the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 08 Oct 2025
| 08 Oct 2025
Atmospheric mercury: recent advances in theoretical, computational, experimental, observational, and isotopic understanding to decipher its redox transformations in the upper and lower atmosphere and interactions with Earth surface reservoirs
Xinyu Shi
Xueling Tang
Guangyi Sun
Che-Jen Lin
Xinbin Feng
Mercury is a volatile heavy element with no known biological function. It is present in trace amounts (on average, ∼ 80 ppb) but is not geochemically well blended in the Earth's crust. As a result, it occurs in extremely high concentrations (up to a few percent) in certain locations. It is found along tectonic plate faults in deposits of sulfide ores (cinnabar), and it has been extensively mobilized during the Anthropocene. Mercury is currently one of the most targeted global pollutants, with methylmercury compounds being particularly neurotoxic. Over 5000 t of mercury is released into the atmosphere annually through primary emissions and secondary re-emissions. Much of the re-emitted mercury resulting from exchanges with surface reservoirs is related to (legacy) human activities, such as direct releases. Understanding the dynamics of the global Hg cycle is critical for assessing the impact of emission reductions under the UN Minamata Convention, which became legally binding in 2017. This review of atmospheric mercury focuses on fundamental advances in field, laboratory, and theoretical studies, including six stable Hg isotope analytical methods, which have contributed recently to a more mature understanding of the complexity of the atmospheric Hg cycle and its interactions with the Earth's surface ecosystem.
- Article
(10912 KB) - Full-text XML
- BibTeX
- EndNote




Mercury (Hg) is a potent neurotoxin that, via methylmercury (MMHg+) food exposure, poses a global health threat (e.g., IQ decrement and heart attacks) (Zhang et al., 2021b). The atmosphere plays a pivotal role in the Hg biogeochemical cycle, functioning as the most important transient reservoir, a conduit for transport and transformation, and a site rich in redox chemistry. In part due to concerns about global Hg transport, the multilateral UN Environment Convention on Hg was negotiated and entered into force in 2017 with a mandate to reduce the intentional use and emissions of Hg (UNEP, 2018). Research on Hg biogeochemical cycling gained momentum after an outbreak of mass MMHg+ poisoning severely affected the population of Minamata Bay, Japan, in the 1950s and 1960s through the consumption of contaminated seafood, and it became clear that MMHg+ was present at chronically high levels in predatory fish in many lakes, particularly those in the boreal forest belt, through long-range transport and biomagnification (Lindqvist et al., 1991). The earliest known series of measurements of airborne elemental Hg, possibly the first systematic study of its kind, was conducted in Pacific North America during the second half of the 1960s (Williston, 1968). It was recognized as early as the 1970s that Hg circulated globally through the atmosphere (Nriagu, 1979). Somewhat later, Slemr et al. (1985) published an influential paper whose results on the distribution, speciation, and budget of atmospheric Hg reproduce the qualitative features of the atmospheric Hg cycle fairly well, such as atomic vapor (Hg0) dominating the atmospheric pool and showing an interhemispheric difference with higher concentrations in the Northern Hemisphere and being relatively well mixed vertically through the troposphere with an extensive residence time (concept of a “global pollutant”).
Knowledge of the physical and chemical processes that govern the dynamics of Hg in the atmosphere has developed gradually. Over time, through technological leaps (stable isotope sampling in natural probes, refined methods in the theoretical and experimental field, etc.), its full complexity has begun to be appreciated. In earlier research, the prevailing view was that water-phase oxidation by ozone could be the primary mechanism initiating the removal of tropospheric Hg0 (Pleijel and Munthe, 1995; Seigneur et al., 1994). However, newer data have indicated that gaseous oxidized mercury (GOM) could also be present in the atmosphere (Xiao et al., 1997; Lindberg and Stratton, 1998), in addition to the particulate form (PBM). Specifically, the observation that Hg0 was periodically depleted in the planetary mixing layer during the polar spring (Schroeder et al., 1998) prompted a reassessment of Hg chemistry in favor of homogeneous gas-phase chemistry (Hynes et al., 2009). The two-step gas-phase oxidation of Hg0 initiated by Br atoms has emerged as the most important global channel for tropospheric conversion to HgII (Donohoue et al., 2006; Holmes et al., 2010). Gas-phase O3 was previously considered an oxidizing agent for Hg0 to HgII. Although this route was discarded, O3 has been found to effectively oxidize intermediate HgI species (Gómez Martín et al., 2022). This suggests that OH- and less certain I-initiated oxidation of Hg0, which produces less stable intermediates than Br and Cl do, may also be important for Hg turnover in parts of the troposphere and beyond (Dibble et al., 2020; Lee et al., 2024). A novel finding is that major HgI,II species, which are expected to be formed in the atmosphere upon oxidation of Hg0, are themselves photolabile and undergo gas-phase reduction (Francés-Monerris et al., 2020; Saiz-Lopez et al., 2019). The complexity of rapid redox Hg chemistry involving multiple gas-phase oxidation states (0, +1 and +2) is further compounded by the impact of multiphase interactions, including reactive uptake and homogeneous and heterogeneous processes in condensed-phase media, on the dynamics of atmospheric Hg. An indicator of the maturation of our understanding of atmospheric Hg chemistry is the inclusion of bromine chemistry in critically evaluated datasets for use in atmospheric studies (Burkholder et al., 2019). Over the past 2 decades, measurements of Hg stable isotope ratios in natural samples have emerged as valuable tools for gaining insights into the atmospheric Hg cycle. One notable outcome of isotope analysis is the recognition that dry Hg0 deposition exerts a more pronounced influence on a global scale than was previously understood, with wet and dry deposition of the atmospheric HgII fraction being of lesser importance (Jiskra et al., 2018).
Hg in the atmosphere has been the subject of reviews over the past 45 years, with topics including biogeochemical cycling (Lindqvist and Rodhe, 1985; Lindqvist et al., 1991; Schroeder and Munthe, 1998; Selin, 2009; Lyman et al., 2020a), observations (Sprovieri et al., 2010; Dommergue et al., 2010; Fu et al., 2015; Steffen et al., 2015; Mao et al., 2016; Zhang et al., 2019c; Custódio et al., 2022; Bencardino et al., 2024), isotopic observational data (Kwon et al., 2020; Liu et al., 2024), atmospheric measurement techniques (Pandey et al., 2011; Huang et al., 2014; Gustin et al., 2015, 2024; Davis and Lu, 2024), anthropogenic emissions (Carpi, 1997; Zhang et al., 2016; Cheng et al., 2023), natural volcanism (Edwards et al., 2021), physical removal and air–surface exchange (Zhang et al., 2009; Sommar et al., 2013, 2020; Zhu et al., 2016; Agnan et al., 2016; Cooke et al., 2020; Zhou et al., 2021; Liu et al., 2024) with emphasis on global change (Obrist et al., 2018; Sonke et al., 2023), polar atmospheric surface layer mercury depletion events (Steffen et al., 2008), chemical conversion in the atmosphere (Schroeder et al., 1991; Lin and Pehkonen, 1999; Lin et al., 2011; Si and Ariya, 2018), aqueous homogeneous and heterogeneous photoredox chemistry (Zhang, 2006; Si et al., 2022), multiphase atmospheric chemistry (Ariya et al., 2015), assessment of critical atmospheric chemical processes using state-of-the-art experimental and computational chemistry methods (Ariya and Peterson, 2005; Ariya et al., 2008; Hynes et al., 2009), receptor (Cheng et al., 2015), and global models (Lin et al., 2006, 2007; Subir et al., 2011, 2012; Amos et al., 2015; Travnikov et al., 2017). This review is based on the perspective of atmospheric scientists, with synthesis and a comprehensive account of the results of fundamental research, including field, laboratory, and theoretical studies, that have contributed to an understanding of the complexity of the atmospheric Hg cycle and its interactions with the Earth's surface ecosystem at the molecular level. This work does not address several topics related to Hg in the atmosphere. These include anthropogenic and natural emission inventories, corresponding top-down constraints, and inverse modeling from atmospheric observations, accounting for long-term air data series and their temporal and spatial trends, observations of the PBM and its particle size distributions, wet deposition, future scenarios for the effects of regulatory measures (Minamata Convention), ongoing climate change, and many more topics. Our goal is to provide a comprehensive review of the atmospheric chemistry of both inorganic and organic Hg in the lower and upper atmosphere, coupled with a compilation of updated, critically evaluated kinetic, thermochemical, photochemical, and isotopic fractionation data. Where appropriate, we introduce the basic concepts and fundamental aspects of Hg chemistry, including those of condensed phases. In atmospheric Hg isotope chemistry, our approach is comprehensive, encompassing a range of activities from field observations of air and Hg0 gas exchange with natural surfaces to laboratory studies of processes that may be relevant to the atmosphere. We also highlight areas of persistent uncertainty or lack of consensus, such as measurement methods for atmospheric Hg speciation and the partitioning of HgII in atmospheric water between inorganic and organic ligands.
Hg is the only metal that is a liquid at standard temperature and pressure (freezing point of −38.8 °C and boiling point of 356.7 °C), and its vapor is monatomic. Under these conditions, the mixing ratio of neurotoxic Hg vapor in equilibrium with metallic liquid is already at the hazardous level of approximately 1.7 ppm (Huber et al., 2006). Liquid Hg possesses properties that have given it a wide range of applications in the past despite its known toxicity, including exceptional surface tension (nearly 7 times greater than that of water at 25 °C), high specific gravity, high electrical conductivity (a reference substance for measuring the SI unit Ω), low compressibility, and a constant volume of expansion coefficient in the liquid state. Hg forms solid alloys (amalgams) with most metals except iron. This property enables its application in gold panning (HgAu) and dental fillings (HgAg) or as an electrode material in the chloralkali industry (NaHg). The electronic configuration of the mercury atom has filled f- and d-orbitals with a high density of 6s valence electrons near the nucleus ([Xe]4f145d106s2), which is related to a relativistic radial contraction of s- and p-orbitals as the inner electrons approach a significant fraction of the speed of light (which for a Hg 1s electron is 58 %, implying a radial shrinkage of 23 %; Pyykkö, 1988). It also follows that oxidation states 0 and +2 (mercuric, d10 metal ion) are the most stable for Hg. Nevertheless, Hg differs from other metals in its propensity to readily form a polycation in the aqueous phase, the mercurous ion, , which is, however, only metastable in the gaseous phase (Strömberg and Wahlgren, 1990). The solubility of Hg0 in water is limited to 0.3 µM (Sanemasa, 1975), and the gas–water equilibrium is governed by Henry's law. The Henry's law coefficient () for Hg0 is 0.11 M atm−1 at 25 °C (Andersson et al., 2008), whereas the value is more than 7 orders of magnitude greater for the HgCl2 molecule at the same temperature (Sommar et al., 2000). Since the mid-19th century, light production, including sharp lines at 184.9 and 257.3 nm, has been achieved by passing an electric arc through Hg0 vapor in a glass bulb. Conversely, absorption spectroscopy uses these atomic deep-UV lines to analytically detect Hg in samples, as discussed in detail in Sect. 3.1.
3.1 Atmospheric measurements of mercury species
Hg is the only trace gas, other than the noble gases (Burnard, 2013), that exists as free atoms (Hg0) in the atmosphere, making this pollutant exceptional in terms of low detection limits by optical measurement techniques. This makes it possible to measure Hg0 vapor emissions in real time, for example, from mining, chloralkali production and geothermal activities, as has been done in Europe for decades via light detection and ranging (lidar) in differential absorption mode by mobile laser systems (Svanberg, 2002). If the optical path length in the measuring cell of an instrument is sufficiently long (i.e., using multipath techniques such as cavity ring-down), then the conditions exist for continuous measurement of Hg0 in ambient air (at the sub-ppt level, ∼ 5×106 atoms cm−3 in the Northern Hemisphere) via atomic absorption spectroscopy (AAS) with Zeeman background correction (Osterwalder et al., 2020). The application of Zeeman AAS in Hg stable isotope analysis has also been described (Lu et al., 2019). As an alternative to Zeeman splitting of the Hg(63P) level for sensitive, selective detection of Hg0 (Sholupov et al., 2004), sequential two-photon laser-induced fluorescence schemes have been used (Bauer et al., 2002, 2014; Hynes et al., 2017). For initial excitation of the Hg(61S0) → Hg(63P1) transition at 253.7 nm, a light beam from a Hg discharge lamp or the frequency-doubled output of a dye laser pumped by the third harmonic of a Nd:YAG laser is used. As shown in Fig. 1a, further excitation involves the sequential excitation of different atomic transitions by two laser systems, both starting from the Hg(63P1) state, followed by the detection of blue-shifted (Hg(61P1) → Hg(61S0) at 184.9 nm) or red-shifted (e.g., at 578.9 nm) fluorescence. The detection of Hg0 with such a sophisticated apparatus is an exception to typical measurements, which are made via cold vapor atomic fluorescence spectroscopy (CV-AFS) after preconcentration sampling on gold (Ambrose, 2017). Smaller non-Hg0 portions of atmospheric Hg are challenging to speciate because of their low concentrations. Instead, they are fractionated operationally based on their oxidation state (Hg0 versus GOM) or phase state (GOM versus PBM). Since gold does not selectively trap Hg0 but also captures other Hg species (Dumarey et al., 1985; Gačnik et al., 2024), the GOM and PBM must be individually collected upstream of the sample air to accurately measure the triad Hg0–GOM–PBM. KCl-coated annular denuders have been utilized for fractionating ambient GOM by gas-phase diffusion for over 2 decades. Nonetheless, upon the development of techniques to regularly assess its accuracy in measuring ambient air, the method was found to be biased in a non-systematic manner towards lower values (Jaffe et al., 2014; Lyman et al., 2010; McClure et al., 2014). The automated KCl denuder method, with its variable efficiency, can thus assess its accuracy in measuring ambient air regularly; the method was found to be biased in a nonsystematic manner, leading to serious underestimation of the GOM, whereas the refluxing mist chamber method, which is an alternative, carries the risk of cosampling the GOM with the PBM (Gustin et al., 2021). However, the KCl-covered denuder does not have full penetration of PBMs < 2.5 µm, but aerosols of 100 nm or less are increasingly trapped by the salt surface (Ghoshdastidar et al., 2019). When compared, refluxing mist chambers yielded ambient GOM concentrations that were 3 to 4 times higher on average than those obtained with KCl-coated annular denuders (Landis et al., 2002). A decade later, the capture and retention efficiency of the KCl denuder method for GOM was evaluated, which was close to 95 % in synthetic Hg0-free air but decreased drastically to between 20 % and 54 % when exposed to ambient air, where ozone and humidity were found to cause severe reductive losses such as Hg0 (McClure et al., 2014). In fact, ozone gas can heterogeneously reduce particle-bound HgII halides, as recent experiments have shown (Ai et al., 2023). In high-humidity marine applications, KCl denuder technology operates at very low efficiency; for example, He and Mason (2021) reported average losses of 80 % during oceanographic expeditions in the Pacific. By determining total airborne mercury (TAM; Steffen et al., 2002; Slemr et al., 2018) and Hg0 in air, a measure of reactive mercury (RM) is obtained as the sum of GOM + PBM by subtracting Hg0 from TAM. In turn, Hg0 is obtained by passing an air stream through a filter and a cation exchange membrane (CEM) in series, whereas TAM is measured as Hg0 after a pyrolysis unit held at 800 °C converts all Hg in the sample air to elemental vapor (Lyman et al., 2020b). CEM is capable of capturing and retaining HgII quantitatively over long storage periods but has no affinity for Hg0 (Miller et al., 2019). However, when two quantities that are usually close to each other are subtracted, the precision of the RM determination is low. Hynes et al. (2017) used two-photon laser-induced fluorescence as an online detection method for RM (by switching between ambient and pyrolyzed air as the source for the Hg0 analyte) and concluded that the variability in ambient Hg0 severely limits the sensitivity of dual-channel difference RM measurements. For the separation of the semivolatile GOM fraction from the PBM in ambient air, various membranes have been examined but with recognized limitations (Dunham-Cheatham et al., 2023; Gustin et al., 2023). The realization of NIST-traceable GOM calibration systems has recently progressed (Gačnik et al., 2022). Several studies have been carried out with the aim of experimentally deciphering the molecular identities (speciation) of the GOM pool in ambient air. Most methods are based on a preconcentration process of GOM on a substrate, which is then thermodesorbed in a gas stream following a programmed temperature ramp and detected as Hg0 after pyrolysis (Gustin et al., 2015), alternatively focused on a capillary column and analyzed by different types (chemical ionization CI; electron impact ionization) of mass spectrometry (MS) (Deeds et al., 2015; Jones et al., 2016). In the former case, standards are used in the form of a number of commercially available Hg chemicals (such as HgBr2, HgCl2, HgO, Hg(NO3)2, and HgSO4) that are assumed to be representative surrogates for GOM (Huang et al., 2017; Sexauer Gustin et al., 2016). As inferred by Khalizov et al. (2020), this speciation is indirect, as it has not been confirmed that the GOM molecules adsorbed on the substrate can be desorbed in the same chemical form as they are in air.
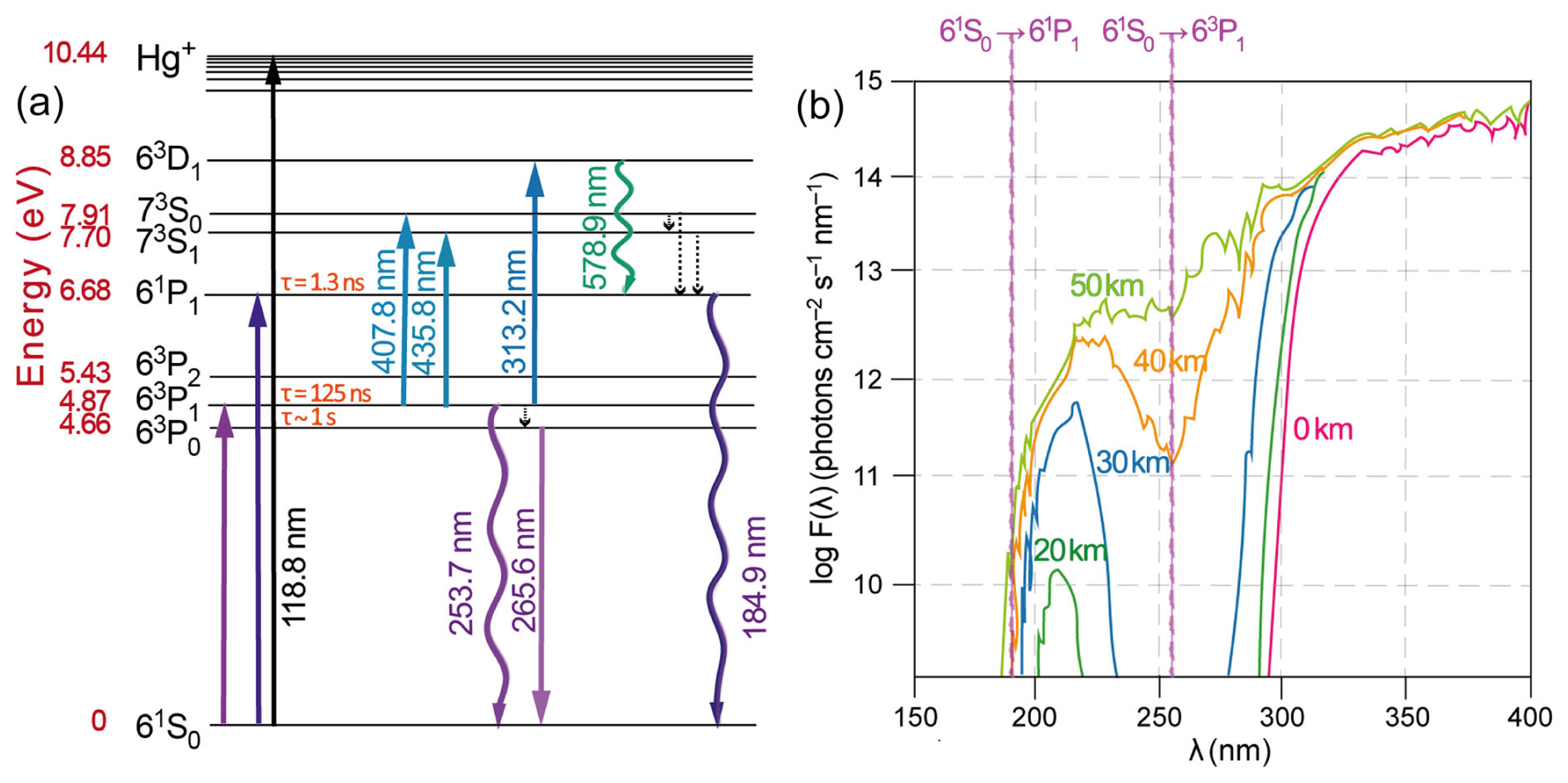
Figure 1(a) Energy level diagram of the Hg atom. The wave-shaped arrows represent resonant radiation. (b) Actinic fluxes as a function of altitude. The wavelengths of the Hg(1S0) → Hg(3P1) and → Hg(1P1) transitions at 253.7 and 184.9 nm, respectively, are given.
In contrast, studies have shown that aerosol reactions lead to the re-speciation of mercuric halides on surfaces (Mao et al., 2021; Mao and Khalizov, 2021). The authors reported that their ion-drift (ID) CI-MS system, which is sensitive enough for detection in laboratory studies, can achieve a limit of detection (LOD) at a 1 amu resolution of (0.8–2.0) × 105 molecules cm−3 toward ambient GOM by switching to multistage atmospheric pressure ID-CI-MS. The feasibility of using proton transfer reaction mass spectrometry (PTR-MS) to study the reaction products (GOM) of Br-initiated Hg0 oxidation has been evaluated by Dibble et al. (2014) but is not recommended because it cannot be applied in multi-stage atmospheric pressure systems (Khalizov et al., 2020). In summary, direct measurements of ambient GOM species have not yet been achieved. No method exists for chemically characterizing the GOM fraction, which is semivolatile and may contain species that are photolytically unstable. Since previous GOM measurements are considered unreliable (Lyman et al., 2020a; Slemr et al., 2016) and emerging RM data (Lyman et al., 2020b; Slemr et al., 2018; Swartzendruber et al., 2009; Gratz et al., 2015; Lyman and Jaffe, 2012) are still too sparse and spatially limited, it is not possible to draw deterministic conclusions on atmospheric HgII. Sampling methods for organic Hg (dimethylmercury; He et al., 2022 and monomethylated HgII species; Lee et al., 2003) in ambient air, as opposed to inorganic Hg species, are more unambiguous. The speciation of Hg in atmospheric waters is discussed in Sect. 4.6. Hg measurement data from air and precipitation, ground-based or aircraft (Slemr et al., 2018, 2016) observations that fall outside the scope of this review, including those reported from continental (Cole et al., 2014, 2013; Schmolke et al., 1999; Wängberg et al., 2001; Gay et al., 2013; Fu et al., 2015) to hemispherical (Bencardino et al., 2024; Szponar et al., 2020; Slemr et al., 2020; Sprovieri et al., 2017, 2016) monitoring networks, some of which have been in operation since before the turn of 2000 (Custodio et al., 2020), have been reviewed elsewhere (Mao et al., 2016; Lyman et al., 2020a; Howard et al., 2017; Angot et al., 2016; Kim et al., 2012; Zhang et al., 2017). In the case of the isotopic characterization of atmospheric Hg, however, we feel justified in compiling, analyzing, and discussing the considerable body of recent observations (Sect. 8.2).
3.2 Stability of atmospheric Hg0
Hg0 represents the primary form of atmospheric mercury in both the troposphere and stratosphere. Considering the spatial variability of Hg0 concentrations1, which depart from a uniform vertical distribution throughout the atmosphere (Slemr et al., 2018), a singular global atmospheric lifetime is not appropriate. A more pertinent measure is the effective lifetime of Hg0, expressed on an annual basis and as a function of its horizontal and vertical location within the atmosphere. The observed disparity in tropospheric Hg0 concentrations between the Northern and Southern Hemispheres of a factor of ∼ 1.41 (Tang et al., 2025), despite anthropogenic emissions in the Northern Hemisphere being approximately 2.5 times greater than those in the Southern Hemisphere (Streets et al., 2019; Sonke et al., 2023), implies that Hg0 has a relatively short effective lifetime in comparison to the interhemispheric air mass exchange time of approximately 1.3 years (Geller et al., 1997). As Hg0 crosses the intertropical convergence zone, it undergoes convective uplift, enabling its transport into the stratosphere (Koenig et al., 2022). Troposphere-to-stratosphere Hg0 transport has been regarded as limited (100–176 Mg yr−1; Lyman and Jaffe, 2012; Horowitz et al., 2017). Nevertheless, recent modeling suggests that the stratosphere is crucial for biogeochemical Hg cycling, acting as the primary pathway for Hg0 exchange between hemispheres and explaining the minor interhemispheric gradient (Saiz-Lopez et al., 2025). As posited by models developed by Shah et al. (2021) and Saiz-Lopez et al. (2025), approximately 17 % of the aggregate atmospheric Hg load is situated within the stratosphere, whereas a previous study reported 12 % (Horowitz et al., 2017). Given that the stratosphere's mass (9.06 × 1017 kg) constitutes approximately 18 % of the total atmospheric air mass (5.13 × 1018 kg; Warneck and Williams, 2012), one might infer that the fraction of mercury present in the stratosphere is comparable to the proportion of stratospheric air relative to the entire atmosphere. However, this scaling is not supported by empirical data. Aerial measurements of Hg in the troposphere and lower stratosphere reveal a steep Hg gradient around and above the tropopause with lower Hg mixing ratios in the upper atmospheric layers (Radke et al., 2007; Talbot et al., 2007; Slemr et al., 2018) linked to a larger contribution of oxidized Hg species partitioned to aerosols (Murphy et al., 1998) from the gas phase.
With respect to the tropospheric Hg budget, there is a relative consensus that the Hg load is close to 4 Gg (Saiz-Lopez et al., 2020; 3.9 ± 1.0, Saiz-Lopez et al., 2025; 3.8, Zhang et al., 2023b; 4.0, Shah et al., 2021; 3.9, Horowitz et al., 2017), with exceptions suggesting that it is closer to 5–6 Gg (Holmes et al., 2010; Zhang et al., 2025) and that anthropogenic emissions, excluding biomass burning, are approximately 2.2–2.6 Gg yr−1 (Horowitz et al., 2017; Shah et al., 2021; Zhang et al., 2023b; Geyman et al., 2024; Saiz-Lopez et al., 2025), with significant reductions across developed countries in the Northern Hemisphere observed in the near term (Custódio et al., 2022; Feinberg et al., 2024). Aircraft-based observations reveal a relatively consistent mixing ratio of Hg0 within the troposphere below the tropopause, encompassing the planetary boundary layer in regions characterized by low primary emissions (Banic et al., 2003; Talbot et al., 2007; Swartzendruber et al., 2008; Weigelt et al., 2016b; Bieser et al., 2017). This uniformity supports the adoption of a steady-state procedure (Seinfeld and Pandis, 2006), where the inverse of the Hg0 lifetime (τtroposphere) is approximated by the sum of its loss rates:
where the indices rxn, ocean, land, wash, and stratosphere are used to represent net oxidation, oceanic uptake, assimilation in land ecosystems, and processes that lead to wet deposition and net transfer to the tropopause/stratosphere, respectively. As discussed subsequently, all the terms in Eq. (1) are subject to significant uncertainties. However, as is the case with many other trace gases, the chemical lifetime (τrxn) undoubtedly plays a controlling role in determining the effective lifetime of Hg0. Representing net oxidation, τrxn encompasses the duration of the initial two-step oxidation to molecular forms and the subsequent redox cycling of the photolabile fraction of these molecules in the gas phase and aerosols prior to deposition. According to the latest redox schemes (Shah et al., 2021; Castro Pelaez et al., 2022; Saiz-Lopez et al., 2025), the extent of bidirectional Hg mass flux by atmospheric chemical conversion (oxidation and reduction, 10.4–13.0 vs. 6.0–6.9 Gg yr−1, respectively) appears to be much greater than previously assumed (e.g., 8.0 vs. 3.7 Gg yr−1, Holmes et al., 2010), which also holds for the bidirectional fluxes (emission and depositional uptake) that occur in the gas exchange of Hg0 between the atmosphere and the land and ocean. Aggregate atmospheric emissions and dry deposition have been approximated at 7.4–11.2 and 2.9–6.8 Gg Hg0 yr−1, respectively (Horowitz et al., 2017; Shah et al., 2021; Sonke et al., 2023; Zhang et al., 2023b), following a tendency of researchers toward augmenting the role of re-emission of legacy Hg from the oceans (3.7–7.2 Gg Hg0 yr−1) and gross biospheric assimilation from the atmosphere (1.2–3.2 Gg Hg0 yr−1; Horowitz et al., 2017; Yuan et al., 2023a; Obrist et al., 2021; Zhou and Obrist, 2021; Feinberg et al., 2022; Wang et al., 2022; Szponar et al., 2025), respectively. To transfer Hg0 from the ocean into the atmosphere, the mass transfer rate is usually parameterized via wind speed dependencies that have been tested for CO2 emissions. However, recent evidence (Osterwalder et al., 2021) suggests that Hg0, which is less soluble than CO2, behaves similarly to O2 and N2, where the impact of bubble-mediated transfer is greater. As a result, ocean emissions play an increased role in the global Hg budget, accounting for approximately 60 % of total Hg emissions to the atmosphere due to a wind speed dependence with a cubic power exponent instead of a quadratic power exponent in model simulations (Zhang et al., 2023b). The greater gross emissions from seawater must be balanced by gross deposition of Hg0, which is, within uncertainties, of comparable magnitude to that of HgII deposition over oceans (Jiskra et al., 2021) and much higher than previously assumed (Soerensen et al., 2010). The global net exchange of Hg0 from the oceans has been estimated at 0.8–4.0 Gg Hg0 yr−1 (Lamborg et al., 2002; Strode et al., 2007; Selin et al., 2008; Holmes et al., 2010; Chen et al., 2014; Horowitz et al., 2017; Shah et al., 2021), and the fraction of Hg0 emissions resulting from HgII reduction in surface waters is at an upper limit of 2.25 ± 0.89 Hg0 Gg yr−1 (Tang et al., 2025). In summary, the latter terms in Eq. (1) correspond to lifetimes, the spans of which are conservatively estimated to exceed 1 year. However, their inverses, referring to Eq. (1), when summed, can shorten τtroposphere by tens of percent beyond what the tropospheric chemical lifetime of Hg0 (τrxn) dictates, taking into account the inherent uncertainties. Currently, Hg0 is estimated to have a τtroposphere of between 3.8 and 7 months (Shah et al., 2016, 2021; Horowitz et al., 2017; Saiz-Lopez et al., 2020, 2025) and an average atmospheric lifetime (troposphere + stratosphere) of 8.2 months (Saiz-Lopez et al., 2025).
The sources of atmospheric HgII are twofold: primary HgII emissions from anthropogenic sources and atmospheric Hg0 oxidation. Compared with that of Hg0, the proportion of HgII in anthropogenic emissions in the troposphere is not well defined. One estimate suggests that 74 % of cumulative anthropogenic Hg emissions into the air are Hg0 (Streets et al., 2017). Currently, East Asia has the most emissions worldwide (Streets et al., 2019); however, compelling evidence indicates that the magnitude of total Hg air emissions in this region has already peaked (Zhang et al., 2023a) and has declined in recent years (Wu et al., 2023a; Feinberg et al., 2024). Nevertheless, a shift in the contributions of distinct source categories, with cement production emerging as the predominant source since 2009 in China (Wu et al., 2016), suggests an increase in the proportion of HgII within Hg emissions (Zhang et al., 2016; Wang et al., 2024). Hg speciation profiles from anthropogenic sources may vary significantly across regions; for example, in continental Europe, the HgII contribution from coal-fired power plants may represent less than 25 % (Weigelt et al., 2016a), whereas in the tropics, artisanal and small-scale gold mining represent a substantial yet largely unconstrained source of atmospheric Hg0 (Obrist et al., 2018). On average, contemporary global models employ 60 % to 65 % Hg0 speciation in current anthropogenic emissions to the atmosphere (Horowitz et al., 2017; Shah et al., 2021; Zhang et al., 2023b). There are significant differences in the estimates of the tropospheric pool of Hg0 (∼ 3.3–4.8 Gg), separated from HgII (0.1–1.0 Gg), within the above-mentioned constrained budgets for the total tropospheric Hg load in contemporary models. Having estimated the atmospheric load of HgII up to 20 km at ∼ 0.36 Gg on the basis of a synthesis of RM measurements at different heights in the atmosphere (Saiz-Lopez et al., 2020), a later contribution (Saiz-Lopez et al., 2025) involving stratospheric transport and chemistry deployed a much larger tropospheric HgII pool (0.51 Gg) associated with downward transport (0.35 Gg yr−1) of mostly photostable HgII from the stratosphere (HgII pool of ∼ 0.2 Gg). The corresponding amount of (wet and dry) HgII deposited on Earth's surface is 6.92 ± 1.70 Gg yr−1, which is outside the previously estimated range of 4.8–6.8 Gg yr−1 HgII (Strode et al., 2007; Zhang et al., 2019b; Feinberg et al., 2022; Sonke et al., 2023). The effective τtroposphere of HgII is a few weeks (Horowitz et al., 2017), whereas HgI species are intermediates (lifetime ≪1 s) in the redox cycle, and their tropospheric mass is negligible (Shah et al., 2021).
Hg0 in the planetary boundary layer can be consumed at a surprisingly high rate, leading to low concentration levels that approach complete depletion. Thus, chemical oxidation by reactive bromine species in a catalytic cycle (“bromine explosion”, Toyota et al., 2014; Gao et al., 2022) can explain atomic Hg depletion events (AMDEs) during the polar spring after sunrise (Schroeder et al., 1998; Sommar et al., 2007; Nerentorp Mastromonaco et al., 2016) and those observed over the Dead Sea (Obrist et al., 2011) (Fig. 2). Br-controlled oxidation via the intermediate •HgIBr is critical for the tropospheric oxidation of Hg0, as described later in the section on gas-phase oxidation. Upon entry into the stratosphere, thermal oxidation with Br• remains important for conversion to HgII but with increasing altitude in the lower stratosphere; Cl chemistry plays the most important role, with OH-directed chemistry in second place at a slow net oxidation rate. With the maximum concentration of the O3 layer (∼ 25 km) as the dividing line, there is a strong dichotomy between the Hg chemistry in the upper and lower stratosphere. The former is UV-C driven (Sun et al., 2022, the UV window > 30 km provides a substantial photon flux at λ=253.7 nm, Fig. 1b), involving optically excited Hg0 states with a strong electrophilic character. The electronic excitation of Hg0 from the ground state (singlet, 1S0) at 253.7 nm is spin-forbidden (leading to a triplet state, 3P1 with a radiative lifetime of ∼ 125 ns; Fig. 1a). The metastable dark Hg(3P0) state cannot be produced directly from Hg(1S0) by light absorption but can be produced by spin–orbit relaxation of Hg(3P1) atoms involving energy transfer to surrounding (air) molecules. In N2, the equilibrium constant between the 3P0 and 3P1 states at room temperature (297 K) is 1.87 × 103 (Callear and Shiundu, 1987), but in the presence of O2, their distribution changes profoundly. Although O2 is a slightly less effective quencher for Hg(3P0) than for Hg(3P1) (Callear, 1987), their effective lifetimes in air at atmospheric pressure differ by only 1 order of magnitude (∼ 1.1 and ∼ 0.2 ns, respectively; Saiz-Lopez et al., 2022). In addition to physical quenching to the ground state, both Hg(3P0) and Hg(3P1) may undergo chemical oxidation to mercury oxide(s) (Callear et al., 1959), although metastable atoms are expected to be less reactive. The chemical conversion of excited Hg atoms by O2 releases HgII, which can further react with more stable species, giving Hg0 in the upper stratosphere a tiny lifetime against oxidation compared with that of transfer to the lower atmosphere (Saiz-Lopez et al., 2022). In the uppermost stratosphere, there appears to be access to deeper UV-C (Fig. 1b) such that at 184.9 nm, a spin-allowed electronic transition from Hg(1S0) to Hg(1P1) occurs, with a light absorption cross-section nearly 2 orders of magnitude greater than that for the Hg(1S0)→ Hg(3P1) transition (Morton, 2000). Like Hg(3P1), the more energetic Hg(1P1) reacts with O2 at a rate approaching the collision frequency, but the HgO product formed in the latter case is so vibrationally hot that it promptly decays into Hg and O atoms. As a result, the chemistry of Hg(1P1) is expected to play a minor role in the turnover of Hg in the upper stratosphere. The calculated lifetime of Hg0 in the middle to upper stratosphere is altitude-dependent, ranging from a fraction to a few hundred hours (Saiz-Lopez et al., 2022), and is most comparable to that of Hg0 during AMDEs. However, the underlying governing physicochemical processes are completely different.
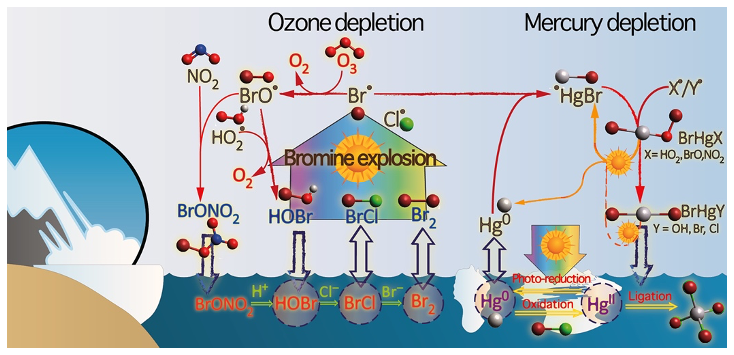
Figure 2The chemistry behind bromine explosion events and related surface layer ozone and mercury depletion events.
4.1 Fundamental kinetics and thermodynamic principles
A chemical process can be decomposed into a sequence of one or more single-step processes as elemental reactions. Elementary processes involve a transition between two atomic or molecular states, separated by a potential energy barrier that represents the activation energy. The rate of a gas-phase reaction depends on the number of collisions between the reactants and the thermodynamics of their interactions (i.e., the change in entropy, ΔS, and enthalpy, ΔH, upon passing through the transition state), whereas for the rate of a reaction in aqueous solution, there are a number of additional factors that can influence the rate, such as solvation, ionic strength, pH, and diffusion rates. Processes that release heat as products and increase the entropy of the system favor the reaction. The balance between ΔH and ΔS is given by the Gibbs free energy equation, where T is the absolute temperature: . If the Gibbs free energy is negative, the reaction is spontaneous from a thermodynamic perspective. The index is used to distinguish the enthalpy of reaction (ΔHR) from, e.g., the enthalpy of formation of a substance (ΔHf). We can calculate the equilibrium constant, K, using ln and determine the ratio of the forward and reverse rate coefficients from . Examples of important types of gas-phase reactions are listed in Table 1.

Termolecular reactions are pressure (M)-dependent at low pressures with an effective rate coefficient (k) of third order but become pressure-independent at high pressures. The transition from third- to second-order behavior is known as the fall-off region. For most atmospheric reactions, we can expect that the rate coefficient is at the low-pressure limit. However, there are exceptions, which are listed in Table 4. While two-body collisions are common in the gas phase, three-body collisions are much less probable, and four-body collisions can be ignored because of their low probability. An overall reaction includes two or more elementary reactions. The temperature dependence of the rate coefficients can be fit over a relatively narrow temperature range via the empirical Arrhenius equation: k(T) = A exp (), where Ea is the activation energy and R is the gas constant. The pre-exponential factor A, a constant in the original Arrhenius equation, is weakly temperature-dependent for most reactions (varying as the square root of T according to collision theory). For a wider temperature range, the modified expression provides a better fit to the experimental data. If the activation energy is high enough, there is a large endothermic barrier that prevents even a reaction with a strongly negative ΔGR from occurring at measurable rates. In select cases, the experimental data show a negative activation energy, suggesting that the reaction proceeds by the addition of reactants to form an intermediate species with excess energy that must be dissipated before decomposing into the final products. The rate constant for termolecular reactions between small molecules in the atmosphere can usually be well approximated by a combination of three parameters k0 (cm6 molecule−2 s−1), k∞ (cm3 molecule−1 s−1) and FC. The first two correspond to the low- and high-pressure limits, and FC is a form factor that describes the transition region.
The temperature dependence of k is expressed by parameterizing k0 and k∞ as a function of temperature with the following expression:
4.2 Surface kinetics
Atmospheric aerosols have a high surface-to-volume ratio that concentrates most of their constituents at the surface. Furthermore, the influence of surface chemistry increases with decreasing particle size. Gas-to-particle reactions, among other heterogeneous reactions, begin with adsorption, which links molecules from the gas phase to the surface of a solid or liquid. This process can be physical, with low adsorption energy (physisorption, van der Waals forces) or chemical (chemisorption) when chemical bonding occurs as molecules approach the surface, overcome the activation energy barrier, and become reactive when the adsorbent reacts with sites on the surface. Importantly, gases and solutes adsorbed at an interface frequently exhibit physicochemical properties that diverge from their bulk properties, including reactivity and spectral shifts. Surface reaction kinetics are often expressed by the uptake probability (γ), which represents the fraction of gas collisions with a substrate surface that yield uptake or reactions. The net uptake of gas γnet is quantified in terms of conductances (Γ), which are normalized to the rate of gas surface collisions:
where Γg, Γrxn, and Γsol represent the processes of gas-phase diffusion to the surface, solubility, and reaction in the bulk liquid phase, respectively, and α represents the (reversible) mass accommodation (“sticking”) across the gas–particle interface. In addition to α, these processes are related to the diffusion constants in gas (Dg) and liquid (Dl) phases, Henry's law coefficient (), and the rate constant of the first-order reaction in the condensed-phase bulk (Finlayson-Pitts and Pitts, 2000). For solids, bulk diffusion is generally too slow to allow bulk solubilities or bulk kinetics to control uptake. To justify the use of the formulation of additive kinetic conductances (Eq. 4) to solve the continuity equation and thus to be sufficient in laboratory studies to measure the net loss of a gas over a condensed phase of known volume and surface area, it is preferable to conduct experiments at low pressure. These experiments are typically performed in a tube reactor (radius r) with fast laminar flow (FF) conditions. To vary the reaction time, a moving injector is employed to change the exposed surface length in this technique. The net flux of the gas X into the condensed phase (Jx) can in this case be expressed as Eq. (5):
where kobs is the experimentally observed first-order rate coefficient and where the indices “g”, ∞, and surf represent the gas, bulk, and surface, respectively. In turn, kobs is approximately related to γnet as shown in Eq. (6):
where Dg is the diffusivity of the gas and where is its mean thermal velocity. The value of γnet changes as the surface is covered by molecules and depends on the concentrations of the reactants and the reaction time. The initial phase is denoted by , whereas the steady-state phase is denoted by . The calculated γnet can be employed to estimate the lifetime of gas X (τX) with respect to the reactive uptake on particles. The following formula has been applied to the uptake of aerosols with a polydisperse distribution (Mao et al., 2021; Sander, 1999; Schwartz, 1986):
The uptake of the only HgII species studied thus far, HgCl2, follows a Hinshelwood–Langmuir mechanism (Mao et al., 2021), where HgCl2(g) must first be adsorbed to a site (∥) on the surface and then react as a surface complex with a reactive center (e.g., anions) R on the surface, forming a product released from ∥, which becomes vacant again:
where K in the above equation is referred to as the Langmuir constant. Deposition velocities and partitioning coefficients constitute an empirical framework for parameterizing heterogeneous atmospheric processes. A coefficient for absorptive partitioning of compound X onto existing aerosols, Kgp, was proposed as in Pankow (2007):
where the index “gp” represents gas–particle partitioning; [X]p and [X]g represent the mass concentrations of compound X in the gas phase and particle phase, respectively, in a unit volume of air; and PM represents the total mass concentration of the particles.
4.3 Aqueous redox equilibria
The Gibbs free energy change (ΔG) presented previously is related to the electrode potential (E) as the equation:
where n is the number of moles of electrons transferred in the reaction and F is the Faraday constant (96 485 C mol−1). The standard potentials for the mercury–mercurous–mercuric free cation couples are as follows:
These positive potentials indicate that the reduction of Hg2 to Hg is favored under standard conditions. It is also evident that Hg0 can be oxidized to rather than to Hg2+(aq) only by agents with potentials ranging from −0.79 to −0.85 V. None of the common oxidizing agents meet this narrow potential range. Therefore, in excess of the oxidizing agent, Hg0 is completely oxidized to Hg2+(aq). Only when the excess Hg0 exceeds 50 % does oxidation lead to (aq). Ligation and hydrolysis have a major impact on standard potentials, including those listed in Reactions (R1)–(R3). For example, Hg(OH ⇄ Hg0 + 2HO−, analogous to Reaction (R3), has an E0 value of 0.206 V.
4.4 Chemical properties of aqueous HgI,II
The Hg2+ aqua ion, , exists only in distinctly acidic aqueous solutions containing a weakly coordinating anion (e.g., ). It readily undergoes hydrolysis at pH > 1 (log , Powell et al., 2005). Owing to its size and stable electron configuration, Hg2+(aq) can be easily polarized by ligands and, therefore, has the potential to form strong covalent bonds. This property allows Hg2+(aq) to interact with organic C to readily form Hg–C bonds through mercury–hydrogen substitution (mercuration), addition (oxy- and amino-mercuration, etc.), and decarboxylation reactions. An example is aniline, which forms a covalent complex with Hg2+ readily in aqueous solution at room temperature.
The formation of organomercurials by mercuration in aqueous solution is generally slow because of the reduced electrophilicity of Hg2+ caused by hydrolysis of the metal center. However, the presence of a polar solvent has little influence on other processes of organomercurial formation, such as decarboxylation. Therefore, abiotic Hg methylation can occur in aqueous solutions with the assistance of, e.g., light carboxylic acids (Deacon et al., 1986). In the case of keto-enolic organic compounds such as acetylacetone (R = H) and malonate (R = OH), the mercuric ion can, in principle, adopt a C bond, an O bond or a chelate structure.
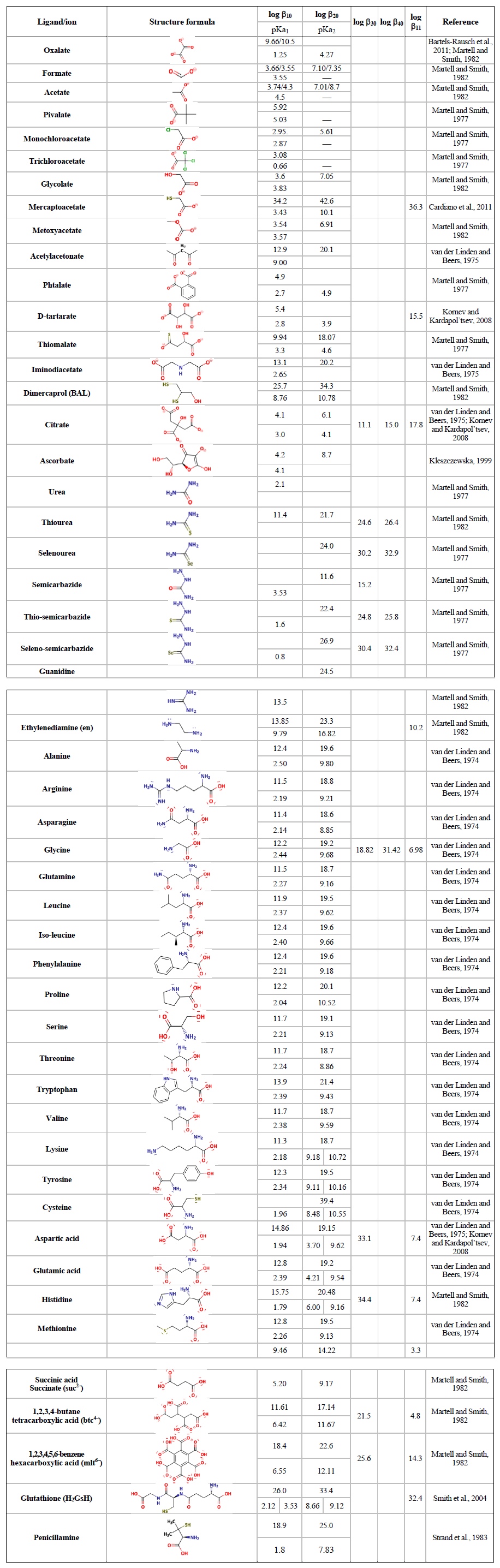
Highly toxic CH3Hg+ (MMHg+) species are by far the most abundant organic Hg in the environment and are formed from inorganic HgII, mainly by the action of FeIII and reducing bacteria. In addition to monomethylation, permethylation can also occur anaerobically (Sommar et al., 1999). (CH3)2Hg (DMHg) is detected mainly in deep-sea waters, but by upwelling waters (Conaway et al., 2009), it may reach the mixed layer, where gas exchange with the atmosphere can occur. DMHg has also been detected in landfills (Lindberg et al., 2005; Feldmann et al., 1994), sewage gas (Sommar et al., 1999), flood plains (Wallschläger et al., 1995), and rice paddies (Wang et al., 2019c). The binding affinity of Hg2+ to ligands is often qualitatively rationalized by Lewis acid–base theory, with the message that mercurials (type B metals) prefer soft ligands such as heavier halides and hydrochalcogenides (e.g., I− and SH−, respectively) to hard ones (e.g., OH− and F−). In fact, Hg2+ is the softest metal ion that acts as a Lewis acid. The preference for low coordination numbers (≤ 4, typically linear two-coordinate) in HgII complexes is related to the fact that relativistic effects come into play for the heaviest elements (Tossell and Vaughan, 1981). The interactions between Hg2+(aq) and inorganic ligands (Table 2) and low-molecular-weight organics (Table 3) are given as stability constants. The tables show that Hg2+ also binds strongly to nitrogenous bases. Interactions with inorganic compounds, such as ammonia, are extensive and complex (Breitinger and Brodersen, 1970). For organic nitrogen ligands, there is a parallel between the basicity of the ligand and the stability of the Hg–ligand complex (e.g., guanidine). Heterocyclic nitrogen compounds, such as histidine, also form strong complexes with mercuric ions. The hard–soft acid–base principle applies only to highly polar solvents, such as aqueous solutions, as a result of solvation (hydrolysis) effects (Riccardi et al., 2013). In the gaseous phase, an inverse relationship prevails (Riccardi et al., 2013) and can be illustrated by the fact that gaseous Hg(OH)2 is a stable molecule, whereas in aqueous solution, Hg2+ and 2 OH− can form the intermediate molecule Hg(OH)2 (Yang et al., 2020b), which eliminates H2O and precipitates solid HgO. Therefore, solid Hg(OH)2 is not known (Wang and Andrews, 2005a). Furthermore, in the aqueous phase, the univalent state (mercurous species) is represented by the metal-metal bound ion , which is ordinarily stable. Like Hg2+(aq), (aq) is a soft Lewis acid.
Hg–ligand complexation is ubiquitous in the environment. This process involves a significant energy shift due to solvation effects, which results in a reduction in the number of solvating water molecules and an increase in the interaction between ligands/anions in the complexes and water. Unlike the dimer cation, the discrete Hg•+ cation is paramagnetic and was detected for the first time via electron spin resonance (Symons and Yandell, 1971). Free Hg•+ is a highly potent reducing agent with a one-electron reduction potential, E0(Hg2+ Hg•+), estimated to be well below −2.0 V (Gårdfeldt and Jonsson, 2003). However, hydrolyzed or ligated forms are less reactive (Gårdfeldt and Jonsson, 2003; Kozin and Hansen, 2013). The dissociation (aq) is considerably less significant than the disproportionation , with a conservative upper bound for the ratio of 10−7 (Moser and Voigt, 1957). Free cation acidity decreases in the order of Hg2+ (pK 3.4), (pK 4.9), and Hg•+ (pK 5.1). has an equilibrium constant of 5.5 × 10−9 M (Moser and Voigt, 1957), which indicates that a solution of initially only in pure water will contain only a single percent Hg2+ in the absence of ligands that form complexes with Hg2+. However, in the presence of ligands that form complexes with Hg2+, disproportionation is rapid, and is consumed. The same applies when Hg0(aq) is removed from the solution, e.g., by a gas stream. can be a major speciation component in heavily polluted waters (Fang et al., 2024) but is insignificant in the atmosphere.
4.5 Chemical equilibria data
For a general complex equilibrium with Hg2+ and the ligand L, , a stability constant βqr is defined as . When the complex is not hydrolyzed, βqr is reduced to . For the equilibrium obtained by adding a ligand (L) to a metal complex in a stepwise manner, Kq is used, which is related to βq by . Tables 2 and 3 present the equilibrium constants for Hg2+ associated with a range of inorganic and organic natural ligands, respectively, without being comprehensive. Quantitative details are available through the open-access AQUAMER database and web server dedicated to Hg, which provides direct speciation results by combining web-based interfaces with a speciation calculator, thermodynamic constant databases, and a computational chemistry toolbox for input to other software to estimate missing constants (Lian et al., 2020).
Table 2Hg2+–inorganic ligand complexes. Omitted in the table are, e.g., interactions with reduced sulfur (HS–, R–S–), which can be found in, e.g., Skyllberg (2011).
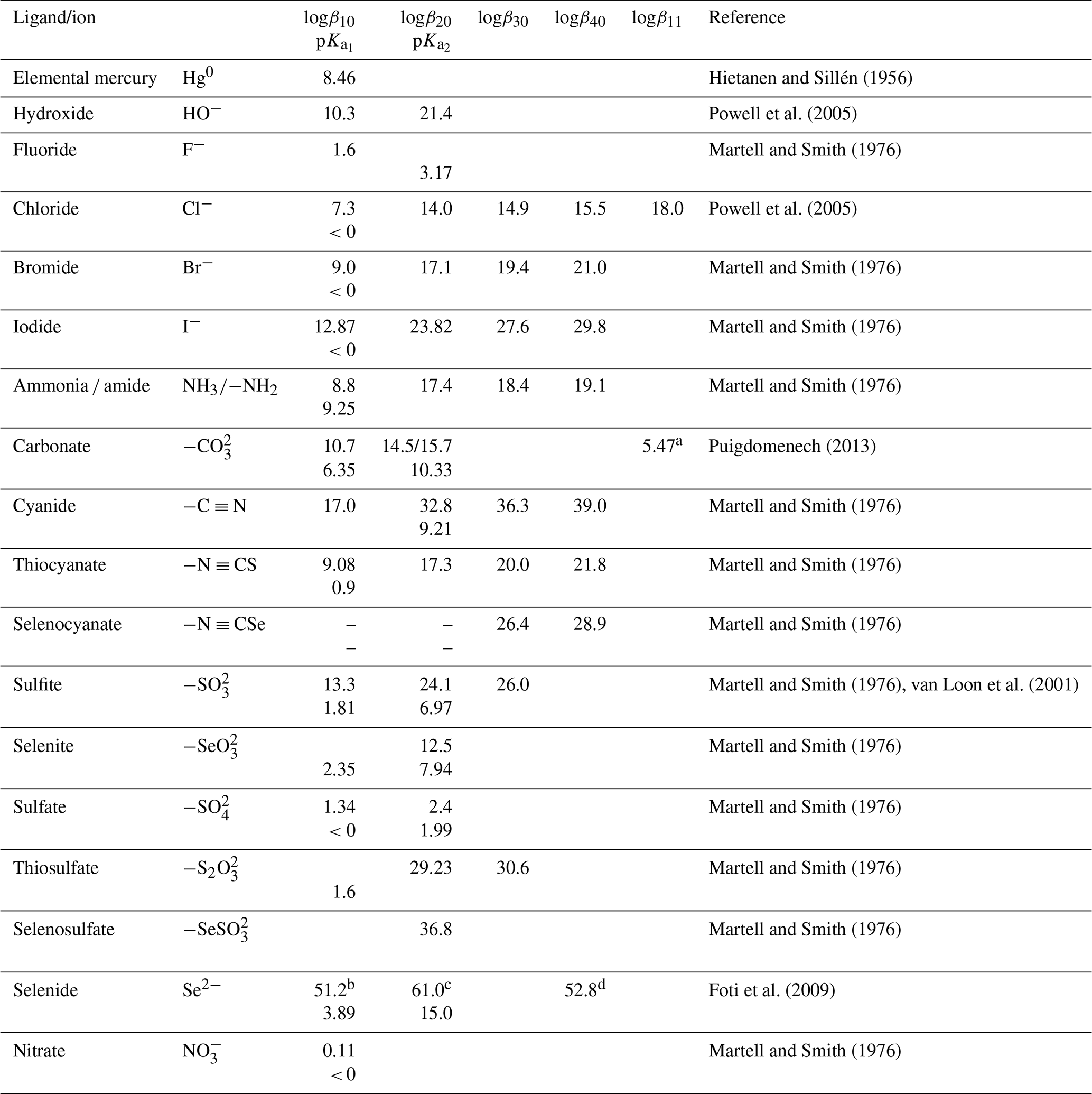
a Hg2+ + HCO. b . c Hg2+ +2 HO +2 HSe ⇄ HgSe. d Hg2+ + HO +2 HSe ⇄ HgHSe.
Table 3Hg2+–organic ligand complexes.
4.6 Speciation of HgII in atmospheric waters
HgII speciation in atmospheric waters such as clouds and fog is governed by interactions with inorganic nucleophiles, low-molecular-weight organics (LMWO), and high-molecular-weight dissolved organic matter (DOM). The identified LMWOs typically make up a smaller mass fraction of the DOM in ambient cloud and fog droplets. Despite its limited abundance (0.5 %–3 % in freshwater), sulfurized DOM exerts control over Hg cycling in terrestrial aquatic systems by forming predominantly strong HgL (logK ∼ 21.9–23.6) and HgL2 (logK ∼ 30.1–31.6) complexes (Dong et al., 2011), where L represents functional groups with reduced sulfur. Although sulfur-containing DOM (with the elemental compositions of CHSO and CHNSO) is also relatively ubiquitous in atmospheric organic matter (AOM), sulfur is present mainly in hexavalent form, with reduced sulfur being rare (Zhao et al., 2013; Bianco et al., 2018; Jiang et al., 2022). In contrast to sub-zero valence S, which is not relevant in this context, conjugate bases of strong oxo acids that are common in AOM, such as organic nitrates and sulfates, form only weak complexes with HgII. Therefore, the application of speciation by equilibrium modeling on a geospherical basis to assess the atmospheric interaction between atmospheric DOM and HgII, as in some studies (Li et al., 2018; Zhen et al., 2023), is questionable. Bittrich et al. (2011) used pH, a confined set of inorganic ions (, , , and Cl−), and LMWO acids to observe dissolved HgII in a study of cloud and fog water. Strongly dependent on pH, at <5, even moderate Cl− levels can control speciation (HgCl2), whereas in more alkaline waters (e.g., influenced by NH3), speciation is represented by Hg(OH)2, Hg(OH)Cl, and to some extent . A more realistic approach is to include DOM in speciation. In this regard, Yang et al. (2019) employed HgII complexation with fulvic acids under conditions of binding to mainly O donors (1:2 complexes with logβ20=5.6, Haitzer et al., 2002) as surrogates for AOM interaction, which, when applied, was found to dominate in the HgII speciation of rainwater samples in rural and urban France. Studies of cloud water in eastern China revealed a marked change in acidity and other chemical compositions in the post-2008 period, where HgII, although the concentration was unchanged over time, in the former acidic environment was mainly bound by DOM (∼ 79 %) (Li et al., 2018) and, in the latter more neutral environment, was more homogeneously distributed in addition to DOM among hydrolyzed and halide (X = Cl, Br)-bound species (Hg(OH)2, HOHgX, and HgX2; Zhen et al., 2023). In conclusion, until the complexation of HgII with AOM is well understood, there is considerable uncertainty regarding the partitioning of aquatic HgII between stable and reduction-labile complexes in the photic atmosphere.
4.7 Chemical reaction data
The subsequent two principal sections address the chemical redox reactions in the gaseous phase (Sect. 5) and in the aqueous phase (Sect. 6). Table 4 summarizes the gas-phase reactions, along with the rate coefficients considered most accurate and the corresponding reaction enthalpies. The reaction numbers are designated with the prefix G (G R1, G R2, etc.). The aqueous-phase reaction numbers are designated with prefix W and are listed in Table 5 with the corresponding rate coefficients. Notably, several chemical reactions that are not labeled with G or W and are not assigned to Tables 4 and 5 appear in the text. This is particularly the case for heterogeneous (multiphase) processes (Sect. 7), such as reactive uptake and reduction on surfaces, which consequently have no prefix and follow sequential numbering throughout the document.
5.1 Inorganic species
5.1.1 Initial reactions of ground-state Hg0
The homogeneous gas-phase oxidation of Hg0 in the electronic ground state is limited to a few reactive species produced photolytically. In the atmosphere, multi-step reactions involving both HgI and HgII species are crucial for Hg transformation. Atmospheric oxidation of Hg0 occurs largely in the gas phase, whereas the rates of aqueous phase reactions in deliquescent aerosols are relatively slower on a unit air volume basis and are inherently limited by the low water solubility of Hg0. The oxidation of Hg0 vapor by closed-shell molecules, such as halogenation chemistry with reference to the gas phase, has been studied in the laboratory at various temperatures (Hall, 1992; Qu et al., 2009; Chi et al., 2009; Ariya et al., 2002; Sumner et al., 2005; Raofie and Ariya, 2004; Raofie et al., 2008; Wilcox, 2009) since Ogg et al. (1936). Direct oxidation by free halogens (X2) via the insertion reaction Hg + X2→ XHgIIX is highly exothermic but very slow under atmospheric conditions due to large energy barriers (Auzmendi-Murua et al., 2014), whereas the abstraction Hg + X2→•HgIX + X• proceeds at significant rates only at high temperatures (Niksa et al., 2001). Thus, free halogen chemistry is important for the conversion of Hg in flue gas from power generation systems (Wilcox, 2009), such as coal-fired (CFPP) systems, but not in the atmosphere. The same applies to the Hg + NO2 reaction, which is barrierless and whose pathway to HgII(ONO)2 shows a negative temperature dependence (Li et al., 2022c). However, reactions that are important only in combustion and flue gas cleaning systems are outside the scope of this review.
Although oxidation of Hg0 vapor by the common atmospheric oxidants O3 (Sumner et al., 2005; Hall, 1995; Pal and Ariya, 2004b; Snider et al., 2008), BrO• (Raofie and Ariya, 2004; Spicer et al., 2002), and (Sommar et al., 1997; Sumner et al., 2005) has been observed in the laboratory, the identity and phase of the product(s) are in doubt. Laboratory studies of gas-phase oxidation of ppb levels of Hg0 (the atmospheric level is sub-ppt) have revealed product particles in the accumulation mode, suggesting that gas-to-particle conversion takes place (Raofie and Ariya, 2004; Sun et al., 2016). These data attributed to the gas phase are almost certainly compromised by complex kinetics, including reactions at the reactor wall (Hynes et al., 2009). In all cases, gas-phase oxidation pathways leading to HgO by O atom transfer are endothermic (Reactions G R5–R7, Table 4). Furthermore, the measured pre-exponential factors for the Hg–O3 reaction, ∼ 10−16–10−18 cm3 molecule−1 s−1 (Hall, 1995; Pal and Ariya, 2004b), are much smaller than expected for simple O atom transfer (Calvert and Lindberg, 2005). Alternative O3 oxidation via a weakly bound (∼ 16 kJ mol−1) adduct, HgO3, lacks exothermic dissociation pathways (i.e., HgO + O2, Reaction G R5a) and is therefore unlikely to occur in the atmosphere. However, in laboratory experiments, HgOx can conceivably diffuse to surfaces and be deposited as solid HgO possibly via oligomerization (Tossell, 2006). Recombination of Hg0 with results in weakly bound •HgINO3 (∼ 27 kJ mol−1), which dissociates in the lower troposphere before oxidation to HgII species of the type O2NOHgO• or O2NOHgY can occur (Edirappulige et al., 2024). Abstractions (e.g., Hg + BrO•→ HgO + Br• or Hg + BrO•→•HgIBr + O, Reactions G R7a and R7b) are endothermic, whereas direct insertion reactions (e.g., Hg + BrO•→ BrHgIIO•, Reaction G R7c) are exothermic (−84 kJ mol−1, Shepler 2006) but affected by large barriers (170 kJ mol−1) and are therefore unlikely to proceed (Balabanov and Peterson, 2003). The remaining exit channels, namely the recombination of Hg and BrO• (Reaction G R7d), leading to the formation of the geometric isomers of BrHgIIO• (•HgIBrO and •HgIOBr), are also inconceivable, as these adducts are thought to be very weakly bound (Shepler, 2006). Stable HgI species of this type have been reported, suggesting that BrO• is important during AMDEs (Raofie and Ariya, 2004). However, other field (Wang et al., 2019a) and model (Xie et al., 2008; Ahmed et al., 2023) studies have shown that the synchronous disappearance of Hg0 and O3 during AMDEs can best be described solely as the action of Br atoms, with an upper limit for kHg + BrO of 1 × 10−15 cm3 molecule−1 s−1, but that the reaction product •HgIBr (Fig. 2) rapidly adds BrO•, presumably mainly to BrHgIIOBr, which is 117 kJ mol−1 more stable than the isomer BrHgIIBrO (Jiao and Dibble, 2017a). Despite its thermal stability, BrHgIIOBr is rapidly photolyzed (Figs. 2 and 4) and therefore does not constitute a significant component of the HgII pool following an AMDE.
Table 4Atmospheric gas-phase reactions. Except where otherwise noted in the reference column, the thermodynamic data have been compiled from the following sources of information: CRC Handbook of Chemistry and Physics (Lide, 2008), Hepler and Olofsson (1975), Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies (Burkholder et al., 2019), Guzman and Bozzelli (2019), Saiz-Lopez et al. (2020, 2022), Balabanov and Peterson (2003, 2004), and Shepler (2006). The photolysis frequencies are calculated via the global annual average photon flux in the troposphere.
a The basics of gas-phase kinetics have been introduced in Sect. 4.1. Unimolecular rate coefficients are in s−1 (photolysis frequencies refer to excitation energies at λ > 290 nm calculated according to , where ϕ is the quantum yield (≤1), ó is the absorption cross section (cm2 molecule−1), F is the photon flux (photons cm2 s−1), λ is the wavelength, and T is absolute temperature), bimolecular reaction rate coefficients are in cm3 molecule−1 s−1 (expressed as a rate constant or as a coefficient with an Arrhenius or other type of temperature dependence), and three-body reactions according to Eq. (2), i.e., , are in cm6 molecule−2 s−1 (where [M] is the number density of air molecules), k0 (cm3 molecule−1 s−1) is the low-pressure limiting rate coefficient, and k8 (cm6 molecule−2 s−1) is the high-pressure limiting rate coefficient. The temperature dependence of k0 and k8 is expressed with Eq. (3). b Refers to the calculated enthalpy (0 K) or to the experimental ditto (298 K). c Equilibrium coefficient (unit: cm3 molecule−1). d Over the interval 333 to 200 K. e Estimated value from CH3O + NO. f Estimated value from CH3O + NO2. g Valid for 395–573 K. h Based on calculation of exit-channel complexes at SC-NEVPT2 level of theory. i Based on ΔfH0(HOHgO•)=63.2 kJ mol−1. j Estimated value from CH3O + NO. k Estimated value from CH3O + NO2. l refers to singlet Hg(OH)2 but is 10 kJ mol−1 endothermic for formation of spin-conserving triplet Hg(OH)2. m Assuming ΔfH0(CH3HgCl) =ΔfH0(CH3HgOH).
In addition to bromine atoms (Br•), hydroxyl radicals (HO•) and, to a lesser extent, chlorine (Cl•) and possibly iodine (I•) atoms have been proposed to initiate the global gas-phase oxidation of Hg0 in the ground state in the atmosphere:
The reaction rates for X = Cl (Reaction G R2, Donohoue et al., 2005; Taylor et al., 2005) and Br (Reaction G R1, Donohoue et al., 2006) have been determined via pulsed laser photolysis–laser-induced fluorescence (PLP-LIF) for a range of pressures and temperatures. The reaction is apparently termolecular; i.e., it shows a linear dependence on pressure (M), a slightly negative temperature dependence, and a significant difference in deactivation efficiency, with N2 and He as third bodies (Donohoue et al., 2005). There are also several experimental static studies of halogen atom reactions carried out at 1 atm pressure, which, with the exception of the studies by Horne et al. (1968) and Greig et al. (1970), have used the relative rate (RR) technique at room temperature (Ariya et al., 2002; Spicer et al., 2002; Sun et al., 2016; Guérette, 2011). The Hg + X• rate expression determined by Donohoue et al. over 0.26−0.79 atm and 243–293 K by the preferred PLP-LIF technique gives rate coefficients of 5.4 × 10−13 cm3 molecule−1 s−1 (Donohoue et al., 2005) and 3.6 × 10−13 cm3 molecule−1 s−1 (Donohoue et al., 2006) at 298 K and 1 atm pressure in air for the Cl• and Br• reactions, respectively. Although the rate constant of the chlorine atom reaction is 50 % greater than that of the bromine atom reaction, the significance of the former is small in the remote troposphere, considering the low concentration of chlorine atoms. Notably, a significant increase in the apparent recombination rate coefficient of Hg + Cl• was observed in the presence of air. This result has been rationalized on the basis that secondarily formed ClOx species may also react rapidly with Hg0 (Donohoue, 2008). A plausible candidate is Hg + ClO2→ •HgICl + O2 (Reaction G R8), which is exothermic (ΔHR = –80 kJ mol−1), but the channel has not been investigated further. Computational studies (Shepler et al., 2007; Goodsite et al., 2004; Goodsite et al., 2012) reported a slightly larger rate constant (∼ 10−12 cm3 molecule−1 s−1) for the Hg + Br• reaction than the absolute PLP-LIF determination at STP. On the other hand, experimental RR studies generally yield rate constants that exceed the limit obtained from theoretical calculations, suggesting complex kinetics, including reactions at the reactor wall.
The reaction with X = OH (Reaction G R3) was studied with PLP-LIF using an excess of Hg0 over •OH (generated from the photolysis of HNO3 at 266 nm) without evidence of a reaction, resulting in an upper rate limit of (<) 1.2 × 10−13 cm3 molecule−1 s−1 (Bauer et al., 2003). The rate constant of Hg +•OH → products determined by Sommar et al. (2001) relative to cyclohexane +•OH → products of 8.7 × 10−14 cm3 molecule−1 s−1 falls below this limit at 295 K and 1 atm air, as does the temperature-resolved kinetic RR study of Pal and Ariya (2004a) extrapolated to 295 K (∼ 1 × 10−13 cm3 molecule−1 s−1). External re-analysis of Pal and Ariya (Calvert and Lindberg, 2005) and Sommar et al. (2001) (Dibble et al., 2020) data via kinetic modeling revealed that •HgIOH under experimental conditions exclusively reacts with NO2 (•HgIOH + NO2→ HOHgIIONO, Reaction G R57) rather than dissociating. The temporal resolution in the PLP-LIF study also allowed a lower-bound estimate of the equilibrium constant of cm3 molecule−1 (Bauer et al., 2003). This equilibrium constant has been estimated via computational studies. Recently, high-level quantum chemical calculations (Dibble et al., 2020) performed at 200–320 K yielded a of ∼ 7 × 10−16 cm3 molecule−1 at 298 K, corresponding to a k13 of cm3 molecule−1 s−1 at 1 atm. In contrast, Saiz-Lopez et al. (2022) reported that was more than an order of magnitude smaller (∼ 5 × 10−17 cm3 molecule−1) at the corresponding temperature.
The kinetics of the reaction between Hg0 and iodine atoms (by photolysis of CH2I2 CF3I) were studied in an early work by monitoring •HgII by absorption spectroscopy at 403–438 K (Greig et al., 1971) and in a later study by following the Hg0 loss by MS at 296 K (Raofie et al., 2008). In the first study, sufficiently high •HgII densities could not be generated to gauge a reaction, for which the rate constant was lower than that of the competing reaction of ∼ 1 × 10−13 cm3 molecule−1 s−1. The latter study lacks conclusive results on the HgII reaction but provides a limit on the rate constant for the reaction of Hg with molecular iodine vapor (≤ cm3 molecule−1 s−1), a reaction that lacks any atmospheric significance. A rate coefficient of 4.0 × 10−13 cm3 molecule−1 s−1 was calculated for the HgII reaction at 1 atm N2 and T between 180 and 400 K via Rice–Ramsperger–Kassel–Markus (RRKM) theory based on the calculated binding energy (46 kJ mol−1) and molecular properties of •HgI(2∑) (Goodsite et al., 2004).
5.1.2 Stability of •HgIX
The first step (termolecular Reactions G1–G3), which is exothermic, produces HgI radical intermediates (•HgIX), which can revert to Hg0 both thermally and photolytically:
Photolytic and thermal dissociation
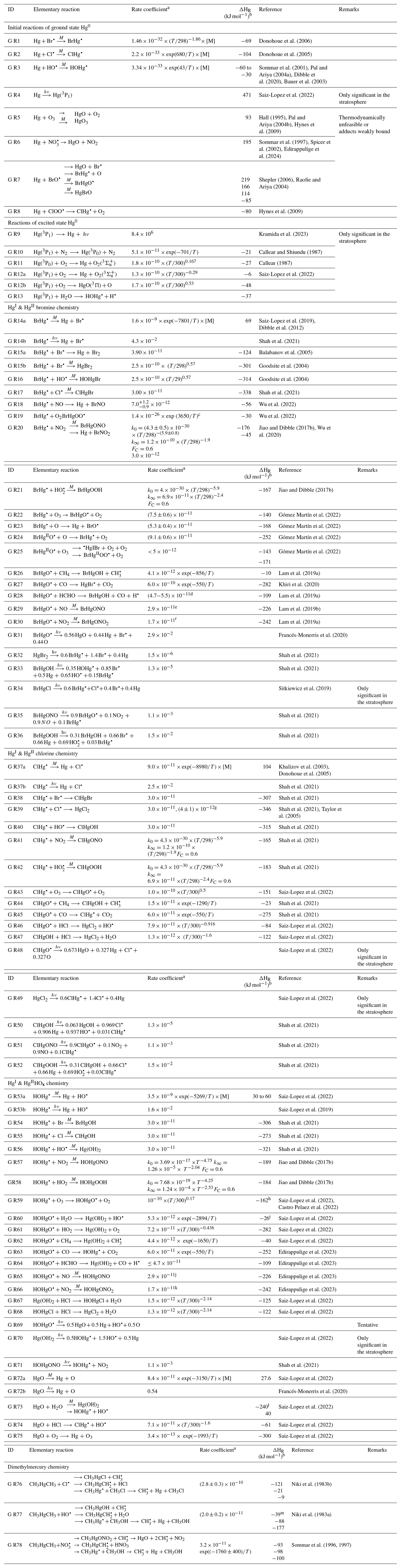
The first excited electronic state of •HgIX (designated A2Π for halogenated radicals) is exclusively repulsive, resulting in dissociation with visible light for wavelengths exceeding ∼ 460 nm, where the absorption maxima are predicted at ∼ 480, ∼ 575, ∼ 650, and ∼ 690 nm for •HgIOH, •HgICl, •HgIBr, and •HgII, respectively (Saiz-Lopez et al., 2019; Fig. 3). While the bond strengths of Hg–Cl and Hg–Br are well defined in relative terms (89.5–98.0 kJ mol−1, Tellinghuisen et al., 1982; Shepler et al., 2005; Saiz-Lopez et al., 2022; Cremer et al., 2008 and 60.2–68.1 kJ mol−1, Goodsite et al., 2004; Shepler et al., 2005; Cremer et al., 2008; Tellinghuisen and Ashmore, 1983, respectively), there is significant variation in the estimates of the bond strengths of •HgII and •HgIOH, ranging from ∼ 33 to 46 kJ mol−1 (Goodsite et al., 2004; Shepler et al., 2005; Cremer et al., 2008; Jordan et al., 1993; Salter et al., 1986) and ∼ 23 to 55 kJ mol−1 (Dibble et al., 2020; Tossell, 2003; Goodsite et al., 2012; Guzman and Bozzelli, 2019; Cremer et al., 2008), respectively. Therefore, the stability of •HgIOH and •HgII is uncertain, and it is debatable whether their thermal lifetimes in the atmosphere are long enough for these radicals to be further oxidized to mercuric species to any significant degree. The question has been raised recently since it was experimentally established that •HgIBr is kinetically oxidized by O3 without a reaction barrier (Reaction G R22), which was also theoretically established to be true at least for •HgICl and •HgIOH (Reactions G R43 and G R59, respectively, Sect. 5.1.3). A study using RRKM theory suggested that the recombination rate coefficients of Hg with I• and HO• are similar in the free troposphere, while the thermal dissociation of •HgII gradually exceeds that of •HgIOH at lower temperatures (Goodsite et al., 2004). •HgII is the •HgIX species with the shortest photolytic lifetime in the troposphere globally (∼ 17 s), according to computational chemistry theory (Saiz-Lopez et al., 2019). Recently, Dibble et al. (2020) estimated the HO–Hg binding energy to be 46 kJ mol−1 using high-level quantum chemical calculations. Compared with a global photolytic lifetime of just over 1 min (Shah et al., 2021), the thermal lifetime of •HgIOH in the lower troposphere is significantly shorter (according to data from Dibble et al., 2020 ∼ 10 ms at the surface up to approximately 10 s at the tropopause). For the lighter mercurous halides (excluding •HgII), the relationship is reversed with respect to the importance of photolytic versus thermal dissociation. The lifetimes of the former channel are ∼ 20 and ∼ 40 s for •HgIBr and •HgICl, respectively, while the thermal decay is slower for •HgIBr above the planetary boundary layer, and •HgICl is much less thermally unstable.
Figure 3Computed absorption spectra of the atmospherically important mercurous chloride, bromide, and hydroxyl radicals. Wavelengths accessible in the troposphere are to the right of the colored area. Data from Saiz-Lopez et al. (2019).
5.1.3 Bimolecular reactions of •HgIX
In addition to thermal and photolytic decomposition, the fate of •HgIX in the atmosphere is controlled by further oxidation to thermally stable mercuric species molecules. Experimental studies of the specific bimolecular reaction kinetics of •HgIX are limited to X = Br and Cl (Reactions G R15 and G R39). Taylor et al. (2005) studied the reactions of •HgICl with Cl2, HCl, and Cl• at temperatures characteristic of post-combustion conditions. The observed reaction with free chlorine to form HgCl2 was rapid (1.2 × 10−11 cm3 molecule−1 s−1) and temperature-independent. Jiao and Dibble (2017b) used computational chemistry to determine the rate constant and product yield for reactions of •HgIBr with abundant atmospheric NO2 (Reaction G R20) and HOO (Reaction G R21) radicals. Analogous to the experimental •HgICl study, these reactions were calculated to be rapid, with the rate constant for oxidation by NO2 being approximately twice that for oxidation by HOO•. This theoretical study indicated that the •HgIBr + NO2 reaction occurs along two competing channels (Reaction G R20a, b), one proceeding via oxidative addition, resulting in BrHgIIONO, and the other operating via reductive displacement, resulting in Hg0 + BrNO2. The dichotomy occurs because •HgIBr (2Σ+) possesses a delocalized electron that spreads more equivalent spin density over the molecule (•HgIBr ↔•HgIBr•), whereas the spin density of the HgOH (1A') radical is most localized on the Hg atom. A reaction with another radical center occurs for •HgIOH when the reactant is oriented toward Hg, leading to addition, while for HgBr, reductive displacement is also possible when the collision involves the Br atom (Castro Pelaez et al., 2022). The existence of a branching ratio was also confirmed by an experimental study of the •HgIBr + NO2 reaction by Wu et al. (2020) using PLP-LIF, who reported that the computed rate coefficients for both reduction and oxidation were greatly overestimated. This study deduced that the importance of the reductive channel increases slowly with increasing altitude from the ground level to the tropopause but is only ∼ 10 % as fast as the oxidation reaction. Wu et al. (2022) also experimentally studied the interaction between NO and •HgIBr, leading to Hg0 + BrNO. •HgIBr (Reaction G R19) is slightly exothermic, while that leading to Hg0 + BrOO• is less feasible due to endothermicity. The •HgIBr + O2 reaction is thus described by •HgIBr , with an equilibrium constant that decreases with increasing temperature (Wu et al., 2022). To the extent that BrHgIIOO• can be attributed significance, it is a reservoir for •HgIBr at low temperatures, with an upper limit of ∼ 50 % stored at 220 K. Wu et al. (2022) argued that BrHgIIOO• behaves like a peroxyl radical (HOO• ROO•) in reactions with atmospheric radicals. Recently, Saiz-Lopez et al. (2020) implied missing oxidation pathways to better reconcile their GEOS-Chem global atmospheric chemistry model simulations with field observations. Suggested by Shepler (2006) and later Lam (2019) as a potential pathway of HgI oxidation, the Saiz-Lopez group has carried out theoretical (Saiz-Lopez et al., 2020) and experimental (Gómez Martín et al., 2022) investigations of the system •HgIBr + O3. In addition, Castro Pelaez et al. (2022) carried out theoretical calculations for rate constants and product yields, including •HgIOH + O3. •HgIX + O3 → XHgIIO• + O2 (Reactions G R22, G R43 and G R59) is highly exothermic (172 kJ mol−1 for X = Br), proceeds without a substantial activation barrier, and is currently considered to be important for the atmospheric oxidation of •HgIX, with XHgIIO• as a key intermediate. As a radical, XHgIIO• is relatively thermally stable with strong Hg–O bond (333 and 294 kJ mol−1 for X = Cl & Br, respectively; Balabanov and Peterson, 2003). Gómez Martin et al. (2022) determined the rate coefficient of the •HgIBr + O3 reaction at 295 K via a PLP-LIF system. To generate •HgIBr (photolysis of HgBr2 at 248 nm by a KrF excimer laser), the introduced O3 would inevitably be photolyzed to some extent before it could react with •HgIBr. This led to complications due to the following potential chemistry:
By performing experiments at different KrF laser energies and ozone concentrations and by numerical modeling of the data, Gómez Martin et al. (2022) isolated k (•HgBr + O3), k(•HgBr + O), and k(BrHgO•+ O) as 7.5, 5.3, and 9.1 (all × 10−11 cm3 molecule−1 s−1), respectively. They presented an upper limit for BrHgIIO•+ O3 ( 10−12 cm3 molecule−1 s−1), which was considered infeasible by theoretical calculations due to steric hindrance. Instead of leading primarily to BrHgIIO•, as is the case for the •HgIBr + O3 reaction, •HgIBr + O results in reductive elimination (Hg0 + BrO•) for all collision geometries. Hg0 is also produced in the rapid reaction between BrHgIIO•+ O. In the lower atmosphere (≤ 25 km), the content of free O atoms is low, and therefore, its role as an oxidant is minor (Calvert et al., 2015). The energetic O(1D), formed primarily by photolysis of O3 by UV light (<340 nm), is rapidly consumed through two competitive channels: deactivation to O(3P) by collision with air molecules or reaction with the ubiquitous water vapor to form OH radicals. O(3P), also formed by the photolysis of NO2 (<430 nm), reacts rapidly and thermally with O2 in the atmosphere to form ozone (Calvert et al., 2015). Importantly, k(•HgBr + O3) is more than twice as fast as k(•HgBr + NO2) when the experimental results are extrapolated to the atmospheric surface layer (1 atm, 295 K). The combination of a high k(•HgBr + O3) and the abundance of ozone relative to other radicals, such as NO2 and HOO, suggests that •HgIBr + O3 is predominant in the conversion of HgI to HgII in the atmosphere. The experimentally determined k(•HgBr + O3) is close to the upper limit of 1 × 10−10 cm3 molecule−1 s−1 estimated by Saiz-Lopez et al. (2020), which excludes steric effects. For an updated chemical mechanism in the global atmospheric model GEOS-Chem, Shah et al. (2021) used a conservative rate constant of 3 × 10−11 cm3 molecule−1 s−1 for the oxidation of •HgIX with O3 (X = Cl, Br and OH). By postulating k(•HgOH + O3)= k(•HgBr + O3), simulations by Shah et al. (2021) revealed that the OH-initiated pathway accounts for one-third of global HgII production. In contrast, by not including •HgIOH + O3 in their model, Dibble et al. (2020) reported that the OH-initiated channel is largely irrelevant, with only some regional significance in areas with high levels of photochemical smog. More recently, Castro Pelaez et al. (2022) compared •HgIBr + O3 and •HgIOH + O3 systems via computational chemistry and reported that the former has a slight tendency (≤ 0.1 %) to undergo reductive elimination (Hg + BrO• + O2) rather than oxidation (BrHgIIO• + O2) when the orientation of the terminal oxygen in ozone is toward the Br atom. There was no such tendency for •HgIOH + O3. It was also found that k(•HgBr + O3) and k(•HgOH + O3) are likely similar at 298 K in the range of (6.6–8.5) × 10−11 cm3 molecule−1 s−1. The positive covariation of O3 and •OH, as opposed to •Br and O3 (O3 titrates •Br, Fig. 2), suggests precedence for OH-initiated Hg oxidation in air with secondary pollutants (Rutter et al., 2012). Field observations of GOM in urban air may suggest radical-initiated Hg0→ HgII gas-phase transformation, which is claimed to be completed by certain radicals (Peleg et al., 2015; Hong et al., 2016; Edirappulige et al., 2024). An interesting case is urban Jerusalem, where episodes of elevated daytime and nighttime gaseous HgII levels covary with O3 (max 250 µg m−3) and NO3 (430 ng m−3), respectively (Peleg et al., 2015). To the east of the city lies the Dead Sea basin, where effective bromine-controlled oxidation of Hg0 has been observed (Tas et al., 2012). Finally, the reactivity of •HgIX toward volatile hydrocarbons is low, as •HgIX does not abstract a hydrogen atom from an alkane (e.g., from CH4), nor does it significantly add to a double bond of an alkene (e.g., to CH2=CH2) (Dibble and Schwid, 2016).
5.1.4 Stability of HgIIXY
Photoreduction and stoichiometric yields
Although atmospheric HgII species are generally more stable than HgI species are, many HgII molecules are still labile, and the atmospheric pool contains mercuric species with different thermal and photolytic stabilities. Most of the atmospherically relevant gas-phase species have well-defined absorption bands in deep UV, in some cases extending into the UV-B and UV-A regions. Early theoretical studies (Strömberg et al., 1989; Strömberg et al., 1991), when knowledge of the atmospheric chemistry of Hg was rudimentary, indicated that the photoreduction of HgCl2 and Hg(CN)2 in actinic light at the Earth's surface was negligible, while that of Hg(OH)2 and Hg(SH)2 was extremely slow. The UV absorption spectra of mercuric halides are increasingly red-shifted as the halogen becomes heavier. HgCl2 vapor absorbs only radiation below 240 nm (Fig. 8a), and HgBr2 absorbs mainly deep-UV light with a tiny tail (< 10−19 cm2 molecule−1, Fig. 8c) into UV-B, while HgI2 has significant absorption in the entire UV region (Maya, 1977; Sitkiewicz et al., 2019). However, binary compounds such as HgBr2 or HgCl2 do not completely dominate the atmospheric HgII(g) speciation. Mixed compounds such as BrHgIIY molecules (Y = ONO, OOH, OH, OCl, OBr, etc.) and XHgIIO radicals (X = Br, OH) are also predicted to be important. Saiz-Lopez et al. (2018) computed the absorption spectra of mixed compounds and found that abundant BrHgIIY molecules absorb in UV-B. The rapidly photolyzed HgII species identified include BrHgONO (Reaction G R35), BrHgOOH (Reaction G R36) and BrHgOBr (with lifetimes of a few minutes to less than a second, Fig. 4a-c), with BrHgOH being comparatively long-lived (> 1 day, Fig. 4d) in terms of photodissociation. In their modeling study, HgCl2 and Hg(OH)2 were estimated to be photolytically stable in the troposphere by Shah et al. (2021), while the photolysis frequency of HgBr2 was calculated to be just over an order of magnitude lower than that of BrHgOH (1.2 × 10−6 and 1.3 × 10−5 s−1, respectively).
Figure 4Computed absorption spectra of the atmospherically important (a) BrHgOBr, (b) BrHgOOH, (c) syn-BrHgONO and (d) BrHgOH. Wavelengths accessible in the troposphere are to the right of the colored area (Francés-Monerris et al., 2020).
The photodissociation mechanism (quantum and product yield) of BrHgIIY has been studied using computer-aided calculations based on 2D potential energy surfaces, with the result that photodynamics lead to different channels in which the Hg-containing products can exhibit +II, +I, and 0 oxidation states (Francés-Monerris et al., 2020; Lam et al., 2019b). Photolysis of BrHgIIONO results in the formation of NO and BrHgIIO• in 90 % of cases, while the remainder reverts to •HgIBr and NO2 (Francés-Monerris et al., 2020). Consistently, a large dominance of the photoproducts BrHgIIO• + NO was predicted by the calculations of Lam et al. (2019b), in contrast to an early work by Saiz-Lopéz et al. (2018) that favored •HgIBr and Hg0 formation. During the photolysis of BrHgIIOOH, the Hg–Br, Hg–O, and O–O bonds can be broken, resulting in three main exit channels:
Thus, the photodissociation of BrHgIIOOH produces Hg0, •HgIBr, and BrHgIIO• to varying degrees (Francés-Monerris et al., 2020). In the case of BrHgIIOH, the photolytic formation of BrHgIIO• is negligible, while in half of the cases (49 %), reduction to elemental Hg occurs, and in the other half, •HgIBr or •HgIOH is formed, with the former being predominant (∼ 70 %) (Francés-Monerris et al., 2020). The photolysis of BrHgIIONO and BrHgIIOOH thus results in significant yields of BrHgIIO•, the radical form of HgII described above as the major product of the rapid reaction between •HgIBr and O3. In this series of reported compositional chemical results, the only YHgIIO• species that has been experimentally characterized is the fluorine analog that is formed along with FOHgIIF when excited Hg atoms react with OF2 (Andrews et al., 2012). Although FHgIIO• has no atmospheric significance, its experimentally determined properties are important benchmarks for other homologs in the series. YHgIIO• has two strong bonds (the dissociation energy for YHg–O is ∼ 250 kJ mol−1) and is thermally stable in the gas phase. However, YHgIIO• is photolabile under UV–VIS light (cf. Fig. 5b) and decomposes photolytically along two channels. The calculated branching ratios for both Y = Cl and Br favor the formation of HgO (67 % and 56 %, respectively, Saiz-Lopez et al., 2022) over splitting into atoms, as shown below:
For HOHgIIO•, there are no stoichiometric calculations for the photoproducts. The main product generated, HgO with a 3Π ground state, as a monomer in the gas phase (Sun et al., 2022), possesses a weak Hg–O bond of disputed magnitude (15–30 kJ mol−1, Tossell, 2006; Balabanov and Peterson, 2003; Cremer et al., 2008; Filatov and Cremer, 2004; Shepler and Peterson, 2003; Peterson et al., 2007), which is only ≤ 10 % as strong as in YHgIIO•. HgO can be reduced to Hg0 by reaction with O2 and by thermal and photo-dissociation:
The HgO + O2 reaction is exothermic but is subject to a barrier, which, using transition state theory, results in a rate coefficient of exp () cm3 molecule−1 s−1 (Saiz-Lopez et al., 2022). The enthalpy of thermal decay of HgO is only weakly endothermic and therefore favored by high temperature, with a dependence of 8.4 × 10−11 exp () cm3 molecule−1 s−1, as calculated by RRKM theory (Saiz-Lopez et al., 2022). In addition, HgO is more photolabile than •HgIOH is, with a calculated global annual mean J(HgO) of 0.54 s−1 for the troposphere (Saiz-Lopez et al., 2018, absorption spectrum in Fig. 5a). These suggest that gas-phase HgO in the troposphere is highly unstable. Although the decay slows at lower temperatures and pressures as the reaction is collisionally activated, the thermal lifetime is still only about 1 ms at 250 K and 0.1 atm. Analogous to the photolysis of HgI compounds, the quantum yield for the photo-dissociation of HgII compounds is assumed to be unity.
Figure 5Computed absorption spectra of (a) HgO (low-lying 3Π and 1Σ states) and (b) ClHgO radical (Saiz-Lopez et al., 2018; Saiz-Lopez et al., 2022). Wavelengths accessible in the troposphere are to the right of the colored area.
Thermochemistry of YHgIIO•
Experimental data on the kinetics and mechanisms of the atmospheric chemistry of YHgIIO• are scarce (the reaction BrHgIIO• + O3 to •HgIBr and O2 has been described above, Gómez Martín et al., 2022). Initially, the focus of computer simulations was on the Br analog and its reactions. Later, the scope was expanded to include the thermochemistry of the OH and Cl analogs, which will be recapitulated below. Dibble and colleagues (Lam et al., 2019b; Khiri et al., 2020; Lam et al., 2019a) concluded that the bimolecular reaction with CH4 is of primary importance for the disappearance of BrHgIIO•: BrHgIIO• + CH4→ BrHgIIOH + •CH3 (Reaction G R26). Unlike •HgIX, BrHgIIO• readily abstracts hydrogen atoms from saturated hydrocarbons and overcomes a modest energy barrier with a rate expression of 4.1 × 10 exp() cm3 molecule−1 s−1 for BrHgIIO• + CH4. Computational modeling suggested that BrHgIIO• mimics the OH radical in terms of reaction selectivity. In addition to BrHgIIO• abstract H from aliphatic hydrocarbons, it adds to the unsaturated bonds of olefins (such as the biogenic isoprene), NO (Reaction G R29), and NO2 (Reaction G R30) and interacts with CO (Reaction G R27). The addition of NO produces BrHgIIONO, which is susceptible to photolytic decomposition to predominantly BrHgIIO•, whereas the addition of NO2 promptly produces peroxynitrites of the BrHgIIOONO type, which are likely isomerized to BrHgIIONO2. Whether bromomercuric nitrate is photolabile in the troposphere is not yet known. Another source of BrHgIIOH is the reaction of BrHgIIO• with aldehydes (e.g HCHO, Reaction G R28). The pathway for the BrHgIIO• + HCHO reaction bifurcates into two processes leading to different products (Khiri et al., 2020). The dominant reaction is H abstraction, leading to BrHgIIOH and a formyl radical. The alternative route involves the addition of the oxygen atom in BrHgIIO• to the carbon center in HCHO to form a methoxy radical, which eliminates a hydrogen atom unimolecularly or in the presence of O2 to form a formate salt (BrHgIIOCHO). Secondary chemistry initiated by O2 after the addition of BrHgIIO• to a carbon double bond (such as in ethene) also involves alkoxy radicals formed after titration of the primary peroxyl radical formed by NO. The atmospheric fate of these mercuric alkoxy and alkyl peroxyl radicals (with one Hg–O bond) is similar to the general characteristics of organic oxidation in the atmosphere described in detail elsewhere (Finlayson-Pitts and Pitts, 2000). However, apart from the CH4 reaction, the interaction between BrHgIIO• and volatile organic compounds (VOCs) is considered limited in the atmosphere.
Analogous to •OH + CO, the reaction between BrHgIIO• and CO is not a simple bimolecular reaction. However, the intermediate BrHgOCO is much less stable than HOCO with respect to the release of CO2. The very weakly bound BrHgOCO promptly dissociates in •HgIBr + CO2 (Khiri et al., 2020). The above reaction is highly exothermic (>280 kJ mol−1); therefore, the •HgIBr product can be chemically activated to the extent that it increasingly decomposes into atoms. The importance of this Hg reduction channel has been identified as difficult to constrain theoretically, as the shape of the potential energy surface is unfavorable for the application of standard kinetic simulation methods. Nevertheless, using an inverse Laplace transformation method, Khiri et al. (2020) calculated the range for the rate coefficient at two temperatures: (9.4–52) × 10−12 cm3 molecule−1 s−1 at 298 K and (3.8–29) × 10−12 cm3 molecule −1 s−1 at 220 K. These data are the basis for the current inclusion of the YBrIIO• + CO →•HgIY + CO2 reaction in chemical models (Shah et al., 2021; Saiz-Lopez et al., 2022), with an average expression of 6.0 × 10 exp() cm3 molecule−1 s−1. With this numerical characterization, the YHgIIO•+ CO reaction becomes profoundly important when implemented in simulations, as it largely counteracts the effect of the •HgIX + O3 reaction, thereby extending the predicted lifetime of Hg0 in the troposphere. However, other candidates have emerged that, like CH4, may react with HOHgIIO• to form the stable Hg(OH)2 molecule, namely water vapor. The reaction HOHgIIO• + H2O → Hg(OH)2 + •OH (Reaction G R60) is nearly thermoneutral due to the stability of Hg(OH)2 ( kJ mol−1; Wang and Andrews, 2005a), and Saiz-Lopez et al. (2022) give a temperature-dependent rate constant expression of 5.3 × 10−12 × exp() cm3 molecule−1 s−1 without further details. Since both the calculated HOHgIIO• + H2O rate coefficient and the H2O(g) mixing ratio vary considerably across the troposphere, the HOHgIIO• loss due to this channel may largely exceed or fall below the more monotonic rate of hydrogen abstraction by HOHgIIO• from CH4, depending on the circumstances. The fate of HOHgIIO• is thus influenced by several exit channels (Edirappulige et al., 2023), none of which have been investigated experimentally. Particularly, the uncertainty of the CO and H2O reactions makes it difficult to determine the importance of OH-initiated oxidation to the atmospheric HgII pool.
Can mercury species nucleate in the atmosphere?
While Hg0 vapor has been observed to nucleate homogeneously in laboratory experiments conducted under high pressures (Martens et al., 1987), neither Hg0 atoms nor GOM species, which are molecular rather than ionic entities, have a vapor pressure that is sufficiently low and a concentration that is sufficiently high in the atmosphere to nucleate new particles by simple condensation (Murphy et al., 1998). However, the concerted action of a foreign gas-phase precursor (e.g., amines, highly oxygenated organics, sulfuric, nitric, and iodic acids; Lehtipalo et al., 2025; He et al., 2021) or heterogeneous condensation on pre-existing nuclei of subcritical or critical size may result in the transfer of GOM species to aerosols (Ariya et al., 2015). Measurements of individual aerosol particles have shown that a significant portion of the aerosols present in the lowest kilometers of the stratosphere contain small yet measurable amounts of HgII. Interestingly, HgII is empirically correlated with bromine and iodine in these organic-sulfate-type particles and has the highest relative concentrations in the stratosphere near the tropopause. However, HgII is rarely observed in the relatively pure sulfuric acid particles characteristic of the main stratospheric aerosol (Junge) layer (Murphy et al., 2006). While bromine and iodine aerosols are also observed throughout the troposphere, no Hg can be detected in these aerosols (Murphy et al., 2006). Both Br and I, with oceans as the primary sources, are injected into the stratosphere, where they account for most of the ozone depletion caused by halogens (Koenig et al., 2020). It is challenging to determine whether there is a causal mechanistic relationship and, if so, what can explain the observed correlation between aerosol Hg, Br, and I. Nevertheless, a plethora of clues can be utilized to assemble a coherent narrative. First, the combination of Br• (Reaction G R14a) and O3 (Reaction G R22) constitutes a significant oxidation pathway for Hg0 to HgII. However, as mentioned above, there is no firm evidence that this reaction pathway is relevant when I• is a substitute for Br•. Second, the gas-phase system I• + O3 + H2O has been identified as a substantial precursor of particle nucleation (as iodine oxoacids) and growth that is highly important within marine (Sipilä et al., 2016) and stratospheric (Koenig et al., 2020) environments. Third, the condensed phases Br− and I− act as robust complexing ligands (Table 2) for the GOM to partition into the aerosol, thereby impeding its recycling back to the gas phase. Presumably, the fundamentals are similar for a particle formation event observed in the context of the polar spring partial AMDE in East Antarctic pack ice by Humphries et al. (2015), where the formation of 3 nm particles lags the phase of gaseous Hg0 loss in the air mass. Observations over a decade in the Canadian High Arctic region clearly show that PBM transmitted by KCl-coated denuders dominated HgII fractionation over the GOM during the early period of AMDEs, where the highest frequencies of depleted Hg0 occurred between –45 and –40 °C, whereas during the late period of higher temperatures and lower particulate concentrations (AMDEs then occurred most frequently between –25 and –20 °C), HgII fractionation shifted to a clear dominance of the GOM (Steffen et al., 2014). The KCl denuder technique used cannot selectively separate nano- to submicron-sized mercuric halide clusters completely from GOM (Ghoshdastidar and Ariya, 2019), which, along with the other nonsystematic bias of the method previously mentioned, makes separation into HgII fractions tentative. The measurement methodology deficiencies make establishing empirical gas–particle partitioning schemes highly uncertain (Amos et al., 2012; Rutter and Schauer, 2007b, a). This complicates the assumption and verification of model parameterization, which relies on accurate atmospheric concentration measurements. For example, the release of HgII from aerosols into the gas phase is assumed to be entirely in the form of the tropospherically stable HgCl2 molecule (Shah et al., 2021). Coupling an oxidized Hg vapor source or a reactor where oxidized Hg is formed by gas-phase oxidation of Hg0 to particle characterization instruments (such as scanning mobility or optical particle sizers) provides conclusive evidence that mercuric halide molecules readily form clusters that undergo particle growth (Ghoshdastidar and Ariya, 2019). In experimental studies of the vapor-phase oxidation of volatile Hg forms Hg0 and (CH3)2Hg, aerosol-phase products have been detected (Raofie et al., 2008; Raofie and Ariya, 2004; Sun et al., 2016; Niki et al., 1983a; Niki et al., 1983b). For example, using a scanning mobility particle sizer, Sun et al. (2016) found that, well below the saturation pressure of HgX2, reaction products from X (Cl and Br)-initiated Hg0 vapor oxidation began to generate particles that grew from the Aitken nuclei range (few tens of nanometers) into the accumulation range (> 100 nm) over the course of a few hours (Fig. 6). Figure 7 summarizes the main elements of the Hg gas-phase chemistry in the troposphere.
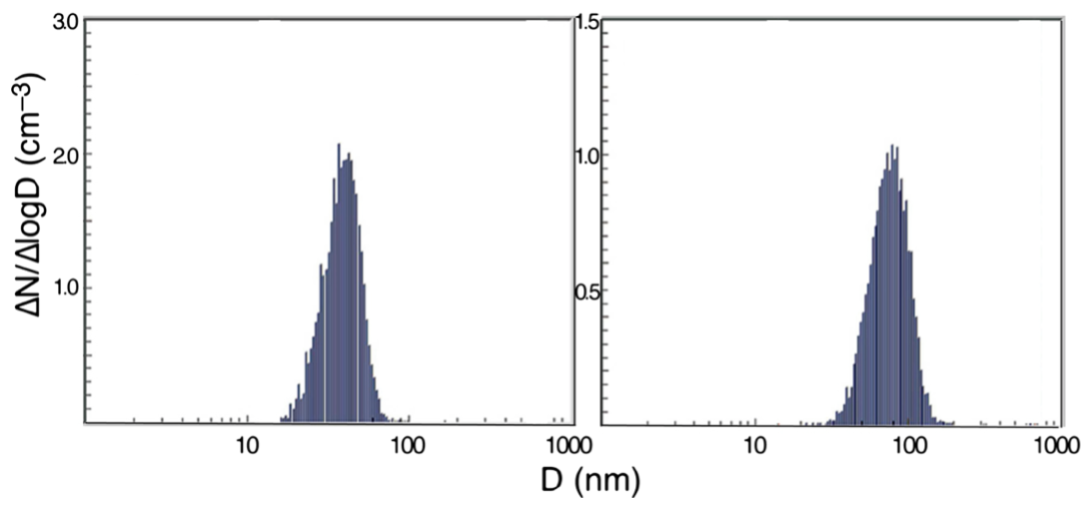
Figure 6Particle growth of the reaction products from the halogen atom (X = Cl, Br)-induced oxidation of Hg0 vapor studied after the same degree of Hg0 conversion (∼ 75 %, 5–8 ppb) but at different reaction times a and b (∼ 45 min, Hg0 + Br and ∼ 4 h, Hg0 + Cl, respectively). Adopted from Sun et al. (2016).
5.1.5 Chemical transformation of Hg in the lower stratosphere
In the lower stratosphere, chlorine atoms and hydroxyl radicals initiate most of the oxidation of Hg0. This is because the concentrations of these species increase with altitude, and the channels in which they are contained produce more photostable products, such as Hg(OH)2 and HgCl2 (Fig. 8a, b). The prediction of these model calculations that Hg0 converts to long-lived (photostable) oxidized forms and thus leads to a higher ratio is supported by hundreds of profile measurements made with an Airbus 340-600 passenger aircraft in intercontinental traffic as an upper troposphere–lowermost stratosphere observatory (Slemr et al., 2018). In addition to the frequently observed higher ratios, a steep decrease in the Hg0 mixing ratio occurs when crossing the tropopause. In the stratosphere, the latter ratio decreases to 0.25–0.7 ng m−3 (STP), measured up to an altitude of 4 km (Slemr et al., 2018). The results of both studies above show a more than 10-fold increase in the lifetime of Hg0 in the lower stratosphere compared with that in the troposphere. The chemical lifetime of Hg0 increases and approaches 10 years as the concentration in the lower stratosphere bound by the ozone layer increases (Saiz-Lopez et al., 2025).
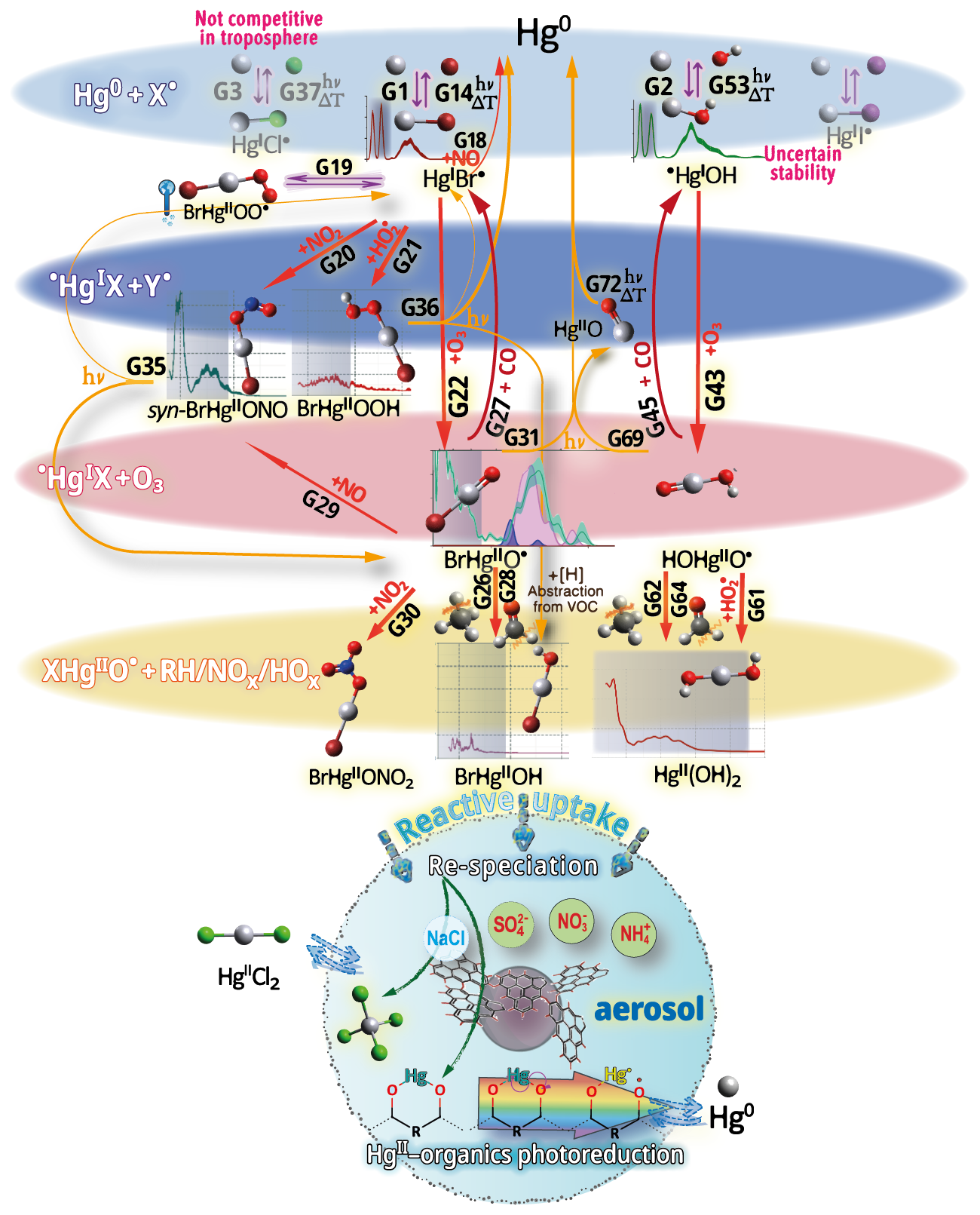
Figure 7Outline of tropospheric gas-phase Hg chemistry. The reactions of the type Hg + XO• (X = O2, Br and NO2) directly leading to mercuric species are, for the (varying) reasons given in the text, impossible processes in the homogeneous gas phase. In the troposphere, gas-phase oxidation of Hg0 in termolecular reactions is initiated by a few radicals in which Br atoms and, with some uncertainty, OH radicals have the greatest importance, leading to the formation of thermally and photolytically labile mercurous radical species (i.e., •HgIBr and •HgIOH). These HgI species are further oxidized to HgII, not only by ozone but also by radicals (in descending order of abundance), such as NO2 > HO2 > BrO ≈ OH. NO cannot efficiently oxidize HgI to HgII but instead induces thermal reduction, e.g., •HgIBr + NO → Hg + BrNO. As O3 is a closed-shell species, it directly oxidizes •HgIBr HgIOH to mercuric radical species YHgIIO•; for example, HOO• and BrO• are added to linear mercuric molecules (e.g., BrHgIIOOH) that are photolytically labile, while those resulting from, e.g., NO2, •OH, and Br• are more photostable. The photolysis of many of the major thermally more stable HgII species, such as syn-BrHgIIONO, BrHgIIOOH, and BrHgIIOH, leads to several species-specific photoproducts (potentially Hg0, HgI species or YHgIIO•) with various yields (Table 4). The remarkably thermally stable YHgO radical exhibits versatile thermochemistry, such as abstracting hydrogen from VOCs, adding to double bonds and being reduced by CO. Some of its bimolecular reactions, such as with CH4, directly form fairly stable HgII compounds such as Hg(OH)2 and BrHgIIOH. When these encounter hydrometeors, they dissolve and are re-speciated by rapid equilibration with major aqueous ligands. This leads to the formation of strong complexes, e.g., by Cl− to chloromercurates HgCl2, HgCl, and HgCl. Thus, molecular HgCl2 is released into the gas phase when the particle dries. HgCl2 is completely photostable and is enriched in the troposphere (a major HgII species), with dry and wet deposition as the only sink processes.
5.1.6 Chemical transformation of Hg in the upper stratosphere
The presence of UV-C radiation above the ozone layer maximum opens completely new reaction pathways for redox cycling of stratospheric Hg. New insights into its conceptual stratospheric chemistry (Saiz-Lopez et al., 2022; 2025) and associated anomalous isotope fractionation (Sun et al., 2022; Fu et al., 2021) have been presented. The gas-phase oxidation of Hg0 is rapid (103–104 times faster than in the troposphere, Saiz-Lopez et al., 2022) and is driven entirely by the oxidation of electronically excited Hg atoms by one of the major constituents of air, O2.
Hg0(3P) reaction with elemental oxygen
Already involved in the discovery of the element oxygen toward the end of the 18th century, the chemistry of the system of Hg + O2 has exhibited intricate complexity. These early observations, made independently in northern and western Europe, address an important aspect of the thermochemistry of the system. A direct combination of liquid Hg and O2 occurs just below the boiling point of Hg to form HgO, but the reaction is reversed above 400 °C. While the reaction of ground-state Hg vapor (Hg(1S)) with O2 is negligibly slow (Hall et al., 1995), deep UV light excitation of singlet to triplet Hg atoms (Hg(3P)) leads to significant homogeneous reactions with O2. In contrast, further excitation of Hg(1P1) in blue and subsequent reaction with O2, as discussed above, is unlikely to result in the net formation of mercury oxides. The gas-phase reactions of Hg(3P) have been studied in the laboratory since 1922 (Cario and Franck, 1922). In particular, O2/air has been used as a route for ozone synthesis since the mid-1920s (Dickinson and Sherrill, 1926). While larger quantities of ozone are produced by Hg photochemistry (photo-sensitization), the elemental vapor is oxidized more slowly, resulting in the deposition of a yellow-brown film of solid HgO on the reactor walls downstream of the irradiation zone (Volman, 1953). However, the Hg(3P) + O2 mechanism is unclear because of the controversy regarding the molecular intermediates, and whether there is a direct route from Hg(3P) to gaseous HgO or oxidation starting from the Hg(1S) state remains undetermined (Callear et al., 1959; Volman, 1953; Hippler et al., 1978; Morand and Nief, 1968). The dark homogeneous reaction Hg(1S) + O3→ HgO + O, supported by early researchers (Callear et al., 1959; Volman, 1953; Pertel and Gunning, 1959) as driving the oxidation in the photochemical experiments, can now be rejected for the reasons discussed above in Sect. 5.1.1. Considering more recent results (Wang and Andrews, 2005a; Hall, 1995), e.g., those obtained by refined computational chemistry, the following mechanism seems to be the most plausible:
Photoexcitation (Reaction G R4) has been discussed, but its reverse (Reaction G R9), i.e., the spontaneous emission of a photon that brings Hg(3P1) to the ground state, is spin-forbidden, and the radiative lifetime is relatively long (0.12 µs corresponding to k9=8.4 × 106 s−1). The quenching of Hg(3P) states (i.e Reactions G R9 and G R10–G R12a) for several gases has been studied, with Hg(3P1) atoms being 21.3 kJ mol−1 more energetic than Hg(3P0) atoms. The two main constituents of air play different roles in the quenching process, with N2 almost exclusively deactivating Hg(3P1) to Hg(3P0) with kG10=5.1 × 10−11 exp () cm3 molecule−1 s−1, while O2 quenches both Hg(3P1) and Hg(3P0) directly to Hg(1S0), with kG12a and kG11 values of 1.3 × 10−10 and 1.8 × 10−10 cm3 molecule −1 s−1, respectively. In the stratosphere (T = 240 K), the ratio is ∼ 50, suggesting that O2 is a much better physical quencher than N2, which is true throughout the atmosphere. Of primary interest here, however, is the spin-conserving Reaction (G R12b), which allows the oxidation of Hg and is overall nearly thermoneutral (exothermic by ∼ 6 kJ mol−1), yielding HgO (3Π) with low vibrational energy, as noted by Saiz-Lopez et al. (2022), which is important for increasing the lifetime of this weakly bound molecule. First tentatively identified as an intermediate in a low-temperature UV-C-irradiated matrix consisting of Hg, O2, and H2 yielding discrete Hg(OH)2 molecules (Wang and Andrews, 2005a), linear OHgIIO as the initial product is calculated to be 275 kJ mol−1 lower in energy than the reactants Hg(3P) + O2 and therefore sufficiently stable over time to participate in barrier rearrangement to Hg(1S) + O2* alongside dissociation to HgO and O. Experimental data suggest that the branching ratio between Reactions (G R12b) and (G R12a) is low, making oxidation the minor process. Sun et al. (2022) reported a quantum yield of up to a few percent for the oxidation step in experiments using synthetic air at 46–88 kPa and 233–298 K. Callear et al. (1959) observed a faster reaction in air than in O2, suggesting that Hg(3P0) may also react with O2 to form HgO, analogous to Hg(3P1).
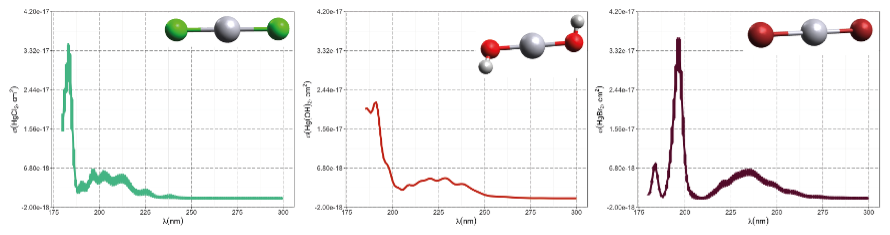
Figure 8Computational absorption spectra of HgCl2, Hg(OH)2, and HgBr2. Data from Saiz-Lopez et al. (2022) and Sitkiewicz et al. (2019).
Chemical turnover of HgO in the stratosphere: formation of HgCl2
The instability of the HgO molecule and its unimolecular decay to elemental Hg was discussed earlier. Produced in greater quantities by rapid photosensitized but nearly thermoneutral oxidation, the initially vibrational cold stratospheric HgO is more likely to survive in the colder part of the upper stratosphere until it can react further into less unstable oxidized forms. The most abundant trace gases in this part of the stratosphere are water vapor, hydrochloric acid, and ozone (Calvert et al., 2015). H2O can oxidize Hg(3P) (Gunning and Strausz, 1963; Gruß et al., 2017) and may react with HgO.
However, the reaction of Hg(3P) + H2O is so exothermic (∼ 200 kJ mol−1) that the product •HgIOH can be expected to be vibrational hot and dissociate rapidly with less time for further bimolecular oxidation. A possible reaction between water vapor and HgO is strongly exothermic if the final product is singlet Hg(OH)2 but weakly endothermic if the triplet form is formed instead. Nevertheless, there is currently no evidence to suggest that HgO can be converted to Hg(OH)2 in a direct reaction with moisture. According to Saiz-Lopez et al. (2022), the reaction between stratospheric HgO and HCl is fast enough (close to the collision limit) to allow some HgII to be converted to •HgICl rather than being reduced to elemental vapor:
As with •HgIOH and •HgIBr, the reaction between •HgICl and O3 is barrierless and rapid; in this case, ClHgIIO• is produced:
Of the versatile tropospheric chemistry presented for YHgIIO•, hydrogen abstraction (Reaction G R44) is still important in the stratosphere and is again dominated by CH4 (which is not photolyzed and reacts with the OH radical as the main sink). The product ClHgIIOH, like ClHgIIO•, is further converted by reaction with HCl to HgCl2 (Reactions G R46 and G R47), which is the most thermally and photolytically stable of the HgII molecules present. The photolytic lifetime of HgCl2 in the upper stratosphere is close to 1 h and about twice that of Hg0, so the oxidized Hg species dominate (of which ≥ 90 % is HgCl2). The HgI concentration increases rapidly above 50 km with increasing UV-C photon flux, so the ratio •HgICl HgIICl2 approaches unity at 60 km. An overview of the gas-phase Hg chemistry in the upper stratosphere is given in Fig. 9.
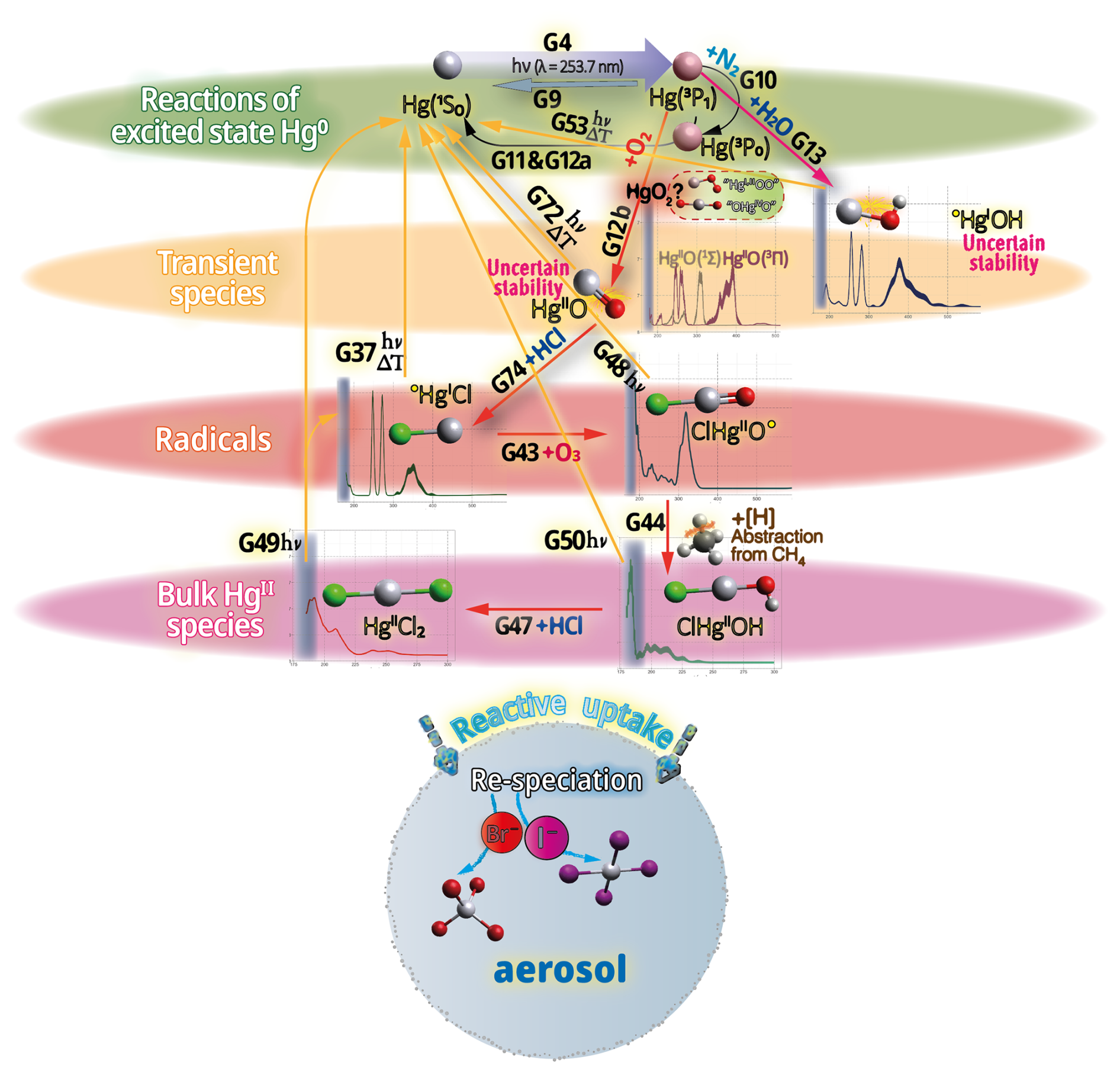
Figure 9At approximately 35 km, the stratosphere begins to contain actinic radiation, which can electronically excite Hg (at 253.7 nm), but below it is absorbed by O3 in the Hartley bands (with a maximum at 254 nm). Electronically excited Hg0 reacts primarily with O2, with one of the exit channels leading to the formation of HgO via the intermediate OHgIVO. Before HgO can fully decompose into elements, it reacts further via secondary HCl-driven fast chemistry to HgCl2, the major constituent of Hg in the middle–upper stratosphere. A 2:1 steady state between HgCl2 and Hg0 occurs because the former photo dissociates more slowly than photosensitized Hg oxidizes, both at significant rates (Saiz-Lopez et al. 2022). The reactive uptake in aerosols, followed by HgII complexation with the heavier halides, reflects the observed situation in the lower stratosphere (Murphy et al. 1998).
5.2 Organic species
5.2.1 Dimethylmercury
In addition to Hg0, “supertoxic” DMHg is another volatile Hg species that exists in nature (Siegler et al., 1999). DMHg has a boiling point below 100 °C, a high vapor pressure, and a Henry's law coefficient equivalent to that of Hg0 (Schroeder and Munthe, 1998). Both DMHg and MMHg+ species have been detected in ambient air (Lee et al., 2003; Bloom et al., 2005; Weiss-Penzias et al., 2018; Baya et al., 2015; Zhang et al., 2019a). DMHg has no known sources in the atmosphere. Its occurrence is due mainly to volatilization from surface waters, where it is transported by upwelling conditions from the deep sea, where it is formed under anoxic conditions (Conaway et al., 2009; Pongratz and Heumann, 1999). Polar sea ice harbors Hg-methylating microbes and is thought to be a source of DMHg that can be degassed as ice melts (Schartup et al., 2020). Recently, DMHg has been measured in marine air and corresponding surface water and has an air-sea gas flux that is of the magnitude of the simultaneously measured Hg0 flux (He et al., 2022). The atmospheric transformation of DMHg is the main source of atmospheric MMHg+ species (Sommar et al., 1997). DMHg vapor does not absorb actinic light (Terenin, 1934; Terenin and Prileshajewa, 1935) and is therefore not photolyzed in the planetary boundary layer, where it is only expected to be found (Sommar et al., 1996). DMHg appears to be prone to rapid gas-phase transformation and, depending on the products formed, could be an important source of atmospheric MMHg+ on a regional scale. However, in addition to MMHg+ species (Niki et al., 1983a; Niki et al., 1983b), inorganic Hg compounds (Thomsen and Egsgaard, 1986; Sommar et al., 1997) have also been reported as products of radical reactions with DMHg. Aware of its acute toxicity (Siegler et al., 1999), it has been more than a quarter of a century since any laboratory kinetic and reaction mechanistic studies of the atmospheric gas-phase chemistry of DMHg have been reported, and, in retrospect, some comments are worth making. There are three thermodynamically accessible bimolecular pathways that can potentially initiate the gas-phase transformation of DMHg, where X• below denotes a radical oxidant:
The existence of the CH3Hg radical formed in the latter reaction was tentatively demonstrated in a matrix isolation study (Snelson, 1970). The small dissociation energy of the methylmercury bond of the radical (Kominar and Price, 1969), together with a predicted barrierless CH3Hg•→•CH3 + Hg reaction (Kallend and Purnell, 1964), suggests rapid decomposition to metallic Hg without time, e.g., a reaction with O2 to form a methylmercury peroxyl radical (CH3HgOO•). In contrast, a composite reaction such as CH3HgCH , which directly produces inorganic Hg, is endothermic and, therefore, less plausible. In a high-pressure study of the gas-phase reaction between atomic F and DMHg of low atmospheric relevance, ∼ 10 % of the reacted DMHg was reported to be converted to CH3F via the above substitution reaction (McKeown et al., 1983). However, a static FT–IR study of the Cl-initiated gas-phase reaction in the presence and absence of O2 at atmospheric pressure revealed the importance of the displacement reaction that generates CH3HgCl. The remaining CH3 group is converted to CH3Cl in N2 as a bath gas in a chain reaction that regenerates Cl atoms, whereas the end products of the group in air can be attributed to the self-reaction of the CH3OO radical. The reaction CH3HgCH3 + •OH was studied with the same static method by photolysis of a mixture of CH3HgCH3, ethyl nitrite, and NO in air, which primarily followed the displacement reaction. The rate constant of ∼ cm3 molecule−1 s−1 indicates that the lifetime of DMHg in the planetary boundary layer with respect to the OH channel active during the day is a few to tens of hours. In the nocturnally active DMHg reaction, studied by fast flow discharge technique with –MS detection (Sommar et al., 1996) and a static long path FT–IR system (Sommar et al., 1997), both CH3 groups contained in DMHg react. A small but significant yield of Hg0 was detected along with a product with = 78 (CH3ONO2) after the reaction of DMHg and under fast flow discharge conditions, indicating that substitution had occurred. In the DMHg + O study, ∼ 95 % of the Hg in the converted DMHg was recovered as HgO downstream of the injector in the fast-flow experiments. In the DMHg + batch reactor study, carbon and nitrogen mass balances ruled out the formation of MMHg+ entities, and no Hg compound other than HgO could be considered an end product. Experimental evidence for the tentative intermediate CH3HgO• (indicated in Fig. 10) is lacking. The rate constant of the nitrate radical reaction (Reaction G R78) evaluated in the temperature range 258–358 K can be described by the Arrhenius expression 3.2 × 10−11 × exp[−(1760 ± 400)/T] cm3 molecule−1 s−1, and the reaction is fast enough to put the lifetime of DMHg during the night in the same time range as that for •OH-initiated degradation during the day (Sommar et al., 1997). To summarize this section, the degradation of DMHg in the atmosphere is illustrated in Fig. 10.
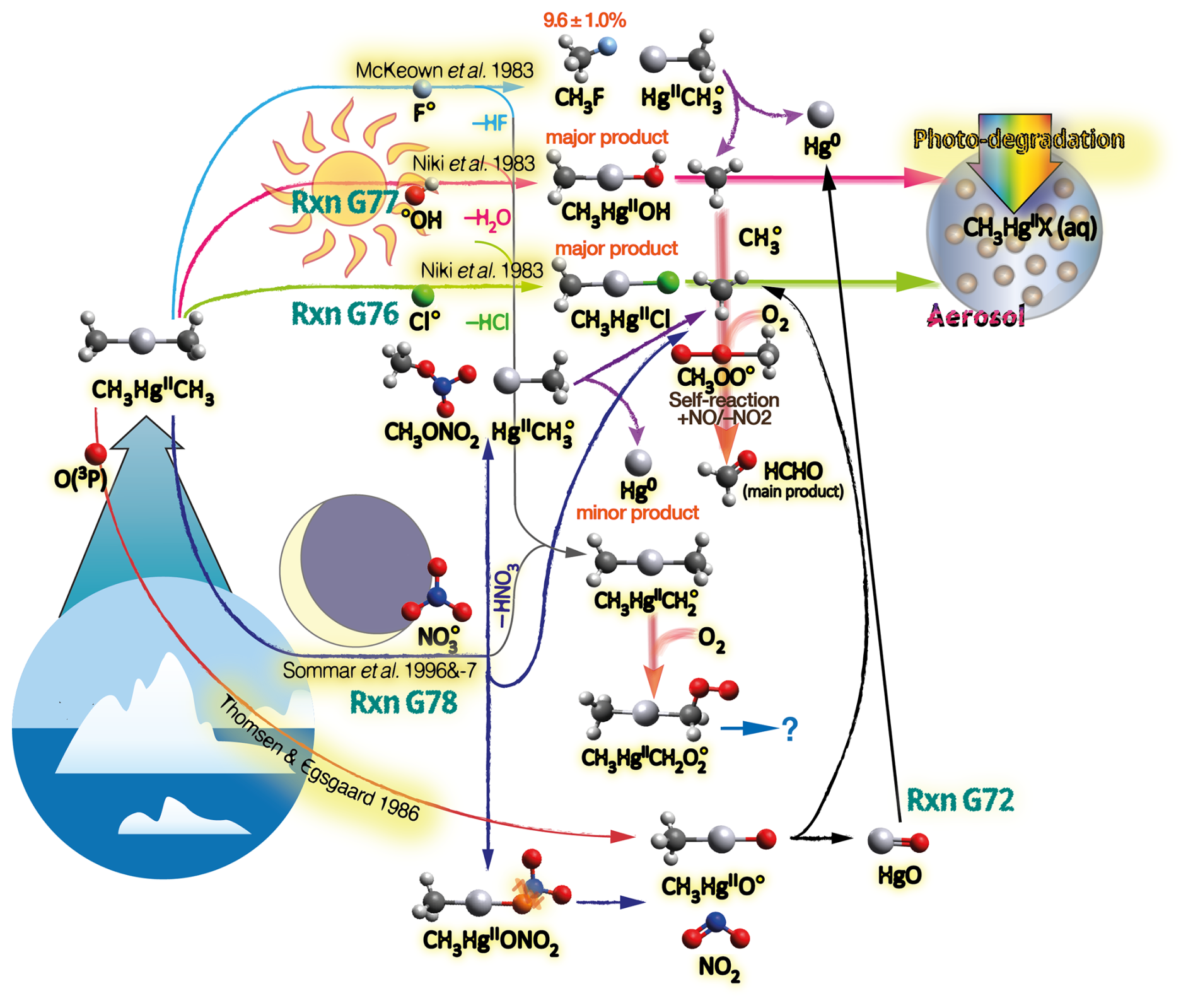
5.2.2 Monomethylmercury species
Although experimental data are lacking, gaseous MMHg+ species (e.g., MMHgCl) are expected to react with atmospheric radicals, which leads to demethylation similar to the process that occurs in the reactions of CH3HgCH3 and radicals. However, the rate constant is likely lower, and the uptake of particles is more important for the atmospheric fate of MMHg+ species.
6.1 Inorganic Hg species
Aqueous redox reactions of HgII complexes can include primary (intramolecular) processes involving direct electron transfer and secondary (intermolecular, usually bimolecular) reactions caused by reactive intermediates. Atmospheric aerosols serve as microreactors for redox Hg reactions (Lin and Pehkonen, 1999). Both oxidation and reduction occur in the aqueous phase. Since Hg2+(aq) has a rapid ligand exchange rate, the formation of HgII hydrated complexes does not limit the redox reaction rates and can therefore be treated separately as chemical equilibria. The aqueous speciation of HgII, where pH is often a critical parameter, is important for the reaction kinetics, not least for the reduction pathways. Thus, aerobic reduction pathways in principle require the formation of specific complexes, since Hg0 cannot be formed from Hg2+ by successive bimolecular (single-electron) reduction steps, since dissolved O2 instantaneously re-oxidizes Hg•+:
The overall forward rate constant for Reaction (W R11) is at the diffusion limit (k∼ 109 M−1 s−1, Nazhat and Asmus, 1973). In contrast, Hg0 can be formed by fragmentation of a ligand bound to HgII (reductive elimination, van Loon et al., 2000). Such photo- or thermolabile HgII complexes are characterized by low-energy ligand-to-metal charge transfer (LMCT) excited states, which tend to induce internal redox processes leading to oxidation of a ligand and reduction of the mercuric ion. There is evidence that Hg2+ complexes can undergo both one-electron and two-electron LMCT. An example is mercuric oxalate, where 2e-LMCT is photoinduced and occurs as part of a concerted series of electron rearrangements (heterolytic cleavage of σ bonds in the complex), resulting in the oxalate ligand being eliminated as two molecules of CO2 and the oxidation state of the metal ion decreasing by two units. This mechanism occurs without any detectable intermediates such as free radicals.
As described in Sect. 8.1 and 8.4, 1e- and 2e-LMCT reactions produce isotopic effects, the specific fractionation of which can be used to identify the reaction mechanism. In addition to the quenching of triplet complex states, the presence of dissolved O2 leads to the scavenging of radicals such as Hg•+ produced by the 1e-LMCT mechanism, resulting in reoxidation to Hg2+ (Reaction W R11, Zhao et al., 2021). As previously noted (Pehkonen and Lin, 1998), in certain laboratory experiments, such as reduction experiments, sufficiently elevated Hg concentrations are employed such that the Hg0 formed exceeds its solubility, thereby existing predominantly in a colloidal form. Table 5 below outlines the potentially significant redox reactions occurring in the aqueous phase, which are then elaborated upon in the subsequent text.
6.1.1 Oxidation channels
The mass transfer (diffusion) of gas-phase Hg0 into typical size regimes of aerosols (radius of 0.1–10 µm) does not limit the rate of aqueous Hg0 oxidation. The concentration of dissolved Hg0 in a droplet is at a steady state governed by Henry's law (Lin and Pehkonen, 1998a).
Elemental mercury
Inorganic oxidants
Reaction W R1: ozone (O3)
The presence of O3 in atmospheric water is due mainly to the scavenging of gaseous O3 ( M atm−1 at 298 K). An early study of the oxidation of Hg0 by O3 in the aqueous phase was carried out by Iverfeldt and Lindqvist (1986) using a flow system in which 70–200 ppb O3 was introduced. Their results suggested a conversion rate of 1 %–4 % h−1 when applied to atmospheric conditions. Munthe and coworkers (McElroy and Munthe, 1991; Munthe, 1992) studied the ozone reaction with the mercurous cation in an acidic solution (pH = 1–3) in a stopped-flow system and with elemental Hg using the relative rate technique (sulfite as a reference compound, pH 5.2–6.2) and obtained pH-independent rate constants of (9.2 ± 0.9) × 106 and (4.7 ± 2.2) × 107 M−1 s−1, respectively.
Table 5Aqueous-phase redox chemistry.
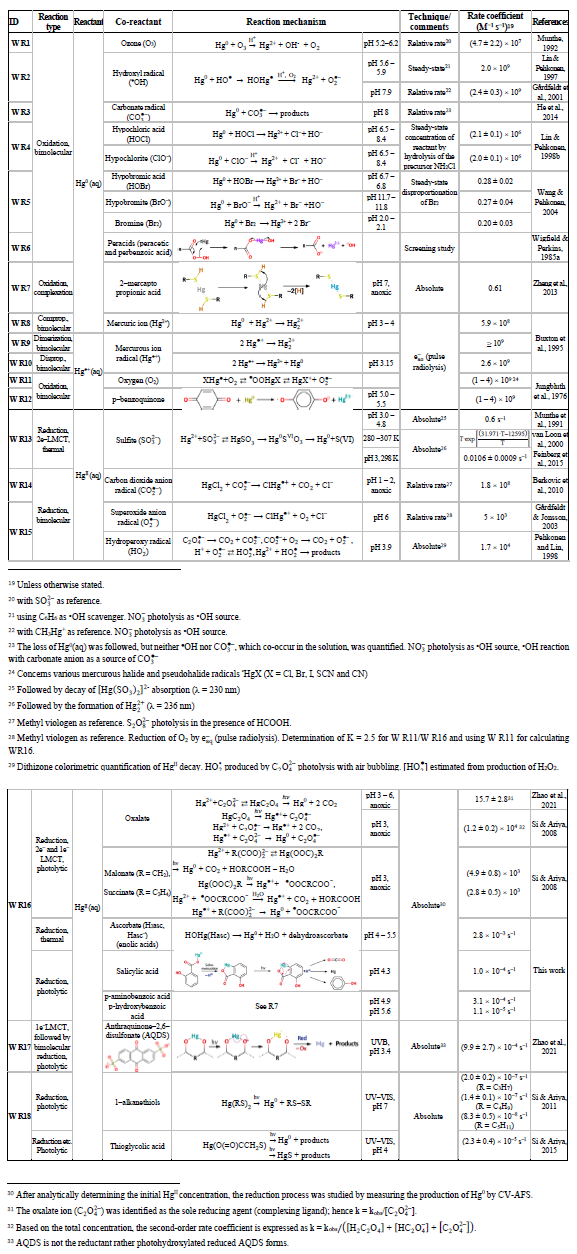
Reaction W R2: hydroxyl radical (•OH)
The OH radical in atmospheric water can come from the air ( M atm−1) or from aqueous-phase production via pathways including photolysis of H2O2, HONO, O3 and NO (Finlayson-Pitts and Pitts, 2000). The reaction rate of Hg0+•OH in the aqueous phase was determined by Lin and Pehkonen (1997) using a steady-state technique with the photolysis of NO as the •OH source and C6H6 as the •OH scavenger to 2.0 × 109 M−1 s−1 at pH 5.6–5.9. Like the first step (Hg0+•OH → •HgIOH), the second step, which is mediated by dissolved O2 (Reaction W R11), is near the diffusion limit. Gårdfeldt et al. (2001) subsequently studied the same reaction at pH 7.9 but with the reaction between CH3Hg+ and •OH as a reference but with similar results (2.4 × 109 M−1 s−1).
Reaction W R3: carbonate radical ()
In water, the carbonate system (HCO and CO) can react with OH radicals to form strongly oxidizing carbonate radicals () in fast reactions (8.5×106 and 3.9 × 108 M−1 s−1, respectively). In a comparative study, He et al. (2014) studied the disappearance of Hg0 in aqueous solutions where NO was photolyzed via UV–VIS in the absence and presence of CO at pH = 8. When both NO (0.23 mM) and CO (2.75 mM) were present in the irradiated solutions (electron paramagnetic resonance spin trapping analysis detected the presence of •OH and ), the rate of oxidation of Hg0(aq) (1.44 h−1) was 8 times faster than that observed when only NO (which produces •OH) was irradiated. The carbonate radical is a single-electron oxidant and reacts according to . In addition to identifying the carbonate radical as an effective oxidant of Hg0 dissolved in water alongside the hydroxyl radical, this study investigated the role of 1Δg O2 (singlet oxygen) as an oxidant for Hg0(aq). However, the latter species, an excited state of O2, does not initiate any measurable oxidation. Notably, the absolute rate constant for Hg0 + remains to be determined.
Reaction W R4: aqueous chlorine (HOCl/ClO−)
Aqueous chlorine is formed mainly by the scavenging of gaseous Cl2 ( 7.61 × 10−2 M atm−1 at 298 K) into the aqueous phase and the oxidation of chloride ions by •OH. Once incorporated into the aqueous phase, it dissociates to form (pKa=7.5) and Cl−, the former being the primary oxidant and increasing the solubility of total chlorine. It is a nocturnal oxidant, as both Cl2 and HOCl are readily photolyzed by solar radiation. The prospects for Hg0 oxidation by aqueous chlorine were investigated by Kobayashi (1987) and Munthe and McElroy (1992). In the former, rapid dissolution of Hg was reported when a gas stream containing Hg0 was passed through a solution containing dissolved chlorine (HClO), while in the latter, Hg(aq) was used as a proxy for Hg0, whose oxidation was observed to be “relatively fast” in a solution containing HClO. A detailed kinetic study (Lin and Pehkonen, 1998b) of the reaction between Hg0 and was carried out using a steady-state method with chloramine as a reservoir of free hypochlorous acid formed by hydrolysis: NH2Cl + H2O → NH3 + HClO. The turnover of Hg0 was studied in the pH range of 6.5–8.5 around the pKa (HClO) to investigate the influence of HClO (aq) and ClO−(aq), which were found to be closely equivalent according to the rate constants for Hg0 + HClO and Hg0 + ClO− of (2.09±0.06) × 106 and (1.99 ± 0.06) × 106 M−1 s−1, respectively. The products of both reactions (2 electrons are transferred) are chloride and hydroxide anions with a stoichiometry of 1:1 together with a mercuric cation, which rapidly forms a strong complex (logβ11= 18.0).
Reaction W R5: aqueous bromine (HOBr/BrOBr2)
Bromine has a higher (0.725 atm M−1) than chlorine does, but the disproportionation of Br2 to (pKa=8.7) and Br− is slow, and the equilibrium is shifted in favor of Br2. In contrast, Br+I is formed by the action of O3 on bromide ions and exists in the presence of Cl− largely as BrCl (Liu and Margerum, 2001). Aqueous bromine (Br2, HOBr) oxidizes Hg0 only slowly (0.2–0.3 M−1 s−1, Wang and Pehkonen, 2004). However, BrCl is likely important, as it is used as an oxidant for Hg in current analytical methods, although the kinetics have not been investigated.
Organic oxidants
Reaction W R6: peroxides
H2O2 cannot oxidize Hg0 (aq) (Kobayashi, 1987) but participates in the metal-catalyzed oxidation of Hg0,, as in Fenton's system. Fenton's reagent itself, Fe2+ + H2O2, produces OH radicals, for which Hg0, Fe2+, and H2O2 compete for oxidation. The latter reaction, H2O2+•OH, produces the HO2 radical, which propagates a chain reaction (Fenton's reaction) supported by Fe3+ acting as a catalyst to decompose H2O2 to O2 and H2O, during which a stable concentration of Fe2+ is produced as a source of •OH. Hg0 oxidation is most pronounced when the ferrous part of the Fenton reaction dominates over the ferric part, corresponding to a higher concentration of OH radicals (Liu, 2011). The –OOH functional group in organic hydroperoxides, like that in hydrogen peroxide, lacks the ability to oxidize Hg0, whereas that in peroxocarboxylic acids (peracetic and perbenzoic acid) seems to possess it, tentatively forming a mercuric carboxylate by a cyclic mechanism (Wigfield and Perkins, 1985b, a).
Reaction W R7: thiocarboxylic acids
Thiol compounds, as substituted carboxylic acids, including cysteine and glutathione, can oxidize Hg0(aq) thermally under anoxic conditions (Gu et al., 2011). For example, Zheng et al. (2013) reported that 2-sulfanylpropanoic acid in greater excess (1000:1) oxidized Hg0 at a rate of 2.18 ± 0.13 h−1. The presence of an electron acceptor (such as a quinone) further increased the reaction rate. The reaction mechanism has been described as oxidative complexation. Hg0, which is polarizable, interacts with a thiol group, leading to ligand-induced oxidative complexation in which hydrogen participates in charge transfer (Cohen-Atiya and Mandler, 2003).
Mercurous radical species (•HgIX)
Inorganic oxidants
Reaction W R11: oxygen (O2)
The reaction between Hg•+ and O2 has been studied for a variety of ligands and over a range of pH values well into the alkaline range using pulse radiolysis (Nazhat and Asmus, 1973; Jungbluth et al., 1976; Fujita et al., 1975; Fujita et al., 1973; Liu et al., 1983; Pikaev et al., 1975). Mercurous species are formed by the reduction of corresponding mercuric species by the action of solvated electrons and H atoms derived from H2O radiolysis: HgX2 + HgIX + X− and HgX2 + H•→•HgIX + H+ + X−. All types of •HgIX species react rapidly (≥1 × 109 M−1 s−1) with O2(aq): , where the equilibrium is very strongly shifted to the right. In one case (X = CN, Jungbluth et al., 1976) the reaction takes place without the clear formation of a peroxyl radical intermediate. In an air-saturated solution (∼ 0.2 mM O2), the lifetime of •HgIX is about 1 µs (Jungbluth et al., 1976).
Organic oxidants
Reaction W R12: quinones
Both and •OOHg+ are rapidly oxidized by benzoquinone (109 and 109 M−1 s−1, Jungbluth et al., 1976), which accepts an electron to form a semiquinone anion. Lalonde et al. (2001) observed that Hg0 is oxidized (∼ 0.6 h−1) in UV-B-irradiated aqueous solutions containing both benzoquinone (32 nM) and chloride ions (0.5 M) without being able to fix the mechanism.
6.1.2 Reduction channels
Mercuric compounds (HgII)
Inorganic reductants
Reaction W R13: sulfite ()
SO2 dissolves in water ( 1.36 M atm−1) to form the weak acid H2SO3 (aq), which can be deprotonated to and . The oxidation of sulfite to sulfate is rapid in the atmosphere and takes a few hours under typical oxygenated conditions in atmospheric droplets. is a soft ligand that forms strong complexes with Hg2+ (Table 2), such as HgSO3 and , the latter completely dominating under natural conditions where the sulfite content greatly exceeds that of Hg2+. The reduction of aqueous HgII by the sulfite system was first investigated by Munthe et al. (1991). is stable, whereas HgSO3 decomposes readily to Hg0 and sulfate with first-order rate constants of < 10−4 and 0.6 s−1, respectively. Scott and co-workers (van Loon et al., 2001, 2000) carried out a thorough re-examination and confirmed that the bis-sulfite complex is thermally stable but that the reduction of HgSO3, which is strongly temperature-dependent (k approximately quadruples with each 10 °C increase in temperature) and weakly pH-dependent, is more than 50 times slower than that reported by Munthe et al. (0.011 vs. 0.6 s−1 at 25 °C). The reaction mechanism is intramolecular with 2e-LMCT and heterolytic cleavage of the Hg–S bond: .
Reaction W R14: carbon dioxide anion radical ()
The carbon dioxide radical () can be formed in nature by the oxidation of carboxylic acids (see above under oxalic acid). It is strongly reducing and occurs in anaerobic environments. Berkovic et al. (2010) studied the -mediated reduction of Hg2+ at low pH by laser flash photolysis of a dilute mixture of HgCl2, formic acid, and sodium peroxydisulfate at 266 nm. The one-electron reaction is exothermic, with a rate constant of 1.8 × 108 M−1 s−1. The Hg•+ formed can only be further reduced to Hg0 in the absence of O2.
Reaction W R15: superoxide anion/hydroperoxy radical ()
(pKa 5.5) is a one-electron reductant of Hg2+ to Hg•+. Gårdfeldt and Jonsson (2003) determined the one-electron reduction potential for the pair HgCl vs. NHE at [Cl−] = 0.05 M to be −0.47 V, which, together with that for O vs. NHE of −0.155 V, gives an equilibrium constant for of at the aforementioned [Cl−]. Given that the rate constant for the reaction •HgICl + O2 is ∼ 109 M−1 s−1 (Reaction W R11), the bimolecular rate constant between HgCl2 and can be estimated to be 5 × 103 M−1 s−1. Pehkonen and Lin (1998) studied the photoreduction of mercuric ions to Hg0 with nitrate or chloride as counterions in the presence of formic, acetic, or oxalic acid at neutral (7.0) and acidic (3.9) pH values in aerated solutions. Only in the presence of oxalic acid does significant photoreduction occur, and as in the later studies by Zhao et al. (2021) and Si and Ariya (2008), an increase in the reaction rate is observed with increasing ratios of oxalic acid to HgII. The reduction is also suppressed in the presence of Cl−. Photoreduction results in an exponential increase in H2O2 formation, which is due to the presence of hydroperoxyl radicals in solution (). In retrospect (see above), this follows from the homolytic decomposition of Hg(η2–C2O4) into radicals in an aerated solution () and does not necessarily mean that can reduce HgII to Hg0.
Organic reductants
In the atmospheric environment, HgII complexation by DOM plays a pivotal role in the redox chemistry of Hg (Åkerblom et al., 2015). The chemical-reducing effect of DOM (humic substances) on HgII has been recognized for nearly 50 years (Alberts et al., 1974). These heterogeneous macromolecular ligands contain not only building blocks that can form complexes with HgII but also redox-active aromatic chromophores that can photolytically convert Hg. The fractions of DOM contributing to HgII photoreduction include fulvic- and flavin-like fractions that contain more quinone and flavin moieties than usual (Yang et al., 2020a). Furthermore, DOM contains several functional groups that can reduce complex-bound mercuric ions to Hg0 by a 2e-LMCT reaction (Table 6).
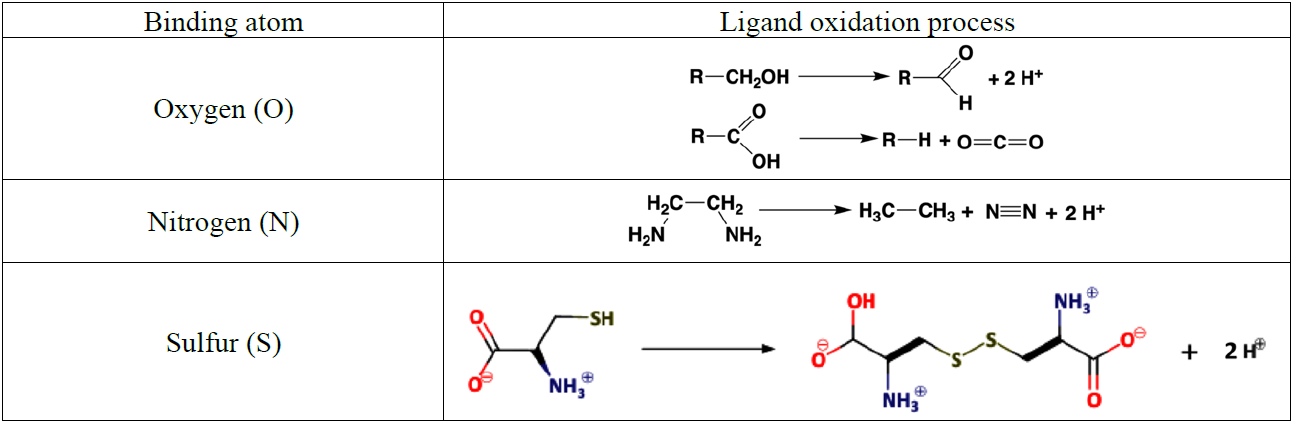
Reaction W R16: organic acids
The low-molecular-weight organic acids present in the atmosphere can reduce HgII to Hg0 in the presence of O2. These include dicarboxylic acids, ortho-substituted aromatic carboxylic acids, and enolic acids. Since 1880 (Eder, 1880), the salt of the lightest dicarboxylic acid, oxalate, has been known to reduce HgII in daylight. Oxalic acid is formed from, e.g., ethylene or acetylene, by atmospheric oxidation over several reaction cycles (chemical aging, Warneck, 2003). Mercuric ions form a complex with oxalate in a 1:1 ratio (Hg(η2-C2O4)), characterized by log β10=9.66, which is most photolabile under UV-B irradiation. Si and Ariya (2008) studied the kinetics and products of the photoreduction of HgII in a series of experiments with different concentrations of the lightest dicarboxylic acids, C2–C4, at an initial pH of 3.0 and a temperature of 296 ± 2 K, while the kinetic, product, and isotopic study of Zhao et al. (2021) involved the system HgII + oxalic acid with as a counterion in the pH range of 2.7–6.3 and a small temperature range of 295–303 K. The pH-resolved experiments show that in the , , and H2C2O4 system, only the oxalate ion reduces Hg2+, with a of 15.7 ± 2.8 M−1 s−1 at 295 ± 1 K. Si and Ariya (2018) reported a much larger bimolecular rate constant between Hg2+ and the total oxalic acid concentration of 1.2 × 104 M−1 s−1 at pH 3.0. The magnitude is surprisingly large and is comparable to the rate constant between Hg2+ and HO radicals (see below). When this higher rate constant, which is based on the total oxalic acid concentration, is implemented in regional air quality models, the impact is significant (Bash et al., 2014). However, with respect to the reaction mechanism, there is more consensus that it follows a branched route. Hg(η2–C2O4) undergoes photolysis followed by partial reductive elimination in one step (insensitive to the presence of O2): Hg(η2-C2O Hg0 + 2 CO2 and, in part, homolysis of a Hg–O bond, which initiates a chain reaction: Hg(η2-C2O4) . Hg0 should form from the reaction of Hg•+ with the bulk ligand , where Hg•+ is reformed from the reaction between bulk Hg2+ and the oxalyl () or carbon dioxide anion () radical. The reduction to Hg0 in the chain reaction is inhibited by O2, which reacts rapidly with both and and re-oxidizes Hg•+ to Hg2+. Like dicarboxylic acids, aqueous solutions of aromatic ortho- and para-substituted carboxylic acids exposed to UV-B can oxidize Hg2+ to Hg0 via elimination of CO2, and Hg Hg0 photoreduction is attenuated but not completely inhibited by the presence of dissolved O2 and competing counterions. Previously, He et al. (2012) studied the aqueous photoreduction of Hg2+ coupled with a series of aromatic carboxylic acid derivatives in the absence of O2 at pH 4.3 and suggested that the reaction proceeded via a radical mechanism. However, studies of the same reactants in our laboratory (unpublished results, shown in Table 5) have shown that Hg0 is formed even in the presence of dissolved O2, suggesting the existence of an additional non-radical reduction pathway. We propose that this channel requires solvo-mercuration to an arylmercurial intermediate followed by photolytically induced 2e-LMCT as part of a concerted series of electron rearrangements, including cleavage of a Hg–C bond yielding Hg0, CO2, and a decarboxylated aromatic as end products. Let us take p-aminobenzoic acid as an example.
Photo-reduction has also been observed in the presence of dissolved O2 when Hg2+ is bound mainly to the amino acid serine (HSer), similar to HgSer2 (Motta et al., 2020b), which can be explained as the result of reductive elimination with CO2 and 2-aminoethanol as by-products in addition to Hg0 and involves an intermediate with a photolabile Hg–C bond (Zhao et al., 2021). In cysteine-mediated photoreduction (Motta et al., 2020b; Zheng and Hintelmann, 2010b), the ligand is converted from a thiol to a disulfide (Table 6). Ascorbic acid, as a representative enolic acid, can readily reduce inorganic divalent Hg in aqueous solutions to Hg0. Studies in our laboratory have shown that the reaction is thermal and not affected by actinic light. When ascorbic acid is in excess () relative to Hg2+, the reaction rate is not significantly affected by increasing ascorbic acid concentration. The reaction rate is highest in the pH range where the hydrogen ascorbate ion (HAsc−) is dominant and the hydrolysis of Hg2+ is not complete, i.e., typical pH values for atmospheric hydrometeors (≤ 5.5). Presumably, HgOH+ (aq) forms a reactive complex with HAsc−, Hg(HAsc)+, which is labile to the elimination of water in a heterolytic process, forming Hg0 and dehydroascorbate as the final products. Enols act as atmospheric intermediates, and it is unclear whether they are present in high concentrations, which makes them interesting reducing agents for atmospheric Hg2+. In any case, is relatively high (∼ 0.17 min−1, Reaction W R16c, Table 5).
Reaction W R17: hydroquinones and polyphenols
The quinonic (Zheng et al., 2012) and fulvic (Yang et al., 2020a) units in DOM act as key redox centers. How this happens at the molecular level is being investigated by studying model compounds that contain redox-active groups but lack other functional groups (Zhao et al., 2021). The simplest quinone forms a redox pair with the corresponding hydroquinone in the half-reaction.
Combined with the half-reaction in Reaction (R3), this gives a ΔE0 > 0 for Hg2+ + C6H4(OH)2→ Hg0 + C6H4(=O)2 + 2 H+, i.e., thermodynamically feasible. Relatively slow reduction of Hg2+ to Hg0 by hydroquinone occurs in the dark in a dilute aqueous solution (8.2 ± 2.4) × 10−5 s−1). These results are consistent with a reaction mechanism involving a hydroxyphenoxymercuric complex or via ipso-mercuration followed by electron shuttling and elimination of Hg0 and H2O.
The aqueous photochemistry of quinones is complicated and can involve both ground and excited state reactions as well as free radicals (Görner, 2019). With respect to the interaction of benzoquinone with Hg under actinic light, one study revealed significant oxidation (∼ 0.6 h−1) of Hg0 in Cl−-enriched water (see above, Lalonde et al., 2001), whereas another study reported photoreduction of HgII→ Hg0 of about the same magnitude (∼ 0.8 h−1) under anaerobic conditions and in the absence of strongly complexing inorganic ligands (Zhao et al., 2021). An anthraquinone (AQ) derivative (AQ-2,6-disulfonate) is an effective electron shuttle that facilitates electron transfer from metal-reducing bacteria (MRB) to HgII (Lee et al., 2018), as well as from Hg0 (aq) to organic thiols (R-SH) during oxidative complexation to form Hg(SR)2 (Zheng et al., 2013). Zheng et al. (2013) reported that AQDS(aq) alone is unable to oxidize Hg0 or reduce HgII under dark and anaerobic conditions. AQDS-assisted biotic HgII reduction by the MRB Shewanella oneidensis MR-1 is associated with negative charge scavenging, which temporarily increases the content of reduced AQDS species, such as AQH2DS and semiquinone radicals (Lee et al., 2018). The reduced species AQH2DS alone is a potent reductant of HgII in the dark. On the other hand, HgII is efficiently reduced to Hg0 in a UV-B-irradiated aqueous solution containing dissolved AQDS (∼ 10−3 M−1 s−1). The reactive species is tentatively photohydrated AQDS (AQH2(OH)DS), which interacts with HgII by forming a photolabile bidentate O–coordinated mercuric complex. In conjunction with a strong isotope effect (Sect. 8.4), the photoreaction is likely to occur via a paramagnetic intermediate (a mercurous semiquinone biradical complex). The reaction rate decreases to ∼ 0.2 h−1 in the presence of dissolved O2 (Zhao et al., 2021). HgII interacts with ortho-QH2 moieties such as those in the natural polyphenols of humic substances and tannins (Jerzykiewicz, 2013). A direct reaction yields redox-active HgI complexes with ligands of semiquinone radical character that may eventually decompose into Hg0 (Jerzykiewicz et al., 2015). Reaction kinetic and mechanistic studies that are more applicable to the environment are not available.
Reaction W R18: thiols
Hg2+ and CH3Hg+ bind extremely strongly to heavier hydrochalcogenide groups (such as RSH and RSeH) and other corresponding groups of reduced chalcogenides, such as sulfides and disulfides (Skyllberg, 2011). Most relevant is that both inorganic (e.g., H2S, CS2) and organic (CH3SH, CH3SCH3) low-molecular-weight reduced sulfur compounds have short lifetimes (Warneck, 1988) and therefore have no effect on aqueous Hg speciation. It is questionable whether reduced sulfur/thiol groups associated with macromolecular organic compounds in aerosols influence internal Hg speciation. The photoreduction of divalent Hg by lighter aliphatic thiols is slow (< 10−7 s−1, Si and Ariya, 2011), whereas that by thioglycolic thiols is slightly faster ( s−1, Si and Ariya, 2015) but hardly significant in the atmosphere.
6.2 Organic mercury
6.2.1 Demethylation channels
Biogenically produced organo-Hg in the environment is almost exclusively methylated Hg, although there are few reports of the presence of ethyl Hg (Wu et al., 2023b), which must be derived from a natural source. However, only methylated Hg has been detected in air. As mentioned above, DMHg is a major source of MMHg+ compounds in the atmosphere through gas-phase degradation. Gaseous MMHg+ species (Lee et al., 2003) can potentially react homogeneously to inorganic Hg, but as MMHg+ species are only semi-volatile and have a high , they are more likely to be rapidly absorbed on aerosols. MMHg+ species have been detected in cloud water (Li et al., 2018; Weiss-Penzias et al., 2018), fog water (Weiss-Penzias et al., 2012), rainwater (Conaway et al., 2010; Won et al., 2019), and snow (St Louis et al., 2007). Photolytic demethylation of dissolved DMHg occurs in pure water (Chen et al., 2024) incubated with sunlight (, ∼ 0.32 ± 0.07 d−1, West et al., 2022). Acidolytic demethylation of DMHg to MMHg+ species is of very minor importance and occurs only at low pH (Maguire and Anand, 1976; Wolfe et al., 1973). A theoretical study of CH3HgOH and CH3HgOH, which dominate the speciation of MMHg+ in natural waters without significant levels of Cl− and reduced sulfur ligands, including DOM, indicated that CH3HgOH can be excited to the triplet state by sunlight and that this state dissociates into CH3 and HgI radicals (Tossell, 1998). A room-temperature study of the photo-degradation of CH3HgOH (aq) when irradiated by a Xe lamp with filter blocking wavelengths < 290 nm reported a rate constant of (2.2 ± 0.2) × 10−4 s−1 (Gårdfeldt et al., 2003). Rapid indirect demethylation of MMHg+ species by a bimolecular process with the OH radical occurs at the limit of what diffusion allows ((9.83 ± 0.66) × 109 M−1 s−1, Chen et al., 2003). In natural water, select reactive oxygen species, such as singlet oxygen (see above, Suda et al., 1993; Zhang and Hsu-Kim, 2010), have been suggested to cause HgII demethylation, but their reactivity has not been directly quantified. Instead, its presence has been suggested based on the results of added scavenger/promoter tests, some of which may yield misleading results for some water compositions (Han et al., 2017). Chen et al. (2003) concluded that OH-initiated demethylation is comparable to the rates of MMHg+ photodegradation reported in situ in natural waters. These researchers reported that, in addition to inorganic HgII, Hg0 was a by-product of OH-initiated degradation in an O2-saturated system, presumably by homolytic substitution.
6.2.2 Methylation channels
The paucity of empirical data renders the budgets of tropospheric MMHg+ species highly uncertain. A recent estimate of the MMHg+ pool size is 5.5 Mg, associated with a lifetime of 1.9 d, of which one of the major sources is inferred to be in-cloud methylation (Wu et al., 2024b). The potential for atmospheric biotic methylation is considered limited, despite the presence of pathogens and bacteria in aerosols and hydrometeors, because Hg-methylating microbes (possessing two important methylation genes, hgcA and hgcB, Parks et al., 2013) usually thrive in anaerobic environments, in contrast to the distinctly oxic environment of atmospheric waters. However, many unknowns about the potential for HgII methylation under oxic conditions need to be resolved (Sonke et al., 2023). There have been extensive studies on the abiotic methylation of Hg2+ (Ullrich et al., 2001). Methylating agents that are important for MMHg+ formation in the atmosphere are oxygenated hydrocarbons containing a methyl group (Yin et al., 2012; Hammerschmidt et al., 2007). Some of them have properties that allow competitive photochemical reduction and methylation of Hg2+ (Yin et al., 2012). Earlier studies have investigated photochemical Hg2+ methylation by deep UV irradiation (Yin et al., 2012; Akagi et al., 1974; Hayashi et al., 1977), making it impossible to generalize these results to the lower atmosphere. The formation of MMHg+ species was observed in the dark in dilute HgII solutions (1 nM) containing an excess of acetic acid (100:1 M M), with an apparent first-order rate constant of 5.4 × 10−6 s−1 in artificial rainwater (pH 4.9 Gårdfeldt et al., 2003). When the system is exposed to sunlight, photo-demethylation occurs, which counteracts MMHg+ formation mediated by acetic acid/acetate, and within hours, the MMHg+ concentration reaches a steady state (∼ 2.5 % of inorganic HgII). Hammerschmidt et al. (2007) noted that the average ratio of MMHg+ to reactive HgII measured in North American continental precipitation (2.5±0.6 %) agrees with the findings of the above laboratory study. Methylation takes place intramolecularly in the acetatic–mercuric complexes present in solution concerted with decarboxylation (Gårdfeldt et al., 2003; Yin et al., 2012; Akagi et al., 1974): .
Multiphase transformations address dynamics and chemistry at interfaces and media, such as aerosol particles and cloud droplets, which interact heterogeneously with gases and solute species. Despite a wealth of studies addressing the multiphase chemical or physical transformation of Hg under processes such as those under simulated post-combustion conditions, which undoubtedly pertain to interactions with certain environmental surfaces, the findings offer limited insight into the surface and heterogeneous atmospheric Hg chemistry. The subsequent chapter addresses the studies that have been identified as contributing meaningfully to the advancement of understanding in this domain.
7.1 Gas–particle partitioning and reactive gas uptake
The behaviors of gaseous Hg0 atoms and HgII molecules in interacting with the atmospheric condensed phase differ. The dominant Hg0 pool has limited water solubility, and the uptake of Hg0 vapor to aerosol surfaces is low, to the limited extent that it has been investigated. Gas-phase HgII molecules, “GOM”, have several orders of magnitude greater than that of Hg0, favoring the liquid phase. The heterogeneous processes that allow GOM to be adsorbed reversibly or irreversibly, modified by ligand exchange, or dissociated to Hg0 by reduction on surfaces, are key parameters that need to be characterized to appropriately parameterize chemical transport models.
7.1.1 HgCl2
Understanding the transformation from GOM to PBM through gas-phase processes (condensation, Sect. 5.1.4) and aerosol surface interaction (Sect. 4.2) is crucial for parameterizing deposition. Since the separation of GOM from PBM with current methods is tentative, the accuracy of studies of HgII distribution between the gas and condensed phases, performed by preconcentration in laboratory experiments with nebulized aerosols (Rutter and Schauer, 2007b, a) and in the field (Amos et al., 2012), is retrospectively ambiguous. Fitting observational data to an equilibrium according to a van't Hoff-type relationship is conducted in models to calculate the volatilization of GOM from atmospheric aerosols, where Kgp (Eq. 9) is weighted by the inverse of the mass concentration of fine particulate matter (PM2.5; Shah et al., 2021). The partitioning expression does not consider that the interaction between GOM and a surface is significantly influenced by the composition of the surface layer. HgCl2(g) partitions among particles consisting of typical alkali metal salts such as chlorides, nitrates, and sulfates (Mao et al., 2021; Malcolm et al., 2009). To compensate, global Hg models treat the uptake of GOM onto sea salt particles separately as an irreversible first-order process parameterized by wind speed and humidity. The equilibrium studies conducted at atmospheric pressure do not provide insights into the dynamics of the system, as the experiments are limited by mass transport, which negates the possibility of obtaining quantitative information on reactive uptake. As an alternative (Liu et al., 2022), partition coefficients have been calculated for individual GOM species based on theoretical predictions of both adsorption and absorption (Wu et al., 2024a). The reactive uptake of HgCl2(g) on surfaces representative of inorganic and organic primary and secondary atmospheric aerosols has recently been studied via the fast flow technique coupled with an ion drift chemical ionization mass spectrometer (ID-CI-MS). The reported data (Mao et al., 2021; Khalizov and Mao, 2023) are summarized in Table 7.
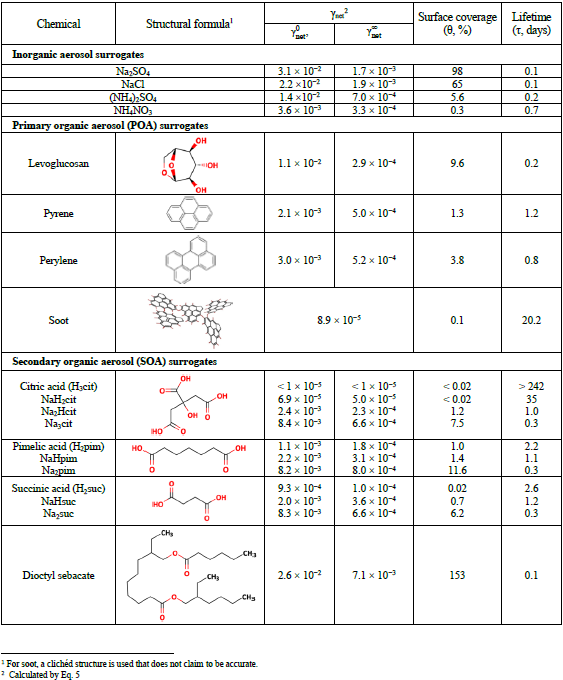
The data in Table 7 are for dry surfaces, where is the initial uptake coefficient, which is relevant throughout the lifetime of the aerosol, as the surface coverage by atmospheric HgCl2 remains unchanged and low. In the presence of sea salt aerosols (> 0.6 µm, initially at pH 8) that dominate in marine air, where NaCl represents > 95 % of its mass, the lifetime of HgCl2(g) is expected to be between 4 and 20 h depending on aerosol loading (Mao et al., 2021). When the relative humidity exceeds ∼ 75 %, a hygroscopic sea salt droplet is formed as the salt deliquesces, and a highly mobile surface phase in which HgII is equilibrated in ionic form as may contribute to more rapid GOM loss in marine air (Holmes et al., 2009). Ammonium salts such as nitrates and sulfates are primarily found in secondary particles, typically in urban and agricultural–rural air. Although HgCl2 uptake is lower here, its lifetime is comparable because of the higher particle number and the large surface area they generally represent. These semi-volatile ammonium salts do not occur in isolation but coexist with oxygenated organics formed through photochemical activity, resulting in the formation of secondary aerosols, which constitute the primary fraction of the atmospheric burden of organic aerosols (OAs, Jimenez et al., 2009). The acidity of secondary organic aerosols (SOAs), a dominant component of PM2.5, affects HgCl2 uptake by controlling the acid–base equilibria of characteristic chemical species such as aliphatic dicarboxylic acids, aromatic polycarboxylic acids, and other oxygenated multi-functional organics in aerosols. For the diprotic acids in Table 7, the reactivity becomes noticeable only after the first deprotonation step at pH 4.5–5.5. For the triprotic citric acid, activation occurs after the second step at pH 6.5. The adsorption of HgCl2 on primary organic aerosol (POA) surfaces is significant in the presence of levoglucosan, an anhydrosugar, which is a fingerprint of fire activity. Nevertheless, the interaction between HgCl2 and polyaromatic hydrocarbons (PAHs) derived from carbonaceous fuel combustion is more constrained, occurring between the electrophile HgCl2 and the π electrons delocalized over the aromatic fused ring skeleton. The observed adsorption on fresh soot, which is porous and graphitic with a high specific surface area, is more than 1 order of magnitude lower than that for the minor type of PAH studied (pyrene, perylene). If morphology affects uptake, so does the state of the surface phase, as a diester of sebacic acid (a close homolog of pimelic acid), octyl sebacate, a lubricant, is more reactive to HgCl2 than the microcrystalline pimelic acid film is. The adsorption of HgCl2 on mineral surfaces (dust aerosols) represented by iron (hydr)oxides has not been studied experimentally, but calculations indicate a partition coefficient (Kgp) for α-Fe2O3 that exceeds that for NaCl by 3 orders of magnitude (Tacey et al., 2018b). The studies listed in Table 7 were performed without observing redox chemistry (i.e., no Hg0 was detected to be emitted from the HgCl2-exposed surfaces when heated to 120 °C), but a combined study using FF-ID-CIMS and Raman spectroscopy revealed that exchange reactions between gaseous mercuric compounds are catalyzed by surfaces such that HgCl2 and HgBr2 molecules in the presence of a deactivated surface produce mixed BrHgCl molecules (Mao and Khalizov, 2021), which are also volatile. Owing to rapid exchange reactions, the prospect of accurately speciating GOMs by pre-concentration on filters and cation exchange membranes, as discussed previously (Sect. 3.1), is unlikely.
7.1.2 Hg0
A challenge in studying gas-phase- or liquid-phase-initiated reactions is the potential for side reactions and phase changes to occur during experiments. Thus, a portion of the loss of gas-phase Hg0 in laboratory experiments designed to study homogeneous oxidation (e.g., by O3, Snider et al., 2008; , Sommar et al., 1997) has been linked to a heterogeneous rate component (ksurf) occurring on new surfaces that form during experiments (product clusters undergoing particle growth in free suspension, Sect. 5.1.4) and/or on initially deactivated existing surfaces (reactor walls) that begin to catalyze Hg0 surface oxidation as deposits form (Sommar et al., 1997; Medhekar et al., 1979). For example, in a series of spherical reactors with varying surface-to-volume ratios (), Pal and Ariya (2004b) reported the loss of Hg0 by reacting with excess O3 in N2 as follows:
where kgas (cm3 molecule−1 s−1) is the gas-phase reaction rate, (cm−1), ksurf (cm4 molecule−1 s−1) is the surface rate loss, and [O3] (molecules cm−3) is the gas-phase O3 concentration. In the range of 0.28−0.93 cm−1, knet increased by 30 % simultaneously with the formation of particles (Snider et al., 2008) during the experiments, which started homogeneously. Using a fluorocarbon film smog chamber (9 m3, = 0.03 cm−1), Rutter et al. (2012) studied the influence of SOA (yielding an ∼ 100-fold increase in the surface area of the system) and secondarily formed •OH (at ambient level due to added scavenger) generated from an irradiated mixture of O3 and various biogenic and anthropogenic VOCs (at a level ∼ 1 order of magnitude greater than ambient) on the oxidation of Hg atoms (at a level ∼ 2 orders of magnitude greater than ambient). Neither Rutter et al. (2012) nor subsequent researchers (Lyman et al., 2022) have been able to identify evidence that interactions with photochemical smog particles significantly contribute to the oxidation of Hg0. Nevertheless, few studies concerning Hg0 uptake have been conducted with a sufficiently rigorous standard, employing techniques used in specific studies of heterogeneous processes to produce a kinetic formalism that can be related to atmospheric models. These studies, which were conducted with a coated-wall laminar flow tube reactor, focused on the light- and moisture-dependent uptake of Hg0 (detected by CV-AFS), which may be photocatalytic, on the major metal oxides (TiO2, Fe2O3, FeOOH, and Al2O3) present in mineral dust aerosols (Kurien et al., 2017; Lee et al., 2022). The first three metal oxides have semiconductor properties with band gaps that allow photoexcitation in the UV-A (≤ 395 nm) and visible (≤ 590 nm) regions, while Al2O3, the second most abundant mineral oxide in the Earth's crust after SiO2, is an insulator but has some thermal conductivity. It has been established for over half a century that Hg0 vapor in the presence of O2 over an irradiated TiO2 surface is consumed by reactive uptake (Kaluza and Boehm, 1971) via the following tentative mechanism:
When excited by light of a wavelength shorter than the band gap energy, the generation of electron–hole pairs (, ) occurs in the conduction and valence bands (Reaction R10a). The electrons and holes transported to the particle surface initiate redox chemistry by reacting with H2O and O2 molecules to form reactive oxygen species (ROS, Reactions R10b, c). The oxidation potential of exceeds +2.27 eV in the TiO2, Fe2O3, and FeOOH cases, which is sufficient to generate hydroxyl radicals from surface water (Reaction R11c) that can oxidize adsorbed Hg0 (Reaction R10d). The reported uptake coefficients are in the range of to > 10−4 (based on the Brunauer–Emmett–Teller surface area), with relative reactivities of Fe2O3 FeOOH < Al2O3 < TiO2, where without irradiation is below the detection limit. The uptake of Hg0 on iron (hydr)oxides is less than 10−8 under both UV and visible light and is inhibited by humidity, as is the case for Al2O3, which shows measurable uptake under UV irradiation ( = 1.2 × 10−8). The photo-initiated uptake of Hg0 on TiO2 is significant, especially under UV light at low humidity ( > , diffusion-controlled limit). However, as with Al2O3, it shows reversibility (desorption of Hg0) in the presence of water vapor during darkness (Lee et al., 2022), whereas Hg0 exhibits almost irreversible binding to iron (hydr)oxides at the temperatures studied (<150 °C, Kurien et al., 2017). Based on limited published data, only under conditions of low humidity and very high mineral dust aerosol loading can the uptake of Hg0 be considered to have any effect on the atmospheric cycling of Hg0. Notably, there are no corresponding experimental data for HgCl2 uptake on mineral dust surrogates.
The uptake of Hg0 on ice, which involves the migration of radioactive Hg isotopes into ice spheres in a packed bed flow tube exposed to a strong temperature gradient, can be described as reversible adsorption without significant solvation. The observations were in accordance with a Langmuir isotherm, where the adsorption equilibrium can be described thermodynamically by Bartels-Rausch et al. (2008):
where K is the Langmuir absorption constant (Eq. 8), R is the gas constant, T is the absolute temperature, and and are the enthalpy and entropy of adsorption, respectively. Compared with for Hg0 (0.18 at 5 °C), the Langmuir adsorption coefficient on ice, which is expressed in a dimensionless way, is much smaller even at temperatures lower than the freezing point of the metal (2.2 × 10−5 at 220 K), which is most relevant for polar regions and the upper troposphere. Therefore, in both atmospheric and polar environments, the uptake of Hg0(g) on ice surfaces is negligible.
7.2 Reduction of mercurial species on surfaces
Computational chemistry studies report that the adsorption of mercuric halides on dry-salt- or mineral-like surfaces reduces the energy required for reduction to Hg0 (Tacey et al., 2016) and that the reduction of HgCl2 and HgBr2 to Hg0 on iron oxide aerosols requires the presence of actinic light (Tacey et al., 2018a). Breaking the first Hg–X bond is possible either thermally or photolytically, while the second requires photons with λ≤ 461 nm. To release Hg0 from the surface, an excitation energy of 2.59 eV (λ≤ 479 nm) is required in a photo-induced charge transfer process between the surface and the adsorbate.
The photoreduction of particle-bound HgII has been the subject of experimental investigations (Tong et al., 2013; Tong et al., 2014). In these experiments, aerosol surrogates doped with HgCl2 were generated and dried in laboratory air and subsequently captured on filters, which were then exposed to light with three spectral options in a flow-through reactor. Photoreduction of NaCl aerosols occurs under actinic light (both UV and visible light, with approximately 2.5 % and 2.0 % of HgII reduced, respectively, during a 30 min exposure, normalized per 100 W m−2 irradiation). However, the presence of iron species (mainly FeIII rather than FeII) has been observed to exert some inhibitory effects (Tong et al., 2013). In contrast, photoreduction on carbon-based synthetic aerosols has been demonstrated to be more significant but also more variable. For example, HgII on adipic acid aerosols is reduced by 8 % (per same time unit and normalization as above), while on levoglucosan, it is less than 2 % (Tong et al., 2014). Notably, however, these experiments were carried out without O2 in the carrier gas stream.
The reduction of HgII in ice in the presence of organics has been studied in an ice-coated flow tube at atmospheric pressure under irradiation with light between 300 and 420 nm (Bartels-Rausch et al., 2011). O2-free ice matrices containing 60 nM Hg were doped with a stoichiometric excess (up to 50:1 M M) of either benzophenone (a strong photosensitizer), oxalic acid oxalate (forming photolabile HgII complexes), or humic acid (ditto photolabile complexes), which, upon irradiation, accelerated the release of Hg0, which was most rapid in the presence of benzophenone at high pH. The presence of O2 (20 % in the gas stream), the introduction of sea-ice-like conditions, or a large drop in temperature (from 270 to 250 K) or pH (to 4) resulted in diminished photoreduction. The mechanism by which HgII reduction is sensitized by benzophenone is challenging to ascertain. One potential mechanism involves the dissociation of an excited state of the major species, Hg(OH)2, which has been reported to be photolabile as a solute in water (Xiao et al., 1994). A controlled laboratory study of light-irradiated natural snow samples at a temperature of −10 °C revealed that the release of Hg0 follows first-order kinetics with a coefficient between 0.18 and 0.25 h−1, corresponding to a natural lifetime of 4–5.6 h (Dommergue et al., 2007). However, no monitoring of HgII in the condensed phase has been conducted. Given that light does not penetrate the entire snowpack, it can be assumed that a HgII gradient toward depletion at the top is established.
Brominated mercurials that are present in the Arctic environment during AMDE may play a role in light-induced Hg re-emission from the cryosphere to the atmosphere (cf. Fig. 2). A computational study (Carmona-Garcia et al., 2025) suggested that, compared with HgBr2 in the gas phase, HgBr2 in solution has an increased absorption cross section for wavelengths longer than 290 nm, whereas bromomercurate anions (Hg and ) have a comparatively greater absorption in actinic light. The low-energy excited states of HgBr2, Hg, and in solution are characterized by electronic transitions in which the electron density is mainly transferred from the Br atoms to the Hg atom, indicating a significant photoreductive character upon light absorption, leading to the generation of HgI species (•HgIBr, •Hg, and ) and a bromine atom. The photoreductive character is also recognized for the aforementioned HgI species in their electronically excited states, which plausibly dissociate via an LMCT mechanism with Hg0 as the product. The predicted peak photolysis constants for the polar spring (March, ∼ 80° N) are 3.9 × 10−6, and 7.9 × 10−5 s−1 for HgBr2, Hg and , respectively.
For pure heterogeneous reduction, there is experimental evidence that SO2(g) can reduce HgO(s) at room temperature via HgSO4 (Zacharewski and Cherniak, 1987) to Hg0, HgS, and HgSO4 as stable products (Scott et al., 2003) and that O3(g) in the presence of actinic light can reduce (s) to mercurous species (Ai et al., 2023, which may tentatively undergo disproportionation). In the latter exploratory study, single-particle reactors, 10–50 µm in size, synthesized from mercuric halides in single-walled carbon nanotubes were prepared to levitate during the experiments via optical tweezers. The turnover of HgX2 by breaking a Hg–X bond was measured by time- and position-resolved Raman spectroscopy, which also showed that the decomposed X atom was bound to the carbon material (X = Cl, Br). Heterogeneous reactions of this type, i.e.,
may explain why KCl-coated denuders do not work as a robust quantitative method for measuring GOM in ambient air (Lyman et al., 2010). Since the gas-phase reaction HgXHgIX + XO• (+ O2) is endothermic (≥66 kJ mol−1) and therefore unlikely, the results of a steady-state study (Tong et al., 2021) claiming gas-phase photoreduction of HgX2 in the presence of O3 and light can instead be attributed to the above-mentioned heterogeneous reactions. Additionally, voltammetry can provide valuable insights into the redox chemistry of mercury. Hg0 is frequently employed as the working electrode and has a high overpotential for the reduction of H3O+ to H2. This enables the utilization of standard potentials as negative as −1 V in acidic solutions and −2 V in basic solutions. The surface of the hanging mercury drop electrode (HMDE) can be readily renewed by extruding a new drop. In a study by Giannakopoulos et al. (2012), the interfacial adsorption mechanism of gallic acid onto HMDE was investigated, and a series of easily reducible HgII complexes with mono-, di-, or tridentate gallic acid ligation were identified.
7.3 Dark oxidation of Hg0 accelerated by freeze-concentration effects
Slow oxidation of dissolved Hg0 by O2 occurs in aquatic systems in the presence of Cl− ions (Amyot et al., 2005; Wang et al., 2023). However, upon freezing, most of the solutes are separated from the forming ice phase and concentrated in the remaining liquid at a significantly reduced pH (Bartels-Rausch et al., 2011). In experimental mimics of the micro-pockets of solutions that occur in ice, experiments in the presence of O2, H2O2, and HONO each result in significant Hg0 oxidation. It has been postulated that protonated forms, and , are responsible for oxidation processes, which can be classified as strongly exothermic on the basis of the provided thermodynamic data (O'Concubhair et al., 2012). Moreover, neither dilute H2O2 (aq) nor HONO (aq) will oxidize Hg0 (aq) to any significant extent at room temperature (Kobayashi, 1987).
7.4 Surface-catalyzed reduction of HgII in aqueous solution
In the presence of a solid phase of ferric (hydr)oxide and dissolved di- or monocarboxylic acids under oxic conditions, the reduction of HgII in aqueous solution to Hg0 occurs upon UV irradiation (Lin and Pehkonen, 1997). The systems studied for the photoreduction of HgII are goethite (α-FeOOH) + oxalate/formate, hematite (α-Fe2O3) + oxalate, and maghemite (γ-Fe2O3) + oxalate. The experiments with filtered Xe light were conducted with 10 µM HgCl2, 1 mM organic acid, and 0.1 g L−1 ferric hydr(oxide) suspension, with a starting pH of 3.9. During some of the experiments, the pH increased substantially, resulting in the dominance of oxalate over hydrogen oxalate. Unlike oxalate, formate alone is not capable of reducing HgII to Hg0 under actinic light. It requires irradiation in the deep UV by processes such as (Leonori and Sturgeon, 2019):
One study described HgII reduction mediated by the carbon dioxide radical anion () generated from formic acid via photo-sensitization by visible light-excited naphthoquinone (Berkovic et al., 2012). Iron(III) complexes with formate and oxalate are photolabile under UV-A and visible, where a fast 1e–LMCT step generates Fe2+ and eventually , which initiates a chain process (Mangiante et al., 2017; Baxendale and Bridge, 1955):
The oxic reaction system described by Reaction (R13) reaction formulas contains a number of ROS with different designations, such as strongly reducing and strongly oxidizing HO•, as extreme cases. One subsystem is Fenton's reagent (Sect. 6.1.1, Reaction W R6), which produces HO•, for which each of the Hg, Fe2+, and H2O2 competes to be oxidized. Except for the heterolytic photolysis of Hg(η2–C2O4), which produces Hg0 from HgII in a single step (Sect. 6.1.2, Reaction W R16), the remaining redox steps involving metals are of the single-electron type. The reduction of HgII occurs via reactions with nucleophiles (Sect. 6.1.2, Reaction W R14), which are both homogeneous and heterogeneous with dissolved and adsorbed ferrous species, respectively. A second-order homogeneous reaction coefficient of ∼ 120–313 M−1 s−1 has been determined in the near-neutral pH range, with Hg(OH)2 and FeOH+ identified as the reactive species in solution (Amirbahman et al., 2013; Schwab et al., 2023). Under anoxic conditions, the rate of Hg0 production derived from surface-catalyzed reduction on hematite and goethite has been described by the expression khet [≡ FeII][Hg(OH)2], with khet values of ∼ 89 and ∼ 78 M−1 s−1, respectively (Amirbahman et al., 2013). In an O2-saturated, non-bubbled solution, a photo-stationary state between HgII and Hg0 occurs, indicating that the reduction pathways (Reaction R13 red.) are gradually balanced by oxidation pathways (Reaction R13 ox., Ababneh et al., 2006). In the absence of Hg0 removal, solubility limitations are easily exceeded during experiments (Lin and Pehkonen, 1997; Ababneh et al., 2006), resulting in the precipitation of colloidal Hg0. The removal of dissolved Hg0 by sorption on hydrous iron oxides, which is relevant here, is also documented (Richard et al., 2016). In the presence of competing anions, such as chloride, the rate of reduction decreases, in part owing to the formation of metastable, poorly soluble dimeric mercurous salts that compete with the disproportionation of HgI to Hg0 and HgII (Pasakarnis et al., 2013).
7.5 Field observations of photoreduction in precipitation, clouds, and fog
In precipitation and clouds, a strong correlation between Hg and total organic carbon was observed (Li et al., 2018; Åkerblom et al., 2015), suggesting that Hg–organic complexes are also important in aerosols. Authentic rain samples, where Hg–organic complexes dominate, present photoreduction rates ranging from 0.02−0.2 h−1 (Yang et al., 2019; Saiz-Lopez et al., 2019; Fu et al., 2021). There have been a handful of measurements of Hg in cloud water (Li et al., 2018; Weiss-Penzias et al., 2018; Malcolm et al., 2003; Gerson et al., 2017; Huang et al., 2016a), but thus far, only a few studies on the photoreduction rate in this category of water exist (Li et al., 2018; Zhen et al., 2023; Gao et al., 2023). Photolysis rates in cloud water samples of 0.07–0.21 h−1 measured in situ under actinic light and in the laboratory under UV (> 290 nm) light are consistent with those observed in precipitation. Whether the photoreduction rates observed in rain or cloud water are representative of atmospheric aerosols is questionable. HgII in snowfall or freshly fallen snow has been reported to be labile for photoreduction (Steffen et al., 2008; Faïn et al., 2013). In temperate urban and pristine rural snow, within 24 h, approximately 50 % (Lalonde et al., 2003; Lalonde et al., 2002) and, within 48 h, up to 90 % (Poulain et al., 2004) of the newly deposited Hg can be effectively recycled back to the atmosphere. The reduction is reportedly strong even under cloudy conditions and is not limited by light (Faïn et al., 2013). In general, less than 5 % of the Hg content of a snowpack is in the elemental form (Hg0), which is concentrated stratigraphically in the first few centimeters. Nevertheless, if the rates are implemented as a mean value (∼ 0.07 h−1), determining the lifetime of atmospheric Hg against wet deposition, then the model-estimated wet deposition underestimates the observations by an average of 25 % globally. Current global chemistry and transport models (GMOS-Chem) consider photoreduction on particles with the pool of HgII complexed with organic ligands as the reactant (Shah et al., 2021).
Natural Hg contains seven stable isotopes with mass numbers of 196, 198, 199, 200, 201, 202, and 204. In the 1920s, significant separation of Hg into its isotopes was achieved through vaporization in a vacuum (Harkins and Mulliken, 1921; Mulliken, 1923; Brønsted and de Hevesy, 1921), a process that is conducted on a preparative scale through electromagnetic (Love, 1973) and photochemical (Vyazovetskii, 2012) methods. When the feed flow is in the form of DMHg, total gram quantities of highly enriched Hg isotopes can be obtained through cascade centrifugation (Babaev et al., 2010). The longest-lived radioisotope is 194Hg at 444 years. Since it does not occur naturally, it cannot be used in the dating typical of 14C. Two additional unstable isotopes (197Hg and 203Hg, with half-lives of 64.1 h and 46.6 d, respectively) are valuable for instrumental neutron activation analysis and radiolabeled Hg compounds because of their decay by emission of readily detectable γ rays. It was not until after the turn of the century, 80 years later, with the development of high-precision analytical instruments such as multi-collector inductively coupled plasma-mass spectrometry (MC-ICP-MS), that it became possible to measure differences in the naturally stable Hg isotopic compositions in the environment (Jackson, 2001; Lauretta et al., 2001). Natural processes, including redox reactions, complexation, sorption, precipitation, dissolution, evaporation, diffusion, and biological processes, can alter the isotopic composition, i.e., cause stable isotope fractionation (cf. Fig. 17). Stable isotope analyses can, therefore, provide a previously untapped source of valuable information on the sources and biogeochemical cycling of natural and anthropogenic Hg. Isotopic fractionation refers to the division of a sample into two (or more) parts with different ratios of “heavy” and “light” isotopes than the original ratio. In isotopic jargon, if one part contains more heavy isotopes, it is said to be “enriched”, while the other part is said to be “depleted”. Hg has extremely large isotopic variation in nature, which, when normalized by the relative mass difference between isotopes, approaches that of traditional light element isotopes (Wiederhold, 2015). However, the overlapping signals from different fractionation processes can be a major challenge in deciphering natural isotopic signatures when tracing sources. It is important to determine the Hg stable isotope fractionation for individual key processes, which can be accomplished, inter alia, through controlled laboratory and field experiments. Stable isotope variations are reported as relative values compared with a reference standard (NIST SRM 3133 Hg solution, Blum and Bergquist, 2007):
where () is the ratio of the isotopes with mass numbers xxx and 198. The prevailing practice of expressing isotope ratios relative to the lightest stable isotope for each element is not applicable to Hg because of the rarity of 196Hg (0.15 % occurrence). The standard unit for δ values is per mill (‰). δ202Hg expresses the total mass-dependent fraction (TMDF, containing contributions from conventional mass-dependent fractionation; hereafter, MDF and nuclear field shift (NFS) are described in Sect. 8.1), while the isotope anomalies caused by mass-independent fractionation, MIF, are expressed by capital deltas; Δ is defined as the difference between the measured δ value and that predicted from the measured δ202Hg value and the scale factor for the kinetic MDF (, where KIE is the kinetic isotope effect; see Sect. 8.1) and is approximated for δ values <10 ‰ according to
which is expressed numerically for each relevant Hg isotope: Δ196Hg 0.508⋅δ202Hg, Δ199Hg =δ199Hg , Δ200Hg =δ200Hg , Δ201Hg =δ201Hg , and Δ204Hg =δ204Hg .
The fractionation between two compounds A and B (assuming that A is a product of a reaction and that B is the remaining reactant) is expressed with the fractionation factor, α, which is defined as the ratio of the isotope ratios in the compounds:
The last term is obtained by substituting Eq. (13) into the first term of Eq. (15). Actual δ values are usually very close to unity. Therefore, it is usually more practical to use an enrichment factor:
The last similarity is valid only for δ values less than 10 ‰. Substitute Eq. (14) into Eq. (16) and obtain
Equation (17) expresses total fractionation during the process A → B, with the first term representing the MIF enrichment factor and the second term representing the total mass-dependent enrichment factor. Thus, the enrichment factor for MIF is written as a capital epsilon:
Many kinetic processes can be described as Rayleigh fractionation, which is an irreversible process in an open system involving the progressive removal of a fraction of a trace substance from a larger reservoir. It is described by the following differential equation:
If the fractionation factor is constant, the differential equation can be integrated directly into the expression:
where is the isotope ratio of the initial reservoir (when fR=1) and where Rxxx is the isotope ratio of the reservoir at a given time when the fraction of initial material remaining in the reservoir is defined by fR. The following expression is often used to evaluate the fractionation factor:
The process tends to enrich the heavier isotopes in the reservoir (α<1, normal kinetic isotope effect, KIE) rather than removing the heavier isotopes from the reservoir more rapidly (α>1, inverse KIE).
8.1 Conventional mass-dependent and mass-independent fractionation
The scaling factor β describes the relationship between the fractionation factors as follows:
where β for mass-dependent equilibrium fractionation (βEIE−MDF) and kinetic fractionation (βKIE−MDF) are as follows (Young et al., 2002):
The equilibrium MDF resulting from the differences in zero-point vibrational energy (ZPE) distances and the kinetic MDF resulting from the differences in dissociation energies between the isotopologues and their respective effects can be expressed in two rules: heavier isotopes are preferentially concentrated in compounds with the highest force constant, where the element is most rigidly bound and has greater potential energy. Conversely, compounds enriched in lighter isotopes have weaker bonds and require less energy to break, so they preferentially enter chemical reactions and are enriched in the product (Criss, 1999). Combining kinetic and equilibrium MDF makes it possible to achieve a limit of approximately 10 ‰ fractionation (Sun et al., 2022).
Properties of nuclei, such as nuclear size and shape or the presence of non-zero nuclear spins, may trigger isotope fractionation that does not follow the expected MDF relationships. The nuclear field shift (NFS, Rosenthal and Breit, 1932) is the interaction of the nuclear volume with electrons (NVE, Schauble, 2007). It is highly relevant for very heavy metals, including Hg, Tl, Pb, and U. NFS involves a shift in the ground electronic energy of an atom or molecule due to differences in nuclear size and shape between isotopes. The shift caused by an odd (neutron number) nucleus scales non-linearly between the shifts of the even isotopes of the next highest and lowest atomic masses. The odd isotope electronic energy level is shifted toward the next lower even nucleus (odd–even staggering). Owing to its smaller size and greater surface charge density, the electronic energy of a light isotope is lower than that of a heavier isotope. The amount of shift is a product of two factors: the electron density at the nucleus and the charge, size, and shape of the nucleus and the change in the latter two between isotopes. Hg orbital electrons significantly overlap with the nucleus, whereas 5p, 5d, and 4f orbitals do not, although f electrons in inner shells have a smaller screening effect on 6s valence electrons (Bigeleisen and Wolfsberg, 1957). The lowest energy of a system occurs when the heavier isotopes of Hg are enriched in chemical species with the fewest s-electrons in the bonding or valence orbital. The largest shifts, therefore, occur when the number of Hg 6s electrons is greatly reduced by the formation of an ionic bond (to an electronegative element), while a covalent bond has less influence. Examples of Hg species in the former category are chloro- or aqua-complexes with high coordination numbers (e.g., [Hg(H2O)6]2+), while the latter includes soft ligands with typical linear bi-coordination (e.g., Hg(SH)2 and (CH3)2Hg). The scale factor of nuclear volume fractionation (βNFS) is defined as follows:
where 〈r2〉 describes the mean-square nuclear charge radii of different isotopes. Coincidentally, MDF and NFS with 198Hg, 200Hg, and 202Hg show almost identical β values, but 199Hg, 201Hg, 204Hg, and, to a lesser extent, 196Hg show distinct non-mass-dependent signatures due to NFS. Only a small proportion of the NFS is mass-independent because it creates a deviation from MDF (Yang and Liu, 2015). The mass-dependent part of the two effects can be synergistic (increasing TMDF) or antagonistic (decreasing TMDE), with the former being dominant for Hg redox chemistry (Hintelmann and Zheng, 2011; Jiskra et al., 2012). The MDF scale is proportional to , whereas the NFS scale is proportional to and is more prominent than MDF for the Hg redox reactions studied (Schauble, 2007). Among the commonly measured isotopes 198–202, a minor to moderate level of MIF has been experimentally observed in the odd isotopes 199 (≤ 0.6 ‰) and 201 as a result of NFS. NFS has been described for equilibrium exchange reactions but has never been extended to kinetic processes. In contrast to the small magnitude observed in natural samples, the possibility has recently been suggested that nonequilibrium isotopic effects of NFS in photodissociation may give rise to a significant magnitude of MIF (Motta et al., 2020b).
The only effect that has been documented to lead to significant odd-mass-number Hg MIF (odd-MIF) in present-day surface ecosystems is the magnetic isotope effect (MgIE). MgIE is a purely kinetic effect triggered by the formation of a long-lived radical pair after a primary process that causes homolysis of a Hg-ligand bond upon photolytic excitation (Fig. 11). Among the stable isotopes of Hg, only 199Hg and 201Hg (odd mass numbers) have non-zero nuclear spin and momentum, with half-integer ( and , respectively) spins. MgIE arises when hyperfine coupling (HFC) acts on a spin-coherent solvent-separated radical pair after dissociation by changing the rate of intersystem crossing from singlet to triplet (S↔T) or vice versa (T↔S) in odd Hg isotopes. Radical pairing and MgIE are suppressed in mercuric complexes with strong spin–orbit coupling (containing bromine and iodine ligands), favoring spin mixing and to the ground state, while S-, Cl-, and C-bonded complexes with generally weak spin–orbit coupling favor strong MgIE (Motta et al., 2020a). If the radical pair is born in the triplet state (lower panel of Fig. 11), HFCs are induced, enriching odd isotopes in the resulting singlet state. The singlet radical pair can then recombine to the ground state, resulting in odd isotope enrichment in the reactant, expressed as (+)MgIE. When the radical pair is in the singlet state (top panel of Fig. 11), the overall effect is to deplete odd isotopes in the reactant, as expressed by (−)MgIE, because mainly the odd isotopes with the majority in the triplet radical pair dissociate into free radicals. A computational study has explained why the photodissociation of monomethyl Hg species in nature is observed to yield only (+)MgIE, whereas the photolysis of inorganic mercuric complexes may yield positive or negative MgIE, depending on the reaction conditions and the degree of complex ligation (Sect. 8.4).
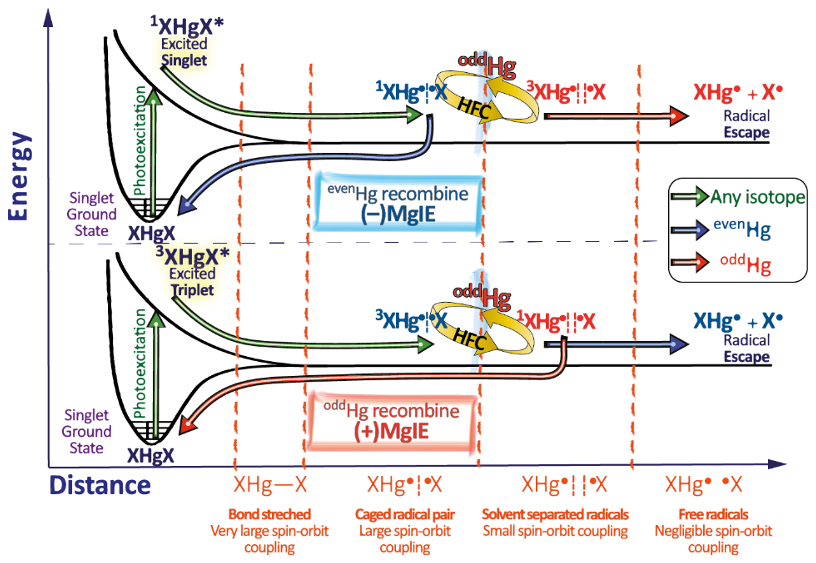
Figure 11General scheme for the photolysis of a molecule to produce a spin-correlated radical pair (RP). The singlet and triplet RPs can be interconverted by intersystem crossing. Both the singlet and triplet RPs can escape from the solvent cage. Only the singlet RP can recombine. Adopted from Turro (1983) and Motta et al. (2020a).
Thus, odd-MIF results from both the MgIE and NFS mechanisms. MgIE is most effective in viscous solvents, where a “solvent cage” environment is possible (Turro, 1983). In addition, a seemingly enigmatic even-mass-number Hg isotope, MIF (even-MIF), has been observed in samples of atmospheric origin or deposition. However, analog atmospheric photochemical anomalous isotope fractionation is well known for the lighter (traditional) elements for which MIF (containing three or more stable isotopes), such as oxygen and sulfur, can be detected. However, understanding the underlying causes of multi-isotope anomalous fractionation is limited because investigations require detailed quantum mechanical calculations at the molecular isotope level, e.g., photodissociation. Recently, rock records have revealed significant even-MIF in the Archean atmosphere, which lacked an ozone (O3) layer to filter UV-C from actinic light, suggesting that contemporary UV-C-induced atmospheric chemistry may be responsible for the coupled changes in even-MIF for both Hg and sulfur (Zerkle et al., 2020). Correlations between these entities have also been observed in marine aerosols in the Southern Hemisphere (Auyang et al., 2022).
MIF signatures as additional tracers
The isotopic measurement of Hg results in up to six useful isotopic signatures (δ202Hg, Δ196Hg, Δ199Hg, Δ200Hg, Δ201Hg and Δ204Hg). In addition, pairs of these signatures have been utilized to distinguish between fractionation mechanisms. The relationship between these signatures is typically illustrated using a three-isotope plot. To interpret the experimental results satisfactorily, specific robust linear regression methods are recommended (Stephan and Trappitsch, 2023). On the basis of early results (Bergquist and Blum, 2007), it was assumed that photoreduction of HgII to Hg0 would result in a ratio of unity. Several investigated photoreactions exhibit just a ratio of 1 within the margin of error (refer to Table 9, Sect. 8.4). However, further data have shown that this is not always the case, as the slope depends on factors such as the complexing ligand and reaction conditions. Clearly, the odd-MIF signature for the photolysis of organomercurials is different from that stipulated for inorganic Hg. The photolytic degradation of MMHg+ species results in a variation in ranging from 1.17 to 1.38 depending on the reaction conditions (Bergquist and Blum, 2007; Chandan et al., 2015; Rose et al., 2015; Malinovsky et al., 2010). Compared with MgIE, NFS generally results in a much weaker MIF, with greater anomalous fractionation of 199Hg than of 201Hg, which approaches a ratio of ∼ 1.6. However, the NFS should be confirmed via alternative methods when the experimentally measured NFS is too limited to determine a definitive odd-MIF ratio (Motta et al., 2020b). Another commonly used parameter is , which describes the degree of odd-MIF in relation to TMDF. The even-MIF signature of Δ200Hg to Δ204Hg, which is negative in natural samples (air, rainfall, and fish), is discussed in Sect. 8.2.4.
8.2 Isotopic characteristics of atmospheric mercury
Figure 13 summarizes the magnitude of the isotopic observations reported in the literature on the main fractions of Hg in the atmosphere, namely gaseous Hg dominated by Hg0, particle-bound Hg, and Hg associated with hydrometeors (rain, snow, and water from clouds and fog). Here, δ202Hg, Δ199Hg, and Δ200Hg are used to describe TMDF, odd-MIF, and even-MIF, respectively. The number of isotopically resolved samples has increased dramatically in recent years. Readers should consult the literature regularly to stay up to date. On the other hand, this development justifies revisiting the topic, even though it has been satisfactorily addressed in the recent past (Kwon et al., 2020). Notably, the spatial distribution of available data is heavily skewed toward North America, Europe, and East Asia, and observations from large parts of the world are missing (Fig. 13). However, as far as marine remote regions are concerned, recent oceanographic expeditions have contributed to an increasing amount of data. Further below, the in situ and laboratory experiments performed thus far to study the gas exchange of Hg0 between air and water, soil, and foliage in terms of isotope fractionation are discussed.
8.2.1 Gaseous Hg
The following part excludes an early series of measurements where the air was not filtered prior to sampling (Rolison et al., 2013). The referenced series of measurements should be considered TAM, not total gaseous mercury (TGM). TGM significantly affects δ202Hg (−3.75 ‰ to 1.52 ‰) and Δ199Hg (−0.62 ‰ to 1.32 ‰) but is more limited to Δ200Hg (−0.22 ‰ to 0.11 ‰). Analogous to HgII deposited in biomass and fossil fuels such as coal (Sun et al., 2014), Hg0 in natural gas (Washburn et al., 2018) and in smoke from spontaneous combustion in coal fields (Sun et al., 2023) has strongly negative δ202Hg values. This differs from the majority of Hg0 in ambient air, which is isotopically heavy (often with positive δ202Hg values). Terrestrial background air (rural, subpolar, and forest in Fig. 13a) has higher δ202Hg0 values because it is modified by vegetation, which preferentially incorporates lighter isotopes of Hg0 into the foliage. Foliar uptake of Hg0 is discussed in more detail in Sect. 8.6.2. The estimate of atmospheric Hg0 dry deposition to vegetation has recently been revised and constrained to approximately 2300 Mg yr−1 (Feinberg et al., 2022), and together with the large negative εfoliage–air of the process, the global atmospheric Hg0 pool is estimated to have a δ202Hg mean of 0.5 ‰, in contrast to that of Hg0 from anthropogenic sources (global bulk mean of −0.7 ‰, Sun et al., 2019). Studies examining the vertical distribution of mercury (Hg0) concentrations from near the ground to above the canopy in different forest types reveal clear gradients averaging 10 % (Wang et al., 2022) and 20 % (Fu et al., 2016b) of ambient Hg0. Under stable conditions, such as during summer nights, Hg0 levels are strongly depleted below the canopy (Fu et al., 2016b; Mao et al., 2008; Poissant et al., 2008; Lan et al., 2012; Fu et al., 2019a). Thus, isotopic measurements of above-canopy air versus in-canopy air (Wang et al., 2022; Fu et al., 2016b) and daytime air versus nighttime air in forests (Kurz et al., 2020) show statistically significant differences (p<0.0) in δ202Hg0. For a deciduous forest in Northeast China, Fu et al. (2019a) reported that δ202Hg0 in biweekly air samples during the growing season was 0.35 ‰ to 0.99 ‰ higher than that during the dormant season. In a subtropical, perennial forest in southwestern China, where there is little seasonal variation in the photosynthetic activity of vegetation, the existing seasonal variation in δ202Hg0 (with an amplitude of 0.4 ‰) can be attributed to the influence of long-range anthropogenic emissions, which primarily occur during the warmer seasons. However, over the last 5–7 years, the air concentrations of Hg0 have decreased significantly in the two mentioned forest reserves due to reduced regional anthropogenic emissions, as evidenced by the median value of δ202Hg0 shifting from 0.42 ‰ to 0.46 ‰ and from 0.17 ‰ to 0.57 ‰. The marine δ202Hg0 data (n=112) are significantly lower (Wilcoxon test, p<0.01) than those from the forest (n=113). Coastal measurements in the Gulf of Mexico show that the marine-influenced air isotopically represents background air modified by Hg0 emitted from the sea after being formed in surface water by photoreduction (Demers et al., 2015). Measurements in the marine boundary layer of the offshore East China Sea indicated that airborne Hg0 is essentially a binary mixture of anthropogenic outflow from mainland China and air masses from the sea (significantly correlated δ202Hg0 and Δ199Hg0 vs. ), with an extrapolated Δ199Hg0 of −0.26 ‰ for the marine component. The extrapolated Δ199Hg0 value agrees well with observations made in Hawaii with passive samplers (Szponar et al., 2020) as well as with the signatures of a larger number of samples from Mauna Loa (3397 m a.s.l.) in the free troposphere (Tate et al., 2023; Yamakawa et al., 2024).
The generally negative signature of Δ199Hg0 in the background air indicates that Hg0 has been added to the pool subsequent to HgII photoreduction (of the variant that induces (+)MgIE in the reactant and complements it by depleting the product Hg0 isotopically for odd isotopes) in oceans and aerosols. This is supported by atmospheric Hg0 exhibiting slopes close to unity (Kwon et al., 2020), similar to aqueous photoreduction of inorganic HgII. However, not all photolytically controlled Hg0 re-emissions from terrestrial ecosystems contribute to negative Δ199Hg0 values in the atmosphere. An analysis of gas exchange in a subtropical beech forest revealed bidirectional fluxes of Hg0, with uptake partially balanced by reemission of previously metabolized HgII. Photoreduction recirculates Hg0, contradicting a retro-flux of deposited Hg0 at the leaf surface (data in Fig. 18a). This re-emission is isotopically distinct in that it is enriched in odd isotopes compared with ambient air (Yuan et al., 2019b), indicating that leaf photoreduction induces (−)MgIE, as reported for HgII bound to organic ligands containing sulfur or nitrogen in low oxidation states (Motta et al., 2020b; Zheng and Hintelmann, 2010b). A mass balance based on isotope measurements indicates that, compared with the uptake of Hg0 from the air, re-emission from beech foliage gradually increases from emergence to senescence, accounting for an average of 30 % (Yuan et al., 2019b). Observations from a temperate deciduous forest revealed 0.06 ‰–0.09 ‰ higher Δ199Hg0 values during the growing season than in winter, suggesting that foliar Hg0 efflux contributes to the atmospheric enrichment of odd Hg isotopes (Fu et al., 2019a).
The large spread of odd-MIF shown by Hg0 in polar air (Araujo et al., 2022; Yu et al., 2021; Sherman et al., 2010, Fig. 13a) is due to the portion of the collected data that includes Hg0 depletion events in the spring and Hg0 enhancement during the summer, when reemissions of Hg0 occur from the cryosphere. Figure 12 shows the isotopic compositions of airborne Hg fractionated into Hg0 and HgII (RM) during the Arctic spring (at three stations) compared with the corresponding data from a background station in the Pyrenees (Fu et al., 2021). With respect to Δ199Hg, a dichotomy between the polar and temperate data is striking for both Hg0 and HgII (RM), in that montane oxidized Hg is enriched in a limited range (0.14 ‰ to 0.77 ‰), whereas polar HgII is depleted in a greater range (−2.15 ‰ to −0.18 ‰), with a complementary relationship existing for Hg0 (−0.31 to −0.16 ‰ versus −0.22 ‰ to 1.32 ‰). This relationship could be caused by surface layer airborne Hg being strongly influenced by the oxidation of Hg0 to HgII, which is controlled by halogen atoms during AMDEs, processes characterized by E199Hg values of −0.37 ‰ and −0.23 ‰ for Cl•-initiated and Br•-initiated oxidation, respectively (Table 8, Sun et al., 2016). In this way, the remaining reactant is driven to a higher Δ199Hg0 and the molecular products assume negative Δ199HgII values. However, this interpretation is not corroborated by the measured Δ199Hg/Δ201Hg ratio of nearly unity in airborne Hg, which is more typically indicative of HgII photo-reduction (−MgIE) occurring in snow. It has been proposed that this process also operates in aerosols of the boundary layer, with Hg0 reemissions providing such a strong positive imprint that the entire boundary layer of the Hg0 pool becomes enriched in odd isotopes (Araujo et al., 2022). Isotopic measurements of HgII(g) separated from HgII(p) using CEM (cf. Sect. 3.1) have commenced and are anticipated to elucidate the mechanisms underlying the pronounced fractionation of odd isotopes in airborne Hg0 and HgII. Several such datasets are currently in preparation for publication. Furthermore, Hg0 in the Arctic during the dark period of the year and from the Antarctic Peninsula throughout the year (Yu et al., 2021) shares a consistently slightly negative Δ199Hg0 with other background air (represented by montane air in Fig. 13a). In the late Arctic summer, minimum Δ199Hg0 values (approaching −0.5 ‰) are observed uniformly without much variation from coastal stations around the Arctic Ocean, which are thought to result from photoreduction of cryospheric HgII, a substrate that has been strongly depleted of odd isotopes during months of long sunshine (Araujo et al., 2022).
Δ200Hg0 is generally negative for non-fossil/anthropogenic sources, while the remainder is significantly shifted to higher values (Wilcoxon t test, e.g., natural gas vs. arid data, p < 0.01). As mentioned above, even-MIF is generated exclusively by atmospheric chemical processes, which may be mainly limited to molecular HgI,II photolysis processes (Sun et al., 2022), of which Hg0 is a product. The marine and polar Δ200Hg0 data have the most negative values. For example, a recently published TGM record from Mauna Loa (not shown in Fig. 13a) in the Pacific Ocean has Δ200Hg values as low as −0.20 ‰ (Yamakawa et al., 2024). The polar pool as a unit significantly shifted toward lower Δ200Hg0 values than the forest pool did (Wilcoxon T test, p<0.05). One can only speculate as to the reason, but it should be mentioned in the context of a halogen-rich environment that any presence of Cl-initiated Hg0 oxidation in the gas phase will result in depletion of 200Hg in the reactant pool (E200Hg ∼ 0.06 ‰, Sun et al., 2016). Owing to its relatively limited range, ambient Δ200Hg0 and Δ204Hg0 are considered conservative tracers of atmospheric Hg0 deposition, and terrestrial surface and water Δ200Hg and Δ204Hg values can constrain the relative contribution of Hg0 to HgII deposition. Throughout, a median value of −0.05 ‰ (IQR −0.02 ‰ to −0.08 ‰) of Δ200Hg0 was used to calculate this contribution to atmospheric transfer to soil (Δ200HgII∼0 ‰, Enrico et al., 2016; Zhou et al., 2021; Zheng et al., 2016) and oceans (Δ200Hg∼ 0.04 ‰, Jiskra et al., 2012). The quantitative AMDEs observed in Alaska are isotopically mass balanced in that the Δ200HgII in snow (−0.06 ‰) corresponds, within the measurement uncertainty, to that in ambient Hg0 (−0.05 ‰).
8.2.2 Aerosol-bound Hg
While Hg0 has a relatively long lifetime and HgII(g) has a short lifetime, the lifetime of particle-bound Hg (PBM, ) reflects that of particles, which varies from days to months due to their size and composition. Isotopic analyses have been performed on airborne PM2.5, PM10, and TSP, as well as on particles in precipitation. Studies of urban air, regionally polluted air, and air associated with anthropogenic emissions (CFPP, traffic and waste incineration, etc.) are well represented and strongly biased toward Asia. As reviewed and discussed in Kwon et al. (2020), attempts to decipher the cause of seasonal variations in urban and industrial air are challenging in environments with a plethora of local and regional emission sources. However, primary particles from fossil fuel and biomass combustion inherit the clearly negative but highly variable δ202HgII(p) and the less negative Δ199HgII(p) of the material. The large range in Δ199HgII(p) (−0.93 ‰ to 1.5 ‰) around the origin depends on HgII(p) photoreduction with (+)MgIE, halogen-atom-initiated Hg0 oxidation or, more speculatively, HgII(p) photoreduction with (–)MgIE, driving the data to extremes. In a series of papers, including field measurements of particle-bound isotopic Hg in regionally polluted air (Huang et al., 2016b; Huang et al., 2019; Qiu et al., 2022; Zhang et al., 2022) and laboratory experiments (Huang et al., 2021; Huang et al., 2015), Chen and colleagues have focused on the effect of (+)MgIE photoreduction, which is accelerated in the presence of a particle surface liquid layer (wet haze) and water-soluble organic carbon as a reducing agent (Zhang et al., 2022). Several peripheral monitoring stations in China, primarily receptors of long-range particle transport, generally measure positive Δ199HgII(p) values (Fu et al., 2019b). A strong anticorrelation between Δ199HgII(p) (up to ∼ 1.2 ‰, but initially at near zero) and the concentration of particle-bound Hg, rationalized as caused by photo-produced Hg0 loss from aerosols, was observed in samples from these stations, with the major potential source area identified as northeastern China and the regions along the lower reaches of the Yangtze River to its mouth (Fu et al., 2019b). The results indicate that the globally modeled tropospheric lifetime of HgII against photoreduction in aerosols and clouds of nearly 2 weeks (Horowitz et al., 2017) is significantly shorter in East Asia, possibly because of a greater fraction of organic aerosols. As shown in Fig. 13b, there is a statistical anomaly in the PBM polar data for all reported isotopic signatures: positively shifted δ202HgII(p), negatively shifted Δ199HgII(p), and negatively shifted Δ200HgII(p). It is represented in both Arctic (Araujo et al., 2022; Zheng et al., 2021) and Antarctic (Auyang et al., 2022; Li et al., 2020a) data in conjunction with AMDEs. In the High Arctic (∼ 83° N), there is good isotopic agreement between ambient Hg0 and PBM associated with nearly complete AMDEs, as would be expected. Moreover, for less quantitative oxidation, PBM is isotopically lighter than Hg0, analogous to kinetic isotope fractionation during oxidation and subsequent uptake of HgII(g) on particles (such as Arctic haze). As described above, halogen atom-driven gas-phase oxidation induces a negative Δ199HgII(Sun et al., 2016; Auyang et al., 2022), which is consistent with the observed signature in PBM. The interpretation of Zheng et al. (2021) that gas-phase oxidation uniquely shapes isotopic fractionation has been challenged by Araujo et al. (2022), who instead consider (–)MgIE photoreduction in aerosols as the imprinting source. The Antarctic coast has shown uniquely high positive δ202HgII(p) values (up to ∼ 3 ‰ and anticorrelated with Δ199HgII(p)) in air masses transported by katabatic winds from the continental shelf, where oxidation of Hg0 persists during summer (Li et al., 2020a). Under precipitation (Sect. 8.2.3), the high Δ200HgII values measured in southern Canada are addressed, noting that this also applies to the particulate fraction in precipitation, which is included in the rural PBM category (Fig. 13b).
8.2.3 Hg in precipitation
Measurements of Hg isotopes in precipitation samples (including fog and cloud water) have been reported at sites in the Northern Hemisphere (map in Fig. 13c), mostly in North America. Compared with the Hg0 and PBM samples, the precipitation samples presented the greatest scatter in both Δ199Hg and Δ200Hg. Nevertheless, the isotopic distribution pattern in precipitation water is generally similar to that of PBM, which is scavenged in precipitation during rainout and washout processes. Precipitation in the vicinity of anthropogenic emission sources (such as CFPPs) tends to be isotopically distinct, with particularly negative δ202HgII values (Sherman et al., 2012). Precipitation from more pristine areas has a δ202HgII that is shifted in a positive direction (significant for marine, polar and rural categories, Wilcoxon t test, p < 0.01) compared with urban precipitation and precipitation near point sources. The general differences between Hg0 and precipitation/PBM in terms of MIF signatures (negative Δ199Hg0 and Δ201Hg0 vs. positive Δ199HgII and Δ201HgII and negative Δ200Hg0 and positive Δ204Hg0 vs. positive Δ200HgII and negative Δ204HgII, respectively) are explained by atmospheric redox processes (Auyang et al., 2022; Kwon et al., 2020). In the case of even-MIF, chlorine-atom-initiated gas-phase oxidation is known to induce a limited positive Δ200HgII in the product. However, its observed magnitude cannot explain the highest Δ200HgII measured in precipitation (Kurz et al., 2021; Chen et al., 2012; Yuan et al., 2022) in North America. Cai and Chen (2015) reported a trend toward increasing Δ200HgII in background precipitation as moving northward along the mid-latitudes of the Northern Hemisphere (∼ 20–45° N), but only with data from a unique station anomalous with greater statistical significance. A 1-year measurement north of Lake Ontario (Chen et al., 2012), separated by a full decade from measurements at the same site limited to the colder parts of the year (Yuan et al., 2022), has shown that precipitation in winter often contains high values of Δ200HgII (and, at the same time, strongly negative Δ204HgII values). During the full-year measurement in 2010, filtered precipitation samples presented a Δ200HgII in the range of 0.21 ‰ to 1.24 ‰, whereas during the colder months around the turn of the year 2020–21, the same category of samples contained between 0.25 ‰ and 1.19 ‰ and between −1.97 ‰ and 0.37 ‰ for Δ200HgII and Δ204HgII, respectively. During the last campaign, isotopic analysis was also performed on precipitation particles, which presented significantly lower positive Δ200HgII values (up to 0.37 ‰) and less negative Δ204HgII values (down to −0.84 ‰). Intermittently, the particle phase has the opposite sign to the solute phase in the same precipitation sample with respect to both odd- and even-MIF. This, together with a time series of unrelated odd-MIF and even-MIF trends during events with large fluctuations in these values, has been interpreted as the influence of the circumpolar vortex with varying contributions of tropospheric and stratospheric air, with the transport of the latter air masses explaining more extreme even-MIF values (Yuan et al., 2022). Compared with snow samples from the Canadian station north of Lake Ontario, rain samples from the Canadian station north of Lake Ontario generally have more moderately positive Δ200HgII values, which is consistent with precipitation observations in the mid-latitudinal USA (Kurz et al., 2021; Demers et al., 2013; Gratz et al., 2010; Sherman et al., 2015), Europe (Fu et al., 2021; Enrico et al., 2016), the Tibetan Plateau (Yuan et al., 2015), and the Pacific Ocean (Motta et al., 2019; Washburn et al., 2021). Although cloud water (Fu et al., 2021; Zhen et al., 2024) and fog water (Washburn et al., 2021) have been isotopically analyzed, there are no apparent differences between them or significant differences from rain samples. In cloud water, Hg speciation with increasing complexation with DOM has been shown to correlate with odd-MIF values (Zhen et al., 2024), which is consistent with the view that these mercuric complexes are photolabile. Polar precipitation samples (only those from AMDEs are reported in the literature, Araujo et al., 2022; Sherman et al., 2012; Zheng et al., 2021) consistently have slightly negative Δ200HgII values, which differ from those of precipitation samples from all other provenances, which have positive median values. The reason for these observations is plausibly that oxidation is so advanced during these AMDEs that the HgII scavenged by precipitation approaches the same isotopic values as the Hg0 in the polar air before the AMDE.
8.2.4 Even-MIF (Δ200Hg/Δ204Hg) ratios in atmospheric samples
Early studies by Gratz et al. (2010) and Chen et al. (2012) revealed that MIF anomalies of even mass number isotope 200Hg are regularly present in atmospheric precipitation. Later, measurements (Demers et al., 2013) were also made at Δ204Hg, which is more challenging due to the limitations of ion beam collector designs (Blum and Johnson, 2017). The anomaly of Δ204Hg was generally larger and opposite to that of Δ200Hg. The Δ200Hg/Δ204Hg ratio has been calculated based on spatial averages and exclusively on precipitation samples, which are usually above measurement uncertainty. For example, a slope of −0.5 was previously reported (Blum and Johnson, 2017) and later adjusted to −0.4 (Kwon et al., 2020) using this method as more data became available. However, when all individual precipitation data up to 2020 were combined, Kwon et al. (2020) obtained a significantly lower regression slope of −0.24. Figure 14 shows the even-MIF data (Δ200Hg vs. Δ204Hg) binned into geographical regions (categorized as Hg0, rain/mist/cloud, PBM, RM, and snowfall samples). Linear regression of York-type Δ200Hg against Δ204Hg yields slopes between −0.07 and −0.53 for data grouped by site and category for data of statistical significance (p ≤ 0.05, indicated by *). When the global data grouped by sample type are analyzed separately, significant (p < 0.001) slopes of −0.51 ± 0.02 (n = 45, Kurz et al., 2021; Yuan et al., 2022), −0.41 ± 0.03 (n = 108, Fu et al., 2021; Demers et al., 2015; Enrico et al., 2016; Sherman et al., 2012; Yuan et al., 2022; Demers et al., 2013; Donovan et al., 2013; Motta et al., 2019; Washburn et al., 2021), −0.29 ± 0.06 (n = 58, Fu et al., 2019b), and −0.11 ± 0.02 (n = 295, Fu et al., 2021; Kurz et al., 2020; Demers et al., 2015; Tate et al., 2023; Araujo et al., 2022; Enrico et al., 2016; Kurz et al., 2021; Demers et al., 2013; Yamakawa et al., 2017; Jiskra et al., 2019; Fu et al., 2016a; Wu et al., 2023a) are obtained for snowfall, rain and fog, particulate matter, and Hg0 respectively. The reaction mechanism triggering even-MIF could be photodissociation in the gas phase (Sun et al., 2022) or on surfaces (Fu et al., 2021). This should lead to varying degrees of fractionation depending on the species undergoing decomposition. As a result, the fractionation of atmospheric HgI and HgII species differs from one another, possibly explaining the divergent Δ200Hg Hg values for HgII(aq), HgII(p), and Hg0(g).
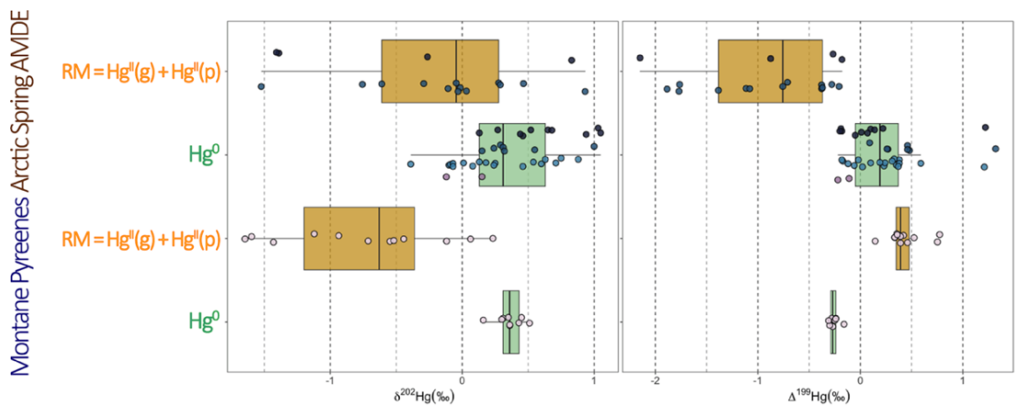
Figure 12A comparison of the TMDF and odd-MIF signatures for atmospheric Hg0 and RM, measured during Arctic AMDEs and in the Pyrenees during winter, reveals notable contrasts.
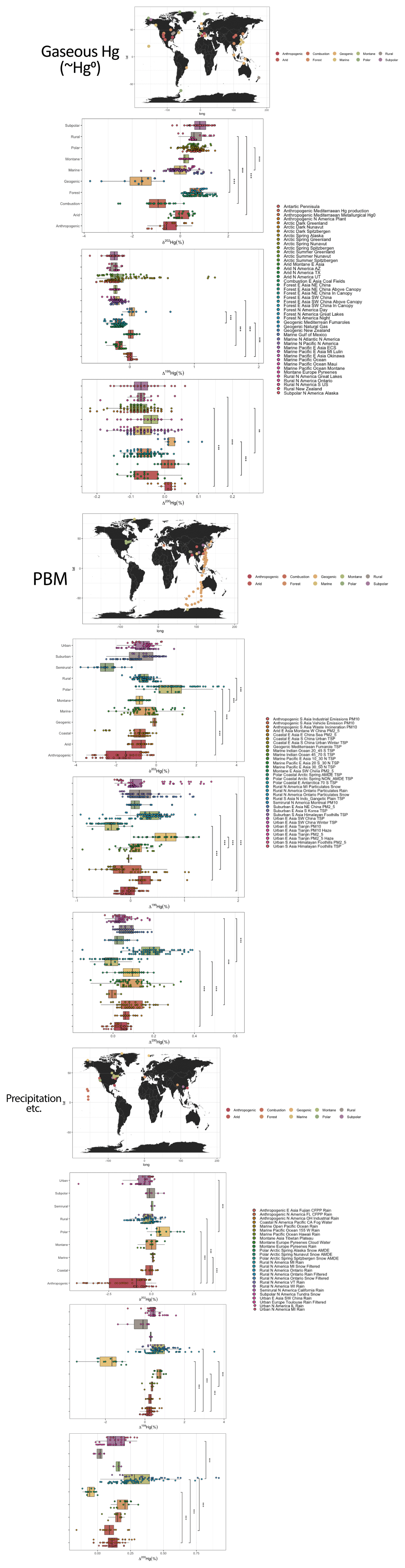
Figure 13Global isotopic observations of gaseous Hg (∼ Hg0) (a), particulate Hg (PBM) (b), and Hg in precipitation (c) divided into δ202Hg (top), Δ199Hg (middle), and Δ200Hg (bottom).
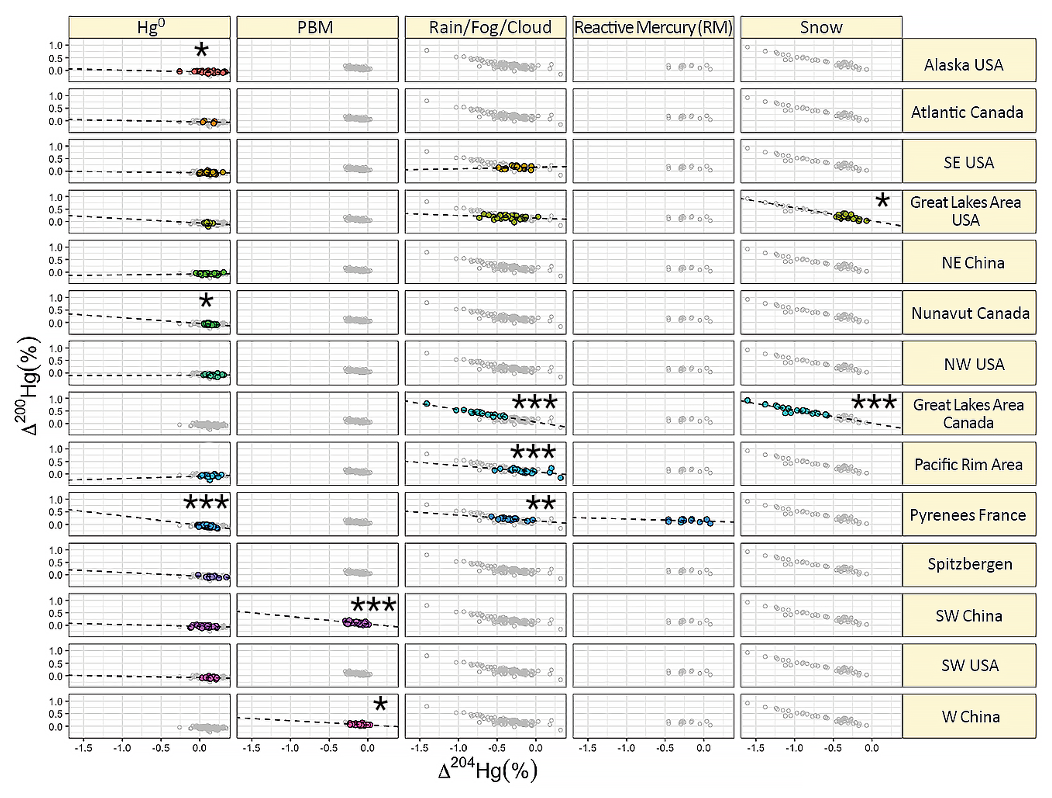
Figure 14Global observations of even-MIF (Δ200Hg vs. Δ204Hg) by, from left, Hg0, PBM, rain/fog/cloud, reactive mercury (RM), and snow.
8.3 Isotope fractionation during gas-phase oxidation
Data on the stable isotopic fractionation of Hg during gas-phase chemical reactions are limited. However, in addition to the published studies on fractionation during the oxidation of Hg0 initiated by Cl and Br atoms (Sun et al., 2016) and during the oxidation of electronically excited Hg0 in the presence of synthetic air (Sun et al., 2022), the corresponding thesis provides additional data (Sun, 2018), which are highlighted here.
8.3.1 Ground-state Hg0 oxidation in air
Isotope fractionation during the oxidation of Hg0 vapor in the ground state has been studied for reactions initiated by Cl•/Br•/•OH/O3/BrO• in air at 750 Torr and 298 K, as listed in Table 8. Figure 15 shows that the Br• and •OH reactions produce a lighter isotope enrichment in the reactant Hg0, unlike the other reactions that follow KIE. This deviation from KIE occurs because the Hg0 to HgI step (Reactions G R1–G R3, Table 4) in the overall Hg0 to HgII oxidation is reversible. EIE is especially notable for the Br• and •OH channels being affected by thermal and photolytic dissociation (Reactions G R14 and G R53), creating a cyclic replenishment of Hg0 at higher temperatures, as discussed in Sect. 5.1.2. EIE predicts the enrichment of heavier isotopes in species with a stronger bonding environment (e.g., HgBr2, Hg(OH)2, Schauble, 2007). However, at temperatures in the upper atmosphere and during AMDEs in polar regions, the rate of Reaction G R14a and G R53a becomes much lower, and the oxidation mechanism moves toward irreversibility, potentially leading to the dominance of KIE at lower temperatures. The chlorine-atom-initiated reaction already displays a KIE at 298 K, which is related to the relative thermal stability of the HgCl intermediate. All the atmospherically relevant reactions investigated (Cl•, Br•, and •OH) give rise to (+) odd-MIF, which is most pronounced for the Cl-initiated reaction (E199Hg 0.37 ‰) compared with the other reactions (E199Hg 0.23 ‰ and −0.18 ‰ for the Br and OH reactions, respectively). Analogous to •OH +•OH recombination, which yields H2O2 in the gas phase (Velivetskaya et al., 2016; Velivetskaya et al., 2018), odd-MIF plausibly occurs due to MgIE triggered by radical–radical (•HgIX + Y•) interactions that occur during reactions, leading to the formation of XHgIIY species. The diagnostic ratio of Δ199Hg/Δ201Hg ∼ 1.9, which is observed for the Hg0 + Cl• system, differs significantly from the ratios reported for the photoreduction of Hg2+ complexes in water (Sect. 8.4.1).
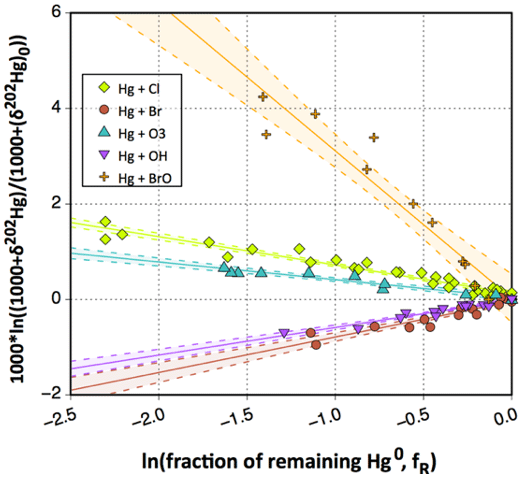
Figure 15Linearized Rayleigh diagram for δ202Hg in Hg0 during Cl, Br, OH, O3, and “BrO” oxidation experiments at ∼ 298 K showing normal and inverse KIEs. Each point represents a single experiment.
Table 8Experimental fractionation factors determined in gas-phase oxidation studies. n/a = not applicable
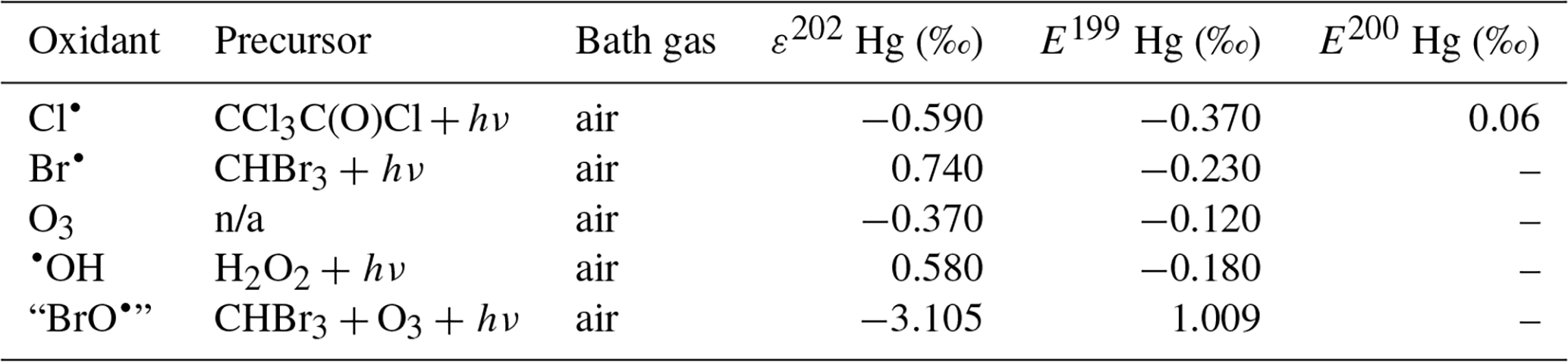
8.3.2 Hg0 oxidation initiated by photosensitized reactions
Ancient rock samples show a significant occurrence of even-MIF in the Archean atmosphere (∼ 2.5 Ga, Zerkle et al., 2020), which lacked an O3 layer to filter out deep UV light in the actinic zone. However, in the modern atmosphere, even MIF does not appear to occur significantly in Hg redox processes at the Earth's surface. The current atmospheric budget reveals notable imbalances between Δ200Hg in Hg emissions from and deposition to the Earth's surface (0.025 ± 0.032 ‰ vs. 0.073 ± 0.019 ‰, Fu et al., 2021). To maintain a steady state, even-MIF sources in the atmosphere are necessary. Studies have shown that UV-C-induced Hg0 vapor in the electronically excited state, Hg(3P1), undergoes chemical transformation under both artificial (Mead et al., 2013) and modern (Sun et al., 2022) atmospheres, resulting in a large MIF of both odd and even Hg isotopes. There are claims (Blum and Johnson, 2017; Mead et al., 2013) that the ratios found in nature are similar to those present in the glass housing of compact fluorescent lamps (CFLs). However, the Δ199HgII, Δ200HgII, and Δ204HgII values in the CFL housing exhibit opposite signs to those observed in nature (cf. Figs. 13 and 14). Laboratory experiments have shown that the net oxidation of Hg0 by the reaction between excited-state Hg0 and atmospheric O2, which is identical to the driving photosensitized reaction for the turnover of Hg0 in the upper stratosphere (Reaction G R12b counteracted by Reaction G R72, Table 4), scrambles the systematics of all Hg isotopes in an entirely mass-independent manner. These laboratory experiments and atmospheric samples show similar observations for the ratio, suggesting that photodissociation is a potential chemical mechanism for triggering even-MIF in the atmosphere (Sun et al., 2022). This review outlines new findings on atmospheric Hg chemistry, supporting the fundamental importance of photodissociation processes (Sect. 5.1.2 and 5.1.4). In addition to the gas-phase, surface-mediated photolysis of mercurous halide species has also been proposed as a mechanism for generating even-MIF (Fu et al., 2021). However, theoretical challenges still need to be solved at the quantum mechanics level to generically expand our understanding of anomalous isotope effects for traditional and non-traditional elements (Lin and Thiemens, 2024). Further field and laboratory research in this area should be encouraged.
8.4 Isotope fractionation during aqueous-phase redox transformation
Hg transformation in the aqueous phase has been reviewed extensively, including stable Hg isotope studies (Hintelmann and Zheng, 2011). The present study does not focus on biotic processes, such as microbial reduction, methylation and demethylation, or phototrophic microbial reduction. Kritee et al. (2013) and Tsui et al. (2020) provide overviews of this field. The focus is on abiotic processes, excluding those involving coordination with macromolecular heterogeneous ligands such as DOM or fractions, and instead on low-molecular-weight ligands, including those with N–, O–, S–, or (pseudo)halide donors. This includes inorganic and organic ligands and oxidizing and reducing processes (Sect. 6).
8.4.1 Reduction
To recapitulate Sect. 8.1, in addition to MDF, isotopic effects in NFS and MgIE occur for Hg during chemical transformation in the aqueous phase. MDF and NFS are present in all reactions to varying magnitudes and in all mechanisms and have a thermodynamic nature. In contrast, MgIE is a kinetic effect and is indicative of spin-selective reactions involving a paramagnetic intermediate. Therefore, MgIE is the only isotope effect that detects the reaction mechanism. MgIE can be both thermally and photolytically induced and can be two-dimensional (+ or − depending on the reaction conditions, Zheng and Hintelmann, 2010b) or one-dimensional (exclusively +), depending on the identity of the HgII complex (Motta et al., 2020a). In cases where the spin-selective reaction can be induced thermally, the radical pair is generated almost exclusively as a singlet (Buchachenko, 2018), which is spin-forbidden to react (dissociate) further into products. For a singlet spin forbidden reaction compared to a triplet spin allowed reaction, the magnitude of the MgIE-MIF is more limited. However, many HgII complexes have a narrow energy separation of a variety of excited states, indicating that the intermediate radical pair can evolve into a triplet or singlet state. Studies of Hg2+ photoreduction in the presence of organic ligands (which consistently follow a pseudo-first-order kinetic pathway) have shown that, depending on the degree of Hg2+(aq) turnover, weak MIF is initially induced by NFS, and then, when most of the Hg2+(aq) has been converted, there is a shift to strong MIF induced by MgIE, the onset of which coincides with strong suppression of MDF (Motta et al., 2020b; Zheng and Hintelmann, 2010b). One explanation for why MgIE first appears closer to complete HgII reduction is, at least in part, that the termination radical–radical step when Hg0 is split off (in a bimolecular reaction, such as in the photoreduction of Hg(η2–C2O4)) is favored by a decreasing concentration ratio of oxidized Hg to bulk ligand (Zhao et al., 2021). As shown in Fig. 11, (−)MgIE is induced when the radical pair is generated in a singlet state, and (+)MgIE is induced when the excitation occurs in a triplet state. Ligand field strength, in combination with atomic orbital hybridization theory, has been used to illustrate MgIE in the (photo)reduction of HgII complexes. This phenomenon has been suggested to vary as a function of, among other things, the arrangement of the ligands around Hg2+, the coordination strength of the ligands, and the presence/absence of light along with its wavelength (Epov, 2011a, b). As discussed in Sect. 4.4, reduced S- and reduced N-containing groups are soft (strong field) ligands, whereas O-donating groups are hard (weak field) ligands. Epov (2011a) rationalized mercuric complexes with strong field ligands such as cysteine () and ethylenediamine (Hg(en)2+) as bright singlets (i.e., in the presence of light) with sp hybridization at the central Hg atom in two binding orbitals. To undergo singlet–triplet evolution by hyperfine coupling between magnetic nuclei (199Hg and 201Hg) and electrons to a paramagnetic state, the orbital hybridization of Hg must change from sp-linear to sp2d-planar square so that the transfer of electrons from the soft ligand to Hg can be accomplished.
Figure 16Mechanism proposed by Epov (2011b) for the photoreduction of Hg(en)2+ via a bright singlet excited complex undergoing intersystem crossing preferentially for the odd isotope to the closest triplet state, which can dissociate following a complex reaction.
Figure 16 shows the schematic for Hg(en)2+ + light, with (–)MgIE. It is postulated that mercuric complexes with O-binding ligands possess a bright triplet state that is more likely (spin allowed) to undergo Hg reduction via 1e-LMCT with an imprint of (+)MgIE.
For a change in the spin state to occur, spin–orbit coupling (SOC) must be induced, but if the SOC is elevated, spin relaxation or phosphorescence can be induced, which prevents the formation of a separating radical pair during dissociation, making MgIE less relevant (cf. Fig. 11). Coupling constants are known experimentally for only a few Hg-containing radicals (CH3Hg•, Karakyriakos and McKinley, 2004; •HgF, Knight et al., 1981; •HgCN, Knight and Lin, 1972; •HgH/•HgD, Stowe and Knight, 2002). Recently, published theoretical electronic structure simulations have been performed on environmentally interesting Hg halides (Cl, Br, I) and pseudohalides (methanethiol). The study (Motta et al., 2020a) reported that the coupling for reactions involving •HgIBr and •HgI is so high that radical pair formation is inhibited, whereas for •HgICl and CH3SHgI•, coupling is sufficient in the caged pair as well as at a low level in the separated pair geometries, allowing MgIE to form. Depending on the identity of the Hg–ligand bond that undergoes homolysis to a radical pair, either quadruple (X = Cl, S) or double (Y = C) degeneracy can occur between the low-lying electronically excited levels and the ground state in the HgX2 and HgXY compounds, respectively, allowing the photoreduction of HgX2 to exhibit (+)MgIE or (–)MgIE while that of HgXY (i.e., MMHgX) exhibits only (+)MgIE. This is based on the premise that the photolysis of MMHgX is exclusively by cleavage of the weaker Hg–C bond rather than the stronger Hg–X bond. (+)MgIE is most evident for the photoreduction of MMHg+ species, as its 3σσ* 1e-LMCT state is energetically separated from other excited states in the paramagnetic intermediate, leading to the maximization of MgIE (Motta et al., 2020a). Stable Hg isotopes provide insight into the dynamics and metabolism of inorganic and methylated Hg in biota. Exposure to the former results in subtle odd-MIF, with a Δ199Hg Δ201Hg ratio close to unity at sampling, while for the latter, this ratio is greater (∼ 1.3) with a large odd-MIF (up to ∼ 5 ‰ in fish, Li et al., 2022b).
From Table 9, which summarizes the isotopic effects quantitatively observed in aqueous-phase laboratory studies, MgIE can have different signs for the same reactant depending on the reaction conditions, as exemplified by the Hg–cysteine–light system. Depending on the degree of photoconversion, the reduction of HgII in the presence of water-soluble diesel soot (aromatic polyacids and humic-like structures) exhibits swings in the direction of MgIE (Huang et al., 2021). Another example of the impact of pH/complexation on the evolution of MgIE can be seen in the UV-C photodegradation of MMHg+ in acidic and alkaline (adjusted with NH3) solutions. In the former, (+)MgIE is significant, but it is limited in the latter. For traditional elements with the same reaction mechanism, the strength development of MgIE depends on various factors, including viscosity, triplet sensitizer, and excited-state quenchers (Turro, 1983; Buchachenko, 2013). As seen in the laboratory experiments, both (+) and (–) net Hg MgIE were observed in samples related to the natural atmosphere, as previously reported in Section 8.2. The reaction conditions also affect the degree of turnover of the Hg reactant at which the onset of MgIE occurs, which incidentally does not correlate with a change in the overall reduction rate. To better interpret odd-MIF signatures and systematically elaborate the roles of reaction parameters (pH, presence of O2, light wavelength, etc., Rose et al., 2015) in excited-state kinetic isotope effects, experimental research is needed. For example, dissolved O2 is a well-known quencher of excited triplet states, but radical-O2 reactions have also been described to induce significant MgIE (Pliss et al., 2019). For the photoreduction of Hg in the presence of multifunctional ligands (such as DOM), the stoichiometry (Hg:L ratio) has been shown to play an important role in the magnitude of MgIE induced. Zheng and Hintelmann (2009) observed an E199Hg optimum (5 %) in anoxic photo-experiments with the DOM fraction from Dorset Lake, Ontario. This optimum is associated with a ligation mode in which all S-bonding functional groups are saturated by Hg2+ cations, increasing the proportion of Hg–O bonds and the ratio of bright triplets to bright singlets, thus making the MgIE increasingly positive. As the Hg:L ratio is further increased, the reduction rate (driven by Hg–O complexes) is significantly affected. The triplet–singlet spin evolution is limited to fewer HgL radical pairs, resulting in a lower −E199Hg (Epov, 2011a). In contrast to freshwater DOM, photo experiments with HgII in the presence of DOM extracted from marine phytoplankton yield (–)MgIE during reduction (Kritee et al., 2018).
Table 9Experimental fractionation factors determined for a variety of Hg redox transformations.
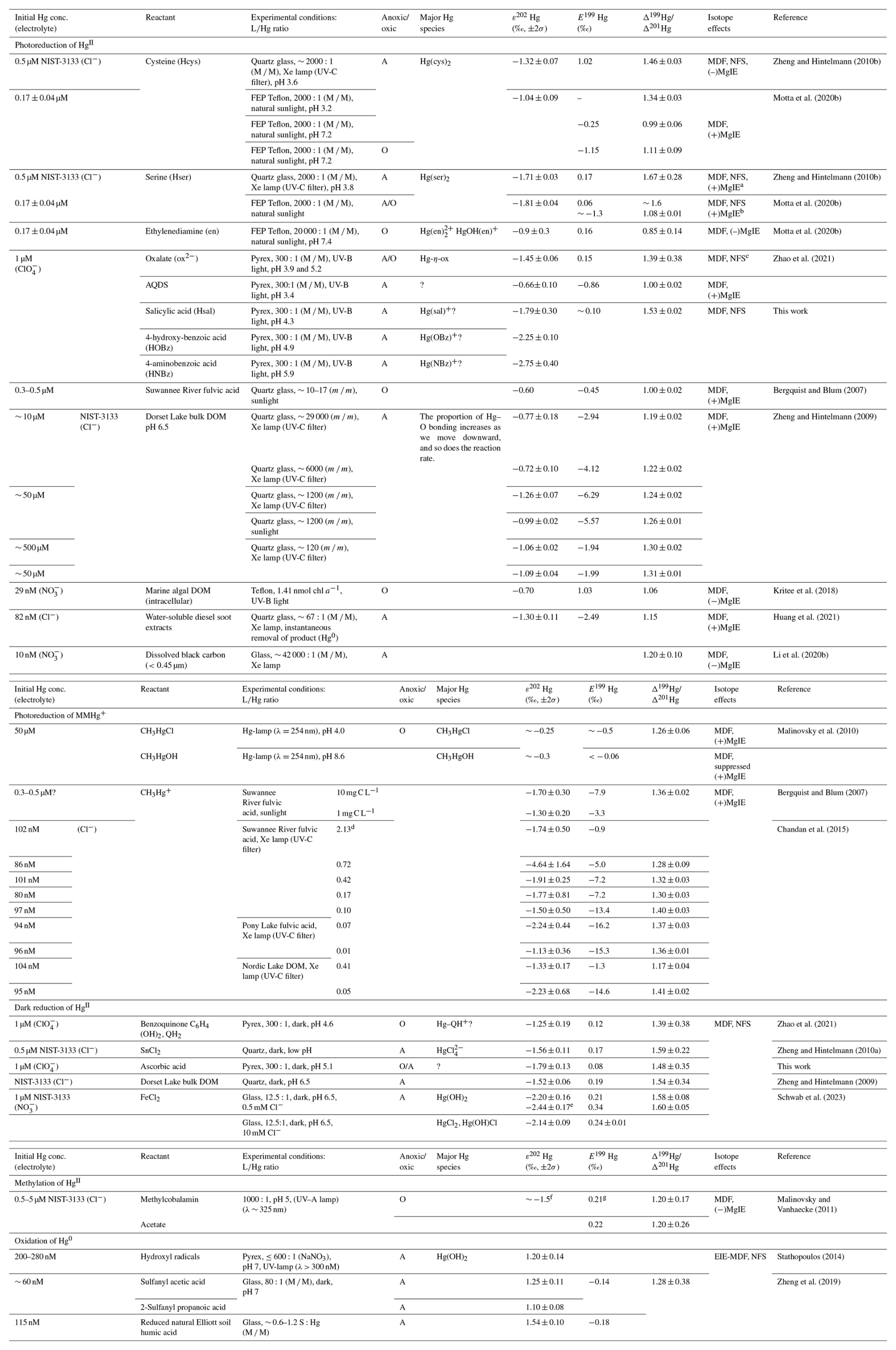
a Appears at 4 h photoreduction and beyond with a Hg of 1.10–1.18. b Onset of (+)MgIE at fR= 0.40−0.76 depending on reaction conditions. c A single experiment (anoxic, pH 6) on oxalate indicates (+)MgIE at fR=0.11. d MMHg/organic bound reduced sulfur (M M). e Refers to a closed system. f Applies to dark conditions; under UV-A irradiation demethylation gradually counteracts MMHg+ formation. g Potentially explained by photodegradation of MMHg+.
As shown in Table 9, photoreduction of Hg2+ often, but by no means always, is associated with high odd-MIF. For macromolecular entities such as DOM and fulvic acids and a selection of smaller organic ligands that use O–, N–, and S– donor atoms to complex with Hg2+, MgIE is initially induced in the photoreduction process, whereas for the amino acid serine, MgIE is triggered only after a significant turnover of Hg2+, the onset of which varies significantly depending on the reaction conditions (Zheng and Hintelmann, 2010b; Motta et al., 2020b). The experimental Δ199Hg/δ202Hg data are described to follow the same trajectory, regardless of when MgIE kicks in during serine-assisted photo-reduction. When oxalic acid was screened with a single light experiment (anoxic, pH 6, fR=0.11), (+)MgIE was observed; anoxic time series experiments with UV-B irradiation at pH 3.9 and 5.2 revealed no evidence of MgIE in the range investigated down to fR= 0.01. This is evidence that Hg oxalate complexes can be directly photodegraded by homolysis (Hg(η2−C2O) as well as heterolysis (Hg(η2−C2O Hg + 2 CO2). Heterolytic photoreduction does not induce MgIE but results in NFS with limited (−)odd-MIF, as is the case for ligation with the substituted aromatic carboxylic acids shown in the table. This also applies to thermal (dark) reduction by a unimolecular (e.g., Hg−QH Hg0 + Q + H+) or bimolecular (e.g., Hg2+ + Sn Hg0 + SnIV) processes. Although NFS is a general isotopic effect, its magnitude depends on the shift in the 6s orbital electron density, which is greater for a redox reaction than for ligand exchange or evaporation. In turn, ionic Hg complexes have greater NFS than more covalent complexes upon reduction to Hg0. NFS typically produces a characteristic Δ199Hg/Δ201Hg slope of ∼ 1.54 to 1.66, as determined from experimental studies and theoretical calculations. However, the application of linear regression to NFS odd-MIF data (Δ199Hg vs. Δ201Hg) is limited in several cases because the observations are distributed over such a small range that they approach the scale of the corresponding analytical precision. Table 9 gives 2 standard deviations of the slope of the linear fits using York's regression, and the uncertainty is so large that it does not allow a definitive Δ199Hg/Δ201Hg ratio to be determined. In these cases, it has been suggested that a better indicator of NFS is instead to confirm that the patterns of Hg isotope fractionation observed mimic the odd–even staggering pattern of nuclear charge radii (Motta et al., 2020b). The description of NFS is limited to equilibrium fractionation (Eq. 25) and predicts, similar to EIE-MDF (Eq. 23), the enrichment of heavier isotopes in the oxidized fraction of the red–ox pair. Calculations performed for a series of HgII complexes, both binary and heterogeneous, containing simple hard and soft ligands relative to Hg0, show that NFS makes the most significant contribution to ε202Hg (ranging in total from 46 to 85 % at 25 °C; Jiskra et al., 2012). The expected mass-independent enrichment E199HgNFS can be calculated based on the calculation of ε202HgNFS, using the scale factors βKIE−MDF and βNFS (Jiskra et al., 2012):
Reduction by FeII and p-substituted benzoic acids results in one of the highest magnitudes of experimentally observed kinetic MDF (Table 9). The former system has been studied anoxically as both an open and closed system (Schwab et al., 2023), where the fractionation is of the Rayleighian model (kinetic) and equilibrium type, respectively. The closed system permits overprinting with the signature of isotopic equilibrium fractionation between Hg0 and hydrolyzed Hg2+, which has been consistently determined in two independent studies to be −2.63 ‰ (Wang et al., 2021) and −2.44 ‰ (Schwab et al., 2023), respectively. As demonstrated below, the magnitude of the equilibrium isotope enrichment factor (ε202Hg) between Hg0 and thiol-bound HgII is significantly lower (1.1–1.6 ‰), which is related to the lower vibrational energy of Hg–S bonds than that of Hg–O/Cl bonds.
8.4.2 Oxidation
To the extent that isotopic effects in aqueous-phase Hg0 oxidation have been studied in the laboratory, it has been observed that oxidized Hg becomes isotopically heavier than the reactant. The observed fractionation does not conform to the Rayleigh model, but it is consistent with EIE in a closed system. Consequently, the isotope ratio of the product(s) linearly approaches that of the reactant at the beginning of the reaction. An example of atmospherically relevant oxidation is the rapid reaction with •OH (Reaction W R2, generated by photolysis of ) with ε202Hg = 1.20 ± 0.14 ‰ (Stathopoulos, 2014). Experiments with thiol-substituted carboxylic acids in the dark produced similar fractionation results (Table 9, Reaction W R7). Additionally, NFS produces a small odd-MIF signal that consistently acts in the opposite direction of mass-dependent fractionation (Zheng et al., 2019). The reason for observing EIE despite the continuous oxidation of Hg0 without any indication of reversibility in the form of back reactions has been attributed to the rapid exchange of Hg isotopes between the remaining Hg0 and the formed HgII complexes (Wang et al., 2020). There is currently debate surrounding the mechanism by which this exchange occurs (Wang et al., 2020, 2021; Zheng et al., 2019). In the presence of humic acid, the oxidation of dissolved Hg0 exhibits two kinetic regimes where the EIE is not fully established in the initial regime (Zheng et al., 2019). KIE-MDF during dark reduction in the presence of DOM and EIE-MDF during dark oxidation caused by humic acid results in fractionation in the same direction and magnitude, so unmasking the controlling redox process from isotopic measurements can be difficult.
8.5 Isotope fractionation during complexation, sorption, and surface-catalyzed reduction
8.5.1 Processes interfacing the aqueous phase
Theoretical computations of EIE based on the MDF and NFS generally agree with experimentally determined fractionation factors for complexation. Competitive complexation of HgII between one of the typical hard ligands HO− and Cl− and a soft ligand in the form of a thiol resin results in a lighter isotopic signature of the sulfur-bound HgII pool (ε202Hg values of −0.62 ‰ and −0.53 ‰, respectively), which is related to increased covalent bonding and electron density in the 6s Hg orbital (Wiederhold et al., 2010). For the sorption of dissolved HgII on α-FeOOH, the observed isotopic fractionation (ε202Hg ∼ −0.4 ‰) is exclusively determined by the process in solution, where a vanishingly small pool (<0.1 %) of isotopically lighter cations is in equilibrium with a bulk of neutral HgII molecules, with only the former being sorption active (Jiskra et al., 2012). Equilibration and kinetic fractionation have been reported to describe the precipitation process of β-HgS and HgO, respectively, from an initially acidic solution, with ε202Hg values between the precipitate and the supernatant being −0.63 ‰ and −0.32 ‰, respectively (Smith et al., 2015). Like adsorption on goethite, the observed fractionation during the precipitation of metacinnabar is interpreted as an effect of solution chemistry, in this case, a transition from O– to S– bonding for HgII. In addition to the homogeneous-phase reduction of HgII by FeII in aqueous solutions (Table 9), the heterogeneous-phase reduction of HgII by surface-bound (adsorbed FeII on goethite/boehmite) or structural FeII (magnetite , Schwab et al., 2023, and siderite/green rust FeCO3, Wang et al., 2021) has been studied isotopically. As shown in Table 10, the isotopic fractionation in heterogeneous reduction is closely related in magnitude to that of homogeneous fractionation by FeII (Table 9), except in the case of magnetite (whose iron structure is present in different oxidation states), which has a much more limited TMDF and MIF (ε202Hg = −1.38 ‰ and E199Hg = 0.13 ‰, respectively). All these processes, when determined with confidence, demonstrate Δ199Hg/Δ201Hg ratios within the range of 1.56 to 1.62, which indicates that the observed MIF (E199Hg in the range of 0.13 ‰ to 0.34 ‰) is caused by NFS.
8.5.2 Processes interfacing the gas phase
Section 8.4.1 and Table 9 refer to a study of HgII photoreduction of aqueous diesel soot, which includes experiments with a stationary soot phase mixed with HgCl2 on a quartz plate over which a slow flow of Ar gas passes, as discussed below (Huang et al., 2021). In comparison, photoreduction in aqueous- and solid-phase diesel soot shows equivalent enrichment of heavier isotopes in the Hg reactant of 1.26 ‰–1.75 ‰. This value overlaps with the values typical of Hg redox chemistry (Table 9). In contrast to the aqueous phase, the photoreduction in the solid phase shows a continuous strong MIF (this time, positive MgIE induced in the Hg0 product) throughout the reaction, whereas in the latter case, a large MIF of the opposite sign occurs after only ∼ 60 % of the reaction. Furthermore, the reduction rate increases with increasing carrier gas humidity. The photo-triggered MgIE is highest when the carrier gas is dehumidified but decreases rapidly as the RH increases (Table 10).
Table 10Experimental fractionation factors determined for HgII complexation, sorption, surface-catalyzed reduction, and processes interfacing the gas phase.
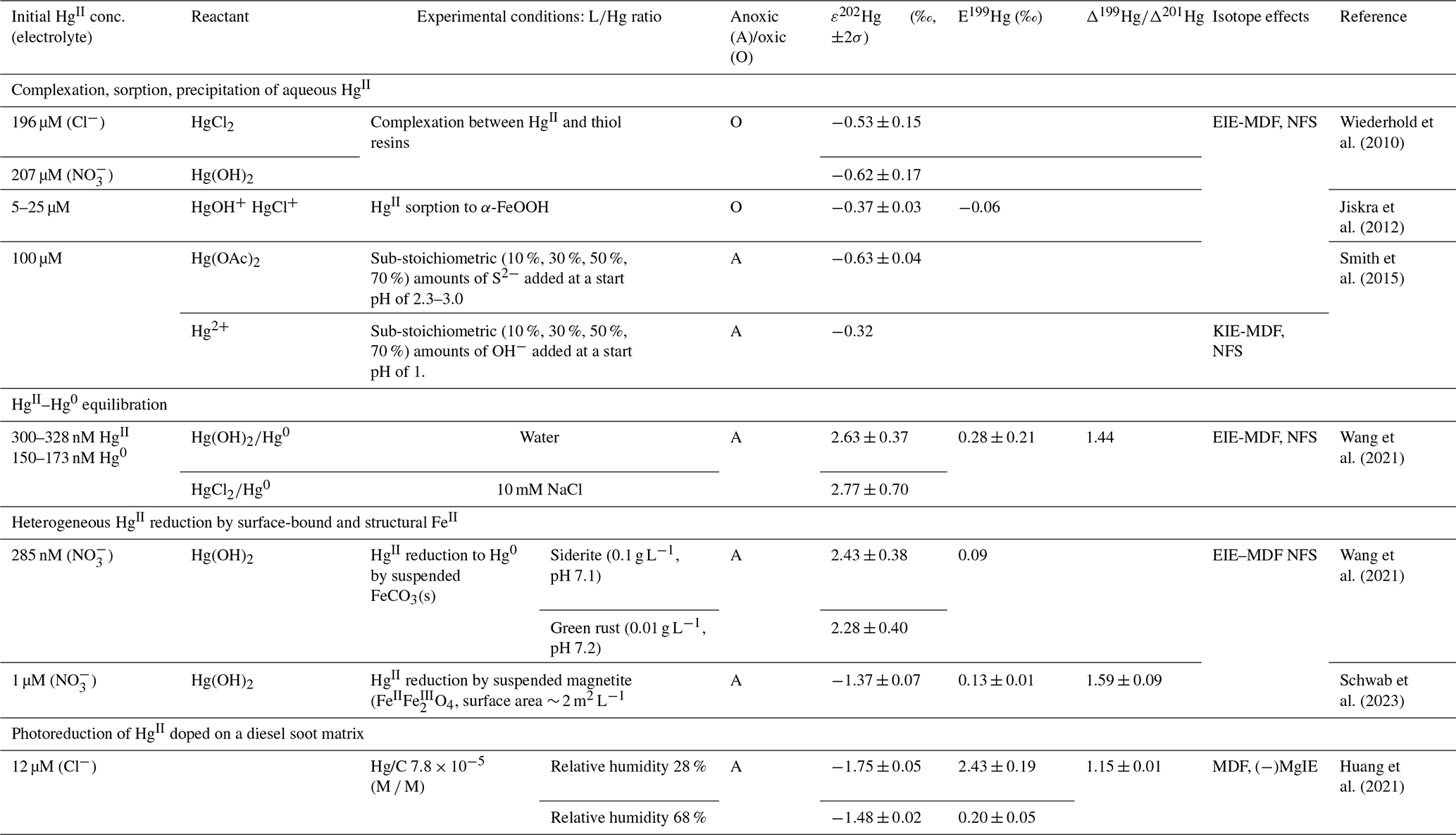
8.6 Isotopic fractionation during air–surface Hg0 gas exchange
The interaction between atmospheric Hg and the Earth's reservoirs has been discussed only briefly in Sect. 3.2, as this area has recently been covered by a literature review (Sommar et al., 2020). Importantly, the gas exchange of volatile Hg is bidirectional. Consequently, the net flux of Hg over an ecosystem may represent a delicate balance between opposing processes, including deposition/uptake versus re-emission. The end members of Hg exchange between the surface (biosphere, pedosphere, lithosphere, hydrosphere, and cryosphere) and atmosphere are all isotopically distinguishable (Liu et al., 2024). A combination of bulk measurements and analysis of stable Hg isotopic compositions enables separation of the contributions from atmospheric HgII and Hg0 deposition, as well as local partitioning between Hg0 deposition and re-emission. The isotopic composition of atmospheric Hg is presented and discussed in Sect. 8.2. In addition to the data, an updated compilation of complementary isotopic Hg data for reservoirs that are in contact with the atmosphere and thus can undergo gas exchange has been produced during the preparation of this review (Liu et al., 2024). In the following, we express absolute deposition with negative values and vice versa for emission throughout.
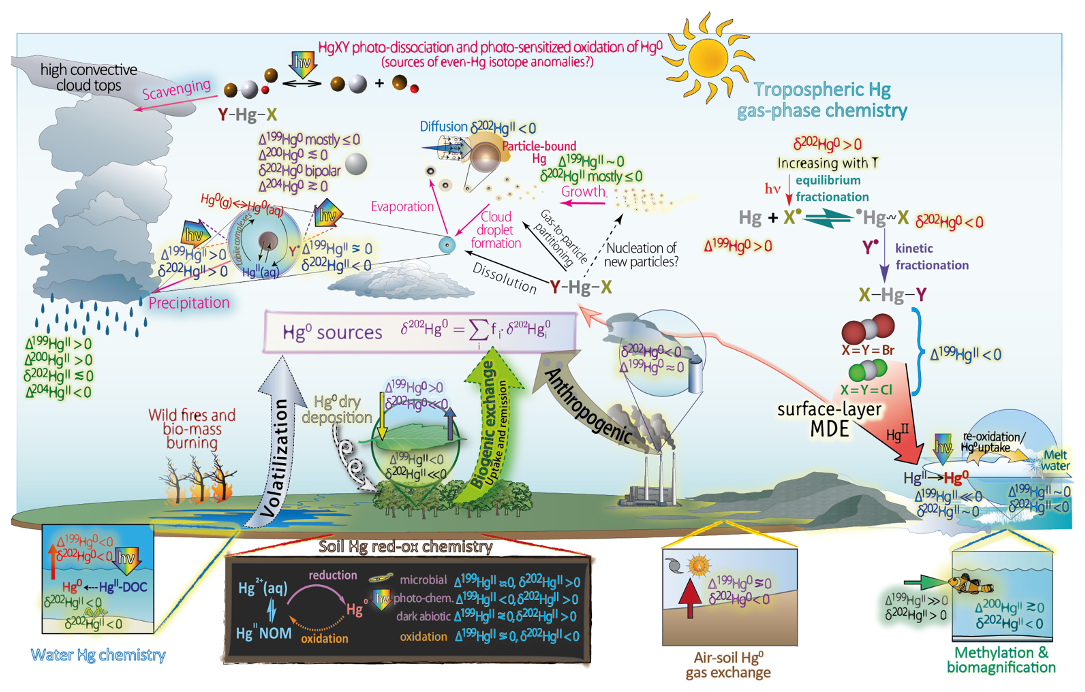
Figure 17Schematic illustration showing key biogeochemical Hg processes in Earth's surface reservoirs and their associated Hg isotope fractionation along with corresponding isotopic composition observations focused on the atmosphere. Section 8.2 addresses the isotopic characteristics of atmospheric mercury. The existing isotopic information on the gas-phase and water-phase redox transformations of mercury is presented in Sect. 8.3 and 8.4, respectively. Section 8.5 and 8.6 describe isotopic fractionation in heterogeneous chemical processes and processes involving Hg0 gas exchange between the atmosphere and the Earth's surface reservoirs, respectively.
8.6.1 Mixing and fractionation modeling of Hg0 deposition and post-depositional processes
Deposition
Isotope-based modeling by binary (e.g., Eq. 27) and ternary mixing with MDF, odd-MIF, and even-MIF signatures of atmospheric Hg0 and other Hg pools as end members has been applied to distinguish the fraction of Hg0 deposition via vegetated surfaces (Wang et al., 2020b, 2019b; Enrico et al., 2016; Obrist et al., 2017; Li et al., 2022a, 2023a, 2023b), soil (Zheng et al., 2016; Obrist et al., 2017; Wang et al., 2019b, 2020a), water (Jiskra et al., 2021; Zhang et al., 2023a), throughfall (Wang et al., 2020b), and snow run-off (Douglas and Blum, 2019), estimated to be 60 %–90 %, 32 %–105 %, 50 %–85 %, 34 %–82 %, and >75 % of total deposition, respectively. As a proxy for atmospheric Hg0, foliage/litter Hg has been used as an end-member in mixing modeling of Hg0 inputs to soil (Demers et al., 2013; Jiskra et al., 2015; Zhang et al., 2013), runoff (Jiskra et al., 2017), and stream water (Woerndle et al., 2018), which may introduce bias because a significant fraction of the gross air Hg0 incorporated as HgII in foliage is re-emitted after photoreduction (Yuan et al., 2019b). The contribution of Hg0 deposition to vegetation Hg uptake is greatest in foliage, followed by branches, bark, stems, and roots (Wang et al., 2020b; Liu et al., 2021a; Sun et al., 2017). The new Hg isotope evidence has demonstrated that Hg throughfall via the canopy and along stems, which was previously assumed to be derived mainly from wet and dry deposition of atmospheric RM (Wright et al., 2016), contains a larger proportion of Hg excreted from biomass, where it originated mainly from Hg0 uptake followed by translocation. The isotope mixing formula is used to determine the proportions of different isotope sources in a mixture, the simplest form of which is as follows:
Post-deposition
Isotopic and concentration measurements of Hg0 jointly in near-surface air and surface pore air/water, in addition to other isotopic data, allow the inference of processes by mass balance or Rayleigh-type models at the air–soil interface and in the surface soil (Jiskra et al., 2019; Li et al., 2023a; Yuan et al., 2021; Chen et al., 2023). For poorly drained boreal organic soil horizons (Histosols), in contrast to Podzols, mixing modeling indicates significant reductive loss (24 %–33 %) to the atmosphere by abiotic reduction (Jiskra et al., 2015). A further multi-process model is presented here, which is designed to elucidate the dynamic evolution of post-depositional Hg (>90 % from litterfall) on the subtropical forest floor over a 500-year period (Yuan et al., 2020). The results indicate that photolytic and microbial reduction processes exert an influence during the initial few years but are subsequently superseded by dark redox processes (exhibiting NFS) in the compost, where HgII finally becomes inert at depths of > 10 cm in the horizon after approximately 420 years. Studies of forest soils in different climatic zones have shown that microbial reduction (ε202Hg = −0.4 ‰, E199Hg ≈ 0, Kritee et al., 2007) plays a dominant role (Yuan et al., 2021; Chen et al., 2023), which, for rainforests, can explain up to 90 % of the HgII reduction in the upper soil horizon (Yuan et al., 2023b). In an open boreal peatland, photoreduction dominated the post-depositional process, accounting for the transformation of 30 % of the annually deposited Hg (Li et al., 2023a).
8.6.2 Enclosure and related flux measurements
Experimental investigations employing dynamic flux chambers (DFCs) have been conducted in both ambient and controlled environments with the objective of elucidating the isotopic dynamics of Hg0 exchange between the atmosphere and vegetation at the branch level (Yuan et al., 2019b; Chen et al., 2023), as well as between air and soil (Yuan et al., 2021; Chen et al., 2023; Zhu et al., 2022; Zhang et al., 2020), water (Zhang et al., 2023a), and snow (Sherman et al., 2010). For this application, in addition to traditional chambers (Demers et al., 2013; Chen et al., 2023; Zhu et al., 2024), a type was used that produces a uniform surface friction velocity over flat ground to couple with ambient shear conditions to scale to the ambient flux (Yuan et al., 2021, 2023b; Lin et al., 2012). The surface–atmosphere Hg0 flux is the result of complicated bidirectional processes, including Hg0 efflux from the surface and direct atmospheric Hg0 deposition.
Deposition and sink processes
When direct Hg0 deposition is measured absolutely and isotopically with a DFC, enrichment factors for TMDF (ε202Hg) and odd-MIF (E199Hg) may be calculated via a linearized Rayleigh fractionation model (Zhu et al., 2022; Mariotti et al., 1981):
where c represents the concentration, and the indices air and DFC refer to the air entering and exiting the DFC, respectively. Alternatively, Eq. (28) is applied to extract ε202Hgsurface–air using measurements of and δ202Hg0 at two pristine sites with and without vegetation (Enrico et al., 2016) or using daytime vs. nighttime segregated ambient air data at the same site (Jiskra et al., 2019). When direct deposition is measured isotopically with a DFC, the residual Hg0 in the chamber outlet shifts to be preferentially isotopically heavier, with a large but variable discrimination observed over soils (ε202Hgsoil–air = ∼ 0 ‰ to −5.8 ‰, Chen et al., 2023; Yuan et al., 2023b; Zhu et al., 2022) and over vegetation (ε202Hg −1 ‰ to −4.2 ‰, Yuan et al., 2019b; Enrico et al., 2016; Demers et al., 2013; Jiskra et al., 2019; Chen et al., 2023). Deposition in contact with any surface does not result in a significant change in Δ199Hg0, unlike the situation with δ202Hg0.
Information on the sink processes of Hg0 in the soil can be obtained by pursuing measurements of isotopic Hg0 in the soil pore air under sub-ambient concentration regimes. In tundra (Jiskra et al., 2019) and peatlands (Li et al., 2023a), the isotopic differences between ambient Hg0 and pore gas Hg0, whose concentrations is sub-ambient (∼ 0.4–∼ 0.6 and ∼ 0.2–∼ 0.7 ng m−3) and therefore mediate Hg0 net diffusion into the substrate via Eq. (28), have been linked to DOM-driven anaerobic oxidation in soil water exhibiting EIE (Zheng et al., 2019). Investigations of the Hg0 level in the pore air of forest soils provide a mixed picture, ranging from sites with highly depleted air (Obrist et al., 2014) to sites with up to 10 times enriched pore air (Yuan et al., 2019a) compared with the ambient concentrations above. In subtropical (Yuan et al., 2019a) and subalpine (Chen et al., 2023) forest soils, the concentration of Hg0 in pore air is typically higher than that in near-surface ambient air and shows seasonal isotopic variations (TMDF and odd-MIF), suggesting complexity in Hg0 gas exchange between air and soil. In tropical forest soils, pore air shifts from being nearly ambient during the rainy season to being markedly sub-ambient during the dry season (Yuan et al., 2023b). To resolve Hg0 flux partitioning here, a combination of DFC measurements of net fluxes and forced unidirectional efflux, soil pore air, and Hg isotopic composition in forest soil depth profiles are employed as inputs into isotope mass balance models based on odd-MIF (Yuan et al., 2021). Net fluxes measured by DFC are interpreted as a ternary mixing of deposition, Hg0 losses from the surface soil via HgII photoreduction, and a term generated by Hg redox processes (dark/microbial reduction vs. oxidation) in the organic soil horizon. Although associated with considerable uncertainties, the estimated gross deposition to the forest floor is between −7.8 and −1.8 ng m−2 h−1 for the subtropical site (Yuan et al., 2021); between −6.7 and −4.4 ng m−2 h−1 for the tropical site (Yuan et al., 2023b), depending on the season; and between −4.9 and −2.0 ng m−2 h−1 for the subalpine site, depending on the type of forest floor (Chen et al., 2023).
Figure 18Statistical summary of observations from isotopic studies of Hg0 exchange between the atmosphere and various groups of surface reservoirs on Earth. In the air–foliage group, data were taken from Yuan et al. (2019b) and Chen et al. (2023). For the air–snow group, data were taken from Sherman et al. (2010) and Douglas and Blum (2019). For the air–water group, data were taken from Zhang et al. (2023a, 2021a). For the air–soil group, data were taken from Zhang et al. (2020), Zhu et al. (2022), Yuan et al. (2021, 2023b), and Chen et al. (2023).
Foliar oxidation of Hg0 drives its reactive uptake and is the most important step in the accumulation of initially Hg0 uptake by plants (Liu et al., 2021b). Direct bio-oxidation from Hg0 to HgII has been traced to heme enzymes that catalyze the degradation of H2O2, specifically to a ferryl (O = FeIV) catalase radical cation complex (Ogata and Aikoh, 1984) that swiftly oxidizes Hg0 (1.4 × 104 M−1 s−1, Wigfield and Tse, 1986):
where E represents the heme group attached to the enzyme, which can provide an electron, reducing the formal oxidation number of iron from 5 to 4. Divalent Hg readily binds to soft functional groups on the enzyme as soon as it is formed. MDF fractionation during oxidation of the absorbed isotopically light Hg0 causes the product pool to be heavier than the reactant, which is consistent with observations that HgII incorporated into leaf shoots is only slightly lighter than Hg0 in ambient air. Notably, in contrast to the Hg pool in the leaf shoots, the Hg in the growing foliage of the current year shifted rapidly in the first months toward clearly negative δ202HgII signatures, the causes of which have been discussed elsewhere (Yuan et al., 2019b).
In contrast to the observations regarding the air modified by interactions with soil and foliage, the residual Hg0 in the outgoing air is significantly lighter than that in the incoming air ( ‰), as observed by DFC, for deposition regimes over freshwater surfaces (Zhang et al., 2023a). This may be interpreted as the dissolved Hg0(aq) being consumed by oxidation, whereby the rapid exchange of Hg isotopes between the remaining Hg0 and the formed HgII (Sect. 8.4.2) causes the former, which is partially returned to the gas phase, to exhibit a more negative δ202Hg0. In surface waters, photolytic re-reduction is also possible, which can be used for determining the isotopic composition of dissolved Hg0 (Zhang et al., 2021a).
During colder seasons with limited solar radiation, there is a small but persistent net Hg0 dry deposition over the snow-covered Arctic interior tundra (Obrist et al., 2017), whose interstitial snow air has sub-ambient concentrations (0.69 vs. 1.07 ng m−3) with comparatively more positive δ202Hg0 values (1.08 vs. 0.77 ‰, Jiskra et al., 2019). Using the exclusion method, this trajectory may reflect Hg0 uptake by ground lichens (Olson et al., 2019). Compared with the hinterland snowpack (∼ 50 ng m−2), the Arctic coastal snowpack has regionally much higher HgII pools (>2000 ng m−2), which are characteristically released as an ionic pulse in the runoff during snowmelt. High HgII concentrations in the coastal marine cryosphere are partially explained by AMDEs (described in Sect. 3.2, Douglas et al., 2017). However, coastal AMDE deposition is mostly re-emitted as Hg0 to the atmosphere before snow melts (see below). In contrast, the pulse in runoff appears to be related mainly to the reactive uptake of Hg0 on marine snow, which is rich in halogen compounds and other reactive species (see Sect. 7.3) (Douglas et al., 2017). In support of the significant reactive uptake of Hg0 on salt-laden snow, analogous odd-MIF signatures between ambient air Hg0 and snowmelt HgII have been reported (Δ199Hg values documented in Fig. 18b, Douglas and Blum, 2019).
Net exchange, source processes, and flux partitioning
Owing to the length of time (typically a few days) required to accumulate sufficient Hg to perform isotopic analysis, samples from a DFC measurement are a composite of periods of net emission and net deposition, unless the Hg0 concentration in the inlet is manipulated so that emission or deposition becomes persistent within the chamber. The TMDF and odd-MIF signatures from DFC measurements in ambient air (“net Hg0 exchange”) are calculated as follows (Zhu et al., 2022):
In the special case of using Hg-free air (zero air) to feed the DFC, and can be determined. The enrichment factors during net Hg0 exchange and emission are calculated using the following set of equations:
In a series of light-, temperature-, and substrate-moisture-controlled laboratory experiments with untilled (forest) and tilled (agricultural) soils, both with elevated Hg levels, enclosed in a DFC fed with Hg-free air, large Hg0 fluxes (≥ 500 ng m−2 h−1) were unanimously associated with the most negative values (−2.9 ‰ to −2.2 ‰ and −4.4 ‰ to −4.2 ‰ for agricultural and forest soils, respectively) when substrates were exposed to elevated temperatures in the dark (100–130 vs. 40 °C), while treatments with light, moisture, or a combination of both at room temperature produced more moderately negative values (−2.1 ‰ to −1.6 ‰ and −3.3 ‰ to −2.6 ‰ for agricultural and forest soils, respectively, Zhang et al., 2020). E199Hgemission of agricultural and forest soils displays a value of approximately 0.2 ‰, and was ∼ 1.55 for the temperature controls, suggesting that the treatment caused Hg0 loss propelled by the thermally driven reduction in HgII in the dark (Sect. 8.4.1). In the light and light–moisture exposure controls, the substrates differed in terms of the observed E199Hgemission, which for agricultural soils was 0.67 ‰ to 0.76 ‰ (mean) and for forest soils of a small magnitude both positive and negative (−0.03 ‰ to 0.18 ‰, mean). The E199Hgemission dichotomy may be interpreted as derived from a composite with Δ199Hg contributions from both (−)MgIE- and (+)MgIE-induced HgII photoreduction pathways, almost completely dominated by (−)MgIE processes (HgII bound to, e.g., N- and S-containing ligands) for agricultural soils and for forest soils with a larger contribution from (+)MgIE processes (HgII bound to, e.g., O-containing ligands), balancing odd-MIF fractionation from (−)MgIE processes. However, the agricultural soil placed under water (rice paddy) photoemits Hg0 characterized by a negative Δ199Hg0 ( ‰, Zhang et al., 2024), which is indicative of all observed HgII photoreduction in natural freshwaters studied in the laboratory as well as in situ.
A field study with DFC of cultivated or managed soils measured exchange fluxes (an MDF Rayleigh model yielded a 10 %–27 % contribution from deposition), which revealed net Hg0 emissions (fraction) associated with average ε202Hgexchange of −1.1 % to −0.1 ‰ and −1.6 % to −0.2 ‰ and average E199Hgex values of −0.27 % to −0.13 ‰ and 0.00 % to 0.14 ‰ for rural and urban soils, respectively. The above enrichment factors and E199Hgexchange ≈ E201Hgexchange indicate that the emitted Hg0 comes mainly from the pool produced by photoreduction. The air concentration positively influences the magnitude of deposition in soils so that at a critical concentration level (compensation point), the net flux tends to change direction. This is reflected in the apparent ε202Hgexchange, which varies with the ambient Hg0 concentration (Zhu et al., 2022). Analogous to laboratory experiments, in situ experiments on the subtropical forest floor have revealed that soil emissions of Hg0 are strongly negative (mean −3.0 ‰, Yuan et al., 2021), while the magnitude of for the tropical rainforest floor is much smaller but still negative (mean −0.7 ‰, Yuan et al., 2023b). E199Hgemission for subtropical forest soils exhibits positive values for all seasons over a considerable range (mean 0.1 ‰–0.7 ‰), whereas for rainforests, E199Hgemission is consistently positive, albeit to a lesser extent (mean 0.2 ‰–0.3 ‰). Limited negative values (mean values of −0.26 ‰, −0.54 ‰, −0.07 ‰ and −0.09 ‰) and consistently positive E199Hgexchange values (mean values of 0.42 ‰, 0.23 ‰, 0.39 ‰ and 0.30 ‰) are observed in net Hg0 gas exchange experiments over subtropical (Yuan et al., 2021), tropical (Yuan et al., 2023b), subalpine (Chen et al., 2023), and temperate (Demers et al., 2013) forest soils, respectively. In conclusion, bare or cultivated soils result in a greater degree of MDF isotope fractionation associated with Hg0 gas exchange with the atmosphere than forest soils do, where the effects of photic and thermal processes are limited by canopy shading. Temporally extensive chamber measurements conducted globally over the forest floor indicate net emissions (Yuan et al., 2019a). For the first three forest soil studies mentioned above, the DFC dataset also contains sufficient isotope data to enable the modeling of net flux partitioning into gross emission and gross deposition.
Re-emissions of Hg0 from perennial foliage of three beech species show an average positive ε202Hgemission and E199Hgemission of 0.6 ‰ and 0.3 ‰, respectively. The studied net exchange of Hg0 between foliage and air for montane evergreen deciduous (Yuan et al., 2019b) and spruce (Chen et al., 2023) forests is mostly on the uptake side, which indicates that is generally more positive than that of ambient air (Fig. 18a, mean shift of 0.72 ‰ for the latter site). The presence of bidirectional fluxes is, however, reflected in the observation that the E199Hgexchange for both sites is consistently positive (mean 0.08 ‰ and 0.13 ‰, respectively), albeit modestly, due to a contribution from Hg0 emissions resulting from (−)MgIE-induced photoreduction.
Isotopic studies of air–snow Hg0 interactions and post-depositional processes have typically been conducted in the Arctic (Araujo et al., 2022; Sherman et al., 2010; Zheng et al., 2021; Jiskra et al., 2019; Obrist et al., 2017; Douglas and Blum, 2019), with occasional studies at mid-latitudes (Kurz et al., 2021; Yuan et al., 2022). Hg in aging snowpacks exhibits by far the most extensive distribution of Δ199HgII among Earth's surface reservoirs, with observations of Δ199Hg progression reported in both positive (Kurz et al., 2021) and negative (Sherman et al., 2010; Zheng et al., 2021; Douglas and Blum, 2019) directions relative to fresh snow. As discussed in Sect. 8.2.1, the larger Δ199Hg spread observed in polar airborne Hg (Hg0 and RM) than in, for instance, high-altitude air from mid-latitudes can be attributed to the influence of AMDEs (during spring after sunrise and during summer) on a significant proportion of the collected polar data. Snow(fall) during the polar night is characterized by positive or near-zero Δ199Hg signatures, as is the case for most global precipitation data (Fig. 13c), while the Δ199Hg values of polar Hg0 for the same period are all slightly negative, which is consistent with the global Hg0 background pool (Fig. 13a). Only sporadic isotopic DFC measurements have been conducted over snow, yet ample measurements of polar air and snow as end-members still offer an understanding of air–surface Hg0 exchange following HgII deposition associated with AMDE. A seminal set of isotope data (Sherman et al., 2010) demonstrating a substantial odd-MIF triggered because of HgII photoreduction in snow was obtained from samples collected during a 9 d AMDE at the Alaskan Arctic coastline in conjunction with periods of minimal snowfall carrying high concentrations of scavenged HgII (0.5 ± 0.4 µg L−1; Johnson et al., 2008). Fresh snow, surface snow, and drifting snow presented, in order, rapidly increasing negative Δ199HgII values of −0.95 ‰ to −1.20 ‰, −2.41 ‰ to −2.63 ‰, and −3.84 ‰ to −5.08 ‰, which, according to Rayleigh fractionation, can correspond to 5 %–30 %, 35 %–50 %, and 65 %–75 % photoreduced HgII, respectively. A chamber measurement was conducted on AMDE-impacted drifting snow that had undergone substantial photoreduction (Δ199Hg ∼ −5.0 ‰) for 10.5 h of sunlight. The total DFC throughput, including the Hg0 emissions corresponding to 6 % of the total HgII in the snow plot (whose Δ199HgII dropped to ∼ −5.4 ‰), exhibited a Δ199Hg0 of −1.87 ‰. Mid-latitude snow (MI, USA), derived from polar vortex-transported air masses originating in AMDE-affected subarctic regions, shows, when Δ199Hg is plotted against δ202Hg, a regression of −3.32 ± 1.19 (Kurz et al., 2021), which, given the uncertainty in the line fit, appears to agree well with the corresponding regression of data from the Alaska DFC snow experiment of −3.44 ± 0.70 (Sherman et al., 2010).
Perennial data from the Canadian High Arctic show that HgII deposited on snow during the most frequent phase of AMDEs just after polar sunrise until early May, which is partly characterized by low temperatures and Arctic haze, has a significantly greater susceptibility to photoreduction and loss as Hg0 (up to 60 %) than that deposited later (<20 %, Zheng et al., 2021). As previously stated in Sect. 5.1.4, airborne HgII originating from High Arctic AMDEs undergoes rapid conversion to the particle phase between March and April, whereas unconverted GOM remains the dominant form between May and June. The cause of the reactivity of deposited HgII is unclear (Sherman et al., 2010; Kurz et al., 2021). It has been speculated that components of Arctic haze, such as black carbon, that cause photoreactivity of particulate HgII are the cause of the observed (−)MgIE signature (Zheng et al., 2021), which is supported by water-phase experiments with HgII and dissolved black carbon (Table 9, Li et al., 2020b). Concurrently, the restricted HgII reduction observed in Arctic snow toward the conclusion of spring is consistent with concurrent observations of substantial reactive uptake of Hg0 (see above; Douglas and Blum, 2019), indicating that the snowpack then contains species with a predominant oxidative capacity. However, during snowmelt on the inland tundra, net Hg0 deposition is disrupted by shifts in the isotopic signatures of snow interstitial air to those indicative of photoreduction, with Δ199Hg values decreasing to −1.37 ‰ in snow and −0.62 ‰ in snow interstitial air, which are consistently lower than those in ambient air (−0.23 ± 0.06 ‰). In contrast to Arctic snow, snow sampling in the U.S. Great Lakes area (with the exceptions noted above) generally results in increasing positive Δ199HgII values (up to 3.51 ‰) in aging snow (Kurz et al., 2021). Indicative of (−) and (+) MgIE triggering photoreduction, respectively, the snow data from coastal Alaska (Sherman et al., 2010; Douglas and Blum, 2019) and the Great Lakes region (Kurz et al., 2021) show steeper Δ199Hg δ202Hg trajectories than is the case for any of the well-studied HgII complex photoreductions in the laboratory (Tables 9 and 10), leaving the question of which snow HgII complexes are involved.
The mean MIF values (Δ199HgII and Δ200HgII) in the pools of fresh and seawater are between the mean values of global atmospheric Hg0 and wet precipitation. However, the variation is particularly pronounced for Δ199HgII in coastal seawater, lakes, and river water (Liu et al., 2024). After three different categories of lakes with DFC were studied, a Δ200Hg isotope mass balance model was used to partition the overall net emission fluxes into gross emission and deposition fluxes, which ranged from 2.1 to 4.2 ng m−2 h−1 and from −2.3 to −1.2 ng m−2 h−1, depending on the lake (Zhang et al., 2023a). Hg0 gross deposition exceeds the measured wet deposition across these lakes and accounts for 56 %–85 % of the total deposition (Feng et al., 2022). The anomalous observation of preferential deposition of heavier Hg isotopes over water has already been discussed. The results of the volatilization experiments of dissolved Hg0 in water indicate an MDF enrichment factor (ε202Hg) of −0.45 ‰ and a negligible E199Hg (Zheng et al., 2007). Emission-controlled experiments for one of the lakes yielded E199Hgemission of −0.38 ‰ and ε202Hgemission of −0.31 ‰, which are subject to large uncertainties, with a resulting E199HgHgemission trajectory of 1.26 ± 0.72, which is within the margin of error for HgII photoreduction mediated by fulvic acids (1.15 ± 0.07, Bergquist and Blum, 2007). The isotopic tracing of the formation of dissolved Hg0 in peat-covered groundwater from HgII in rainwater (1.24 ± 0.68) has also suggested that this process is the same type of photoreduction (Li et al., 2023a). The E199Hgexchange was between −0.76 ‰ and −0.32 ‰, with the highest absolute value for a clear mountain lake fed mainly glacial water, indicating that (+)MgIE photoreduction plays an important role, as has been shown early in laboratory experiments on natural freshwater (Bergquist and Blum, 2007; Zheng and Hintelmann, 2009). The observed substantial positive Δ199HgII shift of the sampled lake surface waters relative to HgII in precipitation can be interpreted as an effect of partial photoreduction of HgII. However, other sources, including MMHg photodegradation, have been suggested (Chen et al., 2016). As discussed in Sect. 4.2, Hg0 emissions from the ocean represent a primary source of Hg in the atmosphere. However, the isotopic signatures of this emission source remain largely unknown. In the absence of in situ sampling, photoexperiments with HgII in the presence of DOM extracted from marine phytoplankton produce (−)MgIE during reduction, in contrast to freshwater DOM (Kritee et al., 2018).
9.1 Theoretical chemistry contributions and challenges
This examination of the advancements made in our comprehension of the mercury cycle in the troposphere and stratosphere reveals iterative interactions among three distinct branches of atmospheric chemistry (modeling, field measurements, and laboratory measurements). Advances in computational chemistry have made seminal contributions to our understanding of gas-phase Hg(I,II) molecules in terms of their geometries, energies, UV–VIS spectra, and reaction kinetics. The treatment of strong relativistic effects, which largely determine the chemistry of Hg-containing species, is crucial for accurate results. Ab initio thermochemical calculations for atmospheric Hg species are performed at a higher level of theory, which incorporates core–valence electron correlation and coupled-cluster methods. This approach yields a significantly improved accuracy of ≤ 4 kJ mol−1, in accordance with high-quality experimental data. However, significant uncertainties in the estimates of the binding strength and thermal and photolytic stability of •HgII (Sect. 5.1.2) remain, limiting the ability to assess the occurrence and significance of iodine-induced Hg0 oxidation in the troposphere and lower stratosphere, as has been suggested from atmospheric observations (Murphy et al., 2006; Lee et al., 2024).
Compared with ab initio thermodynamics, the calculation of ab initio kinetics is a much more challenging task, for which transition state theory (TST) and RRKM theory are often used for barrier and non-barrier bimolecular reactions, respectively. More flexible methods (e.g., variational TST) are now applied to optimize the position of the transition state (TS) by varying it along the reaction coordinate to minimize the free activation energy, which more accurately estimates the rate than traditional TST, which assumes a single, fixed TS that irreversibly leads to products. The calculation of TS energies is more challenging than the calculation of energies of relative minima (metastable species) because of the involvement of extended bonds where the electronic wave function is less dominated by a single electronic configuration. Obtaining a correct barrier energy is crucial for calculating reliable rate constants, as a bias of ∼ 4 kJ mol−1 in the barrier height can lead to an error of nearly an order of magnitude in the resulting rate constant (Ariya and Peterson, 2005; Ariya et al., 2009).
For gas-phase reactions (Sect. 5), calculated rate constants have been presented and compared with those experimentally determined in the laboratory. The level of agreement varies from relatively good (≤ 30 % as Reactions G R1–G R3) to inconsistent (Reactions G R20a, b and G R22). Owing to the complex shape of their potential energy surfaces, the rates of assumed key reactions such as Reactions (G R27), (G R45), and (G R63) are inherently difficult to constrain theoretically (Sect. 5.1.4) and thus require empirical verification, preferably using PLP-LIF or similar techniques. A direct reaction between water vapor and YHgIIO• has recently been proposed for Y = OH (Reaction G R60, Saiz-Lopez et al., 2022). If this reaction is realized with the given rate expression in models, it will result in the conversion of essentially all HOHgIIO• to the completely stable Hg(OH)2 in the tropics. This type of reaction also requires empirical verification and should be given priority in laboratory experiments.
9.2 Laboratory measurement techniques and limitations
The absolute determination of rate constants experimentally with pulsed laser-assisted methods (reaction times typically <0.1 ms), such as PLP-LIF, is more easily facilitated when secondary reactions are negligible and therefore does not contribute to the measured values. In general, absolute determination is conducted by obtaining pseudo-first-order conditions, whereby the more stable reactant is present in a density more than 10-fold that of the other reactant. However, for Hg, this method is viable only for studies that are conducted at elevated temperatures (typically ≥100 °C). At atmospheric temperatures, the relatively low vapor pressure of Hg0 (in comparison to, for instance, DMHg) precludes the possibility of such experiments. Despite the challenges for Hg0, a flow PLP-LIF system has many advantages, including the ability to measure the rate coefficient over a wide range of temperatures and pressures and to test the effect of a change in the bath gas (third body). Nevertheless, to exploit these advantages, alternative methods have been used in which the Hg species is not in excess, but in which the excess is X = Cl and Br when the reaction Hg + X•+M is studied, while it is instead Y = O3, NO2, NO, and O2 when the interaction between •HgIBr and Y is studied. In the study of the former reaction type, X• is present in excess, but its concentration decreases over time owing to the rapid three-body recombination of the species into X2 and M. This results in additional Hg0 exponential decay. To achieve a fit to the observed Hg0 time profiles, rate coefficients must be obtained through numerical integration. This requires monitoring both the X• and Hg0 time profiles using LIF, with the absolute concentration of X atoms known with precision. The experimental measurements of the rate coefficients for the Hg + X•+M reaction by Donohoue et al. (2005, 2006) are in accordance with the findings of theoretical computational studies.
The conversion of •HgIBr by bimolecular elimination reduction (Reactions G R18 and G R20b; Wu et al., 2020; Wu et al., 2022), addition (oxidation assisted by M [Reaction G R20a]; Wu et al., 2020), or abstraction (Reaction G R22, Gómez Martín et al., 2022) is constrained by the capacity to generate sufficiently high densities of •HgIBr through the gas-phase photolysis of HgBr2 in deep UV. Because the vapor pressure of HgBr2 is low (less than one-tenth that of Hg0), it is necessary to keep the HgBr2 source at least 30 °C and the flow tube reactor at least 10 °C higher to prevent vapor condensation. A higher temperature increases the thermal dissociation of •HgIBr; therefore, a large excess of Y is required for the •HgIBr + Y reaction to dominate the conversion of •HgIBr. In the context of laboratory experiments necessitating deep UV irradiation, it is essential to consider that oxygen atoms are formed through the partial photolysis of O3 and NO2, thereby enabling O(3P) to react with •HgIBr (O + HgBr• → Hg + BrO•, Reaction G R23). Experiments to study the reactions of •HgIBr with NO2 and O3 will inevitably result in the observation of a partially reversible oxidation process. This is due to the occurrence of secondary chemistry, including Reactions (G R14), (G R23), (G R24), and (G R29), which take place concurrently with the title Reactions (G R20) and (G R22). Furthermore, to elucidate the influence of secondary chemistry on the observed •HgIBr disappearance, a comprehensive series of experiments must be conducted, with pressure, temperature, [•HgIBr], [Y], and [O] as variables. This necessitates numerical modeling to isolate the individual rate constants. While the laboratory study of •HgIBr + O3 gives an experimental rate constant for the Reaction (G R22) that is in good agreement with computational predictions (Castro Pelaez et al., 2022), experimental kinetic data for •HgIBr + NO2 (Reaction G R20), which must be decoupled into termolecular oxidation (Reaction G R20a) and reduction (Reaction G R20b) reactions, respectively, indicate that computational methods overestimate the rate constants for both channels (Wu et al., 2020). Later, experimental investigations revealed that Reaction (G R20) cannot fully account for observations but that significant losses of •HgIBr must occur via side reactions, probably involving Reaction (G R23), which was unexplored at the time. These intractable shortcomings present a challenge to validating a majority of the proposed reaction steps by computational quantum chemistry in the atmospheric Hg redox cycle, including YHgIIO• chemistry, through experimental means. As requested by theoretical chemists (Edirappulige et al., 2023) and modelers (Shah et al., 2021), better rate constants are needed for YHgO•+ CH4 and YHgO•+ CO reactions, especially for Y = Br and OH, to better assess the atmospheric fate of YHgO•, i.e., whether YHgO• will be mainly reduced or form closed-shell HgII compounds under different atmospheric conditions. As a workaround in the absence of experimentally determined rate constants, Khiri et al. (2020) proposed efforts to perform molecular dynamics simulations via computationally more sophisticated variational TST with multidimensional tunneling.
Many of the proposed key gas-phase Hg species lack experimental characterization (such as spectral proofs). The main method for studying such gas-phase molecules has been spectroscopy after preparation by matrix isolation, which has thus far been used to study the products of photochemical reactions of excited Hg atoms, e.g., O3 (Butler et al., 1979), O2 (Andrews et al., 2023), H2 (Wang and Andrews, 2005b), H2O (Wang and Andrews, 2005a), F2 (Wang et al., 2007), and OF2 (Andrews et al., 2012), in a matrix host of solid Ar and Ne at a cold (typically 4–7 K) surface. Section 5.1.4 has already described some of the isolated molecules of interest, namely Hg(OH)2 (Wang and Andrews, 2005a) and the fluorine analog of YHgIIO• (Andrews et al., 2012). Other studies involve mercury halide molecules (Loewenschuss et al., 1969) and their adducts (Tevault et al., 1977). The reaction mechanism for the formation of Hg(OH)2 tentatively involves insertion as a first step: Hg(3P) + O2 + H2→ (OHgO)* + H2→ HOHgOH, where OHgO () is implicitly indicated as a reactive intermediate (cf. Reaction G R12, although, unlike the analogous complexes for the other Group 12 metals, OZnO and OCdO, it has yet to be identified by IR spectra, Chertihin and Andrews, 1997). Apart from MS experiments of the laser desorption ionization and time-of-flight type with solid HgO as the source and detection of (HgO)x clusters in the gas phase (Jayasekharan and Sahoo, 2014), there is one early (Butler et al., 1979) and one recent (Andrews et al., 2023) matrix study of the products of the Hg(3P) + O2 system, where both 16O2 and 18O2 were used as reagents. The former experiments required co-deposition of Hg with 0.5 % to 5 % O3 in excess of Ar under deep UV photolysis for oxidation to occur, while the latter experiments used laser-ablated Hg atoms energetic enough to form oxygen atoms when deposited in a cryogenic matrix doped with 0.3 % 16O2 or 18O2, which reacts upon annealing to form O3 and a series of HgOx species (x=1 to 3). The observed fundamental harmonic vibrational frequencies in different cryogenic matrices for the simple oxide Hg–O are in the range of 500–600 cm−1, as predicted by high-level calculations (Shepler and Peterson, 2003; Peterson et al., 2007), indicating the presence of a weakly ionic molecule. This is also true for HgO2 and HgO3, which have superoxide (Hg) and ozonide (Hg) characteristics, respectively (Andrews et al., 2023). Notably, the study did not isolate linear mercury dioxide, OHgO, and evidence for this species remains weak. Nevertheless, this species is included as a metastable adduct in the Reaction (G R12) scheme, the key reaction for Hg0 turnover in the stratosphere, whose complex potential energy surface forms the basis of ab initio kinetic calculations. In these calculations (Saiz-Lopez et al., 2022), the energy of the Hg–O bond is assumed to be 27.3 kJ mol−1, which is significantly higher than the most recently published high-level calculation values (Peterson et al., 2007; Cremer et al., 2008). Increased activity and innovation in advanced experimental studies characterizing key species and reactions are needed to verify models of atmospheric Hg chemistry that currently appear overly reliant on computational chemistry. The innovation could be, for example, finding a laboratory method to capture the temporal behavior of HgO (perhaps generated by the spin-allowed Hg(1S) + O(1D) reaction or by reacting DMHg with O(3P)) in the presence of gas-phase coreactants (with reference to Reactions G R73 and G R74), performing a detailed study of Reactions (G R8) and (G R12), or finding a synthetic route to matrix-isolate species such as BrHgO• from laser-ablated Hg atoms.
9.3 Model validation and observational gaps
Current limitations and challenges in accurately measuring speciated atmospheric mercury (Gustin et al., 2024; Sect. 3.1) mean that the basis for verifying models in detail is insufficient, despite reliable measurements of Hg0 in air and HgII in wet deposition. Nevertheless, some models have been developed by including KCl-denuder-based HgII measurements in the reference material (Shah et al., 2021; Fu et al., 2024), which are known to suffer from low bias, and others (Saiz-Lopez et al., 2020, 2025), which seem to stick strictly to RM data for validation or include KCl-denuder-based HgII measurements corrected according to Marusczak et al. (2017); consequently, the evaluation is qualitative (Shah et al., 2021) and consistently fails to simulate the magnitude of recurring episodes of highly oxidized mercury originating in the free troposphere (underestimation by up to several hundred percent, Elgiar et al., 2025; Gustin et al., 2023). In Sect. 3.2, we highlighted the discrepancies that exist in terms of the atmospheric budget and the fluxes into and out of it. Particularly, new model results concerning the importance of the stratosphere are inconsistent with existing empirical data and require further elaboration, as does the stratospheric chemistry discussed above. Recent observations have shown that there are abundant anthropogenic emissions of reactive halogens (e.g., Br2 and BrCl) over continental, densely populated areas of South and East Asia that also have high Hg emissions. This is now beginning to be modeled as a key component of the regional atmospheric Hg redox cycle (Fu et al., 2024), but more field measurements are needed for confirmation. The artisanal and small-scale gold mining sector is currently the largest source of anthropogenic Hg0 emissions to the atmosphere globally. However, emission estimates using bottom-up approaches, such as emissions inventories, are highly unconstrained (Cheng et al., 2023). Independent techniques, such as top-down constraints in the form of inverse modeling of atmospheric observations, are plausibly necessary to ensure the accuracy of these estimates (Sommar et al., 2020).
The difficulties of accurately determining the speciation of HgII in atmospheric water through equilibrium modeling and thus identifying the pool of reducible complexes have been described (Sect. 4.3). Additionally, the potential for simulating the gas–particle distribution of atmospherically oxidized Hg has been explored (Sect. 7.1.1). Stable isotope data have been analyzed to constrain Hg redox chemistry in the atmosphere (Song et al., 2024; Zhen et al., 2024), but there are profound knowledge gaps that require state-of-the-art theoretical and experimental investigations. Section 8.2 describes the isotopic composition of atmospheric samples, a pool generally consisting of filtered air divided into PBM and gaseous mercury (∼ Hg0) and precipitation samples, including those of cloud and fog water. The isotope measurements on RM, which were also performed, are briefly presented here (Fu et al., 2021). With a measuring line consisting of a CEM (HgII(g)), a filter (HgII(p)) and a trap consisting of halogen-impregnated activated carbon (Hg0) in series, the analysis shows that the three groups are clearly isotopically separated from each other and that the resulting samples can thus provide further insights into atmospheric processes (Xuewu Fu, personal communication, 2025). The discovery, made over a decade ago, that atmospheric samples contain a significant level of the even-mass-number isotope MIF with seasonal and geospatial variations has been a source of both benefit and puzzlement for scientists (Sect. 8.2.3 and 8.2.4). As even Hg MIF variation is limited to samples from a few localities thus far (compare Figs. 13 and 14), Δ200Hg and Δ204Hg in the environment are considered conservative tracers because of their generally narrow range, and values of Δ200Hg and Δ204Hg on the land surface and in water confine the relative contribution of Hg0 to the HgII exchange process with the atmosphere. Nevertheless, the underlying chemical processes that give rise to anomalous MIF and the atmospheric conditions that facilitate its occurrence remain to be elucidated in greater detail. In addition to laboratory-based investigations, future field experiments that report vertical profiles of isotopic Hg0 and HgII in the atmosphere may prove invaluable in further constraining the sources of even-MIF.
Data supporting this study are openly available from Zenodo at https://doi.org/10.5281/zenodo.17079781.
Writing (original draft preparation): JOS. Data curation: JOS, XS, XT, and GS. Analysis and visualization: JOS, XS, and XT. Writing (review and editing): JOS, GS, CJL, and XF.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
We would like to express our sincere gratitude to the executive editor board and the editor for agreeing to publish this lengthy review. We would also like to thank the two excellent reviewers, whose comments greatly improved the quality of the paper.
This research has been supported by the National Natural Science Foundation of China (grant nos. 42150710535 and 42373068).
This paper was edited by Andreas Hofzumahaus and reviewed by two anonymous referees.
Ababneh, F. A., Scott, S. L., Al-Reasi, H. A., and Lean, D. R. S.: Photochemical reduction and reoxidation of aqueous mercuric chloride in the presence of ferrioxalate and air, Sci. Total Environ., 367, 831–839, https://doi.org/10.1016/j.scitotenv.2006.02.018, 2006.
Agnan, Y., Le Dantec, T., Moore, C. W., Edwards, G. C., and Obrist, D.: New constraints on terrestrial surface-atmosphere fluxes of gaseous elemental mercury using a global database, Environ. Sci. Technol., 50, 507–520, https://doi.org/10.1021/acs.est.5b04013, 2016.
Ahmed, S., Thomas, J. L., Angot, H., Dommergue, A., Archer, S. D., Bariteau, L., Beck, I., Benavent, N., Blechschmidt, A. M., Blomquist, B., Boyer, M., Christensen, J. H., Dahlke, S., Dastoor, A., Helmig, D., Howard, D., Jacobi, H. W., Jokinen, T., Lapere, R., Laurila, T., Quelever, L. L. J., Richter, A., Ryjkov, A., Mahajan, A. S., Marelle, L., Pfaffhuber, K. A., Posman, K., Rinke, A., Saiz-Lopez, A., Schmale, J., Skov, H., Steffen, A., Stupple, G., Stutz, J., Travnikov, O., and Zilker, B.: Modeling the coupled mercury-halogen-ozone cycle in the central Arctic during spring, Elementa-Science of the Anthropocene, 11, 00129, https://doi.org/10.1525/elementa.2022.00129, 2023.
Ai, Y., Wang, C., Videen, G., and Pan, Y.-L.: Optically levitated, single-particle reactor for the study of surface and heterogeneous chemistry-reactions of particulate-bound mercury with ozone in air, Chem. Phys. Lett., 817, 140428, https://doi.org/10.1016/j.cplett.2023.140428, 2023.
Akagi, H., Fujita, Y., and Tkabatake, E.: Photochemical methylation of inorganic mercuric compounds in aqueous acetic acid solutions, Nippon Kagaku Kaishi, 1974, 1180–1184, https://doi.org/10.1246/nikkashi.1974.1180, 1974.
Alberts, J. J., Schindler, J. E., Miller, R. W., and Nutter, D. E.: Elemental mercury evolution mediated by humic acid, Science, 184, 895–896, 1974.
Ambrose, J. L.: Improved methods for signal processing in measurements of mercury by Tekran® 2537A and 2537B instruments, Atmos. Meas. Tech., 10, 5063–5073, https://doi.org/10.5194/amt-10-5063-2017, 2017.
Amirbahman, A., Kent, D. B., Curtis, G. P., and Marvin-DiPasquale, M. C.: Kinetics of homogeneous and surface-catalyzed mercury(II) reduction by iron(II), Environ. Sci. Technol., 47, 7204–7213, https://doi.org/10.1021/es401459p, 2013.
Amos, H. M., Jacob, D. J., Holmes, C. D., Fisher, J. A., Wang, Q., Yantosca, R. M., Corbitt, E. S., Galarneau, E., Rutter, A. P., Gustin, M. S., Steffen, A., Schauer, J. J., Graydon, J. A., Louis, V. L. St., Talbot, R. W., Edgerton, E. S., Zhang, Y., and Sunderland, E. M.: Gas-particle partitioning of atmospheric Hg(II) and its effect on global mercury deposition, Atmos. Chem. Phys., 12, 591–603, https://doi.org/10.5194/acp-12-591-2012, 2012.
Amos, H. M., Sonke, J. E., Obrist, D., Robins, N., Hagan, N., Horowitz, H. M., Mason, R. P., Witt, M., Hedgecock, I. M., Corbitt, E. S., and Sunderland, E. M.: Observational and modeling constraints on global anthropogenic enrichment of mercury, Environ. Sci. Technol., 49, 4036–4047, https://doi.org/10.1021/es5058665, 2015.
Amyot, M., Morel, F. M. M., and Ariya, P. A.: Dark oxidation of dissolved and liquid elemental mercury in aquatic environments, Environ. Science Technol., 39, 110–114, https://doi.org/10.1021/es035444k, 2005.
Andersson, M. E., Gårdfeldt, K., Wängberg, I., and Strömberg, D.: Determination of Henry's law constant for elemental mercury, Chemosphere, 73, 587–592, https://doi.org/10.1016/j.chemosphere.2008.05.067, 2008.
Andrews, L., Wang, X., Gong, Y., Schloeder, T., Riedel, S., and Franger, M. J.: Spectroscopic observation of a group 12 oxyfluoride: A matrix-isolation and quantum-chemical investigation of mercury oxyfluorides, Angew. Chem. Int. Edit, 51, 8235–8238, https://doi.org/10.1002/anie.201204331, 2012.
Andrews, L. S., Tsegaw, Y. A., Cho, H.-G., and Riedel, S.: Observation and characterization of the Hg-O diatomic molecule: A matrix-isolation and quantum-chemical investigation, Chemistry – A Eur. J., 29, e202202740, https://doi.org/10.1002/chem.202202740, 2023.
Angot, H., Dastoor, A., De Simone, F., Gårdfeldt, K., Gencarelli, C. N., Hedgecock, I. M., Langer, S., Magand, O., Mastromonaco, M. N., Nordstrøm, C., Pfaffhuber, K. A., Pirrone, N., Ryjkov, A., Selin, N. E., Skov, H., Song, S., Sprovieri, F., Steffen, A., Toyota, K., Travnikov, O., Yang, X., and Dommergue, A.: Chemical cycling and deposition of atmospheric mercury in polar regions: review of recent measurements and comparison with models, Atmos. Chem. Phys., 16, 10735–10763, https://doi.org/10.5194/acp-16-10735-2016, 2016.
Araujo, B. F., Osterwalder, S., Szponar, N., Lee, D., Petrova, M. V., Pernov, J. B., Ahmed, S., Heimburger-Boavida, L. E., Laffont, L., Teisserenc, R., Tananaev, N., Nordstrom, C., Magand, O., Stupple, G., Skov, H., Steffen, A., Bergquist, B., Pfaffhuber, K. A., Thomas, J. L., Scheper, S., Petaja, T., Dommergue, A., and Sonke, J. E.: Mercury isotope evidence for Arctic summertime re-emission of mercury from the cryosphere, Nat. Commun., 13, 4956, https://doi.org/10.1038/s41467-022-32440-8, 2022.
Ariya, P. and Peterson, K. A.: Chemical transformation of gaseous elemental mercury in the atmosphere, in: Dynamics of Mercury Pollution on Regional and Global Scales: Atmospheric Processes and Human Exposures Around the World, edited by: Pirrone, N., and Mahaffey, K. R., Springer, 261–294, https://doi.org/10.1007/0-387-24494-8_12, 2005.
Ariya, P. A., Khalizov, A., and Gidas, A.: Reactions of gaseous mercury with atomic and molecular halogens: Kinetics, product studies, and atmospheric implications, J. Phys. Chem. A, 106, 7310–7320, 2002.
Ariya, P. A., Peterson, K., Snider, G., and Amyot, M.: Mercury chemical transformations in the gas, aqueous and heterogeneous phases: state-of-the-art science and uncertainties, in: Mercury fate and transport in the global atmosphere, Emissions, measurements and models, edited by: Mason, R., and Pirrone, N., Springer, 459–501, https://doi.org/10.1007/978-0-387-93958-2_15, 2009.
Ariya, P. A., Skov, H., Grage, M. M. L., and Goodsite, M. E.: Gaseous elemental mercury in the ambient atmosphere: Review of the application of theoretical calculations and experimental studies for determination of reaction coefficients and mechanisms with halogens and other reactants, in: Advances in Quantum Chemistry: Applications of Theoretical Methods to Atmospheric Science, edited by: Sabin, J. R. and Brandas, E. J., Academic Press, 43–55, https://doi.org/10.1016/s0065-3276(07)00204-3, 2008.
Ariya, P. A., Amyot, M., Dastoor, A., Deeds, D., Feinberg, A., Kos, G., Poulain, A., Ryjkov, A., Semeniuk, K., Subir, M., and Toyota, K.: Mercury physicochemical and biogeochemical transformation in the atmosphere and at atmospheric interfaces: A review and future directions, Chem. Rev., 115, 3760–3802, https://doi.org/10.1021/cr500667e, 2015.
Auyang, D., Chen, J., Zheng, W., Zhang, Y., Shi, G., Sonke, J., Cartigny, P., Hongming, C., Yuan, W., Liu, L., Gai, P., and Liu, C.: South-hemispheric marine aerosol Hg and S isotope compositions reveal different oxidation pathways, National Science Open, 1, 20220014, https://doi.org/10.1360/nso/20220014, 2022.
Auzmendi-Murua, I., Castillo, Á., and Bozzelli, J. W.: Mercury oxidation via chlorine, bromine, and iodine under atmospheric conditions: Thermochemistry and kinetics, J. Phys. Chem. A, 118, 2959–2975, https://doi.org/10.1021/jp412654s, 2014.
Åkerblom, S., Meili, M., and Bishop, K.: Organic matter in rain: An overlooked influence on mercury deposition, Environ. Sci. Technol. Lett., 2, 128–132, https://doi.org/10.1021/acs.estlett.5b00009, 2015.
Babaev, N. S., Cheltsov, A. N., Sazykin, A. A., Sosnin, L. Y., and Kuchelev, A. P.: Centrifugal enrichment of mercury isotopes, Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment, 613, 473–476, https://doi.org/10.1016/j.nima.2009.10.006, 2010.
Balabanov, N. B. and Peterson, K. A.: Mercury and reactive halogens: The thermochemistry of Hg + [Cl2, Br2, BrCl, ClO, and BrO], J. Phys. Chem. A, 107, 7465–7470, 2003.
Balabanov, N. B. and Peterson, K. A.: Accurate theoretical near-equilibrium potential energy and dipole moment surfaces of HgClO and HgBrO, J. Chem. Phys., 120, 6585–6592, 2004.
Balabanov, N. B., Shepler, B. C., and Peterson, K. A.: Accurate global potential energy surface and reaction dynamics for the ground state of HgBr2, J. Phys. Chem. A, 109, 8765–8773, 2005.
Banic, C. M., Beauchamp, S. T., Tordon, R. J., Schroeder, W. H., Steffen, A., Anlauf, K. A., and Wong, H. K. T.: Vertical distribution of gaseous elemental mercury in Canada, J. Geophys. Res.-Atmos., 108, 4264, https://doi.org/10.1029/2002JD002116, 2003.
Bartels-Rausch, T., Huthwelker, T., Jori, M., Gaggeler, H. W., and Ammann, M.: Interaction of gaseous elemental mercury with snow surfaces: laboratory investigation, Environ. Res. Lett., 3, 045009, https://doi.org/10.1088/1748-9326/3/4/045009, 2008.
Bartels-Rausch, T., Krysztofiak, G., Bernhard, A., Schläppi, M., Schwikowski, M., and Ammann, M.: Photoinduced reduction of divalent mercury in ice by organic matter, Chemosphere, 82, 199–203, 2011.
Bash, J. O., Carlton, A. G., Hutzell, W. T., and Bullock, O. R.: Regional air quality model application of the aqueous-phase photo reduction of atmospheric oxidized mercury by dicarboxylic acids, Atmosphere, 5, 1–15, https://doi.org/10.3390/atmos5010001, 2014.
Bauer, D., Campuzano-Jost, P., and Hynes, A. J.: Rapid, ultra-sensitive detection of gas phase elemental mercury under atmospheric conditions using sequential two-photon laser induced fluorescence, J. Environ. Monit., 4, 339–343, 2002.
Bauer, D., D'Ottone, L., Campuzano-Jost, P., and Hynes, A. J.: Gas phase elemental mercury: A comparison of LIF detection techniques and study of the kinetics of reaction with the hydroxyl radical, J. Photoch. Photobio. A, 157, 247–256, 2003.
Bauer, D., Everhart, S., Remeika, J., Tatum Ernest, C., and Hynes, A. J.: Deployment of a sequential two-photon laser-induced fluorescence sensor for the detection of gaseous elemental mercury at ambient levels: fast, specific, ultrasensitive detection with parts-per-quadrillion sensitivity, Atmos. Meas. Tech., 7, 4251–4265, https://doi.org/10.5194/amt-7-4251-2014, 2014.
Baxendale, J. H. and Bridge, N. K.: Photoreduction of ferric compounds in aqueous solution, J. Phys. Chem., 59, 783–788, https://doi.org/10.1021/j150530a022, 1955.
Baya, P. A., Gosselin, M., Lehnherr, I., St Louis, V. L., and Hintelmann, H.: Determination of monomethylmercury and dimethylmercury in the Arctic marine boundary layer, Environ. Sci. Technol., 49, 223–232, 2015.
Bencardino, M., D'Amore, F., Angot, H., Angiuli, L., Bertrand, Y., Cairns, W., Diéguez, M. C., Dommergue, A., Ebinghaus, R., Esposito, G., Komínková, K., Labuschagne, C., Mannarino, V., Martin, L., Martino, M., Neves, L. M., Mashyanov, N., Magand, O., Nelson, P., Norstrom, C., Read, K., Sholupov, S., Skov, H., Tassone, A., Vítková, G., Cinnirella, S., Sprovieri, F., and Pirrone, N.: Patterns and trends of atmospheric mercury in the GMOS network: Insights based on a decade of measurements, Environ. Pollut., 363, 125104, https://doi.org/10.1016/j.envpol.2024.125104, 2024.
Bergquist, B. A. and Blum, J. D.: Mass-dependent and -independent fractionation of Hg isotopes by photoreduction in aquatic systems, Science, 318, 417–420, 2007.
Berkovic, A. M., Bertolotti, S. G., Villata, L. S., Gonzalez, M. C., Pis Diez, R., and Mártire, D. O.: Photoinduced reduction of divalent mercury by quinones in the presence of formic acid under anaerobic conditions, Chemosphere, 89, 1189–1194, 2012.
Berkovic, A. M., Gonzalez, M. C., Russo, N., Del Carmen Michelini, M., Diez, R. P., and Mártire, D. O.: Reduction of mercury(II) by the carbon dioxide radical anion: A theoretical and experimental investigation, J. Phys. Chem. A, 114, 12845–12850, 2010.
Bianco, A., Deguillaume, L., Vaïtilingom, M., Nicol, E., Baray, J.-L., Chaumerliac, N., and Bridoux, M.: Molecular characterization of cloud water samples collected at the Puy de Dôme (France) by Fourier transform ion cyclotron resonance mass spectrometry, Environ. Sci. Technol., 52, 10275–10285, https://doi.org/10.1021/acs.est.8b01964, 2018.
Bieser, J., Slemr, F., Ambrose, J., Brenninkmeijer, C., Brooks, S., Dastoor, A., DeSimone, F., Ebinghaus, R., Gencarelli, C. N., Geyer, B., Gratz, L. E., Hedgecock, I. M., Jaffe, D., Kelley, P., Lin, C.-J., Jaegle, L., Matthias, V., Ryjkov, A., Selin, N. E., Song, S., Travnikov, O., Weigelt, A., Luke, W., Ren, X., Zahn, A., Yang, X., Zhu, Y., and Pirrone, N.: Multi-model study of mercury dispersion in the atmosphere: vertical and interhemispheric distribution of mercury species, Atmos. Chem. Phys., 17, 6925–6955, https://doi.org/10.5194/acp-17-6925-2017, 2017.
Bigeleisen, J. and Wolfsberg, M.: Theoretical and experimental aspects of isotope effects in chemical kinetics, in: Advances in Chemical Physics, 15–76, https://doi.org/10.1002/9780470143476.ch2, 1957.
Bittrich, D. R., Chadwick, S. P., Babiarz, C. L., Manolopoulos, H., Rutter, A. P., Schauer, J. J., Armstrong, D. E., Collett, J., and Herckes, P.: Speciation of mercury (II) and methylmercury in cloud and fog water, Aerosol Air Qual. Res., 11, 161–U181, https://doi.org/10.4209/Aaqr.2010.08.0067, 2011.
Bloom, N. S., Grout, A. K., and Prestbo, E. M.: Development and complete validation of a method for the determination of dimethyl mercury in air and other media, Anal. Chim. Acta, 546, 92–101, 2005.
Blum, J. D. and Bergquist, B. A.: Reporting of variations in the natural isotopic composition of mercury, Anal. Bioanal. Chem., 388, 353–359, 2007.
Blum, J. D. and Johnson, M. W.: Recent developments in mercury stable isotope analysis, Rev. Mineral. Geochem., 82, 733–757, 2017.
Breitinger, D. and Brodersen, K.: Development of and problems in the chemistry of mercury-nitrogen compounds, Angewandte Chemie International Edition in English, 9, 357–367, https://doi.org/10.1002/anie.197003571, 1970.
Brønsted, J. N. and de Hevesy, G.: The separation of the isotopes of mercury, Nature, 106, 144–144, 1921.
Buchachenko, A. L.: Mass-independent isotope effects, J. Phys. Chem. B, 117, 2231–2238, https://doi.org/10.1021/jp308727w, 2013.
Buchachenko, A. L.: Mercury isotopes in Earth and environmental chemistry, Russ. J. Phys. Chem. B, 12, 635–644, https://doi.org/10.1134/s1990793118040048, 2018.
Burkholder, J. B., Sander, S. P., Abbatt, J., Barker, J. R., Cappa, C., Crounse, J. D., Dibble, T. S., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Percival, C. J., Wilmouth, D. M., and Wine, P. H.: Chemical kinetics and photochemical data for use in atmospheric studies, Evaluation No. 19, Jet Propulsion Laboratory, Pasadena, https://jpldataeval.jpl.nasa.gov/, 2019.
Burnard, P.: The noble gases as geochemical tracers, Springer, https://doi.org/10.1007/978-3-642-28836-4, 2013.
Butler, R., Katz, S., Snelson, A., and Stephens, J. B.: Identification of HgOx species by matrix-isolation spectroscopy, J. Phys. Chem., 83, 2578–2580, 1979.
Buxton, G. V., Mulazzani, Q. G., and Ross, A. B.: Critical review of rate constants for reactions of transients from metal ions and metal complexes in aqueous solution, J. Phys. Chem. Ref. Data, 24, 1055–1349, https://doi.org/10.1063/1.555966, 1995.
Cai, H. and Chen, J.: Mass-independent fractionation of even mercury isotopes, Sci. Bull., 61, 1–9, https://doi.org/10.1007/s11434-015-0968-8, 2015.
Callear, A. B.: Excited mercury complexes, Chem. Rev., 87, 335–355, 1987.
Callear, A. B. and Shiundu, P. M.: Temperature-dependence of the Hg 6s6p(3P0→3P1) transition induced by nitrogen, Chem. Phys. Lett., 136, 342–345, https://doi.org/10.1016/0009-2614(87)80263-4, 1987.
Callear, A. B., Patrick, C. R., and Robb, J. C.: Reaction of excited mercury (Hg 63P1) with oxygen, T. Faraday Soc., 55, 280–287, 1959.
Calvert, J. G. and Lindberg, S. E.: Mechanisms of mercury removal by O3 and OH in the atmosphere, Atmos. Environ., 39, 3355–3367, 2005.
Calvert, J. G., Orlando, J. J., Stockwell, W. R., and Wallington, T. J.: The mechanisms of reactions influencing atmospheric ozone, Oxford University Press, https://doi.org/10.1093/oso/9780190233020.001.0001, 2015.
Cardiano, P., Cucinotta, D., Foti, C., Giuffrè, O., and Sammartano, S.: Potentiometric, calorimetric, and 1H NMR investigation on Hg2+-mercaptocarboxylate interaction in aqueous solution, J. Chem. Eng. Data, 56, 1995–2004, https://doi.org/10.1021/je101007n, 2011.
Cario, G. and Franck, J.: Über Zerlegung von Wasserstoffmolekülen durch angeregte Quecksilberatome, Z. Phys., 11, 161–166, https://doi.org/10.1007/BF01328410, 1922.
Carmona-Garcia, J., Saiz-Lopez, A., Mahajan, A. S., Wang, F., Borrego-Sanchez, A., Acuna, A. U., Cuevas, C. A., Davalos, J. Z., Feinberg, A., Spolaor, A., Ruiz-Lopez, M. F., Francisco, J. S., and Roca-Sanjuan, D.: Photoreduction of mercuric bromides in polar ice, P. Natl. Acad. Sci. USA, 122, e2422885122, https://doi.org/10.1073/pnas.2422885122, 2025.
Carpi, A.: Mercury from combustion sources: A review of the chemical species emitted and their transport in the atmosphere, Water Air Soil Pollut., 98, 241–254, 1997.
Castro Pelaez, P., Kellö, V., Cernusak, I., and Dibble, T.: Together, not separately, OH and O3 oxidize Hg(0) to Hg(II) in the atmosphere, J. Phys. Chem. A, 126, 8266–8279, https://doi.org/10.1021/acs.jpca.2c04364, 2022.
Chandan, P., Ghosh, S., and Bergquist, B. A.: Mercury isotope fractionation during aqueous photo-reduction of monomethylmercury in the presence of dissolved organic matter, Environ. Sci. Technol., 49, 259–267, https://doi.org/10.1021/es5034553, 2015.
Chen, C., Huang, J.-H., Li, K., Osterwalder, S., Yang, C., Waldner, P., Zhang, H., Fu, X., and Feng, X.: Isotopic characterization of mercury atmosphere – foliage and atmosphere – soil exchange in a Swiss subalpine coniferous forest, Environ. Sci. Technol., 57, 15892–15903, https://doi.org/10.1021/acs.est.3c03576, 2023.
Cheng, I., Xu, X., and Zhang, L.: Overview of receptor-based source apportionment studies for speciated atmospheric mercury, Atmos. Chem. Phys., 15, 7877–7895, https://doi.org/10.5194/acp-15-7877-2015, 2015.
Chen, J., Pehkonen, S. O., and Lin, C. J.: Degradation of monomethylmercury chloride by hydroxyl radicals in simulated natural waters, Water Res., 37, 2496–2504, 2003.
Chen, J., Hintelmann, H., Feng, X., and Dimock, B.: Unusual fractionation of both odd and even mercury isotopes in precipitation from Peterborough, ON, Canada, Geochim. Cosmochim. Acta, 90, 33–46, https://doi.org/10.1016/j.gca.2012.05.005, 2012.
Chen, J., Hintelmann, H., Zheng, W., Feng, X., Cai, H., Wang, Z., Yuan, S., and Wang, Z.: Isotopic evidence for distinct sources of mercury in lake waters and sediments, Chem. Geol., 426, 33–44, https://doi.org/10.1016/j.chemgeo.2016.01.030, 2016.
Chen, L., Wang, H. H., Liu, J. F., Tong, Y. D., Ou, L. B., Zhang, W., Hu, D., Chen, C., and Wang, X. J.: Intercontinental transport and deposition patterns of atmospheric mercury from anthropogenic emissions, Atmos. Chem. Phys., 14, 10163–10176, https://doi.org/10.5194/acp-14-10163-2014, 2014.
Chen, Y., Zhang, Q., Zhang, L., Liu, X., Li, Y., Liu, R., Wang, Y., Song, Y., Li, Y., Yin, Y., and Cai, Y.: Light-induced degradation of dimethylmercury in different natural waters, J. Hazard. Mater., 470, 134113, https://doi.org/10.1016/j.jhazmat.2024.134113, 2024.
Cheng, Y. C., Watari, T., Seccatore, J., Nakajima, K., Nansai, K., and Takaoka, M.: A review of gold production, mercury consumption, and emission in artisanal and small-scale gold mining (ASGM), Resour. Policy, 81, 3370–3370, https://doi.org/10.1016/j.resourpol.2023.103370, 2023.
Chertihin, G. V. and Andrews, L.: Reactions of laser-ablated Zn and Cd atoms with O2: Infrared spectra of ZnO, OZnO, CdO, and OCdO in solid argon, J. Chem. Phys., 106, 3457–3465, https://doi.org/10.1063/1.473441, 1997.
Chi, Y., Yan, N. Q., Qu, Z., Qiao, S. H., and Jia, J. P.: The performance of iodine on the removal of elemental mercury from the simulated coal-fired flue gas, J. Hazard. Mater., 166, 776–781, https://doi.org/10.1016/j.jhazmat.2008.11.130, 2009.
Cohen-Atiya, M. and Mandler, D.: Studying thiol adsorption on Au, Ag and Hg surfaces by potentiometric measurements, J. Electroanal. Chem., 550, 267–276, https://doi.org/10.1016/S0022-0728(02)01145-2, 2003.
Cole, A. S., Steffen, A., Pfaffhuber, K. A., Berg, T., Pilote, M., Poissant, L., Tordon, R., and Hung, H.: Ten-year trends of atmospheric mercury in the high Arctic compared to Canadian sub-Arctic and mid-latitude sites, Atmos. Chem. Phys., 13, 1535–1545, https://doi.org/10.5194/acp-13-1535-2013, 2013.
Cole, A. S., Steffen, A., Eckley, C. S., Narayan, J., Pilote, M., Tordon, R., Graydon, J. A., St Louis, V. L., Xu, X. H., and Branfireun, B. A.: A survey of mercury in air and precipitation across Canada: patterns and trends, Atmosphere, 5, 635–668, 2014.
Conaway, C. H., Black, F. J., Weiss-Penzias, P., Gault-Ringold, M., and Flegal, A. R.: Mercury speciation in Pacific coastal rainwater, Monterey Bay, California, Atmos. Environ., 44, 1788–1797, 2010.
Conaway, C. H., Black, F. J., Gault-Ringold, M., Pennington, J. T., Chavez, F. P., and Flegal, A. R.: Dimethylmercury in coastal upwelling waters, Monterey Bay, California, Environ. Sci. Technol., 43, 1305–1309, https://doi.org/10.1021/es802705t, 2009.
Cooke, C. A., Martínez-Cortizas, A., Bindler, R., and Gustin, M. S.: Environmental archives of atmospheric Hg deposition – A review, Sci. Total Environ., 709, 134800, https://doi.org/10.1016/j.scitotenv.2019.134800, 2020.
Cremer, D., Kraka, E., and Filatov, M.: Bonding in mercury molecules described by the normalized elimination of the small component and coupled cluster theory, Phys. Chem. Chem. Phys., 9, 2510–2521, 2008.
Criss, R. E.: Principles of stable isotope distribution, Oxford University Press, 248 pp., https://doi.org/10.1093/oso/9780195117752.001.0001, 1999.
Custodio, D., Ebinghaus, R., Spain, T. G., and Bieser, J.: Source apportionment of atmospheric mercury in the remote marine atmosphere: Mace Head GAW station, Irish western coast, Atmos. Chem. Phys., 20, 7929–7939, https://doi.org/10.5194/acp-20-7929-2020, 2020.
Custódio, D., Pfaffhuber, K. A., Spain, T. G., Pankratov, F. F., Strigunova, I., Molepo, K., Skov, H., Bieser, J., and Ebinghaus, R.: Odds and ends of atmospheric mercury in Europe and over the North Atlantic Ocean: temporal trends of 25 years of measurements, Atmos. Chem. Phys., 22, 3827–3840, https://doi.org/10.5194/acp-22-3827-2022, 2022.
Davis, M. and Lu, J. L.: Calibration sources for gaseous oxidized mercury: A review of source design, performance, and operational parameters, Crit. Rev. Anal. Chem., 54, 1748–1757, https://doi.org/10.1080/10408347.2022.2131373, 2024.
Deacon, G. B., Faulks, S. J., and Pain, G. N.: The synthesis of organometallics by decarboxylation reactions, in: Advances in Organometallic Chemistry, edited by: Stone, F. G. A. and West, R., Academic Press, 237–276, https://doi.org/10.1016/S0065-3055(08)60576-6, 1986.
Deeds, D. A., Ghoshdastidar, A., Raofie, F., Guérette, É.-A., Tessier, A., and Ariya, P. A.: Development of a particle-trap preconcentration-soft ionization mass spectrometric technique for the quantification of mercury halides in air, Anal. Chem., 87, 5109–5116, https://doi.org/10.1021/ac504545w, 2015.
Demers, J. D., Blum, J. D., and Zak, D. R.: Mercury isotopes in a forested ecosystem: Implications for air-surface exchange dynamics and the global mercury cycle, Global Biogeochem. Cy., 27, 222–238, 2013.
Demers, J. D., Sherman, L. S., Blum, J. D., Marsik, F. J., and Dvonch, J. T.: Coupling atmospheric mercury isotope ratios and meteorology to identify sources of mercury impacting a coastal urban-industrial region near Pensacola, Florida, USA, Global Biogeochem. Cy., 29, 1689–1705, https://doi.org/10.1002/2015GB005146, 2015.
Dibble, T. S. and Schwid, A. C.: Thermodynamics limits the reactivity of BrHg radical with volatile organic compounds, Chem. Phys. Lett., 659, 289–294, https://doi.org/10.1016/j.cplett.2016.07.065, 2016.
Dibble, T. S., Zelie, M. J., and Jiao, Y.: Quantum chemistry guide to PTRMS studies of as-yet undetected products of the bromine-atom initiated oxidation of gaseous elemental mercury, J. Phys. Chem. A, 118, 7847–7854, https://doi.org/10.1021/jp5041426, 2014.
Dibble, T. S., Zelie, M. J., and Mao, H.: Thermodynamics of reactions of ClHg and BrHg radicals with atmospherically abundant free radicals, Atmos. Chem. Phys., 12, 10271–10279, https://doi.org/10.5194/acp-12-10271-2012, 2012.
Dibble, T. S., Tetu, H. L., Jiao, Y. G., Thackray, C. P., and Jacob, D. J.: Modeling the OH-initiated oxidation of mercury in the global atmosphere without violating physical laws, J. Phys. Chem. A, 124, 444–453, https://doi.org/10.1021/acs.jpca.9b10121, 2020.
Dickinson, R. G. and Sherrill, M. S.: Formation of ozone by optically excited mercury vapor, P. Natl. Acad. Sci. USA, 12, 175–178, 1926.
Dommergue, A., Bahlmann, E., Ebinghaus, R., Ferrari, C., and Boutron, C.: Laboratory simulation of Hg0 emissions from a snowpack, Anal. Bioanal. Chem., 388, 319–327, 2007.
Dommergue, A., Sprovieri, F., Pirrone, N., Ebinghaus, R., Brooks, S., Courteaud, J., and Ferrari, C. P.: Overview of mercury measurements in the Antarctic troposphere, Atmos. Chem. Phys., 10, 3309–3319, https://doi.org/10.5194/acp-10-3309-2010, 2010.
Dong, W., Bian, Y., Liang, L., and Gu, B.: Binding constants of mercury and dissolved organic matter determined by a modified ion exchange technique, Environ. Sci. Technol., 45, 3576–3583, 2011.
Donohoue, D. L.: Kinetic studies of the oxidation pathways of gaseous elemental mercury, Ph. D. Thesis, Department of Marine and Atmospheric Chemistry, University of Miami, Coral Gables, Florida, 261 pp., http://scholarlyrepository.miami.edu/oa_dissertations/113, 2008.
Donohoue, D. L., Bauer, D., and Hynes, A. J.: Temperature and pressure dependent rate coefficients for the reaction of Hg with Cl and the reaction of Cl with Cl: A pulsed laser photolysis-pulsed laser induced fluorescence study, J. Phys. Chem. A, 109, 7732–7741, 2005.
Donohoue, D. L., Bauer, D., Cossairt, B., and Hynes, A. J.: Temperature and pressure dependent rate coefficients for the reaction of Hg with Br and the reaction of Br with Br: A pulsed laser photolysis-pulsed laser induced fluorescence study, J. Phys. Chem. A, 110, 6623–6632, 2006.
Donovan, P. M., Blum, J. D., Yee, D., Gehrke, G. E., and Singer, M. B.: An isotopic record of mercury in San Francisco Bay sediment, Chem. Geol., 349, 87–98, 2013.
Douglas, T. A. and Blum, J. D.: Mercury isotopes reveal atmospheric gaseous mercury deposition directly to the Arctic coastal snowpack, Environ. Sci. Technol. Lett., 6, 235–242, https://doi.org/10.1021/acs.estlett.9b00131, 2019.
Douglas, T. A., Sturm, M., Blum, J. D., Polashenski, C., Stuefer, S., Hiemstra, C., Steffen, A., Filhol, S., and Prevost, R.: A Pulse of mercury and major ions in snowmelt runoff from a small Arctic Alaska watershed, Environ. Sci. Technol., 51, 11145–11155, https://doi.org/10.1021/acs.est.7b03683, 2017.
Dumarey, R., Dams, R., and Hoste, J.: Comparison of the collection and desorption efficiency of activated-charcoal, silver, and gold for the determination of vapor-phase atmospheric mercury, Anal. Chem., 57, 2638–2643, 1985.
Dunham-Cheatham, S. M., Lyman, S., and Gustin, M. S.: Comparison and calibration of methods for ambient reactive mercury quantification, Sci. Total Environ., 856, 159219, https://doi.org/10.1016/j.scitotenv.2022.159219, 2023.
Eder, J. M.: Ein neues chemisches Photometer mittelst Quecksilberoxalat zur Bestimmung der Intensität der ultravioletten Strahlen des Tageslichtes und Beiträge zur Photochemie des Quecksilberchlorides, Ber. Dtsch Chem. Ges., 13, 166–168, https://doi.org/10.1002/cber.18800130150, 1880.
Edirappulige, D. T. H., Cheng, L., Castro Pelaez, P., and Dibble, T.: Nitrate radical cannot initiate oxidation of Hg(0) to Hg(II) in the laboratory or at ground level in the atmosphere, Environ. Sci. Technol. (preprint), https://doi.org/10.26434/chemrxiv-2024-qwh3w-v2, submitted, 2024.
Edirappulige, D. T. H., Kirby, I. J., Beckett, C. K., and Dibble, T. S.: Atmospheric chemistry of HOHgIIO? mimics that of a hydroxyl radical, J. Phys. Chem. A, 127, 8392–8403, https://doi.org/10.1021/acs.jpca.3c04159, 2023.
Edwards, B. A., Kushner, D. S., Outridge, P. M., and Wang, F.: Fifty years of volcanic mercury emission research: Knowledge gaps and future directions, Sci. Total Environ., 757, 143800–143800, https://doi.org/10.1016/j.scitotenv.2020.143800, 2021.
Elgiar, T. R., Gratz, L., Hallar, A. G., Volkamer, R., and Lyman, S. N.: Underestimation of atmospheric oxidized mercury at a mountaintop site by the GEOS-Chem chemical transport model, EGUsphere [preprint], https://doi.org/10.5194/egusphere-2025-977, 2025.
Enrico, M., Le Roux, G., Marusczak, N., Heimbürger, L. E., Claustres, A., Fu, X. W., Sun, R. Y., and Sonke, J. E.: Atmospheric mercury transfer to peat bogs dominated by gaseous elemental mercury dry deposition, Environ. Sci. Technol., 50, 2405–2412, 2016.
Epov, V.: Mechanisms of oxidation-reduction reactions can be predicted by the magnetic isotope effect, Adv. Phys. Chem., 450325, https://doi.org/10.1155/2011/450325, 2011a.
Epov, V. N.: Magnetic isotope effect and theory of atomic orbital hybridization to predict a mechanism of chemical exchange reactions, Phys. Chem. Chem. Phys., 13, 13222–13231, https://doi.org/10.1039/c1cp21012b, 2011b.
Faïn, X., Helmig, D., Hueber, J., Obrist, D., and Williams, M. W.: Mercury dynamics in the Rocky Mountain, Colorado, snowpack, Biogeosciences, 10, 3793–3807, https://doi.org/10.5194/bg-10-3793-2013, 2013.
Fang, Y., Liu, G., Wang, Y., Liu, Y., Yin, Y., Cai, Y., Mebel, A. M., and Jiang, G.: Transformation of mercurous [HgI] species during laboratory standard preparation and analysis: Implication for environmental analysis, Environ. Sci. Technol., 58, 6825–6834, https://doi.org/10.1021/acs.est.4c00718, 2024.
Feinberg, A., Dlamini, T., Jiskra, M., Shah, V., and Selin, N. E.: Evaluating atmospheric mercury (Hg) uptake by vegetation in a chemistry-transport model, Environmental Science: Processes & Impacts, 24, 1303–1318, https://doi.org/10.1039/D2EM00032F, 2022.
Feinberg, A., Selin, N. E., Braban, C. F., Chang, K.-L., Custodio, D., Jaffe, D. A., Kyllonen, K., Landis, M. S., Leeson, S. R., Luke, W., Molepo, K. M., Murovec, M., Mastromonaco, M. G. N., Pfaffhuber, K. A., Ruediger, J., Sheu, G.-R., and St. Louis, V. L.: Unexpected anthropogenic emission decreases explain recent atmospheric mercury concentration declines, P. Natl. Acad. Sci. USA, 121, e2401950121, https://doi.org/10.1073/pnas.2401950121, 2024.
Feldmann, J., Grumping, R., and Hirner, A. V.: Determination of volatile metal and metalloid compounds in gases from domestic waste deposits with GC ICP-MS, Fresen. J. Anal. Chem., 350, 228–234, 1994.
Feng, X., Li, P., Fu, X., Wang, X., Zhang, H., and Lin, C.-J.: Mercury pollution in China: implications on the implementation of the Minamata Convention, Environmental Science: Processes & Impacts, 24, 634–648, https://doi.org/10.1039/D2EM00039C, 2022.
Filatov, M. and Cremer, D.: Revision of the dissociation energies of mercury chalcogendies – Unusual types of mercury bonding, Chem. Phys. Phys. Chem., 5, 1547–1557, https://doi.org/10.1002/cphc.200301207, 2004.
Finlayson-Pitts, B. J. and Pitts, J. N.: Chemistry of the upper and lower atmosphere: Theory, experiments and applications, 1st Ed., Academic Press, San Diego, https://doi.org/10.1016/B978-0-12-257060-5.X5000-X, 2000.
Foti, C., Giuffre, O., Lando, G., and Sammartano, S.: Interaction of inorganic mercury(II) with polyamines, polycarboxylates, and amino acids, J. Chem. Eng. Data, 54, 893–903, https://doi.org/10.1021/je800685c, 2009.
Francés-Monerris, A., Carmona-García, J., Acuña, A. U., Dávalos, J. Z., Cuevas, C. A., Kinnison, D. E., Francisco, J. S., Saiz-Lopez, A., and Roca-Sanjuán, D.: Photodissociation mechanisms of major mercury(II) species in the atmospheric chemical cycle of mercury, Angewandte Chemie International Edition, 59, 7605–7610, 2020.
Fu, X., Marusczak, N., Wang, X., Gheusi, F., and Sonke, J. E.: Isotopic composition of gaseous elemental mercury in the free troposphere of the Pic du Midi observatory, France, Environ. Sci. Technol., 50, 5641–5650, https://doi.org/10.1021/acs.est.6b00033, 2016a.
Fu, X., Zhang, H., Liu, C., Zhang, H., Lin, C.-J., and Feng, X.: Significant seasonal variations in isotopic composition of atmospheric total gaseous mercury at forest sites in China caused by vegetation and mercury sources, Environ. Sci. Technol., 53, 13748–13756, https://doi.org/10.1021/acs.est.9b05016, 2019a.
Fu, X., Zhang, H., Feng, X., Tan, Q., Ming, L., Liu, C., and Zhang, L.: Domestic and transboundary sources of atmospheric particulate bound mercury in remote areas of China: Evidence from mercury isotopes, Environ. Sci. Technol., 53, 1947–1957, 2019b.
Fu, X., Jiskra, M., Yang, X., Marusczak, N., Enrico, M., Chmeleff, J., Heimbürger-Boavida, L.-E., Gheusi, F., and Sonke, J. E.: Mass-independent fractionation of even and odd mercury isotopes during atmospheric mercury redox reactions, Environ. Sci. Technol. 55, 10164–10174, 2021.
Fu, X., Zhu, W., Zhang, H., Sommar, J., Yu, B., Yang, X., Wang, X., Lin, C.-J., and Feng, X.: Depletion of atmospheric gaseous elemental mercury by plant uptake at Mt. Changbai, Northeast China, Atmos. Chem. Phys., 16, 12861–12873, https://doi.org/10.5194/acp-16-12861-2016, 2016b.
Fu, X. W., Zhang, H., Yu, B., Wang, X., Lin, C.-J., and Feng, X. B.: Observations of atmospheric mercury in China: a critical review, Atmos. Chem. Phys., 15, 9455–9476, https://doi.org/10.5194/acp-15-9455-2015, 2015.
Fu, X., Sun, X., Travnikov, O., Li, Q., Qin, C., Cuevas, C. A., Fernandez, R. P., Mahajan, A. S., Wang, S., Wang, T., and Saiz-Lopez, A.: Anthropogenic short-lived halogens increase human exposure to mercury contamination due to enhanced mercury oxidation over continents, P. Natl. Acad. Sci. USA, 121, e2315058121, https://doi.org/10.1073/pnas.2315058121, 2024.
Fujita, S., Horii, H., and Taniguchi, S.: Pulse radiolysis of mercuric ion in aqueous solutions, J. Phys. Chem., 77, 2868–2871, https://doi.org/10.1021/j100642a009, 1973.
Fujita, S., Horii, H., Mori, T., and Taniguchi, S.: Pulse radiolysis of mercuric oxide in neutral aqueous solutions, J. Phys. Chem., 79, 960–964, https://doi.org/10.1021/j100577a003, 1975.
Gačnik, J., Zivkovic, I., Guevara, S. R., Kotnik, J., Berisha, S., Nair, S. V., Jurov, A., Cvelbar, U., and Horvat, M.: Calibration approach for gaseous oxidized mercury based on nonthermal plasma oxidation of elemental mercury, Anal. Chem., 94, 8234–8240, https://doi.org/10.1021/acs.analchem.2c00260, 2022.
Gačnik, J., Lyman, S., Dunham-Cheatham, S. M., and Gustin, M. S.: Limitations and insights regarding atmospheric mercury sampling using gold, Anal. Chim. Acta, 1319, 342956, https://doi.org/10.1016/j.aca.2024.342956, 2024.
Gao, Z. Y., Bailey, N., and Wang, F. Y.: Experimental determination of mercury photoreduction rates in cloudwater, J. Geophys. Res.-Atmos., 128, e2022JD038183, https://doi.org/10.1029/2022jd038183, 2023.
Gao, Z., Geilfus, N.-X., Saiz-Lopez, A., and Wang, F.: Reproducing Arctic springtime tropospheric ozone and mercury depletion events in an outdoor mesocosm sea ice facility, Atmos. Chem. Phys., 22, 1811–1824, https://doi.org/10.5194/acp-22-1811-2022, 2022.
Gay, D. A., Schmeltz, D., Prestbo, E., Olson, M., Sharac, T., and Tordon, R.: The Atmospheric Mercury Network: measurement and initial examination of an ongoing atmospheric mercury record across North America, Atmos. Chem. Phys., 13, 11339–11349, https://doi.org/10.5194/acp-13-11339-2013, 2013.
Geller, L. S., Elkins, J. W., Lobert, J. M., Clarke, A. D., Hurst, D. F., Butler, J. H., and Myers, R. C.: Tropospheric SF6: Observed latitudinal distribution and trends, derived emissions and interhemispheric exchange time, Geophysical Research Letters, 24, 675–678, https://doi.org/10.1029/97GL00523, 1997.
Gerson, J. R., Driscoll, C. T., Demers, J. D., Sauer, A. K., Blackwell, B. D., Montesdeoca, M. R., Shanley, J. B., and Ross, D. S.: Deposition of mercury in forests across a montane elevation gradient: Elevational and seasonal patterns in methylmercury inputs and production, J. Geophys. Res.-Biogeosci., 122, 1922–1939, https://doi.org/10.1002/2016JG003721, 2017.
Geyman, B. M., Streets, D. G., Thackray, C. P., Olson, C. L., Schaefer, K., and Sunderland, E. M.: Projecting global mercury emissions and deposition under the shared socioeconomic pathways, Earth's Fut., 12, e2023EF004231, https://doi.org/10.1029/2023EF004231, 2024.
Ghoshdastidar, A. J. and Ariya, P. A.: The existence of airborne mercury nanoparticles, Sci. Rep., 910733, https://doi.org/10.1038/s41598-019-47086-8, 9, 2019.
Ghoshdastidar, J., Ramamurthy, J., Morissette, M., and Ariya, P.: Development of methodology to generate, measure, and characterize the chemical composition of oxidized mercury nanoparticles, Anal. Bioanal. Chem., 412, 691–702, https://doi.org/10.1007/s00216-019-02279-y, 2019.
Giannakopoulos, E., Deligiannakis, Y., and Salahas, G.: Electrochemical interfacial adsorption mechanism of polyphenolic molecules onto Hanging Mercury Drop Electrode surface (HMDE), J. Electroanal. Chem., 664, 117–125, https://doi.org/10.1016/j.jelechem.2011.11.008, 2012.
Gómez Martín, J. C., Lewis, T. R., Douglas, K. M., Blitz, M. A., Saiz-Lopez, A., and Plane, J. M. C.: The reaction between HgBr and O3: kinetic study and atmospheric implications, Phys. Chem. Chem. Phys., 24, 12419–12432, https://doi.org/10.1039/D2CP00754A, 2022.
Goodsite, M., Plane, J. M. C., and Skov, H.: A theoretical study of the oxidation of Hg0 to HgBr2 in the troposphere, Environ. Sci. Technol., 38, 1772–1776, https://doi.org/10.1021/es034680s, 2004.
Goodsite, M. E., Plane, J. M. C., and Skov, H.: A theoretical study of the oxidation of Hg0 to HgBr2 in the troposphere (vol 38, pg 1772, 2004), Environ. Sci. Technol., 46, 5262–5262, https://doi.org/10.1021/es301201c, 2012.
Gratz, L. E., Keeler, G. J., Blum, J. D., and Sherman, L. S.: Isotopic composition and fractionation of mercury in Great Lakes precipitation and ambient air, Environ. Sci. Technol., 44, 7764–7770, 2010.
Gratz, L. E., Ambrose, J. L., Jaffe, D. A., Shah, V., Jaeglé, L., Stutz, J., Festa, J., Spolaor, M., Tsai, C., Selin, N. E., Song, S., Zhou, X., Weinheimer, A. J., Knapp, D. J., Montzka, D. D., Flocke, F. M., Campos, T. L., Apel, E., Hornbrook, R., Blake, N. J., Hall, S., Tyndall, G. S., Reeves, M., Stechman, D., and Stell, M.: Oxidation of mercury by bromine in the subtropical Pacific free troposphere, Geophys. Res. Lett., 2015GL066645, https://doi.org/10.1002/2015GL066645, 2015.
Greig, G., Gunning, H. E., and Strausz, O. P.: Reactions of metal atoms. II. The combination of mercury and bromine atoms and the dimerization of HgBr, J. Chem. Phys., 52, 3684, https://doi.org/10.1063/1.1673544, 1970.
Greig, G., Young, P. J., and Strausz, O. P.: Reactions of metal atoms. IV. The UV Spectra of CdBr, CdI, ZnBr, and ZnI, J. Chem. Phys., 54, 983–991, 1971.
Gruß, A. F., Rodriguez, R., and Mazyck, D. W.: Mercury oxidation by UV irradiation: Effect of contact time, UV wavelength, and moisture content, Indust. Eng. Chem. Res., 56, 6131–6135, https://doi.org/10.1021/acs.iecr.7b00032, 2017.
Gu, B. H., Bian, Y. R., Miller, C. L., Dong, W. M., Jiang, X., and Liang, L. Y.: Mercury reduction and complexation by natural organic matter in anoxic environments, P. Natl. Acad. Sci. USA, 108, 1479–1483, 2011.
Guérette, E.-A.: Kinetic and mass-spectrometric studies of atmospherically relevant mercury-bromine chemistry, M.Sc. thesis, Department of Chemistry, McGill University, Montreal, Canada, 114 pp., https://escholarship.mcgill.ca/concern/theses/xg94hp887, 2011.
Gunning, H. E. and Strausz, O. P.: Isotopic effects and the mechanism of energy transfer in mercury photosensitization, in: Advances in Photochemistry Vol. 1, edited by: Noyes Jr, W. A., Hammond, G. S., and Pitts Jr, J. N., John Wiley & Sons, 209–274, https://doi.org/10.1002/9780470133316.ch7, 1963.
Gustin, M. S., Amos, H. M., Huang, J., Miller, M. B., and Heidecorn, K.: Measuring and modeling mercury in the atmosphere: a critical review, Atmos. Chem. Phys., 15, 5697–5713, https://doi.org/10.5194/acp-15-5697-2015, 2015.
Gustin, M. S., Dunham-Cheatham, S. M., Huang, J. Y., Lindberg, S., and Lyman, S. N.: Development of an understanding of reactive mercury in ambient air: A review, Atmosphere, 12, 73, https://doi.org/10.3390/atmos12010073, 2021.
Gustin, M. S., Dunham-Cheatham, S. M., Allen, N., Choma, N., Johnson, W., Lopez, S., Russell, A., Mei, E. R., Magand, O., Dommergue, A., and Elgiar, T.: Observations of the chemistry and concentrations of reactive Hg at locations with different ambient air chemistry, Sci. Total Environ., 904, 166–184, https://doi.org/10.1016/j.scitotenv.2023.166184, 2023.
Gustin, M. S., Dunham-Cheatham, S. M., Lyman, S., Horvat, M., Gay, D. A., Gačnik, J., Gratz, L., Kempkes, G., Khalizov, A., Lin, C.-J., Lindberg, S. E., Lown, L., Martin, L., Mason, R. P., Macsween, K., Nair, S. V., Nguyen, L. S. P., O'Neil, T., Sommar, J., Zhang, L., Weiss-Penzias, P., and Zivkovic, I.: Measurement of atmospheric mercury: Current limitations and suggestions for paths forward, Environ. Sci. Technol., 58, 12853–12864, https://doi.org/10.1021/acs.est.4c06011, 2024.
Guzman, F. J. and Bozzelli, J.: Thermodynamics of OHgX, XHgOH, XHgOCl, XHgOBr, and HOHgY gaseous oxidized mercury molecules from isodesmic, isogyric, and atomization work reactions (X = halogen, Y = OH, OCl, OBr), J. Phys. Chem. A, 123, 4452–4464, https://doi.org/10.1021/acs.jpca.9b01358, 2019.
Gårdfeldt, K. and Jonsson, M.: Is bimolecular reduction of Hg(II) complexes possible in aqueous systems of environmental importance?, J. Phys. Chem. A, 107, 4478–4482, 2003.
Gårdfeldt, K., Munthe, J., Strömberg, D., and Lindqvist, O.: A kinetic study on the abiotic methylation of divalent mercury in the aqueous phase, Sci. Total Environ., 304, 127–136, https://doi.org/10.1016/S0048-9697(02)00562-4, 2003.
Gårdfeldt, K., Sommar, J., Strömberg, D., and Feng, X. B.: Oxidation of atomic mercury by hydroxyl radicals and photoinduced decomposition of methylmercury in the aqueous phase, Atmos. Environ., 35, 3039–3047, 2001.
Görner, H.: Quinone Photochemistry, in: CRC Handbook of Organic Photochemistry and Photobiology, 3rd Ed., Volume 1, edited by: Griesbeck, A., Oelgemöller, M., and Ghetti, F., CRC Press, 683–714, 2019.
Haitzer, M., Aiken, G. R., and Ryan, J. N.: Binding of mercury(II) to dissolved organic matter: The role of the mercury-to-DOM concentration ratio, Environ. Sci. Technol., 36, 3564–3570, https://doi.org/10.1021/es025699i, 2002.
Hall, B.: An experimental study of mercury reactions in combustion flue gases, Ph. D. Thesis, Department of Inorganic Chemistry, Chalmers University of Technology and Göteborg University, Sweden, 76 pp., http://hdl.handle.net/2077/11386, 1992.
Hall, B.: The gas-phase oxidation of elemental mercury by ozone, Water Air Soil Pollut., 80, 301–315, 1995.
Hall, B., Schager, P., and Weesmaa, J.: The homogeneous gas-phase reaction of mercury with oxygen, and the corresponding heterogeneous reactions in the presence of activated carbon and fly ash, Chemosphere, 30, 611–627, 1995.
Hammerschmidt, C. R., Lamborg, C. H., and Fitzgerald, W. F.: Aqueous phase methylation as a potential source of methylmercury in wet deposition, Atmos. Environ., 41, 1663–1668, https://doi.org/10.1016/j.atmosenv.2006.10.032, 2007.
Han, X. X., Li, Y. B., Li, D., and Liu, C.: Role of free radicals/reactive oxygen species in MeHg photodegradation: Importance of utilizing sppropriate scavengers, Environ. Sci. Technol., 51, 3784–3793, https://doi.org/10.1021/acs.est.7b00205, 2017.
Harkins, W. D. and Mulliken, R. S.: The separation of mercury into isotopes, Nature, 108, 146, https://doi.org/10.1038/108146a0, 1921.
Hayashi, K., Kawai, S., Ohno, T., and Maki, Y.: Photomethylation of inorganic mercury by aliphatic α-amino-acids, J. Chem. Soc. Chem. Comm., 158–159, https://doi.org/10.1039/c39770000158, 1977.
He, F., Zhao, W., Liang, L., and Gu, B.: Photochemical oxidation of dissolved elemental mercury by carbonate radicals in water, Environ. Sci. Technol. Lett., 1, 499–503, 2014.
He, F., Zheng, W., Liang, L. Y., and Gu, B. H.: Mercury photolytic transformation affected by low-molecular-weight natural organics in water, Sci. Total Environ., 416, 429–435, https://doi.org/10.1016/j.scitotenv.2011.11.081, 2012.
He, X.-C., Tham, Y. J., Dada, L., Wang, M., Finkenzeller, H., Stolzenburg, D., Iyer, S., Simon, M., Kürten, A., Shen, J., Rörup, B., Rissanen, M., Schobesberger, S., Baalbaki, R., Wang, D. S., Koenig, T. K., Jokinen, T., Sarnela, N., Beck, L. J., Almeida, J., Amanatidis, S., Amorim, A., Ataei, F., Baccarini, A., Bertozzi, B., Bianchi, F., Brilke, S., Caudillo, L., Chen, D., Chiu, R., Chu, B., Dias, A., Ding, A., Dommen, J., Duplissy, J., El Haddad, I., Gonzalez Carracedo, L., Granzin, M., Hansel, A., Heinritzi, M., Hofbauer, V., Junninen, H., Kangasluoma, J., Kemppainen, D., Kim, C., Kong, W., Krechmer, J. E., Kvashin, A., Laitinen, T., Lamkaddam, H., Lee, C. P., Lehtipalo, K., Leiminger, M., Li, Z., Makhmutov, V., Manninen, H. E., Marie, G., Marten, R., Mathot, S., Mauldin, R. L., Mentler, B., Möhler, O., Müller, T., Nie, W., Onnela, A., Petäjä, T., Pfeifer, J., Philippov, M., Ranjithkumar, A., Saiz-Lopez, A., Salma, I., Scholz, W., Schuchmann, S., Schulze, B., Steiner, G., Stozhkov, Y., Tauber, C., Tomé, A., Thakur, R. C., Väisänen, O., Vazquez-Pufleau, M., Wagner, A. C., Wang, Y., Weber, S. K., Winkler, P. M., Wu, Y., Xiao, M., Yan, C., Ye, Q., Ylisirniö, A., Zauner-Wieczorek, M., Zha, Q., Zhou, P., Flagan, R. C., Curtius, J., Baltensperger, U., Kulmala, M., Kerminen, V.-M., Kurtén, T., Donahue, N. M., Volkamer, R., Kirkby, J., Worsnop, D. R., and Sipilä, M.: Role of iodine oxoacids in atmospheric aerosol nucleation, Science, 371, 589–595, https://doi.org/10.1126/science.abe0298, 2021.
He, Y. P. and Mason, R. P.: Comparison of reactive gaseous mercury measured by KCl-coated denuders and cation exchange membranes during the Pacific GEOTRACES GP15 expedition, Atmos. Environ., 244, 117973, https://doi.org/10.1016/j.atmosenv.2020.117973, 2021.
He, Y. P., Shi, X. M., Huffman, W. W., Lamborg, C. H., and Mason, R. P.: Description of a dimethylmercury automatic analyzer for the high-resolution measurement of dissolved gaseous mercury species in surface ocean waters, Environ. Sci. Technol., 56, 13076–13084, https://doi.org/10.1021/acs.est.2c02908, 2022.
Hepler, L. G. and Olofsson, G.: Mercury – Thermodynamic properties, chemical equilibria, and standard potentials, Chem. Rev., 75, 585–602, https://doi.org/10.1021/cr60297a003, 1975.
Hietanen, S. and Sillén, L. G.: On the standard potentials of mercury, and the equilibrium Hg Hg0(l) = Hg in nitrate and perchlorate solutions, Arkiv för Kemi, 10, 103–125, 1956.
Hintelmann, H. and Zheng, W.: Tracking geochemical transformations and transport of mercury through isotope fractionation, in: Environmental Chemistry and Toxicology of Mercury, edited by: Liu, G., Cai, Y., and O'Driscoll, N., John Wiley & Sons, 293–328, https://doi.org/10.1002/9781118146644.ch9, 2011.
Hippler, H., Wendt, H. R., and Hunziker, H. E.: Excited intermediates in Hg-photosensitized reaction of O2 detected by energy-transfer, J. Chem. Phys., 68, 5103–5111, 1978.
Holmes, C. D., Jacob, D. J., Mason, R. P., and Jaffe, D. A.: Sources and deposition of reactive gaseous mercury in the marine atmosphere, Atmos. Environ., 43, 2278–2285, https://doi.org/10.1016/j.atmosenv.2009.01.051, 2009.
Holmes, C. D., Jacob, D. J., Corbitt, E. S., Mao, J., Yang, X., Talbot, R., and Slemr, F.: Global atmospheric model for mercury including oxidation by bromine atoms, Atmos. Chem. Phys., 10, 12037–12057, https://doi.org/10.5194/acp-10-12037-2010, 2010.
Hong, Q., Xie, Z., Liu, C., Wang, F., Xie, P., Kang, H., Xu, J., Wang, J., Wu, F., He, P., Mou, F., Fan, S., Dong, Y., Zhan, H., Yu, X., Chi, X., and Liu, J.: Speciated atmospheric mercury on haze and non-haze days in an inland city in China, Atmos. Chem. Phys., 16, 13807–13821, https://doi.org/10.5194/acp-16-13807-2016, 2016.
Horne, D. G., Gosavi, R., and Strausz, O. P.: Reactions of metal atoms. I. The combination of mercury and chlorine atoms and the dimerization of HgCl, J. Chem. Phys., 48, 4758–4764, 1968.
Horowitz, H. M., Jacob, D. J., Zhang, Y., Dibble, T. S., Slemr, F., Amos, H. M., Schmidt, J. A., Corbitt, E. S., Marais, E. A., and Sunderland, E. M.: A new mechanism for atmospheric mercury redox chemistry: implications for the global mercury budget, Atmos. Chem. Phys., 17, 6353–6371, https://doi.org/10.5194/acp-17-6353-2017, 2017.
Howard, D., Nelson, P. F., Edwards, G. C., Morrison, A. L., Fisher, J. A., Ward, J., Harnwell, J., van der Schoot, M., Atkinson, B., Chambers, S. D., Griffiths, A. D., Werczynski, S., and Williams, A. G.: Atmospheric mercury in the Southern Hemisphere tropics: seasonal and diurnal variations and influence of inter-hemispheric transport, Atmos. Chem. Phys., 17, 11623–11636, https://doi.org/10.5194/acp-17-11623-2017, 2017.
Huang, J., Lyman, S. N., Hartman, J. S., and Gustin, M. S.: A review of passive sampling systems for ambient air mercury measurements, Environmental Sciences: Processes and Impacts, 16, 374–392, 2014.
Huang, J., Miller, M. B., Edgerton, E., and Sexauer Gustin, M.: Deciphering potential chemical compounds of gaseous oxidized mercury in Florida, USA, Atmos. Chem. Phys., 17, 1689–1698, https://doi.org/10.5194/acp-17-1689-2017, 2017.
Huang, J., Kang, S., Tian, L., Guo, J., Zhang, Q., Cong, Z., Sillanpää, M., Sun, S., and Tripathee, L.: Influence of long-range transboundary transport on atmospheric water vapor mercury collected at the largest city of Tibet, Sci. Total Environ., 566–567, 1215–1222, https://doi.org/10.1016/j.scitotenv.2016.05.177, 2016a.
Huang, Q., He, X., Huang, W., and Reinfelder, J. R.: Mass-Independent fractionation of mercury isotopes during photoreduction of soot particle bound Hg(II), Environ. Sci. Technol., 55, 13783–13791, https://doi.org/10.1021/acs.est.1c02679, 2021.
Huang, Q., Liu, Y., Chen, J., Feng, X., Huang, W., Yuan, S., Cai, H., and Fu, X.: An improved dual-stage protocol to pre-concentrate mercury from airborne particles for precise isotopic measurement, J. Anal. Atomic Spectrom., 30, 957–966, https://doi.org/10.1039/C4JA00438H, 2015.
Huang, Q., Chen, J., Huang, W., Reinfelder, J. R., Fu, P., Yuan, S., Wang, Z., Yuan, W., Cai, H., Ren, H., Sun, Y., and He, L.: Diel variation in mercury stable isotope ratios records photoreduction of PM2.5-bound mercury, Atmos. Chem. Phys., 19, 315–325, https://doi.org/10.5194/acp-19-315-2019, 2019.
Huang, Q., Chen, J., Huang, W., Fu, P., Guinot, B., Feng, X., Shang, L., Wang, Z., Wang, Z., Yuan, S., Cai, H., Wei, L., and Yu, B.: Isotopic composition for source identification of mercury in atmospheric fine particles, Atmos. Chem. Phys., 16, 11773–11786, https://doi.org/10.5194/acp-16-11773-2016, 2016b.
Huber, M. L., Laesecke, A., and Friend, D. G.: Correlation for the vapor pressure of mercury, Ind. Eng. Chem. Fund, 45, 7351–7361, https://doi.org/10.1021/ie060560s, 2006.
Humphries, R. S., Schofield, R., Keywood, M. D., Ward, J., Pierce, J. R., Gionfriddo, C. M., Tate, M. T., Krabbenhoft, D. P., Galbally, I. E., Molloy, S. B., Klekociuk, A. R., Johnston, P. V., Kreher, K., Thomas, A. J., Robinson, A. D., Harris, N. R. P., Johnson, R., and Wilson, S. R.: Boundary layer new particle formation over East Antarctic sea ice – possible Hg-driven nucleation?, Atmos. Chem. Phys., 15, 13339–13364, https://doi.org/10.5194/acp-15-13339-2015, 2015.
Hynes, A. J., Donohoue, D. L., Goodsite, M. E., and Hedgecock, I. M.: Our current understanding of major chemical and physical processes affecting mercury dynamics in the atmosphere and at the air-water/terrestrial interfaces, in: Mercury Fate and Transport in the Global Atmosphere – Emissions, Measurements and Models, edited by: Mason, R. P. and Pirrone, N., Springer, 427–457, https://doi.org/10.1007/978-0-387-93958-2_14, 2009.
Hynes, A. J., Everhart, S., Bauer, D., Remeika, J., and Tatum Ernest, C.: In situ and denuder-based measurements of elemental and reactive gaseous mercury with analysis by laser-induced fluorescence – results from the Reno Atmospheric Mercury Intercomparison Experiment, Atmos. Chem. Phys., 17, 465–483, https://doi.org/10.5194/acp-17-465-2017, 2017.
Iverfeldt, Å. and Lindqvist, O.: Atmospheric oxidation of elemental mercury by ozone in the aqueous phase, Atmos. Environ., 20, 1567–1573, https://doi.org/10.1016/0004-6981(86)90245-3, 1986.
Jackson, T. A.: Reply: Variations in the isotope composition of mercury in a freshwater sediment sequence and food web, Can. J. Fish. Aquat. Sci., 00058, 2312–2317, 2001.
Jaffe, D. A., Lyman, S., Amos, H. M., Gustin, M. S., Huang, J., Selin, N. E., Levin, L., ter Schure, A., Mason, R. P., Talbot, R., Rutter, A., Finley, B., Jaegle, L., Shah, V., McClure, C., Arnbrose, J., Gratz, L., Lindberg, S., Weiss-Penzias, P., Sheu, G.-R., Feddersen, D., Horvat, M., Dastoor, A., Hynes, A. J., Mao, H., Sonke, J. E., Slemr, F., Fisher, J. A., Ebinghaus, R., Zhang, Y., and Edwards, G.: Progress on understanding atmospheric mercury hampered by uncertain measurements, Environ. Sci. Technol., 48, 7204–7206, https://doi.org/10.1021/es5026432, 2014.
Jayasekharan, T. and Sahoo, N. K.: Mercury mono oxide cluster ions (HgO by laser desorption ionization time of flight mass spectrometry, J. Mass Spectrom., 49, 248–250, 2014.
Jerzykiewicz, M.: The effect of Hg(II) ions on the free radicals of humic substances and their model compounds, Chemosphere, 92, 445–450, https://doi.org/10.1016/j.chemosphere.2013.01.048, 2013.
Jerzykiewicz, M., Witwicki, M., and Jezierska, J.: pH-dependent formation of Hg(II)-semiquinone complexes from natural phenols, Chemosphere, 138, 233–238, https://doi.org/10.1016/j.chemosphere.2015.06.006, 2015.
Jiang, H., Li, J., Tang, J., Cui, M., Zhao, S., Mo, Y., Tian, C., Zhang, X., Jiang, B., Liao, Y., Chen, Y., and Zhang, G.: Molecular characteristics, sources, and formation pathways of organosulfur compounds in ambient aerosol in Guangzhou, South China, Atmos. Chem. Phys., 22, 6919–6935, https://doi.org/10.5194/acp-22-6919-2022, 2022.
Jiao, Y. and Dibble, T. S.: Structures, vibrational frequencies, and bond energies of the BrHgOX and BrHgXO species formed in atmospheric mercury depletion events, J. Phys. Chem. A, 121, 7976–7985, https://doi.org/10.1021/acs.jpca.7b06829, 2017a.
Jiao, Y. G. and Dibble, T. S.: First kinetic study of the atmospherically important reactions BrHg•+ NO2 and BrHg•+ HOO, Phys. Chem. Chem. Phys., 19, 1826–1838, https://doi.org/10.1039/c6cp06276h, 2017b.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., E., Dunlea, J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of organic aerosols in the atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Jiskra, M., Wiederhold, J. G., Bourdon, B., and Kretzschmar, R.: Solution speciation controls mercury isotope fractionation of Hg(II) sorption to Goethite, Environ. Sci. Technol., 46, 6654–6662, https://doi.org/10.1021/es3008112, 2012.
Jiskra, M., Sonke, J. E., Agnan, Y., Helmig, D., and Obrist, D.: Insights from mercury stable isotopes on terrestrial–atmosphere exchange of Hg(0) in the Arctic tundra, Biogeosciences, 16, 4051–4064, https://doi.org/10.5194/bg-16-4051-2019, 2019.
Jiskra, M., Wiederhold, J. G., Skyllberg, U., Kronberg, R.-M., and Kretzschmar, R.: Source tracing of natural organic matter bound mercury in boreal forest runoff with mercury stable isotopes, Environ. Sci.-Proc. Imp., 19, 1235–1248, https://doi.org/10.1039/c7em00245a, 2017.
Jiskra, M., Wiederhold, J. G., Skyllberg, U., Kronberg, R. M., Hajdas, I., and Kretzschmar, R.: Mercury deposition and re-emission pathways in boreal forest soils investigated with Hg isotope signatures, Environ. Sci. Technol., 49, 7188–7196, 2015.
Jiskra, M., Heimbürger-Boavida, L.-E., Desgranges, M.-M., Petrova, M. V., Dufour, A., Ferreira-Araujo, B., Masbou, J., Chmeleff, J., Thyssen, M., Point, D., and Sonke, J. E.: Mercury stable isotopes constrain atmospheric sources to the ocean, Nature, 597, 678–682, https://doi.org/10.1038/s41586-021-03859-8, 2021.
Jiskra, M., Sonke, J. E., Obrist, D., Bieser, J., Ebinghaus, R., Myhre, C. L., Pfaffhuber, K. A., Wängberg, I., Kyllönen, K., Worthy, D., Martin, L. G., Labuschagne, C., Mkololo, T., Ramonet, M., Magand, O., and Dommergue, A.: A vegetation control on seasonal variations in global atmospheric mercury concentrations, Nat. Geosci., 11, 244–251, https://doi.org/10.1038/s41561-018-0078-8, 2018.
Johnson, K. P., Blum, J. D., Keeler, G. J., and Douglas, T. A.: Investigation of the deposition and emission of mercury in arctic snow during an atmospheric mercury depletion event, J. Geophys. Res.-Atmos., 113, D17304, https://doi.org/10.1029/2008jd009893, 2008.
Jones, C. P., Lyman, S. N., Jaffe, D. A., Allen, T., and O'Neil, T. L.: Detection and quantification of gas-phase oxidized mercury compounds by GC/MS, Atmos. Meas. Tech., 9, 2195–2205, https://doi.org/10.5194/amt-9-2195-2016, 2016.
Jordan, K. J., Bascal, H. A., Lipson, R. H., and Melchior, M.: The B X2Σ+ transition of HgI, J. Mol. Spectrosc., 159, 144–155, https://doi.org/10.1006/jmsp.1993.1113, 1993.
Jungbluth, H., Beyrich, J., and Asmus, K. D.: Reduction of mercuric halides and pseudohalides in aqueous solution. Formation and some physicochemical properties of HgCl, HgBr, HgI, HgSCN, and HgCN radical molecules, J. Phys. Chem., 80, 1049–1053, https://doi.org/10.1021/j100551a004, 1976.
Kallend, A. S. and Purnell, J. H.: Gas-Phase thermal decomposition of dimethyl mercury .2. Homogeneous reaction, T. Faraday Soc., 60, 103–118, https://doi.org/10.1039/tf9646000103, 1964.
Kaluza, U. and Boehm, H. P.: Titanium dioxide catalyzed photooxidation of mercury, J. Catalys., 22, 347–358, https://doi.org/10.1016/0021-9517(71)90206-5, 1971.
Karakyriakos, E. and McKinley, A. J.: Matrix isolated HgCH3 radical: An ESR investigation, J. Phys. Chem. A, 108, 4619–4626, https://doi.org/10.1021/jp0400925, 2004.
Khalizov, A. and Mao, N.: Heterogeneous reaction of gaseous mercuric chloride with atmospherically relevant organic films, ACS Earth and Space Chemistry, 7, 2593−2602, https://doi.org/10.1021/acsearthspacechem.3c00268, 2023.
Khalizov, A., Guzman, F., Cooper, M., Mao, N., Antley, J., and Bozzelli, J.: Direct detection of gas-phase mercuric chloride by ion drift – Chemical ionization mass spectrometry, Atmos. Environ., 238, 117687, https://doi.org/10.1016/j.atmosenv.2020.117687, 2020.
Khalizov, A. F., Viswanathan, B., Larregaray, P., and Ariya, P. A.: A theoretical study on the reactions of Hg with halogens: Atmospheric implications, J. Phys. Chem. A, 107, 6360–6365, 2003.
Khiri, D., Louis, F., Cernusak, I., and Dibble, T. S.: BrHgO• + CO: Analogue of OH plus CO and reduction path for Hg(II) in the atmosphere, ACS Earth and Space Chemistry, 4, 1777–1784, 2020.
Kim, P. R., Han, Y. J., Holsen, T. M., and Yi, S. M.: Atmospheric particulate mercury: Concentrations and size distributions, Atmos. Environ., 61, 94–102, 2012.
Kleszczewska, E.: The spectrophotometry determination of chelate complex: L-ascorbic acid with cuprum (II) and mercury (II) in alkaline solution, Pol. J. Environ. Stud., 8, 313–318, 1999.
Kobayashi, T.: Oxidantion of metallic mercury in aqueous solution by hydrogen peroxide and chlorine, Journal of Japan Society of Air Pollution, 22, 230–236, https://doi.org/10.11298/taiki1978.22.230, 1987.
Knight Jr., L. B. and Lin, K. C.: ESR spectroscopy and chemical bonding in CdCN and HgCN molecules, J. Chem. Phys., 56, 6044–6049, https://doi.org/10.1063/1.1677153, 1972.
Knight Jr., L. B., Fisher, T. A., and Wise, M. B.: Photolytic codeposition generation of the HgF radical in an argon matrix at 12 K: An ESR investigation, The J. Chem. Phys., 74, 6009–6013, https://doi.org/10.1063/1.441040, 1981.
Koenig, A., Sonke, J., Magand, O., Andrade, M., Moreno R, C. I., Velarde, F., Forno, R., Gutierrez, R., Blacutt, L., Laj, P., Ginot, P., Bieser, J., Zahn, A., Slemr, F., and Dommergue, A.: Evidence for interhemispheric mercury exchange in the Pacific Ocean upper troposphere, J. Geophys. Res.-Atmos., 127, https://doi.org/10.1029/2021JD036283, 2022.
Koenig, T. K., Baidar, S., Campuzano-Jost, P., Cuevas, C. A., Dix, B., Fernandez, R. P., Guo, H., Hall, S. R., Kinnison, D., Nault, B. A., Ullmann, K., Jimenez, J. L., Saiz-Lopez, A., and Volkamer, R.: Quantitative detection of iodine in the stratosphere, P. Natl. Acad. Sci. USA, 117, 1860–1866, https://doi.org/10.1073/pnas.1916828117, 2020.
Kominar, R. J. and Price, S. J.: Determination of bond dissociation energy D(CH3Hg-CH3) by toluene carrier method, Can. J. Chem., 47, 991–994, https://doi.org/10.1139/v69-156, 1969.
Kornev, V. I. and Kardapol'tsev, A. A.: Heteroligand mercury(II) complexes with aspartic, tartaric, and citric acids, Russ. J. Coord. Chem+, 34, 896–900, https://doi.org/10.1134/s107032840812004x, 2008.
Kozin, L. F. and Hansen, S. C.: Mercury Handbook: Chemistry, Applications and Environmental Impact, Royal Society of Chemistry, London, UK, https://doi.org/10.1039/9781849735155, 2013.
Kramida, A., Ralchenko, Y., Reader, J., and Team, N. A.: NIST Atomic Spectra Database (version 5.11), National Institute of Standards and Technology, U.S. Department of Commerce, https://doi.org/10.18434/T4W30F, 2023.
Kritee, K., Blum, J. D., Reinfelder, J. R., and Barkay, T.: Microbial stable isotope fractionation of mercury: A synthesis of present understanding and future directions, Chem. Geol., 336, 13–25, https://doi.org/10.1016/j.chemgeo.2012.08.017, 2013.
Kritee, K., Blum, J. D., Johnson, M. W., Bergquist, B. A., and Barkay, T.: Mercury stable isotope fractionation during reduction of Hg(II) to Hg(0) by mercury resistant microorganisms, Environ. Sci. Technol., 41, 1889–1895, 2007.
Kritee, K., Motta, L. C., Blum, J. D., Tsui, M. T. K., and Reinfelder, J. R.: Photomicrobial visible light-induced magnetic mass independent fractionation of mercury in a marine microalga, ACS Earth and Space Chemistry, 2, 432–440, https://doi.org/10.1021/acsearthspacechem.7b00056, 2018.
Kurien, U., Hu, Z., Lee, H., Dastoor, A. P., and Ariya, P. A.: Radiation enhanced uptake of Hg0(g) on iron (oxyhydr)oxide nanoparticles, RSC Advances, 7, 45010–45021, https://doi.org/10.1039/C7RA07401H, 2017.
Kurz, A., Blum, J., Johnson, M., Nadelhoffer, K., and Zak, D.: Isotopic composition of mercury deposited via snow into mid-latitude ecosystems, Sci. Total Environ., 784, 147252, https://doi.org/10.1016/j.scitotenv.2021.147252, 2021.
Kurz, A. Y., Blum, J. D., Gratz, L. E., and Jaffe, D. A.: Contrasting controls on the diel isotopic variation of Hg0 at two high elevation sites in the Western United States, Environ. Sci. Technol., 54, 10502–10513, 2020.
Kwon, S. Y., Blum, J. D., Yin, R., Tsui, M. T.-K., Yang, Y. H., and Choi, J. W.: Mercury stable isotopes for monitoring the effectiveness of the Minamata Convention on Mercury, Earth-Sci. Rev., 203, 103–111, 2020.
Lalonde, J. D., Poulain, A. J., and Amyot, M.: The role of mercury redox reactions in snow on snow-to-air mercury transfer, Environ. Sci. Technol., 36, 174–178, 2002.
Lalonde, J. D., Amyot, M., Doyon, M. R., and Auclair, J. C.: Photo-induced Hg(II) reduction in snow from the remote and temperate Experimental Lakes Area (Ontario, Canada), J. Geophys. Res.-Atmos., 108, 4200, https://doi.org/10.1029/2001JD001534, 2003.
Lalonde, J. D., Amyot, M., Kraepiel, A. M. L., and Morel, F. M. M.: Photooxidation of Hg(0) in artificial and natural waters, Environ. Sci. Technol., 35, 1367–1372, 2001.
Lam, K. T.: Chemistry and implications of a previously unknown intermediate in the atmospheric mercury oxidation, Ph. D. thesis, College of Environmental Science and Forestry, State University of New York, Syracuse, NY, https://digitalcommons.esf.edu/etds/85, 2019.
Lam, K. T., Wilhelmsen, C. J., and Dibble, T. S.: BrHgO• + C2H4 and BrHgO• + HCHO in atmospheric oxidation of mercury: determining rate constants of reactions with prereactive complexes and bifurcation, J. Phys. Chem. A, 123, 6045–6055, https://doi.org/10.1021/acs.jpca.9b05120, 2019a.
Lam, K. T., Wilhelmsen, C. J., Schwid, A. C., Jiao, Y., and Dibble, T. S.: Computational study on the photolysis of BrHgONO and the reactions of BrHgO• with CH4, C2H6, NO, and NO2: Implications for formation of Hg(II) compounds in the atmosphere, J. Phys. Chem. A, 123, 1637–1647, 2019b.
Lamborg, C. H., Fitzgerald, W. F., O'Donnell, J., and Torgersen, T.: A non-steady-state compartmental model of global-scale mercury biogeochemistry with interhemispheric atmospheric gradients, Geochim. Cosmochim. Ac., 66, 1105–1118, 2002.
Lan, X., Talbot, R., Castro, M., Perry, K., and Luke, W.: Seasonal and diurnal variations of atmospheric mercury across the US determined from AMNet monitoring data, Atmos. Chem. Phys., 12, 10569–10582, https://doi.org/10.5194/acp-12-10569-2012, 2012.
Landis, M. S., Stevens, R. K., Schaedlich, F., and Prestbo, E. M.: Development and characterization of an annular denuder methodology for the measurement of divalent inorganic reactive gaseous mercury in ambient air, Environ. Sci. Technol., 36, 3000–3009, https://doi.org/10.1021/es015887t, 2002.
Lauretta, D. S., Klaue, B., Blum, J. D., and Buseck, P. R.: Mercury abundances and isotopic compositions in the Murchison (CM) and Allende (CV) carbonaceous chondrites, Geochim. Cosmochim. Ac., 65, 2807–2818, https://doi.org/10.1016/S0016-7037(01)00630-5, 2001.
Lee, C. F., Elgiar, T., David, L. M., Wilmot, T. Y., Reza, M., Hirshorn, N., McCubbin, I. B., Shah, V., Lin, J. C., Lyman, S. N., Hallar, A. G., Gratz, L. E., and Volkamer, R.: Elevated tropospheric iodine over the central continental United States: Is iodine a major oxidant of atmospheric mercury?, Geophys. Res. Lett., 51, e2024GL109247, https://doi.org/10.1029/2024GL109247, 2024.
Lee, H., Kurien, U., and Ariya, P. A.: Uptake of Hg0(g) on TiO2, Al2O3, and Fe2O3 nanoparticles: Importance in atmospheric chemical and physical processes, J. Phys. Chem. A, 126, 6953–6962, https://doi.org/10.1021/acs.jpca.2c03428, 2022.
Lee, S., Kim, D.-H., and Kim, K.-W.: The enhancement and inhibition of mercury reduction by natural organic matter in the presence of Shewanella oneidensis MR-1, Chemosphere, 194, 515–522, https://doi.org/10.1016/j.chemosphere.2017.12.007, 2018.
Lee, Y. H., Wängberg, I., and Munthe, J.: Sampling and analysis of gas-phase methylmercury in ambient air, Sci. Total Environ., 304, 107–113, 2003.
Lehtipalo, K., Nieminen, T., Schobesberger, S., Ehn, M., Kulmala, M., and Kerminen, V.-M.: How the understanding of atmospheric new particle formation has evolved along with the development of measurement and analysis methods, J. Aerosol Sci., 184, 106494, https://doi.org/10.1016/j.jaerosci.2024.106494, 2025.
Leonori, D. and Sturgeon, R. E.: A unified approach to mechanistic aspects of photochemical vapor generation, J. Anal. Atom. Spectrom., 34, 636–654, https://doi.org/10.1039/c8ja00354h, 2019.
Li, C., Enrico, M., Magand, O., Araujo, B., Le Roux, G., Osterwalder, S., Dommergue, A., Bertrand, Y., Brioude, J., Vleeschouwer, F., and Sonke, J.: A peat core Hg stable isotope reconstruction of Holocene atmospheric Hg deposition at Amsterdam Island (37.8° S), Geochim. Cosmochim. Ac., 341, 62-74, https://doi.org/10.1016/j.gca.2022.11.024, 2022a.
Li, C., Jiskra, M., Nilsson, M., Osterwalder, S., Zhu, W., Mauquoy, D., Skyllberg, U., Enrico, M., Haijun, P., Song, Y., Björn, E., and Bishop, K.: Mercury deposition and redox transformation processes in peatland constrained by mercury stable isotopes, Nat. Commun., 14, 7389, https://doi.org/10.1038/s41467-023-43164-8, 2023a.
Li, C., Chen, J., Angot, H., Zheng, W., Shi, G., Ding, M., Du, Z., Zhang, Q., Ma, X., Kang, S., Xiao, C., Ren, J., and Qin, D.: Seasonal variation of mercury and its isotopes in atmospheric particles at the coastal Zhongshan station, eastern Antarctica, Environ. Sci. Technol., 54, 11344–11355, https://doi.org/10.1021/acs.est.0c04462, 2020a.
Li, L., Wang, X., Fu, H., Qu, X., Chen, J., Tao, S., and Zhu, D.: Dissolved black carbon facilitates photoreduction of Hg(II) to Hg(0) and reduces mercury uptake by lettuce (Lactuca sativa L.), Environ. Sci. Technol., 54, 11137–11145, https://doi.org/10.1021/acs.est.0c01132, 2020b.
Li, M.-L., Kwon, S. Y., Poulin, B. A., Tsui, M. T.-K., Motta, L. C., and Cho, M.: Internal dynamics and metabolism of mercury in biota: A review of insights from mercury stable isotopes, Environ. Sci. Technol., 56, 9182–9195, https://doi.org/10.1021/acs.est.1c08631, 2022b.
Li, T., Wang, Y., Mao, H., Wang, S., Talbot, R. W., Zhou, Y., Wang, Z., Nie, X., and Qie, G.: Insights on chemistry of mercury species in clouds over northern China: Complexation and adsorption, Environ. Sci. Technol., 52, 5125–5134, https://doi.org/10.1021/acs.est.7b06669, 2018.
Li, X., Wang, X., Zhang, H., and Lu, Z.: Mosses and lichens enhance atmospheric elemental mercury deposition in a subtropical montane forest, Environ. Chem., 20, 105–113, https://doi.org/10.1071/EN22124, 2023b.
Li, X. S., Yan, M., Dibble, T. S., and Zhang, L. Q.: Reaction mechanism and kinetics of the important but neglected reaction of Hg with NO2 at low temperature, Chem. Eng. J., 432, 134373, https://doi.org/10.1016/j.cej.2021.134373, 2022c.
Lian, P., Guo, L., Devarajan, D., Parks, J. M., Painter, S. L., Brooks, S. C., and Smith, J. C.: The AQUA-MER databases and aqueous speciation server: A web resource for multiscale modeling of mercury speciation, J. Comput. Chem., 41, 147–155, https://doi.org/10.1002/jcc.26081, 2020.
Lide, D. R. (Ed.): CRC handbook of chemistry and physics, 89th Ed., CRC Press/Taylor and Francis, Boca Raton, FL, 2736 pp., 2008.
Lin, C.-J., Singhasuk, P., and Pehkonen, S. O.: Atmospheric chemistry of mercury, in: Environmental chemistry and toxicology of mercury, edited by: Liu, G., Cai, Y., and O'Driscoll, N., John Wiley & Sons, 113–154, https://doi.org/10.1002/9781118146644.ch4, 2011.
Lin, C.-J., Zhu, W., Li, X., Feng, X., Sommar, J., and Shang, L.: Novel dynamic flux chamber for measuring air-surface exchange of Hg0 from soils, Environ. Sci. Technol., 46, 8910–8920, https://doi.org/10.1021/es3012386, 2012.
Lin, C. J. and Pehkonen, S. O.: Aqueous free radical chemistry of mercury in the presence of iron oxides and ambient aerosol, Atmos. Environ., 31, 4125–4137, 1997.
Lin, C. J. and Pehkonen, S. O.: Two-phase model of mercury chemistry in the atmosphere, Atmos. Environ., 32, 2543–2558, 1998a.
Lin, C. J. and Pehkonen, S. O.: Oxidation of elemental mercury by aqueous chlorine (HOCl/OCl−): Implications for tropospheric mercury chemistry, J. Geophys. Res.-Atmos., 103, 28093–28102, 1998b.
Lin, C. J. and Pehkonen, S. O.: The chemistry of atmospheric mercury: A review, Atmos. Environ., 33, 2067–2079, 1999.
Lin, C. J., Pongprueksa, P., Lindberg, S. E., Pehkonen, S. O., Byun, D., and Jang, C.: Scientific uncertainties in atmospheric mercury models I: Model science evaluation, Atmos. Environ., 40, 2911–2928, 2006.
Lin, C. J., Pongprueksa, P., Russell Bullock Jr, O., Lindberg, S. E., Pehkonen, S. O., Jang, C., Braverman, T., and Ho, T. C.: Scientific uncertainties in atmospheric mercury models II: Sensitivity analysis in the CONUS domain, Atmos. Environ., 41, 6544–6560, 2007.
Lin, M. and Thiemens, M. H.: 40 years of theoretical advances in mass-independent oxygen isotope effects and applications in atmospheric chemistry: A critical review and perspectives, Appl. Geochem., 161, 105860, https://doi.org/10.1016/j.apgeochem.2023.105860, 2024.
Lindberg, S. E. and Stratton, W. J.: Atmospheric mercury speciation: Concentrations and behavior of reactive gaseous mercury in ambient air, Environ. Sci. Technol., 32, 49–57, 1998.
Lindberg, S. E., Southworth, G., Prestbo, E. M., Wallschlager, D., Bogle, M. A., and Price, J.: Gaseous methyl- and inorganic mercury in landfill gas from landfills in Florida, Minnesota, Delaware, and California, Atmos. Environ., 39, 249–258, 2005.
Lindqvist, O. and Rodhe, H.: Atmospheric Mercury – A Review, Tellus B, 37, 136–159, 1985.
Lindqvist, O., Johansson, K., Aastrup, M., Andersson, A., Bringmark, L., Hovsenius, G., Håkanson, L., Iverfeldt, Å., Meili, M., and Timm, B.: Mercury in the Swedish environment – Recent research on causes, Consequences and corrective methods, Water Air Soil Pollut., 55, 1–261, 1991.
Liu, E., Makarov, I. E., and Pikaev, A. K.: Pulse-radiolysis of alkaline aqueous-solutions of divalent mercury compounds, High Energ. Chem+, 17, 41–46, 1983.
Liu, K., Wu, Q., Wang, S., Chang, X., Tang, Y., Wang, L., Liu, T., Zhang, L., Zhao, Y., Wang, Q., and Chen, J.: Improved atmospheric mercury simulation using updated gas-particle partition and organic aerosol concentrations, J. Environ. Sci., 119, 106–118, https://doi.org/10.1016/j.jes.2022.04.007, 2022.
Liu, Q. and Margerum, D. W.: Equilibrium and kinetics of bromine chloride hydrolysis, Environ. Sci. Technol., 35, 1127–1133, 2001.
Liu, T.: Oxidation of aqueous elemental mercury through the Fenton reaction, M Sc. Thesis, Tennessee Technological University, USA, 106 pp., https://www.proquest.com/docview/924155273?sourcetype=Dissertations & Theses, 2011.
Liu, Y., Lin, C.-J., Yuan, W., Lu, Z., and Feng, X.: Translocation and distribution of mercury in biomasses from subtropical forest ecosystems: evidence from stable mercury isotopes, Acta Geochim., 40, 42–50, https://doi.org/10.1007/s11631-020-00441-3, 2021a.
Liu, Y., Liu, H., Guo, Y., Lu, D., Hou, X., Shi, J., Yin, Y., Cai, Y., and Jiang, G.: Atmospheric Hg(0) dry deposition over environmental surfaces: Insights from mercury isotope fractionation, Eco-Environ. Health, 3, 543–555, https://doi.org/10.1016/j.eehl.2024.04.009, 2024.
Liu, Y., Tao, H., Wang, Y., Fang, Y., Xiang, Y., Liu, G., Guo, Y., Liu, J., Yin, Y., Cai, Y., and Jiang, G.: Gaseous elemental mercury [Hg(0)] oxidation in poplar leaves through a two-step single-electron transfer process, Environ. Sci. Technol. Lett., 8, 1098–1103, https://doi.org/10.1021/acs.estlett.1c00735, 2021b.
Loewenschuss, A., Ron, A., and Schnepp, O.: Vibrational spectra of group IIB halides. II. The halides of cadmium and mercury, J. Chem. Phys., 50, 2502–2512, https://doi.org/10.1063/1.1671408, 1969.
Love, L. O.: Electromagnetic separation of isotopes at Oak Ridge, Science, 182, 343–352, https://doi.org/10.1126/science.182.4110.343, 1973.
Lu, X., Zhao, J., Liang, X., Zhang, L., Liu, Y., Yin, X., Li, X., and Gu, B.: The application and potential artifacts of Zeeman cold vapor atomic absorption spectrometry in mercury stable isotope analysis, Environ. Sci. Technol. Lett., 6, 165–170, https://doi.org/10.1021/acs.estlett.9b00067, 2019.
Lyman, S. and Jaffe, D.: Formation and fate of oxidized mercury in the upper troposphere and lower stratosphere, Nat. Geosci., 5, 114–117, 2012.
Lyman, S. N., Jaffe, D. A., and Gustin, M. S.: Release of mercury halides from KCl denuders in the presence of ozone, Atmos. Chem. Phys., 10, 8197–8204, https://doi.org/10.5194/acp-10-8197-2010, 2010.
Lyman, S. N., Cheng, I., Gratz, L. E., Weiss-Penzias, P., and Zhang, L.: An updated review of atmospheric mercury, Sci. Total Environ., 707, 135575, https://doi.org/10.1016/j.scitotenv.2019.135575, 2020a.
Lyman, S. N., Gratz, L. E., Dunham-Cheatham, S. M., Gustin, M. S., and Luippold, A.: Improvements to the accuracy of atmospheric oxidized mercury measurements, Environ. Sci. Technol., 54, 13379–13388, https://doi.org/10.1021/acs.est.0c02747, 2020b.
Lyman, S. N., Elgiar, T., Gustin, M. S., Dunham-Cheatham, S. M., David, L. M., and Zhang, L.: Evidence against rapid mercury oxidation in photochemical smog, Environ. Sci. Technol., 56, 11225–11235, https://doi.org/10.1021/acs.est.2c02224, 2022.
Maguire, R. J. and Anand, S.: Kinetics of acidolysis of dimethyl mercury, J. Inorg. Nucl. Chem., 38, 1167–1169, 1976.
Malcolm, E. G., Keeler, G. J., Lawson, S. T., and Sherbatskoy, T. D.: Mercury and trace elements in cloud water and precipitation collected on Mt. Mansfield, Vermont, J. Environ. Monit., 5, 584–590, 2003.
Malcolm, E. G., Ford, A. C., Redding, T. A., Richardson, M. C., Strain, B. M., and Tetzner, S. W.: Experimental investigation of the scavenging of gaseous mercury by sea salt aerosol, J. Atmos. Chem., 63, 221–234, 2009.
Malinovsky, D. and Vanhaecke, F.: Mercury isotope fractionation during abiotic transmethylation reactions, Int. J. Mass Spectrom., 307, 214–224, 2011.
Malinovsky, D., Latruwe, K., Moens, L., and Vanhaecke, F.: Experimental study of mass-independence of Hg isotope fractionation during photodecomposition of dissolved methylmercury, J. Anal. Atom. Spectrom., 25, 950–956, 2010.
Mangiante, D. M., Schaller, R. D., Zarzycki, P., Banfield, J. F., and Gilbert, B.: Mechanism of ferric oxalate photolysis, ACS Earth and Space Chemistry, 1, 270–276, https://doi.org/10.1021/acsearthspacechem.7b00026, 2017.
Mao, H., Cheng, I., and Zhang, L.: Current understanding of the driving mechanisms for spatiotemporal variations of atmospheric speciated mercury: a review, Atmos. Chem. Phys., 16, 12897–12924, https://doi.org/10.5194/acp-16-12897-2016, 2016.
Mao, H., Talbot, R. W., Sigler, J. M., Sive, B. C., and Hegarty, J. D.: Seasonal and diurnal variations of Hg° over New England, Atmos. Chem. Phys., 8, 1403–1421, https://doi.org/10.5194/acp-8-1403-2008, 2008.
Mao, N. and Khalizov, A.: Exchange reactions alter molecular speciation of gaseous oxidized mercury, ACS Earth and Space Chemistry, 5, 1842–1853, https://doi.org/10.1021/acsearthspacechem.1c00178, 2021.
Mao, N., Antley, J., Cooper, M., Shah, N., Kadam, A., and Khalizov, A.: Heterogeneous chemistry of mercuric chloride on inorganic salt surfaces, J. Phys. Chem. A, 125, 3943-−3952, https://doi.org/10.1021/acs.jpca.1c02220, 2021.
Mariotti, A., Germon, J. C., Hubert, P., Kaiser, P., Letolle, R., Tardieux, A., and Tardieux, P.: Experimental determination of nitrogen kinetic isotope fractionation – Some principles – Illustration for the denitrification and nitrification processes, Plant Soil, 62, 413–430, https://doi.org/10.1007/Bf02374138, 1981.
Martell, A. and Smith, R. M.: Critical stability constants: Vol. 3 Other Organic Ligands, Springer Science, 495 pp., https://doi.org/10.1007/978-1-4757-1568-2, 1977.
Martell, A. E. and Smith, R. M.: Critical stability constants: Vol. 4 Inorganic complexes, Springer Science, 257 pp., https://doi.org/10.1007/978-1-4757-5506-0, 1976.
Martell, A. E. and Smith, R. M.: Critical stability constants: Vol. 5 First Supplement, Springer Science, 604 pp., https://doi.org/10.1007/978-1-4615-6761-5, 1982.
Martens, J., Uchtmann, H., and Hensel, F.: Homogeneous nucleation of mercury vapor, J. Phys. Chem., 91, 2489–2492, https://doi.org/10.1021/j100294a010, 1987.
Marusczak, N., Sonke, J. E., Fu, X., and Jiskra, M.: Tropospheric GOM at the Pic du Midi observatory – Correcting bias in denuder based observations, Environ. Sci. Technol., 51, 863–869, https://doi.org/10.1021/acs.est.6b04999, 2017.
Maya, J.: Ultraviolet-absorption cross-sections of Hgl2, HgBr2, and tin(II) halide vapors, J. Chem. Phys., 67, 4976–4980, 1977.
McClure, C. D., Jaffe, D. A., and Edgerton, E. S.: Evaluation of the KCl denuder method for gaseous oxidized mercury using HgBr2 at an in-service AMNet site, Environ. Sci. Technol., 48, 11437–11444, 2014.
McElroy, W. J. and Munthe, J.: The oxidation of mercury(I) by ozone in acidic aqueous solutions, Acta Chem. Scand., 45, 254–257, https://doi.org/10.3891/acta.chem.scand.45-0254, 1991.
McKeown, F. P., Iyer, R. S., and Rowland, F. S.: Methyl fluoride formation from thermal 18F reaction with dimethylmercury, J. Phys. Chem., 87, 3972–3975, https://doi.org/10.1021/j100243a035, 1983.
Mead, C., Lyons, J. R., Johnson, T. M., and Anbar, A. D.: Unique Hg stable isotope signatures of compact fluorescent lamp-sourced Hg, Environ. Sci. Technol., 47, 2542–2547, 2013.
Medhekar, A. K., Rokni, M., Trainor, D. W., and Jacob, J. H.: Surface catalyzed reaction of Hg + Cl2, Chem. Phys. Lett., 65, 600–604, 1979.
Miller, M. B., Dunham-Cheatham, S. M., Gustin, M. S., and Edwards, G. C.: Evaluation of cation exchange membrane performance under exposure to high Hg0 and HgBr2 concentrations, Atmos. Meas. Tech., 12, 1207–1217, https://doi.org/10.5194/amt-12-1207-2019, 2019.
Morand, J.-P. and Nief, G.: Oxydation isotopique du mercure par l'oxygène moléculaire sous l'influence de la radiation 2357 Å, J. Chim. Phys., 65, 2058–2068, https://doi.org/10.1051/jcp/1968652058, 1968.
Morton, D. C.: Atomic-data for resonance absorption lines. II. Wavelengths longward of the Lyman limit for heavy elements, Astrophys. J. Supplement Series, 130, 403–436, 2000.
Moser, H. C. and Voigt, A. F.: Dismutation of the mercurous dimer in dilute solutions, J. Am. Chem. Soc., 79, 1837–1839, https://doi.org/10.1021/ja01565a019, 1957.
Motta, L. C., Chien, A. D., Rask, A. E., and Zimmerman, P. M.: Mercury magnetic isotope effect: A plausible photochemical mechanism, J. Phys. Chem. A, 124, 3711–3719, 2020a.
Motta, L. C., Kritee, K., Blum, J. D., Tsz-Ki Tsui, M., and Reinfelder, J. R.: Mercury isotope fractionation during the photochemical reduction of Hg(II) coordinated with organic ligands, J. Phys. Chem. A, 124, 2842–2853, 2020b.
Motta, L. C., Blum, J. D., Johnson, M. W., Umhau, B. P., Popp, B. N., Washburn, S. J., Drazen, J. C., Benitez-Nelson, C. R., Hannides, C. C. S., Close, H. G., and Lamborg, C. H.: Mercury cycling in the north Pacific subtropical gyre as revealed by mercury stable isotope ratios, Global Biogeochem. Cy., 33, 777–794, 2019.
Mulliken, R. S.: The separation of isotopes application of systematic fractionation to mercury in a high-speed evaporation-diffusion apparatus, J. Am. Chem. Soc., 45, 1592–1604, https://doi.org/10.1021/ja01660a003, 1923.
Munthe, J.: The aqueous oxidation of elemental mercury by ozone, Atmos. Environ. A, 26, 1461–1468, https://doi.org/10.1016/0960-1686(92)90131-4, 1992.
Munthe, J. and McElroy, W. J.: Some aqueous reactions of potential importance in the atmospheric chemistry of mercury, Atmos. Environ. A, 26, 553–557, https://doi.org/10.1016/0960-1686(92)90168-k, 1992.
Munthe, J., Xiao, Z. F., and Lindqvist, O.: The aqueous reduction of divalent mercury by sulfite, Water Air Soil Pollut., 56, 621–630, 1991.
Murphy, D. M., Thomson, D. S., and Mahoney, M. J.: In situ measurements of organics, meteoritic material, mercury, and other elements in aerosols at 5 to 19 kilometers, Science, 282, 1664–1669, 1998.
Murphy, D. M., Hudson, P. K., Thomson, D. S., Sheridan, P. J., and Wilson, J. C.: Observations of mercury-containing aerosols, Environ. Sci. Technol., 40, 3163–3167, 2006.
Nazhat, N. B. and Asmus, K. D.: Reduction of mercuric chloride by hydrated electrons and reducing radicals in aqueous-solutions – Formation and reactions of HgCl, J. Phys. Chem., 77, 614–620, 1973.
Nerentorp Mastromonaco, M., Gårdfeldt, K., Jourdain, B., Abrahamsson, K., Granfors, A., Ahnoff, M., Dommergue, A., Méjean, G., and Jacobi, H. W.: Antarctic winter mercury and ozone depletion events over sea ice, Atmos. Environ., 129, 125–132, https://doi.org/10.1016/j.atmosenv.2016.01.023, 2016.
Niki, H., Maker, P. D., Savage, C. M., and Breitenbach, L. P.: A long-path Fourier transform infrared study of the kinetics and mechanism for the HO-radical initiated oxidation of dimethylmercury, J. Phys. Chem., 87, 4978–4981, 1983a.
Niki, H., Maker, P. S., Savage, C. M., and Breitenbach, L. P.: A Fourier transform infrared study of the kinetics and mechanism for the reaction Cl + CH3HgCH3, J. Phys. Chem., 87, 3722–3724, 1983b.
Niksa, S., Helble, J. J., and Fujiwara, N.: Kinetic modeling of homogeneous mercury oxidation: The importance of NO and H2O in predicting oxidation in coal- derived systems, Environ. Sci. Technol., 35, 3701–3706, 2001.
Nriagu, J. (Ed.): The biogeochemistry of mercury in the environment, Elsevier/North- Holland Biomedical Press, 671 pp., ISBN 0444801103, ISBN 9780444801104, 1979.
O'Concubhair, R., O'Sullivan, D., and Sodeau, J. R.: Dark oxidation of dissolved gaseous mercury in polar ice mimics, Environ. Sci. Technol., 46, 4829–4836, https://doi.org/10.1021/es300309n, 2012.
Obrist, D., Pokharel, A. K., and Moore, C.: Vertical profile measurements of soil air suggest immobilization of gaseous elemental mercury in mineral soil, Environ. Sci. Technol., 48, 2242–2252, 2014.
Obrist, D., Tas, E., Peleg, M., Matveev, V., Fain, X., Asaf, D., and Luria, M.: Bromine-induced oxidation of mercury in the mid-latitude atmosphere, Nat. Geosci., 4, 22–26, 2011.
Obrist, D., Agnan, Y., Jiskra, M., Olson, C. L., Colegrove, D. P., Hüber, J., Moore, C. W., Sonke, J. E., and Helmig, D.: Tundra uptake of atmospheric elemental mercury drives Arctic mercury pollution, Nature, 547, 201–204, https://doi.org/10.1038/nature22997, 2017.
Obrist, D., Kirk, J. L., Zhang, L., Sunderland, E. M., Jiskra, M., and Selin, N. E.: A review of global environmental mercury processes in response to human and natural perturbations: Changes of emissions, climate, and land use, Ambio, 47, 116-140, https://doi.org/10.1007/s13280-017-1004-9, 2018.
Obrist, D., Roy, E. M., Harrison, J. L., Kwong, C. F., Munger, J. W., Moosmüller, H., Romero, C. D., Sun, S., Zhou, J., and Commane, R.: Previously unaccounted atmospheric mercury deposition in a midlatitude deciduous forest, P. Natl. Acad. Sci. USA, 118, e2105477118, https://doi.org/10.1073/pnas.2105477118, 2021.
Ogata, M. and Aikoh, H.: Mechanism of metallic mercury oxidation in vitro by catalase and peroxidase, Biochem. Pharmacol., 33, 490–493, https://doi.org/10.1016/0006-2952(84)90246-6, 1984.
Ogg, R. A., Martin, H. C., and Leighton, P. A.: Kinetics of the vapor phase reaction of mercury and halogens, J. Am. Chem. Soc., 58, 1922–1924, https://doi.org/10.1021/ja01301a026, 1936.
Olson, C. L., Jiskra, M., Sonke, J. E., and Obrist, D.: Mercury in tundra vegetation of Alaska: Spatial and temporal dynamics and stable isotope patterns, Sci. Total Environ., 660, 1502–1512, https://doi.org/10.1016/j.scitotenv.2019.01.058, 2019.
Osterwalder, S., Eugster, W., Feigenwinter, I., and Jiskra, M.: Eddy covariance flux measurements of gaseous elemental mercury over a grassland, Atmos. Meas. Tech., 13, 2057–2074, https://doi.org/10.5194/amt-13-2057-2020, 2020.
Osterwalder, S., Nerentorp, M., Zhu, W., Jiskra, M., Nilsson, E., Nilsson, M. B., Rutgersson, A., Soerensen, A. L., Sommar, J., Wallin, M. B., Wängberg, I., and Bishop, K.: Critical observations of gaseous elemental mercury air-sea exchange, Global Biogeochem. Cy., 35, e2020GB006742, https://doi.org/10.1029/2020GB006742, 2021.
Pal, B. and Ariya, P. A.: Gas-phase HO-initiated reactions of elemental mercury: Kinetics, product studies, and atmospheric implications, Environ. Sci. Technol., 38, 5555–5566, 2004a.
Pal, B. and Ariya, P. A.: Studies of ozone initiated reactions of gaseous mercury: kinetics, product studies, and atmospheric implications, Phys. Chem. Chem. Phys., 6, 572–579, 2004b.
Pandey, S. K., Kim, K. H., and Brown, R. J. C.: Measurement techniques for mercury species in ambient air, TrAC – Trends in Analytical Chemistry, 30, 899–917, 2011.
Pankow, J. F.: An absorption model of the gas/aerosol partitioning involved in the formation of secondary organic aerosol, Atmos. Environ., 41, 75–79, https://doi.org/10.1016/j.atmosenv.2007.10.060, 2007.
Parks, J. M., Johs, A., Podar, M., Bridou, R., Hurt, R. A., Smith, S. D., Tomanicek, S. J., Qian, Y., Brown, S. D., Brandt, C. C., Palumbo, A. V., Smith, J. C., Wall, J. D., Elias, D. A., and Liang, L.: The genetic basis for bacterial mercury methylation, Science, 339, 1332–1335, https://doi.org/10.1126/science.1230667, 2013.
Pasakarnis, T., Boyanov, M., Kemner, K., Mishra, B., O'Loughlin, E., Parkin, G., and Scherer, M.: Influence of chloride and Fe(II) content on the reduction of Hg(II) by magnetite, Environ. Sci. Technol., 47, 6987−-6994, https://doi.org/10.1021/es304761u, 2013.
Pehkonen, S. O. and Lin, C. J.: Aqueous photochemistry of mercury with organic acids, J. Air Waste Manage. Assoc., 48, 144–150, 1998.
Peleg, M., Tas, E., Obrist, D., Matveev, V., Moore, C., Gabay, M., and Luria, M.: Observational evidence for involvement of nitrate radicals in nighttime oxidation of mercury, Environ. Sci. Technol., 49, 14008–14018, https://doi.org/10.1021/acs.est.5b03894, 2015.
Pertel, R. and Gunning, H. E.: Photochemical separation of mercury isotopes 2. The reaction of 202Hg(63P1) atoms, photoexcited in natural mercury vapor, with water vapor and other HgO-forming substrates, Can. J. Chem., 37, 35–42, 1959.
Peterson, K. A., Shepler, B. C., and Singleton, J. M.: The group 12 metal chalcogenides: an accurate multireference configuration interaction and coupled cluster study, Mol. Phys., 105, 1139–1155, 2007.
Pikaev, A. K., Sibirskaya, G. K., and Spitsyn, V. I.: Pulse radiolysis of aqueous solutions of bivalent mercury compounds, Doklady Akademii Nauk SSSR, 224, 638–641, 1975.
Pleijel, K. and Munthe, J.: Modeling the atmospheric chemistry of mercury, Water Air Soil Pollut., 80, 317324, https://doi.org/10.1007/BF01189681, 1995.
Pliss, E. M., Grobov, A. M., Kuzaev, A. K., and Buchachenko, A. L.: Magnetic field effect on the oxidation of organic substances by molecular oxygen, J. Phys. Org. Chem., 32, e3915, https://doi.org/10.1002/poc.3915, 2019.
Poissant, L., Pilote, M., Yumvihoze, E., and Lean, D.: Mercury concentrations and foliage/atmosphere fluxes in a maple forest ecosystem in Quebec, Canada, J. Geophys. Res.-Atmos., 113, D10307, https://doi.org/10.1029/2007jd009510, 2008.
Pongratz, R. and Heumann, K. G.: Production of methylated mercury, lead, and cadmium by marine bacteria as a significant natural source for atmospheric heavy metals in polar regions, Chemosphere, 39, 89–102, 1999.
Poulain, A. J., Lalonde, J. D., Amyot, M., Shead, J. A., Raofie, F., and Ariya, P. A.: Redox transformations of mercury in an Arctic snowpack at springtime, Atmos. Environ., 38, 6763–6774, https://doi.org/10.1016/j.atmosenv.2004.09.013, 2004.
Powell, K. J., Brown, P. L., Byrne, R. H., Gajda, T., Hefter, G. T., Sjöberg, S., and Wanner, H.: Chemical speciation of environmentally significant heavy metals with inorganic ligands. Part 1: The Hg2+– Cl−, OH−, CO, SO, and PO aqueous systems, Pure Appl. Chem., 77, 739–800, 2005.
Puigdomenech, I.: SPANA and DATABASE (modified JAVA versions of earlier software MEDUSA, HYDRA and INPUT-SED-PREDOM) Dept. of Inorganic Chemistry, Royal Institute of Technology (KTH), https://www.kth.se/che/medusa/downloads-1.386254 (last access: 26 September 2025), 2013.
Pyykkö, P.: Relativistic Effects in Structural Chemistry, Chem. Rev., 88, 563–594, https://doi.org/10.1021/cr00085a006, 1988.
Qiu, Y., Gai, P., Yue, F., Zhang, Y., He, P., Kang, H., Yu, X., Chen, J., and Xie, Z.: Potential factors impacting PM2.5-Hg during haze evolution revealed by mercury isotope: Emission sources and photochemical processes, Atmos. Res., 277, 106318, https://doi.org/10.1016/j.atmosres.2022.106318, 2022.
Qu, Z., Yan, N. Q., Liu, P., Chi, Y. P., and Jia, J.: Bromine chloride as an oxidant to improve elemental mercury removal from coal-fired flue gas, Environ. Sci. Technol., 43, 8610–8615, https://doi.org/10.1021/es901803s, 2009.
Radke, L. F., Friedli, H. R., and Heikes, B. G.: Atmospheric mercury over the NE Pacific during spring 2002: Gradients, residence time, upper troposphere lower stratosphere loss, and long-range transport, J. Geophys. Res.-Atmos., 112, D19305, https://doi.org/10.1029/2005jd005828, 2007.
Raofie, F. and Ariya, P. A.: Product study of the gas-phase BrO-initiated oxidation of Hg0: evidence for stable Hg1+ compounds, Environ. Sci. Technol., 38, 4319–4326, 2004.
Raofie, F., Snider, G., and Ariya, P. A.: Reaction of gaseous mercury with molecular iodine, atomic iodine, and iodine oxide radicals - Kinetics, product studies, and atmospheric implications, Can. J. Chem., 86, 811–820, https://doi.org/10.1139/v08-088, 2008.
Riccardi, D., Guo, H.-B., Parks, J. M., Gu, B., Summers, A. O., Miller, S. M., Liang, L., and Smith, J. C.: Why mercury prefers soft ligands, J. Phys. Chem. Lett., 4, 2317–2322, https://doi.org/10.1021/jz401075b, 2013.
Richard, J. H., Bischoff, C., Ahrens, C. G. M., and Biester, H.: Mercury (II) reduction and co-precipitation of metallic mercury on hydrous ferric oxide in contaminated groundwater, Sci. Total Environ., 539, 36–44, https://doi.org/10.1016/j.scitotenv.2015.08.116, 2016.
Rolison, J. M., Landing, W. M., Luke, W., Cohen, M., and Salters, V. J. M.: Isotopic composition of species-specific atmospheric Hg in a coastal environment, Chem. Geol., 336, 37–49, 2013.
Rose, C. H., Ghosh, S., Blum, J. D., and Bergquist, B. A.: Effects of ultraviolet radiation on mercury isotope fractionation during photo-reduction for inorganic and organic mercury species, Chem. Geol., 405, 102–111, https://doi.org/10.1016/j.chemgeo.2015.02.025, 2015.
Rosenthal, J. E. and Breit, G.: The isotope shift in hyperfine structure, Phys. Rev., 41, 459–470, https://doi.org/10.1103/PhysRev.41.459, 1932.
Rutter, A. P. and Schauer, J. J.: The impact of aerosol composition on the particle to gas partitioning of reactive mercury, Environ. Sci. Technol., 41, 3934–3939, 2007a.
Rutter, A. P. and Schauer, J. J.: The effect of temperature on the gas-particle partitioning of reactive mercury in atmospheric aerosols, Atmos. Environ., 41, 8647–8657, https://doi.org/10.1016/J.Atmosenv.2007.07.024, 2007b.
Rutter, A. P., Shakya, K. M., Lehr, R., Schauer, J. J., and Griffin, R. J.: Oxidation of gaseous elemental mercury in the presence of secondary organic aerosols, Atmos. Environ., 59, 86–92, https://doi.org/10.1016/j.atmosenv.2012.05.009, 2012.
Saiz-Lopez, A., Sitkiewicz, S. P., Roca-Sanjuán, D., Oliva-Enrich, J. M., Dávalos, J. Z., Notario, R., Jiskra, M., Xu, Y., Wang, F., Thackray, C. P., Sunderland, E. M., Jacob, D. J., Travnikov, O., Cuevas, C. A., Acuña, A. U., Rivero, D., Plane, J. M. C., Kinnison, D. E., and Sonke, J. E.: Photoreduction of gaseous oxidized mercury changes global atmospheric mercury speciation, transport and deposition, Nat. Commun., 9, 4796, https://doi.org/10.1038/s41467-018-07075-3, 2018.
Saiz-Lopez, A., Ulises Acuna, A., Trabelsi, T., Carmona-Garcia, J., Davalos, J. Z., Rivero, D., Cuevas, C. A., Kinnison, D. E., Sitkiewicz, S. P., Roca-Sanjuan, D., and Francisco, J. S.: Gas-phase photolysis of Hg(I) radical species: A new atmospheric mercury reduction process, J. Am. Chem. Soc., 141, 8698–8702, https://doi.org/10.1021/jacs.9b02890, 2019.
Saiz-Lopez, A., Travnikov, O., Sonke, J. E., Thackray, C. P., Jacob, D. J., Carmona-Garcia, J., Frances-Monerris, A., Roca-Sanjuan, D., Acuna, A. U., Davalos, J. Z., Cuevas, C. A., Jiskra, M., Wang, F., Bieser, J., Plane, J. M. C., and Francisco, J. S.: Photochemistry of oxidized Hg(I) and Hg(II) species suggests missing mercury oxidation in the troposphere, P. Natl. Acad. Sci. USA, 117, 30949–30956, 2020.
Saiz-Lopez, A., Acuña, A., Mahajan, A., Dávalos, J., Wuhu, F., Roca-Sanjuán, D., Carmona-García, J., Cuevas, C. A., Kinnison, D., Gomez Martin, J. C., Francisco, J., and Plane, J.: The chemistry of mercury in the stratosphere, Geophys. Res. Lett., 49, e2022GL097953, https://doi.org/10.1029/2022GL097953, 2022.
Saiz-Lopez, A., Cuevas, C. A., Acuña, A. U., Añel, J. A., Mahajan, A. S., de la Torre, L., Feng, W., Dávalos, J. Z., Roca-Sanjuán, D., Kinnison, D. E., Carmona-García, J., Fernandez, R. P., Li, Q., Sonke, J. E., Feinberg, A., Martín, J. C. G., Villamayor, J., Zhang, P., Zhang, Y., Blaszczak-Boxe, C. S., Travnikov, O., Wang, F., Bieser, J., Francisco, J. S., and Plane, J. M. C.: Role of the stratosphere in the global mercury cycle, Sci. Adv., 11, eads1459, https://doi.org/10.1126/sciadv.ads1459, 2025.
Salter, C., Tellinghuisen, P. C., Ashmore, J. G., and Tellinghuisen, J.: The emission spectrum of 200Hg127I, J. Molec. Spectrosc., 120, 334–358, https://doi.org/10.1016/0022-2852(86)90009-3, 1986.
Sander, R.: Modeling Atmospheric Chemistry: Interactions between gas-phase species and liquid cloud/aerosol particles, Surv. Geophys., 20, 1–31, https://doi.org/10.1023/A:1006501706704, 1999.
Sanemasa, I.: Solubility of elemental mercury-vapor in water, B. Chem. Soc. JPN, 48, 1795–1798, https://doi.org/10.1246/bcsj.48.1795, 1975.
Schartup, A. T., Soerensen, A. L., and Heimburger-Boavida, L. E.: Influence of the Arctic Sea-ice regime shift on sea-ice methylated mercury trends, Environ. Sci. Technol. Lett., 7, 708–713, https://doi.org/10.1021/acs.estlett.0c00465, 2020.
Schauble, E. A.: Role of nuclear volume in driving equilibrium stable isotope fractionation of mercury, thallium, and other very heavy elements, Geochim. Cosmochim. Acta, 71, 2170–2189, 2007.
Schmolke, S. R., Schroeder, W. H., Kock, H. H., Schneeberger, D., Munthe, J., and Ebinghaus, R.: Simultaneous measurements of total gaseous mercury at four sites on a 800 km transect: spatial distribution and short-time variability of total gaseous mercury over central Europe, Atmos. Environ., 33, 1725–1733, 1999.
Schroeder, W. H. and Munthe, J.: Atmospheric mercury – An overview, Atmos. Environ., 32, 809–822, 1998.
Schroeder, W. H., Yarwood, G., and Niki, H.: Transformation processes involving mercury species in the atmosphere – Results from a literature survey, Water Air Soil Pollut., 56, 653–666, 1991.
Schroeder, W. H., Anlauf, K. G., Barrie, L. A., Lu, J. Y., Steffen, A., Schneeberger, D. R., and Berg, T.: Arctic springtime depletion of mercury, Nature, 394, 331–332, 1998.
Schwab, L., Gallati, N., Reiter, S. M., Kimber, R. L., Kumar, N., McLagan, D. S., Biester, H., Kraemer, S. M., and Wiederhold, J. G.: Mercury isotope fractionation during dark abiotic reduction of Hg(II) by dissolved, surface-bound, and structural Fe(II), Environ. Sci. Technol., 57, 15243–15254, https://doi.org/10.1021/acs.est.3c03703, 2023.
Schwartz, S. E.: Mass-transport considerations pertinent to aqueous phase reactions of gases in liquid-water clouds, in: Chemistry of multiphase atmospheric systems, edited by: Jäschke, W., NATO ASI Series, G6, Springer-Verlag Berlin Heidelberg, 415–471, https://doi.org/10.1007/978-3-642-70627-1_16 1986.
Scott, S. L., Yusuf, H., Lahoutifard, N., and Maunder, K.: Homogeneous and heterogeneous reactions of atmospheric mercury(II) with sulfur(IV), Journal de Physique IV, 107, 1201–1204, 2003.
Seigneur, C., Wrobel, J., and Constantinou, E.: A chemical kinetic mechanism for atmospheric inorganic mercury, Environ. Sci. Technol., 28, 1589–1597, 1994.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics: from air pollution to climate change, Wiley, Hoboken, 3rd edition, 1152 pp., ISBN 978-1-118-94740-1, 2006.
Selin, N. E.: Global biogeochemical cycling of mercury: A review, Environ. Resour., 34, 43–63, https://doi.org/10.1146/annurev.environ.051308.084314, 2009.
Selin, N. E., Jacob, D. J., Yantosca, R. M., Strode, S., Jaegle, L., and Sunderland, E. M.: Global 3-D land-ocean-atmosphere model for mercury: Present-day versus preindustrial cycles and anthropogenic enrichment factors for deposition, Global Biogeochemical Cycles, 22, GB2011, https://doi.org/10.1029/2007gb003040, 2008.
Sexauer Gustin, M., Pierce, A. M., Huang, J., Miller, M. B., Holmes, H. A., and Loria-Salazar, S. M.: Evidence for different reactive Hg sources and chemical compounds at adjacent valley and high elevation locations, Environ. Sci. Technol., 50, 12225–12231, https://doi.org/10.1021/acs.est.6b03339, 2016.
Shah, V., Jacob, D., Thackray, C., Wang, X., Sunderland, E., Dibble, T., Saiz-Lopez, A., Cernusak, I., Kellö, V., Castro, P., Wu, R., and Wang, C.: Improved mechanistic model of the atmospheric redox chemistry of mercury, Environ. Sci. Technol., 55, 14445–14456, 2021.
Shah, V., Jaeglé, L., Gratz, L. E., Ambrose, J. L., Jaffe, D. A., Selin, N. E., Song, S., Campos, T. L., Flocke, F. M., Reeves, M., Stechman, D., Stell, M., Festa, J., Stutz, J., Weinheimer, A. J., Knapp, D. J., Montzka, D. D., Tyndall, G. S., Apel, E. C., Hornbrook, R. S., Hills, A. J., Riemer, D. D., Blake, N. J., Cantrell, C. A., and Mauldin III, R. L.: Origin of oxidized mercury in the summertime free troposphere over the southeastern US, Atmos. Chem. Phys., 16, 1511–1530, https://doi.org/10.5194/acp-16-1511-2016, 2016.
Shepler, B. C.: Ab Initio Investigation of the thermochemistry, spectroscopy and dynamics of reactions between mercury and reactive halogen species, Ph. D. thesis, Department of Chemistry, Washington State University, Pullman, WA, 289 pp., https://hdl.handle.net/2376/560 , 2006.
Shepler, B. C. and Peterson, K. A.: Mercury Monoxide: A systematic investigation of its ground electronic state, J. Phys. Chem. A, 107, 1783–1787, 2003.
Shepler, B. C., Balabanov, N. B., and Peterson, K. A.: Ab initio thermochemistry involving heavy atoms: An investigation of the reactions Hg + IX (X = I, Br, Cl, O), J. Phys. Chem. A, 109, 10363–10372, 2005.
Shepler, B. C., Balabanov, N. B., and Peterson, K. A.: Hg + Br → HgBr recombination and collision-induced dissociation dynamics, J. Chem. Phys., 127, 164304, https://doi.org/10.1063/1.27771422007, 2007.
Sherman, L. S., Blum, J. D., Dvonch, J. T., Gratz, L. E., and Landis, M. S.: The use of Pb, Sr, and Hg isotopes in Great Lakes precipitation as a tool for pollution source attribution, Sci. Total Environ., 502, 362–374, 2015.
Sherman, L. S., Blum, J. D., Keeler, G. J., Demers, J. D., and Dvonch, J. T.: Investigation of local mercury deposition from a coal-fired power plant using mercury isotopes, Environ. Sci. Technol., 46, 382–390, 2012.
Sherman, L. S., Blum, J. D., Johnson, K. P., Keeler, G. J., Barres, J. A., and Douglas, T. A.: Mass-independent fractionation of mercury isotopes in Arctic snow driven by sunlight, Nat. Geosci., 3, 173–177, 2010.
Sholupov, S., Pogarev, S., Ryzhov, V., Mashyanov, N., and Stroganov, A.: Zeeman atomic absorption spectrometer RA-915+for direct determination of mercury in air and complex matrix samples, Fuel Process. Technol., 85, 473–485, 2004.
Si, L. and Ariya, P.: Recent advances in atmospheric chemistry of mercury, Atmosphere, 9, 76, https://doi.org/10.3390/atmos9020076, 2018.
Si, L. and Ariya, P. A.: Reduction of oxidized mercury species by dicarboxylic acids (C2−C4): Kinetic and product studies, Environ. Sci. Technol., 42, 5150–5155, https://doi.org/10.1021/es800552z, 2008.
Si, L. and Ariya, P. A.: Aqueous photoreduction of oxidized mercury species in presence of selected alkanethiols, Chemosphere, 84, 1079–1084, 2011.
Si, L. and Ariya, P. A.: Photochemical reactions of divalent mercury with thioglycolic acid: Formation of mercuric sulfide particles, Chemosphere, 119, 467–472, https://doi.org/10.1016/j.chemosphere.2014.07.022, 2015.
Si, L., Branfireun, B. A., and Fierro, J.: Chemical oxidation and reduction pathways of mercury relevant to natural waters: A review, Water, 14, 1891, https://doi.org/10.3390/w14121891, 2022
Siegler, R. W., Nierenberg, D. W., and Hickey, W. F.: Fatal poisoning from liquid dimethylmercury: A neuropathologic study, Hum. Pathol., 30, 720–723, https://doi.org/10.1016/s0046-8177(99)90101-6, 1999.
Sipilä, M., Sarnela, N., Jokinen, T., Henschel, H., Junninen, H., Kontkanen, J., Richters, S., Kangasluoma, J., Franchin, A., Peräkylä, O., Rissanen, M. P., Ehn, M., Vehkamäki, H., Kurten, T., Berndt, T., Petäjä, T., Worsnop, D., Ceburnis, D., Kerminen, V.-M., Kulmala, M., and O'Dowd, C.: Molecular-scale evidence of aerosol particle formation via sequential addition of HIO3, Nature, 537, 532–534, https://doi.org/10.1038/nature19314, 2016.
Sitkiewicz, S. P., Rivero, D., Oliva-Enrich, J. M., Saiz-Lopez, A., and Roca-Sanjuán, D.: Ab initio quantum-chemical computations of the absorption cross sections of HgX2 and HgXY (X, Y = Cl, Br, and I): Molecules of interest in the Earth's atmosphere, Phys. Chem. Chem. Phys., 21, 455–467, https://doi.org/10.1039/C8CP06160B, 2019.
Skyllberg, U.: Chemical speciation of mercury in soil and sediment, in: Advances in environmental chemistry and toxicology of mercury edited by: Liu, G., Cai, Y., and O'Driscoll, N., John Wiley & Sons, 218–258, https://doi.org/10.1002/9781118146644.ch7, 2011.
Slemr, F., Schuster, G., and Seiler, W.: Distribution, speciation, and budget of atmospheric mercury, J. Atmos. Chem., 3, 407–434, 1985.
Slemr, F., Martin, L., Labuschagne, C., Mkololo, T., Angot, H., Magand, O., Dommergue, A., Garat, P., Ramonet, M., and Bieser, J.: Atmospheric mercury in the Southern Hemisphere – Part 1: Trend and inter-annual variations in atmospheric mercury at Cape Point, South Africa, in 2007–2017, and on Amsterdam Island in 2012–2017, Atmos. Chem. Phys., 20, 7683–7692, https://doi.org/10.5194/acp-20-7683-2020, 2020.
Slemr, F., Weigelt, A., Ebinghaus, R., Kock, H. H., Bödewadt, J., Brenninkmeijer, C. A. M., Rauthe-Schöch, A., Weber, S., Hermann, M., Becker, J., Zahn, A., and Martinsson, B.: Atmospheric mercury measurements onboard the CARIBIC passenger aircraft, Atmos. Meas. Tech., 9, 2291–2302, https://doi.org/10.5194/amt-9-2291-2016, 2016.
Slemr, F., Weigelt, A., Ebinghaus, R., Bieser, J., Brenninkmeijer, C. A. M., Rauthe-Schöch, A., Hermann, M., Martinsson, B. G., van Velthoven, P., Bönisch, H., Neumaier, M., Zahn, A., and Ziereis, H.: Mercury distribution in the upper troposphere and lowermost stratosphere according to measurements by the IAGOS-CARIBIC observatory: 2014–2016, Atmos. Chem. Phys., 18, 12329–12343, https://doi.org/10.5194/acp-18-12329-2018, 2018.
Smith, R. S., Wiederhold, J. G., and Kretzschmar, R.: Mercury isotope fractionation during precipitation of metacinnabar (β-HgS) and montroydite (HgO), Environ. Sci. Technol., 49, 4325–4334, 2015.
Snelson, A.: Infrared matrix isolation spectrum of methyl radical produced by pyrolysis of methyl iodide and dimethyl mercury, J. Phys. Chem., 74, 537–544, https://doi.org/10.1021/j100698a011, 1970.
Snider, G., Raofie, F., and Ariya, P. A.: Effects of relative humidity and CO(g) on the O3-initiated oxidation reaction of Hg0(g): Kinetic & product studies, Phys. Chem. Chem. Phys., 10, 5616–5623, https://doi.org/10.1039/b801226a, 2008.
Soerensen, A. L., Sunderland, E. M., Holmes, C. D., Jacob, D. J., Yantosca, R. M., Skov, H., Christensen, J. H., Strode, S. A., and Mason, R. P.: An improved global model for air-sea exchange of mercury: High concentrations over the North Atlantic, Environ. Sci. Technol., 44, 8574–8580, https://doi.org/10.1021/es102032g, 2010.
Sommar, J. O.: Published data used to create the plots in Figure 12, Figure 13a, Figure 13b, Figure 13c & Figure 14, Zenodo [data set], https://doi.org/10.5281/zenodo.17079781, 2025.
Sommar, J., Feng, X. B., and Lindqvist, O.: Speciation of volatile mercury species present in digester and deposit gases, Appl. Organomet. Chem., 13, 441–445, 1999.
Sommar, J., Hallquist, M., and Ljungstrom, E.: Rate of reaction between the nitrate radical and dimethyl mercury in the gas phase, Chem. Phys. Lett., 257, 434–438, 1996.
Sommar, J., Lindqvist, O., and Strömberg, D.: Distribution equilibrium of mercury (II) chloride between water and air applied to flue gas scrubbing, J. Air Waste Manage. Assoc., 50, 1663–1666, 2000.
Sommar, J., Osterwalder, S., and Zhu, W.: Recent advances in understanding and measurement of Hg in the environment: Surface-atmosphere exchange of gaseous elemental mercury (Hg0), Sci. Total Environ., 721, 137648, https://doi.org/10.1016/j.scitotenv.2020.137648, 2020.
Sommar, J., Gårdfeldt, K., Strömberg, D., and Feng, X. B.: A kinetic study of the gas-phase reaction between the hydroxyl radical and atomic mercury, Atmos. Environ., 35, 3049–3054, 2001.
Sommar, J., Hallquist, M., Ljungstrom, E., and Lindqvist, O.: On the gas phase reactions between volatile biogenic mercury species and the nitrate radical, J. Atmos. Chem., 27, 233–247, 1997.
Sommar, J., Wängberg, I., Berg, T., Gårdfeldt, K., Munthe, J., Richter, A., Urba, A., Wittrock, F., and Schroeder, W. H.: Circumpolar transport and air-surface exchange of atmospheric mercury at Ny-Ålesund (79° N), Svalbard, spring 2002, Atmos. Chem. Phys., 7, 151–166, https://doi.org/10.5194/acp-7-151-2007, 2007.
Sommar, J., Zhu, W., Lin, C. J., and Feng, X.: Field approaches to measure Hg exchange between natural surfaces and the atmosphere – A review, Crit. Rev. Env. Sci. Tec., 43, 1657–1739, 2013.
Song, Z. C., Huang, S. J., Zhang, P., Yuan, T. F., and Zhang, Y. X.: Isotope data constrains redox chemistry of atmospheric mercury, Environ. Sci. Technol., 58, 13307–13317, https://doi.org/10.1021/acs.est.4c02600, 2024.
Sonke, J. E., Angot, H., Zhang, Y., Poulain, A., Björn, E., and Schartup, A.: Global change effects on biogeochemical mercury cycling, Ambio, 52, 853–876, https://doi.org/10.1007/s13280-023-01855-y, 2023.
Spicer, C. W., Satola, J., Abbgy, A. A., Plastridge, R. A., and Cowen, K. A.: Kinetics of gas-phase elemental mercury reactions with halogen species, ozone, and nitrate radical under atmospheric conditions, in: Final Report to Florida Department of Environmental Protection under Contract AQ174, 1–20, Battelle: Columbus, OH, USA, 2002.
Sprovieri, F., Pirrone, N., Ebinghaus, R., Kock, H., and Dommergue, A.: A review of worldwide atmospheric mercury measurements, Atmos. Chem. Phys., 10, 8245–8265, https://doi.org/10.5194/acp-10-8245-2010, 2010.
Sprovieri, F., Pirrone, N., Bencardino, M., D'Amore, F., Angot, H., Barbante, C., Brunke, E.-G., Arcega-Cabrera, F., Cairns, W., Comero, S., Diéguez, M. D. C., Dommergue, A., Ebinghaus, R., Feng, X. B., Fu, X., Garcia, P. E., Gawlik, B. M., Hageström, U., Hansson, K., Horvat, M., Kotnik, J., Labuschagne, C., Magand, O., Martin, L., Mashyanov, N., Mkololo, T., Munthe, J., Obolkin, V., Ramirez Islas, M., Sena, F., Somerset, V., Spandow, P., Vardè, M., Walters, C., Wängberg, I., Weigelt, A., Yang, X., and Zhang, H.: Five-year records of mercury wet deposition flux at GMOS sites in the Northern and Southern hemispheres, Atmos. Chem. Phys., 17, 2689–2708, https://doi.org/10.5194/acp-17-2689-2017, 2017.
Sprovieri, F., Pirrone, N., Bencardino, M., D'Amore, F., Carbone, F., Cinnirella, S., Mannarino, V., Landis, M., Ebinghaus, R., Weigelt, A., Brunke, E.-G., Labuschagne, C., Martin, L., Munthe, J., Wängberg, I., Artaxo, P., Morais, F., Barbosa, H. D. M. J., Brito, J., Cairns, W., Barbante, C., Diéguez, M. D. C., Garcia, P. E., Dommergue, A., Angot, H., Magand, O., Skov, H., Horvat, M., Kotnik, J., Read, K. A., Neves, L. M., Gawlik, B. M., Sena, F., Mashyanov, N., Obolkin, V., Wip, D., Feng, X. B., Zhang, H., Fu, X., Ramachandran, R., Cossa, D., Knoery, J., Marusczak, N., Nerentorp, M., and Norstrom, C.: Atmospheric mercury concentrations observed at ground-based monitoring sites globally distributed in the framework of the GMOS network, Atmos. Chem. Phys., 16, 11915–11935, https://doi.org/10.5194/acp-16-11915-2016, 2016.
St Louis, V. L., Hintelmann, H., Graydon, J. A., Kirk, J. L., Barker, J., Dimock, B., Sharp, M. J., and Lehnherr, I.: Methylated mercury species in Canadian high arctic marine surface waters and snowpacks, Environ. Sci. Technol., 41, 6433–6441, https://doi.org/10.1021/es070692s, 2007.
Stathopoulos, D.: Fractionation of mercury isotopes in an aqueous environment: Chemical oxidation, M. Sc. Thesis, Faculty of Arts and Science, Trent University, Peterborough, Canada, 77 pp., https://batadora.trentu.ca/objects/etd-308, 2014.
Steffen, A., Schroeder, W., Bottenheim, J., Narayan, J., and Fuentes, J. D.: Atmospheric mercury concentrations: Measurements and profiles near snow and ice surfaces in the Canadian Arctic during Alert 2000, Atmos. Environ., 36, 2653–2661, 2002.
Steffen, A., Bottenheim, J., Cole, A., Ebinghaus, R., Lawson, G., and Leaitch, W. R.: Atmospheric mercury speciation and mercury in snow over time at Alert, Canada, Atmos. Chem. Phys., 14, 2219–2231, https://doi.org/10.5194/acp-14-2219-2014, 2014.
Steffen, A., Douglas, T., Amyot, M., Ariya, P., Aspmo, K., Berg, T., Bottenheim, J., Brooks, S., Cobbett, F., Dastoor, A., Dommergue, A., Ebinghaus, R., Ferrari, C., Gardfeldt, K., Goodsite, M. E., Lean, D., Poulain, A. J., Scherz, C., Skov, H., Sommar, J., and Temme, C.: A synthesis of atmospheric mercury depletion event chemistry in the atmosphere and snow, Atmos. Chem. Phys., 8, 1445–1482, https://doi.org/10.5194/acp-8-1445-2008, 2008.
Steffen, A., Lehnherr, I., Cole, A., Ariya, P., Dastoor, A., Durnford, D., Kirk, J., and Pilote, M.: Atmospheric mercury in the Canadian Arctic. Part I: A review of recent field measurements, Sci. Total Environ., 509, 3–15, 2015.
Stephan, T. and Trappitsch, R.: Reliable uncertainties: Error correlation, rotated error bars, and linear regressions in three-isotope plots and beyond, Int. J. Mass Spectrom., 491, 117053, https://doi.org/10.1016/j.ijms.2023.117053, 2023.
Stowe, A. C. and Knight, L. B.: Electron spin resonance studies of 199HgH, 201HgH, 199HgD and 201HgD isolated in neon and argon matrices at 4K: an electronic structure investigation, Mol. Phys., 100, 353–360, https://doi.org/10.1080/00268970110096443, 2002.
Strand, R., Lund, W., and Aaseth, J.: Complex formation of zinc, cadmium, and mercury with penicillamine, J. Inorg. Biochem., 19, 301–309, https://doi.org/10.1016/0162-0134(83)80003-8, 1983.
Streets, D. G., Horowitz, H. M., Jacob, D. J., Lu, Z., Levin, L., ter Schure, A. F. H., and Sunderland, E. M.: Total Mercury Released to the Environment by Human Activities, Environmental Science & Technology, 51, 5969–5977, https://doi.org/10.1021/acs.est.7b00451, 2017.
Streets, D. G., Horowitz, H. M., Lu, Z. F., Levin, L., Thackray, C. P., and Sunderland, E. M.: Five hundred years of anthropogenic mercury: spatial and temporal release profiles, Environmental Research Letters, 14, ARTN 084004, https://doi.org/10.1088/1748-9326/ab281f, 2019.
Strode, S. A., Jaegle, L., Selin, N. E., Jacob, D. J., Park, R. J., Yantosca, R. M., Mason, R. P., and Slemr, F.: Air-sea exchange in the global mercury cycle, Global Biogeochem. Cy., 21, GB1017, https://doi.org/10.1029/2006gb002766, 2007.
Strömberg, D., Gropen, O., and Wahlgren, U.: Non-relativistic and relativistic calculations on some Zn, Cd and Hg complexes, Chem. Phys., 133, 207–219, https://doi.org/10.1016/0301-0104(89)80202-2, 1989.
Strömberg, D. and Wahlgren, U. First-order relativistic calculations on Au2 and , Chem. Phys. Lett., 169, 109–115, https://doi.org/10.1016/0009-2614(90)85174-B, 1990.
Strömberg, D., Strömberg, A., and Wahlgren, U.: Relativistic quantum calculations on some mercury sulfide molecules, Water Air Soil Pollut., 56, 681–695, https://doi.org/10.1007/BF00342309, 1991.
Subir, M., Ariya, P. A., and Dastoor, A. P.: A review of uncertainties in atmospheric modeling of mercury chemistry I. Uncertainties in existing kinetic parameters – Fundamental limitations and the importance of heterogeneous chemistry, Atmos. Environ., 45, 5664–5676, 2011.
Subir, M., Ariya, P. A., and Dastoor, A. P.: A review of the sources of uncertainties in atmospheric mercury modeling II. Mercury surface and heterogeneous chemistry – A missing link, Atmos. Environ., 46, 1–10, 2012.
Suda, I., Suda, M., and Hirayama, K.: Degradation of methyl and ethyl mercury by singlet oxygen generated from sea-water exposed to sunlight or ultraviolet-light, Arch. Toxicol., 67, 365–368, https://doi.org/10.1007/Bf01973709, 1993.
Sumner, A. L., Spicer, C. W., Satola, J., Mangaraj, R., Cowen, K. A., Landis, M. S., Stevens, R. K., and Atkeson, T.: Environmental chamber studies of mercury reactions in the atmosphere, in: Dynamics of Mercury Pollution on Regional and Global Scales: Atmospheric Processes and Human Exposures Around the World, edited by: Pirrone, N. and Mahaffey, K. R., Springer US, 193–212, https://doi.org/10.1007/b105709, 2005.
Sun, G.: Isotope fractionation during major chemical transformations of atmospheric mercury, Ph D. thesis, Institute of Geochemistry, Chinese Academy of Sciences, Guiyang, 107 pp., 2018.
Sun, G., Feng, X., Yin, R., Wang, F., Lin, C.-J., Li, K., and Sommar, J. O.: Dissociation of mercuric oxides drives anomalous isotope fractionation during net photo-oxidation of mercury vapor in air, Environ. Sci. Technol., 55, 13428–13438, https://doi.org/10.1021/acs.est.2c02722, 2022.
Sun, G., Sommar, J., Feng, X., Lin, C.-J., Ge, M., Wang, W., Yin, R., Fu, X., and Shang, L.: Mass-dependent and -independent fractionation of mercury isotope during gas-phase oxidation of elemental mercury vapor by atomic Cl and Br, Environ. Sci. Technol., 50, 9232–9241, 2016.
Sun, L., Lu, B., Yuan, D., Hao, W., and Zheng, Y.: Variations in the isotopic composition of stable mercury isotopes in typical mangrove plants of the Jiulong estuary, SE China, Environ. Sci. Pollut. Res., 24, 1459–1468, https://doi.org/10.1007/s11356-016-7933-1, 2017.
Sun, R., Sonke, J. E., Heimbürger, L.-E., Belkin, H. E., Liu, G., Shome, D., Cukrowska, E., Liousse, C., Pokrovsky, O. S., and Streets, D. G.: Mercury stable isotope signatures of world coal deposits and historical coal combustion emissions, Environ. Sci. Technol., 48, 7660–7668, https://doi.org/10.1021/es501208a, 2014.
Sun, R. Y., Jiskra, M., Amos, H. M., Zhang, Y. X., Sunderland, E. M., and Sonke, J. E.: Modelling the mercury stable isotope distribution of Earth surface reservoirs: Implications for global Hg cycling, Geochim. Cosmochim. Ac., 246, 156–173, 2019.
Sun, R. Y., Cao, F., Dai, S. F., Shan, B., Qi, C. C., Xu, Z. J., Li, P. F., Liu, Y., Zheng, W., and Chen, J. B.: Atmospheric mercury isotope shifts in response to mercury emissions from underground coal fires, Environ. Sci. Technol., 57, 8638–8649, https://doi.org/10.1021/acs.est.2c08637, 2023.
Svanberg, S.: Geophysical gas monitoring using optical techniques: Volcanoes, geothermal fields and mines, Opt. Laser. Eng., 37, 245–266, 2002.
Swartzendruber, P. C., Chand, D., Jaffe, D. A., Smith, J., Reidmiller, D., Gratz, L., Keeler, J., Strode, S., Jaegle, L., and Talbot, R.: Vertical distribution of mercury, CO, ozone, and aerosol scattering coefficient in the Pacific Northwest during the spring 2006 INTEX-B campaign, J. Geophys. Res.-Atmos., 113, D10305, https://doi.org/10.1029/2007jd009579, 2008.
Swartzendruber, P. C., Jaffe, D. A., and Finley, B.: Development and first results of an aircraft-based, high time resolution technique for gaseous elemental and reactive (oxidized) gaseous mercury, Environ. Sci. Technol., 43, 7484–7489, https://doi.org/10.1021/es901390t, 2009.
Symons, M. C. R. and Yandell, J. K.: Unstable intermediates. Part LXXXVII. The dipositive radical cations of gallium, thallium, and mercury derived from aqueous solutions of gallium(III), thallium(III), and mercury(II) salts at 77 K by γ-irradiation, J. Chem. Soc. A, 6, 760–762, https://doi.org/10.1039/J19710000760, 1971.
Szponar, N., McLagan, D. S., Kaplan, R. J., Mitchell, C. P. J., Wania, F., Steffen, A., Stupple, G. W., Monaci, F., and Bergquist, B. A.: Isotopic characterization of atmospheric gaseous elemental mercury by passive air sampling, Environ. Sci. Technol., 54, 10533–10543, https://doi.org/10.1021/acs.est.0c02251, 2020.
Szponar, N., Vega, C. M., Gerson, J., McLagan, D. S., Pillaca, M., Delgado, S., Lee, D., Rahman, N., Fernandez, L. E., Bernhardt, E. S., Kiefer, A. M., Mitchell, C. P. J., Wania, F., and Bergquist, B. A.: Tracing Atmospheric Mercury from Artisanal and Small-Scale Gold Mining, Environmental Science & Technology, 59, 5021–5033, https://doi.org/10.1021/acs.est.4c10521, 2025.
Tacey, S. A., Xu, L., Mavrikakis, M., and Schauer, J. J.: Heterogeneous reduction pathways for Hg(II) species on dry aerosols: A first-principles computational study, J. Phys. Chem. A, 120, 2106–2113, https://doi.org/10.1021/acs.jpca.5b12769, 2016.
Tacey, S. A., Szilvási, T., Xu, L., Schauer, J. J., and Mavrikakis, M.: The role of iron-oxide aerosols and sunlight in the atmospheric reduction of Hg(II) species: A DFT+U study, Appl. Catal. B–Environ., 234, 347–356, https://doi.org/10.1016/j.apcatb.2018.04.049, 2018a.
Tacey, S. A., Xu, L., Szilvási, T., Schauer, J. J., and Mavrikakis, M.: Quantum chemical calculations to determine partitioning coefficients for HgCl2 on iron-oxide aerosols, Sci. Total Environ., 636, 580–587, https://doi.org/10.1016/j.scitotenv.2018.04.289, 2018b.
Talbot, R., Mao, H., Scheuer, E., Dibb, J., and Avery, M.: Total depletion of Hg degrees in the upper troposphere-lower stratosphere, Geophys. Res. Lett., 34, L23804, https://doi.org/10.1029/2007gl031366, 2007.
Tang, K., Song, Z., Fu, X., Zhang, Y., Zhang, H., Sun, Y., Zhang, H., Wu, X., Deng, Q., Zhang, L., Suratman, S., Seng, T. H., and Feng, X.: An interhemispheric difference in atmospheric gaseous elemental mercury isotopes reveals a new insight in oceanic mercury emissions, J. Geophys. Res.-Atmos., 130, e2024JD042178, https://doi.org/10.1029/2024jd042178, 2025.
Tas, E., Obrist, D., Peleg, M., Matveev, V., Faïn, X., Asaf, D., and Luria, M.: Measurement-based modelling of bromine-induced oxidation of mercury above the Dead Sea, Atmos. Chem. Phys., 12, 2429–2440, https://doi.org/10.5194/acp-12-2429-2012, 2012.
Tate, M. T., Janssen, S. E., Lepak, R. F., Flucke, L., and Krabbenhoft, D. P.: National-scale assessment of total gaseous mercury isotopes across the United States, J. Geophys. Res.-Atmos., 128, e2022JD038276, https://doi.org/10.1029/2022JD038276, 2023.
Taylor, P. H., Mallipeddi, R., and Yamada, T.: LP/LIF study of the formation and consumption of mercury (I) chloride: Kinetics of mercury chlorination, Chemosphere, 61, 685–692, 2005.
Tellinghuisen, J. and Ashmore, J. G.: Mixed representations for diatomic spectroscopic data: application to mercury(I) bromide, Chem. Phys. Lett., 102, 10–16, 1983.
Tellinghuisen, J., Tellinghuisen, P. C., Davies, S. A., Berwanger, P., and Viswanathan, K. S.: B → X transitions in HgCl and HgI, Appl. Phys. Lett., 41, 789–791, https://doi.org/10.1063/1.93704, 1982.
Terenin, A.: Photo-dissociation of the vapors of organo-metallic compounds, J. Chem. Phys., 2, 441–442, https://doi.org/10.1063/1.1749505, 1934.
Terenin, A. and Prileshajewa, N.: Photodissociation of the vapors of some organo-metallic compounds, Acta Physicochim. URSS, 1, 759–776, 1935.
Tevault, D., Strommen, D. P., and Nakamoto, K.: Reactions of mercury halides with carbon monoxide, nitric oxide, molecular nitrogen, and unsaturated hydrocarbons in argon matrices, J. Am. Chem. Soc., 99, 2997–3003, https://doi.org/10.1021/ja00451a024, 1977.
Thomsen, E. L. and Egsgaard, H.: Rate of reaction of dimethylmercury with oxygen atoms in the gas phase, Chem. Phys. Lett., 125, 378–382, 1986.
Tong, Y., Eichhorst, T., Olson, M. R., Rutter, A. P., Shafer, M. M., Wang, X., and Schauer, J. J.: Comparison of heterogeneous photolytic reduction of Hg(II) in the coal fly ashes and synthetic aerosols, Atmos. Res., 138, 324–329, https://doi.org/10.1016/j.atmosres.2013.11.015, 2014.
Tong, Y., Eichhorst, T., Olson, M. R., McGinnis, J. E., Turner, I., Rutter, A. P., Shafer, M. M., Wang, X., and Schauer, J. J.: Atmospheric photolytic reduction of Hg(II) in dry aerosols, Environ. Sci.-Proc. Imp., 15, 1883–1888, https://doi.org/10.1039/c3em00249g, 2013.
Tong, Y. D., Zhang, H. F., Lin, H. M., de Foy, B., Chen, L., Zhang, W., Wang, X. J., and Guan, C. F.: A potential route for photolytic reduction of HgCl2 and HgBr2 in dry air and analysis about the impacts from ozone, Atmos. Res., 249, 105310, https://doi.org/10.1016/j.atmosres.2020.105310, 2021.
Tossell, J. A.: Theoretical study of the photodecomposition of methyl Hg complexes, J. Phys. Chem. A, 102, 3587–3591, https://doi.org/10.1021/jp980244u, 1998.
Tossell, J. A.: Calculation of the energetics for oxidation of gas-phase elemental Hg by Br and BrO, J. Phys. Chem. A, 107, 7804–7808, 2003.
Tossell, J. A.: Calculation of the energetics for the oligomerization of gas phase HgO and HgS and for the solvolysis of crystalline HgO and HgS, J. Phys. Chem. A, 110, 2571–2578, 2006.
Tossell, J. A. and Vaughan, D. J.: Relationships between valence orbital binding-energies and crystal-structures in compounds of copper, silver, gold, zinc, cadmium, and mercury, Inorg. Chem., 20, 3333–3340, https://doi.org/10.1021/ic50224a038, 1981.
Toyota, K., Dastoor, A. P., and Ryzhkov, A.: Air–snowpack exchange of bromine, ozone and mercury in the springtime Arctic simulated by the 1-D model PHANTAS – Part 2: Mercury and its speciation, Atmos. Chem. Phys., 14, 4135–4167, https://doi.org/10.5194/acp-14-4135-2014, 2014.
Travnikov, O., Angot, H., Artaxo, P., Bencardino, M., Bieser, J., D'Amore, F., Dastoor, A., De Simone, F., Diéguez, M. D. C., Dommergue, A., Ebinghaus, R., Feng, X. B., Gencarelli, C. N., Hedgecock, I. M., Magand, O., Martin, L., Matthias, V., Mashyanov, N., Pirrone, N., Ramachandran, R., Read, K. A., Ryjkov, A., Selin, N. E., Sena, F., Song, S., Sprovieri, F., Wip, D., Wängberg, I., and Yang, X.: Multi-model study of mercury dispersion in the atmosphere: atmospheric processes and model evaluation, Atmos. Chem. Phys., 17, 5271–5295, https://doi.org/10.5194/acp-17-5271-2017, 2017.
Tsui, M. T.-K., Blum, J. D., and Kwon, S. Y.: Review of stable mercury isotopes in ecology and biogeochemistry, Sci. Total Environ., 716, 135386, https://doi.org/10.1016/j.scitotenv.2019.135386, 2020.
Turro, N. J.: Influence of nuclear spin on chemical reactions: Magnetic isotope and magnetic field effects (A review), P. Natl. Acad. Sci. USA, 80, 609–621, 1983.
Ullrich, S. M., Tanton, T. W., and Abdrashitova, S. A.: Mercury in the aquatic environment: A review of factors affecting methylation, Crit. Rev. Env. Sci. Tec., 31, 241–293, 2001.
UNEP: Global Mercury Assessment 2018: Sources, Emissions, Releases and Environmental Transport, UNEP Chemicals Branch, Geneva, Switzerland, ISBN 978-92-807-3744-8, https://www.unep.org/resources/publication/global-mercury-assessment-2018, 2018.
van der Linden, W. E. and Beers, C.: Determination of composition and stability-constants of complexes of mercury(II) with amino-acids, Anal. Chim. Acta, 68, 143–154, https://doi.org/10.1016/s0003-2670(01)85155-2, 1974.
van der Linden, W. E. and Beers, C.: Formation constants of mercury(II) with some buffer/masking agents and the formation of mixed-ligand complexes, Talanta, 22, 89–92, https://doi.org/10.1016/0039-9140(75)80148-2, 1975.
van Loon, L. L., Mader, E. A., and Scott, S. L.: Reduction of the aqueous mercuric ion by sulfite: UV spectrum of HgSO3 and its intramolecular redox reaction, J. Phys. Chem. A, 104, 1621–1626, 2000.
van Loon, L. L., Mader, E. A., and Scott, S. L.: Sulfite stabilization and reduction of the aqueous mercuric ion: Kinetic determination of sequential formation constants, J. Phys. Chem. A, 105, 3190–3195, 2001.
Velivetskaya, T. A., Ignatiev, A. V., Yakovenko, V. V., and Vysotskiy, S. V.: Experimental studies of the oxygen isotope anomalies (Δ17O) of H2O2 and their relation to radical recombination reactions, Chem. Phys. Lett., 693, 107–113, https://doi.org/10.1016/j.cplett.2018.01.012, 2018.
Velivetskaya, T. A., Ignatiev, A. V., Budnitskiy, S. Y., Yakovenko, V. V., and Vysotskiy, S. V.: Mass-independent fractionation of oxygen isotopes during H2O2 formation by gas-phase discharge from water vapour, Geochim. Cosmochim. Acta, 193, 54–65, https://doi.org/10.1016/j.gca.2016.08.008, 2016.
Volman, D. H.: Reaction of optically excited mercury vapor with oxygen, J. Chem. Phys., 21, 2086–2087, https://doi.org/10.1063/1.1698769, 1953.
Vyazovetskii, Y. V.: Photochemical production of the 204Hg isotope, Tech. Phys+, 57, 603–607, 2012.
Wallschläger, D., Hintelmann, H., Evans, R. D., and Wilken, R. D.: Volatilization of demethylmercury and elemental mercury from river Elbe floodplain soils, Water Air Soil Pollut., 80, 1325–1329, https://doi.org/10.1007/BF01189798, 1995.
Wang, B., Yuan, W., Wang, X., Li, K., Lin, C.-J., Li, P., Lu, Z., Feng, X., and Sommar, J.: Canopy-level flux and vertical gradients of Hg0 stable isotopes in remote evergreen broadleaf forest show year-around net Hg0 deposition, Environ. Sci. Technol., 56, 5950–5959, https://doi.org/10.1021/acs.est.2c00778, 2022.
Wang, C., Yang, S. C., Li, R. L., Yan, J. Y., Hu, Y. X., Lai, C. Y., Li, Z. G., Li, P., Zhang, L. M., and Feng, X. B.: Atmospheric mercury concentrations and isotopic compositions impacted by typical anthropogenic mercury emissions sources, Environ. Sci. Technol., 58, 16855–16866, https://doi.org/10.1021/acs.est.4c07649, 2024.
Wang, Q., Zhang, L., Liang, X., Yin, X., Zhang, Y., Zheng, W., Pierce, E. M., and Gu, B.: Rates and dynamics of mercury isotope exchange between dissolved elemental Hg(0) and Hg(II) bound to organic and inorganic ligands, Environ. Sci. Technol., 54, 15534–15545, https://doi.org/10.1021/acs.est.0c06229, 2020.
Wang, S., McNamara, S. M., Moore, C. W., Obrist, D., Steffen, A., Shepson, P. B., Staebler, R. M., Raso, A. R. W., and Pratt, K. A.: Direct detection of atmospheric atomic bromine leading to mercury and ozone depletion, P. Natl. Acad. Sci. USA, 116, 14479–14484, https://doi.org/10.1073/pnas.1900613116, 2019a.
Wang, X., Yuan, W., Lin, C.-J., Zhang, L., Zhang, H., and Feng, X.: Climate and vegetation as primary drivers for global mercury storage in surface soil, Environ. Sci. Technol., 53, 10665–10675, https://doi.org/10.1021/acs.est.9b02386, 2019b.
Wang, X., Luo, J., Yuan, W., Lin, C.-J., Wang, F., Liu, C., Wang, G., and Feng, X.: Global warming accelerates uptake of atmospheric mercury in regions experiencing glacier retreat, P. Natl. Acad. Sci. USA, 117, 2049–2055, https://doi.org/10.1073/pnas.1906930117, 2020a.
Wang, X., Yuan, W., Lin, C.-J., Luo, J., Wang, F., Feng, X., Fu, X., and Liu, C.: Underestimated sink of atmospheric mercury in a deglaciated forest chronosequence, Environ. Sci. Technol., 54, 8083–8093, https://doi.org/10.1021/acs.est.0c01667, 2020b.
Wang, X. F. and Andrews, L.: Infrared spectrum of Hg(OH)2 in solid neon and argon, Inorg. Chem., 44, 108–113, 2005a.
Wang, X. F. and Andrews, L.: Mercury dihydride forms a covalent molecular solid, Phys. Chem. Chem. Phys., 7, 750–759, 2005b.
Wang, X. F., Andrews, L., Riedel, S., and Kaupp, M.: Mercury is a transition metal: The first experimental evidence for HgF4, Angew. Chem. Int. Edit., 46, 8371–8375, https://doi.org/10.1002/anie.200703710, 2007.
Wang, Y., Liu, G., Fang, Y., Liu, P., Liu, Y., Guo, Y., Shi, J., Hu, L., Cai, Y., Yin, Y., and Jiang, G.: Dark oxidation of mercury droplet: Mercurous Hg(I) species controls transformation kinetics, Water Res., 244, 120472, https://doi.org/10.1016/j.watres.2023.120472, 2023.
Wang, Y. W., Bartov, G., Wang, T., Reinfelder, J. R., Johnson, T. M., and Yee, N.: Rapid attainment of isotopic equilibrium after mercury reduction by ferrous iron minerals and isotopic exchange between Hg(II) and Hg(0), ACS Earth Space Chem., 5, 1384–1394, https://doi.org/10.1021/acsearthspacechem.1c00026, 2021.
Wang, Z. and Pehkonen, S. O.: Oxidation of elemental mercury by aqueous bromine: atmospheric implications, Atmos. Environ., 38, 3675–3688, 2004.
Wang, Z., Sun, T., Driscoll, C. T., Zhang, H., and Zhang, X.: Dimethylmercury in floodwaters of mercury contaminated rice paddies, Environ. Sci. Technol., 53, 9453–9461, https://doi.org/10.1021/acs.est.8b07180, 2019c.
Warneck, P. (Ed.): Sulfur compounds in the atmosphere, in: Chemistry of the Natural Atmosphere, International Geophysics, Vol. 71, Academic Press, 484–542, https://doi.org/10.1016/S0074-6142(08)60637-3, 1988.
Warneck, P.: In-cloud chemistry opens pathway to the formation of oxalic acid in the marine atmosphere, Atmos. Environ., 37, 2423–2427, https://doi.org/10.1016/s1352-2310(03)00136-5, 2003.
Warneck, P. and Williams, J.: The atmospheric chemist's companion: numerical data for use in the atmospheric sciences, Springer Netherlands, New York, https://doi.org/10.1007/978-94-007-2275-0, 2012.
Washburn, S. J., Blum, J. D., Johnson, M. W., Tomes, J. M., and Carnell, P. J.: Isotopic characterization of mercury in natural gas via analysis of mercury removal unit catalysts, ACS Earth Space Chem., 2, 462–470, https://doi.org/10.1021/acsearthspacechem.7b00118, 2018.
Washburn, S. J., Blum, J. D., Motta, L. C., Bergquist, B. A., and Weiss-Penzias, P.: Isotopic composition of Hg in fogwaters of coastal California, Environ. Sci. Technol. Lett., 8, 3–8, 2021.
Weigelt, A., Slemr, F., Ebinghaus, R., Pirrone, N., Bieser, J., Bödewadt, J., Esposito, G., and van Velthoven, P. F. J.: Mercury emissions of a coal-fired power plant in Germany, Atmos. Chem. Phys., 16, 13653–13668, https://doi.org/10.5194/acp-16-13653-2016, 2016a.
Weigelt, A., Ebinghaus, R., Pirrone, N., Bieser, J., Bödewadt, J., Esposito, G., Slemr, F., van Velthoven, P. F. J., Zahn, A., and Ziereis, H.: Tropospheric mercury vertical profiles between 500 and 10000 m in central Europe, Atmos. Chem. Phys., 16, 4135–4146, https://doi.org/10.5194/acp-16-4135-2016, 2016b.
Weiss-Penzias, P., Sorooshian, A., Coale, K., Heim, W., Crosbie, E., Dadashazar, H., MacDonald, A. B., Wang, Z., and Jonsson, H.: Aircraft measurements of total mercury and monomethyl mercury in summertime marine stratus cloudwater from coastal California, USA, Environ. Sci. Technol., 52, 2527–2537, https://doi.org/10.1021/acs.est.7b05395, 2018.
Weiss-Penzias, P. S., Ortiz, C., Acosta, R. P., Heim, W., Ryan, J. P., Fernandez, D., Collett, J. L., and Flegal, A. R.: Total and monomethyl mercury in fog water from the central California coast, Geophys. Res. Lett., 39, L03804, https://doi.org/10.1029/2011gl050324, 2012.
West, J., Gindorf, S., and Jonsson, S.: Photochemical degradation of dimethylmercury in natural waters, Environ. Sci. Technol., 56, 5920–5928, https://doi.org/10.1021/acs.est.1c08443, 2022.
Wiederhold, J. G.: Metal stable isotope signatures as tracers in environmental geochemistry, Enviro. Sci. Technol., 49, 2606–2624, 2015.
Wiederhold, J. G., Cramer, C. J., Daniel, K., Infante, I., Bourdon, B., and Kretzschmar, R.: Equilibrium mercury isotope fractionation between dissolved Hg(II) species and thiol-bound Hg, Environ. Sci. Technol., 44, 4191–4197, https://doi.org/10.1021/es100205t, 2010.
Wigfield, D. C. and Perkins, S. L.: Oxidation of elemental mercury by hydroperoxides in aqueous solution, Can. J. Chem., 63, 275–277, https://doi.org/10.1139/v85-045, 1985a.
Wigfield, D. C. and Perkins, S. L.: Oxidation state analysis of mercury: evidence of the formation of mercurous ion in the oxidation of mercury by peracetic acid, J. Appl. Toxicol., 5, 339–341, https://doi.org/10.1002/jat.2550050514, 1985b.
Wigfield, D. C. and Tse, S.: The mechanism of biooxidation of mercury, J. Appl. Toxicol., 6, 73–74, https://doi.org/10.1002/jat.2550060115, 1986.
Wilcox, J.: A Kinetic investigation of high-temperature mercury oxidation by chlorine, J. Phys. Chem. A, 113, 6633–6639, https://doi.org/10.1021/jp901050d, 2009.
Williston, S. H.: Mercury in atmosphere, J. Geophys. Res., 73, 7051–7055, https://doi.org/10.1029/JB073i022p07051, 1968.
Woerndle, G. E., Tsui, M. T. K., Sebestyen, S. D., Blum, J. D., Nie, X. P., and Kolka, R. K.: New insights on ecosystem mercury cycling revealed by stable isotopes of mercury in water flowing from a headwater peatland catchment, Environ. Sci. Technol., 52, 1854–1861, https://doi.org/10.1021/acs.est.7b04449, 2018.
Wolfe, N. L., Zepp, R. G., Gordon, J. A., and Baughman, G. L.: Chemistry of methylmercurials in aqueous solution, Chemosphere, 2, 147–152, 1973.
Won, A. Y., Kim, M. K., and Zoh, K. D.: Characteristics of total and methyl mercury in precipitation in Seoul, Korea, Atmos. Pollut. Res., 10, 493–500, https://doi.org/10.1016/j.apr.2018.10.001, 2019.
Wright, L. P., Zhang, L., and Marsik, F. J.: Overview of mercury dry deposition, litterfall, and throughfall studies, Atmos. Chem. Phys., 16, 13399–13416, https://doi.org/10.5194/acp-16-13399-2016, 2016.
Wu, L., Mao, H., Ye, Z., Dibble, T. S., Saiz-Lopez, A., and Zhang, Y.: Improving simulation of gas-particle partitioning of atmospheric mercury using CMAQ-newHg-Br v2, J. Adv. Model. Earth Syst., 16, e2023MS003823, https://doi.org/10.1029/2023MS003823, 2024a.
Wu, P., Song, Z., Zhang, P., Huang, S., Yuan, T., and Zhang, Y.: Atmospheric monomethylmercury: Inferred sources constrained by observations and implications for human exposure, Environ. Int., 193, 109–127, https://doi.org/10.1016/j.envint.2024.109127, 2024b.
Wu, Q., Wang, S., Li, G., Liang, S., Lin, C.-J., Wang, Y., Cai, S., Liu, K., and Hao, J.: Temporal trend and spatial distribution of speciated atmospheric mercury emissions in China during 1978–2014, Environ. Sci. Technol., 50, 13428–13435, https://doi.org/10.1021/acs.est.6b04308, 2016.
Wu, R., Wang, C., and Dibble, T. S.: First experimental kinetic study of the atmospherically important reaction of BrHg + NO2, Chem. Phys. Lett., 759, 137928, https://doi.org/10.1016/j.cplett.2020.137928, 2020.
Wu, R. R., Castro, P. J., Gaito, C., Beiter, K., Dibble, T. S., and Wang, C. J.: Combined experimental and computational kinetics studies for the atmospherically important BrHg radical reacting with NO and O2, J. Phys. Chem. A, 126, 3914–3925, https://doi.org/10.1021/acs.jpca.2c02531, 2022.
Wu, X., Fu, X. W., Zhang, H., Tang, K. H., Wang, X., Zhang, H., Deng, Q. W., Zhang, L. M., Liu, K. Y., Wu, Q. R., Wang, S. X., and Feng, X. B.: Changes in atmospheric gaseous elemental mercury concentrations and isotopic compositions at Mt. Changbai during 2015–2021 and Mt. Ailao during 2017–2021 in China, J. Geophys. Res.-Atmos., 128, e2022JD037749, https://doi.org/10.1029/2022jd037749, 2023a.
Wu, Y. R., Mao, Y. X., Liu, G. L., Li, Y. B., Guo, Y. Y., Liu, Y. W., Yin, Y. G., Cai, Y., and Jiang, G. B.: Analytical methods, occurrence, fate, and toxicity of ethylmercury in the environment: Review and outlook, Rev. Environ. Contam. T., 261, 9, https://doi.org/10.1007/s44169-023-00037-x, 2023b.
Wängberg, I., Munthe, J., Pirrone, N., Iverfeldt, Å., Bahlman, E., Costa, P., Ebinghaus, R., Feng, X., Ferrara, R., Gårdfeldt, K., Kock, H., Lanzillotta, E., Mamane, Y., Mas, F., Melamed, E., Osnat, Y., Prestbo, E., Sommar, J., Schmolke, S., Spain, G., Sprovieri, F., and Tuncel, G.: Atmospheric mercury distribution in Northern Europe and in the Mediterranean region, Atmos. Environ., 35, 3019–3025, 2001.
Xiao, Z., Munthe, J., Strömberg, D., and Lindqvist, O.: Photochemical behaviour of inorganic mercury compounds in aqueous solution, in: Mercury pollution: Integration and synthesis, edited by: Watras, C. J., and Huckabee, CRC Press, 581–594, 1994.
Xiao, Z., Sommar, J., Wei, S., and Lindqvist, O.: Sampling and determination of gas phase divalent mercury in the air using a KCl coated denuder, Fresen. J. Anal. Chem., 358, 386–391, 1997.
Xie, Z.-Q., Sander, R., Pöschl, U., and Slemr, F.: Simulation of atmospheric mercury depletion events (AMDEs) during polar springtime using the MECCA box model, Atmos. Chem. Phys., 8, 7165–7180, https://doi.org/10.5194/acp-8-7165-2008, 2008.
Yamakawa, A., Moriya, K., and Yoshinaga, J.: Determination of isotopic composition of atmospheric mercury in urban-industrial and coastal regions of Chiba, Japan, using cold vapor multicollector inductively coupled plasma-mass spectrometry, Chem. Geol., 448, 84–92, https://doi.org/10.1016/j.chemgeo.2016.11.010, 2017.
Yamakawa, A., Luke, W., Kelley, P., Ren, X., and Iaukea-Lum, M.: Unraveling atmospheric mercury dynamics at Mauna Loa through the isotopic analysis of total gaseous mercury, Ecotox. Environ. Safe., 284, 116993, https://doi.org/10.1016/j.ecoenv.2024.116993, 2024.
Yang, J., Kim, J., Soerensen, A. L., Lee, W., and Han, S.: The role of fluorescent dissolved organic matter on mercury photoreduction rates: A case study of three temperate lakes, Geochim. Cosmochim. Ac., 277, 192–205, 2020a.
Yang, M., Rasche, B., and Compton, R. G.: Acoustic cavitation generates molecular mercury(II) hydroxide, Hg(OH)2, from biphasic water/mercury mixtures, Chem. Sci., 11, 556–560, https://doi.org/10.1039/C9SC04743C, 2020b.
Yang, S. and Liu, Y.: Nuclear volume effects in equilibrium stable isotope fractionations of mercury, thallium and lead, Sci. Rep., 5, 12626, https://doi.org/10.1038/srep12626, 2015.
Yang, X., Jiskra, M., and Sonke, J. E.: Experimental rainwater divalent mercury speciation and photoreduction rates in the presence of halides and organic carbon, Sci. Total Environ., 697, 133821, https://doi.org/10.1016/j.scitotenv.2019.133821, 2019.
Yin, Y., Chen, B., Mao, Y., Wang, T., Liu, J., Cai, Y., and Jiang, G.: Possible alkylation of inorganic Hg(II) by photochemical processes in the environment, Chemosphere, 88, 8–16, 2012.
Young, E. D., Galy, A., and Nagahara, H.: Kinetic and equilibrium mass-dependent isotope fractionation laws in nature and their geochemical and cosmochemical significance, Geochim. Cosmoch. Ac., 66, 1095–1104, https://doi.org/10.1016/S0016-7037(01)00832-8, 2002.
Yu, B., Yang, L., Liu, H., Yang, R., Fu, J., Wang, P., Li, Y., Xiao, C., Liang, Y., Hu, L., Zhang, Q., Yin, Y., Shi, J., and Jiang, G.: Katabatic wind and sea-ice dynamics drive isotopic variations of total gaseous mercury on the Antarctic coast, Environ. Sci. Technol., 55, 6449–6458, https://doi.org/10.1021/acs.est.0c07474, 2021.
Yuan, S., Chen, J., Hintelmann, H., Cai, H., Yuan, W., He, S., Zhang, K., Zhang, Y., and Liu, Y.: Event-based atmospheric precipitation uncovers significant even and odd Hg isotope anomalies associated with the circumpolar vortex, Environ. Sci. Technol., 56, 12713–12722, https://doi.org/10.1021/acs.est.2c02613, 2022.
Yuan, S., Zhang, Y., Chen, J., Kang, S., Zhang, J., Feng, X., Cai, H., Wang, Z., Wang, Z., and Huang, Q.: Large variation of mercury isotope composition during a single precipitation event at Lhasa City, Tibetan Plateau, China, Proced. Earth Plan. Sci., 13, 282–286, https://doi.org/10.1016/j.proeps.2015.07.066, 2015.
Yuan, T. F., Zhang, P., Song, Z. C., Huang, S. J., Wang, X., and Zhang, Y. X.: Buffering effect of global vegetation on the air-land exchange of mercury: Insights from a novel terrestrial mercury model based on CESM2-CLM5, Environ. Int., 174, 107904, https://doi.org/10.1016/j.envint.2023.107904, 2023a.
Yuan, W., Wang, X., Lin, C.-J., Sommar, J., Lu, Z., and Feng, X.: Process factors driving dynamic exchange of elemental mercury vapor over soil in broadleaf forest ecosystems, Atmos. Environ., 219, 117047, https://doi.org/10.1016/j.atmosenv.2019.117047, 2019a.
Yuan, W., Wang, X., Lin, C.-J., Sommar, J. O., Wang, B., Lu, Z., and Feng, X.: Quantification of atmospheric mercury deposition to and legacy re-emission from a subtropical forest floor by mercury isotopes, Environ. Sci. Technol., 55, 12352–12361, https://doi.org/10.1021/acs.est.1c02744, 2021.
Yuan, W., Wang, X., Lin, C.-J., Song, Q., Zhang, H., Wu, F., Liu, N., Lu, H., and Feng, X.: Deposition and re-emission of atmospheric elemental mercury over the tropical forest floor, Environ. Sci. Technol., 57, 10686–10695, https://doi.org/10.1021/acs.est.3c01222, 2023b.
Yuan, W., Wang, X., Lin, C.-J., Wu, C., Zhang, L., Wang, B., Sommar, J., Lu, Z., and Feng, X.: Stable mercury isotope transition during postdepositional decomposition of biomass in a forest ecosystem over five centuries, Environ. Sci. Technol., 54, 8739–8749, https://doi.org/10.1021/acs.est.0c00950, 2020.
Yuan, W., Sommar, J., Lin, C.-J., Wang, X., Li, K., Liu, Y., Zhang, H., Lu, Z., Wu, C., and Feng, X.: Stable isotope evidence shows re-emission of elemental mercury vapor occurring after reductive loss from foliage, Environ. Sci. Technol., 53, 651–660, https://doi.org/10.1021/acs.est.8b04865, 2019b.
Zacharewski, T. R. and Cherniak, E. A.: FTIR investigation of the heterogeneous reaction of HgO (s) with SO2(g) at ambient temperature, Atmos. Environ., 21, 2327–2332, 1987.
Zerkle, A. L., Yin, R., Chen, C., Li, X., Izon, G. J., and Grasby, S. E.: Anomalous fractionation of mercury isotopes in the Late Archean atmosphere, Nat. Commun., 11, 1709, 2020.
Zhang, H.: Photochemical redox reactions of mercury, in: Structure and Bonding 120, edited by: Atwood, D. A., Springer, Berlin Heidelberg, 37–79, https://doi.org/10.1007/430_015, 2006.
Zhang, H., Fu, X. W., Wang, X., and Feng, X. B.: Measurements and distribution of atmospheric particulate-bound mercury: A review, B. Environ. Contam. Tox., 103, 48–54, https://doi.org/10.1007/s00128-019-02663-5, 2019c.
Zhang, H., Fu, X., Wu, X., Deng, Q., Tang, K., Zhang, L., Sommar, J., Sun, G., and Feng, X.: Using mercury stable isotopes to quantify bidirectional water-atmosphere Hg(0) exchange fluxes and explore controlling factors, Environ. Sci. Technol., 57, 10675–10685, https://doi.org/10.1021/acs.est.3c01273, 2023a.
Zhang, H., Tan, Q., Zhang, L., Fu, X., and Feng, X.: A laboratory study on the isotopic composition of Hg(0) emitted from Hg-enriched soils in Wanshan Hg mining area, J. Geophys. Res.-Atmos., 125, e2020JD032572, https://doi.org/10.1029/2020JD032572, 2020.
Zhang, H., Wang, Z. W., and Zhang, X. S.: Methylmercury concentrations and potential sources in atmospheric fine particles in Beijing, China, Sci. Total Environ., 681, 183–190, https://doi.org/10.1016/j.scitotenv.2019.05.128, 2019a.
Zhang, H., Wu, X., Deng, Q., Zhang, L., Fu, X., and Feng, X.: Extraction of ultratrace dissolved gaseous mercury and reactive mercury in natural freshwater for stable isotope analysis, J. Anal. Atom. Spectrom., 36, 1921–1932, https://doi.org/10.1039/D1JA00212K, 2021a.
Zhang, H., Yin, R. S., Feng, X. B., Sommar, J., Anderson, C. W. N., Sapkota, A., Fu, X. W., and Larssen, T.: Atmospheric mercury inputs in montane soils increase with elevation: Evidence from mercury isotope signatures, Sci. Rep., 3, 3322, https://doi.org/10.1038/srep03322, 2013.
Zhang, K., Pu, Q., Liu, J., Hao, Z., Zhang, L., Zhang, L., Fu, X., Meng, B., and Feng, X.: Using mercury stable isotopes to quantify directional soil–atmosphere Hg(0) exchanges in rice paddy ecosystems: Implications for Hg(0) emissions to the atmosphere from land surfaces, Environ. Sci. Technol., 58, 11053–11062, https://doi.org/10.1021/acs.est.4c02143, 2024.
Zhang, K., Zheng, W., Sun, R., He, S., Shuai, W., Fan, X., Yuan, S., Fu, P., Deng, J., Li, X., Wang, S., and Chen, J.: Stable isotopes reveal photoreduction of particle-bound mercury driven by water-soluble organic carbon during severe haze, Environ. Sci. Technol., 56, 10619–10628, https://doi.org/10.1021/acs.est.2c01933, 2022.
Zhang, L., Wright, L. P., and Blanchard, P.: A review of current knowledge concerning dry deposition of atmospheric mercury, Atmos. Environ., 43, 5853–5864, 2009.
Zhang, L., Wang, S., Wu, Q., Wang, F., Lin, C.-J., Zhang, L., Hui, M., Yang, M., Su, H., and Hao, J.: Mercury transformation and speciation in flue gases from anthropogenic emission sources: a critical review, Atmos. Chem. Phys., 16, 2417–2433, https://doi.org/10.5194/acp-16-2417-2016, 2016.
Zhang, L., Lyman, S., Mao, H., Lin, C.-J., Gay, D. A., Wang, S., Sexauer Gustin, M., Feng, X., and Wania, F.: A synthesis of research needs for improving the understanding of atmospheric mercury cycling, Atmos. Chem. Phys., 17, 9133–9144, https://doi.org/10.5194/acp-17-9133-2017, 2017.
Zhang, T. and Hsu-Kim, H.: Photolytic degradation of methylmercury enhanced by binding to natural organic ligands, Nat. Geosci., 3, 473–476, https://doi.org/10.1038/ngeo892, 2010.
Zhang, Y., Horowitz, H., Wang, J., Xie, Z., Kuss, J., and Soerensen, A. L.: A coupled global atmosphere-ocean model for air-sea exchange of mercury: Insights into wet deposition and atmospheric redox chemistry, Environ. Sci. Technol., 53, 5052-5061, https://doi.org/10.1021/acs.est.8b06205, 2019b.
Zhang, Y., Song, Z., Huang, S., Zhang, P., Peng, Y., Wu, P., Gu, J., Dutkiewicz, S., Zhang, H., Wu, S., Wang, F., Chen, L., Wang, S., and Li, P.: Global health effects of future atmospheric mercury emissions, Nat. Commun., 12, 3035, https://doi.org/10.1038/s41467-021-23391-7, 2021b.
Zhang, Y., Xu, Z., Han, G., Chu, Z., Zhou, Q., Chen, Q., Wu, G., Shi, G., Wang, X., and Chen, L.: Improved mechanistic modeling on reproducing particle-bound mercury in the marine atmosphere, Environ. Sci. Technol., 59, 2611–2622, https://doi.org/10.1021/acs.est.4c09481, 2025.
Zhang, Y., Zhang, L., Cao, S., Liu, X., Jin, J., and Zhao, Y.: Improved anthropogenic mercury emission inventories for China from 1980 to 2020: Toward more accurate effectiveness evaluation for the Minamata convention, Environ. Sci. Technol., 57, 8660–8670, https://doi.org/10.1021/acs.est.3c01065, 2023a.
Zhang, Y. X., Zhang, P., Song, Z. C., Huang, S. J., Yuan, T. F., Wu, P. P., Shah, V. R., Liu, M. D., Chen, L., Wang, X. J., Zhou, J., and Agnan, Y.: An updated global mercury budget from a coupled atmosphere-land-ocean model: 40 % more re-emissions buffer the effect of primary emission reductions, One Earth, 6, 316–325, https://doi.org/10.1016/j.oneear.2023.02.004, 2023b.
Zhao, H., Meng, B., Sun, G., Lin, C.-J., Feng, X., and Sommar, J.: Chemistry and isotope fractionation of divalent mercury during aqueous reduction mediated by selected oxygenated organic ligands, Environ. Sci. Technol., 55, 13376–13386, 2021.
Zhao, Y., Hallar, A. G., and Mazzoleni, L. R.: Atmospheric organic matter in clouds: exact masses and molecular formula identification using ultrahigh-resolution FT-ICR mass spectrometry, Atmos. Chem. Phys., 13, 12343–12362, https://doi.org/10.5194/acp-13-12343-2013, 2013.
Zhen, J., Li, T., Cai, H., Nie, X., He, S., Meng, M., Wang, Y., and Chen, J.: Photoreduction and origin of dissolved and particulate mercury in cloud water: Insights from stable mercury isotopes, J. Hazard. Mater., 474, 134654, https://doi.org/10.1016/j.jhazmat.2024.134654, 2024.
Zhen, J., Li, T., Xu, X., Du, P., Song, Y., Nie, X., Liu, X., Liu, H., Bi, Y., Wang, X., Xue, L., and Wang, Y.: Changed mercury speciation in clouds driven by changing cloud water chemistry and impacts on photoreduction: Field evidence at Mt. Tai in eastern China, Water Res., 244, 120402, https://doi.org/10.1016/j.watres.2023.120402, 2023.
Zheng, W. and Hintelmann, H.: Mercury isotope fractionation during photoreduction in natural water is controlled by its Hg/DOC ratio, Geochim. Cosmoch. Ac., 73, 6704–6715, 2009.
Zheng, W. and Hintelmann, H.: Nuclear field shift effect in isotope fractionation of mercury during abiotic reduction in the absence of light, J. Phys. Chem. A, 114, 4238–4245, https://doi.org/10.1021/jp910353y, 2010a.
Zheng, W. and Hintelmann, H.: Isotope fractionation of mercury during its photochemical reduction by low-molecular-weight organic compounds, J. Phys. Chem. A, 114, 4246–4253, 2010b.
Zheng, W., Foucher, D., and Hintelmann, H.: Mercury isotope fractionation during volatilization of Hg(0) from solution into the gas phase, J. Anal. Atom. Spectrom., 22, 1097–1104, https://doi.org/10.1039/B705677j, 2007.
Zheng, W., Liang, L. Y., and Gu, B. H.: Mercury reduction and oxidation by reduced natural organic matter in anoxic environments, Environ. Sci. Technol., 46, 292–299, 2012.
Zheng, W., Obrist, D., Weis, D., and Bergquist, B. A.: Mercury isotope compositions across North American forests, Global Biogeochem. Cy., 30, 2015GB005323, https://doi.org/10.1002/2015GB005323, 2016.
Zheng, W., Lin, H., Mann, B. F., Liang, L., and Gu, B.: Oxidation of dissolved elemental mercury by thiol compounds under anoxic conditions, Environ. Sci. Technol., 47, 12827–12834, https://doi.org/10.1021/es402697u, 2013.
Zheng, W., Demers, J. D., Lu, X., Bergquist, B. A., Anbar, A. D., Blum, J. D., and Gu, B. H.: Mercury stable isotope fractionation during abiotic dark oxidation in the presence of thiols and natural organic matter, Environ. Sci. Technol., 53, 1853–1862, https://doi.org/10.1021/acs.est.8b05047, 2019.
Zheng, W., Chandan, P., Steffen, A., Stupple, G., de Vera, J., Mitchell, C., Wania, F., and Bergquist, B.: Mercury stable isotopes reveal the sources and transformations of atmospheric Hg in the high Arctic, Appl. Geochem., 131, 105002, https://doi.org/10.1016/j.apgeochem.2021.105002, 2021.
Zhou, J. and Obrist, D.: Global mercury assimilation by vegetation, Environ. Sci. Technol., 55, 14245–14257, https://doi.org/10.1021/acs.est.1c03530, 2021.
Zhou, J., Obrist, D., Dastoor, A., Jiskra, M., and Ryjkov, A.: Vegetation uptake of mercury and impacts on global cycling, Nat. Rev. Earth Environ., 2, 269–284, https://doi.org/10.1038/s43017-021-00146-y, 2021.
Zhu, W., Lin, C.-J., Wang, X., Sommar, J., Fu, X., and Feng, X.: Global observations and modeling of atmosphere–surface exchange of elemental mercury: a critical review, Atmos. Chem. Phys., 16, 4451–4480, https://doi.org/10.5194/acp-16-4451-2016, 2016.
Zhu, W., Fu, X., Zhang, H., Liu, C., Skyllberg, U., Sommar, J., Yu, B., and Feng, X.: Mercury isotope fractionation during the exchange of Hg(0) between the atmosphere and land surfaces: Implications for Hg(0) exchange processes and controls, Environ. Sci. Technol., 56, 1445–1457, https://doi.org/10.1021/acs.est.1c05602, 2022.
Zhu, W., Li, Z., Li, P., Sommar, J., Fu, X., Feng, X., Yu, B., Zhang, W., Reis, A. T., and Pereira, E.: Legacy mercury re-emission and subsurface migration at contaminated sites constrained by Hg isotopes and chemical speciation, Environ. Sci. Technol., 58, 5336–5346, https://doi.org/10.1021/acs.est.3c07276, 2024.
There is a widespread practice in the Hg research community to report Hg0 air concentrations in ng m−3 referenced to one atmosphere (101.325 kPa) and 0 °C (STP). By that, the unit represents a mixing ratio not an absolute (mass) concentration.
- Abstract
- Introduction
- Physical chemistry of elemental mercury
- Atmospheric environment
- Kinetics, thermodynamics, and general chemistry
- Gas-phase atmospheric Hg chemistry
- Redox transformations in the aqueous phase
- Multiphase transformations
- Mercury isotope systematics and fractionation
- Future perspectives
- Appendix A
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Abstract
- Introduction
- Physical chemistry of elemental mercury
- Atmospheric environment
- Kinetics, thermodynamics, and general chemistry
- Gas-phase atmospheric Hg chemistry
- Redox transformations in the aqueous phase
- Multiphase transformations
- Mercury isotope systematics and fractionation
- Future perspectives
- Appendix A
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References