the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 05 Nov 2018
| 05 Nov 2018
Understanding mercury oxidation and air–snow exchange on the East Antarctic Plateau: a modeling study
Hélène Angot
Noelle E. Selin
Hubert Gallée
Francesca Sprovieri
Nicola Pirrone
Detlev Helmig
Joël Savarino
Olivier Magand
Aurélien Dommergue
Distinct diurnal and seasonal variations of mercury (Hg) have been observed in near-surface air at Concordia Station on the East Antarctic Plateau, but the processes controlling these characteristics are not well understood. Here, we use a box model to interpret the Hg0 (gaseous elemental mercury) measurements in thes year 2013. The model includes atmospheric Hg0 oxidation (by OH, O3, or bromine), surface snow HgII (oxidized mercury) reduction, and air–snow exchange, and is driven by meteorological fields from a regional climate model. The simulations suggest that a photochemically driven mercury diurnal cycle occurs at the air–snow interface in austral summer. The fast oxidation of Hg0 in summer may be provided by a two-step bromine-initiated scheme, which is favored by low temperature and high nitrogen oxides at Concordia. The summertime diurnal variations of Hg0 (peaking during daytime) may be confined within several tens of meters above the snow surface and affected by changing mixed layer depths. Snow re-emission of Hg0 is mainly driven by photoreduction of snow HgII in summer. Intermittent warming events and a hypothesized reduction of HgII occurring in snow in the dark may be important processes controlling the mercury variations in the non-summer period, although their relative importance is uncertain. The Br-initiated oxidation of Hg0 is expected to be slower at Summit Station in Greenland than at Concordia (due to their difference in temperature and levels of nitrogen oxides and ozone), which may contribute to the observed differences in the summertime diurnal variations of Hg0 between these two polar inland stations.
- Article
(1512 KB) - Full-text XML
-
Supplement
(1940 KB) - BibTeX
- EndNote





Mercury (Hg) is an environmental concern due to its health effects on humans and wildlife (Mergler et al., 2007). This trace element undergoes long-range transport in the atmosphere and is readily cycled at the Earth's surfaces (Selin, 2009), and thus even the remote Antarctic Plateau, a vast (about 5×106 km2) and elevated (about 3 km above sea level) region of snow-covered ice, receives significant mercury inputs (Dommergue et al., 2010).
Over the past decade, field studies have investigated mercury in air and/or snow at a few inland Antarctic stations, i.e., Concordia Station (Dome C, 75∘ S, 123∘ E), Dome Argus (80∘ S, 77∘ E), Dome Fuji (77∘ S, 40∘ E), and the South Pole (90∘ S), as well as along several transects on the plateau (Brooks et al., 2008; Dommergue et al., 2012; Han et al., 2014; Li et al., 2014; Angot et al., 2016b, c; Wang et al., 2016; Han et al., 2017; Spolaor et al., 2018). Most of these studies only measured atmospheric mercury in austral summer, whereas Angot et al. (2016c) reported a year-round observational record at Dome C. All these measurements suggest that in summer (November–February), a photochemical mercury cycle occurs between the atmospheric boundary layer and surface snowpack, including the oxidation of gaseous elemental mercury (Hg0) in air, the deposition of oxidized mercury (HgII) onto snow, the photoreduction of snow HgII, and the re-emission of Hg0 from the snowpack surface. A clear diurnal cycle of Hg0 (peaking at midday and decreasing to a minimum around midnight) was observed in near-surface air and has been attributed to enhanced Hg0 re-emission in the daytime as a result of increasing solar radiation (Dommergue et al., 2012; Angot et al., 2016c; Wang et al., 2016). The summertime photochemical mechanism of Hg0 oxidation in air is unknown but has been related to the high oxidizing capacity of the plateau, which is characterized by high concentrations of NOx, OH, and other oxidants within the Antarctic mixed layers (Eisele et al., 2008; Helmig et al., 2008a, b; Neff et al., 2008; Kukui et al., 2014; Frey et al., 2015). Interestingly, such summertime diurnal variations of Hg0 have not been seen at the polar inland Summit Station atop the Greenland ice sheet (Brooks et al., 2011). As for other seasons, observations at Dome C showed high atmospheric Hg0 in fall (March–April), exceeding those measured at the Antarctic coast and southern hemispheric midlatitude sites. Such seasonal cycles were repeatedly measured in 2012–2015 at Dome C (Angot et al., 2016a). Moreover, in fall, the concentrations of Hg0 peaked during the night. In winter (May–August), as expected, the diurnal cycle of Hg0 disappeared, and a gradual decline of Hg0 was seen in near-surface air.
Overall, these observed seasonal and diurnal features of atmospheric mercury on the plateau are not well understood and not reproduced by global chemical transport models, likely due to their imperfect representations of boundary layer dynamics and chemical reaction pathways (Angot et al., 2016a) and to the singularity of their longitude–latitude grid at the poles. Here, we present detailed box model calculations to interpret observational data collected at Dome C in 2013, and to explore important chemical and physical processes controlling diurnal and seasonal variations of atmospheric mercury. A better knowledge of these characteristics is helpful for evaluating the potential influence of the Antarctic Plateau on the coastal environment (Bargagli, 2016) and for understanding processes occurring in other polar regions.
We have built a multiple-layer box model to account for mercury chemistry and transport in the lower troposphere and surface snow, and the exchange between them. Details on the model setup are given in this section. The modeling results are mainly compared with the measurement data of Hg0 in the year 2013. Briefly, Hg0 concentrations were measured at three inlets (25, 210, and 1070 cm above the surface) of a meteorological tower located in the “clean area” of Dome C (where snow is kept undisturbed). Hg0 concentrations were also measured in the near-surface air and snow interstitial air with multi-inlet snow sampling manifolds (the so-called “snow towers”). The mercury measurements were performed using a Tekran 2537A automated analyzer (Tekran Inc., Toronto, Canada). The experimental details have been described in Angot et al. (2016c).
Table 1Major assumptions and simplifications made in the mercury model.
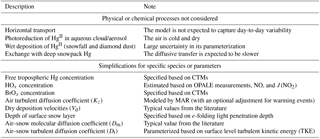
2.1 Model overview
The model accounts for vertical transport using outputs from a regional climate model (Sect. 2.2). As shown in Fig. 1, Hg0 can be oxidized to HgII by different gas-phase chemical schemes (Sect. 2.3). The photoreduction of HgII in aqueous clouds and aerosols is not considered in the model because its mechanism is poorly understood, and also because the air above the plateau is cold and dry. The vertical resolution is ∼2 m near the surface and gradually decreases with height above the surface, and there are 33 atmospheric layers in total below 500 m. In the free troposphere, Hg0 and HgII concentrations are prescribed (Sect. 2.4). Hg0 and HgII are transferred from air to snow through dry deposition (Sect. 2.5). Wet deposition is not considered due to low snow accumulation rates and large uncertainty in parameterizing this process (France et al., 2011; Palerme et al., 2017). Note that Spolaor et al. (2018) have recently suggested that frequent snowfall and diamond dust (tiny ice crystals) events in summer may lead to quick mercury deposition. However, a quantitative parameterization for this process has not been available, and it is thus not included in this model. The model tracks Hg0 and HgII in a surface snow reservoir, in which HgII may be reduced to Hg0 photolytically or in the dark (Sect. 2.5). The depth of the surface snow layer is set to 20 cm, equivalent to one to two e-folding light penetration depths at Dome C (France et al., 2011). The exchange of mercury between the surface snowpack and the deeper snowpack is not considered in the model because the photochemistry in the deeper snowpack is less active, and also because the diffusive transfer of Hg0 between these two snow layers should be slower. Our model calculations are not expected to capture day-to-day variations, since horizontal transport is ignored, and are thus compared with the average monthly and diurnal observations at Dome C as reported in Angot et al. (2016c). Major assumptions and simplifications made in the model are summarized in Table 1.
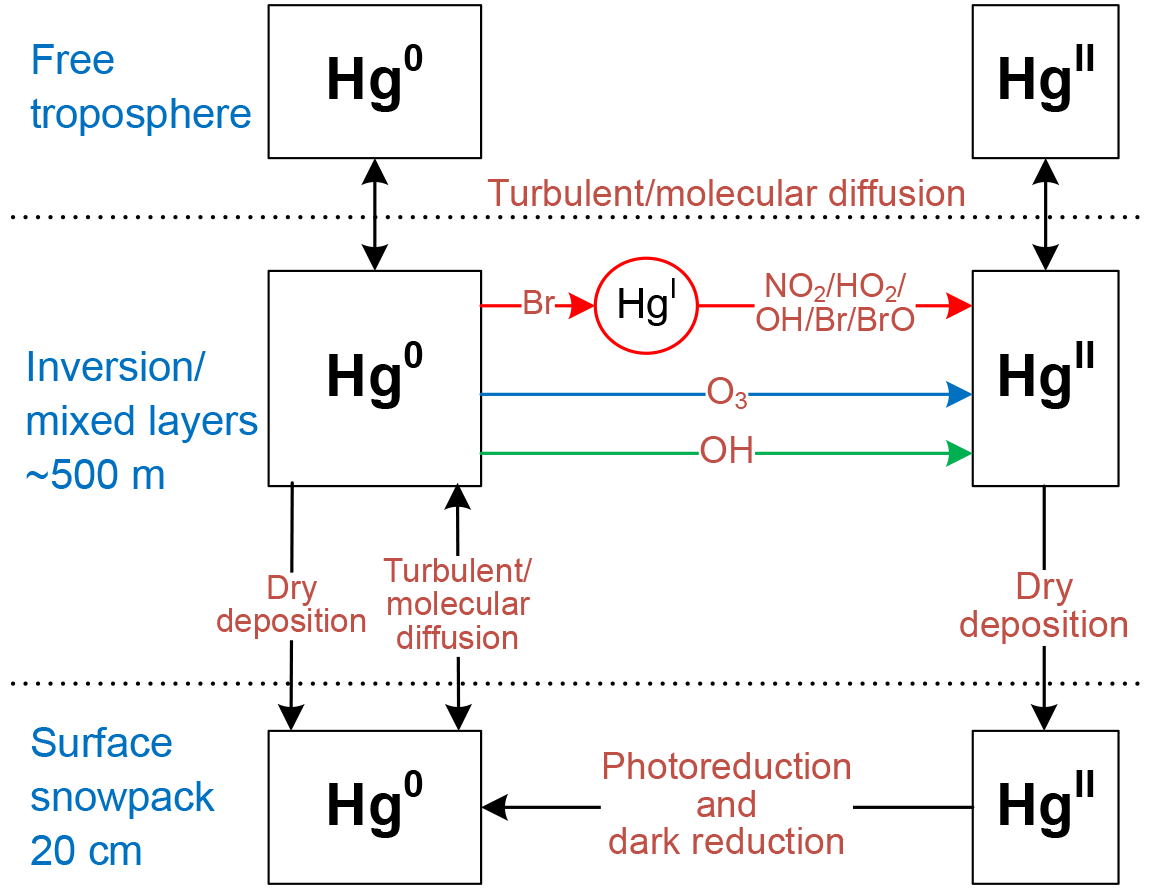
Figure 1Chemical and physical processes represented in the mercury box model. Hg0 can be oxidized to HgII by three different gas-phase chemical schemes (OH, O3, or a two-step Br-initiated scheme). Note that the concentrations of the intermediate HgI in the two-step Br-initiated oxidation mechanism are not tracked since its lifetime is short, and thus effective reaction rates are used to describe the oxidation of Hg0 to HgII for this mechanism (Sect. 2.3). The dark reduction of surface snow HgII may be only important for the non-summer period (Sect. 2.5).
2.2 Meteorology
A surface-based temperature inversion layer exists at Dome C for most of the year, mainly due to radiation imbalance, while a convective mixed layer up to several hundred meters in depth develops during the daytime in summer in response to surface heating (see the Supplement, Sect. S1) (Pietroni et al., 2014). Here, the depth of the inversion/mixed layers is specified as ∼500 m in our model, and the air above is regarded as the free troposphere. The vertical atmospheric transport is represented with turbulent diffusion coefficients (Kz) from the polar-oriented regional climate model MAR (Modèle Atmosphérique Régional) (Sect. S1). The MAR data have been used to simulate several other atmospheric species (e.g., NOx and HONO) in the 2011–2012 summer Oxidant Production in Antarctic Lands and Export (OPALE) campaign at Dome C (Legrand et al., 2014; Frey et al., 2015; Preunkert et al., 2015). In general, MAR simulations agree well with meteorological observations at Dome C (Gallée and Gorodetskaya, 2010; Gallée et al., 2015), whereas the intermittent warming events occurring primarily during the non-summer period, which decrease temperature inversion strength and strongly enhance vertical turbulence (leading to large Kz values), may not be well represented. The vertical temperature gradients measured at a meteorological tower at Dome C indicate that the actual intensities of warming events should be weaker than results from MAR (Genthon et al., 2010). This is likely related to the cloud microphysical scheme in MAR, which is responsible for estimating the cloud cover and thus affects the estimation of surface temperature and buoyant forcing of turbulence. For example, in the wintertime, when the cloudiness is overestimated by the model, the downward infrared radiation is also overestimated. This overestimation limits surface cooling and subsequently the inhibition of turbulence, which is essentially generated by the wind shear. An accurate estimate of the warming events is challenging, and here we tentatively adjust MAR-modeled Kz values during warming events using a rough empirical relationship between the temperature gradients and Kz, resulting in weaker exchange between the surface layers and free troposphere. It is important to note that such an adjustment is subject to large uncertainties and tends to underestimate the strength of vertical turbulence (Sect. S1). Thus, due to uncertainties in estimating warming events and their effects on the vertical transport of mercury in the non-summer period, both original and adjusted Kz values are used to drive the mercury model in this study.
Table 2Gas-phase mercury reactions used in the mercury model.
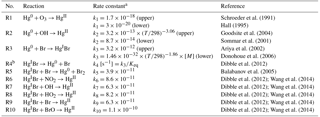
a Rate constants are in cm3 molecule−1 s−1 unless otherwise stated. T represents temperature in K. [M] is the number density of air in molecule cm−3. The “upper” and “lower” indicate the highest and lowest reaction rate constants determined by different kinetic studies (for a review, see Ariya et al., 2015), respectively. The uncertainty ranges of reaction rate constants of R4–R10 are unknown as only computational kinetic data are available for these reactions (Jiao and Dibble, 2017). b R3 and R4 are a pair of reversible reactions. is the equilibrium constant estimated by Dibble et al. (2012), which is very close to the value of cm3 molecule−1 calculated by Goodsite et al. (2012).
2.3 Atmospheric mercury chemistry
In the model, Hg0 is oxidized in the atmosphere to HgII, while the oxidants, chemical kinetics, and oxidant concentrations are all uncertain. As shown in Table 2, the rate constants of Hg0 reactions with O3 (R1), OH (R2), and Br (R3) from existing theoretical and experimental studies may vary by factors of about 60, 8, and 4, respectively. While used in several chemical transport models, O3- and OH-based chemical mechanisms are unlikely as pure gas-phase reactions since the formation of HgO is endothermic (Subir et al., 2011). The two-step Br-initiated scheme (R3–R10) can explain polar atmospheric mercury depletion events (Sprovieri et al., 2005; Steffen et al., 2008) and is likely the dominant Hg0 oxidation pathway globally (Holmes et al., 2006; Horowitz et al., 2017; Ye et al., 2018). The recombination of Hg0 and Br forms unstable HgIBr, which either dissociates or is oxidized to HgII by NO2, HO2, OH, Br, or BrO. The effective oxidation rate constant of this two-step scheme is expressed in Eq. (1), assuming a steady state of HgIBr, as it forms slowly by R3, and is oxidized readily by R6–R10, where terms in brackets refer to concentrations, and k3−k10 are reaction rates of R3–R10. The gas-phase oxidations of Hg0 by other species and the aqueous and heterogeneous processes are not considered here (Sect. S2) (Lin and Pehkonen, 1999; Subir et al., 2011; Ariya et al., 2015).
Concentrations of chemical species, including O3, HOx (OH, HO2), BrOx (Br, BrO), and NOx (NO, NO2), are prescribed based on the available measurements and global chemical transport model (CTM) simulations (details in Sect. S3). Monthly and diurnal averages are computed. The temporal variations of O3 and NOx are specified based on in situ measurements in near-surface air (Angot et al., 2016c; Legrand et al., 2016a; Helmig et al., 2018), and a uniform O3 vertical profile within the inversion/mixed layers is assumed, consistent with aircraft observations on the plateau (Slusher et al., 2010; Legrand et al., 2016a). The NOx vertical profile has not been measured and is estimated assuming an exponential decay with height starting at the surface (Slusher et al., 2010). The previously reported potential bias in the measurement ratios of [NO]∕[NO2] (Frey et al., 2015) does not significantly affect our model results, as suggested by a sensitivity test. The HOx concentrations in summer are set based on measurements from the OPALE campaign, and their values in other seasons are estimated using relationships with J(NO2) and NO (Kukui et al., 2014). The uncertainties in O3 and OH concentrations are assumed to be 2 % and 50 %, respectively, as inferred from in situ measurements at Dome C (Kukui et al., 2014).
For BrO concentrations, due to lack of measurements, we rely on two global CTMs, GEOS-Chem and p-TOMCAT (Yang et al., 2005; Sherwen et al., 2016). We assume no diurnal and vertical variations of BrO (Stutz et al., 2011; Legrand et al., 2016b). The modeled BrO mixing ratios from these two CTMs are similar: less than 0.1 pptv in winter and ∼0.4 pptv in other seasons (Fig. S8 in the Supplement). The modeled BrO is likely at the lower limits of its uncertainty range, as suggested by the comparison of the modeled tropospheric BrO columns and their values retrieved from the Global Ozone Monitoring Experiment-2 (GOME-2) satellite (Sherwen et al., 2016). Legrand et al. (2016b) measured total inorganic gaseous bromine concentrations at Dome C and suggested that the upper limit of BrO is ∼1 pptv. Based on the above information, the uncertainty of BrO concentrations is set as a factor of 2.5. It is important to note that the seasonal patterns of the modeled BrO by the CTMs may have biases, as indicated by the total inorganic bromine measurements at Dome C (Legrand et al., 2016b). The modeled BrO is likely biased high in fall and spring, which affects Hg0 concentrations simulated by the mercury model (Sect. 3.4). The concentrations of Br are estimated assuming a photochemical steady state: (Holmes et al., 2010), where JBrO is the BrO photolysis frequency, and kBrO+NO and are rate constants for and , respectively (Sander et al., 2011).
2.4 Mercury concentrations in the free troposphere
Due to lack of measurements, we rely on two global CTMs, GEOS-Chem (version 9-02) and the Global European Monitoring and Evaluation Programme (EMEP) Multi-media Modelling System (GLEMOS), to specify the free tropospheric mercury concentrations (Angot et al., 2016a; Travnikov et al., 2017). The former uses a Br oxidation scheme, whereas the latter assumes OH and O3 to be the oxidants of Hg0. Monthly Hg0 and HgII concentrations at 500 m above ground level in the Dome C grid box are extracted from these two CTMs. Studies have identified that the CTMs show significant seasonal biases in modeled mercury concentrations when compared to mercury observations at two southern hemispheric background stations, Amsterdam Island (38∘ S, 78∘ E) and Cape Point (34∘ S, 18∘ E) (Angot et al., 2014; Song et al., 2015; Horowitz et al., 2017; Martin et al., 2017), implying potential biases in modeled mercury budgets for the Southern Hemisphere. Hence, we adjust the modeled free tropospheric mercury concentrations using the scaling factors estimated by model–observation comparisons for these two background stations: , where represents the average mercury concentrations, and i and j indicate each month and model, respectively. The two CTMs predict similar total gaseous mercury () concentrations with annual means of ∼1.0 ng m−3, whereas the modeled HgII concentrations during the sunlit period are much higher in GEOS-Chem than in GLEMOS due to their different chemical mechanisms (Fig. S9). In our simulations, the free tropospheric mercury data are chosen from either GEOS-Chem or GLEMOS according to the chemical oxidation scheme (O3, OH, or Br) used in each model scenario, for consistency. For example, the GEOS-Chem free tropospheric mercury data are used when the Br scheme is assumed in the box model simulation. Both CTMs use reaction rate constants at the lower limits. When the upper-limit reaction rates are assumed in the model scenarios, we expect more mercury should exist in its oxidized form, HgII, in the free troposphere, and thus we adjust free tropospheric concentrations of Hg0 and HgII according to this equation: , where R is the ratio between the upper- and lower-limit reaction rate constants, whereas the total HgT concentrations remain unchanged.
2.5 Air–snow mercury exchange and snow mercury transformation
Dry deposition fluxes of Hg0 and HgII are determined by their concentrations at the atmospheric ground level and prescribed deposition velocities (Vd). The effects of wind speeds and snow properties on Vd are not included here. As indicated by previous studies (Lindberg et al., 2002; Brooks et al., 2006; Skov et al., 2006), the values of Vd for Hg0 and HgII are set to and 1 cm s−1, respectively (Zhang et al., 2009). These Vd parameters are not well constrained, but we find that varying the values of Vd by a factor of 2 does not change the main findings of this study. For Hg0, the bidirectional fluxes between surface snow and air are considered and estimated by Hg0 concentration differences and the turbulent and molecular diffusion coefficients in the snow interstitial air. Following Durnford et al. (2012), the molecular diffusion coefficient (Dm) in our model is set to m2 s−1. The turbulent diffusion coefficients (Dt) can be estimated by an explicit representation of the vertical wind pumping within the snowpack, which include several uncertain parameters, such as the height and wavelength of sastrugi (snow-eroded grooves or ridges) and the permeability of surface snowpack (Cunningham and Waddington, 1993; Thomas et al., 2011; Zatko et al., 2013; Toyota et al., 2014b). The estimated values of Dt using this approach and the air and snow properties at Dome C may vary from the order of 10−6 to 10−4 m2 s−1 for the surface snowpack with a depth of 20 cm. Here, a more simple approach is adopted following Durnford et al. (2012), in which Dt is set proportional to the atmospheric ground-level turbulent kinetic energy (TKE) obtained from the MAR model: Dt= TKE (m2 s−2) s. Dt varies by season and by time of day and has an annual median value of m2 s−1. The choice of the scaling factors ( s by default in the model) is found to affect the modeled Hg0 concentrations in the snow interstitial air (Sect. 3.2). A more explicit consideration of the influence of air and snow properties on air–snow exchange is recommended for future mercury modeling studies.
Previous studies have suggested that HgII can be reduced both photolytically and in the dark, and the photolytic and dark oxidation of Hg0 may also occur, but the reaction rates and reductants/oxidants of individual pathways are largely unknown (for a review, see Durnford and Dastoor, 2011). Sunlight, in particular UV-B (280–320 nm) radiation, greatly enhances the formation of Hg0 (Poulain et al., 2004; Dommergue et al., 2007; Johnson et al., 2008). Similar to previous models (Durnford et al., 2012; Toyota et al., 2014a), we include a first-order photoreduction of HgII in the surface snowpack and scale its rate by J(O(1D)), the photolysis frequency of O3. In doing so, we assume that the supply of reductants is ample and that all HgII is reducible (Durnford and Dastoor, 2011). The photoreduction rate is poorly constrained, with a corresponding lifetime (denoted as τPR) from a few days to several weeks (Toyota et al., 2014a). We also include dark reduction of snow HgII (the corresponding lifetime denoted as τDR) in our model simulations for the non-summer period (Sect. 3.4).
3.1 Atmospheric Hg0 oxidation rates
We have computed ranges of atmospheric Hg0 oxidation rates for different schemes (O3, OH, and two-step Br), using the low (i.e., lower-limit) and high (i.e., upper-limit) rate constants listed in Table 2 and uncertainties of oxidant concentrations (Sect. 2.3). As shown in Fig. 2, the Hg0 oxidation rates for these schemes in the inversion/mixed layers have large uncertainty ranges. Since the OH and Br concentrations are largely determined by the amount of solar radiation, the oxidation rates of Hg under these schemes exhibit strong seasonal and diurnal variations, while the O3 scheme does not. In austral summer (November–February), the two-step Br oxidation scheme (corresponding Hg0 oxidation lifetimes denoted as τOX ∼1.7–22 days) is more efficient than the O3 (τOX ∼19–1300 days) and OH (τOX ∼17–350 days) oxidation schemes. We find that the fast two-step Br oxidation is favored by low ambient temperature, high concentrations of NOx, and low concentrations of O3 at Dome C. This is because the thermal dissociation rates of the intermediate HgIBr decrease rapidly at a lower temperature, and because the concentrations of Br are influenced by the concentrations of NOx and O3 (Sect. 2.3). In austral winter (May–August), by contrast, the O3 oxidation scheme (τOX ∼13–900 days) is usually more efficient than the others. A series of combinations of oxidation schemes, oxidant concentrations, and chemical kinetics are tested in our model simulations.
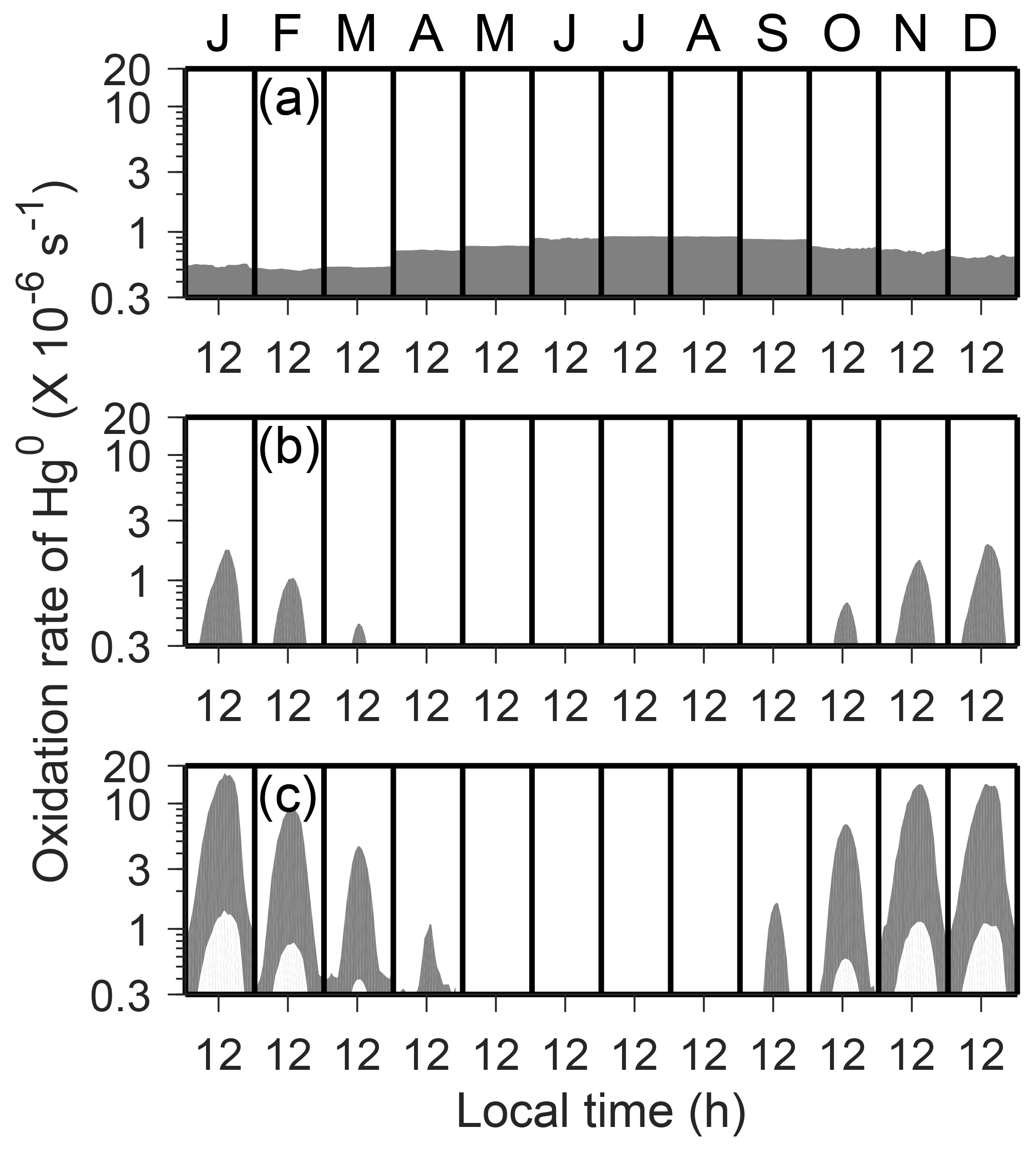
Figure 2Uncertainty ranges of atmospheric Hg0 oxidation rates within the inversion/mixed layers: (a) O3, (b) OH, and (c) Br. Monthly and diurnal variations in the year 2013 are shown in the shaded regions. Note that the y axis is in log scale.
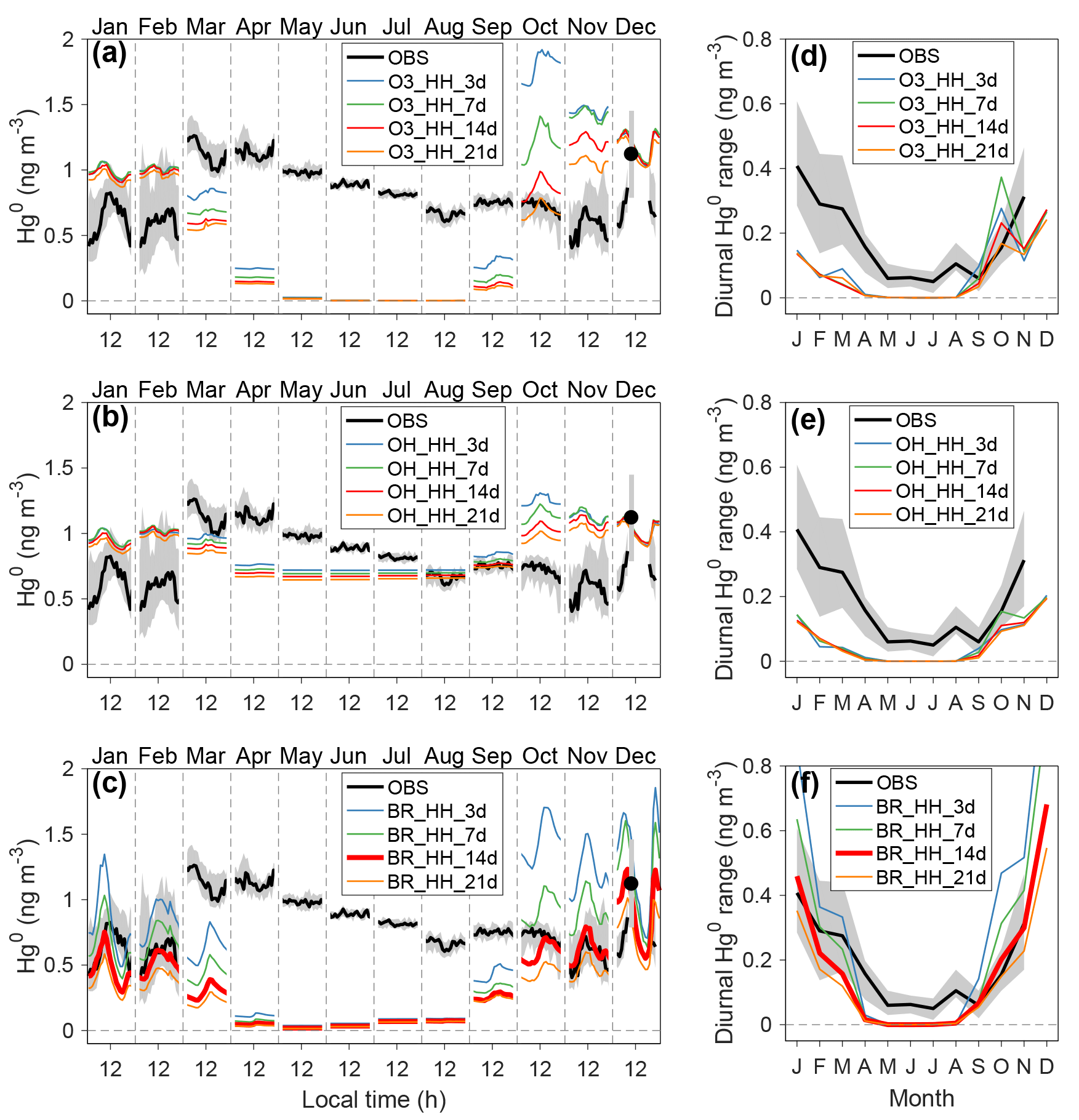
Figure 3Comparison of seasonal and diurnal variations of near-surface atmospheric Hg0 concentrations between observations and model. Panels (a)–(c) show monthly and diurnal Hg0 observations in the year 2013 and modeling results from different scenarios. Panels (d)–(f) show diurnal Hg0 ranges calculated from the maximum and minimum hourly concentrations in each month. The shaded regions indicate 25 % and 75 % percentiles in observations. Observations were conducted at 25 cm above the snow surface at Dome C. The name of each scenario reflects the atmospheric oxidant, its concentration levels, chemical reaction rates (H indicates “high” or “upper”; L indicates “low” or “lower”), and the photoreduction rates of snow mercury (in days). For example, the scenario with name “O3_HH_3d” assumes O3 as the oxidant, and high oxidant concentrations and high reaction rates are applied, and τPR is set to 3 days.
3.2 Strong photochemistry in summer
During the summer months, the observed Hg0 concentrations in near-surface Dome C air show a pronounced diurnal pattern, which usually peaks in the daytime and is minimized at night, as shown in Figs. 3 and S10. The amplitudes of diurnal variations of observed Hg0 reach ∼0.4 ng m−3 in January and ∼0.3 ng m−3 in February and November, respectively, which are higher than those during other seasons. This characteristic has been attributed to enhanced re-emissions of Hg0 in the daytime (Angot et al., 2016c; Wang et al., 2016), highlighting a dynamic Antarctic surface snowpack. The solar zenith angle has a diurnal cycle during summer, and a convective layer develops in the daytime as a response to surface heating, enhancing strengths of vertical mixing and snow ventilation. Previous studies have suggested rapid recurring cycles of oxidation and re-emission of Hg0 in summer, but chemical mechanisms have not been well defined (Angot et al., 2016c; Wang et al., 2016). As photochemical processes in the air and surface snow are of obvious importance for summer, we have conducted a series of mercury model sensitivity simulations by varying atmospheric oxidants (O3, OH, or Br), their concentrations (high or low) and chemical reaction rate constants (upper or lower), and surface snow HgII photoreduction rates (τPR from 3 days to 3 weeks). In total, we ran 24 model sensitivity scenarios (Table S1 in the Supplement). Through comparing modeling results to observations, key atmospheric Hg0 oxidants may be identified, and surface snow HgII photoreduction rates may be constrained. Some of these scenarios have large biases compared to observations for the non-summer months, which is likely due to several factors in these simulations that will be discussed in detail in Sect. 3.4: (1) the adjusted Kz values during the warming events are used, which tends to underestimate the mercury vertical transport from the free troposphere, (2) the Br concentrations used in the model calculations are likely overestimated in the non-summer period, and/or (3) the dark reduction of snow HgII, which may be important in the non-summer period, is not included.
The modeled Hg0 concentrations in near-surface air from various scenarios are compared to observations in Fig. 3 and in Sect. S4 (only the data collected at 25 cm above the surface are shown, and the model–observation comparison results for the data at 210 and 1070 cm are similar). We find, during summer, that model scenarios using either OH or O3 oxidation schemes do not reproduce the diurnal variations of Hg0, and tend to overestimate atmospheric Hg0 concentrations, even when high oxidant concentrations and upper-limit reaction rates are assumed (resulting in τOX∼20 days). Among the scenarios with the bromine oxidation scheme, BR_ HH_14d (using high Br concentrations and upper-limit reaction rate constants; τOX∼2 days and τPR of 2 weeks in summer) best reproduces the concentrations of atmospheric Hg0 and its diurnal patterns during the summer months (calculated normalized root mean square errors of < 20 %; Sect. S4). This scenario shows larger Hg0 diurnal variations in January–December than February–November, consistent with observations (Angot et al., 2016c; Spolaor et al., 2018). The differences in solar radiation in these summer months are expected to influence the strength of photochemical activities (such as Br concentration and photoreduction rates of snow HgII). Therefore, these sensitivity simulations suggest that a fast oxidation for atmospheric Hg0 occurs in the surface layers at Dome C in summer, and that the fast oxidation of Hg0 may be provided by a two-step Br scheme with its upper-limit reaction rates.
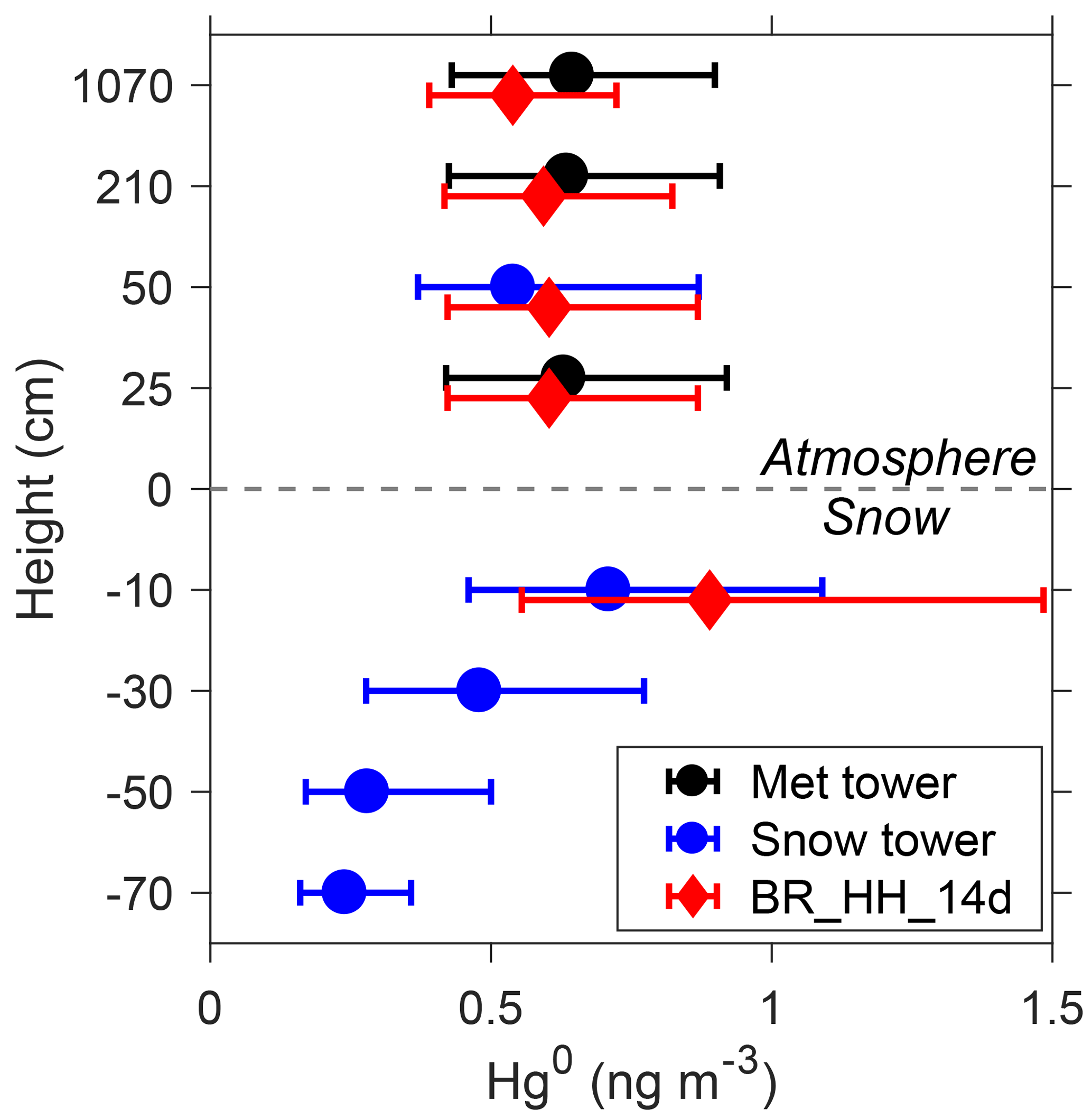
Figure 4Summertime average Hg0 concentrations at different heights from observations and model. The observations include the meteorological tower (25, 210, and 1070 cm above the snow surface) and snow tower (50 cm above the snow surface and 10, 30, 50, and 70 cm below the snow surface). Model results from the BR_ HH_14d scenario are shown. Measurement data are from the snow tower no. 1 as reported in Angot et al. (2016c). Error bars indicate 25 % and 75 % percentiles.
The summertime average Hg0 concentrations modeled by the BR_HH_14d scenario are also compared with those measured at different sampling heights, as shown in Fig. 4. The snow tower measurements indicate that Hg0 concentrations in the surface snow interstitial air (10 cm below the surface) are about 0.2 ng m−3 higher than those in the air (50 cm above the surface). The model predicts a similar Hg0 difference of about 0.3 ng m−3. These results suggest the snow-to-air transport of Hg0 and the production of Hg0 in the surface snowpack. It is noted that the modeled difference in Hg0 concentrations depends on the assumed turbulent diffusion coefficients (Dt). Larger Dt implies faster vertical mixing of Hg0 and thus corresponds to smaller differences between the surface snowpack and atmosphere (Fig. S12). The measured Hg0 concentrations in the interstitial air of the deeper snowpack are lower than those in the surface snowpack, suggesting that the production of Hg0 may mainly occur in the snow near the surface. In the model, the production of Hg0 in surface snow arises from the photoreduction of HgII during summer. The photoreduction rates of surface snow (top 20 cm) HgII in BR_HH_14d (τPR of 2 weeks) agree well with observations at the South Pole in Brooks et al. (2008), who estimated a lifetime of surface snow mercury (assumed to be the top 15 cm) of ∼16 days. The surface snow mercury concentrations modeled by BR_HH_14d are ∼20 ng L−1 (Fig. S13). The available measurements suggest that surface snow mercury concentrations were highly variable, ranging from ∼3 to 50 ng L−1 (Angot et al., 2016c; Spolaor et al., 2018).
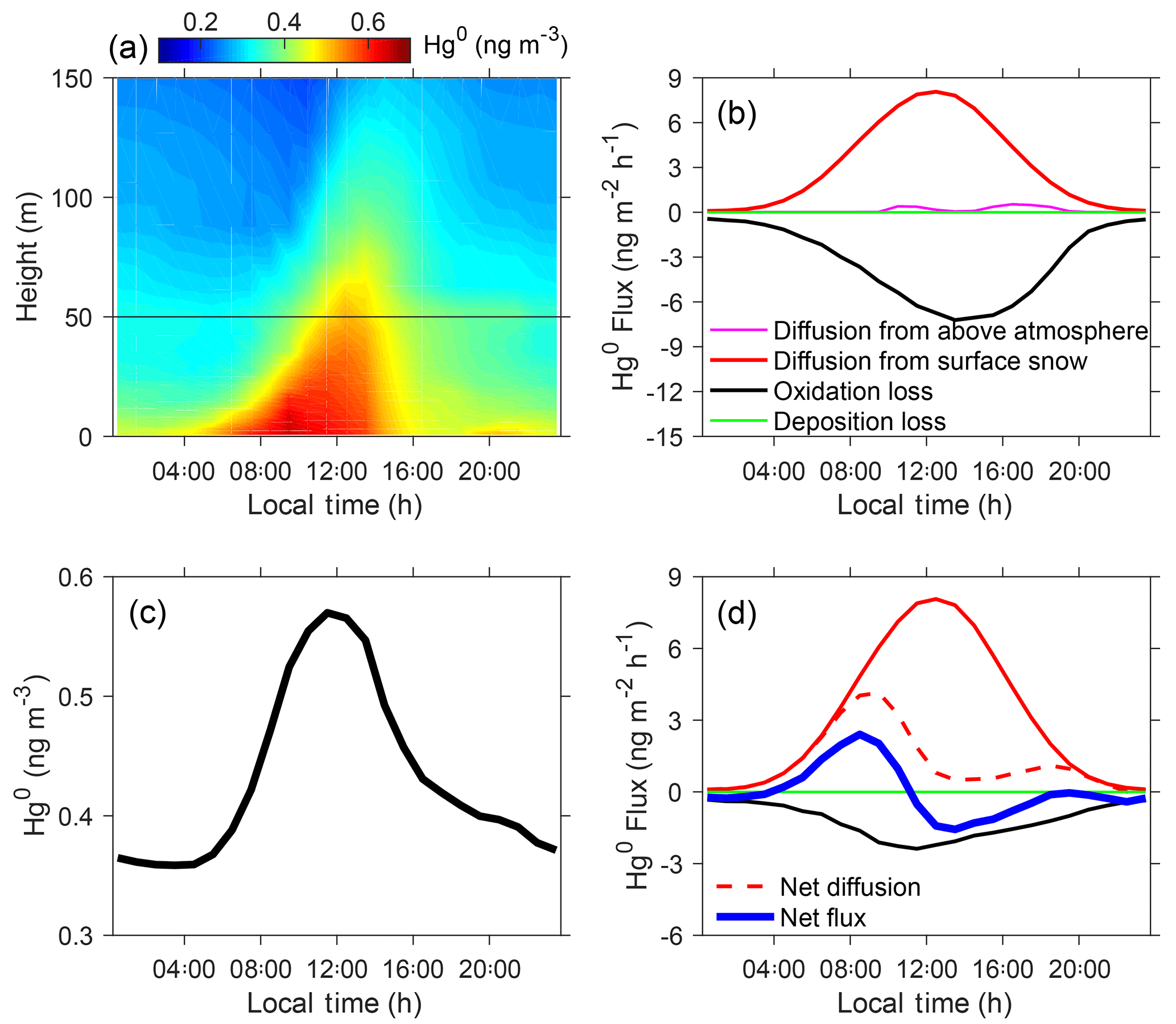
Figure 5Summertime diurnal cycles of Hg0 concentrations and fluxes. Panel (a) shows the modeled vertical distributions of Hg0 concentrations in near-surface air, (b) the modeled Hg0 fluxes in the inversion/mixed layers, (c) the modeled Hg0 concentration averaged for 0–50 m above the snow surface, and (d) the modeled Hg0 fluxes for the air in 0–50 m above the snow surface. Model results from the BR_HH_14d scenario are shown.
The summertime vertical and diurnal profiles of modeled Hg0 concentrations in near-surface air are shown in Fig. 5a. Model results are from the BR_HH_14d scenario (using high Br concentrations and upper-limit reaction rates; τOX ∼2 days and τPR of 2 weeks), which best reproduces the observed Hg0 in summer. We find that the diurnal variation ranges of Hg0 are greater than 0.2 ng m−3 only for near-surface levels from snow to about 50 m above. As shown in Fig. 5b, the summertime Hg0 cycles in the inversion/mixed layers are primarily driven by diffusion from snow and oxidation loss. The dry deposition and transport from the free troposphere are insignificant. The amplitude of Hg0 oxidation loss increases during the daytime due to enhanced photochemical activities. Diffusion of Hg0 from surface snow is controlled by the rate of snow HgII photoreduction, which also peaks in the daytime. The diurnal profiles of the modeled Hg0 fluxes from simulations using the O3 and OH oxidation schemes are given in Fig. S14. As expected, the amplitudes of their fluxes are much smaller than this bromine oxidation model scenario. In order to elucidate the drivers of strong diurnal variations of Hg0 in near-surface vertical levels in summer, we calculated the diurnal cycles of Hg0 concentrations and all related fluxes for 0–50 m above the snow (Fig. 5c and d). The net diffusion of Hg0 refers to difference in its diffusion from snow and to upper levels. The latter is controlled by the changing mixed layer heights, which are low at night (< 50 m) and strongly increased during the daytime (Angot et al., 2016c). Thus, at night, all Hg0 diffused from snow remains inside the shallow mixed layer, while in the daytime a large fraction is transferred to the air above 50 m. The net Hg0 flux, the derivative of its diurnal variation, is determined by the net diffusion and oxidation loss of Hg0. As shown in Fig. 5d, the net flux is positive in the morning but becomes negative in the afternoon, thus leading to the Hg0 maximum around noon. Overall, the diurnal variations of Hg0 in near-surface levels in summer are determined by the changes in the Hg0 oxidation loss, snow HgII photoreduction, and mixed layer depth, all of which are in turn controlled by the strong photochemical activity during this time period at Dome C.
Furthermore, our model results suggest that the air above Dome C is enriched in HgII during summer, consistent with its strong photochemical activity. As shown in Fig. 6, the predicted HgII by the BR_HH_14d scenario increases with height, from ∼0.1 near the surface to ∼0.5 ng m−3 at 500 m. Such HgII concentrations are comparable to the levels identified in the upper free troposphere for the midlatitudes (Bieser et al., 2017). A diurnal pattern of HgII with higher concentrations in the afternoon is predicted in near-surface air by the model. These characteristics should be verified by future measurement studies. Preliminary field sampling using polyethersulfone cation-exchange membranes in a 2014–2015 summer campaign obtained HgII of about 0.4 ng m−3 (average concentration from three filter samples) (Angot, 2016).
3.3 Comparison with summertime data at Summit Station, Greenland
Dome C (75∘ S, 123∘ E; 3 km above sea level) and Summit Station, Greenland (73∘ N, 38∘ W; 3.2 km above sea level), are both located in high altitude and far from the ocean (hundreds of kilometers). As a result, their meteorological and chemical conditions have similarities. In summer, both stations have shallow boundary layers that are stable at night but convective during the day (Helmig et al., 2002; Cohen et al., 2007; Van Dam et al., 2013). Active bromine chemistry was found to occur at Summit Station in summer (Thomas et al., 2011), and the average BrO mixing ratios in near-surface air were 0.9–1.5 pptv (Liao et al., 2011; Stutz et al., 2011), comparable to the 1 pptv upper limit at Dome C (Legrand et al., 2016b). Thus, it is expected that these two stations may have similar mercury variabilities in near-surface air. Brooks et al. (2011) measured atmospheric mercury concentrations in the summer of 2007–2008 at Summit Station but did not observe a significant diurnal cycle of Hg0 peaking at noon as was seen at Dome C. Based on our model analysis, we can identify several potential factors that can contribute to differences in the diurnal cycles of Hg0 between these two inland polar locations.
First, although BrO concentrations at Summit Station are comparable or higher than at Dome C, the concentrations of Br at Summit Station, the primary oxidant of Hg0, may be much lower. As described in Sect. 2.3, the [Br]∕[BrO] ratios are positively related to the concentrations of NO and negatively related to the concentrations of O3. Reported summertime NOx concentrations at Summit Station (∼20 pptv) are lower than at Dome C (∼300 pptv), whereas O3 at Summit Station (∼50 ppbv) is approximately 2 times that at Dome C (∼25 ppbv) (Helmig et al., 2008a; Frey et al., 2015; Kramer et al., 2015; Van Dam et al., 2015; Huang et al., 2017). The larger NOx concentrations at Dome C have been suggested to arise in part from larger NOx emissions from surface snow, which are in turn driven by the photolysis of nitrate in the surface snowpack (Frey et al., 2015). A back-of-the-envelope calculation shows, assuming the same BrO concentrations, that Br concentrations at Dome C would be on average a factor of 6 higher than at Summit Station. Second, the thermal dissociation rate of the intermediate HgIBr at Summit Station should be 1 order of magnitude greater than that at Dome C. This is because this rate strongly depends on temperature (Table 2), and the ambient temperature at Summit Station is about 15 K higher than at Dome C. Third, the oxidation of HgIBr by NO2 (the dominant second step oxidant) is significantly slower at Summit Station than at Dome C, due to their different concentrations of NO2. In fact, the rates of oxidation by NO2 and dissociation of HgIBr are comparable at Summit Station. This is in contrast with Dome C, where the oxidation by NO2 can easily outcompete the thermal dissociation of HgIBr. All in all, we expect that the Br-initiated oxidation of Hg0 should be slower at Summit Station than at Dome C, leading to weaker oxidation/re-emission cycling of Hg0 during summer. It is also noted that atmospheric circulation on Greenland may be influenced by stronger synoptic-scale events than over the Antarctic Plateau, because the air is thicker over the Greenland ice sheet (leading to a weaker decrease of relative vorticity when a large-scale eddy propagates from the ice sheet margin towards the center). However, the impact of this circulation pattern on the diurnal cycle of Hg0 is unclear.
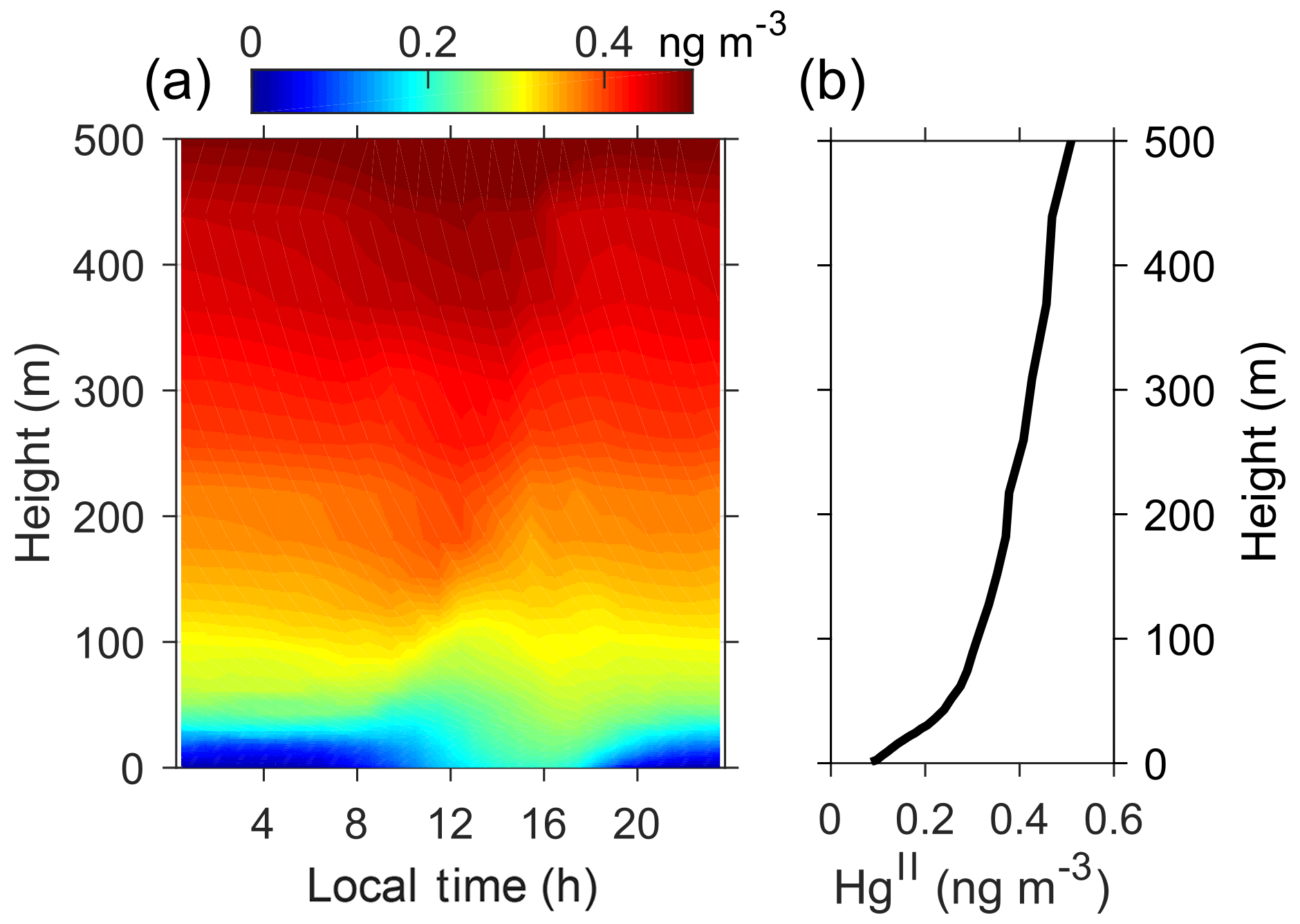
Figure 6Summertime diurnal and vertical profiles of atmospheric HgII concentrations. Panel (a) shows both diurnal and vertical distributions, and (b) only shows the average vertical profile. Model results from the BR_HH_14d scenario are shown.
3.4 Non-summer period
We showed above that the model simulations including the photoreduction of snow HgII and a fast bromine oxidation of atmospheric Hg0 could reasonably explain the observed atmospheric mercury variations during summer. However, these simulations strongly underestimate Hg0 concentrations in the non-summer months (Fig. 3), when solar radiation is weakened or completely absent. Based on our understanding of air and snow mercury cycling (Fig. 1), such model–observation discrepancies may imply, for the non-summer period, that in the model the vertical transport of mercury from the free troposphere is underestimated, the reduction of snow HgII is underestimated, and/or the oxidation of atmospheric Hg0 is overestimated. All these processes are poorly constrained in the non-summer period in part because previous studies have mainly focused on the summer season. The model performance can be improved by modifying the representation of these processes.
First of all, it is important to note in the above simulations that the adjusted Kz values in the warming events are used to drive the mercury model, which tends to underestimate the transport of mercury from the free troposphere. We therefore conducted a sensitivity simulation (BR_S1) to examine the possible effects of warming events on modeling results. The difference between BR_S1 and BR_HH_14d (using high Br concentrations and upper-limit reaction rates; τOX∼2 days and τPR of 2 weeks in summer) is that the original MAR-modeled Kz values are used in BR_S1, which may overestimate the transport of mercury from the free troposphere. As shown in Fig. 7a, in the non-summer months, near-surface air Hg0 concentrations by BR_S1 are close to the prescribed Hg0 concentrations in the free troposphere and are significantly higher than those from BR_HH_14d. However, the BR_S1 scenario cannot reproduce the high atmospheric Hg0 concentrations of ∼1.2 ng m−3 in fall (exceeding its levels at the Antarctic coastal regions and the southern hemispheric midlatitude sites) and the diurnal cycles of Hg0 in fall peaking in the night. This result indicates that Hg0 may be produced below the atmospheric mixed layers at Dome C. In addition, surface snow Hg concentrations by BR_S1 exhibit an increase during the non-summer period (Fig. 7b), as a result of HgII transport in warming events from the free troposphere (Fig. 7c). The deposited HgII is accumulated in surface snow (photoreduction of HgII is weak in the non-summer period). Such an enhancement of snow mercury was not measured at Dome C (Angot et al., 2016c). Therefore, we postulate that the existence of warming events during the non-summer period can significantly enhance Hg0 concentrations in near-surface air but is unlikely to be the only reason for the observed mercury variations.
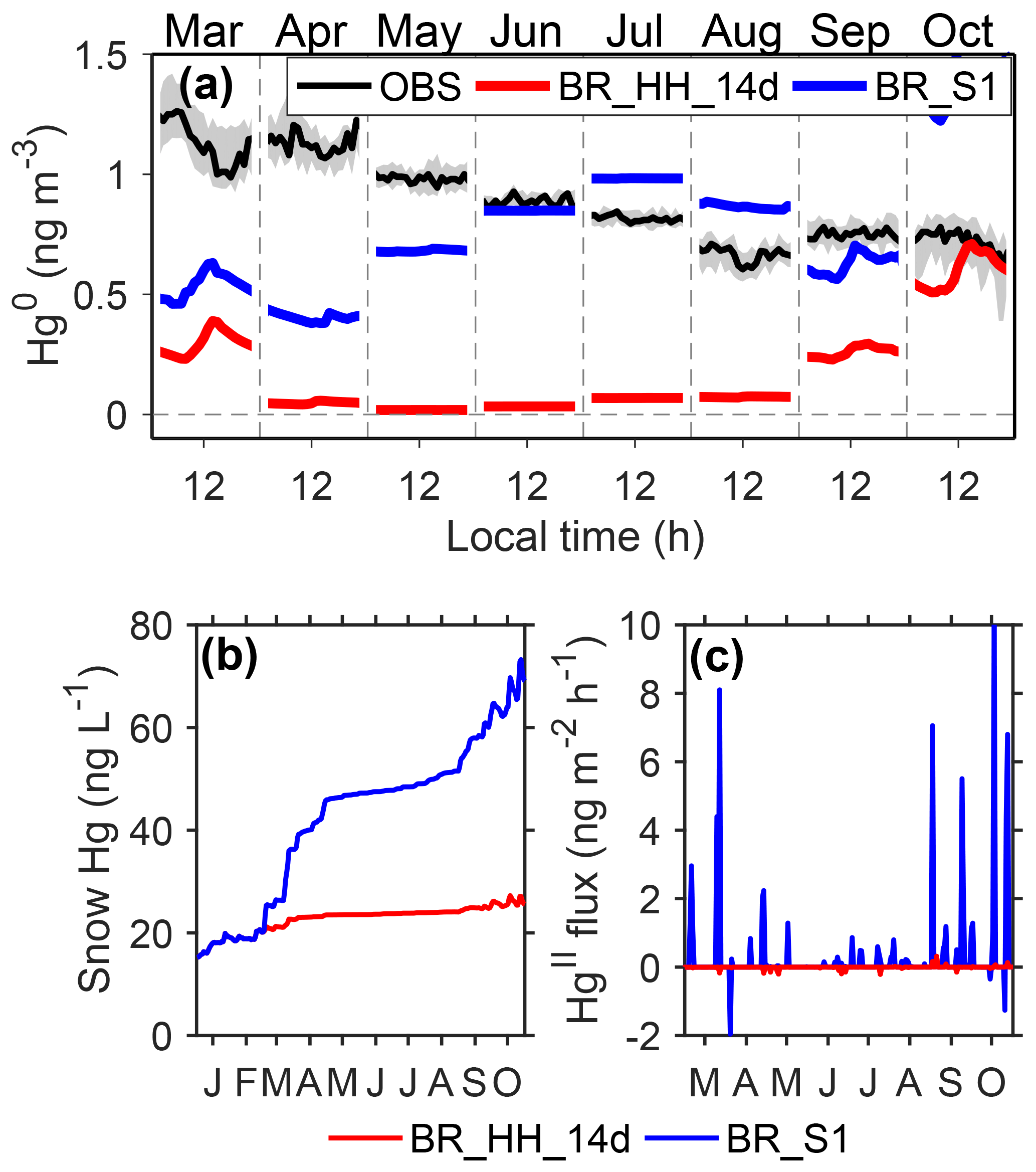
Figure 7Possible impacts of warming events on mercury concentrations in the non-summer period. Panel (a) shows Hg0 observations at 25 cm above the snow at Dome C, and the shaded regions indicate 25 % and 75 % percentiles. The modeled Hg0 concentrations from BR_ HH_14d and BR_S1 are also shown. Panel (b) shows surface snow mercury concentrations from BR_HH_ 14d and BR_S1. Panel (c) shows the exchange fluxes of HgII from the free troposphere modeled by BR_HH_14d and BR_S1.
Second, the reduction of snow HgII might occur in the dark, which would produce Hg0 and sustain atmospheric concentrations of Hg0 through snow-to-air diffusion and convective transport. The possibility of the presence of dark reduction has been reported in previous laboratory and field studies (Lalonde et al., 2003; Ferrari et al., 2004; Dommergue et al., 2007; Faïn et al., 2007), although actual mechanisms remain unclear. The reduction might be a continuation of photolytically initiated reactions or through reactions requiring no insolation at all (Durnford and Dastoor, 2011). The HO2 radical produced in the dark surface snowpack may serve as a potential HgII reductant (Dommergue et al., 2003; Ferrari et al., 2004). The dark reduction rates estimated in these studies are much lower than the photoreduction rates of HgII. Some observational evidence at Dome C supports the hypothesis of snow HgII dark reduction. Near-surface air Hg0 concentrations peaked in the night in fall, and Hg0 concentrations in snow interstitial air were higher than air Hg0 in fall and winter (Angot et al., 2016c). Thus, we have conducted a sensitivity simulation, BR_S2, which added a first-order dark reduction of snow HgII based on BR_HH_14d, in order to examine the possible effects of dark reduction on model results. The reaction rate corresponds to an average τDR of ∼1 year for the non-summer period and is scaled by NOx concentrations since this process is likely related to nitrogen chemistry. As shown in Fig. 8, the hypothesized snow HgII dark reduction process leads to a small increase in the snow-to-air diffusive fluxes of Hg0 (< 0.5 ng m−2 h−1), which can increase the concentrations of atmospheric Hg0 in the non-summer period, especially in winter. This scenario also better reproduces the diurnal variation of Hg0 in the fall months.
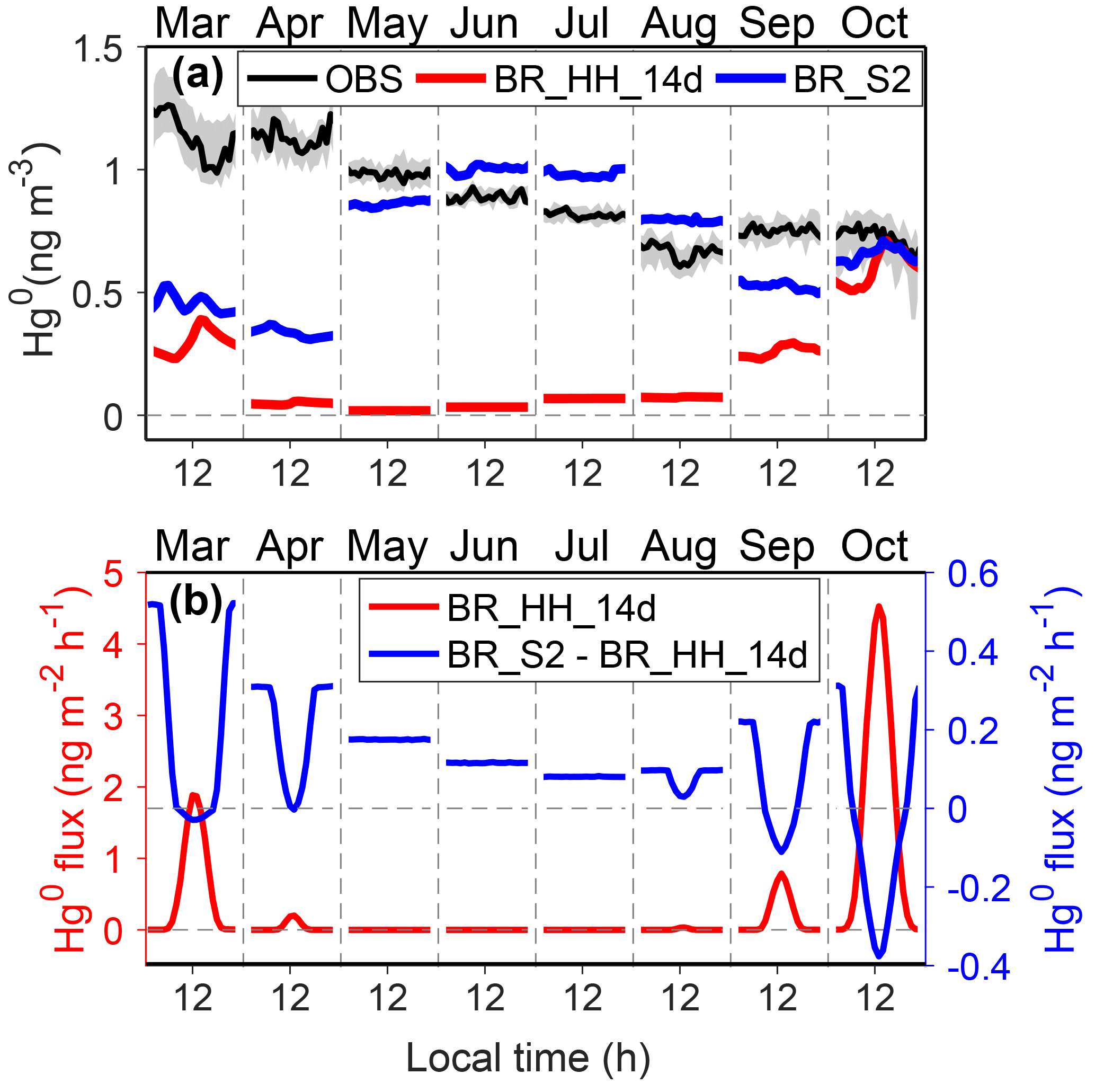
Figure 8Possible impacts of snow mercury dark reduction on Hg0 concentrations and fluxes in the non-summer period. Panel (a) shows Hg0 observations at 25 cm above the snow surface at Dome C, and the shaded regions indicate 25 % and 75 % percentiles. The modeled Hg0 concentrations from BR_HH_14d and BR_S2 are also shown. Panel (b) shows the modeled Hg0 snow-to-air diffusion fluxes from BR_HH_14d (left axis) and the difference of snow-to-air diffusive fluxes between BR_S2 and BR_HH_14d (right axis).
Third, oxidation of atmospheric Hg0 may be overestimated in our model in the non-summer period. As described in Sect. 2.3, the modeled BrO concentrations by the CTMs may have seasonal biases. Total inorganic bromine measurements at Dome C (Legrand et al., 2016b) have suggested that the modeled BrO is likely biased high by up to a factor of 3 in fall and spring. The reasons remain unknown but are probably related to several factors, including depositions of Br-containing species, snow re-emission or long-distance transport of Br2/BrCl, and photochemical Br reactions (Xin Yang, British Antarctic Survey, personal communication, 2017). In order to qualitatively evaluate this potential bias in BrO (and Br) concentrations, we have conducted a sensitivity simulation that reduces BrO (and thus Br) concentrations in fall by a factor of 3. We find that reducing BrO in fall could increase the modeled air Hg0 concentrations during the fall and winter months (Fig. S15).
Based on the above sensitivity analysis, we find that the all these three processes (intermittent warming events, dark reduction of snow mercury, and overestimation of bromine oxidation) can help explain the observed high mercury concentrations in the non-summer period. Their relative contributions, however, are difficult to constrain since the understanding of these processes is limited.
We have conducted box model calculations to explore important chemical and physical processes controlling the diurnal and seasonal variations of mercury at Dome C. The atmospheric Hg0 oxidation rates of the OH, O3, and the two-step Br-initiated schemes all have large uncertainty ranges due to uncertain chemical kinetics and oxidants concentrations. In austral summer, the Br oxidation scheme, favored by low ambient temperature and high concentrations of NOx, is more efficient than the OH and O3 schemes. The model simulations support the hypothesis that rapid recurring cycles of oxidation and re-emission of Hg0 occur in summer. Among the model scenarios tested, the simulations using the Br oxidation scheme (with upper-limit reaction rates) can best match mercury observations in summer. The modeling results indicate that strong diurnal variations of Hg0 in summer may be confined within several tens of meters above the snow surface and are primarily determined by changes in Hg0 oxidation loss, snow HgII photoreduction, and mixed layer depths. For the non-summer period, the model–observation comparisons at Dome C suggest the intermittent warming events and a hypothesized dark reduction of snow HgII may be important processes controlling the mercury variations, but their relative importance is uncertain. The Br-initiated oxidation of Hg0 is expected to be slower at Summit Station, Greenland, because of high temperatures, high O3, and low NOx conditions, which might contribute to the observed differences in the summertime diurnal variations of Hg0 between these two polar inland locations.
In order to obtain a better understanding of mercury cycling over the East Antarctic Plateau, we suggest several areas for future research. (1) It is essential to better constrain the concentration levels of bromine species, especially BrOx, through more field experiments and modeling studies. (2) It is important to reduce uncertainties in existing chemical kinetic parameters of bromine oxidation mechanisms. The rate constant of Hg0 reaction with Br from existing theoretical and experimental studies varies by a factor of 4. (3) Our modeling indicates relatively high atmospheric HgII concentrations in summer, which remains to be verified by additional field measurements. (4) A better characterization of atmospheric vertical transport during the non-summer period is needed, in particular the role of intermittent warming events. (5) The chemical mechanisms and reaction rates for snow mercury processes, including photoreduction and dark reduction, should be further investigated. Future modeling work should also improve the representation of those processes (e.g., diamond dust) shown in Table 2.
Given the rapid exchange of mercury between the surface snowpack and the above atmosphere (especially during summer), regional modeling studies should be conducted in the future in order to understand the total and speciated mercury budgets over the entire Antarctic Plateau and the influence of the plateau on the coastal environments.
The mercury box model is available at http://github.com/shaojiesong/Hg_DomeConcordia (last access: 30 October 2018). The mercury measurement data at Dome C are available upon request at http://sdi.iia.cnr.it/geoint/publicpage/GMOS/gmos_historical.zul (GMOS, 2018). The ozone and NOx measurement data at Dome C are available upon request to the authors.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-15825-2018-supplement.
SS, NES, and AD initialized and designed the study. SS, HA, and NES developed the model. HG provided MAR data. HA, FS, NP, OM, and AD conducted Hg observations, and DH and JS conducted NOx observations used in the model. SS wrote the paper with input from all the authors.
The authors declare that they have no conflict of interest.
The authors acknowledge the MISTI Global Seed Funds (MIT-France). The model
work at MIT is supported by the US NSF Atmospheric Chemistry Program
no. 1053648. Mercury measurements at Concordia Station were initiated with
the FP7 GMOS project and are supported by IPEV GMOstral Program 1028. Detlev Helmig
acknowledges logistical and financial support by the French Polar Institute
(Program 1011, SUNITEDC) and support from the US NSF Office of Polar
Programs through grant no. 1142145. Aurélien Dommergue and Joël Savarino thank LEFE/INSU for their
financial support. Shaojie Song thanks the MIT Henry Houghton Fund, and Hélène Angot the
Univ. Grenoble Alpes Doctoral School for Earth, Planetary and Environmental
Sciences for travel support. We thank Tomás Sherwen for providing
GEOS-Chem bromine model results, Xin Yang for p-TOMCAT bromine model results,
Oleg Travnikov for GLEMOS mercury model results, Michel Legrand for ozone observational data,
Christophe Genthon for the meteorological observational data, and Jack Dibb for access to
the measurement data from Summit Station. We thank Susan Solomon, Ronald Prinn,
Daniel Jaffe, Jennie Thomas, Warren Cairns, Jeroen Sonke, Roberto Grilli,
Michel Legrand, and Christopher Holmes for comments and/or helpful
discussions.
Edited by: Carlo Barbante
Reviewed by: two anonymous referees
Angot, H.: Cycle atmosphérique du mercure dans des zones reculées de l'Hémisphère Sud: cas de la couche limite marine subantarctique et du continent Antarctique, PhD thesis, Université Grenoble Alpes, 2016.
Angot, H., Barret, M., Magand, O., Ramonet, M., and Dommergue, A.: A 2-year record of atmospheric mercury species at a background Southern Hemisphere station on Amsterdam Island, Atmos. Chem. Phys., 14, 11461–11473, https://doi.org/10.5194/acp-14-11461-2014, 2014.
Angot, H., Dastoor, A., De Simone, F., Gårdfeldt, K., Gencarelli, C. N., Hedgecock, I. M., Langer, S., Magand, O., Mastromonaco, M. N., Nordstrøm, C., Pfaffhuber, K. A., Pirrone, N., Ryjkov, A., Selin, N. E., Skov, H., Song, S., Sprovieri, F., Steffen, A., Toyota, K., Travnikov, O., Yang, X., and Dommergue, A.: Chemical cycling and deposition of atmospheric mercury in polar regions: review of recent measurements and comparison with models, Atmos. Chem. Phys., 16, 10735–10763, https://doi.org/10.5194/acp-16-10735-2016, 2016a.
Angot, H., Dion, I., Vogel, N., Legrand, M., Magand, O., and Dommergue, A.: Multi-year record of atmospheric mercury at Dumont d'Urville, East Antarctic coast: continental outflow and oceanic influences, Atmos. Chem. Phys., 16, 8265–8279, https://doi.org/10.5194/acp-16-8265-2016, 2016b.
Angot, H., Magand, O., Helmig, D., Ricaud, P., Quennehen, B., Gallée, H., Del Guasta, M., Sprovieri, F., Pirrone, N., Savarino, J., and Dommergue, A.: New insights into the atmospheric mercury cycling in central Antarctica and implications on a continental scale, Atmos. Chem. Phys., 16, 8249–8264, https://doi.org/10.5194/acp-16-8249-2016, 2016c.
Ariya, P. A., Khalizov, A., and Gidas, A.: Reactions of gaseous mercury with atomic and molecular halogens:? Kinetics, product studies, and atmospheric implications, J. Phys. Chem. A, 106, 7310–7320, https://doi.org/10.1021/jp020719o, 2002.
Ariya, P. A., Amyot, M., Dastoor, A., Deeds, D., Feinberg, A., Kos, G., Poulain, A., Ryjkov, A., Semeniuk, K., Subir, M., and Toyota, K.: Mercury physicochemical and biogeochemical transformation in the atmosphere and at atmospheric interfaces: A review and future directions, Chem. Rev., 115, 3760–3802, https://doi.org/10.1021/cr500667e, 2015.
Balabanov, N. B., Shepler, B. C., and Peterson, K. A.: Accurate global potential energy surface and reaction dynamics for the ground state of HgBr2, J. Phys. Chem. A, 109, 8765–8773, https://doi.org/10.1021/jp053415l, 2005.
Bargagli, R.: Atmospheric chemistry of mercury in Antarctica and the role of cryptogams to assess deposition patterns in coastal ice-free areas, Chemosphere, 163, 202–208, https://doi.org/10.1016/j.chemosphere.2016.08.007, 2016.
Bieser, J., Slemr, F., Ambrose, J., Brenninkmeijer, C., Brooks, S., Dastoor, A., DeSimone, F., Ebinghaus, R., Gencarelli, C. N., Geyer, B., Gratz, L. E., Hedgecock, I. M., Jaffe, D., Kelley, P., Lin, C.-J., Jaegle, L., Matthias, V., Ryjkov, A., Selin, N. E., Song, S., Travnikov, O., Weigelt, A., Luke, W., Ren, X., Zahn, A., Yang, X., Zhu, Y., and Pirrone, N.: Multi-model study of mercury dispersion in the atmosphere: vertical and interhemispheric distribution of mercury species, Atmos. Chem. Phys., 17, 6925–6955, https://doi.org/10.5194/acp-17-6925-2017, 2017.
Brooks, S., Arimoto, R., Lindberg, S., and Southworth, G.: Antarctic polar plateau snow surface conversion of deposited oxidized mercury to gaseous elemental mercury with fractional long-term burial, Atmos. Environ., 42, 2877–2884, https://doi.org/10.1016/j.atmosenv.2007.05.029, 2008.
Brooks, S., Moore, C., Lew, D., Lefer, B., Huey, G., and Tanner, D.: Temperature and sunlight controls of mercury oxidation and deposition atop the Greenland ice sheet, Atmos. Chem. Phys., 11, 8295–8306, https://doi.org/10.5194/acp-11-8295-2011, 2011.
Brooks, S. B., Saiz-Lopez, A., Skov, H., Lindberg, S. E., Plane, J. M. C., and Goodsite, M. E.: The mass balance of mercury in the springtime arctic environment, Geophys. Res. Lett., 33, L13812, https://doi.org/10.1029/2005GL025525, 2006.
Cohen, L., Helmig, D., Neff, W. D., Grachev, A. A., and Fairall, C. W.: Boundary-layer dynamics and its influence on atmospheric chemistry at Summit, Greenland, Atmos. Environ., 41, 5044–5060, https://doi.org/10.1016/j.atmosenv.2006.06.068, 2007.
Cunningham, J. and Waddington, E. D.: Air flow and dry deposition of non-sea salt sulfate in polar firn: Paleoclimatic implications, Atmos. Environ., 27, 2943–2956, https://doi.org/10.1016/0960-1686(93)90327-U, 1993.
Dibble, T. S., Zelie, M. J., and Mao, H.: Thermodynamics of reactions of ClHg and BrHg radicals with atmospherically abundant free radicals, Atmos. Chem. Phys., 12, 10271–10279, https://doi.org/10.5194/acp-12-10271-2012, 2012.
Dommergue, A., Ferrari, C. P., Poissant, L., Gauchard, P.-A., and Boutron, C. F.: Diurnal cycles of gaseous mercury within the snowpack at Kuujjuarapik/Whapmagoostui, Québec, Canada, Environ. Sci. Technol., 37, 3289–3297, https://doi.org/10.1021/es026242b, 2003.
Dommergue, A., Bahlmann, E., Ebinghaus, R., Ferrari, C., and Boutron, C.: Laboratory simulation of Hg0 emissions from a snowpack, Anal. Bioanal. Chem., 388, 319–327, https://doi.org/10.1007/s00216-007-1186-2, 2007.
Dommergue, A., Sprovieri, F., Pirrone, N., Ebinghaus, R., Brooks, S., Courteaud, J., and Ferrari, C. P.: Overview of mercury measurements in the Antarctic troposphere, Atmos. Chem. Phys., 10, 3309–3319, https://doi.org/10.5194/acp-10-3309-2010, 2010.
Dommergue, A., Barret, M., Courteaud, J., Cristofanelli, P., Ferrari, C. P., and Gallée, H.: Dynamic recycling of gaseous elemental mercury in the boundary layer of the Antarctic Plateau, Atmos. Chem. Phys., 12, 11027–11036, https://doi.org/10.5194/acp-12-11027-2012, 2012.
Donohoue, D. L., Bauer, D., Cossairt, B., and Hynes, A. J.: Temperature and pressure dependent rate coefficients for the reaction of Hg with Br and the reaction of Br with Br: A pulsed laser photolysis-pulsed laser induced fluorescence study, J. Phys. Chem. A, 110, 6623–6632, https://doi.org/10.1021/jp054688j, 2006.
Durnford, D. and Dastoor, A.: The behavior of mercury in the cryosphere: A review of what we know from observations, J. Geophys. Res.-Atmos., 116, D06305, https://doi.org/10.1029/2010JD014809, 2011.
Durnford, D., Dastoor, A., Ryzhkov, A., Poissant, L., Pilote, M., and Figueras-Nieto, D.: How relevant is the deposition of mercury onto snowpacks? – Part 2: A modeling study, Atmos. Chem. Phys., 12, 9251–9274, https://doi.org/10.5194/acp-12-9251-2012, 2012.
Eisele, F., Davis, D. D., Helmig, D., Oltmans, S. J., Neff, W., Huey, G., Tanner, D., Chen, G., Crawford, J., Arimoto, R., Buhr, M., Mauldin, L., Hutterli, M., Dibb, J., Blake, D., Brooks, S. B., Johnson, B., Roberts, J. M., Wang, Y., Tan, D., and Flocke, F.: Antarctic Tropospheric Chemistry Investigation (ANTCI) 2003 overview, Atmos. Environ., 42, 2749–2761, https://doi.org/10.1016/j.atmosenv.2007.04.013, 2008.
Faïn, X., Grangeon, S., Bahlmann, E., Fritsche, J., Obrist, D., Dommergue, A., Ferrari, C. P., Cairns, W., Ebinghaus, R., Barbante, C., Cescon, P., and Boutron, C.: Diurnal production of gaseous mercury in the alpine snowpack before snowmelt, J. Geophys. Res.-Atmos., 112, D21311, https://doi.org/10.1029/2007JD008520, 2007.
Ferrari, C. P., Dommergue, A., Boutron, C. F., Skov, H., Goodsite, M., and Jensen, B.: Nighttime production of elemental gaseous mercury in interstitial air of snow at Station Nord, Greenland, Atmos. Environ., 38, 2727–2735, https://doi.org/10.1016/j.atmosenv.2004.02.023, 2004.
France, J. L., King, M. D., Frey, M. M., Erbland, J., Picard, G., Preunkert, S., MacArthur, A., and Savarino, J.: Snow optical properties at Dome C (Concordia), Antarctica; implications for snow emissions and snow chemistry of reactive nitrogen, Atmos. Chem. Phys., 11, 9787–9801, https://doi.org/10.5194/acp-11-9787-2011, 2011.
Frey, M. M., Roscoe, H. K., Kukui, A., Savarino, J., France, J. L., King, M. D., Legrand, M., and Preunkert, S.: Atmospheric nitrogen oxides (NO and NO2) at Dome C, East Antarctica, during the OPALE campaign, Atmos. Chem. Phys., 15, 7859–7875, https://doi.org/10.5194/acp-15-7859-2015, 2015.
Gallée, H. and Gorodetskaya, I. V.: Validation of a limited area model over Dome C, Antarctic Plateau, during winter, Clim. Dynam., 34, 61–72, https://doi.org/10.1007/s00382-008-0499-y, 2010.
Gallée, H., Preunkert, S., Argentini, S., Frey, M. M., Genthon, C., Jourdain, B., Pietroni, I., Casasanta, G., Barral, H., Vignon, E., Amory, C., and Legrand, M.: Characterization of the boundary layer at Dome C (East Antarctica) during the OPALE summer campaign, Atmos. Chem. Phys., 15, 6225–6236, https://doi.org/10.5194/acp-15-6225-2015, 2015.
Genthon, C., Town, M. S., Six, D., Favier, V., Argentini, S., and Pellegrini, A.: Meteorological atmospheric boundary layer measurements and ECMWF analyses during summer at Dome C, Antarctica, J. Geophys. Res.-Atmos., 115, D05104, https://doi.org/10.1029/2009JD012741, 2010.
Global Mercury Observation System (GMOS): Land-based Monitoring Sites, available at: http://sdi.iia.cnr.it/geoint/publicpage/GMOS/gmos_historical.zul, last access: 30 October 2018.
Goodsite, M. E., Plane, J. M. C., and Skov, H.: A Theoretical Study of the Oxidation of Hg0 to HgBr2 in the Troposphere, Environ. Sci. Technol., 38, 1772–1776, https://doi.org/10.1021/es034680s, 2004.
Goodsite, M. E., Plane, J. M. C., and Skov, H.: Correction to a theoretical study of the oxidation of Hg0 to HgBr2 in the troposphere, Environ. Sci. Technol., 46, 5262–5262, https://doi.org/10.1021/es301201c, 2012.
Hall, B.: The gas phase oxidation of elemental mercury by ozone, in: Mercury as a Global Pollutant: Proceedings of the Third International Conference held in Whistler, British Columbia, 10–14 July 1994, edited by: Porcella, D. B., Huckabee, J. W., and Wheatley, B., Springer Netherlands, Dordrecht, 301–315, 1995.
Han, Y., Huh, Y., Hong, S., Hur, S. D., and Motoyama, H.: Evidence of air-snow mercury exchange recorded in the snowpack at Dome Fuji, Antarctica, Geosci. J., 18, 105–113, https://doi.org/10.1007/s12303-013-0054-7, 2014.
Han, Y., Huh, Y., Hur, S. D., Hong, S., Chung, J. W., and Motoyama, H.: Net deposition of mercury to the Antarctic Plateau enhanced by sea salt, Sci. Total Environ., 583, 81–87, https://doi.org/10.1016/j.scitotenv.2017.01.008, 2017.
Helmig, D., Boulter, J., David, D., Birks, J. W., Cullen, N. J., Steffen, K., Johnson, B. J., and Oltmans, S. J.: Ozone and meteorological boundary-layer conditions at Summit, Greenland, during 3–21 June 2000, Atmos. Environ., 36, 2595–2608, https://doi.org/10.1016/S1352-2310(02)00129-2, 2002.
Helmig, D., Johnson, B., Oltmans, S. J., Neff, W., Eisele, F., and Davis, D. D.: Elevated ozone in the boundary layer at South Pole, Atmos. Environ., 42, 2788–2803, https://doi.org/10.1016/j.atmosenv.2006.12.032, 2008a.
Helmig, D., Johnson, B. J., Warshawsky, M., Morse, T., Neff, W. D., Eisele, F., and Davis, D. D.: Nitric oxide in the boundary-layer at South Pole during the Antarctic Tropospheric Chemistry Investigation (ANTCI), Atmos. Environ., 42, 2817–2830, https://doi.org/10.1016/j.atmosenv.2007.03.061, 2008b.
Helmig, D., Liptzin, D., Hueber, J., and Savarino, J.: Seasonal cycle and snow and atmospheric chemistry of ozone and nitrogen oxides at Concordia Station, manuscript in preparation, 2018.
Holmes, C. D., Jacob, D. J., and Yang, X.: Global lifetime of elemental mercury against oxidation by atomic bromine in the free troposphere, Geophys. Res. Lett., 33, L20808, https://doi.org/10.1029/2006GL027176, 2006.
Holmes, C. D., Jacob, D. J., Corbitt, E. S., Mao, J., Yang, X., Talbot, R., and Slemr, F.: Global atmospheric model for mercury including oxidation by bromine atoms, Atmos. Chem. Phys., 10, 12037–12057, https://doi.org/10.5194/acp-10-12037-2010, 2010.
Horowitz, H. M., Jacob, D. J., Zhang, Y., Dibble, T. S., Slemr, F., Amos, H. M., Schmidt, J. A., Corbitt, E. S., Marais, E. A., and Sunderland, E. M.: A new mechanism for atmospheric mercury redox chemistry: implications for the global mercury budget, Atmos. Chem. Phys., 17, 6353–6371, https://doi.org/10.5194/acp-17-6353-2017, 2017.
Huang, Y., Wu, S., Kramer, L. J., Helmig, D., and Honrath, R. E.: Surface ozone and its precursors at Summit, Greenland: comparison between observations and model simulations, Atmos. Chem. Phys., 17, 14661–14674, https://doi.org/10.5194/acp-17-14661-2017, 2017.
Jiao, Y. and Dibble, T. S.: First kinetic study of the atmospherically important reactions BrHg + NO2 and BrHg + HOO, Phys. Chem. Chem. Phys., 19, 1826–1838, https://doi.org/10.1039/C6CP06276H, 2017.
Johnson, K. P., Blum, J. D., Keeler, G. J., and Douglas, T. A.: Investigation of the deposition and emission of mercury in arctic snow during an atmospheric mercury depletion event, J. Geophys. Res.-Atmos., 113, D17304, https://doi.org/10.1029/2008JD009893, 2008.
Kramer, L. J., Helmig, D., Burkhart, J. F., Stohl, A., Oltmans, S., and Honrath, R. E.: Seasonal variability of atmospheric nitrogen oxides and non-methane hydrocarbons at the GEOSummit station, Greenland, Atmos. Chem. Phys., 15, 6827–6849, https://doi.org/10.5194/acp-15-6827-2015, 2015.
Kukui, A., Legrand, M., Preunkert, S., Frey, M. M., Loisil, R., Gil Roca, J., Jourdain, B., King, M. D., France, J. L., and Ancellet, G.: Measurements of OH and RO2 radicals at Dome C, East Antarctica, Atmos. Chem. Phys., 14, 12373–12392, https://doi.org/10.5194/acp-14-12373-2014, 2014.
Lalonde, J. D., Amyot, M., Doyon, M.-R., and Auclair, J.-C.: Photo-induced Hg(II) reduction in snow from the remote and temperate Experimental Lakes Area (Ontario, Canada), J. Geophys. Res.-Atmos., 108, 4200, https://doi.org/10.1029/2001JD001534, 2003.
Legrand, M., Preunkert, S., Frey, M., Bartels-Rausch, Th., Kukui, A., King, M. D., Savarino, J., Kerbrat, M., and Jourdain, B.: Large mixing ratios of atmospheric nitrous acid (HONO) at Concordia (East Antarctic Plateau) in summer: a strong source from surface snow?, Atmos. Chem. Phys., 14, 9963–9976, https://doi.org/10.5194/acp-14-9963-2014, 2014.
Legrand, M., Preunkert, S., Savarino, J., Frey, M. M., Kukui, A., Helmig, D., Jourdain, B., Jones, A. E., Weller, R., Brough, N., and Gallée, H.: Inter-annual variability of surface ozone at coastal (Dumont d'Urville, 2004–2014) and inland (Concordia, 2007–2014) sites in East Antarctica, Atmos. Chem. Phys., 16, 8053–8069, https://doi.org/10.5194/acp-16-8053-2016, 2016a.
Legrand, M., Yang, X., Preunkert, S., and Theys, N.: Year-round records of sea salt, gaseous, and particulate inorganic bromine in the atmospheric boundary layer at coastal (Dumont d'Urville) and central (Concordia) East Antarctic sites, J. Geophys. Res.-Atmos., 121, 997–1023, https://doi.org/10.1002/2015JD024066, 2016b.
Li, C., Kang, S., Shi, G., Huang, J., Ding, M., Zhang, Q., Zhang, L., Guo, J., Xiao, C., Hou, S., Sun, B., Qin, D., and Ren, J.: Spatial and temporal variations of total mercury in Antarctic snow along the transect from Zhongshan Station to Dome A, Tellus B, 66, 25152, https://doi.org/10.3402/tellusb.v66.25152, 2014.
Liao, J., Huey, L. G., Tanner, D. J., Brough, N., Brooks, S., Dibb, J. E., Stutz, J., Thomas, J. L., Lefer, B., Haman, C., and Gorham, K.: Observations of hydroxyl and peroxy radicals and the impact of BrO at Summit, Greenland in 2007 and 2008, Atmos. Chem. Phys., 11, 8577–8591, https://doi.org/10.5194/acp-11-8577-2011, 2011.
Lin, C.-J. and Pehkonen, S. O.: The chemistry of atmospheric mercury: a review, Atmos. Environ., 33, 2067–2079, https://doi.org/10.1016/S1352-2310(98)00387-2, 1999.
Lindberg, S. E., Brooks, S., Lin, C. J., Scott, K. J., Landis, M. S., Stevens, R. K., Goodsite, M., and Richter, A.: Dynamic oxidation of gaseous mercury in the Arctic troposphere at polar sunrise, Environ. Sci. Technol., 36, 1245–1256, https://doi.org/10.1021/es0111941, 2002.
Martin, L. G., Labuschagne, C., Brunke, E.-G., Weigelt, A., Ebinghaus, R., and Slemr, F.: Trend of atmospheric mercury concentrations at Cape Point for 1995–2004 and since 2007, Atmos. Chem. Phys., 17, 2393–2399, https://doi.org/10.5194/acp-17-2393-2017, 2017.
Mergler, D., Anderson, H. A., Chan, L. H. M., Mahaffey, K. R., Murray, M., Sakamoto, M., and Stern, A. H.: Methylmercury exposure and health effects in humans: A worldwide concern, Ambio, 36, 3–11, https://doi.org/10.1579/0044-7447(2007)36[3:MEAHEI]2.0.CO;2, 2007.
Neff, W., Helmig, D., Grachev, A., and Davis, D.: A study of boundary layer behavior associated with high NO concentrations at the South Pole using a minisodar, tethered balloon, and sonic anemometer, Atmos. Environ., 42, 2762–2779, https://doi.org/10.1016/j.atmosenv.2007.01.033, 2008.
Palerme, C., Claud, C., Dufour, A., Genthon, C., Wood, N. B., and L'Ecuyer, T.: Evaluation of Antarctic snowfall in global meteorological reanalyses, Atmos. Res., 190, 104–112, https://doi.org/10.1016/j.atmosres.2017.02.015, 2017.
Pietroni, I., Argentini, S., and Petenko, I.: One year of surface-based temperature inversions at Dome C, Antarctica, Bound.-Lay. Meteorol., 150, 131–151, https://doi.org/10.1007/s10546-013-9861-7, 2014.
Poulain, A. J., Lalonde, J. D., Amyot, M., Shead, J. A., Raofie, F., and Ariya, P. A.: Redox transformations of mercury in an Arctic snowpack at springtime, Atmos. Environ., 38, 6763–6774, https://doi.org/10.1016/j.atmosenv.2004.09.013, 2004.
Preunkert, S., Legrand, M., Frey, M. M., Kukui, A., Savarino, J., Gallée, H., King, M., Jourdain, B., Vicars, W., and Helmig, D.: Formaldehyde (HCHO) in air, snow, and interstitial air at Concordia (East Antarctic Plateau) in summer, Atmos. Chem. Phys., 15, 6689–6705, https://doi.org/10.5194/acp-15-6689-2015, 2015.
Sander, S. P., Abbatt, J., Barker, J. R., Burkholder, J. B., Friedl, R. R., Golden, D. M., Huie, R. E., Kolb, C. E., Kurylo, M. J., Moortgat, G. K., Orkin, V. L., and Wine, P. H.: Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 17, JPL Publication 10-6, Jet Propulsion Laboratory, Pasadena, available at: https://jpldataeval.jpl.nasa.gov (last access: 30 October 2018), 2011.
Schroeder, W. H., Yarwood, G., and Niki, H.: Transformation processes involving mercury species in the atmosphere – results from a literature survey, Water Air Soil Pollut., 56, 653–666, https://doi.org/10.1007/bf00342307, 1991.
Selin, N. E.: Global biogeochemical cycling of mercury: A review, Annu. Rev. Environ. Resour., 34, 43–63, https://doi.org/10.1146/annurev.environ.051308.084314, 2009.
Sherwen, T., Schmidt, J. A., Evans, M. J., Carpenter, L. J., Großmann, K., Eastham, S. D., Jacob, D. J., Dix, B., Koenig, T. K., Sinreich, R., Ortega, I., Volkamer, R., Saiz-Lopez, A., Prados-Roman, C., Mahajan, A. S., and Ordóñez, C.: Global impacts of tropospheric halogens (Cl, Br, I) on oxidants and composition in GEOS-Chem, Atmos. Chem. Phys., 16, 12239–12271, https://doi.org/10.5194/acp-16-12239-2016, 2016.
Skov, H., Brooks, S. B., Goodsite, M. E., Lindberg, S. E., Meyers, T. P., Landis, M. S., Larsen, M. R. B., Jensen, B., McConville, G., and Christensen, J.: Fluxes of reactive gaseous mercury measured with a newly developed method using relaxed eddy accumulation, Atmos. Environ., 40, 5452–5463, https://doi.org/10.1016/j.atmosenv.2006.04.061, 2006.
Slusher, D. L., Neff, W. D., Kim, S., Huey, L. G., Wang, Y., Zeng, T., Tanner, D. J., Blake, D. R., Beyersdorf, A., Lefer, B. L., Crawford, J. H., Eisele, F. L., Mauldin, R. L., Kosciuch, E., Buhr, M. P., Wallace, H. W., and Davis, D. D.: Atmospheric chemistry results from the ANTCI 2005 Antarctic plateau airborne study, J. Geophys. Res.-Atmos., 115, D07304, https://doi.org/10.1029/2009JD012605, 2010.
Sommar, J., Gårdfeldt, K., Strömberg, D., and Feng, X.: A kinetic study of the gas-phase reaction between the hydroxyl radical and atomic mercury, Atmos. Environ., 35, 3049–3054, https://doi.org/10.1016/S1352-2310(01)00108-X, 2001.
Song, S., Selin, N. E., Soerensen, A. L., Angot, H., Artz, R., Brooks, S., Brunke, E.-G., Conley, G., Dommergue, A., Ebinghaus, R., Holsen, T. M., Jaffe, D. A., Kang, S., Kelley, P., Luke, W. T., Magand, O., Marumoto, K., Pfaffhuber, K. A., Ren, X., Sheu, G.-R., Slemr, F., Warneke, T., Weigelt, A., Weiss-Penzias, P., Wip, D. C., and Zhang, Q.: Top-down constraints on atmospheric mercury emissions and implications for global biogeochemical cycling, Atmos. Chem. Phys., 15, 7103–7125, https://doi.org/10.5194/acp-15-7103-2015, 2015.
Spolaor, A., Angot, H., Roman, M., Dommergue, A., Scarchilli, C., Vardè, M., Del Guasta, M., Pedeli, X., Varin, C., Sprovieri, F., Magand, O., Legrand, M., Barbante, C., and Cairns, W. R. L.: Feedback mechanisms between snow and atmospheric mercury: Results and observations from field campaigns on the Antarctic plateau, Chemosphere, 306–317, https://doi.org/10.1016/j.chemosphere.2017.12.180, 2018.
Sprovieri, F., Pirrone, N., Landis, M. S., and Stevens, R. K.: Oxidation of gaseous elemental mercury to gaseous divalent mercury during 2003 polar sunrise at Ny-Alesund, Environ. Sci. Technol., 39, 9156–9165, https://doi.org/10.1021/es050965o, 2005.
Steffen, A., Douglas, T., Amyot, M., Ariya, P., Aspmo, K., Berg, T., Bottenheim, J., Brooks, S., Cobbett, F., Dastoor, A., Dommergue, A., Ebinghaus, R., Ferrari, C., Gardfeldt, K., Goodsite, M. E., Lean, D., Poulain, A. J., Scherz, C., Skov, H., Sommar, J., and Temme, C.: A synthesis of atmospheric mercury depletion event chemistry in the atmosphere and snow, Atmos. Chem. Phys., 8, 1445–1482, https://doi.org/10.5194/acp-8-1445-2008, 2008.
Stutz, J., Thomas, J. L., Hurlock, S. C., Schneider, M., von Glasow, R., Piot, M., Gorham, K., Burkhart, J. F., Ziemba, L., Dibb, J. E., and Lefer, B. L.: Longpath DOAS observations of surface BrO at Summit, Greenland, Atmos. Chem. Phys., 11, 9 899–9910, https://doi.org/10.5194/acp-11-9899-2011, 2011.
Subir, M., Ariya, P. A., and Dastoor, A. P.: A review of uncertainties in atmospheric modeling of mercury chemistry I. Uncertainties in existing kinetic parameters – Fundamental limitations and the importance of heterogeneous chemistry, Atmos. Environ., 45, 5664–5676, https://doi.org/10.1016/j.atmosenv.2011.04.046, 2011.
Thomas, J. L., Stutz, J., Lefer, B., Huey, L. G., Toyota, K., Dibb, J. E., and von Glasow, R.: Modeling chemistry in and above snow at Summit, Greenland – Part 1: Model description and results, Atmos. Chem. Phys., 11, 4899–4914, https://doi.org/10.5194/acp-11-4899-2011, 2011.
Toyota, K., Dastoor, A. P., and Ryzhkov, A.: Air-snowpack exchange of bromine, ozone and mercury in the springtime Arctic simulated by the 1-D model PHANTAS – Part 2: Mercury and its speciation, Atmos. Chem. Phys., 14, 4135–4167, https://doi.org/10.5194/acp-14-4135-2014, 2014a.
Toyota, K., McConnell, J. C., Staebler, R. M., and Dastoor, A. P.: Air-snowpack exchange of bromine, ozone and mercury in the springtime Arctic simulated by the 1-D model PHANTAS – Part 1: In-snow bromine activation and its impact on ozone, Atmos. Chem. Phys., 14, 4101–4133, https://doi.org/10.5194/acp-14-4101-2014, 2014b.
Travnikov, O., Angot, H., Artaxo, P., Bencardino, M., Bieser, J., D'Amore, F., Dastoor, A., De Simone, F., Diéguez, M. D. C., Dommergue, A., Ebinghaus, R., Feng, X. B., Gencarelli, C. N., Hedgecock, I. M., Magand, O., Martin, L., Matthias, V., Mashyanov, N., Pirrone, N., Ramachandran, R., Read, K. A., Ryjkov, A., Selin, N. E., Sena, F., Song, S., Sprovieri, F., Wip, D., Wängberg, I., and Yang, X.: Multi-model study of mercury dispersion in the atmosphere: atmospheric processes and model evaluation, Atmos. Chem. Phys., 17, 5271–5295, https://doi.org/10.5194/acp-17-5271-2017, 2017.
Van Dam, B., Helmig, D., Neff, W., and Kramer, L.: Evaluation of boundary layer depth estimates at Summit Station, Greenland, J. Appl. Meteorol. Clim., 52, 2356–2362, https://doi.org/10.1175/jamc-d-13-055.1, 2013.
Van Dam, B., Helmig, D., Toro, C., Doskey, P., Kramer, L., Murray, K., Ganzeveld, L., and Seok, B.: Dynamics of ozone and nitrogen oxides at Summit, Greenland: I. Multi-year observations in the snowpack, Atmos. Environ., 123, 268–284, https://doi.org/10.1016/j.atmosenv.2015.09.060, 2015.
Wang, F., Saiz-Lopez, A., Mahajan, A. S., Gómez Martín, J. C., Armstrong, D., Lemes, M., Hay, T., and Prados-Roman, C.: Enhanced production of oxidised mercury over the tropical Pacific Ocean: a key missing oxidation pathway, Atmos. Chem. Phys., 14, 1323–1335, https://doi.org/10.5194/acp-14-1323-2014, 2014.
Wang, J., Zhang, L., and Xie, Z.: Total gaseous mercury along a transect from coastal to central Antarctic: Spatial and diurnal variations, J. Hazard. Mater., 317, 362–372, https://doi.org/10.1016/j.jhazmat.2016.05.068, 2016.
Yang, X., Cox, R. A., Warwick, N. J., Pyle, J. A., Carver, G. D., O'Connor, F. M., and Savage, N. H.: Tropospheric bromine chemistry and its impacts on ozone: A model study, J. Geophys. Res.-Atmos., 110, D23311, https://doi.org/10.1029/2005JD006244, 2005.
Ye, Z., Mao, H., Driscoll, C. T., Wang, Y., Zhang, Y., and Jaeglé, L.: Evaluation of CMAQ coupled with a state-of-the-art mercury chemical mechanism (CMAQ-newHg-Br), J. Adv. Model. Earth Syst., 10, 668–690, https://doi.org/10.1002/2017MS001161, 2018.
Zatko, M. C., Grenfell, T. C., Alexander, B., Doherty, S. J., Thomas, J. L., and Yang, X.: The influence of snow grain size and impurities on the vertical profiles of actinic flux and associated NOx emissions on the Antarctic and Greenland ice sheets, Atmos. Chem. Phys., 13, 3547–3567, https://doi.org/10.5194/acp-13-3547-2013, 2013.
Zhang, L., Wright, L. P., and Blanchard, P.: A review of current knowledge concerning dry deposition of atmospheric mercury, Atmos. Environ., 43, 5853–5864, https://doi.org/10.1016/j.atmosenv.2009.08.019, 2009.