the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 30 Aug 2018
| 30 Aug 2018
The diverse chemical mixing state of aerosol particles in the southeastern United States
Amy L. Bondy
Daniel Bonanno
Ryan C. Moffet
Bingbing Wang
Alexander Laskin
Aerosols in the atmosphere are chemically complex with thousands of chemical species distributed in different proportions across individual particles in an aerosol population. An internal mixing assumption, with species present in the same proportions across all aerosols, is used in many models and calculations of secondary organic aerosol (SOA) formation, cloud activation, and aerosol optical properties. However, many of these effects depend on the distribution of species within individual particles, and important information can be lost when internal mixtures are assumed. Herein, we show that – as found during the Southern Oxidant and Aerosol Study (SOAS) in Centreville, Alabama, at a rural, forested location – aerosols frequently are not purely internally mixed, even in the accumulation mode (0.2–1.0 µm). A range of aerosol sources and the mixing state were determined using computer-controlled scanning electron microscopy with energy-dispersive X-ray spectroscopy (CCSEM-EDX) and scanning transmission X-ray microscopy–near-edge X-ray absorption fine structure spectroscopy (STXM-NEXAFS). Particles that were dominated by SOA and inorganic salts (e.g., ammonium sulfate) were the majority of particles by number fraction from 0.2 to 5 µm with an average of 78 % SOA in the accumulation mode. However, during certain periods contributions by sea spray aerosol (SSA) and mineral dust were significant to accumulation (22 % SSA and 26 % dust) and coarse-mode number concentrations (38 % SSA and 63 % dust). The fraction of particles containing key elements (Na, Mg, K, Ca, and Fe) were determined as a function of size for specific classes of particles. Within internally mixed SOA/sulfate particles < 5 % contained Na, Mg, K, Ca, or Fe, though these nonvolatile cations were present in particles from the other sources (e.g., SSA and dust). Mass estimates of the aerosol elemental components were used to determine the extent of internal versus external mixing by calculating the mixing state index (χ). The aerosol population was more externally mixed than internally mixed during all time periods analyzed. Accumulation mode aerosol ranged from more internally mixed during SOA periods to mostly externally mixed during dust periods. Supermicron aerosols were most externally mixed during SOA time periods, when more SOA particles added a distinct supermicron class, and more internally mixed when dominated by a single particle type (e.g., SSA or dust). These results emphasize that neither external nor internal mixtures fully represent the mixing state of atmospheric aerosols, even in a rural, forested environment, which has important implications for air quality and climate modeling.
- Article
(5559 KB) - Full-text XML
-
Supplement
(634 KB) - BibTeX
- EndNote





The southeastern United States has experienced neutral to cooling shifts in regional climate over the past century (Portmann et al., 2009; Saxena and Yu, 1998), in contrast to warming observed in the rest of the United States. This has been partially attributed to increased formation of secondary organic aerosol (SOA) with largely cooling effects due to efficient light scattering and activity in cloud formation (Goldstein et al., 2009; Portmann et al., 2009). Regionally, the main SOA source is oxidation of biogenic volatile organic compounds (BVOCs), followed by condensation or reactive uptake onto preexisting particles containing sulfate, nitrate, and ammonium (Anttila et al., 2007; Boyd et al., 2015; Budisulistiorini et al., 2015a; Carlton et al., 2010; Chameides et al., 1988; Hodas et al., 2014; Lee et al., 2010; Nguyen et al., 2015; Weber et al., 2007; Xu et al., 2015a). Most studies of aerosol climate impacts in the southeast have focused on the effects of SOA, as this region has high concentrations of organic carbon, which, combined with ammonium sulfate, contribute 60 %–90 % of fine particulate matter (PM2.5) (Attwood et al., 2014; Boone et al., 2015; Cerully et al., 2015; Nguyen et al., 2014). However, despite the importance of SOA, the mixing of secondary species (SOA, sulfate, nitrate, etc.) with primary particles is not fully known, particularly for forested locations impacted by regional anthropogenic emissions. The distribution of chemical species in individual particles across a population, i.e., mixing state, is critical for climate-relevant properties including light scattering, water uptake, particle acidity, and aerosol chemistry, such as metals dissolution (Artaxo and Orsini, 1987; Ault et al., 2010; Cong et al., 2010; Craig and Ault, 2018; Fang et al., 2017; Kunkel et al., 2012; Metternich et al., 1986; Violaki and Mihalopoulos, 2010; J. Z. Xu et al., 2015). Therefore, it is important to identify the sources of aerosol particles present in the southeastern United States, as well as their size and mixing state, in order to accurately assess their impact on aerosol direct and indirect effects (Li et al., 2016b; Moise et al., 2015; Posfai and Buseck, 2010).
Mixing state is described in terms of external and internal mixtures: an external mixture consists of particles that contain only one pure species per particle, while an internal mixture describes particles that contain equal amounts of all chemical species (Ault and Axson, 2017; Posfai and Buseck, 2010; Riemer and West, 2013). The mixing states of ambient aerosol populations are complex and can vary as a function of size, altitude, and particle age (Ault et al., 2013a, 2009; Fu et al., 2012; Healy et al., 2014a; Moffet et al., 2010b; Pratt and Prather, 2010). Aging – or atmospheric processing such as coagulation, condensation of secondary species, and heterogeneous reactions – leads to internal mixing, while freshly emitted particles are more externally mixed (Schutgens and Stier, 2014; Weingartner et al., 1997). Here, mixing state is used to describe the distribution of chemical species in a population and is purely based on composition (Ault and Axson, 2017). This does not including particle morphology, phase, coatings, or other physical properties, which can subsequently impact composition (Y. Zhang et al., 2018). Although the representation of mixing state in models is still an open research question (Riemer and West, 2013), an appropriate description of mixing state is critical for modeling optical properties (Chung and Seinfeld, 2005; Jacobson, 2001) and cloud condensation nuclei (CCN) activity (Zaveri et al., 2010).
Riemer and West (2013) introduced the mixing state index (χ) to quantify aerosol mixing state. This parameterization uses single-particle mass fractions of individual components to calculate the average particle-specific diversity and the bulk population diversity, from which the mixing state index can then be determined. This methodology has been applied to a handful of laboratory and field studies, to date. In the laboratory, Dickau et al. (2016) used aerosol sizing and mass instrumentation to quantify the volatile mixing state of soot. Single-particle mass spectrometry data from field studies in London (Giorio et al., 2015) and as part of the MEGAPOLI campaign in Paris (Healy et al., 2014b) found mixing state was dependent upon both time of day and air mass origin. Similarly, mixing state parameters were applied to computer-controlled scanning electron microscopy with energy-dispersive X-ray spectroscopy (CCSEM-EDX) and scanning transmission X-ray microscopy–near-edge X-ray absorption fine structure spectroscopy (STXM-NEXAFS) during the Carbonaceous Aerosol and Radiative Effects Study (CARES) in the Central Valley of California (O'Brien et al., 2015) and in the Amazon during the GoAmazon campaign (Fraund et al., 2017). These studies found changes in mixing state were associated with a buildup of organic matter and particle clusters were less diverse at remote sites, respectively. However, additional studies are needed to quantify the chemical mixing state of aerosols, particularly for rural locations.
In this study, we analyzed individual atmospheric particles collected at a rural location influenced by regional pollution in the southeastern United States during the 2013 Southern Oxidant and Aerosol Study (SOAS) to identify their size-resolved chemical composition and mixing state. CCSEM-EDX was used to determine the size, elemental composition, and number fraction of particles containing metal (nonvolatile) cations. STXM-NEXAFS was used to characterize chemical bonding of carbonaceous components, specifically distinguishing soot from organic carbon. Mass estimates of particle elemental composition from CCSEM-EDX were calculated using a modified version of the method from O'Brien et al. (2015) to quantify the mixing state parameters for both submicron and supermicron particles during time periods dominated by SOA/sulfate, dust, and sea spray aerosol (SSA). Additionally, the variability in the mixing state index during these three time periods of interest showed that submicron aerosol was most internal when SOA particles dominated the aerosol population. However, supermicron particles were most internally mixed when a single source (e.g., SSA or dust) dominated the population and most externally mixed when SOA was present, along with SSA and dust. The variety of particle classes, varying extent of secondary processing, and diverse chemical mixing states at this rural, forested site may impact climate-relevant properties of aerosols in the southeastern United States.
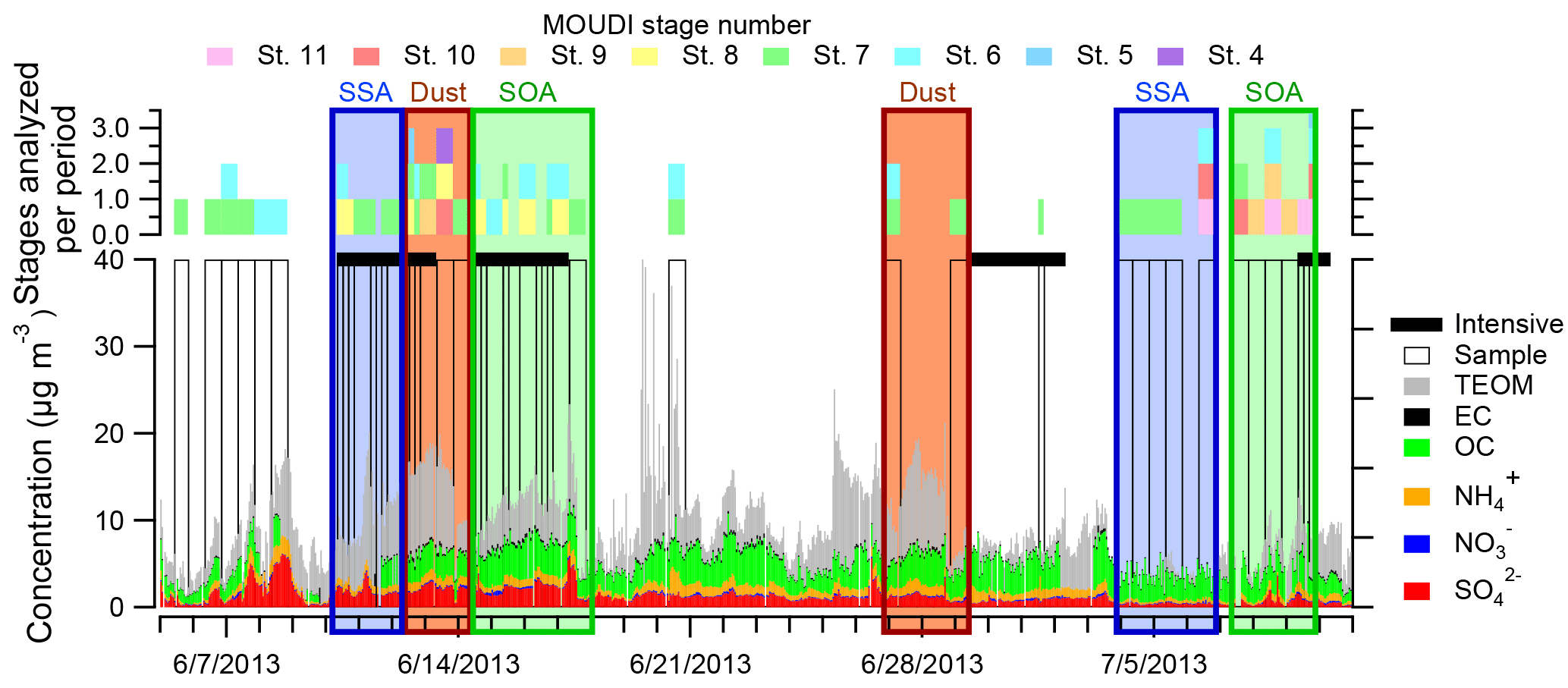
Figure 1SEARCH filter sample data for Centreville, AL, during SOAS with purple boxes overlaid for time periods in which CCSEM was run and the corresponding MOUDI stages that were analyzed. SOA-rich periods denoted with green boxes (14–17 June and 7–11 July 2013) were studied by Xiong et al. (2015), Pye et al. (2015), Xu et al. (2015b), Hu et al. (2015), and Rattanavaraha et al. (2016); dust-rich periods marked with brown boxes (12–13 June and 26–28 June 2013) were identified by Allen et al. (2015); and SSA-rich periods marked with blue boxes (10–11 June and 3–6 July 2013) were identified by Bondy et al. (2017b). Time periods without samples analyzed are due to sample damage and identification of mutually exclusive time periods. Based on periods identified in previous studies, only 29 % of SOA-dominant periods, 40 % of dust periods, and 51 % of SSA periods were analyzed here.
2.1 Aerosol sample collection
Samples of atmospheric particles were collected at the SOAS Centreville, AL, site (32.9030∘ N, 87.2500∘ W; 242 m a.m.s.l.) between 5 June and 11 July 2013 (Bondy et al., 2017b; Carlton et al., 2018; Hidy et al., 2014). The site was located in a rural, forested region near Talladega National Forest, at a location that was part of the SouthEastern Aerosol Research and Characterization Network (SEARCH). Meteorological and filter sample data analyzed from the SEARCH network were used to aid selection of samples for analysis (Figs. 1 and S1 in the Supplement). Particles were collected near ground level (1 m) using a micro-orifice uniform deposit impactor (MOUDI, MSP Corp. Model 110) sampling at 30 L min−1 with a PM10 cyclone (URG Model 786) to exclude particles larger than 10 µm. The 50 % size cut points for the MOUDI used in this analysis had aerodynamic diameters (Da) of 3.2, 1.8, 1.00, 0.56, 0.32 0.18, 0.10, and 0.056 µm (Marple et al., 1991). Throughout SOAS, particles were impacted onto Cu 200 mesh transmission electron microscopy (TEM) grids with Carbon Type B thin film (Ted Pella Inc.) for analysis with SEM-EDX and STXM-NEXAFS. Substrates in the MOUDI were collected daily from 8:00 to 19:00 and 20:00 to 7:00 CST (with 1 h for substrate exchange), except during intensive periods from 10 to 12 June, 14 to 16 June, 29 June to 1 July, and 7 to 9 July, when the sampling schedule was 8:00–11:00, 12:00–15:00, 16:00–19:00, and 20:00–7:00 CST (Table S1). Samples were collected more frequently during intensive time periods, which were determined by meteorological and gas phase concentrations (Budisulistiorini et al., 2015b). In Fig. 1, the MOUDI stages analyzed using CCSEM are noted for each sample. After collection, all substrates were sealed in Teflon-wrapped petri dishes within Ziploc bags and stored at −22 ∘C. Upon arrival at the University of Michigan, Ziploc bags containing samples were removed from the freezer and thawed to room temperature, and TEM grids were transferred to grid storage boxes wrapped with Parafilm. Parafilm-wrapped grid boxes were then stored in Ziploc bags in the freezer until imaging. Prior to imaging the wrapped grid boxes were removed from the freezer and allowed to reach room temperature before being opened. Although all samples were stored at low temperatures after collection to minimize chemical reactions, the loss of semivolatile species (e.g., water and organics) in the MOUDI and in the SEM under vacuum may have led to the EDX results suggesting particles contained slightly less organic carbon than in the ambient atmosphere, which might make them appear slightly more externally mixed.
2.2 CCSEM-EDX analysis
Particles on MOUDI stages 3–10 (Da=0.056–1.8 µm, Fig. 1) were analyzed using CCSEM (FEI Quanta environmental SEM) equipped with a field emission gun operating at 20 kV and a high-angle annular dark-field (HAADF) detector (Laskin et al., 2002, 2006, 2012). The SEM was equipped with an EDX spectrometer (EDAX, Inc.), which was used to quantify X-rays of elements with atomic numbers ). A total of ∼34 000 particles were analyzed during time periods denoted in Table S2, which constitute a representative cross section of the campaign. CCSEM analysis captured particle physical parameters including projected area diameter, projected area, and perimeter. Projected area diameter, which is equivalent to the diameter of a circle with the same area as the particle silhouette, is typically larger than aerodynamic diameters measured by other analytical techniques (Bondy et al., 2017a; Hinds, 1999). For a more accurate representation of particle size, projected area diameters were converted to volume-equivalent diameter using a conversion factor of 0.49 for SOA/sulfate and biomass burning particles and 0.66 for SSA, determined from atomic force microscopy (AFM) volume calculations of particles from SOAS (Tables S3 and S4). EDX spectra from individual particles were analyzed to determine the relative abundance of 14 elements: C, N, O, Na, Mg, Al, Si, P, S, Cl, K, Ca, Ti, and Fe. Note that the Cu signal in the EDX spectra is primarily due to the Cu grid from the substrate and was not included in CCSEM-EDX analysis.
The CCSEM-EDX data sets were analyzed using k-means clustering of the elemental composition following the method described in Ault et al. (2012) using codes written in MATLAB R2013b (MathWorks, Inc.). Clusters were grouped into source-based classes by elemental composition based on previous studies, including mineral dust (Axson et al., 2016b; Coz et al., 2009; Creamean et al., 2016; Laskin et al., 2005; Sobanska et al., 2003), SSA (Ault et al., 2013a; Bondy et al., 2017b; Hopkins et al., 2008; Laskin et al., 2002; Prather et al., 2013), SOA/sulfate (Moffet et al., 2013; O'Brien et al., 2015; Sobanska et al., 2003), biomass burning aerosol (Li et al., 2003; Posfai et al., 2003), fly ash/metals (Ault et al., 2012; Chen et al., 2012; Shen et al., 2016), biological particles (Huffman et al., 2012), and fresh soot (Li et al., 2003). Soot forms fractal aggregates of graphitic carbon (C) which contain tens to hundreds of small spherical aggregates (Li et al., 2003). However, the intense carbon signal from the carbon film substrate made chemical identification of soot difficult, initially resulting in false positives from the substrate. Because of their unique morphology, the size distribution of fresh soot particles without a large secondary organic carbon coating altering the fractal morphology was manually determined. Then, a scaling factor based on the SEARCH network elemental carbon mass concentrations was applied to the size distribution, and this factor was used in the subsequent analysis. More information on this correction for soot can be found in the Supplement, specifically Table S5.
Mass calculations and mixing state parameters
Mole percent of elements analyzed using CCSEM-EDX was converted to mass fractions using the method described by O'Brien et al. (2015) and detailed in the Supplement. Briefly, particle volumes were calculated from volume-equivalent diameters (calculated from projected area diameters using a conversion factor determined from atomic force microscopy height images) assuming the volume of a hemisphere. Particle masses were then calculated (μi= density × volume) assuming the following densities for each class: 1.3 g cm−3 for SOA/sulfate, biomass burning aerosol, and primary biological particles (Li et al., 2016a; Manninen et al., 2014; Nakao et al., 2013); 2.0 g cm−3 for SSA particles (O'Brien et al., 2015); 2.6 g cm−3 for dust particles (Wagner et al., 2009); and 3.0 g cm−3 for fly ash particles (Buha et al., 2014). To calculate the mass of each element, the elemental mole percent was converted to a weight percent, which was multiplied by the estimated particle mass. There are uncertainties in this conversion related to the material densities and shapes assumed, as well as loss of semi-volatiles during analysis (water and some organics), which lead to mass uncertainties on the order of 5 %–10 % on a per-particle basis.
Diversity parameters were calculated using two different methods in this work: elemental diversity was calculated from CCSEM-EDX results similar to O'Brien et al. (2015), and mixing state parameters due to aging were calculated as described below (which use only two diversity species: the mass fraction of elements associated with externally mixed particles and the mass fraction of secondary species). To calculate elemental diversity parameters, the mixing entropy of each particle (Hi) and average particle mixing entropy (Hα) were calculated for each particle class as described in detail by Riemer and West (2013):
where pi is the mass fraction of particle i in the population and is the mass fraction of element a in particle i. The particle diversity (Di) was then calculated by taking the exponent of the particle-specific entropy Hi, and the average particle-specific diversity (Dα) was calculated by taking the exponent of Hα. Dα was used as an indicator of elemental diversity for each particle class: SOA/sulfate, biomass burning particles, fly ash, dust, SSA, and biological particles.
In addition to elemental diversity, diversity parameters were calculated to quantify the extent of particle aging. To calculate the mixing state aging parameters for the three time periods of interest, two final mass values were calculated: the mass of single particles in a class based on the sum of elements characteristic to that class, and the mass of secondary species. The elemental mass fractions as a function of size are depicted in Fig. S3. Due to the semi-quantitative nature of the lower Z elements (Laskin et al., 2006) and substrate interferences, we excluded C, N, and O from mixing state calculations, similar to O'Brien et al. (2015). The mass associated with SOA/sulfate was solely accounted for by S (if present) and therefore was either ignored or severely underestimated. Fresh biomass particles consisted of K and Cl; fly ash particles contained Al and Si; unreacted dust particles consisted of Na, Mg, Al, Si, K, Ca, Ti, and Fe; fresh SSA particles contained Na, Mg, Cl, K, and Ca; and biological particles contained P, Cl, and K. As a metric for aging, all sulfur was assumed to be secondary within particles, though trace primary sulfur is present in SSA and may be in other classes. Thus, for the purposes of this mixing state analysis each particle contained between one and two components: a primary source-based composition and secondary aging due to sulfur. Using the mass fractions of only these two components, Hi, Hα, and the population bulk mixing entropy (Hγ) were calculated for each particle class:
where pa is the mass fraction of element a in the population. The bulk population diversity (Dγ) was then calculated by taking the exponent of Hγ. The mixing state index (χ) is then defined as
where χ can range from 0 % for an external mixture to 100 % for an internal mixture.
2.3 STXM-NEXAFS analysis
STXM-NEXAFS measurements of two MOUDI samples (stage 8, 50 % size cut point of 0.18 µm, 100–200 particles analyzed per sample), 10 June and 7 July 2013, were performed at the carbon K-absorption edge (280–320 eV) to characterize chemical bonding of carbonaceous components, specifically distinguishing soot from organic carbon. An example STXM image, a map, and NEXAFS spectra for a biological particle are provided in the Supplement. STXM-NEXAFS images and spectra of SOA will be the focus of a future publication. STXM was conducted at the Advanced Light Source at Lawrence Berkeley National Laboratory on beamline 5.3.2. The operation of the microscope has been explained in detail by Kilcoyne et al. (2003). The software programs MATLAB and Axis 2000 were both used for spectral analysis of the STXM-NEXAFS data as described by Moffet et al. (2010a, 2016). Stacks of images taken at sequentially increasing photon energies were used to obtain spatially resolved spectroscopic data at the carbon K-edge. For organic identification, pixels were identified where the post-edge minus the pre-edge (optical density (OD) at 320 eV minus OD 278 eV) was greater than zero. For the inorganic component, particles with a ratio of the pre-edge to the post-edge (OD 278 / OD 320) greater than 0.5 were identified. To identify soot inclusions within particles, individual pixels of STXM images were analyzed; if a pixel contained 35 % or greater C = C, a peak which was identified as soot using graphitic carbon as a standard, then that pixel was identified as a soot region. Additional details on identification of SOA-containing soot inclusions are provided in the Supplement.
3.1 Descriptions of particle classes during SOAS
Although SOAS took place in a rural, forested region frequently dominated by SOA/sulfate, a variety of particle classes were observed. Based on the chemical composition, seven main particle classes were identified: SOA/sulfate, biomass burning particles, soot, and fly ash, which are typically present in the submicron (< 1 µm) regime, and mineral dust, SSA, and primary biological particles with characteristic sizes > 1 µm. Figure 2 shows SEM images of representative particles from each class and their corresponding EDX spectra. SOA/sulfate particles were identified by the elemental composition of C and O, along with S, N, or both S and N. As all SOA particles contained inorganic species (e.g., ammonium sulfate) in addition to organic carbon (based on STXM ODpre / ODpost=0.5, ∼20 % by weight), this class is referred to as SOA/sulfate. EDX spectra of SOA/sulfate particles on Si wafers (Fig. S4) confirmed C and O in the particles, as did STXM, as a check due to the carbon film substrate on TEM grids that contributes to the signal for C and O in the CCSEM-EDX analysis (Kirpes et al., 2018). The presence of S and/or N in addition to C and O is likely or , based on Raman microspectroscopy (Ault et al., 2013b; Craig et al., 2017; Ebben et al., 2013), or organonitrate or organosulfate compounds, which are ubiquitous in the southeastern US (Ayres et al., 2015; Carlton et al., 2009; Froyd et al., 2010; Hatch et al., 2011). SOA/sulfate particles were typically circular (circularity ranging from 0.95 to 1, where 1 is perfectly circular; equation in Supplement), though some SOA/sulfate exhibited liquid–liquid phase separation (LLPS), such as core-shell or more complex morphologies, which will be explained in a future publication. Biomass burning aerosol particles were often circular as well (0.96 circularity), with high concentrations of K and frequently S and Cl, in addition to C and O (organic carbon) (Li et al., 2003; Posfai et al., 2003). Fly ash particles were identified primarily by their spherical morphology (0.93 circularity) since fly ash is generated through high-temperature processes (Ault et al., 2012; Chen et al., 2012), in addition to high EDX signals from O along with either Si or Al, likely in the form of SiO2 or Al2O3, respectively. A final class comprised primarily of submicron particles was soot. Fresh soot particles were identified primarily by their morphology consisting of agglomerated spheres, which had substantially smaller diameters than fly ash (Li et al., 2003). However, fresh soot was not very prevalent during SOAS and was typically present within other particles such as SOA, which will be detailed below.

Figure 2SEM images and corresponding EDX spectra for each of the main particle classes identified during SOAS within the submicron (a, SOA; b, biomass burning aerosol particles; c, fly ash; d, soot) and supermicron (e, dust; f, SSA; g, primary biological) sizes. Note the elements with an asterisk are not quantitative due to interference from the substrate or detector.
In addition to submicron classes, three classes of particles predominantly in the supermicron size range were observed during SOAS. Dust particles were identified by strong signals from O, Al, and Si (aluminosilicates), often along with other elements such as Na, Mg, K, Ca, Ti, and Fe (Coz et al., 2009; Laskin et al., 2005; Sobanska et al., 2003). EDX spectra of SSA particles contained a strong Na signal (Na+) and weaker Mg signal (Mg2+) in a ratio, as found in seawater (Pilson, 1998); small contributions from K (K+) and Ca (Ca2+); and were clearly distinguishable from freshwater aerosol (Axson et al., 2016a; May et al., 2016, 2018b). Counter-ion elements such as N, O, S, or Cl (, , or Cl−) depended on whether the SSA was fresh or aged after transport (Bondy et al., 2017b; May et al., 2018a). Finally, biological particles typically contained primarily C due to organic macromolecules, along with lesser amounts of N (likely in the form of amines/amino acids), O, P (), and K (K+) (Huffman et al., 2012), as seen in Fig. 2. Overall, numerous particle classes were detected during SOAS using CCSEM-EDX based on unique chemical composition, morphology, and size.
STXM-NEXAFS was used to investigate carbonaceous particles since these particles were prevalent during SOAS. The carbon K-edge was probed using this technique, and high-spatial-resolution information was obtained regarding sp2 C inclusions within SOA/sulfate, which were identified as soot at 285 eV (Moffet et al., 2010a). In two samples analyzed using STXM-NEXAFS, 6.9 % and 9.9 % of particles by number contained sp2 C inclusions, suggesting that a small fraction of SOA/sulfate contained soot. In comparison, organic carbon–elemental carbon (OC-EC) bulk analysis by the SEARCH network detected ∼2 % elemental carbon by mass, suggesting that, although little soot was present overall, a sizeable fraction was present as small inclusions within SOA/sulfate. It is important to consider the mixing state of aerosols when modeling radiative forcing in the region, because internally mixed particles behave differently than pure components. For example, soot coated with secondary organic material may have an enhanced absorption compared to fresh soot or soot-less SOA, though recent work has suggested that optical properties of coated soot are challenging and nonlinear (Healy et al., 2015; Moffet et al., 2009; Ramanathan and Carmichael, 2008; Zhang et al., 2008). These spectra highlight that, although seven main particle classes were identified, many particles, such as SOA and soot, were partially internally mixed.
3.2 Chemical diversity observed within particle classes
Using SEM-EDX elemental mapping, morphology and the spatial distribution of species within individual particles were examined. In Fig. 3, particles (a–d) were identified as dust based on their morphology and elemental composition. However, only (b–d) are aluminosilicate dust particles; (a) contains high concentrations of Ca and S instead. Based on its chemical composition, this dust particle is most likely gypsum (CaSO4 ⋅ 2H2O) (Hashemi et al., 2011). The elemental map highlights that elements present within the dust class are not homogeneously distributed among all particles. Rather, the dust class consists of externally mixed particles with varying compositions. In addition to dust, two other particle classes are represented in the elemental map in Fig. 3. Particles (e–f) were identified as aged SSA due to the high concentration of Na and Mg along with S and N (likely and ) (Bondy et al., 2017b), and particle (g) is a primary biological particle, possibly coagulated with a small calcium oxide particle based on the morphology and distinctly different elemental compositions of the two components. In addition to differentiating particles among the seven particle classes identified, SEM-EDX mapping allowed investigation into whether coagulation or chemical aging of particles has occurred within particles due to the presence of localized regions of elements or surface-layer coatings (Conny and Norris, 2011). As seen in Fig. 3, very few of the particles have a homogeneous distribution of elements, though vacuum analysis and drying can modify the internal distribution of species within particles. Rather, Na, Ca, S, and Cl often appear in only a few distinct regions within particles, which can likely be attributed to heterogeneous reactions, limited diffusion, or other non-ideal behavior (Y. Zhang et al., 2018). The aluminosilicate dust particle (b) in particular has localized regions of Ca and S () on the edges of the particle, signifying that this particle has undergone aging, resulting in a more diverse physicochemical mixing state (Ault and Axson, 2017). Complex mixing states like this have been observed previously for SSA and dust, showing that these classes of particles can be externally mixed or have surface coatings and inclusions leading to internal mixing, thereby altering their physical and chemical properties (Deboudt et al., 2012; Fitzgerald et al., 2015; Gantt and Meskhidze, 2013; Kandler et al., 2011; Kim and Park, 2012; Sobanska et al., 2012; Sullivan et al., 2007, 2009).

Figure 3SEM image (dark field) and EDX elemental maps of particles indicated that these particle classes had various mixing states. Each of the elemental map panels corresponds to two elements overlaid to show the elemental distributions from the SEM image. The following particle classes are shown: (a–d) dust, (e–f) aged SSA, and (g) primary biological.
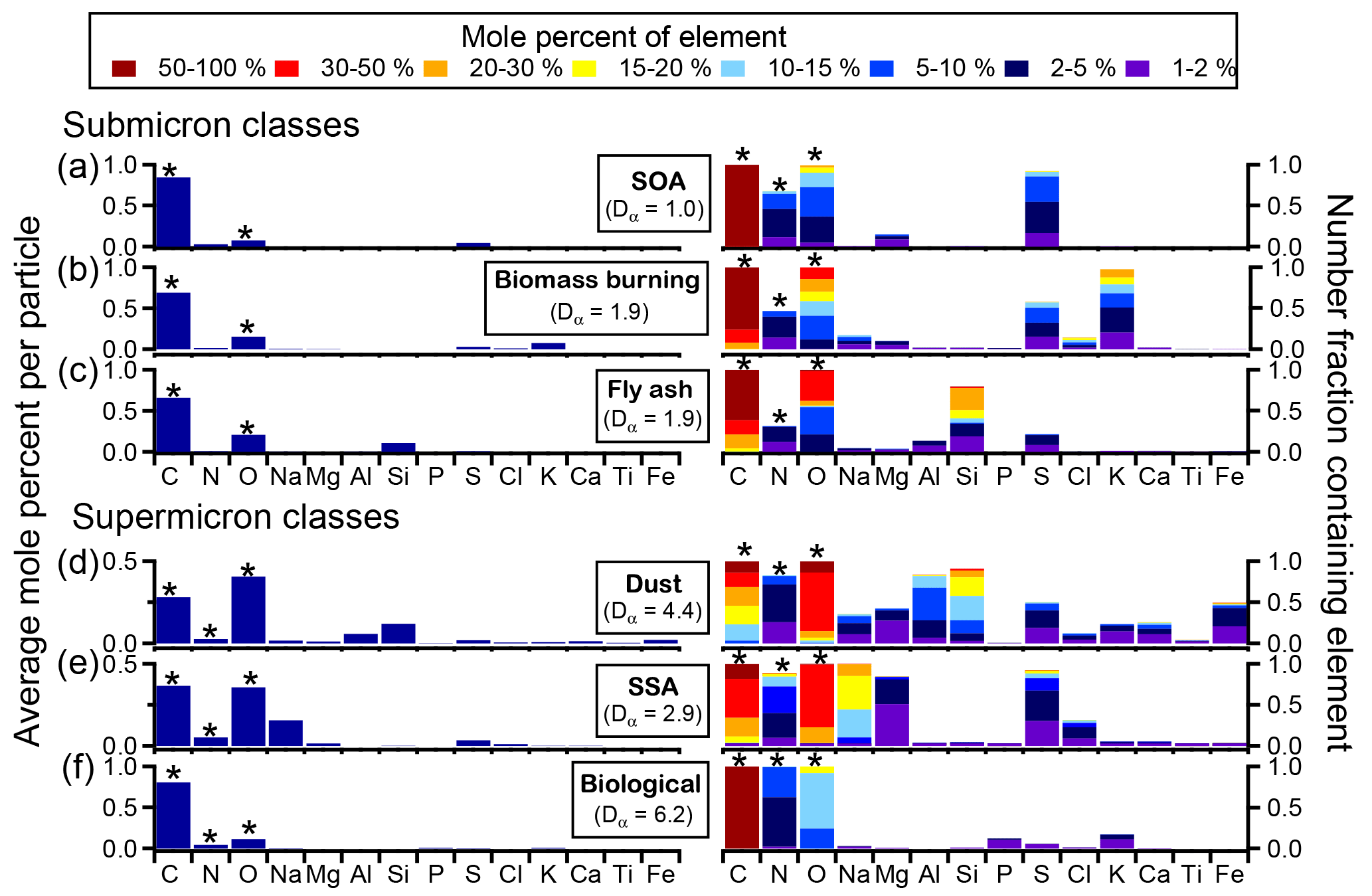
Figure 4Average histograms and digital color histograms of different particle classes from SOAS: (a) SOA, (b) biomass burning, (c) fly ash, (d) dust, (e) SSA, and (f) primary biological. Average spectra are shown on the left as the average mole percent of each element analyzed by CCSEM-EDX (C, N, O, Na, Mg, Al, Si, P, S, Cl, K, Ca, and Fe). On the right, the digital color histogram heights represent the number fraction of particles containing a specific element, and the colors represent the mole percent of that element. The average particle specific diversity (Dα), representing the average number of elements in each particle, is calculated for each submicron and supermicron class and has an uncertainty of 5 %–10 %. Note the elements with an asterisk are not quantitative due to interference from the substrate or detector and are not included in Dα.
To probe the chemical diversity of each particle class, Fig. 4 shows the average EDX elemental percentages for each particle class. The digital color histogram height shows the number fraction of particles in a class containing a specific element, while the color represents the mole percent of the element. For example, 100 % of SOA/sulfate by number contain between 5 % and 100 % C (mole %). To quantify elemental diversity of particles, Dα, representing the average number of elements within particles in each class, was calculated. Dα ranges from 1 (when a particle contains only one element) to A number of elements. Note that, due to interference from the substrate or detector, C, N, and O were not included in Dα calculations. CCSEM-EDX results suggest that SOA/sulfate particles were elementally the least diverse, as the primary quantifiable element was S, leading to a Dα=1.0. However, other studies from SOAS that used an aerosol mass spectrometer (AMS) (Guo et al., 2015; Xu et al., 2015b) or ultra-performance liquid chromatography/electrospray ionization high-resolution quadrupole time-of-flight mass spectrometry (UPLC/ESI-HR-QTOFMS) (Budisulistiorini et al., 2015a; Riva et al., 2016) discovered that a wealth of sources contribute to SOA, resulting in hydrocarbon-like organic aerosol, isoprene-derived organic aerosol, and more-oxidized and less-oxidized oxygenated organic aerosol (Zhang et al., 2018). Their analyses also showed that is the most abundant component aside from organic carbon, with significant concentrations of followed by , consistent with our observations of 92 % of SOA by number containing S and 68 % containing N (a challenging element for EDX) (Budisulistiorini et al., 2015a; Guo et al., 2015; Riva et al., 2016; Xu et al., 2017, 2015b).
The composition of biomass burning particles was elementally more diverse than SOA (Dα=1.9), with large contributions from K+ (1 %–30 % by mole percent), as well as organic carbon (20 %–100 % C and 2 %–50 % O by mole percent). However, in addition to these three components, approximately 60 % of particles by number also contained (1 %–15 % S by mole percent), 45 % contained / (1 %–10 % N by mole percent), and 15 % by number contained 1 %–30 % Cl (mole percent). The presence of Cl suggests that some of the biomass burning particles were fresh. However, because sulfate and nitrate, which are indicative of aging (Li et al., 2003), were present more frequently, biomass burning particles detected during SOAS likely had sufficient time during transport for multiphase reactions to occur (Washenfelder et al., 2015). The final submicron particle class, fly ash (Dα=1.9), showed two distinct compositions in addition to aging: fly ash from SOAS consisted of primarily SiO2, although approximately 15 % by number contained aluminum oxides with silicates. Low concentrations of and (1 %–5 % S and N by mole percent) suggest acidic species, such as sulfuric and nitric acid, reacted with 25 % of fly ash by number.
Within the supermicron particle classes, a range of elemental compositions were observed for dust and SSA (Fig. 4b). Dust was primarily composed of aluminosilicates (10 %–100 % O, 1 %–50 % Si, and 1 %–50 % Al by mole percent), with minor contributions from other chemical species, including or organic coatings (5 %–100 % C by mole percent), (1 %–10 % N by mole percent), (1 %–10 % S by mole percent), Mg2+ (1 %–10 % by mole percent), K+ (1 %–5 % by mole percent), Na+ (1 %–10 % by mole percent), Ca2+ (1 %–10 % by mole percent), and Fe2+/Fe3+ (1 %–30 % by mole percent). The frequency of these minor elements in dust particles varied widely, resulting in a high average particle species diversity (Dα=4.4), with nitrate present in approximately 75 % of the dust population by number and titanium oxides present in less than 5 %. The diversity of dust indicates various sources and processing throughout SOAS, which likely contributed to time periods with distinct dust compositions due to wind speed, wind direction, and pollution levels. Allen et al. (2015) detected two high-nitrate events in the coarse-mode during SOAS, the first corresponding to high levels of SSA and dust, and the second primarily dust. The first event exhibited a higher percent of Na+, not all of which was attributed to SSA due to the low Mg2+ / Na+ molar ratio, while the second event had a higher percent composition of Ca2+. Back trajectory analysis of the air mass origin during the two coarse-particle events indicates that, although the overall pattern in wind trajectories was similar, slight differences in wind patterns at the beginning of each event may have contributed to the observed differences in composition of the aerosol, suggesting a relatively local origin of the dust during the second event. The elements of SSA were more homogeneously distributed throughout the population than dust (Dα=2.9), with 100 % of particles by number containing C, O, and Na; 75 % by number containing S; and ∼70 % by number containing > 1 % (mole %) N and Mg. SSA particles also showed various degrees of aging with respect to the anions, since chloride can be liberated through multi-phase reactions with acidic species such as HNO3, H2SO4, and organic acids (Bondy et al., 2017b). Partially aged SSA comprised approximately 20 % of particles by number, indicated by Cl− (1–10 % Cl by mole percent) in addition to nitrate and sulfate. Complete chloride depletion and aging by nitrate (1 %–30 % N by mole percent) and sulfate (1 %–30 % S by mole percent) was more ubiquitous though, with each secondary species present in ∼90 % of SSA by number. A thorough discussion of the degree of reactive processing of SSA transported inland to Centreville can be found in Bondy et al. (2017b).
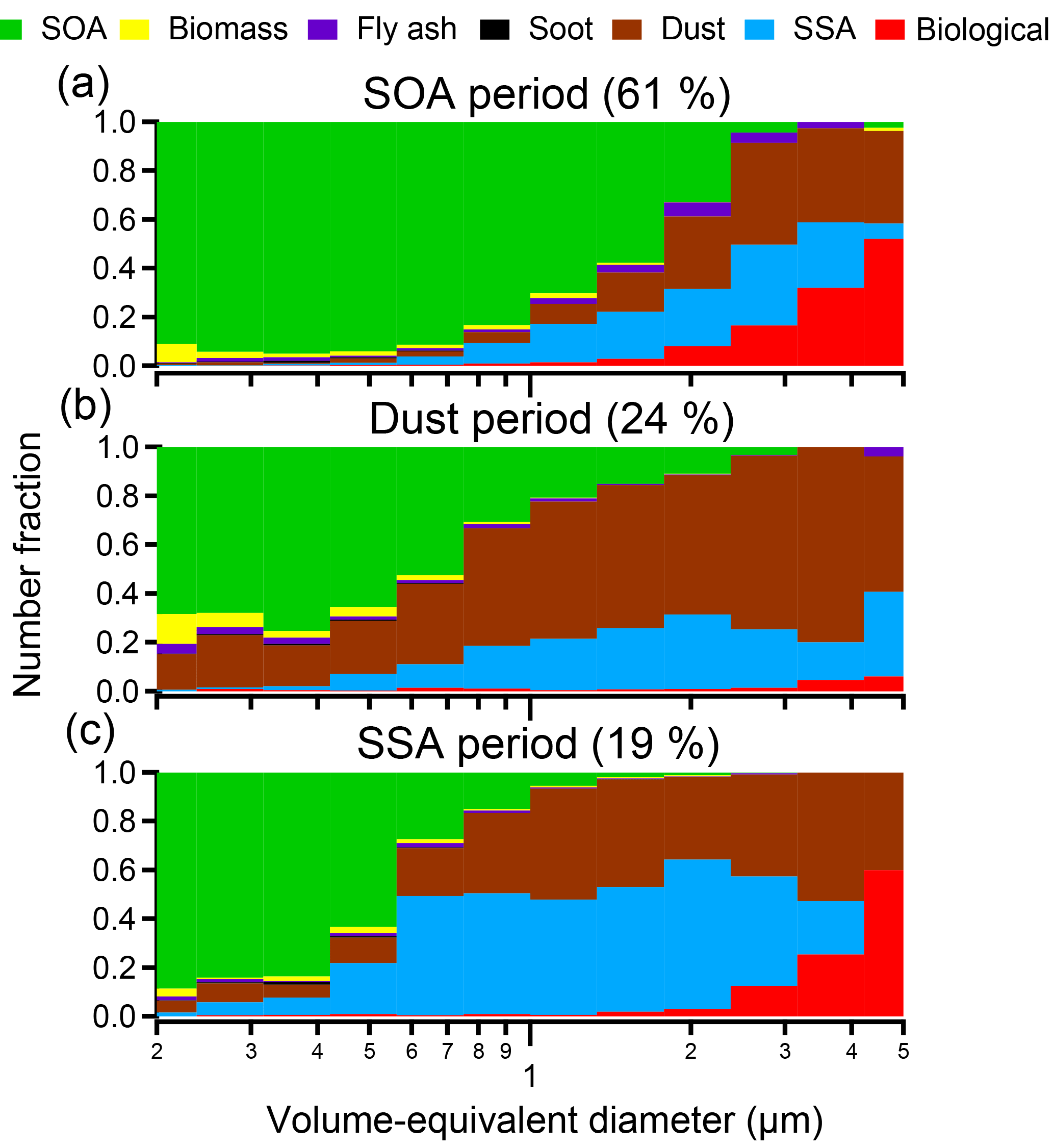
Figure 5Size distributions for specific particle-rich time periods: (a) SOA-rich periods (14–17 June and 7–11 July 2013), (b) dust-rich periods (12–13 June and 26–28 June 2013), and (c) SSA-rich periods (10–11 June and 3–6 July 2013). SOA periods were dominant throughout the times when samples were analyzed during SOAS (61 %), high-dust periods (24 %) (Allen et al., 2015), and SSA periods (19 %) of the time. SSA periods were defined in Bondy et al. (2017). * Literature-identified SSA periods and dust periods overlap from 11 to 13 June 2013; thus the percentage of SOA, dust, and SSA periods is greater than 100 % due to double counting of that time period. Only particles with volume-equivalent diameters between 0.2 and 5 µm are shown due to too few particles present at larger sizes for statistical analysis.
Primary biological particles contained primarily organic carbon (50 %–100 % C and 5 %–20 % O by mole percent) with minor contributions from , , and K+, in addition to other minor elements (Dα=6.2). The minor constituents (P, K, S) were not detected in all particle (20 % by number). The absence of these minor constituents from EDX spectra is likely the result of low concentrations compared to carbon, and signal below the 1 % detection threshold. Although sulfate is typically an indication of aging by H2SO4 in aerosol particles, it is also naturally present in biological particles. Furthermore, because the sulfur signal intensity is on the same scale as the other minor constituents, it is not necessarily from secondary processes. Overall, throughout both the submicron and supermicron particle regimes, particle diversity varied, indicating sources of long- and short-range transport, and various degrees of aging of particles within each class.
3.3 Variations in particle classes observed during key SOAS events
Three main time periods (SOA, dust, and SSA) were identified during SOAS that had distinctly different sources and processing (Fig. 5). Figure 5a depicts the size-resolved chemical composition averaged over two SOA-dominated time periods (14–17 June and 7–11 July 2013), two dust events (11–13 and 26–28 June 2013), and two SSA events (10–11 June 2013 and 3–6 July 2013), though only select MOUDI stages were analyzed for each sampling period. During each time period depicted, SOA/sulfate averaged > 60 % of accumulation mode (0.2–1.0 µm) and 2 % of the supermicron (1.0–5.0 µm) particles by number fraction. However, the number fraction of SOA/sulfate was highly variable between the SOA, dust, and SSA periods. During the two periods dominated by SOA/sulfate depicted in Fig. 5a, the number fraction of SOA/sulfate reached up to 95 % in the accumulation mode and up to 70 % of supermicron particles. Because Centreville, AL, is a forested site and BVOC emissions, such as isoprene, are high in this region, it is not surprising that SOA/sulfate dominated throughout the majority of the campaign, particularly at small particle sizes. However, the fraction of SOA/sulfate > 1 µm is noteworthy, as SOA/sulfate particles are typically considered submicron in size.
Dust was the dominant particle source during two coarse-mode nitrate events (Fig. 5b) detailed previously by Allen et al. (2015) and defined more narrowly herein as 11–13 and 26–28 June 2013 to differentiate from SSA transport time periods and account for available CCSEM data. During the dust-dominated time periods analyzed, dust constituted > 55 % of supermicron particles (1.0–5.0 µm) by number but also contributed, on average, 26 % of accumulation mode particles (0.2–1.0 µm) by number.
Similar to dust, SSA contributed significantly to the overall particle population multiple times throughout the study, comprising approximately 35 % of particles, by number, analyzed during an event in the middle of June (10–11 June 2013) and at the beginning of July (3–6 July 2013). Both of these SSA events were also characterized by high number fractions of dust, as observed in Fig. 5c. During these SSA-rich periods, SSA particles were predominately larger than 1 µm (38 % by number), although notable contributions to accumulation mode number fractions of SSA were also observed (22 % by number from 0.2 to 1.0 µm). During these two events the degree of atmospheric processing varied, with a considerable number fraction of partially aged SSA present during the second event compared to the first event, which was primarily fully aged SSA (Bondy et al., 2017b).
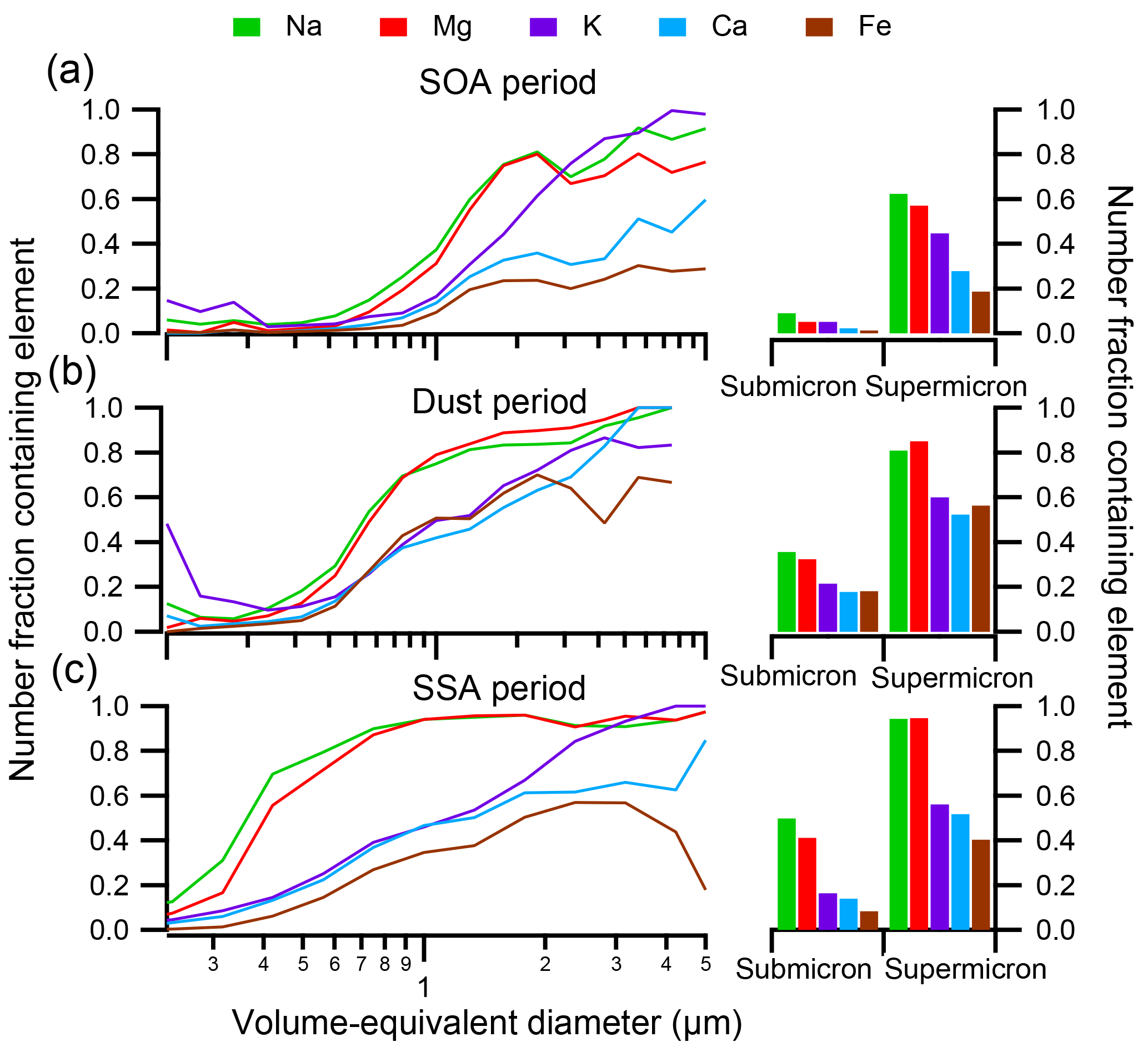
Figure 6(Left) Size-resolved compositions indicate the number fraction of particles containing nonvolatile cations Na, Mg, K, Ca, and Fe during the (a) SOA period, (b) dust period, and (c) SSA period. (Right) The number fraction of submicron and supermicron particles during each period containing each nonvolatile cation.
3.4 Nonvolatile cations during SOAS
Recently, the potential for soluble nonvolatile cations such as Na+, Mg2+, K+, and Ca2+ to improve thermodynamic modeling results of aerosol acidity when included as inputs has been suggested, assuming all species are internally mixed (Guo et al., 2017). As CCSEM-EDX can readily detect metals within individual particles, the number fraction of particles containing Na, Mg, K, Ca, and Fe at sub- and supermicron sizes during the SOA, dust, and SSA events is shown in Fig. 6. In addition to these metals, Mn was detected within < 3 % particles by number during SOAS at > 2 % in a particle, and, given its low fraction, Mn was not included in further analysis. During all events, the number fraction of particles containing nonvolatile cations increased as a function of particle size, with a higher number fraction of metal-containing particles at supermicron sizes (19 %–94 %) compared to submicron sizes (1 %–50 %). During all the time periods depicted, Na was present most frequently, closely followed by Mg, indicative of SSA particles. Fewer particles contained K and Ca by comparison, and Fe was present within the lowest number fraction of particles, except for during the dust period when Fe was more frequent. The number fraction of metal-containing particles was not consistent throughout SOAS but varied dramatically between the SOA, dust, and SSA periods. In general, particles during the dust and SSA events contained higher number fractions of all nonvolatile cations, particularly Na and Mg. However, the variation between specific metals was largely dependent on the dominant particle class during each period.
Figure 7 focuses on the SOA time period and shows the number fraction of particles within each particle class containing Na, Mg, K, Ca, or Fe (dust and SSA periods are shown in Fig. S5). Within both submicron and supermicron particles, the nonvolatile cations within each class are consistent, though a marginally larger number fraction of supermicron particles contained nonvolatile cations, likely due to detection limits for smaller particles. Less than 5 % of SOA/sulfate particles by number contained any Na, Mg, K, Ca, or Fe. Conversely, all other particle classes contained metals within a substantial number of particles. Specifically, all biomass burning particles contained K. Fly ash, on the other hand, most frequently contained Na, though most fly ash contained Al or Si instead (Fig. 4). Additionally, a considerable fraction of dust particles contained Na, Mg, K, Ca, or Fe; all SSA contained Na and many contained Mg; and most primary biological particles contained Na, Mg, and K. When the nonvolatile cations are compared within each class during the SOA, dust, and SSA periods (Figs. 7 and S4), the number fractions of metal-containing particles are consistent for each particle class, suggesting that an internal mixing assumption for nonvolatile cations and their presence in SOA/sulfate particles does not reflect overall particle composition.
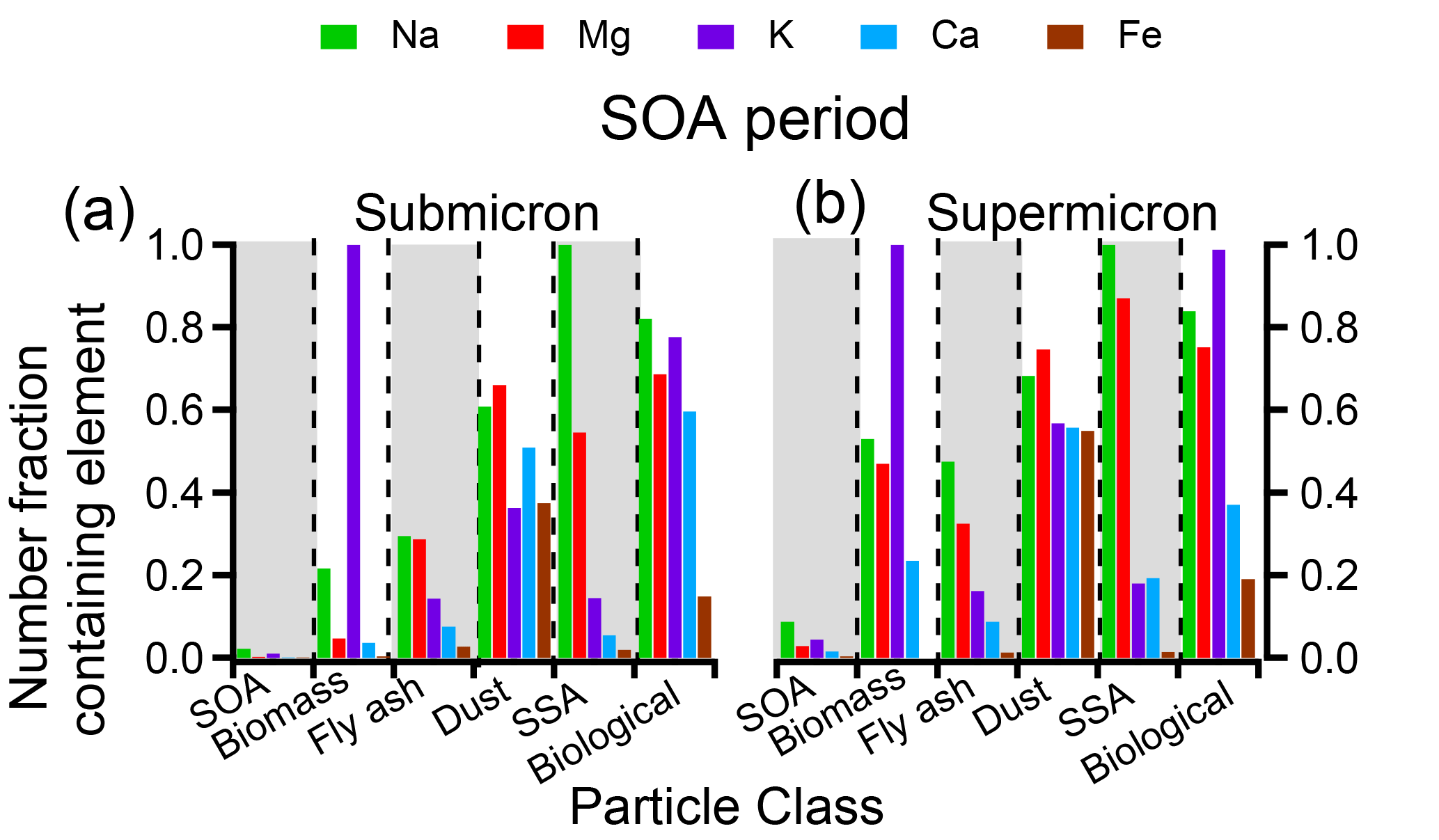
Figure 7Size-resolved particle class compositions indicate the number fraction of particles in each class containing nonvolatile cations Na, Mg, and Fe during the SOA period in the (a) submicron and (b) supermicron size range.
3.5 Particle aging
In contrast to nonvolatile cations, the contribution of secondary components within each class varied drastically throughout SOAS. Due to atmospheric reactions and transport of gases from nearby cities, many of the particles analyzed from SOAS were likely not “fresh” from their source but had undergone secondary processing by species such as HNO3, SO2∕H2SO4, or organic acids. Secondary processing of particles is important because changing their chemical composition can impact light scattering and CCN properties (Chang et al., 2010; Chi et al., 2015; Ghorai et al., 2014; Giordano et al., 2015; Hiranuma et al., 2013; Lu et al., 2011; Moise et al., 2015; Robinson et al., 2013; Sedlacek et al., 2012; Tang et al., 2016). As the chemical composition of particles varied over time, the mixing state index was used to quantify the degree of aging. The degree of secondary processing for each particle class was calculated as the average mass fraction of sulfur per particle; see Fig. S6 and details in the Supplement. Only sulfur was used as an indicator of aging in this study since carbon had interference from the background and nitrogen is only semi-quantitative with CCSEM-EDX (Laskin et al., 2006).
From STXM-NEXAFS, we know that most SOA particles are mixtures of organic and inorganic (mostly ammonium sulfate) components, and there are almost no externally mixed organic or ammonium sulfate particles present. As such, based on elemental composition shown in Fig. 4 and the fact that C, O, and N could not be quantified in this study, particles containing only S in our mixing state analysis are presumed to be SOA/sulfate. A large fraction of sulfur in SOA was likely in the form of sulfate, since sulfate was identified as the major inorganic component within SOA (24 % wt in fine aerosol) (Budisulistiorini et al., 2015b). Additionally, IEPOX-derived organosulfates and other organosulfates, known to contribute to the organic aerosol fraction in Centreville (Bondy et al., 2018; Boone et al., 2015; Budisulistiorini et al., 2015b; Riva et al., 2016; Xu et al., 2015a), also contributed to the EDX sulfur content of SOA. The other five particle classes contained substantially less sulfur than SOA: SSA (20 % S–30 wt % S), biomass burning particles (15 % S–25 wt % S), dust (5 % S–15 wt % S), fly ash (2 % S–10 wt % S), and biological particles (15 % S–25 wt % S). SSA and biomass burning particles are both readily aged by sulfuric acid forming Na2SO4 and K2SO4, respectively (Chen et al., 2017; Hopkins et al., 2008; Li et al., 2003), although up to 8 % of sulfate in SSA may have marine origins (Pilson, 1998). Aluminosilicate dust, the most common type of mineral dust detected during SOAS, is also aged by sulfuric acid (Fitzgerald et al., 2015; Perlwitz et al., 2015; Song et al., 2007; Sullivan et al., 2007). Fly ash detected during SOAS did not contain much sulfur, indicating that it was relatively fresh (Shen et al., 2016), or was aged more by other species such as organics, relative to sulfuric acid (Li et al., 2017). Primary biological particles also contained low mass fractions of sulfur. However, as heterogeneous chemistry of this class of particles has not been explored as extensively as the other classes and the sulfur mass fractions did not follow the same trends for the three time periods (Fig. S6), the sulfur content in biological particles may have been, but was not necessarily, the result of aging (Estillore et al., 2016).
In addition to differences in aging by sulfur for each particle class, the average mass fraction of sulfur within each class varied during the SOA-rich, dust-rich, and SSA-rich time periods (Fig. S6). Specifically, the average mass fraction of sulfur was significantly higher during the SOA-dominated time period compared to the dust and SSA periods at the 95 % confidence interval for all particle classes aside from biological (Tables S6–S7). However, the mass fraction of sulfur was not statistically different between the dust and SSA periods for any particle classes (Table S8). Stagnant air masses, indicated by slower average wind speed at Centreville during the SOA/sulfate period (1.6±0.7) compared to the dust period (2.3±0.9) and SSA period (2.1±0.9), may have led to more aging during the SOA events.
3.6 Quantification of mixing state using aging diversity measures
To quantify the differences in aging during three described events, the mixing state index was calculated for the SOA, dust, and SSA time periods (Fig. 8). Because the particle classes present at sub- and supermicron sizes vary dramatically, mixing state indices were calculated separately for the two size ranges. From calculations of the average particle diversity and the bulk diversity (Fig. 8a), the mixing state index, a ratio measuring how close the population is to an external or internal mixture, could be determined for each time period (Fig. 8b). The mixing state indices for supermicron particles were generally the highest (χ=19 %, 15 %, and 11 % during the SSA, dust, and SOA periods, respectively), signifying that supermicron particles were less diverse than submicron particles. The supermicron SOA period is more externally mixed than the SSA or dust periods for two reasons: (1) it contains the most individual particle classes (largest bulk diversity, ∼5), and (2) the particle-specific diversity is highest as well, indicating that this period has a lot of aging by sulphur (and likely organic carbon), contributing to the relatively high mixing state index. The mixing state indices for accumulation mode particles during the SOA and SSA periods were comparable (χ=10 % and 9 %, respectively), while during the dust time period many more elements were present in separate particles, leading to the most external mixture observed. The SSA time period had a more external accumulation mixing state index than the supermicron mode, which is logical as SSA dominated the supermicron during SSA periods, but the accumulation mode still had distinct classes (e.g., SOA/sulfate). For the SOA time periods, the accumulation mode had the lowest bulk diversity and particle-specific diversity, while the supermicron mode had both high particle diversity and greater particle-specific diversity, which led to them having similar mixing state indices. Overall, Fig. 8a demonstrates that time periods with low bulk diversity, which contain fewer particle classes, have mixing state indices closer to 100 % (more internally mixed).

Figure 8(a) Mixing state diagram showing the bulk diversity and average particle-specific diversity and (b) mixing state indices for sub- and supermicron particles during the SOA, dust, and SSA periods. For submicron particles, contributions by different sources impact mixing state.
Mixing state indices of this work are lower than previous reports by Fraund et al. (2017) and O'Brien et al. (2015) (χ > 80 % and χ > 40 %, respectively), who used CCSEM-EDX and STXM-NEXAFS to analyze particles collected in the Amazon and Central Valley, CA. In both studies, calculations using STXM-NEXAFS resulted in low diversity and high mixing state indices, likely due to the inclusion of organic carbon, which increased particle homogeneity. O'Brien et al. also calculated mixing state indices using solely CCSEM-EDX, similar to our study. From this method, O'Brien found lower mixing state indices using CCSEM-EDX (χ=41 %–90 %) compared to mixing state index calculations from STXM-NEXAFS results (χ > 60 %). However, the mixing state index in this previous work increased during high-SSA periods and periods characterized by increased mass fractions of K, Ca, Zn, and Al, suggesting that periods with higher average particle-specific diversity were more homogeneous, simply because they contained more elements than periods dominated by carbonaceous particles. To address this inherent challenge associated with quantifying mixing state using CCSEM-EDX, in the current study we calculate mixing state based on the number of particle classes and secondary species (in this study, sulfur) rather than the number of elements within particles. Quantifying mixing state parameters using this approach is consistent with our concept of atmospheric aging since the mixing state index increases as bulk diversity decreases and the mass fraction of secondary species increases, signifying aging increases the degree of internal mixing in a population. This method of quantifying aerosol mixing state using single-particle methods can be used to show the varying impact of sources and aging between different air masses at the same location and will be expanded upon in future work.
Even at rural locations, a variety of particle classes with complex chemical compositions can contribute to the aerosol population and mixing state, impacting climate direct and indirect effects. During the SOAS field campaign in Centreville, Alabama, CCSEM-EDX analysis identified the following particle classes: biological, mineral dust, SSA, fly ash, biomass burning aerosol particles, SOA/sulfate, and fresh soot. Although SOA/sulfate dominated the overall measured aerosol population, especially in the accumulation mode (0.2–1.0 µm), it was found to be present at supermicron sizes as well. Additionally, while biological particles, mineral dust, and SSA dominated the supermicron regime, mineral dust and SSA were also observed as significant particle fractions in the accumulation mode. While some of the particle classes indicate nearby regional sources, such as fly ash transported from nearby cities and SOA/sulfate formed from the interaction of biogenic VOCs and anthropogenic pollutants, other classes point toward longer-range transport, such as SSA transported from the Gulf of Mexico. From the single-particle chemical analysis conducted, complex chemical mixing states of particles with secondary processing by sulfur were observed. Finally, even though the sampling site in Centreville was located in a relatively remote region, long- and short-range transport of particles was evident based not only on the wide variety of particle classes and degrees of aging within each class, but also on the variation in number concentration over time. The average mass fraction of sulfur within every particle class, aside from primary biological particles, was greater during more stagnant conditions, leading to more internally mixed-particle populations.
These findings quantify the diversity in particle composition and their mixing states characteristic of the southeastern United States, suggesting that many factors and classes of particles beyond SOA/sulfate contribute to the atmospheric aerosol in this region. Submicron mineral dust and SSA may be underappreciated sources of CCN in this region, as they are sometimes not considered when non-refractory or soot particles are the focus of measurements. Although not highly prevalent, ∼8 % of SOA by number was found to contain soot inclusions, which could have very different optical properties than fresh soot due to lensing effects from coatings and compaction from a fractal to spherical shape. Additionally, since most of the particles in this region have been chemically aged with sulfuric acid/SO2, their hygroscopicity and propensity to form CCN will be altered compared to their fresh counterparts. With this information detailing the particle classes and the mixing states during SOAS, further studies can be conducted and inputs for models can be generated to more accurately assess effects of aerosols on climate in this unique region.
Data are available by contacting the corresponding authors.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-12595-2018-supplement.
ALB and APA prepared the figures and wrote the manuscript. APA organized and led the field sampling efforts. DB and RCM conducted the STXM-NEXAFS analysis. BW and AL assisted with CCSEM-EDX analysis.
The authors declare that they have no conflict of interest.
This work was supported by startup funds from the University of Michigan.
The authors wish to acknowledge EPA grant no. R835409 and NSF grant no. AGS1228496, awarded
to collaborators Steve Bertman (Western Michigan University),
Paul Shepson (Purdue University), and Kerri Pratt (University of
Michigan) for providing funding and logistics support for Ault Group SOAS
sampling. The CCSEM-EDX research was performed at the Environmental
Molecular Sciences Laboratory (EMSL), a science user facility located at the
Pacific Northwest National Laboratory (PNNL), sponsored by the Office of
Biological and Environmental Research of the US Department of Energy (DOE). PNNL
is operated for DOE by Battelle Memorial Institute under contract number
DE-AC06-76RL0 1830. Travel funds to PNNL were provided by UM Rackham
Graduate School and the UM Office of the Provost. The STXM/NEXAFS particle
analysis was performed at beamline 5.3.2 at the Advanced Light Source (ALS)
at Lawrence Berkeley National Laboratory. Additionally, we thank Ann Marie Carlton (Rutgers University, now UC Irvine), Lindsay Yee (UC Berkeley),
Allen Goldstein (UC Berkeley), and Jason Surratt (UNC Chapel Hill) for
organizing SOAS and filter sampling and Manelisi V. Nhliziyo for
assistance collecting samples. Karsten Baumann and the SOAS ground team at
Centreville are acknowledged for logistical assistance.
Edited by: Hang Su
Reviewed by: two anonymous referees
Allen, H. M., Draper, D. C., Ayres, B. R., Ault, A., Bondy, A., Takahama, S., Modini, R. L., Baumann, K., Edgerton, E., Knote, C., Laskin, A., Wang, B., and Fry, J. L.: Influence of crustal dust and sea spray supermicron particle concentrations and acidity on inorganic aerosol during the 2013 Southern Oxidant and Aerosol Study, Atmos. Chem. Phys., 15, 10669–10685, https://doi.org/10.5194/acp-15-10669-2015, 2015.
Anttila, T., Kiendler-Scharr, A., Mentel, T. F., and Tillmann, R.: Size dependent partitioning of organic material: evidence for the formation of organic coatings on aqueous aerosols, J. Atmos. Chem., 57, 215–237, 2007.
Artaxo, P. and Orsini, C.: PIXE and receptor models applied to remote aerosol source apportionment in Brazil, Nucl. Instrum. Methods, Sect. B, 22, 259–263, 1987.
Attwood, A. R., Washenfelder, R. A., Brock, C. A., Hu, W., Baumann, K., Campuzano-Jost, P., Day, D. A., Edgerton, E. S., Murphy, D. M., Palm, B. B., McComiskey, A., Wagner, N. L., de Sá, S. S., Ortega, A., Martin, S. T., Jimenez, J. L., and Brown, S. S.: Trends in sulfate and organic aerosol mass in the Southeast U.S.: Impact on aerosol optical depth and radiative forcing, Geophys. Res. Lett., 41, 7701–7709, 2014.
Ault, A. P. and Axson, J. L.: Atmospheric Aerosol Chemistry: Spectroscopic and Microscopic Advances, Anal. Chem., 89, 430–452, 2017.
Ault, A. P., Moore, M. J., Furutani, H., and Prather, K. A.: Impact of emissions from the Los Angeles port region on San Diego air quality, Environ. Sci. Technol., 43, 3500–3506, 2009.
Ault, A. P., Gaston, C. J., Wang, Y., Dominguez, G., Thiemens, M. H., and Prather, K. A.: Characterization of the single particle mixing state of individual ship plume events measured at the Port of Los Angeles, Environ. Sci. Technol., 44, 1954–1961, 2010.
Ault, A. P., Peters, T. M., Sawvel, E. J., Casuccio, G. S., Willis, R. D., Norris, G. A., and Grassian, V. H.: Single-particle SEM-EDX analysis of iron-containing coarse particulate matter in an urban environment: Sources and distribution of iron within Cleveland, Ohio, Environ. Sci. Technol., 46, 4331–4339, 2012.
Ault, A. P., Moffet, R. C., Baltrusaitis, J., Collins, D. B., Ruppel, M. J., Cuadra-Rodriguez, L. A., Zhao, D., Guasco, T. L., Ebben, C. J., Geiger, F. M., Bertram, T. H., Prather, K. A., and Grassian, V. H.: Size-dependent changes in sea spray serosol composition and properties with different seawater conditions, Environ. Sci. Technol., 47, 5603–5612, 2013a.
Ault, A. P., Zhao, D., Ebben, C. J., Tauber, M. J., Geiger, F. M., Prather, K. A., and Grassian, V. H.: Raman microspectroscopy and vibrational sum frequency generation spectroscopy as probes of the bulk and surface compositions of size-resolved sea spray aerosol particles, Phys. Chem. Chem. Phys., 15, 6206–6214, 2013b.
Axson, J. L., May, N. W., Colon-Bernal, I. D., Pratt, K. A., and Ault, A. P.: Lake Spray Aerosol: A Chemical Signature from Individual Ambient Particles, Environ. Sci. Technol., 50, 9835–9845, 2016a.
Axson, J. L., Shen, H. R., Bondy, A. L., Landry, C. C., Welz, J., Creamean, J. M., and Ault, A. P.: Transported mineral dust deposition case study at a hydrologically sensitive mountain site: Size and composition shifts in ambient aerosol and snowpack, Aerosol Air Qual. Res., 16, 555–567, 2016b.
Ayres, B. R., Allen, H. M., Draper, D. C., Brown, S. S., Wild, R. J., Jimenez, J. L., Day, D. A., Campuzano-Jost, P., Hu, W., de Gouw, J., Koss, A., Cohen, R. C., Duffey, K. C., Romer, P., Baumann, K., Edgerton, E., Takahama, S., Thornton, J. A., Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Wennberg, P. O., Nguyen, T. B., Teng, A., Goldstein, A. H., Olson, K., and Fry, J. L.: Organic nitrate aerosol formation via NO3 + biogenic volatile organic compounds in the southeastern United States, Atmos. Chem. Phys., 15, 13377–13392, https://doi.org/10.5194/acp-15-13377-2015, 2015.
Bondy, A. L., Kirpes, R. M., Merzel, R. L., Pratt, K. A., Banaszak Holl, M. M., and Ault, A. P.: Atomic Force Microscopy-Infrared Spectroscopy of Individual Atmospheric Aerosol Particles: Subdiffraction Limit Vibrational Spectroscopy and Morphological Analysis, Anal. Chem., 89, 8594–8598, 2017a.
Bondy, A. L., Wang, B., Laskin, A., Craig, R. L., Nhliziyo, M. V., Bertman, S. B., Pratt, K. A., Shepson, P. B., and Ault, A. P.: Inland Sea Spray Aerosol Transport and Incomplete Chloride Depletion: Varying Degrees of Reactive Processing Observed during SOAS, Environ. Sci. Technol., 51, 9533–9542, 2017b.
Bondy, A. L., Craig, R. L., Zhang, Z., Gold, A., Surratt, J. D., and Ault, A. P.: Isoprene-Derived Organosulfates: Vibrational Mode Analysis by Raman Spectroscopy, Acidity-Dependent Spectral Modes, and Observation in Individual Atmospheric Particles, J. Phys. Chem. A, 122, 303–315, 2018.
Boone, E. J., Laskin, A., Laskin, J., Wirth, C., Shepson, P. B., Stirm, B. H., and Pratt, K. A.: Aqueous Processing of Atmospheric Organic Particles in Cloud Water Collected via Aircraft Sampling, Environ. Sci. Technol., 49, 8523–8530, 2015.
Boyd, C. M., Sanchez, J., Xu, L., Eugene, A. J., Nah, T., Tuet, W. Y., Guzman, M. I., and Ng, N. L.: Secondary organic aerosol formation from the β-pinene+NO3 system: effect of humidity and peroxy radical fate, Atmos. Chem. Phys., 15, 7497–7522, https://doi.org/10.5194/acp-15-7497-2015, 2015.
Budisulistiorini, S. H., Li, X., Bairai, S. T., Renfro, J., Liu, Y., Liu, Y. J., McKinney, K. A., Martin, S. T., McNeill, V. F., Pye, H. O. T., Nenes, A., Neff, M. E., Stone, E. A., Mueller, S., Knote, C., Shaw, S. L., Zhang, Z., Gold, A., and Surratt, J. D.: Examining the effects of anthropogenic emissions on isoprene-derived secondary organic aerosol formation during the 2013 Southern Oxidant and Aerosol Study (SOAS) at the Look Rock, Tennessee ground site, Atmos. Chem. Phys., 15, 8871–8888, https://doi.org/10.5194/acp-15-8871-2015, 2015a.
Buha, J., Mueller, N., Nowack, B., Ulrich, A., Losert, S., and Wang, J.: Physical and chemical characterization of fly ashes from swiss waste incineration plants and determination of the ash fraction in the nanometer range, Environ. Sci. Technol., 48, 4765–4773, 2014.
Carlton, A. G., Pinder, R. W., Bhave, P. V., and Pouliot, G. A.: To what extent can biogenic SOA be controlled?, Environ. Sci. Technol., 44, 3376–3380, 2010.
Carlton, A. G., Wiedinmyer, C., and Kroll, J. H.: A review of Secondary Organic Aerosol (SOA) formation from isoprene, Atmos. Chem. Phys., 9, 4987–5005, https://doi.org/10.5194/acp-9-4987-2009, 2009.
Carlton, A. G., Gouw, J. D., Jimenez, J. L., Ambrose, J. L., Attwood, A. R., Brown, S., Baker, K. R., Brock, C., Cohen, R. C., Edgerton, S., Farkas, C. M., Farmer, D., Goldstein, A. H., Gratz, L., Guenther, A., Hunt, S., Jaeglé, L., Jaffe, D. A., Mak, J., McClure, C., Nenes, A., Nguyen, T. K., Pierce, J. R., Sa, S. D., Selin, N. E., Shah, V., Shaw, S., Shepson, P. B., Song, S., Stutz, J., Surratt, J. D., Turpin, B. J., Warneke, C., Washenfelder, R. A., Wennberg, P. O., and Zhou, X.: Synthesis of the Southeast Atmosphere Studies: Investigating Fundamental Atmospheric Chemistry Questions, B. Am. Meteorol. Soc., 99, 547–567, 2018.
Cerully, K. M., Bougiatioti, A., Hite Jr., J. R., Guo, H., Xu, L., Ng, N. L., Weber, R., and Nenes, A.: On the link between hygroscopicity, volatility, and oxidation state of ambient and water-soluble aerosols in the southeastern United States, Atmos. Chem. Phys., 15, 8679–8694, https://doi.org/10.5194/acp-15-8679-2015, 2015.
Chameides, W. L., Lindsay, R. W., Richardson, J., and Kiang, C. S.: The role of biogenic hydrocarbons in urban photochemical smog: Atlanta as a case study, Science, 241, 1473–1475, 1988.
Chang, R. Y.-W., Slowik, J. G., Shantz, N. C., Vlasenko, A., Liggio, J., Sjostedt, S. J., Leaitch, W. R., and Abbatt, J. P. D.: The hygroscopicity parameter (κ) of ambient organic aerosol at a field site subject to biogenic and anthropogenic influences: relationship to degree of aerosol oxidation, Atmos. Chem. Phys., 10, 5047–5064, https://doi.org/10.5194/acp-10-5047-2010, 2010.
Chen, H., Laskin, A., Baltrusaitis, J., Gorski, C. A., Scherer, M. M., and Grassian, V. H.: Coal Fly Ash as a Source of Iron in Atmospheric Dust, Environ. Sci. Technol., 46, 2112–2120, 2012.
Chen, J., Li, C., Ristovski, Z., Milic, A., Gu, Y., Islam, M. S., Wang, S., Hao, J., Zhang, H., He, C., Guo, H., Fu, H., Miljevic, B., Morawska, L., Thai, P., Lam, Y. F., Pereira, G., Ding, A., Huang, X., and Dumka, U. C.: A review of biomass burning: emissions and impacts on air quality, health and climate in China, Sci. Total Environ., 579, 1000–1034, 2017.
Chi, J. W., Li, W. J., Zhang, D. Z., Zhang, J. C., Lin, Y. T., Shen, X. J., Sun, J. Y., Chen, J. M., Zhang, X. Y., Zhang, Y. M., and Wang, W. X.: Sea salt aerosols as a reactive surface for inorganic and organic acidic gases in the Arctic troposphere, Atmos. Chem. Phys., 15, 11341–11353, https://doi.org/10.5194/acp-15-11341-2015, 2015.
Chung, S. H. and Seinfeld, J. H.: Climate response of direct radiative forcing of anthropogenic black carbon, J. Geophys. Res.-Atmos., 110, D11102, https://doi.org/10.1029/2004JD005441, 2005.
Cong, Z., Kang, S., Dong, S., Liu, X., and Qin, D.: Elemental and individual particle analysis of atmospheric aerosols from high Himalayas, Environ. Monit. Assess., 160, 323–335, 2010.
Conny, J. M. and Norris, G. A.: Scanning Electron Microanalysis and Analytical Challenges of Mapping Elements in Urban Atmospheric Particles, Environ. Sci. Technol., 45, 7380–7386, 2011.
Coz, E., Gómez-Moreno, F. J., Pujadas, M., Casuccio, G. S., Lersch, T. L., and Artíñano, B.: Individual particle characteristics of North African dust under different long-range transport scenarios, Atmos. Environ., 43, 1850–1863, 2009.
Craig, R. L. and Ault, A. P.: Aerosol Acidity: Direct Measurement from a Spectroscopic Method, in: Multiphase Environmental Chemistry in the Atmosphere, edited by: Hunt, S. W., Laskin, A., and Nizkorodov, S., ACS Books Special Symposium Series, 2018.
Craig, R. L., Bondy, A. L., and Ault, A. P.: Computer-controlled Raman microspectroscopy (CC-Raman): A method for the rapid characterization of individual atmospheric aerosol particles, Aerosol Sci. Technol., 51, 1099–1112, 2017.
Creamean, J. M., Axson, J. L., Bondy, A. L., Craig, R. L., May, N. W., Shen, H., Weber, M. H., Pratt, K. A., and Ault, A. P.: Changes in precipitating snow chemistry with location and elevation in the California Sierra Nevada, J. Geophys. Res.-Atmos., 121, 7296–7309, 2016.
Deboudt, K., Gloter, A., Mussi, A., and Flament, P.: Red-ox speciation and mixing state of iron in individual African dust particles, J. Geophys. Res.-Atmos., 117, D12307, https://doi.org/10.1029/2011JD017298, 2012.
Dickau, M., Olfert, J., Stettler, M. E. J., Boies, A., Momenimovahed, A., Thomson, K., Smallwood, G., and Johnson, M.: Methodology for quantifying the volatile mixing state of an aerosol, Aerosol Sci. Technol., 50, 759–772, 2016.
Ebben, C. J., Ault, A. P., Ruppel, M. J., Ryder, O. S., Bertram, T. H., Grassian, V. H., Prather, K. A., and Geiger, F. M.: Size-Resolved Sea Spray Aerosol Particles Studied by Vibrational Sum Frequency Generation, J. Phys. Chem. A, 117, 6589–6601, 2013.
Estillore, A. D., Trueblood, J. V., and Grassian, V. H.: Atmospheric chemistry of bioaerosols: heterogeneous and multiphase reactions with atmospheric oxidants and other trace gases, Chem. Sci., 7, 6604–6616, 2016.
Fang, T., Guo, H. Y., Zeng, L. H., Verma, V., Nenes, A., and Weber, R. J.: Highly Acidic Ambient Particles, Soluble Metals, and Oxidative Potential: A Link between Sulfate and Aerosol Toxicity, Environ. Sci. Technol., 51, 2611–2620, 2017.
Fitzgerald, E., Ault, A. P., Zauscher, M. D., Mayol-Bracero, O. L., and Prather, K. A.: Comparison of the mixing state of long-range transported Asian and African mineral dust, Atmos. Environ., 115, 19–25, 2015.
Fraund, M., Pham, D., Bonanno, D., Harder, T., Wang, B., Brito, J., de Sá, S., Carbone, S., China, S., Artaxo, P., Martin, S., Pöhlker, C., Andreae, M., Laskin, A., Gilles, M., and Moffet, R.: Elemental mixing state of aerosol particles collected in Central Amazonia during GoAmazon2014/15, Atmosphere, 8, 173, https://doi.org/10.3390/atmos8090173, 2017.
Froyd, K. D., Murphy, S. M., Murphy, D. M., de Gouw, J. A., Eddingsaas, N. C., Wennberg, P. O., and Thiemens, M. H.: Contribution of isoprene-derived organosulfates to free tropospheric aerosol mass, P. Natl. Acad. Sci. USA, 107, 21360–21365, 2010.
Fu, H., Zhang, M., Li, W., Chen, J., Wang, L., Quan, X., and Wang, W.: Morphology, composition and mixing state of individual carbonaceous aerosol in urban Shanghai, Atmos. Chem. Phys., 12, 693–707, https://doi.org/10.5194/acp-12-693-2012, 2012.
Gantt, B. and Meskhidze, N.: The physical and chemical characteristics of marine primary organic aerosol: a review, Atmos. Chem. Phys., 13, 3979–3996, https://doi.org/10.5194/acp-13-3979-2013, 2013.
Ghorai, S., Wang, B., Tivanski, A., and Laskin, A.: Hygroscopic properties of internally mixed particles composed of NaCl and water-soluble organic acids, Environ. Sci. Technol., 48, 2234–2241, 2014.
Giordano, M., Espinoza, C., and Asa-Awuku, A.: Experimentally measured morphology of biomass burning aerosol and its impacts on CCN ability, Atmos. Chem. Phys., 15, 1807–1821, https://doi.org/10.5194/acp-15-1807-2015, 2015.
Giorio, C., Tapparo, A., Dall'Osto, M., Beddows, D. C. S., Esser-Gietl, J. K., Healy, R. M., and Harrison, R. M.: Local and regional components of aerosol in a heavily trafficked street canyon in central London derived from PMF and cluster analysis of single-particle ATOFMS spectra, Environ. Sci. Technol., 49, 3330–3340, 2015.
Goldstein, A. H., Koven, C. D., Heald, C. L., and Fung, I. Y.: Biogenic carbon and anthropogenic pollutants combine to form a cooling haze over the southeastern United States, P. Natl. Acad. Sci. USA, 106, 8835–8840, 2009.
Guo, H., Xu, L., Bougiatioti, A., Cerully, K. M., Capps, S. L., Hite Jr., J. R., Carlton, A. G., Lee, S.-H., Bergin, M. H., Ng, N. L., Nenes, A., and Weber, R. J.: Fine-particle water and pH in the southeastern United States, Atmos. Chem. Phys., 15, 5211–5228, https://doi.org/10.5194/acp-15-5211-2015, 2015.
Guo, H., Nenes, A., and Weber, R. J.: The underappreciated role of nonvolatile cations on aerosol ammonium-sulfate molar ratios, Atmos. Chem. Phys. Discuss., https://doi.org/10.5194/acp-2017-737, in review, 2017.
Hashemi, S. S., Baghernejad, M., and Khademi, H.: Micromorphology of gypsum crystals in southern Iranian soils under different moisture regimes, J. Agric. Sci. Technol., 13, 273–288, 2011.
Hatch, L. E., Creamean, J. M., Ault, A. P., Surratt, J. D., Chan, M. N., Seinfeld, J. H., Edgerton, E. S., Su, Y., and Prather, K. A.: Measurements of Isoprene-Derived Organosulfates in Ambient Aerosols by Aerosol Time-of-Flight Mass Spectrometry – Part 1: Single Particle Atmospheric Observations in Atlanta, Environ. Sci. Technol., 45, 5105–5111, 2011.
Healy, R. M., Evans, G. J., Murphy, M., Juranyi, Z., Tritscher, T., Laborde, M., Weingartner, E., Gysel, M., Poulain, L., Kamilli, K. A., Wiedensohler, A., O'Connor, I. P., McGillicuddy, E., Sodeau, J. R., and Wenger, J. C.: Predicting hygroscopic growth using single particle chemical composition estimates, J. Geophys. Res.-Atmos., 119, 9567–9577, 2014a.
Healy, R. M., Riemer, N., Wenger, J. C., Murphy, M., West, M., Poulain, L., Wiedensohler, A., O'Connor, I. P., McGillicuddy, E., Sodeau, J. R., and Evans, G. J.: Single particle diversity and mixing state measurements, Atmos. Chem. Phys., 14, 6289–6299, https://doi.org/10.5194/acp-14-6289-2014, 2014b.
Healy, R. M., Wang, J. M., Jeong, C. H., Lee, A. K. Y., Willis, M. D., Jaroudi, E., Zimmerman, N., Hilker, N., Murphy, M., Eckhardt, S., Stohl, A., Abbatt, J. P. D., Wenger, J. C., and Evans, G. J.: Light-absorbing properties of ambient black carbon and brown carbon from fossil fuel and biomass burning sources, J. Geophys. Res.-Atmos., 120, 6619–6633, 2015.
Hidy, G. M., Blanchard, C. L., Baumann, K., Edgerton, E., Tanenbaum, S., Shaw, S., Knipping, E., Tombach, I., Jansen, J., and Walters, J.: Chemical climatology of the southeastern United States, 1999–2013, Atmos. Chem. Phys., 14, 11893–11914, https://doi.org/10.5194/acp-14-11893-2014, 2014.
Hinds, W. C.: Aerosol Technology Properties, Behavior, and Measurement of Airborne Particles, John Wiley & Sons, Inc., New York, 1999.
Hiranuma, N., Brooks, S. D., Moffet, R. C., Glen, A., Laskin, A., Gilles, M. K., Liu, P., Macdonald, A. M., Strapp, J. W., and McFarquhar, G. M.: Chemical characterization of individual particles and residuals of cloud droplets and ice crystals collected on board research aircraft in the ISDAC 2008 study, J. Geophys. Res.-Atmos., 118, 6564–6579, 2013.
Hodas, N., Sullivan, A. P., Skog, K., Keutsch, F. N., Collett, J. L., Decesari, S., Facchini, M. C., Carlton, A. G., Laaksonen, A., and Turpin, B. J.: Aerosol liquid water driven by anthropogenic nitrate: implications for lifetimes of water-soluble organic gases and potential for secondary organic aerosol formation, Environ. Sci. Technol., 48, 11127–11136, 2014.
Hopkins, R. J., Desyaterik, Y., Tivanski, A. V., Zaveri, R. A., Berkowitz, C. M., Tyliszczak, T., Gilles, M. K., and Laskin, A.: Chemical speciation of sulfur in marine cloud droplets and particles: Analysis of individual particles from the marine boundary layer over the California current, J. Geophys. Res.-Atmos., 113, D04209, https://doi.org/10.1029/2007JD008954, 2008.
Hu, W. W., Campuzano-Jost, P., Palm, B. B., Day, D. A., Ortega, A. M., Hayes, P. L., Krechmer, J. E., Chen, Q., Kuwata, M., Liu, Y. J., de Sá, S. S., McKinney, K., Martin, S. T., Hu, M., Budisulistiorini, S. H., Riva, M., Surratt, J. D., St. Clair, J. M., Isaacman-Van Wertz, G., Yee, L. D., Goldstein, A. H., Carbone, S., Brito, J., Artaxo, P., de Gouw, J. A., Koss, A., Wisthaler, A., Mikoviny, T., Karl, T., Kaser, L., Jud, W., Hansel, A., Docherty, K. S., Alexander, M. L., Robinson, N. H., Coe, H., Allan, J. D., Canagaratna, M. R., Paulot, F., and Jimenez, J. L.: Characterization of a real-time tracer for isoprene epoxydiols-derived secondary organic aerosol (IEPOX-SOA) from aerosol mass spectrometer measurements, Atmos. Chem. Phys., 15, 11807–11833, https://doi.org/10.5194/acp-15-11807-2015, 2015.
Huffman, J. A., Sinha, B., Garland, R. M., Snee-Pollmann, A., Gunthe, S. S., Artaxo, P., Martin, S. T., Andreae, M. O., and Pöschl, U.: Size distributions and temporal variations of biological aerosol particles in the Amazon rainforest characterized by microscopy and real-time UV-APS fluorescence techniques during AMAZE-08, Atmos. Chem. Phys., 12, 11997–12019, https://doi.org/10.5194/acp-12-11997-2012, 2012.
Jacobson, M. Z. Z.: Strong radiative heating due to the mixing state of black carbon in atmospheric aerosols, Nature, 409, 695–697, 2001.
Kandler, K., Lieke, K., Benker, N., Emmel, C., Küpper, M., Müller-Ebert, D., Ebert, M., Scheuvens, D., Schladitz, A., Schütz, L., and Weinbruch, S.: Electron microscopy of particles collected at Praia, Cape Verde, during the Saharan Mineral Dust Experiment: particle chemistry, shape, mixing state and complex refractive index, Tellus B, 63, 475–496, https://doi.org/10.1111/j.1600-0889.2011.00550.x, 2011.
Kilcoyne, A. L. D., Tyliszczak, T., Steele, W. F., Fakra, S., Hitchcock, P., Franck, K., Anderson, E., Harteneck, B., Rightor, E. G., Mitchell, G. E., Hitchcock, A. P., Yang, L., Warwick, T., and Ade, H.: Interferometer-controlled scanning transmission X-ray microscopes at the Advanced Light Source, J. Synchrotron Radiat., 10, 125–136, 2003.
Kim, J.-S. and Park, K.: Atmospheric aging of Asian dust particles during long range transport, Aerosol Sci. Technol., 46, 913–924, 2012.
Kirpes, R. M., Bondy, A. L., Bonanno, D., Moffet, R. C., Wang, B., Laskin, A., Ault, A. P., and Pratt, K. A.: Secondary sulfate is internally mixed with sea spray aerosol and organic aerosol in the winter Arctic, Atmos. Chem. Phys., 18, 3937–3949, https://doi.org/10.5194/acp-18-3937-2018, 2018.
Kunkel, D., Lawrence, M. G., Tost, H., Kerkweg, A., Jockel, P., and Borrmann, S.: Urban emission hot spots as sources for remote aerosol deposition, Geophys. Res. Lett., 39, L01808, https://doi.org/10.1029/2011GL049634, 2012.
Laskin, A., Iedema, M. J., and Cowin, J. P.: Quantitative time-resolved monitoring of nitrate formation in sea salt particles using a CCSEM/EDX single particle analysis, Environ. Sci. Technol., 36, 4948–4955, 2002.
Laskin, A., Iedema, M. J., Ichkovich, A., Graber, E. R., Taraniuk, I., and Rudich, Y.: Direct observation of completely processed calcium carbonate dust particles, Faraday Discuss., 130, 453–468, 2005.
Laskin, A., Cowin, J. P., and Iedema, M. J.: Analysis of individual environmental particles using modern methods of electron microscopy and X-ray microanalysis, J. Electron Spectrosc., 150, 260–274, 2006.
Laskin, A., Moffet, R. C., Gilles, M. K., Fast, J. D., Zaveri, R. A., Wang, B., Nigge, P., and Shutthanandan, J.: Tropospheric chemistry of internally mixed sea salt and organic particles: Surprising reactivity of NaCl with weak organic acids, J. Geophys. Res.-Atmos., 117, D15302, https://doi.org/10.1029/2012JD017743, 2012.
Lee, S., Wang, Y. H., and Russell, A. G.: Assessment of secondary organic carbon in the southeastern United States: A review, J. Air Waste Manage. Assoc., 60, 1282–1292, 2010.
Li, C., Hu, Y., Chen, J., Ma, Z., Ye, X., Yang, X., Wang, L., Wang, X., and Mellouki, A.: Physiochemical properties of carbonaceous aerosol from agricultural residue burning: Density, volatility, and hygroscopicity, Atmos. Environ., 140, 94–105, 2016.
Li, J., Posfai, M., Hobbs, P. V., and Buseck, P. R.: Individual aerosol particles from biomass burning in southern Africa: 2, Compositions and aging of inorganic particles, J. Geophys. Res.-Atmos., 108, 8484, https://doi.org/10.1029/2002JD002310, 2003.
Li, W., Shao, L., Zhang, D., Ro, C.-U., Hu, M., Bi, X., Geng, H., Matsuki, A., Niu, H., and Chen, J.: A review of single aerosol particle studies in the atmosphere of East Asia: Morphology, mixing state, source, and heterogeneous reactions, J. Clean. Prod., 112, 1330–1349, 2016.
Li, W., Xu, L., Liu, X., Zhang, J., Lin, Y., Yao, X., Gao, H., Zhang, D., Chen, J., Wang, W., Harrison, R. M., Zhang, X., Shao, L., Fu, P., Nenes, A., and Shi, Z.: Air pollution–aerosol interactions produce more bioavailable iron for ocean ecosystems, Sci. Adv., 3, e1601749, https://doi.org/10.1126/sciadv.1601749, 2017.
Lu, J. W., Flores, J. M., Lavi, A., Abo-Riziq, A., and Rudich, Y.: Changes in the optical properties of benzo[a]pyrene-coated aerosols upon heterogeneous reactions with NO2 and NO3, PCCP, 13, 6484–6492, 2011.
Manninen, H. E., Back, J., Sihto-Nissila, S. L., Huffman, J. A., Pessi, A. M., Hiltunen, V., Aalto, P. P., Hidalgo, P. J., Hari, P., Saarto, A., Kulmala, M., and Petaja, T.: Patterns in airborne pollen and other primary biological aerosol particles (PBAP), and their contribution to aerosol mass and number in a boreal forest, Boreal Environ. Res., 19, 383–405, 2014.
Marple, V. A., Rubow, K. L., and Behm, S. M.: A microorifice uniform deposit impactor (MOUDI)-description, calibration and use, Aerosol Sci. Technol., 14, 434–446, 1991.
May, N. W., Axson, J. L., Watson, A., Pratt, K. A., and Ault, A. P.: Lake spray aerosol generation: a method for producing representative particles from freshwater wave breaking, Atmos. Meas. Tech., 9, 4311–4325, https://doi.org/10.5194/amt-9-4311-2016, 2016.
May, N. W., Gunsch, M. J., Olson, N. E., Bondy, A. L., Kirpes, R. M., Bertman, S. B., China, S., Laskin, A., Hopke, P. K., Ault, A. P., and Pratt, K. A.: Unexpected Contributions of Sea Spray and Lake Spray Aerosol to Inland Particulate Matter, Environ. Sci. Tech. Let., 5, 405–412, 2018a.
May, N. W., Olson, N. E., Panas, M., Axson, J. L., Tirella, P. S., Kirpes, R. M., Craig, R. L., Gunsch, M. J., China, S., Laskin, A., Ault, A. P., and Pratt, K. A.: Aerosol Emissions from Great Lakes Harmful Algal Blooms, Environ. Sci. Technol., 52, 397–405, 2018b.
Metternich, P., Georgii, H. W., and Groeneveld, K. O.: Application of particle induced X-ray emission to aerosol long-range transport studies – transport pathways of pollution aerosol over the Atlantic ocean and Europe, Atmos. Res., 20, 3–9, 1986.
Moffet, R. C., Prather, K. A., and Thiemens, M. H.: In-situ measurements of the mixing state and optical properties of soot with implications for radiative forcing estimates, P. Natl. Acad. Sci. USA, 106, 11872–11877, 2009.
Moffet, R. C., Henn, T., Laskin, A., and Gilles, M. K.: Automated chemical analysis of internally mixed aerosol particles using X-ray spectromicroscopy at the carbon K-edge, Anal. Chem., 82, 7906–7914, 2010a.
Moffet, R. C., Henn, T. R., Tivanski, A. V., Hopkins, R. J., Desyaterik, Y., Kilcoyne, A. L. D., Tyliszczak, T., Fast, J., Barnard, J., Shutthanandan, V., Cliff, S. S., Perry, K. D., Laskin, A., and Gilles, M. K.: Microscopic characterization of carbonaceous aerosol particle aging in the outflow from Mexico City, Atmos. Chem. Phys., 10, 961–976, https://doi.org/10.5194/acp-10-961-2010, 2010b.
Moffet, R. C., Rödel, T. C., Kelly, S. T., Yu, X. Y., Carroll, G. T., Fast, J., Zaveri, R. A., Laskin, A., and Gilles, M. K.: Spectro-microscopic measurements of carbonaceous aerosol aging in Central California, Atmos. Chem. Phys., 13, 10445–10459, https://doi.org/10.5194/acp-13-10445-2013, 2013.
Moffet, R. C., O'Brien, R. E., Alpert, P. A., Kelly, S. T., Pham, D. Q., Gilles, M. K., Knopf, D. A., and Laskin, A.: Morphology and mixing of black carbon particles collected in central California during the CARES field study, Atmos. Chem. Phys., 16, 14515–14525, https://doi.org/10.5194/acp-16-14515-2016, 2016.
Moise, T., Flores, J. M., and Rudich, Y.: Optical Properties of Secondary Organic Aerosols and Their Changes by Chemical Processes, Chem. Rev., 115, 4400–4439, 2015.
Nakao, S., Tang, P., Tang, X., Clark, C. H., Qi, L., Seo, E., Asa-Awuku, A., and Cocker Iii, D.: Density and elemental ratios of secondary organic aerosol: Application of a density prediction method, Atmos. Environ., 68, 273–277, 2013.
Nguyen, T. K. V., Petters, M. D., Suda, S. R., Guo, H., Weber, R. J., and Carlton, A. G.: Trends in particle-phase liquid water during the Southern Oxidant and Aerosol Study, Atmos. Chem. Phys., 14, 10911–10930, https://doi.org/10.5194/acp-14-10911-2014, 2014.
Nguyen, T. K. V., Capps, S. L., and Carlton, A. G.: Decreasing aerosol water is consistent with OC trends in the southeast U.S, Environ. Sci. Technol., 49, 7843–7850, 2015.
O'Brien, R. E., Wang, B. B., Laskin, A., Riemer, N., West, M., Zhang, Q., Sun, Y. L., Yu, X. Y., Alpert, P., Knopf, D. A., Gilles, M. K., and Moffet, R. C.: Chemical imaging of ambient aerosol particles: Observational constraints on mixing state parameterization, J. Geophys. Res.-Atmos., 120, 9591–9605, 2015.
Perlwitz, J. P., Pérez García-Pando, C., and Miller, R. L.: Predicting the mineral composition of dust aerosols – Part 1: Representing key processes, Atmos. Chem. Phys., 15, 11593–11627, https://doi.org/10.5194/acp-15-11593-2015, 2015.
Pilson, M. E. Q.: An introduction to the chemistry of the sea, Prentice Hall, 1998.
Portmann, R. W., Solomon, S., Hegerl, G. C., and Held, I. M.: Spatial and seasonal patterns in climate change, temperatures, and precipitation across the United States, P. Natl. Acad. Sci. USA, 106, 7324–7329, 2009.
Posfai, M. and Buseck, P. R.: Nature and climate dffects of individual tropospheric aerosol particles, in: Annual Review of Earth and Planetary Sciences, Vol. 38, edited by: Jeanloz, R. and Freeman, K. H., Annual Review of Earth and Planetary Sciences, 2010.
Posfai, M., Simonics, R., Li, J., Hobbs, P. V., and Buseck, P. R.: Individual aerosol particles from biomass burning in southern Africa: 1. Compositions and size distributions of carbonaceous particles, J. Geophys. Res.-Atmos., 108, 8483, https://doi.org/10.1029/2002JD002291, 2003.
Prather, K. A., Bertram, T. H., Grassian, V. H., Deane, G. B., Stokes, M. D., DeMott, P. J., Aluwihare, L. I., Palenik, B. P., Azam, F., Seinfeld, J. H., Moffet, R. C., Molina, M. J., Cappa, C. D., Geiger, F. M., Roberts, G. C., Russell, L. M., Ault, A. P., Baltrusaitis, J., Collins, D. B., Corrigan, C. E., Cuadra-Rodriguez, L. A., Ebben, C. J., Forestieri, S. D., Guasco, T. L., Hersey, S. P., Kim, M. J., Lambert, W. F., Modini, R. L., Mui, W., Pedler, B. E., Ruppel, M. J., Ryder, O. S., Schoepp, N. G., Sullivan, R. C., and Zhao, D.: Bringing the ocean into the laboratory to probe the chemical complexity of sea spray aerosol, P. Natl. Acad. Sci. USA, 110, 7550–7555, 2013.
Pratt, K. A. and Prather, K. A.: Aircraft measurements of vertical profiles of aerosol mixing states, J. Geophys. Res.-Atmos., 115, D11305, https://doi.org/10.1029/2009JD013150, 2010.
Pye, H. O. T., Luecken, D. J., Xu, L., Boyd, C. M., Ng, N. L., Baker, K. R., Ayres, B. R., Bash, J. O., Baumann, K., Carter, W. P. L., Edgerton, E., Fry, J. L., Hutzell, W. T., Schwede, D. B., and Shepson, P. B.: Modeling the current and future roles of particulate organic nitrates in the southeastern United States, Environ. Sci. Technol., 49, 14195–14203, 2015.
Ramanathan, V. and Carmichael, G.: Global and regional climate changes due to black carbon, Nat. Geosci., 1, 221–227, 2008.
Rattanavaraha, W., Chu, K., Budisulistiorini, S. H., Riva, M., Lin, Y.-H., Edgerton, E. S., Baumann, K., Shaw, S. L., Guo, H., King, L., Weber, R. J., Neff, M. E., Stone, E. A., Offenberg, J. H., Zhang, Z., Gold, A., and Surratt, J. D.: Assessing the impact of anthropogenic pollution on isoprene-derived secondary organic aerosol formation in PM2.5 collected from the Birmingham, Alabama, ground site during the 2013 Southern Oxidant and Aerosol Study, Atmos. Chem. Phys., 16, 4897–4914, https://doi.org/10.5194/acp-16-4897-2016, 2016.
Riemer, N. and West, M.: Quantifying aerosol mixing state with entropy and diversity measures, Atmos. Chem. Phys., 13, 11423–11439, https://doi.org/10.5194/acp-13-11423-2013, 2013.
Riva, M., Budisulistiorini, S. H., Zhang, Z., Gold, A., and Surratt, J. D.: Chemical characterization of secondary organic aerosol constituents from isoprene ozonolysis in the presence of acidic aerosol, Atmos. Environ., 130, 5–13, 2016.
Robinson, C. B., Schill, G. P., Zarzana, K. J., and Tolbert, M. A.: Impact of organic coating on optical growth of ammonium sulfate particles, Environ. Sci. Technol., 47, 13339–13346, 2013.
Saxena, V. K. and Yu, S. C.: Searching for a regional fingerprint of aerosol radiative forcing in the southeastern US, Geophys. Res. Lett., 25, 2833–2836, 1998.
Schutgens, N. A. J. and Stier, P.: A pathway analysis of global aerosol processes, Atmos. Chem. Phys., 14, 11657–11686, https://doi.org/10.5194/acp-14-11657-2014, 2014.
Sedlacek III, A. J., Lewis, E. R., Kleinman, L., Xu, J., and Zhang, Q.: Determination of and evidence for non-core-shell structure of particles containing black carbon using the Single-Particle Soot Photometer (SP2), Geophys. Res. Lett., 39, L06802, https://doi.org/10.1029/2012GL050905, 2012.
Shen, H., Peters, T. M., Casuccio, G. S., Lersch, T. L., West, R. R., Kumar, A., Kumar, N., and Ault, A. P.: Elevated Concentrations of Lead in Particulate Matter on the Neighborhood-Scale in Delhi, India As Determined by Single Particle Analysis, Environ. Sci. Technol., 50, 4961–4970, 2016.
Sobanska, S., Coeur, C., Maenhaut, W., and Adams, F.: SEM-EDX characterisation of tropospheric aerosols in the Negev desert (Israel), J. Atmos. Chem., 44, 299–322, 2003.
Sobanska, S., Hwang, H., Choel, M., Jung, H.-J., Eom, H.-J., Kim, H., Barbillat, J., and Ro, C.-U.: Investigation of the chemical mixing state of individual Asian dust particles by the combined use of electron probe X-ray microanalysis and Raman microspectrometry, Anal. Chem., 84, 3145–3154, 2012.
Song, C. H., Kim, C. M., Lee, Y. J., Carmichael, G. R., Lee, B. K., and Lee, D. S.: An evaluation of reaction probabilities of sulfate and nitrate precursors onto East Asian dust particles, J. Geophys. Res.-Atmos., 112, D18206, https://doi.org/10.1029/2006JD008092, 2007.
Sullivan, R. C., Guazzotti, S. A., Sodeman, D. A., and Prather, K. A.: Direct observations of the atmospheric processing of Asian mineral dust, Atmos. Chem. Phys., 7, 1213–1236, https://doi.org/10.5194/acp-7-1213-2007, 2007.
Sullivan, R. C., Moore, M. J. K., Petters, M. D., Kreidenweis, S. M., Roberts, G. C., and Prather, K. A.: Effect of chemical mixing state on the hygroscopicity and cloud nucleation properties of calcium mineral dust particles, Atmos. Chem. Phys., 9, 3303–3316, https://doi.org/10.5194/acp-9-3303-2009, 2009.
Tang, M. J., Cziczo, D. J., and Grassian, V. H.: Interactions of Water with Mineral Dust Aerosol: Water Adsorption, Hygroscopicity, Cloud Condensation, and Ice Nucleation, Chem. Rev., 116, 4205–4259, 2016.
Violaki, K. and Mihalopoulos, N.: Water-soluble organic nitrogen (WSON) in size-segregated atmospheric particles over the Eastern Mediterranean, Atmos. Environ., 44, 4339–4345, 2010.
Wagner, F., Bortoli, D., Pereira, S., Costa, M. J., Silva, A. M., Weinzierl, B., Esselborn, M., Petzold, A., Rasp, K., Heinold, B., and Tegen, I. N. A.: Properties of dust aerosol particles transported to Portugal from the Sahara desert, Tellus B, 61, 297–306, 2009.
Washenfelder, R. A., Attwood, A. R., Brock, C. A., Guo, H., Xu, L., Weber, R. J., Ng, N. L., Allen, H. M., Ayres, B. R., Baumann, K., Cohen, R. C., Draper, D. C., Duffey, K. C., Edgerton, E., Fry, J. L., Hu, W. W., Jimenez, J. L., Palm, B. B., Romer, P., Stone, E. A., Wooldridge, P. J., and Brown, S. S.: Biomass burning dominates brown carbon absorption in the rural southeastern United States, Geophys. Res. Lett., 42, 2014GL062444, https://doi.org/10.1002/2014GL062444, 2015.
Weber, R. J., Sullivan, A. P., Peltier, R. E., Russell, A., Yan, B., Zheng, M., de Gouw, J., Warneke, C., Brock, C., Holloway, J. S., Atlas, E. L., and Edgerton, E.: A study of secondary organic aerosol formation in the anthropogenic-influenced southeastern United States, J. Geophys. Res.-Atmos., 112, D13302, https://doi.org/10.1029/2007JD008408, 2007.
Weingartner, E., Burtscher, H., and Baltensperger, U.: Hygroscopic properties of carbon and diesel soot particles, Atmos. Environ., 31, 2311–2327, 1997.
Xiong, F., McAvey, K. M., Pratt, K. A., Groff, C. J., Hostetler, M. A., Lipton, M. A., Starn, T. K., Seeley, J. V., Bertman, S. B., Teng, A. P., Crounse, J. D., Nguyen, T. B., Wennberg, P. O., Misztal, P. K., Goldstein, A. H., Guenther, A. B., Koss, A. R., Olson, K. F., de Gouw, J. A., Baumann, K., Edgerton, E. S., Feiner, P. A., Zhang, L., Miller, D. O., Brune, W. H., and Shepson, P. B.: Observation of isoprene hydroxynitrates in the southeastern United States and implications for the fate of NOx, Atmos. Chem. Phys., 15, 11257–11272, https://doi.org/10.5194/acp-15-11257-2015, 2015.
Xu, J. Z., Zhang, Q., Wang, Z. B., Yu, G. M., Ge, X. L., and Qin, X.: Chemical composition and size distribution of summertime PM2.5 at a high altitude remote location in the northeast of the Qinghai-Xizang (Tibet) Plateau: insights into aerosol sources and processing in free troposphere, Atmos. Chem. Phys., 15, 5069–5081, https://doi.org/10.5194/acp-15-5069-2015, 2015.
Xu, L., Guo, H., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., Knote, C., Olson, K., Koss, A., Goldstein, A. H., Hering, S. V., de Gouw, J., Baumann, K., Lee, S.-H., Nenes, A., Weber, R. J., and Ng, N. L.: Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, P. Natl. Acad. Sci. USA, 112, 37–42, 2015a.
Xu, L., Suresh, S., Guo, H., Weber, R. J., and Ng, N. L.: Aerosol characterization over the southeastern United States using high-resolution aerosol mass spectrometry: spatial and seasonal variation of aerosol composition and sources with a focus on organic nitrates, Atmos. Chem. Phys., 15, 7307–7336, https://doi.org/10.5194/acp-15-7307-2015, 2015b.
Xu, L., Guo, H., Weber, R. J., and Ng, N. L.: Chemical characterization of water-soluble organic aerosol in contrasting rural and urban environments in the southeastern United States, Environ. Sci. Technol., 51, 78–88, 2017.
Zaveri, R. A., Barnard, J. C., Easter, R. C., Riemer, N., and West, M.: Particle-resolved simulation of aerosol size, composition, mixing state, and the associated optical and cloud condensation nuclei activation properties in an evolving urban plume, J. Geophys. Res.-Atmos., 115, D17210, https://doi.org/10.1029/2009JD013616, 2010.
Zhang, H., Yee, L. D., Lee, B. H., Curtis, M. P., Worton, D. R., Isaacman-VanWertz, G., Offenberg, J. H., Lewandowski, M., Kleindienst, T. E., and Beaver, M. R.: Monoterpenes are the largest source of summertime organic aerosol in the southeastern United States, P. Natl. Acad. Sci. USA, 115, 2038–2043, 2018.
Zhang, R., Khalizov, A. F., Pagels, J., Zhang, D., Xue, H., and McMurry, P. H.: Variability in morphology, hygroscopicity, and optical properties of soot aerosols during atmospheric processing, P. Natl. Acad. Sci. USA, 105, 10291–10296, 2008.
Zhang, Y., Chen, Y., Lambe, A. T., Olson, N. E., Lei, Z., Craig, R. L., Zhang, Z., Gold, A., Onasch, T. B., Jayne, J. T., Worsnop, D. R., Gaston, C. J., Thornton, J. A., Vizuete, W., Ault, A. P., and Surratt, J. D.: Effect of Aerosol-Phase State on Secondary Organic Aerosol Formation from the Reactive Uptake of Isoprene-Derived Epoxydiols (IEPOX), Environ. Sci. Tech. Let., 5, 167–174, 2018.