the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 25 May 2020
| 25 May 2020
A new marine biogenic emission: methane sulfonamide (MSAM), dimethyl sulfide (DMS), and dimethyl sulfone (DMSO2) measured in air over the Arabian Sea
Christof Stönner
Eva Y. Pfannerstill
Matias Berasategui
David Walter
John N. Crowley
Jos Lelieveld
Jonathan Williams
We present the first ambient measurements of a new marine emission methane sulfonamide (MSAM: CH5NO2S), along with dimethyl sulfide (DMS) and dimethyl sulfone (DMSO2) over the Arabian Sea. Two shipborne transects (W → E, E → W) were made during the AQABA (Air Quality and Climate Change in the Arabian Basin) measurement campaign. Molar mixing ratios in picomole of species per mole of air (throughout this paper abbreviated as ppt) of DMS were in the range of 300–500 ppt during the first traverse of the Arabian Sea (first leg) and 100–300 ppt on the second leg. On the first leg DMSO2 was always below 40 ppt and MSAM was close to the limit of detection. During the second leg DMSO2 was between 40 and 120 ppt and MSAM was mostly in the range of 20–50 ppt with maximum values of 60 ppt. An analysis of HYSPLIT back trajectories combined with calculations of the exposure of these trajectories to underlying chlorophyll in the surface water revealed that most MSAM originates from the Somalia upwelling region, known for its high biological activity. MSAM emissions can be as high as one-third of DMS emissions over the upwelling region. This new marine emission is of particular interest as it contains both sulfur and nitrogen, making it potentially relevant to marine nutrient cycling and marine atmospheric particle formation.
- Article
(6389 KB) - Full-text XML
-
Supplement
(5999 KB) - BibTeX
- EndNote





The ocean plays an important role in the atmospheric chemistry of many trace gases and profoundly influences the global sulfur and nitrogen cycles (Brimblecombe, 2014; Sievert et al., 2007; Bentley and Chasteen, 2004; Fowler et al., 2013, 2015). Dimethyl sulfide (DMS) emitted from the ocean accounts for roughly half of the natural global atmospheric sulfate burden. The global DMS flux to the atmosphere was recently estimated to be 28.1 (17.6–34.4) Tg S yr−1, equivalent to 50 % of the anthropogenic sulfur inputs (Webb et al., 2019). Nitrogen is often a limiting nutrient for phytoplankton growth in the ocean (Voss et al., 2013). Nonetheless, ocean emissions of organic nitrogen do occur in the form of amines (R-NH2) (Ge et al., 2011; Gibb et al., 1999) and in inorganic forms such as nitrous oxide (N2O) (Arévalo-Martínez et al., 2019) and ammonia (Gibb et al., 1999; Johnson et al., 2008; Paulot et al., 2015), particularly in upwelling regions (Carpenter et al., 2012).
Upwelling regions of the ocean are those where nutrient-rich waters from depths of 100 to 300 m are brought to the surface (Voss et al., 2013; Kämpf and Chapman, 2016). Upwelling leads to nutrient-richer zones in the surface ocean and therefore to regions of high phytoplankton activity, resulting in strong carbon dioxide uptake and the release of various volatile organic compounds including sulfur-, halogen-, and alkene-containing trace gases (Arnold et al., 2010; Colomb et al., 2008; Bonsang et al., 2010; Lai et al., 2011; Yassaa et al., 2008). In the Arabian Sea, the location of this study, the Somalian coastal upwelling is a major feature. It is considered the fifth-largest upwelling system in the world (deCastro et al., 2016; Ajith Joseph et al., 2019).
Here we present trace gas measurements taken on a shipborne circumnavigation of the Arabian Peninsula. Relatively few measurements have been made in this region due to political tensions and piracy. Transects of the Arabian Sea (the most southerly section of the route) showed high levels of sulfur-containing gases. These include DMS, dimethyl sulfone (DMSO2), and methane sulfonamide (MSAM), a new marine emission that unusually contains both sulfur and nitrogen atoms.
DMS is known to stem from biochemical reactions within phytoplankton that produce its precursor dimethylsulfoniopropionate (DMSP) (Kiene et al., 2000). Although only a small fraction of the DMS produced within the ocean is released into the atmosphere (Vila-Costa et al., 2006), it is still the most abundant form of oceanic sulfur emission (Kloster et al., 2006; Quinn and Bates, 2011; Lana et al., 2011; Liss et al., 2014).
The oxidation mechanism of DMS in the atmosphere is complex and still not fully understood (Mardyukov and Schreiner, 2018; Barnes et al., 2006; Ayers and Gillett, 2000; Chen et al., 2018). DMSO2, the second sulfur-containing species measured in this study, is a product of DMS oxidation by the OH radical (Arsene et al., 2001; Barnes et al., 2006). It can be formed directly from DMS, via the intermediate dimethyl sulfoxide (DMSO) and from the oxidation of DMSO with BrO and NO3 (Barnes et al., 2006). Even though the oxidation of DMS in the atmosphere is still not fully understood, reaction with the hydroxyl radical (OH) is considered the dominant loss pathway (Khan et al., 2016).
Significantly DMS oxidation ultimately yields sulfates which may act as cloud condensation nuclei (see Fig. 1). In the case of MSAM, there are no previously reported measurements of this species. The MSAM data are assessed here through comparison with the better-known DMS and DMSO2 species and with respect to air mass back trajectories and chlorophyll exposure, in particular in relation to the upwelling region. In summary, we examine the provenance, distribution and fate of DMS, DMSO2, and the new marine emission MSAM in the region of the Arabian Sea.
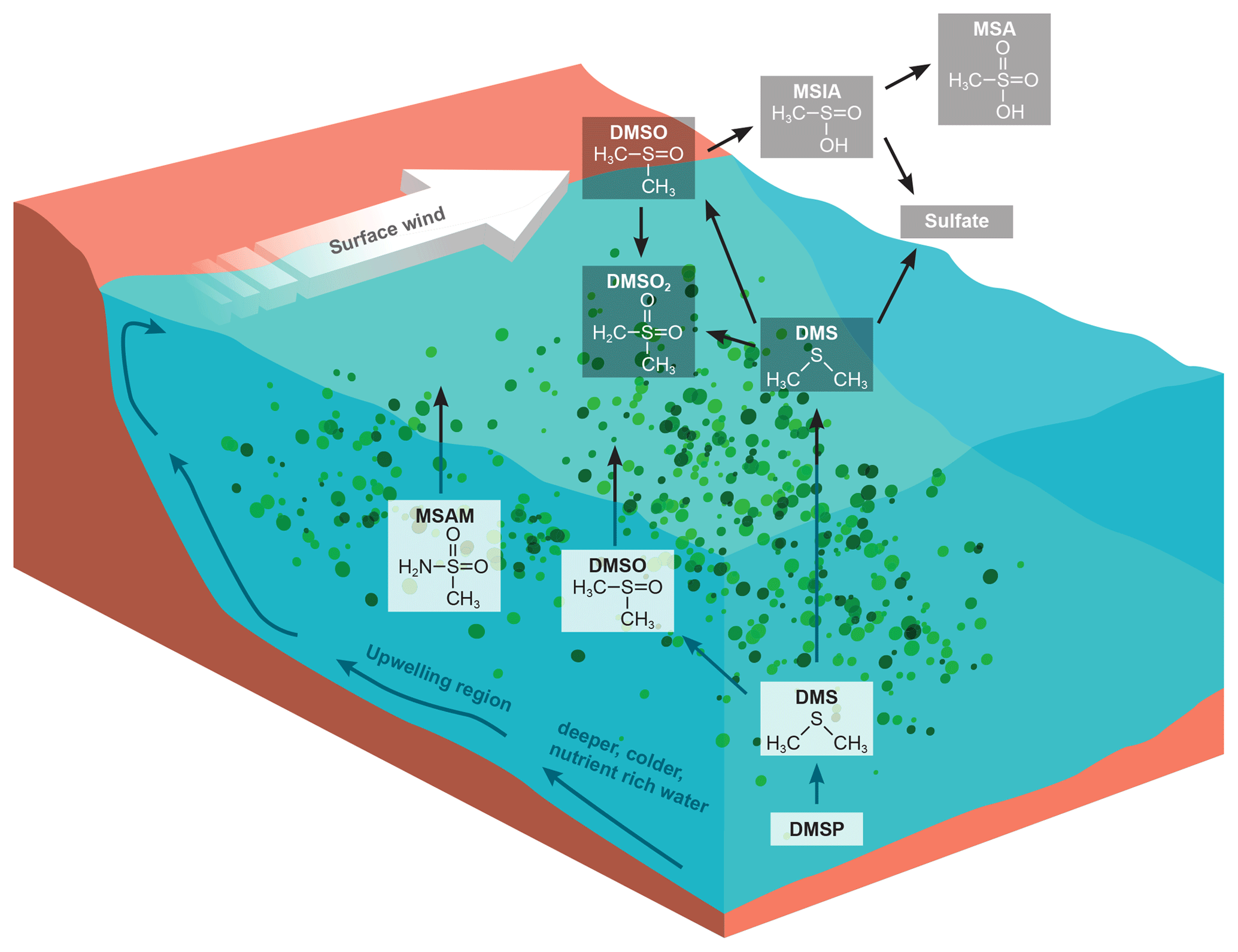
Figure 1DMS oxidation scheme focusing on the trace gases discussed (Barnes et al., 2006). DMSP production within phytoplankton yields DMS in the surface water where it can be oxidize directly to DMSO. A small fraction of DMS is emitted to the atmosphere. Where it is predominantly oxidized by the OH radical yielding methane sulfonic acid (MSA) and sulfates. Additionally we sketched a possible formation pathway for DMSO2. We suggest that MSAM could be formed in the water as a result of microbial activity, and parts of it are then emitted to the atmosphere. The bottom part of the figure illustrates the principle of the Somalia upwelling. Wind blowing along the coast displaces surface water and leads to the upwelling of cold nutrient-rich water, which can support a phytoplankton bloom (Kämpf and Chapman, 2016).
2.1 AQABA campaign
From 25 June to 3 September 2017, the Air Quality and Climate Change in the Arabian Basin (AQABA) cruise took place on the research vessel Kommandor Iona. The first leg of the cruise started from La-Seyne-sur-mer near Toulon (France) and headed through the Suez Canal and around the Arabian Peninsula and ended in Kuwait. The second leg took the same route back (see Fig. 2). Onboard the ship there were a weather station and four laboratory containers equipped with instrumentation for on- and offline measurement of a large suite of trace gases, particles, and radicals (Bourtsoukidis et al., 2019, 2020; Wang et al., 2020).
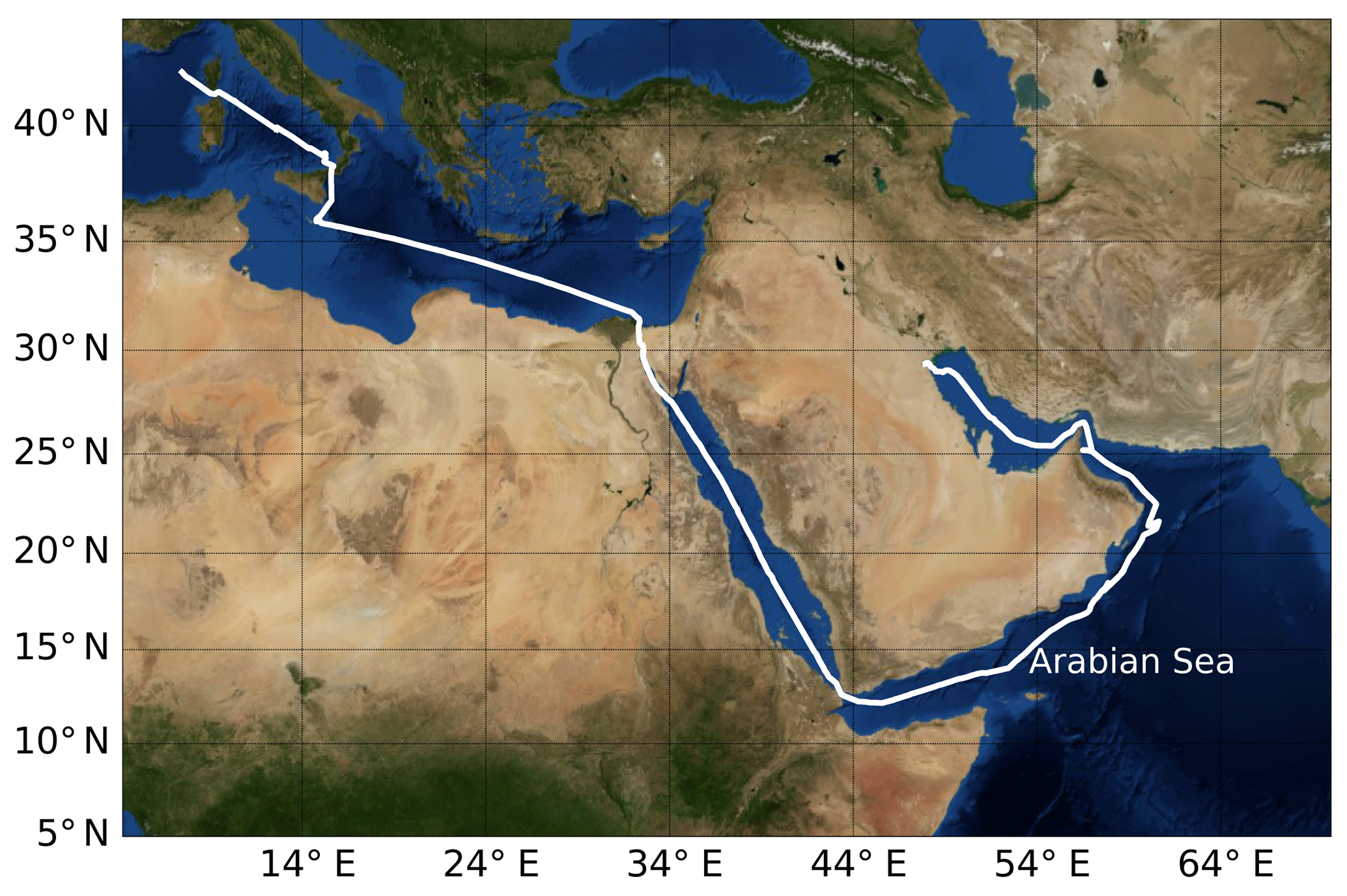
Figure 2Ship track of AQABA cruise. Beginning in July 2017, the campaign started in the south of France near Toulon; the ship arrived in Kuwait at the end of July, started its return back to France at the beginning of August, and was back at its starting point at the beginning of September 2017. On the way to Kuwait it entered the Arabian Sea on 19 July and left it on 24 July. On the way back it entered the Arabian Sea on 11 August and left it on 15 August. Credit: NASA Earth Observatory.
2.2 Sampling
A 10 m high (above sea level) high-volume flow inlet (HUFI) (diameter 15 cm) was used to draw ambient air down to the containers at a flow rate of 10 m3 min−1. The HUFI was situated between the four containers on the foredeck so that when the ship headed into the wind, no interference from the vessel's smokestack or indoor ventilation was measured. From the center of the HUFI, air was drawn continuously at a rate of ca. 5 slpm (standard liter per minute) (first leg) or 3 slpm (second leg) into an air-conditioned laboratory container via an insulated FEP (fluorinated ethylene propylene) tube ( cm o.d., length ca. 10 m). The tube was heated to 50–60 ∘C to avoid condensation inside the air-conditioned container. To prevent sampling of sea spray and particles, a routinely changed PTFE (polytetrafluorethylene) filter was installed in the inlet line before it entered the container. This inlet system was employed for the measurements of VOCs and total OH reactivity (Pfannerstill et al., 2019) simultaneously. The inlet residence time for the VOC measurements was determined by a spiking test with acetone, and it was 12 s during the first leg and 26 s during the second leg.
2.3 Volatile organic compound (VOC) measurements
Online VOC measurements were performed using a proton transfer reaction time-of-flight mass spectrometer (PTR-ToF-MS 8000; manufacturer: Ionicon Analytik GmbH, Innsbruck, Austria). Detailed descriptions of the instrument can be found in Jordan et al. (2009), Graus et al. (2010), and Veres et al. (2013). Drift pressure was maintained at 2.2 mbar and the drift voltage at 600 V (E/N 137 Td). For mass scale calibrations, 1,3,5-trichlorobenzene was continuously fed into the sample stream. The PTR-ToF-MS was calibrated at the beginning, during, and at the end of the campaign (in total five humidity dependent calibrations were conducted as described by Derstroff et al., 2017). Calibrations were performed by using a standard gas mixture (Apel-Riemer Environmental inc., Broomfield, USA) of several VOCs with gravimetrically determined mixing ratios. The VOCs included in the calibration gas were methanol, acetonitrile, acetaldehyde, acetone, dimethyl sulfide, isoprene, methyl vinyl ketone, methacrolein, methyl ethyl ketone, benzene, toluene, o-xylene, 1,3,5-trimethylbenzene, and α-pinene. Clean synthetic air was measured every 3 h for 10 min to determine the instrument background. Synthetic air was supplied to the instrument only and not the whole inlet. The time resolution of the measurement was 1 min and the mass range extended to 450 amu. Mass resolution (full width half maximum) at mass 96 amu was ca. 3500 during the first leg and >4500 during the second leg.
The total uncertainty of the DMS measurement was <30 % (main sources of uncertainty: standard gas mixture 5 %, flow meter 1 %, calibration ≈10 %) and the precision <5 %. DMSO2 and MSAM were not present in the calibration gas. Calculation of the mixing ratio was therefore conducted based on the rate coefficients for proton transfer (Su and Chesnavich, 1982; Chesnavich et al., 1980), the knowledge of transmission factors, the amount of H3O+ ions, and the parameters of the drift region (Lindinger et al., 1998). Applying this method results in a greater uncertainty than for compounds included in the calibration gas mixture of approximately 50 %. The limit of detection (LOD: 3× standard deviation of background) was 20 ppt for DMS, 25 ppt for DMSO2, and 5 ppt for MSAM.
2.4 Discussion of inlet effects
Semi-volatile and especially low-volatile compounds partition reversibly from the gas phase to the walls in Teflon tubing (Pagonis et al., 2017; Liu et al., 2019; Deming et al., 2019). Teflon tubing acts approximately as a chromatography column for these compounds (Pagonis et al., 2017). This leads to a smearing of the time profile of these compounds, which affects the measured concentrations (Deming et al., 2019). To evaluate this effect we measured a step concentration change from a stable MSAM concentration to a zero MSAM concentration (at same humidity and flow). This resulting decay does not necessarily follow a single exponential decay (Liu et al., 2019). In our case we found the best fit was obtained with a triple exponential decay. The e-folding times (time it takes for the signal to decrease by the factor 1∕e) were from minutes to hours (for more details, see Sect. S5). This leads to considerable smoothing of the MSAM signal for concentration changes on timescales of minutes up to hours.
The partitioning of MSAM to the inside wall of the Teflon tubing raises the question of whether the observed MSAM could be generated there on surfaces. No inlet test was done during the campaign to address this issue since this discovery was a surprise. We do not know of a pathway to production in the inlet (see Sect. S6 for a discussion), but it remains a possibility that cannot yet be ruled out.
2.5 HYSPLIT back trajectories
Air mass back trajectories were calculated to investigate the origin of air masses encountered. The Hybrid Single-Particle Lagrangian Integrated Trajectory model (HYSPLIT, version 4, 2014), a hybrid between a Lagrangian and an Eulerian model for tracing small imaginary air parcels forward or back in time (Draxler and Hess, 1998), was used to derive 3-D back trajectories from a starting height of 200 m a.s.l. (above sea level), going 216 h back in time on an hourly grid beginning at the ship's position.
This study focuses on the two crossings of the Arabian Sea during the AQABA campaign. The Arabian Sea was generally characterized by low values of VOCs, especially VOCs related to anthropogenic activities (Bourtsoukidis et al., 2019, 2020; Pfannerstill et al., 2019; Wang et al., 2020). The three molecules presented in this study are the exception. They were higher in the Arabian Sea region than in the other regions. So the absence or very low concentrations of the other molecules plus the high values (compared to other regions) of DMS, DMSO2, and MSAM characterize this part of the cruise as mostly influenced by clean marine air.
3.1 Dimethyl sulfide (DMS)
Measurements of DMS (m∕z 63.0263) during AQABA showed elevated mixing ratios when the vessel traversed the Arabian Sea during both legs (brown shaded region in Fig. 3a). During the first leg over the Arabian Sea (Fig. 3b), DMS mixing ratios were generally in the range of 300–500 ppt, with occasional peaks of 800 ppt. During the second leg (Fig. 3c), the DMS mixing ratios over the Arabian Sea were significantly lower, in the range of 100–300 ppt, again with elevated peaks of short duration (around 2 h).
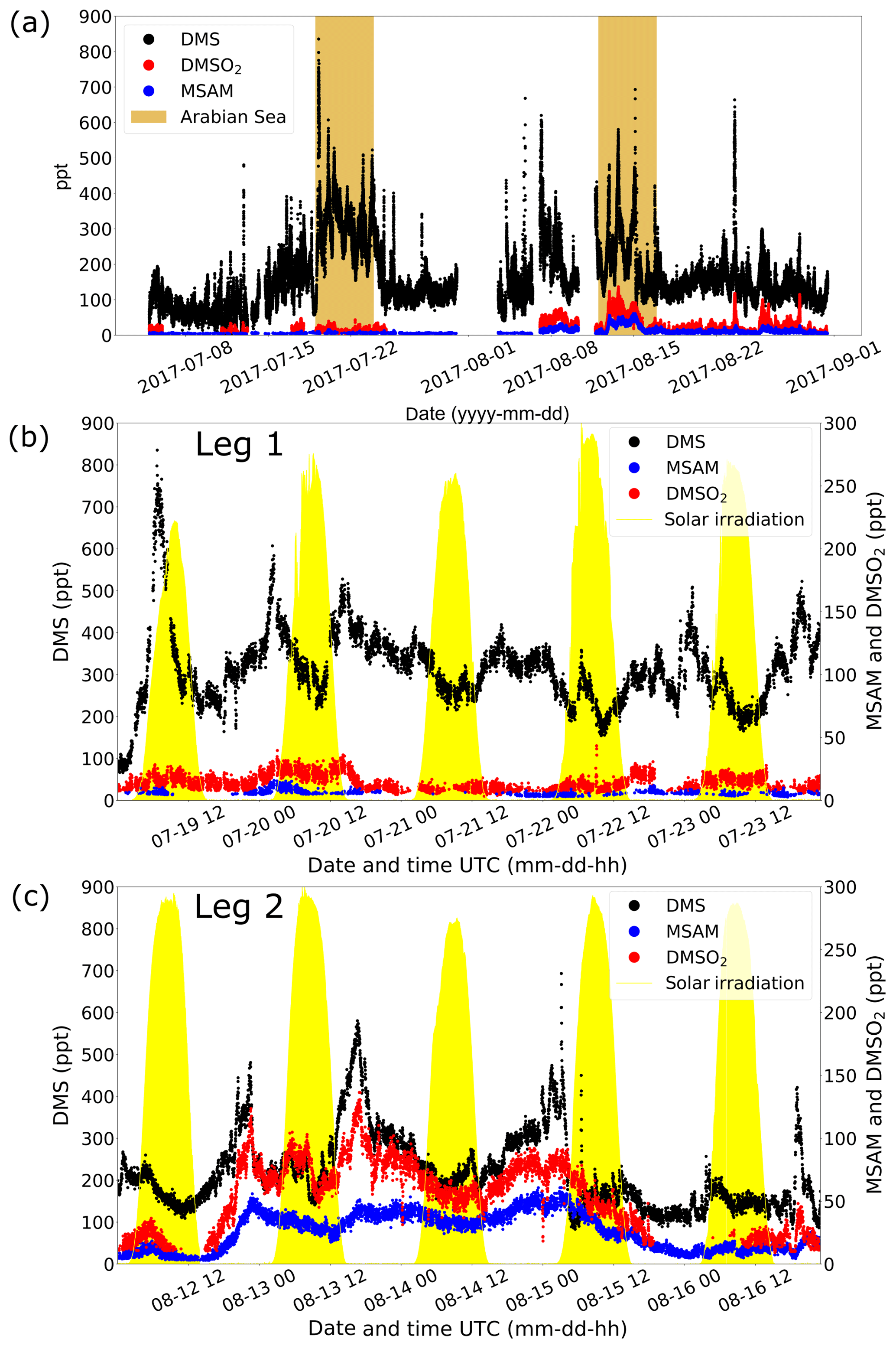
Figure 3In (a) the mixing ratios of for DMS, DMSO2, and MSAM during the whole AQABA cruise are displayed. The Arabian Sea parts of legs 1 and 2 are marked in brown. A zoom-in on measurements over the Arabian Sea is given for leg 1 in (b) and for leg 2 in (c). The yellow filled curves in (b) and (c) show the solar irradiation in arbitrary units.
3.2 Dimethyl sulfone (DMSO2)
DMSO2 was measured by the PTR-ToF-MS at m∕z 95.0161. Within the range of uncertainty for that mass, we did not find another plausible molecular structure. Additionally, the head space of the pure compound (TCI Deutschland GmbH, purity > 99 %) was sampled yielding a peak at the same position as found in ambient air. Measurements of DMSO2 in the Arabian Sea region showed elevated levels between 40 and 120 ppt during the second leg (Fig. 3c) but more modest levels (<40 ppt) on leg 1 (Fig. 3b). To our knowledge, there have been no measurements of DMSO2 performed in this region previously.
3.3 New atmospheric trace gas: MSAM
At m∕z 96.0144, a signal was observed which displayed a strong correlation with DMSO2 (Pearson correlation coefficient: r around 0.8) over the Arabian Sea during the second leg (see Fig. 4). This mass corresponded to MSAM, which has a similar structure to DMSO2, with an amine group substituted for a methyl group (see Fig. 1 for the chemical structures of the molecules). This molecule has not previously been measured in ambient air. To confirm the assignment of mass m∕z 96.0144 to MSAM, the head space of the pure substance MSAM (Alfa Aesar, purity > 98 %) was sampled by the PTR-ToF-MS. The analysis of the pure compound MSAM by PTR-ToF-MS matched the mass found in ambient air. No other plausible molecular structures could be found for this mass within the uncertainty for that mass. Based on the correlation of mass m∕z 96.0144 to DMSO2 in ambient data, the mass spectral match to the pure compound, and the absence of alternative structures at that exact mass, we identify the measured signal as MSAM. In order to test whether MSAM can be observed outgassing from seawater, we flushed the headspace of solutions of 4.2, 0.05, and 0.0005 mol L−1 MSAM in artificial seawater with 100, 50, and 25 mL min−1 of synthetic air (Air Liquide, Krefeld, Germany) each. The resulting mixing ratios measured ranged from 650 ppt (lowest concentration and lowest flow rate) to 130 000 ppt (highest concentration and highest flow rate). Up to a certain flow value the increase in dilution due to an increase in flow is overcompensated for through an enhanced emission of the substance from the seawater. Therefore we measured the highest MSAM mixing ratios at the highest flow rate. During the Arabian Sea section of the second leg, values of up to 60 ppt were measured, but mostly it was found to be in the range of 20–50 ppt. On the first leg, MSAM was occasionally detected in the Arabian Sea, but concentrations were generally close to the LOD.
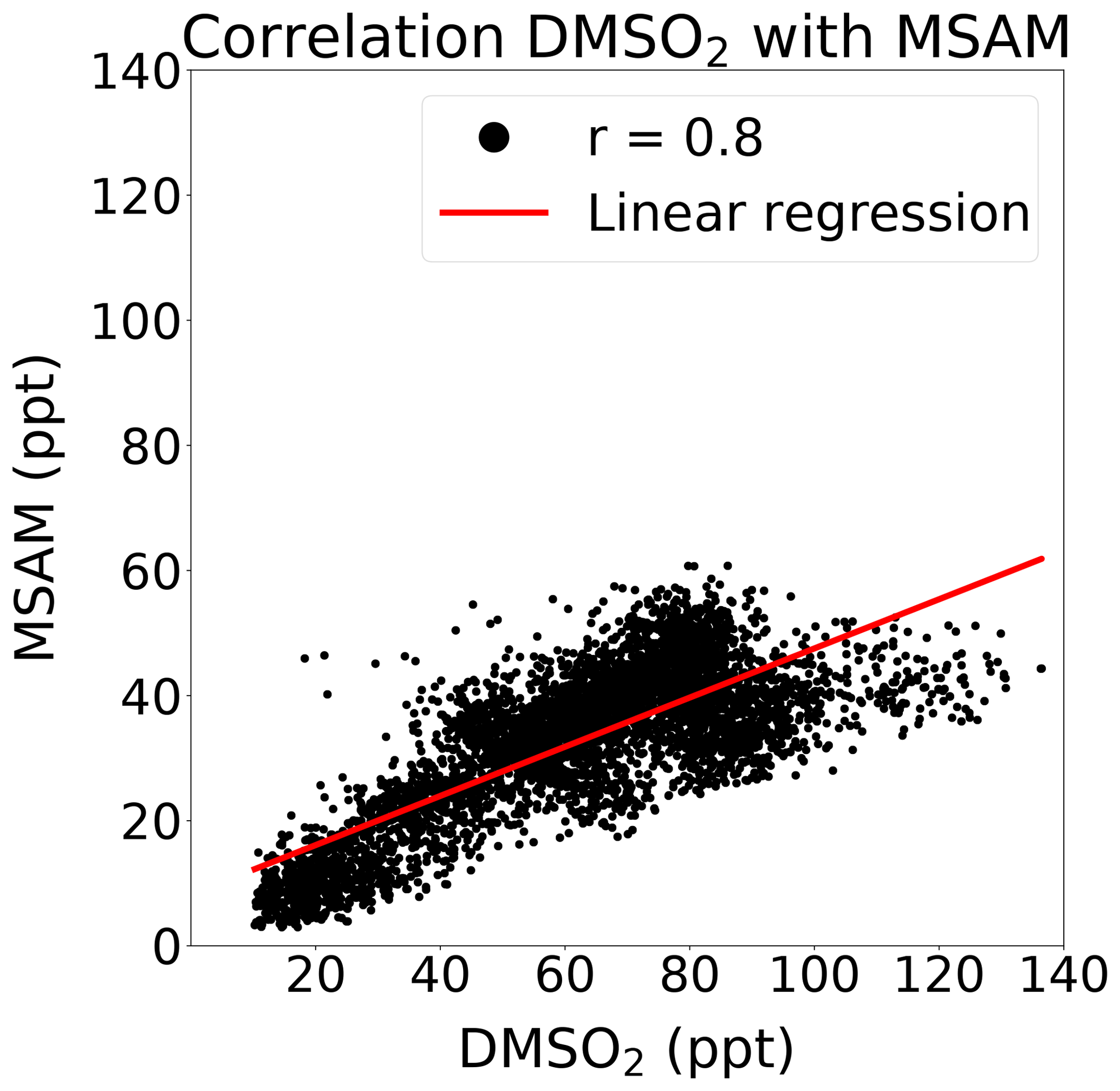
Figure 4Correlation of MSAM with DMSO2 during the second leg in the Arabian Sea. Displayed in red the is the linear regression ([MSAM] = ppt). The Pearson correlation coefficient is 0.8.
Here we discuss DMS, DMSO2, and MSAM measurements in air from a rarely sampled region, the Arabian Sea. First we discuss the difference in DMS abundance between the two legs. Secondly we evaluate the source regions of these trace gases based on knowledge of their atmospheric lifetimes and chlorophyll exposure. Then, finally, we address the implications of these measurements to marine boundary layer chemistry.
4.1 DMS
We observed higher values of DMS in July (leg 1) than in August (leg 2) over the Arabian Sea (see Sect. 3.1). This finding is consistent with DMS flux predictions by Lana et al. (2011) for this region. Sea surface DMS concentrations can be used to estimate DMS fluxes to the atmosphere, and a global climatology of DMS surface water concentrations has been derived by Lana et al. (2011) from over 47 000 seawater measurements worldwide. Lana et al. (2011) predict strong fluxes of DMS in the Arabian Sea region, particularly in June, July, and August, coincident with the AQABA campaign. Seawater concentration data for DMS from the Lana climatology relevant for AQABA has been plotted in Fig. 5 for July and August. In the regions south of the Arabian Peninsula, higher concentrations of DMS in seawater are expected in July than in August. Therefore the measured higher mixing ratios of DMS in July than August support the results of the climatology for this region.
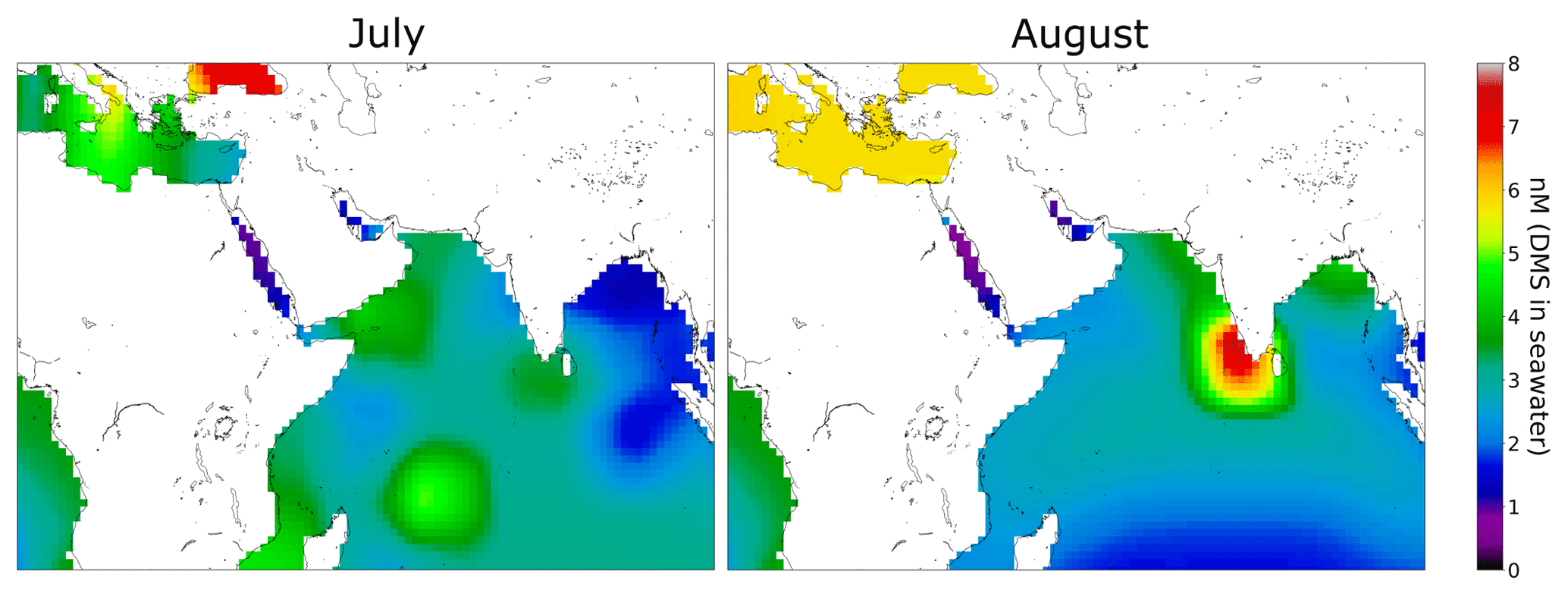
Figure 5Climatology for DMS surface water concentrations in July and August (Lana et al., 2011). Over 47 000 DMS seawater measurements were used together with interpolation or extrapolation techniques in order to obtain a monthly DMS surface water concentration of the whole earth. In the regions south of the Arabian Peninsula the climatology estimates higher values in July than in August.
The highest DMS mixing ratios occurred during the first leg over the Gulf of Aden with around 800 ppt. The peak values are likely related to the ship crossing directly through patches of phytoplankton as evidenced by the observation of strong bioluminescence around the ship during the night. The DMS mixing ratio values of up to 300 ppt during the second leg can be compared to measurements made previously in that region. DMS values of up to 250 ppt associated with upwelling in the Gulf of Aden were reported during a ship cruise in April 2000 (Warneke and de Gouw, 2001). More recent measurements during a ship cruise in July and August 2018 in the western tropical Indian Ocean reported values of up to 300 ppt DMS (Zavarsky et al., 2018). The DMS measurements presented here are therefore consistent with the very limited previous measurements in this region.
4.2 Atmospheric lifetimes of DMS, DMSO2, and MSAM
The lifetime of DMS in the atmosphere with respect to the primary oxidant OH is around 1.3 d (bimolecular rate constant = cm3 molec.−1 s−1 (Albu et al., 2006)). For all lifetimes with respect to OH we use the global average concentration [OH] = 1.1×106 molec. cm−3 (Prinn et al., 2005). In some regions of the marine boundary layer, BrO may also contribute to the oxidation of DMS leading to shorter DMS lifetimes (Breider et al., 2010; Khan et al., 2016; Barnes et al., 2006).
The reaction rate of OH and DMSO2 is cm3 molec.−1 s−1, which leads to a tropospheric lifetime of more than 35 d ([OH] = 1.1×106 molec. cm−3) (Falbe-Hansen et al., 2000), over 20 times longer than DMS. A recent study, Berasategui et al. (2020), measured a rate constant of cm3 molec.−1 s−1 for the reaction of OH with MSAM, which results in a tropospheric lifetime of 75 d ([OH] = 1.1×106 molec. cm−3). As MSAM has a long lifetime with respect to reaction with OH, we must also consider its physical removal by deposition to the ocean surface. We therefore carried out experiments to determine the Henry's law constant for MSAM (for details, see Sect. S2) and found it to be in the range of 3.3×104–6.5×105 M atm−1. DMSO2 has a similarly large Henry's law constant M atm−1 (de Bruyn et al., 1994). A two-layer model can predict the exchange flux between the gas and aqueous phase to derive an estimate of the effective lifetimes (Liss and Slater, 1974; Schwartz, 1992; Yang et al., 2014). For substances with a high Henry's law constant, like MSAM and DMSO2, knowledge of the wind speed and the marine boundary layer height is sufficient to get a prediction of the deposition lifetime (for details, see Sect. S1 in the Supplement). This gives a lifetime of 30.5±23.5 h (marine boundary layer height: 750±250 m and wind speeds 4 to 14 m s−1). The lifetimes for MSAM and DMSO2 are therefore controlled by the deposition rate to the ocean surface and not by OH oxidation. This means that DMS, DMSO2, and MSAM have similar lifetimes. During 12 and 13 August, most of the time air masses from the Somalia upwelling traveled for 10 h up to a day before reaching the ship. On 14 and 15 August, air masses from the Somalia upwelling were around 4 h old (determined from the HYSPLIT back trajectories). A common oceanic source and the similar lifetimes of DMSO2 and MSAM help explain the observed good correlation of MSAM with DMSO2 (see Fig. 4).
4.3 Chlorophyll exposure of HYSPLIT back trajectories
MSAM and DMSO2 were close to the LOD during the first leg, despite the fact that DMS mixing ratios were even higher than during the second leg. This observation excludes a simple relationship between the emissions of DMS and DMSO2 or MSAM. DMSO2 is known to be an oxidation product of DMS and is therefore linked to marine biogenic activity (Barnes et al., 2006). We hypothesize that the newly detected trace gas MSAM is also linked to marine biogenic activity. This is based on the observation that MSAM displays the highest values when influenced mainly by remote marine air without recent contact with land; it correlates well with DMSO2 (see Fig. 4) and is similar in chemical structure to DMS and DMSO2. A good indicator of marine biogenic activity is phytoplankton. Phytoplankton in the water can be detected from space via the chlorophyll a pigment used for photosynthesis. Satellite images of regional chlorophyll can be exploited to investigate emission areas of marine biogenic VOCs. In the following sections, and with the help of HYSPLIT back trajectories and chlorophyll a water content we will investigate where the source of MSAM and DMSO2 is located.
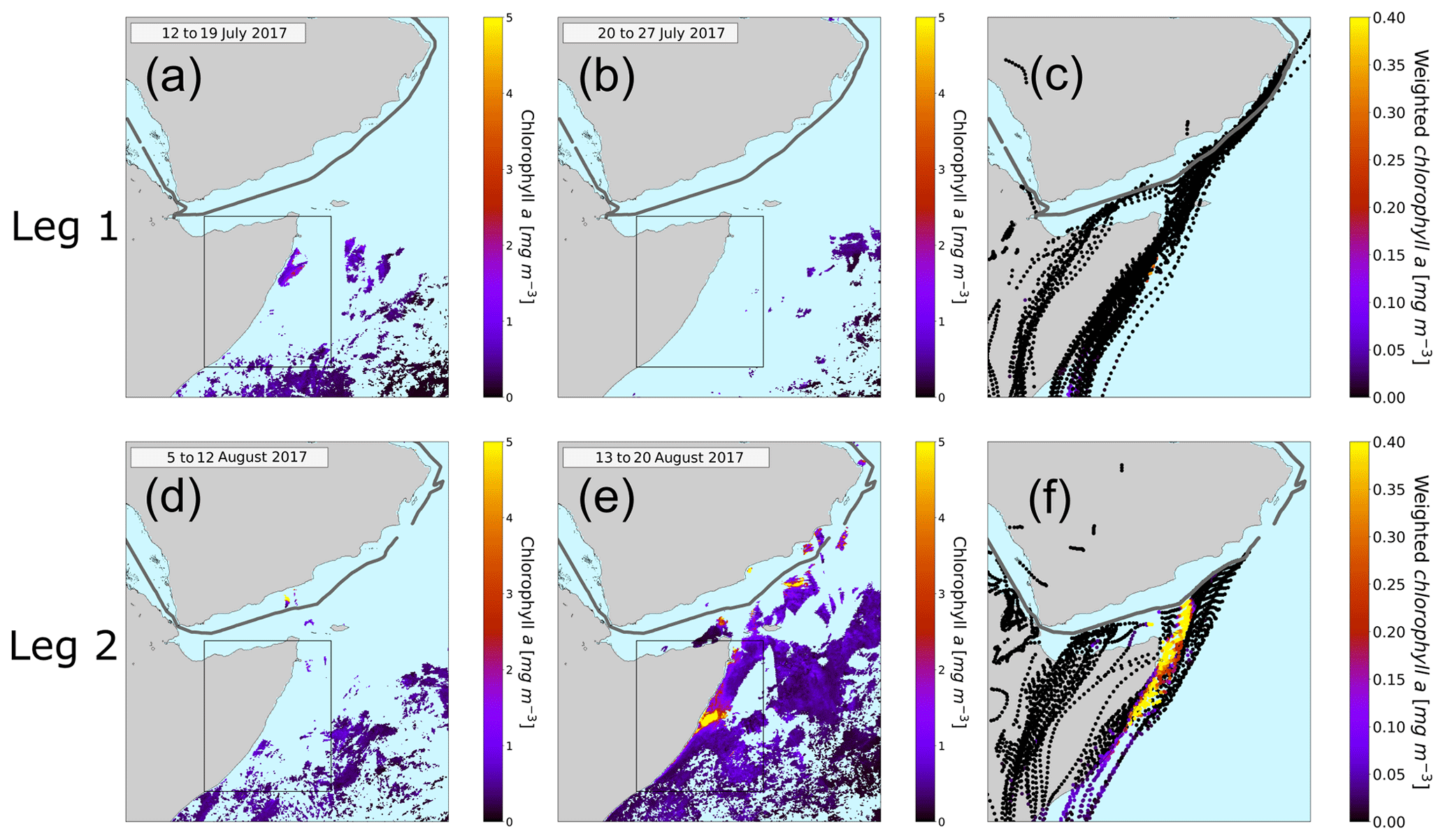
Figure 6Eight-day averaged chlorophyll a concentration in the Arabian Sea. The relevant time periods for the first leg (a, b) and for the second leg (d, e) are pictured here. Panel (a) represents the average chlorophyll a concentration from 12 to 19 July, (b) from 20 to 27 July, (d) from 5 to 12 August, and (e) from 13 to 20 August. The black rectangle represents the region of the Somalia coast upwelling. The ship track is plotted in gray. The light blue means no chlorophyll a content. Panels (c) and (f) display the HYSPLIT back trajectories. The color code of the trajectories represents the weighted amount of chlorophyll a concentration in the water underneath the trajectory.
4.3.1 Chlorophyll a water content
We used data from the satellite MODIS-Aqua (Jackson et al., 2019). In Fig. 6a, b and d, e the chlorophyll a concentration averaged over 8 d is plotted for the time periods relevant for our measurements. During the first leg (Fig. 6a and b) the ship entered the Arabian Sea region on 19 July and left it around 23 July 2017. Figure 6a displays the average chlorophyll distribution from 12 to 19 July, since air masses reaching the ship from 19 July onwards will have traveled over chlorophyll a regions before the time of observation. For the second leg (Fig. 6d and e) (12 to the 16 August 2017) we used the average chlorophyll a content from 5 to 12 August and from 13 to 20 August. The highest chlorophyll concentrations in the region are found off the Horn of Africa or Somalia coast, a strong upwelling region.
4.3.2 Back-trajectory chlorophyll analysis
A trajectory analysis was carried out to investigate whether air masses traveling over regions of high chlorophyll are associated with higher atmospheric levels of DMSO2 and MSAM. To investigate the provenance of air masses sampled at the ship in relation to the chlorophyll distributions shown above, HYSPLIT back trajectories were calculated (9 d back) for every full hour of the cruise. For each point of the trajectory the underlying chlorophyll a content was weighted with respect to the arrival time at the ship to account for oxidation and dispersion effects (see Fig. 6c and f). These values were then added up for each trajectory individually in order to determine quantitatively to what extent the air sampled had passed over areas of high chlorophyll content (indicator for phytoplankton in the water). Only time points when the trajectory was within the marine boundary layer, as calculated from the HYSPLIT model, were considered. Changing the weighting did not materially affect the results. For details on the weighting procedure, see Sect. S3.
The results of these calculations are displayed in Fig. 7. The graphs show the total chlorophyll exposure and the exposure of chlorophyll specifically from the region of the Somalia upwelling (see Fig. 6, the region in the black rectangle). On the first leg (Fig. 7a), when both DMSO2 and MSAM mixing ratios were low, air reaching the ship did not cross chlorophyll-a-rich waters in the previous 1 to 2 d. This is the case for the total exposure as well as for the exposure to chlorophyll in the Somalia upwelling region.
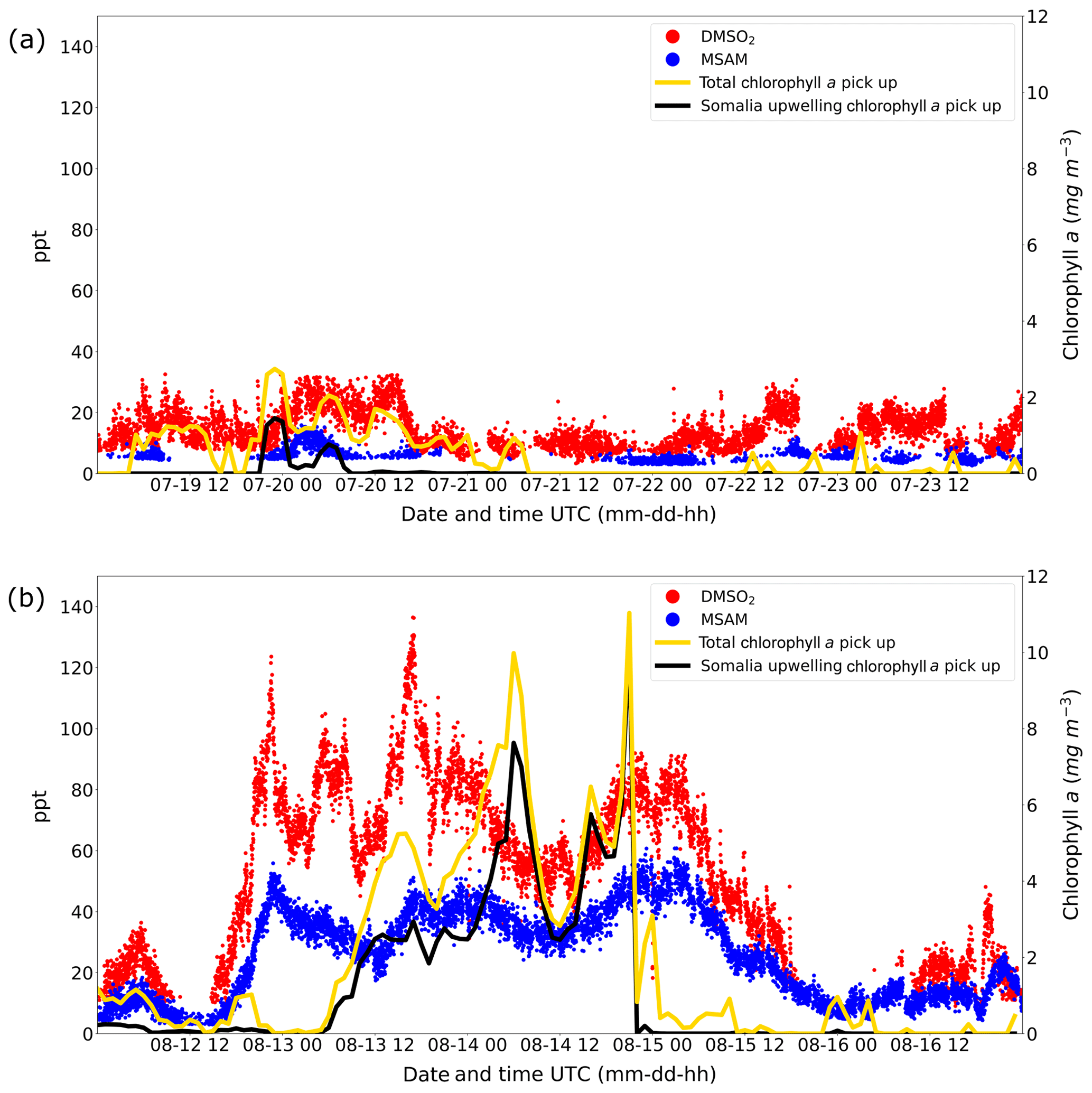
Figure 7Weighted amount of chlorophyll a trajectories crossed over before arrival at the ship for leg 1 (a) and leg 2 (b). The total chlorophyll a exposure (yellow line) and the chlorophyll a exposure originating from the Somalia upwelling region (black line) are plotted. The corresponding y axis for the chlorophyll a exposure for both graphs (a, b) is displayed on the right side. Mixing ratios in ppt for DMSO2 and MSAM are plotted in red and blue with the corresponding y axis on the left side.
However, during the second leg (Fig. 7b), when DMSO2 and MSAM levels were high, the air measured had traveled over extensive chlorophyll-a-rich waters. In general, the exposure in the Somalia upwelling region always constituted the majority of the total exposure, except for one peak in the beginning (13 August from 12:00 to 19:00 UTC) where chlorophyll patches closer to the ship constituted roughly half of the total chlorophyll exposure. From these calculations we can conclude that the occurrence of DMSO2 and MSAM is related to marine emissions in the Somalia upwelling region. MSAM and DMSO2 mixing ratios started to increase around 12:00 UTC of 12 August but the chlorophyll exposure only started to increase around 06:00 UTC on 13 August (Fig. 7b). A possible explanation for the delay in chlorophyll exposure could be that the bloom already started on 12 August and not just on 13 August as indicated in Fig. 6d and e. Possibly the earlier start of the bloom escaped detection by MODIS-Aqua as it sees every point on earth every 1–2 d. Around the Equator some regions escape detection on any given day. An inspection of daily data from MODIS-Aqua revealed that parts of the Somalia upwelling were in a blind spot of the satellite on 12 August. The chlorophyll exposure fell sharply to zero at the beginning of 15 August, roughly 8 h before DMSO2 and MSAM values start to decline as well (Fig. 7b). In this case the calculated HYSPLIT back trajectories no longer pass over the Somalia upwelling but cross Somalia before arriving at the ship. Our measurements thus indicate that we were impacted by the Somalian upwelling region for longer than calculated from the trajectories. This is not unexpected as meteorological data for this region are sparse and the trajectories therefore correspondingly uncertain.
4.4 DMSO2, DMSO, MSIA, and MSA
In the following we will discuss briefly why we did not detect DMSO, methane sulfinic acid (MSIA), and methane sulfonic acid (MSA). These are important oxidation products from DMS.
DMSO is known to be an important intermediate in the oxidation of DMS with OH (Hoffmann et al., 2016). The reaction rate of DMSO with OH is 15 times faster than that of DMS with OH, making it a potentially important sink in the remote marine atmosphere (Barnes et al., 2006). A model study of the sulfur cycle in the global marine atmosphere suggested values of around 10 ppt for DMSO in the region of the Arabian Sea (Chen et al., 2018). This is below the limit of detection (LOD) of around 15 ppt for DMSO in our instrument and probably the reason why we do not observe it in this dataset. Measurements of DMSO made on Amsterdam Island ranged from 0.36 to 11.6 ppt (Sciare et al., 2000).
MSIA has a very high reaction rate of cm3 molec.−1 s−1 with OH radicals (Burkholder et al., 2015; Kukui et al., 2003; Hoffmann et al., 2016). In the model study mentioned above, this leads to concentrations over the Arabian Sea of around 2 ppt which again is below the LOD for MSIA with our instrument (around 20 ppt) (Chen et al., 2018).
MSA, on the other hand, is predicted to be around 20–40 ppt over the Arabian Sea (gas phase and aqueous phase combined), which is above its LOD, but almost all of it will be in the aqueous phase (Chen et al., 2018; Hoffmann et al., 2016). In the gas phase, the maximum MSA values reported to date are below 1 ppt, which is far too low to be measured with our setup (LOD around 15 ppt) (Eisele and Tanner, 1993; Chen et al., 2016; Berresheim, 2002).
We observed DMSO2 mixing ratios during the second leg between 40 and 120 ppt, which is high compared to previous measurements of 0.2–11 ppt (Berresheim et al., 1998; Davis et al., 1998) made in Antarctica. In the following we will briefly describe and discuss formation pathways for DMSO2. This will be done for formation via OH, BrO, and NO3.
4.4.1 OH
DMSO2 formation from OH oxidation, via the intermediate DMSO, has been observed in laboratory studies (Falbe-Hansen et al., 2000). However, it has to be noted that newer studies indicate that mainly MSIA and not DMSO2 is formed (Barnes et al., 2006; Kukui et al., 2003; Hoffmann et al., 2016). Another pathway is formation from the OH–DMS adduct followed by sequential reactions with NO and O2 (Arsene et al., 2001; Barnes et al., 2006). Due to the low NOx of the remote marine atmosphere, this pathway seems unlikely as well. There are speculations about a possible formation from the initially formed OH–DMS adduct by O2 addition and a subsequent complex process which is not yet fully understood (Berndt and Richters, 2012; Arsene et al., 1999; Turnipseed et al., 1996; Barnes et al., 2006).
4.4.2 BrO
BrO can form DMSO2 from DMSO. But these reactions seems rather unlikely because of the slow reaction rate compared to the fast reaction rate of OH and DMSO. This reaction, therefore, would only play a role for much higher concentrations of BrO and not for concentrations of 2 ppt for BrO, which have been proposed to be ubiquitous in the marine troposphere (Read et al., 2008; Platt and Hönninger, 2003).
4.4.3 NO3
Most studies show no formation of DMSO2 from NO3 oxidation (Barnes et al., 2006). NO3 oxidation of the intermediate DMSO is known to only yield DMSO2 (Falbe-Hansen et al., 2000). However, NO3 oxidation is not thought to produce DMSO (Barnes et al., 2006). Maybe the increase in DMSO2 after sunset (see Fig. 3c) is an indication that NO3 is oxidizing the remaining DMSO from the OH oxidation of DMS produced during the daytime.
With the data presented here it is not possible to decide if one or some of the abovementioned mechanisms are responsible for the observed DMSO2 values. A diel analysis of DMS, MSAM, and DMSO2 was made. But due to the fact that we only have 2 consecutive days with elevated DMSO2 on a moving platform, the results must be viewed with caution since variation may come from source or removal process variation. Nevertheless, for completeness, the plots and description of this diel variability are found in Sect. S4.
4.5 MSAM
We are not aware of a possible formation pathway for MSAM in the gas phase. Therefore, we consider it rather unlikely that it is formed via DMS gas phase oxidation. A microbial formation from DMS or DMS products in the water of the highly biological active upwelling region with subsequent emission into the atmosphere seems plausible (see Fig. 1).
To our knowledge there have been no reports of MSAM occurring or being produced in biological systems. MSAM belongs to the class of sulfonamides which is known for its antibacterial properties, and it has therefore been used in antibacterial drugs (Sköld, 2010). The only mention of MSAM in this context was as a metabolite of a drug detected in human urine (Anacardio et al., 2009).
From the dataset presented in this paper, the ocean is expected to be a sink for MSAM. This is shown through our calculations of the lifetime (a few hours to a few days), which are dominated by deposition. The ocean can only become a source of MSAM to the atmosphere if the concentration of MSAM in surface seawater is so large that emission locally dominates over deposition. Our measurements indicate that this was the case in the region of the Somalia upwelling. Although no seawater measurements were made in that region to confirm this, the trajectory data presented here indicate that biologically active areas are able to produce sufficiently large MSAM concentrations.
To our knowledge, no measurements of MSAM have been reported in the atmosphere so far, and thus no information about the potential role it could play there is available. SO2 is an oxidation product of MSAM (Berasategui et al., 2020) and a precursor for sulfuric acid (H2SO4). H2SO4 is known to be a key contributor to new particle formation (Li et al., 2018; Kulmala et al., 2014; Almeida et al., 2013; Sipilä et al., 2010; Weber et al., 1996, 2001; Chen et al., 2016). However, due to the slow reaction of MSAM with OH, the contribution of MSAM to SO2 is negligible (Berasategui et al., 2020). Acid–base reactions (e.g., H2SO4 with ammonia or amines) are very important in new particle formation (Chen and Finlayson-Pitts, 2017; Almeida et al., 2013). MSAM is an acid and therefore could participate in acid–base reactions, but since MSAM is only a weak acid (pKa=10.8; Junttila and Hormi, 2009) its role as an acid in these reactions is probably limited.
Studies indicate that the dominant driving force in new particle formation and growth are the hydrogen bonds formed between common atmospheric nucleation precursors (Xie et al., 2017; Cheng et al., 2017; Li et al., 2018). The newly found trace gas MSAM is very intriguing because it contains a sulfonamide group, which is a sulfonyl group connected to an amine group. The sulfonyl and the amine group both support hydrogen bonding, giving MSAM a high hydrogen-bonding capacity, potentially enabling nucleation.
Because of the comparable lifetimes of MSAM and DMS, we can estimate the relative emission of MSAM to DMS from the ratio of the concentrations [MSAM] ∕ [DMS]. We only included ratios observed in the afternoons of 14 and 15 August, when the ship was in closest proximity to the Somalia upwelling. The afternoon was chosen to make sure that both MSAM and DMS have roughly the same atmospheric lifetime when estimating the relative emission. Deposition to the ocean surface will happen all the time for MSAM; however, OH, the main loss process for DMS, is only present during the day. We derived ratios ranging from 0.1 to 0.27, meaning that emissions of MSAM over the Somalia upwelling can be almost a third of the DMS emissions. Therefore, MSAM could play an important role in particle formation and/or growth over and downwind of upwelling regions. To verify these possibilities, further experiments regarding particle growth and formation with MSAM need to be performed.
During the AQABA campaign we made the first measurements of MSAM in ambient air. Back-trajectory chlorophyll analyses suggest that it is a marine biogenic emission from the highly productive upwelling region off Somalia. During the first leg of the AQABA campaign the ship encountered mostly biogenic emissions from sources located along the ship route when crossing the Arabian Sea. The enhanced DMS values observed there could be attributed to seasonally enhanced DMS fluxes. No oxidation products or other organosulfur compounds were detected in substantial amounts from the local emissions. In contrast, during the second leg, not only DMS but also DMSO2 and MSAM were measured. DMSO2, like MSAM, was shown to originate from the Somalia upwelling region. DMSO2 mixing ratios of up to 120 ppt were measured during the second leg, which is quite substantial considering that previous studies indicate it to be a minor or negligible product in DMS gas phase oxidation. The main loss mechanism for MSAM and DMSO2 is deposition to the ocean surface with lifetimes of a few hours to a few days. MSAM is a molecule which, to our knowledge, has never been reported in the atmosphere. We detected it in concentrations up to 60 ppt during the second leg in the Arabian Sea. Emissions of MSAM over the Somalia upwelling can reach close to a third of the DMS emissions.
A marine emission containing a nitrogen atom is somewhat surprising since under most circumstances primary productivity in the ocean is nitrogen limited. The emission of a nitrogen-containing compound may be related to the abundance of reactive nitrogen provided by the upwelling. Due to its sulfonyl and amine group, MSAM has a high hydrogen-bonding capacity enabling hydrogen bonding to other atmospheric nucleation precursors. These hydrogen bonds are known to be a critical factor in particle growth and formation (Li et al., 2018; Xie et al., 2017; Cheng et al., 2017). Therefore MSAM could prove to be of importance for particle formation and/or growth over upwelling regions. The mechanisms in which gas phase precursors lead to new particle formation are an active research area in atmospheric chemistry because they are subject to large uncertainties (Li et al., 2018; Chen et al., 2016, 2018; Carslaw et al., 2013).
Data available via https://doi.org/10.17617/3.3o (Edtbauer, 2020).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-6081-2020-supplement.
AE and CS were responsible for VOC measurements and data. AE analyzed the data and drafted the article. EYP contributed laboratory experiments concerning Henry's law constant. MB and JNC contributed to data interpretation. DW calculated the back trajectories. JL designed and realized the campaign. JW supervised the study. All authors contributed to manuscript writing and revision and read and approved the submitted version.
The authors declare that they have no conflict of interest.
We thankfully acknowledge the cooperation with the Cyprus Institute (CyI), the King Abdullah University of Science and Technology (KAUST), and the Kuwait Institute for Scientific Research (KISR). We thank Hays Ships Ltd., Captain Pavel Kirzner, and the ship crew for their support on-board the Kommandor Iona. We would like to express our gratitude to the whole AQABA team, particularly Hartwig Harder for daily management of the campaign, and Marcel Dorf, Claus Koeppel, Thomas Klüpfel, and Rolf Hofmann for logistical organization and help with preparation and setup. We are thankful to Jan Schuladen for the J-value measurements, Ulrike Weis for providing artificial seawater, and Tom Jobson and Franziska Köllner for helpful discussions.
The article processing charges for this open-access publication were covered by the Max Planck Society.
This paper was edited by Steven Brown and reviewed by Eric Saltzman and one anonymous referee.
Ajith Joseph, K., Jayaram, C., Nair, A., George, M. S., Balchand, A. N., and Pettersson, L. H.: Remote Sensing of Upwelling in the Arabian Sea and Adjacent Near-Coastal Regions, in: Remote sensing of the Asian Seas, vol. 92, edited by: Barale, V. and Gade, M., Springer, Cham, 467–483, Switzerland, https://doi.org/10.1007/978-3-319-94067-0_26, 2019. a
Albu, M., Barnes, I., Becker, K. H., Patroescu-Klotz, I., Mocanu, R., and Benter, T.: Rate coefficients for the gas-phase reaction of OH radicals with dimethyl sulfide: temperature and O2 partial pressure dependence, Phys. Chem. Chem. Phys., 8, 728–736, https://doi.org/10.1039/B512536G, 2006. a
Almeida, J., Schobesberger, S., Kürten, A., Ortega, I. K., Kupiainen-Määttä, O., Praplan, A. P., Adamov, A., Amorim, A., Bianchi, F., Breitenlechner, M., David, A., Dommen, J., Donahue, N. M., Downard, A., Dunne, E., Duplissy, J., Ehrhart, S., Flagan, R. C., Franchin, A., Guida, R., Hakala, J., Hansel, A., Heinritzi, M., Henschel, H., Jokinen, T., Junninen, H., Kajos, M., Kangasluoma, J., Keskinen, H., Kupc, A., Kurtén, T., Kvashin, A. N., Laaksonen, A., Lehtipalo, K., Leiminger, M., Leppä, J., Loukonen, V., Makhmutov, V., Mathot, S., McGrath, M. J., Nieminen, T., Olenius, T., Onnela, A., Petäjä, T., Riccobono, F., Riipinen, I., Rissanen, M., Rondo, L., Ruuskanen, T., Santos, F. D., Sarnela, N., Schallhart, S., Schnitzhofer, R., Seinfeld, J. H., Simon, M., Sipilä, M., Stozhkov, Y., Stratmann, F., Tomé, A., Tröstl, J., Tsagkogeorgas, G., Vaattovaara, P., Viisanen, Y., Virtanen, A., Vrtala, A., Wagner, P. E., Weingartner, E., Wex, H., Williamson, C., Wimmer, D., Ye, P., Yli-Juuti, T., Carslaw, K. S., Kulmala, M., Curtius, J., Baltensperger, U., Worsnop, D. R., Vehkamäki, H., and Kirkby, J.: Molecular understanding of sulphuric acid–amine particle nucleation in the atmosphere, Nature, 502, 359–363, https://doi.org/10.1038/nature12663, 2013. a, b
Anacardio, R., Mullins, F. G. P., Hannam, S., Sheikh, M. S., O'Shea, K., Aramini, A., D'Anniballe, G., D'Anteo, L., Ferrari, M. P., and Allegretti, M.: Development and validation of an LC-MS/MS method for determination of methanesulfonamide in human urine, J. Chromatogr. B, 877, 2087–2092, https://doi.org/10.1016/j.jchromb.2009.05.051, 2009. a
Arévalo-Martínez, D. L., Steinhoff, T., Brandt, P., Körtzinger, A., Lamont, T., Rehder, G., and Bange, H W.: N2O Emissions From the Northern Benguela Upwelling System, Geophys. Res. Lett., 46, 3317–3326, https://doi.org/10.1029/2018GL081648, 2019. a
Arnold, S. R., v. Spracklen, D., Gebhardt, S., Custer, T., Williams, J., Peeken, I., and Alvain, S.: Relationships between atmospheric organic compounds and air-mass exposure to marine biology, Environ. Chem., 7, 232–241, https://doi.org/10.1071/EN09144, 2010. a
Arsene, C., Barnes, I., and Becker, K. H.: FT-IR product study of the photo-oxidation of dimethyl sulfide: Temperature and O2 partial pressure dependence, Phys. Chem. Chem. Phys., 1, 5463–5470, https://doi.org/10.1039/a907211j, 1999. a
Arsene, C., Barnes, I., Becker, K. H., and Mocanu, R.: FT-IR product study on the photo-oxidation of dimethyl sulphide in the presence of NOx – temperature dependence, Atmos. Environ., 35, 3769–3780, https://doi.org/10.1016/S1352-2310(01)00168-6, 2001. a, b
Ayers, G. P. and Gillett, R. W.: DMS and its oxidation products in the remote marine atmosphere: implications for climate and atmospheric chemistry, J. Sea Res., 43, 275–286, 2000. a
Barnes, I., Hjorth, J., and Mihalopoulos, N.: Dimethyl sulfide and dimethyl sulfoxide and their oxidation in the atmosphere, Chem. Rev., 106, 940–975, https://doi.org/10.1021/cr020529, 2006. a, b, c, d, e, f, g, h, i, j, k, l
Bentley, R. and Chasteen, T. G.: Environmental VOSCs – formation and degradation of dimethyl sulfide, methanethiol and related materials, Chemosphere, 55, 291–317, https://doi.org/10.1016/j.chemosphere.2003.12.017, 2004. a
Berasategui, M., Amedro, D., Edtbauer, A., Williams, J., Lelieveld, J., and Crowley, J. N.: Kinetic and mechanistic study of the reaction between methane sulfonamide (CH3S(O)2NH2) and OH, Atmos. Chem. Phys., 20, 2695–2707, https://doi.org/10.5194/acp-20-2695-2020, 2020. a, b, c
Berndt, T. and Richters, S.: Products of the reaction of OH radicals with dimethyl sulphide in the absence of NOx: Experiment and simulation, Atmos. Environ., 47, 316–322, https://doi.org/10.1016/j.atmosenv.2011.10.060, 2012. a
Berresheim, H.: Gas-aerosol relationships of H2SO4, MSA, and OH: Observations in the coastal marine boundary layer at Mace Head, Ireland, J. Geophys. Res., 107, 24191, https://doi.org/10.1029/2000JD000229, 2002. a
Berresheim, H., Huey, J. W., Thorn, R. P., Eisele, F. L., Tanner, D. J., and Jefferson, A.: Measurements of dimethyl sulfide, dimethyl sulfoxide, dimethyl sulfone, and aerosol ions at Palmer Station, Antarctica, J. Geophys. Res., 1998, 1629–1637, 1998. a
Bonsang, B., Gros, V., Peeken, I., Yassaa, N., Bluhm, K., Zoellner, E., Sarda-Esteve, R., and Williams, J.: Isoprene emission from phytoplankton monocultures: the relationship with chlorophyll-a, cell volume and carbon content, Environ. Chem., 7, 554–563, https://doi.org/10.1071/EN09156, 2010. a
Bourtsoukidis, E., Ernle, L., Crowley, J. N., Lelieveld, J., Paris, J.-D., Pozzer, A., Walter, D., and Williams, J.: Non-methane hydrocarbon (C2–C8) sources and sinks around the Arabian Peninsula, Atmos. Chem. Phys., 19, 7209–7232, https://doi.org/10.5194/acp-19-7209-2019, 2019. a, b
Bourtsoukidis, E., Pozzer, A., Sattler, T., Matthaios, V. N., Ernle, L., Edtbauer, A., Fischer, H., Könemann, T., Osipov, S., Paris, J.-D., Pfannerstill, E. Y., Stönner, C., Tadic, I., Walter, D., Wang, N., Lelieveld, J., and Williams, J.: The Red Sea Deep Water is a potent source of atmospheric ethane and propane, Nat. Commun., 11, 447, https://doi.org/10.1038/s41467-020-14375-0, 2020. a, b
Breider, T. J., Chipperfield, M. P., d. Richards, N. A., Carslaw, K. S., Mann, G. W., and v. Spracklen, D.: Impact of BrO on dimethylsulfide in the remote marine boundary layer, Geophys. Res. Lett., 37, L02807, https://doi.org/10.1029/2009GL040868, 2010. a
Brimblecombe, P.: The Global Sulfur Cycle, in: Treatise on geochemistry, edited by: Holland, H. D., Elsevier, Amsterdam, 559–591, https://doi.org/10.1016/B978-0-08-095975-7.00814-7, 2014. a
Burkholder, J. B., Sander, S. P., Abbatt, J., Barker, J. R., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Wilmouth, D. M., and Wine, P. H.: Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation Number 18, JPL Publication 15-10, Jet Propulsion Laboratory, Pasadena, https://doi.org/10.13140/RG.2.1.2504.2806, 2015. a
Carpenter, L. J., Archer, S. D., and Beale, R.: Ocean-atmosphere trace gas exchange, Chem. Soc. Rev., 41, 6473–6506, https://doi.org/10.1039/c2cs35121h, 2012. a
Carslaw, K. S., Lee, L. A., Reddington, C. L., Pringle, K. J., Rap, A., Forster, P. M., Mann, G. W., v. Spracklen, D., Woodhouse, M. T., Regayre, L. A., and Pierce, J. R.: Large contribution of natural aerosols to uncertainty in indirect forcing, Nature, 503, 67–71, https://doi.org/10.1038/nature12674, 2013. a
Chen, H. and Finlayson-Pitts, B. J.: New Particle Formation from Methanesulfonic Acid and Amines/Ammonia as a Function of Temperature, Environ. Sci. Technol., 51, 243–252, https://doi.org/10.1021/acs.est.6b04173, 2017. a
Chen, H., Varner, M. E., Gerber, R. B., and Finlayson-Pitts, B. J.: Reactions of Methanesulfonic Acid with Amines and Ammonia as a Source of New Particles in Air, J. Phys. Chem. B, 120, 1526–1536, https://doi.org/10.1021/acs.jpcb.5b07433, 2016. a, b, c
Chen, Q., Sherwen, T., Evans, M., and Alexander, B.: DMS oxidation and sulfur aerosol formation in the marine troposphere: a focus on reactive halogen and multiphase chemistry, Atmos. Chem. Phys., 18, 13617–13637, https://doi.org/10.5194/acp-18-13617-2018, 2018. a, b, c, d, e
Cheng, S., Tang, S., Tsona, N. T., and Du, L.: The Influence of the Position of the Double Bond and Ring Size on the Stability of Hydrogen Bonded Complexes, Scient. Rep., 7, 11310, https://doi.org/10.1038/s41598-017-11921-7, 2017. a, b
Chesnavich, W. J., Su, T., and Bowers, M. T.: Collisions in a noncentral field: A variational and trajectory investigation of ion–dipole capture, J. Chem. Phys., 72, 2641–2655, https://doi.org/10.1063/1.439409, 1980. a
Colomb, A., Yassaa, N., Williams, J., Peeken, I., and Lochte, K.: Screening volatile organic compounds (VOCs) emissions from five marine phytoplankton species by head space gas chromatography/mass spectrometry (HS-GC/MS), J. Environ. Monit., 10, 325–330, https://doi.org/10.1039/b715312k, 2008. a
Davis, D., Chen, G., Kasibhatla, P., Jefferson, A., Tanner, D., Eisele, F., Lenschow, D., Neff, W., and Berresheim, H.: DMS oxidation in the Antarctic marine boundary layer: Comparison of model simulations and held observations of DMS, DMSO, DMSO2H2SO4(g), MSA(g), and MSA(p), J. Geophys. Res., 1998, 1657–1678, 1998. a
de Bruyn, W. J., Shorter, J. A., Davidovits, P., Worsnop, D. R., Zahniser, M. S., and Kolb, C. E.: Uptake of gas phase sulfur species methanesulfonic acid, dimethylsulfoxide, and dimethyl sulfone by aqueous surfaces, J. Geophys. Res.-Atmos., 99, 16927–16932, https://doi.org/10.1029/94JD00684, 1994. a
deCastro, M., Sousa, M. C., Santos, F., Dias, J. M., and Gómez-Gesteira, M.: How will Somali coastal upwelling evolve under future warming scenarios?, Scient. Rep., 6, 30137, https://doi.org/10.1038/srep30137, 2016. a
Deming, B. L., Pagonis, D., Liu, X., Day, D. A., Talukdar, R., Krechmer, J. E., de Gouw, J. A., Jimenez, J. L., and Ziemann, P. J.: Measurements of delays of gas-phase compounds in a wide variety of tubing materials due to gas–wall interactions, Atmos. Meas. Tech., 12, 3453–3461, https://doi.org/10.5194/amt-12-3453-2019, 2019. a, b
Derstroff, B., Hüser, I., Bourtsoukidis, E., Crowley, J. N., Fischer, H., Gromov, S., Harder, H., Janssen, R. H. H., Kesselmeier, J., Lelieveld, J., Mallik, C., Martinez, M., Novelli, A., Parchatka, U., Phillips, G. J., Sander, R., Sauvage, C., Schuladen, J., Stönner, C., Tomsche, L., and Williams, J.: Volatile organic compounds (VOCs) in photochemically aged air from the eastern and western Mediterranean, Atmos. Chem. Phys., 17, 9547–9566, https://doi.org/10.5194/acp-17-9547-2017, 2017. a
Draxler, R. R. and Hess, G.: An Overview of the HYSPLIT_4 Modelling System for Trajectories, Dispersion, and Deposition, Aust. Meteorol. Mag., 47, 295–308, 1998. a
Edtbauer, A.: BVOCs measurements around the Arabian Peninsula (AQABA 2017), https://doi.org/10.17617/3.3o, 2020. a
Eisele, F. L. and Tanner, D. J.: Measurement of the gas phase concentration of H2SO4 and methane sulfonic acid and estimates of H2SO4 production and loss in the atmosphere, J. Geophys. Res., 98, 9001–9010, https://doi.org/10.1029/93JD00031, 1993. a
Falbe-Hansen, H., Sørensen, S., Jensen, N. R., Pedersen, T., and Hjorth, J.: Atmospheric gas-phase reactions of dimethylsulphoxide and dimethylsulphone with OH and NO3 radicals, Cl atoms and ozone, Atmos. Environ., 34, 1543–1551, https://doi.org/10.1016/S1352-2310(99)00407-0, 2000. a, b, c
Fowler, D., Coyle, M., Skiba, U., Sutton, M. A., Cape, J. N., Reis, S., Sheppard, L. J., Jenkins, A., Grizzetti, B., Galloway, J. N., Vitousek, P., Leach, A., Bouwman, A. F., Butterbach-Bahl, K., Dentener, F., Stevenson, D., Amann, M., and Voss, M.: The global nitrogen cycle in the twenty-first century, Philos. T. Roy. Soc. Lond. B, 368, 20130164, https://doi.org/10.1098/rstb.2013.0164, 2013. a
Fowler, D., Steadman, C. E., Stevenson, D., Coyle, M., Rees, R. M., Skiba, U. M., Sutton, M. A., Cape, J. N., Dore, A. J., Vieno, M., Simpson, D., Zaehle, S., Stocker, B. D., Rinaldi, M., Facchini, M. C., Flechard, C. R., Nemitz, E., Twigg, M., Erisman, J. W., Butterbach-Bahl, K., and Galloway, J. N.: Effects of global change during the 21st century on the nitrogen cycle, Atmos. Chem. Phys., 15, 13849–13893, https://doi.org/10.5194/acp-15-13849-2015, 2015. a
Ge, X., Wexler, A. S., and Clegg, S. L.: Atmospheric amines – Part I. A review, Atmos. Environ., 45, 524–546, https://doi.org/10.1016/j.atmosenv.2010.10.012, 2011. a
Gibb, S. W., Mantoura, R. F. C., and Liss, P. S.: Ocean-atmosphere exchange and atmospheric speciation of ammonia and methylamines in the region of the NW Arabian Sea, Global Biogeochem. Cy., 13, 161–178, https://doi.org/10.1029/98GB00743, 1999. a, b
Graus, M., Müller, M., and Hansel, A.: High resolution PTR-TOF: quantification and formula confirmation of VOC in real time, J. Am. Soc. Mass Spectrom., 21, 1037–1044, https://doi.org/10.1016/j.jasms.2010.02.006, 2010. a
Hoffmann, E. H., Tilgner, A., Schrödner, R., Bräuer, P., Wolke, R., and Herrmann, H.: An advanced modeling study on the impacts and atmospheric implications of multiphase dimethyl sulfide chemistry, P. Natl. Acad. Sci. USA, 113, 11776–11781, https://doi.org/10.1073/pnas.1606320113, 2016. a, b, c, d
Jackson, T., Chuprin, A., Sathyendranath, S., Grant, M., Zühlke, M., Dingle, J., Storm, T., Boettcher, M., and Fomferra, N.: Ocean Colour Climate Change Initiative: version 4.0, available at: ftp://ftp.rsg.pml.ac.uk/occci-v4.0/geographic/netcdf/8day/chlor_a/2017/, last access: 28 August 2019. a
Johnson, M. T., Liss, P. S., Bell, T. G., Lesworth, T. J., Baker, A. R., Hind, A. J., Jickells, T. D., Biswas, K. F., Woodward, E. M. S., and Gibb, S. W.: Field observations of the ocean-atmosphere exchange of ammonia: Fundamental importance of temperature as revealed by a comparison of high and low latitudes, Global Biogeochem. Cy., 22, GB1019, https://doi.org/10.1029/2007GB003039, 2008. a
Jordan, A., Haidacher, S., Hanel, G., Hartungen, E., Märk, L., Seehauser, H., Schottkowsky, R., Sulzer, P., and Märk, T. D.: A high resolution and high sensitivity proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF-MS), Int. J. Mass Spectrom., 286, 122–128, https://doi.org/10.1016/j.ijms.2009.07.005, 2009. a
Junttila, M. H. and Hormi, O. O. E.: Methanesulfonamide: a cosolvent and a general acid catalyst in sharpless asymmetric dihydroxylations, J. Org. Chem., 74, 3038–3047, https://doi.org/10.1021/jo8026998, 2009. a
Kämpf, J. and Chapman, P.: Upwelling Systems of the World, Springer International Publishing, Cham, https://doi.org/10.1007/978-3-319-42524-5, 2016. a, b
Khan, M., Gillespie, S., Razis, B., Xiao, P., Davies-Coleman, M. T., Percival, C. J., Derwent, R. G., Dyke, J. M., Ghosh, M. V., Lee, E., and Shallcross, D. E.: A modelling study of the atmospheric chemistry of DMS using the global model, STOCHEM-CRI, Atmos. Environ., 127, 69–79, https://doi.org/10.1016/j.atmosenv.2015.12.028, 2016. a, b
Kiene, R. P., Linn, L. J., and Bruton, J. A.: New and important roles for DMSP in marine microbial communities, J. Sea Res., 43, 209–224, https://doi.org/10.1016/S1385-1101(00)00023-X, 2000. a
Kloster, S., Feichter, J., Maier-Reimer, E., Six, K. D., Stier, P., and Wetzel, P.: DMS cycle in the marine ocean-atmosphere system – a global model study, Biogeosciences, 3, 29–51, https://doi.org/10.5194/bg-3-29-2006, 2006. a
Kukui, A., Borissenko, D., Laverdet, G., and Le Bras, G.: Gas-Phase Reactions of OH Radicals with Dimethyl Sulfoxide and Methane Sulfinic Acid Using Turbulent Flow Reactor and Chemical Ionization Mass Spectrometry, J. Phys. Chem A, 107, 5732–5742, https://doi.org/10.1021/jp0276911, 2003. a, b
Kulmala, M., Petäjä, T., Ehn, M., Thornton, J., Sipilä, M., Worsnop, D. R., and Kerminen, V.-M.: Chemistry of atmospheric nucleation: on the recent advances on precursor characterization and atmospheric cluster composition in connection with atmospheric new particle formation, Annu. Rev. Phys. Chem., 65, 21–37, https://doi.org/10.1146/annurev-physchem-040412-110014, 2014. a
Lai, S. C., Williams, J., Arnold, S. R., Atlas, E. L., Gebhardt, S., and Hoffmann, T.: Iodine containing species in the remote marine boundary layer: A link to oceanic phytoplankton, Geophys. Res. Lett., 38, L20801, https://doi.org/10.1029/2011GL049035, 2011. a
Lana, A., Bell, T. G., Simó, R., Vallina, S. M., Ballabrera-Poy, J., Kettle, A. J., Dachs, J., Bopp, L., Saltzman, E. S., Stefels, J., Johnson, J. E., and Liss, P. S.: An updated climatology of surface dimethlysulfide concentrations and emission fluxes in the global ocean, Global Biogeochem. Cy., 25, GB1004, https://doi.org/10.1029/2010GB003850, 2011. a, b, c, d, e
Li, H., Zhang, X., Zhong, J., Liu, L., Zhang, H., Chen, F., Li, Z., Li, Q., and Ge, M.: The role of hydroxymethanesulfonic acid in the initial stage of new particle formation, Atmos. Environ., 189, 244–251, https://doi.org/10.1016/j.atmosenv.2018.07.003, 2018. a, b, c, d
Lindinger, W., Hansel, A., and Jordan, A.: On-line monitoring of volatile organic compounds at pptv levels by means of proton-transfer-reaction mass spectrometry (PTR-MS) medical applications, food control and environmental research, Int. J. Mass Spectrom. Ion Proc., 173, 191–241, https://doi.org/10.1016/S0168-1176(97)00281-4, 1998. a
Liss, P. S. and Slater, P. G.: Flux of Gases across the Air-Sea Interface, Nature, 247, 181–184, https://doi.org/10.1038/247181a0, 1974. a
Liss, P. S., Marandino, C. A., Dahl, E. E., Helmig, D., Hintsa, E. J., Hughes, C., Johnson, M. T., Moore, R. M., Plane, J. M. C., Quack, B., Singh, H. B., Stefels, J., von Glasow, R., and Williams, J.: Short-Lived Trace Gases in the Surface Ocean and the Atmosphere, in: Ocean-Atmosphere Interactions of Gases and Particles, Springer Earth System Sciences, edited by: Liss, P. and Johnson, M. T., pp. 1–54, Springer-Verlag GmbH, https://doi.org/10.1007/978-3-642-25643-1_1, 2014. a
Liu, X., Deming, B., Pagonis, D., Day, D. A., Palm, B. B., Talukdar, R., Roberts, J. M., Veres, P. R., Krechmer, J. E., Thornton, J. A., de Gouw, J. A., Ziemann, P. J., and Jimenez, J. L.: Effects of gas–wall interactions on measurements of semivolatile compounds and small polar molecules, Atmos. Meas. Tech., 12, 3137–3149, https://doi.org/10.5194/amt-12-3137-2019, 2019. a, b
Mardyukov, A. and Schreiner, P. R.: Atmospherically Relevant Radicals Derived from the Oxidation of Dimethyl Sulfide, Account. Chem. Res., 51, 475–483, https://doi.org/10.1021/acs.accounts.7b00536, 2018. a
Pagonis, D., Krechmer, J. E., de Gouw, J., Jimenez, J. L., and Ziemann, P. J.: Effects of gas–wall partitioning in Teflon tubing and instrumentation on time-resolved measurements of gas-phase organic compounds, Atmo. Meas. Tech., 10, 4687–4696, https://doi.org/10.5194/amt-10-4687-2017, 2017. a, b
Paulot, F., Jacob, D. J., Johnson, M. T., Bell, T. G., Baker, A. R., Keene, W. C., Lima, I. D., Doney, S. C., and Stock, C. A.: Global oceanic emission of ammonia: Constraints from seawater and atmospheric observations, Global Biogeochem. Cy., 29, 1165–1178, https://doi.org/10.1002/2015GB005106, 2015. a
Pfannerstill, E. Y., Wang, N., Edtbauer, A., Bourtsoukidis, E., Crowley, J. N., Dienhart, D., Eger, P. G., Ernle, L., Fischer, H., Hottmann, B., Paris, J.-D., Stönner, C., Tadic, I., Walter, D., Lelieveld, J., and Williams, J.: Shipborne measurements of total OH reactivity around the Arabian Peninsula and its role in ozone chemistry, Atmos. Chem. Phys., 19, 11501–11523, https://doi.org/10.5194/acp-19-11501-2019, 2019. a, b
Platt, U. and Hönninger, G.: The role of halogen species in the troposphere, Chemosphere, 52, 325–338, https://doi.org/10.1016/S0045-6535(03)00216-9, 2003. a
Prinn, R. G., Huang, J., Weiss, R. F., Cunnold, D. M., Fraser, P. J., Simmonds, P. G., McCulloch, A., Harth, C., Reimann, S., Salameh, P., O'Doherty, S., Wang, R. H. J., Porter, L. W., Miller, B. R., and Krummel, P. B.: Evidence for variability of atmospheric hydroxyl radicals over the past quarter century, Geophys. Res. Lett., 32, L07809, https://doi.org/10.1029/2004GL022228, 2005. a
Quinn, P. K. and Bates, T. S.: The case against climate regulation via oceanic phytoplankton sulphur emissions, Nature, 480, 51–56, https://doi.org/10.1038/nature10580, 2011. a
Read, K. A., Mahajan, A. S., Carpenter, L. J., Evans, M. J., Faria, B. V. E., Heard, D. E., Hopkins, J. R., Lee, J. D., Moller, S. J., Lewis, A. C., Mendes, L., McQuaid, J. B., Oetjen, H., Saiz-Lopez, A., Pilling, M. J., and Plane, J. M. .: Extensive halogen-mediated ozone destruction over the tropical Atlantic Ocean, Nature, 453, 1232–1235, https://doi.org/10.1038/nature07035, 2008. a
Schwartz, S. E.: Factors Governing Dry Deposition of Gases to Surface-Water, in: Precipitation Scavenging And Atmosphere–Surface Exchange, Vols. 1–3, edited by: Schwartz, S. E. and SLINN, W. G., Hemisphere Publ. Corp., New York, 789–801, 1992. a
Sciare, J., Kanakidou, M., and Mihalopoulos, N.: Diurnal and seasonal variation of atmospheric dimethylsulfoxide at Amsterdam Island in the southern Indian Ocean, J. Geophys. Res., 105, 17257–17265, 2000. a
Sievert, S., Kiene, R., and Schulz-Vogt, H.: The Sulfur Cycle, Oceanography, 20, 117–123, https://doi.org/10.5670/oceanog.2007.55, 2007. a
Sipilä, M., Berndt, T., Petäjä, T., Brus, D., Vanhanen, J., Stratmann, F., Patokoski, J., Mauldin, R. L., Hyvärinen, A.-P., Lihavainen, H., and Kulmala, M.: The Role of Sulfuric Acid in Atmospheric Nucleation, Science, 327, 1243–1246, https://doi.org/10.1126/science.1180315, 2010. a
Sköld, O.: Sulfonamides and trimethoprim, Exp. Rev. Anti-infect. Therapy, 8, 1–6, https://doi.org/10.1586/eri.09.117, 2010. a
Su, T. and Chesnavich, W. J.: Parametrization of the ion–polar molecule collision rate constant by trajectory calculations, J. Chem. Phys., 76, 5183–5185, https://doi.org/10.1063/1.442828, 1982. a
Turnipseed, A. A., Barone, S. B., and Ravishankara, A. R.: Reaction of OH with Dimethyl Sulfide. 2. Products and Mechanisms, J. Phys. Chem., 100, 14703–14713, https://doi.org/10.1021/jp960867c, 1996. a
Veres, P. R., Faber, P., Drewnick, F., Lelieveld, J., and Williams, J.: Anthropogenic sources of VOC in a football stadium: Assessing human emissions in the atmosphere, Atmos. Environ., 77, 1052–1059, https://doi.org/10.1016/j.atmosenv.2013.05.076, 2013. a
Vila-Costa, M., Simó, R., Harada, H., Gasol, J. M., Slezak, D., and Kiene, R. P.: Dimethylsulfoniopropionate uptake by marine phytoplankton, Science, 314, 652–654, https://doi.org/10.1126/science.1131043, 2006. a
Voss, M., Bange, H. W., Dippner, J. W., Middelburg, J. J., Montoya, J. P., and Ward, B.: The marine nitrogen cycle: recent discoveries, uncertainties and the potential relevance of climate change, Philos. T. Roy. Soc. Lond. B, 368, 20130121, https://doi.org/10.1098/rstb.2013.0121, 2013. a, b
Wang, N., Edtbauer, A., Stönner, C., Pozzer, A., Bourtsoukidis, E., Ernle, L., Dienhart, D., Hottmann, B., Fischer, H., Schuladen, J., Crowley, J. N., Paris, J.-D., Lelieveld, J., and Williams, J.: Measurements of carbonyl compounds around the Arabian Peninsula indicate large missing sources of acetaldehyde, Atmos. Chem. Phys. Discuss., https://doi.org/10.5194/acp-2020-135, in review, 2020. a, b
Warneke, C. and de Gouw, J. A.: Organic trace gas composition of the marine boundary layer over the northwest Indian Ocean in April 2000, Atmos. Environ., 35, 5923–5933, https://doi.org/10.1016/S1352-2310(01)00384-3, 2001. a
Webb, A. L., van Leeuwe, M. A., den Os, D., Meredith, M. P., Venables, J. H., and Stefels, J.: Extreme spikes in DMS flux double estimates of biogenic sulfur export from the Antarctic coastal zone to the atmosphere, Scient. Rep., 9, 2233, https://doi.org/10.1038/s41598-019-38714-4, 2019. a
Weber, R. J., MARTI, J. J., McMurry, P. H., Eisele, F. L., Tanner, D. J., and Jefferson, A.: Measured Atmospheric New Particle Formation Rates: Implications For Nucleation Mechanisms, Chem. Eng. Commun., 151, 53–64, https://doi.org/10.1080/00986449608936541, 1996. a
Weber, R. J., Chen, G., Davis, D. D., Mauldin, R. L., Tanner, D. J., Eisele, F. L., Clarke, A. D., Thornton, D. C., and Bandy, A. R.: Measurements of enhanced H2SO4 and 3–4 nm particles near a frontal cloud during the First Aerosol Characterization Experiment (ACE 1), J. Geophys. Res.-Atmos., 106, 24107–24117, https://doi.org/10.1029/2000JD000109, 2001. a
Xie, H.-B., Elm, J., Halonen, R., Myllys, N., Kurtén, T., Kulmala, M., and Vehkamäki, H.: Atmospheric Fate of Monoethanolamine: Enhancing New Particle Formation of Sulfuric Acid as an Important Removal Process, Environ. Sci. Technol., 51, 8422–8431, https://doi.org/10.1021/acs.est.7b02294, 2017. a, b
Yang, M., Beale, R., Liss, P., Johnson, M., Blomquist, B., and Nightingale, P.: Air–sea fluxes of oxygenated volatile organic compounds across the Atlantic Ocean, Atmos. Chem. Phys., 14, 7499–7517, https://doi.org/10.5194/acp-14-7499-2014, 2014. a
Yassaa, N., Peeken, I., Zöllner, E., Bluhm, K., Arnold, S., Spracklen, D., and Williams, J.: Evidence for marine production of monoterpenes, Environ. Chem., 5, 391–401, https://doi.org/10.1071/EN08047, 2008. a
Zavarsky, A., Booge, D., Fiehn, A., Krüger, K., Atlas, E., and Marandino, C.: The Influence of Air-Sea Fluxes on Atmospheric Aerosols During the Summer Monsoon Over the Tropical Indian Ocean, Geophys. Res. Lett., 45, 418–426, https://doi.org/10.1002/2017GL076410, 2018. a