the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 30 Jan 2024
| 30 Jan 2024
Chamber studies of OH + dimethyl sulfoxide and dimethyl disulfide: insights into the dimethyl sulfide oxidation mechanism
The oxidation of dimethyl sulfide (DMS) in the marine atmosphere represents an important natural source of non-sea-salt sulfate aerosol, but the chemical mechanisms underlying this process remain uncertain. While recent studies have focused on the role of the peroxy radical isomerization channel in DMS oxidation, this work revisits the impact of the other channels (OH addition and OH abstraction followed by bimolecular RO2 reaction) on aerosol formation from DMS. Due to the presence of common intermediate species, the oxidation of dimethyl sulfoxide (DMSO) and dimethyl disulfide (DMDS) can shed light on these two DMS reaction channels; they are also both atmospherically relevant species in their own right. This work examines the OH oxidation of DMSO and DMDS, using chamber experiments monitored by chemical ionization mass spectrometry and aerosol mass spectrometry to study the full range of sulfur-containing products across a range of NO concentrations. The oxidation of both compounds is found to lead to rapid aerosol formation (which does not involve the intermediate formation of SO2), with a substantial fraction (14 %–47 % S yield for DMSO and 5 %–21 % for DMDS) of reacted sulfur ending up in the particle phase and the highest yields observed under elevated NO conditions. Aerosol is observed to consist mainly of sulfate, methanesulfonic acid, and methanesulfinic acid. In the gas phase, the NOx dependence of several products, including SO2 and S2-containing organosulfur species, suggest reaction pathways not included in current mechanisms. Based on the commonalities with the DMS oxidation mechanism, DMSO and DMDS results are used to reconstruct DMS aerosol yields; these reconstructions roughly match DMS aerosol yield measurements from the literature but differ in composition, underscoring remaining uncertainties in sulfur chemistry. This work indicates that both the abstraction and addition channels contribute to rapid aerosol formation from DMS and highlights the need for more study into the fate of small sulfur radical intermediates (e.g., CH3S, CH3SO2, and CH3SO3) that are thought to play central roles in the DMS oxidation mechanism.
Please read the corrigendum first before continuing.
-
Notice on corrigendum
The requested paper has a corresponding corrigendum published. Please read the corrigendum first before downloading the article.
-
Article
(2697 KB)
- Corrigendum
-
Supplement
(5894 KB)
-
The requested paper has a corresponding corrigendum published. Please read the corrigendum first before downloading the article.
- Article
(2697 KB) - Full-text XML
- Corrigendum
-
Supplement
(5894 KB) - BibTeX
- EndNote





Dimethyl sulfide (DMS; CH3SCH3) represents an important biogenic contribution to atmospheric sulfur. Through its oxidation in the troposphere, it acts as the dominant source of non-sea-salt sulfate aerosol over the oceans and, as such, may affect the climate system through direct (aerosol–radiation) and indirect (aerosol–cloud) effects. Thus, understanding DMS-derived aerosol formation and properties is important for understanding the natural background climate state (Carslaw et al., 2013; Fung et al., 2022), as well as forecasting climate changes in the future. The detailed chemistry of DMS oxidation determines the yield of aerosol and the ultimate fate of the sulfur, but despite decades of research (Yin et al., 1990a; Barnes et al., 2006; Hoffmann et al., 2016) and notable recent breakthroughs (Wu et al., 2015; Berndt et al., 2019; Veres et al., 2020), the underlying chemical mechanism is not fully understood.
The impacts of DMS-derived aerosol are affected by several chemical factors, including the total aerosol yield, the timescale of aerosol formation, and the aerosol composition. All of these factors may affect the net aerosol radiative impact (Fung et al., 2022), and all are directly controlled by secondary chemistry, much of which remains uncertain. Sulfate from gas-phase DMS oxidation is thought to form through several channels. One involves the formation and subsequent oxidation of SO2, which is relatively slow: SO2 + OH lifetime ≈ 12 d (assuming [OH] = 106 molec. cm−3, 1 atm, and 298 K; Burkholder et al., 2020); for context, SO2 lifetime to all atmospheric losses ≈ 1.4 d (Fung et al., 2022). Alternatively, some channels may lead to the direct formation of SO3, which rapidly converts to sulfuric acid in the presence of water vapor, providing a potentially faster path to sulfate aerosol. This direct-formation route has been known for decades (Bandy et al., 1992; Lucas and Prinn, 2002), is regularly included in chemical mechanisms describing DMS oxidation (Saunders et al., 2003; Barnes et al., 2006; Wollesen de Jonge et al., 2021; Fung et al., 2022), and has been demonstrated in a number of laboratory studies (Shen et al., 2022; Ye et al., 2022; Berndt et al., 2023). We refer to this pathway as “rapid aerosol formation”, defined as aerosol formation that does not involve SO2 as an intermediate species. The variability in timescale for aerosol formation may affect the spatial distribution and amount of secondary sulfate aerosol in the atmosphere and may, as a result, affect radiative impacts (Fung et al., 2022). Sulfate can also be produced in the aqueous phase, and so the balance between gas- and aqueous-phase sulfate-formation pathways may impact total new particle formation (Hodshire et al., 2019). Mechanisms also control aerosol composition, additionally influencing aerosol properties and impact. Aerosol-phase products of DMS consist mostly of sulfate/sulfuric acid and methanesulfonic acid (MSA) (Barnes et al., 2006), and while both can contribute to new particle formation (Hodshire et al., 2019), these species are likely to nucleate at different rates (Chen et al., 2016; Hodshire et al., 2019).
The oxidation of DMS by OH is characterized by three main pathways: OH addition, OH abstraction followed by bimolecular reaction of the RO2 radical, and OH abstraction followed by RO2 isomerization (referred to from here on as addition, abstraction, and isomerization, respectively). These are shown in Fig. 1, which features a simplified oxidation mechanism for DMS. Recent work has focused largely on the isomerization channel (Wu et al., 2015; Berndt et al., 2019; Veres et al., 2020; Ye et al., 2021, 2022; Novak et al., 2021; Jernigan et al., 2022; Assaf et al., 2023), since it represents a major revision of the traditional oxidation mechanism, accounting for 30 %–46 % of the total DMS fate globally (Veres et al., 2020; Novak et al., 2021; Fung et al., 2022). However, the major product of the isomerization channel, hydroperoxymethyl thioformate (HPMTF), is thought not to contribute to rapid aerosol formation and is instead thought to oxidize mainly to SO2 or be lost to clouds (Vermeuel et al., 2020; Novak et al., 2021).
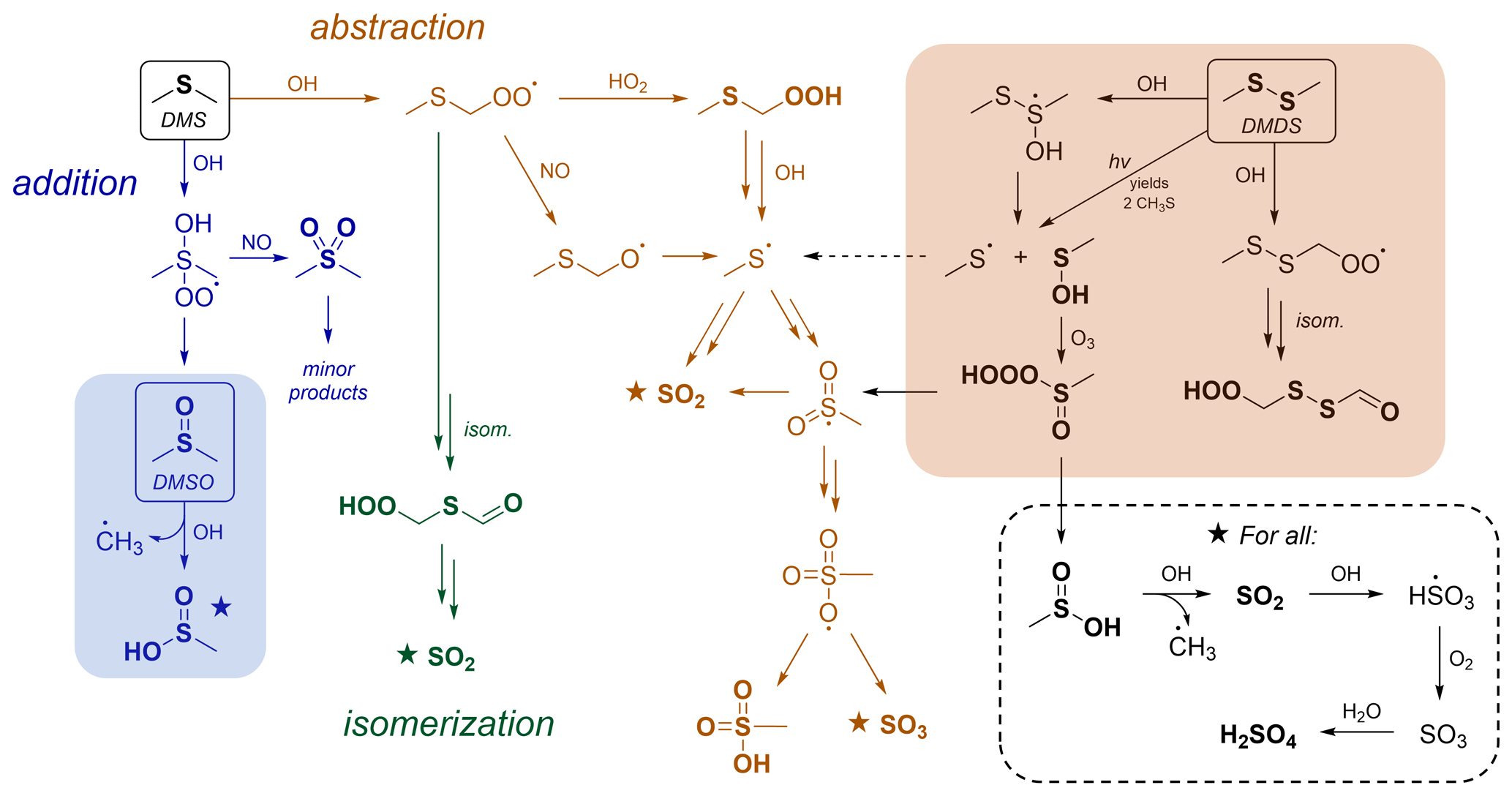
Figure 1Simplified gas-phase oxidation scheme for DMS, DMSO, and DMDS. From the top left: DMS oxidation (Barnes et al., 2006; Wu et al., 2015; Veres et al., 2020), in which three major channels (addition, abstraction, and isomerization shown in blue, orange, and green, respectively) control product distributions. The shaded blue box shows the oxidation of DMSO (Burkholder et al., 2020), which represents an important intermediate in the DMS OH addition channel. The shaded orange box shows the oxidation of DMDS (Berndt et al., 2020), which overlaps with DMS oxidation through the formation of CH3S, a key radical intermediate in the DMS OH abstraction channel. Further reaction of species marked with a star is shown in the dashed box. Compounds in bold represent closed-shell species. Under this scheme, rapid aerosol formation (which does not involve the intermediate formation of SO2) occurs only via the abstraction channel. More complete schemes are given in Barnes et al. (2006), Hoffmann et al. (2016), Ye et al. (2022), and Berndt et al. (2023), as well as in Figs. 4 and S15.
In this study, we focus on the other two channels (abstraction and addition) for which significant uncertainties remain, particularly with respect to their relative contributions to rapid aerosol formation. Under the scheme from the Master Chemical Mechanism (MCM 3.3.1) (Saunders et al., 2003) and the Jet Propulsion Laboratory (JPL) kinetics recommendations (Burkholder et al., 2020), the abstraction channel is almost solely responsible for rapid aerosol formation (Fig. 1). In our recent work, we showed that a modified version of the MCM scheme accurately predicts total aerosol yields as measured in chamber experiments but dramatically underpredicts measured MSA (Ye et al., 2022). Other studies have also noted discrepancies in MSA production between measurements and model predictions (Lucas and Prinn, 2002; von Glasow and Crutzen, 2004; Wollesen de Jonge et al., 2021; Shen et al., 2022). This has led to some suggested changes in the mechanism, most notably a modification to the oxidation of methanesulfinic acid (MSIA), leading to the formation of a radical intermediate (MSIA + OH → CH3SO2 + H2O), which can then react further to generate MSA (Lucas and Prinn, 2002; von Glasow and Crutzen, 2004; Barnes et al., 2006; Wollesen de Jonge et al., 2021; Ye et al., 2022; Shen et al., 2022). This change allows for rapid aerosol formation from the addition channel and improves the model–mechanism agreement substantially in some cases (Wollesen de Jonge et al., 2021; Shen et al., 2022) but not others (Ye et al., 2022). Despite these developments, the relative importance of the abstraction and addition channels for aerosol formation remains poorly constrained.
Here, we investigate the above uncertainties via the oxidation of two related compounds, dimethyl sulfoxide (DMSO; CH3S(O)CH3) and dimethyl disulfide (DMDS; CH3SSCH3). These each have reaction channels in common with the addition and abstraction branches of the DMS mechanism (shaded areas in Fig. 1). DMSO is a key intermediate in the DMS addition channel, and so its oxidation (shown in blue in Fig. 1) provides insight into that channel's product formation and aerosol formation. Similarly, DMDS oxidation (shown in orange in Fig. 1) forms the CH3S radical as a major intermediate. This radical is thought to be a key intermediate in the DMS abstraction channel, leading to the formation of SO2, MSA, and sulfate. These two precursors therefore allow relatively independent access to two of the major branches of the DMS oxidation mechanism, allowing us to investigate product formation, including rapid aerosol production, from each branch. Beyond their direct relevance to DMS, both species are important in their own right. DMDS is emitted directly from marine (Kilgour et al., 2022), biomass burning (Berndt et al., 2020), and agricultural sources (Filipy et al., 2006; Trabue et al., 2008; Rumsey et al., 2014) and is estimated to represent a few percent of biogenic sulfur emissions (Tyndall and Ravishankara, 1991), while DMSO has been observed in measurable concentrations in the marine boundary layer (Berresheim et al., 1993; Bandy et al., 1996; Nowak et al., 2001).
Past experimental study of DMSO oxidation has shown significant variability in product distributions, with relatively little study of aerosol formation. Most prior studies were carried out before the widespread adoption of the aerosol mass spectrometer (AMS) or chemical ionization mass spectrometer (CIMS) and generally apply spectroscopic methods (Barnes et al., 1989; Sørensen et al., 1996; Urbanski et al., 1998; Arsene et al., 2002; Librando et al., 2004) or offline ion chromatography (IC) (Sørensen et al., 1996; Arsene et al., 2002; Librando et al., 2004; Chen and Jang, 2012). While studies generally agree that MSIA is the dominant first-generation oxidation product (Arsene et al., 2002; Barnes et al., 2006), the yields of other products have been inconsistent, with SO2 reported as a major (Sørensen et al., 1996; Kukui et al., 2003; Librando et al., 2004; Chen and Jang, 2012) or a minor (Arsene et al., 2002) product and highly variable yields of MSA (<0.5 %–34 %) (Sørensen et al., 1996; Arsene et al., 2002; Librando et al., 2004; Chen and Jang, 2012) and dimethyl sulfone (DMSO2, 2.9 %–33 %) (Sørensen et al., 1996; Arsene et al., 2002; Librando et al., 2004; Chen and Jang, 2012). The wide variability in the reported product yields may be due to several factors: high starting concentrations (>1 ppm, parts per million) (Barnes et al., 1989; Sørensen et al., 1996; Arsene et al., 2002; Librando et al., 2004) may favor RO2-RO2 reactions; setups that do not allow for aerosol measurements (Barnes et al., 1989; Urbanski et al., 1998; Kukui et al., 2003) may underestimate the yields of more oxidized products; and experiments carried out in nitrogen atmospheres (Kukui et al., 2003) may not promote RO2 chemistry. While offline IC methods (Arsene et al., 2002; Librando et al., 2004; Chen and Jang, 2012) detected aerosol products, to our knowledge only two previous studies (Chen and Jang, 2012; Van Rooy et al., 2021a) have examined aerosol production from DMSO using real-time techniques.
Similar to DMSO, relatively few recent studies have examined the products from DMDS oxidation, and only one study has characterized aerosol-phase products using online measurements. Early work (Yin et al., 1990b; Barnes et al., 1994) reports SO2 as the major product (∼80 %–90 % yield under low NOx and lower at high NOx); MSA and H2SO4 are reported as minor products (0 %–11 %, increasing with increasing NOx) (Yin et al., 1990b). These findings are in agreement with newer studies that find that aerosol concentrations increase with increasing NOx and that the ratio of MSA to H2SO4 depends on the oxidant and relative humidity (Van Rooy et al., 2021a, b). Recently, CIMS studies by Berndt et al. (2020, 2023) found low yields of MSA and MSIA, evidence of gas-phase formation of H2SO4, and evidence of a minor (∼2 %) OH abstraction channel, leading to the formation of HOOCH2SSCHO via isomerization (right side of Fig. 1). While prior studies have established a mechanism that largely explains laboratory observations of gas-phase products (Berndt et al., 2020), the mechanism of aerosol formation has yet to be thoroughly explored.
In this work, we conduct chamber experiments to study the OH oxidation of DMSO and DMDS under different NOx conditions (lower NO and higher NO), measuring the products with an AMS and CIMS. This study seeks not only to assess the relative aerosol yield and composition from DMSO and DMDS oxidation but also to evaluate these results in the context of DMS oxidation to better understand the role of the abstraction and addition channels in rapid aerosol formation.
All experiments were run in a 7.5 m3 environmental chamber (Hunter et al., 2014) operated in “semi-batch” mode, in which clean air was added to replace air sampled by the instruments (chamber dilution lifetime ≈ 8.9 h). Ultraviolet lights centered at ∼340 nm illuminated the chamber ( min−1); only 50 % of lights were used for the OH oxidation of DMDS to slow down oxidation chemistry. All experiments were run at 20 ∘C and <5 % relative humidity, providing conditions that should prevent multiphase chemistry. This allows this work to focus on gas-phase oxidation processes and facilitates comparison with prior studies, most of which were also carried out under dry, room-temperature conditions.
For each experiment, dry sodium nitrate seed particles were atomized into the chamber using an aerosol generator (TSI model 3076) and a diffusion dryer (Brechtel), providing condensation nuclei that can be easily distinguished from secondary sulfate. For DMDS experiments, the seed solution was washed with dichloromethane to remove any organic compounds from the solution. To additionally probe the influence of dichloromethane for DMSO oxidation, 600 ppb (parts per billion) dichloromethane was added to a single experiment (expt 1) at t=1.92 h and was not observed to affect product formation. For lower-NO experiments (defined as experiments with no added source of NOx; estimated background NO ≈ 10 ppt (parts per trillion) in the presence of H2O2 and UV light (Ye et al., 2022)), the OH precursor hydrogen peroxide (H2O2) was added via a direct injection of a known volume of 30 % H2O2 solution into the main chamber dilution airflow. Lower-NO experiments were run first in each series of experiments to reduce the influence of possible residual NOx. For higher-NO experiments (defined as experiments with an added source of NOx; total [NOx] > 20 ppb, with [NO] varying over the course of the experiment; see the Supplement), the OH precursor nitrous acid (HONO) was generated by mixing 10 mL 0.06 M sodium nitrite with 10 mL 0.05 M sulfuric acid and introduced to the chamber by flowing a stream of clean air through the headspace for 20–50 s. Additional NO is introduced to the chamber as a byproduct of this reaction. The flask containing the sulfuric acid and NaNO2 solution was left connected to the chamber after the airflow was stopped, allowing for slow continued diffusion of HONO into the chamber; the degree of diffusion varied between experiments (see the Supplement for NOx data). Previous chamber experiments suggest that reaction with O(3P) can contribute to the oxidation of reduced sulfur compounds (Van Rooy et al., 2021a); however, this is likely to be negligible under the lower-NO2 and lower-UV conditions used here (see the Supplement). DMDS and DMSO (Sigma-Aldrich; >99.0 %) were introduced through the heated inlet (80 and 150 ∘C, respectively) via syringe injection. For some experiments (1 and 5), NOx conditions were perturbed by the addition of NO or HONO after several hours of oxidation. Acetonitrile (0.07 µL; 4.5 ppb) was added to the chamber for use as a dilution tracer, since its loss due to reaction with OH is negligible on the timescale of these experiments. Conditions for each experiment are shown in Table 1.
Table 1Summary of experimental conditions.
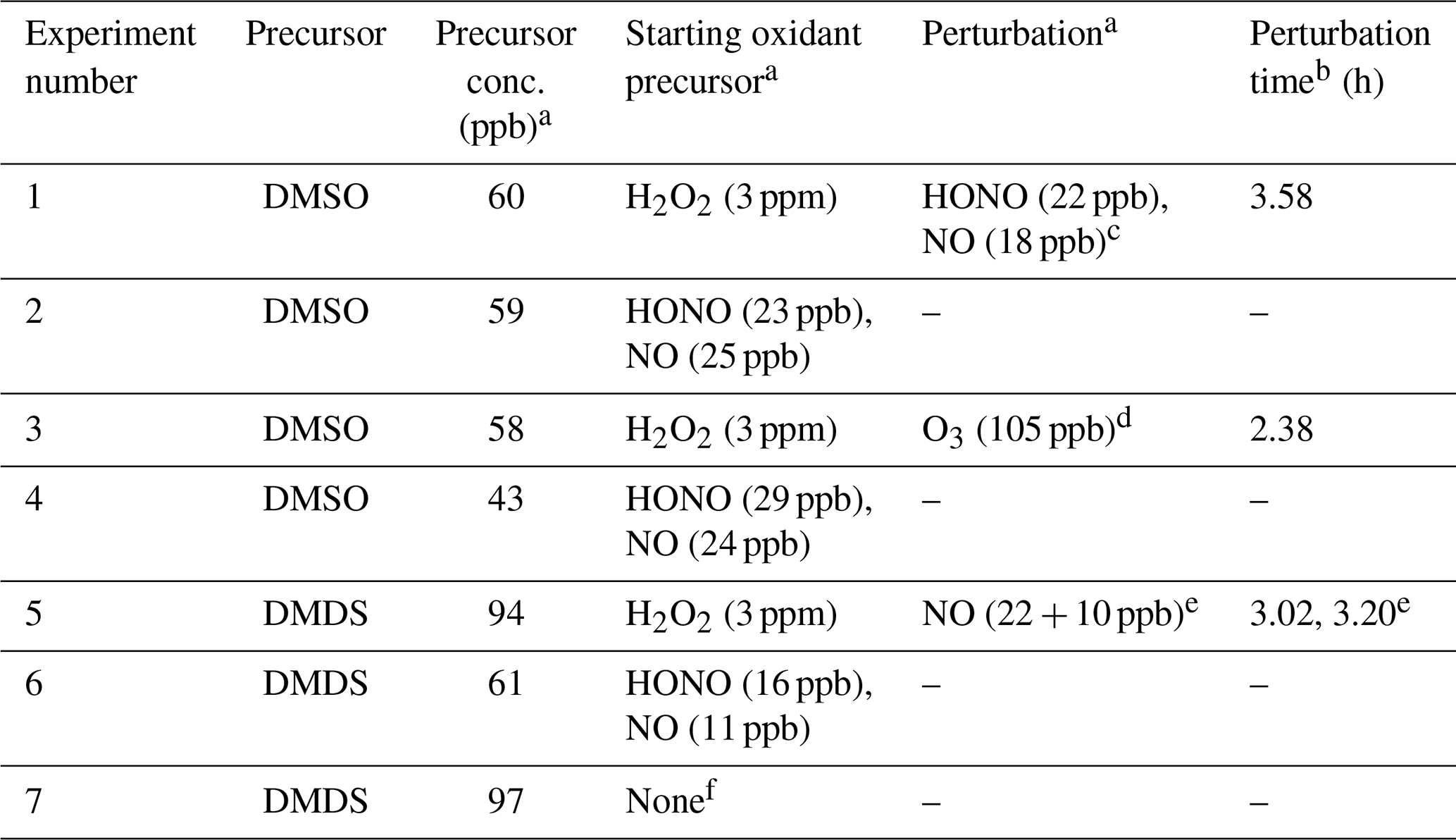
a Concentrations are reported at t=0 or at the time of perturbation. The concentration of H2O2 is reported as the total amount added to the chamber. The HONO concentration is measured using the NO2 channel of the NOx monitor. This represents an upper limit, since [NO2] is assumed to be 0 ppb at t=0 (see the Supplement). b Relative to lights-on time (t=0). c 600 ppb dichloromethane was also added during this experiment at t=1.92 h but was not observed to affect product formation. d O3 was added to investigate the influence of CH3SO2 + O3 chemistry on product distribution. e NO was added in two subsequent additions 11 min apart (see Fig. S5; for simplicity, only the time of the first addition is shown in most plots). f No oxidant precursor added; the experiment measured photolysis only.
Concentrations of precursors and products were monitored via a suite of online instrumentation. DMDS was monitored using a gas chromatograph with flame ionization detection (GC-FID; SRI Instruments). DMSO, acetonitrile, and oxidized gas-phase products were measured using an ammonium chemical ionization mass spectrometer (NH CIMS; modified PTR3; see Zaytsev et al., 2019). For DMSO experiments, the initial DMSO addition was found to overwhelm the primary ion in the NH CIMS. This was avoided by diluting the flow into the CIMS by a factor of ∼14. This dilution factor was quantified by adding the acetonitrile tracer to the chamber before the dilution flow was started and measuring the change in the acetonitrile signal. Particle-phase products were quantified using an aerosol mass spectrometer (Aerodyne Research Inc., HR-ToF-AMS; abbreviated as AMS from here on) and scanning mobility particle sizer (SMPS; TSI models 3080 and 3775). Additional gas monitors measured sulfur dioxide (Teledyne T100), ozone (2BTech Model 202), and NO/NO2 (Thermo Scientific Model 42i). Initial HONO concentration was estimated based on the NO2 channel in the NOx monitor; since NO2 may have also been present, this represents an upper limit.
The concentrations of gas-phase species were calculated based on direct calibration where possible and voltage scanning where reference standards were not available. For DMSO, the NH CIMS sensitivity was directly calibrated using a liquid calibration unit (Ionicon Analytik). One experiment (expt 4) was carried out 2 weeks before the calibration, and the sensitivity was re-scaled based on the change in the primary ion concentration. While most oxidized products showed smooth time series, the DMSO signal (C2H6SO(NH) was somewhat unstable, suggesting inconsistent detection, which may introduce additional uncertainty into this measurement. The sensitivity of the GC-FID to DMDS was calculated based on known volumes added to the chamber. For all other gas-phase organics detected by the NH CIMS, concentrations were derived using voltage scanning, following the methods described in Zaytsev et al. (2019). Gas-phase quantification methods are described in further detail in the Supplement.
Quantification of particle-phase products using the AMS followed a new method developed to distinguish different S-containing aerosol components (sulfate, methanesulfonate, and methanesulfinate). In brief, reference AMS spectra were taken for ammonium methanesulfonate and sodium methanesulfinate atomized directly into the AMS. Organosulfur peaks from the experimental AMS data are fit as a linear combination of the same organosulfur peaks from the two reference spectra. These two factors explain the experimental organosulfur peaks well (median r2≈0.95; Fig. S2). Based on this, MSIA and MSA factors are subtracted out, leaving a residual sulfate signal and a small organic residual. These factors are converted to mass using the relative ionization efficiencies (RIEs) of the respective species. RIE values are directly calculated for sulfate and MSA (2.06; from the ammonium balance method; Hodshire et al., 2019); MSIA is assumed to have the same RIE as MSA, since it cannot be directly calculated via the same method without the ammonium MSIA salt. As discussed below, there is some ambiguity in the particle-phase MSIA assignment, especially for the DMDS experiments; given this uncertainty, we denote this species MSIA*. This assignment, and the AMS quantification methods generally, are described in greater detail in the Supplement.
All gas-phase species were corrected for dilution loss by dividing by a normalized exponential fit of the acetonitrile time series. Aerosol-phase products are corrected for dilution, wall loss, and any changes in the collection efficiency over time by normalizing to the high-resolution nitrate time series from the seed particles (Eq. 4 from Wang et al., 2018). The wall- and dilution-corrected AMS signal is then scaled, such that the initial seed aerosol concentration matches that measured by the SMPS.
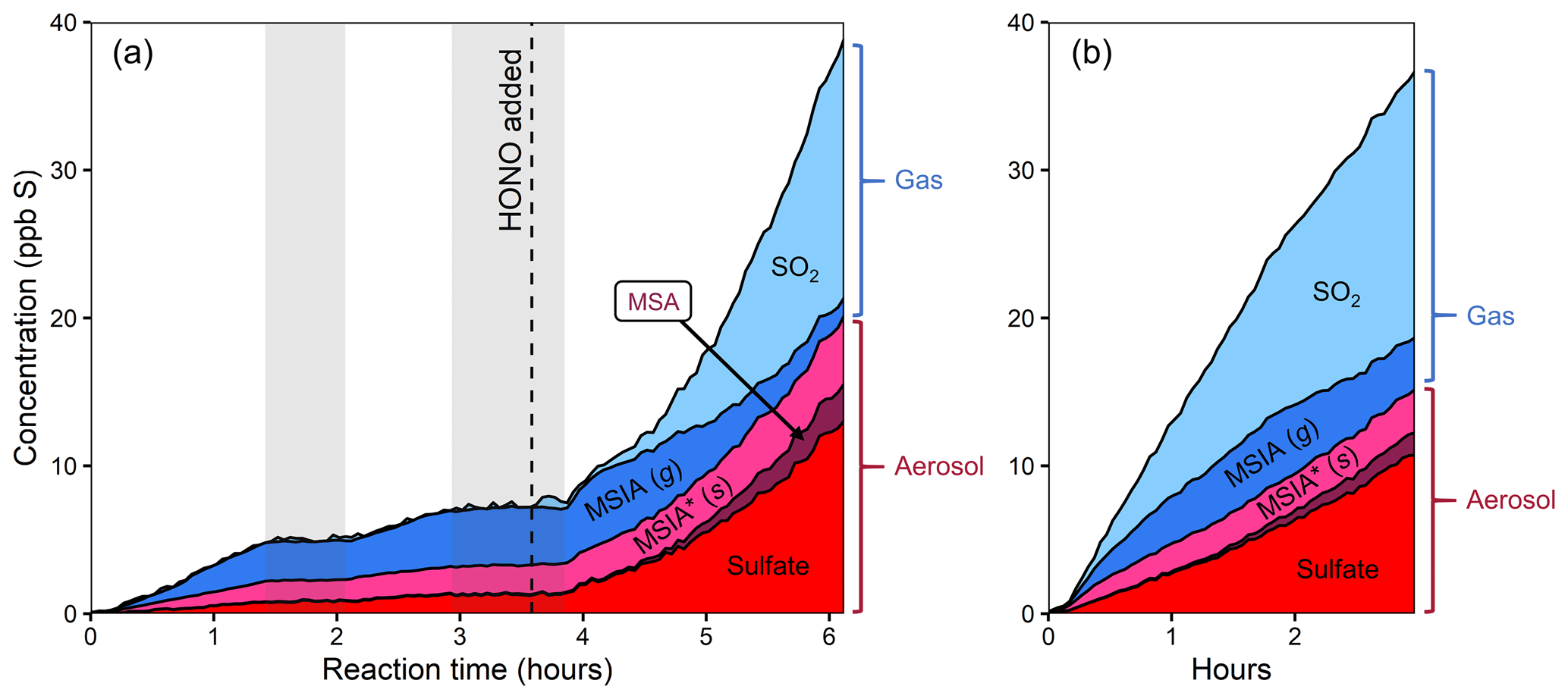
Figure 2Stacked product time series from the oxidation of DMSO, using different oxidant precursors. (a) Experiment 1, with H2O2 followed by HONO addition after several hours. (b) Experiment 2, with HONO. Production of particle-phase products increases dramatically in the presence of NOx. The light gray bars in panel (a) indicate when the chamber lights were turned off for diagnostic purposes. The lower-NO period is dominated by MSIA production, while the higher-NO conditions show large increases in the concentrations of SO2, MSA, and sulfate. The product distribution is comparable in both higher-NO conditions.
3.1 DMSO oxidation experiments
Figure 2 shows stacked time series of oxidation products for two DMSO experiments. In experiment 1 (Fig. 2a), DMSO is initially oxidized with H2O2 as the oxidant precursor and no added NOx. Halfway through the experiment, HONO is added, substantially increasing both total NOx and OH concentrations (see Fig. S4 for NOx time series). In experiment 2 (Fig. 2b), DMSO is oxidized with only HONO as an oxidant precursor. Due to some uncertainty in the DMSO time series, these plots focus only on the product composition; plots that include the DMSO time series are included in the Supplement. While sulfur closure appears complete in some experiments (Fig. S6), total sulfur drops over time during experiments using H2O2 as an oxidant precursor and briefly dips during HONO experiments. Incomplete sulfur closure may be due to a number of factors including the presence of unmeasured products, the loss of species via wall loss or other loss processes, error in CIMS sensitivity values (especially for DMSO), error in absolute particle-phase measurements, or error in the speciation of AMS data; as such, our discussion focuses primarily on trends in product formation and composition.
Under lower-NO conditions (first 3.5 h of expt 1; Fig. 2a), MSIA is the dominant product in the gas phase, and MSIA* the dominant product in the particle phase, with sulfate formed in low but nonzero yield. Notably, no SO2 or MSA are formed under these conditions (replicated in expt 3; Fig. S8a). Under higher-NO conditions, either from adding HONO to the ongoing experiment (last 2.5 h of expt 1; Fig. 2a) or from using HONO as the sole oxidant precursor (expt 2; Fig. 2b), the product distribution is dramatically different, with substantial production of MSA and sulfate in the particle phase and SO2 in the gas phase. All higher-NO experiments (expts 1, 2, and 4) exhibit consistent product distributions (see also Fig. S8b).
The use of HONO in experiments 1, 2, and 4 shifts the chemistry in two primary ways: the increase in NO changes the product branching ratios (i.e., by increasing RO2 + NO), and the increase in HONO and NO increases the OH concentration (directly through HONO photolysis and indirectly through HOx cycling). To distinguish these two effects, the product time series are plotted against the amount of DMSO that has reacted away (Fig. 3), effectively normalizing for differing OH concentrations and allowing comparisons among experiments. To reduce the noise in these plots, the DMSO time series used as the basis for the x axes are smoothed using a penalized spline (see the Supplement). Any uncertainties in [DMSO] from unstable detection in the NH CIMS and possible run-to-run variability in the calibration factor manifest as uncertainty in the x axis in these plots; this likely explains the majority of the x offset in the duplicate experiments (red traces) (see also Fig. S12). As such, these plots cannot distinguish small changes in product yields but should still show major differences in yields.
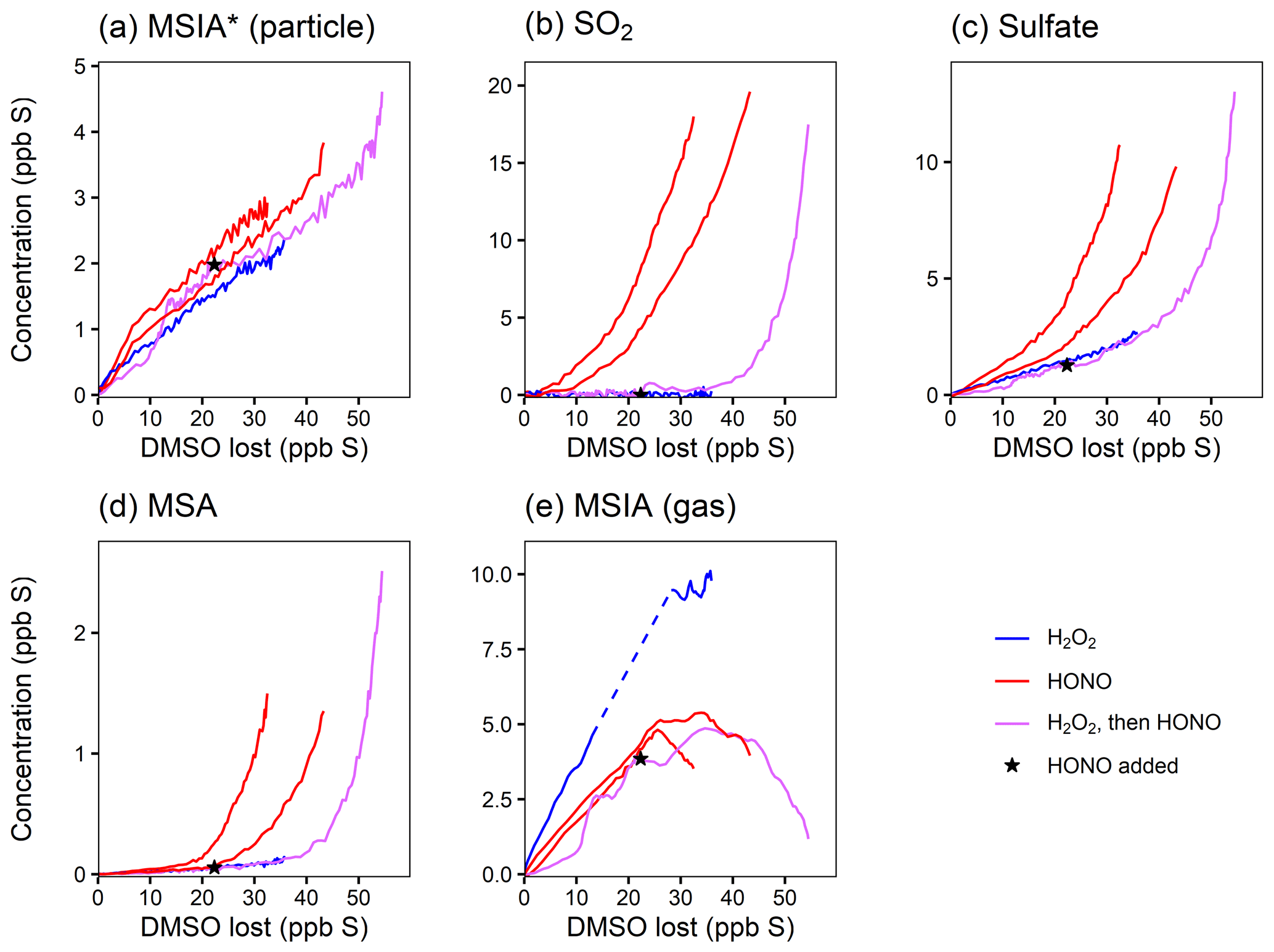
Figure 3Yield plots for DMSO oxidation products. Major products are plotted against the loss of DMSO to normalize for changing OH concentrations and allow for comparisons among experiments 1–4. Colors refer to the oxidant precursor. For experiment 1 (pink), the NOx regime is switched by adding HONO, as marked by the star. The dashed blue line indicates missing data. Note the differing y axes. Where traces lie on top of each other (e.g., for MSIA*; panel a), the addition of NOx does not influence the chemistry. Where traces are distinct (e.g., for SO2; panel b), the product formation is influenced by NOx.
Figure 3a shows that the MSIA* yield is unchanged by the different experimental conditions, suggesting that its formation from DMSO + OH is independent of NOx. This is consistent with the literature mechanism, which involves OH addition followed by loss of the CH3 radical (Fig. 1). This mechanism suggests that MSIA should form in 100 % yield in the first generation of oxidation, which should involve an initial total MSIA slope of 1; the lower slope seen here (Fig. 3a and e) may be a result of incomplete sulfur closure (Fig. S6) and possible uncertainty in the speciation ascribed to AMS data. In contrast to MSIA*, SO2 (Fig. 3b) shows a large shift in yield at a given OH exposure for higher vs. lower NOx, suggesting that NOx plays a role in its formation; this is inconsistent with the literature mechanisms (Fig. 1). Sulfate and MSA (Fig. 3c and d) are intermediate cases; barring significant error in the DMSO calibration (factor of ∼1.5–2), they appear moderately dependent on NOx concentrations. Gas-phase MSIA concentrations start to decrease (Fig. 3e), even as particle-phase MSIA* concentrations continue to grow (Fig. 3a); this suggests that MSIA may experience slower oxidation in the particle phase under these conditions, such that aerosol particles serve as a reservoir for this species.
In addition to using HONO to perturb the system, one lower-NO experiment (expt 3) is perturbed by the addition of O3 to investigate the impact of the CH3SO2 + O3 → CH3SO3 + O2 reaction (Barnes et al., 2006) (Fig. S8a). Since CH3SO3 is thought to be a major intermediate leading to the formation of sulfate and MSA, the addition of ozone is expected to influence the formation of particle-phase products. That no change in product distribution is observed upon the addition of O3 suggests that the CH3SO2 + O3 reaction is slow or that CH3SO2 is not formed from the reaction under these conditions.
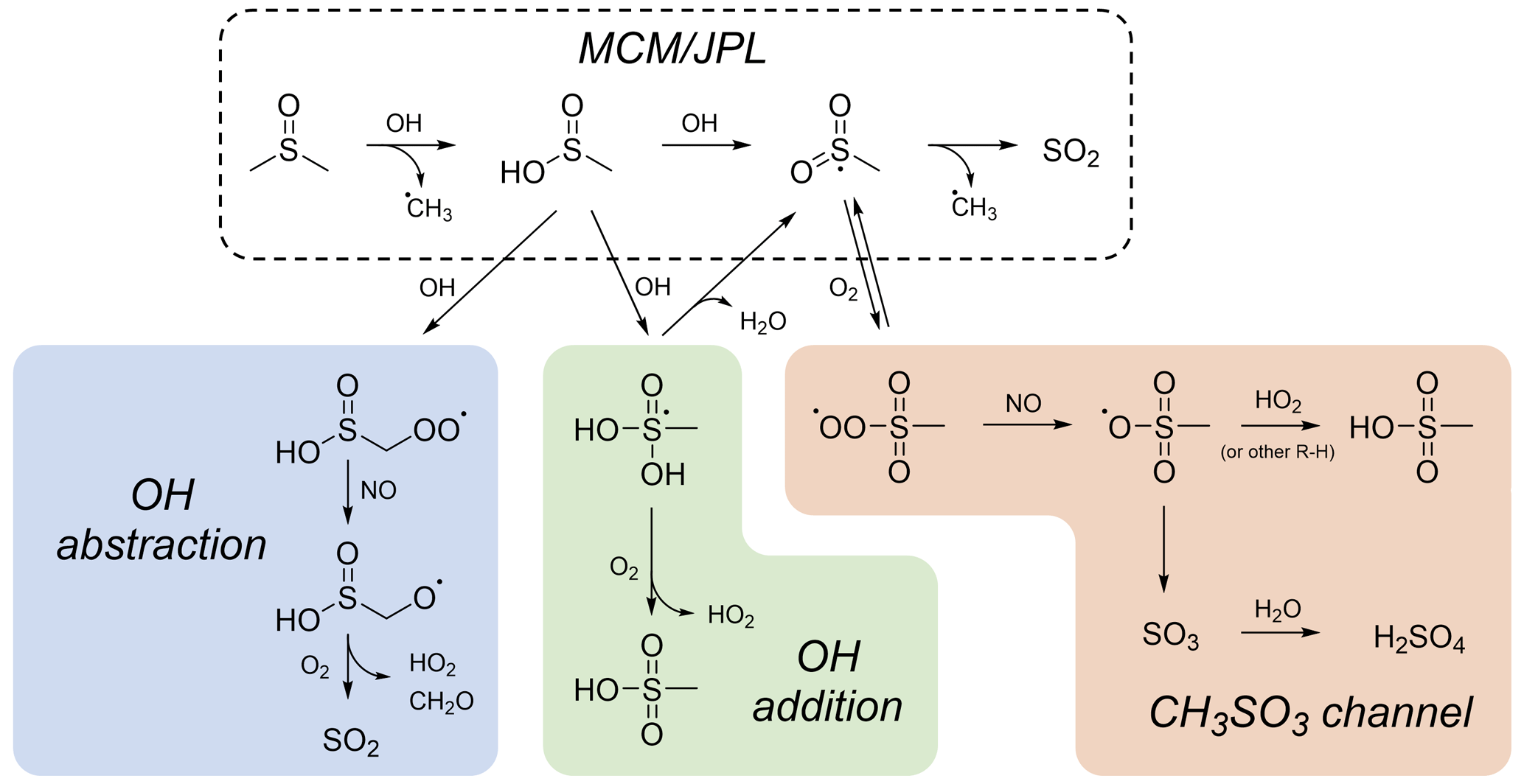
Figure 4Proposed mechanisms for DMSO and MSIA oxidation. The mechanism recommended by JPL and used in the MCM (dashed box) involves the formation of SO2 only. The OH abstraction pathway (blue) proceeds via OH abstraction of a methyl hydrogen from MSIA, leading to the formation of SO2. The OH addition pathway (green) proceeds via OH addition to the S atom of MSIA, leading to the formation of MSA. The CH3SO3 channel (orange) proceeds via O2 addition to the CH3SO2 radical and leads to the formation both MSA and sulfate via the CH3SO3 radical. Estimated rates for these reactions and box model simulation results are included in the Supplement.
While the range of products detected (SO2, MSIA/MSIA*, MSA, and sulfate) is broadly consistent with those found in previous DMSO oxidation studies (Barnes et al., 1989; Sørensen et al., 1996; Urbanski et al., 1998; Arsene et al., 2002; Librando et al., 2004; Chen and Jang, 2012), differences in NOx dependence and aerosol composition stand out. The strong increase in SO2 formation with increased NOx has not been reported in previous studies, possibly due to the range of NOx concentrations used. While some studies (Barnes et al., 1989; Sørensen et al., 1996) were run with ppm levels of NOx, exceptions include Librando et al. (2004), whose low-NOx case was <20 ppb, which may not be sufficiently low to see evidence of this chemistry, and Arsene et al. (2002), who used synthetic air to obtain low-NO conditions and saw a minor shift in the SO2 yield. Previous studies on the dependence of MSA formation on NOx levels are inconsistent, with some (Sørensen et al., 1996; Arsene et al., 2002) showing no dependence and others (Chen and Jang, 2012) showing an increase in MSA with higher initial NO concentrations. The results from Chen and Jang (2012) are in better agreement with our measurements, though their reported MSA sulfate ratio is substantially different (this work reports 0.14:1 to 0.19:1 at elevated NOx; Chen and Jang, 2012, find to at elevated NOx), possibly influenced by their higher-NO concentrations and higher-RH conditions (fostering aqueous chemistry). While MSIA has been measured as a major first-generation product, it has not previously been measured in the particle phase, though the exact speciation of aerosol-phase compounds detected by the AMS carries some uncertainty (see the Supplement). Sulfate, with yields ranging from ∼6 % in lower-NO conditions to ∼27 % in higher-NO conditions, has been quantified in only one other study (Chen and Jang, 2012), where it is seen in lower yield (∼2 %–4 %). Under the conditions in our chamber (dry; [OH] = 3.7×105 to 2.7×106 molec. cm−3), the SO2 lifetime to OH oxidation is >100 h, and heterogeneous oxidation of SO2 is unlikely, implying that the observed sulfate is not formed from SO2. This indicates that our observed formation of sulfate formation is via a rapid aerosol-formation mechanism, likely involving the direct formation of SO3.
In contrast to some previous studies (Sørensen et al., 1996; Arsene et al., 2002; Librando et al., 2004; Chen and Jang, 2012), we did not observe DMSO2 as a product. A small DMSO2 signal appeared when DMSO was added to the chamber, but it did not grow with oxidation and so was likely an impurity in the DMSO or an artifact from the CIMS detection of DMSO. Most previous studies that detected DMSO2 as a product were run at ppm levels of DMSO (Sørensen et al., 1996; Arsene et al., 2002; Librando et al., 2004) and so may have been influenced by bimolecular reactions such as DMSO + RO2 reactions (Arsene et al., 2002), which are less likely to occur under lower-concentration conditions. Similar to DMSO2, methanesulfonyl peroxynitrate (MSPN; CH3S(O)2OONO2), which has previously been detected (Sørensen et al., 1996; Arsene et al., 2002; Librando et al., 2004), was not observed. This might be because MSPN is not detectable with NH CIMS or because of the lower-NOx levels used; in our experiments, total NOx was ∼50 ppb, far lower than the >1 ppm levels used in some previous studies (Arsene et al., 2002; Librando et al., 2004). No other products were observed in the NH CIMS. This supports prior assertions that OH abstraction from the methyl groups of DMSO is too slow to compete (González-García et al., 2006), since we observed no products that would be expected from the resulting peroxy radicals (e.g., from RO2 + HO2).
The observations above suggest a need to revise the standard DMSO oxidation mechanism, as recommended by JPL (Burkholder et al., 2020) and included in the MCM (Saunders et al., 2003). Figure 4 shows this mechanism (dashed box) in addition to other possible mechanisms. In the JPL/MCM mechanism, DMSO reacts with OH to form MSIA, which reacts with OH to form SO2 in unit yield. However, this is inconsistent with our observation of rapid sulfate and MSA formation and the lack of SO2 formation at lower NOx. The shaded boxes in Fig. 4 show three possible alternative pathways, all of which involve modification to the MSIA oxidation mechanism. Pathways that do not involve MSIA formation have been shown to be unlikely (González-García et al., 2006); this is consistent with our lack of detection of products such as DMSO2 or CH3S(O)CH2OOH. Estimated rates for reaction pathways shown in Fig. 4, as well as box model simulations that demonstrate the effects of these pathways using the Framework for 0-D Atmospheric Modeling (F0AM; Wolfe et al., 2016), can be found in the Supplement (Table S2 and Fig. S16).
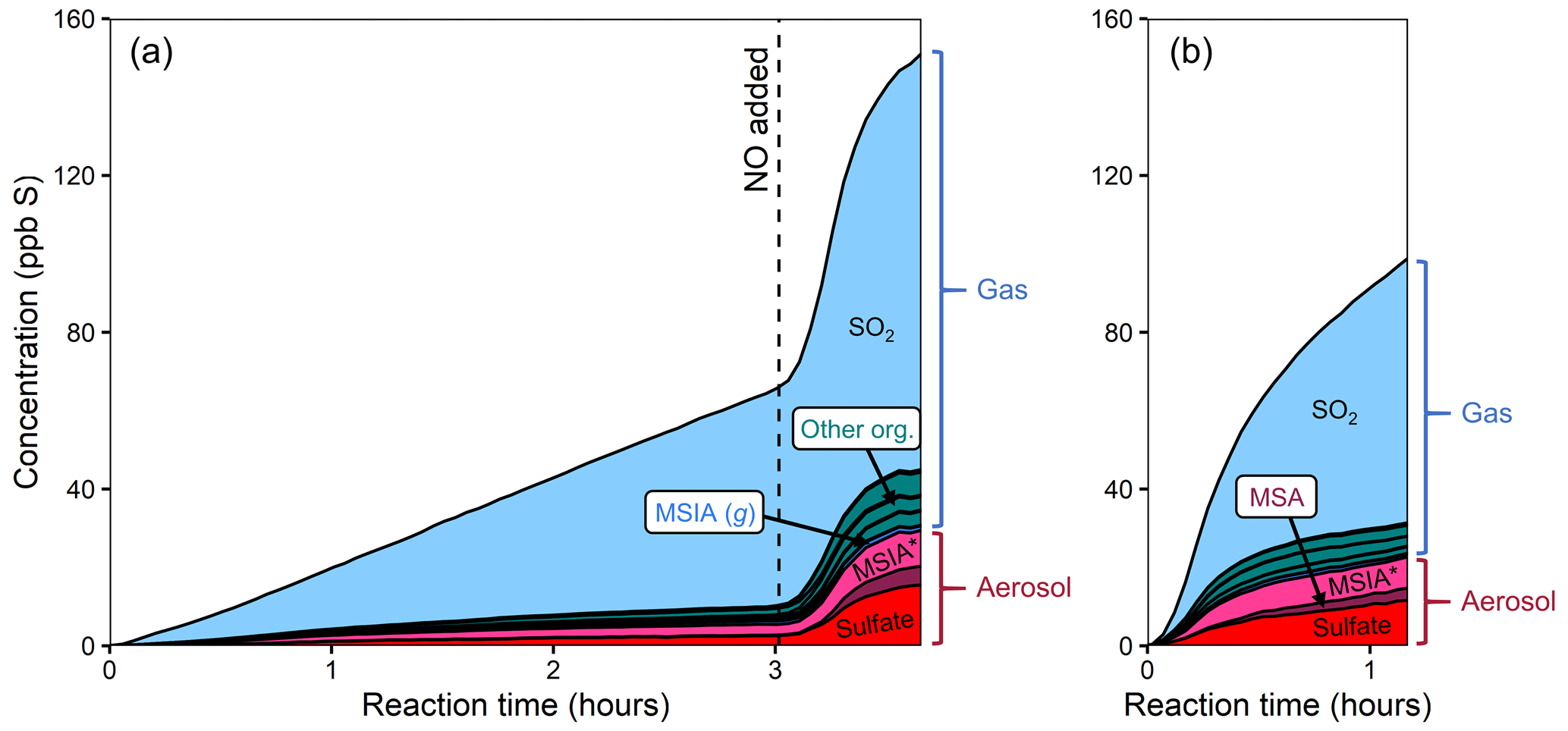
Figure 5Stacked product time series from the oxidation of DMDS, using different oxidant precursors. (a) Experiment 5, with H2O2 followed by NO addition after several hours. (b) Experiment 6, with HONO. All gas-phase organic compounds detected by the NH CIMS, other than MSIA (g), are shown in green and shown in greater detail in Fig. 7. SO2 is the major product formed in both experiments, but other species increase under higher NO. Product distributions are similar under both higher-NO cases (right side of panel a; b).
In the CH3SO3 channel, the CH3SO2 intermediate (formed from abstraction of the acidic hydrogen of MSIA) does not fall apart to CH3 and SO2, as in the JPL/MCM mechanism, but rather reacts with O2 to form more oxidized products (Lucas and Prinn, 2002). This channel has recently received renewed attention (Wollesen de Jonge et al., 2021; Shen et al., 2022; Ye et al., 2022), since it provides a pathway to both MSA and sulfate. However, under higher-NO conditions, where measured MSA and sulfate yields are highest, the HO2 concentration is suppressed. Since HO2 + CH3SO3 is the final reaction leading to MSA, this mechanism can sometimes underpredict MSA (Ye et al., 2022). However, recent experimental evidence (Berndt et al., 2023) supports earlier hypotheses (Yin et al., 1990a; Barnes et al., 2006) that other hydrocarbons may serve as an H atom source for the CH3SO3 → CH3SO3H reaction. This could explain high MSA yields from chamber experiments, where the hydrocarbon concentration is typically much higher than in the atmosphere.
The other pathways shown, OH abstraction and OH addition, stem from possible products of the OH + MSIA reaction. The OH abstraction pathway represents a plausible explanation for the observation of SO2 formation at higher NO; however, OH abstraction of the methyl hydrogens is believed to be too slow to compete (Yin et al., 1990a; González-García et al., 2007). The OH addition channel represents a straightforward pathway to MSA (Shen et al., 2022) but is inconsistent with our observation that MSA forms in greatest yield at elevated [NOx]. While computational studies support this OH addition step as a minor pathway (Tian et al., 2007; González-García et al., 2007), they have not investigated the possibility of reaction with O2 to form MSA.
Several additional pathways to MSA (not shown) have been hypothesized but seem unlikely to be the major sources of MSA in our chamber experiments. Production of MSA via CH3SO2 + OH (Kukui et al., 2003; González-García et al., 2007) does not explain the observed NOx dependence and seems unlikely due to low concentrations of both species. In addition, the disproportionation reaction of CH3SO2OO + RO2 may lead to MSA (Berndt et al., 2023), but this pathway is significant only when RO2 concentrations are sufficiently high to outcompete other pathways. In our chamber, this reaction can occur under lower-NO conditions, where a small amount of MSA is formed, but it is likely only a minor contributor to MSA production under higher-NO conditions, when the majority of MSA is formed (see modeling results in the Supplement). Based on observations of NOx and humidity dependence, Van Rooy et al. (2021b) suggest that CH3SO3 may react with NO or NO2 to form CH3S(O)2ONO or CH3S(O)2ONO2 before reacting with water to form MSA and HNO3 or HONO. We did not observe the nitrite or nitrate compound, and the subsequent hydrolysis step is unlikely under the dry conditions in our chamber.
The observed trends in product formation, particularly the formation of MSA and sulfate and the lack of SO2 formation under lower-NOx conditions, make clear that the commonly used JPL/MCM mechanism of DMSO oxidation is inadequate; however, none of the above-proposed mechanisms discussed above is fully consistent with computational and laboratory results. In box model simulations of these pathways (see Sect. S9), we are unable to reproduce all of the experimental results presented here (especially the NOx dependence of SO2 formation). More computational and experimental studies on the fate of MSIA and radical intermediates (e.g., CH3SO2 and CH3SO3) are thus necessary to better constrain this oxidation mechanism.
3.2 DMDS oxidation experiments
Figure 5 shows stacked time series for the products of two DMDS oxidation experiments. In Fig. 5a (experiment 5), DMDS is oxidized using H2O2 as the OH precursor (lower-NO conditions); after 3 h, NO is added, increasing total NOx and OH concentrations. Figure 5b shows the products of experiment 6, where DMDS was oxidized using HONO as an oxidant precursor. Plots that include the DMDS time series are included in the Supplement.
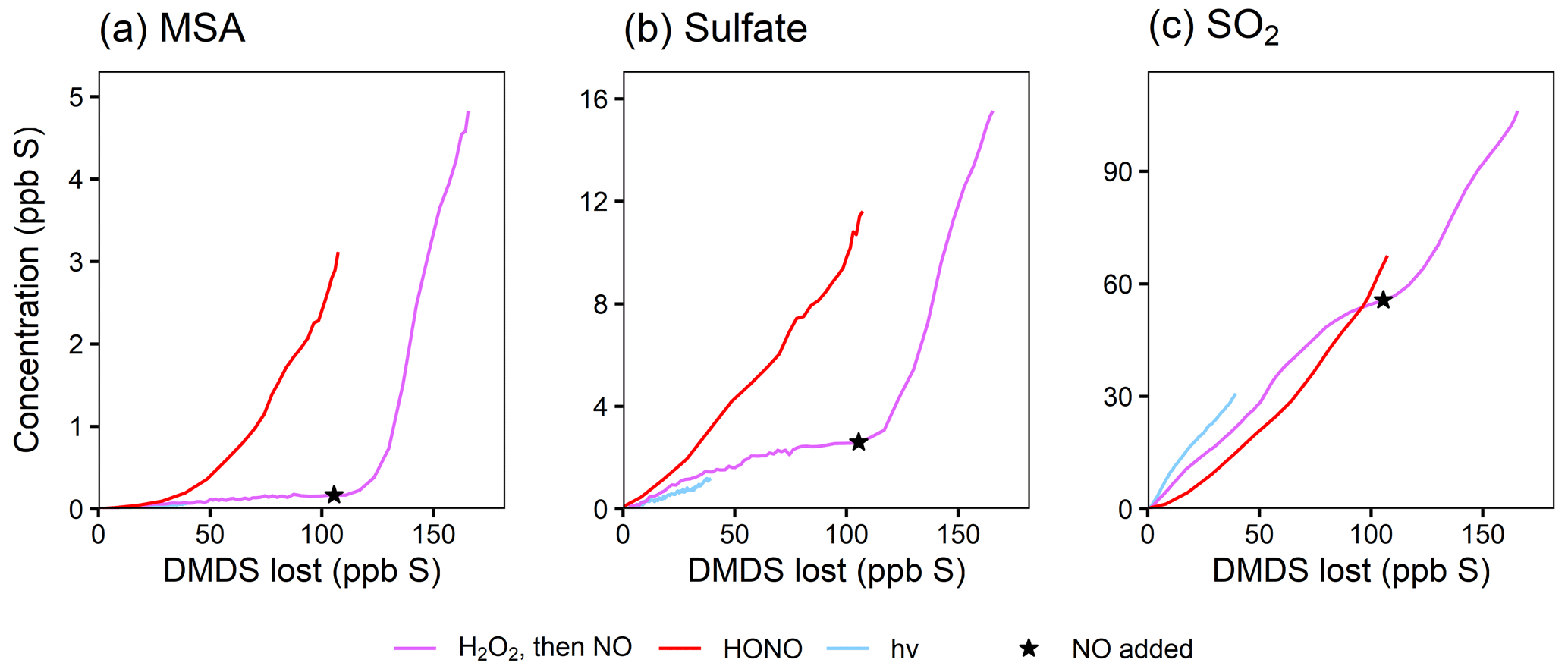
Figure 6Yield plots for selected DMDS oxidation products. MSA, sulfate, and SO2 are plotted against the loss of DMDS to normalize for changing OH concentrations and therefore allow comparisons among experiments 5–7. Colors denote experimental conditions. For one experiment (expt 5; pink trace), the NOx regime is switched by adding NO, as marked by the star. Note the differing y axes. Where traces lie on top of each other (e.g., for SO2), the addition of NOx does not influence the chemistry. Where traces are distinct (e.g., for MSA and sulfate), the product formation is influenced by NOx. See Fig. S13 for similar plots of other products.
In both higher- and lower-NO conditions, oxidation products (Fig. 5) are dominated by SO2, though a range of other gas- and particle-phase products are also formed. As in the DMSO experiments, aerosol formation increases substantially in the presence of NOx, and MSA is formed only after the addition of NOx. Increased NOx also increases the production of organic products detected by the NH CIMS. The product distributions of the two higher-NO cases (expts 5 and 6) are consistent. Direct photolysis of DMDS also occurs to some extent during each experiment. To explore this, DMDS was exposed to twice the light intensity of the other DMDS experiments (expt 7; Fig. S9) and formed almost entirely SO2, suggesting that this may bias SO2 yields from OH oxidation of DMDS. Based on the SO2 yield from photolysis, photolytically derived SO2 is estimated to make up 6 %–20 % of the SO2 generated in the OH oxidation experiments.
One clear difference between the DMSO and DMDS product distributions is the apparent partitioning of MSIA/MSIA* between the gas and particle phase (for DMSO 36±13 % (1σ) particle-phase; for DMDS 91±8 % (1σ) particle-phase; see Figs. 2 and 5). The reason for this difference is not clear. Different particle-phase acidity could affect partitioning, with lower pH driving more MSIA to the gas phase. The discrepancy may also be a result of ambiguity in the AMS spectra, where some organosulfur species, including those with two sulfur atoms, are likely to contribute to the same AMS peaks as MSIA.
As done previously for DMSO, selected DMDS products for experiments 5–7 are plotted against DMDS loss to normalize for changing [OH] and allow for direct comparisons among experiments (Fig. 6). These plots demonstrate dramatic increases in yield for particle-phase species (MSA, sulfate, and MSIA*; see the Supplement) under high-NOx conditions. This is consistent with recent measurements of increased production of gas-phase MSA and H2SO4 (Berndt et al., 2023) and increased production of particle-phase products when NOx is added (Van Rooy et al., 2021a, b); though, in previous work (Van Rooy et al., 2021a), MSA formation was not observed under dry conditions. In contrast to the trends in particle-phase products, SO2 yields are relatively consistent among experiments, and exhibit no obvious dependence on NOx concentrations, suggesting that the pathway leading to SO2 is different from that found in DMSO oxidation. These major products are largely consistent with literature mechanisms (Saunders et al., 2003; Barnes et al., 2006), where a high yield of CH3S provides multiple efficient routes to SO2 via O2 addition and rearrangement. The CH3SO2 radical, which can also form SO2, is in equilibrium with the CH3S(O)2OO radical, which can be diverted towards particle-phase products (MSA and sulfate) by reaction with NO, explaining the elevated aerosol yields at high NOx (see Figs. 1 and 4). This might also explain the slightly lower SO2 yields in the HONO experiment. For the photolysis experiment (expt 7), the SO2 yield is slightly higher, likely due to the greater yield of CH3S radicals per molecule of DMDS.
Thus the major products of DMDS oxidation, including SO2, sulfate, and MSA, are explained reasonably well by known DMDS chemistry (Berndt et al., 2020) and CH3S chemistry, as understood from the DMS oxidation mechanism (Fig. 1). However, the detection of minor gas-phase organosulfur compounds, many containing two sulfur atoms, suggest additional minor reaction pathways. The time series of these “other organics” (shown in green in Fig. 5) are presented in Fig. 7. While S2 products are formed in low yield (∼1 %–3 %), they may influence aerosol formation from DMDS due to their greater molecular weight and might contribute to the observed MSIA* product seen in the AMS.
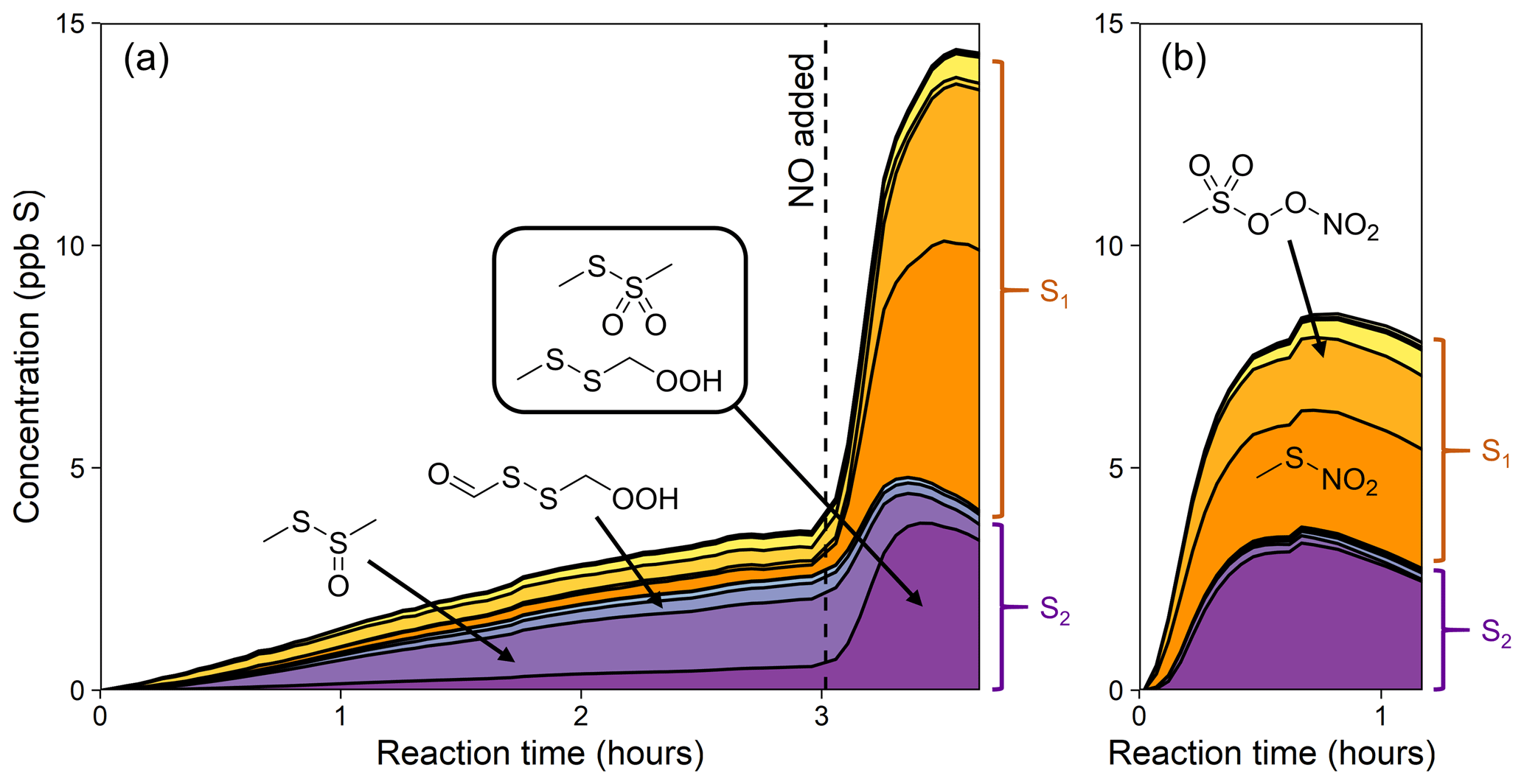
Figure 7Stacked time series of minor gas-phase organosulfur products of DMDS oxidation for experiment 5 (a; H2O2 followed by NO) and experiment 6 (b; HONO). These are the products shown as “Other org.” in Fig. 5. Products are sorted into S1 (orange) and S2 (purple) compounds, and suggested structures of the most abundant products are shown. See Fig. S14 for full results.
Many of the observed organosulfur products are analogous to those formed in DMS oxidation, and include several previously unreported compounds, providing evidence of new DMDS reaction pathways. C2H6S2O is favored at lower NO and decays away after the addition of NO (Figs. 7a and S13b). Since the formation of an alcohol seems unlikely, this product is best explained by the structure CH3SS(O)CH3, a molecule analogous to DMSO and likely formed via the OH adduct (which is usually assumed to fragment into CH3S and CH3SO; Berndt et al., 2020). A complementary product, C2H6S2O2, forms mostly at higher NO (Figs. 7a–b and S13c). This is unlikely to be the hydroperoxide CH3SSCH2OOH, since that would likely be formed only at lower NO. Instead the product is better explained by the structure CH3SS(O)2CH3, which is similar to DMSO2 and likely also formed from the OH adduct. Together, these two compounds appear almost exactly analogous in structure and mechanism to the formation of DMSO and DMSO2 from the DMS–OH adduct and so represent a minor new oxidation pathway for DMDS. Also among the minor organosulfur products is C2H4S2O3, first detected by Berndt et al. (2020) and attributed to the isomerization product of the DMDS abstraction pathway (HOOCH2SSCHO; Figs. 1 and 7). This product is observed to form in greater yield at longer RO2 bimolecular lifetimes. At higher NOx, we observe CH3SO6N, likely methanesulfonyl peroxynitrate formed from CH3S(O)2OO and NO2, and CH3SNO2, likely formed from the reaction of CH3S and NO2. CH4SO4, postulated by Berndt et al. (2020) to be a source of MSIA, was not observed. The total mass spectrometric signal of gas-phase organics decreases slightly at the end of experiments, likely a result of further oxidation leading to fragmentation and/or condensation onto particles or chamber walls. A more detailed product time series figure (Fig. S14), hypothesized reaction mechanism (Fig. S15), and discussion of these species are given in the Supplement.
These chamber studies demonstrate several new observations of DMDS oxidation chemistry. The OH oxidation of DMDS leads to substantial rapid aerosol formation, with strong dependence on the NOx regime (5 %–6 % S yield at lower NOx; 17 %–21 % S yield at higher NOx). In addition to the major products (SO2, sulfate, MSA, and MSIA), this work demonstrates that S2 species, formed through both OH abstraction and stabilization of the OH adduct, may represent a small but non-negligible fraction of the total product distribution, with a measured yield of ∼3 % under higher-NOx conditions.
3.3 Implications for DMS oxidation
As discussed in the introduction, the oxidation mechanisms of DMSO and DMDS overlap substantially with the DMS addition and abstraction channels, respectively, and can therefore be used to help interpret the contributions of these channels to aerosol formation from DMS. Our measurements show that DMSO and DMDS both produce aerosol in lower yield (final S yields of 14 %–15 % and 5 %–6 %, respectively) at lower NO and relatively high yield (final S yields of 34 %–47 % and 17 %–21 %) at higher NO, suggesting that both the addition and abstraction channels can be important contributors to rapid aerosol formation from DMS oxidation.
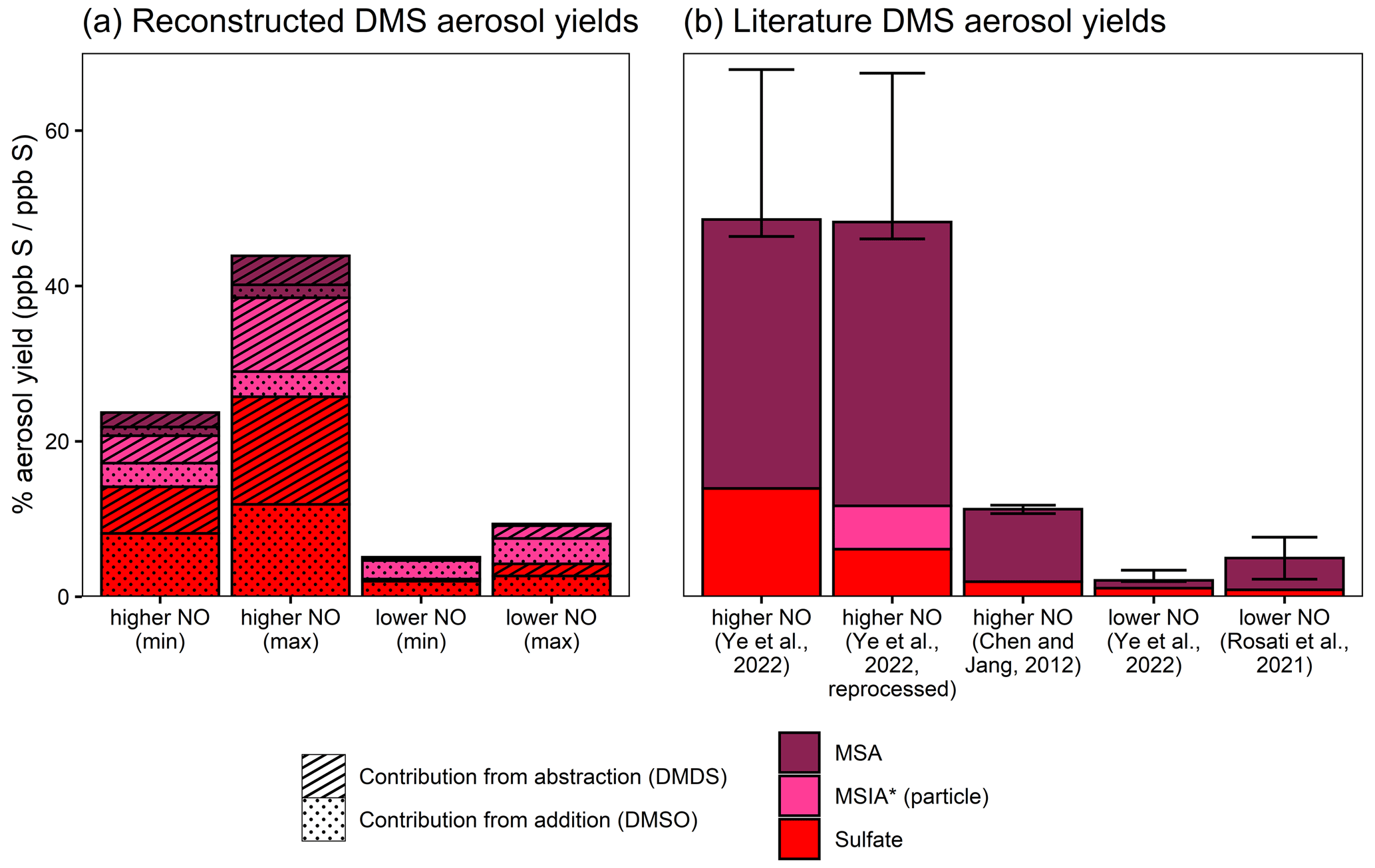
Figure 8Aerosol yields from DMS as reconstructed from DMSO and DMDS results (a) and from the literature measurements (Ye et al., 2022; Chen and Jang, 2012; Rosati et al., 2021) (b). Aerosol yields are shown as product formed divided by DMS reacted (ppb S/ppb S) and only consider rapid aerosol formation. Reconstructed yields shown in the left panel are calculated from DMSO- and DMDS-derived aerosol measurements, as described in the text. In addition to the literature yields, panel (b) includes data from Ye et al. (2022), which are reprocessed using the same AMS quantification methods used in this work (see the text and the Supplement for further details).
We can extrapolate the observations from DMSO and DMDS experiments based on the literature branching ratios to try to explain the rapid aerosol yields from DMS oxidation. Based on the JPL-recommended rates for abstraction and addition at 293 K (Burkholder et al., 2020), OH abstraction contributes 64 % of the DMS + OH reaction, while OH addition contributes the remaining 36 %. Within the addition channel, ∼80 %–100 % of the total sulfur passes through DMSO, depending on the NO concentration. If we assume that NO is relatively high (e.g., 10 ppb), then the isomerization channel is negligible (∼1 %–4 % of CH3SCH2OO fate) (Ye et al., 2022; Assaf et al., 2023), such that all sulfur in the abstraction channel passes through CH3S. Under lower-NO conditions, competition with isomerization lowers this fraction to ∼17 %–41 % (assuming 10 ppt NO and 100 ppt HO2 (Ye et al., 2022); isomerization rate = 0.039–0.13 s−1 (Ye et al., 2022; Assaf et al., 2023); and bimolecular rates taken from MCM (Saunders et al., 2003)). Based on these assumptions, the addition and abstraction channels can therefore be reasonably represented by DMSO and DMDS chemistry, allowing us to reconstruct DMS aerosol yields using the yields measured in this study and appropriate correction factors based on the literature branching ratios.
Figure 8 shows reconstructed DMS aerosol yields from DMSO and DMDS (Fig. 8a) in comparison with the literature DMS aerosol yields (Fig. 8b). These yields only consider rapid aerosol formation and do not include the influence of sulfate formed through SO2 oxidation. Reconstructed yields are calculated by multiplying DMSO and DMDS aerosol yields by the appropriate DMS branching fraction for the addition and abstraction channels (36 % and 64 %, respectively). For lower-NO conditions, DMDS aerosol yields are also multiplied by 17 %–41 % to reflect competition with isomerization. For aerosol yields calculated from DMSO, the minimum and maximum values are calculated from the range of yields observed in our experiments. For those calculated from DMDS, the lower bound is based on the total aerosol yield from DMDS, while the upper bound assumes that only 50 % of DMDS sulfur yields CH3S and that all aerosol is derived from CH3S.
Reconstructed aerosol from DMSO, representing the addition channel, and DMDS, representing the abstraction channel, predicts total DMS aerosol yields of 24 %–44 % at higher NO and 5 %–9 % at lower NO (Fig. 8a). Contributions from the DMSO and DMDS experiments are roughly equal (38 %–88 % from DMSO; 12 %–62 % from DMDS), providing evidence that both abstraction and addition channels represent substantial sources of rapidly formed aerosol.
For comparison, Fig. 8b shows previous measurements of aerosol formation yields from DMS oxidation. At higher NO, reconstructed yields fall slightly below those measured for DMS oxidation by Ye et al. (2022) (experiments performed in the same chamber and under similar conditions at 42–53 ppb NO). However they are substantially greater than values measured by Chen and Jang (2012); those experiments were performed at comparable NO levels (21–117 ppb) but featured higher humidity (28 %–60 %) and did not use seed particles to reduce and account for losses of oxidized products to the chamber walls. At lower NO, reconstructed yields are somewhat greater than those observed in Ye et al. (2022) (∼10 ppt NO) and roughly consistent with measurements reported by Rosati et al. (2021) (dry chamber; 1–2 ppb background NOx). While the general trend of higher aerosol yields at higher NO is qualitatively consistent across the reconstructed and literature results, differences in the experimental conditions and wall loss correction methods likely influence the discrepancies in total observed aerosol yields.
While reconstructed yields are largely similar to the measured yields, the differences in composition are substantial. The majority of aerosol from DMS experiments is in the form of MSA (47 %–83 % of total aerosol), while MSA makes up only 2 %–13 % of the total reconstructed yields. The large discrepancy in aerosol composition might be explained by assumptions in the reconstruction of DMS yields. The reconstruction of DMS yields leaves out the possible formation of aerosol from DMSO2 or the isomerization pathway. But even if these channels were to form MSA in 100 % yield, their effect on composition under elevated NO conditions would be minor, since they only make up ∼4 %–7 % total sulfur at 10 ppb NO (Saunders et al., 2003; Burkholder et al., 2020; Ye et al., 2022; Assaf et al., 2023).
Another possible explanation for the discrepancies in composition could be the use of different AMS quantification techniques. When the MSA/MSIA linear combination method from this work is applied to data from Ye et al. (2022), MSIA* is found to be a minor but non-negligible contributor (10 % of total particulate sulfur), while the fraction of MSA actually increases at the expense of sulfate (Fig. 8b; also see the Supplement). This increases the discrepancy between the aerosol composition as measured for DMS and the reconstructed aerosol composition. While the application of this method to older DMS data is imperfect without contemporaneous reference spectra, it demonstrates that it could be a useful technique in field and laboratory studies under conditions where MSA and MSIA are expected to dominate the particle-phase organosulfur composition.
The differences in aerosol composition are most likely due to subtle chemical dependencies that affect branching between SO2, MSA, and sulfate. As noted previously, it is possible that high hydrocarbon concentrations in atmospheric chambers relative to the real atmosphere may allow a CH3SO3 + R–H reaction that increases MSA yields. If the DMS hydrogen is more labile than that of DMSO or DMDS, as is suggested by (somewhat uncertain) OH abstraction rates (Burkholder et al., 2020; González-García et al., 2006), then this may favor MSA production in DMS experiments. The inconsistencies in yield and composition might also be the result of detailed chemistry of simple sulfur radicals (e.g., CH3S, CH3SO, and CH3SO2), which could be highly dependent on reaction conditions (e.g., through reactions with HO2, NO, NO2, and O3). Higher relative MSA yields from DMSO seen by Chen and Jang (2012) may, for instance, be influenced by sulfur radical branching caused by the higher-NO concentrations used in that study. While recent work has made important advances in the understanding of these reactions (Chen et al., 2023; Berndt et al., 2023), many remain poorly understood, with mechanisms often relying on basic parameterizations (Saunders et al., 2003) or approximate rate estimates (Yin et al., 1990a); these represent an opportunity for further experimental and computational study.
In this study, we conducted experiments examining the OH oxidation of DMSO and DMDS. These results are among the first to focus on the amount and composition of aerosol formed from these two compounds and as such identify both agreement with the literature mechanisms and areas where known mechanisms do not describe the observed products. Major products from DMSO oxidation include MSIA, SO2, and MSA, and sulfate, while DMSO2 is not observed to form. MSA and sulfate yields increase with increasing NOx, while SO2 is observed to form only in the presence of NOx. These observations, particularly the trend in SO2 formation, cannot be fully explained by current mechanisms. While the major MSA and sulfate formation pathways remain somewhat unclear, these results clearly identify DMSO as a precursor of rapid sulfate aerosol formation, in contrast to standard mechanisms for DMSO and MSIA oxidation. We observe rapid sulfate aerosol formation from DMDS oxidation as well, again with a substantial increase in aerosol yield with increasing NOx. Several S2 products are observed for the first time, suggesting that the stabilization of an OH adduct may represent a minor but viable route to further oxidation chemistry.
Based on the overlap with the DMS mechanism (Fig. 1), these results provide insight into the mechanisms of aerosol production from DMS oxidation. While the total aerosol yield can be roughly explained by the upper bound of the combination of DMSO and DMDS results, the previously measured DMS aerosol composition is substantially different, with a much greater MSA component than can be explained by DMSO and DMDS results (Fig. 8). We hypothesize that discrepancies in aerosol composition may be controlled by the chemistry of small sulfur radical intermediates (e.g., CH3S, CH3SO2, and CH3SO3). This chemistry is poorly constrained, and the reactions of these species under variable chemical conditions (e.g., changing NO, NO2, HO2, O3, or hydrocarbon concentration) represent important targets for future work.
Despite uncertainties in the exact contributions of the addition and abstraction channels to aerosol yield and composition, our results demonstrate that both channels contribute appreciably to rapid aerosol formation from DMS oxidation, especially under elevated NO conditions. While this work highlights necessary changes to DMS oxidation mechanisms, additional laboratory and computational studies that focus on key intermediates and that further explore the influence of environmental parameters (e.g., RH and T) are needed in order to develop a mechanism that can fully explain the observed aerosol formation from the oxidation of DMS under the full range of atmospheric conditions.
Chamber data and species concentrations for all experiments have been archived and are available via the Kroll Group publication website at http://krollgroup.mit.edu/publications.html (Kroll Group, 2023) and at the Index of Chamber Atmospheric Research in the United States (ICARUS; https://icarus.ucdavis.edu/experimentset/266, Goss and Kroll, 2023).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-24-1299-2024-supplement.
MBG collected and analyzed the data and wrote the paper. JHK edited the paper and provided project guidance.
The contact author has declared that neither of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
The authors thank Frank Keutsch (Harvard University) for the use of the NH CIMS and Yaowei Li (Harvard University) for his help with maintenance of the instrument. The authors also thank David Hagan and Eben Cross (QuantAQ) for the use of the SO2 monitor. Finally, the authors thank Colette Heald (MIT/ETH Zurich) and Torsten Berndt (Leibniz Institute for Tropospheric Research) for useful discussions.
This research has been supported by the Division of Atmospheric and Geospace Sciences (grant no. AGS-2129835) and the Biological and Environmental Research (grant no. DE-SC0022017).
This paper was edited by Theodora Nah and reviewed by two anonymous referees.
Arsene, C., Barnes, I., Becker, K. H., Schneider, W. F., Wallington, T. T., Mihalopoulos, N., and Patroescu-Klotz, I. V.: Formation of Methane Sulfinic Acid in the Gas-Phase OH-Radical Initiated Oxidation of Dimethyl Sulfoxide, Environ. Sci. Technol., 36, 5155–5163, https://doi.org/10.1021/es020035u, 2002.
Assaf, E., Finewax, Z., Marshall, P., Veres, P. R., Neuman, J. A., and Burkholder, J. B.: Measurement of the Intramolecular Hydrogen-Shift Rate Coefficient for the CH3SCH2OO Radical between 314 and 433 K, J. Phys. Chem. A, 127, 2336–2350, https://doi.org/10.1021/acs.jpca.2c09095, 2023.
Bandy, A. R., Scott, D. L., Blomquist, B. W., Chen, S. M., and Thornton, D. C.: Low yields of SO2 from dimethyl sulfide oxidation in the marine boundary layer, Geophys. Res. Lett., 19, 1125–1127, https://doi.org/10.1029/92GL01041, 1992.
Bandy, A. R., Thornton, D. C., Blomquist, B. W., Chen, S., Wade, T. P., Ianni, J. C., Mitchell, G. M., and Nadler, W.: Chemistry of dimethyl sulfide in the equatorial Pacific atmosphere, Geophys. Res. Lett., 23, 741–744, https://doi.org/10.1029/96GL00779, 1996.
Barnes, I., Bastian, V., Becker, K. H., and Martin, D.: Fourier Transform IR Studies of the Reactions of Dimethyl Sulfoxide with OH, NO3, and Cl Radicals, in: Biogenic Sulfur in the Environment, vol. 393, edited by: Saltzman, E. S. and Cooper, W. J., American Chemical Society, Washington, DC, 476–488, https://doi.org/10.1021/bk-1989-0393, 1989.
Barnes, I., Becker, K. H., and Mihalopoulos, N.: An FTIR product study of the photooxidation of dimethyl disulfide, J. Atmos. Chem., 18, 267–289, https://doi.org/10.1007/BF00696783, 1994.
Barnes, I., Hjorth, J., and Mihalopoulos, N.: Dimethyl Sulfide and Dimethyl Sulfoxide and Their Oxidation in the Atmosphere, Chem. Rev., 106, 940–975, https://doi.org/10.1021/cr020529+, 2006.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Hoffmann, E. H., Tilgner, A., Hyttinen, N., Prisle, N. L., Hansel, A., and Herrmann, H.: Fast Peroxy Radical Isomerization and OH Recycling in the Reaction of OH Radicals with Dimethyl Sulfide, J. Phys. Chem. Lett., 10, 6478–6483, https://doi.org/10.1021/acs.jpclett.9b02567, 2019.
Berndt, T., Chen, J., Møller, K. H., Hyttinen, N., Prisle, N. L., Tilgner, A., Hoffmann, E. H., Herrmann, H., and Kjaergaard, H. G.: SO2 formation and peroxy radical isomerization in the atmospheric reaction of OH radicals with dimethyl disulfide, Chem. Commun., 56, 13634–13637, https://doi.org/10.1039/D0CC05783E, 2020.
Berndt, T., Hoffmann, E. H., Tilgner, A., Stratmann, F., and Herrmann, H.: Direct sulfuric acid formation from the gas-phase oxidation of reduced-sulfur compounds, Nat. Commun., 14, 4849, https://doi.org/10.1038/s41467-023-40586-2, 2023.
Berresheim, H., Eisele, F. L., Tanner, D. J., McInnes, L. M., Ramsey-Bell, D. C., and Covert, D. S.: Atmospheric sulfur chemistry and cloud condensation nuclei (CCN) concentrations over the northeastern Pacific Coast, J. Geophys. Res., 98, 12701,https://doi.org/10.1029/93JD00815, 1993.
Burkholder, J., Sander, S. P., Abbatt, J. P. D., Barker, J. R., Cappa, C., Crounse, J. D., Dibble, T. S., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Percival, C. J., Wilmouth, D. M., and Wine, P. H.: Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, NASA Jet Propulsion Laboratory, https://jpldataeval.jpl.nasa.gov/pdf/NASA-JPL Evaluation 19-5.pdf (last access: 19 May 2021), 2020.
Carslaw, K. S., Lee, L. A., Reddington, C. L., Pringle, K. J., Rap, A., Forster, P. M., Mann, G. W., Spracklen, D. V., Woodhouse, M. T., Regayre, L. A., and Pierce, J. R.: Large contribution of natural aerosols to uncertainty in indirect forcing, Nature, 503, 67–71, https://doi.org/10.1038/nature12674, 2013.
Chen, H., Varner, M. E., Gerber, R. B., and Finlayson-Pitts, B. J.: Reactions of Methanesulfonic Acid with Amines and Ammonia as a Source of New Particles in Air, J. Phys. Chem. B, 120, 1526–1536, https://doi.org/10.1021/acs.jpcb.5b07433, 2016.
Chen, J., Lane, J. R., and Kjaergaard, H. G.: Reaction of Atmospherically Relevant Sulfur-Centered Radicals with RO2 and HO2, J. Phys. Chem. A, 127, 2986–2991, https://doi.org/10.1021/acs.jpca.3c00558, 2023.
Chen, T. and Jang, M.: Chamber simulation of photooxidation of dimethyl sulfide and isoprene in the presence of NOx, Atmos. Chem. Phys., 12, 10257–10269, https://doi.org/10.5194/acp-12-10257-2012, 2012.
Filipy, J., Rumburg, B., Mount, G., Westberg, H., and Lamb, B.: Identification and quantification of volatile organic compounds from a dairy, Atmos. Environ., 40, 1480–1494, https://doi.org/10.1016/j.atmosenv.2005.10.048, 2006.
Fung, K. M., Heald, C. L., Kroll, J. H., Wang, S., Jo, D. S., Gettelman, A., Lu, Z., Liu, X., Zaveri, R. A., Apel, E. C., Blake, D. R., Jimenez, J.-L., Campuzano-Jost, P., Veres, P. R., Bates, T. S., Shilling, J. E., and Zawadowicz, M.: Exploring dimethyl sulfide (DMS) oxidation and implications for global aerosol radiative forcing, Atmos. Chem. Phys., 22, 1549–1573, https://doi.org/10.5194/acp-22-1549-2022, 2022.
González-García, N., González-Lafont, À., and Lluch, J. M.: Variational Transition-State Theory Study of the Dimethyl Sulfoxide (DMSO) and OH Reaction, J. Phys. Chem. A, 110, 798–808, https://doi.org/10.1021/jp054424x, 2006.
González-García, N., González-Lafont, À., and Lluch, J. M.: Methanesulfinic Acid Reaction with OH: Mechanism, Rate Constants, and Atmospheric Implications, J. Phys. Chem. A, 111, 7825–7832, https://doi.org/10.1021/jp0722455, 2007.
Goss, M. B. and Kroll, J. H.: Experiment Set: DMSO and DMDS oxidation experiments, ICARUS [data set], https://icarus.ucdavis.edu/experimentset/266 (last access: 17 November 2023), 2023.
Hodshire, A. L., Campuzano-Jost, P., Kodros, J. K., Croft, B., Nault, B. A., Schroder, J. C., Jimenez, J. L., and Pierce, J. R.: The potential role of methanesulfonic acid (MSA) in aerosol formation and growth and the associated radiative forcings, Atmos. Chem. Phys., 19, 3137–3160, https://doi.org/10.5194/acp-19-3137-2019, 2019.
Hoffmann, E. H., Tilgner, A., Schrödner, R., Bräuer, P., Wolke, R., and Herrmann, H.: An advanced modeling study on the impacts and atmospheric implications of multiphase dimethyl sulfide chemistry, P. Natl. Acad. Sci. USA, 113, 11776–11781, https://doi.org/10.1073/pnas.1606320113, 2016.
Hunter, J. F., Carrasquillo, A. J., Daumit, K. E., and Kroll, J. H.: Secondary Organic Aerosol Formation from Acyclic, Monocyclic, and Polycyclic Alkanes, Environ. Sci. Technol., 48, 10227–10234, https://doi.org/10.1021/es502674s, 2014.
Jernigan, C. M., Fite, C. H., Vereecken, L., Berkelhammer, M. B., Rollins, A. W., Rickly, P. S., Novelli, A., Taraborrelli, D., Holmes, C. D., and Bertram, T. H.: Efficient Production of Carbonyl Sulfide in the Low-NOx Oxidation of Dimethyl Sulfide, Geophys. Res. Lett., 49, e2021GL096838, https://doi.org/10.1029/2021GL096838, 2022.
Kilgour, D. B., Novak, G. A., Sauer, J. S., Moore, A. N., Dinasquet, J., Amiri, S., Franklin, E. B., Mayer, K., Winter, M., Morris, C. K., Price, T., Malfatti, F., Crocker, D. R., Lee, C., Cappa, C. D., Goldstein, A. H., Prather, K. A., and Bertram, T. H.: Marine gas-phase sulfur emissions during an induced phytoplankton bloom, Atmos. Chem. Phys., 22, 1601–1613, https://doi.org/10.5194/acp-22-1601-2022, 2022.
Kroll Group: Chamber data and species concentrations, http://krollgroup.mit.edu/publications.html (last access: 17 November 2023), 2023.
Kukui, A., Borissenko, D., Laverdet, G., and Le Bras, G.: Gas-Phase Reactions of OH Radicals with Dimethyl Sulfoxide and Methane Sulfinic Acid Using Turbulent Flow Reactor and Chemical Ionization Mass Spectrometry, J. Phys. Chem. A, 107, 5732–5742, https://doi.org/10.1021/jp0276911, 2003.
Librando, V., Tringali, G., Hjorth, J., and Coluccia, S.: OH-initiated oxidation of DMS/DMSO: reaction products at high NOx levels, Environ. Pollut., 127, 403–410, https://doi.org/10.1016/j.envpol.2003.08.003, 2004.
Lucas, D. D. and Prinn, R. G.: Mechanistic studies of dimethylsulfide oxidation products using an observationally constrained model, J. Geophys. Res., 107, 4201, https://doi.org/10.1029/2001JD000843, 2002.
Novak, G. A., Fite, C. H., Holmes, C. D., Veres, P. R., Neuman, J. A., Faloona, I., Thornton, J. A., Wolfe, G. M., Vermeuel, M. P., Jernigan, C. M., Peischl, J., Ryerson, T. B., Thompson, C. R., Bourgeois, I., Warneke, C., Gkatzelis, G. I., Coggon, M. M., Sekimoto, K., Bui, T. P., Dean-Day, J., Diskin, G. S., DiGangi, J. P., Nowak, J. B., Moore, R. H., Wiggins, E. B., Winstead, E. L., Robinson, C., Thornhill, K. L., Sanchez, K. J., Hall, S. R., Ullmann, K., Dollner, M., Weinzierl, B., Blake, D. R., and Bertram, T. H.: Rapid cloud removal of dimethyl sulfide oxidation products limits SO2 and cloud condensation nuclei production in the marine atmosphere, P. Natl. Acad. Sci. USA, 118, e2110472118, https://doi.org/10.1073/pnas.2110472118, 2021.
Nowak, J. B., Davis, D. D., Chen, G., Eisele, F. L., Mauldin, R. L., Tanner, D. J., Cantrell, C., Kosciuch, E., Bandy, A., Thornton, D., and Clarke, A.: Airborne observations of DMSO, DMS, and OH at marine tropical latitudes, Geophys. Res. Lett., 28, 2201–2204, https://doi.org/10.1029/2000GL012297, 2001.
Rosati, B., Christiansen, S., Wollesen de Jonge, R., Roldin, P., Jensen, M. M., Wang, K., Moosakutty, S. P., Thomsen, D., Salomonsen, C., Hyttinen, N., Elm, J., Feilberg, A., Glasius, M., and Bilde, M.: New Particle Formation and Growth from Dimethyl Sulfide Oxidation by Hydroxyl Radicals, ACS Earth Space Chem., 5, 801–811, https://doi.org/10.1021/acsearthspacechem.0c00333, 2021.
Rumsey, I. C., Aneja, V. P., and Lonneman, W. A.: Characterizing reduced sulfur compounds emissions from a swine concentrated animal feeding operation, Atmos. Environ., 94, 458–466, https://doi.org/10.1016/j.atmosenv.2014.05.041, 2014.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Shen, J., Scholz, W., He, X.-C., Zhou, P., Marie, G., Wang, M., Marten, R., Surdu, M., Rörup, B., Baalbaki, R., Amorim, A., Ataei, F., Bell, D. M., Bertozzi, B., Brasseur, Z., Caudillo, L., Chen, D., Chu, B., Dada, L., Duplissy, J., Finkenzeller, H., Granzin, M., Guida, R., Heinritzi, M., Hofbauer, V., Iyer, S., Kemppainen, D., Kong, W., Krechmer, J. E., Kürten, A., Lamkaddam, H., Lee, C. P., Lopez, B., Mahfouz, N. G. A., Manninen, H. E., Massabò, D., Mauldin, R. L., Mentler, B., Müller, T., Pfeifer, J., Philippov, M., Piedehierro, A. A., Roldin, P., Schobesberger, S., Simon, M., Stolzenburg, D., Tham, Y. J., Tomé, A., Umo, N. S., Wang, D., Wang, Y., Weber, S. K., Welti, A., Wollesen de Jonge, R., Wu, Y., Zauner-Wieczorek, M., Zust, F., Baltensperger, U., Curtius, J., Flagan, R. C., Hansel, A., Möhler, O., Petäjä, T., Volkamer, R., Kulmala, M., Lehtipalo, K., Rissanen, M., Kirkby, J., El-Haddad, I., Bianchi, F., Sipilä, M., Donahue, N. M., and Worsnop, D. R.: High Gas-Phase Methanesulfonic Acid Production in the OH-Initiated Oxidation of Dimethyl Sulfide at Low Temperatures, Environ. Sci. Technol., 56, 13931–13944, https://doi.org/10.1021/acs.est.2c05154, 2022.
Sørensen, S., Falbe-Hansen, H., Mangoni, M., Hjorth, J., and Jensen, N. R.: Observation of DMSO and CH3S(O)OH from the gas phase reaction between DMS and OH, J. Atmos. Chem., 24, 299–315, https://doi.org/10.1007/BF00210288, 1996.
Tian, Y., Tian, Z.-M., Wei, W.-M., He, T.-J., Chen, D.-M., and Liu, F.-C.: Ab initio study of the reaction of OH radical with methyl sulfinic acid (MSIA), Chem. Phys., 335, 133–140, https://doi.org/10.1016/j.chemphys.2007.04.009, 2007.
Trabue, S., Scoggin, K., Mitloehner, F., Li, H., Burns, R., and Xin, H.: Field sampling method for quantifying volatile sulfur compounds from animal feeding operations, Atmos. Environ., 42, 3332–3341, https://doi.org/10.1016/j.atmosenv.2007.03.016, 2008.
Tyndall, G. S. and Ravishankara, A. R.: Atmospheric oxidation of reduced sulfur species, Int. J. Chem. Kinet., 23, 483–527, https://doi.org/10.1002/kin.550230604, 1991.
Urbanski, S. P., Stickel, R. E., and Wine, P. H.: Mechanistic and Kinetic Study of the Gas-Phase Reaction of Hydroxyl Radical with Dimethyl Sulfoxide, J. Phys. Chem. A, 102, 10522–10529, https://doi.org/10.1021/jp9833911, 1998.
Van Rooy, P., Purvis-Roberts, K. L., Silva, P. J., Nee, M. J., and Cocker, D.: Characterization of secondary products formed through oxidation of reduced sulfur compounds, Atmos. Environ., 256, 118148, https://doi.org/10.1016/j.atmosenv.2020.118148, 2021a.
Van Rooy, P., Drover, R., Cress, T., Michael, C., Purvis-Roberts, K. L., Silva, P. J., Nee, M. J., and Cocker, D.: Methanesulfonic acid and sulfuric acid Aerosol Formed through oxidation of reduced sulfur compounds in a humid environment, Atmos. Environ., 261, 118504, https://doi.org/10.1016/j.atmosenv.2021.118504, 2021b.
Veres, P. R., Neuman, J. A., Bertram, T. H., Assaf, E., Wolfe, G. M., Williamson, C. J., Weinzierl, B., Tilmes, S., Thompson, C. R., Thames, A. B., Schroder, J. C., Saiz-Lopez, A., Rollins, A. W., Roberts, J. M., Price, D., Peischl, J., Nault, B. A., Møller, K. H., Miller, D. O., Meinardi, S., Li, Q., Lamarque, J.-F., Kupc, A., Kjaergaard, H. G., Kinnison, D., Jimenez, J. L., Jernigan, C. M., Hornbrook, R. S., Hills, A., Dollner, M., Day, D. A., Cuevas, C. A., Campuzano-Jost, P., Burkholder, J., Bui, T. P., Brune, W. H., Brown, S. S., Brock, C. A., Bourgeois, I., Blake, D. R., Apel, E. C., and Ryerson, T. B.: Global airborne sampling reveals a previously unobserved dimethyl sulfide oxidation mechanism in the marine atmosphere, P. Natl. Acad. Sci. USA, 117, 4505–4510, https://doi.org/10.1073/pnas.1919344117, 2020.
Vermeuel, M. P., Novak, G. A., Jernigan, C. M., and Bertram, T. H.: Diel Profile of Hydroperoxymethyl Thioformate: Evidence for Surface Deposition and Multiphase Chemistry, Environ. Sci. Technol., 54, 12521–12529, https://doi.org/10.1021/acs.est.0c04323, 2020.
von Glasow, R. and Crutzen, P. J.: Model study of multiphase DMS oxidation with a focus on halogens, Atmos. Chem. Phys., 4, 589–608, https://doi.org/10.5194/acp-4-589-2004, 2004.
Wang, N., Jorga, S. D., Pierce, J. R., Donahue, N. M., and Pandis, S. N.: Particle wall-loss correction methods in smog chamber experiments, Atmos. Meas. Tech., 11, 6577–6588, https://doi.org/10.5194/amt-11-6577-2018, 2018.
Wolfe, G. M., Marvin, M. R., Roberts, S. J., Travis, K. R., and Liao, J.: The Framework for 0-D Atmospheric Modeling (F0AM) v3.1, Geosci. Model Dev., 9, 3309–3319, https://doi.org/10.5194/gmd-9-3309-2016, 2016.
Wollesen de Jonge, R., Elm, J., Rosati, B., Christiansen, S., Hyttinen, N., Lüdemann, D., Bilde, M., and Roldin, P.: Secondary aerosol formation from dimethyl sulfide – improved mechanistic understanding based on smog chamber experiments and modelling, Atmos. Chem. Phys., 21, 9955–9976, https://doi.org/10.5194/acp-21-9955-2021, 2021.
Wu, R., Wang, S., and Wang, L.: New Mechanism for the Atmospheric Oxidation of Dimethyl Sulfide. The Importance of Intramolecular Hydrogen Shift in a CH3SCH2OO Radical, J. Phys. Chem. A, 119, 112–117, https://doi.org/10.1021/jp511616j, 2015.
Ye, Q., Goss, M. B., Isaacman-VanWertz, G., Zaytsev, A., Massoli, P., Lim, C., Croteau, P., Canagaratna, M., Knopf, D. A., Keutsch, F. N., Heald, C. L., and Kroll, J. H.: Organic Sulfur Products and Peroxy Radical Isomerization in the OH Oxidation of Dimethyl Sulfide, ACS Earth Space Chem., 5, 2013–2020, https://doi.org/10.1021/acsearthspacechem.1c00108, 2021.
Ye, Q., Goss, M. B., Krechmer, J. E., Majluf, F., Zaytsev, A., Li, Y., Roscioli, J. R., Canagaratna, M., Keutsch, F. N., Heald, C. L., and Kroll, J. H.: Product distribution, kinetics, and aerosol formation from the OH oxidation of dimethyl sulfide under different RO2 regimes, Atmos. Chem. Phys., 22, 16003–16015, https://doi.org/10.5194/acp-22-16003-2022, 2022.
Yin, F., Grosjean, D., and Seinfeld, J. H.: Photooxidation of dimethyl sulfide and dimethyl disulfide. I: Mechanism development, J. Atmos. Chem., 11, 309–364, https://doi.org/10.1007/BF00053780, 1990a.
Yin, F., Grosjean, D., Flagan, R. C., and Seinfeld, J. H.: Photooxidation of dimethyl sulfide and dimethyl disulfide. II: Mechanism evaluation, J. Atmos. Chem., 11, 365–399, https://doi.org/10.1007/BF00053781, 1990b.
Zaytsev, A., Breitenlechner, M., Koss, A. R., Lim, C. Y., Rowe, J. C., Kroll, J. H., and Keutsch, F. N.: Using collision-induced dissociation to constrain sensitivity of ammonia chemical ionization mass spectrometry (NH CIMS) to oxygenated volatile organic compounds, Atmos. Meas. Tech., 12, 1861–1870, https://doi.org/10.5194/amt-12-1861-2019, 2019.
The requested paper has a corresponding corrigendum published. Please read the corrigendum first before downloading the article.
- Article
(2697 KB) - Full-text XML
- Corrigendum
-
Supplement
(5894 KB) - BibTeX
- EndNote