the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 08 Jan 2025
| 08 Jan 2025
Derivation of atmospheric reaction mechanisms for volatile organic compounds by the SAPRC mechanism generation system (MechGen)
William P. L. Carter
Jia Jiang
John J. Orlando
Kelley C. Barsanti
This paper describes the methods that are used in the SAPRC mechanism generation system, MechGen, to estimate rate constants and derive mechanisms for gas-phase reactions of volatile organic compounds (VOCs) in the lower atmosphere. Versions of this system have been used for over 20 years in the development of the SAPRC mechanisms for air quality models, but this is the first complete documentation of the scientific basis for the chemical mechanisms it derives. MechGen can be used to derive explicit gas-phase mechanisms for most compounds with C, H, O, or N atoms. Included are reactions of organic compounds with hydroxy (OH) and nitrate (NO3) radicals, O3, and O3P; photolysis or unimolecular reactions; and the reactions of the radicals they form in the presence of O2 and oxides of nitrogen (NOx) at lower-atmospheric temperatures and pressures. Measured or theoretically calculated rate constants and branching ratios are used when data are available, but in most cases rate constants and branching ratios are estimated using various structure–reactivity or other estimation methods. Types of reactions include initial reactions of organics with atmospheric oxidants or by photolysis; unimolecular and bimolecular reactions of carbon-centered, alkoxy, and peroxy radicals; and those of Criegee and other intermediates that are formed. This paper documents the methods, assignments, and estimates currently used to derive these reactions and provides examples of MechGen predictions. Many of the estimation methods discussed here have not been published previously, and others have not been used previously in developing comprehensive mechanisms. Our knowledge of atmospheric reactions of organic compounds rapidly and continuously evolves, and therefore mechanism generation systems such as MechGen also need to evolve to continue to represent the current state of the science. This paper points out areas where MechGen may need to be modified when the system is next updated. This paper concludes with a summary of the major areas of uncertainty where further experimental, theoretical, or mechanism development research is most needed to improve predictions of atmospheric reaction mechanisms of volatile organic compounds.
- Article
(1024 KB) - Full-text XML
-
Supplement
(1434 KB) - BibTeX
- EndNote





1.1 Background
When most volatile organic compounds (VOCs) are introduced into the atmosphere in the presence of sunlight, they can react to form various radicals that then further react to form oxidized organic products, including gas-phase toxics and secondary organic aerosol (SOA). In the presence of oxides of nitrogen (NOx) from combustion sources, these radicals also promote the formation of ozone (O3) and oxidized nitrogen compounds that further affect air quality. Many hundreds of types of organic compounds are emitted, from both anthropogenic and biogenic sources. The atmospheric reaction mechanisms for these compounds are complex, and for larger molecules they can involve an extremely large number of reactive intermediates and result in a large number of stable oxidized organic products. Most of these organic products also react in the atmosphere, forming even more intermediates and products. In most cases these mechanisms involve reactions whose rate constants and, in some cases, mechanisms are unknown and thus have to be estimated. Because of the complexity, it is necessary either to greatly simplify the mechanisms for most VOCs, to use extensive lumping or condensations in representations of VOCs, or to use an automated chemical mechanism generation system to derive the mechanisms.
In the case of tropospheric reaction mechanisms of VOCs, existing automated chemical mechanism generation systems include the Generator for Explicit Chemistry and Kinetics of Organics in the Atmosphere (GECKO-A) developed by Aumont and co-workers (Aumont et al., 2005) and the SAPRC mechanism generation system, MechGen (Carter, 2024a), which is the subject of this paper. GECKO-A has been used in a number of studies of varying chemical complexity (e.g., Aumont et al., 2005; Camredon et al., 2007; Lee-Taylor et al., 2011; Aumont et al., 2012; Lannuque et al., 2018; Afreh et al., 2021) and is designed primarily to derive and carry out model simulations using multi-generation mechanisms of selected compounds and all of their oxidation products. The SAPRC MechGen system was developed primarily to support the development of versions of the SAPRC atmospheric gas-phase chemical mechanisms (Carter, 2000, 2010a, b, 2016, 2020) and has been focused on single-generation reactions of individual compounds, with mechanisms for representative organic oxidation products being derived separately. Therefore, the objectives and operations of the two systems are somewhat different. In addition, although the two systems employ similar structure–activity relationships (SARs) and can give very similar predictions, there are some differences in the mechanisms they derive, reflecting both different treatments of uncertainties and also different areas where updates are needed to incorporate recent results. For example, GECKO-A does not yet predict autoxidation reactions of peroxy radicals that are believed to occur (and are predicted by MechGen), while GECKO-A employs more detailed and updated estimates for bimolecular reactions of peroxy radicals and photolysis reactions of larger molecules.
Perhaps the most important difference between GECKO-A and MechGen is that GECKO-A is described in the peer-reviewed literature (e.g., Aumont et al., 2005), while the description of MechGen is largely incomplete in the literature, with only an abbreviated description in applied studies (Jiang et al., 2020; Li et al., 2022). Different versions of MechGen, incorporating our changing and evolving understanding and ability to estimate the underlying chemistry, have been used in the development of SAPRC-99 (Carter, 2000), SAPRC-07 (Carter, 2010a, b), SAPRC-11 (Carter and Heo, 2013), SAPRC-16 (Carter, 2016; Venecek et al., 2018), and SAPRC-18 (Carter, 2020). Additional updates to the system have been made since the release of SAPRC-18. The lack of a stable and peer-reviewed version has inhibited the use of MechGen for atmospheric chemistry research beyond the development of SAPRC mechanisms, despite its potential utility as an alternative to or for comparison with GECKO-A or the semi-explicit Master Chemical Mechanism (Jenkin et al., 1997, 2003; Bloss et al., 2005; MCM, 2023). The purpose of this paper is to document the chemical basis of the estimates and assignments as it currently exists, so it can be appropriately cited and more widely used. The version of MechGen described here has been used in the preparation of the recently completed SAPRC-22 mechanisms (Carter, 2023). This paper can also be used to as a starting point for future updates and to illustrate areas of uncertainty where experimental or theoretical studies are needed.
1.2 Scope
MechGen is capable of generating fully explicit mechanisms for the atmospheric reactions of most types of organic compounds emitted into the atmosphere and the intermediate radicals they form. It is designed to generate mechanisms for lower-tropospheric modeling only, so its mechanisms are applicable primarily for temperatures at or near 300 K and pressures at or near 1 atm. Although temperature-dependent rate constants are assigned or estimated for many reactions, for others the rate constants or branching ratios are only applicable for 300 K and 1 atm. In particular, MechGen is not currently designed for estimating mechanisms for combustion modeling or for low-temperature or low-pressure systems.
Table 1 lists the types of stable compounds and the categories of initial atmospheric reactions that can be generated for those compounds. Table 2 lists the types of reactions within each category that can be generated, including reactions of intermediate radicals and reactions of stable compounds. These include H-atom abstractions from stable compounds by OH, NO3, and Cl radicals; additions to double bonds by these radicals and by O3 and O3P; and photolyses at various groups. The types of radicals that can be generated include carbon-centered radicals that in most cases react primarily with O2; peroxy radicals that in most cases react with NO, NO2, NO3, HO2, or other peroxy radicals and in some cases also have unimolecular reactions; alkoxy radicals that can react with O2 or by various types of unimolecular reactions; and excited and stabilized Criegee intermediates (CIs). More information about their reactions and how they are generated is given below, with additional detail given in the Supplement as referenced below.
Table 1Types of stable compounds whose reactions are supported by MechGen.
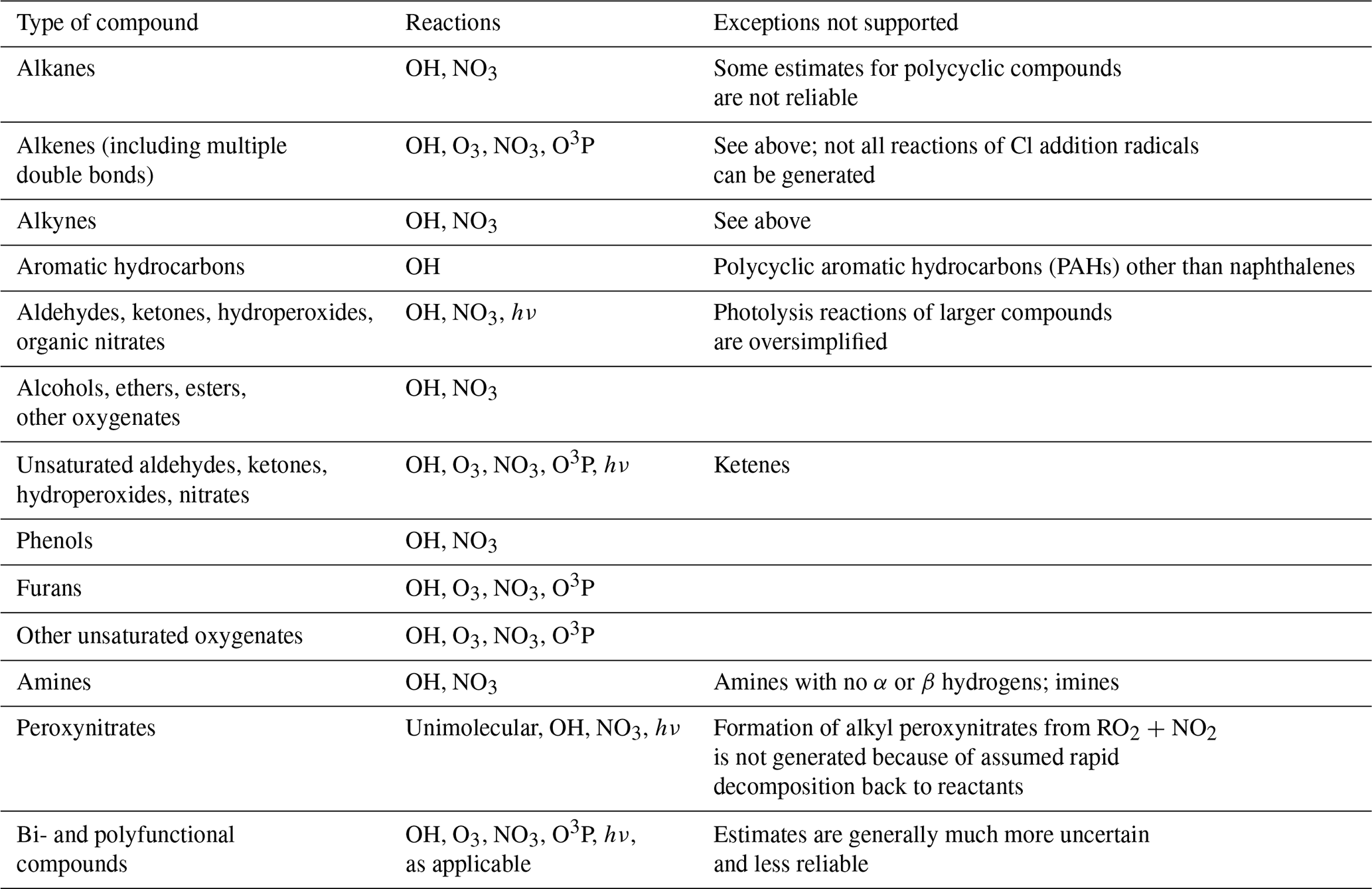
Table 2Summary of types of reactions supported by MechGen. VOC indicates any stable compound supported by the system (see Table 1).
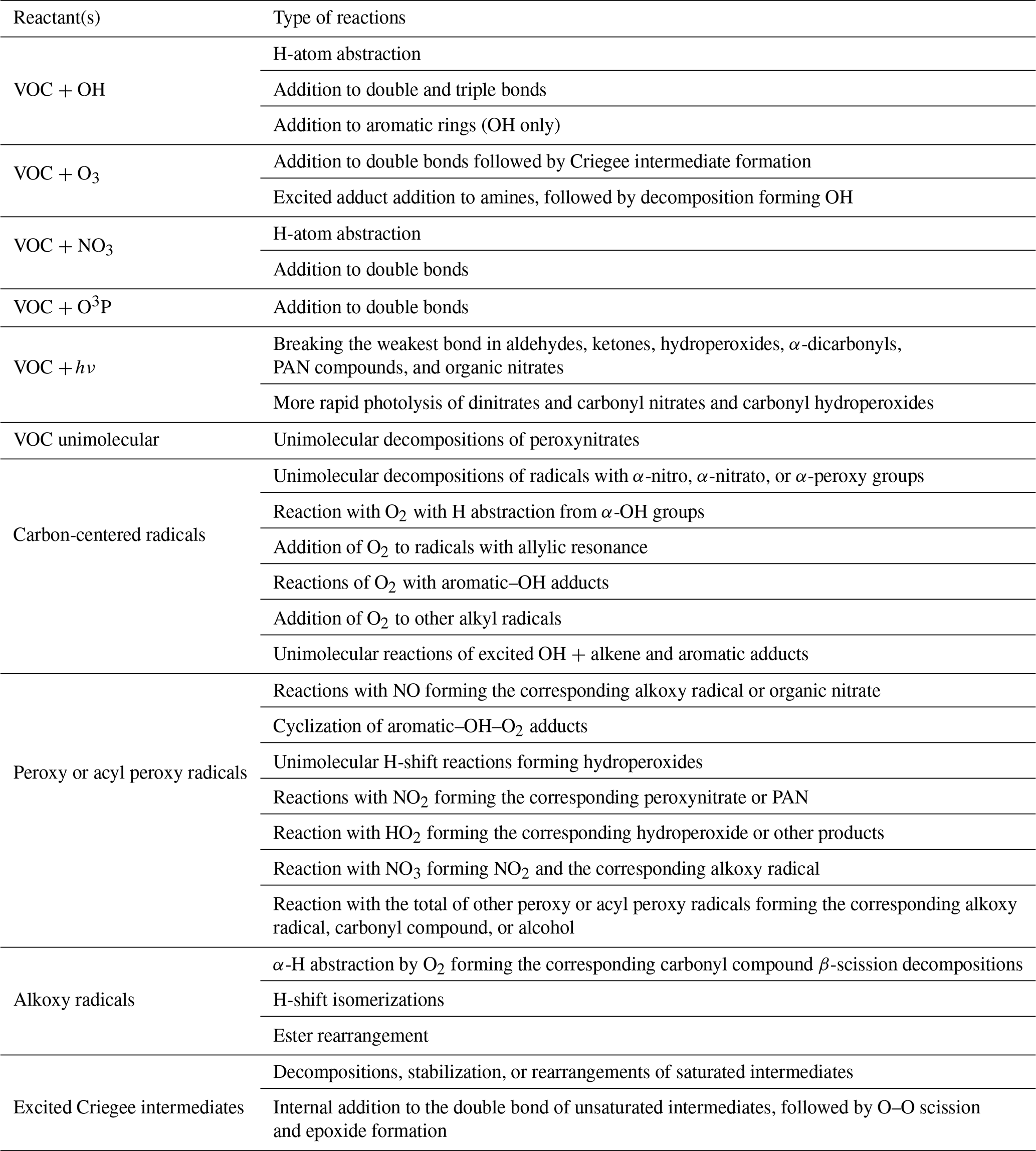
Although the MechGen system can represent chemical structures containing halogen atoms and estimate some types of reactions of compounds with Cl atoms, not all types of reactions of halogen-containing compounds are currently supported. Therefore, a discussion of reactions of halogen-containing compounds is not included here.
The operations and capabilities of MechGen as a software system are described in a separate paper and user's manual (Carter et al., 2024), while this paper focuses on the chemical basis for the derived mechanisms.
1.3 Designation of structures
Information on how to input and designate structures for MechGen is given in the software description and user's manual (Carter et al., 2024). Briefly, the structure of an organic reactant or radical is specified by giving the “groups” in the molecule or radical and indicating the groups each are bonded to, the type of bond, and in some cases (e.g., cis or trans isomers) the orientation of groups around the bonds. Groups are parts of molecules that are treated as units in the system and are used to determine which types of reactions can occur and what products are formed and used by the SARs or other methods to estimate rate constants. These groups contain no more than one carbon or nitrogen atom and also contain zero to three hydrogen atoms and zero to three oxygen atoms. Structure designations are summarized in Sect. S1.1 of the Supplement.
The generated reactions and estimated rate constants depend not only on the group(s) where the reaction occurs but also on the immediate neighbor group and in some cases groups some distance away in the molecule. In many cases, different groups or a combination of groups is estimated to react similarly or have similar effects on reactions at neighboring groups, so this documentation uses designations that refer to such combinations. Examples include –CXxH– to refer to carbon-centered groups with at least one hydrogen (where X designates either H or any neighboring group bonded to the carbon with a single bond) and –CHx–OH to refer to any carbon-centered group bonded to an OH group. G refers to any group, –G–G– refers to chains of groups, etc. Some group designations used in the text will be somewhat different than those used by MechGen in order to be more familiar to chemists. Generally the group designations are noted in cases where they may not be obvious.
2.1 Bimolecular reactions with atmospheric oxidants
The methods MechGen uses to estimate rate constants for reactions of organic compounds with OH, NO3, O3, O3P, and Cl have been documented by Carter (2021), with updates as discussed below and in the Supplement. In most cases the rate constant estimates are made separately for each reaction route, with the total rate constant being the sum of the estimated rate constants for each route. The estimates are made for the various types of reactions using SARs based on the groups in the molecule, the neighboring groups bonded to them (in some cases the second neighbor as well), and the structure of the molecule around the group(s) where the reaction occurs (e.g., groups in a ring). The equations and parameters used for estimating the rate constants are summarized in Table 3, and the parameter values are given in various tables in Sect. S1.2. Most adjustable parameters needed for mechanism generation were derived by optimizations to fit the measured OH, NO3, O3, and Cl rate constants tabulated by McGillen et al. (2020), with the rate constants for most O3P reactions taken from Calvert et al. (2000). There were insufficient data to derive all parameters using this approach, so some parameters were estimated based on assumed relationships with the adjustable parameters.
Table 3Structure–reactivity equations and parameters used to estimate rate constants for reactions of VOCs with OH, NO3, Cl, or O3P. Parameter values are given in the Supplement. Rate constants are per group unless otherwise indicated. The table numbers in the Supplement giving the assigned parameter values are also indicated.

The derivation or estimation of most of the parameters and the ability of the estimates to predict the experimental data were given by Carter (2021). However, parameters for predicting rate constants for reactions of phenols with OH and NO3 were recently updated to improve the ability to model environmental-chamber experiments with generated mechanisms, as discussed in the Supplement. Rate constants for most hydrocarbons and monofunctional compounds can be estimated to within ±30 %, though predictions are not as good for multifunctional compounds and predictions for ∼ 15 % of the rate constants are off by more than a factor of 2. Estimates are more uncertain in the case of NO3 and O3 reactions compared to OH, Cl, and O3P. This reflects the current state of the science in estimating these rate constants with empirical structure–reactivity methods. Note that our estimates perform comparably to those developed for the GECKO-A system by Jenkin et al. (2018a, b) for OH and Kerdouci et al. (2014) for O3, which were developed independently using similar methods (see Carter, 2021).
The following sections describe the mechanisms generated for the various types of reactions and how branching ratios, if applicable, are derived. Note that some compounds may have more than one reaction for a given oxidant, so branching ratios are estimated using ratios of estimated rate constants, unless the branching ratios have been assigned explicitly for the compound and oxidant, as indicated in the tables in Sect. S1.2. If only the total rate constant is assigned, the branching ratios are unaffected because they are derived from ratios of estimated rate constants. Note that additional branching-ratio estimates are needed for additions of O3 or O3P to double- or triple-bond systems, as discussed below.
2.1.1 H-atom abstractions by OH or NO3
OH and NO3 radicals can react with groups containing H atoms as follows, with rate constants estimated as indicated in Table 3a and the parameters as indicated in Table S3 in the Supplement.
Here, G is any group where H is removed, X is OH or NO3, and aG refers to groups in a resonance structure (bonded to alternating double and single bonds). Separate estimates are made for each H-containing group in the molecule, so the branching ratio can be derived from the ratio of estimated rate constants to the sum of all the estimated rate constants for the VOC, including addition reactions (see below) where applicable. The subsequent reactions of the carbon-centered radicals are discussed in Sect. 3.
2.1.2 Additions of OH or NO3 to groups with unsaturated bonds
The OH and NO3 radicals can also add to groups with double, triple, or aromatic bonds as follows, with rate constants estimated as indicated in Table 3b for alkenes and alkynes and Table 3c for aromatics. The parameters are given in the Supplement as indicated in Table S3.
Here, G refers to any group with an unsaturated bond, X refers to the reacting radical, {excited} indicates vibrational excitation, and * indicates ring closure. Separate estimates are made for addition to each group so these can also be used to make the branching-ratio estimates. Most of the excited carbon-centered radicals are stabilized and react with O2 as discussed in Sect. 3.1, but some have fast unimolecular reactions, as discussed in Sect. 3.3 for radical + alkene and alkyne adducts and Sect. 3.4 for radical and aromatic adducts.
2.1.3 Additions of OH or NO3 to amines
Both OH and NO3 radicals are known to react rapidly with amines (McGillen et al., 2020; Carter, 2021), but the fact that the reaction with tertiary amines is about as rapid as reaction with secondary or primary amines indicates that the reaction is due to initial addition of the radical to the amino group, rather than direct H abstraction from the amino group (see discussion in Sect. S1.9 of Carter, 2021). Based on this, MechGen assumes that the rate-determining step is addition of the radical to the amino group, forming an addition complex that rapidly decomposes. An example is shown below.
In the case of tertiary amines, where reactions analogous to Reaction (R1) are not possible, the adduct is assumed to decompose by abstraction of an α-hydrogen if one is present, forming the same products that would be formed if the radical abstracted from the α-hydrogen directly. Both decomposition modes are possible for primary or secondary amines with α-hydrogens, but the formation of the α-amino carbon-centered radical (analogous to Reaction R2) is estimated to be more exothermic and is assumed to dominate. If there is no α-hydrogen, then only the reaction forming the amino radical (analogous to Reaction R1) is generated.
Neither of these decomposition modes are possible for tertiary amines with no α-hydrogens (e.g., tri-t-butyl amine), so MechGen assumes that the adduct decomposes back to reactants and there is no net reaction at the amino group. We are aware of no measurements or calculations of rate constants for these reactions, but they would be interesting to study.
2.1.4 Additions of O3 to unsaturated bonds
O3 is assumed to react only by additions to double- or triple-bond systems as follows, with rate constants estimated as indicated in Table 3d and with parameters as indicated in Table S3. Reactions of O3 with aromatics or cumulated double bonds are assumed to be negligible based on low measured rate constants (McGillen et al., 2020). Note that only total rate constants for additions to unsaturated bond systems are estimated, so it is necessary to assume branching ratios for reactions of the initially formed adducts or, in the case of additions to conjugated bond systems, for the initial reaction. The branching ratios used are shown in brackets where applicable.
Here, α and 1−α are branching ratios assigned for the additions to separated double bonds, G(=O) refers to a carbonyl (–CO– or –CHO) group, G[OO]{excited} refers to an excited Criegee intermediate (CI) whose subsequent reactions are discussed in Sect. 7, and * indicates rings. The additions to double bonds are assumed to proceed via initial formation of excited primary ozonides, which decompose to the products as shown above, while additions to alkynes are assumed to form the excited CIs directly. The assumption that the Criegee intermediates are formed entirely in the excited state is an approximation that is made to simplify the estimations of their subsequent reactions, as discussed in Sect. 7. In most cases it is assumed that formation of all the possible Criegee and carbonyl combinations are equally likely, as indicated above. However, following the recommendations of Jenkin et al. (2020), we estimate that α = 0.1 if G3 is –HCO, –CO–, –O–, or –OH and no such group is bonded to G1 or 0.5 otherwise (or if G3 is absent). In other words, formation of the carbonyl bonded to the oxygenated group is favored.
In the case of alkynes, the primary ozone adduct is assumed to directly form excited CIs with α-carbonyl substituents, with the two possible CIs formed in equal yields. However, it is assumed that the intermediates can rapidly interconvert by O shifting from the Criegee to the carbonyl group, so reactions of both of the isomers in this equilibrium need to be considered. The Criegee reactions are discussed in Sect. 7.
2.1.5 Additions of O3P to unsaturated bonds
O3P is assumed to react only by additions to double bonds, with rate constants estimated as indicated in Table 3d for isolated or conjugated double bonds and in Table 3e for additions to cumulated double bonds. Addition reactions involving O3P are expected to initially form a highly excited oxirane compound or biradical, which can stabilize, rearrange, or decompose to radicals. Based on model simulation results of high-NOx environmental-chamber experiments performed when developing the SAPRC mechanisms for alkenes (e.g., Carter, 2000, 2010b), it is assumed that radical formation from reactions of O3P with C3+ organics are negligible, so only stabilization or rearrangements forming stable compounds are assumed to occur (as shown below). This is assumed for conjugated alkenes and alkynes as well, though this assumption is more uncertain. Although it is assumed that additions occur for both sides of the double bond at the same time, it is necessary to assign branching ratios in cases where there are different possible rearrangements of the exited adducts. The general mechanisms are assumed as follows, with branching ratios assumed shown to the right in brackets.
The only type of rearrangement considered for the initially formed adducts is H shifts from one carbon to another, forming a carbonyl compound. In the case of additions to double bonds, it is assumed that stabilization occurs 50 % of the time if at least one rearrangement is possible (i.e., , ) and all of the time if it is not (i.e., and ). If more than one rearrangement is possible, they are assumed to be equally likely (). In the case of additions to triple bonds, it is assumed that stabilization (not shown) is unlikely and that there is no net reaction if the rearrangement shown is not possible (α6 = 0) and no back decomposition of the adduct otherwise (α6 = 1).
These assumptions are uncertain, but they have relatively little effect on model simulations of most atmospheric systems because reactions of O3P are generally not important unless NO2 is very high, such as in high-NOx environmental-chamber simulations or in combustion/biomass burning plumes.
2.1.6 Reactions of O3 and O3P with amines
The reactions of amines with O3 or O3P are assumed to form excited N oxides, with rate constants estimated as shown in Table 3e and parameters for other addition reactions as indicated in Table S3.
In the case of primary or secondary amines, these excited N oxides are assumed to rapidly rearrange and then stabilize to form –N–OH compounds (α7 = 1). In the case of tertiary amines, it is assumed that the N oxide is stabilized (α7 = 0). The current system does not handle reactions of stabilized N oxides, so it treats them as unreactive. Their reactions and mechanisms are unknown.
2.2 Photolysis reactions
Compounds with the groups –CHO, –CO–, and –ONO2 or the group pairs –O–OH and –O–O– are expected to undergo photolysis at non-negligible rates under lower-tropospheric conditions. These groups or group pairs are referred to as “photoreactive groups” hereafter. Compounds not containing these photoreactive groups may photolyze with UV wavelengths less than the atmospheric cutoff of ∼ 290 nm, but photolyses at this low-wavelength regime are not currently considered in MechGen. Photolysis of radicals and CIs is also ignored because it is assumed that loss of these intermediates by other reactions will dominate over photolysis under atmospheric conditions.
Rate constants for photolysis reactions depend on the compound, reaction, and lighting environment and can be calculated by
where J is the first-order rate constant for a photolysis reaction in a particular lighting environment, photolysis set is a set of wavelength-dependent absorption cross-sections and quantum yields assigned to various types of reactions, and φoverall is an overall wavelength-dependent quantum yield used for the reaction of interest. If needed, λ is the wavelength; σλ and ϕλ are the wavelength-dependent absorption cross-sections and quantum yields at wavelength λ in the photolysis set; and Fλ is the intensity of the light source at wavelength λ, which is a function only of the environment, not the reaction.
MechGen makes no assumptions about the light source (other than having no significant intensity at wavelengths below ∼ 290 nm) and therefore does not output estimated or assigned photolysis rate constants when generating photolysis reactions. Instead, it outputs the name of the photolysis set and the value of overall quantum yield (ϕoverall), if applicable, assigned to the reaction. The photolysis sets assigned by MechGen are a subset of those incorporated in the SAPRC-16, SAPRC-18, and SAPRC-22 mechanisms (Carter, 2016, 2020, 2023) and are summarized in the Supplement in Table S5. That table also indicates how they were derived and gives half-lives (with ϕoverall = 1) with direct overhead sun using the actinic fluxes in the reactivity scenarios of Carter (1994) and orders them by increasing half-life. The files containing the data for these photolysis sets are available online at the SAPRC website (Carter, 2024b). Photolysis rate constants can be calculated using these data and are required for model application of mechanisms generated using MechGen. Photolysis rate constants should be calculated for the light conditions of the experimental or atmospheric conditions of interest. The mechanisms, photolysis sets, and (where applicable) overall quantum yields for the various types of photolysis reactions are summarized below. The photolyses of compounds with only a single photolysis group are described first, followed by a discussion of treatments of photolyses of compounds with more than one such group. Note that if there is more than one reaction, the branching ratios shown are implemented as overall wavelength-independent quantum yields and are assumed not to depend on the spectrum of the light source.
In most cases the estimated mechanisms, absorption cross-sections, and quantum yields for compounds with photoreactive groups are based on those derived for the smallest C3+ or C4+ compound with the photoreactive group(s), with all the other groups being alkyl (–CHx–) groups. This is clearly an oversimplification, especially since the structure and size of the molecule, as well as the presence of non-alkyl substituents, can affect the absorption cross-sections or quantum yields for at least some of the different types of photoreactive compounds (Calvert et al., 2011). Although MechGen currently does not take these differences into account, this needs to be considered in future versions.
2.2.1 Compounds with a single aldehyde group
Aldehydes with a single –CHO group that are not directly bonded to any unsaturated or photoreactive group, other than acetaldehyde and glycolaldehyde for which photolysis assignments are given explicitly (see Supplement), are assumed to photolyze as follows:
where R is any carbon-centered group other than methyl that does not contain a double bond on the group next to the –CHO. The absorption cross-sections used are those recommended by IUPAC (2023) for propionaldehyde and are given in photolysis set C2CHOabs. IUPAC makes no specific recommendations concerning quantum yields or branching ratios, but it appears likely that the quantum yield •CHO formation for λ≳ 295 nm is near unity, based on the data of Chen and Zhu (2001). MechGen assumes that this is applicable for higher aldehydes of this type as well.
Aldehydes where the –CHO group is bonded to a C=C double-bond group are assumed to photolyze with a mechanism based on that derived for acrolein, which is taken as representative of all α-unsaturated aldehydes, including those with conjugated or cumulated double bonds, and with non-alkyl (but non-photoreactive) substituents. The photolysis set giving the absorption cross-sections and quantum yields used for acrolein is ACROL-16, while MACR-15 (based on methacrolein) is used for the others, though the acrolein mechanism is used in all cases to predict the subsequent reactions that occur. This gives half-lives of ∼ 50 and ∼ 60 h, respectively, for loss by photolysis via direct overhead sunlight, which means that photolysis is relatively slow but not necessarily negligible. The assumed mechanism is as follows.
Here, x = 0–2; x′ = 0–1; and [] indicates a carbene radical group, whose subsequent reactions are discussed in Sect. 6.3. These branching ratios are based on the data of Gardner et al. (1987) for acrolein, as shown in Fig. IX-C-7 of Calvert et al. (2011) for air at 1 atm. These are assumed to apply to photolyses of other unsaturated aldehydes as well.
Aldehydes where the –CHO is bonded to aromatic groups are assumed to photolyze with the same rate constant and a similar mechanism as estimated for benzaldehyde, regardless of substituents. The photolysis set used is BALD-11, where absorption cross-sections are those recommended for benzaldehyde by Calvert et al. (2002). In addition, φoverall = 0.06 is used to approximately fit consumption rates of benzaldehyde in chamber experiments (Carter, 2000).
Benzaldehyde acts as an efficient radical terminator when added to environmental-chamber experiments (Carter et al., 1982; Carter, 2000), suggesting that photolyses of aromatic aldehydes do not form radicals. (The reaction with OH radicals is also predicted to be radical terminating, but that would not be sufficient to compensate for radical initiation due to photolysis if the quantum yield for radical production were non-negligible.) For mechanism generation purposes, these are represented as follows:
where –aCx– refers to either –aC– or –aCH– and * indicates ring closure. The actual photolysis reaction is highly uncertain, and it is likely that other products are actually formed, since we know of no evidence that aromatic hydrocarbons are formed in high yields from the photolyses of aromatic aldehydes.
Aldehydes where the –CHO is bonded to an alkyne group, e.g., –C≡C–CHO, are assumed not to photolyze. We could not find data concerning absorption cross-sections or photolyses of these compounds and assume that they either do not photolyze or do so relatively slowly. Data are needed to test this assumption.
2.2.2 Compounds with a single ketone group
C4+ ketones with a single –CO– group that is not directly bonded to any unsaturated or photoreactive group are assigned photolysis sets, giving absorption cross-sections and overall quantum yields as indicated in Table 4. In the case of acetone, the wavelength-dependent quantum yields recommended by IUPAC (2023) for atmospheric temperature and pressure are used. In the case of the higher ketones, the photolysis set only has absorption cross-sections, and overall quantum yield factors are derived so that the SAPRC mechanism simulates environmental-chamber data for ketones (Carter, 2010b). These data suggest that the photolysis rate constants and therefore overall quantum yield factors decrease with the carbon number for C4+ ketones. The structure of the ketone and the presence of non-alkyl but non-photoreactive substituents, other than –O– groups next to the carbonyl (i.e., esters), are assumed not to affect the absorption cross-sections or quantum yields.
Table 4Assignments used to estimate photolysis rate constants for compounds with single ketone groups based on the numbers of carbons in the ketone.
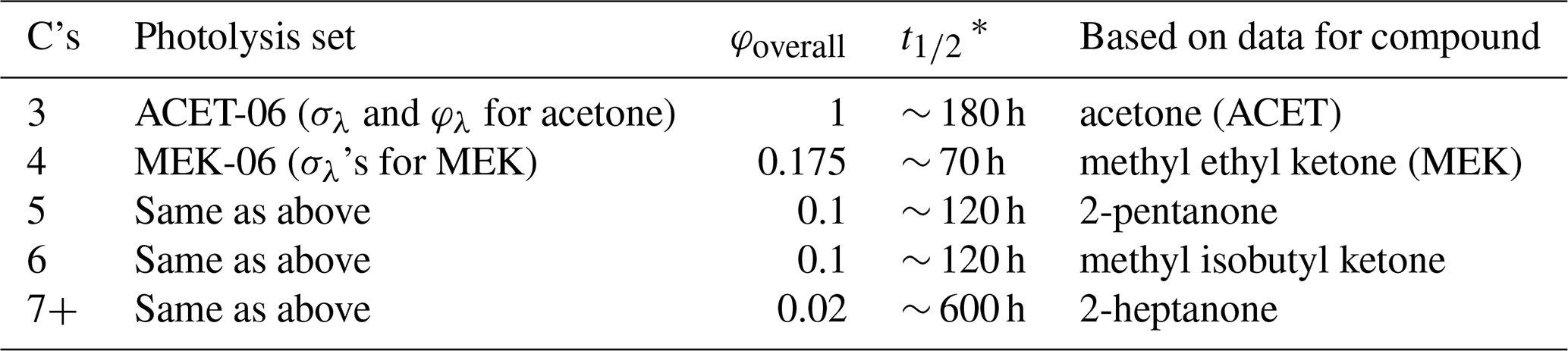
* Half-life relative to the photolysis rate constant for ketone photolysis calculated for direct overhead sunlight (z = 0) using the actinic fluxes used in the reactivity scenarios of Carter (1994).
The photolysis of these ketones is assumed to proceed only by breaking one of the two C–CO bonds, forming an acyl and an alkyl radical.
Here, G is any non-photoreactive group and α1 is the branching ratio for the formation of radical G1•. The relative branching ratios for breaking CO–alkyl bonds are assumed to depend on whether the radical formed is a methyl, secondary, primary, or tertiary, with equal branching ratios if both radicals are of the same type. Based on IUPAC (2023) data for methyl ethyl ketone, we assume branching ratios of methyl vs. ethyl of 15 % and 85 %, respectively, for that compound. Extrapolating this to other higher compounds, we use the following branching ratios, where α = 0.5 is used if both are of the same type.
Note that this is somewhat arbitrary and uncertain (except for methyl ethyl ketone) and is assumed not to be affected by the presence of non-alkyl substituents, which is even more uncertain.
If the ketone group is in a ring, the reaction would form excited biradicals that should re-form the starting ketone if stabilized. In this case, it is assumed that the excited biradical primarily decomposes by CO elimination, i.e.,
where * indicates closure for any size ring.
Esters, carbonates, and acids, i.e., compounds with –CO–O– or –CO–OH group combinations, do not appear to have significant absorption in the atmospheric wavelength region and are assumed not to photolyze.
Estimations of photolyses of ketones with an adjacent double-bond group are based on data for methyl vinyl ketone (MVK). These reactions are assigned the photolysis set MVK-16, which contains IUPAC (2023) recommendations for absorption cross-sections and quantum yields of methyl vinyl ketone at 1 atm. This gives an estimated half-life of ∼ 36 h for photodecompositions of these unsaturated ketones with direct overhead sunlight and gives reasonably good fits to NO oxidation rates in MVK–NOx chamber experiments.
The photolysis reactions for ketones with only a single adjacent double bond are assumed to be as follows.
Here, x = 0–2, x′ = 0–1, and G is any non-photoreactive group with only saturated bonds. This is based on IUPAC (2023) recommendations for MVK but is assumed to apply to all compounds of this type.
Ketones with double-bond groups on both sides of the carbonyl group are estimated to photolyze with the same absorption cross-sections and quantum yields as those bonded to only one double-bond group, but it is assumed that formation of vinylic radicals is not as favorable based on thermochemical estimates. These reactions are assumed to occur only via CO formation, as shown in the second reaction above. This is also assumed for unsaturated ketones where the –CO– group is in a ring, where the first reaction is not expected to occur for the reasons discussed above.
Estimations of photolyses of ketones with an adjacent aromatic or triple-bond group are not supported by MechGen, so no photolysis reactions are generated for these compounds. The need to represent photolyses of these compounds has not been investigated, but it is expected that photodecompositions of aromatic ketones will be relatively slow, and ketones with triple bonds are not expected to be important in atmospheric systems.
2.2.3 Organic nitrates, carbonyl nitrates, and carbonyl peroxynitrates
Compounds with a single organic nitrate group and no other photoreactive groups are assumed to photolyze entirely by scission of the weak O–NO2 bond, forming NO2 and an alkoxy or acyloxy radical whose reactions are discussed in Sect. 5.
These reactions are assigned the IC3ONO2 photolysis set with quantum yields of 1 at all wavelengths, which gives a half-life of ∼ 50 h for loss due to photolysis with direct overhead sunlight. This is based on the IUPAC (2023) recommendation for isopropyl nitrate, but it is assumed to apply to all organic nitrates without other photoreactive groups, regardless of structure, the presence of non-alkyl substituents, and the presence of unsaturated bonds. This is an approximation because isopropyl nitrate has stronger absorptions than for n-propyl and smaller nitrates but is taken as representative. Although the accuracy of this approximation is uncertain in some cases, loss by photolysis is relatively unimportant compared to other reactions.
In the case of acyl peroxynitrates (e.g., PANs), the photolyses are assigned the PAN-11 or PPN-11 photolysis sets, with the former used for PAN itself and the latter used for all other PAN analogues based on peroxypropionyl nitrate (PPN). These contain only absorption cross-sections, since unit quantum yields are assumed. They give approximate photodecomposition half-lives of ∼ 200 and ∼ 100 h, respectively, for direct overhead sunlight. The assumed mechanism is as follows.
This is based on Sander et al. (2006) recommendations for PAN but is applied to all PAN analogues regardless of substituents or the presence of multiple bonds.
In the case of non-acyl (e.g., alkyl) peroxynitrates, photolysis is assumed to proceed primarily by NO2 formation,
with the same rate constant assignments as used for simple organic nitrates. However photolysis is not important for these compounds because they are assumed to undergo rapid thermal unimolecular decomposition, forming the same products, as discussed in Sect. 2.3.
2.2.4 Compounds with a single set of peroxide groups
Compounds with peroxy or hydroperoxy groups and no other photoreactive group are assumed to photolyze entirely by scission of the weak O–O bond, forming alkoxy radicals and (for hydroperoxides) OH.
The reactions are assumed to have unit quantum yields at all wavelengths and are assigned the photolysis set COOH, which contains the absorption cross-section of methyl hydroperoxide (IUPAC, 2023) and gives a half-life of ∼ 30 h for loss by photolysis with direct overhead sun. Note that it is assumed that organic peroxides (which tend to be relatively less important than hydroperoxides in atmospheric systems) photolyze with approximately the same rate constant as hydroperoxides and that the size, structure, and presence of non-photoreactive substituents or unsaturated bonds do not have a significant effect. The most questionable assumption in this case is the assumption that the presence of adjacent unsaturated bonds has no effect on hydroperoxide or peroxide photolysis.
2.2.5 Compounds with more than one photoreactive group
The photolysis sets, quantum yields, and photolysis reactions used for compounds with various combinations of photoreactive groups are summarized in Table 5. If the φoverall column is blank, then no overall quantum yield correction is applied and the photolysis rate constant is the same as that calculated for the photolysis set. The half-lives relative to photodecomposition for direct overhead sun, which incorporate the overall quantum yield corrections if applicable, are also shown in the table. No corrections are made for non-alkyl substituents or unsaturated bonds on the molecule except as indicated in the table. The considerations used when making these assignments are discussed below.
Table 5Summary of photolysis sets, overall quantum yields, approximate photolysis half-lives, and products formed for compounds with more than one photoreactive group.
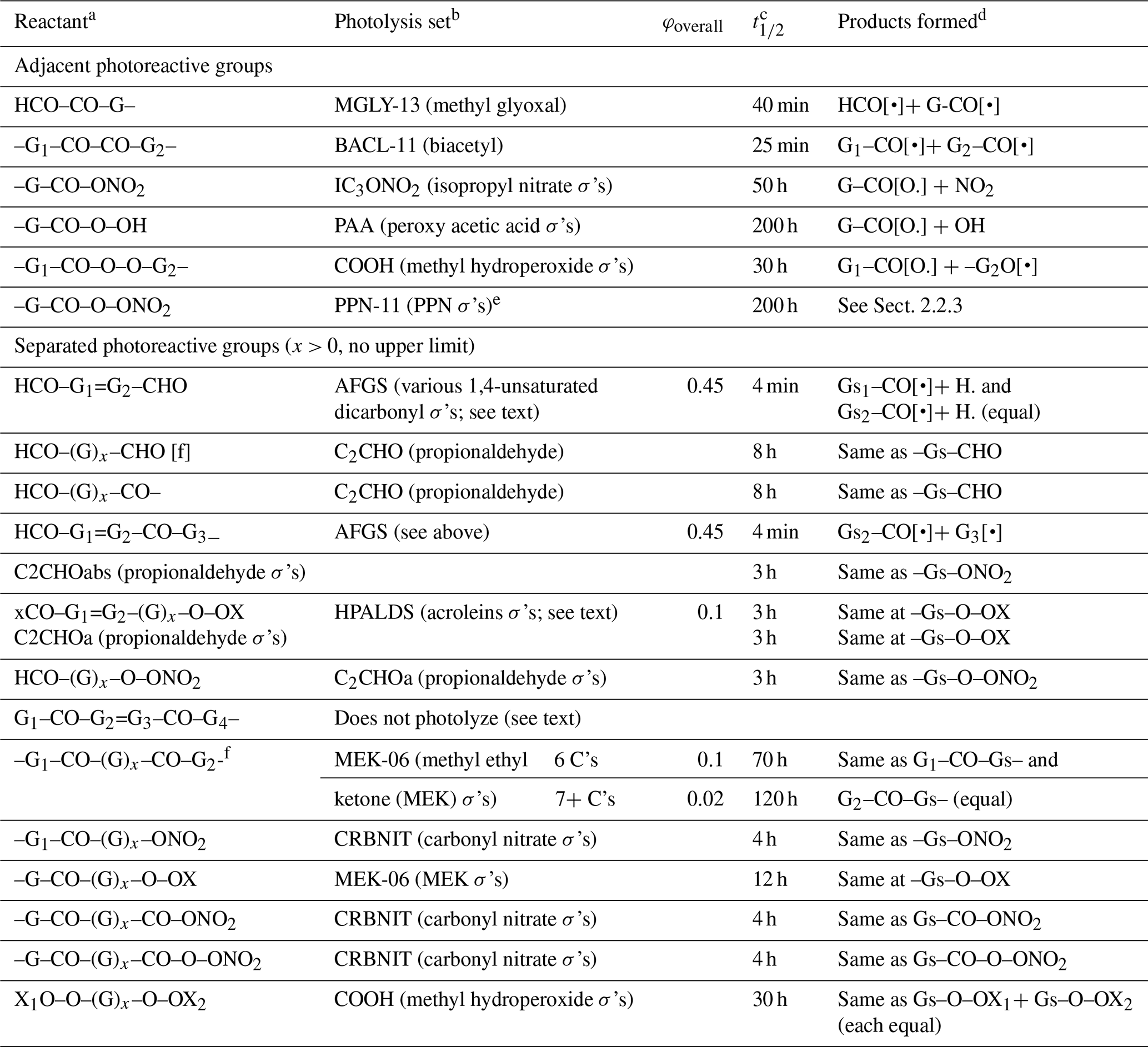
a Group combinations that give unique compounds for which specific assignments are used (e.g., glyoxal and HNO3) are not shown. –OX is –O– or –OH. xCO– is HCO– or –CO–.
b The derivations of the photolysis sets are given in Table S5. σ's means that the photolysis set contains only absorption cross-sections, and (equal) means that both possible reactions occur with equal probability.
c Approximate half-lives relative to loss of the compound by photolysis for direct overhead sunlight, including the contribution by the assigned wavelength-independent quantum yield, if applicable.
d Gs refers to a combination of other groups on the molecule (other than G, G1, or G2), including the other photoreactive group, which is assumed not to be transformed by the reaction.
e Photolysis set PAN-11, giving an approximate photolysis half-life of 100 h for direct overhead sun, is used for PAN itself (G = CH3).
f Not applicable if –(G)x– is –CH = CH, as shown for the previous reactant.
The assignments for some (but not all) types of reactants with adjacent photoreactive groups are made based on available data for the simplest or representative molecule of the types, i.e., methyl glyoxal, biacetyl, peroxy acetic acid, PANs, carbonyl nitrates, dinitrates, and various unsaturated 1,4-dicarbonyls. The applicability to higher-molecular-weight compounds, compounds with non-alkyl substituents, or compounds with multiple bonds is uncertain but not as uncertain as the assignments for the reactants in the table where data for representative compounds either are not available or were not considered.
It is assumed that –CO–O– groups are not photoreactive, so the presence of this group combination is not considered when making photolysis assignments. The exception is compounds with –CO–O–ONO2 group combinations (PAN analogues), where assignments are made based on data for PAN and peroxypropionyl nitrate (PPN) as discussed above.
The unsaturated 1,4-dicarbonyls with at least one aldehyde group are highly photoreactive aromatic-ring-opening products (Calvert et al., 2011; see also Sect. 3.4, below) that account for the relatively high reactivity of aromatic hydrocarbons observed in environmental-chamber experiments. The AFGS photolysis file is used to give absorption cross-sections for all these compounds, with an overall wavelength-independent quantum yield adjusted to give satisfactory fit to results of aromatic–NOx chamber experiments, which are highly sensitive to this photolysis (e.g., Carter, 2010b; Carter and Heo, 2013). The SAPRC-22 mechanism (Carter, 2023) that was derived using this version of MechGen best fit the data, using effective quantum yields close to 25 %. However, the quantum yields in the AFGS file do not take into account absorption cross-sections reported for 4-oxo-2-pentenal reported by Xiang et al. (2007), which are the basis of the current IUPAC (2023) recommendations for this compound. This is because these quantum yields, which are much lower than the previous data for these compounds, could not be used as a basis for a predictive mechanism for aromatics because fitting the chamber data required using an effective quantum yield of greater than 3. Therefore, either there are additional sources of radicals in aromatic photooxidation systems that are not taken into account in the current estimated mechanisms for aromatics, the absorption cross-sections of these 1,4-dicarbonyls are highly dependent on structure, or the data of Xiang et al. (2007) have systemic issues. Until this situation is resolved and MechGen can be updated to derive a more predictive mechanism, we will continue to use the present cross-sections in AFGS to derive aromatics mechanisms for airshed models.
It is assumed that unsaturated 1,4-diketones do not photolyze sufficiently rapidly in ambient sunlight in a manner necessary to include their photolysis in atmospheric models. This is consistent with the discussion of photolyses studies of 3-hexene-2,5-dione given by Calvert et al. (2011), which indicate that radical formation is relatively minor compared to isomerization. This is also consistent with the fact that para-substituted dialkyl benzenes, whose reactions can form these compounds, tend to be less reactive than other isomers, all else being equal (Carter and Heo, 2013).
If a reactant has more than one non-adjacent photoreactive group of the same type, it is assumed that the photolysis reaction at each group occurs with equal probability, with the mechanism the same as if there were no other photoreactive group. However, the total photolysis rate constant for reactions at both groups is estimated to be the same as if the compound had only one group since the same photolysis set is used. This is uncertain.
If a reactant has more than one non-adjacent photoreactive group of different types, it is assumed that the absorption cross-sections can be approximated by those of compounds with the single group that has the highest absorption cross-section at atmospheric wavelengths. On the other hand, the mechanisms (e.g., photolysis products) are assumed to be those resulting from reaction at the group with the weakest bond. This is based on the assumption that once the photon is absorbed the energy is rapidly distributed around the molecule. This is consistent with the data of Wolfe et al. (2012), which suggest that α-unsaturated carbonyls with hydroperoxide groups photolyze at rates consistent with those calculated using absorption cross-sections of α-unsaturated carbonyls but with unit quantum yields and with the reaction breaking the peroxy bond forming OH. This gives a photolysis rate that is about 100 times faster than simple hydroperoxides. However isoprene and 1,3-butadiene NOx experiments are not well simulated with this high of a photolysis rate for α-unsaturated hydroperoxy carbonyls (Carter et al., unpublished results), so we arbitrarily cut the rate by a factor of ∼ 10 by using an effective quantum yield of 0.1 for these compounds. The current system assigns unit quantum yields for saturated hydroperoxy or nitrate-substituted carbonyls, though this is highly uncertain and the estimates are probably upper limits.
Despite the differences in assigned overall quantum yields, the photolysis rate constants calculated for saturated carbonyls with other photoreactive groups are about the same as for those that are unsaturated (see Table 5). This is due to the higher absorption cross-sections assigned to unsaturated aldehydes.
2.3 Unimolecular reactions
2.3.1 Peroxynitrates
Peroxynitrates are formed from the reactions of peroxy or acyl peroxy radicals with NO2, but the O–ONOx bond is weak enough that these can thermally decompose back to reactants at significant rates at atmospheric temperatures.
Here, G is any saturated or unsaturated group and the rate constants are per second, T is the temperature in kelvin, and R is K mol kcal−1. The presence of non-alkyl substituents, unsaturated bonds, or other structural factors is assumed not to affect these rate constant estimates or decomposition mechanisms. The OO–NO2 bonds are currently believed to be the only bonds in compounds formed in atmospheric systems to be weak enough to undergo simple scission at non-negligible rates at atmospheric temperatures. (The weak O–O bonds in peroxy compounds are at least 10 kcal mol−1 stronger.)
The decompositions of the non-acyl peroxynitrates are estimated to have a half-life of ∼ 5 s−1 at 298 K, which is sufficiently fast for the formation of these compounds to be rapidly reversed. Therefore, the default is for MechGen to ignore the formation and therefore the decomposition of these compounds when multi-step mechanisms are generated for atmospheric conditions. However, they do show up when single step mechanisms are generated (Carter, In Prep.). On the other hand, the acyl peroxynitrates (PANs) decompose much more slowly and are observed as photooxidation products in atmospheric systems, so their formation cannot be ignored. It cannot be ruled out that some α substituents other than –CO– may slow down the decomposition sufficiently so that peroxynitrate formation for some non-acyl peroxynitrates may be non-negligible. However, this is not currently considered in MechGen.
2.3.2 Aromatic formation from cyclohexadienones
The other type of non-radical unimolecular reaction that is currently considered involves formation of aromatic rings from 2,4-cyclohexadienone compounds containing a –CHX–CO– group pair in the ring, where an H shift from the –CHX to the carbonyl group is expected to be highly favorable energetically.
MechGen predicts that cyclohexadienones are formed in high yields in the reactions of phenolic compounds if this reaction does not occur (see Sect. 3.4), but they are not observed as products. On the other hand, catechols (the products of this type of isomerization reaction) are observed in high yields in the reactions of OH with phenols (e.g., Olariu et al., 2002; Berndt and Böge, 2003), which would not be expected if this reaction did not occur (Xu and Wang, 2013). The nature of the X group should not have an obvious effect on how fast this reaction occurs, so if this is fast, then it is reasonable to expect compounds with other X substituents, including X=alkyl or H, are also fast. MechGen assumes that this is the case. The rate constant is unknown, but it is assumed to be high in order to account for the observed formations of catechols from phenols. This assumption is implemented by treating such compounds like rapidly reacting radicals when the reactions are generated.
It is possible that this reaction may require the presence of surfaces and/or water to occur at sufficient rates. The H shift involves a strained four-member ring transition state, which may be catalyzed by surfaces, while the involvement of water would result in the same H shift but with a six-member ring transition state. Given surface- or water-free environments rarely occur in the ambient atmosphere, it is probably better for these reactions to be included for modeling applications than to be neglected. For example, the predicted formation of catechols from phenols due to this reaction has implications for predictions of SOA from aromatics.
2.3.3 Other compounds
Decompositions of peroxynitrates and rearrangements of cyclohexadienones are currently the only types of non-radical unimolecular reaction considered by MechGen. In particular, MechGen does not consider other types of water- or surface-catalyzed conversion of non-radical products that could affect the environmental fate of multifunctional products. For example, geminal diols, compounds with HO–CX2–OH groups, are predicted to be formed in some cases and would be expected to be in equilibrium with H2O and XC(=O)X in the environment. It is uncertain whether this or similar reactions would occur at significant rates in the gas phase, and MechGen currently assumes that this is not the case. However, ignoring these potentially heterogeneous or H2O-catalyzed reactions may also have implications for SOA predictions.
Carbon-centered radicals are formed in the initial reactions of most VOCs with atmospheric oxidants, from H-shift isomerization reactions of peroxy and alkoxy radicals and from most alkoxy radical decompositions. Under lower-atmospheric conditions, their major fate is expected to be a reaction with O2, which is estimated to occur with a pseudo-unimolecular rate constant of ∼ 3.8×107 s−1 (see Table S6 for measured radical + O2 rate constants and associated text in the Supplement). However, rapid unimolecular reactions are estimated to dominate for some types of carbon-centered radicals formed in generated mechanisms, as discussed below. Note that there are currently no cases where MechGen assumes that both O2 and unimolecular reactions of carbon-centered radicals occur at competitive rates – one or the other is assumed to dominate. This means that estimates of carbon-centered radical + O2 rate constants are not currently needed or used in the generated mechanism, though estimates of these rate constants are used when considering when unimolecular reactions may dominate.
3.1 Reaction with O2
Most carbon-centered radicals are assumed to primarily add O2 and form the corresponding peroxy radical, whose reactions are discussed in Sect. 4.
However, the peroxy radicals formed from α-OH-substituted radicals are assumed to rapidly decompose to form HO2 and the corresponding carbonyl.
MechGen treats these as concerted processes when reactions of α-OH-substituted carbon-centered radicals are generated, since stabilization and other reactions are expected to be unimportant.
If the carbon-centered radical is adjacent to one or more double-bond groups, it is assumed to be a resonance structure where O2 can add to any of the radical positions, e.g., the following.
Here, α(Ga,Gb) is the fraction of adding to the Ga position when Gb is the other position on the radical. Additions to radicals with more than one double bond are assumed to form only those adducts with the most conjugated structures, e.g., the following.
Because of lack of data indicating otherwise, we assume that the branching ratios, α, depend only on the nature of the radical groups where the addition occurs, regardless of the number of double-bond groups in the radical. Although this has not been examined comprehensively, we believe that all allylic radical structures that may arise in generated mechanisms have only two radical centers where O2 addition forms the most conjugated structures. This is supported by the fact that we have not found examples of where this is not the case when generating many mechanisms.
Available information and estimates concerning branching for allylic additions is discussed in Sect. S1.4.2. There are very limited data, but what is known does not support the assumption that additions at each position are equally likely. Product data for the reactions of 1,3-butadiene and isoprene with OH indicate that in radicals with both primary (–aCH2[•]) and secondary (–aCH[•]–) radical positions, the addition occurs ∼ 65 % of the time at the secondary position. This is consistent with the fact that thermochemical analysis indicates that addition to the secondary radical is ∼ 2 kcal mol−1 more favorable (see the Supplement). In addition, theoretical calculations of Xu and Wang (2013) on phenol mechanisms and of Yuan et al. (2017) on furan mechanisms both indicate that O2 preferentially adds to radical centers with –OH or –OR substituents, instead of those with unsubstituted secondary radical centers. However, if this is assumed, then the mechanisms significantly underpredict reactivities observed in environmental-chamber experiments with phenolic compounds, and better fits to the data are obtained if it is assumed that addition to non-OH-substituted secondary radical centers occur approximately 15 % of the time (Carter et al., unpublished results). This is highly uncertain, but it provides an additional data point for deriving general estimation method incorporating -OH-substituted radicals.
As discussed in Sect. S1.4.2, we assume that the branching ratios can be estimated based on the differences in heats of addition of O2 to the different radical positions, with a correction included if one of the radical centers is -OH- or -OR-substituted. The estimate used is as follows:
where
and ΔΔHr(G) is the difference in heats of reaction for O2 addition to G compared to a primary radical center in a radical that has both, β is a parameter assigned a value of 0.33 to be consistent with the product data for isoprene and 1,3-butadiene, and Ocorr is set at 5.78 kcal mol−1 if G is –OH- or –OR-substituted to be consistent with modeling results for phenols or 0 if there are no such substituents. The weighting factors so derived are as follows (see Sect. S1.4.2 for derivations):
Note that use of this methodology to estimate branching ratios in general is highly uncertain because only two data points are available to derive the two parameters used (β and Ocorr), with no data available to test this estimation method. However, this is the best that can be done at the present time.
The above discussion is not applicable to radicals that are estimated to have rapid unimolecular reactions as discussed in Sect. 3.2. If such a rapid reaction is possible for one of the radical centers in an allylic radical, it is assumed that all the reaction goes by this route unless a rapid reaction is possible at the other radical center, in which case it is assumed that they occur with equal probability. The above discussion is also only applicable to stabilized radicals and not necessarily for excited adducts formed when OH or NO3 adds to double bonds. In most cases excited adducts formed following additions to double bonds are assumed to be primarily stabilized and then react as discussed above, but that is not always the case, as discussed in Sect. 3.3.
A different mechanism is assumed for the addition of O2 to vinylic radicals where the radical center is on an unsaturated carbon. In this case, the reaction with O2 is expected to involve the O2 adding to each side of the bond, with the bonds rearranging to form a carbonyl product and a carbonyl radical, i.e.,
This is based on data of Slagle et al. (1984) for vinyl and Slagle and Gutman (1988) for methylvinyl and i-C4H9•. More recently, Matsugi and Miyoshi (2014) directly measured the yield of HCO radicals from vinyl and observed that the yields were only about 0.2, independent of pressure, with the remainder of the reaction being prompt dissociation to H + CO. This is assumed to be applicable to additions to radicals of any size, though it is possible that rapid energy distribution following a reaction of larger vinyl species with O2 could lead to stabilized vinylperoxy radicals. However, insufficient data are currently available to quantitatively assess the degree to which this occurs, so MechGen assumes that the stabilization is not important. The issue of whether the HCO promptly decomposes is not relevant to atmospheric modeling, since the same products (HO2 and CO) are formed under atmospheric conditions in either case. Therefore, MechGen represents the reactions of CX2=CH• as forming H• + CO alone, since this is the major overall process.
However, the issue of prompt dissociation is relevant in the case of RC(=O)•, since the stabilized radical is expected to primarily add O2 to form an acyl peroxy radical, which can give different products than if RC(=O) decomposes to R• + CO. Decomposition is expected to be more favorable because the lower estimated heat of reaction, though the rate of stabilization of the RC(=O)• should also be greater than for HC(=O)•. MechGen assumes that the energetics is the larger factor in the case of additions of O2 to vinylic radicals; assumes that the prompt dissociation dominates; and also represents the overall reaction as forming the carbonyl, CO, and R• as a single process.
3.2 Unimolecular reactions of stabilized radicals
Several types of carbon-centered radicals are expected to undergo unimolecular reactions sufficiently rapidly that unimolecular reaction is assumed to dominate over reaction with O2, even for thermalized radicals. Note that if these reactions are assumed to be fast for thermalized radicals, they are necessarily assumed to be fast when the radicals are excited.
3.2.1 Rapid α-scission decompositions
Carbon-centered radicals with adjacent hydroperoxy, peroxide, nitrate, or dicarbonyl groups are expected to rapidly undergo decompositions by the simple scission of an adjacent bond, forming products that are more energetically favorable.
We expect that there should not be large entropy or energy barriers to these reactions (e.g., Green et al., 1990; Vereecken et al., 2004; Vereecken, 2008), so MechGen assumes they all dominate over O2 addition whenever radicals of these types are formed. Note that if the peroxy, nitrate, or carbonyl substituent is next to an allylic radical center, the bond scission reaction is still expected to dominate over O2 addition,
so there is no O2 addition to the other radical centers. Reactions of allylic radicals with peroxy, nitrate, or dicarbonyl groups are treated similarly.
3.2.2 Cyclopropyl ring-opening decompositions
Carbon-centered radicals with the radical center in a cyclopropyl ring can decompose by breaking the bond opposite the radical center, relieving the ring strain and forming an energetically favorable allylic stabilized radical.
This reaction is not expected to have large or any barriers, so it is assumed to dominate over O2 addition. On the other hand, the analogous reactions when the radical center is in a cyclobutane ring is estimated to be much less energetically favorable and is only expected to occur in chemically activated radicals, as discussed in Sect. 3.3. The reactions for larger rings are estimated to be endothermic and are assumed to be unimportant relative to stabilization and/or O2 addition even for excited radicals.
3.2.3 Cyclizations forming five-member ring cyclic esters
As discussed in Sect. S1.4.3, Curran et al. (1998) provided estimates for the Arrhenius parameters for decompositions of various QOOH radicals of the type
as a function of cyclic ether ring size. These can be used as a basis for estimating the approximate importance of these and similar reactions in atmospheric systems. In general, both the estimated Arrhenius A factors and activation energies decreased with ring size, with the net result of these opposing factors being that formation of five-member cyclic ethers is generally estimated to be most favorable for atmospheric conditions. Nevertheless, these cyclization reactions are estimated to be too slow for such radicals with only alkyl or hydroperoxide groups to compete with reaction with O2 at atmospheric temperatures.
However, as discussed in Sect. S1.4.4, extrapolating the relation between the rate constant and heats of reaction derived from the results of Curran et al. (1998), in which estimates that cyclizations of carbonyl-containing radicals forming five-member ring esters are ∼ 10 times faster than O2 addition for atmospheric conditions.
In addition, analogous cyclizations forming cyclic esters can also occur with carbon-centered radicals with peroxy, nitrate, or peroxynitrate substitution in the δ position and are also estimated to be faster than O2 addition, i.e.,
where X is G or NO2 and XO is G-O• or NO3.
However, as also discussed in Sect. S1.4.3, Miyoshi (2011) reports results of theoretical calculations of hydroperoxyalkyl rate constants that extrapolate to much lower rate constants at atmospheric temperatures, suggesting that the cyclizations forming five-member ring esters may not be as fast as estimated using the results of Curran et al. (1998). On the other hand, Vereecken and Nozière (2020) indicated that some rate coefficients given by Miyoshi (2011) may not be accurate, and other theoretical work may yield higher rate constants when extrapolated to room temperature (e.g., Ali and Saswathy, 2023). These reactions may still be important for excited radicals. This will need to be re-examined in a subsequent version of MechGen, but presently this reaction is still assumed to be fast even for thermalized radicals.
These reactions are assumed to be fast for mechanism generation purposes if (1) there is a carbonyl group either at the radical center or next to the peroxy oxygen that becomes bonded to the radical center; (2) the cyclic ester, carbonate, or anhydride has a five-member ring; and (3) the radical is not stabilized by resonance. O2 addition is assumed to dominate if not all of these conditions are met. For simplicity and to avoid the necessity of making highly uncertain quantitative rate constant estimates, we assume that either decomposition or O2 addition dominates, with the other being negligible. This assumption can be revisited in the future if the ability to estimate these decomposition rate constants for atmospheric conditions improves.
Although it is not likely in reactions of most VOCs and their oxidation products, in some cases carbon-centered radicals may have more than one peroxy group in positions where decompositions are estimated to be rapid. In these cases, it is assumed that the reactions occur at equal rates.
3.3 Unimolecular reactions of chemically activated radicals
Carbon-centered radicals formed by the addition of OH to double bonds are initially formed with internal excitation and in some cases might undergo unimolecular reactions that may not be important for stabilized radicals. Reactions of the chemically activated radicals formed following additions to aromatic rings are discussed separately in Sect. 3.4. Reactions that are assumed to be fast even for thermalized radicals were discussed in the previous section, and they are also assumed to dominate for excited radicals as well. Reactions that are assumed to dominate for radicals excited by additions of OH radicals to double bonds but which not be important for stabilized radicals are listed in Table 6 and are discussed in more detail in Sect. S1.4.3 and S1.4.4 in the Supplement.
Table 6Summary of types of unimolecular reactions that are assumed to be non-negligible for chemically activated carbon-centered radicals formed following additions of OH to double bonds.

a The section in the Supplement where the reaction and the estimated branching ratios are discussed is shown in parentheses.
b * indicates ring closure, {excited} indicates excitation, {OHadd} indicates excitation by OH addition only, + M indicates stabilization, X is H or any group, and •OX is OH or G–O•.
c Branching ratio assumed for 1 atm pressure (the default) if stabilization is assumed to be non-negligible. Note that MechGen allows for changes in atmospheric pressure, in which case this ratio would change assuming that the stabilization rate constant is proportional to pressure.
Note that additions of NO3 to double bonds also result in the formation of excited radicals, but the excitation energy is estimated to be ∼ 10 kcal mol−1 less than OH addition. Although this is uncertain, we currently assume that stabilization is correspondingly more important for NO3 adducts, and the unimolecular reactions shown in Table 6 are assumed not to occur when mechanisms for reactions of NO3 with alkenes are generated.
3.4 Reactions of radicals formed by additions to aromatic rings
OH radicals can react by adding to various positions around the aromatic ring, with the branching ratios derived using SARs for OH reactions with aromatics, as discussed in Sect. 2.1.2 and by Carter (2021). These excited OH–aromatic adducts can either decompose back to aromatic–OH or be stabilized and then react with O2. The effect of the back decomposition is taken into account in the SARs for OH addition, which are based on total rate constants, so only reaction with O2 is considered. O2 reaction can occur either by abstracting the H atom on the carbon where the OH added; forming HO2 and a phenol; or adding to the ring, forming an OH–aromatic–O2 adduct. As discussed below, one of the possible reactions of this adduct is decomposition to form the same products as the abstraction reaction; for simplicity we assume that this is the major pathway for formation of phenolic products, so only the addition reaction is considered.
The OH–aromatic adduct is an allylic carbon-centered radical with three possible radical centers where O2 can add. Although these adducts have some excitation, we assume that the branching ratios for O2 addition to the different positions are the same as estimated for stabilized allylic radicals, as discussed in Sects. 3.1 and S1.4.2. This means that all the addition is assumed to occur at the ortho positions since this forms the adducts with the most conjugation; that addition to an alkyl-substituted ortho position is ∼ 60 % less likely than adding to the other ortho position; and that addition to an -OH-substituted ortho position (as occurs in reactions with phenols) is ∼ 7 times more likely, assuming that the other ortho position is unsubstituted.
The aromatic–OH–O2 adducts are expected to be formed with some excitation and are assumed to undergo various types of unimolecular reactions as discussed below. Although peroxy radical reactions are discussed separately in Sect. 4, MechGen incorporates the rapid aromatic–OH–O2 isomerization reactions into the overall reactions generated for excited OH–aromatic adducts, so these reactions are discussed in this section.
The subsequent reactions of the excited adducts are assumed to be as shown in Fig. 1, which uses benzene as an example. This is based on the scheme recommended by Jenkin et al. (2018b) and incorporated in the MCM (Bloss et al., 2005; MCM, 2023). It shows the initial formation of the OH–aromatic adducts, their subsequent reactions with O2, and the fast unimolecular reactions assumed for the excited OH–aromatic–O2 adducts. The codes for the branching ratios, which depend on the substituents about the aromatic ring, are also shown. Three types of overall reactions are assumed. These include formation of a phenol + HO2 via a six-member ring transition state where the H transfers from the -OH to the peroxy group, followed by elimination of HO2, with the branching ratio designated fPhen; cyclization to form a bicyclic allylic-stabilized carbon-centered radical with branching ratio fBcyc; and the third involving a different type of cyclization where a peroxy O transfers to the opposite double bond, forming an epoxide and an alkoxy radical, with branching ratio fEpox. The alkoxy radical can form two different products depending on where the O2 added and the substituents about the aromatic ring, which are assumed to be equally likely. Therefore, up to four overall reactions can be generated following addition of OH to aromatic rings.
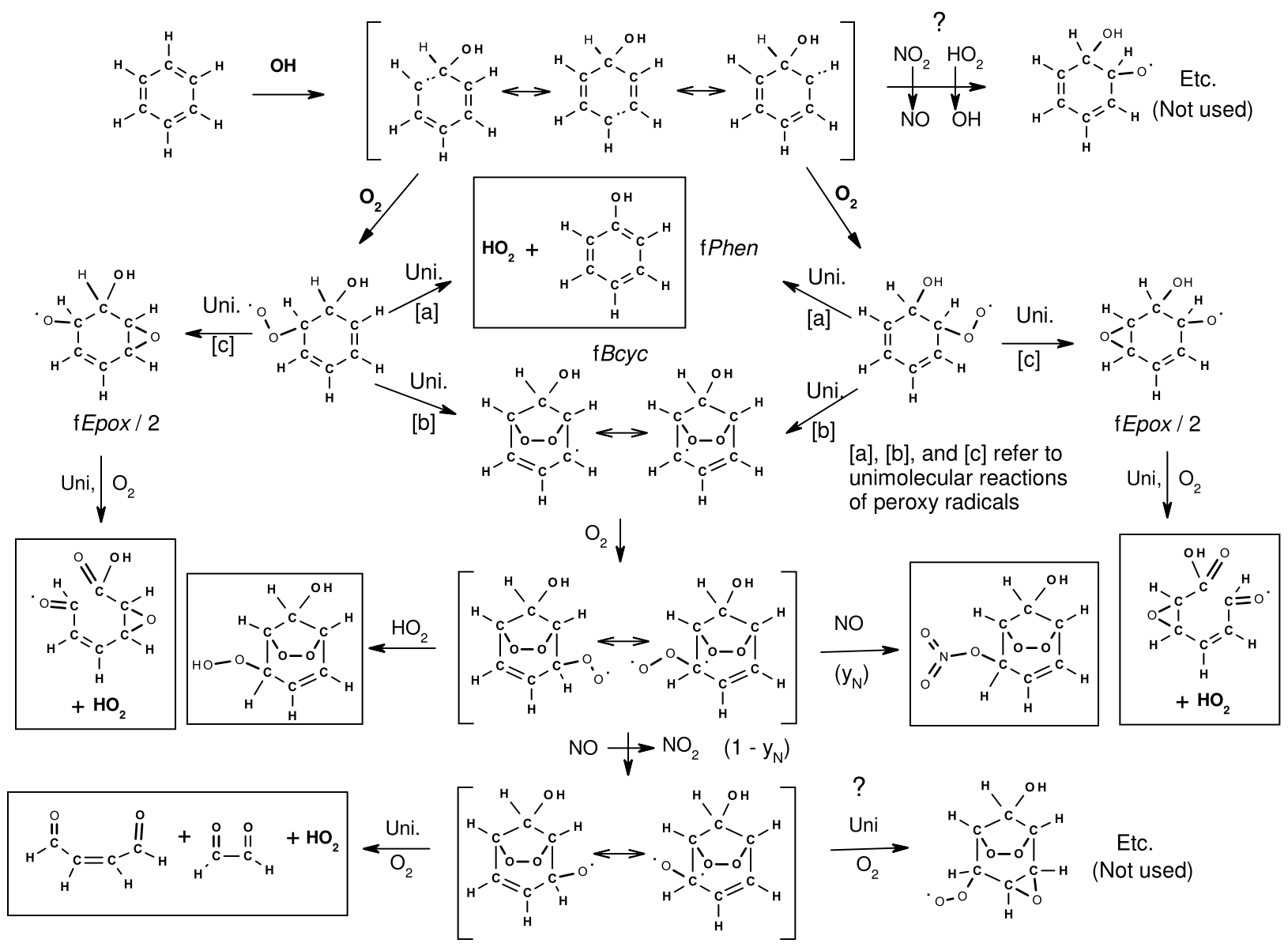
Figure 1Initial reactions following the addition of OH to aromatic rings, using benzene as an example, showing the formation of the OH–aromatic adduct, followed by unimolecular reactions assumed for the OH–aromatic–O2 adduct. Major products assumed to be formed are indicated, and additional pathways that may occur but are not currently used are also shown.
The Phen and Bcyc pathways in aromatic mechanisms (routes [a] and [b] in Fig. 1) are well supported by existing product data (Calvert et al., 2002) and theoretical calculations (e.g., Calvert et al., 2002; Vereecken, 2018, 2019). However, the existence of the Epox pathway (route [c]) is speculative and much more uncertain. It is included in the mechanism to account for carbon balance and is consistent with reactivities of aromatics observed in environmental-chamber experiments (Carter, 2010b; Carter and Heo, 2013). This pathway is also assumed in the aromatics mechanisms recommended by Jenkin et al. (2018b) and is incorporated in the MCM (Bloss et al., 2005; MCM, 2023). However, theoretical calculations discussed by Vereecken (2018) indicate that the direct epoxide formation from aromatic–OH–O2 adducts (route [c]) is unlikely to be important. Vereecken (2018) suggests alternative pathways that are shown on the top and bottom right side of Fig. 1 as alternatives that appear to be more consistent with theoretical calculations and recent experimental data (Xu et al., 2020). These alternatives will be considered when MechGen is updated. The possible alkoxy cyclization reaction shown on the bottom right of Fig. 1 is discussed in Sect. 5.6.
The mechanisms for the subsequent reactions of the carbon-centered and alkoxy radicals formed are estimated using the same procedures discussed elsewhere in this paper. The major stable products predicted to be formed are also indicated by the boxes in Fig. 1. Note that as discussed in Sect. 2.2.5, the unsaturated 1,4-dicarbonyl aldehydes or aldehyde–ketones are predicted to be highly photoreactive, so the yields of these products significantly affect predictions of reactivity in aromatic hydrocarbon–NOx environmental-chamber experiments (e.g., Carter and Heo, 2013).
The branching ratios were derived based on reported yields of phenolic products and α-dicarbonyls from the reactions of OH with benzene and various alkylbenzenes, as well as simulations of environmental-chamber experiments. Values of fPhen were derived based on phenolic yields, fBcyc values are based on 1,2-dicarbonyl yields and modeling environmental-chamber experiments, and fEpox values are obtained as 1 − fPhen − fBcyc. It was sufficient to assume that the branching ratios depend only on the presence of substituents in the ipso and ortho positions, with substituents in the meta or para positions assumed not to have an effect on these branching ratios. The branching ratios used for the various substituent cases are summarized in Table 7. Note that fPhen is necessarily 0 if there is an ipso substituent, and the fBcyc fEpox ratios for such compounds are estimated to be the same as those for compounds without ipso substituents but with the magnitudes increased so they sum up to 1. The ratios in Table 7 are used regardless of substituents, though estimates for compounds with substituents other than alkyl or -OH and therefore generated mechanisms for such compounds should be considered to be unreliable and probably should not be used for mechanism development.
Table 7Branching ratios derived for the reactions of aromatic–OH and aromatic–OH–O2 adducts, based on the presence or absence of substituents in the ipso and ortho positions.
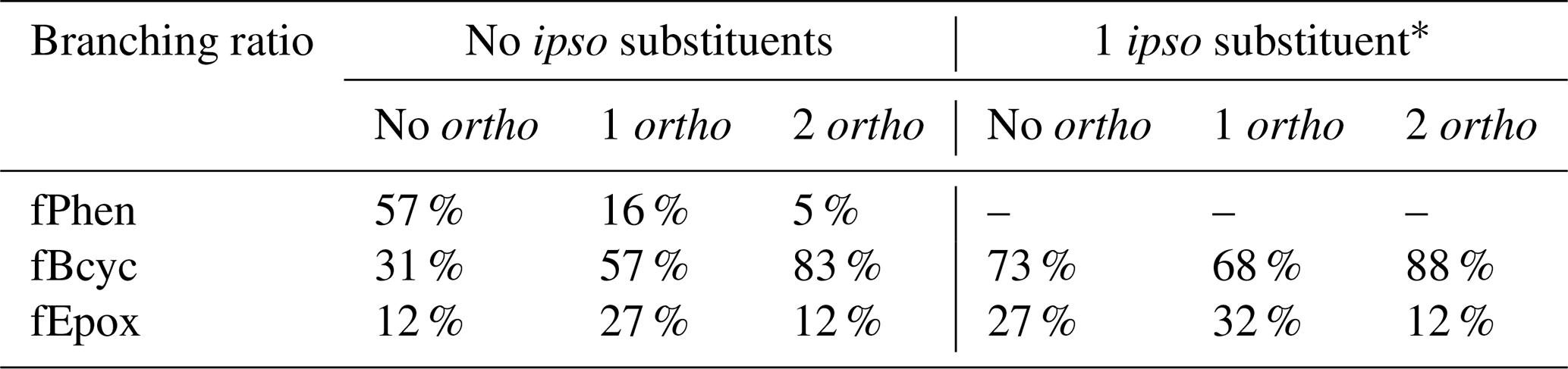
* Derived from fBcyc and fEpox assigned for adducts with no ipso substituents.
The above discussion is applicable only to reactions following additions of OH radicals to aromatic rings, not to additions by NO3 radicals. As discussed by Carter (2021), the rate constants for these additions are estimated to be slow, based on the relatively low total rate constant reactions for aromatic + NO3 reactions. MechGen currently assumes that all aromatic–NO3 adducts rapidly decompose back to reactants, resulting in no net reaction. However, this cannot strictly speaking be true, since the experimental rate constants give SARs that predict nonzero, though low, rate constants for these reactions (Carter, 2021; see also Sect. 2.1.2). However, the predicted rate constants are too low to be important in atmospheric systems, so this oversimplification has no practical consequences for atmospheric mechanisms.
The reactions of radicals formed by the additions of OH radicals to the aromatic rings in phenols are expected to be analogous to those discussed above, but the presence of OH substitution affects branching ratios for the addition to the allylic OH + phenol adduct as discussed in Sect. 3.1, and some of the OH + phenolic + O2 adducts form cyclohexadienones, which are assumed to undergo rapid unimolecular reactions to form catechols as discussed in Sect. 2.3.2. The mechanisms assumed for OH + phenolic adducts are discussed in Sect. S1.8.
Peroxy radicals are formed primarily by the reactions of alkyl radicals with O2 and are critical intermediates in atmospheric-oxidation mechanisms. Peroxy radicals can be classified alkyl peroxy, –G[OO.] or RO2, or acyl peroxy, –G–CO[OO.] or RCO3 (where G is any group that does not have an α-carbonyl substituent). In the following discussion, we will use the term “acyl” peroxy radical to refer to any peroxy radical bonded to a carbonyl group and “alkyl” peroxy radicals otherwise, including peroxy radicals with non-alkyl groups or substituents.
Peroxy radicals can react with NO, NO2, NO3, HO2, and other peroxy radicals, and some can also undergo unimolecular reactions at significant rates under atmospheric conditions. The rate constants assigned when generating bimolecular rate constants are summarized in Table 8, and their derivations are summarized in the following subsections where the mechanisms for the different types of reactions are discussed. More details concerning the derivations of the bimolecular rate are given in Sect. S1.5.1. The unimolecular reactions are discussed separately below.
Table 8Summary of bimolecular rate constants used when generating reactions of peroxy radicals under atmospheric conditions.
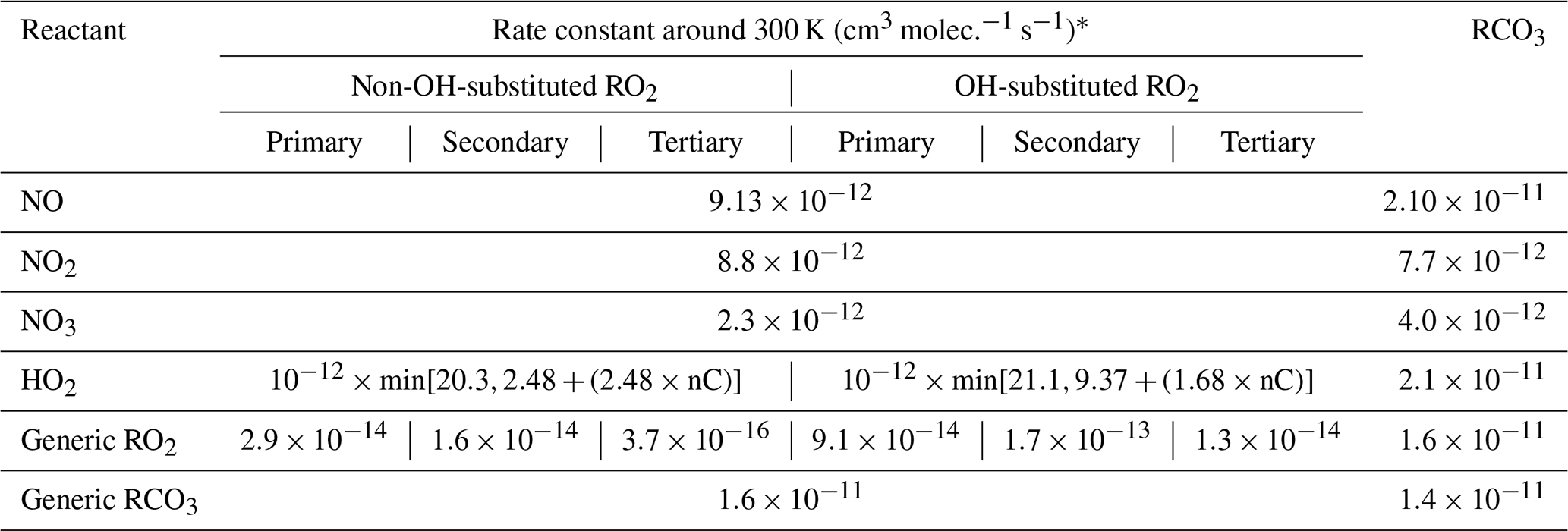
* See Table S45 for the sources of the rate constants used. Note that the same rate constant is used for reactions of alkyl peroxy radicals with NOx species and with acyl peroxy radicals, while the rate constants depend on the carbon number for reactions with HO2 and on the structure of the radical for reactions with other alkyl peroxy radicals (see Sect. 4.4 and 4.5, respectively).
4.1 Reaction with NO
The major atmospheric loss process of most peroxy radicals when formed in the presence of NOx is a reaction with NO. Available data on the rate constants for peroxy + NO reactions are given in Table S45, and the rate constants measured for ethyl peroxy and propionyl peroxy are estimated to apply to all peroxy radicals for mechanism generation. There is some variability in the measured rate constants for the alkyl peroxy radicals, but the data are insufficient to derive estimates for dependences of the rate constant on the structure or size of the radicals. As indicated in Table 8, although different rate constants are used for reactions of acyl peroxy vs. peroxy radicals with NO, because of insufficient information we assume that other structural effects have no effects on the rate constant.
The reactions of peroxy radicals with NO can occur via two possible routes, either forming NO2 and the corresponding alkoxy radical or forming a stable organic nitrate in the presence of a bath gas.
Both reactions are known to be important in the case of alkyl peroxy radicals, and the nitrate yield, yN = , is an important parameter affecting model predictions in atmospheric systems since the conversion of NO to NO2 in Reaction (R3) contributes to O3 formation and the formation of alkoxy radicals results in radical propagation, while nitrate formation in Reaction (R4) causes radical termination and is also a NOx sink, at least for the intermediate term.
Available data and estimates concerning nitrate yields from various peroxy radicals are discussed in Sect. S1.5.2. There are extensive measurements of nitrate yields from various unsubstituted peroxy radicals formed from the reactions of alkanes, including data at various temperatures and pressures, allowing for a parameterization to be derived that fits these data as a function of temperature, pressure, and the carbon number. The parameterization used is similar to that employed previously (e.g., Atkinson et al., 1983), updated based on the mechanism considerations discussed by Zhang et al. (2004) and re-optimized to fit available data as discussed in the Supplement. The predicted nitrate yields at around 298 K and 1 atm pressure are given in Table 9 and are plotted against the carbon number in Fig. 2, where they are compared to available data as discussed below.
Table 9Estimated nitrate yields as a function of the carbon number calculated for ∼ 298K and 1 atm pressure.

Figure 2 shows that the parameterization derived to fit measured nitrate yields for unsubstituted peroxy radicals from alkanes (black line) fits those data reasonably well, but its applicability for substituted radicals from other VOCs is much more uncertain. The limited available measurements of nitrate yields from β-OH-substituted radicals are inconsistent. Data obtained from Shepson's (Muthuramu et al., 1993; Shepson et al., 1985; O'Brien et al., 1998) and Ziemann's (Matsunaga and Ziemann, 2009, 2010) groups gave yields that are about a factor of 2 lower than from unsubstituted radicals, while more recent data from Teng et al. (2015) gave yields that agree with those from the unsubstituted radicals, at least at carbon numbers < 10. The estimated nitrate yields from the OH-substituted radicals significantly affect results of model simulations of alkene–NOx chamber experiments (Carter, 2000, 2010b), and we found that some data are better fit using the higher yields estimated for radicals from alkanes, while some are better fit using the yields that are a factor of ∼ 2 lower (see Sect. S1.5.2).
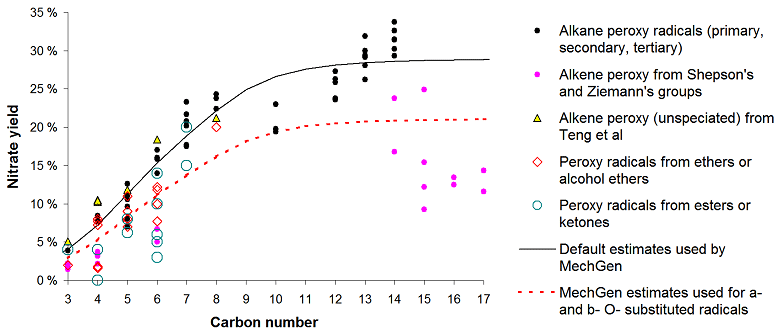
Figure 2Measured and estimated nitrate yields from reactions of various types of peroxy radicals with NO against numbers of carbon atoms in the radicals.
Figure 2 also shows scatter in the available data concerning nitrate yields from peroxy radicals formed from ethers, esters, and ketones, though they tend to be lower than observations for unsubstituted peroxy radicals (Jenkin et al., 2019, and references therein). As discussed in the Supplement, these yields are subject to greater uncertainties because they are derived either from unspeciated total nitrate measurements in complex chemical systems or by fits to environmental-chamber data, where uncertainties are even greater.
Until more information is available to resolve these discrepancies and provide a better basis for making nitrate yield estimates for non-alkyl-substituted peroxy radicals, MechGen uses a parameterization (dotted red line in Fig. 2) that predicts yields halfway between those derived for radicals from alkanes and the lower yields for the β-OH-substituted radicals, when estimating nitrate yields for peroxy radicals bonded to oxygen groups in the α or β positions, e.g., –OH, ether, ester, peroxides, or nitrates. This is similar to the approach used in the nitrate yield estimation method developed by Jenkin et al. (2019) for alcohols, ethers, and esters. Based on the limited data, Jenkin et al. (2019) also assumed carbonyl groups in the α or β positions caused even lower nitrate yields, but the current version of MechGen does not yet incorporate corrections for these cases.
The nitrate yields predicted using this parameterization, derived as discussed in Sect. S1.5.2, are included in Table 9. Note that we assume that substituents farther away than the β position are too far away from the reaction site to affect the nitrate yield. However, the estimates for nitrate yields from substituted peroxy radicals are uncertain by at least a factor of 2 and are more likely to be biased high rather than low.
There is no evidence for nitrate formation in the case of acyl peroxy radicals, so the formation of NO2 and the corresponding acyl oxy radical is assumed to be the only pathway of significance (see, e.g., IUPAC, 2023).
As discussed above, the acyl oxy radicals are expected to rapidly decompose to form CO2 and the corresponding alkyl radical, making the overall process for acyl + NO reactions as indicated above.
4.2 Reaction with NO2
Alkyl and acyl peroxy radicals also react with NO2 under atmospheric conditions, and the rate constants assigned to them are included in Table 8. The assigned rate constants are based on measured high-pressure rate constants for ethyl peroxy and ethyl acyl peroxy radicals, since we could not find data for larger peroxy radicals (see Table S45). Although the reactions of ethyl and ethyl acyl peroxy radicals are not in the high-pressure limit under atmospheric conditions, most of the radicals are larger and their rate constants should be closer or at the high-pressure limit, so the high-pressure rate constant is used for general estimates.
The reactions for both alkyl and acyl peroxy radicals with NO2 are assumed to result entirely in the formation of the corresponding alkyl or acyl peroxynitrate.
Reactions forming NO3 + alkoxy or acyloxy radicals are estimated to be endothermic and are assumed not to be important. The peroxynitrate compounds formed are thermally unstable at atmospheric temperatures, decomposing back to NO2 and the peroxy radical, as discussed in Sect. 2.3. It is assumed that the presence of non-alkyl substituents on the R does not affect either the formation or decomposition rate constants, but this is uncertain.
4.3 Reaction with NO3
Reactions with NO3 can be a potentially important sink for peroxy radicals at nighttime and during potentially other conditions when NO concentrations are low. The rate constants used for mechanism generation (Table 8) are those measured for ethyl peroxy and acyl peroxy radicals because we could not find data for larger radicals (see Table S45).
Peroxy radicals react with NO3 to form the corresponding alkoxy radical, NO2, and O2, with no evidence of other reaction routes or products being reported (e.g., IUPAC, 2023).
Note that the same organic products are formed in this reaction as in the reaction of peroxy radicals with NO, other than the lack of the nitrate formation route in the NO3 reaction. Although nitrate formation in this reaction is energetically possible, there is no evidence that it occurs under atmospheric conditions. The presence of non-alkyl substituents on R is assumed not to affect the rate constants or mechanisms.
4.4 Reaction with HO2
Reactions with HO2 can be the major sink for peroxy radicals under conditions when NOx is low, being generally much more important than reactions with other peroxy radicals. Information concerning measurements and estimates for mechanisms and rate constants for the reactions of peroxy radicals with HO2 are discussed in Sect. S1.5.3. In the case of alkyl peroxy radicals, the data suggest that the rate constants depend on the size of the radical and the presence of OH substitution on the radical, with the following equations being derived for estimation purposes:
where O–RO2 refers to alkyl peroxy with OH substituents anywhere on the radical and RO2 refers to those without. Note that there is some inconsistency in the measured rate constants for OH-substituted radicals (see the Supplement), so there is some uncertainty in these estimates.
The only data we found for reactions of acyl peroxy radicals with HO2 concerned acetyl peroxy radicals, so the rate constant measured for that reaction (on Table S45) is used for estimation purposes. This is uncertain, especially in view of the variability in the measured rate constants for the alkyl peroxy radicals.
Available information concerning the products of the reactions of various types of peroxy radicals with HO2 has been evaluated by IUPAC (2023), and their recommendations of branching ratios for methyl and acetyl peroxy radicals and for methyl peroxy substituted with –CH3, –OH, –OCH3, and –C(O)CH3 are given in Table S12. Their recommendations include the following four routes.
The ozone route is only one considered for acyl peroxy radicals. The carbonyl route occurs via an H shift from an α-hydrogen to the HO2 via a six-member ring transition state and is not possible for tertiary alkyl or acyl peroxy radicals. Although there are data only for the C1 or C2 peroxy radicals, they are assumed to be applicable to all alkyl peroxy radicals with the same types of α substituents for mechanism generation purposes and are the basis for the branching ratios that are summarized in Table 10. However, the applications of these recommendations to the many types of larger peroxy radicals predicted in generated mechanisms are highly uncertain.
Table 10Branching ratios assigned for the four possible routes for the reactions of HO2 with various types of peroxy radicals.
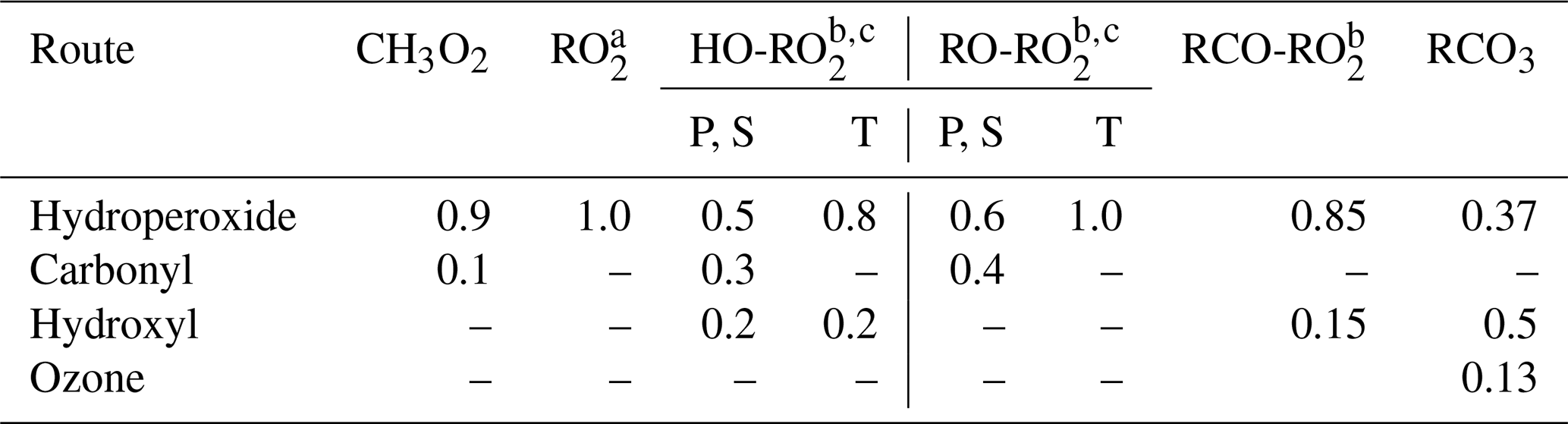
a RO2 refers to peroxy radicals with no -OH, -OR, or carbonyl substituents in the α position. Note that they can have these and other non-alkane substituents or groups elsewhere on the molecule.
b HO-, RO-, and RCO- refer to the substituent in the α position only. RCO- refers to either CHO or -CO-, though data are only available for -CO- substitution.
c P, S, and T refer to primary, secondary, and tertiary radicals, respectively. Since the carbonyl route is not possible for tertiary radicals, the branching-ratio estimate for this route for primary or secondary radicals is added to the estimate for the hydroperoxy route for such radicals.
4.5 Reactions with other peroxy radicals
Organic peroxy radicals can also be consumed under atmospheric conditions by reactions with other organic peroxy radicals. Although this is generally a less important loss process under atmospheric conditions than reactions with NOx or HO2, such reactions can be non-negligible under relatively high-VOC and low-NOx atmospheric conditions and also in some laboratory experiments. Available information concerning measured rate constants for these reactions is given in Table S45. The rate constants vary by orders of magnitude, with the most important factor being whether the alkyl peroxy radicals are primary, secondary, or tertiary. However, the presence of α-OH substitution also appears to affect the rate constant, and this is also taken into account. Table S45 includes a summary of the averages or estimated rate constants for various types of radicals that can be derived from the experimental data, and the rate constants used by MechGen when generating mechanisms for peroxy radicals are included in Table 8.
Because it is not practical to represent all of the peroxy + peroxy combinations in large mechanisms, MechGen does not assign rate constants or generate mechanisms for all combinations of the different types of radicals but instead represents the radicals as reacting with a generic alkyl (RO2) and a generic acyl (RCO3) peroxy radical. The rate constants and also mechanism assignments for the generic RO2 are based on those estimated for secondary peroxy radicals because they are intermediate in reactivity and are also expected to be most frequently formed. The generic acyl peroxy radical is based on acetyl or propionyl peroxy radicals, the only type for which data are available. Although this representation can be inaccurate because it lumps reactions whose rate constants vary by orders of magnitude, it is considered to be relatively unbiased and is better than the alternative of ignoring these reactions altogether. Fortunately this is a relatively unimportant loss process for peroxy radicals under most conditions, so this is not considered to be a significant problem compared to other uncertainties.
Available information concerning the mechanisms of peroxy + peroxy reactions is discussed in Sect. S1.5.4, and recommended and assigned branching is given in Table S14. Three types of generic reactions are considered.
The products formed from the generic radicals are not shown because they are not generated because they are represented separately by the reactions of the individual radicals. (This is an approximation because it undercounts the contributions of self-reactions in the kinetic differential equations.) Note that the carbonyl and alcohol routes are two instances of the disproportionation reaction, where an α-H migrates from the carbon next to the peroxide group on one of the radicals to a peroxy oxygen on the other radical. Formation of carbonyl products is not possible for acyl radicals and tertiary alkyl radicals, and the alcohol route is not possible for generic acyl radicals because they lack the necessary α-hydrogen.
The branching ratios used when generating reactions of the various types of peroxy radicals are summarized in Table 11. These are based roughly on the branching ratios for individual reactions given in Table S14 and are extrapolated to other types of radicals where necessary as discussed in Sect. S1.5.4. These assignments are uncertain when applied to larger radicals and radicals with non-alkyl substituents or groups, but the effects of this uncertainty are probably less than the uncertainty and necessary inaccuracies when estimating the rate constants.
Table 11Branching ratios used when generating mechanisms of peroxy + peroxy reactions.
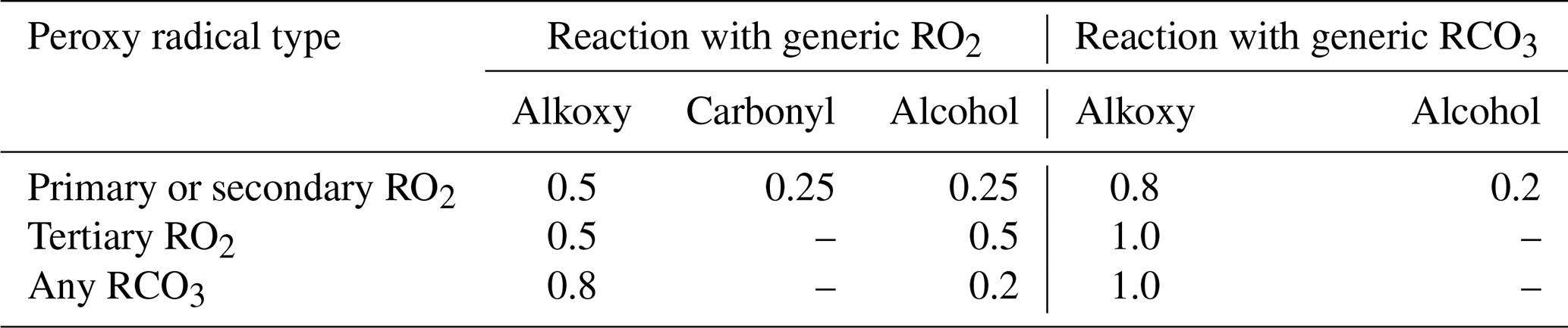
4.6 H-shift isomerizations
Organic peroxy radicals can undergo H-shift isomerizations where the radical center abstracts a hydrogen from elsewhere in the molecule, via a cyclic transition state, forming a hydroperoxide and a carbon-centered radical.
This reaction has been known to occur at higher temperatures in combustion systems, where the α-hydroperoxy carbon-centered radicals formed are designated QOOH, though they had not been expected to be important at atmospheric temperatures until recently. Recent experimental data indicate that these reactions occur at measurable rates and may be non-negligible under atmospheric conditions for certain radicals (e.g., Crounse et al., 2012; Teng et al., 2015; Praske et al., 2018, 2019; Nozière and Vereecken, 2019). This has been supported by quantum-theoretical calculations of rate constants for a wide variety of such reactions (e.g., Davis and Francisco, 2010; Møller et al., 2019; Vereecken and Nozière, 2020, and references therein), whose results are reasonably consistent with the available experimental data and indicate that in some cases these isomerizations would be the dominant fate of such radicals.
Vereecken and Nozière (2020) reviewed the available theoretical and experimental rate constant data for peroxy H-shift isomerizations for a wide variety of peroxy radicals and provided recommended SARs for predicting their rate constants for automated mechanism generation applications. These consist of lookup tables giving the rate constant for various pairs of peroxy radical types and H groups (the groups with the H that is transferred) involved in the H shift, giving the rate constants as a function of transition state ring size, with correction factors for a few types of substituents around the H group. The effects of –OH, –OOH, –OR, carbonyl groups, and double bonds at or near the H group were incorporated into the lookup tables; separate rate constant recommendations were given for reactions of CH2OO•, >CH(OO•)–, and –C(OO•)< radicals, and rate constants were given for ring sizes up to eight or nine. The recommended correction factors covered the effects of α-ONO2, β-OH, β-endo oxo and β-exo oxo substituents on the H group. These recommendations were adopted as the starting point for use in this work. However, the recommended lookup tables did not cover all the types of peroxy radicals that might be formed in atmospheric photooxidation systems, including reactions of acyl peroxy radicals, radicals with both β double bonds and α-OH groups, abstractions from an HCO group with a β double bond, H shifts with transition state rings larger than eight or nine, and several other cases. Therefore, it was necessary in this work to extrapolate or extend the work of Vereecken and Nozière (2020) to allow for predictions for these other types of radicals.
The adaptation of the peroxy H-shift SARs for use in this work is described in detail in Sect. S1.5.5. In order to provide a basis for estimating rate constants, the various factors affecting the rate constants are split up into components that are somewhat more straightforward to estimate when expanding the lookup tables to cover the other cases. The expression used for estimation purposes was
where nH is the number of equivalent abstractable hydrogens; A(n,ts) is an Arrhenius A factor assumed to be dependent only on the size of the transition state ring, n, and the type of transition state, ts; Ea0(H,R) is the activation energy in cases where there is no strain or substituent corrections and is assumed to be dependent only on the type of H group, H, and the type of radical, R; Eastrain(H,R,n) is the ring strain that depends on the ring size as well as the types of groups involved; and Eacorr(subs,n) is corrections for substituents on the H group, some of which depend on ring size. The temperature-dependence recommendations given by Vereecken and Nozière (2020) were used to derive approximate A factors which were then used, in conjunction with the recommended rate constants in the lookup tables, to create lookup tables for Ea0 and for Eastrain for the various ring sizes. These were then used to estimate Ea0 and Eastrain for cases not covered by the original SARs. The recommended substituent correction factors were used to derive Eacorr to use in Eq. (1), where applicable. No new types of substituent correction factors were added in this work.
The range of magnitudes of estimated rate constants for the peroxy H-shift isomerization for the various types of radicals is presented in Fig. 3, which shows the rate constants for the transition state ring size (generally six to eight) that gives the highest rate constant. These maximum rate constants for the different types of reaction range in magnitude from ∼ to ∼ 7×106 s−1, and the lookup tables cover 25 types of H groups and transition states, as indicated on the x axis of the figure. The figure also has lines giving the upper and lower limits of these rate constants used by MechGen when generating peroxy radical reactions – reactions faster than the upper limit are assumed to be the only fate of the peroxy radical, while those that are below the lower limit are treated as negligible and not generated. For those with rate constants between these limits there is a competition between unimolecular and bimolecular reactions of these radicals, so the estimated rate constant would have an impact on predicted mechanisms.
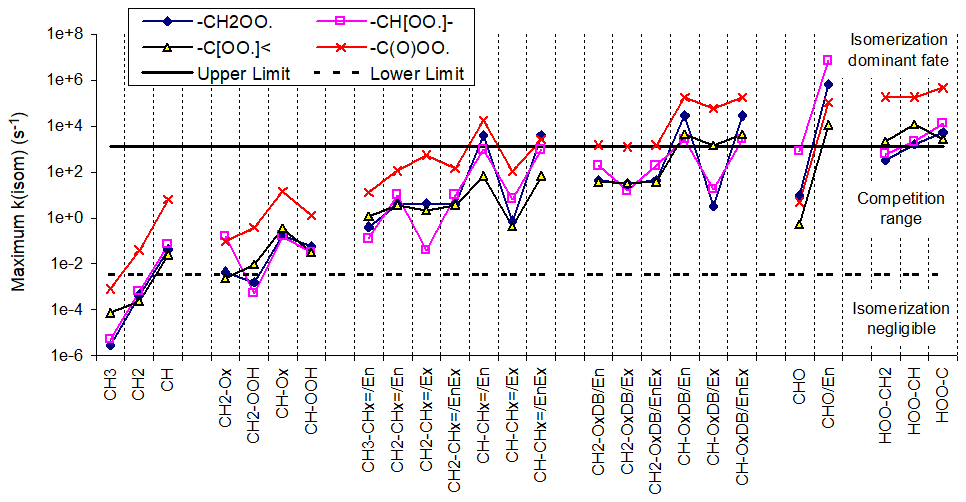
Figure 3Estimated peroxy H-shift isomerization rate constants for the most favorable ring size for various H and R group pairs without β-substitution corrections. Also shown are upper and lower rate constant limits used during the mechanism generation. The nomenclature is as follows. H group with substituents (if applicable), where -Ox = α-OH or -OR substituents. -CHx=/En: β double bond by the H group, with at least one double-bond group in the transition state ring. -CHx/Ex: β double bond not in the transition state. -OxDB: both α-Ox and a double-bond substituent. /EnEx: two β double bonds by the H group.
The H-shift reaction from unsubstituted –CH3 and –CH2– groups in alkyl peroxy radicals was generally negligible, while abstractions from hydroperoxy groups and aldehyde groups and abstractions of allylic hydrogens by acyl peroxy groups were generally fast. Most of the other types were in the intermediate range where there is a competition between unimolecular and bimolecular reactions of these peroxy radicals. This indicates that this is an important process for many radicals that must be considered when implementing mechanisms into models.
The rate constant estimates that were derived directly from the SARs of Vereecken and Nozière (2020) are based on results of quantum-theoretical calculations and may be good to within a factor of ∼ 10, and they can predict the experimental 298 K rate constants to within a factor of 3, as shown in Fig. S12 in the Supplement. Those that are based on our extrapolations or estimates are much more uncertain, though many of these are estimated to be high enough that the actual fate of the peroxy radical may not be sensitive to this estimate. However, experimental and theoretical data are needed to verify or improve our estimated extensions of the Vereecken and Nozière (2020) SARs as discussed in Sect. 1.5.5, as well as more experimental data to validate the extensive set of theoretical calculations that form the primary basis for the SARs that are currently employed.
No attempt was made to make separate estimates of rate constants for H-shift isomerizations of cyclic peroxy radicals either by Vereecken and Nozière (2020) or in this work. However, Vereecken et al. (2021) do include calculations for cyclic peroxy radicals that could be used when MechGen is updated but give insufficient data to derive SARs at this time. For the purpose of making general estimates, we assume that the isomerization reaction does not occur if three or more of the atoms in the transition state ring are in another ring, since they would usually (but not necessarily always) result in a highly strained structure. Although these are not unreasonable assumptions, some isomerizations will be overestimated and others will be overlooked. The practical effects of these uncertainties have not been assessed, but such an assessment may give an indication of priorities in future theoretical or experimental studies of unimolecular reactions of peroxy radicals formed from terpenes and other cycloalkenes.
4.7 Ring closure reactions of unsaturated peroxy radicals
The current estimated mechanisms for aromatics, discussed above in Sect. 3.4, assume two additional types of unimolecular reactions of OH–aromatic–O2 adducts to account for observed aromatic product formation and reactivities, both involving ring formation. These are shown as reactions labeled [b] and [c] in Fig. 1. These reactions are accounted for in the process of generating reactions of OH–aromatic adducts as discussed in Sect. 3.4, and thus their rate constants do not need to be estimated to generate mechanisms for aromatics. However, these reactions may also be non-negligible in non-aromatic systems, since neither the reactants and products nor the transition states have aromatic rings. The reactions shown in Fig. 1 can be generalized as follows.
Here, X is H or any group and * indicates ring closure. The reactions labeled [b] involve the peroxy group adding to a double bond forming a cyclic ether and an allylic carbon-centered radical, while those labeled [c] have a similar cyclic transition state but the O–O bond breaks, with one O adding to the double bond forming an epoxide and the other becoming an alkoxy radical center. Note that the reactions labeled [b5] and [c5] involve five-member ring products or transition states, while those labeled [b7] and [c7] involve a seven-member ring. Since these reactions apparently dominate over bimolecular reactions when formation or intermediacy of both five- and seven-member rings is involved, one would reasonably expect them to be even faster in acyclic systems where only one new ring is involved.
Since six- or seven-member rings tend to have less ring strain than five-member rings (see discussion of peroxy H-shift reactions in Sect. 1.5.5 of the Supplement), one would expect the analogue involving formation or intermediacy of six-member rings also to be fast, e.g., the following.
Therefore, to be consistent with the aromatics mechanisms, the above six types of reactions are generated for peroxy radicals with conjugated double bonds in the α, β, or γ positions, relative to the carbon bonded to the peroxy group, and are assumed to dominate over bimolecular reactions and H-shift isomerizations. Because of this, it is not necessary to estimate their total rate constants, only the branching ratios for the [b] and [c] reaction routes or the fEpox fBcyc ratio using the terminology of Sect. 3.4 in the context of the aromatic system. The fractions reacting via the [b] or Bcyc route used when generating aromatics mechanisms ranged from 68 % to 88 %, depending on the number of ortho substituents in the OH–aromatic adduct. It is highly uncertain whether this branching ratio is an appropriate basis for estimations for acyclic peroxy radicals with conjugated double bonds, but no information is available to justify other assumptions. Therefore, when generating mechanisms for peroxy radicals with conjugated double bonds in the α, β, or γ positions, we assume that
where the fractions are approximately the averages of those given in Table 7. This is highly uncertain, but at least it is reasonably consistent with our treatment of aromatic-ring addition reactions. However, formation of these types of peroxy radicals is probably relatively rare in atmospheric systems, so the practical effect of this uncertainty in atmospheric mechanisms may be relatively small.
Note that, as discussed in Sect. 3.4, the epoxide-forming cyclization reaction may not be as important in aromatic systems as currently estimated, and if this is the case, then their analogues (reactions labeled [c] above) may also not occur in acyclic systems. This will need to be examined when MechGen is updated.
It is also possible that ring closure involving only single double bonds may be non-negligible. However, if such reactions were to be significant, they should be important in the isoprene + OH reaction system, resulting in the formation of different products than what are observed (Calvert et al., 2011, and references therein). Therefore, we currently assume that these cyclizations are only important in radicals with conjugated double bonds.
Alkoxy radicals are formed in atmospheric-oxidation mechanisms primarily from the reactions of peroxy radicals with NO and NO3 and to a lesser extent by their reactions with other peroxy radicals, as discussed above. These can undergo a variety of reactions in atmospheric systems, and this leads to much of the complexity in atmospheric reactions of organic compounds. The types of alkoxy radical reactions that MechGen considers and how their rate constants are estimated are summarized in Table 12. These reactions are discussed further below.
Table 12Summary and examples of reactions of alkoxy radicals as derived using MechGen.
a The following codes are used for unspecified groups: X is any non-radical group except –H; X is –H or any non-radical group; Y is any stable compound group that is bonded to at least two other groups, except =C=, ≡C–, and –aCx; and Y is as above but excludes =CH– and =C<.
b This is assumed to dominate over competing reactions of this radical, so the rate constant does not need to be estimated.
c Vinoxy radicals are expected to exist in resonance with a carbon-centered radical form, e.g., X2C=CXO• ↔ X2C[•]C(O)X, which rapidly adds O2 under atmospheric conditions.
Note that alkoxy radicals formed in the reactions of peroxy radicals with NO could be formed with up to ∼ 12 kcal mol−1 of excitation energy, which would affect estimates for unimolecular reactions that are not highly thermochemically favorable (Orlando et al., 2003). This is not taken into account in current estimates of unimolecular reactions of these radicals but will need to be considered for future updates of the system.
5.1 Bimolecular reactions
Primary and secondary alkoxy radicals can react with O2 via abstraction of an α-hydrogen to form HO2 and the corresponding carbonyl compound, as shown for the first three reactions in Table 12. Available data concerning rate constants for this reaction are discussed in Sect. S1.7.1. These data suggest that the rate constant may only depend on whether the radical is primary or secondary, and the following rate constants are assigned for estimation purposes:
where the rate constant is given for 298 K and kuni is the pseudo-first-order rate constant for 1 atm of air. Note that separate assignments are given for rate constants for reactions of O2 with methoxy or ethoxy radicals (see Table S56), but there is no indication of effects of radical size for larger radicals, though this cannot be ruled out for C6+ radicals. There are also no data on effects of non-alkyl substituents or groups on this rate constant, so these estimates are highly uncertain for non-alkyl radicals.
Although alkoxy radicals can also react with NOx and radical species with relatively high rate constants, the atmospheric levels of NOx are many orders of magnitude less than those of O2 and the rate constants are not sufficiently high for these reactions to be competitive. Therefore, reaction with O2 is the only type of bimolecular reaction generated for alkoxy radicals. The only exceptions for the types of radicals handled by MechGen are tertiary alkoxy radicals, where β-scission decompositions are estimated to be favorable, or phenoxy radicals, which are discussed separately in Sect. 6.1.
5.2 β-scission decompositions
Most alkoxy radicals can undergo β-scission reactions, where a bond on the carbon bonded to the oxy radical center breaks, forming a carbonyl group and a fragment radical. These types of reactions can be represented as
where the R's are any group next to the alkoxy radical group except –H. Decompositions of acyl oxy radicals (Reaction R9) are expected to be fast and thus are the only fate of these radicals considered in MechGen. However, this is not the case for alkoxy radicals in general (Reactions R6–R8), where, depending on the structure of the radical, these reactions can be very fast and dominate over other reactions or they can be negligible compared to a reaction with O2 or other types of unimolecular reactions such as H shifts. Note that the relative importance of the decomposition reactions affects the extent to which the reactions of a VOC leads to fragmentation to lower-molecular-weight products, as opposed to becoming more oxidized and forming lower-volatility products that may contribute to SOA formation.
Available information concerning rate constants for β-scission reactions and methods for their estimation are discussed in Sect. S1.7.2. These include experimentally measured rate constants given by Orlando et al. (2003), theoretically calculated rate constant parameters from Vereecken and Peeters (2009), and rate constants derived from results of experimental product studies. Based on the data and estimates in these resources, MechGen uses the following equations to estimate rate constants for β-scission reactions of alkoxy radicals.
Here, k is the unimolecular rate constant at temperature T per second; n is the number of equivalent R1 groups on the radical; 1.0×1014 s−1 is the estimated A factor for all β-scission reactions; Ea is the calculated activation energy that depends on the reaction; and EaR, EaP, EaRc, EaPc, and R.Corr are estimated or adjustable parameters that depend on the reaction. In addition, Rad refers to the radical R1• formed in the reaction; ProdType refers to whether the carbonyl product formed is formaldehyde, a higher aldehyde, or a ketone (Reactions R6–R8, respectively); R.Subst refers to substituents on R1; P.Subst refers to substituents R2 or R3, if applicable; and Ring refers to the presence of rings on the alkoxy radical at or adjacent to the radical center. The derived or estimated values of these parameters are given in various tables in Sect. S1.7.2. Values of representative parameters are given in Table 13.
Table 13Representative values of parameters used to estimate activation energy of β-scission reactions.
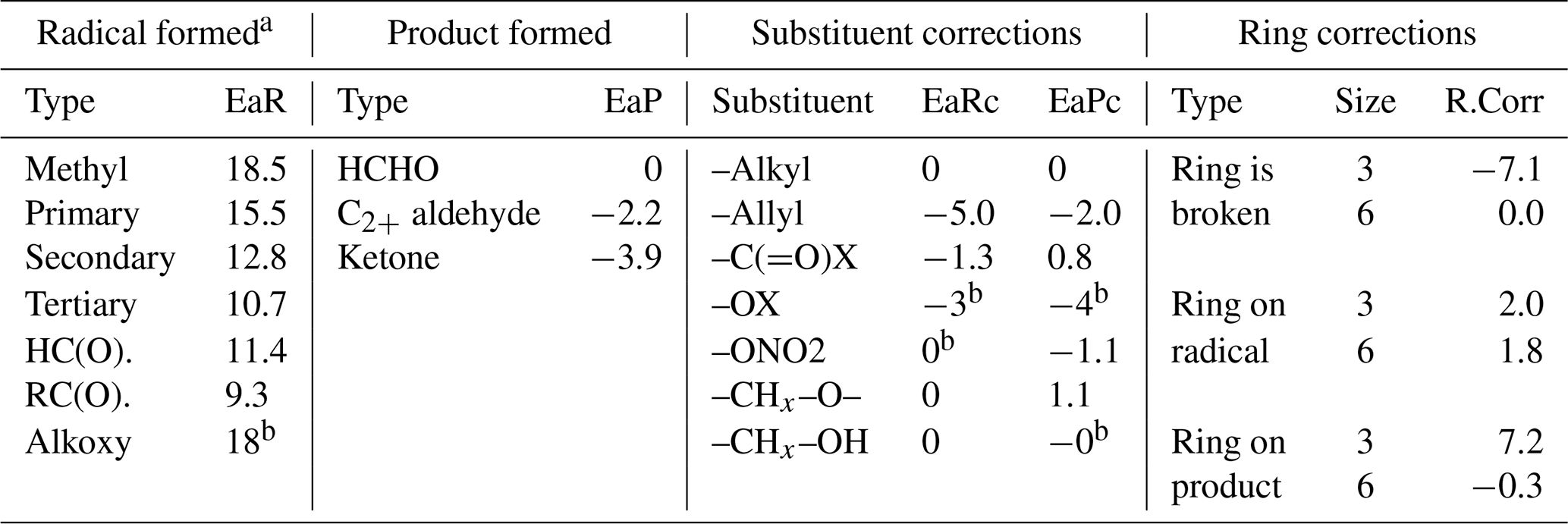
a Parameters are all in units of kilocalories per mole (kcal mol−1). X refers to –H or any other substituent.
b The value of this parameter varies somewhat depending on the specific groups involved, and the average value is given. See the tables in Sect. S1.7.2 for the specific values.
From Table 13 it can be seen that the estimated activation energies range from near 0 to ∼ 20 kcal mol−1 or higher. Note that an activation energy of ∼ 12 kcal mol−1 would give a rate constant that is comparable to the pseudo-unimolecular rate constant for the reactions of primary or secondary alkoxy radicals with O2, so reactions with higher activation energies would generally be negligible, while those with lower Ea values could potentially be the dominant process unless another type of unimolecular reaction (e.g., a β scission involving a different group or an H shift) is also estimated to be fast. Decompositions are usually important for tertiary alkoxy radicals not only because reactions with O2 are not possible but also because their β-scission decompositions are generally estimated to be relatively fast. Decompositions can also be important for primary or secondary radicals.
As discussed in the Supplement, there are sufficient data on rate constants for decompositions of alkoxy radicals formed from alkane systems for cases whose estimates are not considered to be highly uncertain. However, if non-alkyl groups are present, data to derive estimates are highly limited or lacking and thus needed. One uncertainty is the lack of measured or theoretically calculated rate constants for decompositions forming alkoxy radicals, which might occur in atmospheric photooxidation mechanisms of ethers and esters and compete with other reactions discussed below.
5.3 H-elimination decompositions
Primary and secondary alkoxy radicals can also undergo β-scission reactions by H elimination, forming a carbonyl product and a hydrogen atom. Available information and estimates for the rate constants for these reactions are discussed in Sect. S1.7.3. The rate constants for these reactions appear to be enhanced by the presence of α-oxygen substituents, so three types of these reactions are considered.
Here, X is -H or any group and R is any group except -OX. The data are limited but sufficient to derive the following expression to estimate the rate constant:
where Ea1, Ea2, and Ea3 refer to the activation energies for reactions of types 1, 2, and 3, respectively. The estimate for Ea3 is uncertain and is strictly speaking an upper limit; i.e., rate constants for H-elimination reactions of alkoxy radicals with two –OX may be higher than estimated.
Note that under atmospheric conditions, the H-elimination reactions yield exactly the same products as the reactions of the alkoxy radicals with O2 because the eliminated H• rapidly forms HO2. These estimates indicate that the H-elimination reaction is negligible compared to the O2 reaction if there are no α-O substituents, non-negligible but slower than O2 reaction if there is only one, and faster than the O2 reaction if there are two such substituents. Therefore, this reaction is only important in the third case, so only estimates for Ea3 have a practical effect on generated mechanisms.
5.4 H-shift isomerizations
Alkoxy radicals with sufficiently long chains (≈ C4 or larger) can also undergo H-shift isomerization reactions, where the alkoxy group abstracts an H from elsewhere in the molecule via a cyclic transition state, forming a carbon-centered radical with an –OH substituent. These are similar to the H-shift isomerizations that were previously discussed for peroxy radicals (Sect. 4.6), but in this case the reactions are much more exothermic, and their importance in atmospheric systems has been recognized for some time. Methods to estimate their rate constant have been developed for previous versions of the SAPRC mechanism, with the version used for developing SAPRC-99 being the most completely documented (Carter, 2000). The method used for SAPRC-99 has been subsequently updated for SAPRC-18 to incorporate evaluated experimental data (Atkinson, 2007; IUPAC, 2023), results of theoretical quantum chemistry calculations of Vereecken and Peeters (2010), and upper-limit rate constants derived from various product studies, as given in Table S50. This updated method is documented in the Supplement in Sect. S1.6.3.
The rate constants for alkoxy H-shift reactions are estimated in the same way as for peroxy radicals, using Eq. (3) in Sect. 4.6, which is based on the types of groups from which the H is abstracted, their substituents, and also substituents elsewhere in the transition state ring. Because of a more limited amount of theoretically calculated data available at the time, this method was last updated in the mid-2010s; only a single set of ring-size-dependent A factors and Eastrain values were derived, and Ea0 values were derived using only four types of H groups (–CH3, –CH2–, –CH<, and –OOH), with no distinction being made between primary, secondary, or tertiary alkoxy radicals. To compensate for this, a larger number of substituent correction factors were employed. The A factors and strain energies used are given in Table S25; the Ea0 values are given in Table S26; and the Eacorr values are given in Tables S16, S27, and S28. Footnotes to these tables indicate how these were derived or estimated.
Estimated 298 K rate constants for H-shift isomerizations of various unsubstituted alkoxy radicals are listed in Table 14, which also shows the ratios of these rate constants to the pseudo-unimolecular rate constant for the reaction of secondary alkoxy radicals with O2 in the atmosphere. It can be seen that 1,4-H-shift isomerizations, with six-member ring transition states, are ∼ 10–350 times faster than an O2 reaction, while those involving 1,5-H shifts are ∼ 4–130 times faster, while 1,2-H, 1,3-H, and 1,8+-H shifts are negligible to minor.
Table 14Representative 298 K rate constants for H-shift isomerizations of unsubstituted alkoxy radicals.
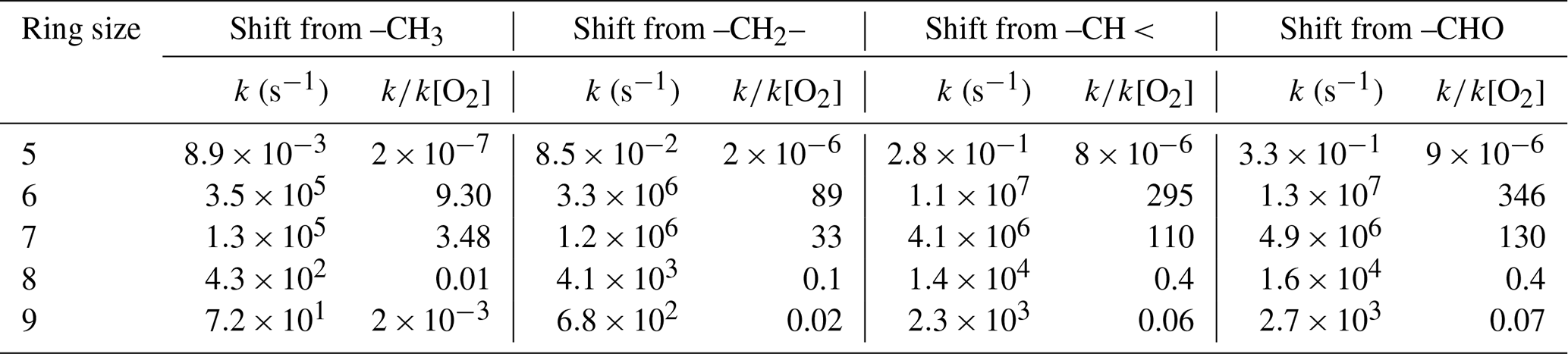
Note that the current estimation methods do not consider effects of functional groups that are between the radical center and the H atom being transferred. This may be considered in the future as more experimental or theoretical data become available.
Although there are uncertainties in these estimates, particularly if non-alkyl groups or substituents are present, their effects on generated mechanisms would be minor if these reactions are estimated to be either very slow or very fast compared to competing processes. Uncertainties will affect generated mechanisms primarily when there are competing β-scission decompositions with comparable estimated rate constants.
5.5 Ester and nitrate rearrangements
Alkoxy radicals with an α-hydrogen and either an ester or nitrate group in the α position can undergo a second type of H-shift isomerization, where the α-hydrogen transfers to the carbonyl or nitrate oxygen and the bonds rearrange to form a carbonyl radical and a carboxylic or nitric acid, via a five-member ring transition state, e.g.,
Evidence for these reactions and their measured or estimated rate constants are discussed in Sect. S1.7.4. Based on limited data, the following rate constants are estimated.
This predicts that the ester rearrangement of primary alkoxy radicals with ester substituents occurs at a similar rate as reaction with O2 under atmospheric conditions, while it is the dominant fate for most secondary alkoxy radicals with ester substituents, being predicted to be faster than possible competing H-shift or β-scission decompositions, as well as the reaction with O2. On the other hand, the nitrate rearrangement is predicted to be slower than the O2 reaction, so it is not expected to be important in atmospheric systems.
Formation of α-nitrato alkoxy radicals that might undergo the nitrate rearrangement is not expected to be as important in atmospheric-oxidation systems because α-nitrato carbon-centered radicals formed in reactions of organic nitrates that could be precursors to these radicals rapidly decompose by eliminating NO2, as discussed above in Sect. 3.2.1, rather than reacting to form the alkoxy radical. Therefore, any uncertainties in estimating rate constants for this reaction are not expected to be important.
Radicals that can undergo the ester rearrangement are predicted to be formed in the atmospheric oxidation of esters, so this reaction is a factor in predicting their atmospheric mechanisms. The uncertainties in the estimated rate constant have a significant effect on predictions for esters forming primary alkoxy radicals because this reaction is competitive with the O2 reaction but is not as important for those forming secondary radicals because the ester rearrangement is predicted to dominate.
5.6 Cyclization reactions
Unsaturated alkoxy radicals could possibly undergo cyclization reactions where the alkoxy radical adds to the double bond, e.g., the following.
These reactions are not considered in the current version of MechGen because they are not consistent with available product data on the reactions of OH with isoprene (Calvert et al., 2000), where the observation of methacrolein and methyl vinyl ketone as major products indicates that these reactions do not compete with the β-scission reaction forming α-hydroxy alkyl radicals. However, theoretical calculations and experimental data suggest that the epoxide formation reaction of β-unsaturated alkoxy radicals (process (a), above) is important following the addition of NO3 radicals with isoprene; Carlsson et al. (2023) and Vereecken (2018) suggested that this reaction may also account for the missing mass balance in aromatic systems (see Sect. 3.4). This epoxide-forming reaction being non-negligible in aromatic systems but not in the case of isoprene oxidation could be attributed to β-scission decompositions forming α-nitrato- or α-peroxy-substituted alkyl radicals in the aromatic systems being much faster than decompositions forming α-hydroxy radicals formed in the isoprene systems. However, our current estimates discussed in Sect. 5.2 and the Supplement are based on the isoprene system. Note, however, that the competing cyclization process (b) is estimated to be somewhat more thermochemically favored, and cyclizations forming larger rings (e.g., (c) and (d), above) are even more thermochemically favored, though they have not been suggested in published mechanisms to our knowledge.
None of these alkoxy cyclization reactions are currently considered by MechGen. However, based on recent results discussed above, they will need to be considered when MechGen is updated.
The previous sections focused on the reactions of unsubstituted and substituted alkyl radicals and the peroxy and alkoxy radicals formed from them in the presence of O2 and NOx, which are the major types of radicals formed in atmospheric-oxidation systems. Other types of radicals are also formed in some circumstances, and the treatments of these are discussed in the subsections below. Note that CIs are discussed separately in Sect. 7.
6.1 Phenoxy radicals
Phenoxy radicals are formed in the reactions of phenols, aromatic aldehydes, and potentially other aromatic compounds. They cannot react with O2 or undergo any of the unimolecular alkoxy radical reactions discussed in Sect. 5. Therefore, their only atmospheric fate is expected to be reactions with NOx species or other radicals. MechGen assumes that their major sinks in atmospheric systems are a reaction with either O3, NO2, or HO2. Reactions with NO are ignored because they are expected to form nitroso species or nitrites that would rapidly photolyze to re-form the reactants and because the concentrations of NO are usually lower than NO2 in the atmosphere.
MechGen assumes that the mechanisms for the reactions of phenoxy with O3 and HO2 are as expected for general alkoxy systems and involve simple O or H transfers, as follows.
The rate constants are in cm3 molec.−1 s−1; the one for O3 is from Tao and Li (1999), and that for HO2 is estimated to be the same as for methoxy + HO2 (Assaf et al., 2018). The O3 reaction is not a net sink for phenoxy radicals because they can be regenerated by reactions of phenyl peroxy radicals with NO. This reaction will also convert NO to NO2, which can also regenerate O3 under atmospheric conditions. On the other hand, the reaction with HO2 is a net sink for both HO2 and phenoxy, though it will regenerate the phenol whose reactions are the source of the phenoxy radicals.
The reactions of phenoxy radicals with NO2 are expected to be the main sink for these radicals in the presence of NOx. Alkoxy radicals are expected to react with NO2, forming the corresponding nitrate, but for most alkoxy radicals this is minor compared to reaction with O2 or unimolecular reactions, so these reactions can be ignored. In the case of phenoxy, the analogous formation of benzyl nitrate is not expected to be an overall sink for the phenoxy radical because the BzO–NO2 bond is so weak (Batiha et al., 2012) that the phenyl nitrate would be expected to rapidly decompose to reactants resulting in no net reaction, analogous to the formation of alkyl peroxynitrates discussed in Sect. 2.3. Instead, the reaction is assumed to occur as follows:
where the rate constant is from Platz et al. (1998). If the initially formed product has an H atom on the same carbon where the NO2 added it will undergo a fast unimolecular rearrangement to a nitrophenol as shown above and discussed in Sect. 2.3.2. This is consistent with theoretical studies (e.g., Batiha et al., 2012) and products formed in reactions of phenols with NO3 (e.g., Calvert et al., 2002, 2011). However, this fast reaction is not possible if NO2 adds to a carbon with another substituent, in which case a 2-nitro-3,5-cyclohexadienone with a substituent in the 2 position is predicted to be the final product.
The presence of substituents on the aromatic rings is assumed not to affect the rate constants or mechanisms of the bimolecular reactions of phenoxy radicals. Note that the ortho NO2 addition can occur in two different positions for unsymmetrical molecules, and currently MechGen assumes that the reaction at each position is equally likely, regardless of substituents.
6.2 Nitrogen-centered radicals
Nitrogen-centered radicals can be formed in the atmospheric reactions of some amines, as discussed in Sect. 2.1.3 above. Those with α-hydrogens are assumed to react primarily with O2, forming HO2 and the corresponding amide,
while those lacking α-hydrogens are assumed to undergo only bimolecular reactions, as follows:
where the rate constants are in units of cm3 molec.−1 s−1 and are at the high-pressure limit where applicable. The high-pressure rate constants for the reactions with NO and NO2 are estimated to be the same as the IUPAC (2023) recommendation for their reactions with ethoxy radicals, while the rate constant for the reaction with HO2 is estimated to be approximately the same as given in the NIST kinetics database for methoxy + HO2 (Sander et al., 2009). MechGen does not generate reactions of amino radicals without α-hydrogens, but they would only be formed from reactions of amines with more than one tertiary substituent, which do not appear to be important in current atmospheric emissions.
Note that this mechanism predicts that formation of N-centered radicals in the reactions of amines without α-hydrogens is a radical-terminating process, which means that such amines tend to be radical inhibitors when they react in the atmosphere. This is consistent with results of environmental-chamber experiments with t-butyl amine and 2-amino-2-methyl-1-propanol (Carter, 2008). This is not expected to be the case with other amines, where results of chamber experiments do not indicate significant radical inhibition in their reactions.
6.3 Carbenes
Carbenes are predicted to form in some photolysis reactions, so their reactions also need to be considered. It is assumed that their major fate is the addition of O2 to form highly excited Criegee intermediates, e.g.,
where {excited} indicates excitation. Reactions of excited and stabilized CIs are discussed in the following section. Note that CIs formed from reactions of O2 with carbenes are expected to have much higher levels of excitation than those formed from the reactions of O3 with alkenes, which affects estimates of stabilization in some cases.
Criegee intermediates (CIs) are highly reactive intermediates with a general structure of X2COO that are formed primarily in the reactions of O3 with alkenes. These can have either zero, one, or two substituents, and those with one or two different substituents can exist in either the syn or anti form, depending on the configuration of the substituent(s) relative to the two oxygens:
They are also formed to a lesser extent from the reactions of O3 with alkynes and in the reactions of O2 with carbenes formed in some photolysis reactions. In all cases they are expected to be formed in a vibrationally excited state, with the amount of excitation depending on the formation reaction, though the amount of excitation is expected to be variable because, except for reactions of O3 with cyclic compounds, the available energy is distributed among two fragments. In some cases the excitation may be sufficient to allow for interconversions between syn and anti, though Vereecken et al. (2017, 2022, and references therein) calculated that the isomerization barriers are high, and interconversions of excited CIs may be slow in many cases. In any case, the interconversion between syn and anti is assumed to be slow once the intermediate is stabilized. The excited intermediates can undergo unimolecular decompositions or be collisionally stabilized, and the stabilized intermediates can also undergo unimolecular reactions or react with water, though reactions of stabilized intermediates involving interactions between the substituent and the Criegee oxygens requires that the substituent be in the syn configuration.
For estimation purposes, we make the assumptions that CIs are initially formed in excited states and that the unimolecular reactions of the excited CIs do not depend on syn or anti configurations. Their configurations are determined only after they are stabilized. This is an oversimplification because the excited CIs may actually have different configurations and may not always interconvert rapidly compared to decompositions. However, available experimental and theoretical data are insufficient to derive the additional parameters required to make separate configuration-dependent estimates for excited as well as stabilized CIs. In many cases the estimates are constrained by experimental or theoretical results, and this informs the parameters used in the estimates. Thus while the parameters may have been different if the configuration were taken into account for excited CI reactions, the predictions would be similar. However, when applied to systems for which no data are available, the parameters and the predictions may be different and are therefore more uncertain.
Information and estimates about the reactions of excited and stabilized CIs are discussed in Sect. S1.9. Briefly, the mechanisms adopted for MechGen are based largely on the IUPAC (2023) reviews of the reactions of O3 with ethene and alkenes with methyl substituents and on the theoretical calculations of Vereecken et al. (2017) on the possible unimolecular reactions of the intermediates. Note that Vereecken et al. (2022) provided a more recent update on the reactions of oxygenated and unsaturated CIs, but these have not yet been taken into account in the current estimates. Table 15 lists the types of intermediates and the reactions and branching ratios considered for them. The types of reactions considered are summarized below.
Table 15Summary of types of Criegee intermediates and the reactions generated for them.
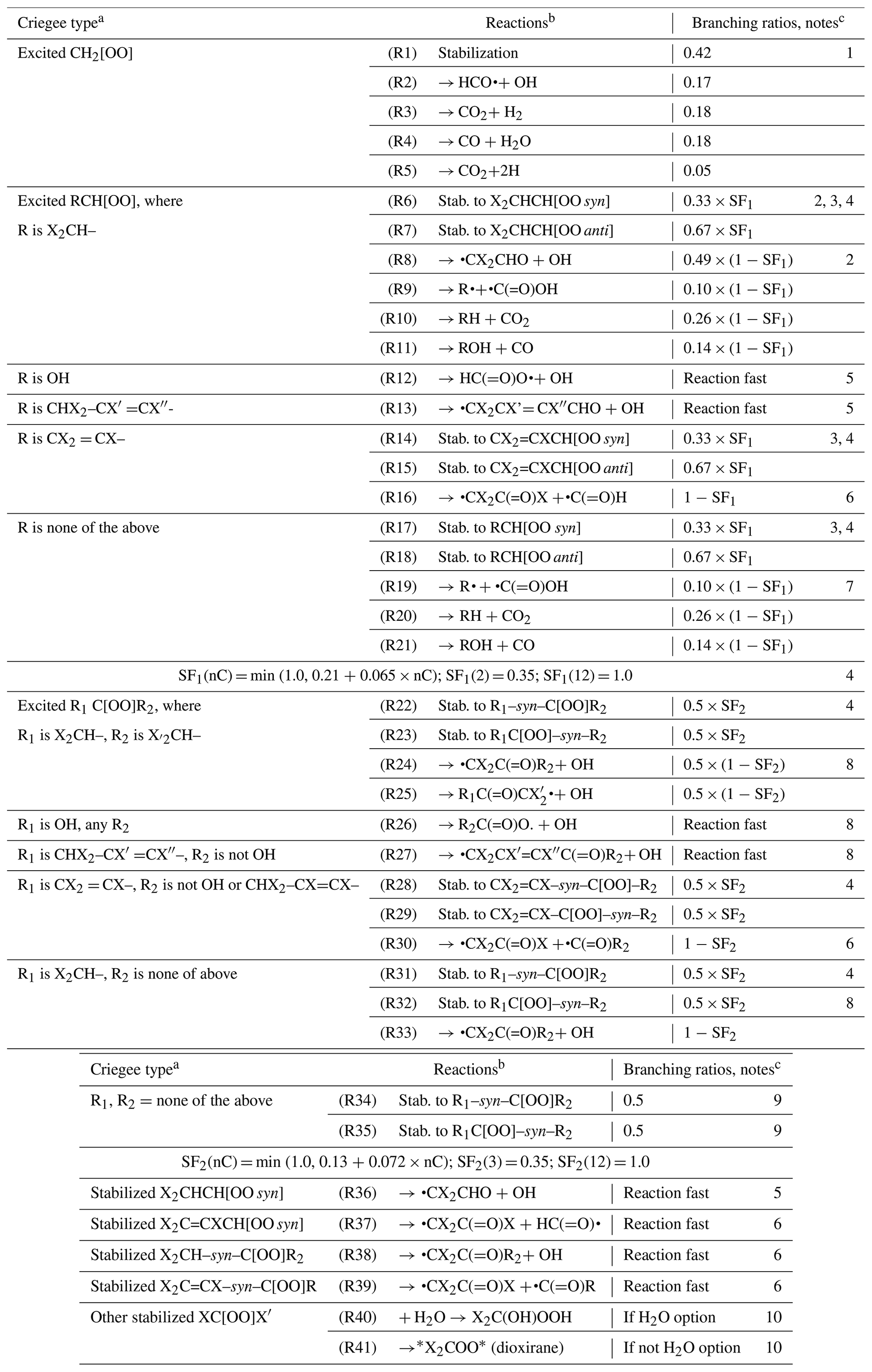
a X is H or any group, R is any group other than –H or as indicated below, syn and anti refer to the orientation of the group with respect to the C–OO bond, * refers to ring closure, SF is the stabilization fraction, and nC is the number of carbons in the intermediate.
b The first column gives the reaction numbers, which are referenced in the discussion in the text.
c Notes for the derivations of the branching ratios are as follows.
1 The reactions and branching ratios of excited CH2[OO] are based on the recommendations of IUPAC (2023) in their review of ethene + O3 reactions.
2 The reactions and branching ratios of excited CH3CH[OO] are based primarily on the recommendations of IUPAC (2023) in their review of the reactions of O3 with propene and the 2-butenes, with some adjustments as discussed in the Supplement.
3 The assumption that the anti isomer is favored is necessary to improve simulations of OH yields from some alkenes. The assumption that the syn isomer is formed ∼ of the time gives predictions that are reasonably consistent with the data.
4 The stabilization fractions are assumed to increase linearly with the size of the intermediate from 35 % for methyl-substituted intermediates to 100 % for intermediates with 16 carbons. This improves model predictions but is somewhat arbitrary, and other methods for estimating size dependence may perform as well or better. It is also assumed that the stabilization fraction is not affected by how the CIs are formed, except that SF is assumed to be 0 for the highly excited intermediates formed from O2 + carbene reactions.
5 These reactions are assumed to be fast based on the calculations of Vereecken et al. (2017) and are assumed to dominate over stabilization or competing reactions of the excited intermediates when they are possible.
6 These reactions are assumed to be fast for stabilized as well as excited intermediates based on the calculations of Vereecken et al. (2017), though they are not so fast that they dominate over stabilization of excited radicals where these are possible. Vereecken et al. (2017) also calculated that they are more rapid than the vinyl hydroperoxide (VHP) rearrangement, so they dominate in excited disubstituted radicals where both are possible.
7 The branching ratios for the hot-acid decompositions are derived from those used for methyl-substituted intermediates, with the yield of the VHP reaction set to 0 and the yields of the others adjusted upward to take this into account.
8 The VHP reaction is assumed to be the only fate of this excited intermediate other than stabilization. If two different reactions are possible, they are assumed to have equal probability; otherwise the only possible VHP reaction dominates.
9 Stabilization is assumed to dominate if the VHP, hot-acid, or fast decomposition reactions are not possible.
10 Rate constants for these reactions are not estimated for this version, so the H2O option determines the major fate assumed for unreactive stabilized intermediates. Note that this ignores loss by photolysis, which may be non-negligible.
Excited intermediates with an alkyl group with α-hydrogens and also stabilized intermediates with such a group in the syn position are assumed to undergo the vinyl hydroperoxide (VHP) rearrangement, where the α-hydrogen transfers to the OO group, forming an excited VHP, which then decomposes to OH and an α-carbonyl radical (e.g., Reactions R8, R24, R25–R27, R36, and R38 in Table 15). Vereecken et al. (2017) calculated these were relatively fast even for stabilized intermediates.
Vereecken et al. (2017) calculated that intermediates with –OH substituents, rare in the atmosphere, will very rapidly rearrange by the O from the –OH group transferring to the [OO] group and then decomposing to OH and an acyl oxy radical (which then decomposes to form CO2 and a radical) (Reactions R12 and R26 in Table 15). These reactions are assumed to be so rapid that they dominate over stabilization.
Excited intermediates with a double-bond group in the α position and also stabilized intermediates with such a group in the syn position can react in one of two ways, depending on whether there is an abstractable hydrogen in the γ position. If there is such a hydrogen, it is assumed that the Criegee group abstracts the γ-hydrogen to form a highly excited allylic hydroperoxide, which then rapidly decomposes to form OH and an allylic carbonyl; this is shown as a single process in Table 15 and Reactions (R13) and (R27). Vereecken et al. (2017) calculated this to be extremely fast, and we assume that this is sufficiently fast that stabilization does not have a chance to occur. If there are no γ-hydrogens, it is assumed that the Criegee group adds to the double bond, forming a highly excited unsaturated peroxy ether, which then rearranges and decomposes to ultimately form the ring-opening products shown as Reactions (R16), (R30), (R37), and (R39) in Table 15. Vereecken et al. (2017) calculates that internal cyclization reactions are more rapid than the VHP rearrangement, so it is assumed that cyclization is the major process in excited disubstituted radicals, where both are possible. Note, however, that Vereecken et al. (2017) only calculated rate constants for the formation of the excited unsaturated peroxy ether, but we assume that the subsequent rearrangement and decomposition is also fast (see Sect. S1.9.2).
Excited intermediates with zero or one substituent can undergo “hot-acid” rearrangements, where an H bonded to the Criegee group is involved in a rearrangement, forming a highly excited acid, which then decomposes in several ways, forming either radicals or stable compounds (e.g., Reactions R1–R5, R9–R11, and R19–R21 in Table 15). These reactions are assumed only to occur when the intermediate is excited. These estimates are based on data for simple alkenes and may not correctly predict products formed from more complex CIs with a single substituent. Other hot acids may not decompose in this way or may be mostly stabilized. These pathways will need to be re-examined when MechGen is updated.
MechGen assumes that excited CIs with α-carbonyl groups are in rapid equilibrium with the primary ozonide and the isomer where the O is transferred to the other group, forming a different carbonyl-substituted intermediate if the groups bonded to them are different, i.e.,
It further assumes that the most reactive isomer determines the subsequent reactions or that a reaction of either form is equally likely if they undergo the same types of reactions. Although Vereecken et al. (2017) discussed a possible reaction forming primary ozonides, they did not include discussion of this particular O-atom rearrangement reaction. However, calculations by Cremer et al. (2001) on the O3 + acetylene system suggest a relatively high barrier to this rearrangement, so this ring closure reaction may be much slower than MechGen assumes. This will need to be re-examined when MechGen is updated.
How MechGen processes stabilized intermediates that cannot undergo the above reactions depends on whether the H2O option is specified in MechGen. If it is, they are assumed to react with H2O, forming an α-hydroxy hydroperoxide (Reaction R40 in Table 15). If not, they are assumed to isomerize to the corresponding dioxirane (Reaction R41). MechGen treats both of these as non-radical products when generating mechanisms. MechGen does not consider effects of substituents in these cases.
MechGen currently does not consider photolysis of stabilized CIs, but loss by photolysis may well be non-negligible for the less reactive intermediates. IUPAC (2023) evaluated data concerning photolyses of the simpler Criegee intermediates and gave recommended cross-sections for unsubstituted and methyl-substituted intermediates. They also recommended assuming unit quantum yields for the photolysis-forming O3P and the corresponding carbonyl. The recommended absorption cross-sections correspond to rate constants for loss by photolysis of ∼ 0.15 s−1 with direct overhead sun, which is faster than the estimated rate constants calculated by Vereecken et al. (2017) for the dioxirane-forming reaction but may not be faster than loss by reaction with H2O, depending on the intermediate and the environment. Therefore, photolysis of the less reactive stabilized CIs may need to be considered in future versions of MechGen.
The stabilization factors and branching ratios are given in Table 15 for the various types of CIs, and footnotes indicate how they were derived. More details are given in Sect. S3.7. The branching ratios used for unsubstituted and methyl-substituted intermediates are based primarily on IUPAC (2023) recommendations and include VHP and hot-acid reactions of the excited intermediate, with the stabilization fraction being 42 % for H2C[OO] and 35 % for the two methyl-substituted intermediates. It is assumed that the VHP reactions also occur with stabilized intermediates if the substituent with the group with the α-hydrogen is in the syn orientation.
Estimates for larger alkyl-substituted intermediates are more uncertain, but the reactions are assumed to be analogous to those of the methyl-substituted intermediates as long as the VHP reaction is possible. If the VHP reaction is not possible, then only the hot-acid reactions are assumed for monosubstituted intermediates and only stabilization is assumed to occur for disubstituted intermediates, where the hot-acid reactions are not possible.
The largest uncertainties concern stabilization fractions for the larger CIs or intermediates formed in reactions where there is not a separate carbonyl co-product, such as reactions of O3 with double bonds in a ring or with triple bonds. As discussed in the Supplement, best fits to the observed OH yields in O3 + alkene reactions (Calvert et al., 2000) and in simulations of NO oxidation and O3 formation rates in some chamber experiments with alkenes (see, e.g., Carter, 2000, 2010b) are obtained if it is assumed that (1) stabilization increases with the size of the molecule; (2) stabilization is the same even if all of the energy is in the intermediate formed; and (3) formation of the anti isomer is favored when monosubstituted Criegee intermediates are stabilized, with the enhancement (somewhat arbitrarily) being assumed to be a factor of 2. The assumption that stabilization energies do not affect stabilization of intermediates formed with higher excitation energies is questionable but is necessary to avoid significantly overpredicting OH yields from cycloalkenes.
On the other hand, we assume that CIs formed from reactions of carbenes with O2 are so highly excited that they react before they are stabilized. In the case of unsubstituted carbenes (Reactions R1–R5 in Table 15), we use a branching ratio of 0 for Reaction R1 and increase the others accordingly. In the case of substituted intermediates, the branching ratios are as shown in S33, calculated using SF1 or SF2 = 0. This is uncertain, but carbene formation is generally not predicted except in some photolysis reactions (see Sect. 2.2.1).
MechGen does not consider all the many possible reactions of CIs or complications considered by Vereecken et al. (2017) or other more recent theoretical or experimental results (e.g., Vereecken et al., 2022), somewhat arbitrary assumptions had to be made in some cases, and treatments of excited or unreactive stabilized intermediates are likely to be overly simplistic. MechGen also does not consider bimolecular reactions with species other than H2O, though such reactions may be non-negligible in some circumstances, such as some laboratory experiments. Therefore, treatments of reactions of CIs must be considered to be uncertain and are an appropriate area to update in future versions of MechGen.
Although application and analysis of mechanisms for individual compounds are beyond the scope of the present paper, examples of generated mechanisms, including numbers of reactions and numbers and types of products, are given for illustrative purposes. We generated mechanisms and product yields at representative atmospheric NOx and radical levels (see Sect. S3) for 38 representative compounds. These include propane; the C4, C8, C12, and C16 n-alkanes; 2-methyl alkanes; 1-alkenes; 2-alkanes; other representative C8 branched alkanes; alkenes; aromatics; oxygenates; and representative terpenes. Note that the generated mechanisms considered only the reactions of the starting compounds and the radicals they form, not the subsequent reaction of the non-radical products formed. Therefore, these are only single generation mechanisms; multi-generation mechanisms, such as those derived using GECKO-A (Aumont et al., 2005), will predict many more products. The specific compounds and selected results for these compounds are given in Table S36.
Selected results of the example mechanism generations by MechGen are given in Table S36 and in Fig. 4. As will be discussed in the corresponding MechGen system paper that is in preparation (Carter et al., 2024), the exact numbers of reactions generated and products predicted depend on the mechanism generation parameters designed to minimize the numbers of very minor reactions and species predicted, with the values given in Table S36 being representative of results when default options are used. The distribution of products also depends on the environments where the compounds are reacted because they are affected by competitions involving bimolecular reactions with atmospheric species such as OH, O3, and NOx species and peroxy radicals. The yield data shown here were calculated based on reactions under urban conditions where O3 formation is equally sensitive to changes in VOC and NOx, which roughly represent conditions of the single-day box model scenarios used to derive EBIR (equal benefit incremental reactivity) ozone reactivity scales (Carter, 1994). More information about how the yields were derived is given in Sect. S3.1.

Figure 4Comparisons of total mole carbon yields of various types of products for generated mechanisms for reactions of representative C8 compounds and terpenes. Notes are as follows. [a] Sum of carbons in products with the indicated groups. [b] Sum of carbons in multifunctional products with two or more different non-alkyl groups on the same carbon. [c] Sum of carbons in products multiplied by the fraction of the product in equilibrium with the condensed phase for atmospheric organic aerosol levels of 50 µg m−3.
Figure 4 shows total yields of compounds containing the types of structural groups formed in the highest yields. Many or most of the products contain more than one such group on the molecule, and some compounds can contribute to total yields of more than one group or (if they have more than one of the same type of group) may contribute more than 1 mol to their group total. The category labeled “multiple groups” gives the total yields of compounds with more than one non-alkyl group bonded to a carbon, which are types of compounds whose mechanisms and estimated rate constants are particularly uncertain. The yields of such multifunctional compounds are the highest for some of the terpenes but are non-negligible for most types of compounds except alkanes.
MechGen has the capability to estimate vapor pressures of generated compounds, using either the SIMPOL (Pankow and Asher, 2008) or EVAPORATION (Compernolle et al., 2011) group contribution methods. The SIMPOL method was used here for illustrative purposes because it provides estimates for a wider range of compounds. Estimated vapor pressures were used to estimate the equilibrium fractions of the first-generation products in the condensed phase at an assumed total organic aerosol level of 50 µg m−3. The estimated total fractions of the products in the condensed phase (sum of product yields times the fraction of each in the condensed phase) are given in Table S36 and included in Fig. 4. More information about the method used is given in Sect. S2.2. Figure 5 shows the fraction of products in the condensed phase as a function of the carbon number for all 38 compounds. As expected, the yields of condensed-phase products increase with the carbon number, with the yields becoming significant for most C12+ compounds, though they are highly variable with the type of compound. These volatility estimates are based on uncertain vapor pressure estimates in many cases (Bilde et al., 2015) and, most importantly, ignore the contributions from products formed in subsequent generation reactions, which tend to form lower-volatility products than first-generation reactions (see, e.g., Aumont et al., 2012, 2013). A comprehensive investigation of the use of MechGen to predict low-volatility products is beyond the scope of this paper, but this is a potentially useful area of future study and comparison with GECKO-A predictions.
Figure 5Estimated fraction of products in the condensed phase at 298 K and with 50 µg m−3 of background organic aerosol as a function of the carbon number for representative compounds.
The MechGen system provides a means to derive nearly fully explicit mechanisms for the reactions of most organic compounds under lower-atmospheric conditions. These can be used as a basis for deriving more condensed mechanisms for use in urban, regional, and global models and serve as standards for developing and evaluating mechanism reduction approaches. The level of chemical detail that generated mechanisms potentially provide may not be necessary for modeling urban O3 formation but may well be necessary for developing or evaluating chemically based models for the formation of SOA in the atmosphere. MechGen, like GECKO-A (Aumont et al., 2005), also provides a potentially useful alternative to the widely used MCM (MCM, 2023) for evaluating chemical systems for laboratory and other studies. Like MCM, it is available to the general user online, but unlike MCM, it can be used to derive mechanisms for compounds whose reactions have not been previously estimated.
Explicit chemical detail in and of itself does not guarantee chemical accuracy or predictive capability. Detailed mechanisms that incorporate incorrect rate constants and branching ratios, incorporate reactions that do not occur, omit important reactions, or incorporate inappropriate assumptions cannot be expected to give reliable predictions and may provide an illusion of accuracy and understanding that does not exist. On the other hand, if the mechanism generation process appropriately incorporates our current knowledge and estimates based on the best available expert judgment, then the generated mechanism provides our best estimate for the reactions of the compound(s) of interest and, if our knowledge is sufficient, has a reasonable chance of predictive capability. However, our current knowledge of atmospheric reactions of organics is not complete, so our best judgment as to the most likely detailed mechanism will evolve over time, and any mechanism generation system will need to evolve as well so its predictions continue to represent the state of the science.
9.1 Summary of areas of mechanism uncertainty
Most of the types of chemical reactions discussed in this paper have at least some areas of significant uncertainty or areas where simplifying assumptions have to be made to avoid excessive complexity either that is impractical to currently implement or that is not justified by available knowledge. The impacts of these uncertainties are expected to vary widely depending on the compounds whose mechanisms are being generated, the environment where the mechanism will be applied, and the objectives of the model application. Table 16 lists what are considered to be the major areas of uncertainty in the current mechanism generation system and gives very brief indications of the associated problems and impacts. Sections of this paper and Supplement where the issues are discussed in more detail are also given. This can serve both as a guideline for areas of research that could be most beneficial for improving detailed mechanisms and also areas of MechGen that need to be the focus of future updates. These are certainly not the only areas of uncertainty in developing comprehensive mechanisms but appear based on this work to be priority areas of research. However, the greatest uncertainty may be reactions or reaction mechanisms that are not currently considered and are therefore omitted entirely in atmospheric mechanisms or models.
Table 16Summary of major areas of uncertainty in the current mechanism generation system.
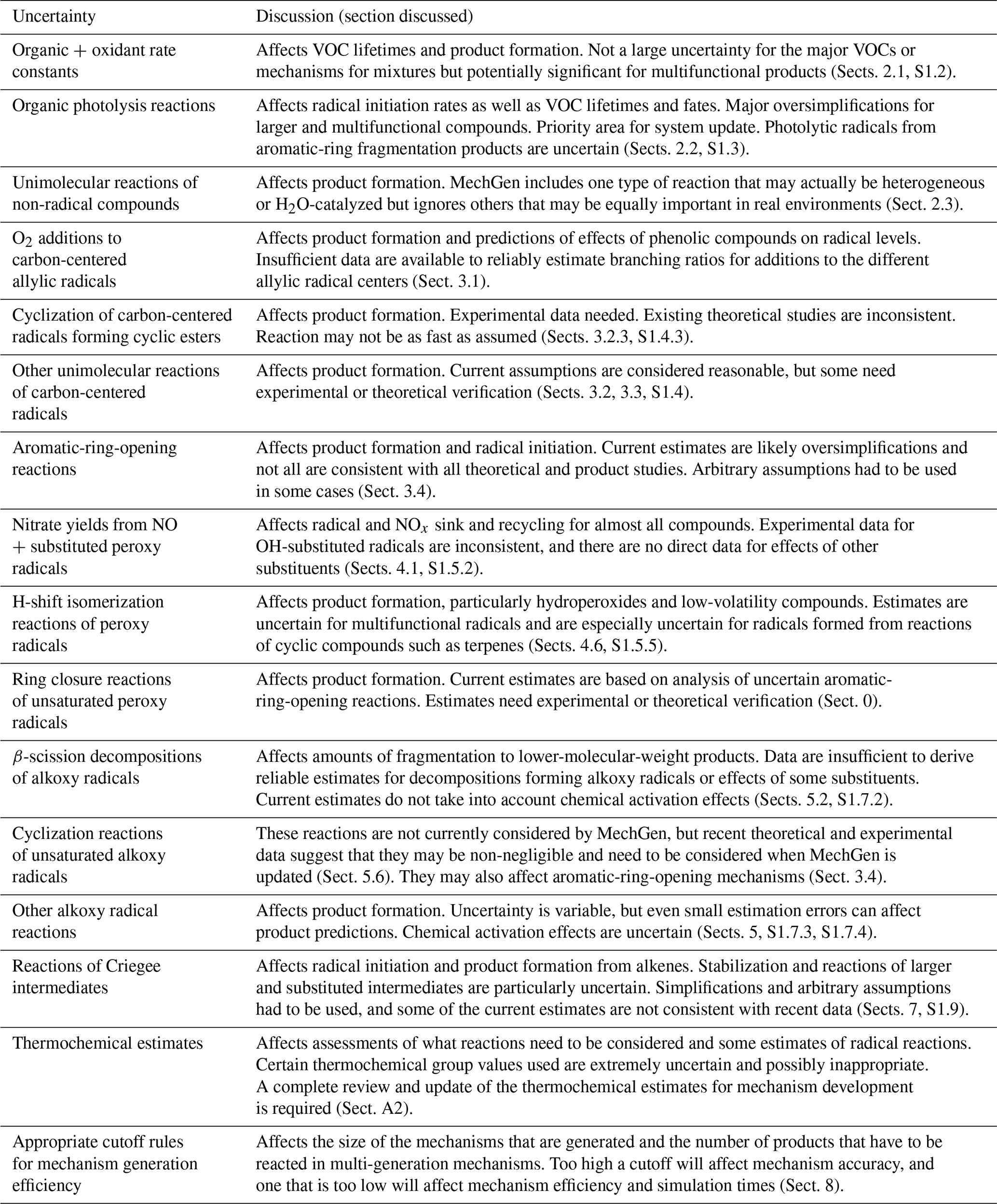
9.2 Recommendations
Mechanism generation systems such as MechGen provide an important link between basic kinetic and mechanistic data and theories and applications such as air quality modeling for research and regulatory applications and analysis of laboratory and field data. With regard to regulatory modeling, Kaduwela et al. (2015) proposed an approach for developing atmospheric chemical mechanisms for the future, in which the development and applications of detailed chemical mechanism generation systems played an essential role. Although generated mechanisms are much too large for full three-dimensional atmospheric modeling, they can serve as a basis for deriving more condensed mechanisms for modeling application that still have a direct link to the underlying basic data and theories and where the effects of the simplifications and condensations can be systematically characterized. Without these links, research and regulatory modeling may not benefit from the emerging science in atmospheric chemistry.
Collection and evaluation of relevant laboratory and theoretical data are essential to detailed mechanism development, both for direct incorporation into the mechanisms and to support the development of the many types of SARs needed for comprehensive mechanism development. The ongoing IUPAC (2023) and NASA (Sander et al., 2006, 2009) evaluations provide an essential role in this effort, as have the books by Calvert et al. (2000, 2002, 2008, 2011, 2015) on atmospheric chemistry, and efforts like these need to continue. Recently McGillen et al. (2020) made available a comprehensive collection of rate constants for reactions of organics with OH, O3, NO3, and Cl atoms, and this data collection effort is continuing, and updates will be made available at https://data.eurochamp.org/data-access/kin/ (last access: 23 June 2023). Similar collections are needed for other types of reactions, particularly data to support SAR development for the many types of radical reactions. The NIST kinetics database (National Institute of Standards and Technology (NIST) Chemical Kinetics Database, 2023) is a useful resource, but recommendations are not provided, and it is limited to reactions of simpler molecules and radicals. Quantum theory has become an increasingly important tool in developing SARs for radical reactions, since experimental data are insufficient for this purpose. Compilations and evaluations of available theoretical data are needed to take full advantage of this ongoing work.
Vereecken et al. (2018) reviewed the current status and research needs for the development of SARs for detailed chemical mechanisms. In addition to discussing data needs and the importance of data collection, they also discussed the need to evaluate and improve existing SARs, particularly with regard to multifunctional compounds. Many of the SARs used in this work may not incorporate all the relevant data and need to be updated or possibly replaced by newer versions. Work on SAR development and improvement needs to be ongoing for chemical mechanisms to continue to represent the state of the science and retain their link to laboratory data and theories.
The predictive capabilities of atmospheric chemical mechanisms, whether detailed or condensed, need to be evaluated for comparing their predictions against laboratory data. In some cases, MechGen had to be modified so that developed mechanisms gave predictions of NO oxidation and O3 formation consistent with results of environmental-chamber experiments. Consistency with O3 formation in well-characterized environmental-chamber data needs to be a priority for mechanisms used for regulatory modeling, though this is not a sufficient evaluation for detailed mechanisms. Comparison of experimental vs. predicted product yields is necessary for a comprehensive evaluation, but this is a major effort with many components and experimental difficulties. This would include comparisons with ambient data as well as results of laboratory and environmental-chamber studies. Doing this comprehensively is a multi-year effort, but it needs to be carried out.
Many if not most of the areas of uncertainty in generated mechanisms may not be reduced significantly in the near term. These uncertainties have variable effects on model predictions of interest, with some highly uncertain or questionable estimates having almost no effects on predictions and, in some cases, only moderate uncertainties in rate constants having large effects. A systematic study of the effects of the various types of uncertainties in predictions of interest is needed not only to prioritize areas where basic research is needed but also to prioritize which SARs and estimates used in systems like MechGen should be given the highest priorities for future work.
The research needs do not end once a detailed mechanism is developed and its predictive capabilities have been evaluated. Mechanisms output by systems such as MechGen or GECKO-A are far too large and complex for most modeling applications and need to be reduced to be useful. The reduction method will depend on the intended application, with, for example, detailed product predictions being a relatively low priority in O3 models but a high priority in models for SOA and toxics. Further discussion of reduction issues is beyond the scope of this work, though it should be noted that MechGen as documented in this paper has been used to develop updated versions of the SAPRC mechanisms for use in airshed models for predicting O3 and toxics, designated SAPRC-22 (Carter, 2023). It can also be used as a basis for developing future versions of the mechanism for predicting SOA.
The MechGen code is available at the MechGen website at https://intra.engr.ucr.edu/~carter/MechGen/ (Carter, 2024a). The version incorporating the methods and assignments documented here is dated 9 July 2024.
The data used in this paper consist of the various parameters used in the estimation methods, experimental or calculated data to derive these parameters, and the assigned rate constants or branching ratios. These are all given in tables in the main text and the Supplement.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-25-199-2025-supplement.
This paper largely represents the body of work on chemical mechanism development led by WPLC, and he is the main contributor to this publication. JJO provided consultation on the methods, including rate constant and branching-ratio estimates, and the resulting mechanism predictions. JJO and KCB contributed to the writing and editing of the paper, and JJ contributed to the editing of the paper and checking of the IUPAC references for accuracy.
At least one of the (co-)authors is a member of the editorial board of Atmospheric Chemistry and Physics. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
Although this publication has been funded in part by the U.S. EPA and the California Air Resources Board (CARB), it has not been formally reviewed by either agency. The views expressed in this document are solely those of the authors and do not necessarily reflect those of these agencies. The EPA and CARB do not endorse any products or commercial services mentioned in this publication.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
This work was supported in part by the California Air Resources Board (CARB; primarily through contract no. 11-761) and in part by the University of California Retirement Plan system. William P. L. Carter wishes to thank Ajith Kaduwela, the CARB project officer, for his support and helpful discussions. We also thank Luc Vereecken of Forschungszentrum Jülich for his helpful and thorough review of this work and Zhizhao Wang of University of California, Riverside, for assistance in preparing this paper.
This work has also been supported by a grant from the U.S. Environmental Protection Agency (U.S. EPA) Science to Achieve Results (STAR) program. This publication was developed in part under an assistance agreement awarded by the U.S. EPA (no. 84000701).
This research has been supported by the U.S. Environmental Protection Agency (grant no. 84000701) and the California Air Resources Board (contract no. 11-761).
This paper was edited by Qiang Zhang and reviewed by Luc Vereecken and one anonymous referee.
Afreh, I. K., Aumont, B., Camredon, M., and Barsanti, K. C.: Using GECKO-A to derive mechanistic understanding of secondary organic aerosol formation from the ubiquitous but understudied camphene, Atmos. Chem. Phys., 21, 11467–11487, https://doi.org/10.5194/acp-21-11467-2021, 2021.
Ali, M. A. and Saswathy, R.: Temperature-and pressure-dependent branching ratios for 2,6-dimethylheptyl radicals (C9H19) + O2 reaction: An ab initio and RRKM/ME approach on a key component of bisabolane biofuel, Fuel, 351, 128969, https://doi.org/10.1016/j.fuel.2023.128969, 2023.
Assaf, E., Schoemaecker, C., Vereecken, L., and Fittschen, C.: The reaction of fluorine atoms with methanol: yield of and rate constant of the reactions CH3O + CH3O and CH3O + HO2, Phys. Chem. Chem. Phys., 20, 10660–10670, https://doi.org/10.1039/C7CP05770A, 2018.
Atkinson, R.: Rate constants for the atmospheric reactions of alkoxy radicals: An updated estimation method, Atmos. Environ., 41, 8468–8485, https://doi.org/10.1016/j.atmosenv.2007.07.002, 2007.
Atkinson, R., Carter, W. P. L., and Winer, A. M.: Effects of temperature and pressure on alkyl nitrate yields in the nitrogen oxide (NOx) photooxidations of n-pentane and n-heptane, J. Phys. Chem., 87, 2012–2018, https://doi.org/10.1021/j100234a034, 1983.
Aumont, B., Szopa, S., and Madronich, S.: Modelling the evolution of organic carbon during its gas-phase tropospheric oxidation: development of an explicit model based on a self generating approach, Atmos. Chem. Phys., 5, 2497–2517, https://doi.org/10.5194/acp-5-2497-2005, 2005.
Aumont, B., Valorso, R., Mouchel-Vallon, C., Camredon, M., Lee-Taylor, J., and Madronich, S.: Modeling SOA formation from the oxidation of intermediate volatility I-alkanes, Atmos. Chem. Phys., 12, 7577–7589, https://doi.org/10.5194/acp-12-7577-2012, 2012.
Aumont, B., Camredon, M., Mouchel-Vallon, C., La, S., Ouzebidour, F., Valorso, R., Lee-Taylor, J., and Madronich, S.: Modeling the influence of alkane molecular structure on secondary organic aerosol formation, Faraday Discuss., 165, 105–122, https://doi.org/10.1039/C3FD00029J, 2013.
Batiha, M., Al-Muhtaseb, A. H., and Altarawneh, M.: Theoretical study on the reaction of the phenoxy radical with O2, OH, and NO2, Int. J. Quantum Chem., 112, 848–857, https://doi.org/10.1002/qua.23074, 2012.
Berndt, T. and Böge, O.: Gas-phase reaction of OH radicals with phenol, Phys. Chem. Chem. Phys., 5, 342–350, https://doi.org/10.1039/B208187C, 2003.
Bilde, M., Barsanti, K., Booth, M., Cappa, C. D., Donahue, N. M., Emanuelsson, E. U., McFiggans, G., Krieger, U. K., Marcolli, C., Tropping, D., Ziemann, P., Barley, M., Clegg, S., Dennis-Smither, B., Hallquist, M., Hallquist, A. M., Khlystov, A., Kulmala, M., Mogensen, D., Percival, C. J., Pope, F., Reid, J. P., da Silva, M. A. V. R., Rosenoern, T., Salo, K., Soonsin, V. P., Yli-Juuti, T., Prisle, N. L., Pagels, J., Rarey, J., Zardini, A. A., and Riipinen, I.: Saturation Vapor Pressures and Transition Enthalpies of Low-Volatility Organic Molecules of Atmospheric Relevance: From Dicarboxylic Acids to Complex Mixtures, Chem. Rev., 115, 4115–4156, https://doi.org/10.1021/cr5005502, 2015.
Bloss, C., Wagner, V., Jenkin, M. E., Volkamer, R., Bloss, W. J., Lee, J. D., Heard, D. E., Wirtz, K., Martin-Reviejo, M., Rea, G., Wenger, J. C., and Pilling, M. J.: Development of a detailed chemical mechanism (MCMv3.1) for the atmospheric oxidation of aromatic hydrocarbons, Atmos. Chem. Phys., 5, 641–664, https://doi.org/10.5194/acp-5-641-2005, 2005.
Calvert, J. G., Atkinson, R., Kerr, J. A., Madronich, S., Moortgat, G. K., Wallington, T. J., and Yarwood, G.: The Mechanisms of Atmospheric Oxidation of the Alkenes, Oxford University Press, Oxford, New York, 560 pp., ISBN 0-19-513177-0, 2000.
Calvert, J. G., Atkinson, R., Becker, K. H., Kamens, R. M., Seinfeld, J. H., Wallington, T. H., and Yarwood, G.: The Mechanisms of Atmospheric Oxidation of the Aromatic Hydrocarbons, Oxford University Press, Oxford, New York, 566 pp., ISBN 0-19-514628-X, 2002.
Calvert, J. G., Derwent, R. G., Orlando, J. J., Tyndall, G. S., and Wallington, T. J.: Mechanisms of Atmospheric Oxidation of the Alkanes, Oxford University Press, Oxford, New York, 1008 pp., ISBN 978-0-19-536581-8, 2008.
Calvert, J. G., Mellouki, A., Orlando, J., Pilling, M., and Wallington, T.: Mechanisms of Atmospheric Oxidation of the Oxygenates, Oxford University Press, Oxford, New York, 1634 pp., ISBN 978-0-19-976707-6, 2011.
Calvert, J. G., Orlando, J. J., Stockwell, W. R., and Wallington, T. J.: The Mechanisms of Reactions Influencing Atmospheric Ozone, Oxford University Press, Oxford, New York, 608 pp., ISBN 978-0-19-023302-0, 2015.
Camredon, M., Aumont, B., Lee-Taylor, J., and Madronich, S.: The SOA/VOC/NOx system: an explicit model of secondary organic aerosol formation, Atmos. Chem. Phys., 7, 5599–5610, https://doi.org/10.5194/acp-7-5599-2007, 2007.
Carlsson, P. T. M., Vereecken, L., Novelli, A., Bernard, F., Brown, S. S., Brownwood, B., Cho, C., Crowley, J. N., Dewald, P., Edwards, P. M., Friedrich, N., Fry, J. L., Hallquist, M., Hantschke, L., Hohaus, T., Kang, S., Liebmann, J., Mayhew, A. W., Mentel, T., Reimer, D., Rohrer, F., Shenolikar, J., Tillmann, R., Tsiligiannis, E., Wu, R., Wahner, A., Kiendler-Scharr, A., and Fuchs, H.: Comparison of isoprene chemical mechanisms under atmospheric night-time conditions in chamber experiments: evidence of hydroperoxy aldehydes and epoxy products from NO3 oxidation, Atmos. Chem. Phys., 23, 3147–3180, https://doi.org/10.5194/acp-23-3147-2023, 2023.
Carter, W. P. L.: Development of Ozone Reactivity Scales for Volatile Organic Compounds, J. Air Waste Manage., 44, 881–899, https://doi.org/10.1080/1073161X.1994.10467290, 1994.
Carter, W. P. L.: Documentation of the SAPRC-99 Chemical Mechanism for VOC Reactivity Assessment, Version v1, Zenodo, https://doi.org/10.5281/zenodo.12600705, 2000.
Carter, W. P. L.: Reactivity Estimates for Selected Consumer Product Compounds, Zenodo, https://doi.org/10.5281/zenodo.13777103, 2008.
Carter, W. P. L.: Development of the SAPRC-07 Chemical Mechanism, Atmos. Environ., 44, 5324–5335, https://doi.org/10.1016/j.atmosenv.2010.01.026, 2010a.
Carter, W. P. L.: Development of the SAPRC-07 Chemical Mechanism and Updated Ozone Reactivity Scales, Version v1, Zenodo, https://doi.org/10.5281/zenodo.12601346, 2010b.
Carter, W. P. L.: Preliminary Documentation of the SAPRC-16 Mechanism, Version v1, Zenodo, https://doi.org/10.5281/zenodo.12601416, 2016.
Carter, W. P. L.: Documentation of the SAPRC-18 Mechanism, Version v1, Zenodo, https://doi.org/10.5281/zenodo.12601475, 2020.
Carter, W. P. L.: Estimation of Rate Constants for Reactions of Organic Compounds under Atmospheric Conditions, Atmosphere, 12, 1250, https://doi.org/10.3390/atmos12101250, 2021.
Carter, W. P. L.: Documentation of the SAPRC-22 Mechanisms, Version v1, Zenodo, https://doi.org/10.5281/zenodo.12601488, 2023.
Carter, W. P. L.: SAPRC Mechanism Generation System for the Atmospheric Reactions of Volatile Organic Compounds in the Presence of NOx, https://intra.cert.ucr.edu/~carter/MechGen/ (last access: 10 December 2024), 2024a.
Carter, W. P. L.: SAPRC Chemical Mechanisms, Test Simulations, and Environmental Chamber Simulation Files, https://intra.cert.ucr.edu/~carter/SAPRC/SAPRCfiles.htm (last access: 22 June 2024), 2024b
Carter, W. P. L. and Heo, G.: Development of revised SAPRC aromatics mechanisms, Atmos. Environ., 77, 404–414, https://doi.org/10.1016/j.atmosenv.2013.05.021, 2013.
Carter, W. P. L., Atkinson, R., Winer, A. M., and Pitts Jr., J. N.: Experimental investigation of chamber-dependent radical sources, Int. J. Chem. Kinet., 14, 1071–1103, https://doi.org/10.1002/kin.550141003, 1982.
Carter, W. P. L., Jiang, J., Wang, Z., and Barsanti, K. C.: The SAPRC atmospheric chemical mechanism generation system (MechGen), in preparation, 2024.
Chen, Y. and Zhu, L.: The Wavelength Dependence of the Photodissociation of Propionaldehyde in the 280−330 nm Region, J. Phys. Chem. A, 105, 9689–9696, https://doi.org/10.1021/jp011445s, 2001.
Compernolle, S., Ceulemans, K., and Müller, J.-F.: EVAPORATION: a new vapour pressure estimation methodfor organic molecules including non-additivity and intramolecular interactions, Atmos. Chem. Phys., 11, 9431–9450, https://doi.org/10.5194/acp-11-9431-2011, 2011.
Cremer, D., Crehuet, R., and Anglada, J.: The Ozonolysis of Acetylene: A Quantum Chemical Investigation, J. Am. Chem. Soc., 123, 6127–6141, https://doi.org/10.1021/ja010166f, 2001.
Crounse, J. D., Knap, H. C., Ørnsø, K. B., Jørgensen, S., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric Fate of Methacrolein. 1. Peroxy Radical Isomerization Following Addition of OH and O2, J. Phys. Chem. A, 116, 5756–5762, https://doi.org/10.1021/jp211560u, 2012.
Curran, H. J., Gaffuri, P., Pitz, W. J., and Westbrook, C. K.: A Comprehensive Modeling Study of n-Heptane Oxidation, Combust. Flame, 114, 149–177, https://doi.org/10.1016/S0010-2180(97)00282-4, 1998.
Davis, A. C. and Francisco, J. S.: Ab Initio Study of Hydrogen Migration in 1-Alkylperoxy Radicals, J. Phys. Chem. A, 114, 11492–11505, https://doi.org/10.1021/jp1042393, 2010.
Gardner, E. P., Sperry, P. D., and Calvert, J. G.: Photodecomposition of acrolein in oxygen-nitrogen mixtures, J. Phys. Chem., 91, 1922–1930, https://doi.org/10.1021/j100291a048, 1987.
Green, M., Yarwood, G., and Niki, H.: FTIR study of the Cl-atom initiated oxidation of methylglyoxal, Int. J. Chem. Kinet., 22, 689–699, 1990.
IUPAC (International Union of Pure and Applied Chemistry): https://iupac.aeris-data.fr/en/home-english/, last access: 26 September 2023.
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997.
Jenkin, M. E., Saunders, S. M., Wagner, V., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part B): tropospheric degradation of aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 181–193, https://doi.org/10.5194/acp-3-181-2003, 2003.
Jenkin, M. E., Valorso, R., Aumont, B., Rickard, A. R., and Wallington, T. J.: Estimation of rate coefficients and branching ratios for gas-phase reactions of OH with aliphatic organic compounds for use in automated mechanism construction, Atmos. Chem. Phys., 18, 9297–9328, https://doi.org/10.5194/acp-18-9297-2018, 2018a.
Jenkin, M. E., Valorso, R., Aumont, B., Rickard, A. R., and Wallington, T. J.: Estimation of rate coefficients and branching ratios for gas-phase reactions of OH with aromatic organic compounds for use in automated mechanism construction, Atmos. Chem. Phys., 18, 9329–9349, https://doi.org/10.5194/acp-18-9329-2018, 2018b.
Jenkin, M. E., Valorso, R., Aumont, B., and Rickard, A. R.: Estimation of rate coefficients and branching ratios for reactions of organic peroxy radicals for use in automated mechanism construction, Atmos. Chem. Phys., 19, 7691–7717, https://doi.org/10.5194/acp-19-7691-2019, 2019.
Jenkin, M. E., Valorso, R., Aumont, B., Newland, M. J., and Rickard, A. R.: Estimation of rate coefficients for the reactions of O3 with unsaturated organic compounds for use in automated mechanism construction, Atmos. Chem. Phys., 20, 12921–12937, https://doi.org/10.5194/acp-20-12921-2020, 2020.
Jiang, J., Carter, W. P. L., Cocker III, D. R., and Barsanti, K. C.: Development and Evaluation of a Detailed Mechanism for Gas-Phase Atmospheric Reactions of Furans, ACS Earth and Space Chemistry, 4, 1254–1268, https://doi.org/10.1021/acsearthspacechem.0c00058, 2020.
Kaduwela, A., Luecken, D., Carter, W., and Derwent, R.: New directions: Atmospheric chemical mechanisms for the future, Atmos. Environ., 122, 609–610, https://doi.org/10.1016/j.atmosenv.2015.10.031, 2015.
Kerdouci, J., Picquet-Varrault, B., and Doussin, J.-F.: Structure–activity relationship for the gas-phase reactions of NO3 radical with organic compounds: Update and extension to aldehydes, Atmos. Environ., 84, 363–372, https://doi.org/10.1016/j.atmosenv.2013.11.024, 2014.
Lannuque, V., Camredon, M., Couvidat, F., Hodzic, A., Valorso, R., Madronich, S., Bessagnet, B., and Aumont, B.: Exploration of the influence of environmental conditions on secondary organic aerosol formation and organic species properties using explicit simulations: development of the VBS-GECKO parameterization, Atmos. Chem. Phys., 18, 13411–13428, https://doi.org/10.5194/acp-18-13411-2018, 2018.
Lee-Taylor, J., Madronich, S., Aumont, B., Baker, A., Camredon, M., Hodzic, A., Tyndall, G. S., Apel, E., and Zaveri, R. A.: Explicit modeling of organic chemistry and secondary organic aerosol partitioning for Mexico City and its outflow plume, Atmos. Chem. Phys., 11, 13219–13241, https://doi.org/10.5194/acp-11-13219-2011, 2011.
Li, Q., Jiang, J., Afreh, I. K., Barsanti, K. C., and Cocker III, D. R.: Secondary organic aerosol formation from camphene oxidation: measurements and modeling, Atmos. Chem. Phys., 22, 3131–3147, https://doi.org/10.5194/acp-22-3131-2022, 2022.
Matsugi, A. and Miyoshi, A.: Yield of Formyl Radical from the Vinyl + O2 Reaction, Int. J. Chem. Kinet., 46, 260–274, https://doi.org/10.1002/kin.20823, 2014.
Matsunaga, A. and Ziemann, P. J.: Yields of β-Hydroxynitrates and Dihydroxynitrates in Aerosol Formed from OH Radical-Initiated Reactions of Linear Alkenes in the Presence of NOx, J. Phys. Chem. A, 113, 599–606, https://doi.org/10.1021/jp807764d, 2009.
Matsunaga, A. and Ziemann, P. J.: Yields of β-hydroxynitrates, dihydroxynitrates, and trihydroxynitrates formed from OH radical-initiated reactions of 2-methyl-1-alkenes, P. Natl. Acad. Sci. USA, 107, 6664–6669, https://doi.org/10.1073/pnas.0910585107, 2010.
McGillen, M. R., Carter, W. P. L., Mellouki, A., Orlando, J. J., Picquet-Varrault, B., and Wallington, T. J.: Database for the kinetics of the gas-phase atmospheric reactions of organic compounds, Earth Syst. Sci. Data, 12, 1203–1216, https://doi.org/10.5194/essd-12-1203-2020, 2020.
MCM (Master Chemical Mechanism): http://chmlin9.leeds.ac.uk/MCM/roots.htt, last access: 26 September 2023.
Miyoshi, A.: Systematic Computational Study on the Unimolecular Reactions of Alkylperoxy (RO2), Hydroperoxyalkyl (QOOH), and Hydroperoxyalkylperoxy (O2QOOH) Radicals, J. Phys. Chem. A, 115, 3301–3325, https://doi.org/10.1021/jp112152n, 2011.
Møller, K. H., Bates, K. H., and Kjaergaard, H. G.: The Importance of Peroxy Radical Hydrogen-Shift Reactions in Atmospheric Isoprene Oxidation, J. Phys. Chem. A, 123, 920–932, https://doi.org/10.1021/acs.jpca.8b10432, 2019.
Muthuramu, K., Shepson, P. B., and O'Brien, J. M.: Preparation, analysis, and atmospheric production of multifunctional organic nitrates, Environ. Sci. Technol., 27, 1117–1124, https://doi.org/10.1021/es00043a010, 1993.
National Institute of Standards and Technology (NIST) Chemical Kinetics Database: https://kinetics.nist.gov/kinetics/, last access: 26 September 2023.
Nozière, B. and Vereecken, L.: Direct Observation of Aliphatic Peroxy Radical Autoxidation and Water Effects: An Experimental and Theoretical Study, Angew. Chem. Int. Edit., 58, 13976–13982, https://doi.org/10.1002/anie.201907981, 2019.
O'Brien, J. M., Czuba, E., Hastie, D. R., Francisco, J. S., and Shepson, P. B.: Determination of the Hydroxy Nitrate Yields from the Reaction of C2–C6 Alkenes with OH in the Presence of NO, J. Phys. Chem. A, 102, 8903–8908, https://doi.org/10.1021/jp982320z, 1998.
Olariu, R. I., Klotz, B., Barnes, I., Becker, K. H., and Mocanu, R.: FT–IR study of the ring-retaining products from the reaction of OH radicals with phenol, o-, m-, and p-cresol, Atmos. Environ., 36, 3685–3697, https://doi.org/10.1016/S1352-2310(02)00202-9, 2002.
Orlando, J. J., Tyndall, G. S., and Wallington, T. J.: The Atmospheric Chemistry of Alkoxy Radicals, Chem. Rev., 103, 4657–4690, https://doi.org/10.1021/cr020527p, 2003.
Pankow, J. F. and Asher, W. E.: SIMPOL.1: a simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds, Atmos. Chem. Phys., 8, 2773–2796, https://doi.org/10.5194/acp-8-2773-2008, 2008.
Platz, J., Nielsen, O. J., Wallington, T. J., Ball, J. C., Hurley, M. D., Straccia, A. M., Schneider, W. F., and Sehested, J.: Atmospheric Chemistry of the Phenoxy Radical, C6H5O(•): UV Spectrum and Kinetics of Its Reaction with NO, NO2, and O2, J. Phys. Chem. A, 102, 7964–7974, https://doi.org/10.1021/jp982221l, 1998.
Praske, E., Otkjær, R. V., Crounse, J. D., Hethcox, J. C., Stoltz, B. M., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric autoxidation is increasingly important in urban and suburban North America, P. Natl. Acad. Sci. USA, 115, 64–69, https://doi.org/10.1073/pnas.1715540115, 2018.
Praske, E., Otkjær, R. V., Crounse, J. D., Hethcox, J. C., Stoltz, B. M., Kjaergaard, H. G., and Wennberg, P. O.: Intramolecular Hydrogen Shift Chemistry of Hydroperoxy-Substituted Peroxy Radicals, J. Phys. Chem. A, 123, 590–600, https://doi.org/10.1021/acs.jpca.8b09745, 2019.
Sander, S. P., Friedl, R. R., and Ravishankara, A. R.: Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation Number 15, 2006.
Sander, S. P., Friedl, R. R., and Barker, J. R.: Supplement to Evaluation 15: Update of Key Reactions, 2009.
Shepson, P. B., Edney, E. O., Kleindienst, T. E., Pittman, J. H., and Namie, G. R.: Production of organic nitrates from hydroxide and nitrate reaction with propylene, Environ. Sci. Technol., 19, 849–854, https://doi.org/10.1021/es00139a014, 1985.
Slagle, I. R. and Gutman, D.: Kinetics of the reaction of C4H3 with molecular oxygen from 298–900 K, Symposium (International) on Combustion, 21, 875–883, 1988.
Slagle, I. R., Park, J.-Y., Heaven, M. C., and Gutman, D.: Kinetics of polyatomic free radicals produced by laser photolysis. 3. Reaction of vinyl radicals with molecular oxygen, J. Am. Chem Soc., 106, 4356–4361, 1984.
Tao, Z. and Li, Z: A kinetics study on reactions of C6H5O with C6H5O and O3 at 298 k, Int. J. Chem. Kinet., 31, 65–72, https://doi.org/10.1002/(SICI)1097-4601(1999)31:1<65::AID-KIN8>3.0.CO;2-J, 1999.
Teng, A. P., Crounse, J. D., Lee, L., St. Clair, J. M., Cohen, R. C., and Wennberg, P. O.: Hydroxy nitrate production in the OH-initiated oxidation of alkenes, Atmos. Chem. Phys., 15, 4297–4316, https://doi.org/10.5194/acp-15-4297-2015, 2015.
Venecek, M. A., Cai, C., Kaduwela, A., Avise, J., Carter, W. P. L., and Kleeman, M. J.: Analysis of SAPRC16 chemical mechanism for ambient simulations, Atmos. Environ., 192, 136–150, https://doi.org/10.1016/j.atmosenv.2018.08.039, 2018.
Vereecken, L.: Computational study of the stability of a-nitroxy-substituted alkyl radicals, Chem. Phys. Lett., 466, 127–130, 2008.
Vereecken, L.: Interactive comment on “Estimation of rate coefficients and branching ratios for gas-phase reactions of OH with aromatic organic compounds for use in automated mechanism construction” by Michael E. Jenkin et al., https://doi.org/10.5194/acp-2018-146-SC1, 2018.
Vereecken, L.: Reaction mechanisms for the atmospheric oxidation of monocyclic aromatic compounds, in: Advances in Atmospheric Chemistry: Vol. 2: Organic Oxidation and Multiphase Chemistry, edited by: Barker, J. R., Steiner, A. L., and Wallington, T. J., World Scientific Publishing Co. Pte. Ltd., Singapore, 377–527, https://doi.org/10.1142/9789813271838_0006, 2019.
Vereecken, L. and Nozière, B.: H migration in peroxy radicals under atmospheric conditions, Atmos. Chem. Phys., 20, 7429–7458, https://doi.org/10.5194/acp-20-7429-2020, 2020.
Vereecken, L. and Peeters, J.: Decomposition of substituted alkoxy radicals–part I: a generalized structure–activity relationship for reaction barrier heights, Phys. Chem. Chem. Phys., 11, 9062–9074, https://doi.org/10.1039/B909712K, 2009.
Vereecken, L. and Peeters, J.: A structure–activity relationship for the rate coefficient of H-migration in substituted alkoxy radicals, Phys. Chem. Chem. Phys., 12, 12608–12620, https://doi.org/10.1039/C0CP00387E, 2010.
Vereecken, L., Nguyen, T. L., Hermans, I., and Peeters, J.: Computational study of the stability of a-hydroperoxyl- or a-alkylperoxyl substituted alkyl radicals, Chem. Phys. Lett., 393, 432–436, 2004.
Vereecken, L., Novelli, A., and Taraborrelli, D.: Unimolecular decay strongly limits the atmospheric impact of Criegee intermediates, Phys. Chem. Chem. Phys., 19, 31599–31612, https://doi.org/10.1039/C7CP05541B, 2017.
Vereecken, L., Aumont, B., Barnes, I., Bozzelli, J. W., Goldman, M. J., Green, W. H., Madronich, S., Mcgillen, M. R., Mellouki, A., Orlando, J. J., Picquet-Varrault, B., Rickard, A. R., Stockwell, W. R., Wallington, T. J., and Carter, W. P. L.: Perspective on Mechanism Development and Structure-Activity Relationships for Gas-Phase Atmospheric Chemistry, Int. J. Chem. Kinet., 50, 435–469, https://doi.org/10.1002/kin.21172, 2018.
Vereecken, L., Vu, G., Wahner, A., Kiendler-Scharr, A., and Nguyen, H. M. T.: A structure activity relationship for ring closure reactions in unsaturated alkylperoxy radicals, Phys. Chem. Chem. Phys., 23, 16564–16576, https://doi.org/10.1039/d1cp02758a, 2021.
Vereecken, L., Novelli, A., Kiendler-Scharr, A., and Wahner, A.: Unimolecular and water reactions of oxygenated and unsaturated Criegee intermediates under atmospheric conditions, Phys. Chem. Chem. Phys., 24, 6428–6443, https://doi.org/10.1039/D1CP05877K, 2022.
Wolfe, G. M., Crounse, J. D., Parrish, J. D., Clair, J. M. S., Beaver, M. R., Paulot, F., Yoon, T. P., Wennberg, P. O., and Keutsch, F. N.: Photolysis, OH reactivity and ozone reactivity of a proxy for isoprene-derived hydroperoxyenals (HPALDs), Phys. Chem. Chem. Phys., 14, 7276–7286, https://doi.org/10.1039/C2CP40388A, 2012.
Xiang, B., Zhu, L., and Tang, Y.: Photolysis of 4-Oxo-2-pentenal in the 190–460 nm Region, J. Phys. Chem. A, 111, 9025–9033, https://doi.org/10.1021/jp0739972, 2007.
Xu, C. and Wang, L.: Atmospheric Oxidation Mechanism of Phenol Initiated by OH Radical, J. Phys. Chem. A, 117, 2358–2364, https://doi.org/10.1021/jp308856b, 2013.
Xu, L., Møller, K. H., Crounse, J. D., Kjaergaard, H. G., and Wennberg, P. O.: New Insights into the Radical Chemistry and Product Distribution in the OH-Initiated Oxidation of Benzene, Environ. Sci. Technol., 54, 13467–13477, https://doi.org/10.1021/acs.est.0c04780, 2020.
Yuan, Y., Zhao, X., Wang, S., and Wang, L.: Atmospheric Oxidation of Furan and Methyl-Substituted Furans Initiated by Hydroxyl Radicals, J. Phys. Chem. A, 121, 9306–9319, https://doi.org/10.1021/acs.jpca.7b09741, 2017.
Zhang, J., Dransfield, T., and Donahue, N. M.: On the Mechanism for Nitrate Formation via the Peroxy Radical + NO Reaction, J. Phys. Chem. A, 108, 9082–9095, https://doi.org/10.1021/jp048096x, 2004.
- Abstract
- Introduction
- Initial reactions of organic compounds
- Reactions of carbon-centered radicals
- Reactions of peroxy radicals
- Reactions of alkoxy radicals
- Reactions of other types of radicals
- Reactions of Criegee intermediates
- Examples of results
- Discussion and conclusions
- Code availability
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement
- Abstract
- Introduction
- Initial reactions of organic compounds
- Reactions of carbon-centered radicals
- Reactions of peroxy radicals
- Reactions of alkoxy radicals
- Reactions of other types of radicals
- Reactions of Criegee intermediates
- Examples of results
- Discussion and conclusions
- Code availability
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement