the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 09 Apr 2026
| 09 Apr 2026
Rapid formation of secondary aerosol precursors from the autoxidation of C5–C8 n-aldehydes
Avinash Kumar
Prasenjit Seal
Siddharth Iyer
Long chain aldehydes are common atmospheric constituents, and their gas-phase oxidation form low volatility condensable products leading to secondary organic aerosol. Although the oxidation of n-aldehydes initiated by OH radicals is dominated by aldehydic hydrogen abstraction, the non-aldehydic hydrogen abstractions tend to become competitive with the increase of aldehyde carbon chain length. Here, we experimentally investigated the oxidation of C5–C8 n-aldehydes in variable reaction times (1–13 s) in a flow tube reactor coupled to a nitrate ion time-of-flight chemical ionization mass spectrometer (-ToF-CIMS). Octanal produced highly oxygenated organic molecules (HOMs – low volatility products) with up to 7 O atoms within 1.0 s while the same level of oxygenation was acquired by pentanal within 2.3 s. In long reaction time (11–13 s) experiments, we observed HOMs with progressively more O atoms and higher product yields with the increase of carbon atoms in the precursor aldehydes. Our experiments in the presence of high NO concentrations (2 ppb to 1 ppm) showed the formation of prominent highly oxygenated organonitrates along with the suppression of HOM accretion products. However, some enhancement in the monomeric HOMs even with 6 O atoms were seen under variable NO conditions. Results from hydrogen to deuterium () exchange experiments showed that the studied n-aldehydes undergo similar autoxidation mechanisms, but the reactivity and HOM formation potential increase with increasing carbon chain length.
- Article
(1276 KB) - Full-text XML
-
Supplement
(1538 KB) - BibTeX
- EndNote




Secondary organic aerosol (SOA) refers to the aerosol material that is mainly formed by the atmospheric gas-phase oxidation of volatile organic compounds (VOCs) (Kroll and Seinfeld, 2008; Ziemann and Atkinson, 2012; Seinfeld and Pandis, 2016). Atmospheric oxidation of VOCs increases the oxygen to carbon ratios (O:C) in the oxidation products and can form highly oxygenated organic molecules (HOMs). These low volatility products are found to play a key role in the formation and growth of SOA (Ehn et al., 2014; Öström et al., 2017; Bianchi et al., 2019; Brean et al., 2019, 2020). However, heterogeneous and multiphase chemistry involving the reactions of organic compounds directly onto solid particles or inside liquid particles can also be an important contributor to SOA mass (Ervens et al., 2011, 2014; Kuang et al., 2020; Gu et al., 2023). The SOA is a dominant component of tropospheric fine particulate matter (Hallquist et al., 2009; Spracklen et al., 2011; Huang et al., 2014), influences oxidative capacity, local and global air quality, climate change, and human health (Jacobson et al., 2000; Hansen and Sato, 2001; Kanakidou et al., 2005; Zhang et al., 2014). Despite having significant attention due to its importance on SOA, its sources and formation processes are yet to be fully understood.
Aldehydes are common emissions in natural and polluted environments and have both biogenic and anthropogenic sources (Lipari et al., 1984; Carlier et al., 1986; Ciccioli et al., 1993; Schauer et al., 1999a, b, 2001) and are also formed by chemical transformation of other VOCs, especially ozonolysis of alkenes (Calogirou et al., 1999). Atmospheric degradation of aldehydes is mainly governed by photolysis and the reaction with OH radicals in the daytime (Mellouki et al., 2003; Calvert et al., 2011; Mellouki et al., 2015). During nighttime, the reactions with NO3 radicals are the dominant sink of aldehydes (Calvert et al., 2011). Although there are prior kinetic studies of reactions of n-aldehydes with OH radicals available in the literature (Albaladejo et al., 2002; Cassanelli et al., 2005; Iuga et al., 2010; Castañeda et al., 2012; Wang et al., 2015; Aguirre et al., 2025), they are limited to the initial steps of oxidation except recent studies (Barua et al., 2023; Yang et al., 2024) showing further oxidation steps leading to the formation of more functionalized products including HOMs.
It is well understood that the reaction of aldehydes with OH radicals is predominantly initiated by the abstraction of aldehydic hydrogen atom due to its weaker bond strength. However, with the increase of carbon chain length, the abstraction of other hydrogen atoms distant from the aldehydic moiety can also contribute to the overall oxidation process. The aldehydic H abstraction can lead to the cleavage of that carbon (C1) by CO loss from acyl (RC(O)) intermediate (Rissanen et al., 2014; Barua et al., 2023). Alternatively, it leads to the acyl peroxy (RC(O)OO) and, in high NOx (NO + NO2) condition, subsequently to acyloxy (RC(O)O) intermediate followed by CO2 loss ultimately forming Cn−1 aldehyde, Cn−1 alkyl nitrate, and Cn−1 alkoxy isomerization products (Vereecken and Peeters, 2009; Chacon-Madrid et al., 2010). Besides, it can also produce Cn peroxyacyl nitrates (PAN) via RC(O)OO + NO2 reaction (Mellouki et al., 2003; Chacon-Madrid et al., 2010; Calvert et al., 2011; Mellouki et al., 2015), a reservoir species for long-range transport of NOx in the free troposphere. Additionally, Cn peroxy acids can be formed by the reaction of RC(O)OO intermediate and HO2 (Barua et al., 2023).
Chacon-Madrid et al. (2010) have conducted a comparative study of the SOA yields in OH-initiated oxidation of n-aldehydes and n-alkanes under high NOx conditions. They reported near identical SOA yields from n-tridecanal and n-dodecane where the precursor alkane is having one less carbon than the precursor aldehyde. The finding was attributed to the formation of C12 alkoxy radical intermediate from both precursors undergoing similar subsequent reactions leading to SOA. This indicates that the dominant fate of aldehyde oxidation by OH leads to the fragmentation of its carbon backbone losing one carbon atom rather than producing functionalized products with the same number of carbon atoms as the parent molecule.
In low NOx conditions, previous experimental studies have shown that the abstraction of the aldehydic hydrogen can also lead to molecular functionalization forming HOMs (Ehn et al., 2014; Rissanen et al., 2014; Tröstl et al., 2016; Wang et al., 2021) and potentially promote the SOA yields. Recently, Barua et al. (2023) have studied hexanal OH oxidation reaction in detail using high level quantum chemical computations as well as experimental mass spectrometry technique. They showed that both the aldehydic and non-aldehydic H abstraction pathways can contribute to the functionalization of hexanal resulting in rapid formation of HOMs via autoxidation. Autoxidation refers to chain radical processes, generally starting with an oxygen-centered radical that undergoes unimolecular isomerization reaction leading to a carbon-centered radical species whose dominant fate is to add additional molecular oxygen and thus increases product O:C ratios (Crounse et al., 2013; Jokinen et al., 2014; Rissanen et al., 2014; Berndt et al., 2015, 2016; Mentel et al., 2015; Rissanen et al., 2015). Along the aldehydic H abstraction reaction route, the fastest isomerization (1,6 H-shift rate coefficient, k = 0.2 s−1) of the RC(O)OO intermediate was shown to be the key for autoxidation reaction chain propagation and competitive with any bimolecular reaction mediated RC(O)O fragmentation (Barua et al., 2023). Thus, the RC(O)OO isomerization reaction (Seal et al., 2023) keeps the carbon backbone of the precursor aldehyde intact. A non-aldehydic H abstraction from C4 was also seen to be competitive and its corresponding RO2 was shown to undergo a 1,6 H-shift reaction with the aldehydic H atom at a higher rate (k = 0.9 s−1) than the H-shifts in RC(O)OO radical. The fast aldehydic H-shift to the peroxy group is consistent with other carbonyl systems reported previously (Da Silva, 2011; Crounse et al., 2012; Møller et al., 2016, 2019). Moreover, the kinetic modelling simulation on OH-initiated oxidation of hexanal conducted by Barua et al. (2023) showed that a detectable concentration (1.3 × 104 ) of O7 HOMs were produced even in the presence of 1 ppb NO. Because the non-aldehydic H abstraction pathways are likely less prone to fragmentation and promote functionalization, the effect of carbon chain length of n-aldehydes on HOM yields is of great interest.
In this work, we experimentally studied the OH initiated autoxidation of C5–C8 n-aldehydes in variable reaction times under atmospheric pressure and room temperature using state-of-the-art mass spectrometry technique. In addition, the reactions were studied in the presence of variable concentrations of NO to examine the effect of NO on the oxidation process. The study shows how the length of carbon chain in linear aldehydes directly affects the reactivity and functionalization of the molecules during their oxidation initiated by OH radicals.
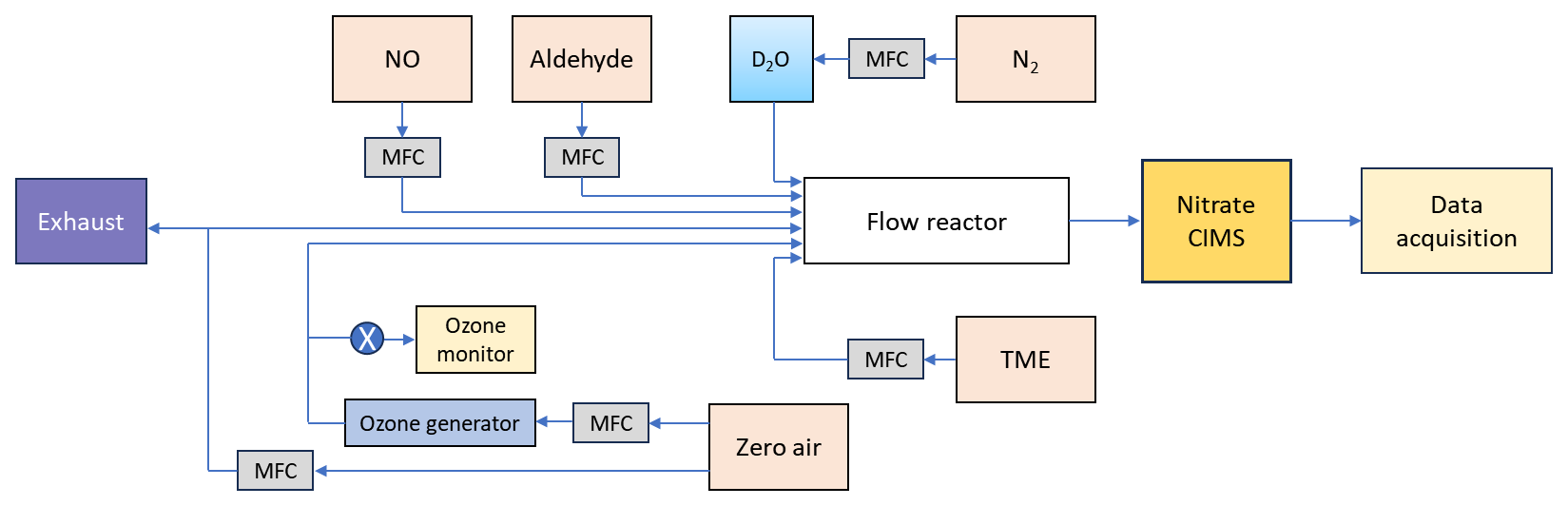
Figure 1Schematic representation of a flow reactor setup showing a nitrate () based chemical ionization mass spectrometer coupled to ambient pressure flow reactor. TME = tetramethylethylene (C6H12). The oxidant OH radical was produced in situ by TME + O3 reaction. MFC = mass flow controller.
2.1 Experimental setup
The gas-phase oxidation reactions of n-aldehydes with OH radicals were conducted in a flow reactor setup in the laboratory as shown in Fig. 1. All the experiments were conducted at room temperature and 1 atm pressure of air. The precursor aldehydes were introduced into the reactor from their individual gas cylinders. The oxidant OH radicals were produced in situ by the reaction of tetramethylethylene (TME, C6H12) with ozone. We used an ozone generator that photolyzes zero-air by a mercury lamp (UVP, Analytik Jena) to provide ozone to the reactor while TME was supplied from a gas cylinder. The zero-air was produced by feeding compressed clean air to a zero-air generator (AADCO-737-15) which was also used as bath gas in the reactor maintaining a total sample flow of 8–10 slpm. The initial concentrations of reactant precursors were determined by controlling the individual gas flows using calibrated mass flow controllers (Alicat Scientific). An ozone analyzer (2B Technologies model 205) was used to measure the ozone concentrations. Further details regarding reactant concentration measurements, as well as specifications for the chemicals and gas cylinders, are provided in Sects. S1 and S2 in the Supplement.
Table 1The experimental conditions for OH initiated oxidation of studied C5–C8 n-aldehydes.
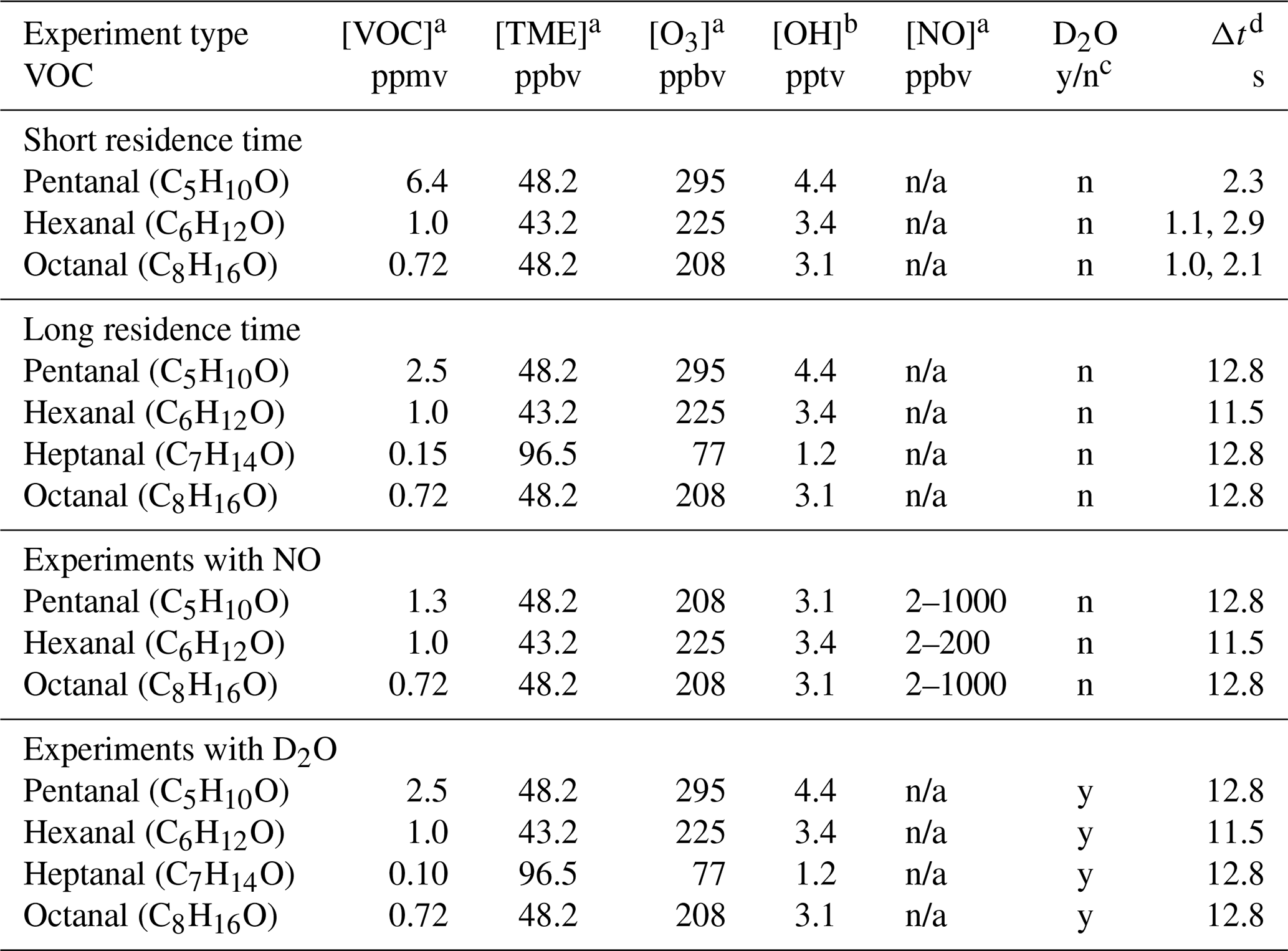
a Initial reactant concentrations. b The initial OH concentrations were calculated using bimolecular rate coefficients = 1.5 × 10−15 , kOH−TME = 1.0 × 10−10 (Manion et al., 2015), and the expression . n/a = not applicable. c D2O added = y, not added = n. d Reaction time (Δt).
We studied the reactions over a range of reaction times (short: 1–3 s, and long: 11–13 s). The experimental conditions are presented in Table 1. Long reaction time experiments were conducted using a borosilicate flow reactor (length: 100 cm, and i.d.: 4.7 cm) while a quartz flow reactor (length: 100 cm, and i.d.: 2.2 cm) was used for the short reaction time experiments. We utilized the full volume of the reactor to achieve a long reaction time. However, the short reaction times were achieved by controlling the distance between the mass spectrometer orifice and the position where the precursor aldehyde meets the oxidant OH inside the reactor. This was done by using a movable injector that brings the aldehyde of interest at variable positions inside the reactor. The shortest possible reaction time of an individual aldehyde was chosen by the detection of any HOMs from its oxidation initiated by OH radicals. In short reaction time experiments, the highest concentration of VOC (6.4 ppmv) was used for pentanal while the concentrations of other aldehydes were up to 1 ppmv. The VOC concentrations of 0.2–2.5 ppmv were used in long reaction time experiments. The other reactants including 43–97 ppbv of TME, and 77–295 ppbv of ozone were maintained nearly constant with respect to individual VOC in all experiment types (see Table 1). Among all the studied n-aldehyde systems, the lowest level of aldehyde and O3 concentrations were maintained for heptanal oxidation experiment. This is because higher concentrations led to irrelevant products likely originating from the ozonolysis of heptanal stabilizers (see Fig. S2). To observe the effect of NOx on the OH induced oxidation of n-aldehydes, we conducted the experiments with variable NO concentrations (2–1000 ppbv). Additionally, one more set of experiments, hydrogen to deuterium () exchange, were conducted by the addition of D2O flow to the n-aldehyde OH oxidation reaction. These experiments give an estimate of the number of functional groups with labile H atoms (e.g., OH, OOH, and C(O)OOH) in the oxidation products. A near complete conversion was confirmed by monitoring the reagent ion signals of and fully converting to and , respectively (see Fig. S14). The time series of reactive species, VOC, OH, RO2, O3, and NO under different reaction conditions are shown in Figs. S16 and S17.
The oxidation products were detected using a nitrate ion time-of-flight chemical ionization mass spectrometer (-ToF-CIMS) as their adducts. A zero-air sheath flow of 20 slpm was provided to the chemical ionization inlet. The ions were produced from gas-phase HNO3 flow under soft X-ray exposure while being carried by N2 to the inlet. The mass spectrometric data processing, including averaging, baseline removal, mass axis calibration, and peak integration were done using the tofTools v6.03 package for MATLAB.
2.2 Kinetic simulation
We estimated the concentrations of reactive species in the flow reactor during different n-aldehyde oxidation experiments using a kinetic simulator Kinetiscope (version 1.1.1136.x64) (Hinsberg and Houle, 2022). These include the average concentrations of OH radicals and initial RO2 radicals (i.e., CnH2n−1O3, the first peroxy radicals formed in the oxidation process) produced in the experiments. In the simulations, a single-reactor model with constant volume, pressure, and temperature was employed. The temperature was set to 298.15 K. The simulation setting parameters such as total number of particles (1 × 109) and random number seed (12947) were kept constant for consistency, while the maximum simulation time was matched to that of individual experiments. Details of all the simulations are provided in Sect. S13.
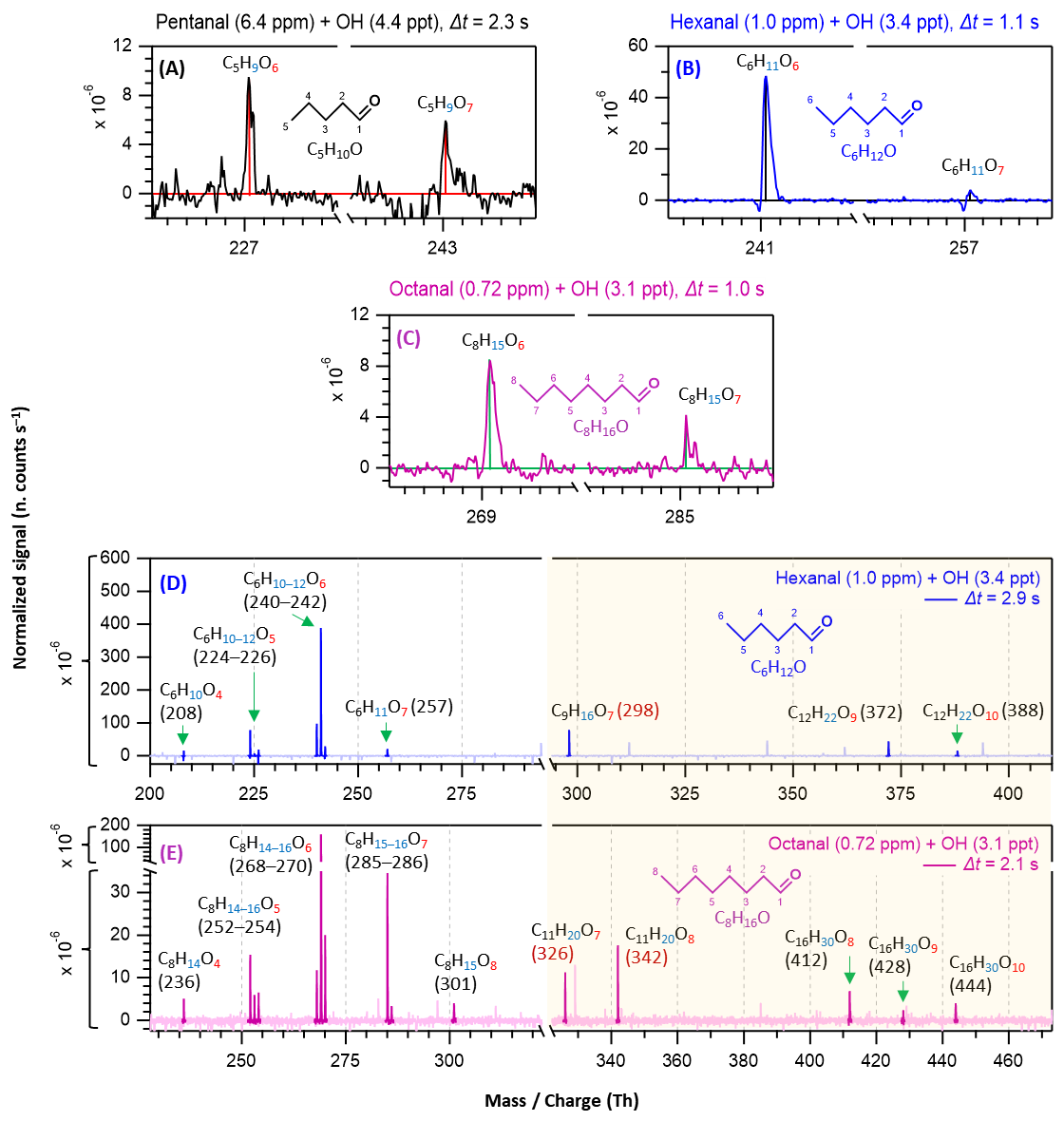
Figure 2Nitrate chemical ionization mass spectra of OH initiated oxidation of n-aldehydes showing the formation of HOMs in different reaction times (Δt): 2.3 s – pentanal in black (a), 1.1 and 2.9 s – hexanal in blue (b, d), and 1.0 and 2.1 s – octanal in purple (c, e). The product peaks are labelled with the exclusion of ion attachment in their compositions. The backgrounds of TME ozonolysis (TME + O3) and aldehyde have been subtracted from all spectra, resulting in several negative peaks in panels (a–e). The accretion product region is highlighted in light gold background. The accretion products labeled with nominal mass/charge in dark red () are related to the TME-derived peroxy radical C3H5O3.
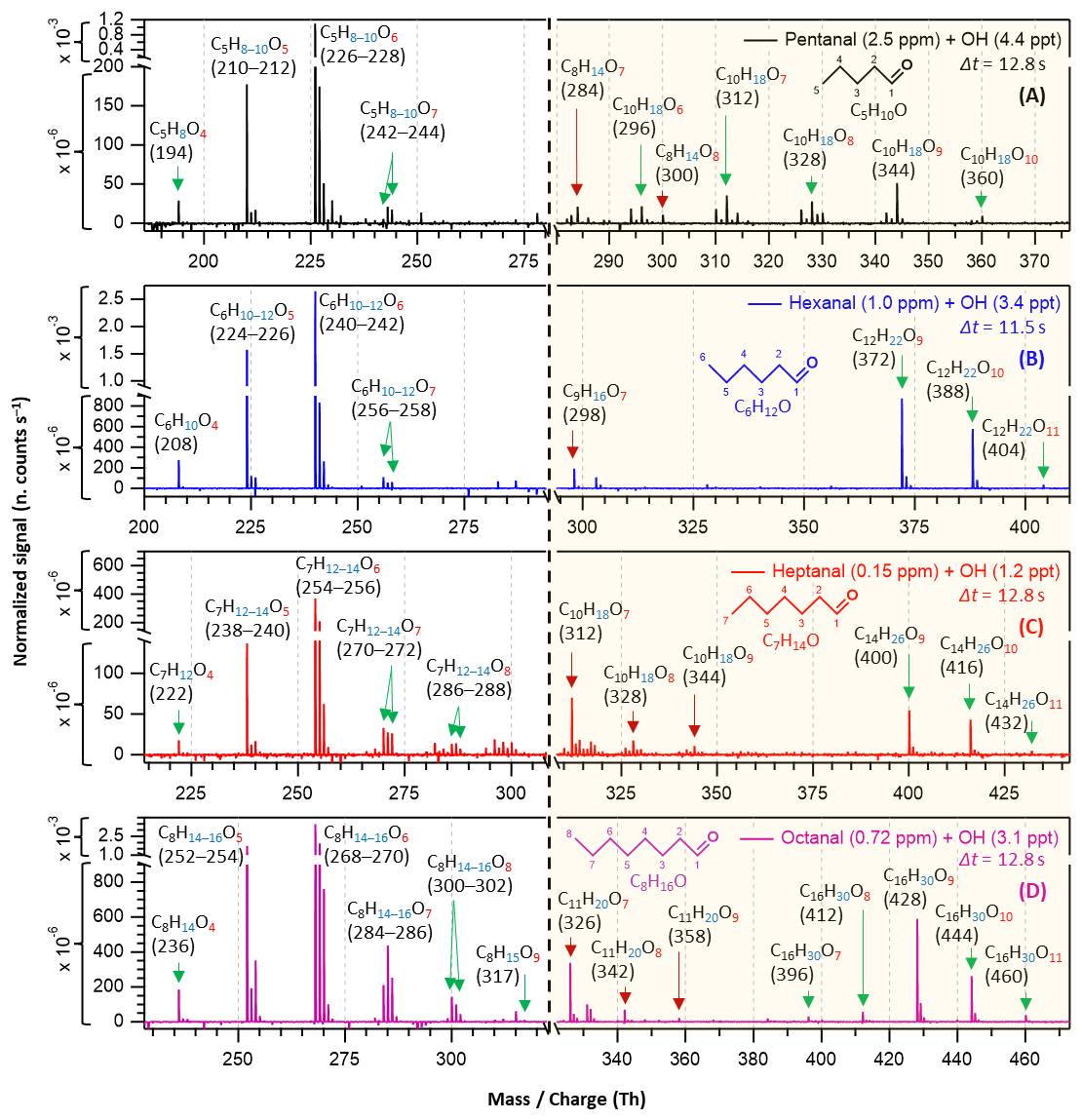
Figure 3Nitrate chemical ionization mass spectra of OH initiated oxidation of n-aldehydes in 11–13 s residence times: pentanal in black (a), hexanal in blue (b), heptanal in red (c), and octanal in purple (d). The product peaks are labelled with the exclusion of ion attachment in their compositions. The backgrounds of TME ozonolysis (TME + O3) and aldehyde have been subtracted from all spectra, resulting in several negative peaks in panels (a–d). The accretion product region is highlighted in light gold background. The accretion products marked by dark red arrows () are related to the TME-derived peroxy radical C3H5O3.
3.1 Detection of HOM in short to long reaction time experiments
In this section, we discuss how early HOMs formed and how they evolved with the progress of reaction time in different n-aldehyde OH oxidation experiments. The hexanal OH oxidation spectra included in Figs. 2 and 3 are reproduced from our previous study (Barua et al., 2023) for comparison with the other aldehydes. Figure 2 shows the results from short reaction time (Δt = 1–3 s) experiments. We observed the formation of O6 and O7 HOMs within 1.0, 1.1, and 2.3 s reaction times in the oxidation experiments of octanal, hexanal, and pentanal, respectively. It is essential to mention that with the increase of number of carbon atoms in the studied aldehydes, the required precursor concentrations for first HOM observation decreased (from 6.4 ppm pentanal, 1 ppm hexanal to 0.72 ppm octanal; corresponding reacted concentrations 1.15, 0.34, and 0.30 ppb, respectively). This shows a clear effect of carbon chain length on the reactivity of linear aldehydes towards HOM formation. Because the oxidation process of n-aldehydes (CnH2nO) with OH is initiated by the abstraction of a H atom (aldehydic or non-aldehydic), the first formed acyl (or alkyl) peroxy radical CnH2n−1O3 contains an odd number of oxygen atoms. If autoxidation outcompetes any other bimolecular reactions (e.g., RO2 + RO2, RO2 + HO2, etc.), the product spectrum will be mostly dominated by odd number of oxygen containing products. In all studied n-aldehyde systems, the intensity of O6 HOM is higher than that of O7 HOM (Figs. 2 and 3). The formation of CnH2n−1O6 peroxy radical indicates that the process certainly involves a bimolecular reaction step. In hexanal OH oxidation, Barua et al. (2023) computationally showed that the formation of C6H11O5 peroxy radical via autoxidation is very fast while the subsequent isomerization reaction leading to C6H11O7 is slower. The same is likely to hold true for other aldehydes and it is expected that the CnH2n−1O5 peroxy radical undergoes a bimolecular reaction converting it to CnH2n−1O4 alkoxy radical, followed by a H-shift, and subsequent O2 addition reactions producing the dominant CnH2n−1O6 HOM (see Figs. S7–S11). As the reaction time increased, we observed the formation of monomeric HOM up to O7 and accretion products up to O10 composition within 2.9 s in hexanal oxidation (see Fig. 2d) with the consumption of ∼ 8.88 × 10−4 of its initial concentration. In the case of octanal, monomeric O8 HOM and accretion products up to O10 formed within 2.1 s reaction time (see Fig. 2e) with a comparable reacted fraction (∼ 8.71 × 10−4) of its initial concentration.
In long reaction time (11–13 s) experiments, we observed higher intensities of product signals (see Fig. 3) in comparison to their intensities in the short reaction time experiments – as expected. For pentanal, Fig. 3a shows that HOM accretion products up to O10 formed within 12.8 s reaction time which were not seen in the short reaction time experiment (2.3 s). A lower precursor concentration, 2.5 ppm of pentanal in long reaction time experiment compared to earlier 6.4 ppm in short reaction time, was sufficient to produce the observed HOMs in this case. A close observation of C6–C8 n-aldehyde oxidation spectra (Fig. 3b–d) reveals that HOM accretion products up to O11 formed within 11–13 s reaction time under the experimental conditions. In all n-aldehyde experiments, we also observed the accretion products resulting from different combinations of aldehyde-derived peroxy radicals and TME-derived peroxy radical C3H5O3 (see Fig. S13 for details) which are marked with dark red arrows. Figure 3d implies that the highest oxygenation (C8H15O9) in the monomeric HOM products is associated with octanal, whereas Fig. 3c shows that the most oxygenated products produced from heptanal are C7H12−14O8. In the case of pentanal and hexanal, monomeric HOMs are limited to seven oxygen atoms (see Fig. 3a and b). All in all, we notice a near identical distribution of oxidation products in the experiments with all C5–C8 n-aldehydes. However, the tendency of oxidation gets faster and advances to higher oxygenated products when the carbon chain length increases.
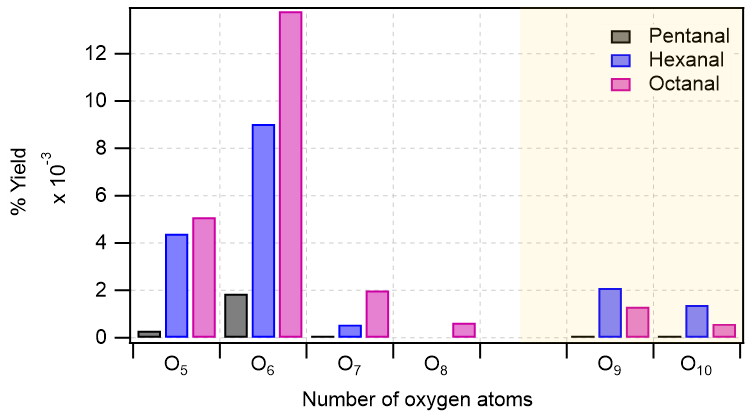
Figure 4Distribution of major oxidation products (O5–O8 in monomeric regime with white background, and O9–O10 in accretion product regime with light gold background) in pentanal, hexanal, and octanal oxidation initiated by OH radical. In y-axis, the numbers are the cumulative sum of yields of products with the same oxygen number. Reaction time, Δt = 11–13 s.
The bar plot (see Fig. 4) compares the yields of major oxidation products from different n-aldehydes. It clearly shows that the yields of higher oxygenated products increase as we move from pentanal to octanal. In the accretion product regime, the yields of O9–O10 products in octanal are lower than that of hexanal which is also reflected in their dimer to monomer ratios with octanal being 8.8 × 10−2 and hexanal being 2.5 × 10−1. The lower ratio for octanal compared to hexanal is observed despite both precursors producing comparable quantities of initial RO2 radicals (9.12 × 109 and 9.37 × 109 from octanal and hexanal, respectively; see Table S1 in the Supplement). This can lie in the variation of RO2 + RO2 reaction rate coefficients forming the accretion products (RO2 + RO2 → ROOR + O2) which is highly dependent on specific RO2 structures (Shallcross et al., 2005; Berndt et al., 2018).
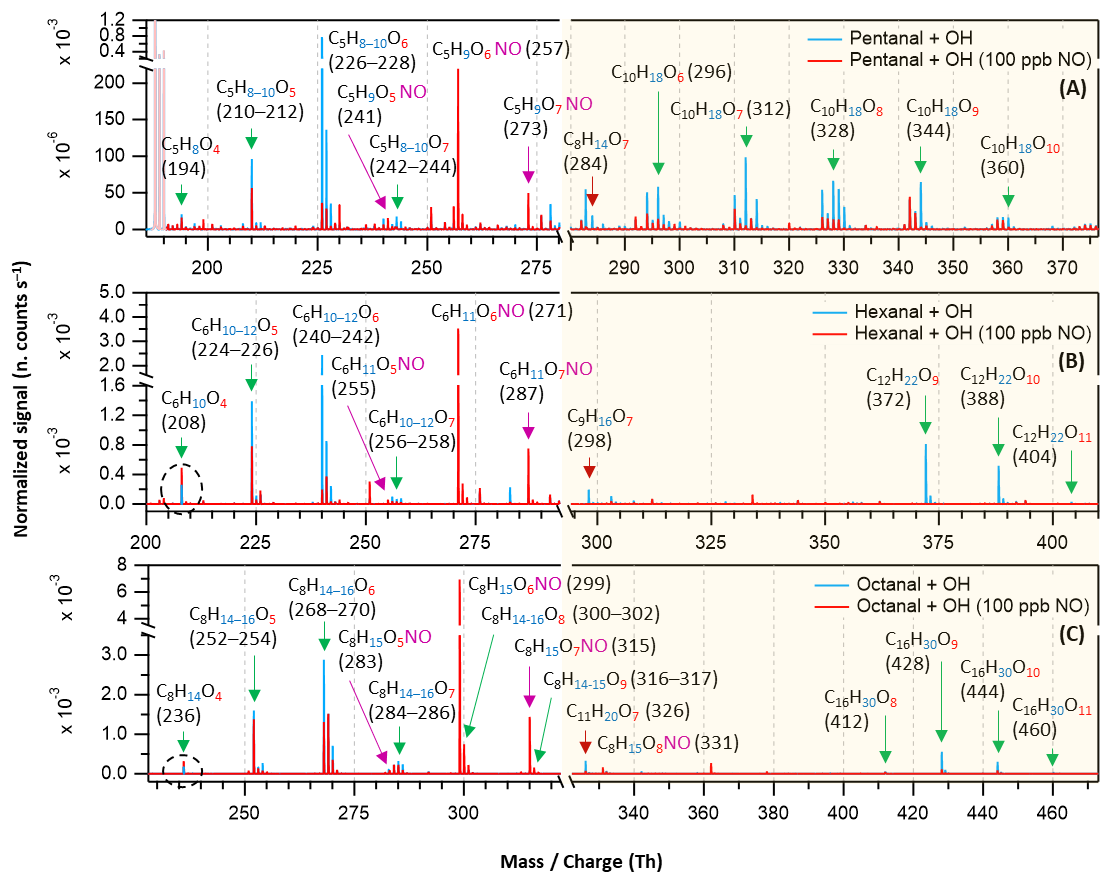
Figure 5Overlaid nitrate chemical ionization mass spectra of OH initiated oxidation of n-aldehydes without (in blue) and with the presence of 100 ppb of initial NO (in red): pentanal (a), hexanal (b), and octanal (c). The product peaks are labelled with the exclusion of ion attachment in their compositions. The accretion product region is highlighted in light gold background. The accretion products marked by dark red arrows () are related to the TME-derived peroxy radical C3H5O3. The organonitrates (in the presence of NO) are labelled with the extension NO and marked with purple arrows. Reaction time, Δt = 11–13 s.
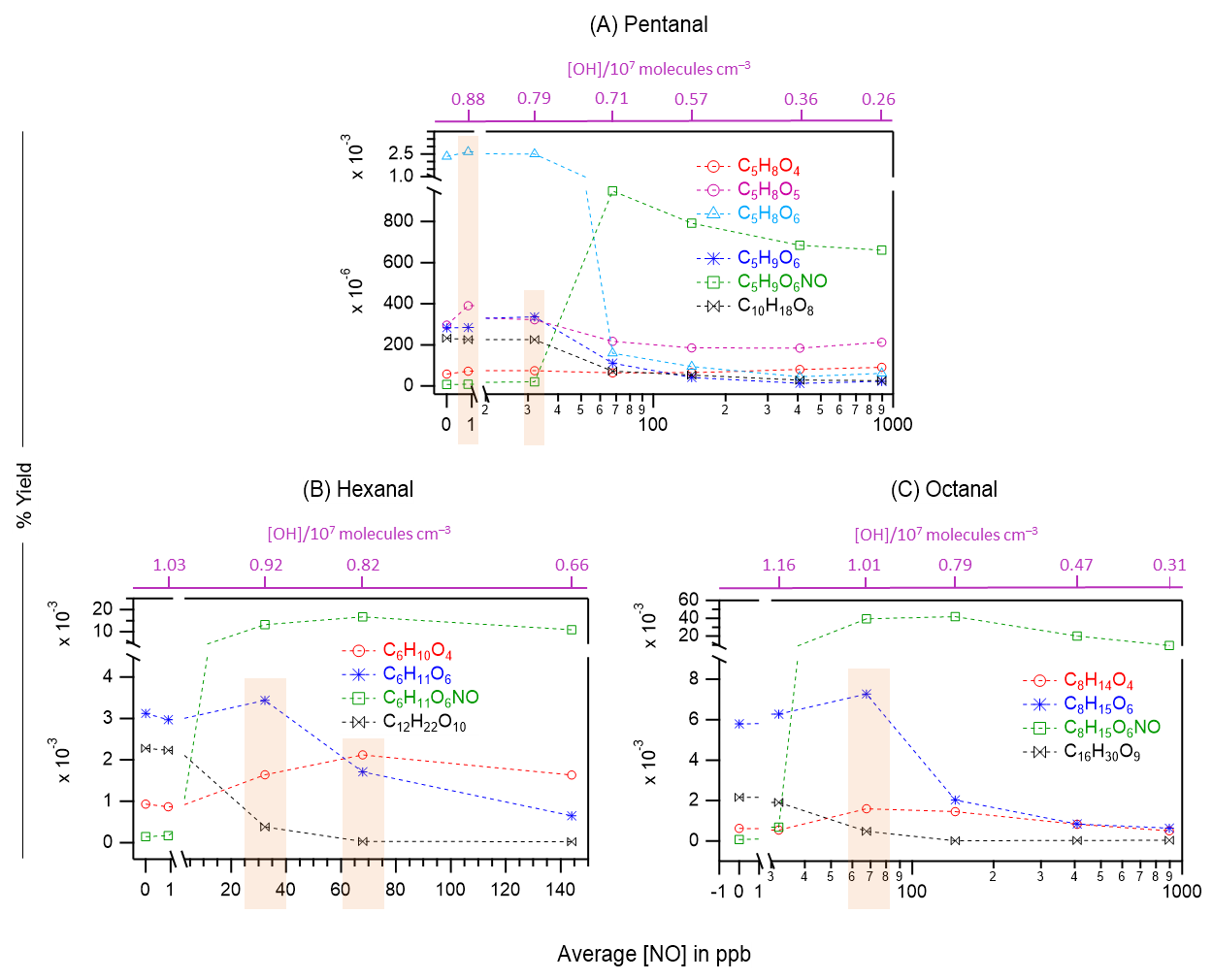
Figure 6The yields of different oxidation products including monomeric HOMs, organonitrates (green markers), and HOM accretion products (black markers) as a function of average NO concentrations in OH initiated oxidation of n-aldehydes: pentanal (a), hexanal (b), and octanal (c). The corresponding average OH radical concentrations in these experiments are shown with purple scales. Note the logarithmic scale (x-axis) in panels (a) and (c). The orange rectangles highlight the enhanced yields of several non-nitrogen containing products: O4–O6 closed-shell products from pentanal under around 1 ppb NO (a), O4 closed-shell products from hexanal and octanal under around 70 ppb NO (b, c), O6 peroxy radical from pentanal and hexanal under around 30 ppb NO (a, b) and the same from octanal under 70 ppb NO (c). Reaction time, Δt = 11–13 s.
3.2 Experiments in the presence of NO
It has been widely acknowledged that the formation of HOM is suppressed in high NOx conditions (Wildt et al., 2014; Praske et al., 2018; McFiggans et al., 2019; Pullinen et al., 2020), thus reducing the SOA yields. In this process, the reduction in SOA yield is largely attributed to the suppression of highly condensable HOM accretion products (RO2 + RO2 → ROOR) (Kirkby et al., 2016; Pullinen et al., 2020). However, other studies have shown that NO can also enhance HOM formation by producing reactive RO radicals (RO2 + NO → RO + NO2) that can propagate autoxidation (Rissanen, 2018; Yan et al., 2020; Wang et al., 2021; Shen et al., 2022; Nie et al., 2023; Barua et al., 2025; Kang et al., 2025). In our different n-aldehyde oxidation experiments in the presence of variable concentrations of NO, we observed the general tendency of dropping of the accretion product signals (C2nH4n−2Oz) and their corresponding yields as expected (see Figs. 5 and 6). Figure 5 represents the mass spectra recorded in the experiments without (in blue) and with the presence of 100 ppb initial NO (in red). At this condition, most of the monomeric products are quenched at different extents alongside the formation of organonitrates while the accretion products are quenched nearly completely. Interestingly, we observed some enhancement in the intensities of O4 products in the case of hexanal and octanal (Fig. 5b and c) under 100 ppb NO. A closer look at Fig. 6a reveals that the yields of closed-shell products C5H8O4−6 somewhat increased under around 1 ppb average NO condition which then started to decrease under higher NO of around 30 ppb and above in pentanal oxidation. In both pentanal and hexanal oxidations, the dominant O6 peroxy radicals (C5H9O6 and C6H11O6, respectively, blue markers) gained higher yields under around 30 ppb of NO (Fig. 6a and b) compared to without NO condition.
On the other hand, in octanal oxidation, the yield of dominant O6 peroxy radical (C8H15O6) increased even under 70 ppb of NO (see Fig. 6c). These observations indicate that the suppressing effect of NO on the yields of HOMs in n-aldehyde oxidation is perturbed with the increase of carbon chain length of the precursor aldehyde. Also, in all the studied n-aldehydes, although highly oxygenated products are suppressed under higher NO concentrations, we see some enhancement in the early oxygenated closed shell products (O4–O5, and even O6 product in pentanal) under relatively lower NO concentrations. It should be noted that the formation of organonitrates with chemical composition CnH2n−1OzNO strongly supports our assignment of reactive peroxy radical intermediates CnH2n−1Oz. Additional insights into the abundance of HOMs under varying NO concentrations are discussed in Sect. S12.
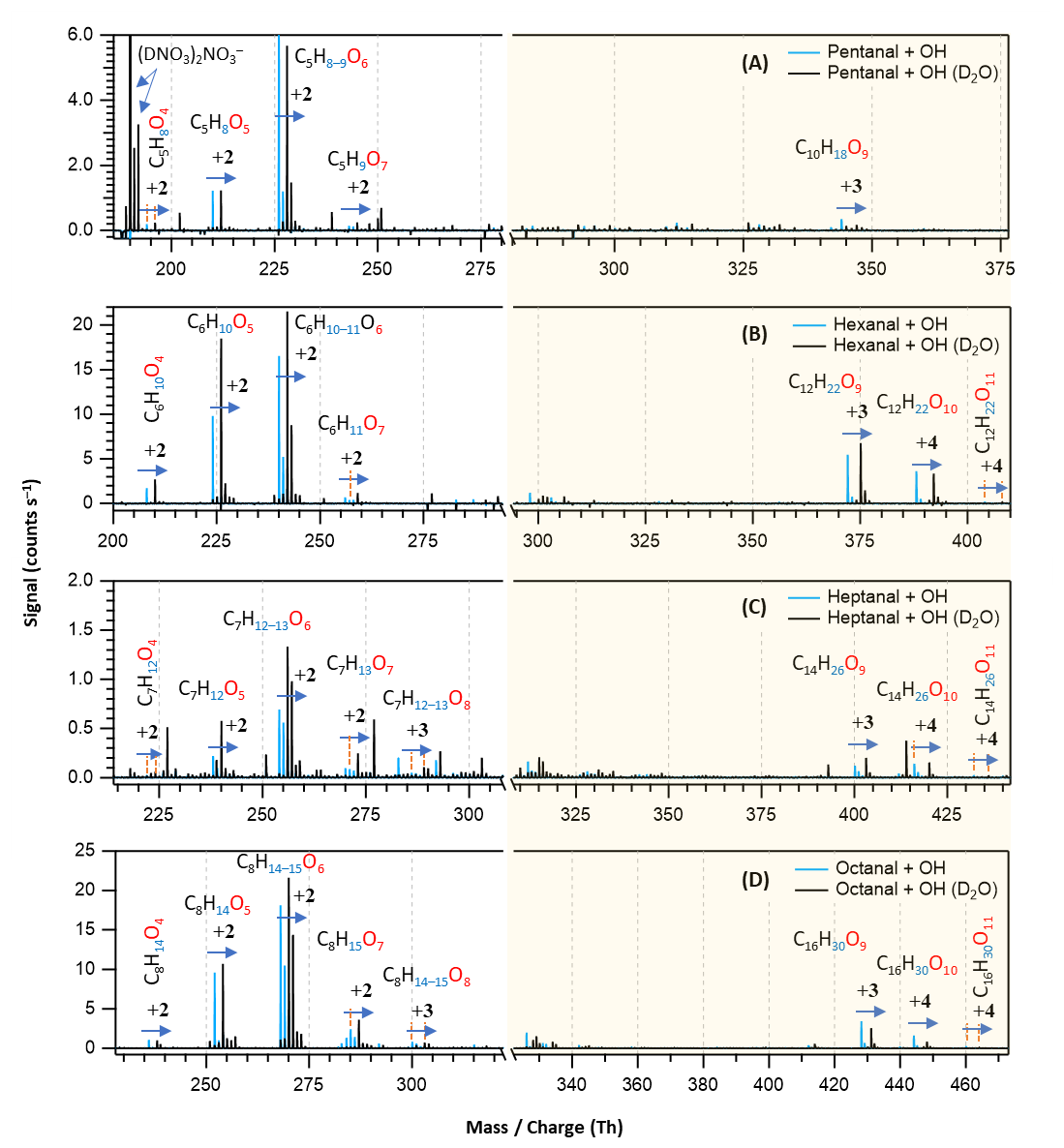
Figure 7Overlaid nitrate chemical ionization mass spectra of OH initiated oxidation of n-aldehydes without (in blue) and with the presence of D2O (in black): pentanal (a), hexanal (b), heptanal (c), and octanal (d). In panel (a), the label is assigned to the deuterated nitric acid trimer reagent signal. The product peaks are labelled with the exclusion of ion attachment in their compositions and the numbers on the blue arrows indicate the individual counts of mass shift during exchange. The backgrounds of TME ozonolysis (TME + O3) and aldehyde have been subtracted from the spectra, resulting in several negative peaks in panels (a) and (b). The accretion product region is highlighted in light gold background.
3.3 D2O experiments
With the addition of D2O in the OH initiated oxidation experiments of n-aldehydes, we observed a shift in individual product signals in the mass spectra equivalent to the number of exchangeable H atoms in the product structures (see Fig. 7). This provides additional insight into the product identities in terms of the total number of OH, OOH, and (or) C(O)OOH groups present in their molecular structures. Figure 7 shows that the oxidation products with same number of oxygen atoms in all the studied n-aldehydes undergo identical mass shifts ( exchange) in the presence of D2O. This observation indicates that the autoxidation mechanism derived by Barua et al. (2023) for hexanal OH oxidation (see Fig. S5) is directly applicable to the other linear aldehydes studied here and thus produces similar product structures. The original mechanism is extended to HOMs up to nine oxygen atoms and presented in Fig. S6. The likely formation process of HOM accretion products () is shown in Fig. S12. The proposed structures of O5 closed-shell products, O6–O8 monomeric HOMs as well as O9−11 HOM accretion products (see Figs. S6–S12) agree with the exchange experiments in terms of their 2–4 units of mass shifts in the respective n-aldehyde mass spectra (see Fig. 7).
3.4 Atmospheric implications
Ambient concentration of individual longer chain aldehyde (≥ C5) can vary from sub-ppb to several ppb depending on time and location (Williams et al., 1996; Duan et al., 2008; Li et al., 2018; Ma et al., 2019). A total concentration of C6–C10 n-aldehydes in Monti Cimini Forest in Italy was measured to be 8.8 ppb (Ciccioli et al., 1993). In indoor air, the concentration can be significantly higher, even around 50 ppb (Birmili et al., 2022). Atmospheric lifetime of n-aldehydes due to their reactivity with OH radicals is generally less than 10 h (Albaladejo et al., 2002; Aguirre et al., 2025). Because of their significant photochemical ozone formation potential (Jenkin et al., 2017; Aguirre et al., 2025), they are good candidates for generating photochemical smog in NOx rich polluted urban atmosphere. On the other hand, previous studies have shown that atmospheric oxidation products of longer chain n-aldehydes are direct contributors to the formation of SOA (Chacon-Madrid et al., 2010; Fan et al., 2024). Here, we demonstrated that the studied C5–C8 n-aldehydes can rapidly form HOM via autoxidation initiated by OH radicals, and the length of carbon chain controls the efficiency of the process. Therefore, with the increase of carbon chain length in n-aldehydes, the fast formation of HOMs is expected to take part in the early stages of gas-to-particle formation and growth contributing to atmospheric SOA. Our experiments in the presence of NO showed a general decreasing trend of HOM accretion products with increasing NO but also formed the corresponding highly oxygenated organic nitrates (HOM-ONs). In our study with the n-aldehydes under 1–70 ppb NO conditions, some yield enhancement with oxidation products up to 6 O atoms was also seen. Pullinen et al. (2020) showed that both HOMs and HOM-ONs originated from the same peroxy radicals with more than six O atoms condensed on particles by about 50 % and those with more than eight O atoms condensed by about 100 % to form SOA in monoterpene photooxidation. Moreover, other reports show that HOM-ONs originated from different VOCs can contribute to low volatility products (Barua et al., 2025) and thereby particle growth and aerosol mass loading (Fry et al., 2014; Lee et al., 2016; Huang et al., 2019).
This study represents the significance of longer chain linear aldehydes (≥ C5), key components of atmospherically abundant oxygenated volatile organic compounds (OVOCs), in rapid formation of HOMs upon atmospheric oxidation and their potential contribution to atmospheric SOA. Among the studied C5–C8 n-aldehydes, the fastest HOM formation is associated with octanal forming O6 and O7 HOMs within 1.0 s reaction time with low precursor loading. Pentanal and hexanal formed HOMs with the same number of oxygen atoms as early as 2.3 and 1.1 s, respectively but with higher precursor loadings (i.e., 6.4 ppm pentanal and 1.0 ppm hexanal compared to 0.72 ppm octanal). The highest oxygenated monomeric HOM with 9 oxygen atoms was formed from octanal whereas the numbers are up to 8 oxygen atoms for heptanal and 7 oxygen atoms for both pentanal and hexanal oxidation initiated by OH radicals within 13 s reaction time. Although the highest precursor concentration (6.4 ppm) was required for the first observation of detectable HOM in short reaction time experiment with pentanal, a lower concentration (2.5 ppm) was used to obtain its observed mass spectrum in the long reaction experiment. The HOM accretion products with up to 11 oxygen atoms were observed in C6–C8 n-aldehyde oxidation experiments while they were limited to maximum 10 oxygen atoms in pentanal case. We also observed the trend of increased oxidation product yields with the increase of carbon chain length. In all studied systems, the dominant product signals are O6 HOMs with the O5 HOMs being the second dominant ones. Previous mechanistic understanding (Barua et al., 2023) as well as current experimental observations reveal that autoxidation process forming O5 RO2 in n-aldehydes is very fast while the subsequent unimolecular rearrangements of the RO2 intermediates are in competition with bimolecular reactions including other RO2, HO2, and NOx. The experiments in the presence of high NO concentrations (30 ppb and above) produced the highest yields of HOM-ONs, compared to neighboring non-nitrogen HOMs, with the expense of HOM accretion products. However, some enhancements with the yields of low oxygenated closed-shell products and O6 peroxy radicals were also seen under 1–70 ppb NO conditions. The results of hydrogen to deuterium () exchange experiments with identical mass shifts in the oxidation products of all studied n-aldehydes imply that the autoxidation mechanism established for hexanal (Barua et al., 2023) is valid for other n-aldehydes. Therefore, accounting for linear aldehydes and their atmospheric oxidation with increasing importance to longer carbon chain length as a direct source of condensable materials even under moderately polluted urban areas is essential.
Details about the experimental setup and mass spectrometry, chemicals and gas cylinders, mechanistic details of the oxidation steps, additional insights into abundance of HOMs, and details of kinetic simulations are provided in the Supplement. An Excel file (.xlsx) with results from mass spectrometry, and two example Kinetiscope (.rxn) files containing kinetic simulation input parameters – one with NO, another without – are available online (https://doi.org/10.5281/zenodo.18894230; Barua, 2026).
The supplement related to this article is available online at https://doi.org/10.5194/acp-26-4711-2026-supplement.
Conceptualization: MR, SB, AK; data curation: SB, AK; formal analysis: SB; investigation: SB, AK, PS, SI, MR; methodology: SB, AK; writing (original draft preparation): SB; writing (review and editing): SB, AK, PS, SI, MR; funding acquisition: SI, MR.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. The authors bear the ultimate responsibility for providing appropriate place names. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
We thank the tofTools team for providing the data analysis program.
This research has been supported by the H2020 European Research Council (grant no. 101002728), the HORIZON EUROPE European Research Council (grant no. 101096133), the Research Council of Finland (grant nos. 331207, 336531, 346373, 347775, 353836, and 355966), and the Doctoral school of the Faculty of Engineering and Natural Sciences of Tampere University.
This paper was edited by Ivan Kourtchev and reviewed by four anonymous referees.
Aguirre, F., Lugo G, P. L., Straccia C, V. G., Teruel, M. A., and Blanco, M. B.: Atmospheric oxidation of long chain aldehydes: OH and Cl reactivity, mechanisms and environmental impact, Atmos. Environ., 360, 121429, https://doi.org/10.1016/j.atmosenv.2025.121429, 2025.
Albaladejo, J., Ballesteros, B., Jiménez, E., Martín, P., and Martínez, E.: A PLP–LIF kinetic study of the atmospheric reactivity of a series of C4–C7 saturated and unsaturated aliphatic aldehydes with OH, Atmos. Environ., 36, 3231–3239, https://doi.org/10.1016/S1352-2310(02)00323-0, 2002.
Barua, S.: Rapid formation of secondary aerosol precursors from the autoxidation of C5–C8 n-aldehydes, Zenodo [data set], https://doi.org/10.5281/zenodo.18894230, 2026.
Barua, S., Iyer, S., Kumar, A., Seal, P., and Rissanen, M.: An aldehyde as a rapid source of secondary aerosol precursors: theoretical and experimental study of hexanal autoxidation, Atmos. Chem. Phys., 23, 10517–10532, https://doi.org/10.5194/acp-23-10517-2023, 2023.
Barua, S., Kumar, A., Seal, P., Bezaatpour, M., Jha, S., Myllys, N., Iyer, S., and Rissanen, M.: Rapid formation of aerosol precursors from the autoxidation of aromatic carbonyls and the remarkable enhancing influence of NO addition, Research Square [preprint], https://doi.org/10.21203/rs.3.rs-7332278/v1, 28 August 2025.
Berndt, T., Richters, S., Kaethner, R., Voigtländer, J., Stratmann, F., Sipilä, M., Kulmala, M., and Herrmann, H.: Gas-Phase Ozonolysis of Cycloalkenes: Formation of Highly Oxidized RO2 Radicals and Their Reactions with NO, NO2, SO2, and Other RO2 Radicals, J. Phys. Chem. A, 119, 10336–10348, https://doi.org/10.1021/acs.jpca.5b07295, 2015.
Berndt, T., Richters, S., Jokinen, T., Hyttinen, N., Kurtén, T., Otkjær, R. V., Kjaergaard, H. G., Stratmann, F., Herrmann, H., Sipilä, M., Kulmala, M., and Ehn, M.: Hydroxyl radical-induced formation of highly oxidized organic compounds, Nat. Commun., 7, 13677, https://doi.org/10.1038/ncomms13677, 2016.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Self- and Cross-Reactions of RO2 Radicals in the Atmosphere, Angew. Chem. Int. Edit., 57, 3820–3824, https://doi.org/10.1002/anie.201710989, 2018.
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Birmili, W., Daniels, A., Bethke, R., Schechner, N., Brasse, G., Conrad, A., Kolossa-Gehring, M., Debiak, M., Hurraß, J., Uhde, E., Omelan, A., and Salthammer, T.: Formaldehyde, aliphatic aldehydes (C2-C11), furfural, and benzaldehyde in the residential indoor air of children and adolescents during the German Environmental Survey 2014–2017 (GerES V), Indoor Air, 32, https://doi.org/10.1111/ina.12927, 2022.
Brean, J., Harrison, R. M., Shi, Z., Beddows, D. C. S., Acton, W. J. F., Hewitt, C. N., Squires, F. A., and Lee, J.: Observations of highly oxidized molecules and particle nucleation in the atmosphere of Beijing, Atmos. Chem. Phys., 19, 14933–14947, https://doi.org/10.5194/acp-19-14933-2019, 2019.
Brean, J., Beddows, D. C. S., Shi, Z., Temime-Roussel, B., Marchand, N., Querol, X., Alastuey, A., Minguillón, M. C., and Harrison, R. M.: Molecular insights into new particle formation in Barcelona, Spain, Atmos. Chem. Phys., 20, 10029–10045, https://doi.org/10.5194/acp-20-10029-2020, 2020.
Calogirou, A., Larsen, B. R., and Kotzias, D.: Gas-phase terpene oxidation products: A review, Atmos. Environ., 33, 1423–1439, https://doi.org/10.1016/S1352-2310(98)00277-5, 1999.
Calvert, J., Mellouki, A., Orlando, J., Pilling, M., and Wallington, T.: Mechanisms of Atmospheric Oxidation of the Oxygenates, Oxford University Press, USA, https://doi.org/10.1093/oso/9780199767076.001.0001, 2011.
Carlier, P., Hannachi, H., and Mouvier, G.: The chemistry of carbonyl compounds in the atmosphere—A review, Atmos. Environ., 20, 2079–2099, https://doi.org/10.1016/0004-6981(86)90304-5, 1986.
Cassanelli, P., Johnson, D., and Anthony Cox, R.: A temperature-dependent relative-rate study of the OH initiated oxidation of n-butane: The kinetics of the reactions of the 1- and 2-butoxy radicals, Phys. Chem. Chem. Phys., 7, 3702, https://doi.org/10.1039/b507137b, 2005.
Castañeda, R., Iuga, C., Álvarez-Idaboy, J. R., and Vivier-Bunge, A.: Rate Constants and Branching Ratios in the Oxidation of Aliphatic Aldehydes by OH Radicals under Atmospheric Conditions, J. Mex. Chem. Soc., 56, 316–324, https://doi.org/10.29356/jmcs.v56i3.296, 2012.
Chacon-Madrid, H. J., Presto, A. A., and Donahue, N. M.: Functionalization vs. fragmentation: n-aldehyde oxidation mechanisms and secondary organic aerosol formation, Phys. Chem. Chem. Phys., 12, 13975, https://doi.org/10.1039/c0cp00200c, 2010.
Ciccioli, P., Brancaleoni, E., Frattoni, M., Cecinato, A., and Brachetti, A.: Ubiquitous occurrence of semi-volatile carbonyl compounds in tropospheric samples and their possible sources, Atmos. Environ. A-Gen., 27, 1891–1901, https://doi.org/10.1016/0960-1686(93)90294-9, 1993.
Crounse, J. D., Knap, H. C., Ørnsø, K. B., Jørgensen, S., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric Fate of Methacrolein. 1. Peroxy Radical Isomerization Following Addition of OH and O2, J. Phys. Chem. A, 116, 5756–5762, https://doi.org/10.1021/jp211560u, 2012.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013.
Da Silva, G.: Kinetics and Mechanism of the Glyoxal + HO2 Reaction: Conversion of HO2 to OH by Carbonyls, J. Phys. Chem. A, 115, 291–297, https://doi.org/10.1021/jp108358y, 2011.
Duan, J., Tan, J., Yang, L., Wu, S., and Hao, J.: Concentration, sources and ozone formation potential of volatile organic compounds (VOCs) during ozone episode in Beijing, Atmos. Res., 88, 25–35, https://doi.org/10.1016/j.atmosres.2007.09.004, 2008.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Ervens, B., Turpin, B. J., and Weber, R. J.: Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies, Atmos. Chem. Phys., 11, 11069–11102, https://doi.org/10.5194/acp-11-11069-2011, 2011.
Ervens, B., Sorooshian, A., Lim, Y. B., and Turpin, B. J.: Key parameters controlling OH-initiated formation of secondary organic aerosol in the aqueous phase (aqSOA), J. Geophys. Res.-Atmos., 119, 3997–4016, https://doi.org/10.1002/2013JD021021, 2014.
Fan, C., Yan, H., Wang, W., Sun, Z., and Ge, M.: Study on the reactions of n-pentanal and n-hexanal with Br atoms: Kinetics, gas-phase products, and SOA formation, Atmos. Environ., 339, 120869, https://doi.org/10.1016/j.atmosenv.2024.120869, 2024.
Fry, J. L., Draper, D. C., Barsanti, K. C., Smith, J. N., Ortega, J., Winkler, P. M., Lawler, M. J., Brown, S. S., Edwards, P. M., Cohen, R. C., and Lee, L.: Secondary Organic Aerosol Formation and Organic Nitrate Yield from NO3 Oxidation of Biogenic Hydrocarbons, Environ. Sci. Technol., 48, 11944–11953, https://doi.org/10.1021/es502204x, 2014.
Gu, Y., Huang, R.-J., Duan, J., Xu, W., Lin, C., Zhong, H., Wang, Y., Ni, H., Liu, Q., Xu, R., Wang, L., and Li, Y. J.: Multiple pathways for the formation of secondary organic aerosol in the North China Plain in summer, Atmos. Chem. Phys., 23, 5419–5433, https://doi.org/10.5194/acp-23-5419-2023, 2023.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Hansen, J. E. and Sato, M.: Trends of measured climate forcing agents, P. Natl. Acad. Sci. USA, 98, 14778–14783, https://doi.org/10.1073/pnas.261553698, 2001.
Hinsberg, W. and Houle, F.: Kinetiscope: A stochastic kinetics simulator, http://hinsberg.net/kinetiscope (last access: 6 November 2023), 2022.
Huang, R.-J., Zhang, Y., Bozzetti, C., Ho, K.-F., Cao, J.-J., Han, Y., Daellenbach, K. R., Slowik, J. G., Platt, S. M., Canonaco, F., Zotter, P., Wolf, R., Pieber, S. M., Bruns, E. A., Crippa, M., Ciarelli, G., Piazzalunga, A., Schwikowski, M., Abbaszade, G., Schnelle-Kreis, J., Zimmermann, R., An, Z., Szidat, S., Baltensperger, U., El Haddad, I., and Prévôt, A. S. H.: High secondary aerosol contribution to particulate pollution during haze events in China, Nature, 514, 218–222, https://doi.org/10.1038/nature13774, 2014.
Huang, W., Saathoff, H., Shen, X., Ramisetty, R., Leisner, T., and Mohr, C.: Chemical Characterization of Highly Functionalized Organonitrates Contributing to Night-Time Organic Aerosol Mass Loadings and Particle Growth, Environ. Sci. Technol., 53, 1165–1174, https://doi.org/10.1021/acs.est.8b05826, 2019.
Iuga, C., Ignacio Sainz-Díaz, C., and Vivier-Bunge, A.: On the OH initiated oxidation of C2–C5 aliphatic aldehydes in the presence of mineral aerosols, Geochim. Cosmochim. Ac., 74, 3587–3597, https://doi.org/10.1016/j.gca.2010.01.034, 2010.
Jacobson, M. C., Hansson, H. -C., Noone, K. J., and Charlson, R. J.: Organic atmospheric aerosols: Review and state of the science, Rev. Geophys., 38, 267–294, https://doi.org/10.1029/1998RG000045, 2000.
Jenkin, M. E., Derwent, R. G., and Wallington, T. J.: Photochemical ozone creation potentials for volatile organic compounds: Rationalization and estimation, Atmos. Environ., 163, 128–137, https://doi.org/10.1016/j.atmosenv.2017.05.024, 2017.
Jokinen, T., Sipilä, M., Richters, S., Kerminen, V., Paasonen, P., Stratmann, F., Worsnop, D., Kulmala, M., Ehn, M., Herrmann, H., and Berndt, T.: Rapid Autoxidation Forms Highly Oxidized RO2 Radicals in the Atmosphere, Angew. Chem. Int. Edit., 53, 14596–14600, https://doi.org/10.1002/anie.201408566, 2014.
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005.
Kang, S., Wildt, J., Pullinen, I., Vereecken, L., Wu, C., Wahner, A., Zorn, S. R., and Mentel, T. F.: Formation of highly oxygenated organic molecules from α-pinene photooxidation: evidence for the importance of highly oxygenated alkoxy radicals, Atmos. Chem. Phys., 25, 15715–15740, https://doi.org/10.5194/acp-25-15715-2025, 2025.
Kirkby, J., Duplissy, J., Sengupta, K., Frege, C., Gordon, H., Williamson, C., Heinritzi, M., Simon, M., Yan, C., Almeida, J., Tröstl, J., Nieminen, T., Ortega, I. K., Wagner, R., Adamov, A., Amorim, A., Bernhammer, A.-K., Bianchi, F., Breitenlechner, M., Brilke, S., Chen, X., Craven, J., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Hakala, J., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Mathot, S., Molteni, U., Onnela, A., Peräkylä, O., Piel, F., Petäjä, T., Praplan, A. P., Pringle, K., Rap, A., Richards, N. A. D., Riipinen, I., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Scott, C. E., Seinfeld, J. H., Sipilä, M., Steiner, G., Stozhkov, Y., Stratmann, F., Tomé, A., Virtanen, A., Vogel, A. L., Wagner, A. C., Wagner, P. E., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Zhang, X., Hansel, A., Dommen, J., Donahue, N. M., Worsnop, D. R., Baltensperger, U., Kulmala, M., Carslaw, K. S., and Curtius, J.: Ion-induced nucleation of pure biogenic particles, Nature, 533, 521–526, https://doi.org/10.1038/nature17953, 2016.
Kroll, J. H. and Seinfeld, J. H.: Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 42, 3593–3624, https://doi.org/10.1016/j.atmosenv.2008.01.003, 2008.
Kuang, Y., He, Y., Xu, W., Yuan, B., Zhang, G., Ma, Z., Wu, C., Wang, C., Wang, S., Zhang, S., Tao, J., Ma, N., Su, H., Cheng, Y., Shao, M., and Sun, Y.: Photochemical Aqueous-Phase Reactions Induce Rapid Daytime Formation of Oxygenated Organic Aerosol on the North China Plain, Environ. Sci. Technol., 54, 3849–3860, https://doi.org/10.1021/acs.est.9b06836, 2020.
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurtén, T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvery, K., Shepson, P. B., Baumann, K., Edgerton, E., Liu, J., Shilling, J. E., Miller, D. O., Brune, W. H., D'Ambro, E. L., and Thornton, J. A.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci. USA, 113, 1516–1521, https://doi.org/10.1073/pnas.1508108113, 2016.
Li, J., Zhai, C., Yu, J., Liu, R., Li, Y., Zeng, L., and Xie, S.: Spatiotemporal variations of ambient volatile organic compounds and their sources in Chongqing, a mountainous megacity in China, Sci. Total Environ., 627, 1442–1452, https://doi.org/10.1016/j.scitotenv.2018.02.010, 2018.
Lipari, Frank., Dasch, J. M., and Scruggs, W. F.: Aldehyde emissions from wood-burning fireplaces, Environ. Sci. Technol., 18, 326–330, https://doi.org/10.1021/es00123a007, 1984.
Ma, Z., Liu, C., Zhang, C., Liu, P., Ye, C., Xue, C., Zhao, D., Sun, J., Du, Y., Chai, F., and Mu, Y.: The levels, sources and reactivity of volatile organic compounds in a typical urban area of Northeast China, J. Environ. Sci., 79, 121–134, https://doi.org/10.1016/j.jes.2018.11.015, 2019.
Manion, J. A., Huie, R. E., Levin, R. D., Burgess Jr, D. R., Orkin, V. L., Tsang, W., McGivern, W. S., Hudgens, J. W., Knyazev, V. D., Atkinson, D. B., Chai, E., Tereza, A. M., Lin, C.-Y., Allison, T. C., Mallard, W. G., Westley, F., Herron, J. T., Hampson, R. F., and Frizzell, D. H.: NIST Chemical Kinetics Database, NIST Standard Reference Database 17, version 7.0 (web version), release 1.6.8, data version 2015.09, National Institute of Standards and Technology, MD, https://kinetics.nist.gov/ (last access: 20 August 2025), 2015.
McFiggans, G., Mentel, T. F., Wildt, J., Pullinen, I., Kang, S., Kleist, E., Schmitt, S., Springer, M., Tillmann, R., Wu, C., Zhao, D., Hallquist, M., Faxon, C., Le Breton, M., Hallquist, Å. M., Simpson, D., Bergström, R., Jenkin, M. E., Ehn, M., Thornton, J. A., Alfarra, M. R., Bannan, T. J., Percival, C. J., Priestley, M., Topping, D., and Kiendler-Scharr, A.: Secondary organic aerosol reduced by mixture of atmospheric vapours, Nature, 565, 587–593, https://doi.org/10.1038/s41586-018-0871-y, 2019.
Mellouki, A., Le Bras, G., and Sidebottom, H.: Kinetics and Mechanisms of the Oxidation of Oxygenated Organic Compounds in the Gas Phase, Chem. Rev., 103, 5077–5096, https://doi.org/10.1021/cr020526x, 2003.
Mellouki, A., Wallington, T. J., and Chen, J.: Atmospheric Chemistry of Oxygenated Volatile Organic Compounds: Impacts on Air Quality and Climate, Chem. Rev., 115, 3984–4014, https://doi.org/10.1021/cr500549n, 2015.
Mentel, T. F., Springer, M., Ehn, M., Kleist, E., Pullinen, I., Kurtén, T., Rissanen, M., Wahner, A., and Wildt, J.: Formation of highly oxidized multifunctional compounds: autoxidation of peroxy radicals formed in the ozonolysis of alkenes – deduced from structure–product relationships, Atmos. Chem. Phys., 15, 6745–6765, https://doi.org/10.5194/acp-15-6745-2015, 2015.
Møller, K. H., Otkjær, R. V., Hyttinen, N., Kurtén, T., and Kjaergaard, H. G.: Cost-Effective Implementation of Multiconformer Transition State Theory for Peroxy Radical Hydrogen Shift Reactions, J. Phys. Chem. A, 120, 10072–10087, https://doi.org/10.1021/acs.jpca.6b09370, 2016.
Møller, K. H., Praske, E., Xu, L., Crounse, J. D., Wennberg, P. O., and Kjaergaard, H. G.: Stereoselectivity in Atmospheric Autoxidation, J. Phys. Chem. Lett., 10, 6260–6266, https://doi.org/10.1021/acs.jpclett.9b01972, 2019.
Nie, W., Yan, C., Yang, L., Roldin, P., Liu, Y., Vogel, A. L., Molteni, U., Stolzenburg, D., Finkenzeller, H., Amorim, A., Bianchi, F., Curtius, J., Dada, L., Draper, D. C., Duplissy, J., Hansel, A., He, X.-C., Hofbauer, V., Jokinen, T., Kim, C., Lehtipalo, K., Nichman, L., Mauldin, R. L., Makhmutov, V., Mentler, B., Mizelli-Ojdanic, A., Petäjä, T., Quéléver, L. L. J., Schallhart, S., Simon, M., Tauber, C., Tomé, A., Volkamer, R., Wagner, A. C., Wagner, R., Wang, M., Ye, P., Li, H., Huang, W., Qi, X., Lou, S., Liu, T., Chi, X., Dommen, J., Baltensperger, U., El Haddad, I., Kirkby, J., Worsnop, D., Kulmala, M., Donahue, N. M., Ehn, M., and Ding, A.: NO at low concentration can enhance the formation of highly oxygenated biogenic molecules in the atmosphere, Nat. Commun., 14, 3347, https://doi.org/10.1038/s41467-023-39066-4, 2023.
Öström, E., Putian, Z., Schurgers, G., Mishurov, M., Kivekäs, N., Lihavainen, H., Ehn, M., Rissanen, M. P., Kurtén, T., Boy, M., Swietlicki, E., and Roldin, P.: Modeling the role of highly oxidized multifunctional organic molecules for the growth of new particles over the boreal forest region, Atmos. Chem. Phys., 17, 8887–8901, https://doi.org/10.5194/acp-17-8887-2017, 2017.
Praske, E., Otkjær, R. V., Crounse, J. D., Hethcox, J. C., Stoltz, B. M., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric autoxidation is increasingly important in urban and suburban North America, P. Natl. Acad. Sci. USA, 115, 64–69, https://doi.org/10.1073/pnas.1715540115, 2018.
Pullinen, I., Schmitt, S., Kang, S., Sarrafzadeh, M., Schlag, P., Andres, S., Kleist, E., Mentel, T. F., Rohrer, F., Springer, M., Tillmann, R., Wildt, J., Wu, C., Zhao, D., Wahner, A., and Kiendler-Scharr, A.: Impact of NOx on secondary organic aerosol (SOA) formation from α-pinene and β-pinene photooxidation: the role of highly oxygenated organic nitrates, Atmos. Chem. Phys., 20, 10125–10147, https://doi.org/10.5194/acp-20-10125-2020, 2020.
Rissanen, M. P.: NO2 Suppression of Autoxidation–Inhibition of Gas-Phase Highly Oxidized Dimer Product Formation, ACS Earth Space Chem., 2, 1211–1219, https://doi.org/10.1021/acsearthspacechem.8b00123, 2018.
Rissanen, M. P., Kurtén, T., Sipilä, M., Thornton, J. A., Kangasluoma, J., Sarnela, N., Junninen, H., Jørgensen, S., Schallhart, S., Kajos, M. K., Taipale, R., Springer, M., Mentel, T. F., Ruuskanen, T., Petäjä, T., Worsnop, D. R., Kjaergaard, H. G., and Ehn, M.: The Formation of Highly Oxidized Multifunctional Products in the Ozonolysis of Cyclohexene, J. Am. Chem. Soc., 136, 15596–15606, https://doi.org/10.1021/ja507146s, 2014.
Rissanen, M. P., Kurtén, T., Sipilä, M., Thornton, J. A., Kausiala, O., Garmash, O., Kjaergaard, H. G., Petäjä, T., Worsnop, D. R., Ehn, M., and Kulmala, M.: Effects of Chemical Complexity on the Autoxidation Mechanisms of Endocyclic Alkene Ozonolysis Products: From Methylcyclohexenes toward Understanding α-Pinene, J. Phys. Chem. A, 119, 4633–4650, https://doi.org/10.1021/jp510966g, 2015.
Schauer, J. J., Kleeman, M. J., Cass, G. R., and Simoneit, B. R. T.: Measurement of Emissions from Air Pollution Sources. 1. C1 through C29 Organic Compounds from Meat Charbroiling, Environ. Sci. Technol., 33, 1566–1577, https://doi.org/10.1021/es980076j, 1999a.
Schauer, J. J., Kleeman, M. J., Cass, G. R., and Simoneit, B. R. T.: Measurement of Emissions from Air Pollution Sources. 2. C1 through C30 Organic Compounds from Medium Duty Diesel Trucks, Environ. Sci. Technol., 33, 1578–1587, https://doi.org/10.1021/es980081n, 1999b.
Schauer, J. J., Kleeman, M. J., Cass, G. R., and Simoneit, B. R. T.: Measurement of Emissions from Air Pollution Sources. 3. C1 -C29 Organic Compounds from Fireplace Combustion of Wood, Environ. Sci. Technol., 35, 1716–1728, https://doi.org/10.1021/es001331e, 2001.
Seal, P., Barua, S., Iyer, S., Kumar, A., and Rissanen, M.: A systematic study on the kinetics of H-shift reactions in pristine acyl peroxy radicals, Phys. Chem. Chem. Phys., 25, 28205–28212, https://doi.org/10.1039/D3CP01833D, 2023.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics: From air pollution to climate change, 3rd edn., John Wiley & Sons, ISBN 9781118947401, 2016.
Shallcross, D., Teresaraventosduran, M., Bardwell, M., Bacak, A., Solman, Z., and Percival, C.: A semi-empirical correlation for the rate coefficients for cross- and self-reactions of peroxy radicals in the gas-phase, Atmos. Environ., 39, 763–771, https://doi.org/10.1016/j.atmosenv.2004.09.072, 2005.
Shen, H., Vereecken, L., Kang, S., Pullinen, I., Fuchs, H., Zhao, D., and Mentel, T. F.: Unexpected significance of a minor reaction pathway in daytime formation of biogenic highly oxygenated organic compounds, Sci. Adv., 8, eabp8702, https://doi.org/10.1126/sciadv.abp8702, 2022.
Spracklen, D. V., Jimenez, J. L., Carslaw, K. S., Worsnop, D. R., Evans, M. J., Mann, G. W., Zhang, Q., Canagaratna, M. R., Allan, J., Coe, H., McFiggans, G., Rap, A., and Forster, P.: Aerosol mass spectrometer constraint on the global secondary organic aerosol budget, Atmos. Chem. Phys., 11, 12109–12136, https://doi.org/10.5194/acp-11-12109-2011, 2011.
Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., Wagner, R., Simon, M., Lehtipalo, K., Williamson, C., Craven, J. S., Duplissy, J., Adamov, A., Almeida, J., Bernhammer, A.-K., Breitenlechner, M., Brilke, S., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Gysel, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Keskinen, H., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lawler, M., Leiminger, M., Mathot, S., Möhler, O., Nieminen, T., Onnela, A., Petäjä, T., Piel, F. M., Miettinen, P., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Sipilä, M., Smith, J. N., Steiner, G., Tomè, A., Virtanen, A., Wagner, A. C., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Carslaw, K. S., Curtius, J., Dommen, J., Kirkby, J., Kulmala, M., Riipinen, I., Worsnop, D. R., Donahue, N. M., and Baltensperger, U.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 533, 527–531, https://doi.org/10.1038/nature18271, 2016.
Vereecken, L. and Peeters, J.: Decomposition of substituted alkoxy radicals—part I: A generalized structure–activity relationship for reaction barrier heights, Phys. Chem. Chem. Phys., 11, 9062, https://doi.org/10.1039/b909712k, 2009.
Wang, S., Davidson, D. F., and Hanson, R. K.: High temperature measurements for the rate constants of C1–C4 aldehydes with OH in a shock tube, Proc. Combust. Inst., 35, 473–480, https://doi.org/10.1016/j.proci.2014.06.112, 2015.
Wang, Z., Ehn, M., Rissanen, M. P., Garmash, O., Quéléver, L., Xing, L., Monge-Palacios, M., Rantala, P., Donahue, N. M., Berndt, T., and Sarathy, S. M.: Efficient alkane oxidation under combustion engine and atmospheric conditions, Commun. Chem., 4, 18, https://doi.org/10.1038/s42004-020-00445-3, 2021.
Wildt, J., Mentel, T. F., Kiendler-Scharr, A., Hoffmann, T., Andres, S., Ehn, M., Kleist, E., Müsgen, P., Rohrer, F., Rudich, Y., Springer, M., Tillmann, R., and Wahner, A.: Suppression of new particle formation from monoterpene oxidation by NOx, Atmos. Chem. Phys., 14, 2789–2804, https://doi.org/10.5194/acp-14-2789-2014, 2014.
Williams, I. D., Revitt, D. M., and Hamilton, R. S.: A comparison of carbonyl compound concentrations at urban roadside and indoor sites, Sci. Total Environ., 189–190, 475–483, https://doi.org/10.1016/0048-9697(96)05248-5, 1996.
Yan, C., Nie, W., Vogel, A. L., Dada, L., Lehtipalo, K., Stolzenburg, D., Wagner, R., Rissanen, M. P., Xiao, M., Ahonen, L., Fischer, L., Rose, C., Bianchi, F., Gordon, H., Simon, M., Heinritzi, M., Garmash, O., Roldin, P., Dias, A., Ye, P., Hofbauer, V., Amorim, A., Bauer, P. S., Bergen, A., Bernhammer, A.-K., Breitenlechner, M., Brilke, S., Buchholz, A., Mazon, S. B., Canagaratna, M. R., Chen, X., Ding, A., Dommen, J., Draper, D. C., Duplissy, J., Frege, C., Heyn, C., Guida, R., Hakala, J., Heikkinen, L., Hoyle, C. R., Jokinen, T., Kangasluoma, J., Kirkby, J., Kontkanen, J., Kürten, A., Lawler, M. J., Mai, H., Mathot, S., Mauldin, R. L., Molteni, U., Nichman, L., Nieminen, T., Nowak, J., Ojdanic, A., Onnela, A., Pajunoja, A., Petäjä, T., Piel, F., Quéléver, L. L. J., Sarnela, N., Schallhart, S., Sengupta, K., Sipilä, M., Tomé, A., Tröstl, J., Väisänen, O., Wagner, A. C., Ylisirniö, A., Zha, Q., Baltensperger, U., Carslaw, K. S., Curtius, J., Flagan, R. C., Hansel, A., Riipinen, I., Smith, J. N., Virtanen, A., Winkler, P. M., Donahue, N. M., Kerminen, V.-M., Kulmala, M., Ehn, M., and Worsnop, D. R.: Size-dependent influence of NOx on the growth rates of organic aerosol particles, Sci. Adv., 6, eaay4945, https://doi.org/10.1126/sciadv.aay4945, 2020.
Yang, X., Wang, H., Lu, K., Ma, X., Tan, Z., Long, B., Chen, X., Li, C., Zhai, T., Li, Y., Qu, K., Xia, Y., Zhang, Y., Li, X., Chen, S., Dong, H., Zeng, L., and Zhang, Y.: Reactive aldehyde chemistry explains the missing source of hydroxyl radicals, Nat. Commun., 15, 1648, https://doi.org/10.1038/s41467-024-45885-w, 2024.
Zhang, X., Cappa, C. D., Jathar, S. H., McVay, R. C., Ensberg, J. J., Kleeman, M. J., and Seinfeld, J. H.: Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol, P. Natl. Acad. Sci. USA, 111, 5802–5807, https://doi.org/10.1073/pnas.1404727111, 2014.
Ziemann, P. J. and Atkinson, R.: Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 41, 6582, https://doi.org/10.1039/c2cs35122f, 2012.