the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 30 Jul 2019
| 30 Jul 2019
Effects of inorganic salts on the heterogeneous OH oxidation of organic compounds: insights from methylglutaric acid–ammonium sulfate
Hoi Ki Lam
Sze Man Shum
James F. Davies
Mijung Song
Andreas Zuend
Man Nin Chan
Atmospheric particles, consisting of inorganic salts, organic compounds and a varying amount of water, can continuously undergo heterogeneous oxidation initiated by gas-phase oxidants at the particle surface, changing the composition and properties of particles over time. To date, most studies focus on the chemical evolution of pure organic particles upon oxidation. To gain more fundamental insights into the effects of inorganic salts on the heterogeneous kinetics and chemistry of organic compounds, we investigate the heterogeneous OH oxidation of 3-methylglutaric acid (3-MGA) particles and particles containing both 3-MGA and ammonium sulfate (AS) in an organic-to-inorganic mass ratio of 2 in an aerosol flow tube reactor at a high relative humidity of 85.0 %. The molecular information of the particles before and after OH oxidation is obtained using the direct analysis in real time (DART), a soft atmospheric pressure ionization source coupled to a high-resolution mass spectrometer. Optical microscopy measurements reveal that 3-MGA–AS particles are in a single liquid phase prior to oxidation at high relative humidity. Particle mass spectra show that C6 hydroxyl and C6 ketone functionalization products are the major products formed upon OH oxidation in the absence and presence of AS, suggesting that the dissolved salt does not significantly affect reaction pathways. The dominance of C6 hydroxyl products over C6 ketone products could be explained by the intermolecular hydrogen abstraction by tertiary alkoxy radicals formed at the methyl-substituted tertiary carbon site. On the other hand, kinetic measurements show that the effective OH uptake coefficient, γeff, for 3-MGA–AS particles (0.99±0.05) is smaller than that for 3-MGA particles (2.41±0.13) by about a factor of ∼2.4. A smaller reactivity observed in 3-MGA–AS particles might be attributed to a higher surface concentration of water molecules and the presence of ammonium and sulfate ions, which are chemically inert to OH radicals, at the particle surface. This could lower the collision probability between the 3-MGA and OH radicals, resulting in a smaller overall reaction rate. Our results suggest that inorganic salts likely alter the overall heterogeneous reactivity of organic compounds with gas-phase OH radicals rather than reaction mechanisms in well-mixed aqueous organic–inorganic droplets at a high humidity, i.e., 85 % relative humidity (RH). It also acknowledges that the effects of inorganic salts on the heterogeneous reactivity could vary greatly, depending on the particle composition and environmental conditions (e.g., RH and temperature). For instance, at lower relative humidities, aqueous 3-MGA–AS droplets likely become more concentrated and more viscous before efflorescence, possibly giving rise to diffusion limitation during oxidation under relatively dry or cold conditions. Further studies on the effects of inorganic salts on the diffusivity of the species under different relative humidities within the organic–inorganic particles are also desirable to better understand the role of inorganic salts in the heterogeneous reactivity of organic compounds.
- Article
(1353 KB) - Full-text XML
-
Supplement
(1002 KB) - BibTeX
- EndNote





Atmospheric particles are chemically complex and are comprised of a large variety of organic compounds, inorganic salts and water. Organic compounds contribute a significant mass fraction (20 %–90 %) of atmospheric particles (Kanakidou et al., 2005; Zhang et al., 2007; Jimenez et al., 2009). Laboratory and modeling studies have revealed that organic compounds present at, or near, the particle surface can be efficiently oxidized by gas-phase oxidants such as hydroxyl (OH), ozone (O3) and nitrate radicals (Rudich et al., 2007; George and Abbatt, 2010; Kroll et al., 2015; Estillore et al., 2016; Chapleski et al., 2016). The effective OH uptake coefficient, γeff, defined as the fraction of OH collisions with organic molecules that yields a reaction of a target organic molecule, has been commonly used to describe the kinetics and has been reported for a variety of pure organic compounds. The γeff in general has a value of ≥0.1 and even ≥1, indicating the occurrence of secondary chemistry (e.g., chain reactions induced by the hydrogen abstraction of organic molecules by alkoxy radicals) (Richards-Henderson et al., 2015). These heterogeneous oxidative processes can continuously alter the surface and bulk composition of the particles (Slade and Knopf, 2013; Li et al., 2018), and thus modify particle properties such as light extinction, hygroscopicity and cloud condensation nuclei activity (Petters et al., 2006; George et al., 2007; Lambe et al., 2007, 2009; Cappa et al., 2011; Slade et al., 2015, 2017).
While the transformation of pure organic particles has become more reasonably understood, the chemical transformation of organic particles in the presence of dissolved inorganic salts is largely unclear. Only a few laboratory studies have investigated the heterogeneous oxidation of organic–inorganic particles (McNeill et al., 2007, 2008; Dennis-Smither et al., 2012). In those studies, hydrophobic organic compounds (e.g., oleic acid and palmitic acid) have been chosen as model compounds. Due to the hydrophobic nature of these compounds, the particles tend to be phase separated with a thin organic layer on the surface of the aqueous inorganic core (e.g., aqueous sodium chloride or ammonium sulfate (AS) phases). For these phase-separated particles, the molecular structure and orientation of organic molecules at the particle surface are observed to alter the reactive uptake of gas-phase oxidants such as O3 and OH radicals. The reaction products formed from the ozonolysis of oleic acid–AS particles are very similar to those found in pure oleic acid particles (McNeill et al., 2007). These observations are consistent with the hypothesis that a thin organic layer effectively shields the aqueous inorganic core from being oxidized at the particle surface.
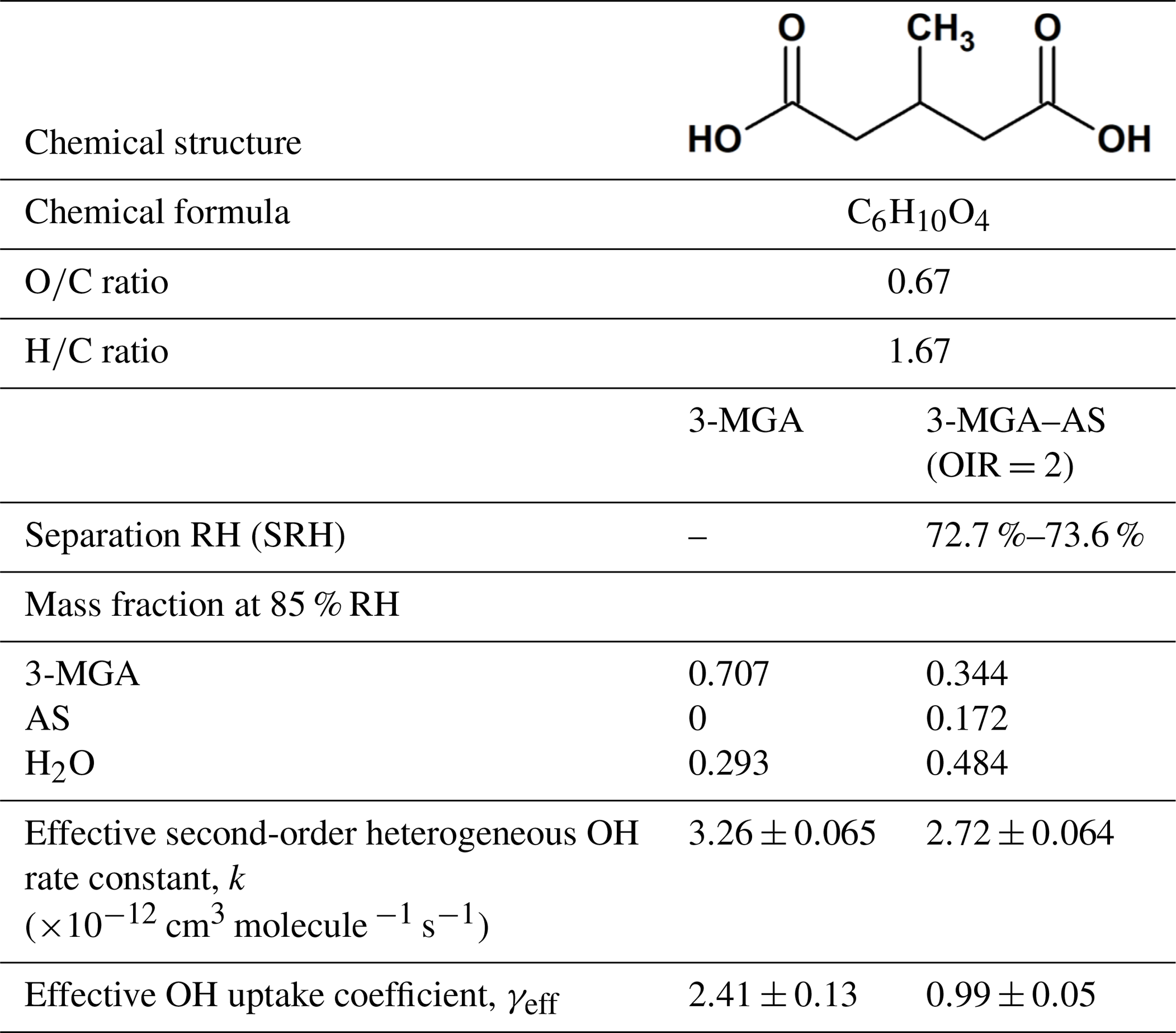
To date, there are still uncertainties on how salts affect the heterogeneous reactivity of organic compounds, in particular the more oxygenated ones, which exhibit moderate to high solubility in water. In this work, experiments were conducted to investigate the evolution of molecular composition of 3-methylglutaric acid (3-MGA) particles and particles containing 3-MGA and AS in an organic-to-inorganic mass ratio (water-free) (OIR) of 2 upon heterogeneous OH oxidation at a relative humidity (RH) of 85.0 % using an aerosol flow tube reactor coupled to the direct analysis in real time (DART) mass spectrometer. Here, 3-MGA is chosen as a model compound for methyl-substituted dicarboxylic acids (Table 1), while AS is chosen as the model inorganic salt. The OIR of 2 used in this work is in the range of the OIR commonly observed in atmospheric particles (Jimenez et al., 2009). The model system allows us to gain more insights into the physics and chemistry of heterogeneous reactions. The physical state of the particle is known to be a key factor in controlling the heterogeneous reactivity (Renbaum and Smith, 2009; Shiraiwa et al., 2011; Chan et al., 2014; Slade and Knopf, 2014; Fan et al., 2015; Marshall et al., 2018). Recent laboratory and modeling studies have shown that in addition to deliquescence and efflorescence, particles containing organic compounds and inorganic salts can undergo phase separation, depending on the particle composition and environmental conditions such as RH and temperature (Ciobanu et al., 2009; Reid et al., 2011; Song et al., 2012a, b; Zuend and Seinfeld, 2012; Qiu and Molinero, 2015; Stewart et al., 2015; You and Bertram, 2015; Freedman, 2017; Losey et al., 2018). Since whether particles are well mixed or phase separated governs the surface composition of particles and thus the heterogeneous reactivity, the phase separation behavior of 3-MGA–AS particles was investigated using an aerosol flow cell coupled to an optical microscope. By assessing the molecular transformation of 3-MGA and 3-MGA–AS particles upon oxidation together with phase separation data obtained from optical microscopy measurements, the effects of AS on the heterogeneous OH kinetics and chemistry of 3-MGA are examined. More recently, we have measured the heterogeneous OH reactivity of pure 2-MGA particles, a structural isomer of 3-MGA (Chim et al., 2017a). Given their similar structures, the results of this work together with 2-MGA data might provide some new aspects on how dissolved inorganic salts would alter the heterogeneous kinetics and chemistry of methyl-substituted dicarboxylic acids.
Table 1Chemical structure, properties, effective heterogeneous OH rate constant, effective OH uptake coefficient of 3-methylglutaric acid (3-MGA) and 3-MGA mixed with ammonium sulfate (AS) in an organic-to-inorganic dry mass ratio (OIR) =2.
2.1 Heterogeneous OH oxidation of 3-MGA and 3-MGA–AS particles
The OH-initiated heterogeneous oxidation of 3-MGA and 3-MGA–AS particles was performed in an aerosol flow tube reactor at 85.0 % RH. Details of the experimental methods have been described elsewhere (Chim et al., 2017a, b). In brief, the particle stream did not pass through a diffusion dryer and was directly mixed with O3, oxygen (O2), dry nitrogen (N2), humidified N2 and hexane before being introduced into the reactor. The RH within the reactor was controlled by varying the dry N2-to-humidified N2 ratio and was measured at the inlet of the reactor. A water jacket around the reactor was used to maintain a constant temperature of 20 ∘C inside the reactor. Inside the reactor, gas-phase OH radicals were generated via the photolysis of O3 using UV lamps at 254 nm. The processes can be described by the following reactions:
The gas-phase concentration of OH radicals was controlled by varying the O3 concentration introduced to the reactor and was determined by measuring the decay of hexane, a gas-phase tracer of OH radicals, using a gas chromatograph coupled with a flame ionization detector (GC–FID). The OH exposure, a quantity defined as the product of OH concentration and particle residence time (∼1.3 min), can be determined by the following equation (Smith et al., 2009):
where kHex is the rate constant for the reaction of OH radicals with hexane ( cm3 molecule−1 s−1), [Hex]0 is the initial hexane concentration entering the reactor and [Hex] is the concentration of hexane leaving the reactor. The OH exposure was varied from 0 to a maximum of 4.06×1011 molecules cm−3 s in 3-MGA experiments and was varied from 0 to a maximum of 3.84×1011 molecules cm−3 s in 3-MGA–AS experiments. The oxidation levels are equivalent to about 3 d in the atmosphere under a moderate to high level of OH concentration (1.5×106 molecules cm−3). Since the OH exposure (and OH concentration) was determined by the in situ measurement of the decay of hexane, the impacts of RH and water uptake by particles inside the reactor on the generation and concentration of gas-phase OH radicals have been taken into account. The competitions between the heterogeneous oxidation and the gas-phase oxidation have also been considered when quantifying OH concentration. Upon exiting the reactor, the particle stream then passed through an annular Carulite® catalyst denuder for O3 removal and an activated charcoal denuder for the removal of organic gas-phase species remaining in the stream. Hence, only particle-phase reaction products were analyzed. It is acknowledged that Carulite® catalyst denuder can slightly decrease the RH of the particle stream. However, this would not have a significant effect on the reaction products analyzed because the decrease in RH after oxidation would not significantly affect the formation of reaction products, which primarily occurred inside the reactor. Size distribution of the particles was determined by sampling a small portion of the particle stream using a scanning mobility particle sizer (SMPS, TSI) after oxidation. The remaining flow was directed to a heater at 250–300 ∘C to fully vaporize the particles. Both 3-MGA and 3-MGA–AS particles were confirmed to be fully vaporized upon heating at 250 ∘C or above by measuring the size distribution of the particles leaving the heater with the SMPS in separate experiments. The resulting gas-phase species were then directed to an ionization region, a narrow open space between the DART ionization source (DART SVP, IonSense Inc.) and the inlet of the high-resolution mass spectrometer (Q Exactive Orbitrap, ThermoFisher) (Chan et al., 2013; Nah et al., 2013).
Details of the DART operation have been described in the work of Cody et al. (2005). The DART ionization source was operated in negative ionization mode with helium (He) as the ionizing gas. The formation of gas-phase ions in the ionization region can be described as below (Cody, 2009):
Atmospheric O2 molecules abstract the electrons (e−) produced by the Penning ionization of metastable He in the DART ionization source to form anionic oxygen ions () which then react with the gas-phase species (M) to form deprotonated molecular ions () by proton abstraction. Previous studies have shown that the acidic proton of the carboxyl group can be abstracted by the ions to generate the , which was observed dominantly in the mass spectra (Cheng et al., 2015; Chim et al., 2017a, b). In this work, it is likely that proton abstraction from the carboxyl group of 3-MGA and its reaction products occurred to produce the . These ions were sampled by the high-resolution mass spectrometer. Mass spectra, over a scan range from m∕z 70 to 400 at a mass resolution of about 140 000, were collected. The mass spectra were analyzed using the Xcalibur software (Xcalibur Software Inc., Herndon, VA, USA). It is acknowledged that thermal composition of reaction products could possibly occur during the thermal desorption process (Stark et al., 2017). The mass spectra of some organic acids and alcohols (e.g., succinic acid, ketosuccinic acid and tartaric acid) are available in the work of Chan et al. (2014), showing insignificant thermal decomposition during the DART analysis. In this study, the thermal decomposition of 3-MGA was found to be insignificant as the deprotonated molecular ion of 3-MGA is the dominant peak before oxidation in the mass spectra (Fig. 1). We acknowledge that reactions between peroxy radicals may yield organic peroxides and oligomers, which may decompose thermally. We cannot completely rule out the possibility of such reactions, but there was no indication of any of the fragment ions expected from the thermal decomposition in the mass spectra. Together, these results suggest that the impact of thermal decomposition on the observed product distribution is likely insignificant. Two control experiments were conducted: one in the presence of O3 without the UV light and another one in the absence of O3 with the UV light on. No compositional changes were observed for 3-MGA and 3-MGA–AS particles in both control experiments, indicating that the reaction of 3-MGA with O3 is insignificant and that 3-MGA is not likely to be photolyzed.
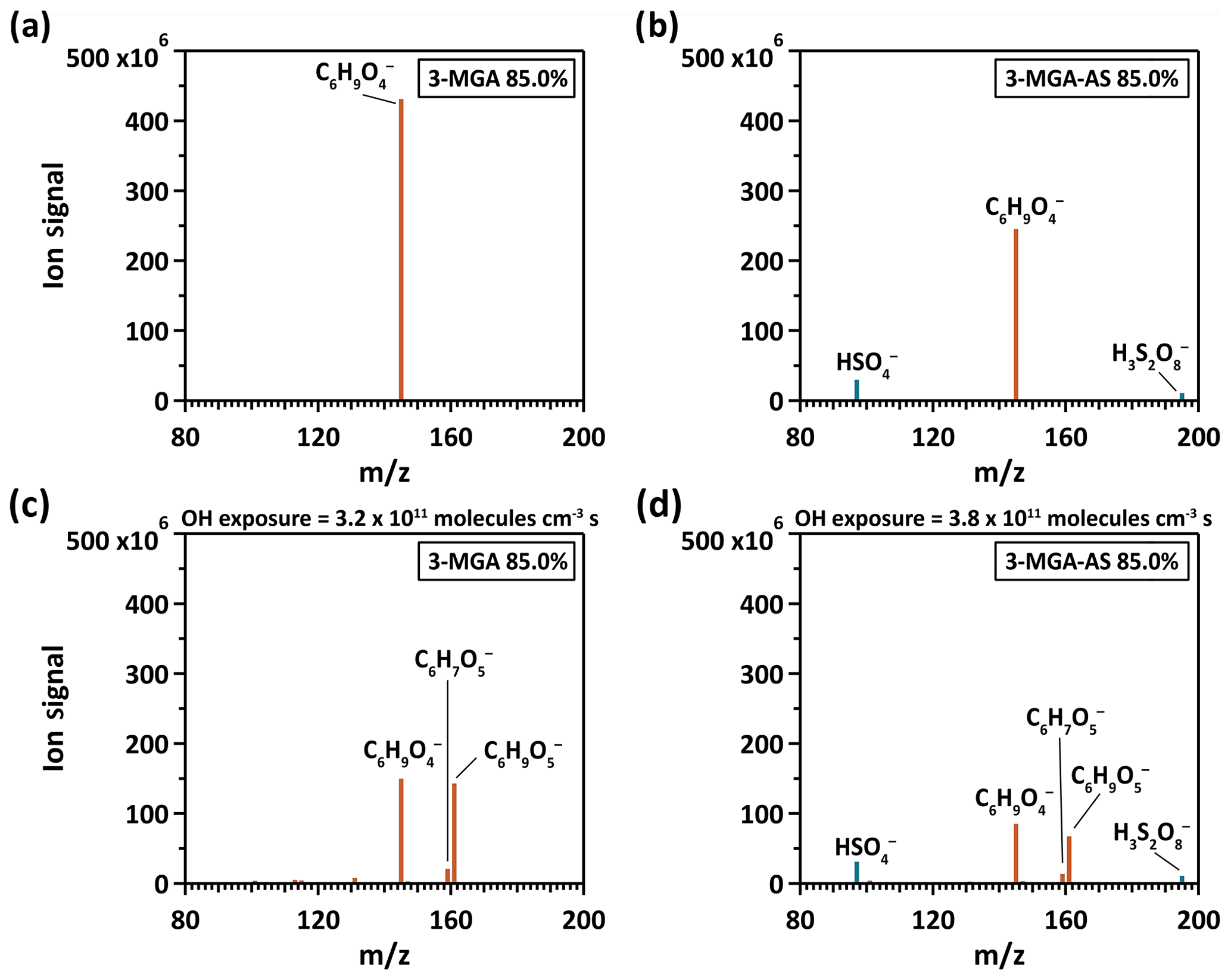
Figure 1The particle mass spectrum of (a, c) 3-MGA and (b, d) 3-MGA–AS before (a, b) and after (c, d) heterogeneous OH oxidation at 85.0 % RH, respectively. The brown color represents organic species and the blue color represents inorganic species.
The hygroscopicity data of 3-MGA have been reported in the work of Marsh et al. (2017) with a growth factor of ∼1.2 at 85 % RH. As shown by the hygroscopicity curve measured, 3-MGA particles absorb and desorb water reversibly as the RH increases or decreases, indicating that they are likely aqueous droplets prior to oxidation. Optical microscopy measurements have been carried out (Fig. S1 in the Supplement) and show that 3-MGA–AS particles are in a single liquid phase prior to oxidation at 85.0 % RH as the particles become phase separated when the RH is below the separation RH (SRH =72.7 %–73.6 %) (Fig. S2). Details of the optical microscopy measurements have been given in the Supplement. Since the particles were always exposed to high humidity and the experiments were carried out at 85.0 % RH, which is higher than the SRH, 3-MGA–AS particles are likely to be single-phase liquid droplets prior to oxidation.
3.1 Particle mass spectra of 3-MGA and 3-MGA–AS before and after OH oxidation
Figure 1 shows the mass spectra of 3-MGA and 3-MGA–AS before and after OH oxidation at 85.0 % RH, respectively. For 3-MGA, a dominant peak is observed before oxidation at , which corresponds to the deprotonated molecular ion of 3-MGA (). After oxidation, two major product peaks evolve, corresponding to two C6 functionalization products (C6 hydroxyl products (C6H10O5) and C6 ketone products (C6H8O5)). A few minor product peaks, such as , , , and , are also observed. Each of these peaks contributes less than 2.5 % of the total ion signal at the maximum OH exposure. The mass spectra of 3-MGA–AS particles in OIR = 2 are very similar to those of 3-MGA particles, except for the two inorganic sulfate peaks that originate from dissolved AS. Before oxidation (Fig. 1), three peaks at , 145 and 195 are observed, corresponding to the bisulfate ion () and the deprotonated molecular ion of 3-MGA and , respectively. One possibility is that is likely the dissolved ion from aqueous AS that became acidified by the evaporative loss of ammonia (NH3) into the gas phase and can be detected via direct ionization in the negative ion mode (Hajslova et al., 2011). has been detected when aqueous AS particles are ionized by the DART ionization source as well as when being a reaction product formed in the heterogeneous OH oxidation of sodium methyl sulfate, sodium ethyl sulfate and methanesulfonic acid particles (Kwong et al., 2018a, b). However, we do not have a clear explanation for the formation of , which is likely an adduct of and H2SO4. After oxidation, the deprotonated ions of C6 hydroxyl () and C6 ketone products () are observed in addition to the unreacted 3-MGA. Some small product peaks are detected (, , , and ), with each contributing less than 2.5 % of the total ion signal.
As shown in Fig. 2, the chemical evolution of the composition of 3-MGA and 3-MGA–AS particles upon oxidation is very similar. At the maximum OH exposure, the C6 hydroxyl products are the most abundant species, which accounts for 38.0 %–48.2 % of the total organic ion signal, followed by unreacted 3-MGA (37.3 %–47.9 %) and the C6 ketone products (7.3 %–7.6 %). For 3-MGA–AS, the intensities of and remain about the same before and after OH oxidation (Fig. S3). This could be attributed to the fact that dissolved AS does not react effectively with gas-phase OH radicals at the particle surface (George and Abbatt, 2010). In general, the same reaction products are observed for both 3-MGA and 3-MGA–AS particles after oxidation, suggesting that AS does not significantly affect the reaction pathways.
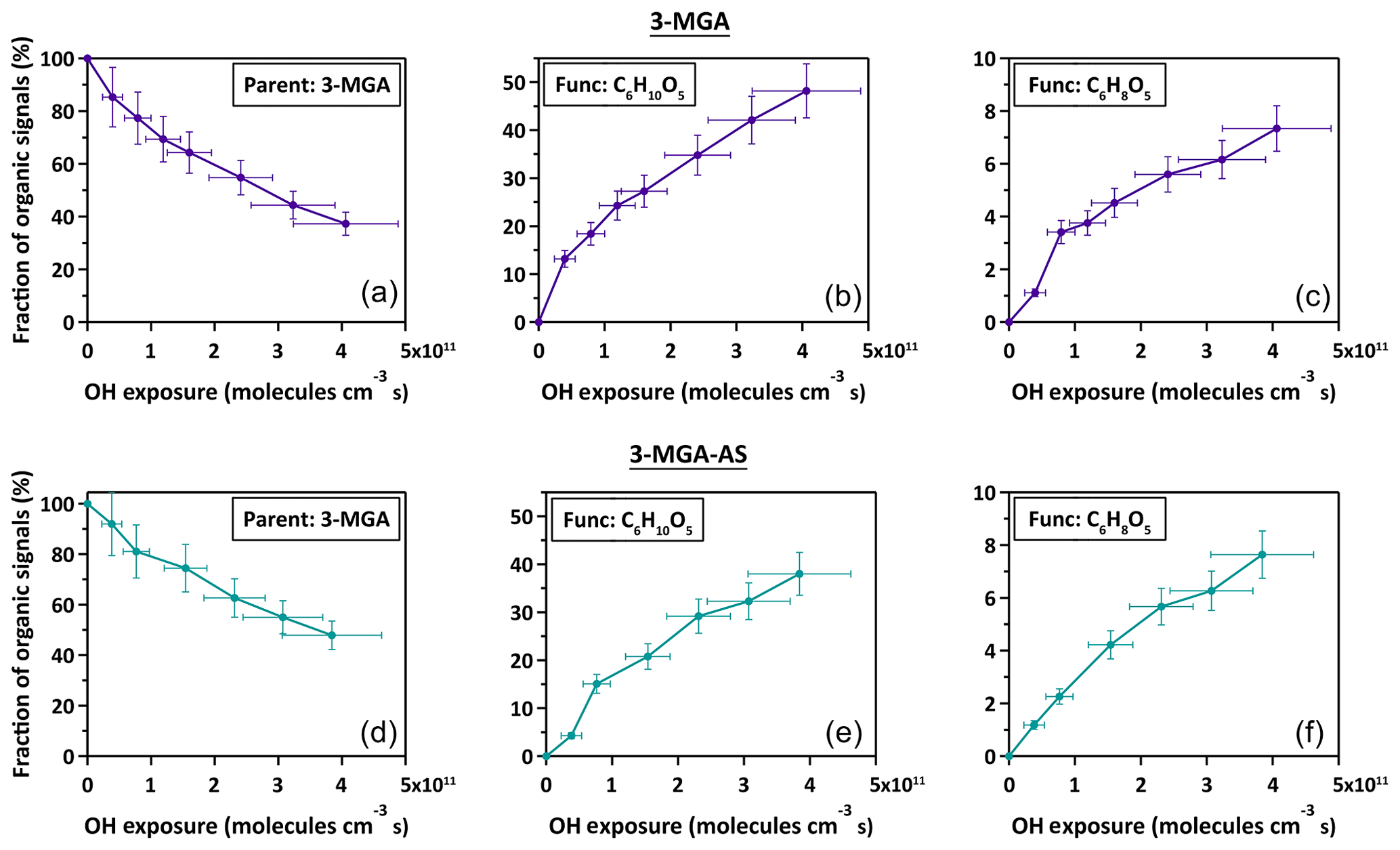
Figure 2(a, b, c) The fraction of the organic ion signal attributed to the parent 3-MGA, the major C6 hydroxyl and C6 ketone products of 3-MGA particles during the heterogeneous OH oxidation shown against OH exposure. (d, e, f) Analogous to the above, but for the case of mixed 3-MGA–AS particles.
3.2 Oxidative kinetics of 3-MGA and 3-MGA–AS
The normalized parent decay in 3-MGA and 3-MGA–AS particles as a function of OH exposure at 85.0 % RH is shown in Fig. 3. For both systems, the OH-initiated decay of 3-MGA follows an exponential trend and can be fit with an exponential function to obtain an effective second-order OH reaction rate constant (k):
where I0 is the ion signals of 3-MGA before oxidation, I is the ion signals of 3-MGA at a given OH exposure, [OH] is the gas-phase concentration of the OH radical and t is the reaction time. The fitted values of k for the 3-MGA and 3-MGA–AS are ( cm3 molecule−1 s−1 and ( cm3 molecule−1 s−1, respectively. Using the fitted k value, the effective OH uptake coefficient, γeff, defined as the fraction of OH collisions with particles that yields a reaction, can be computed by the following equation (Davies and Wilson, 2015):
where ρ0 is the density of particle, D0 is the particle diameter, mfs is the mass fraction of 3-MGA in the particle, Mw is the molecular weight of 3-MGA, NA is Avogadro's number and is the mean speed of gas-phase OH radicals. The mean surface-weighted diameters prior to OH oxidation (203.0 nm for 3-MGA and 200.8 nm for 3-MGA–AS) are used in the calculation of γeff. Upon oxidation, the mean surface-weighted diameter decreases from 203.0 to 170.7 nm for 3-MGA particles and decreases from 200.8 to 187.8 nm for 3-MGA–AS particles (Fig. S5). The decrease in the particle diameter upon oxidation is likely attributed to the formation and volatilization of fragmentation products and the associated evaporative loss of water molecules. Vaden et al. (2011) have discussed that evaporation of highly viscous particles is likely independent of particle size distribution and is unlikely to significantly influence the overall evaporation behavior. As the study of Vaden et al. (2011) focused on highly viscous particles, while the focus of this study is more liquid-like particles, their results may not be applicable in our study. Since 3-MGA–AS particles are more liquid-like particles, the evaporation rate would scale with the total surface area of the polydisperse particle population. Since the spread of the polydisperse particle population is small in this work, the size change is not likely to be substantial, especially in the determination of γeff. In the work of Meng and Seinfeld (1996), the mixing timescales of volatile species were evaluated. Although it was suggested by the study that the timescales may increase with increasing particle size, the difference may not be that significant in our study, as the span of the polydisperse particles is much smaller than the difference between coarse particles and fine particles used in Meng and Seinfeld (1996). We thus postulate that the spread of particle size and the mixing timescale would not play a substantial role in the evaporation of fragmentation products during oxidation. As the change in particle size upon oxidation is not very significant, the change in particle diameter was not accounted for in the γeff calculation. The γeff may thus considered to be an initial uptake coefficient (Chim et al., 2018). We acknowledge that the spread of particle size could potentially affect the uncertainty and determination of γeff, but we could not quantify it since the particles are polydisperse in our study. Future investigations can be carried out to measure the γeff for both monodisperse (size selected) and polydisperse particle populations. The γeff assembled from different monodisperse particle sizes can be compared with that obtained from polydisperse populations using the surface-weighted mean diameter in order to assess how the spread and uncertainty in the particle size distribution of polydisperse particle populations affect the determination of γeff. The mfs values were obtained from equilibrium composition calculations using the Aerosol Inorganic-Organic Mixtures Functional groups Activity Coefficients (AIOMFAC) model available online (https://aiomfac.lab.mcgill.ca, last access: 5 March 2019) (Table 1) (Zuend et al., 2008, 2011). Based on the composition (i.e., mfs), the densities of 3-MGA and 3-MGA–AS particles were estimated using the volume additivity rule with a known density of 3-MGA, AS and water (Chim et al., 2017a). Using Eq. (3), the γeff values for 3-MGA and 3-MGA–AS are calculated to be 2.41±0.13 and 0.99±0.05, respectively. The value of γeff for 3-MGA particles is larger than that for 3-MGA–AS particles by a factor of 2.4. One possible explanation is that the mass fraction of 3-MGA in 3-MGA–AS particles (mfs =0.344) is smaller than that in 3-MGA particles (mfs =0.707) at 85.0 % RH before oxidation (Table 1), and this likely resulted from the presence of AS and the concomitant increase in particle hygroscopicity. A simple analysis shows that the surface coverage of 3-MGA in 3-MGA particles and 3-MGA–AS particles is roughly estimated to be 51.4 % and 21.6 %, respectively (see Supplement). A smaller surface concentration of 3-MGA in 3-MGA–AS particles might reduce the collision probability between 3-MGA and gas-phase OH radicals at the air–particle interface and thus lower the overall reactivity in comparison to 3-MGA particles. With reference to the work of Jungwirth et al. (2003) and Jungwirth and Tobias (2006), it is acknowledged that dissolved inorganic ions (e.g., ) may not be homogeneously distributed in the droplets and may be concentrated in the core, which may increase the surface concentration of 3-MGA. Furthermore, the surface activity of 3-MGA is not known and slight surfactant behavior could drastically alter the surface concentration. Thus the numbers presented here are to serve as a first approximation illustrating the possible effect of AS addition on the surface coverage of 3-MGA. Further investigations on the surfactant properties of 3-MGA and molecular dynamic simulation would be useful to better understand the surface composition of both 3-MGA and 3-MGA–AS particles.
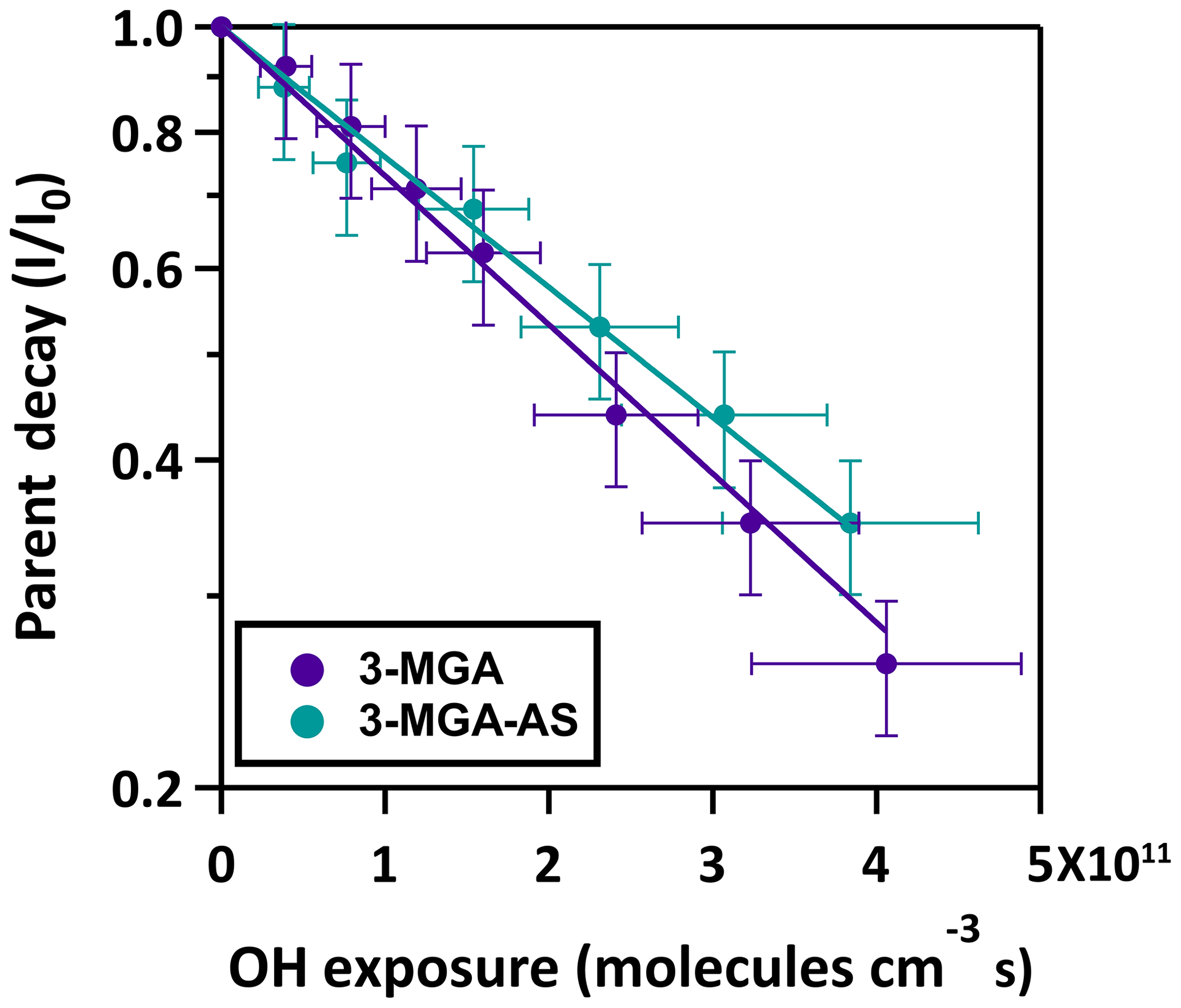
Figure 3The normalized parent decay for the heterogeneous OH oxidation of 3-MGA and 3-MGA–AS particles at 85.0 % RH. Note the logarithmic scale of the ordinate.
A similar result has also been observed for the OH oxidation with methanesulfonic acid (MSA) reported in the literature. Mungall et al. (2017) have investigated the heterogeneous OH oxidation of MSA–AS particles with a mass fraction of MSA = 0.16 at 75 % RH. The γeff was reported to be 0.05±0.03, which is smaller than that of pure MSA particles () measured at a slightly higher RH (90 %) (Kwong et al., 2018b). The results obtained in this work and in the literature suggest that for a given RH, inorganic salts (e.g., AS) might lower the heterogeneous reactivity of organic compounds toward gas-phase OH radicals due to the smaller surface concentration of 3-MGA resulting from the presence of AS and concomitant increase in water uptake. It is acknowledged that ammonium () and sulfate () ions, which are chemically inert to OH radicals, present at or near the surface could lower the overall reaction rates by reducing the surface concentration of organic compounds. However, the additional effects of and ions on the surface activity and configuration of organic molecules, which could play a role in determining the heterogeneous activity, are not yet well understood and warrant further investigations.
Kinetic measurements show that γeff values for both 3-MGA and 3-MGA–AS particles are close to or greater than 1. This indicates that more than one 3-MGA molecule is reacted away per OH radical collision with the particle surface, suggesting the occurrence of secondary chemistry in the particle phase. In the following sections, reaction mechanisms are tentatively proposed and discussed to explain the formation of major products detected in the particles' mass spectra and the reaction pathways likely responsible for the secondary chemistry.
3.3 Reaction mechanisms
As shown in Fig. 2, the reaction products observed in 3-MGA and 3-MGA–AS particles are about the same upon oxidation. A generalized reaction scheme is thus proposed for both systems based on well-known particle-phase reactions previously published in literature (Russell, 1957; Bennett and Summers, 1974; George and Abbatt, 2010). As shown in Scheme 1, the OH oxidation with 3-MGA can be initiated by the hydrogen abstraction at three different carbon sites: tertiary backbone carbon site (Path A), secondary backbone carbon site (Path B) and the primary carbon site of the branched methyl group (Path C). Depending on the initial OH reaction site, a variety of reaction products can be formed and are broadly classified into two groups: functionalization and fragmentation products.
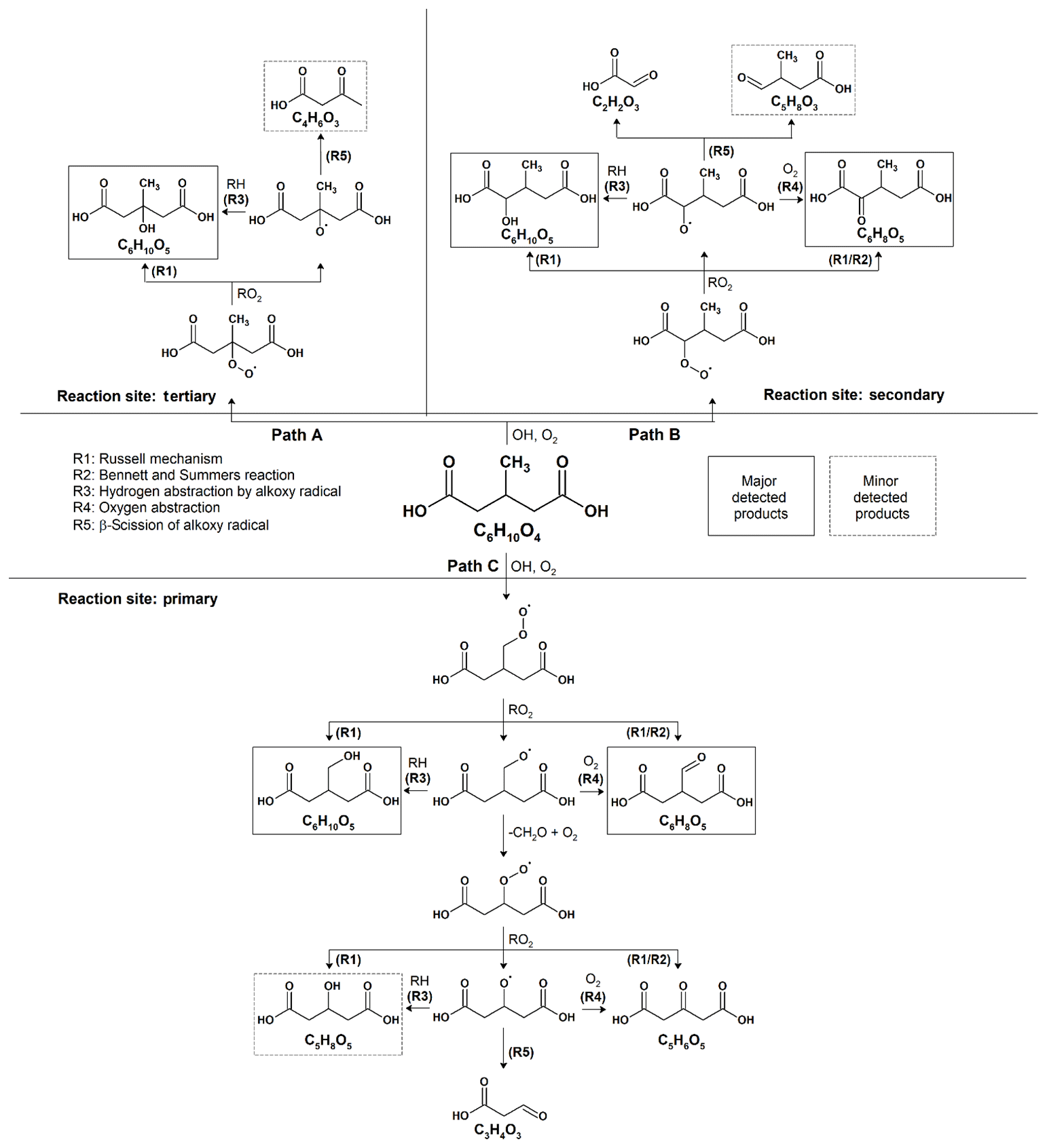
Scheme 1Proposed reaction mechanisms for the heterogeneous OH oxidation of 3-MGA and 3-MGA–AS particles.
3.3.1 Functionalization products
At the first oxidation step, a hydrogen atom is abstracted from a 3-MGA molecule by an OH radical, forming an alkyl radical that reacts quickly with an O2 molecule to form a peroxy radical. The major C6 hydroxyl (C6H10O5) and C6 ketone (C6H8O5) products can be generated from the self-reactions of two peroxy radicals through the Russell mechanism (Reaction R1) and/or Bennett–Summers reactions (Reaction R2). Alternatively, the self-reactions of two peroxy radicals can form two alkoxy radicals which can then abstract hydrogen atoms from the neighboring organic molecules (Reaction R3) to form C6 hydroxyl products, or react with O2 molecules (Reaction R4) to form C6 ketone products. However, when the hydrogen abstraction occurs at the tertiary carbon site (Scheme 1, Path A), only the C6 hydroxyl product can be formed because only a hydroxyl group can be added to the tertiary carbon site. Depending on initial reaction site, structural isomers of these C6 hydroxyl and ketone products are likely formed during oxidation.
3.3.2 Fragmentation products
The fragmentation products can be generated from the decomposition of alkoxy radicals (Reaction R5). For instance, when the initial hydrogen abstraction occurs at the secondary carbon site (Scheme 1, Path B), the decomposition of the secondary alkoxy radical can yield either a C2 (C2H2O3) or a C5 fragmentation product (C5H8O3). On the other hand, a C4 fragmentation product (C4H6O3) can be yielded from the decomposition of the alkoxy radical formed at the tertiary carbon site (Scheme 1, Path A) while oxidation at the primary carbon site (Scheme 1, Path C) can yield a C3 fragmentation product (C3H4O3). For both 3-MGA and 3-MGA–AS, the ion signal intensity of fragmentation products is small (Fig. 1). For example, only a small signal of C4 fragmentation product (C4H6O3), which is formed from the oxidation at the tertiary carbon site (Scheme 1, Path A), is detected. It contributes to less than 2 % of the total ion signal at the maximum OH exposure. The observed low abundances of fragmentation products could be explained by their higher volatilities (Tables S1 and S2) and some (e.g., C5H8O5) may be explained by the preference for the initial OH reaction site as discussed below. It is noted that for 3-MGA–AS particles, the presence of AS increases the activity coefficients of fragmentation products in the particle phase based on the thermodynamic model predictions and thus increases their volatilities in general (Table S3).
3.3.3 Large C6 hydroxyl-to-C6 ketone product ratio: implications for secondary chemistry
From the particle composition data, a large C6 hydroxyl-to-C6 ketone product ratio is observed. At the maximum OH exposure, the relative abundance of C6 hydroxyl products is about 5.0–6.6 times that of C6 ketone products for both 3-MGA and 3-MGA–AS particles. We acknowledge that although the ionization efficiencies are not corrected for these products in this study, the ionization efficiency of C4 hydroxyl products is found to be about the same or even lower than that of C4 ketone products during the DART ionization processes (Chan et al., 2014). The abundance of C6 hydroxyl products might be even larger than that of C6 ketone products after correcting their ionization efficiencies, supporting the statement above. One possible explanation for the dominance of C6 hydroxyl products is that the OH abstraction may preferentially occur at the tertiary carbon site (Scheme 1, Path A) since the tertiary alkyl radicals are more stable than secondary and primary alkyl radicals (Cheng et al., 2015). Only an addition of a hydroxyl group at the tertiary carbon site is allowed via the alkoxy or peroxy radical reactions.
Another possibility is that the branched methyl group may sterically hinder the two peroxy radicals from arranging into a cyclic tetroxide intermediate, which is essential for the formation of hydroxyl and ketone functionalization products through the Russell and the Bennett–Summers mechanisms (Cheng et al., 2015). Alternatively, alkoxy radicals are more likely formed through the self-reaction of two peroxy radicals and can react with neighboring organic molecules (e.g., unreacted 3-MGA) by intermolecular hydrogen abstraction to form the C6 hydroxyl products. Furthermore, as proposed by Peeters et al. (2004) and Vereecken and Peeters (2009), the strong hydrogen bonding among the two terminal carboxyl groups might lower the decomposition rate of the alkoxy radical. This could increase the competitiveness of the intermolecular hydrogen abstraction by the alkoxy radicals. It is worthwhile to note that the intermolecular hydrogen abstraction can regenerate an alkyl radical and eventually produce peroxy radicals. These peroxy radicals can react again with other peroxy radicals to regenerate alkoxy radicals. This allows the chain reactions to propagate and increases the overall reactivity (i.e., more than one 3-MGA molecule can be reacted per initial OH collision via secondary chemistry and, thus, γeff is larger than 1). Overall, the alkoxy radical chemistry, originating from the OH abstraction at the tertiary carbon site at the first oxidation step (Scheme 1, Path A) is likely the important reaction pathway for the OH reactions with 3-MGA.
Atmospheric particles can keep colliding with gas-phase oxidants, allowing continuous oxidation to occur at or near the particle surface. To better understand how dissolved inorganic salts affect the heterogeneous chemistry and kinetics of organic compounds with gas-phase OH radicals, we investigated the kinetics, products and mechanisms of particles comprised of 3-MGA and 3-MGA–AS in an OIR of 2 upon heterogeneous OH oxidation at 85.0 % RH. Optical microscopy measurements for the detection of phase separation reveal that 3-MGA–AS particles exhibit a single liquid phase prior to oxidation. Same major reaction products are formed as a result of heterogeneous OH oxidation with both 3-MGA and 3-MGA–AS particles. These data suggest that the presence of aqueous AS does not significantly affect the formation pathways of major reaction products. On the other hand, in the presence of AS, the heterogeneous reactivity of 3-MGA toward gas-phase OH radicals is slower in 3-MGA–AS particles compared to that in 3-MGA particles. It is likely attributed to a lower concentration of 3-MGA at the surface of 3-MGA–AS particles relative to 3-MGA particles, reducing the collision probability between 3-MGA and gas-phase OH radicals. The results from this work and the literature suggest that the presence of dissolved salts could reduce the overall heterogeneous reactivity of organic compounds with gas-phase OH radicals at the surface by lowering the surface concentration of organic compounds at a given RH and temperature. Until recently, the kinetic parameters (e.g., γeff) reported in the literature were mostly measured based on experiments with salt-free organic particles. The chemical lifetime of organic compounds or chemical tracers against heterogeneous OH reaction in the atmosphere could be longer than expected when the salts are present. Further investigations on how the amount and types of inorganic salts alter heterogeneous kinetics and chemistry are highly desirable.
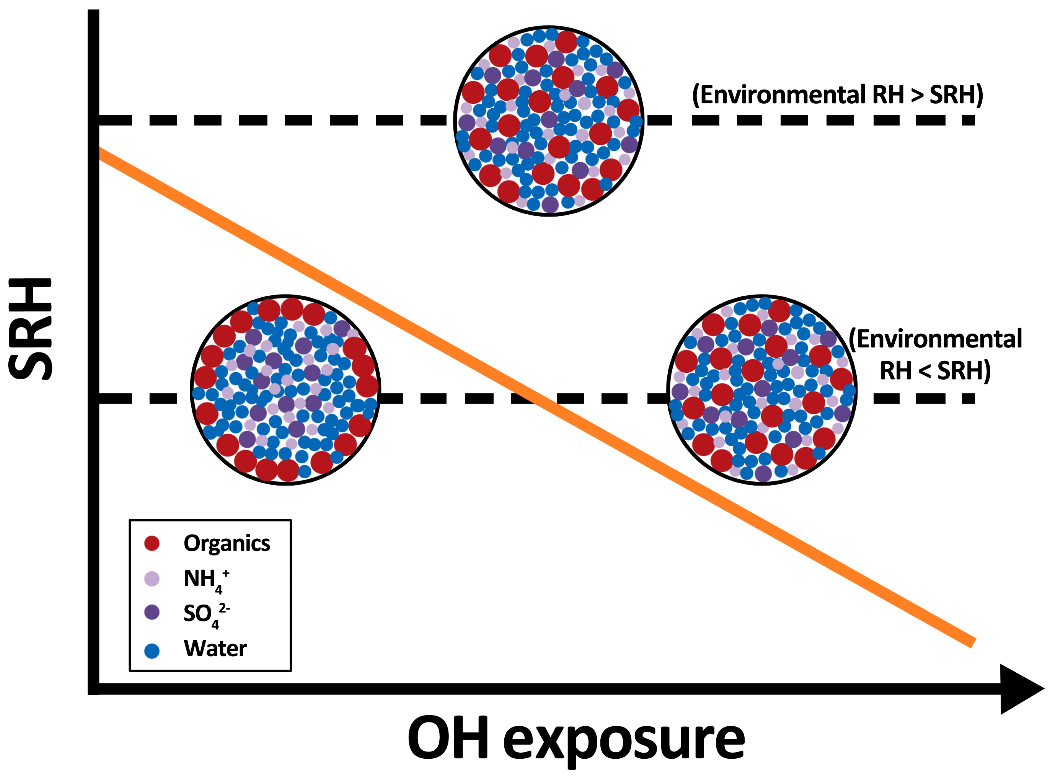
Figure 4A simplified diagram illustrating the change in separation relative humidity (SRH, orange solid line) and phase composition of droplets containing inorganic salts and organic compounds (single liquid phase vs. liquid–liquid phase separated) upon heterogeneous oxidation at two different environmental RHs.
Over the past decade, laboratory and modeling studies have demonstrated that atmospheric particles can undergo phase separation and exhibit different morphologies, which play a role in many atmospheric processes. For example, the inhomogeneous distribution of inorganic and organic species within phase-separated particles can affect the reactive uptake of gas-phase species (e.g., N2O5) (Gaston et al., 2014) and water uptake of organic–inorganic particles (Chan and Chan, 2007; Zuend and Seinfeld, 2012; Hodas et al., 2015). It is still unclear how the occurrence of liquid–liquid phase separation alters the heterogeneous reactivity of organic–inorganic particles over time. As shown in Fig. S2, 3-MGA–AS particles become phase separated when the RH is below the SRH (72.7 %–73.6 %). The phase-separated particles might exhibit different reactivity compared to those in a single liquid phase investigated in this work since the surface composition of the particles is different in these two phases. Furthermore, there is a possibility that the phase separation behavior (e.g., SRH) of the particles may change in response to the change in the particle composition over time. Although the phase of oxidized 3-MGA–AS particles has not been determined experimentally in this work, the overall 〈O∕C〉 is found to increase slightly from 0.67 to ∼0.75 (see Supplement), and the SRH is expected to decrease slightly after oxidation (Bertram et al., 2011; Song et al., 2012b; You et al., 2013, 2014). Since the experimental RH inside the reactor was fixed at 85.0 %, it is very likely that 3-MGA–AS particles remain in a single liquid phase state during oxidation. During oxidation, the degree of aerosol oxidation state (e.g., expressed by 〈O∕C〉) typically increases due to the formation of more oxygenated reaction products and, consequently, the SRH is expected to decrease. As shown in Fig. 4, it is hypothesized that initially phase-separated particles might transition to a homogeneous, single liquid phase state, depending on the extent of oxidation and the environmental thermodynamic conditions. Hence, it is of interest to investigate how the phase separation characteristics of organic–inorganic particles change in response to a change in the composition upon oxidation (Slade et al., 2015, 2017). Moreover, with respect to future work, it would be interesting to understand the dynamic interplay between the particle composition, heterogeneous reactivity, liquid–liquid phase separation and the effects on particle morphology under different environmental conditions and extents of oxidation.
The underlying research data are available upon request from the corresponding author (mnchan@cuhk.edu.hk).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-9581-2019-supplement.
HKL, SMS and MNC designed and ran the experiments. HKL, SMS and MNC prepared the paper. All co-authors provided comments and suggestions on the paper.
The authors declare that they have no conflict of interest.
We would like to thank Kevin Wilson for his insightful comments.
This research has been supported by the Hong Kong Research Grants Council (HKRGC) (project ID: 2191111, ref 24300516).
This paper was edited by Markus Ammann and reviewed by two anonymous referees.
Bennett, J. E. and Summers, R.: Product studies of the mutual termination reactions of sec-alkylperoxy radicals: Evidence for non-cyclic termination, Can. J. Chem., 52, 1377–1379, https://doi.org/10.1139/v74-209, 1974.
Bertram, A. K., Martin, S. T., Hanna, S. J., Smith, M. L., Bodsworth, A., Chen, Q., Kuwata, M., Liu, A., You, Y., and Zorn, S. R.: Predicting the relative humidities of liquid-liquid phase separation, efflorescence, and deliquescence of mixed particles of ammonium sulfate, organic material, and water using the organic-to-sulfate mass ratio of the particle and the oxygen-to-carbon elemental ratio of the organic component, Atmos. Chem. Phys., 11, 10995–11006, https://doi.org/10.5194/acp-11-10995-2011, 2011.
Cappa, C. D., Che, D. L., Kessler, S. H., Kroll, J. H., and Wilson, K. R.: Variations in organic aerosol optical and hygroscopic properties upon heterogeneous OH oxidation, J. Geophys. Res., 116, D15204, https://doi.org/10.1029/2011JD015918, 2011.
Chan, M. N. and Chan, C. K.: Mass transfer effects on the hygroscopic growth of ammonium sulfate particles with a water-insoluble coating, Atmos. Environ., 41, 4423–4433, 2007.
Chan, M. N., Nah, T., and Wilson, K. R.: Real time in-situ chemical characterization of sub-micron organic aerosols using Direct Analysis in Real Time Mass Spectrometry (DART-MS): The effect of aerosol size and volatility, Analyst, 138, 3749–3757, 2013.
Chan, M. N., Zhang, H., Goldstein, A. H., and Wilson, K. R.: Role of water and phase in the heterogeneous oxidation of solid and aqueous succinic acid aerosol by hydroxyl radicals, J. Phys. Chem. C, 118, 28978–28992, 2014.
Chapleski Jr., R. C., Zhang, Y., Troyaa, D., and Morris, J. R.: Heterogeneous chemistry and reaction dynamics of the atmospheric oxidants, O3, NO3, and OH, on organic surfaces, Chem. Soc. Rev., 45, 3731–3746, 2016.
Cheng, C. T., Chan, M. N., and Wilson, K. R.: The role of alkoxy radicals in the heterogeneous reaction of two structural isomers of dimethylsuccinic acid, Phys. Chem. Chem. Phys., 17, 25309–25321, https://doi.org/10.1039/c5cp03791c, 2015.
Chim, M. M., Chow, C. Y., Davie, J. F., and Chan, M. N.: Effects of relative humidity and particle phase water on the heterogeneous OH oxidation of 2-methylglutaric acid aqueous droplets, J. Phys. Chem. A, 121, 1666–1674, 2017a.
Chim, M. M., Cheng, C. T., Davies, J. F., Berkemeier, T., Shiraiwa, M., Zuend, A., and Chan, M. N.: Compositional evolution of particle-phase reaction products and water in the heterogeneous OH oxidation of model aqueous organic aerosols, Atmos. Chem. Phys., 17, 14415–14431, https://doi.org/10.5194/acp-17-14415-2017, 2017b.
Chim, M. M., Lim, C. Y., Kroll, J. H., and Chan, M. N.: Evolution in the reactivity of citric acid toward heterogeneous oxidation by gas-phase OH radicals, ACS Earth Space Chem., 2, 1323–1329, 2018.
Ciobanu, V. G., Marcolli, C., Krieger, U. K., Weers, U., and Peter, T.: Liquid-liquid phase separation in mixed organic/inorganic aerosol particles, J. Phys. Chem. A, 113, 10966–10978, 2009.
Cody, R. B.: Observation of molecular ions and analysis of nonpolar compounds with the direct analysis in real time ion source, Anal. Chem., 81, 1101–1107, 2009.
Cody, R. B., Laramée, J. A., and Durst, H. D., Versatile new ion source for the analysis of materials in open air under ambient conditions, Anal. Chem., 77, 2297–2302, 2005.
Davies, J. F. and Wilson, K. R.: Nanoscale interfacial gradients formed by the reactive uptake of OH radicals onto viscous aerosol surfaces, Chem. Sci., 6, 7020–7027, 2015.
Dennis-Smither, B. J., Miles, R. E. H., and Reid, J. P.: Oxidative aging of mixed oleic acid/sodium chloride aerosol particles, J. Geophys. Res., 117, D20204, https://doi.org/10.1029/2012JD018163, 2012.
Estillore, A. D., Trueblood, J. V., and Grassian, V. H.: Atmospheric chemistry of bioaerosols: Heterogeneous and multiphase reactions with atmospheric oxidants and other trace gases, Chem. Sci., 7, 6604–6616, 2016.
Fan, H., Tinsley, M. R., and Goulay, F.: Effect of relative humidity on the OH-initiated heterogeneous oxidation of monosaccharide nanoparticles, J. Phys. Chem. A, 119, 11182–11190, 2015.
Freedman, M. A.: Phase separation in organic aerosol, Chem. Soc. Rev., 46, 7694–7705, 2017.
Gaston, C. J., Thornton, J. A., and Ng, N. L.: Reactive uptake of N2O5 to internally mixed inorganic and organic particles: the role of organic carbon oxidation state and inferred organic phase separations, Atmos. Chem. Phys., 14, 5693–5707, https://doi.org/10.5194/acp-14-5693-2014, 2014.
George, I. J. and Abbatt, J. P. D.: Heterogeneous oxidation of atmospheric aerosol particles by gas-phase radicals, Nature Chem., 2, 713–722, 2010.
George, I. J., Vlasenko, A., Slowik, J. G., Broekhuizen, K., and Abbatt, J. P. D.: Heterogeneous oxidation of saturated organic aerosols by hydroxyl radicals: uptake kinetics, condensed-phase products, and particle size change, Atmos. Chem. Phys., 7, 4187–4201, https://doi.org/10.5194/acp-7-4187-2007, 2007.
Hajslova, J., Cajka, T., and Vaclavik, L.: Challenging applications offered by direct analysis in real time (DART) in food-quality and safety analysis, TrAC-Trend. Anal. Chem., 30, 204–218, 2011.
Hodas, N., Zuend, A., Mui, W., Flagan, R. C., and Seinfeld, J. H.: Influence of particle-phase state on the hygroscopic behavior of mixed organic–inorganic aerosols, Atmos. Chem. Phys., 15, 5027–5045, https://doi.org/10.5194/acp-15-5027-2015, 2015.
Jimenez, J., Canagaratna, M., Donahue, N., Prevot, A., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., and Ng, N.: Evolution of organic aerosols in the atmosphere, Science, 326, 1525–1529, 2009.
Jungwirth, P. and Tobias, D. J.: Specific ion effects at the air/water interface, Chem. Rev., 106, 1259–1281, 2006.
Jungwirth, P., Curtis, J. E., and Tobias, D. J.: Polarizability and aqueous solvation of the sulfate dianion, Chem. Phys. Lett., 367, 704–710, 2003.
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005.
Kroll, J. H., Lim, C. Y., Kessler, S. H., and Wilson, K. R.: Heterogeneous oxidation of atmospheric organic aerosol: Kinetics of changes to the amount and oxidation of particle-phase organic carbon, J. Phys. Chem. A, 119, 10767–10783, 2015.
Kwong, K. C., Chim, M. M., Davies, J. F., Wilson, K. R., and Chan, M. N.: Importance of sulfate radical anion formation and chemistry in heterogeneous OH oxidation of sodium methyl sulfate, the smallest organosulfate, Atmos. Chem. Phys., 18, 2809–2820, https://doi.org/10.5194/acp-18-2809-2018, 2018a.
Kwong, K. C., Chim, M. M., Hoffmann, E. H., Tilgner, A., Herrmann, H., Davies, J. F., Wilson, K. R., and Chan, M. N.: Chemical transformation of methanesulfonic acid and sodium methanesulfonate through heterogeneous OH oxidation, ACS Earth Space Chem., 2, 895–903, 2018b.
Lambe, A. T., Zhang, J. Y., Sage, A. M., and Donahue, N. M.: Controlled OH radical production via ozone-alkene reactions for use in aerosol aging studies, Environ. Sci. Technol., 41, 2357–2363, 2007.
Lambe, A. T., Miracolo, M. A., Hennigan, C. J., Robinson, A. L., and Donahue, N. M.: Effective rate constants and uptake coefficients for the reactions of organic molecular markers (n-alkanes, hopanes, and steranes) in motor oil and diesel primary organic aerosols with hydroxyl radicals, Environ. Sci. Technol., 43, 8794–8800, 2009.
Li, Z., Smith, K. A., and Cappa, C. D.: Influence of relative humidity on the heterogeneous oxidation of secondary organic aerosol, Atmos. Chem. Phys., 18, 14585–14608, https://doi.org/10.5194/acp-18-14585-2018, 2018
Losey, D. J., Ott, E. J. E., and Freedman, M. A.: Effects of high acidity on phase transitions of an organic aerosol, J. Phys. Chem. A, 122, 3819–3828, 2018.
Marsh, A., Miles, R. E. H., Rovelli, G., Cowling, A. G., Nandy, L., Dutcher, C. S., and Reid, J. P.: Influence of organic compound functionality on aerosol hygroscopicity: dicarboxylic acids, alkyl-substituents, sugars and amino acids, Atmos. Chem. Phys., 17, 5583–5599, https://doi.org/10.5194/acp-17-5583-2017, 2017.
Marshall, F. H., Berkemeier, T., Shiraiwa, M., Nandy, L., Ohm, P. B., Dutcher, C. S., and Reid, J. P.: Influence of particle viscosity on mass transfer and heterogeneous ozonolysis kinetics in aqueous-sucrose-maleic acid aerosol, Phys. Chem. Chem. Phys., 20, 11560–11573, 2018.
McNeill, V. F., Wolfe, G. M., and Thornton, J. A.: The oxidation of oleate in submicron aqueous salt aerosols: Evidence of a surface process, J. Phys. Chem. A, 111, 1073–1083, 2007.
McNeill, V. F., Yatavelli, R. L. N., Thornton, J. A., Stipe, C. B., and Landgrebe, O.: Heterogeneous OH oxidation of palmitic acid in single component and internally mixed aerosol particles: vaporization and the role of particle phase, Atmos. Chem. Phys., 8, 5465–5476, https://doi.org/10.5194/acp-8-5465-2008, 2008.
Meng, Z. Y. and Seinfeld, J. H.: Time scales to achieve atmospheric gas-aerosol equilibrium for volatile species, Atmos. Environ., 30, 2889–2900, 1996.
Mungall, E. L., Wong, J. P. S., and Abbatt, J. P. D.: Heterogeneous Oxidation of Particulate Methanesulfonic Acid by the Hydroxyl Radical: Kinetics and Atmospheric Implications, ACS Earth Space Chem., 2, 48–55, 2017.
Nah, T., Chan, M. N., Leone, S. R., and Wilson, K. R.: Real time in situ chemical characterization of submicrometer organic particles using direct analysis in real time-mass spectrometry, Anal. Chem., 85, 2087–2095, 2013.
Peeters, J., Fantechi, G., and Vereecken, L.: A generalized structure-activity relationship for the decomposition of (substituted) alkoxy radicals, J. Atmos. Chem., 48, 59–80, 2004.
Petters, M., Prenni, A. J., Kreidenweis, S. M., DeMott, P. J., Matsunaga, A., Lim, Y. B., and Ziemann, P. J.: Chemical aging and the hydrophobic-to-hydrophilic conversion of carbonaceous aerosol, Geophys. Res. Lett., 33, L24806, https://doi.org/10.1029/2006GL027249, 2006.
Qiu, Y. and Molinero, V.: Morphology of liquid-liquid phase separated aerosols, J. Am. Chem. Soc., 137, 10642–10651, https://doi.org/10.1021/jacs.5b05579, 2015.
Reid, J. P., Dennis-Smither, B. J., Kwamena, N. O. A., Miles, R. E. H., Hanford, K. L., and Homer, C. J.: The morphology of aerosol particles consisting of hydrophobic and hydrophilic phases, hydrocarbons. alcohols and fatty acids as the hydrophobic component, Phys. Chem. Chem. Phys., 13, 15559–15572, 2011.
Renbaum, L. H. and Smith, G. D.: The importance of phase in the radical-initiated oxidation of model organic aerosols: Reactions of solid and liquid brassidic acid particles, Phys. Chem. Chem. Phys., 11, 2441–2451, 2009.
Richards-Henderson, N. K., Goldstein, A. H., and Wilson, K. R.: Large enhancement in the heterogeneous oxidation rate of organic aerosols by hydroxyl radicals in the presence of nitric oxide, J. Phys. Chem. Lett., 6, 4451–4455, 2015.
Rudich, Y., Donahue, N. M., and Mentel, T. F.: Aging of organic aerosol: bridging the gap between laboratory and field studies, Annu. Rev. Rhys. Chem., 58, 321–352, 2007.
Russell, G. A.: Deuterium-isotope effects in the autoxidation of aralkyl hydrocarbons. mechanism of the interaction of peroxy radicals, J. Am. Chem. Soc., 79, 3871–3877, https://doi.org/10.1021/ja01571a068, 1957.
Shiraiwa, M., Ammann, M., Koop, T., and Pöschl, U.: Gas uptake and chemical aging of semisolid organic aerosol particles, P. Natl. Acad. Sci. USA, 108, 11003–11008, 2011.
Slade, J. H. and Knopf, D. A.: Heterogeneous OH oxidation of biomass burning organic aerosol surrogate compounds: Assessment of volatilization products and the role of OH concentration on the reactive uptake kinetics, Phys. Chem. Chem. Phys., 15, 5898–5915, 2013.
Slade, J. H. and Knopf, D. A.: Multiphase OH oxidation kinetics of organic aerosol: The role of particle phase state and relative humidity, Geophys. Res. Lett., 41, 5297–5306, 2014.
Slade, J. H., Thalman, R., Wang, J., and Knopf, D. A.: Chemical aging of single and multicomponent biomass burning aerosol surrogate particles by OH: implications for cloud condensation nucleus activity, Atmos. Chem. Phys., 15, 10183–10201, https://doi.org/10.5194/acp-15-10183-2015, 2015.
Slade, J. H., Shiraiwa, M., Arangio, A., Su, H., Pöschl, U., Wang, J., and Knopf, D. A.: Cloud droplet activation through oxidation of organic aerosol influenced by temperature and particle physical state, Geophys. Res. Lett., 44, 1583–1591, 2017.
Smith, J. D., Kroll, J. H., Cappa, C. D., Che, D. L., Liu, C. L., Ahmed, M., Leone, S. R., Worsnop, D. R., and Wilson, K. R.: The heterogeneous reaction of hydroxyl radicals with sub-micron squalane particles: a model system for understanding the oxidative aging of ambient aerosols, Atmos. Chem. Phys., 9, 3209–3222, https://doi.org/10.5194/acp-9-3209-2009, 2009.
Song, M., Marcolli, C., Krieger, U. K., Zuend, A., and Peter, T.: Liquid-liquid phase separation and morphology of internally mixed dicarboxylic acids/ammonium sulfate/water particles, Atmos. Chem. Phys., 12, 2691–2712, https://doi.org/10.5194/acp-12-2691-2012, 2012a.
Song, M., Marcolli, C., Krieger, U. K., Zuend, A., and Peter, T.: Liquid-liquid phase separation in aerosol particles: Dependence on O:C, organic functionalities, and compositional complexity, Geophys. Res. Lett., 39, L19801, https://doi.org/10.1029/2012GL052807, 2012b.
Song, M. J., Marcolli, C., Krieger, U. K., Zuend, A., and Peter, T.: Morphologies of mixed organic/inorganic/aqueous aerosol droplets, Faraday Discuss., 165, 289–316, 2013.
Stark, H., Yatavelli, R. L. N., Thompson, S. L., Kang, H., Krechmer, J. E., Kimmel, J. R.,Palm, B. B., Hu, W. W., Hayes, P. L., Day, D. A., Campuzano-Jost, P., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., and Jimenez, J. L.: Impact of Thermal Decomposition on Thermal Desorption Instruments: Advantage of Thermogram Analysis for Quantifying Volatility Distributions of Organic Species, Environ. Sci. Technol., 51, 8491–8500, 2017.
Stewart, D. J., Cai, C., Nayler, J., Preston, T. C., Reid, J. P., Krieger, U. K., Marcolli, C., and Zhang, Y. H.: Liquid-liquid phase separation in mixed organic/inorganic single aqueous aerosol droplets, J. Phys. Chem. A, 119, 4177–4190, 2015.
Vaden, T. D., Imre, D., Beranek, J., Shrivastava, M., and Zelenyuk, A.: Evaporation kinetics and phase of laboratory and ambient secondary organic aerosol, P. Natl. Acad. Sci. USA, 108, 2190–2195, 10.1073/pnas.1013391108, 2011.
Vereecken, L. and Peeters, J.: Decomposition of substituted alkoxy radicals – Part I: A generalized structure–activity relationship for reaction barrier heights, Phys. Chem. Chem. Phys., 11, 9062–9074, 2009.
You, Y. and Bertram, A. K.: Effects of molecular weight and temperature on liquid–liquid phase separation in particles containing organic species and inorganic salts, Atmos. Chem. Phys., 15, 1351–1365, https://doi.org/10.5194/acp-15-1351-2015, 2015.
You, Y., Renbaum-Wolff, L., and Bertram, A. K.: Liquid–liquid phase separation in particles containing organics mixed with ammonium sulfate, ammonium bisulfate, ammonium nitrate or sodium chloride, Atmos. Chem. Phys., 13, 11723–11734, https://doi.org/10.5194/acp-13-11723-2013, 2013.
You, Y., Smith, M. L., Song, M., Martin, S. T., and Bertram, A. K.: Liquid–liquid phase separation in atmospherically relevant particles consisting of organic species and inorganic salts, Int. Rev. Phys. Chem., 33, 43–-77, DOI:10.1080/0144235X.2014.890786, 2014.
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Allan, J. D., Coe, H., Ulbrich, I., Alfarra, M. R., Takami, A., Middlebrook, A. M., Sun, Y. L., Dzepina, K., Dunlea, E., Docherty, K., DeCarlo, P. F., Salcedo, D., Onasch, T., Jayne, J. T., Miyoshi, T., Shimono, A., Hatakeyama, S., Takegawa, N., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Williams, P., Bower, K., Bahreini, R., Cottrell, L., Griffin, R. J., Rautiainen, J., Sun, J. Y., Zhang, Y. M., and Worsnop, D. R.: Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes, Geophys. Res. Lett., 34, L13801, https://doi.org/10.1029/2007GL029979, 2007.
Zuend, A. and Seinfeld, J. H.: Modeling the gas-particle partitioning of secondary organic aerosol: the importance of liquid-liquid phase separation, Atmos. Chem. Phys., 12, 3857–3882, https://doi.org/10.5194/acp-12-3857-2012, 2012.
Zuend, A., Marcolli, C., Luo, B. P., and Peter, T.: A thermodynamic model of mixed organic-inorganic aerosols to predict activity coefficients, Atmos. Chem. Phys., 8, 4559–4593, https://doi.org/10.5194/acp-8-4559-2008, 2008.
Zuend, A., Marcolli, C., Booth, A. M., Lienhard, D. M., Soonsin, V., Krieger, U. K., Topping, D. O., McFiggans, G., Peter, T., and Seinfeld, J. H.: New and extended parameterization of the thermodynamic model AIOMFAC: calculation of activity coefficients for organic-inorganic mixtures containing carboxyl, hydroxyl, carbonyl, ether, ester, alkenyl, alkyl, and aromatic functional groups, Atmos. Chem. Phys., 11, 9155–9206, https://doi.org/10.5194/acp-11-9155-2011, 2011.