The relatively wide range of the glass transition region (3σ; grey shaded area) includes uncertainties in the Gordon–Taylor fits of Fig. 2 as well as those of the temperature dependence of aw stemming from two slightly different fitting procedures. (For more details see Supplement.) Also shown in Fig. 4 is the water-activity-dependent ice-melting-point curve (red line) as well as the homogeneous ice-nucleation-temperature curve (black line) (Koop et al., 2000; Koop and Zobrist, 2009).
The phase diagrams shown in Fig. 4 now allow a more detailed comparison of our experimental laboratory results with the findings from field measurements of Bateman et al. (2015) about the phase state of SOA particles in the Amazonian rain forest. They stated, in agreement with a study by Song et al. (2015) on laboratory-produced isoprene-derived SOA, that said particles were primarily liquid at least near the surface where temperature was almost always above 296 K and relative humidity was above 80 % (i.e., particle water activity is above 0.8) about 70 % and 80 % of the time in the dry and wet season, respectively (Bateman et al., 2015; Song et al., 2015). Our measurements show that tetraol, as a representative compound for SOA substances in the Amazonian rain forest, is indeed always liquid above 273 K at high and even at low relative humidity. Bateman et al. further observed that the relative humidity dropped to values of ∼50 % during some hot afternoons in the dry season. They speculated whether these conditions may lead to semisolid or solid particles, although they deemed this not to be very likely owing to the high temperatures of 300–310 K during these low-RH events. Our measurements show that, at least for pure tetraol aerosol particles, the temperature would indeed have to be below 210 K at a RH of 50 % in order for solid SOA particles to form. Our measurements thus imply that even semisolid SOA particles are unlikely at the observed low-RH conditions in the dry season of the Amazonian rain forest.
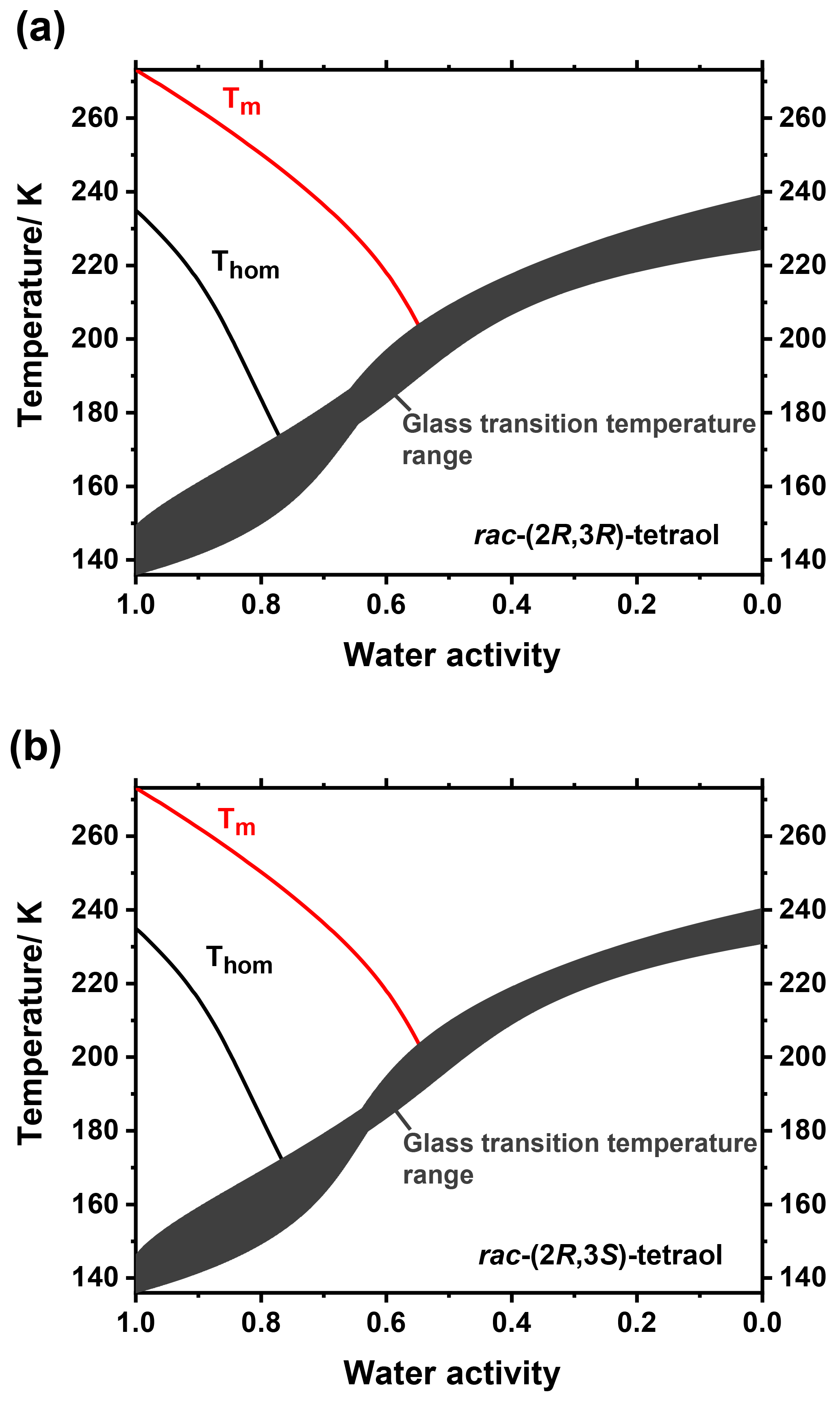
Figure 4Partial phase diagrams of mixtures of rac-(2R,3R)-tetraol with water (a) and rac-(2R,3S)-tetraol with water (b) plotted as a function of water activity. In addition to the ice-melting-point line (Tm; red line) the homogeneous ice nucleation temperatures (Thom; black line) are shown. The dark grey area indicates the glass transition temperatures and their uncertainty; see text for details.
3.4 Water uptake by the tetraol
Tetraol is a hygroscopic compound that takes up water from the ambient air (see discussion above). The kinetics and mechanism of water uptake by hygroscopic aerosol particles are currently an intensely investigated subject. Different ideas on how water uptake into and water diffusion within particles occur are studied experimentally or are being described using different types of modeling approaches (Berkemeier et al., 2013, 2014; Hosny et al., 2013, 2016; Mikhailov et al., 2009; Rothfuss et al., 2018; Zaveri et al., 2014; Zobrist et al., 2011). It has been shown that particularly in semisolid and glassy matrices water diffusion can be strongly inhibited, thereby leading to concentration gradients within the particle matrix (Berkemeier et al., 2014; Hosny et al., 2016; Zobrist et al., 2011). Moreover, in the classical case where the diffusion coefficient of the diffusing tracer molecule (here water) is constant throughout the matrix, the time-dependent concentration profile can be calculated assuming Fickian diffusion, thus resulting in relatively shallow exponential concentration gradients. The situation changes dramatically when the diffusion coefficient of the tracer is concentration dependent. For example, water acts as a plasticizer in many matrices, including tetraol, thus reducing their viscosity and, thereby, increasing its own diffusion coefficient. As a result, during the water uptake of a particle a sharp transition line with a steep concentration gradient develops between two nearly homogeneous domains – a pure organic core with very low water diffusivity and a well-mixed organic–water shell with high water diffusivity (Berkemeier et al., 2014; Hosny et al., 2016; Mikhailov et al., 2009; Zobrist et al., 2011). As time progresses, this transition line moves towards the particle's center until finally one homogeneous liquid particle is formed (see Fig. 5).

Figure 5Schematics of water uptake and diffusion into an aerosol particle exposed to a fixed relative humidity with a sharp transition line distinguishing two homogeneous domains during diffusion: the dark green organic core and the light green organic–water shell that is in equilibrium with the gas-phase humidity.
In a very similar manner, water diffuses into a macroscopic tetraol sample placed inside a DSC pan when the sample is exposed to a humid atmosphere before sealing the pan (see Figs. 6 and 7a). In some preliminary DSC measurements this process was observed to occur when samples were exposed to room atmosphere for more than 10 min before the samples were sealed: instead of a single glass transition step two distinguishable glass transition steps were revealed in the thermogram, indicating the presence of two homogeneous but distinguishable compositional domains within the sample. To illustrate this fact, we performed the following experiment. We filled a DSC sample pan with rac-(2R,3R)-tetraol and placed the pan into an environmental chamber on top of a copper rod partly immersed in a tempered saturated aqueous sodium chloride solution (Fig. 6). The relative humidity over such a solution is practically independent of temperature and amounts to 75 % RH. The temperature of the bath and, therefore, also that of the material in the DSC sample pan was set to 0.5 ∘C, i.e., a temperature at which the diffusion of water into the sample is slow.
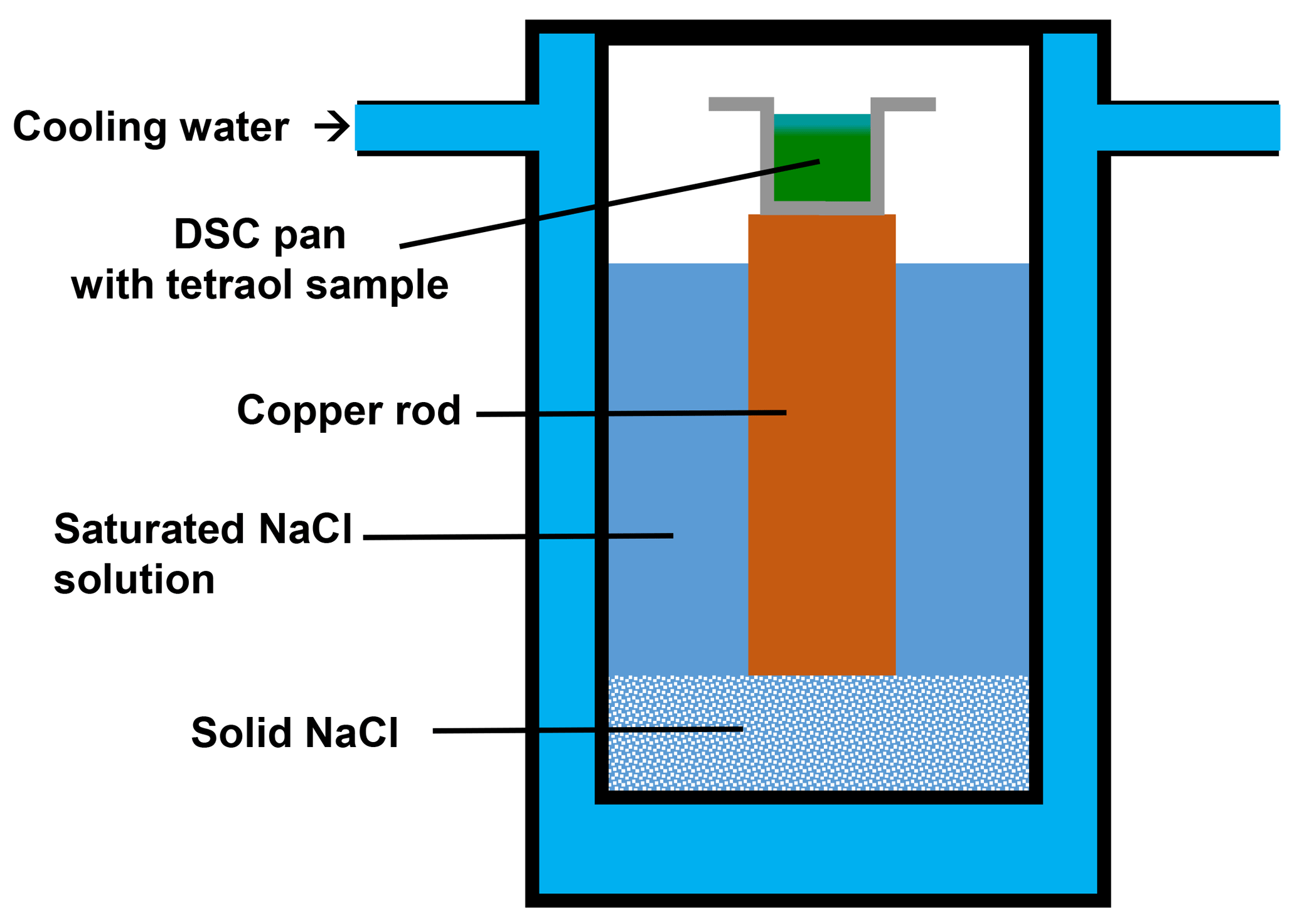
Figure 6Sketch of the environmental chamber used in this study to expose rac-(2R,3R)-tetraol to an atmosphere with 75 % RH. The sample is placed on top of a copper rod immersed in a saturated aqueous NaCl solution inside a temperature-controlled chamber set to 0.5 ∘C.
After 250 min of exposure time the DSC pan was taken out of the chamber, immediately sealed, weighed, and then placed into the DSC. Thereafter, the sample underwent six cooling–heating cycles at rates of 10 K min−1. In each of the first four cycles the sample was first cooled to 153 K and subsequently heated to 303 K with a 20 min holding time at this latter temperature. After the fifth cycle, the sample was heated to 353 K and held at this temperature for 20 min to ensure complete diffusive mixing, because diffusion is significantly faster at higher temperature.
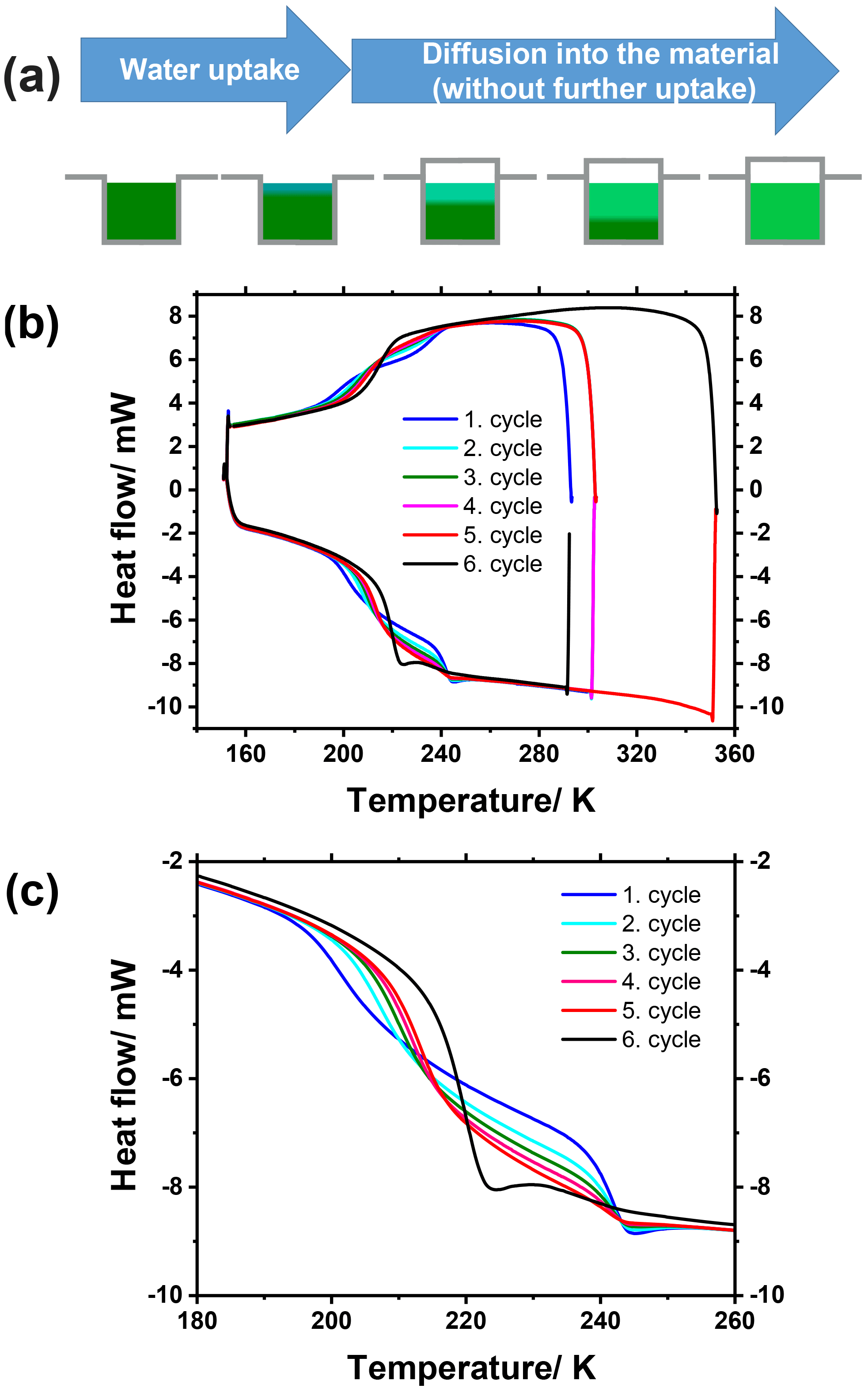
Figure 7(a) Illustration of water uptake by a hygroscopic substance and subsequent water diffusion within it for a sample inside a DSC sample pan. (b, c) DSC thermograms showing several consecutive cooling–heating cycles of wetted rac-(2R,3R)-tetraol (b), with a close-up view of the glass transition region (c). After each of the first four cycles the temperature was held constant at 303.15 K for 20 min before the next cycle was initiated. After the fifth cycle the temperature was raised to 353.15 K and held constant for 20 min before the last sixth cycle was initiated.
Figure 7b shows an overview of the measured thermograms, while Fig. 7c is a close-up of the glass transition temperature range. The first cycle (blue line) reveals that two glass transitions occur in the sample both upon cooling and upon heating, one at about 197 K and another at about 239 K. The latter, higher Tg can be assigned to the bottom domain of the sample consisting of only rac-(2R,3R)-tetraol (see dark green domain in Fig. 7a), because its glass transition occurs at the same temperature that was measured for spray-dried rac-(2R,3R)-tetraol within the measurement uncertainty. The lower Tg can be assigned to the upper, water-containing domain of the sample (Fig. 7a). The glass transition is at lower temperature because water (Tg of 136 K) acts as a plasticizer as discussed above in the analysis of Figs. 2 and 4. In the subsequent cooling and heating cycles changes can be observed for each of the two glass transitions. First, the temperature of the lower Tg increases from one cycle to the next and concomitantly its step height (i.e., the magnitude of the signal) becomes larger. At the same time, the temperature of the higher Tg does not change from one cycle to the next (238.5 K for the first cycle and 238.6 K for the fifth cycle), but its step height decreases with every cycle. These findings are in full agreement with the anticipated behavior of the sample. While the water diffusion gradient transition line moves deeper into the sample with time, the tetraol core becomes smaller and the aqueous tetraol shell becomes larger (see Fig. 7a), which explains the development of the Tg step heights. During this process, the total amount of water is constant as the DSC sample pan is sealed. Therefore, the concentration of water in the growing aqueous shell becomes smaller, leading to an increase in tetraol concentration and, thus, an increase in the lower Tg. Finally, after five cycles, the sample was heated to 353 K and held at that temperature for 20 min to speed up the mixing process. Indeed, the sample fully equilibrated into one homogeneous phase, as can be seen from the fact that only one single glass transition step at 215 K is observed in the final sixth cooling and heating cycle (black lines in Fig. 7b and c). Because the sample was weighed before and after water uptake, the overall water mass fraction was known. With this information the anticipated Tg of this mixture could be inferred from the Gordon–Taylor fit shown in Fig. 2. The measured Tg and the one derived from the Gordon–Taylor fit both amount to 215 K.
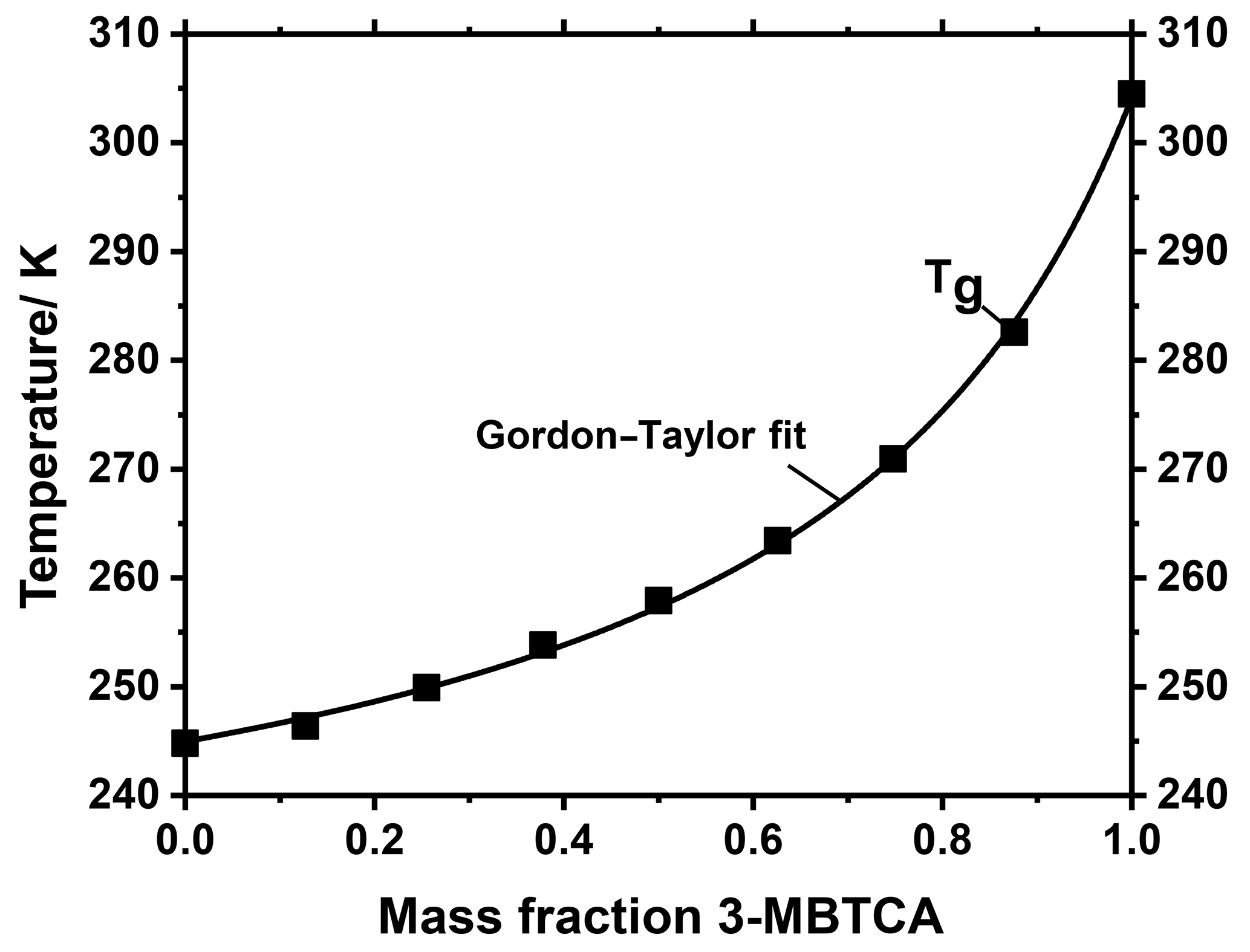
Figure 8Glass transition temperatures (points) of spray-dried mixtures of rac-(2R,3S)-tetraol and 3-MBTCA. The black line represents the Gordon–Taylor fit to the data.
3.5 Glass transition temperatures of mixtures of tetraol with 3-MBTCA
In the atmosphere, it is very unlikely that aerosol particles consist of only one compound such as tetraol. Instead, atmospheric aerosol particles, and in particular (secondary) organic aerosol particles, predominantly consist of complex mixtures of compounds, depending upon their precursor compounds and their state of oxidation (Decesari et al., 2006; Hallquist et al., 2009; Jimenez et al., 2009; Kanakidou et al., 2005; Kroll and Seinfeld, 2008; Li et al., 2016; Nozière et al., 2015; Rogge et al., 1993). Another important representative for secondary organic aerosols is 3-methylbutane-1,2,3-tricarboxylic acid (3-MBTCA). 3-MBTCA is an oxidation product of α-pinene and is often used as a marker compound (Donahue et al., 2012; Szmigielski et al., 2007; Yasmeen et al., 2011). The tetraol studied above is a marker compound for isoprene SOA. Both α-pinene and isoprene are believed to be the most abundant biogenic VOC precursors in tropical forests such as in Amazonia (Bateman et al., 2015). Hence, we further investigated how the glass transition temperatures of tetraol and 3-MBTCA change when these compounds are mixed. We used the MARBLES setup to spray-dry aqueous mixtures of 3-MBTCA and rac-(2R,3S)-tetraol with different mass ratios. The Tg values of the dried samples obtained from DSC experiments are shown in Fig. 8.
These Tg values can be described very well with the Gordon–Taylor equation and a corresponding value of , underpinning the usefulness of the Gordon–Taylor approach for describing glass transitions even in SOA compound mixtures derived from different precursor gases. A value of kGT of 3.79 implies a relatively strong decrease in Tg even at small to moderate tetraol mass fractions, e.g., from 306 K in pure 3-MBTCA to about 270 K at a 3-MBTCA mass fraction of 0.75. The Tg of aerosol particles with this composition would be even lower when water is taken up, suggesting that SOA particles containing predominantly isoprene and α-pinene oxidation products are in a liquid state at atmospheric conditions in the boundary layer of a tropical forest, in agreement with in situ observations (Bateman et al., 2015) and global modeling approaches (Shiraiwa et al., 2017). Such aerosol particles may, however, transform into a glassy state in the upper troposphere, i.e., at low temperature and low to moderate relative humidity. We note that the value of of the binary tetraol–3-MBTCA mixture determined here differs significantly from the value of kGT=1 assumed for multicomponent SOA mixtures in a recent modeling study (Shiraiwa et al., 2017). We also note that to our knowledge this tetraol–3-MBTCA mixture is the second binary mixture of individual SOA compounds investigated so far, with the other being pinonic acid–3-MBTCA (Dette et al., 2014). Hence, it would be premature to conclude that these two binary systems are representative for the entire variety of atmospheric binary mixtures, which may also exhibit kGT values smaller than 1. Therefore, as the different kGT values may compensate for each other in multicomponent mixtures a mean kGT of one is not implausible, although our present study may be a first hint that kGT may be slightly larger than 1.
We described the syntheses of 2-methylbutane-1,2,3,4-tetraol in the form of the two possible racemates and of enantiomerically enriched materials with 65 %–97 % enantiomeric excess in detail with a focus on the isolation of the compounds. A simple purification of the tetraols via their diacetonides is introduced. We investigated the phase states of the individual stereoisomers of the tetraol, their racemic mixtures, and their mixtures with water. All stereoisomers were obtained as a viscous liquid at room temperature. The racemates rac-(2R,3S)-tetraol and rac-(2R,3R)-tetraol transitioned from a liquid to a glass upon cooling at about 238 and 245 K, respectively. The glass transition temperatures Tg for the enantiomerically enriched tetraols were found to be identical with those of the corresponding racemates. The Tg values of mixtures of the two racemates show a small and nearly linear dependence with mass ratio of the racemates.
We also investigated the water uptake and diffusion into a dry tetraol bulk sample by monitoring the sample mass and the glass transition temperature via DSC measurements. Water uptake and diffusion was slow, proceeding on the timescale of hours, just as expected for a semisolid bulk sample. At first two distinct amorphous domains form, which were distinguishable by their individual glass transition temperatures. The equilibration of the two domains into one homogeneous liquid phase was followed by monitoring of the magnitude and temperature of the two glass transitions as a function of time.
We further determined atmospherically relevant phase transition temperatures in tetraol–water mixtures over the entire mass fraction range including ice melting and glass transition temperatures. By determining the water activity of tetraol–water mixtures we were able to construct a phase diagram of these mixtures covering the entire tropospheric temperature and humidity range. These phase diagrams imply that aerosol particles consisting predominantly of tetraol are most likely in a liquid or highly viscous semisolid state in the atmosphere, and not in a glassy or crystalline solid state. Glass transition measurements of tetraol–3-MBTCA mixtures suggest that SOA particles containing oxidation products of isoprene as well as oxidation products of α-pinene are liquid at temperatures of the lower troposphere, but may be semisolid or even glassy at upper tropospheric conditions, particularly at low relative humidity. Our data support using the Gordon–Taylor approach for describing Tg values of SOA compound mixtures (Shiraiwa et al., 2017), even when their components are rather dissimilar in chemical structure as is the case when they derive from different types of volatile precursors.
Source data of the x–y plots shown in Figs. 1–3 and 8 have been deposited in the public institutional repository (PUB) of Bielefeld University (http://dx.doi.org/10.4119/unibi/2931835; Lessmeier et al., 2018).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-15841-2018-supplement.
The study was conceived by all authors, the synthesis was planned and performed by JL and AG, the physical experiments were performed by JL and HPD, and the results were analyzed by JL and HPD and discussed by all authors. The paper was written with contributions from all authors.
The authors declare that they have no conflict of interest.
We acknowledge support for the Article Processing Charge by the Deutsche
Forschungsgemeinschaft and the Open Access Publication Fund of Bielefeld
University. We thank Florian Uthoff for advice at the very early stages of
applying enzymatic racemate resolution and Thomas Berkemeier for helpful discussions.
Edited by: Alexander Laskin
Reviewed by: three anonymous referees
Andreae, M. O.: Atmospheric Aerosols: Biogeochemical Sources and Role in Atmospheric Chemistry, Science, 276, 1052–1058, https://doi.org/10.1126/science.276.5315.1052, 1997.
Angell, C. A.: Liquid Fragility and the Glass Transition in Water and Aqueous Solutions, Chem. Rev., 102, 2627–2650, https://doi.org/10.1021/cr000689q, 2002.
Anthonsen, T., Hagen, S., Kazi, M. A., Shah, S. W., Tagar, S., Norgård, S., Kjøsen, H., and Liaaen-Jensen, S.: 2-C-Methyl-erythritol, a New Branched Alditol from Convolvulus glomeratus, Acta Chem. Scand., 30b, 91–93, https://doi.org/10.3891/acta.chem.scand.30b-0091, 1976.
Anthonsen, T., Hagen, S., and Sallam, M. A. E.: Synthetic and spectroscopic studies of 2-C-methyl-erythritol and 2-C-methyl-threito, Phytochemistry, 19, 2375–2377, https://doi.org/10.1016/S0031-9422(00)91030-6, 1980.
Bateman, A. P., Belassein, H., and Martin, S. T.: Impactor Apparatus for the Study of Particle Rebound: Relative Humidity and Capillary Forces, Aerosol Sci. Tech., 48, 42–52, https://doi.org/10.1080/02786826.2013.853866, 2014.
Bateman, A. P., Gong, Z., Liu, P., Sato, B., Cirino, G., Zhang, Y., Artaxo, P., Bertram, A. K., Manzi, A. O., Rizzo, L. V., Souza, R. A. F., Zaveri, R. A., and Martin, S. T.: Sub-micrometre particulate matter is primarily in liquid form over Amazon rainforest, Nat. Geosci., 9, 34–37, https://doi.org/10.1038/ngeo2599, 2015.
Baustian, K. J., Wise, M. E., Jensen, E. J., Schill, G. P., Freedman, M. A., and Tolbert, M. A.: State transformations and ice nucleation in amorphous (semi-)solid organic aerosol, Atmos. Chem. Phys., 13, 5615–5628, https://doi.org/10.5194/acp-13-5615-2013, 2013.
Berkemeier, T., Huisman, A. J., Ammann, M., Shiraiwa, M., Koop, T., and Pöschl, U.: Kinetic regimes and limiting cases of gas uptake and heterogeneous reactions in atmospheric aerosols and clouds: a general classification scheme, Atmos. Chem. Phys., 13, 6663–6686, https://doi.org/10.5194/acp-13-6663-2013, 2013.
Berkemeier, T., Shiraiwa, M., Pöschl, U., and Koop, T.: Competition between water uptake and ice nucleation by glassy organic aerosol particles, Atmos. Chem. Phys., 14, 12513–12531, https://doi.org/10.5194/acp-14-12513-2014, 2014.
Carslaw, K. S., Lee, L. A., Reddington, C. L., Pringle, K. J., Rap, A., Forster, P. M., Mann, G. W., Spracklen, D. V., Woodhouse, M. T., Regayre, L. A., and Pierce, J. R.: Large contribution of natural aerosols to uncertainty in indirect forcing, Nature, 503, 67–71, https://doi.org/10.1038/nature12674, 2013.
Claeys, M.: Formation of Secondary Organic Aerosols Through Photooxidation of Isoprene, Science, 303, 1173–1176, https://doi.org/10.1126/science.1092805, 2004.
Cole-Filipiak, N. C., O'Connor, A. E., and Elrod, M. J.: Kinetics of the Hydrolysis of Atmospherically Relevant Isoprene-Derived Hydroxy Epoxides, Environ. Sci. Technol., 44, 6718–6723, https://doi.org/10.1021/es1019228, 2010.
Decesari, S., Fuzzi, S., Facchini, M. C., Mircea, M., Emblico, L., Cavalli, F., Maenhaut, W., Chi, X., Schkolnik, G., Falkovich, A., Rudich, Y., Claeys, M., Pashynska, V., Vas, G., Kourtchev, I., Vermeylen, R., Hoffer, A., Andreae, M. O., Tagliavini, E., Moretti, F., and Artaxo, P.: Characterization of the organic composition of aerosols from Rondônia, Brazil, during the LBA-SMOCC 2002 experiment and its representation through model compounds, Atmos. Chem. Phys., 6, 375–402, https://doi.org/10.5194/acp-6-375-2006, 2006.
Dette, H. P. and Koop, T.: Glass Formation Processes in Mixed Inorganic/Organic Aerosol Particles, J. Phys. Chem. A, 119, 4552–4561, https://doi.org/10.1021/jp5106967, 2015.
Dette, H. P., Qi, M., Schröder, D. C., Godt, A., and Koop, T.: Glass-forming properties of 3-methylbutane-1,2,3-tricarboxylic acid and its mixtures with water and pinonic acid, J. Phys. Chem. A, 118, 7024–7033, https://doi.org/10.1021/jp505910w, 2014.
Donahue, N. M., Epstein, S. A., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set: 1. organic-aerosol mixing thermodynamics, Atmos. Chem. Phys., 11, 3303–3318, https://doi.org/10.5194/acp-11-3303-2011, 2011.
Donahue, N. M., Henry, K. M., Mentel, T. F., Kiendler-Scharr, A., Spindler, C., Bohn, B., Brauers, T., Dorn, H. P., Fuchs, H., Tillmann, R., Wahner, A., Saathoff, H., Naumann, K.-H., Möhler, O., Leisner, T., Müller, L., Reinnig, M.-C., Hoffmann, T., Salo, K., Hallquist, M., Frosch, M., Bilde, M., Tritscher, T., Barmet, P., Praplan, A. P., DeCarlo, P. F., Dommen, J., Prévôt, A. S. H., and Baltensperger, U.: Aging of biogenic secondary organic aerosol via gas-phase OH radical reactions, P. Natl. Acad. Sci. USA, 109, 13503–13508, https://doi.org/10.1073/pnas.1115186109, 2012.
Duvold, T., Calí, P., Bravo, J.-M., and Rohmer, M.: Incorporation of 2-C-Methyl-d-erythritol, a Putative Isoprenoid Precursor in the Mevalonate-Independent Pathway, into Ubiquinone and Menaquinone of Escherichia coli, Tetrahedron Lett., 38, 6181–6184, https://doi.org/10.1016/S0040-4039(97)01392-0, 1997.
Ebben, C. J., Strick, B. F., Upshur, M. A., Chase, H. M., Achtyl, J. L., Thomson, R. J., and Geiger, F. M.: Towards the identification of molecular constituents associated with the surfaces of isoprene-derived secondary organic aerosol (SOA) particles, Atmos. Chem. Phys., 14, 2303–2314, https://doi.org/10.5194/acp-14-2303-2014, 2014.
Edney, E. O., Kleindienst, T. E., Jaoui, M., Lewandowski, M., Offenberg, J. H., Wang, W., and Claeys, M.: Formation of 2-methyl tetrols and 2-methylglyceric acid in secondary organic aerosol from laboratory irradiated isoprene/NOx/SO2/air mixtures and their detection in ambient PM2.5 samples collected in the eastern United States, Atmos. Environ., 39, 5281–5289, https://doi.org/10.1016/j.atmosenv.2005.05.031, 2005.
Enders, D., Peiffer, E., and Raabe, G.: Asymmetric Synthesis of (S, S)- and (R, R)-2-Methylthreitol, Synthesis (Stuttg.), 2007, 1021–1026, https://doi.org/10.1055/s-2007-965967, 2007.
Fontana, A., Messina, R., Spinella, A., and Cimino, G.: Simple and versatile synthesis of branched polyols: (+)-2-C-methylerythritol and (+)-2-C-methylthreitol, Tetrahedron Lett., 41, 7559–7562, https://doi.org/10.1016/S0040-4039(00)01296-X, 2000.
Ghosh, S. K., Butler, M. S., and Lear, M. J.: Synthesis of 2-C-methylerythritols and 2-C-methylthreitols via enantiodivergent Sharpless dihydroxylation of trisubstituted olefins, Tetrahedron Lett., 53, 2706–2708, https://doi.org/10.1016/j.tetlet.2012.03.071, 2012.
Giner, J.-L., Ferris, W. V., and Mullins, J. J.: Synthesis of 2-Methyl-d-erythritol via Epoxy Ester-Orthoester Rearrangement, J. Org. Chem., 67, 4856–4859, https://doi.org/10.1021/jo020168y, 2002.
Goldstein, A. H. and Galbally, I. E.: Known and Unexplored Organic Constituents in the Earth's Atmosphere, Environ. Sci. Technol., 41, 1514–1521, https://doi.org/10.1021/es072476p, 2007.
González, N. J. D., Borg-Karlson, A.-K., Redeby, J. P., Nozière, B., Krejci, R., Pei, Y., Dommen, J., and Prévot, A. S. H.: New method for resolving the enantiomeric composition of 2-methyltetrols in atmospheric organic aerosols, J. Chromatogr. A, 1218, 9288–9294, https://doi.org/10.1016/j.chroma.2011.10.069, 2011.
Gordon, H., Sengupta, K., Rap, A., Duplissy, J., Frege, C., Williamson, C., Heinritzi, M., Simon, M., Yan, C., Almeida, J., Tröstl, J., Nieminen, T., Ortega, I. K., Wagner, R., Dunne, E. M., Adamov, A., Amorim, A., Bernhammer, A.-K., Bianchi, F., Breitenlechner, M., Brilke, S., Chen, X., Craven, J. S., Dias, A., Ehrhart, S., Fischer, L., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Hakala, J., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Kim, J., Kirkby, J., Krapf, M., Kürten, A., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Mathot, S., Molteni, U., Monks, S. A., Onnela, A., Peräkylä, O., Piel, F., Petäjä, T., Praplan, A. P., Pringle, K. J., Richards, N. A. D., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Scott, C. E., Seinfeld, J. H., Sharma, S., Sipilä, M., Steiner, G., Stozhkov, Y., Stratmann, F., Tomé, A., Virtanen, A., Vogel, A. L., Wagner, A. C., Wagner, P. E., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Zhang, X., Hansel, A., Dommen, J., Donahue, N. M., Worsnop, D. R., Baltensperger, U., Kulmala, M., Curtius, J., and Carslaw, K. S.: Reduced anthropogenic aerosol radiative forcing caused by biogenic new particle formation, P. Natl. Acad. Sci. USA, 113, 12053–12058, https://doi.org/10.1073/pnas.1602360113, 2016.
Gordon, M. and Taylor, J. S.: Ideal Copolymers and the second-order Transition of synthetic Rubbers. I. Non-crystalline Copolymers, J. Appl. Chem., 2, 493–500, 1952.
Guenther, A., Hewitt, C. N., Erickson, D., Fall, R., Geron, C., Graedel, T., Harley, P., Klinger, L., Lerdau, M., Mckay, W. A., Pierce, T., Scholes, B., Steinbrecher, R., Tallamraju, R., Taylor, J., and Zimmerman, P.: A global model of natural volatile organic compound emissions, J. Geophys. Res., 100, 8873, https://doi.org/10.1029/94JD02950, 1995.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, T. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Haywood, J. and Boucher, O.: Estimates of the direct and indirect radiative forcing due to tropospheric aerosols: A review, Rev. Geophys., 38, 513–543, https://doi.org/10.1029/1999RG000078, 2000.
Hoeffler, J.-F., Grosdemange-Billiard, C., and Rohmer, M.: Synthesis of tritium labelled 2-C-methyl-d-erythritol, a useful substrate for the elucidation of the methylerythritol phosphate pathway for isoprenoid biosynthesis, Tetrahedron Lett., 41, 4885–4889, https://doi.org/10.1016/S0040-4039(00)00726-7, 2000.
Hosny, N. A., Fitzgerald, C., Tong, C., Kalberer, M., Kuimova, M. K., and Pope, F. D.: Fluorescent lifetime imaging of atmospheric aerosols: a direct probe of aerosol viscosity, Faraday Discuss., 165, 343–356, https://doi.org/10.1039/c3fd00041a, 2013.
Hosny, N. A., Fitzgerald, C., Vyšniauskas, A., Athanasiadis, A., Berkemeier, T., Uygur, N., Pöschl, U., Shiraiwa, M., Kalberer, M., Pope, F. D., and Kuimova, M. K.: Direct imaging of changes in aerosol particle viscosity upon hydration and chemical aging, Chem. Sci., 7, 1357–1367, https://doi.org/10.1039/C5SC02959G, 2016.
IPCC: IPCC, 2013: Climate Change 2013: The Physical Science Basis, in: Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, UK and New York, NY, USA, 2013.
Isaacman-VanWertz, G., Yee, L. D., Kreisberg, N. M., Wernis, R., Moss, J. A., Hering, S. V., de Sá, S. S., Martin, S. T., Alexander, M. L., Palm, B. B., Hu, W., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Riva, M., Surratt, J. D., Viegas, J., Manzi, A., Edgerton, E., Baumann, K., Souza, R., Artaxo, P., and Goldstein, A. H.: Ambient Gas-Particle Partitioning of Tracers for Biogenic Oxidation, Environ. Sci. Technol., 50, 9952–9962, https://doi.org/10.1021/acs.est.6b01674, 2016.
Jabrane, S., Létoffé, J. M., and Claudy, P.: Vitrification and crystallization in the R(−)1,2-propanediol-S(+)1,2-propanediol system, Thermochim. Acta, 258, 33–47, https://doi.org/10.1016/0040-6031(94)02235-G, 1995.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of organic aerosols in the atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005.
Kar, A. and Argade, N. P.: A Facile Synthesis of Natural Products Chaetomellic Acid A and 1,7(Z)-Nonadecadiene-2,3-dicarboxylic Acid, J. Org. Chem., 67, 7131–7134, https://doi.org/10.1021/jo020195o, 2002.
Kidd, C., Perraud, V., Wingen, L. M., and Finlayson-Pitts, B. J.: Integrating phase and composition of secondary organic aerosol from the ozonolysis of α-pinene, P. Natl. Acad. Sci. USA, 111, 7552–7557, https://doi.org/10.1073/pnas.1322558111, 2014.
Klimovica, K., Grigorjeva, L., Maleckis, A., Popelis, J., and Jirgensons, A.: C-Quaternary Vinylglycinols by Metal-Catalyzed Cyclization of Allylic Bistrichloroacetimidates, Synlett, 2011, 2849–2851, https://doi.org/10.1055/s-0031-1289537, 2011.
Koop, T. and Zobrist, B.: Parameterizations for ice nucleation in biological and atmospheric systems, Phys. Chem. Chem. Phys., 11, 10839–10850, https://doi.org/10.1039/b914289d, 2009.
Koop, T., Luo, B., Tsias, A., and Peter, T.: Water activity as the determinant for homogeneous ice nucleation in aqueous solutions, Nature, 406, 611–614, https://doi.org/10.1038/35020537, 2000.
Koop, T., Bookhold, J., Shiraiwa, M., and Pöschl, U.: Glass transition and phase state of organic compounds: dependency on molecular properties and implications for secondary organic aerosols in the atmosphere, Phys. Chem. Chem. Phys., 13, 19238–19255, https://doi.org/10.1039/c1cp22617g, 2011.
Kostenidou, E., Karnezi, E., Kolodziejczyk, A., Szmigielski, R., and Pandis, S. N.: Physical and Chemical Properties of 3-Methyl-1,2,3-butanetricarboxylic Acid (MBTCA) Aerosol, Environ. Sci. Technol., 52, 1150–1155, https://doi.org/10.1021/acs.est.7b04348, 2018.
Koumbis, A. E., Kotoulas, S. S., and Gallos, J. K.: A convenient synthesis of 2-C-methyl-d-erythritol 4-phosphate and isotopomers of its precursor, Tetrahedron, 63, 2235–2243, https://doi.org/10.1016/j.tet.2006.12.078, 2007.
Kroll, J. H. and Seinfeld, J. H.: Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 42, 3593–3624, https://doi.org/10.1016/j.atmosenv.2008.01.003, 2008.
Kroll, J. H., Donahue, N. M., Jimenez, J. L., Kessler, S. H., Canagaratna, M. R., Wilson, K. R., Altieri, K. E., Mazzoleni, L. R., Wozniak, A. S., Bluhm, H., Mysak, E. R., Smith, J. D., Kolb, C. E., and Worsnop, D. R.: Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol, Nat. Chem., 3, 133–139, https://doi.org/10.1038/nchem.948, 2011.
Kuhn, U.: Isoprene and monoterpene emissions of Amazônian tree species during the wet season: Direct and indirect investigations on controlling environmental functions, J. Geophys. Res., 107, 8071, https://doi.org/10.1029/2001JD000978, 2002.
Kuwata, M. and Martin, S. T.: Phase of atmospheric secondary organic material affects its reactivity, P. Natl. Acad. Sci. USA, 109, 17354–17359, https://doi.org/10.1073/pnas.1209071109, 2012.
Lessmeier, J., Dette, H. D., Godt, A., and Koop, T.: Supplementary Data to the scientific article “Physical state of 2-methylbutane-1,2,3,4-tetraol in pure and internally mixed aerosols”, Bielefeld University, http://dx.doi.org/10.4119/unibi/2931835, 2018.
Li, Y., Pöschl, U., and Shiraiwa, M.: Molecular corridors and parameterizations of volatility in the chemical evolution of organic aerosols, Atmos. Chem. Phys., 16, 3327–3344, https://doi.org/10.5194/acp-16-3327-2016, 2016.
Li, Y.-J., Liu, P.-F., Bergoend, C., Bateman, A. P., and Martin, S. T.: Rebounding hygroscopic inorganic aerosol particles: Liquids, gels, and hydrates, Aerosol Sci. Tech., 51, 388–396, https://doi.org/10.1080/02786826.2016.1263384, 2017.
Lohmann, U. and Feichter, J.: Global indirect aerosol effects: a review, Atmos. Chem. Phys., 5, 715–737, https://doi.org/10.5194/acp-5-715-2005, 2005.
Maclean, A. M., Butenhoff, C. L., Grayson, J. W., Barsanti, K., Jimenez, J. L., and Bertram, A. K.: Mixing times of organic molecules within secondary organic aerosol particles: a global planetary boundary layer perspective, Atmos. Chem. Phys., 17, 13037–13048, https://doi.org/10.5194/acp-17-13037-2017, 2017.
Martin, S. T.: Phase Transitions of Aqueous Atmospheric Particles, Chem. Rev., 100, 3403–3454, https://doi.org/10.1021/cr990034t, 2000.
Martin, S. T., Andreae, M. O., Artaxo, P., Baumgardner, D., Chen, Q., Goldstein, A. H., Guenther, A., Heald, C. L., Mayol-Bracero, O. L., McMurry, P. H., Pauliquevis, T., Pöschl, U., Prather, K. A., Roberts, G. C., Saleska, S. R., Silva Dias, M. A., Spracklen, D. V., Swietlicki, E., and Trebs, I.: Sources and properties of Amazonian aerosol particles, Rev. Geophys., 48, RG2002, https://doi.org/10.1029/2008RG000280, 2010.
Mikhailov, E., Vlasenko, S., Martin, S. T., Koop, T., and Pöschl, U.: Amorphous and crystalline aerosol particles interacting with water vapor: conceptual framework and experimental evidence for restructuring, phase transitions and kinetic limitations, Atmos. Chem. Phys., 9, 9491–9522, https://doi.org/10.5194/acp-9-9491-2009, 2009.
Moen, A. R., Ruud, K., and Anthonsen, T.: Chemo-Enzymatic Synthesis of All Isomers of 2-Methylbutane-1,2,3,4-tetraol – Important Contributors to Atmospheric Aerosols, Eur. J. Org. Chem., 2007, 1262–1266, https://doi.org/10.1002/ejoc.200600873, 2007.
Moise, T., Flores, J. M., and Rudich, Y.: Optical Properties of Secondary Organic Aerosols and Their Changes by Chemical Processes, Chem. Rev., 115, 4400–4439, https://doi.org/10.1021/cr5005259, 2015.
Nozière, B., González, N. J. D., Borg-Karlson, A.-K., Pei, Y., Redeby, J. P., Krejci, R., Dommen, J., Prevot, A. S. H., and Anthonsen, T.: Atmospheric chemistry in stereo: A new look at secondary organic aerosols from isoprene, Geophys. Res. Lett., 38, L11807, https://doi.org/10.1029/2011GL047323, 2011.
Nozière, B., Kalberer, M., Claeys, M., Allan, J., D'Anna, B., Decesari, S., Finessi, E., Glasius, M., Grgić, I., Hamilton, J. F., Hoffmann, T., Iinuma, Y., Jaoui, M., Kahnt, A., Kampf, C. J., Kourtchev, I., Maenhaut, W., Marsden, N., Saarikoski, S., Schnelle-Kreis, J., Surratt, J. D., Szidat, S., Szmigielski, R., and Wisthaler, A.: The Molecular Identification of Organic Compounds in the Atmosphere: State of the Art and Challenges, Chem. Rev., 115, 3919–3983, https://doi.org/10.1021/cr5003485, 2015.
Pankow, J.: An absorption model of the gas/aerosol partitioning involved in the formation of secondary organic aerosol, Atmos. Environ., 41, 75–79, https://doi.org/10.1016/j.atmosenv.2007.10.060, 2007.
Perraud, V., Bruns, E. a, Ezell, M. J., Johnson, S. N., Yu, Y., Alexander, M. L., Zelenyuk, A., Imre, D., Chang, W. L., Dabdub, D., Pankow, J. F., and Finlayson-Pitts, B. J.: Nonequilibrium atmospheric secondary organic aerosol formation and growth, P. Natl. Acad. Sci. USA, 109, 2836–2841, https://doi.org/10.1073/pnas.1119909109, 2012.
Pöschl, U.: Atmospheric aerosols: composition, transformation, climate and health effects, Angew. Chem. Int. Ed. Engl., 44, 7520–7540, https://doi.org/10.1002/anie.200501122, 2005.
Pöschl, U., Martin, S. T., Sinha, B., Chen, Q., Gunthe, S. S., Huffman, J. A., Borrmann, S., Farmer, D. K., Garland, R. M., Helas, G., Jimenez, J. L., King, S. M., Manzi, A., Mikhailov, E., Pauliquevis, T., Petters, M. D., Prenni, A. J., Roldin, P., Rose, D., Schneider, J., Su, H., Zorn, S. R., Artaxo, P., and Andreae, M. O.: Rainforest Aerosols as Biogenic Nuclei of Clouds and Precipitation in the Amazon, Science, 329, 1513–1516, https://doi.org/10.1126/science.1191056, 2010.
Proteau, P. J., Woo, Y.-H., Williamson, R. T., and Phaosiri, C.: Stereochemistry of the Reduction Step Mediated by Recombinant 1-Deoxy-d-xylulose 5-Phosphate Isomeroreductase, Org. Lett., 1, 921–923, https://doi.org/10.1021/ol990839n, 1999.
Riechers, B., Wittbracht, F., Hütten, A., and Koop, T.: The homogeneous ice nucleation rate of water droplets produced in a microfluidic device and the role of temperature uncertainty, Phys. Chem. Chem. Phys., 15, 5873–5887, https://doi.org/10.1039/c3cp42437e, 2013.
Robinson, T. V., Pedersen, D. S., Taylor, D. K., and Tiekink, E. R. T.: Dihydroxylation of 4-Substituted 1,2-Dioxines: A Concise Route to Branched Erythro Sugars, J. Org. Chem., 74, 5093–5096, https://doi.org/10.1021/jo900669u, 2009.
Rogge, W. F., Mazurek, M. A., Hildemann, L. M., Cass, G. R., and Simoneit, B. R. T.: Quantification of urban organic aerosols at a molecular level: Identification, abundance and seasonal variation, Atmos. Environ. A, 27, 1309–1330, https://doi.org/10.1016/0960-1686(93)90257-Y, 1993.
Roozeboom, H. W. B.: Löslichkeit und Schmelzpunkt als Kriterien für racemische Verbindungen, pseudoracemische Mischkrystalle und inaktive Konglomerate, Z. Phys. Chem., 28U, 494–517, https://doi.org/10.1515/zpch-1899-2832, 1899.
Rothfuss, N. E., Marsh, A., Rovelli, G., Petters, M. D., and Reid, J. P.: Condensation Kinetics of Water on Amorphous Aerosol Particles, J. Phys. Chem. Lett., 9, 3708–3713, https://doi.org/10.1021/acs.jpclett.8b01365, 2018.
Saukko, E., Kuuluvainen, H., and Virtanen, A.: A method to resolve the phase state of aerosol particles, Atmos. Meas. Tech., 5, 259–265, https://doi.org/10.5194/amt-5-259-2012, 2012a.
Saukko, E., Lambe, A. T., Massoli, P., Koop, T., Wright, J. P., Croasdale, D. R., Pedernera, D. A., Onasch, T. B., Laaksonen, A., Davidovits, P., Worsnop, D. R., and Virtanen, A.: Humidity-dependent phase state of SOA particles from biogenic and anthropogenic precursors, Atmos. Chem. Phys., 12, 7517–7529, https://doi.org/10.5194/acp-12-7517-2012, 2012b.
Schill, G. P., De Haan, D. O., and Tolbert, M. A.: Heterogeneous Ice Nucleation on Simulated Secondary Organic Aerosol, Environ. Sci. Technol., 48, 1675–1682, https://doi.org/10.1021/es4046428, 2014.
Seinfeld, J. H., Bretherton, C., Carslaw, K. S., Coe, H., DeMott, P. J., Dunlea, E. J., Feingold, G., Ghan, S., Guenther, A. B., Kahn, R., Kraucunas, I., Kreidenweis, S. M., Molina, M. J., Nenes, A., Penner, J. E., Prather, K. A., Ramanathan, V., Ramaswamy, V., Rasch, P. J., Ravishankara, A. R., Rosenfeld, D., Stephens, G., and Wood, R.: Improving our fundamental understanding of the role of aerosol-cloud interactions in the climate system, P. Natl. Acad. Sci. USA, 113, 5781–5790, https://doi.org/10.1073/pnas.1514043113, 2016.
Sharma, A., Das, P., and Chattopadhyay, S.: Concise asymmetric syntheses of the (+)-2-C-methyltetritol isomers, Tetrahedron: Asymmetry, 19, 2167–2170, https://doi.org/10.1016/j.tetasy.2008.09.002, 2008.
Shiraiwa, M., Pfrang, C., Koop, T., and Pöschl, U.: Kinetic multi-layer model of gas-particle interactions in aerosols and clouds (KM-GAP): linking condensation, evaporation and chemical reactions of organics, oxidants and water, Atmos. Chem. Phys., 12, 2777–2794, https://doi.org/10.5194/acp-12-2777-2012, 2012.
Shiraiwa, M., Li, Y., Tsimpidi, A. P., Karydis, V. A., Berkemeier, T., Pandis, S. N., Lelieveld, J., Koop, T., and Pöschl, U.: Global distribution of particle phase state in atmospheric secondary organic aerosols, Nat. Commun., 8, 15002, https://doi.org/10.1038/ncomms15002, 2017.
Shrivastava, M., Cappa, C. D., Fan, J., Goldstein, A. H., Guenther, A. B., Jimenez, J. L., Kuang, C., Laskin, A., Martin, S. T., Ng, N. L., Petaja, T., Pierce, J. R., Rasch, P. J., Roldin, P., Seinfeld, J. H., Shilling, J., Smith, J. N., Thornton, J. A., Volkamer, R., Wang, J., Worsnop, D. R., Zaveri, R. A., Zelenyuk, A., and Zhang, Q.: Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 55, 509–559, https://doi.org/10.1002/2016RG000540, 2017.
Song, M., Liu, P. F., Hanna, S. J., Li, Y. J., Martin, S. T., and Bertram, A. K.: Relative humidity-dependent viscosities of isoprene-derived secondary organic material and atmospheric implications for isoprene-dominant forests, Atmos. Chem. Phys., 15, 5145–5159, https://doi.org/10.5194/acp-15-5145-2015, 2015.
Surratt, J. D., Chan, A. W. H., Eddingsaas, N. C., Chan, M., Loza, C. L., Kwan, A. J., Hersey, S. P., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Reactive intermediates revealed in secondary organic aerosol formation from isoprene, P. Natl. Acad. Sci. USA, 107, 6640–6645, https://doi.org/10.1073/pnas.0911114107, 2010.
Szmigielski, R., Surratt, J. D., Gómez-González, Y., Van der Veken, P., Kourtchev, I., Vermeylen, R., Blockhuys, F., Jaoui, M., Kleindienst, T. E., Lewandowski, M., Offenberg, J. H., Edney, E. O., Seinfeld, J. H., Maenhaut, W., and Claeys, M.: 3-methyl-1,2,3-butanetricarboxylic acid: An atmospheric tracer for terpene secondary organic aerosol, Geophys. Res. Lett., 34, L24811, https://doi.org/10.1029/2007GL031338, 2007.
Tripp, J. C., Schiesser, C. H., and Curran, D. P.: Stereochemistry of Hexenyl Radical Cyclizations with tert-Butyl and Related Large Groups: Substituent and Temperature Effects, J. Am. Chem. Soc., 127, 5518–5527, https://doi.org/10.1021/ja042595u, 2005.
Urbansky, M., Davis, C. E., Surjan, J. D., and Coates, R. M.: Synthesis of Enantiopure 2-C-Methyl-d-erythritol 4-Phosphate and 2,4-Cyclodiphosphate from d-Arabitol, Org. Lett., 6, 135–138, https://doi.org/10.1021/ol0362562, 2004.
Virtanen, A., Joutsensaari, J., Koop, T., Kannosto, J., Yli-Pirilä, P., Leskinen, J., Mäkelä, J. M., Holopainen, J. K., Pöschl, U., Kulmala, M., Worsnop, D. R., and Laaksonen, A.: An amorphous solid state of biogenic secondary organic aerosol particles, Nature, 467, 824–827, https://doi.org/10.1038/nature09455, 2010.
Virtanen, A., Kannosto, J., Kuuluvainen, H., Arffman, A., Joutsensaari, J., Saukko, E., Hao, L., Yli-Pirilä, P., Tiitta, P., Holopainen, J. K., Keskinen, J., Worsnop, D. R., Smith, J. N., and Laaksonen, A.: Bounce behavior of freshly nucleated biogenic secondary organic aerosol particles, Atmos. Chem. Phys., 11, 8759–8766, https://doi.org/10.5194/acp-11-8759-2011, 2011.
Wexler, A. and Hasegawa, S.: Relative Humidity-Temperature Relationships of Some Saturated Salt Solutions in the Temperature, J. Res. Natl. Bur. Stand. 1934, 53, 19–26, 1954.
Yasmeen, F., Szmigielski, R., Vermeylen, R., Gómez-González, Y., Surratt, J. D., Chan, A. W. H., Seinfeld, J. H., Maenhaut, W., and Claeys, M.: Mass spectrometric characterization of isomeric terpenoic acids from the oxidation of α-pinene, β-pinene, d-limonene, and Δ3-carene in fine forest aerosol, J. Mass Spectrom., 46, 425–442, https://doi.org/10.1002/jms.1911, 2011.
Zarzana, K. J., De Haan, D. O., Freedman, M. A., Hasenkopf, C. A., and Tolbert, M. A.: Optical Properties of the Products of α-Dicarbonyl and Amine Reactions in Simulated Cloud Droplets, Environ. Sci. Technol., 46, 4845–4851, https://doi.org/10.1021/es2040152, 2012.
Zaveri, R. A., Easter, R. C., Shilling, J. E., and Seinfeld, J. H.: Modeling kinetic partitioning of secondary organic aerosol and size distribution dynamics: representing effects of volatility, phase state, and particle-phase reaction, Atmos. Chem. Phys., 14, 5153–5181, https://doi.org/10.5194/acp-14-5153-2014, 2014.
Zhang, Z., Lin, Y.-H., Zhang, H., Surratt, J. D., Ball, L. M., and Gold, A.: Technical Note: Synthesis of isoprene atmospheric oxidation products: isomeric epoxydiols and the rearrangement products cis- and trans-3-methyl-3,4-dihydroxytetrahydrofuran, Atmos. Chem. Phys., 12, 8529–8535, https://doi.org/10.5194/acp-12-8529-2012, 2012.
Zobrist, B., Marcolli, C., Pedernera, D. A., Koop, T., Kob, W., Angell, C. A., Debenedetti, P. G., Stillinger, F. H., Fulcher, G. S., Quintas, M., Brandão, T. R. S., Silva, C. L. M., and Cunha, R. L.: Do atmospheric aerosols form glasses?, Atmos. Chem. Phys., 8, 5221–5244, https://doi.org/10.5194/acp-8-5221-2008, 2008.
Zobrist, B., Soonsin, V., Luo, B. P., Krieger, U. K., Marcolli, C., Peter, T., and Koop, T.: Ultra-slow water diffusion in aqueous sucrose glasses, Phys. Chem. Chem. Phys., 13, 3514–3526, https://doi.org/10.1039/c0cp01273d, 2011.