the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 25 Nov 2025
| 25 Nov 2025
Criegee + HONO reaction: a bimolecular sink of Criegee, and the missing non-photolytic source of OH•
Vishva Jeet Anand
Philips Kumar Rai
One of the most important puzzles in atmospheric chemistry is a mismatch between observed and modelled concentrations of OH•/HO in the presence of high concentration of volatile organic compounds. It is now well established that to fulfill this gap, one needs a reaction that is not only capable of producing OH• but also able to act as a sink of HO. In the present work, we are proposing the Criegee + HONO reaction as a possible solution of this puzzle. Our quantum chemical and kinetic calculations clearly suggest that this reaction can not only be an important source of OH radical but can also act as a sink of HO2 radical. Our study also suggests that HONO has the potential to act as a bimolecular sink of Criegee intermediates, and for some Criegee intermediates under certain atmospheric condition it can even surpass the traditionally known bimolecular sinks such as SO2 and water dimer, even in high humid conditions.
- Article
(1501 KB) - Full-text XML
-
Supplement
(1029 KB) - BibTeX
- EndNote




It is well-known that the atmospheric chemistry is mainly dominated by the radicals (Anderson, 1987; Monks, 2005). Particularly in the troposphere, these radicals are key in degrading various pollutants, a phenomenon as important as the ozone layer for the existence of life (Weinstock, 1969; Lelieveld et al., 2004). The primary radicals responsible for the oxidative power of troposphere come from the HOX (OH•, HO, RO•, RO etc.) family (Prinn, 2003; Ehhalt, 1987; Khan et al., 2018). Among them, OH• is considered as the most important oxidant in the troposphere (Lelieveld et al., 2002, 2016). Although OH• is the most studied radical in the atmosphere, there are still open questions regarding its sources in the atmosphere (Heald and Kroll, 2021; Yang et al., 2024). For a long time, it was believed that OH radicals are mainly formed in daytime via photolysis of tropospheric ozone (O3), and nitrous acid (HONO) (Calvert et al., 1994; Alicke et al., 2003; Griffith et al., 2016; Aumont et al., 2003). But now, with various on-field measurements (Geyer et al., 2003; Ren et al., 2003; Emmerson and Carslaw, 2009), it is well established that OH radicals are also present at night in sufficient amounts. In fact, average nighttime concentration of OH• (∼ 2.6 ×105 molec. cm−3) is only one order of magnitude lower than its average daytime concentration (∼ 1.9 ×106 molec. cm−3) (Emmerson and Carslaw, 2009). As the lifetime of OH• is only ∼ 1 s, this much concentration of OH• during night indicates its in situ generation via non-photolytic sources. The major non-photolytic source of OH• is the recycling of HO radicals (Whalley et al., 2011; Stone et al., 2012; Hofzumahaus et al., 2009; Smith et al., 2006; Hens et al., 2014). Specifically, during the daytime, the primary reaction contributing to this recycling process is NO HO, whereas at night, the key reaction is NO HO (Hall et al., 1988; Mellouki et al., 1988, 1993; Rai and Kumar, 2024). However, compared to photolytic sources, non-photolytic sources of OH• remain less understood in atmospheric chemistry (Brown and Stutz, 2012; Emmerson and Carslaw, 2009). This is evidenced by the fact that, in the atmosphere with a high concentration of volatile organic compounds (VOCs), atmospheric models consistently under-predict the concentration of OH• compared to the observed value (Emmerson and Carslaw, 2009; Stone et al., 2012). This discrepancy is especially pronounced in winter (Harrison et al., 2006; Heard et al., 2004; Slater et al., 2020) and indoor environments (Østerstrøm et al., 2025; Gomez Alvarez et al., 2013; Reidy et al., 2023), where light plays a minimal role. In addition, the discrepancy between measured and observed value of OH• was also found to depend upon NOX concentration. Both under low NOX (Carslaw et al., 2001; Tan et al., 2001; Lelieveld et al., 2008; Tan et al., 2017) as well as high NOX (above 6 ppbv) (Slater et al., 2020), the discrepancy was found to be quite significant. As the primary recycling of HO to OH• occurs via NOX, the under-prediction of OH• by models under low NOX conditions suggests either the presence of another route for recycling or some new non-photolytic source of OH•. This hypothesis is further strengthened by a few combined experimental and modelling studies. For example, Lu et al. (2012) have to introduce an artificial source of OH• ↔ HO inter-conversion (RO X ⟶ HO, HO X ⟶ OH•) in their atmospheric model to match the experimental concentration profile. In an another study, to match the experimental OH concentration with models, Whalley et al. (2011) increased the concentration of VOCs in their model. Although their computed OH• concentration becomes closer to experimental value, the mismatch between observed and measured concentration of HO becomes worse.
There have been various attempts to identify the missing source of OH• in the atmosphere (Paulot et al., 2009; Peeters et al., 2014; Sander et al., 2019). For example, Peeters et al. (2009); Peeters and Müller (2010); Peeters et al. (2014) suggested that the oxidation of isoprene can regenerate HOX radicals in the presence of light via isoprene-peroxy radical interconversion and isomerisation pathways (Leuven Isoprene Mechanism (LIM)). Although the introduction of LIM into chemical models were found to improve the value of modelled OH• concentration, the modelled values still remain under-predicted (Crounse et al., 2011; Teng et al., 2017; Berndt et al., 2019; Novelli et al., 2020; Medeiros et al., 2022). Particularly, the LIM is more effective in regions where biogenic volatile organic compounds (BVOCs) dominate and NOX concentration is ultra low, e.g. rain forest regions (Whalley et al., 2011; Feiner et al., 2016; Lew et al., 2020). In contrast, in regions where sufficient anthropogenic sources of VOCs are present, e.g. in polluted areas, LIM is not effective. In addition, LIM is not fundamentally a HO to OH• interconversion process, rather it is the recycling of VOCs to OH•. In a recent study, Yang et al. (2024) suggested that aldehyde could be an additional source of OH•. Authors proposed that the autoxidation of carbonyl organic peroxy radicals (R(CO)O2) derived from higher aldehydes, can produce OH• through photolysis (RAM mechanism). Though RAM mechanism efficiently predicts OH• production at low NOX concentrations, it still under-predicts the same at high NOX concentrations. Interestingly, when both LIM and RAM are incorporated into a base model in the presence of moderate concentration of NOX, OH• concentration improves significantly, but the discrepancy in the modelled and observed HO remains unresolved. It is also worth mentioning that photolysis is an important part of both, LIM and RAM, and hence, both of these mechanism do not offer any help in improving the model OH• concentration in nocturnal environment. Furthermore, both LIM and RAM are also not directly involved in recycling of HO to OH•. The discrepancy in the model occurs during both day and night (Faloona et al., 2001; Hens et al., 2014; Geyer et al., 2003), and is associated with HO to OH conversion (Whalley et al., 2011; Hofzumahaus et al., 2009). In light of these studies, we believe that the puzzle of missing OH• source is very much alive and the key to this puzzle may be a non-photolytic source capable of HO ↔ OH• recycling.
In the present work, we are proposing reaction of Criegee intermediate with HONO as a source of OH•. Criegee Intermediates (CIs) are formed during the ozonolysis of alkenes (Criegee, 1975; Johnson and Marston, 2008; Taatjes, 2017). In fact, alkene ozonolysis is a highly exothermic reaction produces energized CIs. Some of the energized CIs readily convert into OH• via unimolecular decomposition, while the remaining CIs get collisionally stabilized (sCI) (Horie and Moortgat, 1991; Donahue et al., 2011; Novelli et al., 2014; Alam et al., 2011). sCIs can undergo either a thermal unimolecular dissociation or a bimolecular reaction. Depending upon concentration of the co-reactant and rate constant of such bimolecular reaction, the bimolecular reaction paths can be the main sink of sCI (Osborn and Taatjes, 2015; Lin et al., 2015; Sheps et al., 2014; Vereecken and Francisco, 2012). There are several studies in the literature that suggest CI reacts rapidly with the trace gases present in the atmosphere (Cox et al., 2020; Mallick and Kumar, 2020; Vereecken et al., 2015; Long et al., 2016, 2021). In this work, we are suggesting HONO as a new partner for the bimolecular reaction of Criegee intermediates that is capable of producing OH radicals. The concentration of CI (∼ 104–105 molec. cm−3) in the atmosphere is comparable with Cl• (∼ 5.0 ×104–3.0 ×105 molec. cm−3) and OH• (∼ 1.0 ×105–4.0 ×106 molec. cm−3) (Khan et al., 2018; Novelli et al., 2017). Similarly, nitrous acid (HONO) is also an important trace gas present in the nighttime atmosphere in a considerable amount (Li et al., 2021; Song et al., 2023). The average concentration of HONO is ∼ 8.9 ×1010 molec. cm−3, which can reach as high as ∼,6.9 ×1011 molec. cm−3 during the fog event (Pawar et al., 2024). Although a general wisdom about HONO is, its concentration builds up in nighttime, and in daytime, it decomposes via photolysis to give OH•, HONO itself is a highly reactive molecule and can participate in various bimolecular chemical reactions during night (Anglada and Sole, 2017; Lu et al., 2000; Wallington and Japar, 1989). Moreover, in indoor environments, high concentrations of OH• have been found to strongly correlate with high concentrations of HONO (Gomez Alvarez et al., 2013). It is important to mention that, the reaction of HONO with the simple Criegee intermediate (CH2OO) has already been investigated theoretically (Kumar et al., 2022). In that investigation, the major product was predicted to be hydroperoxymethyl nitrite (HPMN). We will show in the present work that the main product of this reaction is OH• and this newly found path is the dominant path of the title reaction.
2.1 Electronic structure theory
There are two parts of electronic structure theory; optimization and subsequent single-point energy calculations. The criteria behind choosing a method for optimization is; it should be computationally not very demanding and at the same time, it should accurately predict the geometries and frequencies of the species involved in the reaction. Based on these criteria, in the present work, the CCSD(T)/CBS//M062X/aug-cc-pVTZ level of theory was chosen, which is known to give reasonable results in various previous studies (Kumar et al., 2022; Vereecken et al., 2017, 2014; Vereecken, 2017) for reactions involving Criegee intermediates. Gaussian16 software package (Frisch et al., 2016) has been used to carry out all the optimization and single-point energy calculations. To estimate energies at CCSD(T)/CBS level of theory, first, we calculated the single point energies at CCSD(T)/aug-cc-pVDZ, and CCSD(T)/aug-cc-pVTZ level of theory, and then extrapolated these energies to corresponding CBS limit using the method of Varandas and Pansini (Varandas and Pansini, 2014; Pansini et al., 2016) (see Sect. S2 in the Supplement).
2.2 Kinetics
Energetics calculations shed light only on enthalpic requirement of the reaction, for a barrierless process, entropy is an equally important factor. Therefore, to account for both, enthalpy and entropy, we have estimated the rate constant for CH2OO + HONO reaction within a temperature range of 213–320 K. The mechanism of CH2OO + HONO reaction can be represented by following reaction:
To calculate the overall rate constant of the title reaction, we have used the master equation approach as implemented in the MESMER software package. The Reaction (R1) proceeds in three steps. In the first step, the formation of RC occurs via a barrierless association of isolated reactants. MESMER uses the inverse-Laplace-transform (ILT) method to estimate the energy-dependent rate constant, k(E), for this step. This, in turn, requires fitted Arrhenius parameters as input to MESMER, which are obtained using KTOOLS code as implemented in the MultiWell suite of programs (Barker et al., 2021). KTOOLS uses variational transition state theory (VTST) for the barrierless reaction. The inputs for KTOOLS are energies and frequencies calculated on potential energy surface (PES) scans along the coordinate describing the dissociation of RC into isolated reactants. Each point on the PES serves as a trial transition state; KTOOLS searches for the transition state for which the reaction flux is minimized. In the present work, we have obtained this PES scan at CCSD(T)/CBS//M062X/aug-cc-pVTZ level of theory (Table S3 in Sect. S1 of the Supplement contains the energy as well as frequencies at each scan points).
In the next step, RC undergoes unimolecular dissociation to PC via a transition state. MESMER uses Rice-Ramsperger-Kassel-Marcus (RRKM) theory, including tunneling contributions via an unsymmetrical Eckart barrier to compute the unimolecular reaction rate. In the final step, PC spontaneously dissociates to form isolated products. It is important to mention that we do not find any tight transition state for product formation from PC; therefore, we have treated this step also using ILT assuming that rate constants are independent of temperature. The obtained rate constants within 213–320 K were then fitted with Arrhenius equation and supplied to the MESMER.
It is worth noting that the reactant complex (RC) and the transition state (TS) exhibit hindered rotational motions, and multiple conformations may exist due to different torsional angles. To account for this, we have used the HinderedRotorQM1D model in MESMER to compute rate constants. Specifically, we performed a one-dimensional potential energy scan of OH torsion along the N–O bond in both RC and TS at CCSD(T)/CBS//M062X/aug-cc-pVTZ level of theory, that covers the full 0 to 360° range. The resulting energy profiles are used to calculate the hindered rotor partition functions. During this scan, we found local minima in both RC and TS, suggesting that our originally optimized structures correspond to the global minimum conformers. To verify this, we also manually searched for other possible minimum conformers and again found that our original structures are global minimum conformers. The Lennard-Jones (L-J) model is used to calculate the collision frequency between reactants and the bath gas. To obtain the L-J parameters for RC, we performed a PES scan along the reaction coordinate separating bath gas from RC, and fitted the obtained PES with the 12-6 L-J potential expression. The fitted L-J parameters for RC turn out to be, σ=3.1 Å and ϵ=895.5 K. A single-exponential down model is used to describe the collisional energy transfer probability with a maximum energy grain size of 100 cm−1 and ΔEdown=150 cm−1.
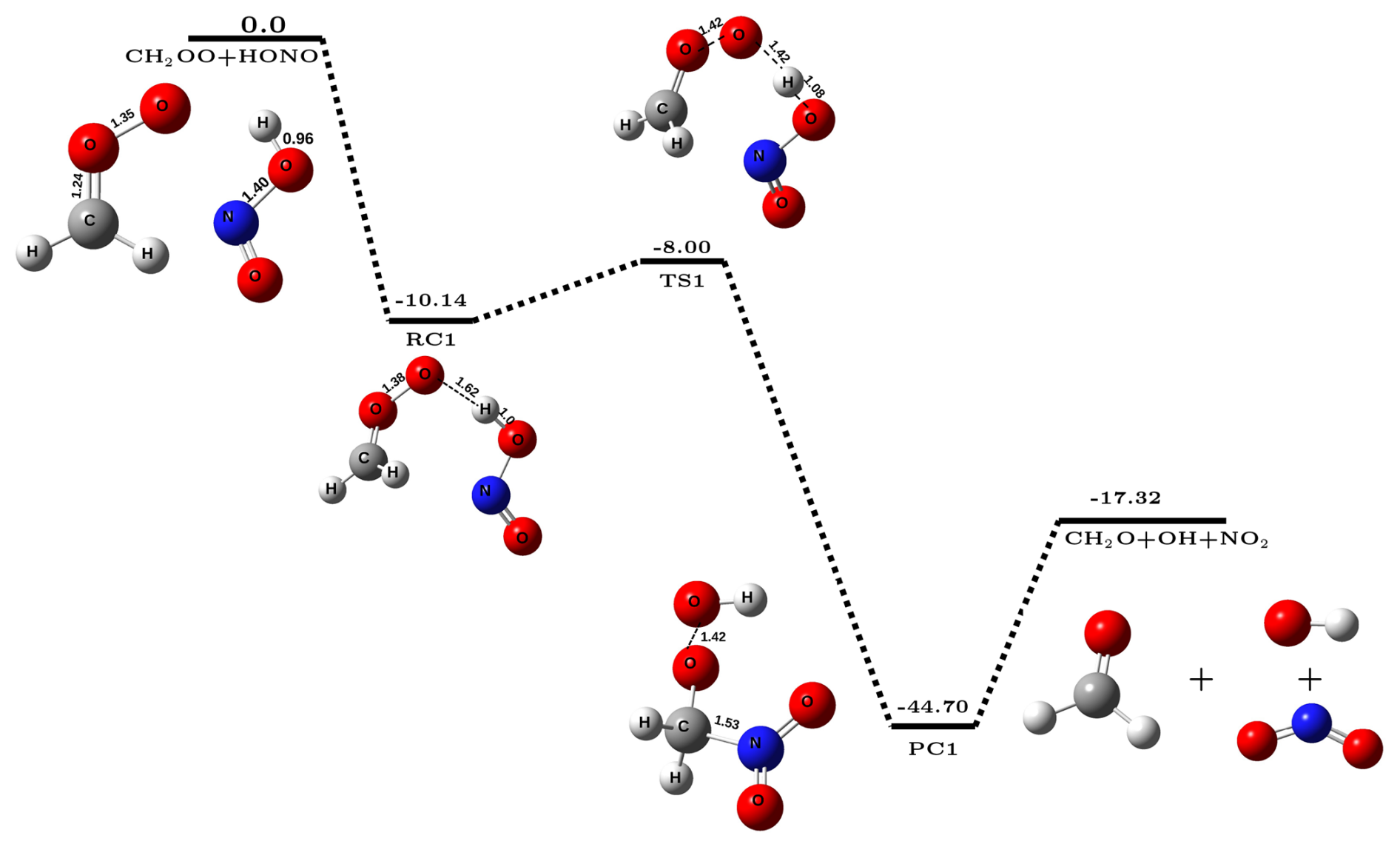
Figure 1The potential energy surface for CH2OO + HONO reaction (in kcal mol−1) obtained at CCSD(T)/CBS//M06-2X/aug-cc-pVTZ level of theory along with optimized geometries of species involved in the reaction.
In the present work, we have investigated the reactions of Criegee intermediates (CIs) with nitrous acid (HONO). It is known that the reactivity of CI is greatly influenced by the substitution group present on carbon center of the CI. Therefore, to account for it, we have studied two types of CIs; the simplest Criegee intermediate (CH2OO) and the dimethyl-substituted Criegee intermediate ((CH3)2COO). Another motivation for choosing (CH3)2COO comes from the fact that in contrast to simple Criegee which is formed only from the ozonolysis of ethene, the dimethyl-substituted Criegee intermediate can be generated from the ozonolysis of many highly abundant alkenes, such as terpenes and mycrene, and hence, the concentration of (CH3)2COO is significantly higher in the atmosphere. In this section, we will first discuss the energetics and kinetics of CH2OO + HONO reaction, followed by (CH3)2COO + HONO reaction. The potential energy surface for CH2OO + HONO reaction is depicted in Fig. 1. It is evident from Fig. 1 that reaction occurs in three steps; in the first step, CH2OO interacts with H atom of HONO via hydrogen bonding and forms a stable reactant-complex (RC1), which is ∼ 10.1 kcal mol−1 stable than isolated reactants. In the next step, RC1 undergoes a unimolecular transformation to form product-complex (PC1) which has stabilization energy of kcal mol−1 with respect to the isolated reactants. This happens via a transition-state (TS1) that is effectively ∼8.0 kcal mol−1 below the isolated reactants. In the last step, PC1 undergoes unimolecular dissociation to form final products, i.e., CH2O, OH•, and NO2. Gibbs free energy at 298 K associated with this conversion of PC1 to isolated products is kcal mol−1 (Sect. S4), which suggests that the formation of OH• via CH2OO + HONO reaction is a spontaneous process.
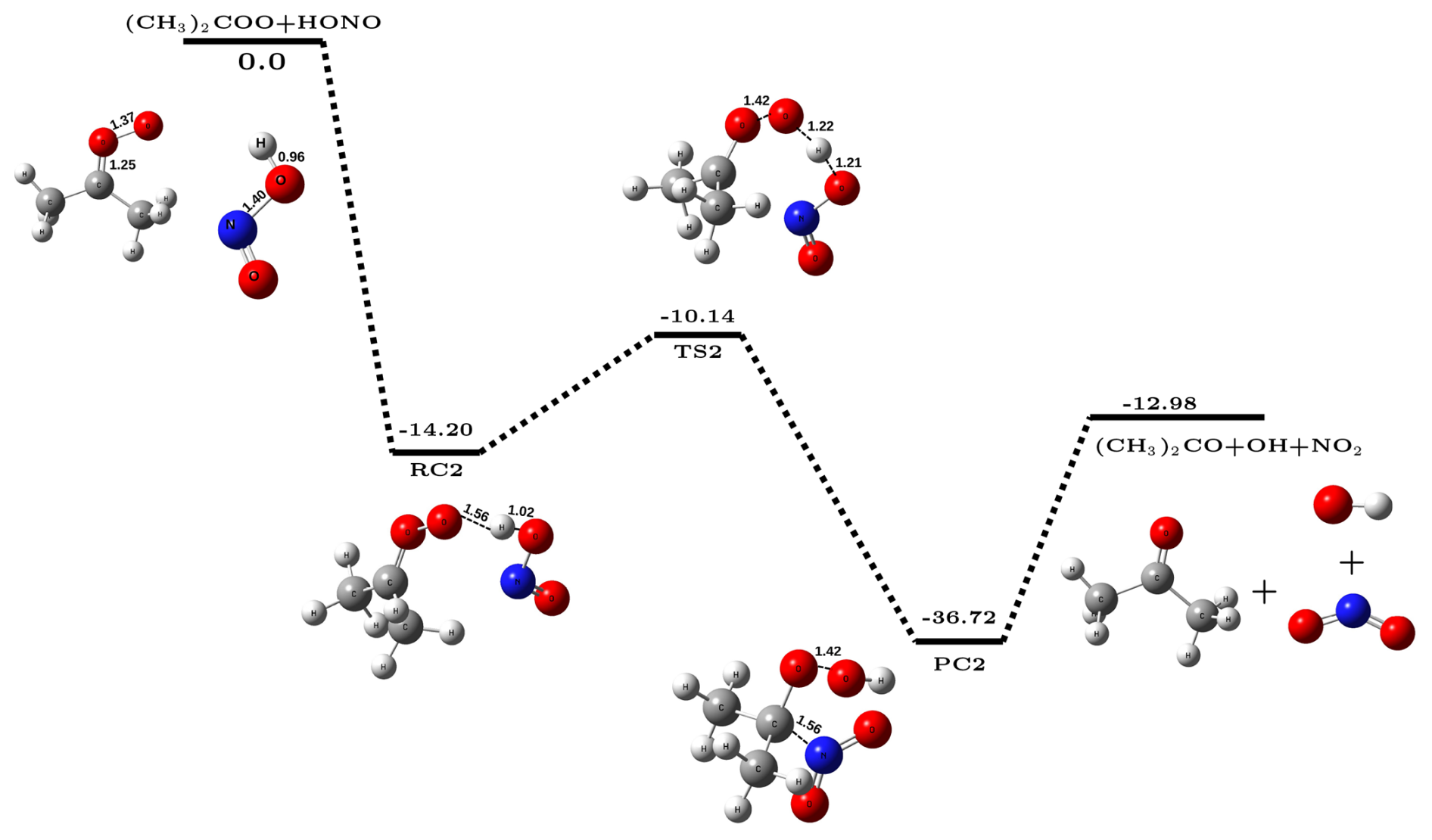
Figure 2The potential energy surface for (CH3)2COO + HONO reaction (in kcal mol−1) obtained at CCSD(T)/CBS//M06-2X/aug-cc-pVTZ level of theory along with optimized geometries of species involved in the reaction.
Table 1Bimolecular rate constants (kbi, in cm3 molec.−1 s−1) for CH2OO/(CH3)2COO + HONO reaction within the temperature range of 213–320 K.

The overall reaction was found to be exothermic by ∼ 17.3 kcal mol−1 that lies close to the experimental value of ∼ 16.9 kcal mol−1 (Ruscic and Bross, 2021), again confirming the adequacy of the methodology used. The computed bimolecular rate constant values () for CH2OO + HONO reaction in the temperature range 213–320 K are given in Table 1. It is evident from Table 1 that the values of slightly decrease with increasing temperature, a typical character of a barrierless process. For example, at 213 K, values of is ∼ 1.17 cm3 molec.−1 s−1 which becomes ∼ 6.3 cm3 molec.−1 s−1 at 320 K. Figure 2 depicts the potential energy surface of (CH3)2COO + HONO reaction. It is evident from Fig. 2 that (CH3)2COO + HONO reaction also proceeds in three steps; in the first step, (CH3)2COO associates with HONO to form a stable reactant-complex (RC2) that is ∼ 14.2 kcal mol−1 more stable than isolated reactants. Next, RC2 transforms into product-complex (PC2) having stabilization energy of kcal mol−1 with respect to isolated reactants. This transformation occurs through a transition state that lies ∼10.1 kcal mol−1 below the isolated reactants. At last, PC2 undergoes unimolecular dissociation to form final products, i.e., (CH3)2CO, OH•, and NO2. Here also, the Gibbs free energy at 298 K associated with the conversion of PC2 to isolated products is kcal mol−1 (Sect. S4), making the overall product formation spontaneous. Using the energetics, we have also computed the rate constant for (CH3)2COO + HONO reaction employing master equation in the same 213–320 K temperature range. The calculated bimolecular rate constants () are listed in Table 1. It is evident from Table 1 that similar to CH2OO + HONO reaction, here also the values of slightly decrease with increasing temperature across the whole range of temperature. But the bimolecular rate constant of (CH3)2COO + HONO reaction becomes ∼ 2.6 to 3.6 times higher compared to the same for CH2COO + HONO reaction at all temperatures considered in the present work. For example, at 298 K, the value of is ∼ 2.03 cm3 molec.−1 s−1, whereas the value of is only ∼ 7.2 ,cm3 molec.−1 s−1.
It is worth noticing that, while computing the bimolecular rate constant, the capture rates of both the reactions are almost same (Table S6 in Sect. S5). The difference in the rate values of the two reactions depends on whether the reactant complex will proceed forward or backward, which further depends on the forward and backward Gibbs free energy barriers of the reactant complex. The Gibbs free energy profile at 298 K is shown in Fig. S2 of Sect. S4. It is evident from Fig. S2 that due to the higher stabilization of RC2 (corresponding to dimethyl-substituted CI), its reverse free energy barrier is high (∼ 2.9 kcal mol−1), while the same is very low for RC1 (corresponding to simplest CI) ( kcal mol−1). Consequently, the relative yields of product are higher for the (CH3)2COO + HONO reaction compared to CH2COO + HONO reaction. Lastly, it is important to discuss the uncertainties associated with the computed rate constant due to limitations in the methodology (Fernández-Ramos et al., 2006). For example, a major source of uncertainty can originate from the fact that Criegee intermediates are known to possess moderate multireference character, and CCSD(T)/CBS sometimes fails in accurately predicting the energetics of such reactions (Rai and Kumar, 2022; Mallick et al., 2019; Mallick and Kumar, 2018). It is worth mentioning that for multireference systems, incorporating higher-level excitations at the coupled-cluster level yield energetics within chemical accuracy (Tajti et al., 2004; Misiewicz et al., 2018; Nguyen et al., 2013; Anand and Kumar, 2023; Rai and Kumar, 2023). To assess the uncertainty in the energetics arising from the multireference character, we have carried out CCSDT(Q)/CBS calculations for the smaller Criegee intermediate reaction, i.e., CH2OO + HONO. We focused on key stationary points; the reactant complex (RC) and the transition state (TS). The various components of the post-CCSD(T) corrections (δT and δT(Q)) are provided in Table S4 of Sect. S2. It is evident from Table S4 that post-CCSD(T) corrections lead to only minor changes in the calculated energetics of CH2OO + HONO reaction. Quantitatively, these corrections reduce the stabilization energy of RC by ∼ 0.54 kcal mol−1, while increasing the barrier height by a similar ∼ 0.67 kcal mol−1. Both variations fall well within the range of chemical accuracy. Furthermore, we have also estimated the capture and bimolecular rate constants using post-CCSD(T) energetics (see Table S9 in Sect. S5), which suggest that at 298 K, the bimolecular rate constants calculated at post-CCSD(T) and CCSD(T)/CBS levels are almost similar (5.53 × 10−12 and 7.21 × 10−12 cm3 molec.−1 s−1, respectively). This supports the reliability and computational efficiency of our chosen level of theory, CCSD(T)/CBS//M06-2X/aug-cc-pVTZ, for studying the title reaction.
Another source of uncertainty in the computed rate constant may arise from the error in estimation of frequency. Such errors in frequency estimation may lead to 2σ (± 2 kcal mol−1) uncertainties in the computed barrier heights. To account for this, we have assumed an uncertainty of ± 2 kcal mol−1 in both well depths and reaction barriers. Using this assumption, we estimated the resulting uncertainty in the rate constants at 213 K and 298 K for the model reaction CH2OO + HONO. Due to ± 2 kcal mol−1 uncertainty in the reaction barriers and well depths, the deviation in the rate constant at 213 K is ∼ 1.17 cm3 molec.−1 s−1 (±2 kcal mol−1 reaction barriers) and ∼ 1.17 cm3 molec.−1 s−1 (±2 kcal mol−1 well depths), respectively. At 298 K, the same becomes cm3 molec.−1 s−1 (±2 kcal mol−1 reaction barriers) and cm3 molec.−1 s−1 (±2 kcal mol−1 well depths), respectively. This study suggests due to 2σ error in the barrier height, there can be an error of a ∼ factor-of-two in the estimated rate constant values. Our analysis also suggests that the uncertainty in the rate constant estimation is much lower at low temperature region compare to high temperature regions. In addition, in the estimation of the partition function, the rigid rotor harmonic oscillator (RRHO) approximation is employed, which again can introduce some error in the final rate constant. For a typical 2σ error, the uncertainty arising from the RRHO approximation can also contribute approximately a factor-of-two uncertainty in the evaluated partition function ratios.
After estimating the energetics and kinetics of title reaction, it is important to discuss the impact of title reaction in the atmospheric chemistry. The importance of title reaction in the atmosphere critically depends on how it competes with other known sinks of Criegee intermediate, i.e., H2O, (H2O)2, NO2, NO, CO, and SO2. The efficiency of a chemical reaction in the atmosphere depends upon two factors; rate of reaction and concentration of co-reactants. The effective rate constant (keff) captures both of these factors as it is defined as the multiplication of bimolecular rate and concentration of co-reactants. Therefore, we have used keff to compare the effectiveness of title reaction compared to other sinks of Criegee intermediates. A list of effective rates for the reaction of CI with H2O, (H2O)2, NO2, NO, CO, and SO2 at 298 K are provided in Table S7 of Sect. S5. To compute keff, the average concentrations of all the sinks have been taken from polluted urban environments. The corresponding rate coefficients of all the sinks are taken from experimental measurements. One can see from Table S7, the effective rate coefficients (keff) of CO, NO, and NO2 are lower compared to those of SO2, H2O, and (H2O)2. For example, keff for the reaction of CI with SO2 is 3.35 s−1, while that for NO2 is only 0.9 s−1. Therefore, in the present work, we have focused our attention on a detailed comparison of the title reaction with SO2, H2O, and (H2O)2.
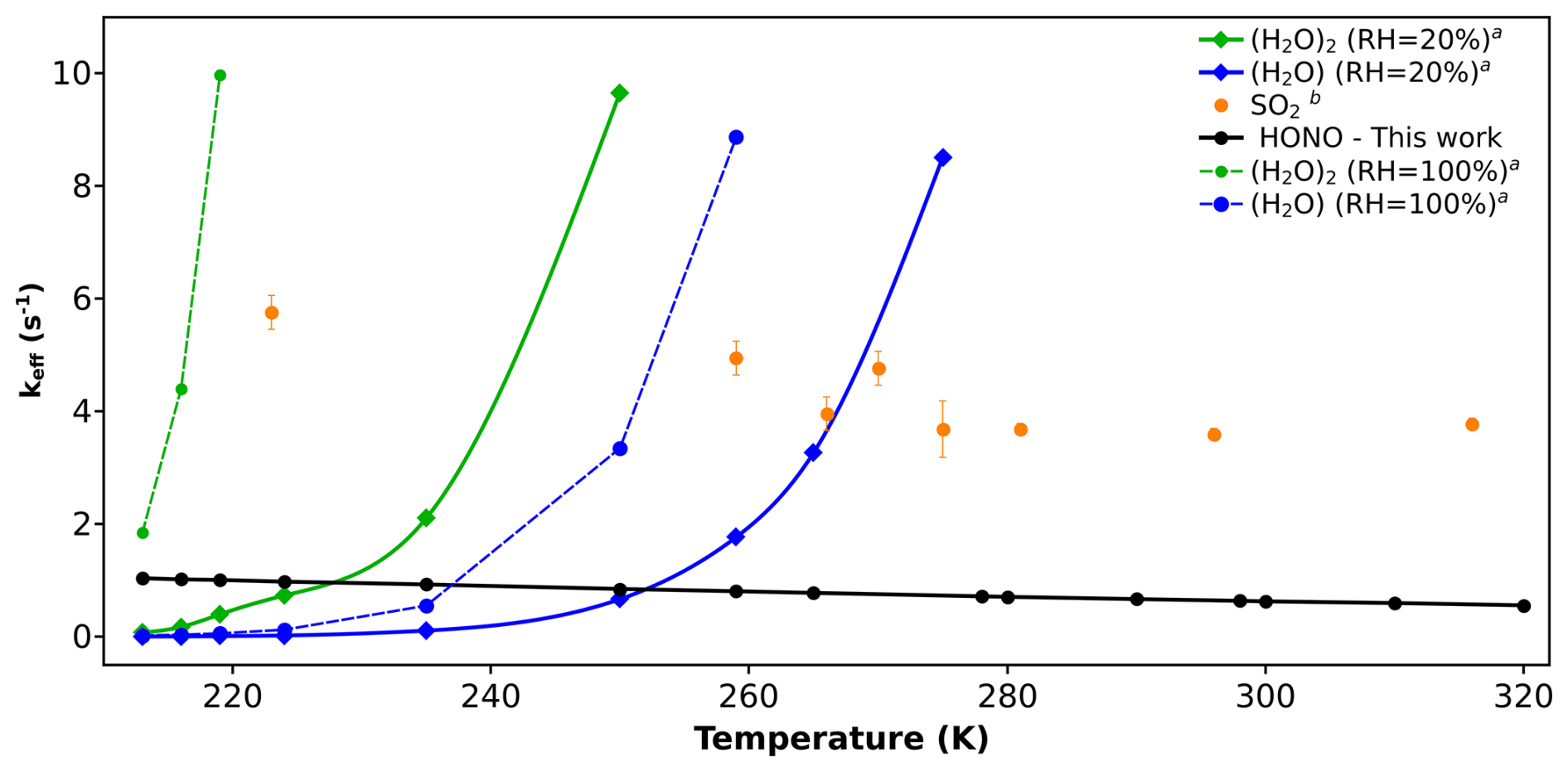
Figure 3Effective rate constant comparison (keff, in s−1) of CH2OO + HONO with the keff of previously known sinks of CH2OO. (a) Values are taken from reference (Lin et al., 2016). (b) Values are taken from reference (Onel et al., 2021).
As far as abundance of HONO is concerned, it is found in both regions; forested as well as polluted in significant amounts (Kim et al., 2015; Acker et al., 2006; Ren et al., 2010; Zhang et al., 2012; He et al., 2006; Su et al., 2008; Ren et al., 2006; Rondon and Sanhueza, 1989; Zhou et al., 2011; Pawar et al., 2024; Vereecken et al., 2012). Among the two, HONO concentrations are comparatively higher in polluted urban areas, such as megacities. Therefore, we expect HONO to play a more effective role as a sink for Criegee intermediates in such regions. Hence, we have used representative concentrations of HONO and SO2 in urban areas for the primary comparison. The concentration of water varies greatly in the atmosphere depending upon saturation vapour pressure and relative humidity (RH) (Anglada et al., 2013; Rai and Kumar, 2025). Therefore, in the case of H2O and (H2O)2, we have taken two concentrations; one calculated at 20 % RH, and the other calculated at 100 % RH. The former serves as lower limits of H2O and (H2O)2 concentrations, whereas the latter serves as the upper limits of H2O and (H2O)2 concentrations. For comparison, we have taken the rate constants reported by Lin et al. (2016) for H2O and (H2O)2, and by Onel et al. (2021) for SO2. In Fig. 3, we have compared the keff of CH2OO + HONO with the keff of CH2OO + H2O/(H2O)2/SO2 reactions.
Figure 3 shows that HONO is a minor sink of simplest Criegee intermediate (CH2OO) compare to SO2, H2O and (H2O)2. In fact, at 100 % RH, keff of CH2OO + (H2O)2 is the dominant reaction across the entire temperature range (213–320 K). At 20 % RH, keff for CH2OO + (H2O)2 and CH2OO + H2O remain dominant at higher temperatures, specifically within 235–320 and 260–320 K, respectively. However, at lower temperatures, keff of CH2OO + HONO becomes dominant, surpassing both, CH2OO + (H2O)2 and CH2OO + H2O in the range of 213–235 and 213–260 K, respectively. Although CH2OO + HONO reaction dominates over CH2OO + (H2O)2 and CH2OO + H2O at low temperature and low humidity, it remains only a minor contributor compared to CH2OO + SO2 reaction at the same conditions. For example, keff values of CH2OO + SO2 reaction are ∼ 5 times higher than that of CH2OO + HONO reaction within the whole temperature range, indicating that CH2OO + HONO reaction is never a dominant sink of CH2OO intermediate.
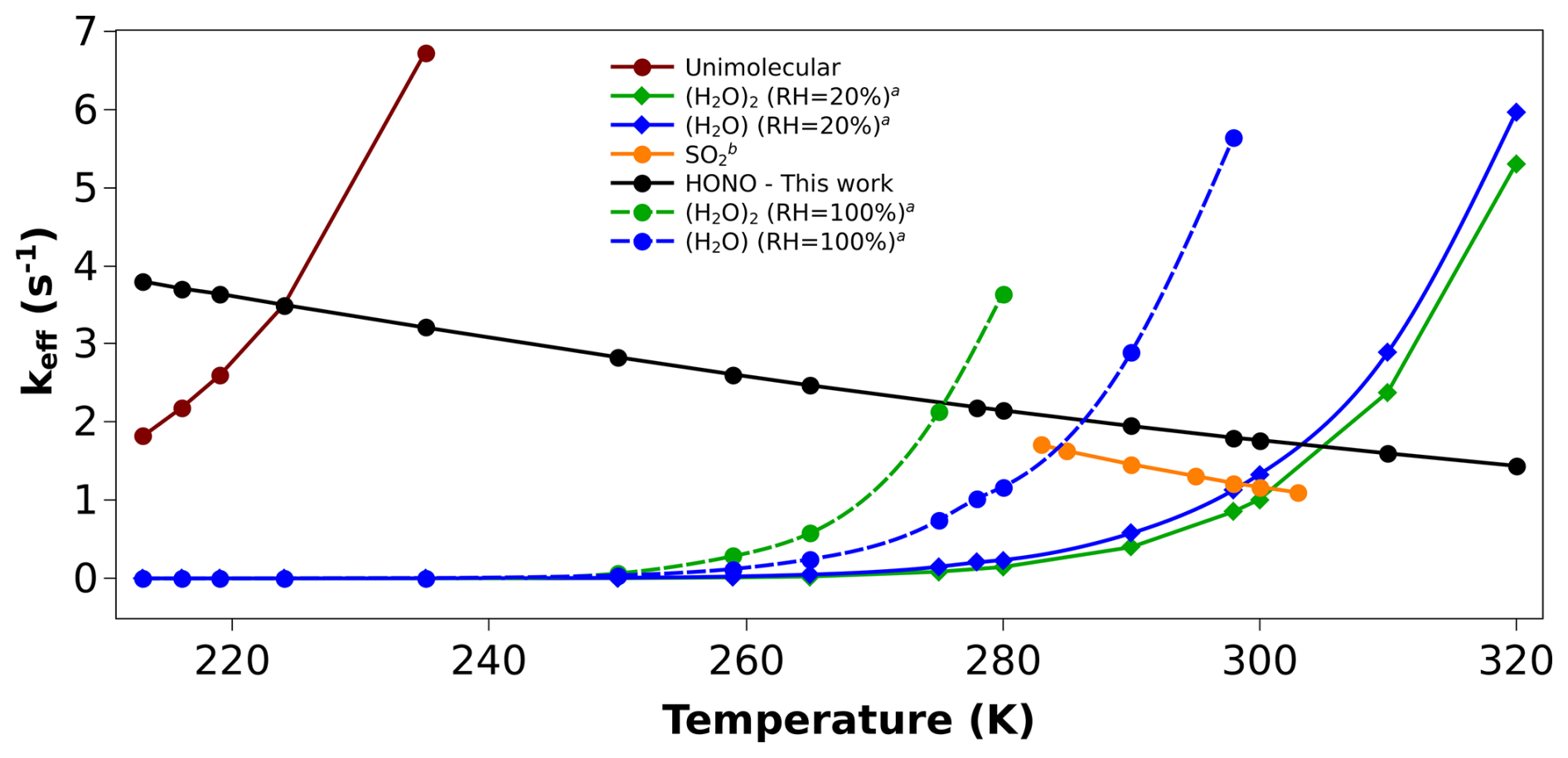
Figure 4Effective rate constant comparison (keff, in s−1) of (CH3)2COO + HONO with the keff of previously known sinks of (CH3)2COO. (a) Values are taken from reference (Vereecken et al., 2017). (b) Values are taken from reference (Smith et al., 2016).
Similarly, we have compared our dimethyl substituted Criegee reaction ((CH3)2COO + HONO) with other known bimolecular reactions of (CH3)2COO. Here also, we have computed keff for the comparison (see Fig. 4). The rate constants of (CH3)2COO + SO2 reaction (Smith et al., 2016) is known in the range of 283–303 K, and hence, we have compared its keff in this temperature range with (CH3)2COO + HONO reaction. Figure 4 shows that unlike CH2OO + HONO reaction, here keff of (CH3)2COO +HONO is ∼ 2 times higher than the same for (CH3)2COO + SO2 reaction within 283–303 K. In addition, it is worth mentioning that under certain atmospheric conditions, concentration of HONO can be quite high compared to SO2. For example, during fog events, it is well known that concentration of SO2 drops significantly (Zhang et al., 2013) while concentration of HONO increases (Pawar et al., 2024), making HONO a potentially major bimolecular sink of Criegee intermediates in fog-like environments. In addition, as SO2 mainly comes from human activities, its concentrations are high in polluted areas and become quite very low in tropical forests and rural areas. In fact, its concentrations fall below detection limits in tropical forest regions (Vereecken et al., 2012). In contrast, although HONO concentration is high in polluted regions compared to a clean environment, due to the various in situ sources, HONO is present in reasonable amounts even in tropical forest areas (Zhang et al., 2012). Therefore, in this region also, HONO is a more effective sink of CI compared to SO2. Moreover, CI + HONO reaction is a hydrogen atom transfer (HAT) process, and hence, the presence of water can effectively catalyze this reaction (Buszek et al., 2012; Viegas and Varandas, 2012; Rai and Kumar, 2025). In contrast, the presence of water, particularly droplets and aerosols, can act as a sink for SO2 (Zhang et al., 2013), and hence, in the presence of water, Criegee + SO2 reaction should be less important compared to CI + HONO reaction. After establishing that compared to SO2, HONO is a more effective sink for (CH3)2COO under most of the conditions, at last, it is important to compare it with (CH3)2COO + H2O/(H2O)2 reactions (Vereecken et al., 2017).
It can be seen from Fig. 4 that even at 100 % RH, keff of (CH3)2COO + HONO can dominate over keff of (CH3)2COO + H2O and (CH3)2COO + (H2O)2 for a relatively wider range of temperatures. For example, the dominant temperature range of (CH3)2COO + HONO is, 213–275 K for (CH3)2COO + (H2O)2 and 213–290 K for (CH3)2COO + H2O. At 20 % RH, keff of (CH3)2COO + HONO becomes dominant over keff of both, (CH3)2COO + H2O and (CH3)2COO + (H2O)2 in almost whole temperature range (213–310 K). For example, at 298 K, keff of (CH3)2COO + HONO is ∼ 1.8 s−1, which is 1.6 times and 2.2 times higher than the same for (CH3)2COO + H2O and (CH3)2COO + (H2O)2, respectively. This suggests that the major bimolecular sink of substituted CI can be its reaction with HONO in the atmosphere even in the presence of high humidity and SO2. At last, it is important to compare the keff of (CH3)2COO + HONO reaction with the unimolecular dissociation rate of (CH3)2COO. Figure 4 also contains the unimolecular dissociation rate of (CH3)2COO. It is evident from Fig. 4 that unimolecular dissociation remains the dominant removal path of (CH3)2COO above 225 K temperature. Only below 225 K temperature, the bimolecular reaction of (CH3)2COO + HONO becomes dominant. To conclude, although HONO is a dominant bimolecular sink for (CH3)2COO, it is still primarily removed by its unimolecular dissociation, particularly at room temperature. For example, the unimolecular dissociation rate of (CH3)2COO is ∼ 276 s−1 at room temperature (Fang et al., 2017) whereas the keff of (CH3)2COO + HONO is only ∼ 1.8 s−1. Interestingly, the unimolecular rate increases rapidly with temperature, while for the bimolecular reaction (CH3)2COO + HONO, keff decreases only slightly. As a result, at lower temperatures, keff may become comparable to the unimolecular dissociation rate of (CH3)2COO. For example, at 213 K, keff and the unimolecular rate constants are 3.80 and 1.82 s−1, respectively. A comparison between keff and the unimolecular dissociation rate constant of (CH3)2COO within 213–320 K is provided in Table S8 of Sect. S5. It is evident from Table S8 that under conditions of high HONO concentration and low temperature, the bimolecular reaction of (CH3)2COO with HONO can compete with its unimolecular dissociation.
Finally, it is important to assess the extent to which the title reaction can contribute in resolving the puzzle of mismatch between measured and modelled OH•/HO concentrations. It is important to mention that during daytime, HONO undergoes rapid photolysis; therefore, its concentration is higher in the absence of light, e.g. at night, indoors, in winter, etc. For example, the photolysis rate of HONO is known to be ∼ 10−3 s−1, which is several orders of magnitude higher than the effective rate constant of its reaction with Criegee intermediates (∼ 10−7–10−6 s−1, computed using maximum Criegee concentration of ∼105 molec. cm−3) (Shabin et al., 2023). Therefore, during the peak of daytime, title reaction does not contribute much to OH• production; rather, it can play a key role in nocturnal atmospheric chemistry, specifically at times when both, concentrations of HONO and CI are high, and, at the same time, the presence of light is minimal.
To understand the efficiency of the title reaction in affecting OH• concentration in a nocturnal environment, we can compare it with NO HO reaction, which is a well-known source of OH• at nighttime. The rate constants for CH2OO + HONO reaction are ∼ 2 times higher compared to NO + HO. For example, at 298 K, the rate value for CH2OO + HONO is cm3 molec.−1 s−1, which is almost double compared to the rate value (Rai and Kumar, 2024) for NO + HO, i.e., cm3 molec.−1 s−1. In the atmosphere, average concentration of both NO and HO are ∼108 molec. cm−3(Bottorff et al., 2023; Brown and Stutz, 2012), thus combined concentration turns out to be ∼1016 molec.2 cm−6. Similarly, the combined concentration will be ∼1015 molec.2 cm−6 for CH2OO + HONO under high concentrations of CI (∼105 molec. cm−3)(Khan et al., 2018) and HONO (∼ 1010 molec. cm−3) (Pawar et al., 2024). It suggests that CH2OO + HONO reaction may be somewhat slower in producing OH•. However, since the rate of (CH3)2COO + HONO reaction is one order of magnitude higher compared to NO HO, we believe both NO HO and title reactions should be of similar importance as far as the production of nighttime OH• is concerned. In other words, title reaction has the potential to serve as a significant contributor to OH• production in nighttime atmospheric chemistry.
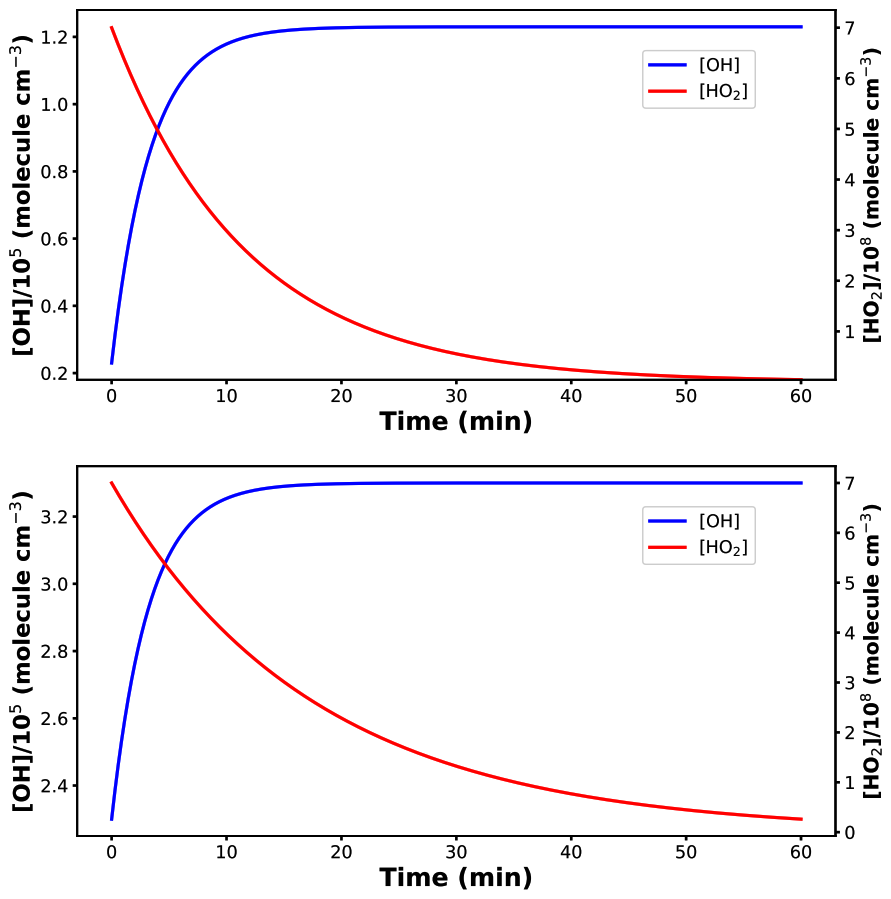
Figure 5Top panel: Concentration profiles of HO and OH• using CH2OO + HONO reaction into the model. Bottom panel: Concentration profiles of HO and OH• using (CH3)2COO + HONO reaction into the model.
Another factor worth noting is, besides OH•, the title reaction produces HCHO/(CH3)2CO, and NO as products. It is well known that both HCHO/(CH3)2CO (Gao et al., 2024; Long et al., 2022; Hermans et al., 2004) and NO (Christensen et al., 2004) can act as sinks for HO2 radicals (corresponding reactions are listed in the box below).
It suggests that title reaction has the potential for recycling of HO ↔ OH• process. To illustrate the ability of title reaction in recycling HO ↔ OH• process, we have developed a kinetic model consisting of the following reactions (see Sect. S3):
This model requires two key components: first, the rate coefficients of the relevant reactions, which have been taken from the recommended literature values (Gao et al., 2024; Hermans et al., 2004; Long et al., 2022; Christensen et al., 2004), and second, a list of realistic initial concentrations of the reactive species involved in HO ↔ OH• recycling process (Table S5 in Sect. S3). We first tracked the change in concentration of OH• and HO using the first kinetic model consisting of CH2OO + HONO reaction, followed by second model consisting of (CH3)2COO + HONO reaction.
Initial concentrations of relevant species (HCHO, HONO, (CH3)2CO, and HO) are chosen based on literature values representing polluted urban conditions (Vereecken et al., 2012; Pawar et al., 2024). Although the average concentration of OH• can vary within ∼104–106 molec. cm−3 in the atmosphere, we have used a modelled value of it in the present work. In CH2OO + HONO reaction model, the initial OH• concentration was set to ∼104 molec. cm−3, while in (CH3)2COO + HONO model, it was set to ∼105 molec. cm−3. This difference was chosen based on how much OH each reaction is expected to produce when no in situ reactions are taking place from the byproducts of the title reaction. Since (CH3)2COO + HONO reaction can generate more OH, starting with a higher initial concentration helps one observe a noticeable change in OH• levels during the simulation. This makes it easier to observe and compare the effect of OH• production between the two reactions. It is important to mention that the maximum concentration of OH• can be taken as ∼105 molec. cm−3 in the kinetic model. This is because the production of OH• is limited by the available concentration of CI which can be as high as ∼ 105 molec. cm−3. Therefore, taking OH• concentration more than ∼ 105 molec. cm−3 would produce no effect on the concentration of OH•. This also reveals the fact that the title reaction is capable of producing OH• in regions where the concentration of OH• is already low. Similarly, the concentration of NO2 can vary within ∼1010–1012 molec. cm−3 in polluted urban regions. However, in the present model, we have kept it at ∼1010 molec. cm−3 in order to observe a clear numerical change in the values of HO. Taking a high concentration of NO2 (∼1012 molec. cm−3) would drastically consume HO, and a gradual change would not be observed.
We have divided the simulation results into two parts; first we will discuss CH2OO + HONO reaction followed by (CH3)2COO + HONO. The model results have been shown in Fig. 5.
It is evident from Fig. 5 that CH2OO + HONO reaction increases OH• concentration while simultaneously reducing HO concentration. Quantitatively, this reaction increases OH• production by five times its initial value while decreasing HO production by more than one order of magnitude. Furthermore, when we consider dimethyl-substituted Criegee intermediate reaction ((CH3)2COO + HONO), OH• production has been found to increase by only a factor of two compared to its initial concentration, while HO production again decreases by the same one order of magnitude (Fig. 5). The difference in OH• production can be attributed to the fact that, in case of (CH3)2COO + HONO, the initial OH• concentration was taken to be ∼105 molec. cm−3 compared to ∼104 molec. cm−3 in case of CH2OO + HONO. This further strengthens the fact that the effect of title reaction on OH• production will be more pronounced in the conditions where OH• concentration is lower in the atmosphere, e.g., at night. The overall simulation results suggest that incorporating title reaction into atmospheric models can improve their accuracy in predicting OH• and HO concentrations. It is important to note that the kinetics model used in the present work is priliminary. However, a more realistic impact of the title reaction on the budget of both OH• and HO, requires a more complete modeling. In order to do so, one needs accurate estimation of the rate constants for the reaction of HONO with various important Criegee intermediates. For bigger Criegee intermediates, computation will be more costly and require a separate study. In addition, being a HAT reaction, the effect of humidity on the title reaction is also important to build a complete model.
In this work, we have studied the energetics and kinetics of bimolecular reaction of simple and dimethyl-substituted Criegee with HONO using high-level electronic structure theory and chemical kinetics. Our quantum chemical calculations suggest that both of the reactions are barrierless and kinetic calculations reveal that reaction of substituted Criegee with HONO is ∼ 2.6–3.6 times faster than simple Criegee + HONO reaction. By comparing it with other known sinks of CI, we have shown that HONO can serve as a major bimolecular sink for bigger Criegee intermediate ((CH3)2COO) and minor contributor at low humidity and low temperature for simple CH2OO. In addition, we have also shown that title reaction can be an important source of OH• in nocturnal atmosphere. In addition, the products of CI + HONO reaction can be a sink for HO2 radicals, and hence this reaction is capable of HO ↔ OH• recycling. Consequently, this reaction can be key in fulfilling the gap between the observed OH radicals and modelled values. Although in urban areas, HONO can be the dominant sink of certain CIs, it is important to notice that larger Criegee intermediates predominantly originate from biogenic volatile organic compounds (BVOCs). On the other hand, HONO concentrations in forested regions are also found to be moderate (∼108 to 1010 molec. cm−3). Therefore, we believe a separate study is required to understand the fate of larger Criegee intermediates in the presence of HONO. At last, we look forward to the experimental verification of our results.
All data supporting the findings of this study are available in the Supplement file.
The supplement related to this article is available online at https://doi.org/10.5194/acp-25-16713-2025-supplement.
VJA: Conducted the investigation, Writing–original draft, Formal analysis, curated the data. PKR: Contributed to partial formal analysis, writing, reviewing, and editing the manuscript. PK: Provided supervision, resources, and methodology; conceptualized the study; acquired funding; and contributed to the review and editing of the manuscript.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
V.J.A. and P.K.R acknowledge MNIT Jaipur for financial assistance. P.K. acknowledges DST, Govt. of India, for the financial support through the sanctioned project No. EEQ/2023/000351.
This research has been supported by the Science and Engineering Research Board (grant no. EEQ/2023/000351).
This paper was edited by Kelvin Bates and reviewed by Stephen Klippenstein and one anonymous referee.
Acker, K., Möller, D., Wieprecht, W., Meixner, F. X., Bohn, B., Gilge, S., Plass-Dülmer, C., and Berresheim, H.: Strong daytime production of OH from HNO2 at a rural mountain site, Geophys. Res. Lett., 33, L02809, https://doi.org/10.1029/2005GL024643, 2006. a
Alam, M. S., Camredon, M., Rickard, A. R., Carr, T., Wyche, K. P., Hornsby, K. E., Monks, P. S., and Bloss, W. J.: Total radical yields from tropospheric ethene ozonolysis, Phys. Chem. Chem. Phys., 13, 11002–11015, 2011. a
Alicke, B., Geyer, A., Hofzumahaus, A., Holland, F., Konrad, S., Pätz, H., Schäfer, J., Stutz, J., Volz-Thomas, A., and Platt, U.: OH formation by HONO photolysis during the BERLIOZ experiment, J. Geophys. Res., 108, https://doi.org/10.1029/2001JD000579, 2003. a
Anand, V. J. and Kumar, P.: Mechanistic insight into the N2O + O(1D, 3P) reaction: role of post-CCSD (T) corrections and non-adiabatic effects, Phys. Chem. Chem. Phys., 25, 33119–33129, 2023. a
Anderson, J. G.: Free Radicals in the Earth's Atmosphere: Their Measurement and Interpretation, Annu. Rev. Phys. Chem., 38, 489–520, 1987. a
Anglada, J. M. and Sole, A.: The atmospheric oxidation of HONO by OH, Cl, and ClO radicals, J. Phys. Chem. A, 121, 9698–9707, 2017. a
Anglada, J. M., Hoffman, G. J., Slipchenko, L. V., M. Costa, M., Ruiz-Lopez, M. F., and Francisco, J. S.: Atmospheric significance of water clusters and ozone–water complexes, J. Phys. Chem. A, 117, 10381–10396, 2013. a
Aumont, B., Chervier, F., and Laval, S.: Contribution of HONO sources to the NOX/HOX/O3 chemistry in the polluted boundary layer, Atmos. Environ., 37, 487–498, 2003. a
Barker, J., Nguyen, T., Stanton, J., Aieta, C., Ceotto, M., Gabas, F., Kumar, T., Li, C., Lohr, L., Maranzana, A., Ortiz, N., Preses, J., Simmie, J., Sonk, J., and Stimac, P.: MultiWell-2021 Software Suite; J. R. Barker, University of Michigan, Ann Arbor, Michigan, USA, https://multiwell.engin.umich.edu/ (last access: 5 March 2025), 2021. a
Berndt, T., Hyttinen, N., Herrmann, H., and Hansel, A.: First oxidation products from the reaction of hydroxyl radicals with isoprene for pristine environmental conditions, Commun. Chem., 2, 21, https://doi.org/10.1038/s42004-019-0120-9, 2019. a
Bottorff, B., Lew, M. M., Woo, Y., Rickly, P., Rollings, M. D., Deming, B., Anderson, D. C., Wood, E., Alwe, H. D., Millet, D. B., Weinheimer, A., Tyndall, G., Ortega, J., Dusanter, S., Leonardis, T., Flynn, J., Erickson, M., Alvarez, S., Rivera-Rios, J. C., Shutter, J. D., Keutsch, F., Helmig, D., Wang, W., Allen, H. M., Slade, J. H., Shepson, P. B., Bertman, S., and Stevens, P. S.: OH, HO2, and RO2 radical chemistry in a rural forest environment: measurements, model comparisons, and evidence of a missing radical sink, Atmos. Chem. Phys., 23, 10287–10311, https://doi.org/10.5194/acp-23-10287-2023, 2023. a
Brown, S. S. and Stutz, J.: Nighttime radical observations and chemistry, Chem. Soc. Rev., 41, 6405–6447, 2012. a, b
Buszek, R. J., Barker, J. R., and Francisco, J. S.: Water effect on the OH + HCl reaction, J. Phys. Chem. A, 116, 4712–4719, 2012. a
Calvert, J., Yarwood, G., and Dunker, A.: An evaluation of the mechanism of nitrous acid formation in the urban atmosphere, Res. Chem. Intermed., 20, 463–502, 1994. a
Carslaw, N., Creasey, D., Harrison, D., Heard, D., Hunter, M., Jacobs, P., Jenkin, M., Lee, J., Lewis, A., Pilling, M., Saunders, S., and Seakins, P.: OH and HO2 radical chemistry in a forested region of north-western Greece, Atmos. Environ., 35, 4725–4737, 2001. a
Christensen, L. E., Okumura, M., Sander, S. P., Friedl, R. R., Miller, C. E., and Sloan, J. J.: Measurements of the Rate Constant of HO2 + NO2 + N2 ⟶ HO2NO2 + N2 Using Near-Infrared Wavelength-Modulation Spectroscopy and UV- Visible Absorption Spectroscopy, J. Phys. Chem. A, 108, 80–91, 2004. a, b
Cox, R. A., Ammann, M., Crowley, J. N., Herrmann, H., Jenkin, M. E., McNeill, V. F., Mellouki, A., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume VII – Criegee intermediates, Atmos. Chem. Phys., 20, 13497–13519, https://doi.org/10.5194/acp-20-13497-2020, 2020. a
Criegee, R.: Mechanism of ozonolysis, Angew. Chem. Internat. Edit., 14, 745–752, 1975. a
Crounse, J. D., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Peroxy radical isomerization in the oxidation of isoprene, Phys. Chem. Chem. Phys., 13, 13607–13613, 2011. a
Donahue, N. M., Drozd, G. T., Epstein, S. A., Presto, A. A., and Kroll, J. H.: Adventures in ozoneland: down the rabbit-hole, Phys. Chem. Chem. Phys., 13, 10848–10857, 2011. a
Ehhalt, D.: Free Radicals in the Atmosphere, Free Radic. Res. Commun., 3, 153–164, 1987. a
Emmerson, K. and Carslaw, N.: Night-time radical chemistry during the TORCH campaign, Atmos. Environ., 43, 3220–3226, 2009. a, b, c, d
Faloona, I., Tan, D., Brune, W., Hurst, J., Barket Jr., D., Couch, T. L., Shepson, P., Apel, E., Riemer, D., Thornberry, T., Carroll, M. A., Sillman, S., Keeler, G. J., Sagady, J., Hooper, D., and Paterson, K.: Nighttime observations of anomalously high levels of hydroxyl radicals above a deciduous forest canopy, J. Geophys. Res. Atmos., 106, 24315–24333, 2001. a
Fang, Y., Barber, V. P., Klippenstein, S. J., McCoy, A. B., and Lester, M. I.: Tunneling Effects in the Unimolecular Decay of (CH3)2COO Criegee Intermediates to OH Radical Products, J. Chem. Phys., 146, 134307, https://doi.org/10.1063/1.4979297, 2017. a
Feiner, P. A., Brune, W. H., Miller, D. O., Zhang, L., Cohen, R. C., Romer, P. S., Goldstein, A. H., Keutsch, F. N., Skog, K. M., Wennberg, P. O., Nguyen, T. B., Teng, A. P., DeGouw, J., Koss, A., Wild, R. J., Brown, S. S., Guenther, A., Edgerton, E., Baumann, K., and Fry, J. L.: Testing atmospheric oxidation in an Alabama forest, J. Atmos. Sci., 73, 4699–4710, 2016. a
Fernández-Ramos, A., Miller, J. A., Klippenstein, S. J., and Truhlar, D. G.: Modeling the kinetics of bimolecular reactions, Chem. Rev., 106, 4518–4584, 2006. a
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Petersson, G. A., Nakatsuji, H., Li, X., Caricato, M., Marenich, A. V., Bloino, J., Janesko, B. G., Gomperts, R., Mennucci, B., Hratchian, H. P., Ortiz, J. V., Izmaylov, A. F., Sonnenberg, J. L., Williams-Young, D., Ding, F., Lipparini, F., Egidi, F., Goings, J., Peng, B., Petrone, A., Henderson, T., Ranasinghe, D., Zakrzewski, V. G., Gao, J., Rega, N., Zheng, G., Liang, W., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Throssell, K., Montgomery, Jr., J. A., Peralta, J. E., Ogliaro, F., Bearpark, M. J., Heyd, J. J., Brothers, E. N., Kudin, K. N., Staroverov, V. N., Keith, T. A., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A. P., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Millam, J. M., Klene, M., Adamo, C., Cammi, R., Ochterski, J. W., Martin, R. L., Morokuma, K., Farkas, O., Foresman, J. B., and Fox, D. J.: Gaussian ˜16 Revision C.01, gaussian Inc. Wallingford CT, 2016. a
Gao, Q., Shen, C., Zhang, H., Long, B., and Truhlar, D. G.: Quantitative kinetics reveal that reactions of HO2 are a significant sink for aldehydes in the atmosphere and may initiate the formation of highly oxygenated molecules via autoxidation, Phys. Chem. Chem. Phys., 26, 16160–16174, 2024. a, b
Geyer, A., Bächmann, K., Hofzumahaus, A., Holland, F., Konrad, S., Klüpfel, T., Pätz, H.-W., Perner, D., Mihelcic, D., Schäfer, H.-J., Volz-Thomas, A., and Platt, U.: Nighttime formation of peroxy and hydroxyl radicals during the BERLIOZ campaign: Observations and modeling studies, J. Geophys. Res. Atmos., 108, https://doi.org/10.1029/2001JD000656, 2003. a, b
Gomez Alvarez, E., Amedro, D., Afif, C., Gligorovski, S., Schoemaecker, C., Fittschen, C., Doussin, J.-F., and Wortham, H.: Unexpectedly high indoor hydroxyl radical concentrations associated with nitrous acid, Proc. Natl. Acad. Sci., 110, 13294–13299, 2013. a, b
Griffith, S. M., Hansen, R. F., Dusanter, S., Michoud, V., Gilman, J. B., Kuster, W. C., Veres, P. R., Graus, M., de Gouw, J. A., Roberts, J., Young, C., Washenfelder, R., Brown, S. S., Thalman, R., Waxman, E., Volkamer, R., Tsai, C., Stutz, J., Flynn, J. H., Grossberg, N., Lefer, B., Alvarez, S. L., Rappenglueck, B., Mielke, L. H., Osthoff, H. D., and Stevens, P. S.: Measurements of hydroxyl and hydroperoxy radicals during CalNex-LA: Model comparisons and radical budgets, J. Geophys. Res., 121, 4211–4232, 2016. a
Hall, I. W., Wayne, R. P., Cox, R. A., Jenkin, M. E., and Hayman, G. D.: Kinetics of the reaction of nitrate radical with hydroperoxo, J. Phys. Chem., 92, 5049–5054, 1988. a
Harrison, R., Yin, J., Tilling, R., Cai, X., Seakins, P., Hopkins, J., Lansley, D., Lewis, A., Hunter, M., Heard, D., Carpenter, L., Creasey, D., Lee, J., Pilling, M., Carslaw, N., Emmerson, K., Redington, A., Derwent, R., Ryall, D., Mills, G., and Penkett, S.: Measurement and modelling of air pollution and atmospheric chemistry in the UK West Midlands conurbation: Overview of the PUMA Consortium project, Sci. Total Environ., 360, 5–25, 2006. a
He, Y., Zhou, X., Hou, J., Gao, H., and Bertman, S. B.: Importance of dew in controlling the air-surface exchange of HONO in rural forested environments, Geophys. Res. Lett., 33, L02813, https://doi.org/10.1029/2005GL024348, 2006. a
Heald, C. L. and Kroll, J. H.: A radical shift in air pollution, Science, 374, 688–689, 2021. a
Heard, D., Carpenter, L., Creasey, D., Hopkins, J., Lee, J., Lewis, A., Pilling, M., Seakins, P., Carslaw, N., and Emmerson, K.: High levels of the hydroxyl radical in the winter urban troposphere, Geophys. Res. Lett., 31, L18112, https://doi.org/10.1029/2004GL020544, 2004. a
Hens, K., Novelli, A., Martinez, M., Auld, J., Axinte, R., Bohn, B., Fischer, H., Keronen, P., Kubistin, D., Nölscher, A. C., Oswald, R., Paasonen, P., Petäjä, T., Regelin, E., Sander, R., Sinha, V., Sipilä, M., Taraborrelli, D., Tatum Ernest, C., Williams, J., Lelieveld, J., and Harder, H.: Observation and modelling of HOx radicals in a boreal forest, Atmos. Chem. Phys., 14, 8723–8747, https://doi.org/10.5194/acp-14-8723-2014, 2014. a, b
Hermans, I., Nguyen, T. L., Jacobs, P. A., and Peeters, J.: Tropopause chemistry revisited: HO-initiated oxidation as an efficient acetone sink, J. Am. Chem. Soc., 126, 9908–9909, 2004. a, b
Hofzumahaus, A., Rohrer, F., Lu, K., Bohn, B., Brauers, T., Chang, C.-C., Fuchs, H., Holland, F., Kita, K., Kondo, Y., Li, X., Lou, S., Shao, M., Zeng, L., Wahner, A., and Zhang, Y.: Amplified trace gas removal in the troposphere, Science, 324, 1702–1704, 2009. a, b
Horie, O. and Moortgat, G.: Decomposition pathways of the excited Criegee intermediates in the ozonolysis of simple alkenes, Atmos. Environ., 25A, 1881–1896, 1991. a
Medeiros, D. J., Blitz, M. A., Seakins, P. W., and Whalley, L. K.: Direct measurements of isoprene autoxidation: Pinpointing atmospheric oxidation in tropical forests, JACS Au, 2, 809–818, 2022. a
Johnson, D. and Marston, G.: The gas-phase ozonolysis of unsaturated volatile organic compounds in the troposphere, Chem. Soc. Rev., 37, 699–716, 2008. a
Khan, M., Percival, C., Caravan, R., Taatjes, C., and Shallcross, D.: Criegee intermediates and their impacts on the troposphere, Environ. Sci.: Process. Impacts, 20, 437–453, 2018. a, b, c
Kim, S., Kim, S.-Y., Lee, M., Shim, H., Wolfe, G. M., Guenther, A. B., He, A., Hong, Y., and Han, J.: Impact of isoprene and HONO chemistry on ozone and OVOC formation in a semirural South Korean forest, Atmos. Chem. Phys., 15, 4357–4371, https://doi.org/10.5194/acp-15-4357-2015, 2015. a
Kumar, A., Mallick, S., and Kumar, P.: Nitrous acid (HONO) as a sink of the simplest Criegee intermediate in the atmosphere, Phys. Chem. Chem. Phys., 24, 7458–7465, 2022. a, b
Lelieveld, J., Peters, W., Dentener, F., and Krol, M.: Stability of tropospheric hydroxyl chemistry, J. Geophys. Res., 107, https://doi.org/10.1029/2002JD002272, 2002. a
Lelieveld, J., Dentener, F. J., Peters, W., and Krol, M. C.: On the role of hydroxyl radicals in the self-cleansing capacity of the troposphere, Atmos. Chem. Phys., 4, 2337–2344, https://doi.org/10.5194/acp-4-2337-2004, 2004. a
Lelieveld, J., Gromov, S., Pozzer, A., and Taraborrelli, D.: Global tropospheric hydroxyl distribution, budget and reactivity, Atmos. Chem. Phys., 16, 12477–12493, https://doi.org/10.5194/acp-16-12477-2016, 2016. a
Lelieveld, J. a., Butler, T. M., Crowley, J. N., Dillon, T. J., Fischer, H., Ganzeveld, L., Harder, H., Lawrence, M. G., Martinez, M., Taraborrelli, D., and Williams, J.: Atmospheric oxidation capacity sustained by a tropical forest, Nature, 452, 737–740, 2008. a
Lew, M. M., Rickly, P. S., Bottorff, B. P., Reidy, E., Sklaveniti, S., Léonardis, T., Locoge, N., Dusanter, S., Kundu, S., Wood, E., and Stevens, P. S.: OH and HO2 radical chemistry in a midlatitude forest: measurements and model comparisons, Atmos. Chem. Phys., 20, 9209–9230, https://doi.org/10.5194/acp-20-9209-2020, 2020. a
Li, Y., Wang, X., Wu, Z., Li, L., Wang, C., Li, H., Zhang, X., Zhang, Y., Li, J., Gao, R., Xue, L., Mellouki, A., Ren, Y., and Zhang, Q.: Atmospheric nitrous acid (HONO) in an alternate process of haze pollution and ozone pollution in urban Beijing in summertime: Variations, sources and contribution to atmospheric photochemistry, Atmos. Res., 260, 105689, https://doi.org/10.1016/j.atmosres.2021.105689, 2021. a
Lin, H.-Y., Huang, Y.-H., Wang, X., Bowman, J. M., Nishimura, Y., Witek, H. A., and Lee, Y.-P.: Infrared identification of the Criegee intermediates syn-and anti-CH3CHOO, and their distinct conformation-dependent reactivity, Nat. Commun., 6, 7012, https://doi.org/10.1038/ncomms8012, 2015. a
Lin, L.-C., Chang, H.-T., Chang, C.-H., Chao, W., Smith, M. C., Chang, C.-H., Jr-Min Lin, J., and Takahashi, K.: Competition between H2O and (H2O)2 reactions with CH2OO/CH3CHOO, Phys. Chem. Chem. Phys., 18, 4557–4568, 2016. a
Long, B., Bao, J. L., and Truhlar, D. G.: Atmospheric Chemistry of Criegee Intermediates: Unimolecular Reactions and Reactions with Water, J. Am. Chem. Soc., 138, 14409–14422, 2016. a
Long, B., Wang, Y., Xia, Y., He, X., Bao, J. L., and Truhlar, D. G.: Atmospheric Kinetics: Bimolecular Reactions of Carbonyl Oxide by a Triple-Level Strategy, J. Am. Chem. Soc., 143, 8402–8413, 2021. a
Long, B., Xia, Y., and Truhlar, D. G.: Quantitative kinetics of HO2 reactions with aldehydes in the atmosphere: High-order dynamic correlation, anharmonicity, and falloff effects are all important, J. Am. Chem. Soc., 144, 19910–19920, 2022. a, b
Lu, K. D., Rohrer, F., Holland, F., Fuchs, H., Bohn, B., Brauers, T., Chang, C. C., Häseler, R., Hu, M., Kita, K., Kondo, Y., Li, X., Lou, S. R., Nehr, S., Shao, M., Zeng, L. M., Wahner, A., Zhang, Y. H., and Hofzumahaus, A.: Observation and modelling of OH and HO2 concentrations in the Pearl River Delta 2006: a missing OH source in a VOC rich atmosphere, Atmos. Chem. Phys., 12, 1541–1569, https://doi.org/10.5194/acp-12-1541-2012, 2012. a
Lu, X., Park, J., and Lin, M.-C.: Gas phase reactions of HONO with NO2, O3, and HCl: Ab initio and TST study, J. Phys. Chem. A, 104, 8730–8738, 2000. a
Mallick, S. and Kumar, P.: Impact of Post-CCSD(T) Corrections on Reaction Energetics and Rate Constants of the OH• + HCl Reaction, J. Phys. Chem. A, 122, 7151–7159, 2018. a
Mallick, S. and Kumar, P.: The reaction of N2O with the Criegee intermediate: A theoretical study, Comput. Theor. Chem., 1191, 113023, https://doi.org/10.1016/j.comptc.2020.113023, 2020. a
Mallick, S., Kumar, A., and Kumar, P.: Revisiting the reaction energetics of the CH3O• + O2 (3Σ−) reaction: the crucial role of post-CCSD(T) corrections, Phys. Chem. Chem. Phys., 21, 6559–6565, 2019. a
Mellouki, A., Le Bras, G., and Poulet, G.: Kinetics of the reactions of nitrate radical with hydroxyl and hydroperoxo, J. Phys. Chem. A, 92, 2229–2234, 1988. a
Mellouki, A., Talukdar, R., Bopegedera, A., and Howard, C. J.: Study of the kinetics of the reactions of NO3 with HO2 and OH, Int. J. Chem. Kinet., 25, 25–39, 1993. a
Misiewicz, J. P., Elliott, S. N., Moore, K. B., and Schaefer, H. F.: Re-examining ammonia addition to the Criegee intermediate: converging to chemical accuracy, Phys. Chem. Chem. Phys., 20, 7479–7491, 2018. a
Monks, P. S.: Gas-phase radical chemistry in the troposphere, Chem. Soc. Rev., 34, 376–395, 2005. a
Nguyen, T. L., Li, J., Dawes, R., Stanton, J. F., and Guo, H.: Accurate determination of barrier height and kinetics for the F + H2 → HF + OH reaction, J. Phys. Chem. A, 117, 8864–8872, 2013. a
Novelli, A., Vereecken, L., Lelieveld, J., and Harder, H.: Direct observation of OH formation from stabilised Criegee intermediates, Phys. Chem. Chem. Phys., 16, 19941–19951, 2014. a
Novelli, A., Hens, K., Tatum Ernest, C., Martinez, M., Nölscher, A. C., Sinha, V., Paasonen, P., Petäjä, T., Sipilä, M., Elste, T., Plass-Dülmer, C., Phillips, G. J., Kubistin, D., Williams, J., Vereecken, L., Lelieveld, J., and Harder, H.: Estimating the atmospheric concentration of Criegee intermediates and their possible interference in a FAGE-LIF instrument, Atmos. Chem. Phys., 17, 7807–7826, https://doi.org/10.5194/acp-17-7807-2017, 2017. a
Novelli, A., Vereecken, L., Bohn, B., Dorn, H.-P., Gkatzelis, G. I., Hofzumahaus, A., Holland, F., Reimer, D., Rohrer, F., Rosanka, S., Taraborrelli, D., Tillmann, R., Wegener, R., Yu, Z., Kiendler-Scharr, A., Wahner, A., and Fuchs, H.: Importance of isomerization reactions for OH radical regeneration from the photo-oxidation of isoprene investigated in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 20, 3333–3355, https://doi.org/10.5194/acp-20-3333-2020, 2020. a
Onel, L., Lade, R., Mortiboy, J., Blitz, M. A., Seakins, P. W., Heard, D. E., and Stone, D.: Kinetics of the gas phase reaction of the Criegee intermediate CH2OO with SO2 as a function of temperature, Phys. Chem. Chem. Phys., 23, 19415–19423, 2021. a
Osborn, D. L. and Taatjes, C. A.: The physical chemistry of Criegee intermediates in the gas phase, Int. Rev. Phys. Chem., 34, 309–360, 2015. a
Pansini, F., Neto, A., and Varandas, A.: Extrapolation of Hartree–Fock and multiconfiguration self-consistent-field energies to the complete basis set limit, Theor. Chem. Acc., 135, 1–6, 2016. a
Paulot, F., Crounse, J. D., Kjaergaard, H. G., Kürten, A., St. Clair, J. M., Seinfeld, J. H., and Wennberg, P. O.: Unexpected epoxide formation in the gas-phase photooxidation of isoprene, Science, 325, 730–733, 2009. a
Pawar, P. V., Mahajan, A. S., and Ghude, S. D.: HONO chemistry and its impact on the atmospheric oxidizing capacity over the Indo-Gangetic Plain, Sci. Total Environ., 174604, https://doi.org/10.1016/j.scitotenv.2024.174604, 2024. a, b, c, d, e
Peeters, J. and Müller, J.-F.: HOX radical regeneration in isoprene oxidation via peroxy radical isomerisations. II: experimental evidence and global impact, Phys. Chem. Chem. Phys., 12, 14227–14235, 2010. a
Peeters, J., Nguyen, T. L., and Vereecken, L.: HOX radical regeneration in the oxidation of isoprene, Phys. Chem. Chem. Phys., 11, 5935–5939, 2009. a
Peeters, J., Muller, J.-F., Stavrakou, T., and Nguyen, V. S.: Hydroxyl radical recycling in isoprene oxidation driven by hydrogen bonding and hydrogen tunneling: The upgraded LIM1 mechanism, J. Phys. Chem. A, 118, 8625–8643, 2014. a, b
Prinn, R. G.: The Cleansing Capacity of the Atmosphere, Annu. Rev. Environ. Resour., 28, 29–57, 2003. a
Rai, P. K. and Kumar, P.: Role of post-CCSD (T) corrections in predicting the energetics and kinetics of the OH• + O3 reaction, Phys. Chem. Chem. Phys., 24, 13026–13032, 2022. a
Rai, P. K. and Kumar, P.: Accurate determination of reaction energetics and kinetics of HO+O3 → OH•+2O2 reaction, Phys. Chem. Chem. Phys., 25, 8153–8160, 2023. a
Rai, P. K. and Kumar, P.: Mechanistic Inside into the Gas-Phase NO3 + HO2 Reaction, J. Phys. Chem. A, 128, 7907–7913, 2024. a, b
Rai, P. K. and Kumar, P.: Influence of Water on the NO3 + HO2 Reaction, J. Phys. Chem. A, 129, 2067–2076, 2025. a, b
Reidy, E., Bottorff, B. P., Rosales, C. M. F., Cardoso-Saldaña, F. J., Arata, C., Zhou, S., Wang, C., Abeleira, A., Hildebrandt Ruiz, L., Goldstein, A. H., Novoselac, A., Kahan, T. F., Abbatt, J. P. D., Vance, M. E., Farmer, D. K., and Stevens, P. S.: Measurements of hydroxyl radical concentrations during indoor cooking events: Evidence of an unmeasured photolytic source of radicals, Environ. Sci. Technol., 57, 896–908, 2023. a
Ren, X., Harder, H., Martinez, M., Lesher, R. L., Oliger, A., Shirley, T., Adams, J., Simpas, J. B., and Brune, W. H.: HOX concentrations and OH reactivity observations in New York City during PMTACS-NY2001, Atmos. Environ., 37, 3627–3637, 2003. a
Ren, X., Brune, W. H., Oliger, A., Metcalf, A. R., Simpas, J. B., Shirley, T., Schwab, J. J., Bai, C., Roychowdhury, U., Li, Y., Cai, C., Demerjian, K. L., He, Y., Zhou, X., Gao, H., and Hou, J.: OH, HO2, and OH reactivity during the PMTACS–NY Whiteface Mountain 2002 campaign: Observations and model comparison, J. Geophys. Res. Atmos., 111, D10S03, https://doi.org/10.1029/2005JD006126, 2006. a
Ren, X., Gao, H., Zhou, X., Crounse, J. D., Wennberg, P. O., Browne, E. C., LaFranchi, B. W., Cohen, R. C., McKay, M., Goldstein, A. H., and Mao, J.: Measurement of atmospheric nitrous acid at Bodgett Forest during BEARPEX2007, Atmos. Chem. Phys., 10, 6283–6294, https://doi.org/10.5194/acp-10-6283-2010, 2010. a
Rondon, A. and Sanhueza, E.: High HONO atmospheric concentrations during vegetation burning in the tropical savannah, Tellus B, 41, 474–477, 1989. a
Ruscic, B. and Bross, D. H.: Active Thermochemical Tables (ATcT) Thermochemical Values ver. 1.122v, https://doi.org/10.17038/CSE/1885921, 2021. a
Sander, R., Baumgaertner, A., Cabrera-Perez, D., Frank, F., Gromov, S., Grooß, J.-U., Harder, H., Huijnen, V., Jöckel, P., Karydis, V. A., Niemeyer, K. E., Pozzer, A., Riede, H., Schultz, M. G., Taraborrelli, D., and Tauer, S.: The community atmospheric chemistry box model CAABA/MECCA-4.0, Geosci. Model Dev., 12, 1365–1385, https://doi.org/10.5194/gmd-12-1365-2019, 2019. a
Shabin, M., Kumar, A., Hakkim, H., Rudich, Y., and Sinha, V.: Sources, sinks, and chemistry of stabilized Criegee intermediates in the indo-gangetic plain, Sci. Total Environ., 896, 165281, https://doi.org/10.1016/j.scitotenv.2023.165281, 2023. a
Sheps, L., Scully, A. M., and Au, K.: UV absorption probing of the conformer-dependent reactivity of a Criegee intermediate CH3CHOO, Phys. Chem. Chem. Phys., 16, 26701–26706, 2014. a
Slater, E. J., Whalley, L. K., Woodward-Massey, R., Ye, C., Lee, J. D., Squires, F., Hopkins, J. R., Dunmore, R. E., Shaw, M., Hamilton, J. F., Lewis, A. C., Crilley, L. R., Kramer, L., Bloss, W., Vu, T., Sun, Y., Xu, W., Yue, S., Ren, L., Acton, W. J. F., Hewitt, C. N., Wang, X., Fu, P., and Heard, D. E.: Elevated levels of OH observed in haze events during wintertime in central Beijing, Atmos. Chem. Phys., 20, 14847–14871, https://doi.org/10.5194/acp-20-14847-2020, 2020. a, b
Smith, M. C., Chao, W., Takahashi, K., Boering, K. A., and Lin, J. J.-M.: Unimolecular decomposition rate of the Criegee intermediate (CH3)2COO measured directly with UV absorption spectroscopy, J. Phys. Chem. A, 120, 4789–4798, 2016. a
Smith, S. C., Lee, J. D., Bloss, W. J., Johnson, G. P., Ingham, T., and Heard, D. E.: Concentrations of OH and HO2 radicals during NAMBLEX: measurements and steady state analysis, Atmos. Chem. Phys., 6, 1435–1453, https://doi.org/10.5194/acp-6-1435-2006, 2006. a
Song, M., Zhao, X., Liu, P., Mu, J., He, G., Zhang, C., Tong, S., Xue, C., Zhao, X., Ge, M., and Mu, Y.: Atmospheric NOX oxidation as major sources for nitrous acid (HONO), npj clim. atmos. sci., 6, 30, https://doi.org/10.1038/s41612-023-00357-8, 2023. a
Stone, D., Whalley, L. K., and Heard, D. E.: Tropospheric OH and HO2 radicals: field measurements and model comparisons, Chem. Soc. Rev., 41, 6348–6404, 2012. a, b
Su, H., Cheng, Y. F., Shao, M., Gao, D. F., Yu, Z. Y., Zeng, L. M., Slanina, J., Zhang, Y. H., and Wiedensohler, A.: Nitrous acid (HONO) and its daytime sources at a rural site during the 2004 PRIDE-PRD experiment in China, J. Geophys. Res. Atmos., 113, D14312, https://doi.org/10.1029/2007JD009060, 2008. a
Taatjes, C. A.: Criegee intermediates: What direct production and detection can teach us about reactions of carbonyl oxides, Annu. Rev. Phys. Chem., 68, 183–207, 2017. a
Tajti, A., Szalay, P. G., Császár, A. G., Kállay, M., Gauss, J., Valeev, E. F., Flowers, B. A., Vázquez, J., and Stanton, J. F.: HEAT: High accuracy extrapolated ab initio thermochemistry, J. Chem. Phys., 121, 11599–11613, 2004. a
Tan, D., Faloona, I., Simpas, J. B., Brune, W., Shepson, P. B., Couch, T. L., Sumner, A. L., Carroll, M. A., Thornberry, T., Apel, E., Riemer, D., and Stockwell, W.: HOX budgets in a deciduous forest: Results from the PROPHET summer 1998 campaign, J. Geophys. Res. Atmos., 106, 24407–24427, 2001. a
Tan, Z., Fuchs, H., Lu, K., Hofzumahaus, A., Bohn, B., Broch, S., Dong, H., Gomm, S., Häseler, R., He, L., Holland, F., Li, X., Liu, Y., Lu, S., Rohrer, F., Shao, M., Wang, B., Wang, M., Wu, Y., Zeng, L., Zhang, Y., Wahner, A., and Zhang, Y.: Radical chemistry at a rural site (Wangdu) in the North China Plain: observation and model calculations of OH, HO2 and RO2 radicals, Atmos. Chem. Phys., 17, 663–690, https://doi.org/10.5194/acp-17-663-2017, 2017. a
Teng, A. P., Crounse, J. D., and Wennberg, P. O.: Isoprene peroxy radical dynamics, J. Am. Chem. Soc., 139, 5367–5377, 2017. a
Varandas, A. and Pansini, F.: Narrowing the error in electron correlation calculations by basis set re-hierarchization and use of the unified singlet and triplet electron-pair extrapolation scheme: Application to a test set of 106 systems, J. Chem. Phys., 141, 224113, https://doi.org/10.1063/1.4903193, 2014. a
Vereecken, L.: The reaction of Criegee intermediates with acids and enols, Phys. Chem. Chem. Phys., 19, 28630–28640, 2017. a
Vereecken, L. and Francisco, J. S.: Theoretical studies of atmospheric reaction mechanisms in the troposphere, Chem. Soc. Rev., 41, 6259–6293, 2012. a
Vereecken, L., Harder, H., and Novelli, A.: The reaction of Criegee intermediates with NO, RO2, and SO2, and their fate in the atmosphere, Phys. Chem. Chem. Phys., 14, 14682–14695, 2012. a, b, c
Vereecken, L., Harder, H., and Novelli, A.: The reactions of Criegee intermediates with alkenes, ozone, and carbonyl oxides, Phys. Chem. Chem. Phys., 16, 4039–4049, 2014. a
Vereecken, L., Rickard, A., Newland, M., and Bloss, W.: Theoretical study of the reactions of Criegee intermediates with ozone, alkylhydroperoxides, and carbon monoxide, Phys. Chem. Chem. Phys., 17, 23847–23858, 2015. a
Vereecken, L., Novelli, A., and Taraborrelli, D.: Unimolecular decay strongly limits the atmospheric impact of Criegee intermediates, Phys. Chem. Chem. Phys., 19, 31599–31612, 2017. a, b
Viegas, L. P. and Varandas, A. J.: Can water be a catalyst on the HO2 + H2O + O3 reactive cluster?, Chem. Phys., 399, 17–22, 2012. a
Wallington, T. J. and Japar, S. M.: Fourier transform infrared kinetic studies of the reaction of HONO with HNO3, NO3 and N2O5 at 295 K, J. Atmos. Chem., 9, 399–409, 1989. a
Weinstock, B.: Carbon monoxide: Residence time in the atmosphere, Science, 166, 224–225, 1969. a
Whalley, L. K., Edwards, P. M., Furneaux, K. L., Goddard, A., Ingham, T., Evans, M. J., Stone, D., Hopkins, J. R., Jones, C. E., Karunaharan, A., Lee, J. D., Lewis, A. C., Monks, P. S., Moller, S. J., and Heard, D. E.: Quantifying the magnitude of a missing hydroxyl radical source in a tropical rainforest, Atmos. Chem. Phys., 11, 7223–7233, https://doi.org/10.5194/acp-11-7223-2011, 2011. a, b, c, d
Yang, X., Wang, H., Lu, K., Ma, X., Tan, Z., Long, B., Chen, X., Li, Y., Qu, K., Xia, Y., Zhang, Y., Li, X., Chen, S., Dong, H., Zeng, L., and Zhang, Y.: Reactive aldehyde chemistry explains the missing source of hydroxyl radicals, Nat. Commun., 15, 1648, https://doi.org/10.1038/s41467-024-45885-w, 2024. a, b
Zhang, N., Zhou, X., Bertman, S., Tang, D., Alaghmand, M., Shepson, P. B., and Carroll, M. A.: Measurements of ambient HONO concentrations and vertical HONO flux above a northern Michigan forest canopy, Atmos. Chem. Phys., 12, 8285–8296, https://doi.org/10.5194/acp-12-8285-2012, 2012. a, b
Zhang, Q., Tie, X., Lin, W., Cao, J., Quan, J., Ran, L., and Xu, W.: Variability of SO2 in an intensive fog in North China Plain: Evidence of high solubility of SO2, Particuology, 11, 41–47, 2013. a, b
Zhou, X., Zhang, N., TerAvest, M., Tang, D., Hou, J., Bertman, S., Alaghmand, M., Shepson, P., Carroll, M., Griffith, S., Dusanter, S., and Stevens, P.: Nitric acid photolysis on forest canopy surface as a source for tropospheric nitrous acid, Nat. Geosci., 4, 440–443, https://doi.org/10.1038/ngeo1164, 2011. a
Østerstrøm, F. F., Carter, T. J., Shaw, D. R., Abbatt, J. P. D., Abeleira, A., Arata, C., Bottorff, B. P., Cardoso-Saldaña, F. J., DeCarlo, P. F., Farmer, D. K., Goldstein, A. H., Ruiz, L. H., Kahan, T. F., Mattila, J. M., Novoselac, A., Stevens, P. S., Reidy, E., Rosales, C. M. F., Wang, C., Zhou, S., and Carslaw, N.: Modelling indoor radical chemistry during the HOMEChem campaign, Environ. Sci.: Process. Impacts, 27, 188–201, https://doi.org/10.1039/D4EM00628C, 2025. a