the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 16 Mar 2020
| 16 Mar 2020
Air quality in the eastern United States and Eastern Canada for 1990–2015: 25 years of change in response to emission reductions of SO2 and NOx in the region
Jian Feng
Elton Chan
Robert Vet
SO2 and NOx are precursors to form sulfate, nitrate, and ammonium particles in the air, which account for more than 50 % of PM2.5 mass in the eastern US (Bell et al., 2007) and are dominant components of PM2.5 during many smog events (Dabek-Zlotorzynska et al., 2011). H2SO4 and HNO3, formed from the oxidation of SO2 and NOx, respectively, are the main sources of acid deposition through wet and dry depositions. NOx is also a precursor to the formation of tropospheric O3, which is an important atmospheric oxidant and is also essential for the formation of other atmospheric oxidants, such as OH and H2O2.
In the past 26 years from 1990 to 2015, emissions of SO2 and NOx in the US were significantly reduced from 23.1 and 25.2 million t yr−1 in 1990 to 3.7 and 11.5 million t yr−1 in 2015, respectively. In Canada, SO2 and NOx were reduced by 63 % and 33 % from 1990 to 2014. In response to the significant reductions of SO2 and NOx emissions, air quality in the eastern US and Eastern Canada improved tremendously during 1990–2015. In this study, we analyzed surface air concentrations of , , , HNO3, and SO2 measured weekly by the Clean Air Status and Trends Network (CASTNET) in the US and measured daily from the Canadian Air and Precipitation Monitoring Network (CAPMoN) in Canada to reveal the temporal and spatial changes in each species during the 25-year period. For the whole eastern US and Eastern Canada, the annual mean concentrations of , , , HNO3, SO2, and TNO3 ( + HNO3, expressed as the mass of equivalent ) were reduced by 73.3 %, 29.1 %, 67.4 %, 65.8 %, 87.6 %, and 52.6 %, respectively, from 1990 to 2015. In terms of percentage, the reductions of all species except were spatially uniform. The reductions of SO2 and HNO3 were similar in the warm season (May–October) and the cold season (November–April), and the reductions of , , and were more significant in the warm season than in the cold season. The reductions of and SO2 mainly occurred in 1990–1995 and 2007–2015 during the warm season and in 1990–1995 and 2005–2015 during the cold season. The reduction of mainly occurred in the Midwest after 2000. Other than in the Midwest, exhibited very little change during the cold season for the period. The reduction of generally followed the reduction trend of ; especially after 2000, the temporal trend of was almost identical to that of . The ratio of S in to total S in plus SO2, as well as the ratio of to TNO3 increased by more than 50 % during the period. This indicates that a notable change in regional chemistry took place from the beginning to the end of the period, with a higher percentage of SO2 being oxidized to and a higher percentage of HNO3 being neutralized to NH4NO3 near the end of the period.
- Article
(7609 KB) - Full-text XML
-
Supplement
(20821 KB) - BibTeX
- EndNote





Gases and particulate matter released into the air through anthropogenic activities can pollute the air and deteriorate the air quality locally, regionally, and continentally. Air pollution, which can decrease lung function, causing the development of asthma, bronchitis, and lung cancer (Kunzli et al., 2000; Heroux et al., 2015; WHO, 2006), is considered a major environmental risk to human health by the World Health Organization (WHO). Air pollution is also linked to stroke and heart disease, and the improvement of air quality can significantly reduce the PM2.5- and O3-related mortality burden (Zhang et al., 2018). When emitted gases and particulate matter or secondary pollutants formed in the air from emissions are brought to the Earth's surface through dry and/or wet deposition, they pose a risk to the established ecosystems through acid rain as well as excessive deposition of nitrogen and sulfur. Air pollution also affects long-term climate through scattering and absorption of solar radiation by directly emitted or secondarily formed aerosols in the air (Haywood and Shine, 1995; Yu et al., 2006). In some heavily polluted regions, even local weather can be affected due to the change in energy budgets in the atmosphere and at the Earth's surface (Kajino et al., 2017).
In order to control air pollution, the US passed the Clean Air Act (CAA) of 1963 (Kuklinska et al., 2015). Major amendments to the law were passed in 1970, 1977, and 1990 (Kuklinska et al., 2015). The amendments to the CAA of 1990 addressed acid deposition, ozone depletion, and toxic air pollution (CAA, 1990). Specifically, Title IV of the 1990 amendments to the CAA, also known as acid deposition control, targeted emission reductions of two acid deposition precursors, SO2 and NOx, which along with CO, O3, Pb, and particulate matter are among the six species designated as criteria pollutants by United States Environmental Protection Agency (US EPA). SO2 and NOx in the air can be oxidized to form acid H2SO4 and HNO3, which in turn can react with NH3 to form fine particulate matter (PM2.5) and with crustal material or sea salts to form coarse particles (Yoshizumi and Hoshi, 1985; Zhuang et al., 1999). NOx, together with volatile organic compounds (VOCs), also participates in the formation of tropospheric O3, which is another criteria pollutant and an important atmospheric oxidant. Title IV of the Clean Air Act 1990 specifically targets SO2 and NOx emissions from stationary fuel combustion facilities. The first phase of Title IV of the 1990 CAA amendment, which was implemented on 1 January 1995, requires 110 power plants to reduce SO2 emissions to a level calculated as the product of an emissions rate of 2.5 lbs (1 lb is equal to 0.4636 kg) of SO2 per million British thermal units (Btu; 1 Btu is equal to 1055.056 J) multiplied by an average of their 1985–1987 fuel use (Lee, 1991). The second phase, which took effect on 1 January 2000, requires approximately 2000 utilities to reduce SO2 emissions to a level of 1.2 lbs of SO2 per million British thermal units multiplied by the average of their 1985–1997 fuel use (Lee, 1991). Since 1990, the national emissions of SO2 in the US have decreased steadily from 23.1 million tons in 1990 to 21.3 million tons in 1994 and dropped significantly to 18.6 million tons in 1995 due to the first phase implementation of Title IV of the 1990 CAA amendments (EPA, 2016, 2019). The SO2 emissions underwent a small increase during 1996–1998, to 18.9 million tons in 1998, and then continued the steady decrease to 14.5 million tons in 2005. From 2005 to 2012, the decrease in the emissions was accelerated with an annual reduction rate of 1.34 million t yr−1 during the period. The emissions of SO2 leveled off during 2012–2015. In 1990, 87.9 % of SO2 emissions was from stationary fuel combustion facilities, 2 % from on-road vehicles, and 2 % from off-road mobile sources. By 2007, SO2 emissions from on-road vehicles were totally eliminated due to cleaner gasoline. In 2014, of the 4.9 million tons of total SO2 emissions, stationary fuel combustion, off-road mobile, and industrial and other processes contributed 4.1, 0.1, and 0.7 million tons, respectively (EPA, 2016).
NOx forms in the air when nitrogen reacts with oxygen under high temperature. Anthropogenic emissions of NOx are mainly due to stationary fuel combustion, on-road vehicles, and off-road mobile operations. Nationwide in the US, they contributed 10.9, 9.6, and 3.8 million tons of the total 25.2 million tons of NOx in 1990 (EPA, 2016). Changes in NOx emissions during the 1990s were relatively small (Butler et al., 2003). Total NOx emissions remained generally constant from 1990 to 1998. From 1999 there was a decrease in NOx emissions from stationary fuel combustion due to the implementation of Title IV of the 1990 CAA amendment as well as the implementation of the NOx Budget Trading Program (NBP). Title IV of the 1990 CAA amendment not only required the reduction of SO2, but also stipulated the reduction of NOx emissions from power plants, and it took effect in 1996. The NBP started in 2003 and was created to reduce NOx emissions from power plants and other large combustion sources in the eastern US during warm months (https://www.epa.gov/airmarkets/nox-budget-trading-program, last access: 13 November 2019). The NBP was replaced by the ozone season NOx program under the Clean Air Interstate Rule in 2009. The NOx emissions from stationary combustion facilities decreased steadily from 10.4 million tons in 1998 to 3.6 million tons in 2012, then remained relatively unchanged thereafter (EPA, 2016). Emissions of NOx from on-road vehicles declined slowly from 1990 until 2001. After 2002, on-road emissions of NOx decreased continuously and steadily. The trend of NOx emissions from off-road mobile operations generally increased during the period 1990–2002, up from 3.8 to 4.9 t, but after that it was reduced gradually to 2.7 t in 2014. Combining the emissions from stationary fuel combustion, on-road vehicles, and off-road mobile operations, the nationwide emissions of NOx in the US changed little during 1990–1998 and decreased during 1998–2001. After 2002, they decreased steeply up to recent years. Note that there was a change in NOx measurement methodology from 2001 to 2002, and it caused a sharp increase in the reported NOx emissions in the US from 2001 to 2002 (EPA, 2019)
In Canada, similar measures were adopted to reduce air pollutant emissions. SO2 emissions in Canada were mainly from three major sectors: ore and mineral industries, the oil and gas industry, and electric utilities. For each sector, the annual SO2 emissions were reduced from 1.5, 0.53, and 0.62 million tons in 1990 to 0.47, 0.28, and 0.27 million tons in 2015 (ECCC, 2019). Nationally, annual SO2 emissions were reduced from 3.1 million tons in 1990 to 1.1 million tons in 2015 (ECCC, 2019). In 1990, the annual emissions of SO2 from Eastern Canada accounted for 59 % of the national annual emissions. NOx emissions in Canada were mainly from transportation (43%) and oil and gas industries (14 %) (ECCC, 2019). Nationally, annual emissions of NOx were reduced by 25 %, from 2.4 million tons in 1990 to 1.8 million tons in 2015. Specifically, annual emissions of NOx in Eastern Canada were reduced by close to 50 %, from 1.2 million tons to 0.64 million tons.
Air quality trends during the past few decades, especially since 1990, are of great interest for both scientific communities and the general public. For the eastern part of the US and Canada, trends of air quality after 1990 have been reported in previous studies for O3 (Chan and Vet, 2010), O3 and nitrate (Butler et al., 2011), particulate (Hand et al., 2012), and air quality and atmospheric deposition (Sickles II and Shadwick, 2007; Sickles II and Shadwick, 2015; Cheng and Zhang, 2017). Sickles II and Shadwick (2007, 2015) compared the 5-year averages of air quality and atmospheric deposition in the eastern US for 1990–2004 and 1990–2009. Cheng and Zhang (2017) reported the temporal trends of the annual concentration of air pollutants from 31 Canadian rural locations, most of which were located in Eastern Canada. Aas et al. (2019) reported global and regional trends of atmospheric sulfur for 1990–2015 and found that North America and East Asia had the largest reductions of sulfur emissions during the late part of the period. In this study, we analyze the surface air concentration data measured weekly by the CASTNET network in the US and measured daily from the CAPMoN network in Canada to reveal the detailed temporal and spatial trends of air quality from 1990 to 2015. These trends are not only important for the assessment of the improvement of air quality due to emissions reductions, but are also essential for the evaluation of chemical transportation models. The analysis will answer the following questions: (1) what are the trends of air pollutants over the eastern US and Eastern Canada following the significant reductions of SO2 and NOx emissions during 1990–2015? (2) What are the physical and chemical mechanisms responsible for the trends? We will look at the air concentrations of gases SO2 and HNO3, as well as particulates , , and , which are either due to direct emissions of SO2 or due to the oxidation of SO2 and NOx, as well as the reaction of these oxidants with NH3.
2.1 Networks of measurement: CASTNET and CAPMoN
The monitoring of background- and regional-level ambient pollutants is essential for assessing regional air quality. In the US and Canada, this long-term monitoring of air quality in rural and remote areas is fulfilled by two monitoring networks: CASTNET and CAPMoN, respectively.
CASTNET is a monitoring network managed and operated by the US EPA in cooperation with several other federal, state, and local partners (Clarke et al., 1997; Bloomer et al, 2010). The network was established under the 1990 CAA to assess the trends of acidic deposition due to emission reduction programs. The network makes weekly integrated measurements of gases (SO2 and HNO3) and particulates (, , , Mg2+, Ca2+, Na+, and Cl−) using filter pack methods, as well as hourly measurements of O3. At selected sites, it also measures hourly concentrations of NO, reactive nitrogen (NOy), SO2, and CO.
CAPMoN is a monitoring network operated by Environment and Climate Change Canada (ECCC). The network began operation in 1983, although one of its two predecessor networks, the Air and Precipitation Network (APN), measured air concentrations as far back as 1978. The network measures 24 h integrated air concentrations of pollutants (from 08:00 to 08:00 LT, local time) through filter pack sampling and 24 h wet deposition by the collection of precipitation samples at the ground level. The daily air concentration measurements by CAPMoN also include gases (SO2 and HNO3) and particulates (, , , Mg2+, Ca2+, Na+, and Cl−), similar to CASTNET's weekly measurements. CAPMoN also measures hourly air concentrations of O3, NOy, and gaseous Hg at selected sites. More details about the CAPMoN dataset can be found in Cheng and Zhang (2017).
2.2 Statistical analysis and methods
As we focus on the long-term trends of air pollutants over the region, annual means for all seasons and seasonal means for the warm and cold seasons were derived for each site from the weekly measurements of CASTNET and daily measurements of CAPMoN. To be precise, the seasonal mean concentrations in this study refer to the mean concentrations calculated for the warm (May–October) and cold (November–April) seasons for each year. In order to avoid the fluctuations of annual or seasonal mean concentrations due to meteorology, 3-year averages were used to represent the mean concentrations at the beginning or the end of a period in calculating changes for that period.
The Mann–Kendall test (MKT) is a nonparametric test to detect the trend of a time series and it does not require the variable of the time series to follow a normal distribution (Mann, 1945; Du et al., 2014). In this study, the MKT was used to detect if an increasing or decreasing trend exists when a time series generally looks flat. The p value and tau coefficient are the two statistical parameters of the MKT, indicating the statistical significance and significance of a monotonic trend, respectively.
To assess the changes in air pollutants in response to emission reductions of SO2 and NOx, we looked at the following for species of , SO2, , , HNO3, and TNO3 ( + HNO3, expressed as equivalent ):
-
temporal and spatial trends in the eastern US and Eastern Canada;
-
10-year and 25-year changes for the periods of 1990–2000 and 1990–2015;
-
differences in trends in cold and warm seasons;
-
time series of the yearly regional means during the warm and cold seasons; and
-
long-term trends derived from polynomial regressions.
We also looked at correlations between and SO2, the ratio of sulfur (RSO4) in to total sulfur in plus SO2 in the air, the ratio of nitrogen (RNO3) in to TNO3, and their changes during the period in order to explain the physical and chemical mechanisms responsible for the trends.
2.3 Region clustering of CASTNET and CAPMoN sites in the eastern US and Eastern Canada
In the eastern US (EUS) and Eastern Canada (EC), there are significant spatial differences in emissions of SO2, NOx, and NH3. This results in distinctive regional patterns of air concentrations of , , , HNO3, and SO2. In this study, we used the cold season (November to April) 3-year mean concentrations of and SO2 of each site, supplemented with the ratio of RNO3, as the criteria to cluster the CASTNET and CAPMoN sites into four different regions. The reasons for selecting the cold season are the following: (1) is mainly in the form of NH4NO3 (Zhang et al., 2008), and it is more thermodynamically stable in the cold season than in the warm season; (2) the oxidation rate of SO2 is much lower in the cold season than in the warm season, and therefore the air concentration of SO2 more reflects the SO2 emission rate of the region; (3) because NH4NO3 is much more thermodynamically stable and much less affected by ambient temperature, RNO3 is mainly determined by the availability of NH3 over the region, and therefore RNO3 during the cold season is an indicator of the abundance of NH3 to form NH4NO3. The mean concentrations at the beginning of the period were used to cluster the sites as the emission rate of SO2 was the highest.
Based on the spatial patterns of the mean air concentration of , SO2, and RNO3 during the cold season of 1989–1991, which are shown in Table S1b and Fig. S1b in the Supplement, four regions in the EUS and EC were clustered.
-
Region 1: sites located north of latitude 40∘ and with a concentration of SO2 less than 6.4 µg m−3 in the cold season.
-
Region 2: sites with a mean concentration of greater than 2.5 µg m−3. Except for site ARE128 at 2.1 µg m−3, the highest air concentration of of all other sites was 1.9 µg m−3. For sites in region 2, RNO3 was greater than 54 %, which was higher than any CASTNET and CAPMoN sites in other regions.
-
Region 3: sites excluded from regions 1 and 2 and with an air concentration of SO2 greater than 15.0 µg m−3 during the cold season.
-
Region 4: all other sites excluded from regions 1, 2, and 3. The highest mean SO2 of sites in region 4 during the cold season was less than 11.7 µg m−3.
The clustering of sites is shown in Fig. 1 with site names. Regions 2, 3, and 4 roughly correspond to the Midwest, Mid-Atlantic, and Southeast regions participating in the NBP (Butler et al., 2011). The site SND152 had a characteristic of region 2, but it is geologically located in region 4; therefore the site SND152 was not included in the study. Characteristics of each region are listed in Table 1. After grouping the sites into each cluster appropriately, annual and seasonal mean concentrations of each species from each site within the cluster show high correlations with the averaged values of the cluster, as shown in Fig. S3 and Table S3. For example, as shown in Table S3 for region 2, the averaged correlation coefficients of the seasonal mean concentrations of each site vs. the averaged values of the cluster during the warm season are 0.98, 0.95, 0.98, 0.97, 0.98, and 0.98 for , , , HNO3, SO2, and TNO3, respectively. For all seasons, the corresponding averaged correlation coefficients are 0.97–0.99, 0.59–0.91, 0.93–0.98, 0.94–0.99, 0.96–0.99, and 0.86–0.98 for regions 1–4.
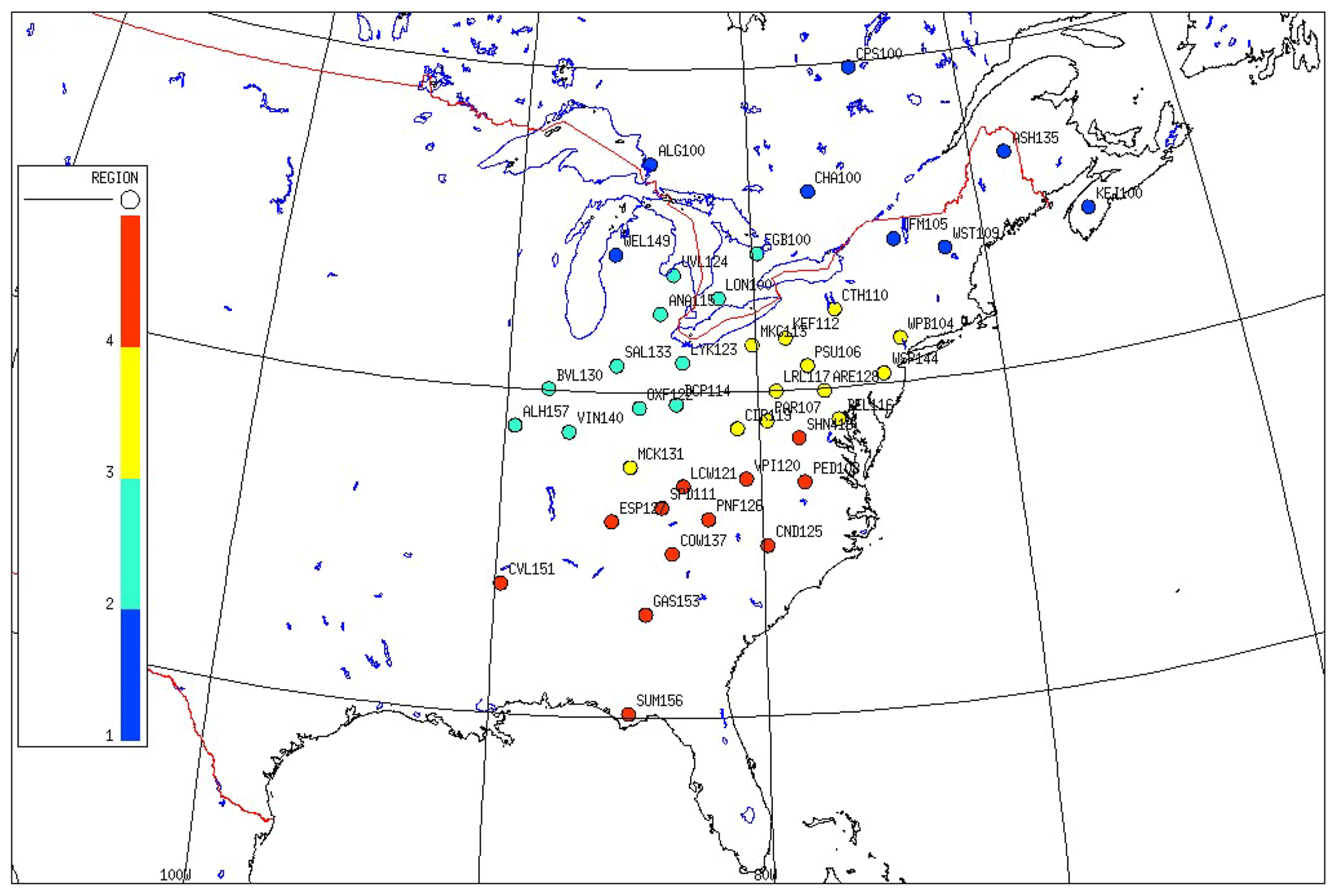
Figure 1Clustered sites of CASTNET and CAPMoN in regions 1–4.
Table 1Characteristics of four regions based on 3-year averages of 1989–1991 during the cold season.

3.1 Air quality in the eastern US and Eastern Canada at the beginning of the study period: 1989–1991
The 3-year averages of the air concentrations of , , , HNO3, SO2, and TNO3, as well as RSO4 and RNO3 for 1989–1991 are used to describe the air quality at the beginning of the study period and are shown in Table S1a and b for the warm and cold seasons. Mapping of 3-year average for each species is also provided as Fig. S1. Among the four regions and both the warm and cold seasons, region 1 had the lowest air concentration of all species, with mean , HNO3, and concentrations of less than 1.0 µg m−3. The mean air concentration of during the warm season was only 0.14 µg m−3. Mean concentrations were 2.9 and 2.3 µg m−3 during the warm and cold seasons, respectively, and SO2 was 1.6 and 3.6 µg m−3 correspondingly.
For regions 2–4, was highest in region 3 and lowest in region 4 for both seasons, varying from 7.6 to 8.2 µg m−3 during the warm season and 3.6 to 4.2 µg m−3 during the cold season. The difference in the regional mean of between region 2 and 3 was less than 0.1 µg m−3 during the cold season. Generally, in regions 2–4 was spatially uniform. For each region, during the warm season was about double that during the cold season. The same as , SO2 was also highest in region 3 and lowest in region 4 for regions 2–4, but SO2 in region 3 was much higher and was about 2.5 times that in region 4. SO2 in regions 3 and 2 during the cold season, being 19.2 and 13.7 µg m−3, respectively, showed the two highest concentrations and the only two concentrations greater than 10.0 µg m−3 among all species in four regions and during the warm and cold seasons. The difference in SO2 between region 3 and 2 was less than 1.0 µg m−3 during the warm season but was more than 5.0 µg m−3 during the cold season. Despite significant differences in SO2 for regions 2–4, the corresponding differences in were small. As an example, during the cold season, the seasonal mean concentration of SO2 in region 3 was higher than that in region 4 by 10.0 µg m−3, but the corresponding difference in was only 0.7 µg m−3. This can be attributed to the fact that the lifetime of (∼5–7 d) in the air is much longer than that of SO2 (∼2 d) (Penner et al., 2001; Pitari et al., 2016). Lee et al. (2011) estimated the mean SO2 lifetime in the eastern US to be 19±7 h in summer and 58±20 h in winter. Comparing SO2 in the cold season to that in the warm season, it was about 2 times higher in regions 3 and 4 and 59 % higher in region 2. In contrast to the pattern of , in regions 2–4 was significantly different from region to region. Region 2 had the highest concentration of at 1.5 µg m−3 during the warm season and 3.6 µg m−3 during the cold season; these values were about triple the value in region 3, which was the second highest. Region 4 had the lowest concentrations among regions 2–4, being 0.3 and 0.6 µg m−3 for the warm and cold seasons, respectively. The lowest value for stations in region 2 during the cold season was 2.5 µg m−3 at ALH157, higher than the highest value of 2.1 µg m−3 at BEL116 in region 3. During the warm season, HNO3 ranged from 1.7 µg m−3 in region 4 to 2.8 and 2.9 µg m−3 in regions 3 and 2. During the cold season, the highest concentration of HNO3 was in region 3 and the lowest in region 2, with values of 2.3 and 1.8 µg m−3, respectively. Considering both seasons, region 3 had the highest concentration of HNO3 among the four regions. Region 2 had the lowest concentration of HNO3 among regions 2–4 during the cold season due to the fact that a large portion of HNO3 was neutralized by NH3 to form NH4NO3. For TNO3, in both seasons, region 2 had the highest concentration, being 4.3 and 5.3 µg m−3 in the warm and cold seasons, mainly because of the significantly higher concentration of than other regions. TNO3 was 3.2 and 3.5 µg m−3 for region 3 and 2.0 and 2.3 µg m−3 for region 4 during the warm and cold seasons. in regions 2–4 varied from 1.9 to 2.7 µg m−3 during the warm season and 1.1 to 2.3 µg m−3 during the cold season, with the highest concentrations in region 2 and the lowest concentrations in region 4 for both seasons. was higher in the warm season than in the cold season for all regions, as much more (NH4)2SO4 formed in the warm season than in the cold season.
In general, region 1 had the lowest concentration of all species among the four regions, and region 4 had the second lowest except HNO3, which was slightly more than in region 2 during the cold season. Regions 2 and 3 were the two most polluted regions in the EUS and ECA. Region 3 had the highest regional concentration of SO2 in both seasons, more than double that in regions 1 and 4; region 2 had the highest concentration of and TNO3. In both seasons, in region 2 was more than 4 times higher than that in regions 1 and 4, and TNO3 was more than double that in regions 1 and 4.
3.2 Time series of seasonal mean concentrations of , SO2, , HNO3, TNO3, and during 1990–2015
Time series of seasonal mean concentrations of , SO2, , HNO3, TNO3, and for each region as well as each site of the region are shown in Fig. S3a for the cold season and Fig. S3b for the warm season. As an example, time series of regional averaged seasonal mean concentrations in regions 2 and 4 are shown in Fig. 2a and b for the cold and warm seasons. Time series of regional averages for regions 1–4 normalized to the year 2000 are presented in Fig. 3a and b for the warm and cold seasons. The year 2000 was chosen for normalization because (1) emissions of NOx changed little during the first 10 years, and (2) 10 and 25 years of change in annual and seasonal mean concentrations of each species will be discussed in Sect. 3.3. As shown in Fig. S3, the time series for the stations within each region were very highly correlated when the stations are properly grouped into four regions. The discussion in this section is based on the time series of seasonal mean concentrations presented in Fig. S3 as well as Fig. 3.
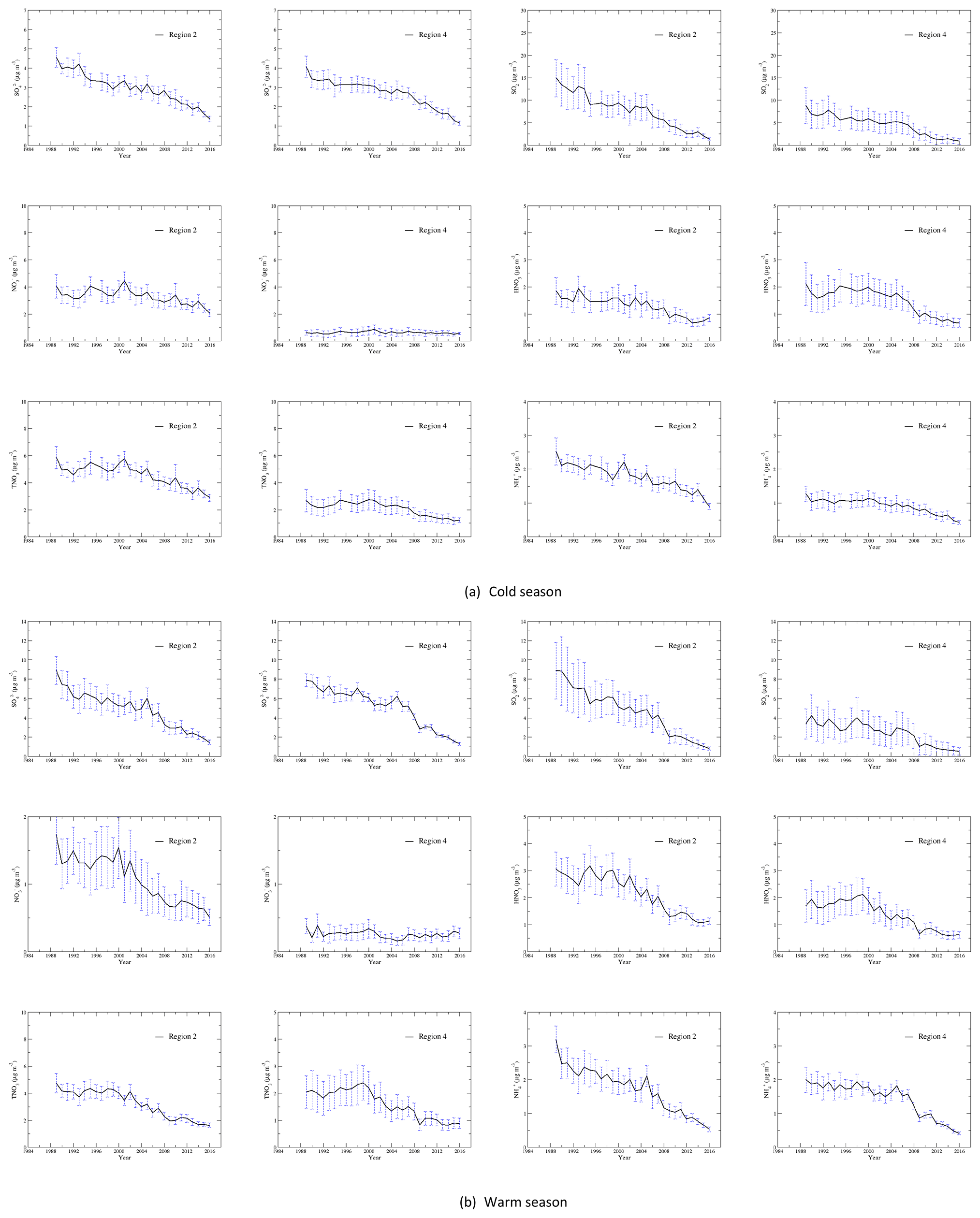
Figure 2Time series of the regionally averaged seasonal mean concentrations during the cold (a) and warm (b) seasons for each species in region 2 (Midwest) and region 4 (Southeast). The vertical dashed line represents the standard deviation of the regionally averaged seasonal mean concentration. The regionally averaged seasonal mean concentrations in regions 2 and 4 during the cold season were not generated for 1996 because of an insufficient number of measurements.
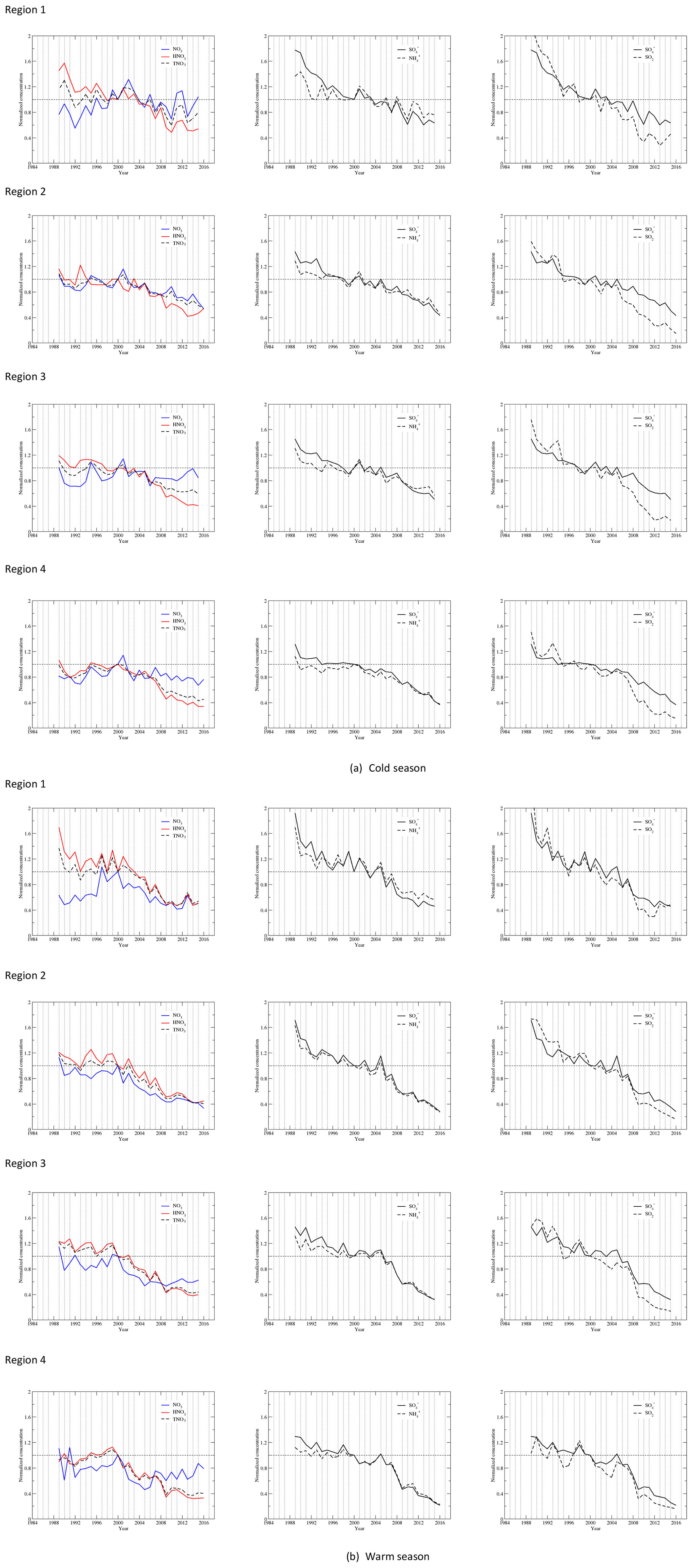
Figure 3Time series of the regional seasonal mean concentrations normalized to the year 2000 for each species during the cold (a) and warm (b) seasons.
3.2.1 and SO2
and SO2 during the cold season
As seen from the time series of the seasonal mean concentrations in Fig. S3, in region 1 had a steady decreasing trend from 1989 to 2000, then a relatively slower decreasing trend from 2001 to 2016. in region 2 had a decreasing trend during 1989–1995 and a slow decreasing trend during 1995–2005. From 2005 to 2016, there was a straight decreasing trend with only small increases in 2008 and 2014. in region 3 generally decreased significantly for the period 1989–1999 and was followed by a slow decreasing period from 2000 to 2008. There was a smooth decreasing trend from 2008 to 2016, and the drop in the seasonal mean concentration was significant in 2008–2011 and 2014–2016. in region 4 had a peak in 1989 for 1988–1990, which was followed by three relatively constant periods of 1990–1993, 1994–2001, and 2002–2007, with the averages of the seasonal mean concentrations of during the periods being 3.4, 3.1, and 2.8 µg m−3, respectively. There were only two major drops between the periods, in 1994 and 2002. From 2007 to 2016, in region 4 had a steep drop, and the concentration was reduced from 2.7 µg m−3 in 2007 to 1.1 µg m−3 in 2016. In general, in the EUS and EC during the cold season can be described by two fast decreasing periods of 1989–1995 and 2005–2016 and a slow decreasing period of 1995–2005. The annual reduction rates during the three periods were 0.14, 0.03, and 0.05 µg m−3 yr−1 in region 1; 0.16, 0.04, and 0.13 µg m−3 yr−1 in region 2; 0.15, 0.05, and 0.15 µg m−3 yr−1 in region 3; and 0.10, 0.04, and 0.14 µg m−3 yr−1 in region 4. The decreasing rates in regions 2 and 3 were close. If all sites within regions 1–4 were combined, the corresponding rates would be 0.14, 0.04, and 0.12 µg m−3 yr−1 for the three periods.
SO2 in regions 2–4 during the cold season had a significant drop in 1989–1995, with a temporary increase in 1993 and 1994. From 1995 to 2005 the decreasing trend was slow, and then there was a very steep reduction from 2005 to 2012. The trend from 2012 to 2016 was relatively flat. The trend of SO2 in region 1 was similar except that there was no obvious increase in 1993–1994.
SO2 exhibited an annual reduction rate of 0.28, 0.06, and 0.12 µg m−3 yr−1 in region 1; 0.83, 0.09, and 0.73 µg m−3 yr−1 in region 2; 1.13, 0.22, and 1.13 µg m−3 yr−1 in region 3; and 0.32, 0.08, and 0.49 µg m−3 yr−1 in region 4 for the periods of 1989–1995, 1995–2005, and 2005–2016. For regions 1–4 combined, the corresponding reduction rates were 0.72, 0.16, and 0.48 µg m−3 yr−1. For the third period, if we only consider 2005–2012, the annual reduction rate was 0.61 µg m−3 yr−1. Although the trend of SO2 during 2012–2016 was generally flat, the decrease in during the period was still significant, especially in terms of percentage.
and SO2 during the warm season
in region 1 had a significant decrease from 1989 to 1996, followed by a leveling-off until 2005 and a decreasing trend from 2005 to 2016. After a significant peak in 1989, in region 2 had a large drop from 1989 to 1993. There was a steady decreasing trend for 1994–2004, followed by a significant peak in 2005, which was captured by all measurement sites within the region. This was followed by a smooth decreasing trend for 2005–2016, with major drops in 2005–2006, 2007–2009, 2012, and 2013–2016. Unlike region 2, in region 3 had no significant peak in 1989 and had a decreasing trend during 1989–1999. There was a leveling-off for 1999–2005, and it was followed by a significant decreasing trend for 2005–2016 with steep drops in 2005–2006, 2007–2009, and 2011–2016. in region 4 had a decreasing trend in 1989–1994 and a slow decreasing trend in 1994–2000. After a drop in 2001, it had another leveling-off in 2001–2007 with a peak in 2005. There was a significant straight decreasing trend for 2007–2016 with a major drop during 2007–2009. at all stations converged to the regional average after 2009. In general, the trend of during the warm season can be characterized by two fast reduction periods of 1989–1995 and 2007–2016 and a slow period of 1995–2007. For the three periods, had a reduction rate of 0.17, 0.01, and 0.14 µg m−3 yr−1 for region 1; 0.39, 0.03, and 0.43 µg m−3 yr−1 for region 2; 0.28, 0.01, and 0.52 µg m−3 yr−1 for region 3; and 0.24, 0.04, and 0.50 µg m−3 yr−1 for region 4. For regions 3 and 4, the most significant decrease occurred in 2007–2009.
SO2 concentrations during the warm season were quite low during 1990–2015 in region 1. There was a slow decreasing trend from 1990 to 2007, a relatively large decreasing trend in 2007–2009, and a level-off at a very low concentration (<1.0 µg m−3) in 2009–2016. SO2 during the warm season in regions 2–4 had similar trends: (1) a fast decreasing period of 1989–1995, with a leveling-off period of 1992–1994 in region 2 and a peak in 1993 in regions 3 and 4; (2) a slowly decreasing period of 1995–2007 with an initial slow increase in 1995–1999 in region 2 and an initial steep increase in 1996–1998 in region 3 and 4; (3) a fast decreasing period of 2007–2016 with very steep decreases in 2007–2009. For the three periods, SO2 had annual reduction rates of 0.12, 0.03, and 0.04 µg m−3 yr−1 for region 1; 0.55, 0.08, and 0.43 µg m−3 yr−1 for region 2; 0.46, 0.11, and 0.48 µg m−3 yr−1 for region 3; and 0.13, 0.05, and 0.22 µg m−3 yr−1 for region 4. For the whole region, the annual reduction rates during the warm season were 0.31, 0.04, and 0.38 µg m−3 yr−1 for and 0.34, 0.08, and 0.30 µg m−3 yr−1 for SO2 for the periods of 1989–1995, 1995–2007, and 2007–2016.
3.2.2 , HNO3, and TNO3
, HNO3, and TNO3 during the cold season
in EUS and EC during the cold season was dominated by in region 2, which was much higher than in other regions. As the trends of during the cold season were not as obvious as other species, the MKT was applied to detect if a monotonic (increasing or decreasing) trend existed. The MKT indicated the following: (1) in region 2 had no obvious trend in the period of 1989–2001 (p=0.45, τ=0.18), but there was a decreasing trend for the period 2001–2016 (p<0.001, ); the trends of in regions 1, 3, and 4 over the whole study period were pretty flat (p=0.17–0.40, ).
Excluding a peak of 1.9 µg m−3 in 1993, the trend of HNO3 in region 2 in the 1990s is flat. From 2003 to 2013, there was a decreasing trend, then a slow increasing trend for 2013–2016 in region 2. HNO3 in region 3 showed a general declining trend by 21.7 % (2004–2005 vs. 1989–1990) during 1989–2005. The declining trend of HNO3 in the 1990s was consistent with the increasing trend of during the period, as more HNO3 was neutralized by additional NH3 made available from decreasing . HNO3 decreased markedly from 2005 to 2009 in region 3, then exhibited a slower decreasing trend for 2009–2016. HNO3 in region 4 had an initial decrease from 1989 to 1991, then an increasing trend of 21.1 % (1999–2000 vs. 1991–1992) for 1991–2000. The region then showed a slow decrease in 2000–2005, a fast decrease in 2005–2009, and a slow decrease in 2009–2016.
TNO3 in the cold seasons over regions 2, 3, and 4 had similar trends. For 1990–2001, the general trend of TNO3 over the regions was from roughly constant to slowly increasing. This is consistent with the NOx emission trend during the period. For 2001–2016, there was a significant decrease in TNO3 over the three regions. For region 3, it occurred mainly during the period 2001–2009.
, HNO3, and TNO3 during the warm season
in region 1 had an increasing trend in 1990–2000, a decreasing trend in 2000–2007, and a flat trend in 2007–2016. Linear regression of the seasonal mean concentration in region 2 during the warm season from 1990 to 1999 showed a slow increasing trend for in the 1990s. After 2002, there was a straight decline of the concentration until 2009 and a steady decreasing from 2011 to 2016. Linear regression of in region 3 shows a flat trend during 1989–2000 and a weakly increasing trend during 1990–1999. Between 2000 and 2005, in region 3 exhibited a clearly declining trend. From 2005 to 2016, the trend was generally flat. The seasonal concentration of in region 4 had a weakly increasing trend for 1992–2000, followed by a decreasing trend for 2000–2005 and a slow increasing trend for 2005–2016. In general, during the warm season had a flat to weakly increasing trend for 1990–2000 and a strong decreasing trend for 2000–2005 for regions 2–4. For 2005–2016, there was a decreasing trend for region 2, a flat trend for region 3, and a weakly increasing trend for region 4.
HNO3 in region 1 was very low (<1.0 µg m−3) in general during the whole study period. There was flat trend in 1990 to 2001, a slow decreasing trend in 2001 to 2009, and a flat trend again in 2009 to 2016. HNO3 in regions 2–4 can be characterized by three periods: a flat (regions 2 and 3) or a weak increasing (region 4) trend for 1989–1999, a significant decreasing trend for 1999–2009, and a generally flat trend for 2009–2016.
HNO3 dominated over in TNO3 during the warm season for all regions, especially in regions 3 and 4 where the ratio of to TNO3 was usually less than 20 %. Therefore, the trend of TNO3 generally followed that of HNO3 during the warm season. In the 1990s (1990–1999) the trend of TNO3 in the warm season over regions 2 and 3 was very flat, and there was a very weakly increasing trend over region 4. The turning point of the trend was in 1999. For the period 1999–2009, all three regions showed significant decreasing trends. For 2009–2016, the trends in the three regions were generally flat.
3.2.3
during the cold season
during the cold season in region 1 had a flat trend in 1990–1999 and a very slow decreasing trend in 1999–2009, followed by a generally flat trend in 2009–2015. The trend of in region 2 during the cold season was affected by both and . It had a near-linear decreasing trend from 1991 to 2009 and was followed by another steeper declining trend for 2009–2016. in region 3 had a steep decrease in 1989–1994 and was followed by a weak decrease and a steep decrease in 1994–2009 and 2009–2016, respectively. in region 4 remained unchanged during 1990–2001. After 2001, decreased steadily until 2016.
during the warm season
in region 1 changed little in 1990–2005 and 2009–2015 during the warm season, but there was a decreasing trend in 2005–2009. Excluding the two significant peaks in 1989 and 2005, in region 2 showed a consistent and steady decreasing trend from 1990 to 2016. Figure 3 shows that the trend of in region 3 generally followed that of closely because in region 3 was dominantly associated with during the warm season. The two trends were almost identical after 2000. From 1995 to 2005, the trend of in region 3 was almost flat. There was a major decrease in in region 3 in 2005–2009 when it decreased by almost 50 % in 4 years. in region 4 changed little from 1990 to 2000. Although during the same period had an obvious decreasing trend, its impact on was offset by the increasing trend of during this period. After a drop in 2000–2001, had a flat trend for 2001–2007, a dramatic decrease in 2007–2009, and a steady decreasing trend for 2010–2016.
Table 2Changes in the regionally averaged air concentrations of pollutants (µg m−3), RSO4 (%), and RNO3 (%) between 1989–1991 and 1999–2001 for regions 1–4.
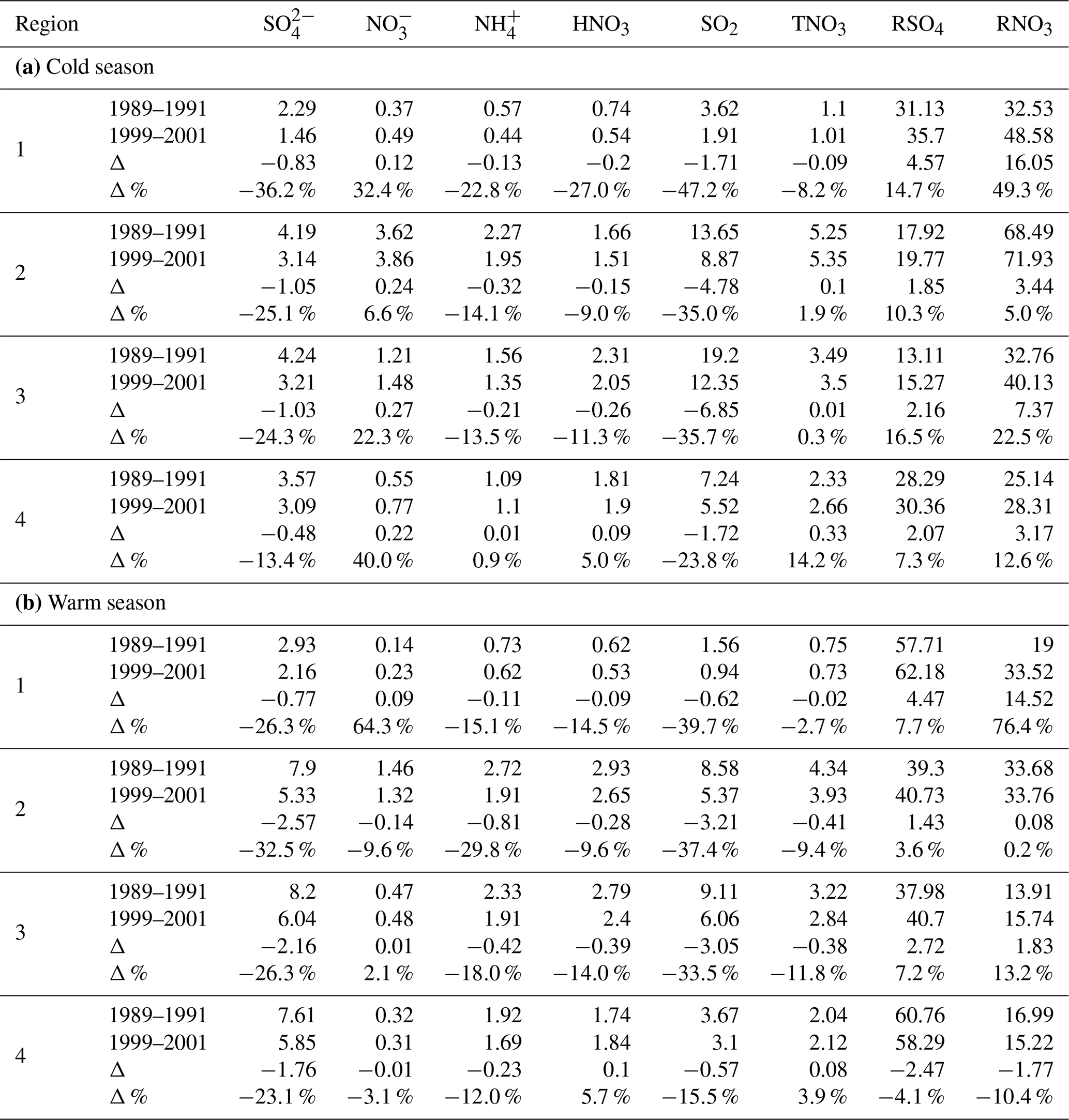
Table 3Changes in the regionally averaged air concentrations of air pollutants (µg m−3), RSO4 (%), and RNO3 (%) between 1989–1991 and 2014–2016 for the eastern US and Eastern Canada. Bold in (a) indicates the 3-year average concentrations still exceeding 1.0 µg m−3 in 2014–2016. Bold in (b) and (c) indicates reduction and increase rates exceeding 50 %.
3.3 10 and 25 years of changes in ambient , SO2, , , HNO3, and TNO3 in the EUS and EC for 1990–2015
As mentioned in Sect. 1, emissions of NOx changed little during the 1990s in the EUS. Correspondingly, the time series of TNO3 in regions 2–4 during the 1990s did not decrease or even increased, as shown in Sect. 3.2. Also, some important metrics, such as RSO4 during the cold season and RNO3 during the warm and cold seasons, only started to have significant changes after the first 10 years. In order to capture how ambient air pollutants responded to emission reductions of SO2 and NOx temporarily, 10 and 25 years of change in ambient , SO2, , , HNO3, and TNO3 during 1990–2015 are presented in Tables 2 and 3 and are summarized in Sect. 3.3.1 and 3.3.2. To avoid the fluctuation of annual ambient concentrations due to the variation of meteorology, the changes are calculated based on 3-year averages centered on 1990, 2000, and 2015.
3.3.1 10 years of changes for the period 1990–2000
During the 10-year period of 1990–2015, air quality in the EUS and EC underwent a number of major changes, which are summarized as follows.
-
During the first 10 years, SO2 concentrations declined in all regions and seasons by more than 25.0 % except for region 4, which had a reduction of 15.5 % during the warm season and 23.8 % during the cold season.
-
showed a similar but less significant decreasing trend as SO2. The reduction was more than 20 % in all regions except for region 4 during the cold season. Region 4 during the warm season had a similar reduction rate as region 3 despite the significant difference in the reduction rates of SO2 in the two regions.
-
increased between 6.6 % and 40.0 % during the cold season for regions 1–4. Changes in during the warm season in regions 3 and 4 were very small and only had a significant reduction of 9.6 % in region 2.
-
TNO3 increased little in region 1, by 0.09 and 0.02 µg m−3 for the cold and warm seasons, respectively. TNO3 in regions 2 and 3 changed very little during the cold season and had a 9.4 % and 11.8 % reduction during the warm season. TNO3 in region 4 increased by 3.9 % during the warm season and by 14.2 % during the cold season.
-
Except for a negligible change in region 4, decreased by 13.5 % to 22.8 % for regions 1–3 during the cold season; during warm season, it decreased by 12.0 % to 29.8 % for regions 1–4.
-
In summary, over the first 10-year period of 1990–2010, SO2, , and declined by 31.6 %, 26.7 %, and 18.5 %, respectively, in the EUS and EC. HNO3 was reduced in regions 1–3. increased in regions 1–4 during the cold season and changed very little (<0.15 µg m−3) during the warm season. Considering both seasons and all regions, increased by 12.7 %, HNO3 declined by 5.6 %, and the change in TNO3 was negligible with the mean concentration being 3.02 µg m−3 for 1989–1990 vs. 3.05 µg m−3 for 1999–2001.
3.3.2 25 years of changes for the period 1990–2015
During the 25-year period of 1990–2015, air quality in the EUS and EC changed significantly and is summarized as follows.
-
Among all species, the most significant reduction during the period was for SO2. The reduction of SO2 in regions 2–4 was similar in percentage, from 83.9 % in the warm season for region 4 to 91.2 % in the warm season for region 3. There were no major differences between the warm and cold seasons in terms of percentage reduction. In terms of absolute value, the biggest reduction was for SO2 in region 3 during the cold season, and the 3-year-averaged seasonal mean concentration was reduced from 19.2 to 2.2 µg m−3.
-
The reduction in concentrations during the cold season was relatively uniform in terms of percentage, ranging from 60.1 % in region 2 to 62.5 % in region 3. The reduction was more significant during the warm season than during the cold season, ranging from 72.7 % in region 1 to 78.7 % in region 4. The reductions in regions 2, 3, and 4 were similar in terms of values in both seasons. The reduction of in terms of percentage was much smaller than SO2 in all regions during both seasons except for during the warm season in region 1.
-
During the warm season, the reduction of was seen in all four regions, ranging from 14.3 % and 15.6 % in regions 1 and 4 to 36.2 % and 57.5 % in regions 3 and 2. The reduction of during the cold season was only observed in region 2 (30.4 %). Although TNO3 was reduced during the cold season in regions 3 and 4, a higher percentage of HNO3 was converted to as more excess NH3 was available to form NH4NO3 due to the reduction of . As a result, the trend of in the two regions during the cold season changed very little. Unlike regions 3 and 4, region 2 did experience a significant reduction of in the cold season, following a 38.3 % reduction of TNO3. This can be explained as region 2 is an NH3-rich region. The formation of NH4NO3 during the cold season in the region is less sensitive to the excess NH3 made available from reduction than in regions 3 and 4. This can also be demonstrated by the least reduction of HNO3 (in terms of percentage) in region 2 during the cold season as well as the correlations of RNO3 vs. shown in Sect. 5.4.
-
The reduction of HNO3 was similar in all four regions during the warm season, ranging from 63.1 % to 68.8 %. During the cold season, region 2 had the lowest percentage reduction at 56.0 %, and region 1 had the highest at 63.5 %. The reduction of HNO3 can be through two paths: a reduction of NOx emissions and an increased neutralization of HNO3 by more excess NH3 due to less formation of (NH4)2SO4 and NH4HSO4. In terms of percentage, the reduction of HNO3 was more significant than TNO3 during the cold season, ranging from 14.4 % more in region 4 to 28.0 % more in region 1.
-
TNO3 had a reduction rate ranging from 35.5 % for the cold season in region 1 to 64 % during the warm season in region 3. The reduction during the warm season was much greater than in the cold season, ranging from 11.4 % higher in region 4 to 23.9 % higher in region 3. The difference was partially due to extra reductions of NOx emissions from power plants and other large combustion sources during the ozone season (May–September) required by the NBP that began in 2003 and the Clean Air Interstate Rule that started in 2009 (Napolitano et al., 2007; Butler et al., 2011; Sickles II and Shadwick, 2015).
-
The reduction of was similar in regions 2, 3, and 4, ranging from 48.9 % to 53.2 % in the cold season and from 74.0 % to 75.7 % in the warm season. The reduction of during the warm season was more significant than in the cold season, over 20 % more in regions 2–4. The reduction of generally followed the trends of , but the reduction rate was much lower than that of during the cold season because a certain percentage of was associated with and the reduction of was not as significant as during the cold season. Region 2 exhibited the largest reduction of (75.7 %) during the warm season, contributed by a 76.8 % reduction of as well as a 57.5 % reduction of .
-
RSO4 increased the most in region 3 during the cold season at 166.3 % and the least in region 1 during the warm season at 0.6 %. During the warm season, RSO4 increased by 54.9 % and 58.4 % in regions 2 and 3, respectively. The increase in RSO4 during the cold season was much higher than in the warm season in terms of percentage, ranging from 48.6 % in region 1 to 166.3 % in region 3.
-
RNO3 increased significantly in regions 1, 3, and 4 in both seasons, ranging from 73.9 % to 94.9 %, but RNO3 only increased by 8.0 % and 12.8 % in the warm and cold seasons for region 2, which was rich in NH3.
-
As presented in Table 3, the following points apply for the whole region: (1) among the five species of , , , HNO3, and SO2, only and SO2 still had regionally averaged annual mean concentrations exceeding 1.0 µg m−3 at the end of the study period. (2) was reduced by 73.3 % for the whole region during the study period, and it was reduced about 15 % more in the warm season than in the cold season in terms of percentage; (3) was reduced more in the warm season than in the cold season in terms of both percentage and absolute value; (4) was reduced by 29.1 % for the whole region. The reduction during the cold season occurred only in region 2, and the reduction during the warm season mainly occurred in regions 2 and 3. The reduction of for the whole region was mainly due to the reductions in region 2 during the warm and cold seasons; (5) RSO4 increased by 97.7 % in the cold season, much higher than 26.2 % in the warm season. RSO4 increased the most in region 3 during the cold season in terms of both absolute value and percentage.
3.4 Air quality at the end of the study period: 2014–2016
The 3-year-averaged air concentrations for 2014–2016 are used to describe the air quality at the end of the study period and are presented in Table S2 and Fig. S2. The air concentration mentioned in this section refers to the 3-year-averaged air concentration for each species, either for the warm or the cold season.
As at the beginning of the period, region 1 had the cleanest air among all regions, with the lowest air concentrations of less than 1.0 µg m−3 for all species and for both the warm and cold seasons. Unlike at the beginning of the period when during the warm season was about double that during the cold season in regions 2–4, at the end of the period had no significant differences between the two seasons. The air concentrations of were less than 2.0 µg m−3 in all regions and both seasons. For regions 2–4 the regional averages ranged from 1.6 to 1.8 µg m−3 during the warm season and from 1.4 to 1.7 µg m−3 during the cold season. SO2 during the warm season was only from 0.6 to 1.0 µg m−3 for regions 2–4. In the cold season, SO2 in regions 2 and 3 was the same at 2.2 µg m−3 and was only 1.1 µg m−3 in region 4. during the warm season varied from 0.5 to 0.7 µg m−3 for regions 2–4. During the cold season, it was 0.5 and 0.8 µg m−3 in regions 4 and 3, respectively, and it was much higher in region 2 with the value of 1.2 µg m−3. The air concentration of during the warm season was very low in regions 3 and 4, with values of 0.3 µg m−3, and it was doubled in region 2, being 0.6 µg m−3. During the cold season, was much higher than during the warm season, being 2.5, 1.3, and 0.5 µg m−3 for regions 2, 3, and 4, respectively. HNO3 in regions 2–4 varied from 0.6 µg m−3 in region 4 to 1.1 µg m−3 in region 2 during the warm season and from 0.7 µg m−3 in region 4 to 0.9 µg m−3 in region 3 during the cold season. There was little difference between the warm and cold seasons in regions 3 and 4. TNO3 was the highest in region 2 in both seasons, being 1.7 and 3.2 µg m−3 for the warm and cold seasons, respectively. Region 3 had the second-highest TNO3 with values of 1.2 and 2.1 µg m−3 for the warm and cold seasons, and the corresponding values for region 4 were 0.9 and 1.3 µg m−3.
In summary, for species of , , , HNO3, and SO2, region 1 had air concentrations of less than 1.0 µg m−3 for all species in both seasons. For regions 2–4, was less than 1.0 µg m−3 for all regions and both seasons except regions 2 and 3 during the cold season, for which the air concentrations of were 2.5 and 1.3 µg m−3, respectively; HNO3 was less than 1.0 µg m−3 except region 2 during the warm season with a value of 1.1 µg m−3; was less than 1.0 µg m−3 for all regions except region 2 during the cold season at 1.2 µg m−3; was greater than 1.0 but less than 2.0 µg m−3 for regions 2–4 and both seasons; SO2 was greater than 1.0 but less than 2.5 µg m−3 for regions 2–4 and both seasons, except regions 3 and 4 during the warm season, being 0.8 and 0.6 µg m−3. Among the four regions, region 2 had the highest air concentration for all species except HNO3 during the cold season. in region 2 was especially high, double the second-highest value in region 3 in both seasons. Also, in region 2 had the highest value (at 2.5 µg m−3) among all species in four regions and both seasons, although it significantly decreased from 3.6 µg m−3 at the beginning of the study period.
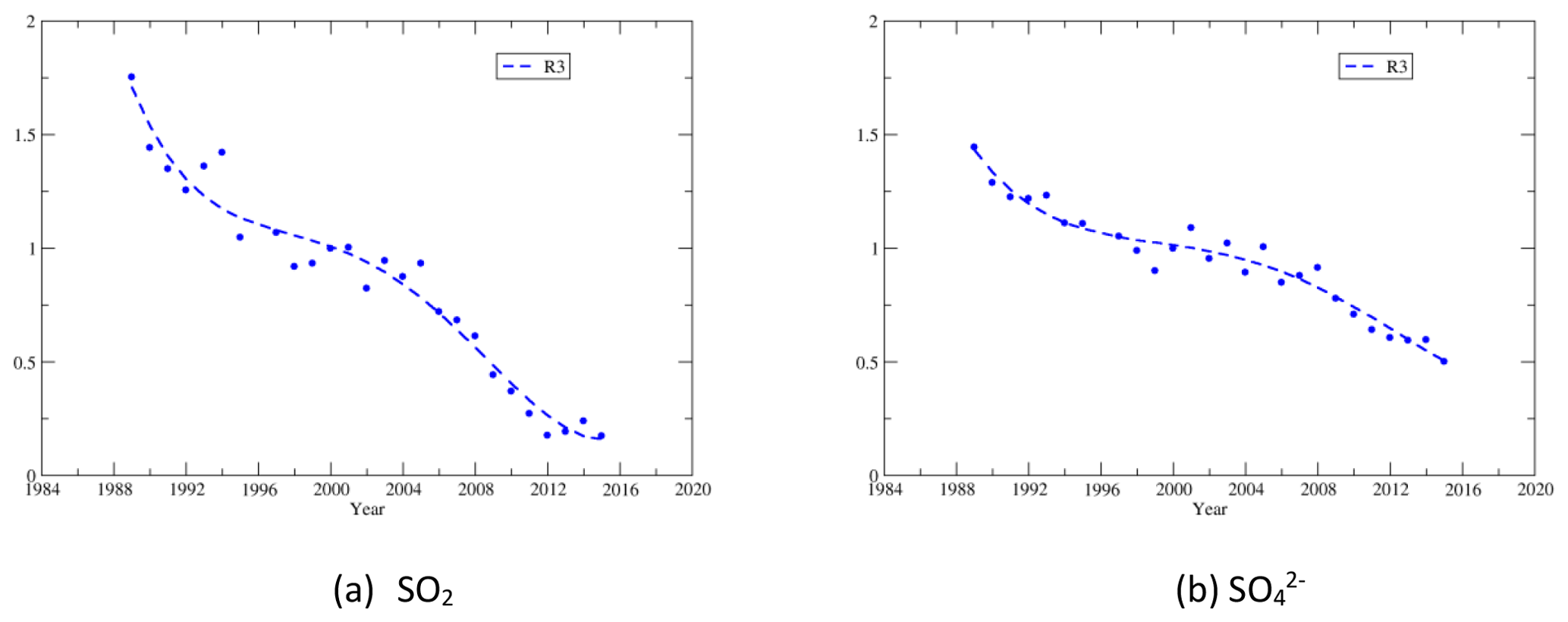
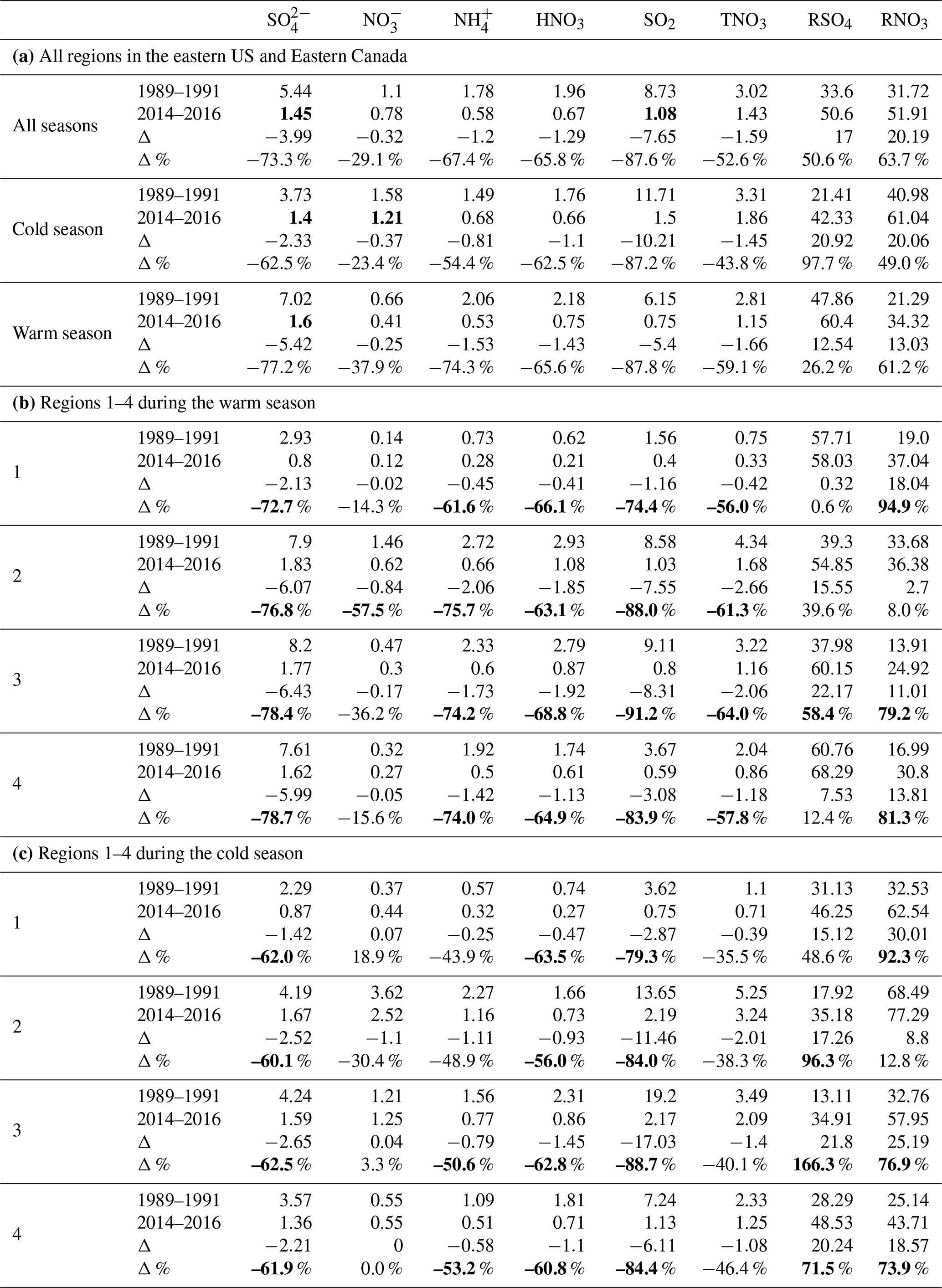
Figure 4Examples of fourth-order polynomial regressions of SO2 (a) and (b) for region 3 during the cold season.
3.5 The long-term trends derived with polynomial regressions
Through trial and error, we found that polynomial regressions can reasonably describe the long-term trends of species for the period. Through these regressions, we can eliminate the relatively short-term variations due to meteorology. Fourth-order polynomial regressions were applied to the normalized seasonal means of , SO2, and during the cold and warm seasons. For , HNO3, and TNO3, we applied fifth-order polynomial regressions to better capture the trends. The regressed trends are normalized to the regressed values of the year 2000 as this was the turning point for the trends of . Examples of the regression for and SO2 in region 3 during the cold season are shown in Fig. 4. Figure 5 shows comparisons between the normalized seasonal means during the cold season in region 3 and the corresponding regressed values.

Figure 5Comparisons of the normalized seasonal mean concentrations with the ones regressed with polynomial regressions for , SO2, , , and HNO3 for region 3 during the cold season. The dotted lines link the seasonal mean concentrations from 1990 to 2016 for species except for to show the temporal trends.
The regressed trends for , SO2, , , HNO3, and TNO3 for the four regions during the cold and warm seasons are shown in Fig. 6a. The regressed trends for regions 1–4 clearly show that (1) the most significant reduction of all species was SO2, and (2) there were significant disparities in the reduction rates between and SO2 during the cold season. There were also disparities during the warm season in regions 1–3, but they were much less significant than the cold season; (3) among all species, the least significant reduction was for during the period. Figure 6b shows the regressed trends for each species during the cold and warm seasons for different regions. For and SO2, there were large differences in the trends during 1990–2000 between regions 1 and 4. The trends of were different for the four regions during 1990–2000 in both seasons and during 2000–2016 in the cold season. In the warm season, the trends of were similar for regions 2–4 during 1990–2000 but were different during 2000–2016. The trends of HNO3 and TNO3 were different for 1990–2000 during the warm season but were similar during 2000–2016.
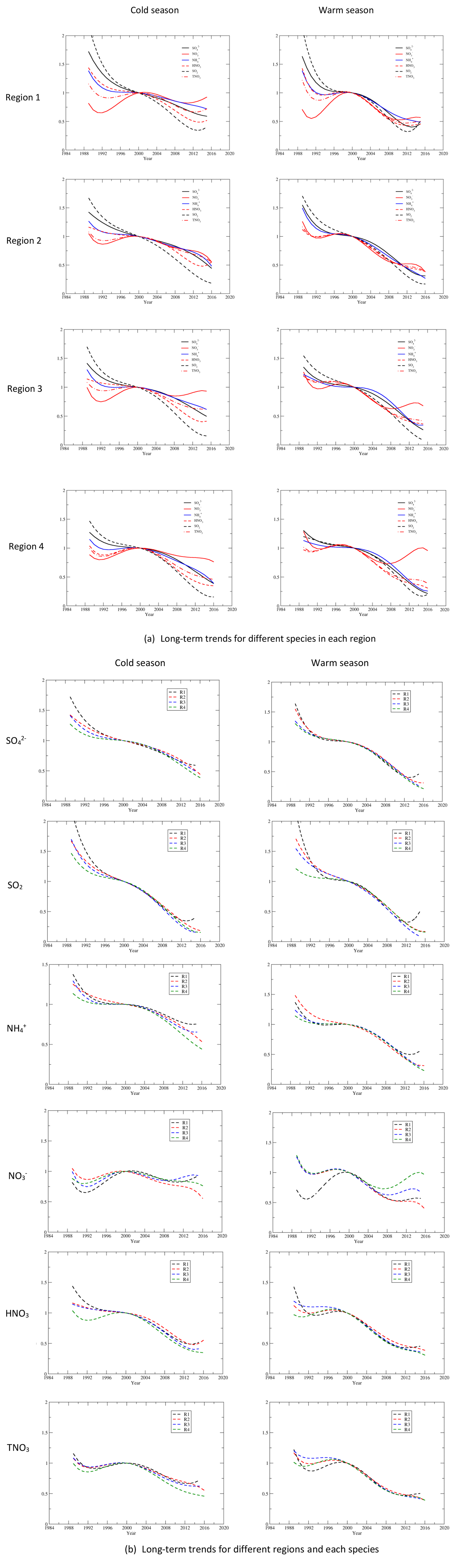
Figure 6Long-term trends derived with fourth-order polynomial regressions for , SO2, and and fifth-order polynomial regressions for , HNO3, and TNO3 during the warm and cold seasons.
4.1 RSO4 and correlations of RSO4 vs. SO2
RSO4 is a metric describing how much sulfur in the air is oxidized from gas SO2 to particulate . A similar metric, the ratio of SO2 mass oxidized below 2 km to SO2 mass emitted, was used by Shah et al. (2018). RSO2, which is 1 – RSO4, was used by Sickles II and Shadwick (2015). RSO4 is also an indicator of the gas–particle partition ratio for sulfur in the air as SO2 and exist in the air as a gas and particle, respectively. This metric depends on a number of factors: the oxidation capacity of the air, the local emission rate of SO2, the transportation of SO2 and from upwind regions, and the time it takes to bring upwind SO2 and to a local site. Generally, the longer it takes to transport an upwind air parcel, the higher the percentage of SO2 oxidized into , and more SO2 is dry-deposited than , so RSO4 is higher. Also, the lower the percentage of SO2 emitted locally and the higher the atmospheric oxidation capacity, the higher RSO4. RSO4 for 1989–1991 and 2014–2016, and the changes in RSO4 between 1989–1991 and 2014–2016 during the warm and cold seasons, are presented in Fig. S4. During the warm season, because more solar photons are available to produce O3 from NOx and VOCs, RSO4 was much higher in the warm season than the cold season. At the beginning of the period, RSO4 in the warm season was about double that in the cold season in regions 1, 2, and 4 and around triple in region 3. RSO4 was much higher in regions 4 and 1 than in regions 2 and 3 because the local emissions of SO2 were much higher in regions 2 and 3. The “freshly” emitted SO2 made RSO4 in regions 2 and 3 relatively smaller. RSO4 in region 3 during the cold season was only 13.1 % in 1989–1991, indicating a very low sulfur gas–particle partitioning ratio. Figure S4 shows that during the cold season, RSO4 increased by more than 40 % at all sites except for VPI120, at which it increased by 18.7 %. During the warm season, RSO4 increased at all sites except for VPI120, ASH135, and WST109, at which it decreased by 25.0 %, 12.5 %, and 3.9 %, respectively. The most significant increase in RSO4 was in region 3 during the cold season, with a regional average of 166.3 %.
Figure 7 shows that RSO4 increased linearly with the year for region 1 and quadratically for regions 2–4 for both seasons. RSO4 increased significantly after 2005 in regions 2–4. Figure 8 shows the correlations of RSO4 vs. SO2 for regions 2–4, and it is clear that RSO4 increased with the decrease in SO2. The increase in RSO4 was relatively slow when the concentration of SO2 was greater than 5 µg m−3 in the cold season and 7.5 µg m−3 in the warm season. RSO4 soared when SO2 was less than 5 µg m−3 in the cold season in regions 2–4 and less than 3 µg m−3 in the warm season in regions 2 and 3. The increase in RSO4 with the decrease in SO2 can be explained as follows: (1) the atmospheric oxidants did not decrease as much as SO2 emissions did. For example, the daily maximum 8 h average O3 only decreased by 14 % for the EUS during the May–September ozone season from 1997 to 2008 (Butler et al., 2011), and it decreased by 4 %–15 % during 1997–2006 for regions 2, 3, and 4 (Chan, 2009). No decreasing trend was found for the EUS during the cold season for 1997–2006 (Chan, 2009). Sickles II and Shadwick (2015) found that O3 in the EUS increased during the cold season for 1990–2010. O3 is an atmospheric oxidant and is a precursor to the formation of other atmospheric oxidants, such as OH and H2O2. Therefore, relative to the significantly reduced SO2, more atmospheric oxidants were available to oxidize SO2, and RSO4 increased significantly during the period. (2) NH3 was relatively unchanged during the period and even increased in some regions (Yao and Zhang, 2016). The decrease in SO2 caused the decrease in H2SO4 formation. Together this made cloud or rain droplets or snow particles less acidic, which was beneficial for the oxidation of SO2 by H2O2 in the aqueous phase (Makar et al., 2009; Jones and Harrison, 2011).


Figure 8Correlations of the seasonal means during the cold and warm seasons: RSO4 vs. SO2 for regions 2–4. Fourth-order polynomial and quadratic regressions were applied for the cold and warm seasons, respectively. R2, 3, and 4 refer to regions 2–4. The dots with circles represent the seasonal means in the first and last 3 years.
The disparity in the reduction of SO2 and in response to emission reductions of SO2, namely the fact that the reduction rate of SO2 was faster than , has been reported and discussed in some previous studies (Lövblad et al., 2004; Reid et al., 2001; Sickles II and Shadwick, 2015; Shah et al., 2018; Aas et al., 2019). The time series of the normalized regional concentrations of and SO2 in Fig. 3 clearly show the disparity during the period of 1990–2015. The significant increase in RSO4 during the period, especially during the cold season, explains why the reduction rate of SO2 was much higher than that of . The reduction of SO2 was not only due to the emission reductions, but also to the fact that a higher fraction of SO2 was converted to . A faster reduction of SO2 was observed for all four regions during the cold season, both before and after the year 2000, and it was more significant after 2000. This can be explained by the fact that the increase in RSO4 with time was nonlinear. As shown in Table 2, in the first 10 years of the study period, the increase in RSO4 was relatively limited. During the cold season, it only increased by 7.3 % in region 4 to 16.5 % in region 3. It was in the last 10 years from 2005 to 2015 when SO2 was further reduced so that RSO4 increased dramatically. As shown in Tables 2 and 3, during the cold season in region 3, RSO4 only increased by 16.5 % in the first 10 years, 1990–2000, but it increased by 149.8 % for the last 15 years of 2000–2015. During the warm season, the disparity in the reduction between SO2 and was much lower, as clearly shown in Fig. 3. This is because the increase in RSO4 during the warm season was much less significant than during the cold season (Table 4). In the first 10 years, RSO4 changed from −4.1 % in region 4 to 7.7 % in region 1. For the period of 1990–2015, RSO4 only increased by 0.6 % and 12.4 % in regions 1 and 4. The disparity in the reduction rate of SO2 vs. for these two regions was only 1.7 % and 5.2 % during the warm season, respectively. This is expected and can be explained as follows: (1) in the warm season more atmospheric oxidants are produced due to more solar photons being available than in the cold season, so the oxidation of SO2 is less limited by the availability of atmospheric oxidants in the warm season; (2) in the cold season, limited atmospheric oxidants are available for the oxidation of SO2. The reduction of SO2 emissions to the air will make more atmospheric oxidants available to each SO2 molecule, increase the oxidation rate of SO2, and result in an increase in RSO4; (3) furthermore, in the EUS, the seasonal mean O3 concentration decreased in the warm season and increased in the cold season for the study period (Sickles II and Shadwick; 2015). This made the overall oxidation capacity of the lower atmosphere in the EUS higher in the cold season and lower in the warm season.
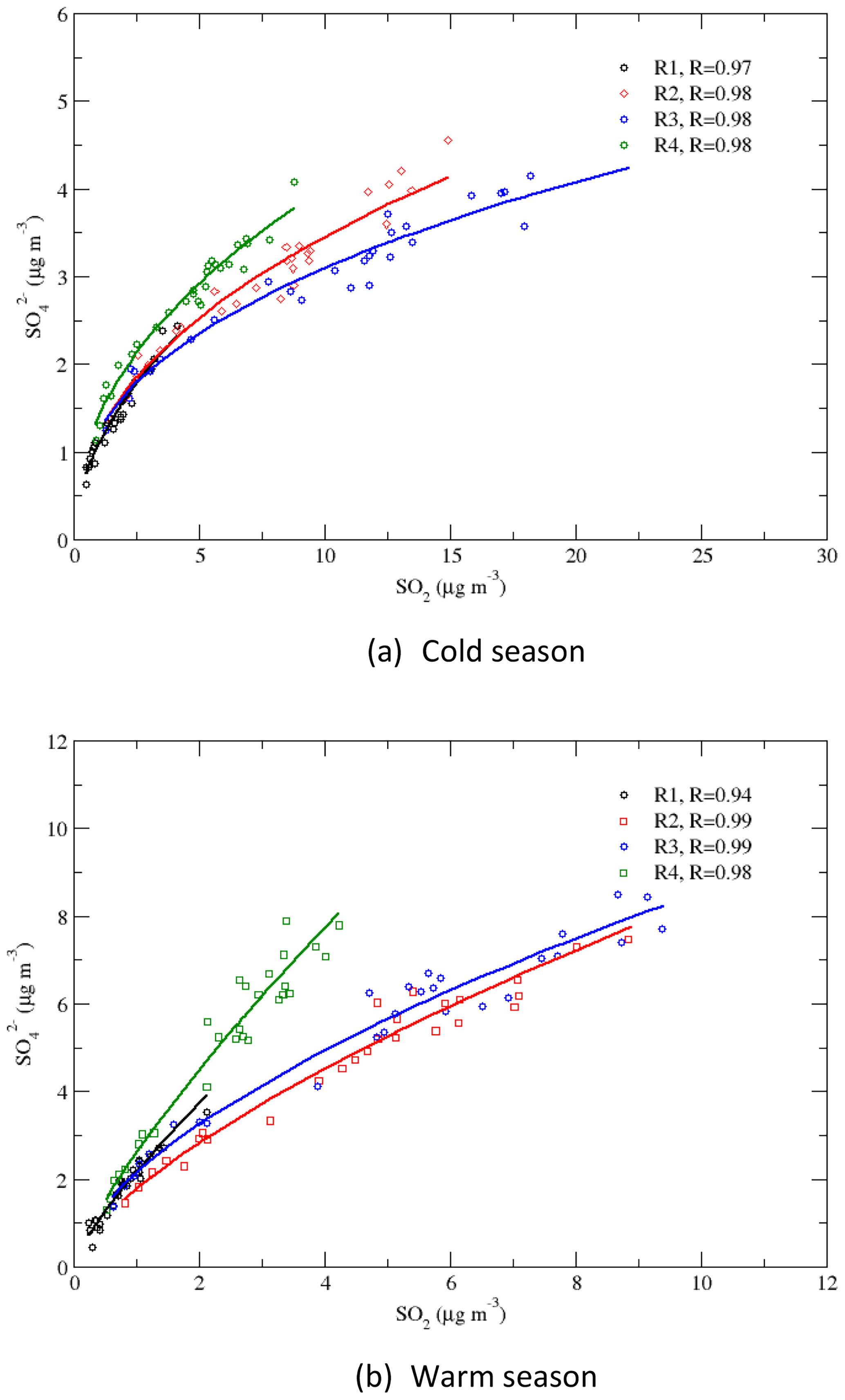
Figure 9Correlations of the seasonal means during the cold and warm seasons: vs. SO2 for regions 1–4. R1, 2, 3, and 4 refer to regions 1–4.
4.2 Correlations of vs. SO2
Correlations between and SO2 are presented in Fig. 9 for regions 1–4 and for the warm and cold seasons. The –SO2 relationships for the period of 1990–2010 can be described by linear regressions (not shown in the graph), with R=0.87–0.98 during the warm season and R=0.96–0.99 during the cold season. During the cold season, region 1 had the highest slope, and it was followed by regions 4, 2, and 3. During the warm season, the slopes for regions 1 and 4 were similar and were higher than the slopes for regions 2 and 3. A linear relationship between the seasonal mean concentrations of and SO2 indicates that there is a linear relationship between the concentration of and the emission of SO2. This is consistent with the relationship of the concentration and the SO2 emission rate from the early 1990s through 2010 revealed in the study of Hand et al. (2012). As RSO4 significantly increased when SO2 was further reduced during 2010–2016, as seen in Fig. 8, the slopes of the linear regression for 2010–2016 were much higher than those for 1990–2010. A power-law regression, which bends a linear regression with a gentle slope to a linear regression with a steep slope, described the –SO2 relationships very well, with R=0.97–0.98 during the cold season and R=0.94–0.99 during the warm season, as shown in Fig. 9. In some previous studies (e.g., Jones and Harrison, 2011), nonlinear power-law relationships have been found for observations at different sites for different seasons and periods. Our results indicate that a linear relationship between and SO2 exists for a subperiod of a long-term period, but generally the correlation of vs. SO2 is a power-law relationship.
4.3 RNO3
Similar to RSO4 being a gas–particle partition indicator for sulfur in the air, RNO3 is a metric indicating the fraction of gas HNO3 that is aerosolized (Sickles II and Shadwick, 2015). In the air, the emitted NOx is oxidized to gas HNO3, which can be aerosolized through two paths: (1) reaction with NH3 to form NH4NO3 and (2) reaction with existing aerosols such as sea salts and crustal materials to form NaNO3, Ca(NO3)2, and Mg(NO3)2. The ratio is significantly sensitive to the air temperature, as NH4NO3, NH3, and HNO3 in the air are in equilibrium and temperature changes can affect the partitioning between gas and particle phases (Doyle et al., 1979; Harrison and Pio, 1982).
RNO3 for 1989–1991 and 2014–2016, as well as the change in RNO3 between the two periods are shown in Fig. S5. At the beginning of the period, (1) RNO3 in the cold season was much higher than the warm season for all regions. RNO3 in the cold season in regions 2 and 3 was more than double that for the warm season; (2) RNO3 in region 2 was much higher than other regions and was more than double that in regions 3 and 4. For the 25-year period of 1990–2015, RNO3 significantly increased by more than 70 % in regions 1, 3, and 4 during both seasons. In region 2, RNO3 only increased by 12.8 % and 8.0 % during the cold and warm seasons, respectively. The significant increase in RNO3 in regions 1, 3, and 4 can be attributed to the significant reduction of during the period, as is explained in Sect. 4.4.
Figure 7 shows that RNO3 had an increasing trend with the year for all regions and both seasons except for region 2 during the warm season. The trends can be described well by linear regressions in regions 1 and 2 and by quadratic regressions in regions 3 and 4. The linear regression shows that RNO3 in region 2 had a decreasing trend for 1990–2010 during the warm season. The exact reason for this is unknown. One hypothesis is that due to the global warming trend in recent years and the significant reductions of sulfate and nitrate aerosols (which cool the atmosphere by reflecting more solar radiation back to space), the near-surface temperature in the Midwest had an increasing trend during the period of 1990–2010 (National Climate Assessment, 2014). As region 2 is rich in NH3, RNO3 is more sensitive to the air temperature than to the availability of NH3. An increasing trend of air temperature in the warm season can cause a decreasing trend of RNO3.
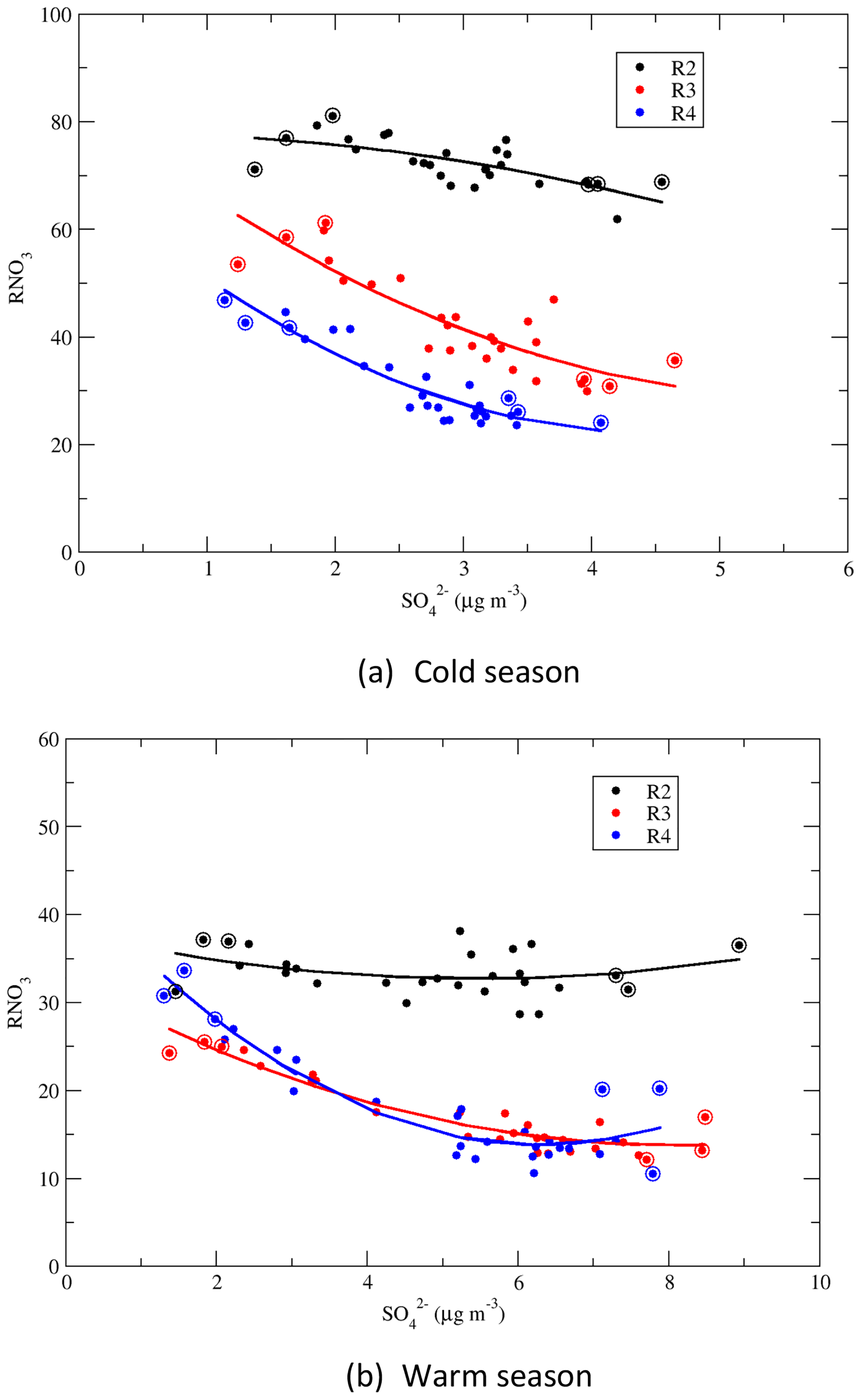
Figure 10Correlations of the seasonal means during the cold and warm seasons: RNO3 vs. for regions 2–4. The dots with circles represent the seasonal means in the first and last 3 years.
4.4 Correlations of RNO3 vs.
Correlations between the seasonal mean RNO3 and the seasonal mean concentration of for regions 2–4 and for the warm and cold seasons are presented in Fig. 10. For NH3-rich region 2, RNO3 increased slightly with the decrease in during the cold season, and there was no obvious trend during the warm season. For regions 3 and 4, which were NH3-limited, RNO3 increased with the decrease in . RNO3 increased steeply when the seasonal mean concentration of was less than 4 µg m−3 during the warm season and less than 3 µg m−3 during the cold season. The increase in RNO3 with the decrease in can be explained as follows: (1) in regions 3 and 4, the formation of NH4NO3 was limited by the availability of NH3; (2) as decreased, some of the NH3 previously forming (NH4)2SO4 ∕ NH4HSO4 was released and was available to react with HNO3 to form NH4NO3. In contrast, RNO3 was much less sensitive to the reduction in region 2 as the emissions of NH3 there were much higher than in regions 3 and 4, as seen in Fig. S6. Thus, in general there was always excess NH3 available to react with HNO3 to form NH4NO3 in region 2, which resulted in a lack of trends in RNO3. This also explains why TNO3 decreased by 40.1 % and 46.4 %, respectively, during the cold season in regions 3 and 4, while the change in was negligible in these two regions.
4.5 Correlations of vs. and
Correlations of vs. and vs. are shown in Fig. 11 for regions 2–4 and for the cold and warm seasons. During the warm season, in regions 3 and 4 changed little in value during 1990–2015, while changed significantly, and this change was mostly linearly associated with the change in . In region 2, the change in was also mostly associated with the change in , but the change in also made a contribution to it. During the cold season, correlated with linearly very well in regions 2–4. In region 2, also changed significantly during the period, and the variation of correlated well with the variation of with R=0.87, indicating that some of the reduction of in region 2 was associated with the reduction of during the period. The variations of in regions 3 and 4 were relatively small, and the correlations between and , with R=0.42 and 0.40, respectively, were much less significant than those for vs. . Figure 11 shows that in the EUS and EC, the reduction of during 1990–2015 was mainly due to the reduction of in regions 2–4, but in region 2 the reduction of also made a substantial contribution to it.
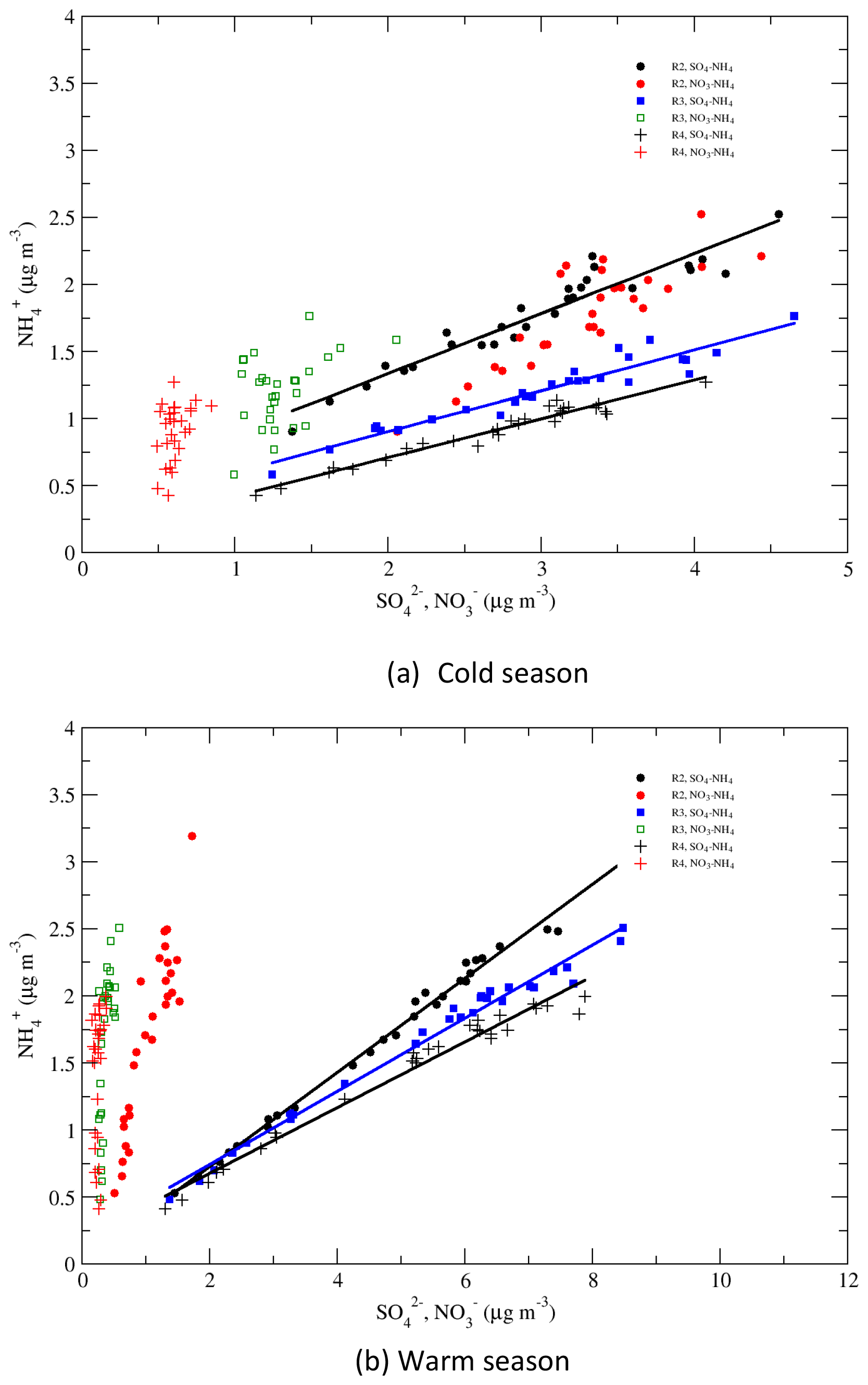
Figure 11Correlations of vs. and vs. during the cold and warm seasons. R2, 3, and 4 refer to regions 2–4.
4.6 Sulfate–nitrate–ammonium (SNA) aerosols
Sulfate, nitrate, and ammonium are the major components of the secondary aerosols in the atmosphere in the EUS and EC (Bell et al., 2007; Dabek-Zlotorzynska et al., 2011). Time series of the seasonal means of the total mass of sulfate–nitrate–ammonium aerosols during the warm and cold seasons are shown in Fig. 12. During the cold season, mainly due to the concentrations of and , SNA had the highest and second-highest seasonal mean concentrations in region 4 and region 3, respectively, and the lowest seasonal mean concentration in region 1. During the warm season, SNA in regions 2 and 3 was comparable and a little higher than that in region 4. The trends in regions 2–4 are similar during the warm season, and the SNA in regions 2–4 is much higher than that in region 1. Figure 13 shows that SNA in region 1 during the warm season was higher than that during the cold season until 2007, and the trend was reversed after that. In region 2, SNA was generally higher during the warm season until 2005, and it was opposite thereafter. SNA in region 3 was significantly higher during the warm season than the cold season until 2007, and the trend was opposite after 2012. Similarly, SNA in region 4 was much higher in the warm season until 2008 and was comparable between the warm and cold seasons after 2012. Two points can be derived from the above trends: (1) in the EUS and EC, SNA during the warm season was mainly due to (NH4)2SO4∕NH4HSO4. When the emission of SO2 over the region decreased significantly, SNA followed the decreasing trend of even though the reduction in was not significant; (2) during the cold season, besides (NH4)2SO4∕NH4HSO4, NH4NO3 also made a considerable contribution to SNA. As decreased significantly in both the warm and cold seasons, but changed little during the cold season except in region 2, the SNA during the cold season became comparable or even higher than the SNA during the warm season. SNA pollution in regions 2 and 3 was more of an issue during the cold season than during the warm season when the emissions of SO2 were further reduced during 2006–2015.
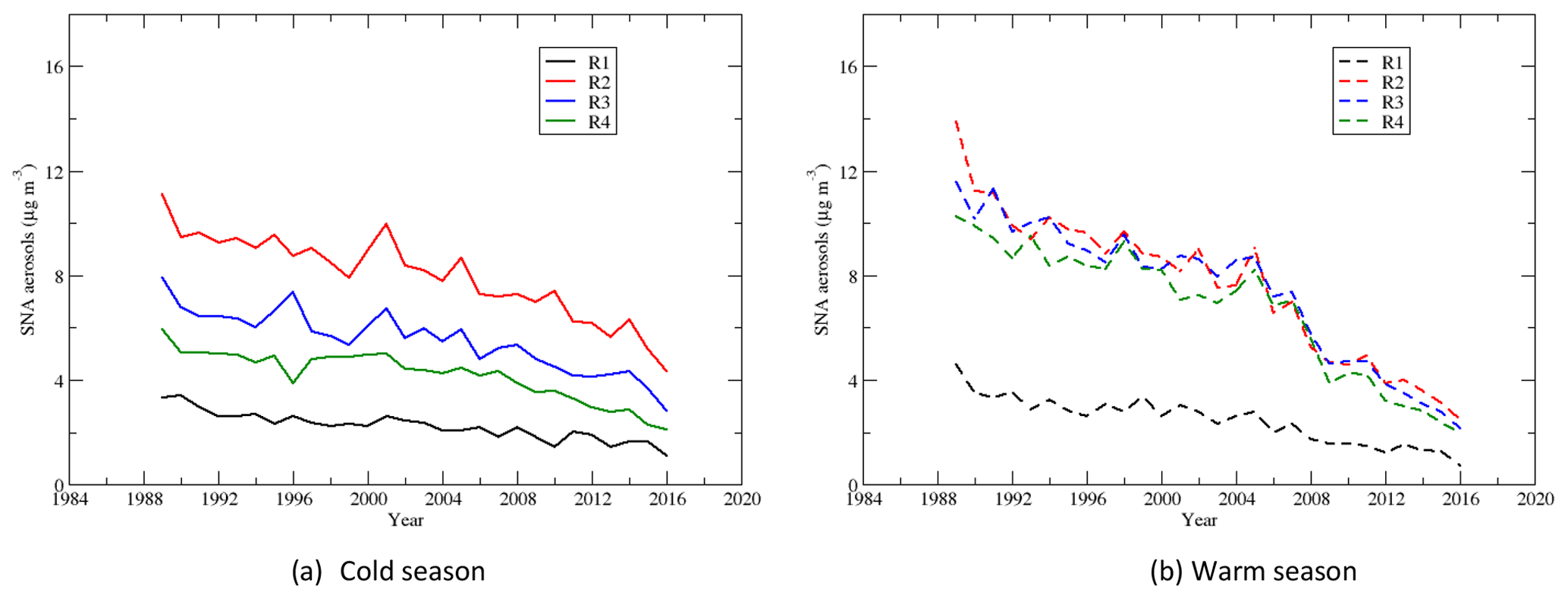
Figure 12Time series of the seasonal means of sulfate–nitrate–ammonium aerosols during the cold and warm seasons.
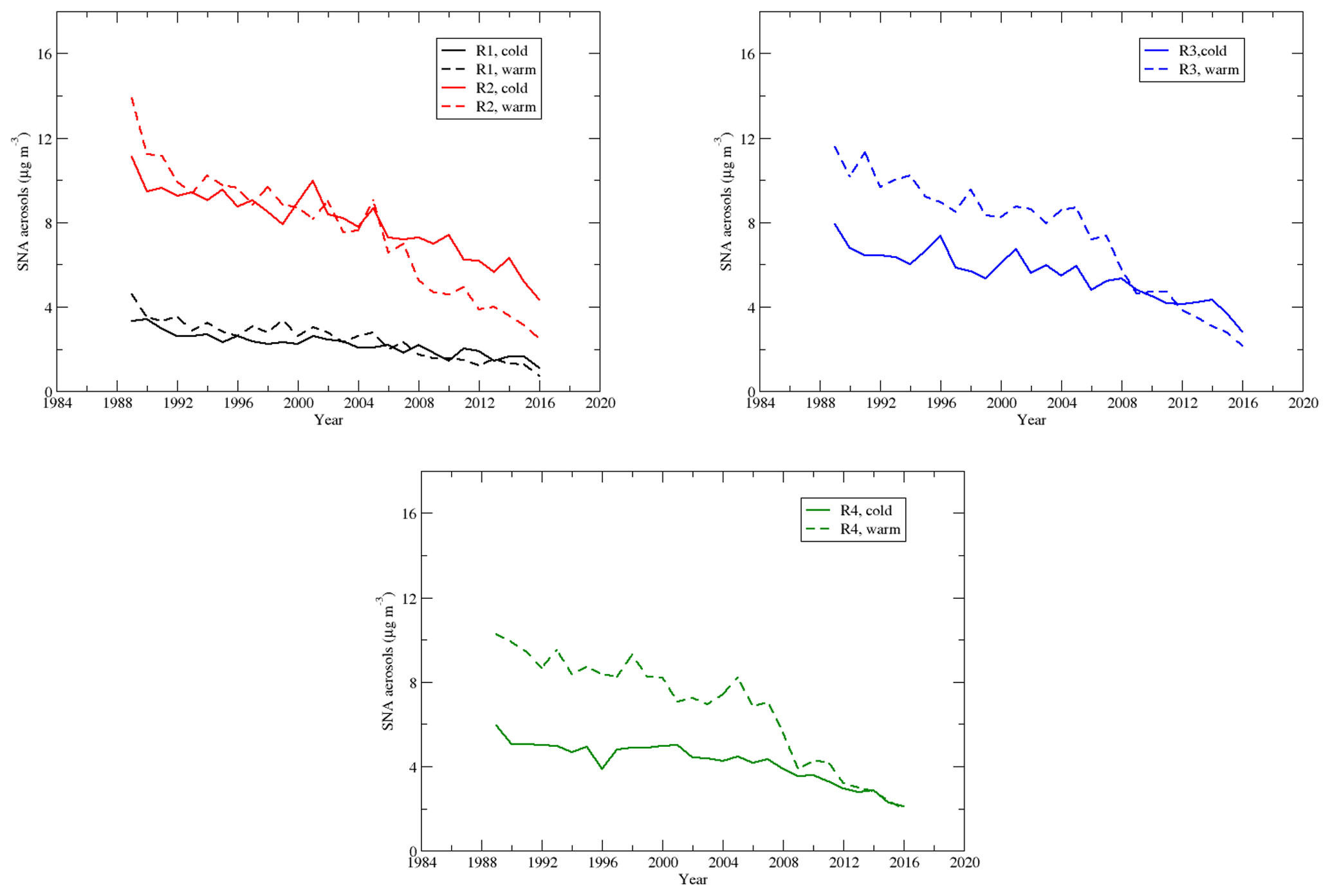
Figure 13Time series of the seasonal means of sulfate–nitrate–ammonium aerosols in regions 1–4.
With the implementation of the Title IV of the 1990 amendments to the CAA in the US in the 1990s, the emissions of SO2 and NOx in the US were reduced from 23.1 million to 3.7 million t yr−1 for SO2 and from 25.2 million to 11.5 million t yr−1 for NOx from 1990 to 2015. In Canada, compared to the emission level in 1990, SO2 and NOx emissions in 2015 were reduced by 65 % and 25 %, respectively. In both the US and Canada, the reduction of emissions was mainly in the eastern regions of the countries. The air concentrations of gases SO2 and HNO3, as well as particles , , and , had a very different nonlinear response, both spatially and temporally, to the significant reductions of SO2 and NOx emissions.
In this study, we analyzed the air concentrations of , , , HNO3, and SO2 measured weekly by the CASTNET network in the US and daily by the CAPMoN network in Canada from 1990 to 2015 to reveal the temporal and spatial changes during the period. Four distinct regions, each with a characteristic pattern of air quality in the EUS and EC, were identified: the northeastern US and Eastern Canada (region 1), the Midwest (region 2), the Mid-Atlantic (region 3), and the southeastern US (region 4). In the first 10-year period of 1990–2000, SO2 and decreased by more than 20 % except for in region 4 during the cold season and SO2 in region 4 during the warm season. declined by 12 % to 29.8 % during both seasons except that region 4 during the cold season had a negligible change. increased in regions 1–4 during the cold season and changed little during the warm season. HNO3 was reduced in regions 1–3 by more than 9 % during both seasons and increased in region 4 by 5 % and 5.7 % during the cold and warm seasons, respectively. In a 25-year period of 1990–2015, the reduction of ranged from 60.1 % in region 2 to 62.5 % in region 3 during the cold season and from 72.7 % in region 1 to 78.7 % in region 4 during the warm season. The reduction of SO2 was the most significant among all species, ranging from 83.9 % during the warm season in region 4 to 91.2 % during the warm season for region 3. During the warm season, the reduction of was seen in all regions, ranging from 14.3 % in region 1 to 57.5 % in region 2. However, during the cold season, the reduction of was only seen in region 2, and it was reduced by 30.4 %. The change in was negligible in regions 3 and 4 during the cold season. The reduction of HNO3 during the warm season was relatively uniform in terms of percentage, ranging from 63.1 % to 68.8 %. During the cold season, region 2 had the lowest percentage of reduction at 56.0 %, and region 1 had the highest at 63.5 %. The reduction of was the most significant during the warm season in terms of both percentage and absolute value. The reduction ranged from 74.0 % to 75.7 % in regions 2–4 during the warm season, which was 20 % more than the corresponding reduction during the cold season. The time series of the seasonal mean concentrations during the warm and cold seasons show that the reduction of each species was not even in time during the period. The reductions of and SO2 mainly occurred during 1990–1995 and 2007–2016 for the warm season and during 1990–1995 and 2005–2016 for the cold season. The reduction of was mainly after the year 2000.
RSO4 is a metric indicating the gas–aerosol partitioning of sulfur in the air, and RNO3 is an indicator of the fraction of HNO3 being aerosolized. RSO4 increased by 48.6 % to 166.3 % during the cold season and by 0.6 % to 58.4 % during the warm season. RSO4 was found to increase quadratically with the decrease in SO2 for regions 2–4 and the two seasons. The significant increase in RSO4 during the cold season explains why the reduction rate of SO2 was much higher than that of during the period, as the reduction of SO2 was not only due to the emission reduction, but also more SO2 being converted to . A faster reduction of SO2 was observed for all four regions during the cold season, both before and after the year 2000, and it was more significant after 2000. During the warm season, the difference in the reduction rate between SO2 and was much lower. This is because the increase in RSO4 during the warm season was much less significant than during the cold season. In regions 1 and 4, RSO4 only increased by 0.6 % and 12.4 % during the warm season. Differences in the reduction rate between SO2 and for these two regions during the warm season were only 1.7 % and 5.2 %, respectively. For regions 1, 3, and 4, RNO3 increased between 79.2 % and 94.9 % during the warm season and between 73.9 % and 92.3 % during the cold season. For region 2, RNO3 for the warm and cold seasons only increased by 8.0 % and 12.8 %, respectively, as NH3 in the region was in excess to neutralize HNO3. RNO3 was found to increase quadratically with the decrease in in regions 3 and 4.
In summary, with the significant reductions of SO2 and NOx emissions in the EUS and EC during 1990–2015, , SO2, , and HNO3 were reduced significantly by 73.3 %, 87.6 %, 67.4 %, and 65.8 % for the whole region. The reduction of was relatively less significant at 29.1 %, and it mainly occurred (1) after the year 2000, (2) in regions 1–4 during the warm season, and (3) in region 2 only during the cold season.
The CASTNET data used in this study can be downloaded from the website of the U.S. Environmental Protection Agency at https://java.epa.gov/castnet/clearsession.do (EPA, 2020). The CAPMoN data used in this study can be accessed through the Open Government Portal of Canada at https://open.canada.ca/data/en/dataset?keywords=CAPMoN (ECCC, 2020) or by contacting the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-3107-2020-supplement.
JF carried out the overall analysis and interpretation of the data as well as writing the paper. EC did the initial data and trend analysis. RV provided supervision for the study and discussed the results.
The authors declare that they have no conflict of interest.
We would like to thank Tom Butler and the anonymous reviewer. Their detailed, insightful, and constructive suggestions and comments greatly improved the quality of the paper. We also thank Irene Cheng for the internal review. The CASTNET and CAPMoN data used in this study were provided by Environment and Climate Change Canada (ECCC) through the Canadian National Atmospheric Chemistry (NAtChem) Particulate Matter and Precipitation Databases.
This paper was edited by Veli-Matti Kerminen and reviewed by two anonymous referees.
Aas, W., Mortier, A., Bowersox, V., Cherian, R., Faluvegi, G., Fagerli, H., Hand, J., Klimont, Z., Galy-Lacaux, C., Lehmann, C. M. B., Myhre, C. L., Myhre, G., Olivié, D., Sato, K., Quaas, J., Rao, P. S. P., Schulz, M., Shindell, D., Skeie, R. B., Stein, A., Takemura, T., Tsyro, S., Vet, R., and Xu, X.: Global and regional trends of atmospheric sulfur, Sci. Rep., 9, 953, https://doi.org/10.1038/s41598-018-37304-0, 2019.
Bell, M. L., Dominici, F., Ebisu, K., Zeger, S. L., and Samet, J. M.: Spatial and temporal variation in PM2.5 chemical composition in the United States for health effects studies, Environ. Health Perspect., 115, 989–995, https://doi.org/10.1289/ehp.9621, 2007.
Bloomer, B. J., Vinnikov, K. Y., Dickerson, R. R.: Changes in seasonal and diurnal cycles of ozone and temperature in the eastern U.S., Atmos. Environ., 44, 2543–2551, https://doi.org/10.1016/j.atmosenv.2010.04.031, 2010.
Butler, T. J., Likens, G. E., Vermeylen, F. M., and Stunder, B. J. B.: The relation between NOx emissions and precipitation in the eastern USA, Atmos. Environ., 37, 2093–2104, https://doi.org/10.1016/S1352-2310(03)00103-1, 2003.
Butler, T. J., Vermeylen, F. M., Rury, M., Likens, G. E., Lee, B., Bowker, G. E., and McCluney, L.: Response of ozone and nitrate to stationary source NOx emission reductions in the eastern USA, Atmos. Environ., 45, 1084–1094, https://doi.org/10.1016/j.atmosenv.2010.11.040, 2011.
CAA – Clean Air Act: The Clean Air Act – Highlights of the 1990 Amendments, available at: https://www.epa.gov/clean-air-act-overview/clean-air-act-highlights-1990-amendments, last access: November 2019.
Chan, E.: Regional ground-level ozone trends in the context of meteorological influences across Canada and the eastern United States from 1997 to 2006, J. Geophys. Res., 114, D05301, https://doi.org/10.1029/2008JD010090, 2009.
Chan, E. and Vet, R. J.: Baseline levels and trends of ground level ozone in Canada and the United States, Atmos. Chem. Phys., 10, 8629–8647, https://doi.org/10.5194/acp-10-8629-2010, 2010.
Cheng, I., and Zhang, L.: Long-term air concentrations, wet deposition, and scavenging ratios of inorganic ions, HNO3, and SO2 and assessment of aerosol and precipitation acidity at Canadian rural locations, Atmos. Chem. Phys., 17, 4711–4730, https://doi.org/10.5194/acp-17-4711-2017, 2017.
Clarke, J.-F., Edgerton, E. S., Martin, B. E.: Dry deposition calculations for the clear air status and trends network, Atmos. Environ., 31, 3667–3678, https://doi.org/10.1016/S1352-2310(97)00141-6, 1997.
Dabek-Zlotorzynska, E., Dann, T. F., Martinelango, P. K., Celo, V., Brook, J. R., Mathieu, D., Ding, L., and Austin, C. C.: Canadian National Air Pollution Surveillance (NAPS) PM2.5 speciation program: Metholodology and PM2.5 chemical composition for the years 2003–2008, Atmos. Environ., 45, 673–686, https://doi.org/10.1016/j.atmosenv.2010.10.024, 2011.
Doyle, G. J., Tuazon, E. C., Graham, R. A., Mischke, T. M, Winer, A. M., and Pitts Jr., J. N.: Simultaneous concentrations of ammonia and nitric acid in a polluted atmosphere and their equilibrium relationship to particulate ammonium nitrate, Environ. Sci. Technol., 13, 1416–1419, https://doi.org/10.1021/es60159a010, 1979.
Du, E., de Vires, W., Galloway, J. N., Hu., X., and Fang, J.: Changes in wet nitrogen deposition in the United States between 1985 and 2012, Environ. Res. Lett., 9, 095004, https://doi.org/10.1088/1748-9326/9/9/095004, 2014.
ECCC – Environment and Climate Change Canada: Canadian Environmental Sustainability Indicators: Air pollutant emissions,available at: https://www.canada.ca/en/environment-climate-change/services/environmental-indicators/air-pollutant-emissions.html, last access: 13 November 2019.
ECCC – Environment and Climate Change Canada: Canadian Air and Precipitation Monitoring Network (CAPMoN) datasets, available at: https://open.canada.ca/data/en/dataset?keywords=CAPMoN, last access: 11 March 2020.
EPA: Our Nation's Air: Status and Trends through 2015, available at: https://gispub.epa.gov/air/trendsreport/2016/ (last access: 13 November 2019), 2016.
EPA: Air Emissions Inventories, available at: https://www.epa.gov/air-emissions-inventories/air-pollutant-emissions-trends-data, last access: 13 November 2019.
EPA: Clean Air Status Trends Network (CASTNET) – Filter Pack Concentrations, available at: https://java.epa.gov/castnet/clearsession.do, last access: 11 March 2020.
Hand, J. L., Schichtel, B. A., Malm, W. C., and Pitchford, M. L.: Particulate sulfate ion concentration and SO2 emission trends in the United States from the early 1990s through 2010, Atmos. Chem. Phys., 12, 10353–10365, https://doi.org/10.5194/acp-12-10353-2012, 2012.
Harrison, R. M. and Pio, C. A.: An investigation of the atmospheric HNO3–NH3–NH4NO3 equilibrium relationship in a cool, humid climate, Tellus B, 35, 155–159, https://doi.org/10.3402/tellusb.v35i2.14795, 1982.
Haywood, J. M. and Shine, K. P.: The effect of anthropogenic sulfate and soot aerosol on the clear sky planetary radiation budget, Geophys. Res. Lett., 22, 603–606, https://doi.org/10.1029/95GL00075, 1995.
Heroux, M.-E., Anderson, H. R., Atkinson, R., Brunekreef, B., Cohen, A., Forastiere, F., Hurley, F., Katsouyanni, K., Krewski, D., Krzyanowski, M., Kunzli, N., Mills, I., Querol, X., Ostro, B., and Walton, H.: Quantifying the health impacts of ambient air pollutants: recommendations of a WHO/Europe project, Int. J. Publ. Health, 60, 619–627, https://doi.org/10.1007/s00038-015-0690-y, 2015.
Jones, A. M. and Harrison, R. M.: Temporal trends in sulphate concentrations at European sites and relationships to sulphur dioxide, Atmos. Environ., 45, 873–882, https://doi.org/10.1016/j.atmosenv.2010.11.020, 2011.
Kajino, M., Ueda, H., Han, Z., Kudo, R., Inomata, Y., and Kaku, H.: Synergy between air pollution and urban meteorological changes through aerosol-radiation-diffusion feedback – A case study of Beijing in January 2013, Atmos. Environ., 171, 98–110, https://doi.org/10.1016/j.atmosenv.2017.10.018, 2017.
Kuklinska, K., Wolska, L., and Namiesnik, J.: Air quality policy in the U.S. and the EU – a review, Atmos. Poll. Res., 6, 129–137, https://doi.org/10.5094/APR.2015.015, 2015.
Kunzli, N., Kaiser, R., Medina, S., Studnicka, M., Chanel, O., Filiger, P., Herry, M., Horak Jr., F., Puybonnieux-Texier, V., Quenel, P., Schneider, J., Seethaler, R., Vergnaud, J.-C., and Sommer, H.: Public-health imact of outdoor and traffic-related air pollution: A European assessment, Lancet, 356, 795–801, https://doi.org/10.1016/S0140-6736(00)02653-2, 2000.
Lee, B.: Highlights of the Clean Air Act Amendments of 1990, J. Air Waste Manage. Assoc., 41, 16–19, https://doi.org/10.1080/10473289.1991.10466820, 1991.
Lee C., Martin, R. V., van Donkellar, A., Lee, H., Dickerson, R. R., Hains, J. C., Krotkov, N., Richter, A., Vinnikov, K., and Schwab, J. J.: SO2 emissions and lifetimes: Estimates from inverse modeling using in situ and global, space-based (SCIAMACHY and OMI) observations, J. Geophys. Res., 116, D06304, https://doi.org/10.1029/2010JD014758, 2011.
Lövblad, G., Tarrason, L., Tørseth, K., and Dutchak, S.: EMEP Assessment, Part I, European Perspective, Norwegian Meterological Institute, Oslo, Norway, available at: http://emep.int/publ/reports/2004/assessment_2004.html (last access: 13 November 2019), 2004.
Makar, P. A., Moran, M. D., Zheng, Q., Cousineau, S., Sassi, M., Duhamel, A., Besner, M., Davignon, D., Crevier, L.-P., and Bouchet, V. S.: Modelling the impacts of ammonia emissions reductions on North America air quality, Atmos. Chem. Phys., 9, 7183–7212, https://doi.org/10.5194/acp-9-7183-2009, 2009.
Mann, H. B.: Non-parametric tests against trend, Econometrica, 13, 245–59, https://doi.org/10.2307/1907187, 1945.
Napolitano, S., Stevens, G., Schreifels, J., and Culigan, K.: The NOx Budget Trading Program: A collaborative, innovative approach to solving a regional air pollution problem, Elec. J., 20, 65–76, https://doi.org/10.1016/j.tej.2007.09.005, 2007.
National Climate Assessment: U.S. Global Change Research Program, available at: https://nca2014.globalchange.gov/report/regions/midwest/graphics/temperatures-are-rising-midwest (last access: 13 November 2019), 2014.
Penner, J., Hegg, D., Andreae, M., Leaitch, D., Pitari, G., Annegarn, H., Murphy, D., Nganga, J., Barrie, L., Feichter, H: IPCC, Climate Change 2001: Aerosols and Indirect Cloud Effects, IPCC Third Assessment Report, Cambridge University Press, Cambridge, UK, 289–348, 2001.
Pitari, G., Visioni, D., Mancini, E., Cionni, I., Di Genova, G., and Gandolfi, I.: Sulfate aerosols from non-explosive volcanoes: Chemical-radiative effects in the troposphere and lower stratosphere, Atmosphere, 7, 85, https://doi.org/10.3390/atmos7070085, 2016.
Reid, N., Misra, P. K., Bloxam, R., Yap, D., Rao, S. T., Civerolo, K., Brankov, E., and Vet, R. J.: Do We Understand Trends in Atmospheric Sulfur Species?, J. Air. Waste. Manage., 51, 1561–1567, https://doi.org/10.1080/10473289.2001.10464384, 2001.
Shah, V., Jaeglé, L., Thornton, J. A., Lopez-Hilfiker, F. D., Lee, B. H., Schroder, J. C., Campuzano-Jost, P., Jimenez, J. L., Guo, H., Sullivan, A. P., Weber, R. J., Green, J. R., Fiddler, M. N., Bililign, S., Campos, T. L., Stell, M., Weinheimer, A. J., Montzka, D. D., and Brown, S. S.: Chemical feedbacks weaken the wintertime response of particulate sulfate and nitrate to emissions reductions over the eastern United States, P. Natl. Acad. Sci. USA, 115, 8110–8115, https://doi.org/10.1073/pnas.1803295115, 2018.
Sickles II, J. E. and Shadwick, D. S.: Changes in air quality and atmospheric deposition in the eastern United States: 1990–2004, J. Geophys. Res., 112, D17301, https://doi.org/10.1029/2006JD007843, 2007.
Sickles II, J. E. and Shadwick, D. S.: Air quality and atmospheric deposition in the eastern US: 20 years of change, Atmos. Chem. Phys., 15, 173–197, https://doi.org/10.5194/acp-15-173-2015, 2015.
WHO – World Health Organization: WHO Air quality guidelines for particulate matter, ozone, nitrogen dioxide and sulfur dioxide, Global update 2005, Summary of risk assessment, Geneva, p. 22, 2006.
Yao, X. and Zhang, L.: Trends in atmospheric ammonia at urban, rural, and remote sites across North America, Atmos. Chem. Phys., 16, 11465–11475, https://doi.org/10.5194/acp-16-11465-2016, 2016.
Yoshizumi, K. and Hoshi, A.: Size distributions of ammonium nitrate and sodium nitrate in atmospheric aerosols, Environ. Sci. Technol., 19, 258–261, https://doi.org/10.1021/es00133a007, 1985.
Yu, H., Kaufman, Y. K., Chin, M., Feingold, G., Remer, L. A., Anderson, T. L., Balkanski, Y., Bellouin, N., Boucher, O., Christopher, S., DeCola, P., Kahn, R., Koch, D., Loeb, N., Reddy, M. S., Schulz, M., Takemura, T., and Zhou, M.: A review of measurement-based assessments of the aerosol direct radiative effect and forcing, Atmos. Chem. Phys., 6, 613–666, https://doi.org/10.5194/acp-6-613-2006, 2006.
Zhang, L., Vet, R., Wiebe, A., Mihele, C., Sukloff, B., Chan, E., Moran, M. D., and Iqbal, S.: Characterization of the size-segregated water-soluble inorganic ions at eight Canadian rural sites, Atmos. Chem. Phys., 8, 7133–7151, https://doi.org/10.5194/acp-8-7133-2008, 2008.
Zhang, Y., West, J. J., Mathur, R., Xing, J., Hogrefe, C., Roselle, S. J., Bash, J. O., Pleim, J. E., Gan, C.-M., and Wong, D. C.: Long-term trends in the ambient PM2.5- and O3-related mortality burdens in the United States under emission reductions from 1990 to 2010, Atmos. Chem. Phys., 18, 15003–15016, https://doi.org/10.5194/acp-18-15003-2018, 2018.
Zhuang, H., Chan, C. K., Fang, M., and Wexler, A. S.: Size distributions of particulate sulfate, nitrate, and ammonium at a coastal site in Hong Kong, Atmos. Environ., 33, 843–853, https://doi.org/10.1016/S1352-2310(98)00305-7, 1999.