the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 21 Oct 2024
| 21 Oct 2024
Ether and ester formation from peroxy radical recombination: a qualitative reaction channel analysis
Marie Camredon
Richard Valorso
Bernard Aumont
Theo Kurtén
The least volatile organic compounds participating in atmospheric new-particle formation are very likely accretion products from self- and cross-reactions of peroxy radicals (RO2). It has long been assumed that the only possible accretion product channel in this reaction is that forming a peroxide (), but it has recently been discovered that a rapid alkoxy radical (RO) decomposition may precede the accretion step of the mechanism, forming slightly fragmented but more stable ether (ROR) or ester (RC′(O)OR) accretion products. In this work, the atmospheric implications of this new reaction channel have been explored further by using a modified version of the Generator for Explicit Chemistry and Kinetics of Organics in the Atmosphere (GECKO-A) software to generate a large amount of representative RO2 + RO2 reactive pairs formed from the oxidation of typical primary hydrocarbons and by applying structure–activity relationships (SARs) to predict the potential accretion products. These data are analysed in terms of the formation of low-volatility products, and new discoveries are presented on what types of RO2 are especially efficient (and which are surprisingly inefficient) at forming accretion products. These findings are discussed in terms of the atmospheric relevance of these new RO2 + RO2 reaction channels. As the generation of these data rests on several simplifications and assumptions, many open questions worthy of later studies are also raised.
- Article
(5378 KB) - Full-text XML
-
Supplement
(2184 KB) - BibTeX
- EndNote





1.1 Atmospheric background
The formation and growth of secondary organic aerosol (SOA) particles in pristine environments is dependent on the gas-phase formation of low-volatility organic molecules, but the exact formation pathways of such organics is only partially known (Kanakidou et al., 2005). The self- and cross-reactions of peroxy radicals (RO2) are assumed to be important sources of such low-volatility molecules (Berndt et al., 2015), as this is one of the rare cases of gas-phase atmospheric reactions where accretion products, products with a larger carbon count than the reactant radicals, can form. The previously known accretion-product-forming pathway of these recombination reactions is a peroxide connecting the carbon skeletons of the reactant RO2 in the reaction . This is, however, not the only possible product of this reaction, and we will therefore briefly review the history of studies on this reaction. The mechanism of RO2 recombination has been known to go through an unstable tetroxide intermediate since the proposed Russell mechanism for its decomposition (Russell, 1957). However, the currently accepted mechanism for the reaction was presented by Ingold (1969), in which the tetroxide ejects an O2 molecule, leaving behind a triplet-state bimolecular complex of two alkoxy radicals, 3(RO…OR):
Recent theoretical studies indicate that, in the case of CH3O2 + CH3O2 recombination, the ejection of one CH3O radical from the (CH3O…O2…OCH3) complex is thermodynamically (but not necessarily kinetically) favourable to that of O2 (Salo et al., 2024). Recent experimental studies also suggest a novel in-complex reaction channel from the self-reaction of the HOC2H4O2 radical (Murphy et al., 2023). Here, however, we will operate under the assumption that these reactions are only possible for very weakly bound systems and that channel (R1) is exclusively the fate of generic RO2 + RO2 reactions. In the mechanism proposed by Ingold (1969), the 3(RO…OR) complex has three further reaction channels: a dissociation pathway (Reaction R2) into two free alkoxy radicals (RO), an intermolecular H-shift (Reaction R3) forming an alcohol and a carbonyl, and an intersystem crossing (ISC) into the singlet state followed by barrierless recombination into a peroxide accretion product (Reaction R4, ROOR). The branching ratios for these three pathways have been studied both experimentally (Orlando and Tyndall, 2012) and computationally (Hasan, 2023). In our previous work (Peräkylä et al., 2023), we found an unexpected fourth reaction channel for an α-pinene-derived 3(C10H15O2O…OC10H15O2) complex, in which one of the alkoxy radicals undergoes a rapid β-scission reaction, thereby producing a C19H28O5 ester accretion product. Assuming these kinds of reactions are possible for other systems as well, we may update the full mechanism of peroxy radical recombination to include the following four pathways:
where R′ denotes that the original organic R group may be fragmented, in which case P is the second fragmentation product (CH2O is the case of the previously mentioned C19H28O5 ester product). The computational results provided by Peräkylä et al. (2023) suggest that the occurrence of channel (R5) depends on the unimolecular decomposition rate of the RO outspeeding reaction channels (R2), (R3), and (R4), on which we already have produced a body of computational work. The rate of the dissociation channel is primarily predicted by the binding energy of the 3(RO…OR) complex, ranging from 2⋅101 to . (Franzon, 2023) Dissociation is therefore likely to be uncompetitive for larger, more functionalized, and thus more strongly binding pairs of RO. An equally unambiguous trend for the H-shift rate has proved difficult to determine (Hasan et al., 2023), but the computed H-shift rates show a weak negative correlation with the binding energy, suggesting that this pathway is also less competitive for larger and more functionalized radicals. These H-shift rates are rarely above 109 s−1. It is also notable that only primary and secondary RO can act as H donors, meaning that this channel does not exist at all if both of the reacting RO2 are either acyl peroxy radicals (RC(O)O2) or tertiary RO2. The ISC rate seems to be systematically on the order of 109 s−1, with the 3(CH3O…OCH3) complex and some stereoisomers of ONO2-substituted RO acting as outliers (Hasan, 2023). In summary, if reaction channels of type (R5) are of any significance, they must outspeed the ISC rate if the 3(RO…OR) is strongly binding and outspeed the dissociation and bimolecular H-shift rates if the complex is weakly binding. Based on the information above, it seems that the RO decay rate must be close to 108 s−1 to be of any importance and preferably above 109 s−1 to be a main channel.
The in-complex RO decomposition we observed (Peräkylä et al., 2023) was a β-scission reaction forming an acyl-centred radical combining with the other RO to form an ester. Similar reactions forming alkyl radicals as decomposition products will presumably result in ether accretion products. Decay rates of RO radicals inside complexes are currently unknown, but, as a first approximation, we may estimate them using the corresponding reaction rates of free RO. This is convenient, as the atmospheric reactions of alkoxy radicals have already been studied widely. The structure–activity relationship (SAR) by Vereecken and Peeters (2009) and Novelli et al. (2021) for β-scission reactions of free RO suggests that there are multiple chemical structures that reach the approximate threshold of 108 s−1, suggesting that this channel may be quite common. We also ought to entertain the possibility that β-scission reactions are not the only kind of unimolecular RO reaction which may occur in-complex. A review on the atmospheric chemistry of alkoxy radicals by Orlando et al. (2003) discussed three additional reaction classes for (non-halogenated) RO: unimolecular H-shifts, ester rearrangement, and O2 addition. The ranges of known reaction rates for each of these are shown in Fig. 1. Upon closer examination, however, only the unimolecular H-shift pathway is reportedly fast enough to cross our importance threshold of 108 s−1 in some known cases, as the ester rearrangement and O2 addition pathways are both limited to (pseudo-unimolecular in the latter case) rates of around 105 s−1. To avoid confusion between H-shift reactions of type (R3) and type (R5), we refer to the former as bimolecular H-shifts and the latter as unimolecular H-shifts.
In this work, we aim to explore the wider atmospheric significance of channel (R5) using an RO2 + RO2 accretion product generator code based on the Generator for Explicit Chemistry and Kinetics of Organics in the Atmosphere (GECKO-A; Aumont et al., 2005) software, which we call GECKO-AP (… of Organic Accretion Products). GECKO-AP makes use of all the RO2 and RO chemistry already included in GECKO-A, combined with simple parametrizations of the knowledge we currently have of the competing reaction channels to generate datasets of all the possible peroxide, ether, and ester accretion products derived from a GECKO-A-generated set of RO2. These datasets of accretion products are then analysed in order to learn which of these reactions are most important, especially when it comes to the formation of low-volatility products. The purpose of this article is not to determine accurate branching ratios for these channels, as this most likely requires both characterization of further ester or ether product from experiments and extensive computations benchmarked against such experiments. Instead, the purpose of this work is to inform future efforts on where to start looking and to assess general qualitative features of accretion products formed by the recently discovered pathways.
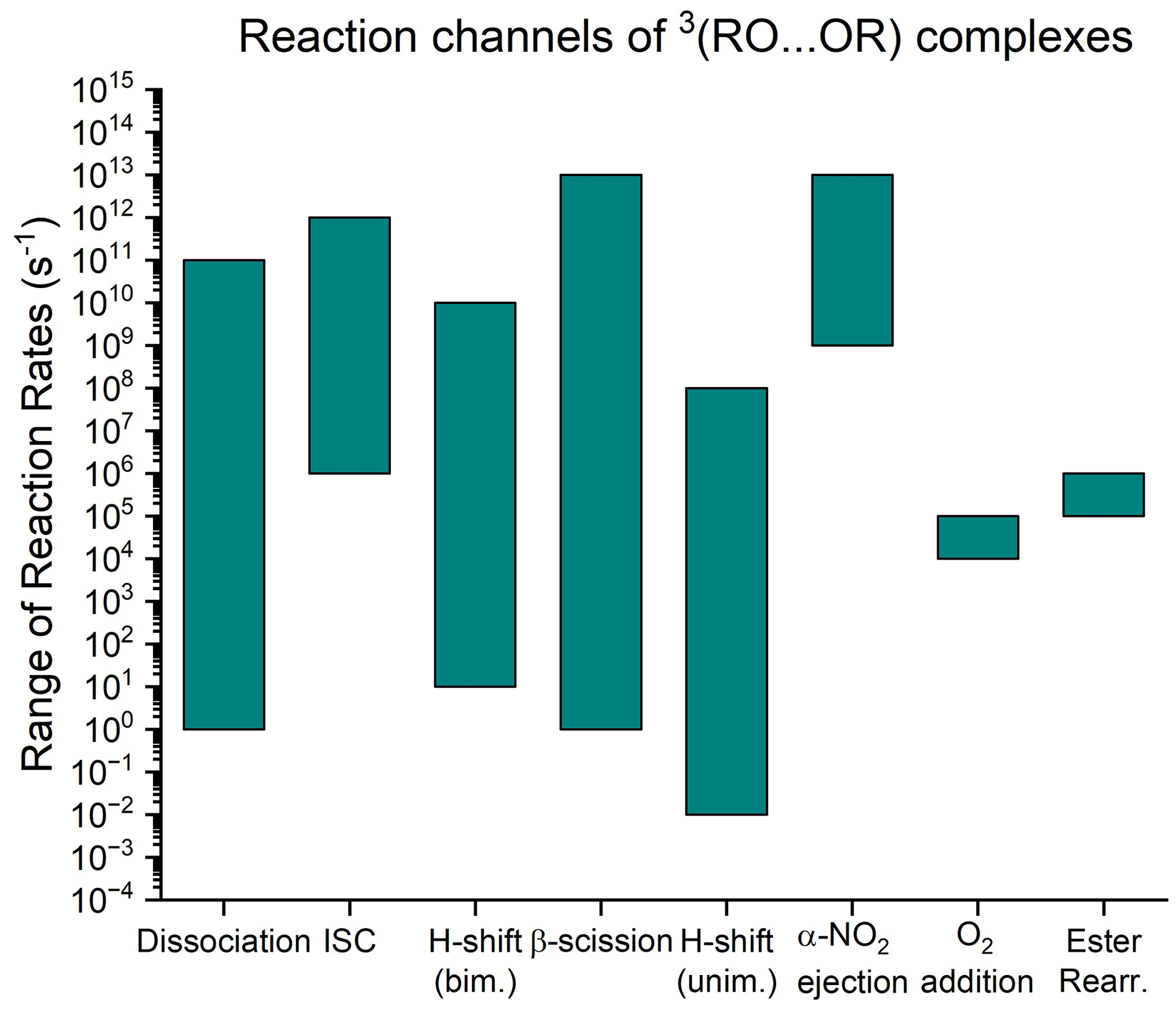
Figure 1A visualization of the range of rates of the three known reaction channels of 3(RO…OR) complexes compared to the unimolecular reaction rates of free RO radicals considered in this work. The range of dissociation rates is from Franzon (2023), the ISC and bimolecular H-shifts are from Hasan (2023), the β-scission and unimolecular H-shift rates are from Vereecken and Peeters (2009) and Vereecken and Peeters (2010), and the O2 addition and α-ester rearrangement are from Orlando et al. (2003). The α-NO2 ejection is discussed in Sect. 2.1.4.
1.2 GECKO-A
In order to place the importance of Reaction (R5) in a wider atmospheric context, a large selection of atmospherically relevant peroxy radicals must be screened to determine the cases where these in-complex decomposition reactions are competitive. For this purpose, GECKO-A was used to generate large numbers of atmospherically relevant RO2. As GECKO-A only generates the products from channels (R2) and (R3) for RO2 recombination reactions according to the parametrization presented in Jenkin et al. (2019), a new code was written to generate all the possible accretion products (both channels R4 and R5) from each individual pair of RO2. The structure of the code is described in detail in Sect. 2.1. Using this code, three large accretion product datasets from representative precursor molecules were produced, which are presented in Sect. 2.2 and analysed in detail in Sect. 3 to determine two things: what the existence of reaction channel (R5) implies for the formation of low-volatility products from RO2 recombination and which aspects of this channel are worth studying in more detail.
The GECKO-A tool generates explicit atmospheric chemical mechanisms from a list of organic precursors provided as input. The chemistry included in the version of GECKO-A used to generate the data was VOC oxidation by OH (Jenkin et al., 2018a, b), alkene oxidation by O3 (Jenkin et al., 2020), VOC oxidation by NO3 (Kerdouci et al., 2014), photolysis for some select chromophores (Aumont et al., 2005), thermal decomposition of peroxy acyl nitrates (−C(O)OONO2) (Jenkin et al., 2019), RO decomposition (Vereecken and Peeters, 2009), (mostly bimolecular) reactions of RO2 (Jenkin et al., 2019), and stabilized Criegee intermediate chemistry (Newland et al., 2022). All generated molecules are grouped by generation based on the minimum number of stable closed-shell species that are produced in the formation of that molecule. For example, CH2OHCH2OOH and CH2(ONO2)CH2OOH are first-generation products of ethene. The molecules produced from the oxidation of these (including the radicals) are second-generation products, and so on. The GECKO-A molecule generator creates all the oxidation pathways up to a specified generation n and above a specified critical saturation vapour pressure pc (Aumont et al., 2005). In this study, the generated chemical mechanisms were used to provide lists of peroxy radicals expected to be representative of typical atmospheric RO2 in their structure but also in their probability to be formed. GECKO-A also calculates a theoretical maximum yield of formation parameter y, which is used to filter out minor reaction channels in the mechanism generation. As the mechanism generation code makes minimal assumptions on environmental conditions such as relative humidity and bimolecular reactant (such as OH, O3, NO3, HO2, NO, NO2, and RO2) concentrations, this parameter does not account for competition between different bimolecular reactions. The maximum yield is therefore “theoretical” in the sense that the y value of (for example) isoprene + NO3 products corresponds to real yields in conditions where isoprene oxidation is completely dominated by NO3. This means the y values have the disadvantage of occasionally over-representing products with high yields from slow reactions, but, from the perspective of our qualitative RO2 + RO2 reaction channel analysis, the environmental independence is an advantage, as it allows us to treat all hypothetically possible atmospheric RO2 + RO2 reactions uniformly without the loss of generality. The y values of the RO2 generated by GECKO-A were thus used as a filter with which to select RO2 + RO2 pairs for further treatment. A comparison of theoretical maximum yields with simulated formation rates and concentrations is performed in Sect. S8.
2.1 Generation of accretion product datasets
As the scope of this work is limited to exploring the potential accretion products, the GECKO-AP code was built to simply generate a list of RO2 + RO2 accretion products from a list of RO2 generated in the GECKO-A mechanism. In this section, the process of creating accretion product datasets from an RO2 list is described in detail. As the potential number of pairings increases combinatorically with the length of the RO2 list (), care was taken to efficiently filter out all of the least important radicals (Sect. 2.1.1), least probable RO2 + RO2 pairs (Sect. 2.1.2 and 2.1.3), and least competitive product channels (Sect. 2.1.5) in the code.
2.1.1 Filtering of individual RO2
The list of RO2 radicals generated in a GECKO-A mechanism was used as input for the GECKO-AP code, with some filtering done to reduce the number of products. Firstly, a yield cutoff yc was chosen such that all RO2 with theoretical maximum yield y<yc were filtered out. Secondly, CH3O2 was systematically left out as a rule from these lists, as all of our previously studied 3(CH3O…OR) systems have had rapid dissociation (Franzon, 2023) and bimolecular H-shift (Hasan et al., 2023) rates. We thus suspect that CH3O2 + RO2 reactions are not a significant source of accretion products in the atmosphere and most certainly are not a source of large low-volatility accretion products. Thirdly, RC(O)O2s with hydroperoxide (OOH) substituents was also excluded, as the rapid H-scrambling reaction into peracid-substituted alkyl peroxy radicals is expected to be effectively irreversible (Knap and Jørgensen, 2017). As GECKO-A currently lacks RO2 H-shift and H-scrambling reactions, a simple exclusion of these radicals from partaking in RO2 + RO2 was seen as a suitable correction.
2.1.2 Filtering of RO2 pairs by probability
Another method of filtering out the least important data is to only treat the RO2 + RO2 pairs above a certain probability threshold, which in the GECKO-AP code is formulated in terms of RO2 + RO2 reaction kinetics. The formation rate of a 3(RO…OR) complex is
Here, [α] and [β] are the concentrations of the two reacting peroxy radicals, and is the recombination rate coefficient. Since the best estimation we have of individual RO2 concentrations in the GECKO-A mechanism generator is the theoretical maximum yield y, we estimate an equivalent recombination yield using these:
where yα and yβ are the theoretical maximum yields of the RO2 α and β. Since a yield must be between 0 and 1 by definition, the rate coefficient krel must also be expressed relative to some maximum, for which we use the RO2 + RO2 rate coefficient for the GECKO-A RO2 class 8 (see Table 3): . The RO2 + RO2 rate coefficient is determined using a slight simplification of the scheme described in the Supplement of Jenkin et al. (2019), where the individually calculated self-reaction rate coefficient is replaced by one of the nine GECKO-A RO2 class rates (Table 1). This way, krel can efficiently be calculated for large numbers of RO2 pairs without using up memory for self-reaction rates. As suggested by Jenkin et al. (2019), cross-reactions between RC(O)O2 and other RO2 are always treated as collision-limited, whereas other rates are determined using a geometric mean of the two self-reaction rates, with an additional factor of 2 for reactions between tertiary and primary or secondary RO2 ().
Table 1The nine RO2 + RO2 rate classes used in GECKO-A, adapted from Jenkin et al. (2019).

For reactions between RC(O)O2 and other radicals, two additional correction factors are used to scale down the yields. Firstly, as GECKO-A lacks unimolecular H-shift (autoxidation) reactions for peroxy radicals, the recombination yields of RC(O)O2 other than CH3C(O)O2 were scaled down based on a uniform autoxidation sink of 1 s−1. Secondly, we must also consider that bimolecular reactions between RC(O)O2 and common atmospheric inorganic radicals (OH, HO2, NOx) are generally faster than the corresponding reactions for alkyl RO2, which means that the relationship between the faster recombination rates of RC(O)O2 and the resulting recombination yields is not directly comparable. Thus, we apply a “rate-to-yield factor” of 0.56 to RO2 + RO2 reactions involving acyl peroxy radicals based on a quick derivation of the relationship between the rates and the yield. More details are given in Sect. S2. To summarize, we may rewrite Eq. (2) in the form used in the GECKO-AP code:
where k8 is the RO2 + RO2 rate coefficient for rate class 8 and f9 is the correction applied to RO2+RC(O)O2 reactions, which is 1 if neither α or β is RC(O)O2, 0.56 if either is CH3C(O)O2, and 0.035 if either is some other RC(O)O2 (see Supplement). To filter out less probable RO2 pairs, a cutoff yield yc is defined below which the RO2 + RO2 pair is not considered. The same value is used to filter out individual RO2, as yα<yc directly implies yαβ<yc.
2.1.3 Filtering of RO2 pairs with rapid dissociation
As we are primarily interested in the accretion-product-forming channels (R4) and (R5), it also makes sense to filter out RO2 pairs for which the branching ratios of channels (R2) and (R3) can be presumed to be high. As experimental data on these branching ratios are still relatively scarce, we are not able to create reliable SAR calculators for the rates of channels (R2) and (R3), let alone branching ratios. However, the results from our previous computational studies (Franzon, 2023; Hasan, 2023) indicate that both of these rates are negatively correlated with the binding energy of the 3(RO…OR) complex. It has been suggested elsewhere (Peräkylä et al., 2023) that the ability of the RO to form intermolecular H-bonds is key to suppressing these two channels, especially channel (R2), and this might offer us a viable approach to filter out RO2 pairs with weakly bound 3(RO…OR) complexes using only the information available to the mechanism generator. For this purpose, −OH, −OOH, −C(O)OH, and −C(O)OOH were treated as H-bond-donating groups, whereas −CHO, −C=O−, −C(O)OH, −C(O)OOH, −NO2, −ONO2, −OONO2, −C(O)OONO2, and the radical oxygen were treated as H-bond-accepting groups. In addition, every C–H bond (aliphatic or aromatic) was treated as a partial H-bond donor, as these may stabilize larger 3(RO…OR) complexes in the presence of H-bond acceptors, as noted in the Supplement of Peräkylä et al. (2023). Using these parameters, an effective H-bond number (HBN) was calculated for each pair of alkoxy radicals:
where D and A refer to donor and acceptor, respectively, and α and β refer to the two RO in the complex. Regarding the specific form of the equation, we emphasize that the purpose of the equation is to mimic the observed trends in 3(RO…OR) binding energies, which do not exclusively depend on the ability to form H-bonds but also on dipole–dipole bonds, dispersion interactions, and Pauli repulsion of the two radical oxygens. Out of these secondary interactions, the dipole–dipole bonding largely depends on the presence of the same functional groups as the H-bonding interactions, at least in the context of gas-phase organic chemistry. This means that we were able to obtain a good agreement (a correlation coefficient of ≈0.92) between the HBNα,β value and the binding energies of 3(RO…OR) complexes presented by Hasan (2023) by adjusting the “partial H-bond donor” value assigned to each C–H bond. The optimal value chosen used in the code was 0.04. Based on the dissociation rates calculated using these binding energies (Franzon, 2023), an HBN cutoff of 1.75 was chosen, below which accretion products are not generated for that specific RO2 pair. The full analysis of how this cutoff was chosen is described in Sect. S1.
2.1.4 In-complex RO reactions
For all RO2 in the input list, a systematic search of decomposition reactions is performed for the corresponding RO. The approach for finding the reactions is very similar to that using the ordinary GECKO-A code (Aumont et al., 2005). As discussed in the Introduction, the following three reaction classes were judged to potentially be competitive in-complex:
-
β-scission, which turns the alkoxy moiety into a carbonyl and the site of the broken bond into an alkyl radical. The search is performed in a similar way to in the base GECKO-A code for free alkoxy radicals, relying mainly on the structure–activity relationship (SAR) of Vereecken and Peeters (Vereecken and Peeters, 2009; Novelli et al., 2021).
-
A unimolecular H-shift to the alkoxy oxygen resulting in the formation of an alkyl radical with one additional OH substituent. This relies mainly on the SAR of Vereecken and Peeters (Vereecken and Peeters, 2010).
-
α-NO2 ejection, resulting in a carbonyl and an NO2 radical. This reaction is set to an arbitrarily high () rate in GECKO-A to conveniently get rid of these compounds. As this reaction happens to have interesting implications for accretion product formation (See Sect. 3.3), computational rate coefficients were calculated for a set of small representative compounds using the ORCA software (Neese, 2022). According to these calculations, the reaction will typically have rates closer to the range, which is still highly competitive in-complex. Computational details are found in Sect. S3.
The rates of all reactions for each individual RO are compared, and all channels found to be competitive enough to at least be minor products (using a branching ratio cutoff of 0.05) next to an assumed universal ISC rate of 109 s−1 are considered for in-complex reaction branching ratio calculations.
For every reaction, the stability of the radical product is checked by running through a list of barrierless decomposition reactions. This was done utilizing a version of the code described in Sect. 3.1 of Aumont et al. (2005), with a shorter list of “immediate” reactions to account for what kind of reactions might actually occur inside the complex. The code starts by identifying if a radical is delocalized or not, and it implements the following list of reactions for either the non-delocalized radical or both Lewis structures of the delocalized radical. All three reactions are barrierless according to Vereecken et al. (2004) and Vereecken (2008).
- 1.
. Barrierless decomposition of peroxides at the radical centre.
- 2.
. Barrierless decomposition of α-hydroperoxy alkyl radicals into a carbonyl and an OH radical.
- 3.
. Barrierless decomposition of α-nitrate alkyl radicals into a carbonyl and a nitro radical.
These may not be the only further decomposition reactions that are possible for the product radicals of in-complex RO decomposition reactions. Low-barrier reactions, such as decomposition of acyl oxy radicals into alkyl radicals and carbon dioxide (; Vereecken and Peeters, 2009) and decomposition of diacyl radicals into an acyl radical and carbon monoxide (; Méreau et al., 2001), were also considered but ultimately not included, as these reactions are not barrierless and may not occur after an endothermic (Orlando et al., 2003) RO decomposition. These downstream decomposition reactions must likely be barrierless to efficiently compete with the association of the radical product and the other RO in the 3(RO…OR) complex.
A separate output is written for all the RO reactions that are considered for in-complex branching to help keep track of the accretion products.
2.1.5 Filtering of accretion products
As discussed in Sect. 1.1, we currently assume that the RO decomposition reaction is generally followed by recombination of the two radicals into an ether or ester. In the Supplement of Peräkylä et al. (2023), this assumption was tested for a small model system, where it turned out that this recombination had a high-energy barrier in the triplet state and thus required an ISC. However, this ISC was faster than the ISC of the 3(RO…OR) complex, being on the order of 1011 s−1. While a fast dissociation of the two radicals post-RO decomposition cannot entirely be ruled out in reality, it is reasonable to assume that the dissociation of the product radical and the remaining RO is less competitive than the dissociation of the two RO in the 3(RO…OR) complex, especially as RO β-scission reactions are typically endothermic. The GECKO-AP code thus operates on the assumption that the RO decomposition always leads to recombination of the radical product with the second RO in the complex.
The final part of the code cycles through all pairs of RO2 not filtered out by either the probability or the HBN criteria. Branching ratios for all available reaction routes are considered by comparing the reaction rates of both RO and the ISC rate, with the latter assumed to be 109 s−1 in all cases. Reaction routes are filtered by two criteria: (1) branching ratio and (2) final yield, which is a combination of recombination yield and branching ratio. Low branching ratios are filtered out using the branching ratio cutoff 0.05, with more tolerance for channels with higher rate uncertainty. The uncertainty factors f used were 1 for β-scissions, 5 for H-shifts, and 50 for spin-flips. The first two of these factors were chosen based on the reported uncertainties in the SARs for these reaction classes: a factor of 2 for the β-scissions (Vereecken and Peeters, 2009), and a factor of 10 for the fastest H-shifts (Vereecken and Peeters, 2010). The relative uncertainty of the ISC rates was determined from the variance in the available computational ISC rates for 3(RO…OR) rates (Hasan, 2023).
In other words, channels were filtered out if
where nX is the number of unimolecular reaction channels found for the RO X, whereas ki and kj are the rates of said channels. Note that the rates of channels (R2) and (R3) are neglected by necessity, as we lack a simple way to estimate them with the information available to the code. As a final criterion, Eqs. (2) and (6) are combined into a single inequality with an additional factor of 10:
where yc is the same cutoff used to filter RO2 pairs by recombination probability. This value was adjusted between the three datasets, as seen in the next section. The role of the factor of 10 is to ensure that minor channels are more heavily filtered out for less likely pairs of RO2. For the reaction channels that pass all filters, the molecular structure, the molecular mass, and the saturation vapour pressure are printed out in the output using two different group additivity methods, SIMPOL (Pankow and Asher, 2008) and Nannoolal (Nannoolal et al., 2004, 2008) (the latter with Compernolle's additional −OOH and −C(O)OOH parameters; Compernolle et al., 2010).
As an additional note on the Nannoolal vapour pressures, a previous computational study on the vapour pressures of large ROOR-type accretion products (Kurtén et al., 2016) suggested that the Nannoolal model produces strange results when applied to these molecules. A comparison was performed on the vapour pressures presented in that study with those predicted by the above implementation of the Nannoolal model (see Sect. S4). Based on this comparison, we conclude that the error is likely in the UManSysProp (Topping, 2015) implementation of Nannoolal utilized by Kurtén et al. (2016), not in the model itself.
2.2 Data generation and curation
2.2.1 Presentation of the datasets
Three datasets of accretion products with different precursor molecules were produced in order to analyse the most important trends in varying atmospheric conditions. For all runs, a critical vapour pressure value of atm was used, meaning that further gas-phase chemistry was not generated for closed-shell molecules with pSat<pc. The maximum generation of oxidation products and the cutoff value for were adjusted for each run to ensure that the datasets were kept at a manageable size and that low-yield downstream products would not be overrepresented in the data. A sensitivity analysis of the yc parameter is found in Sect. S5. These three datasets are presented below:
- 1.
The DTA dataset, including the accretion products produced in the atmospheric oxidation of n-decane, toluene, and α-pinene. The oxidation products generated by GECKO-A for this set of precursor molecules have been studied in detail before (Isaacman-VanWertz and Aumont, 2021; Besel et al., 2023), so a dataset made out of oxidation products previously missing from GECKO-A is a good addition. Accretion products were generated up to the fourth generation, and the yield cutoff yc=0.0045 was used.
- 2.
The terpene dataset, including the accretion products produced in the atmospheric oxidation of isoprene, α-pinene, β-pinene, limonene, β-ocimene, sabinene, Δ-3-carene, and myrcene. These are the eight most common terpene molecules (Sindelarova et al., 2014). A dataset composed of all the RO2 + RO2 cross-products from these precursors should represent accretion product formation occurring in pristine low-NOx forest environments reasonably well. Accretion products were generated up to the second generation, and the yield cutoff yc=0.003 was used.
- 3.
The caryophyllene dataset, including the accretion products from β-caryophyllene, a sesquiterpene for which aerosol particle formation has recently been studied (Dada et al., 2023). Here, products were only generated up to the first generation, as some of the second-generation accretion products proved unmanageably complex for the mechanism generator. The yield cutoff yc=0.001 was used.
The isomer switching code presented by Valorso et al. (2011) was turned off during the runs to ensure the traceability of the RO2 formation mechanisms. RO2+OH reactions were also turned off when generating these datasets, as this leads to the formation of hydrotrioxides (ROOOH) in the current parametrization of GECKO-A. The chemistry of these molecules is not well-known, but, based on known decomposition and OH oxidation rate coefficients for CH3OOOH and isoprene-derived ROOOH (Assaf et al., 2018; Anglada and Solé, 2018; Berndt et al., 2022), we are not able to completely neglect other OH or O3 oxidation pathways of hydrotrioxides, both of which we lack systematic data for. Fortunately, with a typical atmospheric OH concentration of (Wayne, 2000), the RO2+OH channel will often be outcompeted by the other channels, so ignoring them is not a massive loss in chemical accuracy.
2.2.2 Dataset curation
Additional GECKO-A mechanisms without the GECKO-AP code were generated for each set of precursor molecules to supply the datasets with additional metadata. Six mechanisms were generated with a limited set of VOC oxidants (only OH, only O3, only NO3, OH + O3, OH + NO3, and O3 + NO3). Each RO2 in the accretion product datasets was assigned a required combination of atmospheric oxidants based on the RO2 list resulting from these mechanisms. Similarly, for the DTA and terpene datasets, a GECKO-A mechanism was generated for each product generation leading up to the final one to assign a generation to each of the RO2 in the dataset. Finally, for each precursor molecule in the DTA and terpene datasets, a mechanism was generated with only one precursor in to label which of the precursors each of the RO2 is derived from. Based on this RO2 labelling, a final probability-based filtering criterion was applied to all datasets, as OH and NO3 are known to rarely have simultaneously high concentrations: the former is produced by photolysis of O3, while the latter is decomposed by photolysis (Seinfeld and Pandis, 2016). For this reason, the cross-reactions of OH-derived RO2 and NO3-derived RO2, along with all their products, were removed from the data. Care was taken not to accidentally remove radicals produced from both oxidation mechanisms (for example, products of H-abstraction). By the same logic, RO2 with formation mechanisms requiring OH oxidation of a VOC in one generation and NO3 oxidation in the next (or vice versa) was also removed. Admittedly, these mechanisms are more plausible than cross-reactions of OH-derived RO2 and NO3-derived RO2 if the lifetimes of the first-generation closed-shell products exceed the diurnal cycle; nevertheless, it is likely that the GECKO-A mechanism generator overestimates the yields of these radicals relative to all other RO2.
For the purpose of data analysis, four versions of the final data were created for each dataset. The first contains all the reaction channels found by the code, including those removed by the OH + NO3 criteria. These data are not analysed separately in this work, but they are distributed as a reference. In the second version of data, the OH + NO3 products have been removed, but nothing else has. This version is analysed in Sect. 3.3. In the third version, all of the non-accretion products are removed (analysed in Sect. 3.4), and, in the fourth version, all the duplicate accretion products are removed. This last version is only used for the figures in Sects. 3.2 and S6, but the existence of such uniqueness-filtered datasets was regarded as useful for potential follow-up studies, such as the analysis performed by Besel et al. (2023) for the data presented by Isaacman-VanWertz and Aumont (2021).
2.3 Estimation of atmospheric concentrations
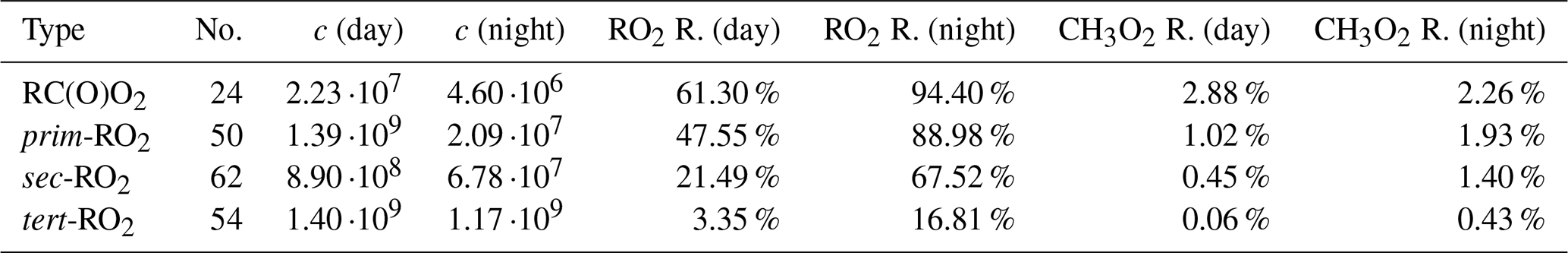
In order to explore the impact of accretion product formation from RO2 + RO2 reactions, RO2 concentrations were calculated for representative conditions of biogenic environments where the RO2 + RO2 reactions are expected to be relatively important. RO2 concentrations were calculated from the numerical solution of the kinetic steady-state equations (Eq. 8) based on the reactions in the GECKO-A mechanism. Furthermore, the mechanism was limited to isoprene oxidation and includes only the first- and second-generation radicals (7581 reactions). Firstly, however, a representative set of bimolecular reactant concentrations was chosen based on balancing a simpler set of steady-state equations, with only CH4, CO, CH2O, and CH3OH present as organic precursors. The criteria used to find the ideal RO2 + RO2 conditions were based on the GEOS-Chem-modelled probability distributions presented by Kenagy et al. (2024), according to which a value of 0.2 corresponds to both an ratio well above 1 and a not-insignificant probability density. Assuming the RO2+NO and RO2+HO2 rates are approximately similar for typical RO2, we thus balanced the steady-state equations for OH, O3, HO2, NO, NO2, and NO3, aiming for a ratio of 0.2.
As the probability distribution of Kenagy et al. (2024) was weighted by isoprene + OH, it likely over-represents daytime conditions, as both isoprene and OH concentrations peak during the day (Wennberg et al., 2018). We thus re-balanced the steady-state equations with no photolysis reactions present, using the diurnal cycles presented by Bey et al. (1997) as reference. The resulting day- and nighttime concentrations of OH, O3, HO2, NO, NO2, and NO3 were treated as constants in the steady-state equations for the isoprene-RO2. A (constant) isoprene concentration of was used for daytime, and was used for nighttime. The temperature T=298 K was used for both. Full details of how the steady-state equations were solved are found in Sect. S8.
3.1 Competitive decomposition channels
Firstly, we use the β-scission and H-shift SARs (Vereecken and Peeters, 2009, 2010) to assess which of the reaction channels described in Sect. 2.1.4 are fast enough to occur in 3(RO…OR) complexes.
Assuming a constant ISC rate of 109 s−1, the β-scission rate must presumably be at least 108 s−1 in order for the product yield to be non-negligible. With Vereecken's suggested constant A-factor of , an activation barrier below 30 kJ mol−1 is needed to reach this rate at 298 K, which means 44.9 kJ mol−1 worth of activating factors from the SAR's base value of 74.9 kJ mol−1. The only activating factors to reach this barrier reduction single-handedly are the α-O group (present in all RC(O)O2+RO2 reactions), the β-NO group (not present in our data), and the opening of three- and four-membered rings (relevant for both pinenes, sabinene, and Δ-3-carene-derived RO2). Other activating factors that are frequently present in our generated RO2 are β (leading to the formation of ester accretion products), β−OOH, β−OH, β, and the opening of five- and six-membered rings. In summary, there is no shortage of chemical structures present in atmospheric RO2 that lead to competitive RO β-scission reactions. Interestingly, all of the most competitive β-scissions seem to be of the ring-opening type, which may partially explain why these reactions have eluded detection for so long. Exocyclic β-scissions have two products, whereas endocyclic scissions have one. Only the former is distinguishable from the ROOR in the mass spectrometric measurements which have thus far been the detection method of choice for highly oxidized organics in atmospheric chemistry (Ehn et al., 2014; Rissanen et al., 2014; Bianchi et al., 2019).
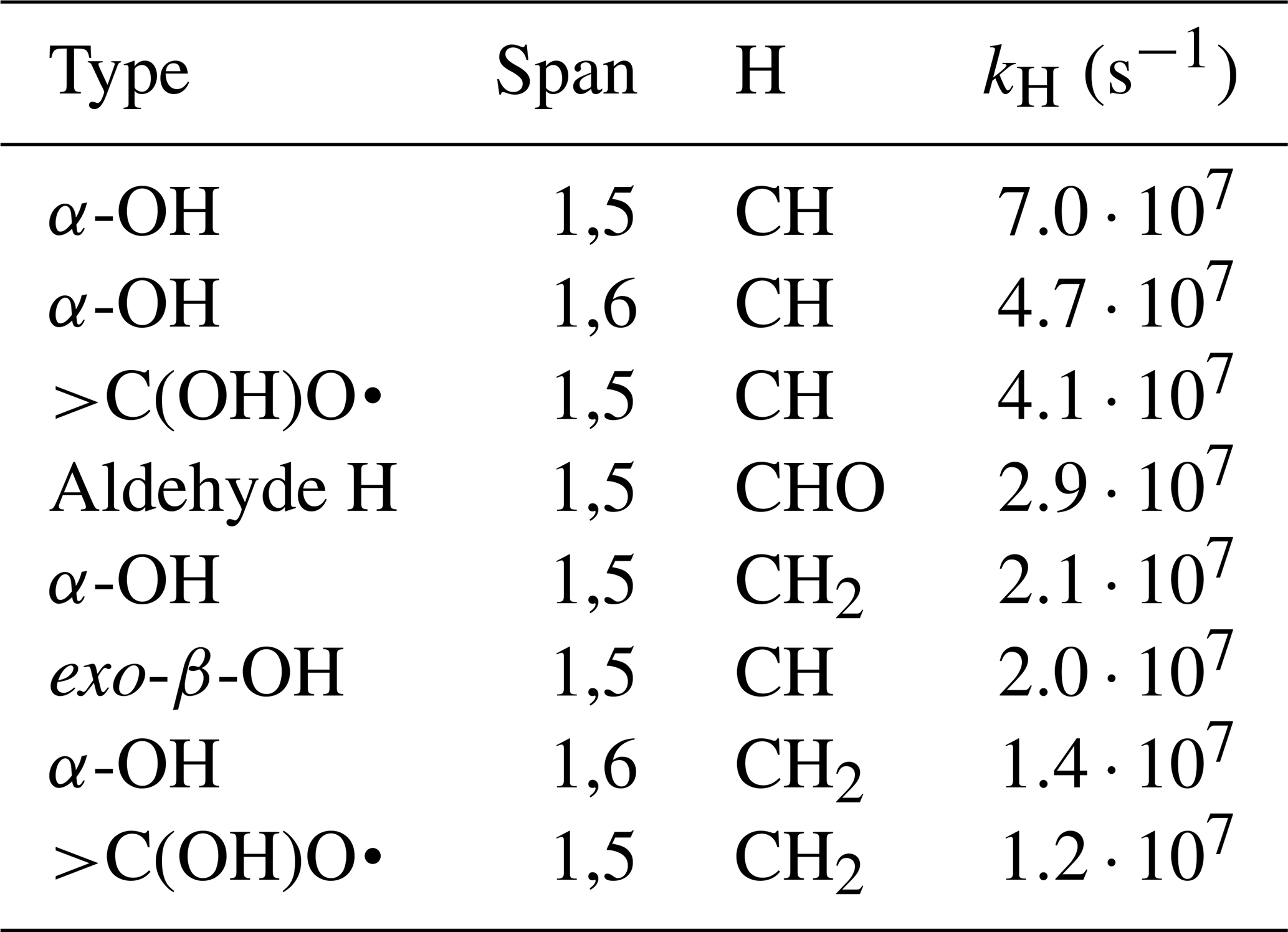
For the unimolecular H-shifts, the list of potentially competitive reactions is considerably shorter, as no H-shift has rates above 108 s−1 in the SAR. However, as these rates are uncertain by a factor of 10 (Vereecken and Peeters, 2010), the reactions with rates above 107 s−1 are still worth considering, especially as rates above 108 s−1 have been reported (Orlando et al., 2003). The unimolecular H-shift reactions above this rate threshold are listed in Table 2.
Table 2A list of the RO unimolecular H-shifts which might be fast enough to occur in 3(RO…OR) complexes according to Vereecken's SAR (Vereecken and Peeters, 2010).
Overall, it is notable that the in-complex decomposition channels outnumber the ISC channels in the data: 142 009 to 79 833 in the DTA dataset and 161 784 to 129 585 in the terpene dataset. The ISC channels also have lower yields on average, accounting for only 22.1 % of the total yield of products from first-generation radicals in the DTA dataset and 35.4 % in the terpene dataset. These yields should be taken with a grain of salt, considering the large number of simplifications done when applying the filtering criteria, but they do offer some indication that these in-complex RO decomposition channels are common and become increasingly competitive with more complex RO intermediates.
3.2 Statistics on molecular properties
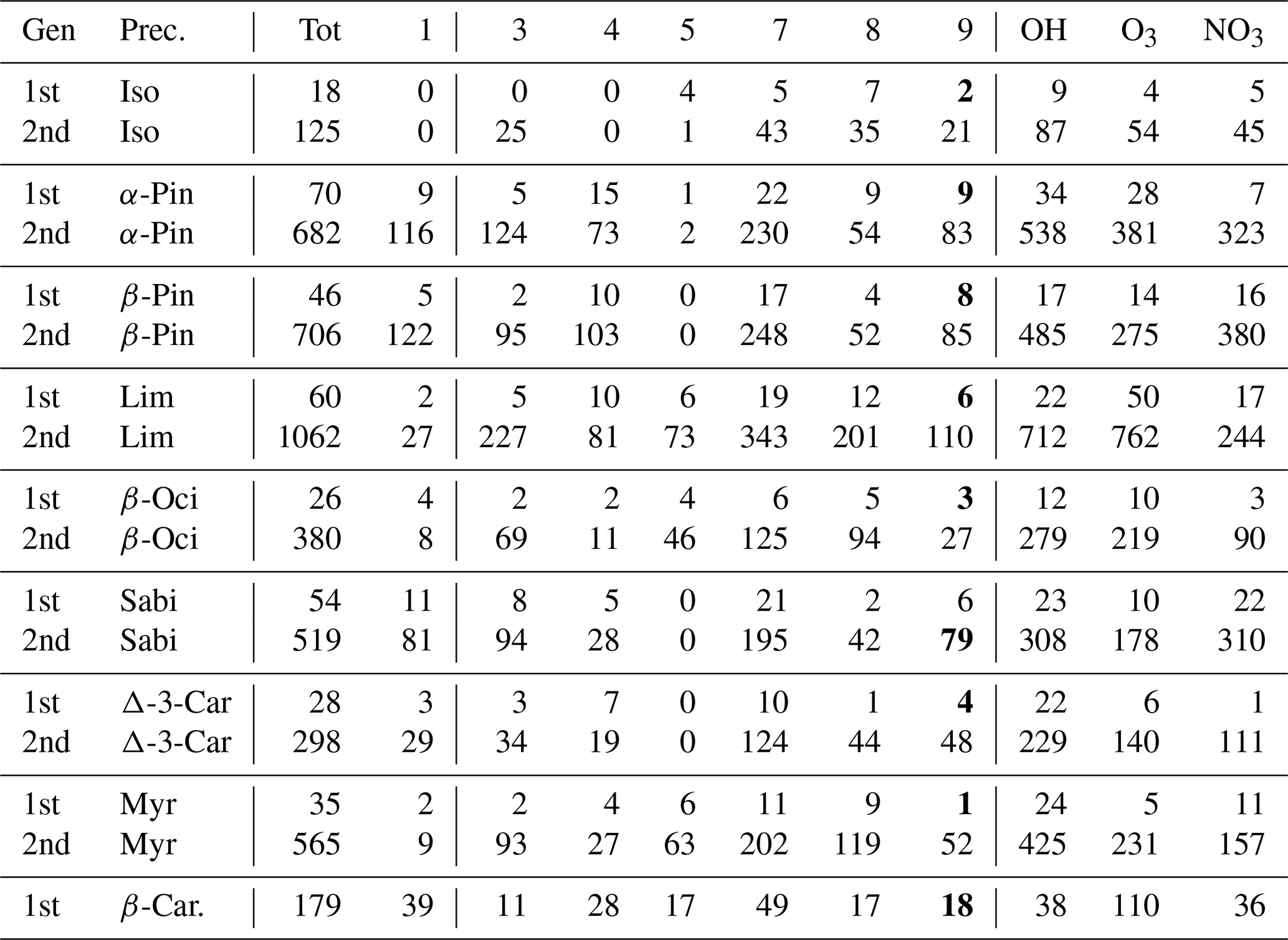
The distribution of RO2 by rate classes are presented in Table 3 for the DTA dataset and in Table 4 for the terpene and β-caryophyllene datasets. One detail of note in the latter is that isoprene seems to almost exclusively generate RO2 with fast RO2 + RO2 rates. This implies that isoprene-derived RO2 contributes more than expected from its concentration when it comes to RO2 recombination yields. Out of the seven monoterpenes treated in the data, limonene clearly produces the largest diversity of RO2, owing to having both an endocyclic and an exocyclic C=C bond. We also see from both tables that the production of tert-RO2 molecules is mostly dependent on the existence of tertiary double-bonded carbons, which is largely unsurprising.
Table 3The number of peroxy radicals per generation, recombination rate class, and oxidant for each precursor molecule in the DTA dataset. For an explanation of the classes, refer to Table 1. The oxidant labels are defined inclusively (H-abstraction RO2 with the label “OH, NO3” is included in both columns). The class-9 number is bolded if it includes CH3C(O)O2.
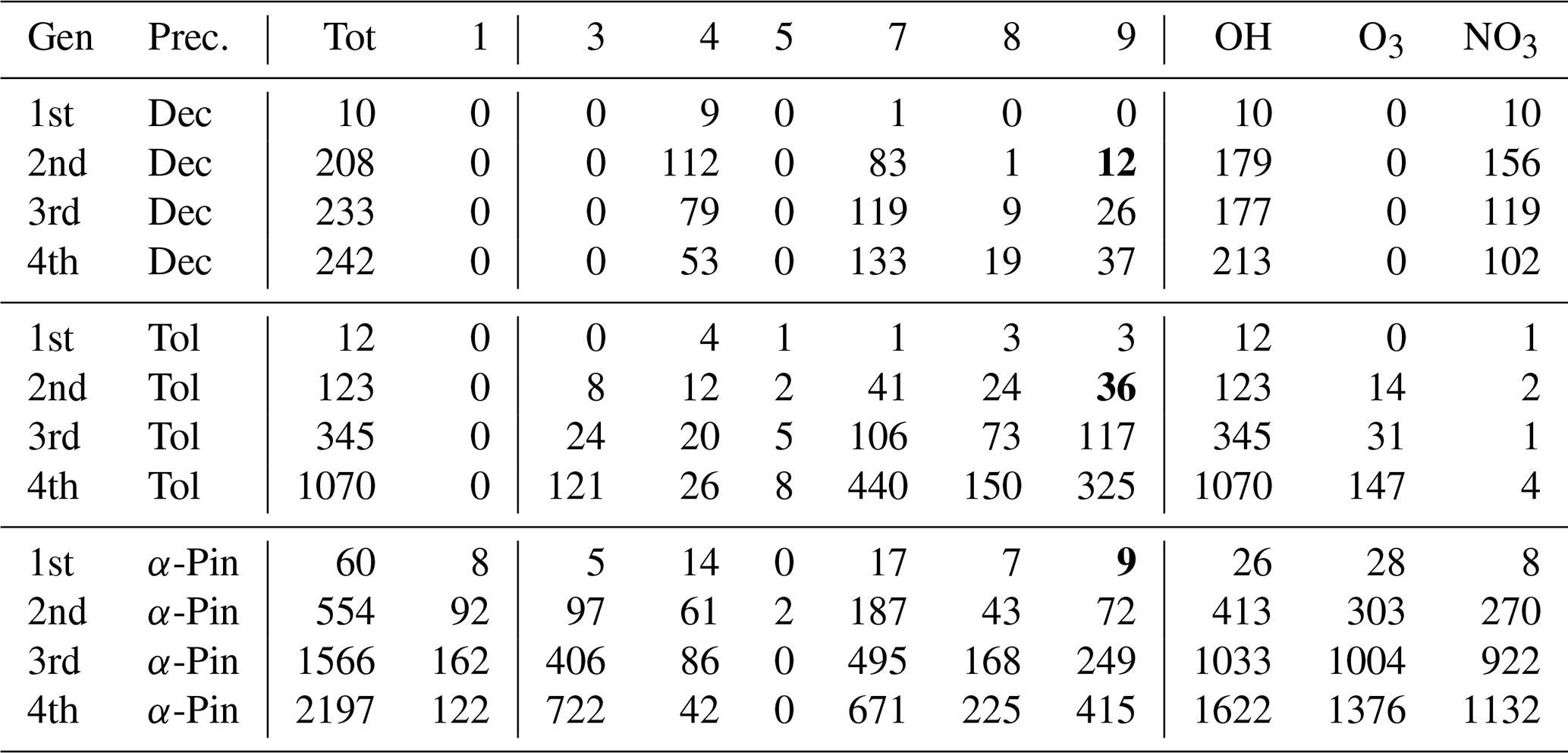
Table 4The number of peroxy radicals per generation, recombination rate class, and oxidant for each precursor molecule in the terpene and β-caryophyllene datasets. For an explanation of the classes, refer to Table 1. The class-9 number is bolded if it includes CH3C(O)O2.
As atmospheric oxidation proceeds, the reactant RO2 gets increasingly fragmented and oxidized. This is showcased for the radicals in the DTA dataset in Fig. 2 and for the terpene and β-caryophyllene datasets in Fig. 3. For the formed accretion products, the distribution of the same properties is presented in Figs. 4 and 5. As our methodology might be overemphasizing accretion products in later generations (see Sect. S5), all RO2 are grouped by the generation where they first form, and all accretion products are labelled “Gen n+m”, where n and m are the generations of the two reacting RO2. We also present histograms where each accretion product is weighted by our best proxy for the formation rate, which is the product of the RO2 + RO2 recombination rate (Eq. 3) and the in-complex branching ratio (Eq. 6). With this weighting, we neglect the relative importance of individual RO2 radicals, as the theoretical maximum yields are not fully comparable. Comparing the branching-ratio-weighted carbon number distributions of accretion product and RO2 in Fig. 5 to the corresponding radicals in Fig. 3, we notice an interesting trend in both the monoterpene and sesquiterpene results: the largest peak of accretion products occurs at C atom numbers only slightly larger than the bulk of the RO2. In the terpene dataset, all of the major first-generation RO2 have 10 C atoms, but the largest peak in accretion product yield is at 11 and 12 C atoms rather than 20. The same is seen in the β-caryophyllene dataset, where the RO2 yield peaks at 13–15 C atoms, whereas the accretion product yield peaks at 14–17. However, the explanations for this phenomenon seem to differ between the two datasets. In the β-caryophyllene data, 73 % of this yield peak comes from cross-reactions of CH3C(O)O2 with the C13–15 radicals; in the terpene dataset, this number is only 30 %. On the other hand, 38 % of this yield comes from fragmentations of larger RO2 + RO2 pairs, and the rest simply comes from recombinations of RO2 with smaller C atom numbers. This underlines the importance of the RC(O)O2 for accretion product formation, as they, at least in the Jenkin et al. (2019) parametrization, react rapidly even with the largest and most sterically hindered tert-RO2.
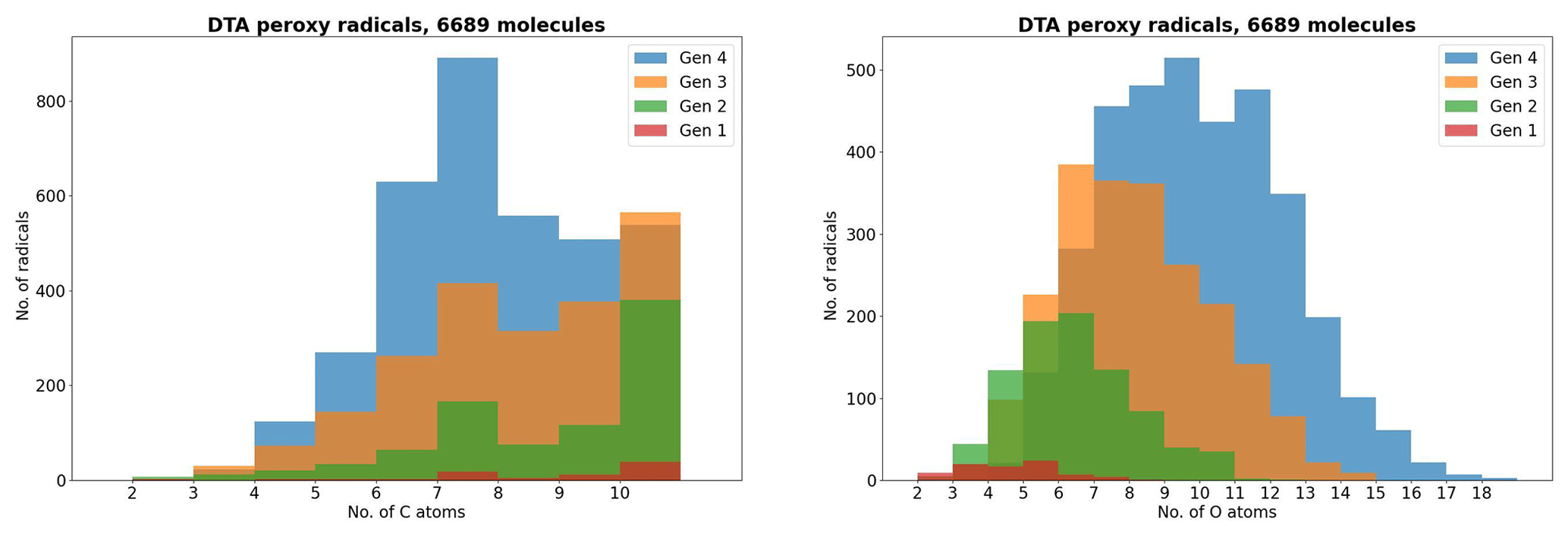
Figure 2The peroxy radicals in the DTA dataset by number of C and O atoms, the latter including the two O atoms in the RO2 functional group.
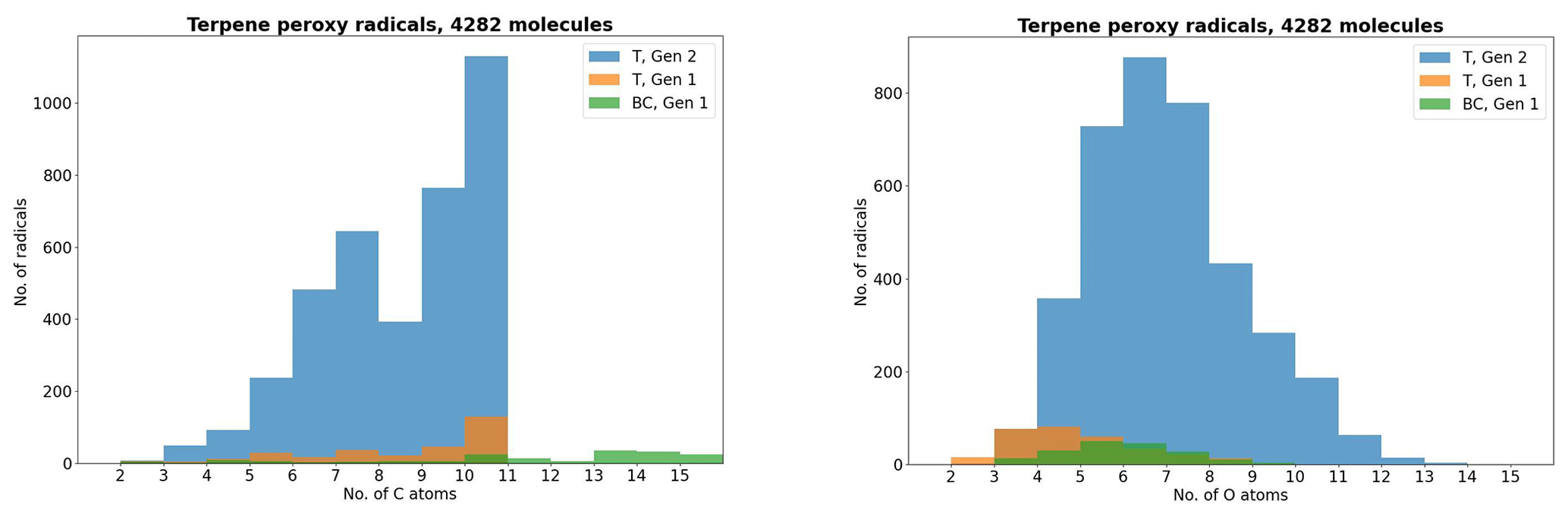
Figure 3The peroxy radicals in the terpene (T) and β-caryophyllene (BC) datasets by number of C and O atoms, the latter including the two O atoms in the RO2 functional group.
The distribution of functional groups in the RO2 and (unique) accretion products is shown in Fig. 6. When comparing the two figures, we see that the number of accretion products with either ether or ester functionalities is less than those with a peroxide functionality, and this is only partially explained by the presence of peroxides in the DTA RO2. This is likely due to the fact that there are more duplicates among the ether and ester products. The average in-complex branching ratio of all the ISC products remaining after filtering is 33.5 % in the DTA dataset and 36.6 % in the terpene dataset, again implying that the total yield of ether and ester products is typically higher. Interestingly, aromatic rings are highly represented in the DTA accretion products. The reason for this seems to be instrumental: the aromatic RO2 in the data all have high yields due to being directly derived from the first H-abstraction product Ar–CH2O2, which has a yield of 100 % from NO3 oxidation of toluene. Of course, H-abstraction from a methyl group by NO3 is a slow reaction (Kerdouci et al., 2010), meaning that toluene + NO3 might in reality not effectively be the dominant reaction even in favourable conditions. This detail is another example of why our results ought to be treated as qualitative rather than quantitative.
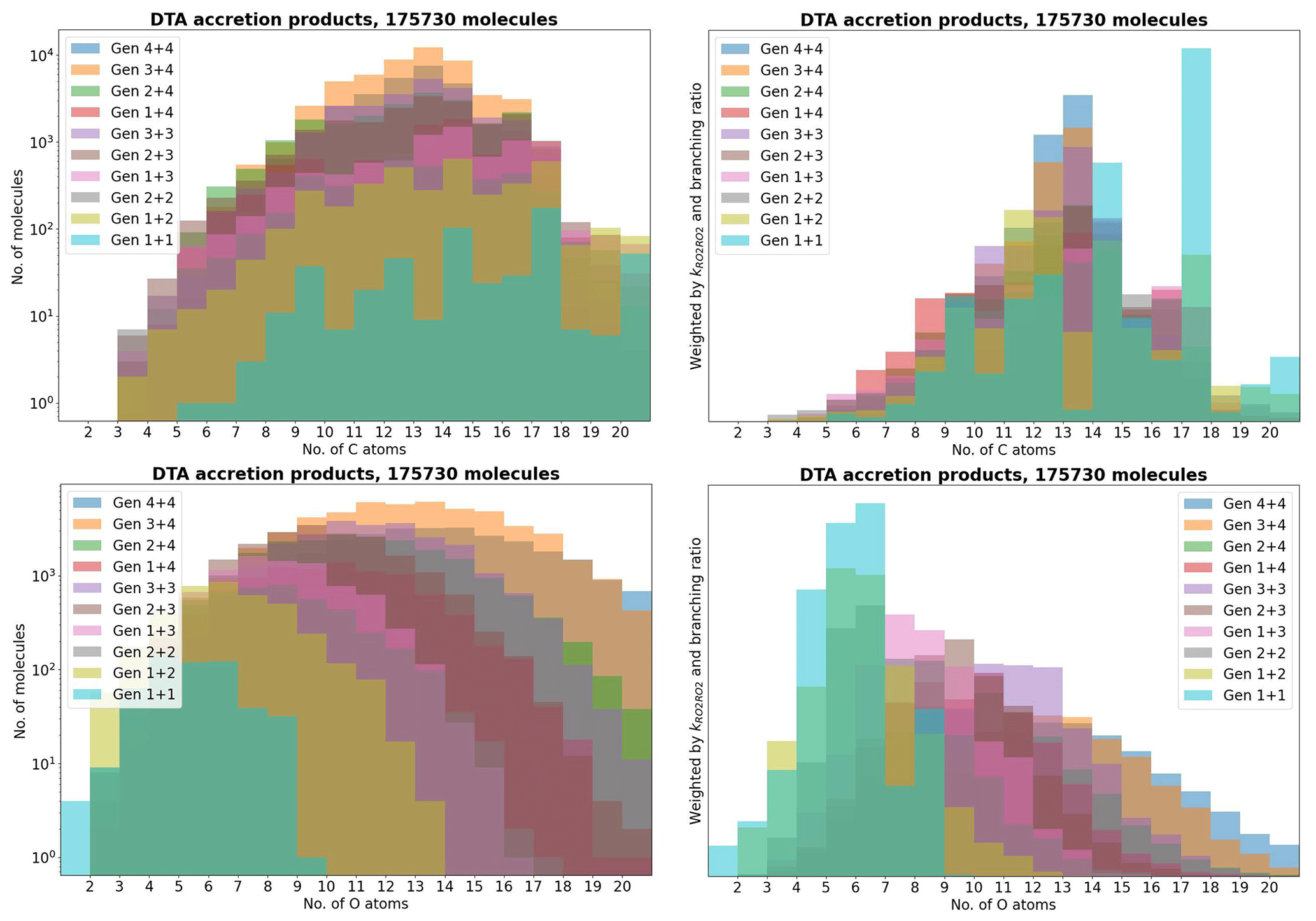
Figure 4The accretion products in the decane, toluene, and α-pinene (DTA) dataset by number of C and O atoms.
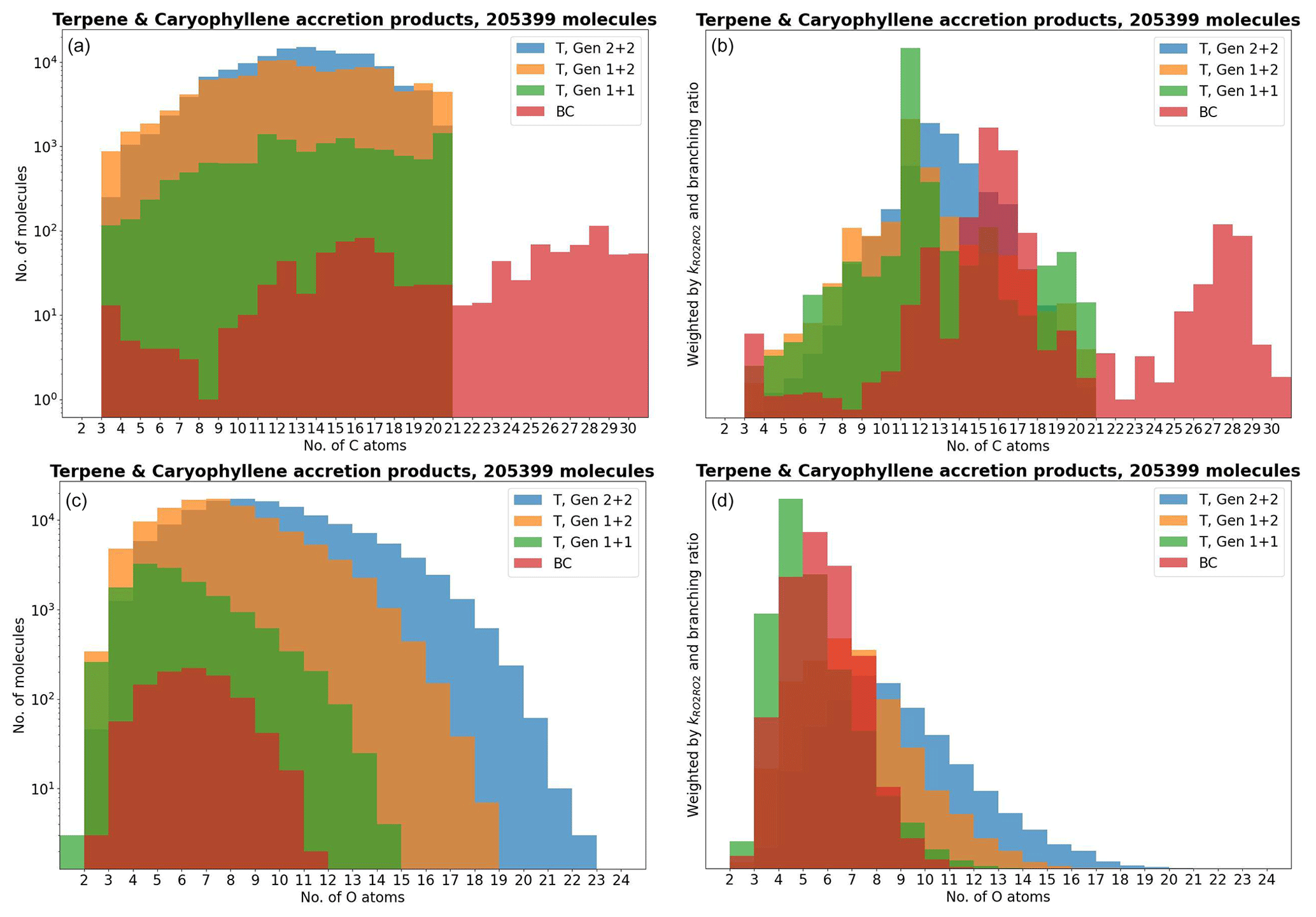
Figure 5The accretion products in the terpene (T) and β-caryophyllene (BC) datasets by number of C and O atoms.
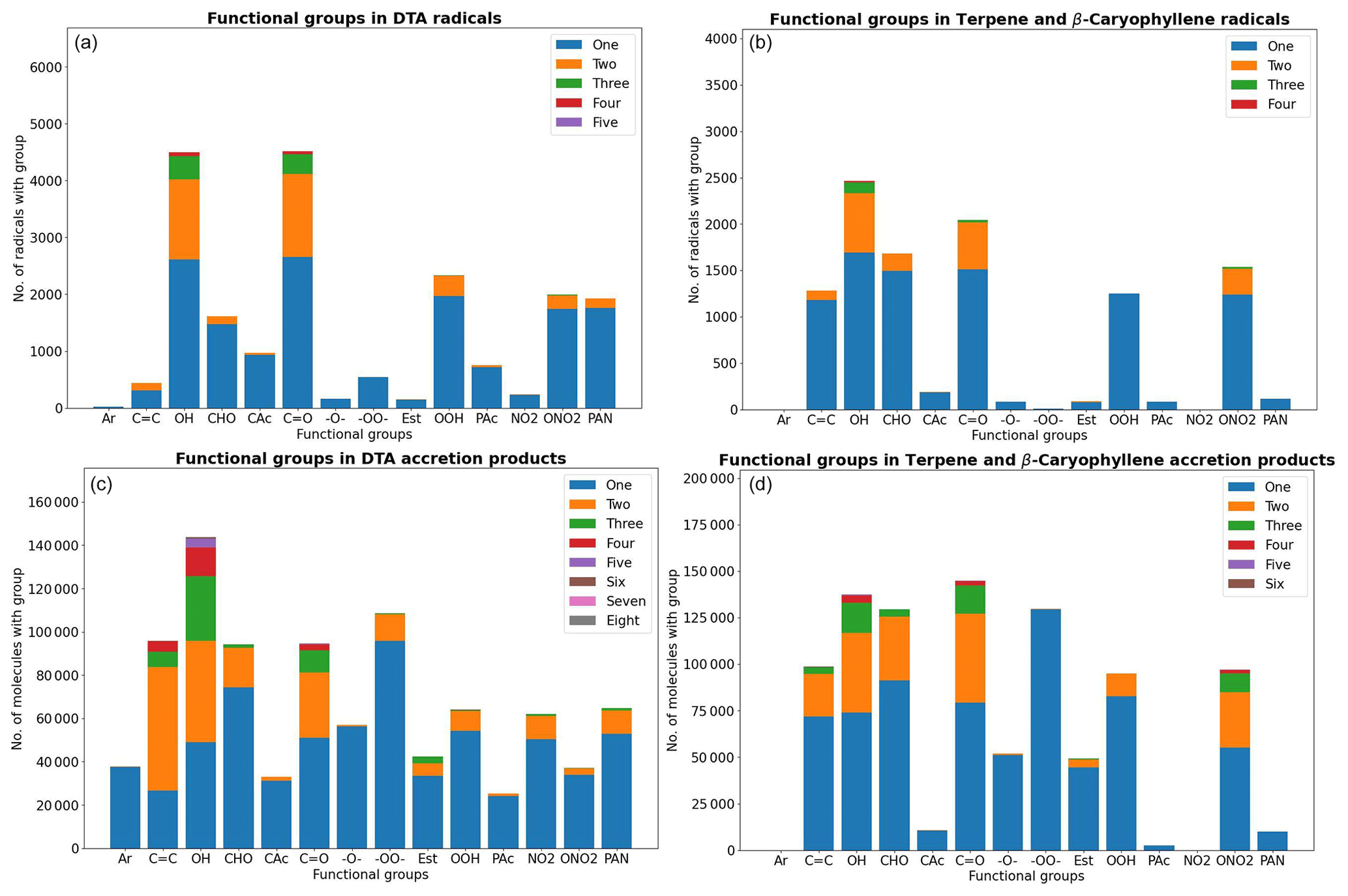
Figure 6The distribution of functional groups in the DTA and terpene datasets for the RO2 (a, b) and for the (unique) accretion products (c, d). CAc: carboxylic acid, C(O)OH. Est: ester, C(O)OC. PAc: peroxy acid, C(O)OOH. PAN: peroxy acyl nitrate, C(O)OONO2.
3.3 Accretion-product-inhibiting reactions
Since we have presented a new channel for accretion product inhibition, namely the alkoxy radical decomposition reaction leading to ejection of either an OH or NO2 radical, we should analyse the extent to which these inhibiting reactions occur in the data. This was done exclusively on the DTA dataset, due to the generally higher oxidation states of its RO2 resulting in more competitive RO decompositions. The simplest type of inhibition reaction is the α-nitro ejection, which in the code ensures that peroxy radicals with an NO2 group in the geminal position never form accretion products. As shown by our computational reaction rates in Sect. S3, this will not necessarily always be the case in reality, but the α-NO2 ejection still ought to be among the most competitive channels. This is our first accretion-product-inhibiting reaction, and it can be treated as a given rule. The reaction is presented in the following scheme:
These α-NO2 radicals all form downstream from toluene oxidation pathways in the DTA dataset. The reaction channel responsible for these is NO2 addition to aryloxy radicals (; Platz et al., 1998); thus these peroxy radicals and the associated (RO…OR) complexes were left out from the following analysis.
The second reaction inhibiting the accretion product formation is the barrierless decomposition of α-hydroperoxy alkyl radicals, a reaction channel that is possible for all peroxy radicals with −OOH substituents. The inhibition is presented in the following scheme, in which ROOH is a OOH-substituted carbon chain:
where the reaction step ROOHO•→R2•OOH may be either a β-scission or a unimolecular H-shift. As a measure of the competitiveness of RO reactions leading to this inhibition channel, the average branching ratio as a function of the combined number of OOH groups in the RO2 pair is shown in Fig. 7, both weighted and unweighted by the pair's recombination yield. As we see, the inhibition of accretion product formation increases significantly with the number of hydroperoxide groups.
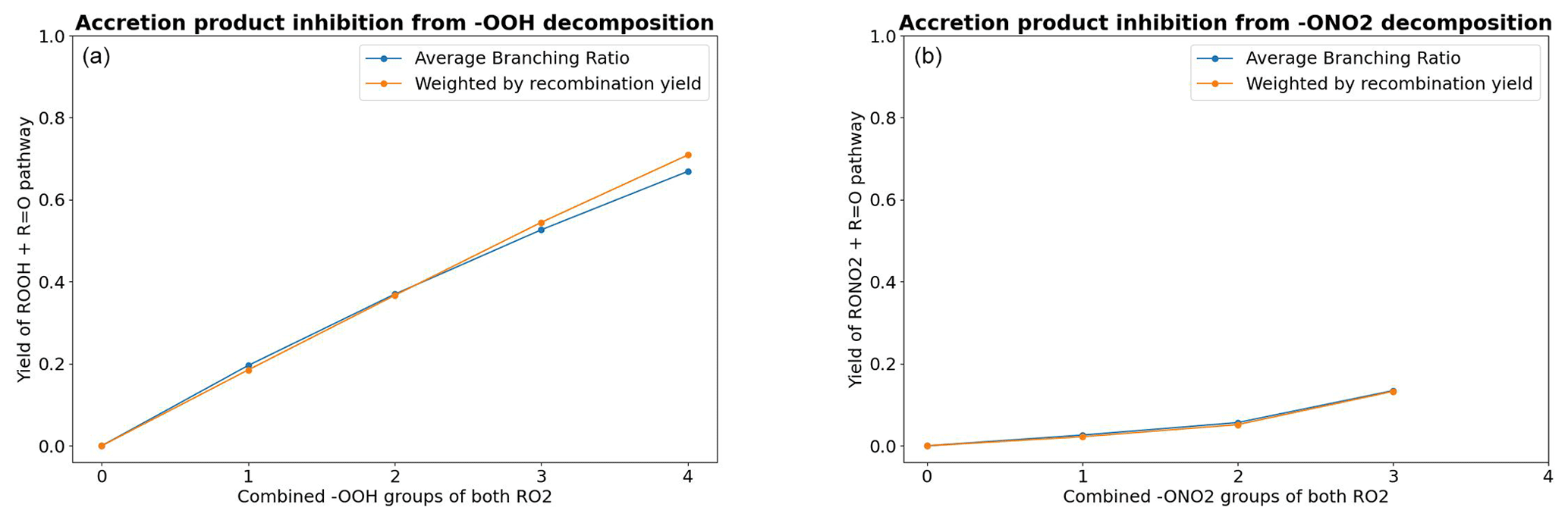
Figure 7The impact of barrierless C•OOH and C•ONO2 decomposition on the formation of accretion products. (a) The average branching ratio of RO reactions leading to C•OOH decomposition by the combined number of −OOH groups in the complex. (b) The same for RO reactions leading to C•ONO2 decomposition. The results for n=4 are excluded because there was only one such RO2 pair in the data.
Our third and final inhibition reaction is the barrierless decomposition of α-nitrate alkyl radicals, a very similar reaction to the corresponding −OOH reaction.
The average branching ratio of these reactions is presented in Fig. 7.
As seen in Fig. 7, the hydroperoxide decomposition channel has a significantly higher inhibition yield compared to the nitrate decomposition channel. As most of the competitive alkoxy radical decomposition channels are β-scissions, the explanation for this observation can be found in the group contribution parameters in the β-scission SAR of Vereecken and Peeters (Vereecken and Peeters, 2009): if the alkoxy radical has a hydroperoxide group in the β-carbon, the activation energy is lowered by 38.9 kJ mol−1. On the other hand, a nitrate group in the β-carbon only lowers the activation energy by 11.7 kJ mol−1. Plugging this difference into an Arrhenius expression () tells us that β-scission reactions leading to the formation of α-OOH alkyl radicals and to the ejection of an OH radical will typically be 104 times faster than those leading to the ejection of an NO2 radical; thus, they are more likely to be competitive. What this means in terms of atmospheric conditions is that highly ONO2-substituted peroxy radicals forming in high-NOx conditions have a slightly lower chance of forming organic accretion products from RO2 recombination reactions compared to other sufficiently oxidized RO2, while highly OOH-substituted peroxy radicals (for example, those forming in high HO2 conditions) have an even lower chance. Even more importantly, peroxy radical autoxidation is also known to form products with multiple −OOH groups (Bianchi et al., 2019). Autoxidation reactions are currently missing from GECKO-A, so it is unclear how well the highly −OOH-substituted RO2 in the data corresponds to those formed from autoxidation reactions. Considering the fact that the inhibition is primarily driven by fast β-scissions in the data, it is particularly notable that the optimal RO2 H-shift span is 1,5 or 1,6 rather than 1,4 (Vereecken and Nozière, 2020). This suggest that −OOH groups originating from autoxidation may be less likely to be in β-position relative to the alkoxy carbon. For autoxidation-derived RO2, the inhibition yield may thus increase less steeply compared to Fig. 7.
3.4 Analysis of vapour pressure distributions
As our main interest for accretion products comes from the formation of large low-volatility organic molecules, it is a worthwhile exercise to analyse the vapour pressure distribution of the predicted products. However, as the amount of data produced is rather large, this section will only discuss and visualize the observed main trends. Sections S6 and S7 feature more figures and tables on the vapour pressure distribution of the products. In addition to grouping the products by generation of radicals, they are also grouped by reaction channel, precursor molecule, and required VOC oxidants (OH, O3, and NO3) to better identify which reaction channels lead to the lowest-volatility products.
3.4.1 Distribution by reaction channel
The vapour pressure distribution of all the accretion products in the DTA and terpene datasets categorized by reaction channel is presented in Fig. 8. A few interesting trends pop up when viewing the data this way. Firstly, the vapour pressure distribution of the exocyclic β-scission products is shifted towards higher pSat values relative to the ISC products. This makes sense, as these products are more fragmented than their ROOR counterparts. This adds an interesting element to the suggestion by Peräkylä et al. (2023) that the ether and ester accretion products contribute more to aerosol particle growth thanks to their higher (thermal) stability. This increased stability often comes at the price of increased volatility, especially if the radical product of the β-scission is relatively small. In other words, these reactions may not always contribute more to aerosol particle growth compared to the ROOR product. On the other hand, the vapour pressure distribution of the endocyclic β-scission products is shifted towards lower pSat values, meaning that the same trade-off between stability and (low) volatility is not present. The reason for this is somewhat obvious: endocyclic β-scissions do not fragment the molecule. The same is true of the unimolecular H-shift products (which in theory ought to be even less volatile, as the reaction gives the product an additional OH group), but these products are less common due to the lower reaction rates (at least in the SAR). As seen when comparing the results between the three datasets, the different molecular structures present do not have a massive impact on the noted differences between the reaction types, but the ratio of exocyclic and endocyclic β-scissions obviously depends on the ring structures and positions of C=C bonds in the precursor VOC.
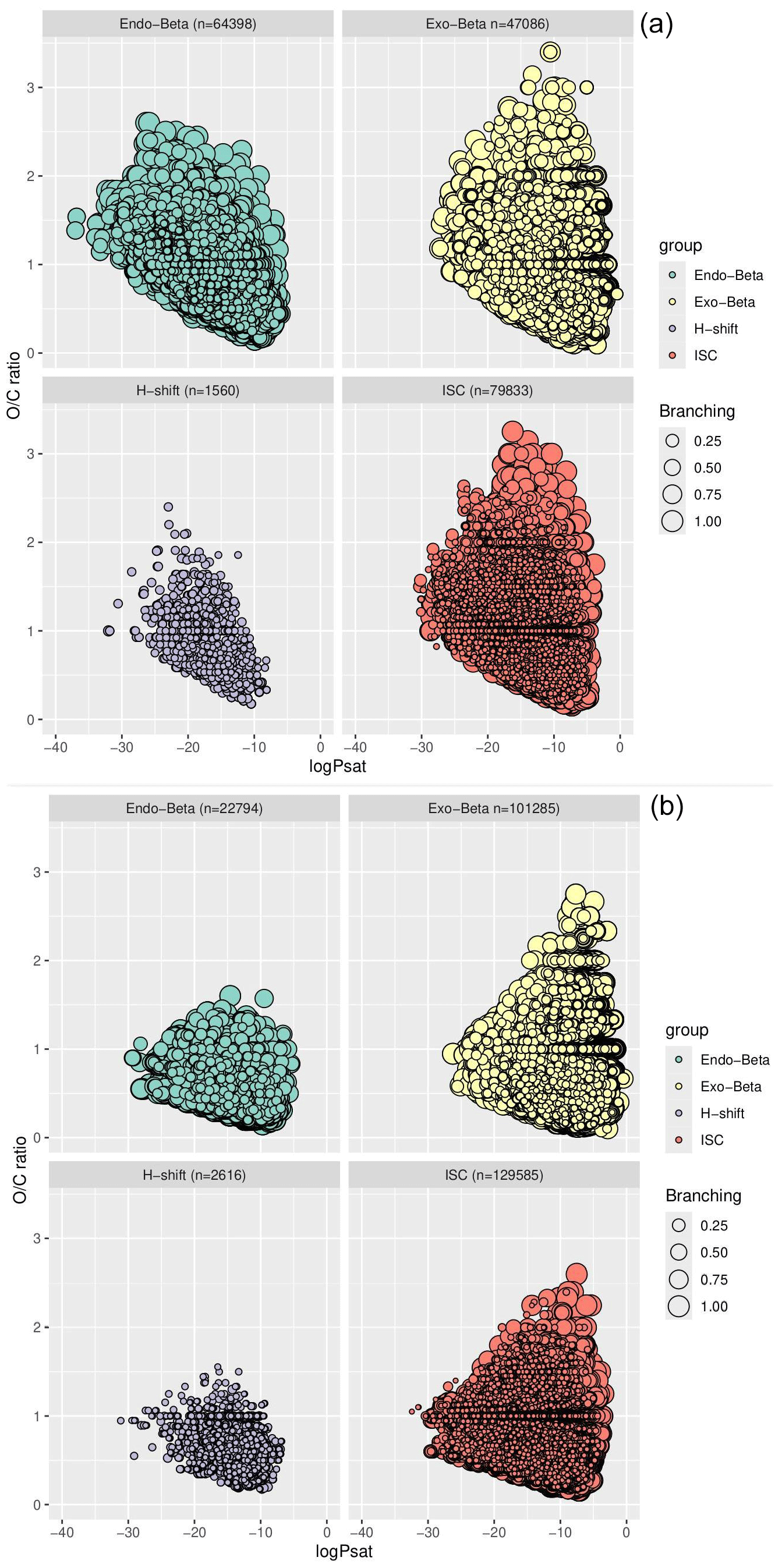
Figure 8A bubble plot of the pSat distribution of all accretion products categorized by reaction channel for (a) the DTA dataset and (b) the terpene dataset. pSat is expressed in atm.
3.4.2 Distribution by precursor molecule
If endocyclic β-scissions are indeed the key to forming low-volatility ether and ester products, then the next question is which chemical structures present in the data are especially efficient at forming endocyclic RO2 in high yields. This is, however, not the only important factor when it comes to the formation of accretion products, as endocyclic RO2 tends to be on the lower end of the scale in terms of self-reaction rates, as it is by definition either secondary or tertiary RO2 (see Table 1). Thus, simultaneously producing large amounts of rapidly reacting RC(O)O2 or primary RO2 with activating groups may also enhance accretion product formation from the endocyclic RO2, as the cross-reactions between these will be faster than the self-reactions of the endocyclic RO2.
Figure 9 shows the early-generation accretion products from the DTA and terpene datasets categorized by precursor molecule, including only RO2 + RO2 pairs where both radicals are produced by the same precursor molecule. From the DTA data, we see rather unsurprisingly that α-pinene and toluene are large sources of low-volatility accretion products, whereas n-decane is not. In the terpene results, we see that the lowest-volatility bins ( atm) are dominated by β-pinene and sabinene, for less obvious reasons. Out of the 132 β-pinene-derived first-generation accretion products in these volatility bins, over half (68) were endocyclic β-scission channels. In all of these channels, the radical undergoing the decomposition is one of seven individual endocyclic RO combined with different pairs in the 3(RO…OR) complex. Recognizable from these seven were three OH- and ONO2-addition-derived radicals each: one with both rings intact and two with only the four-ring intact. The seventh endocyclic RO is the VHP dissociation product of the C9 Criegee intermediate from β-pinene ozonolysis. That these 7 (out of 52 non-filtered first-generation RO2 radicals) dominate the low-volatility accretion products is another example of the key role endocyclic β-scissions play in low-volatility accretion product formation. For sabinene, another interesting trend emerges: only 40 out of the 100 atm accretion products are endocyclic β-scission products. There are 46 ISC products and 10 H-shift products. Upon closer examination, all four of the radicals undergoing the H-shift are primary RO in which sabinene's five-carbon ring has already been broken. In the H-shift classification of Vereecken and Peeters (2010), these reactions are either secondary α-OH or tertiary β-exo-oxo shifts. None of these are major products, as the SAR never predicts H-shift rates above 108 s−1, but these are nevertheless good examples of systems where the unimolecular H-shifts could be a competitive route to accretion product formation.
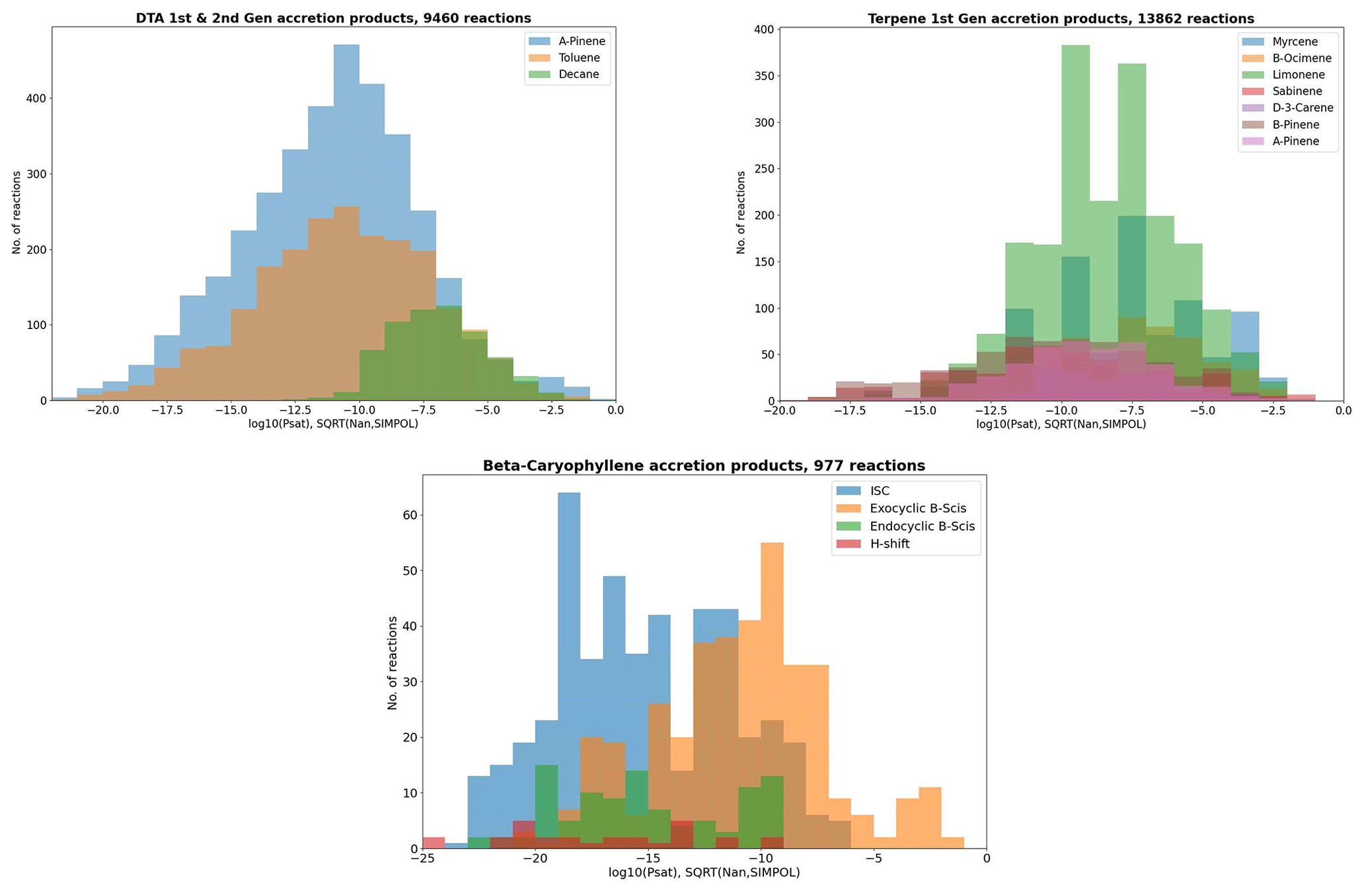
Figure 9pSat distributions of the early-generation accretion products in each dataset categorized by precursor molecule.
Another point worthy of discussion is the detail raised in Sect. 3.2 that isoprene-derived RO2 (iso-RO2) typically has high RO2 + RO2 reaction rates. This implies that cross-reactions between iso-RO2 with the larger but less abundant monoterpene-derived RO2 (mono-RO2) might be atmospherically significant. In Fig. 10, we see that the pSat values of these iso-RO2 + mono-RO2 products are not quite as low as those of mono-RO2 + mono-RO2, but a significant number of them stretch below the 10−13 atm threshold nevertheless. We should also note that, as our filtering method is based on relative product yields rather than product concentrations, we are likely underestimating the atmospheric occurrence of iso-RO2 + mono-RO2 pairs relative to mono-RO2 + mono-RO2 pairs, as global isoprene emissions outnumber total monoterpene emissions 3-fold in terms of concentration according to Sindelarova et al. (2014) (6-fold in terms of mass in the original source), which, roughly estimated, means a 20-fold higher formation rate on average relative to the individual monoterpenes in the dataset. We thus suspect that iso-RO2 + mono-RO2 reactions are an atmospherically significant but understudied pathway to the formation of atm contributing to aerosol particle growth, despite being experimentally less important than mono-RO2 + mono-RO2 for new particle formation according to Dada et al. (2023).
The results for β-caryophyllene are comparable to those of monoterpenes, as seen in Fig. 9c. However, we do see that the vapour pressure gap between the RO-fragmenting exocyclic β-scissions and the non-fragmenting endocyclic β-scission, H-shift, and ISC products is even wider, a fact which is largely explained by the closed-shell products of the exocyclic β-scissions also being larger. What this tells us is that formation of ELVOCs from sesquiterpenes is likely not qualitatively different from ELVOC formation from monoterpenes. Even with larger molecular sizes, the ELVOC formation is probably still dominated by the handful of reaction channels that produce the fastest oxidation without too much fragmentation.
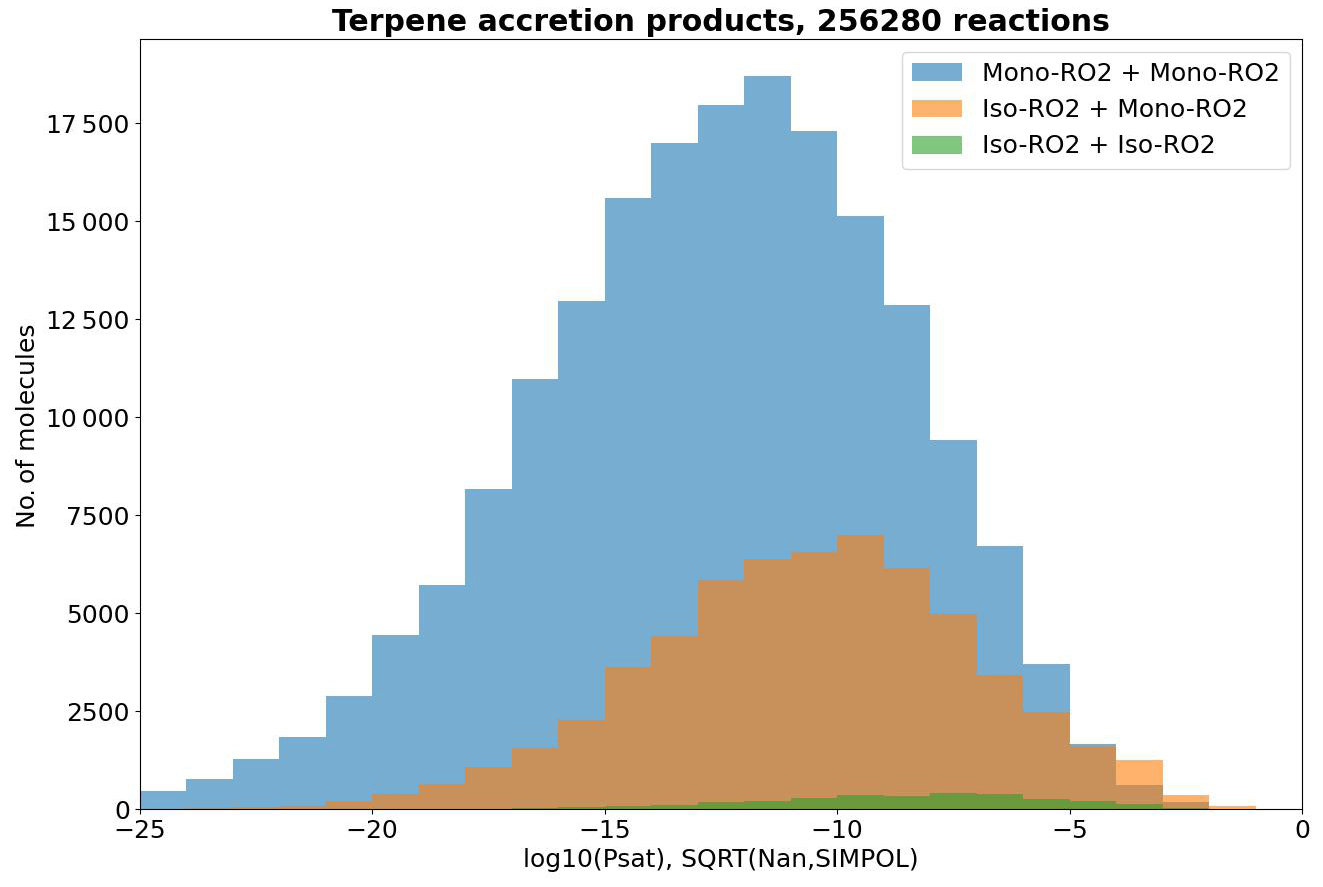
Figure 10pSat distributions of the accretion products in the terpene dataset categorized by type of RO2 pair. CH3C(O)O2-including pairs were separated from the rest due to their abundance and relatively high volatilities and due to CH3C(O)O2 being formed from all precursors in the dataset.
One interesting detail to consider is to which extent the use of SAR reaction rates and lack of RO2 autoxidation misrepresents these trends. For the former, a recent computational study of β-scission rates in NO3-derived monoterpene (Draper et al., 2024) RO provides an interesting point of comparison. For β-pinene in particular, the computational ring-opening activation energies are considerably higher than in the SAR, partially due to stereoelectronic effects arising from the orientation of the exocyclic ONO2 group and partially due to steric repulsion from the four-carbon ring preventing the scission of the C–C bond connected to it. In terms of reaction rates, it seems that none of the SAR-predicted competitive () endocyclic β-scissions of limonene, β-pinene, and Δ-3-carene are competitive anymore once one accounts for these new stereochemical effects. This underlines the limitations of the SAR models when dealing with especially complex molecules. On the impact of the missing autoxidation reactions, the importance of endocyclic RO2 makes it especially notable that GECKO-A lacks most unimolecular RO2 ring closure reactions, which adds competitive routes for forming endocyclic RO2. The only such reaction included in the current version is the bicyclic RO2 formation known from OH oxidation of aromatics (first reaction in Table 14 of Jenkin et al., 2019). However, the work of Vereecken et al. (2021) shows that these reactions produce both endocyclic and exocyclic RO2, of which the latter channel is more competitive in most cases. Nevertheless, the net increase in ring structures likely leads to the formation of endocyclic RO2 in the next generation with some probability. We might also compare our emphasis on endocyclic RO2 formation to the established consensus on monoterpene oxidation. According to Lee et al. (2023), it is the exocyclic RO2 formed after the opening of the four-carbon ring in α- and β-pinene that contributes the most to the formation of low-volatility organics, at least among the OH-oxidation products. While this seems to be in conflict with our findings at first glance, we note that this contribution is due to a rapid H-shift reaction which leads to the formation of a (delocalized) endocyclic alkyl radical that may form two RO2 (Fig. 11) (Møller et al., 2020). Furthermore, applying the SAR to the corresponding RO leads to an endocyclic β-scission rate of in both cases. In other words, the formation of low-volatility organics from this reaction channel is also likely driven by in-complex endocyclic β-scissions, and the lack of RO2 autoxidation in GECKO-A only means that we are missing some of the reactions forming the most oxidized endocyclic RO2.
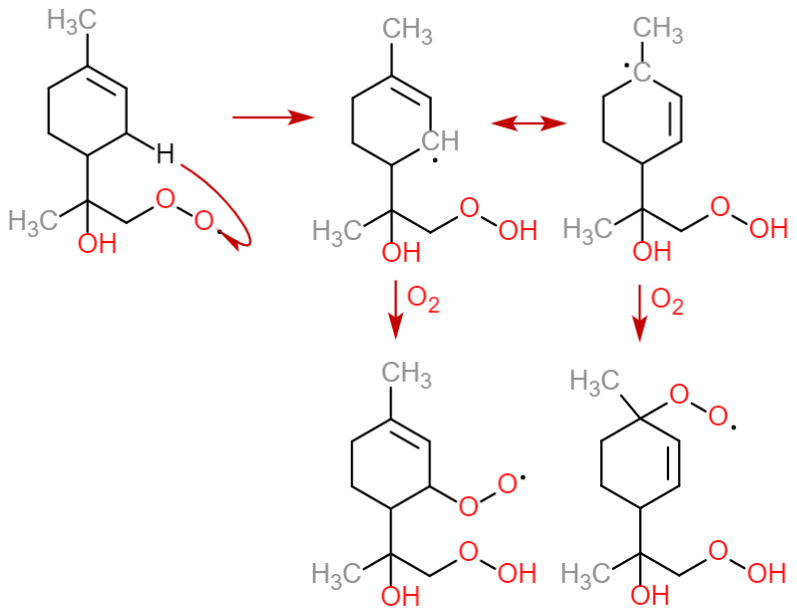
Figure 11An autoxidation scheme for α-pinene-derived endocyclic RO2 with rapid RO β-scission rates, adapted from Møller et al. (2020).
3.5 Atmospheric concentrations
The steady-state equations resulted in a total RO2 concentration of in the daytime and in the nighttime. The daytime HO2 concentration was , which means that our ratio is within the Kenagy et al. (2024) probability distribution we were aiming for. In Table 5, we see how the high RO2 concentration translated to reactivities (Eq. 9a, i.e. the fraction of RO2 reacting with other RO2) for different kinds of RO2. CH3O2 reactivities are also presented, as all of these reactions were neglected in the GECKO-AP code. In Eq. (9a), α is the Jenkin rate class of the RO2 the quantity is calculated for. Once again, we see a strong contrast between the classes of RO2 with fast RO2 + RO2 rates and classes with slow RO2 + RO2 rates in the Jenkin et al. (2019) parametrization. RC(O)O2 and β-substituted primary RO2 react primarily with other RO2 in sufficiently low-NOx conditions, whereas secondary and especially tertiary RO2 reacts primarily with inorganic radicals. We also see greatly elevated RO2 reactivities at night due to lower concentrations of RO2 scavengers NO, HO2, and (to a lesser extent) OH. This suggests that accretion product formation from RO2 + RO2 reactions might be more important in the nighttime, all else being equal. The important caveat here is that all else is not equal in real diurnal cycles, in part due to generally lower RO2 production with lower biogenic emission rates. Box-modelling studies will have to be performed to determine which effect is dominant. Another interesting detail is that CH3O2 was only responsible for a small fraction of the RO2 reactivity, despite CH3O2 being known as the major atmospheric peroxy radical (Wayne, 2000). This may be explained by the fact that CH3O2 has no primary source besides CH4+OH, for which the rate coefficient is over 4 orders of magnitude slower than that of isoprene + OH (Seinfeld and Pandis, 2016), which makes isoprene oxidation the far bigger source of RO2 in these relative concentrations. This is a very promising result for the importance of the accretion product formation studied in this work, but it should be noted that the isoprene concentrations used here are only representative of environments close to emission sources, due to the short atmospheric lifetime (Wennberg et al., 2018). CH3O2 is thus more likely to dominate RO2 + RO2 reactions in environments far away from both alkene emission sources.
Table 5Concentrations and RO2 and CH3O2 reactivities by RO2 type in the steady-state solution.
The estimated RO2 concentrations can further be used to estimate accretion product formation rates for the isoprene-derived products in the terpene dataset, through , by combining the concentrations with Eqs. (4) and (6). This results in a total daytime formation rate of and a nighttime formation rate of for ROOR-type accretion products, whereas, for our RO-decomposition-derived ether- and ester-type accretion products, the daytime formation rate is and the nighttime rate is . Since the latter reactions include an additional fragmentation step, it is worth noting that a majority of the formed ethers and esters (5.37⋅106 and in the day and night, respectively) have more than isoprene's five carbon atoms. We expect the observed difference between daytime and nighttime rates to be mostly isoprene-specific, due to the exceptionally fast OH addition rate and due to the faster recombination rates of the most important daytime isoprene-RO2. The fact that Jenkin's SAR for RO2 self-reactions lacks activation parameters for nitrate groups (Jenkin et al., 2019) may also play a role in the underestimation of important nighttime radicals. The latter daytime value is almost 20 % of the product formation rate from RO2+HO2, assuming an upper-limit rate coefficient of and the daytime concentrations mentioned above, suggesting that these ether and ester accretion products are an important part of RO2 chemistry in low-NOx, high-VOC environments. More analysis of the results can be found in Sect. S8.
In our previous work (Peräkylä et al., 2023), we observed that rapid decomposition of alkoxy radicals in the 3(RO…OR) intermediates of peroxy radical recombination reactions may form ether and ester accretion products in addition to the already-known peroxides. In this work, we looked at a wider range of rapid alkoxy radical decomposition reactions to explore the full atmospheric implications of this new observed channel. While our quantitative data rest on a mountain of assumptions concerning the rates of the competing channels, we can already discern several qualitatively reliable trends:
-
Our previously observed in-complex alkoxy radical decomposition was a β-scission reaction. By systematically looking at all the known reactions of free alkoxy radicals, we have concluded that the β-scission is often the most important in-complex RO decomposition channel.
-
Furthermore, the most competitive β-scission reactions are endocyclic β-scissions, which produce products of the same mass as the known ROOR peroxide product. These were likely produced in many previous experiments but eluded identification due to the use of mass spectrometric detection methods.
-
The products of exocyclic β-scission reactions typically have higher vapour pressures than the corresponding ROOR, which is to be expected, as they are smaller molecules. However, as these are ethers or esters rather than peroxides, they will be more stable towards both photochemical and thermal decomposition, as well as fragmenting oxidation, and thus more likely to contribute to SOA formation and growth.
-
Unimolecular alkoxy radical H-shift reactions are rarely competitive with the β-scissions, but their products typically have lower vapour pressures than any other peroxy radical recombination products. These reactions might be very important for SOA formation from a small specific subset of peroxy radical recombinations.
-
Acyl peroxy radicals are especially interesting for accretion product formation from peroxy radial recombination due to their ability to readily react with large tert-RO2 with otherwise slow RO2 + RO2 rates.
-
It has recently been postulated that the branching ratio of accretion-product-forming channels of peroxy radical recombination will progressively increase with RO2 size and oxidation state (Hasan, 2023). This may not always be the case thanks to alkoxy decomposition reactions, where the products are a closed-shell organic molecule and a small inorganic radical. These reactions grow increasingly likely as the number of hydroperoxide, nitrate, and nitro groups in the reactant peroxy radicals increases.
The datasets generated by the code are available in a Zenodo repository (https://doi.org/10.5281/zenodo.13253425, Franzon, 2024). These data include labelled peroxy radical lists, peroxy radical pair lists, in-complex alkoxy radical reaction lists, and accretion product lists with various levels of filtering. The ORCA output files of the computational results presented in Sect. S3 and the calculated steady-state concentrations for the isoprene-derived radicals and accretion products are also included here.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-24-11679-2024-supplement.
LF wrote the GECKO-AP code, performed the data analysis on the generated datasets, and drafted the paper. MC, RV, and BA provided help and consultation on the usage of the GECKO-A code and advice on how to filter, analyse, and represent the data. The original idea was envisioned by TK. The paper was reviewed and revised by all authors.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
Computational resources for the quantum chemical scans were provided by the Finnish Centre for Scientific Computing (CSC). Lauri Franzon personally thanks Matti Rissanen for useful discussions on the reactivity of acyl peroxy radicals, Siddharth Iyer on toluene oxidation, Vili-Taneli Salo on recent computational findings of tetroxide decomposition, Rashid Valiev for trends and uncertainties on 3(RO…OR) ISC rates, and Emelda Ahongshangbam for ROOOH reactions. William Carter and an anonymous referee are gratefully acknowledged for review and discussion that helped to improve the article, and Thomas Berkemeier is thanked for editing it.
This research has been supported by the Research Council of Finland (grant no. 364224) and the Jane ja Aatos Erkon Säätiö (grant no. 4708470).
Open-access funding was provided by the Helsinki University Library.
This paper was edited by Thomas Berkemeier and reviewed by William Carter and one anonymous referee.
Anglada, J. M. and Solé, A.: Tropospheric oxidation of methyl hydrotrioxide (CH3OOOH) by hydroxyl radical, Phys. Chem. Chem. Phys., 20, 27406–27417, 2018. a
Assaf, E., Schoemaecker, C., Vereecken, L., and Fittschen, C.: Experimental and theoretical investigation of the reaction of RO2 radicals with OH radicals: Dependence of the HO2 yield on the size of the alkyl group, Int. J. Chem. Kinet., 50, 670–680, 2018. a
Aumont, B., Szopa, S., and Madronich, S.: Modelling the evolution of organic carbon during its gas-phase tropospheric oxidation: development of an explicit model based on a self generating approach, Atmos. Chem. Phys., 5, 2497–2517, https://doi.org/10.5194/acp-5-2497-2005, 2005. a, b, c, d, e
Berndt, T., Richters, S., Kaethner, R., Voigtländer, J., Stratmann, F., Sipilä, M., Kulmala, M., and Herrmann, H.: Gas-phase ozonolysis of cycloalkenes: formation of highly oxidized RO2 radicals and their reactions with NO, NO2, SO2, and other RO2 radicals, J. Phys. Chem. A, 119, 10336–10348, 2015. a
Berndt, T., Chen, J., Kjærgaard, E. R., Møller, K. H., Tilgner, A., Hoffmann, E. H., Herrmann, H., Crounse, J. D., Wennberg, P. O., and Kjaergaard, H. G.: Hydrotrioxide (ROOOH) formation in the atmosphere, Science, 376, 979–982, 2022. a
Besel, V., Todorović, M., Kurtén, T., Rinke, P., and Vehkamäki, H.: Atomic structures, conformers and thermodynamic properties of 32k atmospheric molecules, Scientific Data, 10, 450, https://doi.org/10.1038/s41597-023-02366-x, 2023. a, b
Bey, I., Aumont, B., and Toupance, G.: The nighttime production of OH radicals in the continental troposphere, Geophys. Res. Lett., 24, 1067–1070, 1997. a
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, J., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly oxygenated organic molecules (HOM) from gas-phase autoxidation involving peroxy radicals: A key contributor to atmospheric aerosol, Chem. Rev., 119, 3472–3509, 2019. a, b
Compernolle, S., Ceulemans, K., and Müller, J.-F.: Technical Note: Vapor pressure estimation methods applied to secondary organic aerosol constituents from α-pinene oxidation: an intercomparison study, Atmos. Chem. Phys., 10, 6271–6282, https://doi.org/10.5194/acp-10-6271-2010, 2010. a
Dada, L., Stolzenburg, D., Simon, M., Fischer, L., Heinritzi, M., Wang, M., Xiao, M., Vogel, A. L., Ahonen, L., Amorim, A., Baalbaki, R., Baccarini, A., Baltensperger, U., Bianchi, F., Daellenbach, K. R., DeVivo, J., Dias, A., Dommen, J., Duplissy, J., Finkenzeller, H., Hansel, A., He, X.-C., Hofbauer, V., Hoyle, C. R., Kangasluoma, J., Kim, C., Kürten, A., Kvashnin, A., Mauldin, R., Makhmutov, V., Marten, R., Mentler, B., Nie, W., Petäjä, T., Quéléver, L. L. J., Saathoff, H., Tauber, C., Tome, A., Molteni, U., Volkamer, R., Wagner, R., Wagner, A. C., Wimmer, D., Winkler, P. M., Yan, C., Zha, Q., Rissanen, M., Gordon, H., Curtius, J., Worsnop, D. R., Lehtipalo, K., Donahue, N. M., Kirkby, J., Haddad, I. E., and Kulmala, M: Role of sesquiterpenes in biogenic new particle formation, Science Advances, 9, eadi5297, https://doi.org/10.1126/sciadv.adi5297, 2023. a, b
Draper, D., Almeida, T. G., Iyer, S., Smith, J. N., Kurtén, T., and Myllys, N.: Unpacking the diversity of monoterpene oxidation pathways via nitrooxy–alkyl radical ring-opening reactions and nitrooxy–alkoxyl radical bond scissions, J. Aerosol Sci., 179, 106379, https://doi.org/10.1016/j.jaerosci.2024.106379, 2024. a
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I. H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Dal Maso, M., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V. M., Kulmala, M., Worsnop, D. R., Wildt, J. and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, 2014. a
Franzon, L.: Simple Physical Model for the Estimation of Irreversible Dissociation Rates for Bimolecular Complexes, J. Phys. Chem. A, 127, 5956–5966, 2023. a, b, c, d, e
Franzon, L.: Supplementary datasets of ACP article Ether and Ester Formation from Peroxy Radical Recombination, Zenodo [data set], https://doi.org/10.5281/zenodo.13253425, 2024. a
Hasan, G.: Computational Studies of Reaction Channels for 3(RO…OR) Intermediates formed in Peroxy Self and Cross-Reactions, PhD thesis, University of Helsinki, https://helda.helsinki.fi/items/a22e0965-1788-4383-b22b-8af767447789 (last access: 10 October 2024), 2023. a, b, c, d, e, f, g
Hasan, G., Salo, V.-T., Golin Almeida, T., Valiev, R. R., and Kurtén, T.: Computational Investigation of Substituent Effects on the Alcohol+ Carbonyl Channel of Peroxy Radical Self-and Cross-Reactions, J. Phys. Chem. A, 127, 1686–1696, 2023. a, b
Ingold, K. U.: Peroxy radicals, Accounts Chem. Res., 2, 1–9, 1969. a, b
Isaacman-VanWertz, G. and Aumont, B.: Impact of organic molecular structure on the estimation of atmospherically relevant physicochemical parameters, Atmos. Chem. Phys., 21, 6541–6563, https://doi.org/10.5194/acp-21-6541-2021, 2021. a, b
Jenkin, M. E., Valorso, R., Aumont, B., Rickard, A. R., and Wallington, T. J.: Estimation of rate coefficients and branching ratios for gas-phase reactions of OH with aliphatic organic compounds for use in automated mechanism construction, Atmos. Chem. Phys., 18, 9297–9328, https://doi.org/10.5194/acp-18-9297-2018, 2018a. a
Jenkin, M. E., Valorso, R., Aumont, B., Rickard, A. R., and Wallington, T. J.: Estimation of rate coefficients and branching ratios for gas-phase reactions of OH with aromatic organic compounds for use in automated mechanism construction, Atmos. Chem. Phys., 18, 9329–9349, https://doi.org/10.5194/acp-18-9329-2018, 2018b. a
Jenkin, M. E., Valorso, R., Aumont, B., and Rickard, A. R.: Estimation of rate coefficients and branching ratios for reactions of organic peroxy radicals for use in automated mechanism construction, Atmos. Chem. Phys., 19, 7691–7717, https://doi.org/10.5194/acp-19-7691-2019, 2019. a, b, c, d, e, f, g, h, i, j
Jenkin, M. E., Valorso, R., Aumont, B., Newland, M. J., and Rickard, A. R.: Estimation of rate coefficients for the reactions of O3 with unsaturated organic compounds for use in automated mechanism construction, Atmos. Chem. Phys., 20, 12921–12937, https://doi.org/10.5194/acp-20-12921-2020, 2020. a
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005. a
Kenagy, H., Heald, C., Tahsini, N., Goss, M., Matthew, B., and Kroll, J. H.: Can we achieve atmospheric chemical environments in the laboratory? An integrated model-measurement approach to chamber SOA studies, Sci. Adv., 10, eado1482, https://doi.org/10.1126/sciadv.ado1482, 2024. a, b, c
Kerdouci, J., Picquet-Varrault, B., and Doussin, J.-F.: Prediction of Rate Constants for Gas-Phase Reactions of Nitrate Radical with Organic Compounds: A New Structure–Activity Relationship, ChemPhysChem, 11, 3909–3920, 2010. a
Kerdouci, J., Picquet-Varrault, B., and Doussin, J.-F.: Structure–activity relationship for the gas-phase reactions of NO3 radical with organic compounds: Update and extension to aldehydes, Atmos. Environ., 84, 363–372, 2014. a
Knap, H. C. and Jørgensen, S.: Rapid hydrogen shift reactions in acyl peroxy radicals, J. Phys. Chem. A, 121, 1470–1479, 2017. a
Kurtén, T., Tiusanen, K., Roldin, P., Rissanen, M., Luy, J.-N., Boy, M., Ehn, M., and Donahue, N.: α-Pinene autoxidation products may not have extremely low saturation vapor pressures despite high O:C ratios, J. Phys. Chem. A, 120, 2569–2582, 2016. a, b
Lee, B. H., Iyer, S., Kurtén, T., Varelas, J. G., Luo, J., Thomson, R. J., and Thornton, J. A.: Ring-opening yields and auto-oxidation rates of the resulting peroxy radicals from OH-oxidation of α-pinene and β-pinene, Environmental Science: Atmospheres, 3, 399–407, 2023. a
Méreau, R., Rayez, M.-T., Rayez, J.-C., Caralp, F., and Lesclaux, R.: Theoretical study on the atmospheric fate of carbonyl radicals: kinetics of decomposition reactions, Phys. Chem. Chem. Phys., 3, 4712–4717, 2001. a
Møller, K. H., Otkjær, R. V., Chen, J., and Kjaergaard, H. G.: Double bonds are key to fast unimolecular reactivity in first-generation monoterpene hydroxy peroxy radicals, J. Phys. Chem. A, 124, 2885–2896, 2020. a, b
Murphy, S. E., Crounse, J. D., Møller, K. H., Rezgui, S. P., Hafeman, N. J., Park, J., Kjaergaard, H. G., Stoltz, B. M., and Wennberg, P. O.: Accretion product formation in the self-reaction of ethene-derived hydroxy peroxy radicals, Environmental Science: Atmospheres, 3, 882–893, 2023. a
Nannoolal, Y., Rarey, J., Ramjugernath, D., and Cordes, W.: Estimation of pure component properties: Part 1. Estimation of the normal boiling point of non-electrolyte organic compounds via group contributions and group interactions, Fluid Phase Equilibr., 226, 45–63, 2004. a
Nannoolal, Y., Rarey, J., and Ramjugernath, D.: Estimation of pure component properties: Part 3. Estimation of the vapor pressure of non-electrolyte organic compounds via group contributions and group interactions, Fluid Phase Equilibr., 269, 117–133, 2008. a
Neese, F.: Software update: The ORCA program system – Version 5.0, WIREs Computational Molecular Science, 12, 1606, https://doi.org/10.1002/wcms.1606, 2022. a
Newland, M. J., Mouchel-Vallon, C., Valorso, R., Aumont, B., Vereecken, L., Jenkin, M. E., and Rickard, A. R.: Estimation of mechanistic parameters in the gas-phase reactions of ozone with alkenes for use in automated mechanism construction, Atmos. Chem. Phys., 22, 6167–6195, https://doi.org/10.5194/acp-22-6167-2022, 2022. a
Novelli, A., Cho, C., Fuchs, H., Hofzumahaus, A., Rohrer, F., Tillmann, R., Kiendler-Scharr, A., Wahner, A., and Vereecken, L.: Experimental and theoretical study on the impact of a nitrate group on the chemistry of alkoxy radicals, Phys. Chem. Chem. Phys., 23, 5474–5495, 2021. a, b
Orlando, J. J. and Tyndall, G. S.: Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 41, 6294–6317, 2012. a
Orlando, J. J., Tyndall, G. S., and Wallington, T. J.: The atmospheric chemistry of alkoxy radicals, Chem. Rev., 103, 4657–4690, 2003. a, b, c, d
Pankow, J. F. and Asher, W. E.: SIMPOL.1: a simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds, Atmos. Chem. Phys., 8, 2773–2796, https://doi.org/10.5194/acp-8-2773-2008, 2008. a
Peräkylä, O., Berndt, T., Franzon, L., Hasan, G., Meder, M., Valiev, R. R., Daub, C. D., Varelas, J. G., Geiger, F. M., Thomson, R. J., Rissanen, M., Kurtén, T., and Ehn, M.: Large Gas-Phase Source of Esters and Other Accretion Products in the Atmosphere, J. Am. Chem. Soc., 145, 7780–7790, 2023. a, b, c, d, e, f, g, h
Platz, J., Nielsen, O., Wallington, T., Ball, J., Hurley, M., Straccia, A., Schneider, W., and Sehested, J.: Atmospheric chemistry of the phenoxy radical, C6H5O*: UV spectrum and kinetics of its reaction with NO, NO2, and O2, J. Phys. Chem. A, 102, 7964–7974, 1998. a
Rissanen, M. P., Kurtén, T., Sipilä, M., Thornton, J. A., Kangasluoma, J., Sarnela, N., Junninen, H., Jørgensen, S., Schallhart, S., Kajos, M. K., Taipale, R., Springer, M., Mentel, T. F., Ruuskanen, T., Petäjä, T., Worsnop, D. R., Kjaergaard, H. G., and Ehn, M: The formation of highly oxidized multifunctional products in the ozonolysis of cyclohexene, J. Am. Chem. Soc., 136, 15596–15606, 2014. a
Russell, G. A.: Deuterium-isotope effects in the autoxidation of aralkyl hydrocarbons. mechanism of the interaction of peroxy radicals, J. Am. Chem. Soc., 79, 3871–3877, 1957. a
Salo, V.-T., Chen, J., Runeberg, N., Kjaergaard, H. G., and Kurtén, T.: Multireference and Coupled-Cluster Study of Dimethyltetroxide (MeO4Me) Formation and Decomposition, J. Phys. Chem. A, 128, 1825–1836, https://doi.org/10.1021/acs.jpca.3c08043, 2024. a
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics: from air pollution to climate change, John Wiley & Sons, ISBN 9781118947401, 2016. a, b
Sindelarova, K., Granier, C., Bouarar, I., Guenther, A., Tilmes, S., Stavrakou, T., Müller, J.-F., Kuhn, U., Stefani, P., and Knorr, W.: Global data set of biogenic VOC emissions calculated by the MEGAN model over the last 30 years, Atmos. Chem. Phys., 14, 9317–9341, https://doi.org/10.5194/acp-14-9317-2014, 2014. a, b
Topping, D.: UManSysProp Multiphase system online property prediction, http://umansysprop.seaes.manchester.ac.uk (last access: 27 March 2024), 2015. a
Valorso, R., Aumont, B., Camredon, M., Raventos-Duran, T., Mouchel-Vallon, C., Ng, N. L., Seinfeld, J. H., Lee-Taylor, J., and Madronich, S.: Explicit modelling of SOA formation from α-pinene photooxidation: sensitivity to vapour pressure estimation, Atmos. Chem. Phys., 11, 6895–6910, https://doi.org/10.5194/acp-11-6895-2011, 2011. a
Vereecken, L.: Computational study of the stability of α-nitroxy-substituted alkyl radicals, Chem. Phys. Lett., 466, 127–130, 2008. a
Vereecken, L. and Nozière, B.: H migration in peroxy radicals under atmospheric conditions, Atmos. Chem. Phys., 20, 7429–7458, https://doi.org/10.5194/acp-20-7429-2020, 2020. a
Vereecken, L. and Peeters, J.: Decomposition of substituted alkoxy radicals – part I: a generalized structure–activity relationship for reaction barrier heights, Phys. Chem. Chem. Phys., 11, 9062–9074, 2009. a, b, c, d, e, f, g, h
Vereecken, L. and Peeters, J.: A structure–activity relationship for the rate coefficient of H-migration in substituted alkoxy radicals, Phys. Chem. Chem. Phys., 12, 12608–12620, 2010. a, b, c, d, e, f, g
Vereecken, L., Nguyen, T. L., Hermans, I., and Peeters, J.: Computational study of the stability of α-hydroperoxyl-or α-alkylperoxyl substituted alkyl radicals, Chem. Phys. Lett., 393, 432–436, 2004. a
Vereecken, L., Vu, G., Wahner, A., Kiendler-Scharr, A., and Nguyen, H.: A structure activity relationship for ring closure reactions in unsaturated alkylperoxy radicals, Phys. Chem. Chem. Phys., 23, 16564–16576, 2021. a
Wayne, R. P.: Chemistry of Atmospheres, 3rd edn., Oxford University Press, Oxford, England, ISBN 9780198503750, 2000. a, b
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., St Clair, J. M., Teng, A. P., Zhang, X., and Seinfeld, J. H.: Gas-phase reactions of isoprene and its major oxidation products, Chem. Rev., 118, 3337–3390, 2018. a, b